
A Critical Assessment of Exosomes in the Pathogenesis and Stratification of Parkinson’s Disease
Abstract
Extracellular vesicles including exosomes are released by a variety of cell types including neurons and exhibit molecular profiles that reflect normal and disease states. As their content represents a snapshot of the intracellular milieu, they could be exploited as biomarkers of the otherwise inaccessible brain microenvironment. In addition they may contribute to the progression of neurodegenerative disorders by facilitating the spread of misfolded proteins at distant sites or activating immune cells. This review summarizes recent advances in the study of exosomes in Parkinson’s disease pathophysiology and their potential as disease biomarkers.
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disorder affecting 1% of people over the age of 60. Clinically it is characterized primarily by a movement disorder causing resting tremor, bradykinesia, rigidity, postural instability and diverse non-motor symptoms including dementia, which in community-based studies, was reported in up to 80% of patients with long disease duration [1]. This latter finding indicates that PD is a diffuse neurodegenerative disorder. Similarly, detailed neuropathological studies have shown that one of the cardinal histological features, the intraneuronal inclusions called Lewy bodies (LB) that are comprised of misfolded α-synuclein, are detected in numerous cortical areas and often correlate with the extent of cognitive decline [2]. Although, clinically a heterogeneous disorder, at the molecular level defective protein trafficking to endosomes and lysosomes has emerged as a potentially unifying cellular pathway in the pathogenesis of sporadic PD [3, 4]. Because the release of exosomes is tightly linked to intracellular protein trafficking along the endosomal-lysosomal pathway, their biology is likely to be relevant to PD. Secretion of extracellular vesicles (EV) including exosomes is a well-recognized means of communication between cells of the same or different type. As their content represents a snapshot of intracellular states they have attracted considerable attention as potential biomarkers of the otherwise inaccessible microenvironment of the living brain. Exosomes may also represent means of elimination of misfolded proteins that contribute to the progression of neurodegenerative disorders. This review summarizes recent advances in the study of exosomes in PD pathogenesis and their potential as disease biomarkers.
EXOSOME BIOGENESIS
EV are composed of a lipid bilayer containing transmembrane proteins and enclosing soluble proteins and RNA. Depending on their origin, they bear surface molecules that enable their uptake by recipient cells. Once attached to a target cell, EVs induce signaling via receptor-ligand interaction or get internalized by endocytosis and/or phagocytosis or even fuse with the target cell’s membrane to deliver their content into its cytosol, thereby modifying the physiological state of the recipient cell.
It is now clear that cells can release EVs of different types. Large EVs called ectosomes are directly shed from the cell’s plasma membrane and display sizes that range from 100– 1000 nm. In contrast, exosomes are generated within late endosomal compartments called multivesicular bodies (MVB) and secreted when these compartments fuse with the plasma membrane, thus releasing their intraluminal vesicles in the extracellular milieu. This process may be enhanced when there is a block in the fusion of MVB with lysosomes where their content is degraded. Therefore, exosomes are similar in terms of size (i.e. 40– 150 nm in diameter) and content of endosome-associated proteins to the intraluminal vesicles of MVBs. Typically, formation of MVBs and exosomes is driven by the endosomal sorting complex required for transport (ESCRT), which is composed of approximately thirty proteins that assemble into four complexes (ESCRT-0, -I, -II and -III) with associated proteins (VPS4, VTA1, ALIX) that are conserved from yeast to mammals. ESCRT independent mechanisms have also been proposed [5].
The protein content of exosomes has been studied extensively using unbiased methods. A comprehensive database named Vesiclepedia, which is continuously updated by the scientific community is available on line (http://www.microvesicles.org). Initial proteomic studies showed that exosomes contain a specific subset of cellular proteins, some of which are cell-type specific and others generic. The latter include proteins from endosomes, plasma membrane and the cytosol, whereas proteins from the nucleus, mitochondria, endoplasmic reticulum, and the Golgi complex are mostly absent. These observations highlight the specificity of formation of these vesicles and show that they represent a specific subcellular compartment and not random cell fragments. Recent proteomic comparison of subpopulations of EV showed that several classically used exosome markers, such as major histocompatibility complex, flotillin, and heat-shock 70-kDa proteins, are similarly present in all EVs. Instead, syntenin-1 (a key regulator of exosome biogenesis) and TSG101 represent distinct ‘bona fide’ exosome markers [6].
In addition to proteins, EV including exosomes are enriched in small RNA, including mRNA, and miRNA. One of the most interesting outcomes of miRNA discovery in exosomes is that they can modulate gene expression in distant cells. Such a functional transfer has been demonstrated in vitro [7] and in wildtype mice using self-derived dendritic exosomes engineered to express their transmembrane protein Lamp2b fused to a Rabies virus Glycoprotein (RVG) peptide that increased their uptake in the nervous system [8]. Intravenously injected RVG-targeted exosomes delivered siRNA specifically to neurons, microglia and oligodendrocytes in the brain, resulting in a specific gene knockdown of proteins including ones implicated in neurodegeneration. To what extent such RNA-mediated effects have biological relevance in the brain under physiological conditions is currently unclear.
EXOSOMES IN PARKINSON’S DISEASE PATHOBIOLOGY
Because of the central role of α-synuclein in the cause of sporadic PD [9], its potential association with exosomes has attracted early attention. Initial studies in SY5Y cells expressing α-synuclein showed that both monomeric and oligomeric forms of α-synuclein are released both free in the medium and within exosomes the latter in a process that was calcium-dependent [10]. Subsequent studies confirmed these findings in cell lines and also showed that exosomal release of α-synuclein and uptake by recipient cells was increased when the lysosome was inhibited [11, 12]. When compared to free α-synuclein oligomers derived from conditioned media, exosome-associated α-synuclein oligomers were more likely to be taken up by recipient cells and cause toxicity [12].
The exact trigger for the incorporation of α-synuclein into exosomes or their release is currently unknown. α-Synuclein is found in association with endosomes [13, 14] and lysosomes extracted from mouse brains [15]. Mechanistically, α-synuclein is targeted to endosomes by ubiquitination by the E3 ligase Nedd4 [16, 17] and this process is negatively regulated by USP8 [18]. Therefore, the endosomal fraction of α-synuclein could conceivably escape degradation under conditions of lysosomal impairment and be released either by the recycling endosome in a process involving Rab11a and Hsp90 [13, 19] or within exosomes from MVB. Previous work also highlighted the AAA (ATPases Associated with diverse cellular Activities)-ATPase, VPS4, as an important regulator of α-synuclein sorting into MVBs [13]. More recently the covalent conjugation of SUMO (Small Ubiquitin like Modifier) on α-synuclein was also shown to regulate its recruitment to exosomes via a mechanism that involved the interaction of SUMO with phospholipids and required VPS4, Alix and TSG101 [20]. It should be noted that both in earlier studies using SY5Y cells [10] and our own investigation in conditioned media from iPSC-derived dopaminergic neurons from healthy subjects and PD patients with heterozygous GBA mutations [21], less than 2% of α-synuclein is found in association with exosomes. In addition, α-synuclein oligomers were present both in association with the outside membrane of exosomes as well as inside the exosome lumen [12] suggesting that the actual amount of α-synuclein released within exosomes is a minute fraction of total extracellular α-synuclein.
Nevertheless, these early studies lend support to the concept that pathogenic forms of α-synuclein may spread via exosomes and contribute to the progression of pathology in the brain. This is especially relevant to recent evidence that α-synuclein propagation along brain regions may account for disease progression: LB pathology was identified in embryonic neural grafts 12– 16 years after transplantation into the brains of people with PD [22, 23] and in transgenic mice, human α-synuclein can transit to nerve cells grafted into the hippocampus [24]. Direct inoculation of human brain extract of fibrils from patients with MSA or LB pathology led to progressive α-synuclein aggregation and neurodegeneration in connected areas of the brain of animal models including non-human primates [25– 27]. α-Synuclein pathology in the brain was also demonstrated following the systemic administration of α-synuclein assemblies [28]. Therefore, based on these models of disease, it is conceivable that proteopathic assemblies of α-synuclein that are capable of seeding further aggregation are released in exosomes and taken up by interconnected or distant neurons or non-neuronal cells. Accordingly, cerebrospinal fluid exosomes derived from Parkinson’s disease and dementia with Lewy bodies are enriched in α-synuclein species that can induce the oligomerization of α-synuclein in a reporter cell line in a dose-dependent manner [29]. More recently, exosomes derived from brains with Lewy body pathology caused aggregation of the endogenous protein when directly inoculated in the hippocampus of wildtype mice [30]. Whether exosomes contributed to the spread of the pathology at distant brain sites is currently unknown. In addition other exosome-associated factors beyond α-synuclein, may account for such observations. For example, the lipid composition of exosomes was shown to promote the aggregation of recombinant α-synuclein in vitro [31]. It is also unclear whether such “pathogenic” exosomes are primarily released from neurons or other cell types. It is noteworthy that BV-2 microglia exposed to α-synuclein activated exosome release, which increased apoptosis when added to primary cortical neurons [32]. In addition, microglia isolated from adult but not young mice, displayed reduced phagocytosis of exosome-associated α-synuclein oligomers and enhanced TNFα secretion [33]. These studies suggest that microglia in the aging brain may potentiate neurodegeneration by either failing to clear or promoting the release of exosomes which contain toxic forms of α-synuclein or other pathogenic factors.
Other genes that are mutated in familial PD have also been linked to exosome biology. LRRK2 is a multidomain protein with GTPase and kinase functions and a number of protein/protein interaction motifs. Autosomal dominant mutations in LRRK2 cause familial PD [34, 35], and have since been found in up to 2% of apparently sporadic cases [36], indicating that LRRK2 is the commonest gene defect in PD. Mutant LRRK2 acts at multiple points within the endosomal pathway and thus has the potential to modulate exosome biogenesis. For example, LRRK2 has been demonstrated to interact with Rab5b, which regulates endocytic vesicle trafficking within cells [37] and implicated in late endosome maturation and fusion with lysosomes by interacting with Rab7 [38]. More recenlty, LRRK2 was shown to directly phosphorylate a number of Rab proteins, which may account for its role in diverse trafficking pathways [39]. Therefore it is not surprising that LRRK2 is released in exosomes and regulates their biogenesis [40]. Elevated ratio of phosphorylated Ser-1292 LRRK2 to total LRRK2 in urine exosomes predicted LRRK2 mutation status and PD risk among LRRK2 mutation carriers [41]. This phenomenon was also seen in a small cohort of 79 patients with sporadic PD, where increased urinary levels of exosomal Ser(P)-1292 LRRK2 correlated with the severity of cognitive impairment and difficultly in accomplishing activities of daily living [42]. These data suggest that LRRK2-regulated exosome biogenesis may be relevant to sporadic disease. The origin of these urinary exosomes is unknown but may be immune-derived as LRRK2 is highly expressed in cells of the innate immune system, where it becomes autophosphorylated and released in exosomes in response to inflammatory stimuli [43].
The link between endosomal trafficking and exosome biology in the context of parkinsonism is also supported by studies in a rare autosomal recessive condition due to mutations in ATP13A2. ATP13A2 is expressed in dopaminergic neurons of the substantia nigra and patients with loss of function mutations in ATP13A2 develop Kufor-Rakeb syndrome [44], which is characterized by juvenile-onset parkinsonism, pyramidal signs, and cognitive decline. It was previously shown that expression of ATP13A2 suppressed α-synuclein toxicity in primary DA neurons [45]. ATP13A2 localized to multivesicular bodies (MVBs) in the human H4 cell line whereas patient fibroblasts with loss of ATP13A2 function exhibited decreased number of intraluminal vesicles in MVBs and diminished release of exosomes into culture media. This loss of ATP13A2 function resulted in decreased secretion of α-synuclein, increasing its intracellular retention and accumulation [46]. In contrast, overexpressed ATP13A2 promoted secretion of α-synuclein, at least partly in exosomes, by regulating their biogenesis through functional interaction with the endosomal sorting complex required for transport machinery [46, 47]. The E3 ligase parkin, which is the commonest cause of autosomal recessive PD, has also been implicated in exosome biogenesis. The primary function of parkin is in the regulation of mitophagy through the ubiquitination of mitochondrial membrane proteins [48]. However expression of mutant parkin was shown to impair the organization of multivesicular bodies and retromer assembly leading to increased exosome release [49]. Interestingly, parkin was implicated in the trafficking of mitochondrial-derived vesicles to the endosomal compartment [50]. Whether parkin selectively regulates the exosomal release of mitochondrial components is currently unclear.
EXOSOME AS BIOMARKERS IN PARKINSON’S DISEASE
Although PD can be diagnosed on clinical grounds with a high degree of certainty [51], prediction of its rate of progression is what matters most to patients and when testing new therapies. This is currently challenging, as the disease is clinically heterogeneous without an objective marker of cumulative disability. Therefore a disease marker that could aid the stratification of patients is urgently needed.
Because of their mode of production (i.e. within cells), exosomes may provide a means for neurons to jettison excessive, potentially toxic forms of α-synuclein or other disease-associated proteins from the brain into the CSF and the systemic circulation. In support of this notion, a very small fraction of 125I-labelled α-synuclein or tau injected intracerebroventricularly was detected in plasma exosomes expressing the neuronal adhesion molecule L1CAM [52, 53]. Although our own unbiased proteomic approach in total exosome fractions based on ultracentrifugation did not identify α-synuclein enrichment in the serum of PD patients [54], immunoprecipitation and characterization of neuron-specific exosomes in plasma using the neuronal adhesion molecule L1CAM showed that exosomal α-synuclein was increased in PD patients [52]. ROC analysis in a large cohort of 267 PD and 215 matched controls revealed a moderate performance of plasma exosomal α-synuclein (AUC = 0.654, sensitivity = 70.1%, specificity = 52.9%) or when analyzed as a ratio to total α-synuclein (AUC = 0.657, sensitivity = 71.2%, specificity = 50.0%). Interestingly, a significant correlation was observed between the plasma α-synuclein in L1CAM-containing exosomes (r = 0.176, p = 0.004, Pearson correlation) or the plasma exosomal/total α-synuclein ratio (r = 0.130, p = 0.035) with the Unified Parkinson’s Disease Rating Scale motor scores [52]. Tau in L1CAM-positive plasma-derived exosomes was also modestly predictive in distinguishing PD from healthy controls (AUC = 0.607, sensitivity = 57.8%, specificity = 65.1%) and was associated with disease duration but not severity as assessed by Unified Parkinson’s Disease Rating Scale motor scores. In contrast to the prediction of these studies, i.e. that neuron-derived exosomal α-synuclein is increased in PD, assessment of exosomal α-synuclein in the CSF was not consistently increased across cohorts and was actually lower in Dementia with Lewy bodies when compared to PD [29]. However, these authors did observe a positive correlation between exosomal α-synuclein in CSF of patients with dementia with Lewy bodies and cognitive decline or CSF levels of tau, which is a marker of neuronal damage. The sensitivity and specificity of CSF exosomal α-synuclein to distinguish dementia with Lewy bodies from Parkinson’s disease were 85.7% and 80.6% with a positive and negative predictive value of 81.1% and 85.3%, respectively. The sensitivity and specificity for the distinction of dementia with Lewy bodies from progressive supranuclear palsy was 71.4% and 92.0%. Therefore, higher levels of exosomal α-synuclein in advanced stages of Lewy body disease may reflect an increased pathological burden and the higher number of neurons with aggregates disposing exosomal α-synuclein in the CSF. In other words, these clinical correlations suggest that exosomal release of α-synuclein is a late defense mechanism and therefore a potential readout of disease progression.
Beyond α-synuclein and tau, other exosome-associated factors may be used for biomarker development. As described earlier, urinary exosomes where also shown to have increased levels of Ser(P)-1292 LRRK2 in sporadic PD that correlated with the severity of cognitive impairment and difficultly in accomplishing activities of daily living [42]. Our unbiased mass spectrometry indicates that certain exosomal protein cargoes such as syntenin 1 are increased in PD [54]. Similarly, RNA profiling suggests that certain miRNAs are differentially enriched in PD serum-derived exosomes [55]. These differences may account for our observation that when added to stressed primary neurons, serum PD-derived exosomes had a paradoxically beneficial effect compared to controls [54]. These studies provide a first glimpse of what may emerge to be a multitude of circulating exosome subpopulations in PD with distinct properties reflecting an active signaling process between the degenerating brain and other systems. It is worth noting that in general the total number or size of exosomes is not increased in the serum [54] or CSF [29] of PD patients when compared to controls. This is also the case in other neurodegenerative diseases e.g. AD [56], even though one study reported a possible correlation between exosome number and cognitive decline in a small group of PD patients [57].
CONCLUDING REMARKS
The realization that endosomal trafficking is a critical pathway in PD provided a scientific rationale for the relevance of exosome biology to the disease pathogenesis (Fig. 1). Exosomal release of α-synuclein accounts for less than 2% of total extracellular α-synuclein but a detailed understanding of how it is packaged into exosomes for release may be fundamentally linked to cellular defense against toxic forms of this protein or non-cell autonomous mechanisms of disease progression. In this context, accurate measurements of its content in CSF or the systemic circulation may signify more advanced stages of pathology and provide the urgently needed prognostic biomarker. Therefore, improved multiplexed assays with high sensitivity [58] combined with antibodies against pathogenic or modified forms of α-synuclein, tau or LRRK2 in exosomes may improve their utility in PD stratification. On the other end, exosomes generated outside the brain may also convey trophic signals to the degenerating neurons or exert toxic effects due to persistent immune activation. Whether there is a specific marker of a “PD exosome” or multiple subpolulations of EV that are differentially activated in PD remains to be seen. Therefore more unbiased analyses of the composition of exosome subpopulations in CSF and plasma or serum are needed to fully exploit their potential as biomarkers. In addition, functional studies of their effects in cellular and animal models could help to better define their role in the pathobiology of PD.
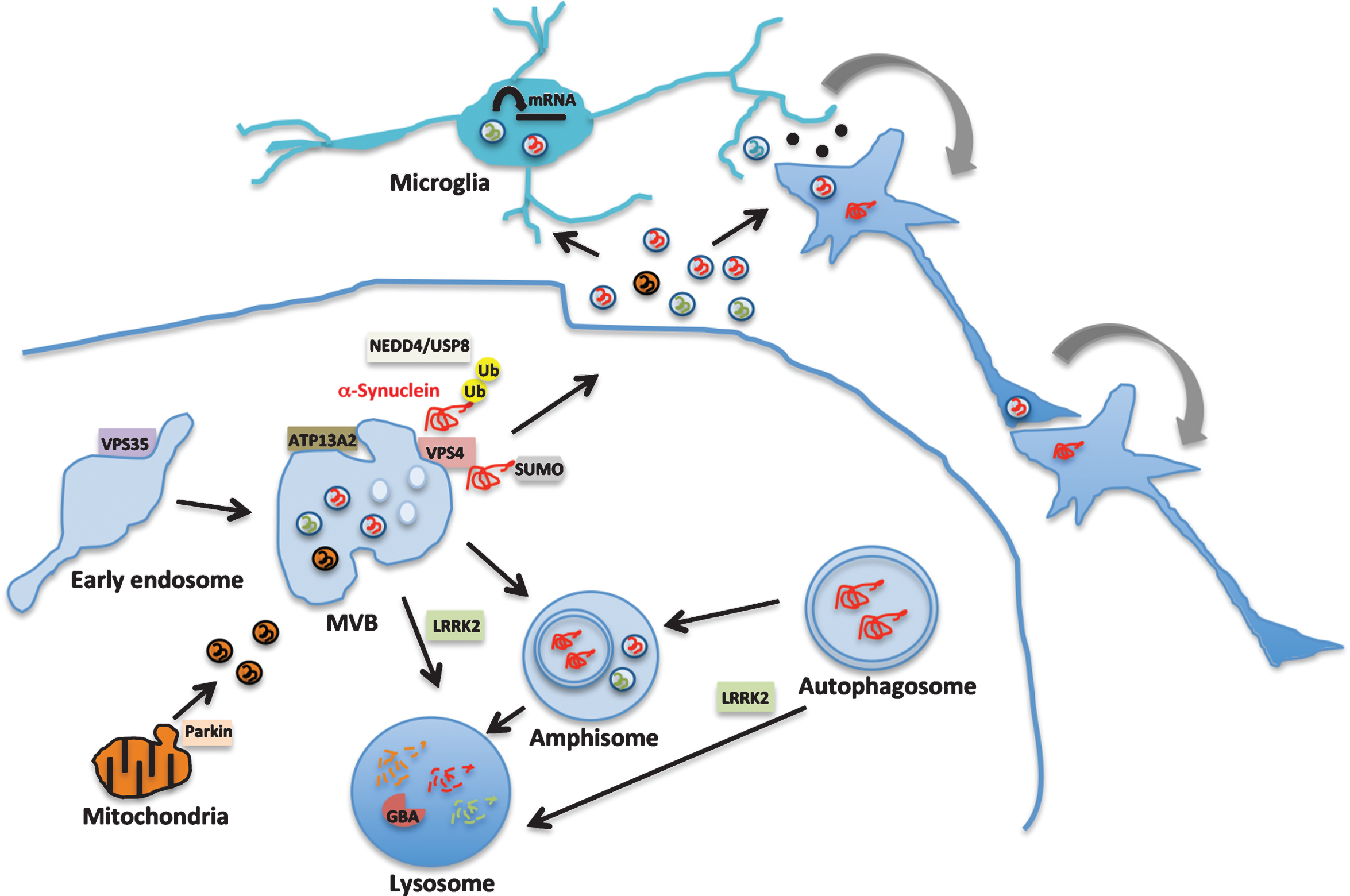
Fig.1
Exosome signaling in Parkinson’s disease pathobiology. Exosomes are generated within the multivesicular bodies (MVB) of the endosomal-lysosomal pathway. Genes that are implicated in familial forms of PD such as VPS35, LRRK2, ATP13A2 and Parkin, function in this pathway as shown in the diagram. Heterozygous mutations in GBA that increase the risk of PD by 6-fold impair lysosomal function. LRRK2 regulates this pathway at multiple steps through its kinase activity and is also an exosomal cargo protein (shown in green). α-Synuclein (red) misfolding is considered a key pathogenic event in sporadic PD. α-Synuclein is trafficked via the endosomal pathway by NEDD4/USP8 regulated ubiquitination (Ub) or sumoylation (SUMO) and released in exosomes. Exosomal α-synuclein, LRRK2 or other proteins may contribute to PD pathology by modulating microglial cell activation, facilitating the propagation of proteopathic α-synuclein assemblies or additional signaling events.
CONFLICT OF INTEREST
The author has no conflict of interest to report.
ACKNOWLEDGMENTS
GKT is funded by a Wellcome Trust Intermediate Clinical Fellowship (097479/Z/11/Z) and the Wellcome Beit Prize fellowship (097479/Z/11/A), European Union’s Horizon 2020 research and innovation programme under grant agreement No. 116060 (IMPRiND), ARUK, BMA, the Oxford Biomedical Research Centre and EPSRC (EP/M006204/1). The opinion expressed and arguments employed herein do not necessarily reflect the official views of these funding bodies.
REFERENCES
[1] | Hely MA , Reid WG , Adena MA , Halliday GM , & Morris JG ((2008) ) The Sydney multicenter study of Parkinson’s disease: The inevitability of dementia at 20 years. Mov Disord 23: , 837–844. |
[2] | Schneider JA , Arvanitakis Z , Yu L , Boyle PA , Leurgans SE , & Bennett DA ((2012) ) Cognitive impairment, decline and fluctuations in older community-dwelling subjects with Lewy bodies. Brain 135: , 3005–3014. |
[3] | Tofaris GK ((2012) ) Lysosome-dependent pathways as a unifying theme in Parkinson’s disease. Mov Disord 27: , 1364–1369. |
[4] | Perrett RM , Alexopoulou Z , & Tofaris GK ((2015) ) The endosomal pathway in Parkinson’s disease. Mol Cell Neurosci 66: , 21–28. |
[5] | Colombo M , Raposo G , & Théry C ((2014) ) Biogenesis, secretion and intracellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 30: , 255–289. |
[6] | Kowal J , Arras G , Colombo M , Jouve M , Morath JP , Primdal-Bengtson B , Dingli F , Loew D , Tkach M , & Théry C ((2016) ) Proteomic comon defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. E968-E. Proc Natl Acad Sci U S A 113: , 977–paris. |
[7] | Pegtel DM , Cosmopoulos K , Thorley-Lawson DA , van Eijndhoven MA , Hopmans ES , Lindenberg JL , de Gruijl TD , Würdinger T , & Middeldorp JM ((2010) ) Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S A 107: , 6328–6333. |
[8] | Alvarez-Erviti L , Seow Y , Yin H , Betts C , Lakhal S , & Wood MJ ((2011) ) Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol 29: , 341–345. |
[9] | Tofaris GK , & Spillantini MG ((2007) ) Physiological and pathological properties of alpha-synuclein. Cell Mol Life Sci 64: , 2194–2201. |
[10] | Emmanouilidou E , Melachroinou K , Roumeliotis T , Garbis SD , Ntzouni M , Margaritis LH , Stefanis L , & Vekrellis K ((2010) ) Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci 30: , 6838–6851. |
[11] | Alvarez-Erviti L , Seow Y , Schapira AH , Gardiner C , Sargent IL , Wood MJ , & Cooper JM ((2011) ) Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis 42: , 360–367. |
[12] | Danzer KM , Kranich LR , Ruf WP , Cagsal-Getkin O , Winslow AR , Zhu L , Vanderburg CR , & McLean PJ ((2012) ) Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener 7: , 42. |
[13] | Hasegawa T , Konno M , Baba T , Sugeno N , Kikuchi A , Kobayashi M , Miura E , Tanaka N , Tamai K , Furukawa K , Arai H , Mori F , Wakabayashi K , Aoki M , Itoyama Y , & Takeda A ((2011) ) The AAA-ATPase VPS4 regulates extracellular secretion and lysosomal targeting of α-synuclein. PLoS One 6: , e29460. |
[14] | Boassa D , Berlanga ML , Yang MA , Terada M , Hu J , Bushong EA , Hwang M , Masliah E , George JM , & Ellisman MH ((2013) ) Mapping the subcellular distribution of α-synuclein in neurons using genetically encoded probes for correlated light and electron microscopy: Implications for Parkinson’s disease pathogenesis. J Neurosci 33: , 2605–2615. |
[15] | Mak SK , McCormack AL , Manning-Bog AB , Cuervo AM , & Di Monte DA ((2010) ) Lysosomal degradation of alpha-synuclein in vivo. J Biol Chem 285: , 13621–13629. |
[16] | Tofaris GK , Kim HT , Hourez R , Jung JW , Kim KP , & Goldberg AL ((2011) ) Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc Natl Acad Sci U S A 108: , 17004–17009. |
[17] | Davies SE , Hallett PJ , Moens T , Smith G , Mangano E , Kim HT , Goldberg AL , Liu JL , Isacson O , & Tofaris GK ((2014) ) Enhanced ubiquitin-dependent degradation by Nedd4 protects against α-synuclein accumulation and toxicity in animal models of Parkinson’s disease. Neurobiol Dis 64: , 79–87. |
[18] | Alexopoulou Z , Lang J , Perrett RM , Elschami M , Hurry ME , Kim HT , Mazaraki D , Szabo A , Kessler BM , Goldberg AL , Ansorge O , Fulga TA , & Tofaris GK ((2016) ) Deubiquitinase Usp8 regulates α-synuclein clearance and modifies its toxicity in Lewy body disease. Proc Natl Acad Sci USA 113: , E4688–E4697. |
[19] | Liu J , Zhang JP , Shi M , Quinn T , Bradner J , Beyer R , Chen S , & Zhang J ((2009) ) Rab11a and HSP90 regulate recycling of extracellular alpha-synuclein. J Neurosci 29: , 1480–1485. |
[20] | Kunadt M , Eckermann K , Stuendl A , Gong J , Russo B , Strauss K , Rai S , Kügler S , Falomir Lockhart L , Schwalbe M , Krumova P , Oliveira LM , Bähr M , Möbius W , Levin J , Giese A , Kruse N , Mollenhauer B , Geiss-Friedlander R , Ludolph AC , Freischmidt A , Feiler MS , Danzer KM , Zweckstetter M , Jovin TM , Simons M , Weishaupt JH , & Schneider A ((2015) ) Extracellular vesicle sorting of α-Synuclein is regulated by sumoylation. Acta Neuropathol 129: , 695–713. |
[21] | Fernandes HJ , Hartfield EM , Christian HC , Emmanoulidou E , Zheng Y , Booth H , Bogetofte H , Lang C , Ryan BJ , Sardi SP , Badger J , Vowles J , Evetts S , Tofaris GK , Vekrellis K , Talbot K , Hu MT , James W , Cowley SA , & Wade-Martins R ((2016) ) ER stress and autophagic perturbations lead to elevated extracellular α-synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Reports 6: , 342–356. |
[22] | Kordower JH , Chu Y , Hauser RA , Freeman TB , & Olanow CW ((2008) ) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med 14: , 504–506. |
[23] | Li JY , Englund E , Holton JL , Soulet D , Hagell P , Lees AJ , Lashley T , Quinn NP , Rehncrona S , Björklund A , Widner H , Revesz T , Lindvall O , & Brundin P ((2008) ) Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med 14: , 501–503. |
[24] | Desplats P , Lee HJ , Bae EJ , Patrick C , Rockenstein E , Crews L , Spencer B , Masliah E , & Lee SJ ((2009) ) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 106: , 13010–13015. |
[25] | Masuda-Suzukake M , Nonaka T , Hosokawa M , Oikawa T , Arai T , Akiyama H , Mann DM , & Hasegawa M ((2013) ) Prion-like spreading of pathological α-synuclein in brain. Brain 136: , 1128–1138. |
[26] | Recasens A , Dehay B , Bové J , Carballo-Carbajal I , Dovero S , Pérez-Villalba A , Fernagut PO , Blesa J , Parent A , Perier C , Fariñas I , Obeso JA , Bezard E , & Vila M ((2014) ) Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 75: , 351–362. |
[27] | Prusiner SB , Woerman AL , Mordes DA , Watts JC , Rampersaud R , Berry DB , Patel S , Oehler A , Lowe JK , Kravitz SN , Geschwind DH , Glidden DV , Halliday GM , Middleton LT , Gentleman SM , Grinberg LT , & Giles K ((2015) ) Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A 112: , E5308–E5317. |
[28] | Peelaerts W , Bousset L , Van der Perren A , Moskalyuk A , Pulizzi R , Giugliano M , Van den Haute C , Melki R , & Baekelandt V ((2015) ) α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522: , 340–344. |
[29] | Stuendl A , Kunadt M , Kruse N , Bartels C , Moebius W , Danzer KM , Mollenhauer B , & Schneider A ((2016) ) Induction of α-synuclein aggregate formation by CSF exosomes from patients with Parkinson’s disease and dementia with Lewy bodies. Brain 139: , 481–494. |
[30] | Ngolab J , Trinh I , Rockenstein E , Mante M , Florio J , Trejo M , Masliah D , Adame A , Masliah E , & Rissman RA ((2017) ) Brain-derived exosomes from dementia with Lewy bodies propagate α-synuclein pathology. Acta Neuropathol Commun 5: , 46. |
[31] | Grey M , Dunning CJ , Gaspar R , Grey C , Brundin P , Sparr E , & Linse S ((2015) ) Acceleration of α-synuclein aggregation by exosomes. J Biol Chem 290: , 2969–2982. |
[32] | Chang C , Lang H , Geng N , Wang J , Li N , & Wang X ((2013) ) Exosomes of BV-2 cells induced by alpha-synuclein: Imortant mediator of neurodegeneration in PD. Neurosci Lett 548: , 190–195. |
[33] | Bliederhaeuser C , Grozdanov V , Speidel A , Zondler L , Ruf WP , & Bayer H ((2016) ) Age-dependent defects of alpha-synuclein oligomer uptake in microglia and monocytes. Acta Neuropathol 131: , 379–391. |
[34] | Paisán-Ruíz C , Jain S , Evans EW , Gilks WP , et al ((2004) ) Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44: , 595–600. |
[35] | Zimprich A , Biskup S , Leitner P , Lichtner P , et al ((2004) ) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44: , 601–607. |
[36] | Berg D , Schweitzer KJ , Leitner P , Zimprich A , Lichtner P , Belcredi P , Brüssel T , Schulte C , Maass S , Nägele T , Wszolek ZK , & Gasser T ((2005) ) Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson’s disease. Brain 128: , 3000–3011. |
[37] | Shin N , Jeong H , Kwon J , Heo HY , Kwon JJ , Yun HJ , Kim CH , Han BS , Tong Y , Shen J , Hatano T , Hattori N , Kim KS , Chang S , & Seol W ((2008) ) LRRK2 regulates synaptic vesicle endocytosis. Exp Cell Res 314: , 2055–2065. |
[38] | Gómez-Suaga P , Rivero-Ríos P , Fdez E , Blanca Ramírez M , Ferrer I , Aiastui A , López De Munain A , & Hilfiker S ((2014) ) LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity. Hum Mol Genet 23: , 6779–6796. |
[39] | Steger M , Tonelli F , Ito G , Davies P , Trost M , Vetter M , Wachter S , Lorentzen E , Duddy G , Wilson S , Baptista MA , Fiske BK , Fell MJ , Morrow JA , Reith AD , Alessi DR , & Mann M ((2016) ) Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 5: , pii: e 12813. |
[40] | Fraser KB , Moehle MS , Daher JP , Webber PJ , Williams JY , Stewart CA , Yacoubian TA , Cowell RM , Dokland T , Ye T , Chen D , Siegal GP , Galemmo RA , Tsika E , Moore DJ , Standaert DG , Kojima K , Mobley JA , & West AB ((2013) ) LRRK2 secretion in exosomes in regulated by 14-3-3. Hum Mol Genet 22: , 4988–5000. |
[41] | Fraser KB , Moehle MS , Alcalay RN , West AB , LRRK2 Cohort Consortium ((2016) ) Urinary LRRK2 phosphorylation predicts parkinsonian phenotypes in G2019S LRRK2 carriers. Neurology 86: , 994–999. |
[42] | Fraser KB , Rawlins AB , Clark RG , Alcalay RN , Standaert DG , Liu N , Parkinson’s Disease Biomarker Program Consortium, & West AB ((2016) ) Ser(P)-1292 LRRK2 in urinary exosomes is elevated in idiopathic Parkinson’s disease. Mov Disord 31: , 1543–1550. |
[43] | Moehle MS , Webber PJ , & Tse T , et al ((2012) ) LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci 32: , 1602–1611. |
[44] | Ramirez A , Heimbach A , Gründemann J , Stiller B , Hampshire D , Cid LP , Goebel I , Mubaidin AF , Wriekat AL , Roeper J , Al-Din A , Hillmer AM , Karsak M , Liss B , Woods CG , Behrens MI , & Kubisch C (Genet) ((2006) ) Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat 38: , 1184–1191. |
[45] | Gitler AD , Chesi A , Geddie ML , Strathearn KE , Hamamichi S , Hill KJ , Caldwell KA , Caldwell GA , Cooper AA , Rochet JC , & Lindquist S ((2009) ) Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet 41: , 308–315. |
[46] | Tsunemi T , Hamada K , & Krainc D ((2014) ) ATP13A2/ PARK9 regulates secretion of exosomes and α-synuclein. J Neurosci 34: , 15281–15287. |
[47] | Kong SM , Chan BK , Park JS , Hill KJ , Aitken JB , Cottle L , Farghaian H , Cole AR , Lay PA , Sue CM , & Cooper AA ((2014) ) Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α-Synuclein externalization via exosomes. Hum Mol Genet 23: , 2816–2833. |
[48] | McWilliams TG , & Muqit MM ((2017) )Curr Opin Cell Biol 45: , 83–91 PINK1 and Park, Emerging themes in mitochondrial homeostasis. |
[49] | Song P , Trajkovic K , Tsunemi T , & Krainc D ((2016) ) Parkin modulates endosomal organization and function of the endo-lysosomal pathway. J Neurosci 36: , 2425–2437. |
[50] | McLelland GL , Lee SA , McBride HM , & Fon EA ((2016) ) Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J Cell Biol 214: , 275–291. |
[51] | Hughes AJ , Daniel SE , Ben-Shlomo Y , & Lees AJ ((2002) ) The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain 125: , 861–870. |
[52] | Shi M , Liu C , Cook TJ , Bullock KM , Zhao Y , Ginghina C , Li Y , Aro P , Dator R , He C , Hipp MJ , Zabetian CP , Peskind ER , Hu SC , Quinn JF , Galasko DR , Banks WA , & Zhang J ((2014) ) Plasma exosomal α-synuclein is likely CNS-derived and increased in Parkinson’s disease. Acta Neuropathol 128: , 639–650. |
[53] | Shi M , Kovac A , Korff A , Cook TJ , Ginghina C , Bullock KM , Yang L , Stewart T , Zheng D , Aro P , Atik A , Kerr KF , Zabetian CP , Peskind ER , Hu SC , Quinn JF , Galasko DR , Montine TJ , Banks WA , & Zhang J ((2016) ) CNS tau efflux via exosomes is likely increased in Parkinson’s disease but not in Alzheimer’s disease. Alzheimers Dement 12: , 1125–1131. |
[54] | Tomlinson PR , Zheng Y , Fischer R , Heidasch R , Gardiner C , Evetts S , Hu M , Wade-Martins R , Turner MR , Morris J , Talbot K , Kessler BM , & Tofaris GK ((2015) ) Identification of distinct circulating exosomes in Parkinson’s disease. Ann Clin Transl Neurol 2: , 353–361. |
[55] | Cao XY , Lu JM , Zhao ZQ , Li MC , Lu T , An XS , & Xue LJ ((2017) ) MicroRNA biomarkers of Parkinson’s disease in serum exosome-like microvesicles. Neurosci Lett 644: , 94–99. |
[56] | Winston CN , Goetzl EJ , Akers JC , Carter BS , Rockenstein EM , Galasko D , Masliah E , & Rissman RA ((2016) ) Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimers Dement 3: , 63–72. |
[57] | Alamri Y , Vogel R , MacAskill M , & Anderson T ((2016) ) Plasma exosome concentration may correlate with cognitive impairment in Parkinson’s disease. Alzheimers Dement 4: , 107–108. |
[58] | Li Q , Tofaris GK , & Davis JJ ((2017) ) Concentration-normalized electroanalytical assaying of exosomal markers. Anal Chem 89: , 3184–3190. |

