
Glucocerebrosidase Mutations in Parkinson Disease
Abstract
Following the discovery of a higher than expected incidence of Parkinson Disease (PD) in Gaucher disease, a lysosomal storage disorder, mutations in the glucocerebrocidase (GBA) gene, which encodes a lysosomal enzyme involved in sphingolipid degradation were explored in the context of idiopathic PD. GBA mutations are now known to be the single largest risk factor for development of idiopathic PD. Clinically, on imaging and pharmacologically, GBA PD is almost identical to idiopathic PD, other than certain features that can be identified in the specialist research setting but not in routine clinical practice. In patients with a known GBA mutation, it is possible to monitor for prodromal signs of PD. The clinical similarity with idiopathic PD and the chance to identify PD at a pre-clinical stage provides a unique opportunity to research therapeutic options for early PD, before major irreversible neurodegeneration occurs. However, to date, the molecular mechanisms which lead to this increased PD risk in GBA mutation carriers are not fully elucidated. Experimental models to define the molecular mechanisms and test therapeutic options include cell culture, transgenic mice and other in vivo models amenable to genetic manipulation, such as drosophilia. Some key pathological pathways of interest in the context of GBA mutations include alpha synuclein aggregation, lysosomal-autophagy axis changes and endoplasmic reticulum stress. Therapeutic agents that exploit these pathways are being developed and include the small molecule chaperone Ambroxol. This review aims to summarise the main features of GBA-PD and provide insights into the pathological relevance of GBA mutations on molecular pathways and the therapeutic implications for PD resulting from investigation of the role of GBA in PD.
INTRODUCTION
Parkinson disease (PD) is the second most common neurodegenerative disease after Alzheimer disease, with a lifetime risk of 4%. The disease is characterized pathologically by loss of dopaminergic neurons in the substantia nigra pars compacta, accumulation and aggregation of alpha-synuclein, and clinically by motor symptoms of tremor, rigidity and bradykinesia, and non-motor symptoms that encompass gastro-intestinal dysfunction, depression, anosmia, cognitive decline, pain and sleep disturbances [1, 2].
Homozygous or compound heterozygous mutations of the glucocerebrosidase gene (GBA), were initially discovered in patients suffering from Gaucher Disease (GD), a lysosomal-storage disease characterised by accumulation of glycolipid substrate due to reduced glucocerebrosidase enzyme (GCase) activity. Gcase normally functions as a lysosomal enzyme that converts glucosylceramide to glucose and ceramide. Patient macrophages engorged with this glycolipid load are referred to as “Gaucher cells” and are a pathological hallmark of the disease. Single heterozygous mutations were initially thought to be non-pathogenic, but, following investigation of early reports of Parkinsonism in GD patients and their heterozygote carrier family members unaffected by GD [3], it was found that heterozygote mutations confer a significant risk for developing PPD [4–6]. Heterozygotes have a 10–30% chance of developing PD by age 80, which constitutes a 20-fold increase compared to non-carriers [7–10], and approximately 5–25% of “idiopathic” PD patients carry GBA mutations, making GBA mutations the greatest risk factor for PD discovered to date [11, 12].
In the routine clinical environment, GBA linked PD is virtually indistinguishable from idiopathic PD, with a marginally earlier age of onset and slightly higher prevalence of cognitive effects the only suggestive features in this setting [13, 14]. There are more subtle features associated with GBA PD, which may be elicited in research settings and are discussed in the clinical section of this review. No differences are observed in imaging, pharmacological response or pathology between idiopathic and GBA mutation linked PD [15].
To date, the molecular mechanisms which lead to an increased PD risk in GBA mutation carriers have not been fully elucidated. It has been suggested that both a chronic loss of GCase enzyme activity, as well as a possible toxic gain-of-function of the mutated GCase results in lysosomal dysfunction and endoplasmic reticulum (ER) stress, which could contribute to disease pathogenesis [16]. Interestingly, reduced GCase activity has been found in post mortem samples of PD patients without GBA mutations, suggesting a reduction in activity in PD patients may contribute to PD pathogenesis [17]. As a consequence, targeting of this pathway may provide disease therapy for all PD patients, not just GBA mutation carriers [18].
This review aims to summarise the main features of GBA-PD and provide insights into the pathological relevance of GBA mutations and the therapeutic implications for PD resulting from investigation of the role of GBA in PD.
GENETICS
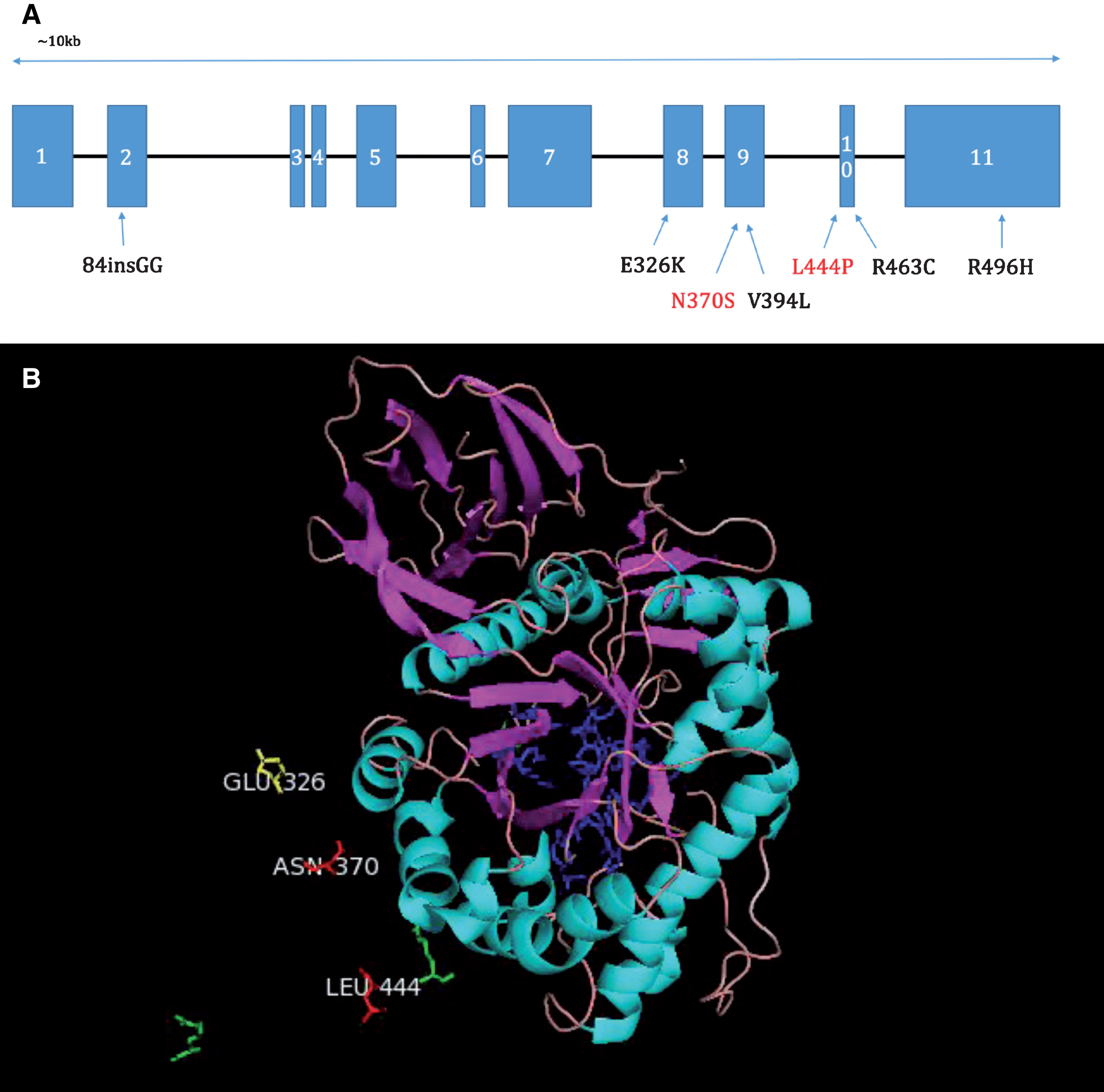
The GBA1 gene is found on chromosome 1q21 and consists of 11 exons and 10 introns. It is approximately 7.6 kb in length. A pseudogene of GBA (GPAP) exists 16 kb downstream of GBA and is highly homologous (96%). Over 300 mutations of the gene have been discovered to date, including frameshift mutations, insertions, deletions and splice site mutations. However, the two most common mutations are N370S and L444P, point mutations in exon 9 and 10 respectively. The total frequency of mutations and the prevalence of specific mutations varies widely between ethnicities, for example in the Ashkenazi Jewish population approximately 1 in 14–18 individuals is a carrier of a mutation, compared with other populations where just 1% are carriers. Sequencing is complicated by the presence of the pseudogene which naturally contains some of the disease-causing mutations, for example L444P [19].
GCase is a 497 amino acid protein of approximately 62 kDa which can be divided into three domains: Domain 1-an antiparallel B-sheet, Domain 2 containing the active site of the protein a triose phosphate isomerase barrel, and Domain 3, an 8-stranded B-barrel. Mutations have been found throughout the protein (see Fig. 1), and despite the proximity of certain mutations to the active site, mutation location is not a reliable predictor of disease severity [16]. In fact, great variability of symptoms and clinical presentation can be seen between GD patients with the same mutation, suggesting the existence of modifying factors. Interestingly, the E326K mutation increases the risk for PD, but when present in homozygous form does not result in GD [20]. This suggests a specific and perhaps distinct mechanism through which mutations may predispose carriers to PD, separate from a GD-causing mechanism.
Fig.1
A. Diagram of the positions of common mutations in the GBA gene. N370S and L444P shown with red writing are the two most common mutations (Hruska et al., 2008). B. Pymol image showing the positions of the most common mutations L444P, N370S and a mutation which increases the risk of PD, but when present in homozygous form does not produce GD, E326K, relative to the active site (shown in blue). Amino acids labelled in green show the positions of less common mutations. Portions of the full GCase protein have been omitted for clarity.
MODELS OF GBA-PD
The contribution of GBA mutations to PD pathogenesis has been investigated thus far in a number of different disease models. Animal models had been developed for the study of GD, before the risk for PD was appreciated and now additional models have been developed specifically for the study of the relationship of GBA mutations to PD. In addition to this, a large number of cell-based models exist, both primary cells, and patient-derived cell lines, including induced pluripotential stem cells (iPSCs).
Cell models (see Table 1)
Table 1
Cell models
| Type | Modification | Ref |
| Mouse embryonic fibroblasts | GBA1 heterozygous mutation | Magalhaes et al. [23] |
| GBA1 Knock Out | ||
| SH-SY5Y cells human α-syn | Knock down by GBA1 siRNA | |
| Mice neurons | CBE exposure | |
| Mouse neurons and astrocytes | GBA knock out | Osellame et al. [33] |
| Neuroblastoma cells | CBE exposure | Manning-Boğ et al. [25] |
| Fibroblasts from PD patients | Heterozygous GBA N370S or L444P mutations | Sanchez-Martinez et al. [40] |
| Fibroblasts from patients | Gaucher Disease | McNeill et al. [26] |
| Fibroblasts from patients | Heterozygous GBA mutation carriers with and without PD | Ambrosi et al. [84] |
| iPSC-derived midbrain dopaminergic neurons from patients | Gaucher Disease | Schöndorf et al. [28] |
| PD harbouring GBA1 mutations | Yang et al. [85] | |
| iPSC-derived midbrain dopaminergic neurons from patients | Heterozygous GBA N370S mutation in twins discordant for PD | Woodard et al. [29] |
| Fibroblast and iPSC-derived NSC and dopaminergic neurons from patients | Heterozygous GBA: N370S mutation | Momcilovic et al. [30] |
| Yang et al. [85] |
Cell lines such as SH-SY5Y and PC12 are more robust than primary cells and as such can survive harsh genetic techniques such as transfection. Additionally they are cost-effective and free from the ethical concerns that accompany primary cell and stem cell use [21, 22]. In the case of GBA-PD research, cell lines stably expressing alpha-synuclein can be transfected with siRNA to silence GBA expression and thus serve as a robust model of reduced Gcase activity in a PD setting [23].
Primary cell culture can be immensely useful in the study of GBA-PD, for example, chemically-induced models of GBA mutations using the irreversible inhibitor, conduritol B-epoxide (CBE) [24] have shown inhibition of GCase induces accumulation of alpha-synuclein, a key discovery linking GBA activity and PD [25]. Research groups with direct access to patients carrying GBA mutations have developed fibroblast cell lines from skin biopsies taken from these patients, which maintain expression of the mutant GCase as seen in the donor patient. Fibroblast lines may also be derived from GBA mutation-carrying mice. These models allow investigation of the cellular mechanisms by which altered GCase activity may affect patient susceptibility to PD and can serve as an inexpensive model for testing potential drug treatments [23, 26]. These lines, however, are not neuronal, and do not express alpha-synuclein, which can limit the key research questions that can be answered using them.
Recently, developments have been made in creating GBA mutation-carrying iPSC lines, using novel reprogramming technology [27]. These iPSCs can then be differentiated into especially disease-relevant cell types, such as dopaminergic neurons [28–30]. This allows for the characterisation of human dopaminergic neurons carrying GBA mutations. In this way analysis and comparison of mutation carriers versus controls at a cellular and molecular level is possible, including live-cell imaging [28].
Animal models (see Table 2)
Table 2
Animal models
| Type | Modification | Ref |
| Mice | CBE injection | Manning-Boğ et al. [25] |
| Mice | GBA Knock Out | Holleran et al. [31] |
| Mice | GBA Knock Out in all tissues except the skin | Enquist [32] |
| Mice | GBA knock out | Osellame et al. [33] |
| Mice | GBA point mutations (N370S, V394L, D409H, D409V) | Xu et al. [34] |
| Mice | GBA point mutation (N370S and L444P) | Sanders et al. [35] |
| Mice | alpha-synuclein A53T mutation + GBA L444P mutation | Fishbein et al. [36] |
| Mice | A53T-SNCA + CBE administration | Rockenstein et al. [37] |
| Mice | Heterozygous L444P GBA mutation | Migdalska-Richards et al. [38] |
| Drosophila | GBA1 and GBA2 deletion | Davis et al. [39] |
| Drosophila | GBA N370S or L444P mutations | Sanchez-Martinez et al. [40] |
| Zebrafish | 23bp GBA deletion | Keatinge et al. [41] |
| Cynomolgus monkeys | Wild-type | Migdalska-Richards et al. [86] |
CBE, Conduritol-β-epoxide; iPSC, induced pluripotent stem cell; NSC, neural stem cells.
Neonatal lethality limited the usefulness of knockout mice for GD. These animals died primarily due to epidermal permeability defects resulting in excessive water loss [31]. Subsequently models have been produced in which the knockout exists in all tissues except the skin, allowing increased survival rates and improved potential for experimental use [32]. In these models the phenotype of type 1 GD is reliably recapitulated and severe, with accumulation of alpha-synuclein and ubiquitinated proteins, as is seen in the human disease. Investigations using such mouse models are critical for research focused on molecular level analysis of the effects of GCase reduction. Defects in autophagic and proteasomal machinery, as well as dysfunction and fragmentation of mitochondria in neurons and astrocytes lacking GBA have been described in the knockout mice [33].
GBA transgenic mice-point mutations
Transgenic mouse lines have been produced with a number of GBA point mutations introduced into the gene, these include the N370S, L444P, V394L, D409H, D409V [34]. Mouse lines with human GBA introduced also exist with N370S and L444P mutations present [35]. Mouse lines for the most part recapitulate aspects of GD including substrate accumulation, the presence of “Gaucher cells” and inflammation.
PD mice and GD carrier cross
Following the discovery of GBA mutations as risk factors for PD, a mouse line has been created where alpha-synuclein A53T mutation mice (A53T mice) are crossed with mice carrying one of the most common GBA mutations, L444P [36]. Interestingly, augmentation of the PD phenotype was found when the PD line was crossed with L444P heterozygote mice which have just 40% residual GCase activity. A decrease in alpha synuclein degradation was found, as well as worsened motor and gastro-enteric phenotypes. Reciprocal modulation of alpha-synuclein and GCase levels was demonstrated in a mouse model [37].
Interestingly, treatment of one PD mouse model (SNCA/SNCA mice), using a small molecule chaperone known to improve GCase abundance and activity, decreased α-synuclein and phospho-α-synuclein protein levels in the mice, and increased activity in L444P heterozygote mice [38]. This suggests that use of PD mouse models in testing GCase targeted drug treatments can be useful for assessing application of such drugs to PD patients in general [38].
Non-mouse models
Drosophila
One model of GCase impairment was created Gba1del through deletion of the coding sequences of gba1 and gba2, the two homologues of human GBA in flies [39] to assess the effects of GBA knockout in the Drosophila system.
Homozygotes for the deletion displayed reduced GCase activity levels and exhibit a motor deficit. Intriguingly, this phenotype was not significantly altered when human alpha synuclein was introduced to the system, suggesting these deficits develop in an alpha synuclein-independent manner.
Sanchez-Martinez et al., have also generated Drosophila GBA models, however rather than a total deletion of the endogenous genes, they generated a series of fly lines expressing the human wild-type GBA or human mutant GBA genes, carrying the N370S or L444P mutations, most common in patients. Flies carrying such mutations developed ER stress and progressive locomotor deficits, which were alleviated by treatment with small molecular chaperones such as ambroxol and isofagomine [40].
Zebrafish
A GBA mutant Zebrafish line was produced through introduction of a 23bp deletion using TALEN mutagenesis. Homozygotes develop normally, but from 8 weeks onwards disease phenotype becomes apparent with significant weight loss and impaired balance resulting in corkscrew swimming. “Gaucher like” cells were observed from 9 weeks, and a loss of dopaminergic neurons was seen at 12 weeks. Heterozygotes had an intermediate motor phenotype and developed some mild accumulation of “Gaucher like” cells by 12 weeks. Additionally, established GD biomarkers were abnormal with hexosaminidase or chitotriosidase activity, as well as mitochondrial respiratory chain activity impaired [41].
PATHOGENIC MECHANISM/BIOCHEMISTRY IN GBA-PD
The underlying mechanism or mechanisms by which a GBA mutation may predispose the carrier to developing PD remains poorly understood. There are a number of possible mechanisms potentially contributing to an increased risk of PD development, which will now be discussed.
Interaction with alpha synuclein
One of the key pathological hallmarks of PD is the accumulation of alpha synuclein aggregates in the cells of patients. Importantly, a reciprocal relationship between GCase levels and alpha synuclein levels has been described, with decreases in GCase, through CBE treatment, or the introduction of mutations, resulting in increases of intracellular alpha synuclein and conversely, overexpression of alpha synuclein has been shown to reduce GCase levels pointing to a vicious cycle leading to reduced GCase activity and increased alpha synuclein levels [12, 25]. The exact mechanisms facilitating this interaction remains unclear, with both loss and gain-of-function theories suggested and supported by experimental evidence, and crucially these suggestions are not mutually exclusive. A multitude of possible interactions, direct and indirect may contribute to the reciprocal relationship observed [16].
Interaction with the autophagy-lysosomal system
The Autophagy-Lysosomal system consists of three distinct pathways to the lysosome which together control the degradation of unnecessary or dysfunctional proteins. These are: macroautophagy, a non-specific pathway for ubiquitin-tagged proteins, chaperone-mediated autophagy (CMA), a pathway through which proteins bearing a specific motif are recognised by a chaperone protein and transported into the lysosome, and finally microautophagy, which consists of direct invaginations of the lysosome membrane to collect small volumes of soluble proteins from the cytosol. The lysosome is known to be dysfunctional in GD, whereby reduced GCase activity levels result in build-up of its substrate in the lysosome and a “clogging” effect on the autophagy-lysosomal system as a whole. Impairment of the autophagy-lysosomal pathway in heterozygote carriers may contribute to increasing PD risk, although substrate accumulation has not been observed [42]. Interestingly, accumulation of key markers of dysfunction of macroautophagy, p62 and LC3-II has been described in post-mortem PD brains, irrespective of GBA mutation status. Key research has also heavily implicated CMA in the degradation of alpha-synuclein, which contains the specific CMA binding motif, suggesting dysfunction of both CMA and macroautophagy pathways in PD patients generally.
GCase leading to ER stress
Abnormally folded GCase has been found to accumulate in the ER which results in reduced levels of GCase in the lysosome as well as triggers the unfolded protein response and ER-associated degradation (ERAD) [43–45]. Markers of ERAD were indeed found to be increased in the PD-GBA brains (Gegg Ann Neurol 2012). iPSC work confirmed increased ER stress and ERAD in GBA carrierlines [46].
CLINICAL PRESENTATION
GBA related Parkinsonism, when first described, appeared identical to idiopathic PD. Although phenotypically very similar and certainly not distinguishable by routine clinical assessment alone, evidence over the last 5 years suggests unique motor, non-motor and cognitive features of GBA PD in individuals with heterozygous or homozygous GBA mutations. Additionally, the prodromal features associated with idiopathic PD are also found in the GBA population. The advantage of detailed characterization of the signature features of GBA associated PD is to gain insight into pre-clinical and “transitional” PD that would typically go undetected in this population. This facilitates our understanding of pre-symptomatic PD and can inform research into identifying biomarkers of pre-clinical features in order to detect, stratify and treat these patients at an early stage of their disease, then widen these findings to other PD groups.
MOTOR FEATURES
Studies of the presenting motor features in GBA associated PD demonstrated that bradykinesia is a more common initial symptom compared with idiopathic cases and that age of onset in GBA carriers (heterozygotes) and homozygotes is earlier [47–49]. No difference was observed in tremor and dyskinesia between GBA and idiopathic cases in one series [49], although a large European series reported that levodopa induced dyskinesia was more severe in PD with GBA mutations than with controls [48]. Severity of the GBA mutation in terms of its effect on enzyme activity, is associated with earlier age of onset [48, 50].
Longitudinal cohort studies have demonstrated that motor progression is more rapid in the GBA PD group (using UPDRS and H&Y staging) than non GBA mutation PD [51, 52]. Further work on motor function in the GBA group has identified a GBA polymorphism, E236K, which is associated with rapid motor progression, postural and gait instability but not tremor [53], wearing off or dyskinesia [54]. The association between other common GBA polymorphisms, cardinal motor symptoms and rate of progression have not been fully defined.
NON MOTOR FEATURES
Non-motor symptoms have been reported to be more severe in PD patients with GBA mutations than in idiopathic PD [55]. In common with idiopathic PD, non-motor symptoms are the earliest presenting symptoms in GBA associated PD.
Visual dysfunction is PD is an increasingly recognised non-motor feature, with pathological involvement of retinal, brainstem and cortical structures. Retinal dopaminergic neuronal degeneration and alpha synuclein deposition are seen in the brainstem, occipital and parietal cortices, which can explain dysfunction with eye movement, visual perception and visuospatial ability [56].
Olfaction is impaired in GBA mutation carriers and in early PD compared with controls [57, 58]. Early olfactory dysfunction is in keeping with the Braak hypothesis that PD pathogenesis commences in peripheral neuronal populations after an as yet unknown insult and that alpha synuclein migrates retrogradely via the enteric, vagal and olfactory tracts to the brainstem, then into cortical and subcortical regions [59]. The Braak hypothesis explains why impaired olfaction and non-motor features occur in the prodromal (pre-motor) phase of PD and may be useful in detecting pre-clinical disease which it is almost uniquely possible to study in the GBA population.
In the idiopathic PD population, it is well established that gastroparesis and constipation are seen in all stages of PD [60]. This, and other non-motor features are more common in GBA-PD including falls with loss of consciousness and fatigue [61]; sexual dysfunction and constipation [62]; orthostatic symptoms, urinary function, sexual function, bowel function [55]. The finding on orthostatic hypotension was confirmed in one recent study [63], but not in another [64]. GBA positive patients have been found by some groups to demonstrate reduced myocardial 123I-metaiodobenzylguanidine (MIBG) uptake from as early as 1 year of disease duration [65], suggesting either the process began before clinical presentation, or that progression is more rapid in this group. The reduced MIBG uptake was associated with reduction in the heart to mediastinum ratio, the degree of which correlated with the stage of PD and dementia [65]. Other groups, however, have not found cardiac MIBG uptake abnormalities associated with GBA PD [54].
Several studies assessed the difference in cognitive performance between PD patients with and without GBA mutations [10, 55, 58, 61–63, 65–70]; according to a recent meta-analysis, the risk of cognitive impairment is estimated to be 3 times higher for patients with GBA mutations [49]. However, significant differences in cognitive function between PD patients with and without GBA mutations were not confirmed in some studies [13, 62, 71, 72]. It has been reported that there is significantly higher frequency of usage of acetylcholine esterase inhibitors for dementia in GBA PD compared with idiopathic PD [73], and one study found significant differences in self-reported cognitive impairment [74]. PD patients with GBA mutation also showed a more rapid cognitive decline [51, 52, 54], although this evidence was not supported by all studies [69]. It has been suggested that in carriers of “severe” mutations (p.L444P, p.G377S, splicing mutation IVS10+1G>T) the risk of dementia was 5.6 times higher when compared to PD non-carriers, and 2.9 times higher when compared to carriers of “mild” mutations (p.N370S) [63]. There is poor agreement on the role of the GBA polymorphisms E326K on cognition: one study found that both E326K patients and GBA mutation patients have a higher risk of cognitive deterioration [75], another found that the association between GBA and cognitive decline was significant in E326K patients but not in PD patients with GBA mutations [53] and, finally, patients with PD and GBA mutations but not E326K or other polymorphisms progress to dementia more rapidly than non-carriers [52]. Specific cognitive profiles have been associated with GBA mutations in PD patients: one study focused on memory and visuospatial domains [10], while another on abstraction and orientation domains [61]. Impairment in working memory, executive function and visuospatial abilities seemed to be a characteristic of PD both with GBA mutations and E326K variant [75]. Deficits in visual short-term memory seemed to be present both in GBA-positive individuals without PD and GBA-negative patients with PD, but with different characteristics; moreover, GBA-positive individuals who developed PD showed a worse performance overall, leading the authors to hypothesize a “double hit” model [76].
Recent evidence shows significant reductions in regional cerebral blood flow associated with GBA PD compared with idiopathic cases, particularly in the parietal cortices and precuneus [54, 77]. This pattern of cerebral blood flow alternation seen in Lewy body dementia and contributes to neurobiological explanation for early cognitive decline in GBA PD.
Patients with GBA mutations showed a higher risk [54, 63] and an earlier development of psychosis [54] as well as an increased frequency of delirium and hallucinations [65] when compared with PD patients without mutations. GBA-mutation PD patients appeared to have a higher risk of reporting hallucinations when compared to idiopathic PD patients, but no differences between mild and severe mutations were found [73]. A statistically significant higher frequency of hallucinations in PD patients with GBA mutation was not confirmed in all studies [62, 72].
Depression has been reported to be more common in PD patients with GBA mutations [55, 61]. However, in one study, even though depression scores were higher in GBA mutation PD patients than in GBA mutation negative patients, the difference in scoring above the cut-off was not significant [62]; other studies did not find any significant differences in mood disorders [10, 13, 54], and a longitudinal study did not show differences in depression neither at baseline nor during the follow-up [51].
Anxiety [55, 61] and apathy [55, 63] have been reported to be more common in GBA mutation patients when compared to PD patients without GBA mutations, even though not all studies confirmed this finding [62].
PD patients with GBA mutations seem have a higher frequency of non-motor symptoms than PD patients without GBA mutations, in particular:
Two studies reported that pain was more frequent in PD patients with GBA mutations than in PD patients without mutations [61, 78], although a significant difference between these two groups was not confirmed in another study [79].
PRODROMAL SYMPTOMS OF PD IN PATIENTS WITH GBA MUTATIONS
The baseline data from a longitudinal study found that prodromal signs of PD such and impairment of olfaction, cognition, and motor signs were more frequent in Gaucher Disease (GD) patients and GBA mutation carriers without previous diagnosis of PD than in controls, while no difference was found in REM sleep behavior disorder (RBD) or autonomic dysfunction [58]. A two year follow up of these patients showed that olfactory scores and cognitive performance remained stable but significantly worse than controls; when outcomes for both GD and carriers were considered, RBD, depression and motor signs showed a significant deterioration and, when compared to controls, significant differences in smell, autonomic dysfunction, cognition and motor signs were evident [57]. A recent study on GD patients without a diagnosis of PD reported a relatively high prevalence of prodromal symptoms of PD, although no comparisons have been performed [80]. A web-based assessment of PD prodromal markers to estimate the risk of PD showed that the odds ratio of having GBA variant was 9.5 times greater in the “high risk group” than in other groups [81].
GCASE AS A THERAPEUTIC TARGET
The current treatment of PD is symptomatic and no drug or intervention has been shown to slow the progression of the neurodegeneration [82]. The discovery of the link between GBA mutations and PD has opened up a novel area for therapeutic investigation, with the possibility of targeting GCase as an important contributor to autophagy dysfunction and alpha synuclein accumulation in the disease [83]. The similarity in clinical presentation, pathology and disease progression between GBA mutation carriers and non-carriers, suggests that neuroprotective treatments developed on the basis of GBA-PD research could be translated to idiopathic PD.
A number of treatment options may soon become available to PD patients. Specifically, there has been a focus on the development and testing of small molecule chaperones to assist in the correct folding of mutant GCase molecules in the ER, thus aiding their transport to the lysosomes and resulting in increased GCase activity. One such molecule is ambroxol, which has been tested for safety and efficacy in GD patients to date [9] as well as initial testing in PD patient fibroblast lines [26, 84], GBA mutant and PD mouse models [38], Drosophila models of GCase deficiency [40], in GD patient iPSC derived macrophages [85] and its brain penetration has been confirmed in non-human primates [86]. Another molecule which has been tested both in vitro and in vivo and proved efficacious is isofagamine [85, 86]. Both molecules are brain-penetrant and orally active, however further study must be done to assess their ability to improve brain GCase activity and to lower alpha-synuclein levels in humans. More recently, an additional novel compound NCGC607 has been shown to be effective in lowering alpha-synuclein and glycolipid Levels in iPSC-derived dopaminergic neurons from patients with GD and PD [87].
A number of investigations into the use of gene therapy to augment GCase levels in GBA-mutation carrying models and models of PD, through adeno-associated virus-mediated (AAV-mediated) expression of GCase in the CNS via cerebral injections has found hugely beneficial effects [88, 89], which serves as an excellent proof of concept for the use of GCase activity augmentation in the reduction of alpha-synuclein accumulation and transmission [89, 90], and neurodegeneration in PD models [88], which will be an avenue to pursue further with a view to translation to human PD patients.
CONCLUSION
Mutations of the GBA gene are the most significant risk factor for PD, and are the most common genetic mutation so far identified in this disease. The molecular mechanisms by which GBA mutations result in this increased risk are the focus of a significant research effort. It appears that there is a reciprocal relationship between GCase and alpha-synuclein that is common to individuals without as well as those with mutations. This is important as it suggests that manipulation of the GCase-synuclein interaction may be of benefit to all PD patients, as well as potentially of benefit to others with synucleinopathies. There are several methods by which the interaction may be influenced to reduce alpha-synuclein levels. Thus, the GBA-lysosomal-synuclein axis is now the focus of experimental therapeutics with the development of novel drugs to modulate GCAse activity and some of these are currently entering clinical trial.
CONFLICT OF INTEREST STATEMENT
The authors have no conflict of interest to report.
REFERENCES
[1] | Chaudhuri KR , Healy DG , & Schapira AHV ((2006) ) Non-motor symptoms of Parkinson’s disease: Diagnosis and management. Lancet Neurol, 5: , 235–245. |
[2] | Schapira AHV , Chaudhuri KR , & Jenner P ((2017) ) Non-motor features in Parkinson’s disease. Nat Rev Neurosci, in press. |
[3] | Tayebi N , Callahan M , Madike V , Stubblefield BK , Orvisky E , Krasnewich D , Fillano JJ , & Sidransky E ((2001) ) Gaucher disease and parkinsonism: A phenotypic and genotypic characterization. Mol Genet Metab, 73: , 313–321. |
[4] | de Carvalho Guimarães B , Valente Pereira AC , da Costa Rodrigues F , Vaz dos Santos A , Mendonça dos Santos MCJJ , Lima dos Santos F , Zuma de Rosso AL , Nicaretta DH , Pereira JS , José da Silva D , Della Coletta MV , Santos-Rebouças CB , & Gonçalves Pimentel MM ((2012) ) Glucocerebrosidase N370S and L444P mutations as risk factors for Parkinson’s disease in Brazilian patients. Parkinsonism Relat Disord, 18: , 688–689. |
[5] | Wang Y , Liu L , Xiong J , Zhang X , Chen Z , Yu L , Chen C , Huang J , Zhang Z , Mohmed A , Lin Z , Xiong N , & Wang T ((2012) ) Glucocerebrosidase L444P mutation confers genetic risk for Parkinson’s disease in central China. Behav Brain Funct, 8: , 57. |
[6] | Sidransky E , & Lopez G ((2012) ) The link between the GBA gene and parkinsonism. Lancet Neurol, 11: , 986–998. |
[7] | Anheim M , Elbaz A , Lesage S , Durr A , Condroyer C , Viallet F , Pollak P , Bonaïti B , Bonaïti-Pellié C , & Brice A ((2012) ) Penetrance of Parkinson’s disease in glucocerebrosidase gene mutation carriers. Neurology, 78: , 417–420. |
[8] | Rana HQ , Balwani M , Bier L , & Alcalay RN ((2013) ) Age-specific Parkinson’s disease risk in GBA mutation carriers: Information for genetic counseling. Genet Med, 15: , 146–149. |
[9] | Zimran A , Altarescu G , & Elstein D ((2013) ) Pilot study using ambroxol as a pharmacological chaperone in type 1 Gaucher disease. Blood Cells Mol Dis, 50: , 134–137. |
[10] | Alcalay RN , Caccappolo E , Mejia-Santana H , Tang MX , Rosado L , Orbe Reilly M , Ruiz D , Ross B , Verbitsky M , Kisselev S , Louis E , Comella C , Colcher A , Jennings D , Nance M , Bressman S , Scott WK , Tanner C , Mickel S , Andrews H , Waters C , Fahn S , Cote L , Frucht S , Ford B , Rezak M , Novak K , Friedman JH , Pfeiffer R , Marsh L , Hiner B , Siderowf A , Payami H , Molho E , Factor S , Ottman R , Clark LN , & Marder K ((2012) ) Cognitive performance of GBA mutation carriers with early-onset PD: The CORE-PD study. Neurology, 78: , 1434–1440. |
[11] | Neumann J , Bras J , Deas E , O’sullivan SS , Parkkinen L , Lachmann RH , Li A , Holton J , Guerreiro R , Paudel R , Segarane B , Singleton A , Lees A , Hardy J , Houlden H , Revesz T , & Wood NW ((2009) ) Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain, 132: , 1783–1794. |
[12] | Schapira AHV ((2015) ) Glucocerebrosidase and Parkinson’s disease: Recent advances. Mol Cell Neurosci, 66: , 37–42. |
[13] | Nichols WC , Pankratz N , Marek DK , Pauciulo MW , Elsaesser VE , Halter CA , Rudolph A , Wojcieszek J , Pfeiffer RF , Foroud T , & Parkinson Study Group-PROGENI, Investigators ((2009) ) Mutations in GBA are associated with familial Parkinson’s disease susceptibility and age at onset. Neurology, 72: , 310–316. |
[14] | Alcalay RN , Caccappolo E , Mejia-Santana H , Tang MX , Rosado L , Ross BM , Verbitsky M , Kisselev S , Louis ED , Comella C , Colcher A , Jennings D , Nance MA , Bressman SB , Scott WK , Tanner C , Mickel S , Andrews H , Waters C , Fahn S , Cote L , Frucht S , Ford B , Rezak M , Novak K , Friedman JH , Pfeiffer R , Marsh L , Hiner B , Siderowf A , Ottman R , Marder K , & Clark LN ((2010) ) Arch. Neurol, 67: , 1116–1122. |
[15] | McNeill A , Wu RM , Tzen KY , Aguiar PC , Arbelo JM , Barone P , Bhatia K , Barsottini O , Bonifati V , Bostantjopoulou S , Bressan R , Cossu G , Cortelli P , Felicio A , Ferraz HB , Herrera J , Houlden H , Hoexter M , Isla C , Lees A , Lorenzo-Betancor O , Mencacci NE , Pastor P , Pappata S , Pellecchia MT , Silveria-Moriyama L , Varrone A , Foltynie T , & Schapira AH ((2013) ) Dopaminergic neuronal imaging in genetic Parkinson’s disease: Insights into pathogenesis. PLoS One, 8: , e69190. |
[16] | Sardi SP , Cheng SH , & Shihabuddin LS ((2015) ) Gaucher-related synucleinopathies: The examination of sporadic neurodegeneration from a rare (disease) angle. Prog Neurobiol, 125: , 47–62. |
[17] | Alcalay RN , Levy OA , Waters CC , Fahn S , Ford B , Kuo SH , Mazzoni P , Pauciulo MW , Nichols WC , Gan-Or Z , Rouleau GA , Chung WK , Wolf P , Oliva P , Keutzer J , Marder K , & Zhang X ((2015) ) Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain, 138: , 2648–2658. |
[18] | Schapira AHV , & Gegg ME ((2013) ) Glucocerebrosidase in the pathogenesis and treatment of Parkinson’s disease. Proc Natl Acad Sci U S A, 110: , 3214–3215. |
[19] | Hruska KS , LaMarca ME , Scott CR , & Sidransky E ((2008) ) Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat, 29: , 567–583. |
[20] | Horowitz M , Pasmanik-Chor M , Ron I , & Kolodny EH ((2011) ) The enigma of the E326K mutation in acid β-glucocerebrosidase. Mol Genet Metab, 104: , 35–38. |
[21] | Irfan Maqsood M , Matin MM , Bahrami AR , & Ghasroldasht MM ((2013) ) Immortality of cell lines: Challenges and advantages of establishment. Cell Biol Int, 37: , 1038–1045. |
[22] | Kaur G , & Dufour JM ((2012) ) Cell lines: Valuable tools or useless artifacts. Spermatogenesis, 2: , 1–5. |
[23] | Magalhaes J , Gegg ME , Migdalska-Richards A , Doherty MK , Whitfield PD , & Schapira AHV ((2016) ) Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: Relevance to Parkinson’s disease. Hum Mol Genet, 25: , 3432–3445. |
[24] | Kanfer JN , Legler G , Sullivan J , Raghavan SS , & Mumford RA ((1975) ) The Gaucher mouse. Biochem Biophys Res Commun, 67: , 85–90. |
[25] | Manning-Boğ AB , Schüle B , & Langston JW ((2009) ) Alpha-synuclein-glucocerebrosidase interactions in pharmacological Gaucher models: A biological link between Gaucher disease and parkinsonism. Neurotoxicology, 30: , 1127–1132. |
[26] | McNeill A , Magalhaes J , Shen C , Chau KY , Hughes D , Mehta A , Foltynie T , Cooper JM , Abramov AY , Gegg M , & Schapira AHV ((2014) ) Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson’s disease cells. Brain, 137: , 1481–1495. |
[27] | Kiskinis E , & Eggan K ((2010) ) Progress toward the clinical application of patient-specific pluripotent stem cells. J Clin Invest, 120: , 51–59. |
[28] | Schöndorf DC , Aureli M , McAllister FE , Hindley CJ , Mayer F , Schmid B , Sardi SP , Valsecchi M , Hoffmann S , Schwarz LK , Hedrich U , Berg D , Shihabuddin LS , Hu J , Pruszak J , Gygi SP , Sonnino S , Gasser T , & Deleidi M ((2014) ) iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun, 5: , 4028. |
[29] | Woodard CM , Campos BA , Kuo SH , Nirenberg MJ , Nestor MW , Zimmer M , Mosharov EV , Sulzer D , Zhou H , Paull D , Clark L , Schadt EE , Sardi SP , Rubin L , Eggan K , Brock M , Lipnick S , Rao M , Chang S , Li A , & Noggle SA ((2014) ) iPSC-derived dopamine neurons reveal differences between monozygotic twins discordant for Parkinson’s disease. Cell Rep, 9: , 1173–1182. |
[30] | Momcilovic O , Sivapatham R , Oron TR , Meyer M , Mooney S , Rao MS , & Zeng X ((2016) ) Derivation, characterization, and neural differentiation of integration-free induced pluripotent stem cell lines from Parkinson’s disease patients carrying SNCA, LRRK2, PARK2, and GBA mutations. PLoS One, 11: , e0154890. |
[31] | Holleran WM , Ginns EI , Menon GK , Grundmann JU , Fartasch M , McKinney CE , Elias PM , & Sidransky E ((1994) ) Consequences of β-glucocerebrosidase deficiency in epidermis: Ultrastructure and permeability barrier alterations in Gaucher disease. J Clin Invest, 93: , 1756–1764. |
[32] | Enquist IB , Lo Bianco C , Ooka A , Nilsson E , Månsson JE , Ehinger M , Richter J , Brady RO , Kirik D , & Karlsson S ((2007) ) Murine models of acute neuronopathic Gaucher disease. Proc Natl Acad Sci U S A, 104: , 17483–17488. |
[33] | Osellame LD , Rahim AA , Hargreaves IP , Gegg ME , Richard-Londt A , Brandner S , Waddington SN , Schapira AHV , & Duchen MR (Metab) ((2013) ) Mitochondria and quality control defects in a mouse model of Gaucher disease—links to Parkinson’s disease. Cell, 17: , 941–953. |
[34] | Xu YH , Quinn B , Witte D , & Grabowski GA ((2003) ) Viable mouse models of acid beta-glucosidase deficiency: The defect in Gaucher disease. Am J Pathol, 163: , 2093–2101. |
[35] | Sanders A , Hemmelgarn H , Melrose HL , Hein L , Fuller M , & Clarke LA ((2013) ) Transgenic mice expressing human glucocerebrosidase variants: Utility for the study of Gaucher disease. Blood Cells Mol Dis, 51: , 109–115. |
[36] | Fishbein I , Kuo YM , Giasson BI , & Nussbaum RL ((2014) ) Augmentation of phenotype in a transgenic Parkinson mouse heterozygous for a Gaucher mutation. Brain J Neurol, 137: , 3235–3247. |
[37] | Rockenstein E , Clarke J , Viel C , Panarello N , Treleaven CM , Kim C , Spencer B , Adame A , Park H , Dodge JC , Cheng SH , Shihabuddin LS , Masliah E , & Sardi SP ((2016) ) Glucocerebrosidase modulates cognitive and motor activities in murine models of Parkinson’s disease. Hum Mol Genet, 25: , 2645–2660. |
[38] | Migdalska-Richards A , Daly L , Bezard E , & Schapira AHV ((2016) ) Ambroxol effects in glucocerebrosidase and α-synuclein transgenic mice. Ann Neurol, 80: , 766–775. |
[39] | Davis M , Trinh K , Thomas R , Whittley B , Montine T , & Pallanck L ((2015) ) Using a Drosophila GBA deficiency model to understand the role of GBA in Parkinson’s disease. Neurology, 84: , P2.146. |
[40] | Sanchez-Martinez A , Beavan M , Gegg ME , Chau KY , Whitworth AJ , & Schapira AHV ((2016) ) Parkinson’s disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci Rep, 6: , 31380. |
[41] | Keatinge M , Bui H , Menke A , Chen YC , Sokol AM , Bai Q , Ellett F , Da Costa M , Burke D , Gegg M , Trollope L , Payne T , McTighe A , Mortiboys H , de Jager S , Nuthall H , Kuo MS , Fleming A , Schapira AHV , Renshaw SA , Highley JR , Chacinska A , Panula P , Burton EA , O’Neill MJ , & Bandmann O ((2015) ) Glucocerebrosidase 1 deficient Danio rerio mirror key pathological aspects of human Gaucher disease and provide evidence of early microglial activation preceding alpha-synuclein-independent neuronal cell death. Hum Mol Genet, 24: , 6640–6652. |
[42] | Gegg ME , Sweet L , Wang BH , Shihabuddin LS , Sardi SP , & Schapira AHV ((2015) ) No evidence for substrate accumulation in Parkinson brains with GBA mutations. Mov Disord, 30: , 1085–1089. |
[43] | Bendikov-Bar I , Ron I , Filocamo M , & Horowitz M ((2011) ) Characterization of the ERAD process of the L444P mutant glucocerebrosidase variant. Blood Cells Mol Dis, 46: , 4–10. |
[44] | Mu TW , Fowler DM , & Kelly JW ((2008) ) Partial restoration of mutant enzyme homeostasis in three distinct lysosomal storage disease cell lines by altering calcium homeostasis. PLoS Biol, 6: , e26. |
[45] | Ron I , & Horowitz M ((2005) ) ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum Mol Genet, 14: , 2387–2398. |
[46] | Fernandes HJR , Hartfield EM , Christian HC , Emmanoulidou E , Zheng Y , Booth H , Bogetofte H , Lang C , Ryan BJ , Sardi SP , Badger J , Vowles J , Evetts S , Tofaris GK , Vekrellis K , Talbot K , Hu MT , James W , Cowley SA , & Wade-Martins R ((2016) ) ER stress and autophagic perturbations lead to elevated extracellular alpha-synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Rep, 6: , 342–356. |
[47] | Gan-Or Z , Bar-Shira A , Mirelman A , Gurevich T , Kedmi M , Giladi N , & Orr-Urtreger A ((2010) ) LRRK2 and GBA mutations differentially affect the initial presentation of Parkinson’s disease. Neurogenetics, 11: , 121–125. |
[48] | Lesage S , Anheim M , Condroyer C , Pollak P , Durif F , Dupuits C , Viallet F , Lohmann E , Corvol JC , Honoré A , Rivaud S , Vidailhet M , Dürr A , Brice A , & French Parkinson’s Disease Genetics Study Group ((2011) ) Large-scale screening of the Gaucher’s disease-relatedglucocerebrosidase gene in Europeans with Parkinson’s disease. Hum Mol Genet, 20: , 202–210. |
[49] | Zhang Y , Sun QY , Zhao YW , Shu L , Guo JF , Xu Q , Yan XX , & Tang BS ((2015) ) Effect of GBA mutations on phenotype of Parkinson’s disease: A study on Chinese population and a meta-analysis. Parkinsons Dis, 2015: , 916971. |
[50] | Gan-Or Z , Amshalom I , Kilarski LL , Bar-Shira A , Gana-Weisz M , Mirelman A , Marder K , Bressman S , Giladi N , & Orr-Urtreger A ((2015) ) Differential effects of severe vs mild GBA mutations on Parkinson’s disease. Neurology, 84: , 880–887. |
[51] | Brockmann K , Srulijes K , Pflederer S , Hauser AK , Schulte C , Maetzler W , Gasser T , & Berg D ((2014) ) GBA-associated Parkinson’s disease: Reduced survival and more rapid progression in a prospective longitudinal study: GBA status and PD disease progression. Mov Disord, 30: , 407–411. |
[52] | Winder-Rhodes SE , Evans JR , Ban M , Mason SL , Williams-Gray CH , Foltynie T , Duran R , Mencacci NE , Sawcer SJ , & Barker RA ((2013) ) Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain, 136: (Pt 2), 392–399. |
[53] | Davis MY , Johnson CO , Leverenz JB , Weintraub D , Trojanowski JQ , Chen-Plotkin A , Van Deerlin VM , Quinn JF , Chung KA , Peterson-Hiller AL , Rosenthal LS , Dawson TM , Albert MS , Goldman JG , Stebbins GT , Bernard B , Wszolek ZK , Ross OA , Dickson DW , Eidelberg D , Mattis PJ , Niethammer M , Yearout D , Hu SC , Cholerton BA , Smith M , Mata IF , Montine TJ , Edwards KL , & Zabetian CP ((2016) ) Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson’s disease. JAMA Neurol, 73: , 1217–1224. |
[54] | Oeda T , Umemura A , Mori Y , Tomita S , Kohsaka M , Park K , Inoue K , Fujimura H , Hasegawa H , Sugiyama H , & Sawada H ((2015) ) Impact of glucocerebrosidase mutations on motor and nonmotor complications in Parkinson’s disease. Neurobiol Aging, 36: , 3306–3313. |
[55] | Brockmann K , Srulijes K , Hauser AK , Schulte C , Csoti I , Gasser T , & Berg D ((2011) ) GBA-associated PD presents with nonmotor characteristics. Neurology, 77: , 276–280. |
[56] | Weil RS , Schrag AE , Warren JD , Crutch SJ , Lees AJ , & Morris HR ((2016) ) Visual dysfunction in Parkinson’s disease. Brain, doi: 10.1093/brain/aww175 |
[57] | Beavan M , McNeill A , Proukakis C , Hughes DA , Mehta A , & Schapira AH ((2015) ) Evolution of prodromal clinical markers of Parkinson’s disease in a GBA mutation–positive cohort. JAMA Neurol, 72: , 201. |
[58] | McNeill A , Duran R , Proukakis C , Bras J , Hughes D , Mehta A , Hardy J , Wood NW , & Schapira AH ((2012) ) Hyposmia and cognitive impairment in Gaucher disease patients and carriers: Hyposmia in Gaucher disease. Mov Disord, 27: , 526–532. |
[59] | Braak H , Del Tredici K , Rüb U , de Vos RA , Jansen Steur EN , & Braak E ((2003) ) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging, 24: , 197–211. |
[60] | Pellegrini C , Antonioli L , Colucci R , Ballabeni V , Barocelli E , Bernardini N , Blandizzi C , & Fornai M ((2015) ) Gastric motor dysfunctions in Parkinson’s disease: Current pre-clinical evidence, Parkinsonism Relat. Disord, 21: , 1407–1414. |
[61] | McNeill A , Duran R , Hughes DA , Mehta A , & Schapira AHV ((2012) ) A clinical and family history study of Parkinson’s disease in heterozygous glucocerebrosidase mutation carriers. J Neurol Neurosurg Psychiatry, 83: , 853–854. |
[62] | Wang C , Cai Y , Gu Z , Ma J , Zheng Z , Tang BS , Xu Y , Zhou Y , Feng T , Wang T , Chen SD , & Chan P ((2013) ) Clinical profiles of Parkinson’s disease associated with common leucine-rich repeat kinase 2 and glucocerebrosidase genetic variants in Chinese individuals. Neurobiol Aging, 35: , 725.e1–725.e6. |
[63] | Cilia R , Tunesi S , Marotta G , Cereda E , Siri PsyDC , Tesei S , Zecchinelli A , Canesi M , Mariani CB , Meucci N , Sacilotto G , Zini M , Barichella M , Magnani C , Duga S , Asselta R , Soldá G , Seresini A , Seia M , Pezzoli G , & Goldwurm S ((2016) ) Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann Neurol, 80: , 662–673. |
[64] | Sidransky E , Nalls MA , Aasly JO , Aharon-Peretz J , Annesi G , Barbosa ER , Bar-Shira A , Berg D , Bras J , Brice A , Chen CM , Clark LN , Condroyer C , De Marco EV , Dürr A , Eblan MJ , Fahn S , Farrer MJ , Fung HC , Gan-Or Z , Gasser T , Gershoni-Baruch R , Giladi N , Griffith A , Gurevich T , Januario C , Kropp P , Lang AE , Lee-Chen GJ , Lesage S , Marder K , Mata IF , Mirelman A , Mitsui J , Mizuta I , Nicoletti G , Oliveira C , Ottman R , Orr-Urtreger A , Pereira LV , Quattrone A , Rogaeva E , Rolfs A , Rosenbaum H , Rozenberg R , Samii A , Samaddar T , Schulte C , Sharma M , Singleton A , Spitz M , Tan EK , Tayebi N , Toda T , Troiano AR , Tsuji S , Wittstock M , Wolfsberg TG , Wu YR , Zabetian CP , Zhao Y , & Ziegler SG ((2009) ) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med, 361: , 1651–1661. |
[65] | Li Y , Sekine T , Funayama M , Li L , Yoshino H , Nishioka K , Tomiyama H , & Hattori N ((2014) ) Clinicogenetic study of GBA mutations in patients with familial Parkinson’s disease. Neurobiol Aging, 35: , 935.e3–935.e8. |
[66] | Crosiers D , Verstraeten A , Wauters E , Engelborghs S , Peeters K , Mattheijssens M , De Deyn PP , Theuns J , Broeckhoven C , & Cras P ((2016) ) Mutations in glucocerebrosidase are a major genetic risk factor for Parkinson’s disease and increase susceptibility to dementia in a Flanders-Belgian cohort, Neurosci. Lett, 629: , 160–164. |
[67] | Nalls MA , Duran R , Lopez G , Kurzawa-Akanbi M , McKeith IG , Chinnery PF , Morris CM , Theuns J , Crosiers D , Cras P , Engelborghs S , De Deyn PP , Van Broeckhoven C , Mann DM , Snowden J , Pickering-Brown S , Halliwell N , Davidson Y , Gibbons L , Harris J , Sheerin UM , Bras J , Hardy J , Clark L , Marder K , Honig LS , Berg D , Maetzler W , Brockmann K , Gasser T , Novellino F , Quattrone A , Annesi G , De Marco EV , Rogaeva E , Masellis M , Black SE , Bilbao JM , Foroud T , Ghetti B , Nichols WC , Pankratz N , Halliday G , Lesage S , Klebe S , Durr A , Duyckaerts C , Brice A , Giasson BI , Trojanowski JQ , Hurtig HI , Tayebi N , Landazabal C , Knight MA , Keller M , Singleton AB , Wolfsberg TG , & Sidransky E ((2013) ) A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol, 70: , 727. |
[68] | Malec-Litwinowicz M , Rudzińska M , Szubiga M , Michalski M , Tomaszewski T , & Szczudlik A ((2014) ) Cognitive impairment in carriers of glucocerebrosidase gene mutation in Parkinson’s disease patients. Neurol Neurochir Pol, 48: , 258–261. |
[69] | Setó-Salvia N , Pagonabarraga J , Houlden H , Pascual-Sedano B , Dols-Icardo O , Tucci A , Paisán-Ruiz C , Campolongo A , Antón-Aguirre S , Martín I , Muñoz L , Bufill E , Vilageliu L , Grinberg D , Cozar M , Blesa R , Lleó A , Hardy J , Kulisevsky J , & Clarimón J ((2012) ) Glucocerebrosidase mutations confer a greater risk of dementia during Parkinson’s disease course. Mov Disord, 27: , 393–399. |
[70] | Chahine LM , Qiang J , Ashbridge E , Minger J , Yearout D , Horn S , Colcher A , Hurtig HI , Lee VMY , Van Deerlin VM , Leverenz JB , Siderowf AD , Trojanowski JQ , Zabetian CP , & Chen-Plotkin A ((2013) ) Clinical and biochemical differences in patients hng Parkinson’s disease with vs without GBA mutations. JAMA Neurol, 70: , 852–avi. |
[71] | Clark LN , Ross BM , Wang Y , Mejia-Santana H , Harris J , Louis ED , Cote LJ , Andrews H , Fahn S , Waters C , Ford B , Frucht S , Ottman R , & Marder K ((2007) ) Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson’s disease. Neurology, 69: , 1270–1277. |
[72] | Aharon-Peretz J , Badarny S , Rosenbaum H , & Gershoni-Baruch R ((2005) ) Mutations in the glucocerebrosidase gene and Parkinson’s disease: Phenotype-genotype correlation. Neurology, 65: , 1460–1461. |
[73] | Barrett MJ , Shanker VL , Severt WL , Raymond D , Gross SJ , Schreiber-Agus N , Kornreich R , Ozelius LJ , Bressman SB , & Saunders-Pullman R ((2014) ) Cognitive and antipsychotic medication use in monoallelic GBA-related Parkinson’s disease. JIMD Rep, 16: , 31–38. |
[74] | Alcalay RN , Mejia-Santana H , Tang MX , Rakitin B , Rosado L , Ross B , Verbitsky M , Kisselev S , Louis ED , Comella CL , Colcher A , Jennings D , Nance MA , Bressman S , Scott WK , Tanner C , Mickel SF , Andrews HF , Waters CH , Fahn S , Cote LJ , Frucht SJ , Ford B , Rezak M , Novak K , Friedman JH , Pfeiffer R , Marsh L , Hiner B , Siderowf A , Ottman R , Clark LN , Marder KS , & Caccappolo E ((2010) ) Self-report of cognitive impairment and mini-mental state examination performance in PRKN, LRRK2, and GBA carriers with early onset Parkinson’s disease. J Clin Exp Neuropsychol, 32: , 775–779. |
[75] | Mata IF , Leverenz JB , Weintraub D , Trojanowski JQ , Chen-Plotkin A , Van Deerlin VM , Ritz B , Rausch R , Factor SA , Wood-Siverio C , Quinn JF , Chung KA , Peterson-Hiller AL , Goldman JG , Stebbins GT , B. Bernard B , Espay AJ , Revilla FJ , Devoto J , Rosenthal LS , Dawson TM , Albert MS , Tsuang D , Huston H , Yearout D , Hu SC , Cholerton BA , Montine TJ , Edwards KL , & Zabetian CP ((2015) ) GBA Variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Mov Disord, 31: , 95–102. |
[76] | Zokaei N , McNeill A , Proukakis C , Beavan M , Jarman P , Korlipara P , Hughes D , Mehta A , Hu MTM , Schapira AHV , & Husain M ((2014) ) Visual short-term memory deficits associated with GBA mutation and Parkinson’s disease. Brain, 137: , 2303–2311. |
[77] | Goker-Alpan O , Masdeu JC , Kohn PD , Ianni A , Lopez G , Groden C , Chapman MC , Cropp B , Eisenberg DP , Maniwang ED , Davis J , Wiggs E , Sidransky E , & Berman KF ((2012) ) The neurobiology of glucocerebrosidase-associated parkinsonism: A positron emission tomography study of dopamine synthesis and regional cerebral blood flow. Brain, 135: , 2440–2448. |
[78] | Kresojević N , Janković M , Petrović I , Kumar KR , Dragašević N , Dobričić V , Novaković I , Svetel M , Klein C , Pekmezović T , & Kostić VS ((2015) ) Presenting symptoms of GBA-related Parkinson’s disease. Parkinsonism Relat Disord, 21: , 804–807. |
[79] | Gan-Or Z , Giladi N , Rozovski U , Shifrin C , Rosner S , Gurevich T , Bar-Shira T , & Orr-Urtreger A ((2008) ) Genotype-phenotype correlations between GBA mutations and Parkinson’s disease risk and onset. Neurology, 70: , 2277–2283. |
[80] | Gatto EM , Etcheverry JL , Sanguinetti A , Cesarini M , Fernandez Escobar N , & Drelichman G ((2016) ) Prodromal clinical markers of Parkinson’s disease in Gaucher disease individuals. Eur Neurol, 76: , 19–21. |
[81] | Noyce AJ , Mencacci NE , Schrag A , Bestwick JP , Giovannoni G , Lees AJ , & Hardy J ((2015) ) Web-based assessment of Parkinson’s prodromal markers identifies GBA variants. Mov Disord, 30: , 1002–1003. |
[82] | Lynch-day MA , Mao K , Wang K , Zhao M , & Klionsky DJ ((2012) ) The role of autophagy in Parkinson’s disease. Cold Spring Harb Perspect Med, 2: , a009357. |
[83] | Olanow CW , & Schapira AHV ((2013) ) Therapeutic prospects for Parkinson’s disease. Ann Neurol, 74: , 337–347. |
[84] | Ambrosi G , Ghezzi C , Zangaglia R , Levandis G , Pacchetti C , & Blandini F ((2015) ) Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol Dis, 82: , 235–242. |
[85] | Yang SY , Beavan M , Chau KY , Taanman JW , & Schapira AHV ((2017) ) A human neural crest stem cell-derived dopaminergic neuronal model recapitulates biochemical abnormalities in GBA1 mutation carriers. Stem Cell Rep, 8: , 728–742. |
[86] | Migdalska-Richards A , Ko WK , Li Q , Bezard E , & Schapira AHV ((2017) ) Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate. Synapse, doi: 10.1002/syn.21967 |
[87] | Sun Y , Ran H , Liou B , Quinn B , Zamzow M , Zhang W , Bielawski J , Kitatani K , Setchell KDR , Hannun YA , & Grabowski GA ((2011) ) Isofagomine in vivo effects in a neuronopathic gaucher disease mouse. PLoS One, 6: , e19037. |
[88] | Sun Y , Liou B , Xu YH , Quinn B , Zhang W , Hamler R , Setchell KDR , & Grabowski GA ((2012) ) Ex vivo and in vivo effects of isofagomine on acid β-glucosidase variants and substrate levels in Gaucher disease. J Biol Chem, 287: , 4275–4287. |
[89] | Aflaki E , Borger DK , Moaven N , Stubblefield BK , Rogers SA , Patnaik S , Schoenen FJ , Westbroek W , Zheng W , Sullivan P , Fujiwara H , Sidhu R , Khaliq ZM , Lopez GJ , Goldstein DS , Ory DS , Marugan J , & Sidransky E ((2016) ) A new glucocerebrosidase chaperone reduces α-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with Gaucher disease and parkinsonism. J Neurosci, 36: , 7441–7452. |
[90] | Rocha EM , Smith GA , Park E , Cao H , Brown E , Hayes MA , Beagan J , McLean JR , Izen SC , Perez-Torres E , Hallett PJ , & Isacson O ((2015) ) Glucocerebrosidase gene therapy prevents α-synucleinopathy of midbrain dopamine neurons. Neurobiol Dis, 82: , 495–503. |


