
c-Abl and Parkinson’s Disease: Mechanisms and Therapeutic Potential
Abstract
Although the etiology of Parkinson’s disease (PD) is poorly understood, oxidative stress has long been implicated in the pathogenesis of the disease. However, multifaceted and divergent signaling cascades downstream of oxidative stress have posed challenges for researchers to identify a central component of the oxidative stress-induced pathways causing neurodegeneration in PD. Since 2010, c-Abl—a non-receptor tyrosine kinase and an indicator of oxidative stress—has shown remarkable potential as a future promising drug target in PD therapeutics. Although, the constitutively active form of c-Abl, Bcr-Abl, has a long history in chronic myeloid leukemia and acute lymphocytic leukemia, the role of c-Abl in PD and relevant neurodegenerative diseases was completely unknown. Recently, others and we have identified and validated c-Abl as an important pathogenic mediator of the disease, where activated c-Abl emerges as a common link to various PD-related inducers of oxidative stress relevant to both sporadic and familial forms of PD and α-synucleinopathies. This review discusses the role of c-Abl in PD and the latest advancement on c-Abl as a drug target and as a prospective biomarker.
THE NON-RECEPTOR TYROSINE KINASE c-Abl
c-Abl (ABL1; Abelson tyrosine kinase) is a member of Abl family of non-receptor tyrosine kinases, of which the only other member is Arg (ABL2; Abl-related gene) [1, 2]. c-Abl is highly conserved among metazoans and expressed ubiquitously in many subcellular compartments including in the cytoplasm, nucleus, mitochondria and endoplasmic reticulum [1, 2]. c-Abl has diverse functional targets ranging from cell signaling adaptors to kinases and phosphatases with which it interacts and c-Abl functions are implicated in a variety of biological processes including the regulation of cell growth and survival, integrin signaling, actin polymerization and cell migration [1–3]. In the central nervous system (CNS), the major biological functions of c-Abl include regulation of synapse formation, neurite outgrowth, neurogenesis and cerebellar development, although Arg can also complement these functions [2, 4, 5].
Tight regulation of tyrosine kinase activity of c-Abl is pivotal as aberrant c-Abl activation correlates with oncogenic and neurodegenerative potential. Bcr-Abl expression and its link to leukemia have been comprehensively studied over the last 30 years [1, 2]. The oncogenic form of c-Abl – the fusion protein Bcr-Abl – is constitutively overactive and is linked to different forms of leukemia, such as chronic myeloid leukemia (CML) and some forms of acute lymphocytic leukemia (ALL) [1, 2].
There are two splice variants of ABL1 transcripts, 1a and 1b, which differ in their N-terminal sequences [1]. The 1b variant undergoes myristoylation at the N-terminus, whereas the 1a variant is 19-amino acid shorter and lacks the ability to be myristolylated [1]. Structurally, c-Abl consists of N-terminal SH3 and SH2 domains followed by its tyrosine kinase (catalytic) domain, DNA-binding domain, and a c-terminal actin-binding domain [1, 2, 8].
As aberrant c-Abl activation is associated with several deleterious effects, regulation of tyrosine kinase activity of c-Abl is critical to the biological function of cells. Thus, c-Abl exists natively in an autoinhibited state governed by complex intramolecular interactions [8–10]. Autophosphorylation of c-Abl stimulates its catalytic activity (Fig. 1) [8]. In particular, tyrosine (Y) 412 phosphorylation leads to a 9-fold increase in catalytic activity [8], and it is likely that physiological c-Abl activation is achieved through an optimum level of Y412 phosphorylation [8, 11]. Y245 phosphorylation is also known to stimulate c-Abl kinase activity by 2.5 fold [8]. However, c-Abl catalytic activity is markedly reduced in phospho-deficient Y412 mutant (Y412F) c-Abl, but there was only 50% reduction in the phospho-deficient Y245 (Y245F) mutant [8]. This indicates that c-Abl phosphorylation in vivo may happen in a sequential manner with phosphorylation at Y412 occurring first, followed by Y245 phosphorylation [8].
Fig.1
c-Abl activation links diverse pathogenic pathways leading to neuronal death in response to oxidative stress in PD and related α-synucleinopathies. Mitochondrial dysfunction and resulting oxidative stress is the key feature in both sporadic PD and familial PD related α-synucleinopathies. c-Abl activation acts as a sensor of oxidative stress which in turn triggers multiple pathogenic signals primarily leading to inactivation of parkin, activation of p38α, NLRP3-inflammasome-mediated NF-κB activation in microglia, and α-synuclein phosphorylation at Y39. Parkin inactivation causes accumulation of pathogenic parkin substrates PARIS and AIMP2 and subsequently neuronal death. Activation of p38α and microglial activation of NF-κB merge on to cellular death. α-synuclein phosphorylation at Y39 is potentially linked to aggregation and neuronal toxicity.
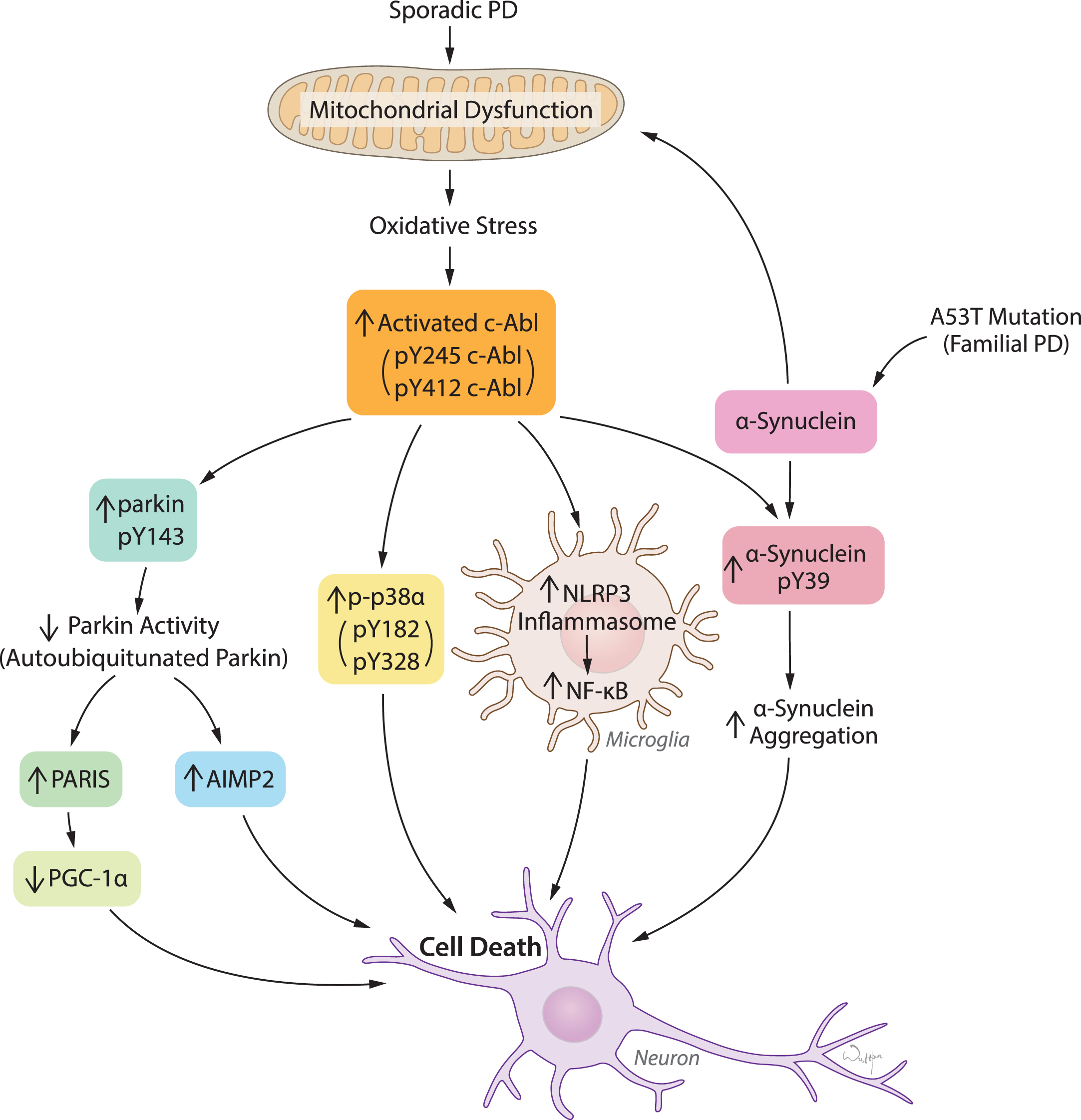
Emerging evidence suggests that overactivation of c-Abl also plays a major role in neurodegeneration in Alzheimer’s disease (AD) and Parkinson’s disease (PD) [5–7]. This review will focus on the role of c-Abl in PD and provide a historical review, as well as current findings including the translational potential of c-Abl inhibitors and c-Abl substrates as biomarkers.
c-Abl ACTIVATION: AN INDICATOR OF OXIDATIVE STRESS
Cellular oxidative stress occurs when reactive oxygen species (ROS) overpower the cellular antioxidant defense system. It contributes to the pathogenesis of a broad range of diseases such as cancer and several age-related neurodegenerative disorders including PD [6, 12–15]. In addition to direct chemical modifications on DNA, proteins and lipids, ROS trigger several signaling cascades, enhance or inhibit protein-protein interactions, and lead to post-translational modifications that eventually cause cellular damage and death [6, 16, 17]. Interestingly, ROS activate c-Abl, which serves as an oxidative stress sensor (Fig. 1) [6, 8, 18, 19]. Studies indicate that oxidative stress activates ataxia-telangiectasia mutated (ATM) kinase [20], which in turn can activate c-Abl [21], thus providing a potential mechanism by which ROS may activate c-Abl. Furthermore, it has been reported that PKCδ, a protein kinase C isoform, phosphorylates and activates c-Abl in response to H2O2 [18]. ROS may also directly activate c-Abl. It is difficult to explain precisely how ROS trigger c-Abl activation. We believe that the cellular oxidant level, cell types and contexts are the key determinants that regulate the molecular cascade of c-Abl activation by ROS. Following the activation by ROS, c-Abl interacts with regulators of the redox state in cells such as catalase, which is one of the enzymes in the antioxidant defense system, phosphorylating it at Y321 and Y386 and inducing its ubiquitination and proteosomal degradation [6, 22]. c-Abl also has a negative regulatory effect on the level of the antioxidant protein peroxiredoxin I (Prx1) [23]. Thus, c-Abl activation in general disrupts antioxidant defense mechanisms driving oxidative injury in a feed-forward mechanism enhancing degeneration.
c-Abl ACTIVATION AND AGING: RELEVANCE TO PARKINSON’S DISEASE
PD is an age-related progressive neurodegenerative disease that affects more than 1% of the population over the age of 55, and 5% of the population over age of 85 [24–26]. Accumulation of high molecular weight α-synuclein species in Lewy bodies (LB) and Lewy neurites (LN), proteinaceous cytoplasmic and neuritic inclusions, respectively, are major pathological hallmarks of the disease. Pathologically aggregated forms of α-synuclein are a major driving force in the degeneration of substantia nigra (SN) dopamine (DA) neurons [24, 27–30]. Increased c-Abl activity, as measured by the relative level of pY245 c-Abl over c-Abl, is observed in PD postmortem brains in the SN and the striatum, which are pathologically affected in PD [4, 14, 31–34]. Minimal c-Abl activation is observed in the cortex, a region which is relatively unaffected in PD [35]. c-Abl activation as measured by pY412 c-Abl is also observed in PD postmortem brains [33]. These findings indicate a correlation between c-Abl activation and PD pathology. Age-dependent activation of c-Abl is observed in human A53T (hA53T) α-synuclein transgenic mice, an animal model of α-synucleinopathy induced degeneration [32]. While at 2 months of age there is minimal c-Abl activation, it increases significantly and progressively at 4 and 6 months of age and remains activated until death. Shortly after the activation of c-Abl is observed, accumulation of pathological α-synuclein is observed, which begins at 7 to 8 months of age and continues until death [32]. This temporal progression of c-Abl activation is followed by accumulation of pathological α-synuclein implicating a possible role of c-Abl in α-synuclein-mediated neurodegeneration (Fig. 1). Interestingly, age-matched control human postmortem brains as well as aged non-transgenic mice do not exhibit overactivation of Y245 c-Abl [4, 31–33]. c-Abl activation was further observed in a number of PD-relevant animal models such as α-synucleinopathies, MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) intoxication and lentiviral delivery of α-synuclein in the SN associated with nigral loss of DA neurons [4, 33, 34, 36–38]. Taken together, these data strongly suggest that c-Abl activation may play a pivotal role in neurodegeneration in PD.
c-Abl ACTIVATION PLAYS A ROLE IN α-SYNUCLEINOPATHY-INDUCED NEURODEGENERATION
Recent studies have revealed that aberrant activation of c-Abl plays a major role in α-synuclein-induced neurodegeneration [32, 33, 39]. A comprehensive study by our group, using two-directional approaches consisting of mutant mouse models of c-Abl overexpression and c-Abl knockout mice with hA53T α-synuclein transgenic mice, has established a role for c-Abl activation in α-synucleinopathy induced degeneration [32]. Overexpression of constitutively active Bcr-Abl exacerbates the behavioral deficits and reduces the life-span of hA53T α-synuclein transgenic mice approximately by 3.5 months. Bcr-Abl also accelerates the α-synuclein pathology and neurodegeneration as measured by accumulation of high molecular weight species of α-synuclein and pS129 α-synuclein (a marker of α-synuclein pathology) [40–42] in non-ionic detergent-insoluble fractions [32]. In addition, Bcr-Abl leads to an enhanced accumulation of ubiquitin (a prominent indicator of pathology), and glial fibrillary acidic protein (GFAP, an indirect indicator of overall neurodegeneration). All these features are absent in age-matched hA53T α-synuclein transgenic littermates [32]. In contrast, conditional neuronal knockout of c-Abl prolongs the survival of hA53T α-synuclein transgenic mice by 3 months and retards behavioral, neurodegenerative and pathological features [32]. More importantly, Bcr-Abl expression causes degeneration of SN DA neurons in wild type mice indicating that c-Abl activation is sufficient to kill SN DA neurons [32].
Recent studies have identified α-synuclein as a substrate of c-Abl. c-Abl interacts with α-synuclein and phosphorylates α-synuclein at Y39 (Fig. 1) [32, 39, 43]. Although Mahul-Mellier et al. reported that in addition to Y39, c-Abl also phosphorylates Y125 of α-synuclein, our study demonstrates that Y39 of α-synuclein is the exclusive phosphorylation target of c-Abl [32, 39]. We provided 100% sequence coverage of α-synuclein by mass spectrometry and phospho-deficient mutational analysis strongly supports our finding, whereas the other group showed only 66% coverage and no peptide coverage was provided for Y125. c-Abl phosphorylation of α-synuclein at Y39 is pivotal for the aggregation of α-synuclein as the phospho-deficient form of Y39 α-synuclein (Y39F α-synuclein) is resistant to aggregation [32]. This also suggests that pY39 α-synuclein might play a role in neurotoxicity. Future studies are required to investigate the possible neurotoxic role of this post-translational modification.
Although the above studies have firmly demonstrated a pivotal role of c-Abl activation in α-synucleinopathy-induced neurodegeneration, one disadvantage of the hA53T α-synuclein transgenic mouse model is the lack of degeneration of SN DA neurons [44]. Notably, a recent report using a mouse model based on lentiviral delivery of α-synuclein in the SN demonstrates c-Abl activation (pY412 c-Abl/c-Abl) in the SN [33].
Emerging evidence suggest that prion-like propagation of α-synuclein plays a significant role in the progressive neurodegeneration in PD [45, 46]. In support of this, the newly characterized recombinant pre-formed fibril (PFF)-based animal model shows progressive nigrostriatal degeneration [45]. Our group has recently identified the neuronal receptor lymphocyte activation gene 3 (Lag3) as an endocytosis and neuronal transmission receptor for pathological α-synuclein [47]. It would be important to know whether activated c-Abl also contributes to nigral degeneration in the propagation-based models.
How aberrant c-Abl activation contributes to accumulation of toxic α-synuclein species is poorly understood. Since c-Abl phosphorylation of α-synuclein at Y39 seems to be critical for aggregation, it would be intriguing to explore how this post-translational modification leads to aggregation of α-synuclein. It might be possible that Y39 phosphorylation of α-synuclein induces conformational changes leading to structural rearrangement favoring aggregation.
Taken together, these observations strongly suggest that c-Abl activation is a key mediator of neurodegeneration in PD and related α-synucleinopathies relevant to both sporadic and familial forms of these diseases. Thus, c-Abl inhibition is a compelling target for development of neuroprotective therapies for these disorders.
c-Abl ACTIVATION IMPAIRS PARKIN ACTIVITY: IMPLICATIONS FOR PARKINSON’S DISEASE
Mutations in parkin are the most common cause of autosomal recessive PD, and the disease resulting from parkin mutations are almost indistinguishable from the sporadic form of the disease [4, 48]. Parkin is an ubiquitin E3 ligase, and familial PD-linked mutations are thought to impair its ubiquitin E3 ligase activity. Parkin enhances mono- and polyubiquitination of substrates via K29, K48 and K63 chains. Polyubiquitination by parkin via K48 chains leads to proteasomal degradation [4, 49–54]. The major biological functions of parkin are maintenance of mitochondrial homeostasis and regulation of substrates that are targeted for degradation via the ubiquitin-proteasome system [55–57]. The mitochondrial kinase PINK1 (PTEN-induced putative kinase 1), which is also mutated in autosomal recessive PD, phosphorylates parkin and ubiquitin at S65 activating parkin in response to mitochondrial depolarization. This leads to parkin recruitment to damaged mitochondria where parkin ubiquitinates outer mitochondrial membrane proteins and thereby facilitates the clearance of damaged mitochondria [56, 57]. For review of PINK1 and parkin-mediated mitochondrial quality control see [55, 58].
Several independent studies have identified parkin as a c-Abl substrate and demonstrated that c-Abl phosphorylates parkin specifically at Y143 (Fig. 1) [4, 31]. c-Abl phosphorylation of parkin impairs its ubiquitin E3 ligase activity and subsequently leads to accumulation of parkin substrates [4, 31]. In vitro ubiquitination assays demonstrate that c-Abl kinase activity is essential for tyrosine phosphorylation of parkin and subsequent impairment of its activity, as evident from loss of parkin autoubiquitination and ubiquitination of parkin’s substrates by the kinase active form of c-Abl, but not the kinase-dead form [4, 31]. The phospho-deficient Y143F parkin mutant (Y143F-parkin) is resistant to loss of autoubiquitination by c-Abl, indicating that phosphorylation of parkin at Y143 negatively regulates parkin activity [4, 31]. Interestingly, c-Abl knockout prevents DA neuron loss in the MPTP mouse model of PD, and this phenomenon is accompanied by significant reductions in parkin tyrosine phosphorylation and accumulation of parkin substrate levels, AIMP2 and FBP-1 in response to MPTP [4]. Furthermore, we observed that c-Abl activation in pathologically affected regions of PD postmortem brain is strongly associated with an increase in parkin tyrosine phosphorylation, decreased autoubiquitinated parkin, and increased levels of parkin substrates [4, 31]. Taken together, it suggests that loss of parkin activity and subsequent accumulation of its substrates may be intimately linked to neurodegeneration.
Two major pathogenic parkin substrates implicated in PD are aminoacyl-tRNA synthetase-interacting multifunctional protein type 2 (AIMP2) (p38/JTV-1), and the PARkin Interacting Substrate/zinc finger protein 746 (PARIS/ZNF746) (Fig. 1) [59–61]. Accumulation of AIMP2 is found in LB inclusions in the SN of PD postmortem brains and it is also elevated in the MPTP intoxication mouse model of PD, parkin knockout mice, as well as PD postmortem brains from patients with parkin mutations and sporadic PD [4, 31, 36, 60, 62, 63]. AIMP2 accumulation plays an important role in PD pathogenesis, as transgenic overexpression of AIMP2 induces an age-dependent progressive and selective loss of DA neurons via parthanatos mediated cell death [60]. Furthermore, PARIS accumulates in animal models of parkin inactivation and also specifically kills dopamine (DA) neurons [59]. Consistent with these findings, impairment of parkin activity and accumulation of PARIS and AIMP2 have been observed in the SN of PD postmortem brains [4, 59, 61]. Furthermore, PARIS is a transcriptional repressor that suppresses peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α), a transcriptional co-activator and a key regulator of mitochondrial biogenesis (Fig. 1). Overexpression of PGC-1α prevents DA neuron loss in animal model of PARIS overexpression indicating that PGC-1α is a target of PARIS in mediating neurodegeneration [59]. Although PARIS and AIMP2-mediated neuronal loss involve distinct molecular pathways, it would be intriguing to know if one regulates another in causing neurodegeneration. Since parkin is inactivated in sporadic PD and there is accumulation of AIMP2 and PARIS, it will be important in future studies to examine the status of parkin activity and the accumulation of parkin substrates in the setting of α-synuclein accumulation.
c-ABL AND c-ABL SUBSTRATES: IMPLICATIONS FOR POTENTIAL BIOMARKERS
The diagnosis of PD is currently made based solely on clinical criteria [64]. However, this approach has several limitations when compared to diagnoses made based on molecular biomarkers. First, it only allows for the diagnosis of patients who are already in a symptomatic stage of the disease, when disease modifying therapies are less likely to be effective. Second, using clinical criteria as the sole means of diagnosis may cause a number of molecularly heterogeneous diseases that have clinically similar symptoms to be grouped as a single disorder. Finally in a clinical trial setting, the lack of molecular markers prevents investigators from evaluating whether the intervention being tested effectively engages its target.
Currently, three primary types of biomarkers exist: 1) diagnostic biomarkers are used to mark disease onset and can supplement or substitute for clinical criteria in diagnosis; 2) prognostic (or predictive) biomarkers can be used to predict clinical course and severity; and, finally, 3) target engagement biomarkers are used to evaluate whether a novel treatment effectively manipulates the molecular pathway of interest [64–66]. For PD diagnosis there is considerable interest in evaluating molecular biomarkers in the cerebrospinal fluid (CSF), as they are likely to best closely reflect the molecular changes that occur inside the brain [66]. Here, in the sections below, we will briefly discuss the prospects of c-Abl and its substrates that warrant testing as CSF biomarkers for PD.
Previous in vitro studies have reported that c-Abl autophosphorylation at Y245 and Y412 in murine and human c-Abl promotes kinase activity and can serve as a marker for c-Abl activation. [67]. As such, it is possible that levels of unphosphorylated and phosphorylated c-Abl in the CSF could be used to directly evaluate c-Abl activation. In hA53T α-synuclein mice, levels of detergent-soluble pY245 c-Abl in pathological brain regions steadily increase early (presymptomatic stage) in disease progression (4-6 months of age) before declining precipitously upon symptomatic onset [32]. Because of the presymptomatic increase in soluble pY245 c-Abl and the dramatic decrease following disease onset, tracking pY245 c-Abl levels may serve as a diagnostic marker for PD. However, it is unclear whether a large cytosolic protein such as c-Abl can be found in CSF at sufficient levels to be a useful biomarker, even under neurodegenerative conditions under which dying neurons may release c-Abl into the extracellular space. To the best of our knowledge, there is only one study that has detected the presence of c-Abl and tyrosine-phosphorylated c-Abl in CSF [68]. While the results of this study suggest that CSF c-Abl may be a target engagement biomarker for c-Abl targeting therapies, the lack of further clinical validation means that it is still unclear whether c-Abl is present in the CSF and can be effectively used as a potential biomarker for PD.
c-Abl phosphorylated α-synuclein may also be an effective biomarker for PD. In PD patients, total CSF α-synuclein level is lower while oligomeric and phosphorylated α-synuclein level is elevated [65]. Nevertheless, it remains unclear whether total CSF α-synuclein is sensitive or reliable enough as a PD biomarker. Investigations on whether α-synuclein can be used as a prognostic marker have yielded inconsistent results [65]. However, most studies evaluating CSF α-synuclein as a PD biomarker do not evaluate the levels of specific post-translational modifications. Thus, it is possible that mechanism-specific modifications may be the more sensitive biomarkers. c-Abl phosphorylated (pY39) α-synuclein accumulates early in hA53T α-synuclein overexpressing mice before soluble levels plummet upon symptomatic onset—paralleling the trend observed with pY245 c-Abl. On the other hand, levels of total and pS129 α-synuclein remained mostly unchanged, though insoluble pS129 and pY39 α-synuclein accumulate upon symptomatic onset [32]. This suggests that the level of pY39 α-synuclein may be a more faithful surrogate for c-Abl activation than α-synuclein phosphorylated at other residues. Moreover, the accumulation of soluble pY39 α-synuclein prior to onset of motor symptoms in hA53T α-synuclein mice establishes it as a potential diagnostic marker for PD. Future investigations are needed to show whether pY39 α-synuclein may be a more sensitive biomarker than total or pS129 α-synuclein.
Another c-Abl substrate that has attracted attention as a biomarker for neurodegenerative disease is tau [66]. c-Abl seems to phosphorylate tau primarily at Y394, and pY394 tau has been found in neurofibrillary tangles in AD patient brains [69, 70]. While Y197F and Y310F tau also show reduced tyrosine phosphorylation, it is unclear whether those two sites are directly phosphorylated by c-Abl or simply facilitate c-Abl phosphorylation of other tyrosine sites [62]. A growing body of evidence suggests that tau may play some role in PD pathogenesis; tau and α-synuclein can promote each other’s aggregation, p-tau sometimes co-localizes with α-synuclein aggregates in PD patient brains, and polymorphisms in MAPT are associated with increased risk of PD [71, 72]. Moreover, CSF levels of tau have been proposed as diagnostic biomarkers in PD, with PD patients showing significantly lower baseline levels of CSF tau and p-tau [73]. While it is unclear whether tau and p-tau levels can reliably be used as prognostic biomarkers in PD, a number of studies have suggested that higher baseline levels of p-tau in PD patients is predictive of more severe progression of motor symptoms and accelerated decline in certain cognitive and neuropsychological evaluations [73, 74].
Similarly to the use of phosphorylated α-synuclein as a biomarker for PD, a major challenge with evaluating p-tau as a biomarker for AD or PD is that most studies look at overall tau phosphorylation rather than the phosphorylation of specific tau residues. While it was found that c-Abl inhibition reduced overall tau phosphorylation in AD mouse models [75], it is doubtful that overall tau phosphorylation will be an accurate proxy of c-Abl activity in PD because tau hyperphosphorylation is not associated with typical PD pathology. Because certain residues may be modified at different stages of disease, evaluating residue-specific phosphorylation may provide information about disease state obscured by nonspecific evaluation of overall phosphorylation. c-Abl, for example, phosphorylates monomeric tau early in the course of AD [69, 76]. As discussed above it is also possible that other tyrosine residues beside Y394 (e.g., Y197 and Y310) may also be phosphorylation targets of c-Abl. As such, future studies that evaluate levels of c-Abl phosphorylated tau (at Y394 and potentially other tyrosine residues) in PD may be able to improve the sensitivity of tau as a biomarker for PD.
Due to inter-patient variation, inconsistencies in study design and execution, and many other factors, it is likely that several biomarkers will be necessary to achieve sufficient sensitivity for effective diagnosis and prognosis of PD [64–66]. We believe that c-Abl and its substrates have high potential for being effective diagnostic biomarkers and therapeutic targets for PD. First, c-Abl plays a central role in PD pathogenesis and thus it and its substrates have direct molecular correlations with disease state. Moreover, two c-Abl substrates (tau and α-synuclein) are already present in the CSF and under study as biomarkers for AD or PD. While future studies must effectively evaluate post-translational modifications that are specific to c-Abl activation, the development of c-Abl and c-Abl substrates as PD biomarkers may bring us closer to standardized evaluation of PD diagnosis, prognosis, and target engagement based on the molecular pathophysiology of the disease.
c-Abl INHIBITORS: THERAPEUTIC POTENTIAL
Current treatments available for PD patients can effectively mitigate many of the motor and non-motor symptoms of the disease, but not all of the symptoms. Moreover, currently, disease modifying therapies that can delay or stop disease progression do not exist. As a major contributor to PD pathogenesis, c-Abl may be an effective target for disease modifying treatment of PD. Currently, a number of c-Abl inhibitors are widely used in the treatment of CML [77]. To the best of our knowledge, only three of these inhibitors (Imatinib, Nilotinib, and Bafetinib) have been evaluated as potential disease modifying PD therapies. Here, we summarize adapting these c-Abl inhibitors for the potential PD therapies and discuss future prospects for development of c-Abl targeting therapies.
A number of early studies explored the potential of Imatinib (STI-571, Gleevec, formerly known as CGP57148)—a first-line drug for CML designed to specifically inhibit the BCR-Abl fusion protein [77]—in cell and animal models of AD and PD. Treatment of primary hippocampal neurons with Imatinib was shown to protect against the toxicity induced by Aβ fibrils [78]. Furthermore, several recent studies have also found that Imatinib prevents c-Abl-mediated phosphorylation and inactivation of parkin following the MPTP treatment in vitro and in vivo. However, based on these studies, it remains unclear whether Imatinib treatment can protect against neurodegeneration [4, 31, 59, 60]. Moreover, because of Imatinib’s poor blood-brain barrier (BBB) penetrance, it is unclear whether or not tolerable doses are sufficient to modify disease course in PD patients [31, 79]. The CNS levels of Imatinib are further reduced through active transport outside of the brain via the efflux transporters ABCG2 and ABCB1 (also known as MDR1 and P-Glycoprotein) [79, 80]. Due to these factors, it seems unlikely that Imatinib can successfully be adapted as a therapy for PD patients.
Nilotinib (AMN107, Tasigna®), a second-generation c-Abl tyrosine kinase inhibitor, is similarly used clinically in the treatment of CML and other blood cancers [81]. Compared to Imatinib, it has several advantages that seemingly make it more attractive as a potential treatment for PD, including increased specificity for c-Abl, potency, and increased BBB penetrance [33, 81, 82]. However, similarly to Imatinib, CNS concentrations of Nilotinib are still limited, in part, due to efflux transport by ABCG2 and ABCB1 [83]. Recent studies in MPTP mouse models have indicated that Nilotinib treatment initiated prior to MPTP administration can inhibit c-Abl activation, reduce pathological phosphorylation of c-Abl substrates, rescue motor deficits, and protect against degeneration of nigral DA neurons [36, 37]. Nilotinib treatment reduces the accumulation of α-synuclein in hA53T α-synuclein mice and mice that overexpresses α-synuclein in the SNpc, potentially by presumably promoting autophagy and reducing phosphorylation at Y39—and rescue motor deficits [33]. In a small cohort of human PD patients, daily Nilotinib treatments led to minor improvements in motor and cognitive performance at 6 months of treatments. However, the lack of a placebo control group renders it impossible to conclude whether Nilotinib modifies disease course. Furthermore, these benefits were completely reversed once 12 weeks had elapsed following cessation of treatment [68]. The fact that clinical benefits reversed so rapidly after treatment cessation renders it questionable whether patient improvement was due to reduced neurodegeneration. More significantly, of the 12 patients enrolled in the study, one patient withdrew due to a life-threatening cardiac event while one other patient experienced QTc prolongation [68], well-known side effects of Nilotinib [84]. Other adverse events experienced by patients on Nilotinib included increased hyper-dopaminergic symptoms (psychosis and dyskinesias) requiring reduction or cessation in DA replacement therapy [68]. Thus, significant concerns remain regarding whether Nilotinib treatment is safe and tolerable in PD patients. While this study does not completely discount the possibility that Nilotinib may still be an effective PD therapy if administered correctly, it is paramount that well-conducted randomized, placebo-controlled trials are carried out to properly evaluate its safety and efficacy in human patients.
Recently, it was found that the second-generation c-Abl inhibitor Bafetinib (INNO-406) seems to reach central nervous system (CNS) concentrations thousands-fold greater than Nilotinib, and can achieve CSF concentrations of 10 percent that of plasma concentrations [34, 36, 85]. In contrast to Imatinib or Nilotinib, which are actively transported outside of the brain, Bafetinib seems to inhibit the transporters ABCG2 and ABCB1, suggesting that its ability to achieve higher CNS concentrations may be due to reduced transporter-mediated efflux [86]. Once inside the brain, it successfully inhibits c-Abl activity and protects against MPTP-induced parkin inactivation, accumulation of parkin substrate AIMP2, and DA neurodegeneration [34]. While these studies still require independent validation, they indicate that Bafetinib may warrant further investigation in additional preclinical models of PD. Furthermore, the fact that an existing c-Abl inhibitor shows relatively high brain penetrance suggests that closer scrutiny of other existing c-Abl inhibitors or development of new c-Abl inhibitors with similar properties may yield c-Abl inhibitors that are more brain-penetrant than either Imatinib or Nilotinib and thus more suitable for the treatment of PD.
While c-Abl inhibition has shown promise in preclinical animal models of PD, many challenges remain for applying existing c-Abl inhibitors for PD treatment. First, the most commonly used c-Abl inhibitors Imatinib and Nilotinib both show limited BBB penetrance. As a result, these drugs would likely need to be administered at dangerously high dosages in order to achieve the necessary CNS concentrations to elicit significant therapeutic benefit. In contrast to cancer, the goal of treatment in PD is to maintain the survival of vulnerable neurons while limiting damage to other cell types across prolonged treatment. However, chronic peripheral administration of current FDA approved c-Abl inhibitors in CML patients is associated with cardiovascular, pulmonary, GI-associated, and liver adverse events [84]. Though Bafetinib may be an attractive alternative as it can reach higher concentrations in the CNS compared to Imatinib and Nilotinib, it still remains to be confirmed whether it can effectively block neurodegeneration across multiple preclinical animal models. Regardless, the success of c-Abl inhibition as a treatment strategy for PD requires the identification or design of brain penetrant c-Abl inhibitors in order to achieve the level of CNS target engagement necessary without inducing dangerous peripheral side effects.
The second major challenge with adapting current c-Abl inhibitors to PD is specificity of kinase inhibition. To the best of our knowledge, currently six small molecular inhibitors of c-Abl exist in clinical trials or already FDA approved to treat numerous peripheral cancers, including: Imatinib, Nilotinib, Bafetinib, Ponatinib, Dasatinib, and Bosutinib [82, 87]. Imatinib, Nilotinib, Ponatinib, and Bafetinib are type-II kinase inhibitors, which bind to a hydrophobic pocket adjacent to the kinase ATP-binding domain and trap the kinase in an inactive conformation [87, 88]. Dasatinib and Bosutinib, on the other hand, are type-I kinase inhibitors that compete with the ATP binding site in the kinase domain. Type-II kinase inhibitors tend to bind more potently and specifically than type-I inhibitors [88]. However, even the two most specific c-Abl inhibitors (Nilotinib and Imatinib) inhibit other tyrosine kinases such as KIT or PDGFR. Other c-Abl kinases listed above are even more promiscuous, binding to many different classes of tyrosine kinases and even some serine/threonine kinases [82, 88, 89]. Bafetinib, for example inhibits numerous kinases other than c-Abl including several from the SRC, KIT/PDFGR, Ephrin receptor families, Zak, and DDR1/2 [90]. Thus, while Bafetinib may be more brain penetrant than Imatinib or Nilotinib, it is possible that its off-target inhibition of other kinases may lead to unacceptable side effects in human patients. The identification or design of more specific c-Abl inhibitors may prove necessary to eliminate deleterious off-target effects.
While a large and growing body of evidence indicates that c-Abl inhibitors such as Imatinib, Nilotinib, and Bafetinib show efficacy in preclinical models of PD, it remains to be convincingly shown that any of these three inhibitors have potential for the chronic treatment of human patients. Many challenges remain in applying c-Abl inhibition as a disease-modifying therapeutic strategy for PD, including low BBB penetrance and inhibition of off-target kinases. Despite these hurdles, based on the growing evidence suggesting that c-Abl plays a critical role in neurodegeneration in PD and the early success of these imperfect c-Abl inhibitors as potential PD therapy; there is a growing need and importance for future development of c-Abl inhibitors that have properties of higher BBB penetrant and target-specificity to treat PD.
CONCLUDING REMARKS
c-Abl is a key regulator of many essential cell functions, and its activity is tightly controlled. It exists in a minimally active state at basal level and its overactivation is an important driver of oncogenesis in CML and ALL. It is thus intriguing that c-Abl appears to be aberrantly activated in postmortem PD patient brain tissue and in multiple mouse models of PD specifically in regions pathologically affected by neurodegeneration. Moreover, c-Abl is also activated by buildup of ROS, a key driver of cell death in both familial and sporadic PD [14, 15], and may aggravate oxidative stress by suppressing antioxidant response system through its kinase activity. As such, c-Abl activation seems to be intimately associated with PD pathological changes in both human patients and mouse models.
A growing body of evidence indicates that c-Abl activation not only results in neurodegenerative changes in PD, but is itself a direct driver of neurodegeneration. Investigations thus far have unveiled two primary pathological processes triggered by c-Abl activation. First, c-Abl directly phosphorylates α-synuclein at Y39, which has been shown to promote α-synuclein aggregation and inhibit autophagic clearance of α-synuclein [32, 33, 39]. It is conceivable that these two mechanisms—increased aggregation and reduced clearance—interact in a powerful pathological circuit to promote formation of toxic α-synuclein aggregates. The second major pathological pathway involves c-Abl phosphorylation of parkin at Y143, which induces inactivation of parkin’s E3 ubiquitin ligase activity [4, 31]. Parkin inactivation leads to the accumulation of toxic substrates PARIS and AIMP2, which leads to degeneration of nigral DA neurons by impairing mitochondrial biogenesis and inducing parthanatos, respectively [59, 60]. It would be intriguing to know how these two pathways interact. c-Abl phosphorylation of parkin and α-synuclein could proceed along parallel pathways where each pathway contributes to loss of SN DA neurons or since aggregation of α-synuclein seems to be driver of PD pathogenesis, parkin inactivation by c-Abl phosphorylation could be downstream of c-Abl phosphorylation of Y39 α-synuclein. Future studies are required to test these possibilities.
Many questions remain regarding the pathways through which c-Abl activation leads to neurodegeneration in PD. Because concerns remain regarding the translatability of α-synuclein overexpression models to the human disease, future studies in a more physiologically accurate model of α-synuclein neurodegeneration, such as the recently developed α-synuclein PFF model [45] are needed to further parse the effect c-Abl activation has on α-synuclein accumulation. Moreover, it is important to further elucidate how different parkin substrates each can contribute to c-Abl driven neurodegeneration, and whether c-Abl inactivation of parkin compromises other reported aspects of parkin-mediated mitochondrial quality control such as transport, fission/fusion dynamics, or mitophagy [55, 58]. Investigations into these mechanisms of c-Abl neurodegeneration are still in the early stages, and it is likely that future studies will shed light on additional c-Abl dependent neurodegenerative pathways. A recent study, for example, has suggested a role for p38α, a member of MAP kinase family, as a c-Abl substrate that is potentially important in MPTP-induced neurotoxicity [38]. Glial mechanisms involving c-Abl activation might also contribute to PD pathogenesis. Interestingly, recent reports by Lawana et al., demonstrate that c-Abl activation mediates lipopolysaccharide and rotenone-induced activation of microglial cells through activation of the NLRP3 inflammasome, thereby suggesting that c-Abl may participates in neuroinflammatory processes in PD (Fig. 1) [91].
Exploration of novel c-Abl substrates also seems particularly important for the advancement of the field. Understanding the diverse c-Abl-mediated pathways that are linked to neurodegeneration may open-up avenues for new drug targets. In addition, screening of additional c-Abl substrates may yield biomarkers that have even greater potential than alpha -synuclein.
Most if not all the c-Abl inhibitors currently in clinical trial or already approved by the FDA lack favorable properties including limited brain penetrance, toxicity and inhibition of off-target kinases severely hampering their long-term success in PD. It is one thing to have an agent approved for a lethal disease such as CML, it is another thing to have an agent approved for a chronic disease like PD. As such, the emphasis should be made on development and use of more selective and safe brain-penetrating c-Abl kinase inhibitors instead of rushing to test current inhibitors. Although the activated form of c-Abl itself maybe a potential candidate biomarker for PD [68], it awaits independent validation as a biomarker for PD using rigorous approaches. Sensitive assay methods need to be developed to accurately detect residue-specific tyrosine phosphorylated c-Abl substrates that can be used as theranostic biomarkers in future c-Abl clinical trials with the new brain-penetrant c-Abl inhibitors that are under development.
CONFLICT OF INTEREST STATEMENT
The research discussed in this article is related to agents manufactured by Inhibikase Therapeutics. Dr. T.M. and V.L. Dawson are consultants to Inhibikase Therapeutics and own stock options in the company. This arrangement has been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies. The value of patents owned by Valted, LLC could be affected by this article. Also, Dr. T.M. and V.L. Dawson are founders of Valted, LLC and hold an ownership equity interest in the company. This arrangement has been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institute of Neurological Disorders and Stroke (NINDS, NIH) (NS38377, the Morris K. Udall Parkinson’s Disease Research Center, and NS082205 and NS098006) and the JPB Foundation. T.M. Dawson is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. The authors acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation and the Diana Helis Henry Medical Research Foundation through their direct engagement in the continuous active conduct of medical research in conjunction with Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the foundations’ Parkinson’s Disease Programs, H-1 and M-2014. I-Hsun Wu generated Fig. 1.
REFERENCES
[1] | Hantschel O , & Superti-Furga G ((2004) ) Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat Rev Mol Cell Biol, 5: , 33–44. |
[2] | Wang JY ((2014) ) The capable ABL: What is its biological function? Mol Cell Biol, 34: , 1188–1197. |
[3] | Moresco EM , Donaldson S , Williamson A , & Koleske AJ ((2005) ) Integrin-mediated dendrite branch maintenance requires Abelson (Abl) family kinases. J Neurosci, 25: , 6105–6118. |
[4] | Ko HS , Lee Y , Shin JH , Karuppagounder SS , Gadad BS , Koleske AJ , Pletnikova O , Troncoso JC , Dawson VL , & Dawson TM ((2010) ) Phosphorylation by the c-Abl protein tyrosine kinase inhibitsparkin’s ubiquitination and protective function. Proc NatlAcad Sci USA, 107: , 16691–16696. |
[5] | Schlatterer SD , Acker CM , & Davies P ((2011) ) c-Abl in neurodegenerative disease. J Mol Neurosci, 45: , 445–452. |
[6] | Gonfloni S , Maiani E , Di Bartolomeo C , Diederich M , & Cesareni G ((2012) ) Oxidative Stress, DNA Damage, and c-Abl Signaling: At the Crossroad in Neurodegenerative Diseases? Int J Cell Biol, 2012: , 683097. |
[7] | Estrada LD , Zanlungo SM , & Alvarez AR ((2011) ) C-Abl tyrosine kinase signaling: A new player in AD tau pathology. Curr Alzheimer Res, 8: , 643–651. |
[8] | Brasher BB , & Van Etten RA ((2000) ) c-Ab1 has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J Biol Chem, 275: , 35631–35637. |
[9] | Barila D , & Superti-Furga G ((1998) ) An intramolecular SH3-domain interaction regulates c-Abl activity. Nat Genet, 18: , 280–282. |
[10] | Wen ST , Jackson PK , & Van Etten RA ((1996) ) The cytostatic function of c-Abl is controlled by multiple nuclear localization signals and requires the p53 and Rb tumor suppressor gene products. EMBO J, 15: , 1583–1595. |
[11] | Plattner R , Kadlec L , DeMali KA , Kazlauskas A , & Pendergast AM ((1999) ) c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev, 13: , 2400–2411. |
[12] | Perry G , Cash AD , & Smith MA ((2002) ) Alzheimer disease and oxidative stress. J Biomed Biotechnol, 2: , 120–123. |
[13] | Swomley AM , & Butterfield DA ((2015) ) Oxidative stress in Alzheimer disease and mild cognitive impairment: Evidence from human data provided by redox proteomics. Arch Toxicol, 89: , 1669–1680. |
[14] | Gaki GS , & Papavassiliou AG ((2014) ) Oxidative stress-induced signaling pathways implicated in the pathogenesis of Parkinson’s disease. Neuromolecular Med, 16: , 217–230. |
[15] | Zhou C , Huang Y , & Przedborski S ((2008) ) Oxidative stress in Parkinson’s disease: A mechanism of pathogenic and therapeutic significance. Ann N Y Acad Sci, 1147: , 93–104. |
[16] | Martindale JL , & Holbrook NJ ((2002) ) Cellular response to oxidative stress: Signaling for suicide and survival. J Cell Physiol, 192: , 1–15. |
[17] | Ditch S , & Paull TT ((2012) ) The ATM protein kinase and cellular redox signaling: Beyond the DNA damage response. Trends Biochem Sci, 37: , 15–22. |
[18] | Sun X , Wu F , Datta R , Kharbanda S , & Kufe D ((2000) ) Interaction between protein kinase C delta and the c-Abl tyrosine kinase in the cellular response to oxidative stress. J Biol Chem, 275: , 7470–7473. |
[19] | Cao C , Ren X , Kharbanda S , Koleske AJ , Prasad KV , & Kufe D ((2001) ) The ARG tyrosine kinase interacts with Siva-1 in the apoptotic response to oxidative stress. J Biol Chem, 276: , 11465–11468. |
[20] | Guo Z , Kozlov S , Lavin MF , Person MD , & Paull TT ((2010) ) ATM activation by oxidative stress. Science, 330: , 517–521. |
[21] | Meltser V , Ben-Yehoyada M , & Shaul Y ((2011) ) c-Abl tyrosine kinase in the DNA damage response: Cell death and more. Cell Death Differ, 18: , 2–4. |
[22] | Cao C , Leng Y , Liu X , Yi Y , Li P , & Kufe D ((2003) ) Catalase is regulated by ubiquitination and proteosomal degradation. Role of the c-Abl and Arg tyrosine kinases. Biochemistry, 42: , 10348–10353. |
[23] | Li B , Wang X , Rasheed N , Hu Y , Boast S , Ishii T , Nakayama K , Nakayama KI , & Goff SP ((2004) ) Distinct roles of c-Abl and Atm in oxidative stress response are mediated by protein kinase C delta. Genes Dev, 18: , 1824–1837. |
[24] | Dawson TM , Ko HS , & Dawson VL ((2010) ) Genetic animal models of Parkinson’s disease. Neuron, 66: , 646–661. |
[25] | Shulman JM , De Jager PL , & Feany MB ((2011) ) Parkinson’s disease: Genetics and pathogenesis. Annu Rev Pathol, 6: , 193–222. |
[26] | Martin I , Dawson VL , & Dawson TM ((2011) ) Recent advances in the genetics of Parkinson’s disease. Annu Rev Genomics Hum Genet, 12: , 301–325. |
[27] | Goedert M , Spillantini MG , Del Tredici K , & Braak H ((2013) ) 100 years of Lewy pathology. Nat Rev Neurol, 9: , 13–24. |
[28] | Goedert M ((2001) ) Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci, 2: , 492–501. |
[29] | Lee VM , & Trojanowski JQ ((2006) ) Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: New targets for drug discovery. Neuron, 52: , 33–38. |
[30] | Maries E , Dass B , Collier TJ , Kordower JH , & Steece-Collier K ((2003) ) The role of alpha-synuclein in Parkinson’s disease: Insights from animal models. Nat Rev Neurosci, 4: , 727–738. |
[31] | Imam SZ , Zhou Q , Yamamoto A , Valente AJ , Ali SF , Bains M , Roberts JL , Kahle PJ , Clark RA , & Li S ((2011) ) Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: Implications for Parkinson’s disease. J Neurosci, 31: , 157–163. |
[32] | Brahmachari S , Ge P , Lee SH , Kim D , Karuppagounder SS , Kumar M , Mao X , Shin JH , Lee Y , Pletnikova O , Troncoso JC , Dawson VL , Dawson TM , & Ko HS ((2016) ) Activation of tyrosine kinase c-Abl contributes to alpha-synuclein-induced neurodegeneration. J Clin Invest, 126: , 2970–2988. |
[33] | Hebron ML , Lonskaya I , & Moussa CE ((2013) ) Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of alpha-synuclein in Parkinson’s disease models. Hum Mol Genet, 22: , 3315–3328. |
[34] | Imam SZ , Trickler W , Kimura S , Binienda ZK , Paule MG , Slikker W Jr , Li S , Clark RA , & Ali SF ((2013) ) Neuroprotective efficacy of a new brain-penetrating C-Abl inhibitor in a murine Parkinson’s disease model. PLoS One, 8: , e65129. |
[35] | Cunnington R , Iansek R , Johnson KA , & Bradshaw JL ((1997) ) Movement-related potentials in Parkinson’s disease. Motor imagery and movement preparation. Brain, 120: (Pt 8), 1339–1353. |
[36] | Karuppagounder SS , Brahmachari S , Lee Y , Dawson VL , Dawson TM , & Ko HS ((2014) ) The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson’s disease. Sci Rep, 4: , 4874. |
[37] | Tanabe A , Yamamura Y , Kasahara J , Morigaki R , Kaji R , & Goto S ((2014) ) A novel tyrosine kinase inhibitor AMN107 (nilotinib) normalizes striatal motor behaviors in a mouse model of Parkinson’s disease. Front Cell Neurosci, 8: , 50. |
[38] | Wu R , Chen H , Ma J , He Q , Huang Q , Liu Q , Li M , & Yuan Z ((2016) ) c-Abl-p38alpha signaling plays an important role in MPTP-induced neuronal death. Cell Death Differ, 23: , 542–552. |
[39] | Mahul-Mellier AL , Fauvet B , Gysbers A , Dikiy I , Oueslati A , Georgeon S , Lamontanara AJ , Bisquertt A , Eliezer D , Masliah E , Halliday G , Hantschel O , & Lashuel HA ((2014) ) c-Abl phosphorylates alpha-synuclein and regulates its degradation: Implication for alpha-synuclein clearance and contribution to the pathogenesis of Parkinson’s disease. Hum Mol Genet, 23: , 2858–2879. |
[40] | Fujiwara H , Hasegawa M , Dohmae N , Kawashima A , Masliah E , Goldberg MS , Shen J , Takio K , & Iwatsubo T ((2002) ) alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol, 4: , 160–164. |
[41] | Sato H , Kato T , & Arawaka S ((2013) ) The role of Ser129 phosphorylation of alpha-synuclein in neurodegeneration of Parkinson’s disease: A review of in vivo models. Rev Neurosci, 24: , 115–123. |
[42] | Lue LF , Walker DG , Adler CH , Shill H , Tran H , Akiyama H , Sue LI , Caviness J , Sabbagh MN , & Beach TG ((2012) ) Biochemical increase in phosphorylated alpha-synuclein precedes histopathology of Lewy-type synucleinopathies. Brain Pathol, 22: , 745–756. |
[43] | Dikiy I , Fauvet B , Jovicic A , Mahul-Mellier AL , Desobry C , El-Turk F , Gitler AD , Lashuel HA , & Eliezer D ((2016) ) semisynthetic and in vitro phosphorylation of alpha-synuclein at Y39 promotes functional partly helical membrane-bound states rebling those induced by PD mutations. ACS Chem Biol, 11: , 2428–2437. |
[44] | Lee MK , Stirling W , Xu Y , Xu X , Qui D , Mandir AS , Dawson TM , Copeland NG , Jenkins NA , & Price DL ((2002) ) Human alpha-synuclein-harboring familial Parkinson’s disease-linked Ala-53 –>Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc NatlAcad Sci USA, 99: , 8968–8973. |
[45] | Luk KC , Kehm V , Carroll J , Zhang B , O’Brien P , Trojanowski JQ , & Lee VM ((2012) ) Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science, 338: , 949–953. |
[46] | Volpicelli-Daley LA , Luk KC , Patel TP , Tanik SA , Riddle DM , Stieber A , Meaney DF , Trojanowski JQ , & Lee VM ((2011) ) Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron, 72: , 57–71. |
[47] | Mao X , Ou MT , Karuppagounder SS , Kam TI , Yin X , Xiong Y , Ge P , Umanah GE , Brahmachari S , Shin JH , Kang HC , Zhang J , Xu J , Chen R , Park H , Andrabi SA , Kang SU , Goncalves RA , Liang Y , Zhang S , Qi C , Lam S , Keiler JA , Tyson J , Kim D , Panicker N , Yun SP , Workman CJ , Vignali DA , Dawson VL , Ko HS , & Dawson TM ((2016) ) Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science, 353: , pii: aah3374. |
[48] | Dawson TM , & Dawson VL ((2010) ) The role of parkin in familial and sporadic Parkinson’s disease. Mov Disord, 25: (Suppl 1), S32–S39. |
[49] | Stevens DA , Lee Y , Kang HC , Lee BD , Lee YI , Bower A , Jiang H , Kang SU , Andrabi SA , Dawson VL , Shin JH , & Dawson TM ((2015) ) Parkinloss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc Natl Acad Sci USA, 112: , 11696–11701. |
[50] | Moore DJ ((2006) ) Parkin: A multifaceted ubiquitin ligase. Biochem Soc Trans, 34: , 749–753. |
[51] | Geisler S , Holmstrom KM , Skujat D , Fiesel FC , Rothfuss OC , Kahle PJ , & Springer W ((2010) ) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol, 12: , 119–131. |
[52] | Olzmann JA , & Chin LS ((2008) ) Parkin-mediated K63-linked polyubiquitination: A signal for targeting misfolded proteins to the aggresome-autophagy pathway. Autophagy, 4: , 85–87. |
[53] | Chung KK , Thomas B , Li X , Pletnikova O , Troncoso JC , Marsh L , Dawson VL , & Dawson TM ((2004) ) S-nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science, 304: , 1328–1331. |
[54] | LaVoie MJ , Ostaszewski BL , Weihofen A , Schlossmacher MG , & & SelkoeDJ ((2005) ) Dopamine covalently modifies and functionally in activates parkin. Nat Med, 11: , 1214–1221. |
[55] | Truban D , Hou X , Caulfield TR , Fiesel FC , & Springer W ((2017) ) PINK1, Parkin, and Mitochondrial Quality Control: What can we Learn about Parkinson’s Disease Pathobiology? J Parkinsons Dis, 7: , 13–29. |
[56] | Wauer T , Simicek M , Schubert A , & Komander D ((2015) ) Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature, 524: , 370–374. |
[57] | Sauve V , Lilov A , Seirafi M , Vranas M , Rasool S , Kozlov G , Sprules T , Wang J , Trempe JF , & Gehring K ((2015) ) A Ubl/ubiquitin switch in the activation of Parkin. EMBO J, 34: , 2492–2505. |
[58] | Scott L , Dawson VL , & Dawson TM ((2017) ) Trumping neurodegeneration: Targeting common pathways regulated by autosomal recessive Parkinson’s disease genes. Exp Neurol, doi: 10.1016/j.expneurol.2017.04.008 |
[59] | Shin JH , Ko HS , Kang H , Lee Y , Lee YI , Pletinkova O , Troconso JC , Dawson VL , & Dawson TM ((2011) ) PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell, 144: , 689–702. |
[60] | Lee Y , Karuppagounder SS , Shin JH , Lee YI , Ko HS , Swing D , Jiang H , Kang SU , Lee BD , Kang HC , Kim D , Tessarollo L , Dawson VL , & Dawson TM ((2013) ) Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat Neurosci, 16: , 1392–1400. |
[61] | Lee Y , Stevens DA , Kang SU , Jiang H , Lee YI , Ko HS , Scarffe LA , Umanah GE , Kang H , Ham S , Kam TI , Allen K , Brahmachari S , Kim JW , Neifert S , Yun SP , Fiesel FC , Springer W , Dawson VL , Shin JH , & &Dawson TM ((2017) ) PINK1 primes Parkin-mediated ubiquitination of PARIS in dopaminergic neuronal survival. Cell Rep, 18: , 918–932. |
[62] | Corti O , Hampe C , Koutnikova H , Darios F , Jacquier S , Prigent A , Robinson JC , Pradier L , Ruberg M , Mirande M , Hirsch E , Rooney T , Fournier A , & Brice A ((2003) ) The p38 subunit of the aminoacyl-tRNA synthetase complex is a Parkin substrate: Linking protein biosynthesis and neurodegeneration. Hum Mol Genet, 12: , 1427–1437. |
[63] | Ko HS , von Coelln R , Sriram SR , Kim SW , Chung KK , Pletnikova O , Troncoso J , Johnson B , Saffary R , Goh EL , Song H , Park BJ , Kim MJ , Kim S , Dawson VL , & Dawson TM ((2005) ) Accumulation of the authentic parkin substrate aminoacyl-tRNA synthetase cofactor, p38/JTV-1, leads to catecholaminergic cell death. J Neurosci, 25: , 7968–7978. |
[64] | Parnetti L , Castrioto A , Chiasserini D , Persichetti E , Tambasco N , El-Agnaf O , & Calabresi P ((2013) ) Cerebrospinal fluid biomarkers in Parkinson disease. Nat Rev Neurol, 9: , 131–140. |
[65] | Blennow K , Biscetti L , Eusebi P , & Parnetti L ((2016) ) Cerebrospinal fluid biomarkers in Alzheimer’s and Parkinson’s diseases-From pathophysiology to clinical practice. Mov Disord, 31: , 836–847. |
[66] | Kim D , Kim YS , Shin DW , Park CS , & Kang JH ((2016) ) Harnessing cerebrospinal fluid biomarkers in clinical trials for treating Alzheimer’s and Parkinson’s diseases: Potential and challenges. J Clin Neurol, 12: , 381–392. |
[67] | Colicelli J ((2010) ) ABL tyrosine kinases: Evolution of function, regulation, and specificity. Sci Signal, 3: , re6. |
[68] | Pagan F , Hebron M , Valadez EH , Torres-Yaghi Y , Huang X , Mills RR , Wilmarth BM , Howard H , Dunn C , Carlson A , Lawler A , Rogers SL , Falconer RA , Ahn J , Li Z , & Moussa C ((2016) ) Nilotinib effects in Parkinson’s disease and dementia with Lewy bodies. J Parkinsons Dis, 6: , 503–517. |
[69] | Derkinderen P , Scales TM , Hanger DP , Leung KY , Byers HL , Ward MA , Lenz C , Price C , Bird IN , Perera T , Kellie S , Williamson R , Noble W , Van Etten RA , Leroy K , Brion JP , Reynolds CH , & Anderton BH ((2005) ) Tyrosine 394 is phosphorylated in Alzheimer’s paired helical filament tau and in fetal tau with c-Abl as the candidate tyrosine kinase. J Neurosci, 25: , 6584–6593. |
[70] | Tremblay MA , Acker CM , & Davies P ((2010) ) Tau phosphorylated at tyrosine 394 is found in Alzheimer’s disease tangles and can be a product of the Abl-related kinase, Arg. J Alzheimers Dis, 19: , 721–733. |
[71] | Moussaud S , Jones DR , Moussaud-Lamodiere EL , Delenclos M , Ross OA , & McLean PJ ((2014) ) Alpha-synuclein and tau: Teammates in neurodegeneration? Mol Neurodegener, 9: , 43. |
[72] | Nalls MA , Pankratz N , Lill CM , Do CB , Hernandez DG , Saad M , DeStefano AL , Kara E , Bras J , Sharma M , Schulte C , Keller MF , Arepalli S , Letson C , Edsall C , Stefansson H , Liu X , Pliner H , Lee JH , & Cheng R ; International Parkinson’s Disease Genomics Consortium (IPDGC); Parkinson’s Study Group (PSG) Parkinson’s Research: The Organized GENetics Initiative (PROGENI); 23andMe; GenePD; NeuroGenetics Research Consortium (NGRC); Hussman Institute of Human Genomics (HIHG); Ashkenazi Jewish Dataset Investigator; Cohorts for Health and Aging Research in Genetic Epidemiology (CHARGE); North American Brain Expression Consortium (NABEC); United Kingdom Brain Expression Consortium (UKBEC); Greek Parkinson’s Disease Consortium; Alzheimer Genetic Analysis Group, Ikram MA , Ioannidis JP , Hadjigeorgiou GM , Bis JC , Martinez M , Perlmutter JS , Goate A , Marder K , Fiske B , Sutherland M , Xiromerisiou G , Myers RH , Clark LN , Stefansson K , Hardy JA , Heutink P , Chen H , Wood NW , Houlden H , Payami H , Brice A , Scott WK , Gasser T , Bertram L , Eriksson N , Foroud T , & Singleton AB ((2014) ) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet, 46: , 989–993. |
[73] | Leaver K , & Poston KL ((2015) ) Do CSF biomarkers predict progression to cognitive impairment in Parkinson’s disease patients? A systematic review. Neuropsychol Rev, 25: , 411–423. |
[74] | Hall S , Surova Y , Ohrfelt A , Zetterberg H , Lindqvist D , & Hansson O ((2015) ) CSF biomarkers and clinical progression of Parkinson disease. Neurology, 84: , 57–63. |
[75] | Cancino GI , Toledo EM , Leal NR , Hernandez DE , Yevenes LF , Inestrosa NC , & Alvarez AR ((2008) ) STI571 prevents apoptosis, tau phosphorylation and behavioural impairments induced by Alzheimer’s beta-amyloid deposits. Brain, 131: , 2425–2442. |
[76] | Lebouvier T , Scales TM , Williamson R , Noble W , Duyckaerts C , Hanger DP , Reynolds CH , Anderton BH , & Derkinderen P ((2009) ) The microtubule-associated protein tau is also phosphorylated on tyrosine. J Alzheimers Dis, 18: , 1–9. |
[77] | Cilloni D , & Saglio G ((2012) ) Molecular pathways: BCR-ABL. Clin Cancer Res, 18: , 930–937. |
[78] | Alvarez AR , Sandoval PC , Leal NR , Castro PU , & Kosik KS ((2004) ) Activation of the neuronal c-Abl tyrosine kinase by amyloid-beta-peptide and reactive oxygen species. Neurobiol Dis, 17: , 326–336. |
[79] | Breedveld P , Pluim D , Cipriani G , Wielinga P , van Tellingen O , Schinkel AH , & Schellens JH ((2005) ) The effect of Bcrp1 (Abcg2) on the in vivo pharmacokinetics and brain penetration of imatinib mesylate (Gleevec): Implications for the use of breast cancer resistance protein and P-glycoprotein inhibitors to enable the brain penetration of imatinib in patients. Cancer Res, 65: , 2577–2582. |
[80] | Dai HQ , Marbach P , Lemaire M , Hayes M , & Elmquist WF ((2003) ) Distribution of STI-571 to the brain is limited by P-glycoprotein-mediated efflux. J Pharmacol Exp Therap, 304: , 1085–1092. |
[81] | Weisberg E , Manley P , Mestan J , Cowan-Jacob S , Ray A , & Griffin JD ((2006) ) AMN107 (nilotinib): A novel and selective inhibitor of BCR-ABL. Br J Cancer, 94: , 1765–1769. |
[82] | Green MR , Newton MD , & Fancher KM ((2016) ) Off-Target Effects of BCR-ABL and JAK2 Inhibitors. Am J Clin Oncol, 39: , 76–84. |
[83] | Shukla S , Skoumbourdis AP , Walsh MJ , Hartz AM , Fung KL , Wu CP , Gottesman MM , Bauer B , Thomas CJ , & Ambudkar SV ((2011) ) Synthesis and characterization of a BODIPY conjugate of the BCR-ABL kinase inhibitor Tasigna (nilotinib): Evidence for transport of Tasigna and its fluorescent derivative by ABC drug transporters. Mol Pharm, 8: , 1292–1302. |
[84] | Pinilla-Ibarz J , Sweet K , Emole J , & Fradley M ((2015) ) Long-term BCR-ABL1 tyrosine kinase inhibitor therapy in chronic myeloid leukemia. Anticancer Res, 35: , 6355–6364. |
[85] | Yokota A , Kimura S , Masuda S , Ashihara E , Kuroda J , Sato K , Kamitsuji Y , Kawata E , Deguchi Y , Urasaki Y , Terui Y , Ruthardt M , Ueda T , Hatake K , Inui K , & Maekawa T ((2007) ) INNO-406, a novel BCR-ABL/Lyn dual tyrosine kinase inhibitor, suppresses the growth of Ph+leukemia cells in the central nervous system, and cyclosporine A augments its in vivo activity. Blood, 109: , 306–314. |
[86] | Zhang YK , Zhang GN , Wang YJ , Patel BA , Talele TT , Yang DH , & Chen ZS ((2016) ) Bafetinib (INNO-406) reverses multidrug resistance by inhibiting the efflux function of ABCB1 and ABCG2 transporters. Sci Rep, 6: , 25694. |
[87] | O’Hare T , Shakespeare WC , Zhu X , Eide CA , Rivera VM , Wang F , Adrian LT , Zhou T , Huang WS , Xu Q , Metcalf CA 3rd , Tyner JW , Loriaux MM , Corbin AS , Wardwell S , Ning Y , Keats JA , Wang Y , Sundaramoorthi R , Thomas M , Zhou D , Snodgrass J , Commodore L , Sawyer TK , Dalgarno DC , Deininger MW , Druker BJ , & Clackson T ((2009) ) AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell, 16: , 401–412. |
[88] | Giansanti P , Preisinger C , Huber KV , Gridling M , Superti-Furga G , Bennett KL , & Heck AJ ((2014) ) Evaluating the promiscuous nature of tyrosine kinase inhibitors assessed in A431 epidermoid carcinoma cells by both chemical- and phosphoproteomics. ACS Chem Biol, 9: , 1490–1498. |
[89] | Tse A , & Verkhivker GM ((2015) ) Molecular determinants underlying binding specificities of the ABL kinase inhibitors: Combining alanine scanning of binding hot spots with network analysis of residue interactions and coevolution. PLoS One, 10: , e0130203. |
[90] | Rix U , Remsing Rix LL , Terker AS , Fernbach NV , Hantschel O , Planyavsky M , Breitwieser FP , Herrmann H , Colinge J , Bennett KL , Augustin M , Till JH , Heinrich MC , Valent P , & Superti-Furga G ((2010) ) A comprehensive target selectivity survey of the BCR-ABL kinase inhibitor INNO-406 by kinase profiling and chemical proteomics in chronic myeloid leukemia cells. Leukemia, 24: , 44–50. |
[91] | Lawana V , Singh N , Sarkar S , Charli A , Jin H , Anantharam V , Kanthasamy AG , & Kanthasamy A ((2017) ) Involvement of c-Abl kinase in microglial activation of NLRP3 inflammasome and impairment in autolysosomal system. J Neuroimmune Pharmacol, doi: 10.1007/s11481-017-9746-5 |


