
Phase IIb Study of Intranasal Glutathione in Parkinson’s Disease
Abstract
Background: Reduced glutathione (GSH) is an endogenously synthesized tripeptide depleted early in the course of Parkinson’s disease (PD) and GSH augmentation has been proposed as a therapeutic strategy in PD.
Objective: This Phase IIb study was designed to evaluate whether a Phase III study of intranasal GSH, (in)GSH, for symptomatic relief is warranted and to determine the most appropriate trial design for a disease-modification study.
Methods: This was a double-blind, placebo-controlled trial of 45 individuals with Hoehn & Yahr Stage 1–3 PD. Participants were randomized to receive intranasal placebo (saline), 100 mg GSH, or 200 mg GSH thrice daily for three months, and were observed over a one-month washout period.
Results: All cohorts improved over the intervention period, including placebo. The high-dose group demonstrated improvement in total Unified PD Rating Scale (UPDRS) (–4.6 (4.7), P = 0.0025) and UPDRS motor subscore (–2.2 (3.8), P = 0.0485) over baseline, although neither treatment group was superior to placebo. One participant in the high-dose GSH cohort developed cardiomyopathy.
Conclusions: Although predicted improvements in PD total and motor scores were observed, these data do not suggest (in)GSH is superior to placebo after a three month intervention. The symptomatic effects are sufficient to warrant a delayed-start or wash-out design study for disease-modification trials. Whether long-term use of (in)GSH leads to clinical improvements that are sustained and significantly different than placebo will require appropriately-powered longer-duration studies in larger cohorts. The improvement in the placebo arm was more robust than has been observed in previous PD studies and warrants further investigation.
INTRODUCTION
Reduced glutathione (GSH, γ-L-glutamyl-L-cysteinylglycine) is a tripeptide that is involved in scavenging of reactive oxygen species (ROS), reduction of hydrogen peroxide, cellular detoxification, and serves as a reservoir for cysteine, glycine, and glutamate. GSH deficiency is thought to interfere with a cell’s ability to clear cellular waste, and to impair defense against reactive oxygen species (ROS), reactive nitrogen species (RNS), and H2O2.
Post mortem analysis of substantia nigra (SN) tissue from individuals with PD vs. controls reveals that GSH is depleted early in the course of the disease [1–3]. A recent cross-sectional study demonstrated a correlation between whole blood glutathione concentrations and PD clinical severity [4].
GSH depletion does not, on its own, result in SN cell loss in rodent models of GSH depletion [5]. However, under conditions of normal aging or when neurons are exposed to the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), depletion of GSH exacerbates SN degeneration [6]. This, combined with evidence for lowered GSH levels in numerous other central nervous system (CNS) diseases [7, 8] suggests that although GSH depletion is not likely the initial step in development of PD, it may occur early in the disease process secondary to an initial environmental or metabolic insult.
Oral GSH is often reported to have low bioavailability [9] and the high GSH requirement by the intestinal mucosal cells and liver decreases the likelihood that GSH delivered by oral bolus will reach CNS target tissue [10]. Intravenous (iv) GSH, which avoids first-pass loss in the intestines and liver, has been administered to individuals with PD in two clinical trials, both of which reported improvements in motor symptoms, although neither was powered for efficacy [11, 12]. In spite of these positive outcomes, the utility of (iv)GSH is limited by cost, inconvenience, and side effects associated with intravenous administration, thus, alternative methods of GSH augmentation are being explored. Nasal delivery, which is non-invasive, relatively convenient, and can result in preferential delivery of certain drugs to the brain, has been proposed as a potentially useful approach for delivering GSH to the brain [13].
Intranasal GSH, (in)GSH, has been in clinical use, without FDA approval, for over 18 years [14]. A phase I/IIa study of (in)GSH in PD demonstrated that the intervention was safe and tolerable over a three-month intervention period [15]. The study was not designed to draw statistical conclusions regarding efficacy, although an improvement in total UPDRS and motor UPDRS scores in active arms was observed over placebo, with symptom worsening occurring during the one month observed washout period. (Fig. 1; Data were replotted from reference 12).
Fig.1
Results demonstrating improvement in clinical measures of PD severity from the Phase I/IIa Study of (in) GSH in PD. The goal of this Phase IIb study was to determine whether these results were reproducible. All study medications were given thrice daily, e.g. 100 mg/ml is equal to 300 mg/ day. Data were re-plotted from [15].
![Results demonstrating improvement in clinical measures of PD severity from the Phase I/IIa Study of (in) GSH in PD. The goal of this Phase IIb study was to determine whether these results were reproducible. All study medications were given thrice daily, e.g. 100 mg/ml is equal to 300 mg/ day. Data were re-plotted from [15].](https://content.iospress.com:443/media/jpd/2017/7-2/jpd-7-2-jpd161040/jpd-7-jpd161040-g001.jpg)
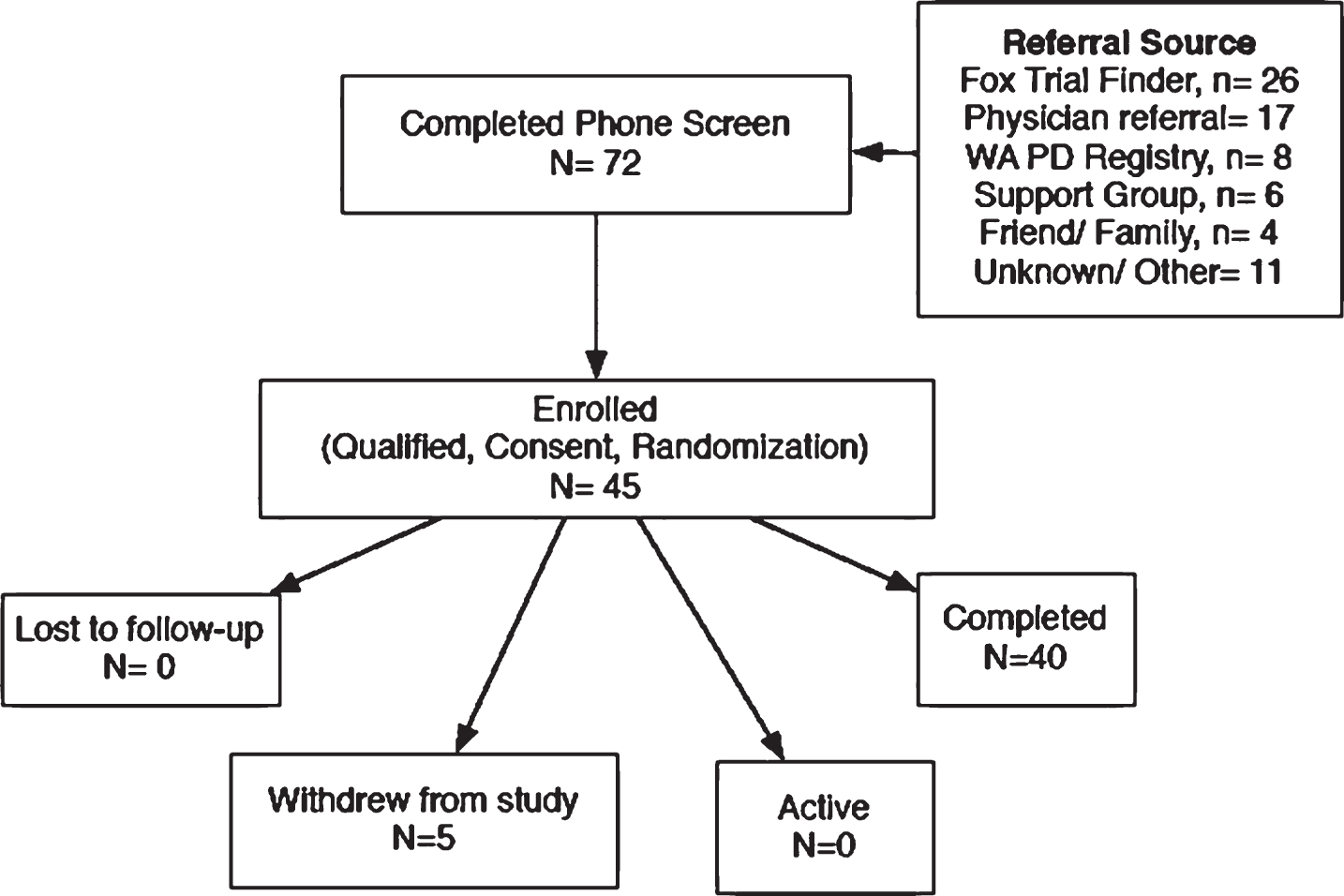
Fig.2
CONSORT flow diagram.
In order to evaluate whether (in)GSH penetrates into brain, magnetic resonance spectroscopy (MRS) was employed to quantify CNS concentrations of glutathione following 200 mg (in)GSH. Over the course of one hour after (in)GSH administration to individuals in a supine position inside the MRI instrument, mean GSH levels increased more than 200% in a region of brain that included the putamen [16]. The demonstration of target validation and the capacity to use MRS as an outcome measure both provided support for further evaluation of (in)GSH as a therapeutic agent in PD.
All three clinical trials of exogenously-administered GSH [11, 12, 15] (as well as anecdotal video case reports posted to social media) indicate that GSH has the potential to improve motor symptoms in PD. Given that GSH acts primarily as a free radical scavenger and to aid cellular detoxification, a commonly hypothesized mechanism of action for GSH in PD is that GSH may delay PD progression by reducing the rate of neurodegeneration. However, since this proposed mechanism would not account for the observed improvement in motor outcomes, mechanisms by which GSH may alter motor function by affecting dopaminergic neurotransmission have also been explored. GSH is a neuromodulator at glutamate NMDA receptors [17], however, it is not known to directly interact with dopamine receptors. Chronic high doses of (iv)GSH have been reported to modestly increase dopamine transporter density (24%), as measured by 123-IFP-CIT single-photon emission computed tomography, in the putamen of individuals with PD [18, 19]. Recently, N-acetyl cysteine (NAC), a GSH precursor, has also been shown to increase dopamine transporter (DAT) density (by 4–8%) in the caudate and putamen of patients with PD, with corresponding improvements in clinical outcomes [20]. While NAC has been shown to raise GSH concentrations, it is also possible the impact on DAT density occurs via a mechanism other than GSH augmentation. While the mechanisms may be poorly-understood, these data provide some insight into the means by which GSH could impact motor outcomes in PD.
This Phase IIb study was designed to evaluate whether a Phase III study of (in)GSH for PD symptomatic relief is warranted (preliminary efficacy), and to determine the most appropriate trial design for disease-modification studies [21]. The primary aim of this study was to determine whether the UPDRS total and motor improvements seen in the Phase I/IIa trial were reproducible. The secondary aim of this study was to evaluate biomarkers and clinical outcome measures (in addition to UPDRS), that may be responsive to GSH augmentation.
MATERIALS AND METHODS
This study was a 12-week randomized, double blind, placebo-controlled Phase IIb study of the safety, tolerability, and preliminary efficacy of (in)GSH in 45 PD patients with Hoehn & Yahr stage 1–3 PD, conducted between June 20, 2015 and Feb 3, 2016. As a Phase II trial, this study was not powered for efficacy, but to further establish safety and tolerability, as well as to generate data that can be used to inform the power calculations for a Phase III trial. Using the data generated from the Phase I/IIa (in)GSH study [15] and G*power 3.1 software [22], a sample size of 45 was the minimum sample size needed to determine whether there was sufficient symptomatic effect to justify subsequent disease-modifications studies employing a delayed start or wash-out design. The a priori statistical endpoints were the mean change from baseline, per cohort, in total and motor UPDRS among matched pairs, given the small sample size. The results from the Phase I/IIa study were used to power this study. The overarching hypothesis is that GSH deficiency contributes to disease progression, and there is urgency within the community to conduct disease-modification trials. Therapies that also reduce symptoms require more complicated and expensive study designs when attempting to evaluate disease-modification, e.g. delayed-start design [23].
The primary objective of the study was to determine whether the (in)GSH improvements seen in the Phase I/IIa study on UPDRS total (4 point) and motor sub-score (2 point) over baseline were reproducible. The study was not powered for efficacy over placebo, although a placebo arm was included to provide information about the duration and extent of the placebo response, essential data for appropriately powering a Phase III efficacy trial, if warranted. The secondary objectives were to describe the systemic absorption characteristics of (in)GSH and the tolerability of (in)GSH in participants with PD. The goal of this study was to enroll enough subjects to detect a 4-point improvement in total UPDRS, if it exists, with a 5% probability of rejecting the true null hypothesis and an 80% power, with an expected 20% dropout rate. Prior to implementation, the study protocol was approved by the Bastyr University and the University of Washington Institutional Review Boards, and was registered on ClinicalTrials.gov (NCT02424708). There were no changes to methodology after trial commencement.
Recruitment efforts utilized the Fox Trial Finder, local physician referrals, the Washington State PD Registry, local support groups, and word-of-mouth recruiting (Fig. 2). Informed consent was obtained from all individuals before initiating participation. All participants were screened within 30 days of baseline to ensure that they were physically independent and otherwise healthy. To qualify, individuals must have been diagnosed with PD in the previous 10 years, be stable on all medications for at least 30 days prior to start of study, and have no exposure to N-acetyl cysteine (GSH precursor) or GSH in any form for the previous 90 days. Study participants were asked to remainstable on their medications throughout the entire study; should their symptoms require medication modification, they were encouraged to follow the advice of their prescribing doctor and assured that they would not be disqualified from study participation. Medications were reviewed at each study visit and dopamine equivalents were calculated [24].
Participants were randomized to 200 mg GSH/ml, 100 mg GSH/ml or placebo (saline) three times daily for 12 weeks, with a four-week washout period. Throughout the study, only the compounding pharmacy was unblinded. The study statistician used Stata 13.1 (College Station, TX) random number generator to assign individuals equally between the three groups, depending on whether or not they were eligible for the MRI portion of the study. Of the 45 enrolled participants, 15 participated in a sub-study in which magnetic resonance spectroscopy (MRS) was performed at baseline and 24 hours following discontinuation of the three month intervention according to previously described procedures [16].
The study medication was pre-packaged into 1 ml syringes and kept refrigerated in two opaque plastic bags. Participants and their caregivers were trained in the administration of the study medication. Mucosal Atomization DevicesTM (MAD)(Teleflex, Morrisville, NC) were used for drug delivery. Participants were asked to rinse MAD tips after each use, let air dry, and reuse throughout the study. MAD tips were replaced monthly. Participants were advised to use the study medicine morning, afternoon, and evening, and to tip head back or lie down during administration, while inhaling deeply.
All clinical evaluations were performed at the Bastyr University Clinical Research Center by the same clinician, who remained blinded for the duration of the study. Because there is a mild sulfur smell to GSH, the study clinician performing evaluations was never in the room with the packaged product. To evaluate whether participants were appropriately blinded, they were asked at each visit to predict the group to which they had been assigned. At each visit, there was an open-ended interview in which participants provided feedback, then participants were asked to complete online case report forms using REDCap (Research Electronic Data Capture) [25]. Participants completed the PROMIS Global, UPDRS questions 1–17, Non-motor Symptom Score, PDQ-39, and PRO-PD on REDCap.
Measures of oxidative stress and defense were performed by Genova Diagnostics (CLIA: #34D0655571, Asheville, NC, USA) according to previously described methods [4]. Because endogenous GSH levels are reported to fluctuate with circadian rhythm and during/after meals, participants were scheduled for study visits at the same time of day for all appointments, (e.g., a participant with a baseline appointment at 11 am had all remaining study visits at 11 am). Participants were asked to maintain their typical diet on the days of study visits. At each visit, participants were given a fresh batch of study medication, sealed in two layers of opaque plastic with an ice pack, as packaged by compounding pharmacy. Participants were frequently reminded to keep the study medication refrigerated and away from light and air.
Proton magnetic resonance spectroscopy (1H-MRS), a non-invasive approach enabling the determination of in vivo GSH concentrations [relative to creatine (Cr)], was used to measure changes in GHS levels in the brain following the 12-week intervention. Of the 15 individuals in each cohort, five underwent scans. Individuals who expressed a willingness to have two 1H-MRS scans were randomized within the 1H-MRS subset. Because GSH levels exhibit circadian fluctuations, pre- and post-treatment scans were always scheduled at the same time of day. Participants were asked to use their last dose of (in)GSH 24 hours before their scheduled follow-up 1H-MRS scan. MEGA-PRESS double-editing for the cysteinyl β-CH2 residue of GSH was used to determine GSH levels, and GSH and creatine peak areas were determined according to methods previously described [16].
Paired t-tests were used evaluate differences within subjects by comparing laboratory and clinical assessments at baseline and after 12 weeks of using study medication. Statistical significance was set at P < 0.05 using two-sided testing for all analyses. Adjustments for multiple comparisons were made using the Bonferroni method for all exploratory analyses. All analyses were performed using Stata version 11 (Statacorp, College Station, TX).
RESULTS
In the placebo arm, one participant withdrew due to chronically-irritated sinuses and headaches attributable to the study intervention. Two individuals withdrew from the 300 mg/d cohort; one due to a fall unrelated to the study resulting in a broken bone, and the other reported puffiness under the eyes, attributable to the study intervention. Two participants withdrew from the 600 mg/d cohort; one encountered logistical problems (unrelated to the study intervention) that prevented attendance at study visits, the other withdrew due to tachycardia and newly-diagnosed cardiomyopathy. This participant had a history of anxiety and tachycardia following a stressful event several years prior to study enrollment; at the screening visit the participant reported that all symptoms had resolved and the participant saw a cardiologist annually in follow-up. The participant’s annual cardiology appointment occurred approximately 5 weeks into the study, at which time the cardiomyopathy was diagnosed. Based on a small body of literature related to reductive stress resulting in insult to cardiomyocytes, the participant’s cardiologist recommended withdrawal from the study. After stopping the study medicine, the participant reported resolution of the tachycardia.
There was equal distribution between genders in the study, with a mean age of 60.9 ± 11.0 and a mean of 3.57 ± 2.15 years since diagnosis (Table 1). In order to evaluate whether participants were able to maintain the blind, they were asked at each study visit to indicate the group to which they thought they had been assigned (Table 2). Participants in all cohorts guessed incorrectly frequently enough to suggest the blind was adequately maintained. The change in dopamine equivalents (DE) remained relatively stable throughout the study (Table 5) across study arms. The greatest change observed was a 90-point reduction in DE in the high-dose (in)GSH arm, although this reduction did not reach statistical significance.
Table 1
Demographics and baseline characteristics of study participants
| N | % | |
| Gender | ||
| Male | 23 | 51% |
| Female | 22 | 49% |
| Age | ||
| Mean Age (SD), years | 60.9 (11.0) | |
| Median Age (Range), years | 64 (29, 78) | |
| Race/ Ethnicity | ||
| White | 45 | 100% |
| Hispanic | 1 | 2% |
| Non-Hispanic | 44 | 98% |
| PD Severity | ||
| Mean years since diagnosis (SD) | 3.5 (2.15) | |
| Hoehn &Yahr Stage | ||
| 1 Unilateral | 9 | 20% |
| 1.5 Unilateral+axial | 6 | 13% |
| 2 Bilateral, intact balance | 18 | 40% |
| 2.5 Bilateral with recovery on pull test | 7 | 16% |
| 3 Postural instability, physically independent | 5 | 11% |
Table 2
Participant capacity to predict group assignment
| I think | I think | I don’t know | |
| I received | I received | ||
| GSH | placebo | ||
| Month 1 | |||
| Placebo | 4 (27%) | 7 (47%) | 4 (27%) |
| 300 mg/d | 3 (21%) | 7 (47%) | 4 (29%) |
| 600 mg/d | 7 (47%) | 1 (7%) | 6 (43%) |
| Month 2 | |||
| Placebo | 6 (43%) | 6 (43%) | 2 (14%) |
| 300 mg/d | 3 (23%) | 7 (54%) | 3 (23%) |
| 600 mg/d | 7 (50%) | 6 (43%) | 1 (7%) |
| Month 3 | |||
| Placebo | 6 (43%) | 6 (43%) | 2 (14%) |
| 300 mg/d | 1 (8%) | 7 (58%) | 4 (33%) |
| 600 mg/d | 7 (54%) | 5 (39%) | 1 (8%) |
| Month 4 (1 month washout) | |||
| Placebo | 7 (50%) | 6 (43%) | 1 (7%) |
| 300 mg/d | 2 (17%) | 6 (50%) | 4 (33%) |
| 600 mg/d | 8 (57%) | 3 (21%) | 3 (21%) |
As was seen in the Phase I/IIa study [15], UPDRS-Part 3 motor scores worsened in the placebo group over the three months of the study, while there was a mild improvement in motor scores in both the low and high dose cohorts, with all cohorts worsening during withdrawal [Fig. 3B, Tables 3 and 4]. Total UPDRS results were also similar to those seen in the Phase I/IIa study, although in this Phase 2b study, the placebo arm had an unusually robust improvement [Fig. 3A, Tables 3 and 4].
Fig.3
Change in mean total UPDRS (A), UPDRS-Motor Subscore (Part 3) (B), and central nervous system (CNS) glutathione (C) over the twelve weeks of the study, and following a four-week washout period. CNS glutathione (as GSH / total creatine peak area ratio) was measured by proton magnetic resonance spectroscopy (1H-MRS) and the volume of interest was a 4×4×5 cm region centered on the left dorsal putamen at the level of the lentiform nucleus. Error bars indicate SD.
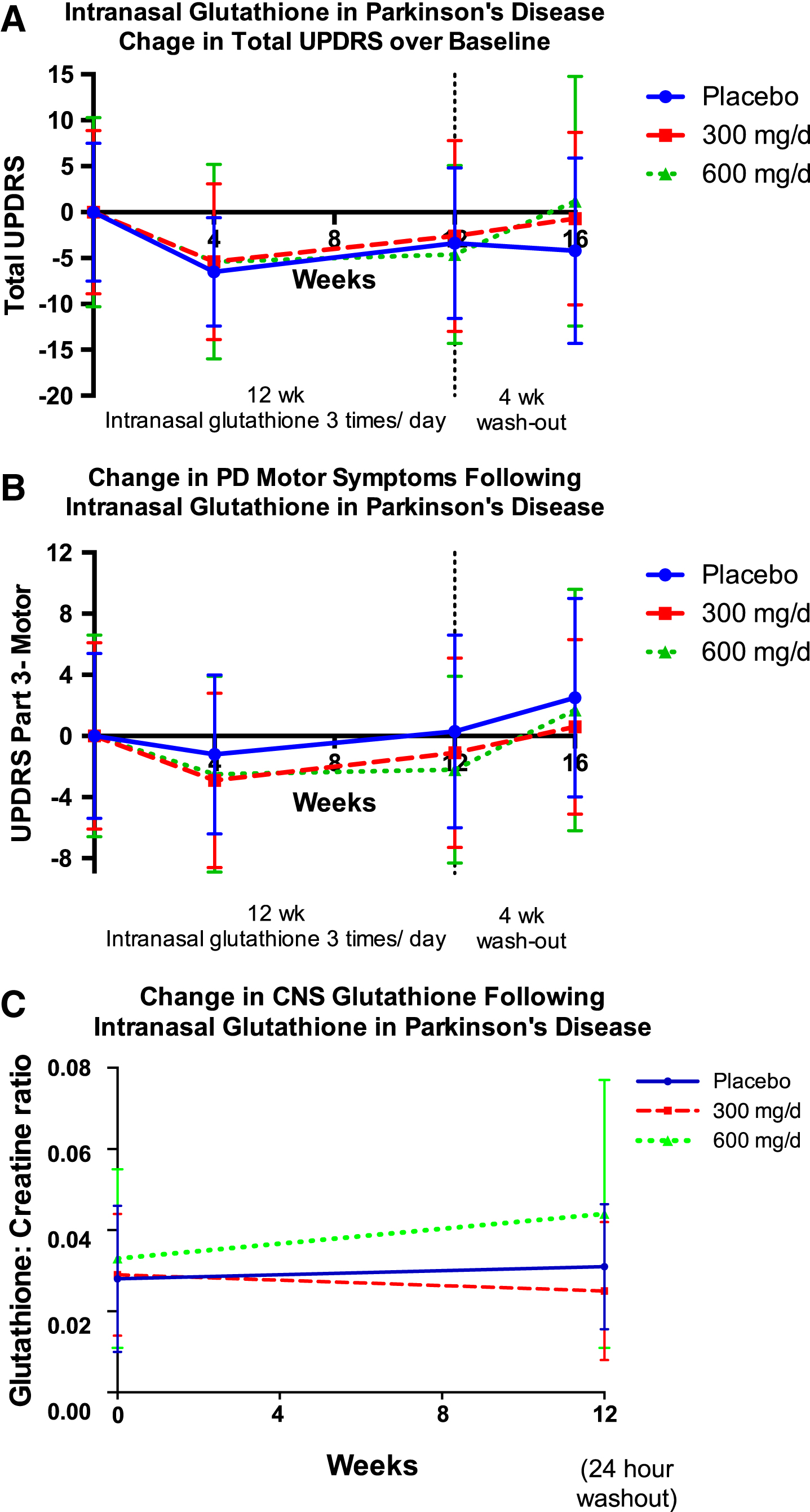
Table 3
Change in UPDRS, by cohort, over the three-month study intervention and following one month washout.
| n | Mean | Mean | Mean | Pre-Post 2 sided | Absolute | Mean 4wk | Week 12 vs | |
| Baseline (SD) | Week 4 (SD) | Week 12 (SD) | P-value | Change (SD) | Washout (SD) | Washout P-value | ||
| Total UPDRS | ||||||||
| Placebo | 14 | 64.9 (7.5) | 58.4 (5.9) | 61.5 (8.2) | 0.0590 | –3.4 (6.2) | 63.71 (10.1) | 0.2029 |
| 300 mg/d | 11 | 66.2 (8.9) | 60.8 (8.5) | 63.6 (10.4) | 0.2618 | –2.6 (7.4) | 66.91 (9.4) | 0.0347 |
| 600 mg/d | 14 | 65.7 (10.3) | 60.3 (10.6) | 61.1 (9.7) | 0.0025 | –4.6 (4.7) | 66.85 (13.6) | 0.0007 |
| Part 1- Mentation | ||||||||
| Placebo | 14 | 3.7 (1.1) | 3.4 (1.9) | 3.2 (1.8) | 0.2362 | –0.5 (1.5) | 2.92 (1.5) | 0.5569 |
| 300 mg/d | 11 | 3.4 (1.1) | 3.8 (1.4) | 3.6 (2.2) | 0.5884 | 0.27 (1.6) | 3.7 (2.1) | 0.8213 |
| 600 mg/d | 14 | 4.0 (2.2) | 3.4 (1.8) | 3.6 (2.2) | 0.3619 | –0.43 (1.7) | 3.18 (2.0) | 0.4890 |
| Part 2- Activities of Daily Living | ||||||||
| Placebo | 14 | 23.4 (3.8) | 20.4 (3.5) | 20.1 (3.7) | 0.0011 | –3.3 (3.0) | 20.6 (3.9) | 0.5930 |
| 300 mg/d | 11 | 22.8 (3.0) | 20.2 (2.5) | 21.0 (3.8) | 0.0663 | –1.8 (2.9) | 22.5 (4.1) | 0.00965 |
| 600 mg/d | 14 | 22.4 (4.5) | 21.0 (4.5) | 21.4 (4.8) | 0.1607 | –0.9 (2.3) | 22.9 (5.5) | 0.0802 |
| Part 3- Motor | ||||||||
| Placebo | 14 | 26.1 (5.4) | 24.9 (5.2) | 26.43 (6.3) | 0.7829 | 0.4 (4.8) | 28.6 (6.5) | 0.0445 |
| 300 mg/d | 11 | 28.5 (6.1) | 25.6 (5.7) | 27.36 (6.2) | 0.3736 | –1.1 (3.9) | 29.1 (5.7) | 0.0977 |
| 600 mg/d | 14 | 27.5 (6.6) | 25.0 (6.4) | 25.29 (6.1) | 0.0485 | –2.2 (3.8) | 29.2 (7.9) | 0.0005 |
| Part 4- Complications of Therapy | ||||||||
| Placebo | 14 | 11.7 (0.8) | 11.2 (0.8) | 11.7 (1.0) | 1.0000 | 0 (1.1) | 11.5 (1.2) | 0.6484 |
| 300 mg/d | 11 | 11.6 (1.4) | 11.1 (0.7) | 11.6 (0.7) | 1.0000 | 0 (1.3) | 11.6 (1.3) | 1.0000 |
| 600 mg/d | 14 | 11.9 (1.9) | 11.4 (1.8) | 10.8 (0.7) | 0.0417 | –1.1 (1.8) | 11.3 (1.0) | 0.0254 |
Comparisons between study visits. P-values lower than 0.05 are in bold, P-values lower than the multiple comparison adjusted cutoff (0.0033) are italicized.
Table 4
Change in UPDRS, by cohort, over the three-month study intervention and following one month washout.
| Mean | Mean | Placebo vs | Mean 600 mg/d | Placebo vs | |
| Placebo (SD) | 300 mg/d (SD) | 300 mg/d (SD) | (SD) | 600 mg/d | |
| P-value | P-value | ||||
| Total UPDRS | |||||
| Baseline | 64.9 (7.5) | 66.2 (8.9) | 0.6603 | 65.7 (10.3) | 0.8161 |
| Week 4 | 58.4 (5.9) | 60.8 (8.5) | 0.3934 | 60.3 (10.6) | 0.5629 |
| Week 12 | 61.5 (8.2) | 63.6 (10.4) | 0.5581 | 61.1 (9.7) | 0.3817 |
| Washout | 63.71 (10.1) | 66.91 (9.4) | 0.3934 | 66.85 (13.6) | 0.4941 |
| Part 1- Mentation | |||||
| Baseline | 3.7 (1.1) | 3.4 (1.1) | 0.4770 | 4.0 (2.2) | 0.6519 |
| Week 4 | 3.4 (1.9) | 3.8 (1.4) | 0.5315 | 3.4 (1.8) | 1.0000 |
| Week 12 | 3.2 (1.8) | 3.6 (2.2) | 0.6030 | 3.6 (2.2) | 0.6030 |
| Washout | 2.92 (1.5) | 3.7 (2.1) | 0.2566 | 3.18 (2.0) | 0.6572 |
| Part 2- Activities of Daily Living | |||||
| Baseline | 23.4 (3.8) | 22.8 (3.0) | 0.6467 | 22.4 (4.5) | 0.5224 |
| Week 4 | 20.4 (3.5) | 20.2 (2.5) | 0.8632 | 21.0 (4.5) | 0.6969 |
| Week 12 | 20.1 (3.7) | 21.0 (3.8) | 0.5310 | 21.4 (4.8) | 0.4295 |
| Washout | 20.6 (3.9) | 22.5 (4.1) | 0.2202 | 22.9 (5.5) | 0.2131 |
| Part 3- Motor Daily Living | |||||
| Baseline | 26.1 (5.4) | 28.5 (6.1) | 0.2804 | 27.5 (6.6) | 0.5444 |
| Week 4 | 24.9 (5.2) | 25.6 (5.7) | 0.7370 | 25.0 (6.4) | 0.9642 |
| Week 12 | 26.43 (6.3) | 27.36 (6.2) | 0.6756 | 25.29 (6.1) | 0.6427 |
| Washout | 28.6 (6.5) | 29.1 (5.7) | 0.8304 | 29.2 (7.9) | 0.8280 |
| Part 4- Complications of Therapy | |||||
| Baseline | 11.7 (0.8) | 11.6 (1.4) | 0.8183 | 11.9 (1.9) | 0.7195 |
| Week 4 | 11.2 (0.8) | 11.1 (0.7) | 0.7277 | 11.4 (1.8) | 0.7071 |
| Week 12 | 11.7 (1.0) | 11.6 (0.7) | 0.7616 | 10.8 (0.7) | 0.0105 |
| Washout | 11.5 (1.2) | 11.6 (1.3) | 0.8342 | 11.3 (1.0) | 0.6359 |
Comparisons between cohorts. P-values lower than 0.05 are in bold.
Table 5
Dopamine equivalents taken by participants during the study, by cohort
| n | Mean | Mean | Mean | Pre-Post | Absolute | Mean 4wk | Week 12 vs | |
| Baseline | Week 4 | Week 12 | 2 sided | Change | Washout | Washout | ||
| (SD) | (SD) | (SD) | P-value | (SD) | (SD) | P-value | ||
| Placebo | 14 | 498.9 (389.8) | 490.6 (393.3) | 425.6 (313.8) | 0.5833 | –8.9 (22.8) | 425.6 (313.8) | 1.0000 |
| 300 mg/d | 13 | 542.3 (411.7) | 552.5 (406.3) | 558.1 (412.9) | 0.9202 | –21.5 (41.2) | 595.4 (385.5) | 0.8179 |
| 600 mg/d | 14 | 617.8 (380.8) | 571.9 (413.9) | 525.5 (342.8) | 0.49977 | –90.7 (207.9) | 532.6 (338.8) | 0.9565 |
The secondary aim of this study was to evaluate other clinical rating scales or biomarkers that may be more sensitive change in response to (in)GSH, relative to the UPDRS. 2-sided P-values were used for all analyses, and adjustments were made for multiple comparisons. There was a statistically significant improvement in the Non-Motor Symptom Score (NMSS) [26] in the 600 mg/d cohort, with a mean 10.17 (13.18) point improvement (P = 0.0217) over baseline. The pre- post change was not statistically significant (P values > 0.05) for Kinetics Objective Measures (Timed-Up-&-Go, Postural Sway, Keyboard Dexterity), Patient-Reported Outcomes in PD (PRO-PD), Montreal Cognitive Assessment (MoCA), PDQ-39, or PROMIS Global Health.
After correcting for multiple comparisons, differences between biological markers (relative to baseline, and between groups) did not reach statistical significance for any of the following measurements: blood cysteine:sulfate ratios, blood cysteine: cystine ratios, blood glutathione peroxidase, blood superoxide dismutase (SOD), blood lipid peroxides, urine lipid peroxides, and urine 8-OHdG. Contrary to predictions, at three months, whole blood total glutathione decreased in all three cohorts, with both the placebo and 600 mg/d cohorts evidencing a statistically-significant decrease relative to baseline (P = 0.0142 and 0.0112, respectively).
There was a trend toward increasing brain GSH concentrations in the 600 mg/d cohort, as measured by 1H-MRS, although this improvement did not reach statistical significance [Fig. 3C].
DISCUSSION
While this is the fourth published report of mild-to-moderate symptomatic improvement following the administration of GSH to individuals with PD [11, 12, 15], none of these studies has been powered to detect improvement over placebo. Unfortunately, the diversity of delivery methods and study designs prevents cohesive interpretation. One was open-label with twice daily interventions requiring the attention of study staff [11], and thus expected to result in a robust placebo response. The most notable difference between the Phase IIb study presented here, where a robust placebo response was observed, and the Phase I/IIa study, where a more modest placebo response was observed, is the increased attention to delivery. During the current study, individuals were instructed to lie down, if possible, and tilt head back while inhaling deeply; it is possible that these instructions inadvertently increased the ritual of administering the treatment and resulted in “placebo efficacy”[27].
A recent publication has demonstrated that the glutathione precursor, NAC, produced a mean 13% improvement in UPDRS scores, and significantly increased DAT density in the caudate and putamen in PD, suggesting that NAC and/or GSH can affect dopaminergic neurotransmission [28]. In addition to having potential as a disease-modifying agent, these data support the idea that (in)GSH may also have symptomatic efficacy. Use of a delayed-start, wash-out, or complete two-period clinical trial design would allow evaluation of symptomatic and disease-modification simultaneously [29].
Only the high-dose cohort exhibited CNS GSH concentrations that increased over the 12-week intervention; the diversity between individuals and the lack of established reference ranges make these data difficult to interpret, although these results suggest doses of at least 600 mg/ d should be used in subsequent trials. It is possible that GSH did not reach the target tissue or that the GSH augmentation provided by (in)GSH is not sustained after a 24-hour washout. Since the identical product and delivery were previously shown to reach target tissue and effectively augment CNS GSH up to 60 minutes [16], the latter explanation is most likely. Subsequent pharmacokinetic studies will be necessary to evaluate the half-life of (in)GSH, which may inform dosing recommendations for future trials.
Contrary to expectations, the whole blood GSH concentrations decreased over the course of the study in all cohorts. The analyses were run in batches and consistency was maintained between scheduled study visits, personnel, collection and shipping procedures, and laboratory methodology in an attempt to minimize error. There were no reported inconsistencies in collection, shipment, or analysis. Whole blood GSH was analyzed in the three-month randomized controlled trial, Phase I study of (in)GSH in PD. In the Phase I study, there was no dose-response effect or trend toward RBC increase in the active arms over the placebo arm [30]. Whole blood GSH concentrations are influenced by diet [31] and even yoga [32, 33], so it is possible that factors unrelated to study participation influenced blood GSH concentrations. It is also possible that some aspect of study participation led to a depletion of whole blood GSH; future trials should determine whether these results are reproducible and whether they are relevant to PD clinicaloutcomes.
There was a lack of racial diversity in the study, although there was an even distribution across gender and HY stage. It is unclear whether the single incident of cardiomyopathy in this study is related to the study medication. Research in rodents suggests that reductive stress (attributable to an overabundance of antioxidants), may result in insult to cardiomyocytes. In a transgenic mouse model generated to overexpress the antioxidant heat shock protein 27 (Hsp27), mice developed cardiomyopathy, with evidence for reductive stress (increased ratio of reduced: oxidized glutathione) decreased levels of reactive oxygen species, upregulated GSH peroxidase 1, and decreased iron content in cardiac tissue relative to controls. Interestingly, inhibition of glutathione peroxidase 1 activity prevented the development of cardiomyopathy in these rodents [34]. These data suggest that future clinical trials of (in)GSH should involve screening for cardiac side effects.
The placebo response in this study deserves more attention. Whereas the placebo cohort in the Phase I/IIa (in)GSH in PD study exhibited a mean 1.1 + 4.1 point reduction in UPDRS scores (on par with other randomized controlled drug trials in PD), the placebo cohort in the current phase IIb study exhibited a mean 3.4 (+6.2) point reduction in UPDRS scores (Fig. 4). As the goal of this study was to determine whether the symptomatic improvements seen in the Phase I/IIa were reproducible, all efforts were made to minimize differences between the two study protocols; interventions, study duration, setting, clinician, etc. were unchanged relative to the phase I/IIa trial. Differences between the current phase IIb and the previous phase I/IIa study included: one less study visit, more direct data entry into computer and more outcome measures, e.g. use of iPhone for Timed-Up-and-Go test, and additional counseling on method of administration. A study of CNS uptake of GSH in PD, which was conducted in the time period between the Phase I/IIa and IIb studies, demonstrated that supine delivery increased measurable GSH concentrations in the putamen [16]. Thus, in the Phase IIb study, participants were encouraged to administer the study medication in a supine position (head tipped back), and a demonstration video was provided. It is possible the emphasis on supine delivery, and staff confidence that this approach improves distribution of the drug to target tissue, may have resulted in increased attention to the ‘ritual of administration’. As has been seen in studies of chronic obstructive pulmonary disease and cystic fibrosis, it is also possible the (in)saline resulted in a physiological response, e.g. greater regional blood flow, clearing of sinuses, etc., which itself led to symptomaticimprovement [35].
![Change in total UPDRS scores from placebo arms of randomized controlled PD trials [15, 36–42].](https://content.iospress.com:443/media/jpd/2017/7-2/jpd-7-2-jpd161040/jpd-7-jpd161040-g004.jpg)
Both study staff expertise and community enthusiasm for this research may have increased in the years between studies; whereas many studies struggle to meet recruitment goals, this study had a waiting list of participants eager to participate. Positive interviewing technique was used throughout study visits and participants were regularly thanked for their contributions to the research. It is possible community enthusiasm resulted in an exaggerated placebo response, exemplified by an email from a study participant, “ Subject: GLUTATHIONE - THE MIRACLE MOLECULE ... LAURIE, I AM EAGER TO READ THE STUDY. IS IT AVAILABLE? AND IF NOT WHEN? I MISS THOSE MONTHLY SESSIONS, YOUR ENTHUSIASM AND ENERGY ARE THERAPUTIC ... BE WELL, KEEP UP THE GOOD WORK”. Given the robust placebo response, which was apparently sustained throughout the three-month intervention, subsequent efficacy studies will need to follow participants for extended periods to give the placebo response time to subside.
Based on biological plausibility, temporality, demonstration of safety, tolerability, and target validation, a disease-modification trial of (in)GSH in PD is warranted. As this is the fourth published study of GSH supplementation in PD, and the fourth to report symptomatic improvement, further research into the capacity of GSH to improve symptoms is warranted. This study suggests that the UPDRS Part III (motor subset) and the Non-Motor Symptom Score are appropriate outcome measures for subsequent symptomatic trials. Alternatives to (in)saline should be considered for use in subsequent controlarms.
FUNDING
Study funding was provided by the Michael J. Fox Foundation.
CONFLICT OF INTEREST
L.K.M. is founder of NeurRx Social Purpose Corporation. The other authors have no conflicts of interest to report.
AUTHOR CONTRIBUTIONS
L.K.M. was responsible for conception, design, and execution of the clinical trial, and manuscript preparation. R.C.L. was responsible for blinded data analysis. E.G.S. and T.K.W. performed acquisition of the MR image, J.M.P. assisted in study design, blinded MRS analysis, and manuscript preparation.
ACKNOWLEDGMENTS
We wish to thank our Research Assistants, Prysilla U. De La Torre and Gisela Medoza, and Dr. Ali Samii for serving as the Medical Monitor. Teleflex generously provided Mucosal Atomization Devices syringe tips for the study.
REFERENCES
[1] | Perry TL , Godin DV , & Hansen S ((1982) ) Parkinson’s disease: A disorder due to nigral glutathione deficiency? Neurosci Lett, 33: , 305–310. |
[2] | Sian J , Dexter DT , Lees AJ , Daniel S , Agid Y , Javoy-Agid F , Jenner P , & Marsden CD ((1994) ) Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol, 36: , 348–355. |
[3] | Sofic E , Lange KW , Jellinger K , & Riederer P ((1992) ) Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson’s disease. Neurosci Lett, 142: , 128–130. |
[4] | Mischley LKSL , Weiss NS , Padowski JM , Kavanagh TJ , White CC , & Rosenfeld ME ((2016) ) Glutathione as a biomarker in Parkinson’s disease: Associations with aging and disease severity. Oxid Med Cell Longev, 2016: , Article ID 9409363. |
[5] | Toffa S , Kunikowska GM , Zeng BY , Jenner P , & Marsden CD ((1997) ) Glutathione depletion in rat brain does not cause nigrostriatal pathway degeneration. J Neural Transm (Vienna), 104: , 67–75. |
[6] | Wullner U , Loschmann PA , Schulz JB , Schmid A , Dringen R , Eblen F , Turski L , & Klockgether T ((1996) ) Glutathione depletion potentiates MPTP and MPP+toxicity in nigral dopaminergic neurones. Neuroreport, 7: , 921–923. |
[7] | Do KQ , Trabesinger AH , Kirsten-Kruger M , Lauer CJ , Dydak U , Hell D , Holsboer F , Boesiger P , & Cuenod M ((2000) ) Schizophrenia: Glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur J Neurosci, 12: , 3721–3728. |
[8] | Saharan S , & Mandal PK ((2014) ) The emerging role of glutathione in Alzheimer’s disease. J Alzheimers Dis, 40: , 519–529. |
[9] | Allen J , & Bradley RD ((2011) ) Effects of oral glutathione supplementation on systemic oxidative stress biomarkers in human volunteers. J Altern Complement Med, 17: , 827–833. |
[10] | Witschi A , Reddy S , Stofer B , & Lauterburg BH ((1992) ) The systemic availability of oral glutathione. Eur J Clin Pharmacol, 43: , 667–669. |
[11] | Sechi G , Deledda MG , Bua G , Satta WM , Deiana GA , Pes GM , & Rosati G ((1996) ) Reduced intravenous glutathione in the treatment of early Parkinson’s disease. Prog Neuropsychopharmacol Biol Psychiatry, 20: , 1159–1170. |
[12] | Hauser RA , Lyons KE , McClain T , Carter S , & Perlmutter D ((2009) ) Randomized, double-blind, pilot evaluation of intravenous glutathione in Parkinson’s disease. Mov Disord, 24: , 979–983. |
[13] | Mischley LK ((2011) ) Glutathione deficiency in Parkinson’s disease: Intranasal administration as a method of augmentation. J Orthomol Med, 26: , 32–36. |
[14] | Mischley LK , Vespignani MF , & Finnell JS ((2013) ) Safety survey of intranasal glutathione. J Altern Complement Med, 19: , 459–463. |
[15] | Mischley LK , Leverenz JB , Lau RC , Polissar NL , Neradilek MB , Samii A , & Standish LJ ((2015) ) A randomized, double-blind phase I/IIa study of intranasal glutathione in Parkinson’s disease. Mov Disord, 30: , 1696–1701. |
[16] | Mischley LKCK , Shankland EG , Kavanagh TJ , Rosenfeld ME , Duda JE , While CC , Wilbur TK , DeLaTorre PU , & Padowski JM ((2016) ) Central nervous system uptake of intranasal glutathione in Parkinson’s disease. npj Parkinson’s Disease, 2: , Article number: 16002. |
[17] | Janáky R , Varga V , Saransaari P , & Oja SS ((1993) ) Glutathione modulates the N-methyl-d-aspartate receptor-activated calcium influx into cultured rat cerebellar granule cells. Neurosci Lett, 156: , 153–157. |
[18] | Sechi GP ((2010) ) Reduced glutathione and Parkinson’s disease. Mov Disord, 25: , 2690–2691. |
[19] | Sechi G , Nuvoli S , Agnetti V , Paulus K , Spanu A , Cocco G , & Madeddu G ((2006) ) Influence of parenteral GSH on striatal dopamine transporter in PD. Mov Disord, 21: (Suppl. 15), S579. |
[20] | Monti DA , Zabrecky G , Kremens D , Liang TW , Wintering NA , Cai J , Wei X , Bazzan AJ , Zhong L , Bowen B , Intenzo CM , Iacovitti L , & Newberg AB ((2016) ) N-acetyl cysteine may support dopamine neurons in Parkinson’s disease: Preliminary clinical and cell line data. PLoS One, 11: , e0157602. |
[21] | McGhee DJ , Ritchie CW , Zajicek JP , & Counsell CE ((2016) ) A review of clinical trial designs used to detect a disease-modifying effect of drug therapy in Alzheimer’s disease and Parkinson’s disease. BMC Neurol, 16: , 92. |
[22] | Faul F , Erdfelder E , Lang AG , & Buchner A ((2007) ) G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods, 39: , 175–191. |
[23] | Olanow CWHR , Jankovic J , Langston W , Lang A , Poewe W , Tolosa E , Stocchi F , Melamed E , Eyal E , & Rascol O ((2008) ) A randomized, double-blind, placebo-controlled, delayed start study to assess rasagiline as a disease modifying therapy in Parkinson’s disease (the ADAGIO study): Rationale, design, and baseline characteristics. Mov Disord, 23: , 2194–2201. |
[24] | Smith C ((2010) ) Levodopa dose equivalency: A systematic review [PowerPoint slides]. Retrived from http://www.birmingham.ac.uk/Documents/college-mds/trials/bctu/PDRehab/Investigators/meetings/2010-2/CSmithLEDReview.pdf |
[25] | Harris PA , Taylor R , Thielke R , Payne J , Gonzalez N , & Conde JG ((2009) ) Research electronic data capture (REDCap)–a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform, 42: , 377–381. |
[26] | Chaudhuri KR , Martinez-Martin P , Brown RG , Sethi K , Stocchi F , Odin P , Ondo W , Abe K , Macphee G , Macmahon D , Barone P , Rabey M , Forbes A , Breen K , Tluk S , Naidu Y , Olanow W , Williams AJ , Thomas S , Rye D , Tsuboi Y , Hand A , & Schapira AH ((2007) ) The metric properties of a novel non-motor symptoms scale for Parkinson’s disease: Results from an international pilot study. Mov Disord, 22: , 1901–1911. |
[27] | Miller FG , Kallmes DF , & Buchbinder R ((2011) ) Vertebroplasty and the placebo response. Radiology, 259: , 621–625. |
[28] | Monti DA , Zabrecky G , Kremens D , Liang TW , Wintering NA , Cai J , Wei X , Bazzan AJ , Zhong L , Bowen B , Intenzo CM , Iacovitti L , & Newberg AB ((2016) ) N-acetyl cysteine may support dopamine neurons in Parkinson’s disease: Preliminary clinical and cell line data. PLoS One, 11: , e0157602. |
[29] | McGhee DJ , Ritchie CW , Zajicek JP , & Counsell CE ((2016) ) A review of clinical trial designs used to detect a disease-modifying effect of drug therapy in Alzheimer’s disease and Parkinson’s disease. BMC Neurol, 16: , 92. |
[30] | Mischley LK ((2016) ) Glutathione in Parkinson’s disease (Doctoral dissertation). Retrieved from https://digital.lib.washington.edu, 89. |
[31] | Jones DP , Coates RJ , Flagg EW , Eley JW , Block G , Greenberg RS , Gunter EW , & Jackson B ((1992) ) Glutathione in foods listed in the National Cancer Institute’s Health Habits and History Food Frequency Questionnaire. Nutr Cancer, 17: , 57–75. |
[32] | Patil SG , Dhanakshirur GB , Aithala MR , Naregal G , & Das KK ((2014) ) Effect of yoga on oxidative stress in elderly with grade-I hypertension: A randomized controlled study. J Clin Diagn Res, 8: , BC04–BC07. |
[33] | Lim SA , & Cheong KJ ((2015) ) Regular yoga practice improves antioxidant status, immune function, and stress hormone releases in young healthy people: A randomized, double-blind, controlled pilot study. J Altern Complement Med, 21: , 530–538. |
[34] | Zhang X , Min X , Li C , Benjamin IJ , Qian B , Zhang X , Ding Z , Gao X , Yao Y , Ma Y , Cheng Y , & Liu L ((2010) ) Involvement of reductive stress in the cardiomyopathy in transgenic mice with cardiac-specific overexpression of heat shock protein 27. Hypertension, 55: , 1412–1417. |
[35] | Dentice RL , Elkins MR , Middleton PG , Bishop JR , Wark PA , Dorahy DJ , Harmer CJ , Hu H , & Bye PT ((2016) ) A randomised trial of hypertonic saline during hospitalisation for exacerbation of cystic fibrosis. Thorax, 71: , 141–147. |
[36] | Parkinson Study Group ((2002) ) A controlled trial of rasagiline in early Parkinson disease: The TEMPO Study. Arch Neurol, 59: , 1937–1943. |
[37] | Hauser RA , Choudhry SD , Eyal A , & Isaacson SE ((2014) ) Randomized, controlled trial of rasagiline as an add-on to dopamine agonists in Parkinson’s disease. Mov Disord, 29: , 1028–1034. |
[38] | Parkinson Study Group ((2005) ) A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: The PRESTO study. Arch Neurol, 62: , 241–248. |
[39] | LeWitt PA ((1991) ) Deprenyl’s effect at slowing progression of parkinsonian disability: The DATATOP study. The Parkinson Study Group. Acta Neurol Scand Suppl, 136: , 79–86. |
[40] | ((1994) ) A controlled trial of lazabemide (Ro 19-6327) in levodopa-treated Parkinson’s disease. Parkinson Study Group. Arch Neurol, 51: , 342–347. |
[41] | Parkinson Study Group QE3 Investigators, Beal MF , Oakes D , Shoulson I , Henchcliffe C , Galpern WR , Haas R , Juncos JL , Nutt JG , Voss TS , Ravina B , Shults CM , Helles K , Snively V , Lew MF , Griebner B , Watts A , Gao S , Pourcher E , Bond L , Kompoliti K , Agarwal P , Sia C , Jog M , Cole L , Sultana M , Kurlan R , Richard I , Deeley C , Waters CH , Figueroa A , Arkun A , Brodsky M , Ondo WG , Hunter CB , Jimenez-Shahed J , Palao A , Miyasaki JM , So J , Tetrud J , Reys L , Smith K , Singer C , Blenke A , Russell DS , Cotto C , Friedman JH , Lannon M , Zhang L , Drasby E , Kumar R , Subramanian T , Ford DS , Grimes DA , Cote D , Conway J , Siderowf AD , Evatt ML , Sommerfeld B , Lieberman AN , Okun MS , Rodriguez RL , Merritt S , Swartz CL , Martin WR , King P , Stover N , Guthrie S , Watts RL , Ahmed A , Fernandez HH , Winters A , Mari Z , Dawson TM , Dunlop B , Feigin AS , Shannon B , Nirenberg MJ , Ogg M , Ellias SA , Thomas CA , Frei K , Bodis-Wollner I , Glazman S , Mayer T , Hauser RA , Pahwa R , Langhammer A , Ranawaya R , Derwent L , Sethi KD , Farrow B , Prakash R , Litvan I , Robinson A , Sahay A , Gartner M , Hinson VK , Markind S , Pelikan M , Perlmutter JS , Hartlein J , Molho E , Evans S , Adler CH , Duffy A , Lind M , Elmer L , Davis K , Spears J , Wilson S , Leehey MA , Hermanowicz N , Niswonger S , Shill HA , Obradov S , Rajput A , Cowper M , Lessig S , Song D , Fontaine D , Zadikoff C , Williams K , Blindauer KA , Bergholte J , Propsom CS , Stacy MA , Field J , Mihaila D , Chilton M , Uc EY , Sieren J , Simon DK , Kraics L , Silver A , Boyd JT , Hamill RW , Ingvoldstad C , Young J , Thomas K , Kostyk SK , Wojcieszek J , Pfeiffer RF , Panisset M , Beland M , Reich SG , Cines M , Zappala N , Rivest J , Zweig R , Lumina LP , Hilliard CL , Grill S , Kellermann M , Tuite P , Rolandelli S , Kang UJ , Young J , Rao J , Cook MM , Severt L , & Boyar K ((2014) ) A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol, 71: , 543–552. |
[42] | Fahn S , Parkinson Study Group. ((2005) ) Does levodopa slow or hasten the rate of progression of Parkinson’s disease? J Neurol, 252: (Suppl 4), IV37–IV42. |


