
The Proteasome Inhibition Model of Parkinson’s Disease
Abstract
The pathological hallmarks of Parkinson’s disease are the progressive loss of nigral dopaminergic neurons and the formation of intracellular inclusion bodies, termed Lewy bodies, in surviving neurons. Accumulation of proteins in large insoluble cytoplasmic aggregates has been proposed to result, partly, from a failure in the function of intracellular protein degradation pathways. Evidence in support for such a hypothesis emerged in the beginning of the years 2000 with studies demonstrating structural and functional deficits in the ubiquitin-proteasome pathway in post-mortem nigral tissue of patients with Parkinson’s disease. These fundamental findings have inspired the development of a new generation of animal models based on the use of proteasome inhibitors to disturb protein homeostasis and trigger nigral dopaminergic neurodegeneration. In this review, we provide an updated overview of the current approaches in employing proteasome inhibitors to model Parkinson’s disease, with particular emphasis on rodent studies. In addition, the mechanisms underlying proteasome inhibition-induced cell death and the validity criteria (construct, face and predictive validity) of the model will be critically discussed. Due to its distinct, but highly relevant mechanism of inducing neuronal death, the proteasome inhibition model represents a useful addition to the repertoire of toxin-based models of Parkinson’s disease that might provide novel clues to unravel the complex pathogenesis of this disorder.
INTRODUCTION
Parkinson’s disease (PD) is an age-related neurodegenerative condition, in which nigral dopamine (DA) producing neurons become dysfunctional and are progressively lost during a protracted period of disease development. The gradual degeneration of the nigrostriatal DA-ergic pathway leads to the manifestation of motor-related symptoms, such as resting tremor, difficulty in initiating movements (akinesia), slowness of movements (bradykinesia), muscular rigidity and loss of balance [1]. The main pathological hallmarks of PD are the depigmentation of the substantia nigra pars compacta (SNc) caused by the progressive loss of nigral DA-ergic neurons, and the presence of intracellular inclusion bodies, termed Lewy bodies (LBs), in surviving neurons [2]. These LBs are proteinaceous aggregates highly enriched in the pre-synaptic protein α-synuclein [3] and their formation indicates that disturbances in protein homeostasis (i.e. proteostasis) are part of the pathomechanisms underlying DA neuron loss in PD. Accordingly, similar to other neurodegenerative disorders such as multiple system atrophy and dementia with Lewy bodies (DLB), PD is classified as a synucleinopathy [4].
Although the precise cause of PD has remained elusive, various pathogenic mechanisms have been associated with nigral cell death, including oxidative stress, mitochondrial dysfunction, neuroinflammation, glutamate excitotoxicity and impairment in protein degradation pathways, such as the ubiquitin-proteasome system [5]. These fundamental discoveries regarding the pathogenesis of PD have been instrumental in our attempts for modeling the disease in vitro and in vivo. Introduced during the early 2000s, following the discovery of structural and functional deficits in the proteasome system in patients with PD, the proteasome inhibition model relies on application of proteasome inhibitors to induce disturbances in proteostasis and trigger DA neurodegeneration. The model has attracted attention from the research community for being able to trigger accumulation and aggregation of endogenous proteins, such as α-synuclein, leading to neurotoxicity and thereby replicating one of the central pathogenic pathways in PD. However, in spite of its contemporary use, certain aspects related to the model are still under research and need to be further defined, such as the validity in replicating PD related changes, the similarities / dissimilarities with other toxin-based or genetic models of PD, the mechanisms of DA neuron death in conditions of proteasome inhibition, and the interaction of this pathway with PD related genetic factors. In this review, we will discuss these issues and provide an updated overview on the proteasome inhibition model of PD, highlighting defining features with regards to molecular and cellular changes, and the implementation of the model both in cell culture and in vivo. In order to aid prospective research groups that would like to implement this model in their laboratories, we also provide a comprehensive guide with the different sites of administration of proteasome inhibitors in rats and mice, and their effects on the nigrostriatal DA-ergic pathway and motor behavior.
THE UBIQUITIN-PROTEASOME SYSTEM AND ITS DYSFUNCTION IN PARKINSON’S DISEASE
The ubiquitin-proteasome system is a major intracellular protein degradation pathway that ensures proteostasis in eukaryotic cells (for review see [6, 7]). The cycle of protein degradation via the proteasome system begins with the attachment of a polyubiquitin chain (typically containing at least four ubiquitin molecules, linked to each other via Lys48-linkages) to the target protein. This process requires ATP and is accomplished in a series of reactions catalyzed by ubiquitin-activating (E1), ubiquitin-conjugating (E2) and ubiquitin-ligase (E3) enzymes. During the first step, E1 activates ubiquitin molecules in an ATP-dependent reaction, followed by the transfer of activated ubiquitin from E1 to E2. Next, ubiquitin is successively conjugated to the target protein by an E3 ubiquitin-ligase enzyme, leading to the covalent attachment of a polyubiquitin chain to the substrate. The tagged protein is subsequently recognized by the 26S proteasome, a large multimeric complex formed by a catalytic core (20S) and a regulatory particle (19S, also known as PA700) that caps the 20S proteasome at either end (Fig. 1). The 19S regulatory particle binds the polyubiquitin chain, cleaves it from the substrate, and unfolds the substrate, allowing it to enter 20S, a core particle that contains 28 subunits organized in two outer rings (α) and two inner rings (β) (Fig. 1). Within the 20S core particle, the substrate is digested into peptide fragments by three types of enzymatic activities (chymotrypsin-like, trypsin-like and caspase-like protease activities) provided by the β subunits. In addition to 26S proteasomes, free (i.e. uncapped) 20S proteasomes also exist in mammalian cells and mediate ubiquitin-independent and ATP-independent protein degradation of partially unfolded proteins (natively unfolded or damaged proteins) (Fig. 1) [8]. The 20S proteasome can become a major degradation pathway in certain conditions (e.g. oxidative stress) [9], and the expression of the 20S proteasome can increase via disassembly of the 26S proteasome into its 20S and 19S components in such situations [10]. While most proteins require polyubiquitination to be directed to and degraded by the 26S proteasome, α-synuclein is an example of a protein that can be directly degraded by the 20S proteasome without requirement of ubiquitination. Both ubiquitin-dependent [11, 12] and ubiquitin-independent [13, 14] degradation via the 26S/20S proteasome have been described forα-synuclein.
An increasing amount of evidence indicates that the proteasome function is impaired in the substantia nigra (SN) of PD patients (Table 1). Post-mortem data obtained from sporadic PD indicate the presence of structural and functional defects in the 26/20S proteasome, including loss of 20S core α-subunits [15, 16], decreased expression of PA700 [17], as well as a general loss of all three peptidase activities of the 20S proteasome [18, 19]. In addition, indirect proof of a failure of the proteasome system in PD was provided from neuropathological studies documenting an accumulation of components of the proteasome system, such as proteasomal subunits and proteasomal activators, heat shock proteins, and ubiquitin and ubiquitinated proteins in LBs [20]. However, more recent evidence seems to suggest that ubiquitin accumulated in LBs is preferentially K63-linked (rather than K48-linked) and that this process may be unrelated to impaired proteasome function [21]. In particular, accumulation of K63-linkedubiquitin in LB disease was found to be partly caused by an increase in the levels of the Usp8, an enzyme that deubiquitinates K63-linked chains on α-synuclein leading to a decrease of its degradation via the lysosomal pathway [21].
The underlying causes of proteasome inhibition in PD have not been elucidated. Interestingly, ageing, the main risk factor for developing PD, has been shown to negatively affect both proteasome structure and function [22–24]. Of note, the SN is particularly vulnerable to age-related decreases in proteasome activity, evidenced by a simultaneous decrease of all three protease activities of the proteasome in the aged SN of rats and mice [25]. In addition, various disease-relevant factors have been demonstrated to negatively influence the function of the proteasome system, including pesticides such as rotenone [26], paraquat [27], dieldrin [28] and maneb [29], as well as the mitochondrial toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [30]. The fact that toxins affecting mitochondrial function also lead to impairment of proteasome degradation is not surprising, given that the proteasome degradation cycle is ATP-dependent. Bioenergetic failure, as occurs in PD, could be a significant contributor to the impairment in proteasome function [31]. A recent study using PD cybrids created by transferring mitochondria of PD patients into recipient mitochondrial DNA-depleted cells (NT2 Rho0 cells), demonstrated that PD-related mitochondrial dysfunction is sufficient to decrease the catalytic activity of the 20S proteasome [32]. Also disease-relevant, α-synuclein, especially in its mutated [33, 34] or aggregated [35, 36] forms, can bind to and inhibit the proteasome. Moreover, the finding that DA [37, 38] or factors intrinsic to nigral DA neurons, such as neuromelanin [39] or the DA metabolite aminochrome [40], can inhibit proteasomal function is intriguing, and might underlie the selective vulnerability of nigral DA neurons to proteasomal impairment in PD.
PROTEASOME INHIBITORS AND THEIR MECHANISM OF ACTION
Proteasome inhibitors can be broadly categorized based on their origin into synthetic or natural compounds. Some of the first synthetic inhibitors designed to target the proteasome were peptide aldehydes that act as substrate analogues and potent transition-state inhibitors, primarily of the chymotrypsin-like activity of the 20S proteasome [41]. These compounds, including carbobenzoxy-L-leucyl-L-leucyl-L-leucinal (MG132), carbobenzoxy-L-leucyl-L-leucyl-L-norvalinal (MG115) and car-bobenzoxy-L-isoleucyl-L-gamma-t-butyl-L-glut-amyl-L-alanyl-L-leucinal (PSI), are cell-permeable and block the proteolytic activity of the 26S proteasome, in a reversible manner. In spite of their potency, one of the drawbacks of these compounds is their decreased specificity, as they also inhibit certain lysosomal cysteine proteases and calpains [41].Actinobacteria have been found to naturally produce proteasome inhibitors such as lactacystin and epoxomicin. In contrast to synthetic peptide aldehydes, these structurally distinct natural inhibitors covalently bind to subunits of the proteasome and irreversibly block the proteolytic activity of the proteasome [42]. Previous in vitro studies have provided detailed insight into the molecular mechanism of action of lactacystin by demonstrating that in aqueous environments, lactacystin undergoes spontaneous hydrolysis to clasto-lactacystin dihydroxic acid and N-acetylcysteine, with the intermediacy of clasto-lactacystin-β-lactone [43]. Subsequent studies have demonstrated that clasto-lactacystin-β-lactone, but not lactacystin, is cell permeable and can enter cells where it interacts with the 20S proteasome [44]. In particular, clasto-lactacystin-β-lactone was found to form an ester-linked adduct with the amino-terminal threonine of the mammalian proteasome subunit X, a β-subunit of the 20S proteasome [45]. By covalently attaching to subunit X, clasto-lactacystin-β-lactone potently inhibits all three peptidase activities of the 20S proteasome [45]. Early studies indicated that lactacystin (via the intermediacy of the β-lactone) is highly specific for the proteasome and does not inhibit serine and cysteine proteases [45] or lysosomal protein degradation [46]. Subsequent studies, however, have highlighted additional intracellular targets besides the 20S proteasome, including cathepsin A [47] and tripeptidyl peptidase II [48], which should be acknowledged when interpreting the biological effects using this compound. Given the widespread use of the lactacystin model (especially for rodent studies), findings obtained using this neurotoxin will be emphasized and supported by studies using structurally distinct inhibitors.
MODELING PARKINSON’S DISEASE USING PROTEASOME INHIBITORS
In order to better understand the relation between proteasome inhibition and pathogenic processes relevant in PD, various studies have assessed the effects of proteasome inhibitors on the viability of DA-ergic neurons in vitro and in vivo. In turn, these findings have led to the development and characterization of the proteasome inhibition model of PD.
In this section, we will separately discuss the findings obtained in different model systems, including results from cell culture, Caenorhabditis elegans(C. elegans), medaka fish, rodent models and non-human primates. Each of these model systems provides their own unique advantages and limitations. Studies from cell cultures (either DA-ergic cell lines or primary neuronal cultures) have provided fundamental insight with regards to the pathways recruited and the cell type specificity of proteasome inhibition-induced cell death. Application of proteasome inhibitors in small animal models, such as C. elegans and medaka fish, provides a unique advantage in modeling this PD-related pathway for high throughput genetic or drug screening. The higher complexity of the nervous system of rodents makes them suited as model species to investigate the DA-ergic neurotoxicity of proteasome inhibitors, in particular as it allows the simultaneous assessment of behavioral features. In addition, the use of genetically engineered mice (such as knock-out mice) can reveal novel mediators of proteasome inhibition-induced neurodegeneration in vivo. Finally, non-human primates represent the best approximation of the human central nervous system and their use is indispensable for both investigating pathogenic mechanisms and validating promising therapeutic drugs before they enter clinical trials. As will be discussed, with small exceptions, application of proteasome inhibitors to various model systems leads to death of DA neurons and formation of intracytoplasmic LB-like inclusion bodies (Table 2).
DA-ergic cell lines
Rat adrenal pheochromocytoma PC12 cells have been shown to synthesize, store, release and metabolize DA, and can be differentiated into a neuron-like phenotype upon treatment with nerve growth factor [49]. Treatment of naive and neuronally differentiated PC12 cells with structurally distinct proteasome inhibitors, including lactacystin, epoxomicin, PSI and MG115, was found to lead to dose-dependent cell death [50–54]. Application of proteasome inhibitors caused mitochondrial-mediated apoptotic cell death, as evidenced by increases in mitochondrial membrane permeabilization, mitochondrial cytochrome c release, caspase-3 and -9 activation [51, 53, 55] as well as morphological assessment by electron microscopy [50]. The fact that cell death induced by proteasome inhibitors is apoptotic was confirmed by the protection offered by caspase inhibitor zVAD-fmk against lactacystin- [54] and PSI-induced cell death [53]. Importantly, Fornai and collaborators reported that treatment of PC12 cells with proteasome inhibitors such as lactacystin or epoxomicin leads to the formation of intracellular LB-like aggregates containing α-synuclein, ubiquitin, parkin and ubiquitin-activating enzyme E1 [50]. Consistent with these findings, treatment of neuronally differentiated PC12 cells with lactacystin, epoxomicin or PSI was shown to induce formation of cytoplasmic fibrillar inclusions containing α-synuclein and ubiquitin [54, 56, 57]. Comparable findings were obtained using the mesencephalic DA-ergic cell line N27. Treatment with MG132 was found to lead to dose-dependent death of cultured N27 cells. Similar to PC12 cells, cell death was apoptotic and initiated by a mitochondrial-mediated cascade involving mitochondrial depolarization, cytochrome c release and activation of caspase-2, -3 and -9 [38, 58, 59].
The human neuroblastoma cell line SH-SY5Y has both DA β-hydroxylase and DA transporter (DAT) activity and has been previously used as a model of DA-ergic neurons [60]. Treatment of this cell line with lactacystin has been found to cause dose-dependent apoptotic cell death, involving loss of mitochondrial membrane potential and cytochrome c release [61, 62]. Furthermore, administration of lactacystin to SH-SY5Y cells leads to formation of cytoplasmic aggregates containing ubiquitin andα-synuclein [63].
Primary neuronal cultures
In accordance with the experiments performed on DA-ergic cell lines, application of structurally distinct proteasome inhibitors including lactacystin, epoxomicin and MG132 leads to dose-dependent cell death of DA-ergic neurons in primary ventral mesencephalic cultures [55, 58, 64–67]. Apoptotic cell death is probably involved, as lactacystin induces mitochondrial release of cytochrome c and activation of caspase-3 [55], while caspase inhibitor BAF protects DA neurons against lactacystin- or epoxomicin-induced cell death [67]. Importantly, application of proteasome inhibitors to primary ventral mesencephalic cultures leads to generation of α-synuclein oligomers [68] and formation of cytoplasmic inclusion bodies containing ubiquitin and α-synuclein in surviving DA neurons [64, 67].
C. elegans
Exposure of C. elegans to proteasome inhibitor MG132 results in impaired ubiquitin-related protein degradation and progressive loss of DA neurons [69]. Interestingly, exposure of C. elegans overexpressing α-synuclein in DA-ergic neurons to MG132 led to a significant increase in the degree of protein accumulation and neurodegeneration compared to control α-synuclein overexpressing worms [70].
Medaka fish
Recently, Matsui and collaborators reported the induction of PD-like features after proteasome inhibition in medaka, a small fish of the Actinopterygii class (to which zebrafish also belong) [71]. The authors found that administration of proteasome inhibitors such as lactacystin or epoxomicin to medaka via the cerebrospinal fluid induces a selective loss of DA-ergic and noradrenergic neurons (with a concomitant decrease in DA and noradrenaline tissue levels), leading to a reduction in spontaneous swimming movements. Interestingly, these effects were accompanied by formation of cytoplasmic inclusions containing ubiquitin in different regions of the brain, including in tyrosine hydroxylase (TH)-positive neurons [71]. The development of a medaka proteasome inhibition model showing DA neuron loss and protein aggregation is of note as it can be amenable to high throughput genetic or drug screening experiments evaluating PD-related pathogenesis or novel therapeutic targets [72].
Rodent models
The use of proteasome inhibitors to study the link between impaired protein degradation and DA neuron degeneration has been extended to rodent studies. Proteasome inhibitors have been administered to rats and mice either intracerebrally (at different sites of the nigrostriatal DA-ergic pathway) (Table 3) or systemically (Table 4).
Stereotaxic infusion in the substantia nigra pars compacta
Administration of proteasome inhibitor lactacystin to the SNc of rats leads to nigrostriatal DA neurodegeneration and motor impairment [73–81]. Loss of nigral DA neurons is dose-dependent [77–79] and was found to be either progressive [73, 80] or non-progressive [74, 78], and in certain cases to spread bilaterally [78]. DA cell death following lactacystin is most likely apoptotic in nature, as observed by ultrastructural assessment using electron microscopy [80]. In addition to nigral neurons, neighboring regions such as the ventral tegmental area (VTA) and substantia nigra pars reticulata (SNr) were also investigated for their susceptibility to intranigral lactacystin. VTA DA neurons were seen to be damaged by toxin administration, but to a smaller degree than nigral DA neurons [74, 78], in line with the pattern of degeneration observed in human PD. Furthermore, intranigral lactacystin leads to a moderate loss of non-DA neurons in the SNr and loss of γ-aminobutyric acid (GABA) tissue levels [75], reflecting a certain degree of non-specific damage to SNr GABA-ergic neurons. At a pathological level, DA neuron degeneration resulting from toxin administration is accompanied by microglial activation [73, 82] and iron accumulation [81]. Notably, intranigral lactacystin administration leads to cytoplasmic accumulation of α-synuclein and formation of α-synuclein-positive aggregates in surviving nigral DA neurons [79–82], indicating that proteasome impairment leads to formation of intracellular inclusion bodies in this model. Motor dysfunction induced by intranigral lactacystin is reversed by administration of apomorphine [79] and L-DOPA [76], reflecting a good degree of predictive validity.
Besides affecting neuronal cell populations located in (or neighboring the) SNc, intranigral administration of lactacystin to rats further involves extra-nigral brain regions. In particular, nigral infusion of lactacystin was found to damage cholinergic [82] and non-cholinergic [83] neurons located in the pedunculopontine nucleus (PPN). This was associated with significant microgliosis in the PPN and presence of α-synuclein aggregates in the surviving PPN neurons [82, 83]. The spread of the toxic effects of lactacystin to the ipsilateral PPN is intriguing and might result from the transport or diffusion of injected lactacystin (or pathogenic proteins induced by lactacystin).
Despite the extensive characterization of the intranigral proteasome inhibition model in rats, its application in mice has been more limited. Sun and collaborators initially reported that intranigral proteasome inhibition using MG132 leads to loss of nigral DA neurons and striatal DA levels [58]. Subsequently, Subramaniam and collaborators found that intranigral injection of epoxomicin in mice leads to significant loss of nigral DA neurons, which followed a caudo-rostral gradient similar to that observed in PD [84]. Interestingly, despite the significant loss of nigral DA neurons (60%), neighboring VTA DA neurons were unaffected. In line with the increased resistance of VTA DA neurons to proteasome inhibition-induced cell death, the authors reported that direct intra-VTA infusion of epoxomicin failed to affect this neuronal population [84]. Similarly, intranigral infusion of lactacystin in mice in a dose that causes partial loss of SNc DA neurons (40%) does not lead to loss of VTA DA neurons [85]. These findings are at odds with the intranigral rat models, where loss of VTA DA neurons was previously reported [74, 78]. However, variations in the dose of proteasome inhibitor or time point when neurodegeneration was assessed might account for these differences. In particular, loss of VTA DA neurons following intranigral proteasome inhibition in rats was shown to be both dose-dependent [78] and progressive [74]. Furthermore, infusion of the toxin could cause only a local effect on VTA DA neurons surrounding the injection tract [81, 85] and might not be easily detectable when counting the structure in its entirety. Pathologically, the degeneration of the nigrostriatal DA-ergic pathway following lactacystin administration to the SNc of mice occurs acutely and does not show a progressive pattern [85]. Furthermore, the lesion is accompanied by nigral accumulation of α-synuclein and Ser129-phosphorylated α-synuclein, a post-translationally modified form enriched in LBs [86] and development of motor impairment [85].
Stereotaxic infusion in the medial forebrain bundle
Similar as for intranigral delivery, administration of lactacystin to the medial forebrain bundle (mfb) of rats leads to nigral DA neuron loss and PD-like motor deficits [55, 87]. Nigral DA neuron loss was found to be either progressive [55] or non-progressive [87]. Pathologically, nigral cell loss was accompanied by accumulation of α-synuclein in the form of cytoplasmic inclusion bodies [87]. Using MRI to characterize morphological changes occurring after intra-mfb delivery of lactacystin in rats, Vernon and collaborators revealed a distinct pattern of volumetric changes in both nigral and extra-nigral regions [87]. The authors found that lactacystin leads to an early and sustained decrease in cortical volume (evident already at one week post lesion), followed by subsequent decreases in the volume of the midbrain and striatum and a hypertrophy of the lateral ventricles (appearing from three weeks onwards). Interestingly, decreased cortical volume was the best predictor of poor motor function in lactacystin-treated rats. Cortical thinning was not associated with gross neuronal cell loss, but instead might have resulted from a loss of DA innervation from the midbrain [87].
Similar as in rats, intra-mfb administration of lactacystin in mice leads to dose-dependent progressive nigrostriatal DA neurodegeneration and subsequent motor impairment [68, 88]. Apoptotic nigral cell death could be revealed by electron microscopy and immunostaining for cleaved caspase-3. In comparison to nigral DA neurons, VTA DA neurons were less affected, suggesting a pattern resembling neurodegenerative changes occurring in PD [88]. Pathologically, nigral cell loss was accompanied by a marked glial (astrocytic and microglial) reaction and increases in iron levels in the SN [88]. Furthermore, lactacystin administration led to nigral α-synuclein accumulation and formation (in a subset of TH-positive neurons) of α-synuclein-positive intracellular aggregates. Interestingly, the decreases in spontaneous motor behavior in lactacystin-treated mice could be reversed using DA receptor agonist pramipexole [88]. In addition, Xiao and collaborators recently reported that ageing increases the susceptibility of mice to intra-mfb administration of lactacystin [89]. In particular, the authors reported that aged (12 months old) mice suffer from increased loss of nigral DA neurons and striatal DA depletion following intra-mfb lactacystin compared with young (5 month old) mice. Furthermore, a sustained activation of microglial cells could be observed in the aged (but not young) SN followinglactacystin [89].
Stereotaxic infusion in the striatum
In addition to the SNc and mfb, the striatum has also been investigated as a possible site of delivery of proteasome inhibitors. In an early study, Fornai and collaborators reported that striatal delivery of proteasome inhibitors, such as lactacystin or epoxomicin, to rats leads to nigral DA neuron loss [50]. Importantly, striatal GABA-ergic neurons were not sensitive to intrastriatal delivery of lactacystin, nor were VTA DA neurons, reflecting the specificity of the lesion. Nigral DA neuron death was apoptotic and accompanied by the formation of LB-like intracellular inclusions in surviving neurons that contained α-synuclein, ubiquitin, parkin and ubiquitin-activating enzyme E1 [50]. In concordance with these findings, Miwa and collaborators reported that intrastriatal administration of lactacystin leads to dose-dependent loss of nigral TH-positive neurons and formation of intracytoplasmic α-synuclein-positive inclusions [90]. Interestingly, they also note that higher concentrations of lactacystin can induce non-specific effects and significantly damage striatal cells. These findings support the notion that lactacystin, once administered intrastriatally, can either be retrogradely transported or initiate retrograde degeneration [91] of nigrostriatal DA neurons, as previously reported with other toxins such as 6-hydroxydopamine (6-OHDA). In contrast with this hypothesis, Lorenc-Koci and collaborators failed to reproduce significant nigrostriatal DA neurodegeneration following intrastriatal administration of lactacystin to rats, even at high doses [77]. The reasons for these discrepancies are currently unclear.
Systemic administration
In 2004, McNaught and collaborators described a model of PD resulting from systemic (subcutaneous) chronic administration of proteasome inhibitor PSI [92]. This model replicated key features of PD, including progressive loss of nigral DA neurons, significant neurodegeneration in extra-nigral regions known to be affected in human PD, and development of a progressive parkinsonian syndrome which was L-DOPA- and apomorphine- responsive. Importantly, LB-like intracytoplasmic protein inclusions containing α-synuclein, ubiquitin, synphilin-1, γ-tubulin and parkin, and reactive to thioflavin-S (demonstrating their fibrillar nature), developed in affected regions [92]. By reproducing key pathological features as well as replicating the slow and chronic neurodegeneration associated with PD, the systemic proteasome inhibition model has attracted significant attention from the research community. The initial promising findings were, however, difficult to reproduce in subsequent studies. While some investigators confirmed nigrostriatal pathology, including nigral DA neurodegeneration, following systemic administration of PSI in rats [93–97] or mice [53], others failed to find any significant effects of systemic proteasome inhibition on the nigrostriatal pathway [98–103], or reproduced only partial features of the model such as transient motor impairment without DA neuron degeneration [104] or modest loss of striatal DA levels without motor impairment [105] (see Table 4). The reasons for these discrepant findings are unclear. Various factors leading to poor reproducibility between laboratories have been proposed, including batch-to-batch or supplier-to-supplier variation in the properties of PSI (such as purity or solubility), variations in the manner of preparing and/or administering PSI, or fluctuations in the housing conditions of rats/mice receiving PSI which might influence the metabolism of the toxin [106]. In addition, the brain bioavailability of PSI is not well understood, although the compound is lipophilic and believed to cross the blood-brain barrier. However, with the exception of the initial publication of McNaught and collaborators (that confirmed a significant decrease in proteasome function in the midbrain following systemic PSI), most of the subsequent studies did not control for the proper brain bioavailability of PSI after administration (Table 4). In fact, using transgenic mice expressing GFPu (a reporter that acts as substrate for ubiquitin-dependent proteasome degradation), Bove and collaborators failed to identify any increase in GFPu levels in the brain following systemic PSI administration [98]. This indicates that the compound might not have reached inhibitory concentrations in the brain, leading to the observed lack of effect. Furthermore, as Bukhatwa and collaborators point out, both the dose as well as the route of administration, can influence the impact of PSI on the nigrostriatal pathway and motor function [94]. In particular, the authors note that increasing the dose of PSI leads to a surprising loss of effect after subcutaneous administration, while subcutaneous and oral, but not intraperitoneal, administration of the same dose of PSI (8 mg/kg) leads to a significant reduction of nigral DA neurons and ensuing motor deficits [94]. The fact that the route of administration influences the outcome further suggests that proper bioavailability of the toxin is critical for successfully creating the model. The loss of effect with increasing doses of PSI, also reported by McNaught and Olanow [106], is interesting and might indicate that at higher doses PSI becomes less soluble, limiting its absorption from the injection site. In an attempt to achieve a more continuous delivery of the toxin, Shin and collaborators investigated the administration of PSI using osmotic minipumps implanted subcutaneously [107]. Using these conditions, PSI induced a progressive deterioration of locomotor activity, which was however unrelated to nigrostriatal degeneration (as nigral DA neurons were not significantly affected by the toxin) [107]. Given the limited reproducibility of the model amongst different laboratories, and until the reasons for these discrepancies are being elucidated, the use of the systemic proteasome inhibition model of PD remains controversial [108].
Non-human primate models
In the only attempt to date to model proteasome inhibition-induced neurodegeneration in non-human primates, cynomolgus monkeys were treated systemically with PSI, using a similar protocol as proposed in rats. Systemic PSI administration to monkeys failed, however, to produce any significant loss of nigral DA neurons [101]. Similar to the rodent systemic PSI model, various factors might explain this lack of effect, including uncontrolled variables related to the pharmacokinetics and brain penetranceof PSI.
Genetic models
The toxin-based models described above employ proteasome inhibitors to target the 20S catalytic core particle, thereby affecting both 26S and 20S protein degradation pathways. In addition, most of the experiments reported on human post-mortem tissue utilize kinetic assays and fluorogenic peptide substrates that are degraded by the 20S proteasome, thereby specifically assessing the proteolytic activities of the 20S core particle (Table 1). Less is known, however, regarding the particular involvement of 26S proteasomal degradation in PD. Experimental evidence in this regard was recently provided by Bedford and collaborators, utilizing the Cre/loxP system to generate mice with a targeted deletion of a key subunit of the 19S regulatory particle, Psmc1, in TH neurons (Psmc1fl/fl; THCre) [109]. Deletion of Psmc1 prevented the correct assembly of the 19S particle with the 20S proteasome, leading to a specific disruption of 26S proteasomal degradation, while 20S degradation remained preserved. Interestingly, in such conditions, the authors observed a striking loss of nigral DA-ergic neurons and striatal DA-ergic fibers, and a substantial depletion of striatal DA content (∼90%) compared to control mice. In addition, surviving neurons developed intracellular eosinophilic inclusions containing α-synuclein, ubiquitin and mitochondria, resembling human pale bodies (precursors to LBs) [109]. This genetic model provides an interesting platform to investigate 26S proteasomal dysfunction in nigral DA neurons and illustrates the possible involvement of this pathway in PD. One of the limitations of the model might be that Psmc1fl/fl; THCre mice die before postnatal day 28 due to autonomic dysfunction [109].
MECHANISMS UNDERLYING PROTEASOME INHIBITION-INDUCED CELL DEATH
Various studies have been performed in an attempt to decipher the pathways mediating neuronal death induced by proteasome inhibitors. As described below, the determinants of cell death have been suggested to involve diverse cellular pathways related to production of reactive oxygen species, disturbances in mitochondrial function, induction of endoplasmic reticulum (ER) stress, and cytoplasmic accumulation of p53, iron and α-synuclein (Fig. 2, upperpanel).
Accumulation of p53
Multiple lines of evidence obtained from in vitro and in vivo experiments suggest that cell death induced by proteasomal inhibitors is apoptotic in nature. One candidate for regulating apoptotic cell death following proteasome inhibition is transcription factor p53. Accumulation of p53 leads to activation of pro-apoptotic pathways, which can be mediated by transcriptional effects, such as increased expression of pro-apoptotic genes including Puma or Bax, or non-transcriptional effects, for instance by direct actions of p53 on mitochondrial permeability. Of note, p53 is primarily degraded via the proteasome [110]. As a consequence, treatment of PC12 cells or primary neuronal cultures with proteasome inhibitors such as PSI, lactacystin or MG115 leads to accumulation of p53 [52, 53, 111]. Remarkably, inhibition of p53 (either chemical or genetic) is sufficient to prevent proteasome inhibition-induced apoptotic cell death of PC12 cells [52, 53] as well as of primary cortical neurons [111]. These findings suggest that accumulation of p53 following proteasome inhibition might be a key event triggering apoptotic cell death, and some evidence suggests that it does so via non-transcriptional mechanisms [53]. Consistent with such a hypothesis is the finding of increased p53 levels in the SNc of mice treated systemically with PSI at a time point preceding DA neuron loss, as well in the SNc of PD patients [53].
Mitochondrial dysfunction
By treating SH-SY5Y cells with continuous low levels of MG115, Sullivan and collaborators could find that chronic proteasome inhibition leads to a marked reduction in mitochondrial complex I and complex II maximal activities, and an increase in the production of mitochondrial reactive oxygen species [112]. Furthermore, genetic depletion of 26S proteasomes in vivo induces the accumulation of mitochondria within intracellular inclusion bodies resembling PD-related pale bodies [109, 113]. These findings suggest that proteasome inhibition influences mitochondrial function andturnover.
Oxidative and nitrosative stress
Treatment of cell lines and primary cell cultures with proteasome inhibitors such as MG132, epoxomicin or lactacystin has been found to lead to glutathione depletion, formation of reactive oxygen species, and increases in levels of protein carbonyls, malondialdehyde, 8-hydroxyguanosine and 8-hydroxy-2’-deoxyguanosine [51, 62, 114–116]. Notably, treatment with antioxidants can prevent proteasome inhibition-induced cell death of PC12 cells [62] as well as of cultured mesencephalic neurons [115], suggesting that this state of oxidative stress can be cytotoxic. Evidence for oxidative stress following proteasome inhibition has also been obtained in vivo, as intrastriatal administration of lactacystin in rats leads to an increase in the oxidative stress marker HO-1 in nigral neurons [90], while genetic depletion of neuronal 26S proteasomes in mice leads to generation of reactive oxygen species and accumulation of malondialdehyde [117]. In addition, proteasome inhibitors epoxomicin and lactacystin have been found to increase the levels of reactive nitrogen species and 3-nitrotyrosine in cell cultures, while nitric oxide synthase inhibitor L-NAME protects against lactacystin-induced toxicity [116]. In turn, reactive oxygen species [118] and oxidative stress end products such as 4-hydroxy-2,3-trans-nonenal [119] can inhibit proteasome function, suggesting a close interaction between the two pathways during neurodegeneration.
Iron dysregulation
Accumulating evidence suggests that iron dysregulation might be a key mechanism underlying proteasome inhibition-induced DA neuron death [120]. Treatment of the MES23.5 DA-ergic cell line with lactacystin has been found to cause a marked increase in labile iron (Fe2+) levels. Accumulation of iron seemed to be cytotoxic, as iron chelator deferoxamine reduced lactacystin-induced neuron death [121]. Similarly, in vivo, iron levels have been found to be increased in the SN following intranigral [81] or intra-mfb [88, 122, 123] administration of lactacystin. Evidence suggesting that such an increase might be detrimental for nigral DA neurons has been provided by the findings that both chemical [123] as well as genetic [122] iron chelation are neuroprotective against lactacystin-induced DA neurotoxicity in vivo. Iron accumulation induces the formation of reactive oxygen species (such as hydroxyl radicals, following the Fenton reaction) as well as the formation of α-synuclein aggregates which can contribute to the demise of nigral DAneurons [124].
Accumulation of α-synuclein
Accumulation of the presynaptic protein α-synuclein plays a central role in PD pathogenesis. Evidence that increased levels of α-synuclein are toxic to nigral DA neurons comes from the findings that duplication or triplication of Snca (the gene encoding α-synuclein) can induce PD in humans with a gene-dosage effect [125], while accumulation and aggregation of α-synuclein in the form of LBs is a pathological hallmark of sporadic PD [3]. Furthermore, overexpression of α-synuclein (for instance via viral vector delivery) leads to nigral DA neurodegeneration [126, 127], while recent in vivo findings support the hypothesis that the process of α-synuclein aggregation is toxic to nigral DA neurons [128, 129]. As such, a detailed understanding of the mechanisms regulating α-synuclein degradation is of critical importance in elucidating its aberrant accumulation in synucleinopathies. A great deal of effort has been invested in deciphering the degradation pathways controlling α-synuclein in neurons [130]. The results of these studies seem to indicate that α-synuclein is a substrate for both proteasomal and autophagic degradation, although whether or not α-synuclein is turned over via the proteasome has remained to a certain degree contentious. Cell-free models in which recombinant α-synuclein was incubated with purified 20S [13, 14, 131–133] or 26S [13] proteasomes provide direct evidence for proteasomal degradation of this protein. Similarly, experiments performed in DA-ergic cell lines such as PC12 and SH-SY5Y have indicated that application of proteasome inhibitors lactacystin and epoxomicin leads to an increase in the levels of α-synuclein [14, 134–137]. The notion that α-synuclein is a substrate for the proteasome was, however, challenged by the failure of various laboratories to replicate significant α-synuclein accumulation in PC12 cells, HEK293 cells and primary mesencephalic cultures following application of structurally distinct proteasome inhibitors such as lactacystin, MG132 or PSI [54, 138–142], or found only modest accumulation of α-synuclein in primary mesencephalic cultures treated with epoxomicin [143]. The reasons for these discrepancies are currently unclear, but may relate to the use of different protocols of administration of proteasome inhibitors (e.g. dosing, incubation time, see [14]) or the use of distinct cell models and/or culture conditions which could lead to different baseline expression levels of α-synuclein. In addition, proteasome inhibition can induce a compensatory activation of autophagy as a means to maintain cellular proteostasis [137, 144–146]. As α-synuclein is a substrate for autophagic degradation [130], this might explain the lack of effect seen in certain models.
In order to address this controversy and critically assess the degradation pathways of α-synuclein, Ebrahimi-Fakhari and collaborators applied multiphoton imaging and cranial windows to study the turnover of α-synuclein in vivo. In a seminal study, the authors reported that topical treatment of the cortex with clasto-lactacystin-β-lactone induced accumulation of endogenous α-synuclein in nontransgenic mice as well as in human α-synuclein transgenic mice, providing direct evidence that the proteasome system regulates α-synuclein turnover in vivo [147]. In contrast, topical application of BafA1, an inhibitor of the autophagy-lysosomal pathway, led to increased levels of α-synuclein in human α-synuclein transgenic mice but not in nontransgenic mice. The authors propose a model in which the proteasome system degrades α-synuclein under both physiological conditions and under increased α-synuclein burden, whereas the autophagy-lysosomal pathway is recruited to degrade α-synuclein only when intracellular levels become elevated [148].
In view of these findings, and considering the toxicity associated with increased α-synuclein expression and aggregation [149], it might be speculated that α-synuclein plays a relevant role in mediating proteasome inhibition-induced cell death. Surprisingly, such a hypothesis has not been investigated in detail. In a recent study, Bir and collaborators reported that accumulation of α-synuclein following lactacystin administration to SH-SY5Y cells plays an important role in mediating proteasome inhibition induced toxicity, as siRNA-mediated knockdown of α-synuclein was able to completely prevent lactacystin-induced cell death [61]. At the same time, using α-synuclein knock-out mice, Paine and collaborators reported that genetic depletion of 26S proteasomes in nigral DA neurons of mice leads to nigral DA neuron death and formation of intracellular inclusion bodies, irrespective of the presence or absence of α-synuclein [113]. This suggests that in certain scenarios inhibition of the proteasome system might cause cellular toxicity independent of α-synuclein. Further studies would be instrumental to define the role played by α-synuclein in proteasome inhibition-induced cell death.
Besides changes in expression, α-synuclein can also undergo post-translational modifications, such as oxidation, nitration, phosphorylation or truncation, which might play important roles in mediating its aggregation and/or toxicity [150, 151]. Previous studies analyzing the composition of LBs revealed high levels of α-synuclein phosphorylated at Ser129 [86, 152], indicating that this post-translational modification might be associated with fibril formation and/or toxicity. Although its role is not completely understood [153], studies evaluating the impact of authentically phosphorylated α-synuclein at Ser129 have revealed gain-of-function toxic roles in both Drosophila [154] and rat [155] α-synuclein-based models. Interestingly, Machiya and collaborators reported a specific increase in Ser129-phosphorylated α-synuclein after treatment of SH-SY5Y cells with proteasome inhibitors lactacystin or MG132 suggesting that Ser129-phopshorylated α-synuclein is a substrate for proteasomal degradation [156]. Similarly, inhibition of the proteasome in vivo using lactacystin leads to accumulation of Ser129-phopshorylated α-synuclein in nigral (DA-ergic) neurons [85]. As for α-synuclein, however, it is currently unknown whether the increase in Ser129-phosphorylated α-synuclein following proteasome inhibition contributes to neuronal toxicity or constitutes an epiphenomenon following lesion.
It is interesting to note that while proteasome inhibition can lead to accumulation of α-synuclein, increased levels of α-synuclein can in turn inhibit proteasomal function. Previous findings indicate that α-synuclein, in particular in its oligomeric form [35, 157], can interact with and inhibit the catalytic activity of the 26S proteasome [34, 36]. This, in turn can lead to further α-synuclein accumulation, initiating a pathological feed-back loop [158, 159]. In a compelling study addressing the relation between α-synuclein aggregation and proteasome dysfunction, Chu and collaborators described a decreased expression of the 20S proteasome in SNc neurons of PD patients, but only in cells that contain α-synuclein inclusions [160]. Remarkably, the expression of the 20S proteasome in nigral DA-ergic neurons without α-synuclein pathology in PD was the same as in healthy controls. In a similar manner, the authors describe a decreased expression of the 20S proteasome in nigral DA-ergic neurons with α-synuclein inclusions (but not in those without α-synuclein inclusions) after AAV6 mediated over-expression of α-synuclein in the rat SNc [160]. This would argue for a close relation between α-synuclein aggregation and proteasome dysfunction during the course of PD. Such a view might seem at odds with the finding of intact proteasome activity in brain regions such as the striatum, hippocampus, or cortex[16, 17, 19, 161] that do develop LB pathology in the course of PD. However, these findings should be interpreted with caution, as in the post-mortem studies performed so far evaluating proteasome function in extra-nigral regions, the neuropathology and Braak staging of the patients investigated was not fully described. Notably, extra-nigral regions such as the striatum, hippocampus, and cortex become pathologically involved during later disease stages (Braak stages 4–6) [162], and possible effects on proteasome function in such regions might have been overlooked if using tissue from patients with a short disease duration (e.g. Braak stage 3, in which the SNc is already affected by LB pathology). Case in point might be the study of Tofaris and collaborators that could reveal a significant decrease in the proteasome activity in the SN, but not frontal, cingulate or occipital cortices from cases with mild pathology and relatively short disease duration (6.3±3 years) [19]. At the same time, in the study of McNaught and collaborators, the proteasome activity in the upper pons at the level of the locus coeruleus (LC) was not affected in patients in which the proteasome activity was found to be decreased in the SNc [17]. This is interesting, as the LC can develop Lewy body pathology at earlier Braak stages (Braak stage 2) than the SNc (Braak stage 3) [162]. This suggests that other factors, besides α-synuclein aggregation, might be required to decrease the proteasome function (e.g. mitochondrial impairment), and the cumulation of such factors occurs in the SNc but not in other regions of the brain. Future investigations are warranted into the integrity of the proteasome system in extra-nigral regions, and ideally controlled for disease staging and LB distribution. Such studies hold the promise to reveal whether the proteasome function in extra-nigral regions succumbs in later stages of disease, and whether this is universally (i.e. outside the SN) linked with the process of LB formation. Alternatively, it may be conceded that proteasome dysfunction and generation of LBs are partly dissociated processes (at least in some brain structures), as evidenced by the lack of proteasome impairment in cingulate and frontal cortices of cases of DLB, regions which stain positive forLBs [19].
Microarray and proteomic studies
In addition to targeted pathway analysis, unbiased screens using microarray or proteomic studies have also revealed insights into mechanisms associated with proteasome inhibition. Microarray studies performed on primary cortical neurons upon exposure to proteasome inhibitor lactacystin revealed a biphasic time-dependent effect, characterized by an initial activation of neuroprotective pathways which was followed, at later stages, by activation of pro-apoptotic pathways [163, 164]. One of the protective responses observed in the early phases was characterized by the upregulation of components of the ubiquitin-proteasome system, such as genes encoding ubiquitins (Ubb), proteasome subunits (e.g. Psma1, Psma7, Psmc1, Psmc3), E2 ubiquitin-conjugating enzyme (Ubc) and ubiquitin protein ligase E3A (Ube3a), possibly as part of a regulatory mechanism attempting to counteract the effects of proteasome inhibition. In addition, genes encoding heat shock proteins and molecular chaperones, such as heat shock protein 70 (Hspa1a), heat shock protein A (Hspa9a), heat shock protein 22 (Cryac), heat shock protein 27 (Hspb8) and heat shock protein 47 (Serpinh1), were upregulated, presumably to prevent accumulation of misfolded/damaged proteins. ER stress-associated genes, such as DNA-damage inducible transcript 3 (Ddit3) (also known as CHOP) and CCAAT/enhancer binding protein (C/EBP) beta (Cebpb), were also found to be increased in expression at an early time point after proteasome inhibition. As both proteins mediate induction of cell death in conditions of ER stress, this suggests an early pro-apoptotic role of ER stress after lactacystin administration [163]. The later stages of proteasome inhibition were associated with neuronal death and characterized by an upregulation of antioxidant genes (e.g. Gclm, Gsta4, MgstI, MtI) and a downregulation of genes involved in cholesterol biosynthesis (e.g. Hmgcr, Cyp51, Dhcr7, Lss) [163, 164]. Similar changes could be observed in proteomic screens of PC12 cells incubated with proteasome inhibitors. PSI application to PC12 cells led to upregulation of GRP94 and GRP78, molecular chaperones residing in the ER (and induced in conditions of ER stress), as well as of heat shock protein 27. In addition, PSI led to an upregulation of aldose reductase and a downregulation of galectin-1 [165]. Incubation of PC12 cells with the structurally distinct proteasome inhibitor lactacystin caused, besides others, an increase in the expression of heat shock proteins Hsc70 and GRP78, and aldose reductase, and a decreased expression of TH [166]. The upregulation of aldose reductase, a NADPH-dependent oxidoreductase mediating the reduction of aldehydes and involved in protection against oxidative stress [167], suggests a compensatory reaction of the cells to limit the detrimental effects after PSI or lactacystin administration. Conversely, the downregulation of galectin-1, a β-galactosidase binding lectin induced by brain injury and associated with neuronal regeneration [168], might reflect a decreased capacity to cope with cellular stress in conditions of proteasome inhibition. Aside from the above mentioned studies, performed at acute time points following incubation with proteasome inhibitors (maximum 24–48 h), Ding and collaborators investigated the effects of low-level, chronic proteasome inhibition on gene expression [169]. Incubation of neural SH-SY5Y cells with MG115 during 12 weeks led to a particular gene expression profile, characterized, besides others, by an increase in genes related to inflammation (e.g. STAT1, ISGF3, NPTX1), apoptosis (e.g. PIDD, PDCD4), nuclear homeostasis (e.g. MAN1) and lipid metabolism (e.g. NSMAF). In addition, the authors note an important downregulation of the synaptic vesicle monoamine transporter, suggesting impaired catecholamine sequestration in synaptic vesicles following chronic proteasomeinhibition [169].
CELL TYPE-SPECIFIC VULNERABILITY TO PROTEASOME INHIBITION-INDUCED DEATH
In contrast to other classical PD toxins which achieve specific DA cell death due to their intrinsic mechanism of action (such as the requirement for DAT for uptake of 6-OHDA or MPP+), the active metabolite of lactacystin, clasto-lactacystin-β-lactone, diffuses freely through the cell membrane, thereby influencing both DA-ergic and non-DA-ergic neurons. It has been argued, however, that DA-ergic neurons might be particularly vulnerable to proteasome-inhibition induced cell death. Petrucelli and collaborators reported that treatment of primary mesencephalic cultures with lactacystin or MG132 leads to a reduction in the number of TH-positive cells, while TH-negative neurons remained unaffected [170]. Similarly, treatment of primary mesencephalic cultures with lactacystin or MG132 induces loss of DA-ergic neurons, with either no [64] or a significantly smaller [65] decrease in viability of GABA-ergic neurons. It has been proposed that the vulnerable nature of DA-ergic neurons could be due to the presence of DA itself. In an elegant study, Fornai and collaborators demonstrated that depleting DA in PC12 cells with reserpine led to a remarkable resistance to lactacystin-induced inclusion body formation and cell death [50]. Conversely, when PC12 cells were exposed to L-DOPA, thereby increasing intracellular DA levels, PC12 cells became more sensitive and demonstrated an increase in inclusion body formation and cell death following lactacystin [50]. Notably, similar results were obtained in vivo. Decreasing DA synthesis by systemic administration of TH inhibitor α-methyl-p-tyrosine significantly protected the nigrostriatal DA-ergic pathway against intrastriatal lactacystin administration. Conversely, systemic administration of L-DOPA prior to intrastriatal lactacystin led to an enhancement of the neurodegenerative effects observed at the level of the nigrostriatal pathway [50]. The reasons for which DA neurons are particularly vulnerable to the effects of proteasome inhibitors are not well understood, but might result from the metabolism of DA that leads to generation of reactive oxygen species and oxidation of proteins, providing an additional burden to proteasomaldegradation.
At the same time, it should be mentioned that other studies performed in primary mesencephalic cultures failed to provide evidence for the specificity of proteasome inhibitors to induce DA cell death [66, 115, 171]. The reasons for these discrepancies are unclear, but might be due in part to differences in culture conditions or the concentration and duration of exposure to proteasome inhibitors. Of note, Biasini and collaborators reported that application of MG132 and lactacystin to primary mesencephalic cultures at low concentration leads to specific loss of DA content, without changes in GABA, whereas at higher concentrations of inhibitor GABA levels were also decreased [140]. Similar findings have been obtained in vivo. Intranigral administration of lactacystin in a dose that causes partial (40%) loss of nigral DA neurons does not damage the GABA-ergic neurons in the surrounding SNr [85]. However, administration of the toxin in a higher dose, causing a severe (90%) loss of nigral DA neurons leads, however, to a partial (27%) loss of non-DA neurons (presumably GABA-ergic) in the SNr and modest (16%) depletion of SN GABA tissue levels [75]. This suggests that, while DA-ergic neurons might be particularly vulnerable to proteasome inhibition, additional effects on non-DA-ergic neurons can also be observed, especially at higher doses of the toxin.
Besides neurons, proteasome inhibitors can diffuse into glial cells. Interestingly, glial cells, especially astrocytes, seem to be particularly resistant to proteasome inhibition-induced toxicity [172]. Cultured astrocytes were found to be resistant to concentrations of proteasome inhibitors that cause neuronal death [173–175]. The mechanisms underlying the resistance of astrocytes to proteasome inhibition-induced toxicity are incompletely understood, but might be related to the high constitutive levels of small heat shock protein HSP25 [176], the intrinsically higher proteasome activity of astrocytes compared to neurons [177] or the increased resistance of astrocytes to proteasome inhibition-induced oxidative stress [114]. However, non-specific effects on glial cells might occur at high doses of proteasome inhibitors and should be carefully considered[79, 175].
Although the indiscriminate mechanism of cell entry of proteasome inhibitors might seem to be a disadvantage, its independence of transport systems, such as DAT (required for the uptake of 6-OHDA and MPP+), or metabolic enzymes, such as monoamine oxidase B (MAO-B) (required for the metabolization of MPTP) represents a distinct feature compared to classical toxin-based models of PD. In particular, the proteasome inhibition model can be employed to assess neuroprotective compounds that are known to (or might) influence the expression of DAT or MAO-B [178]. In addition, the unrestricted mechanism of action of proteasome inhibitors makes them attractive in modeling extra-nigral pathology in PD. Indeed, neurodegeneration in PD is not restricted to the SNc, but can extend to non-DA-ergic neurons, such as cholinergic neurons in the nucleus basalis of Meynert (NBM), dorsal motor nucleus of the vagus (DMNV) and PPN, noradrenergic neurons in the LC, serotonergic neurons in the raphe nuclei (RN) and hypocretin/orexin neurons in the hypothalamus, which contributes to the development of motor and non-motor symptoms [179–184]. Studies using systemic administration of proteasome inhibitors support the presence of extra-nigral pathology, including noradrenergic degeneration in the LC, cholinergic degeneration in the NBM and DMNV, and serotonergic degeneration in the RN following toxin administration (Table 4), as does the evidence of α-synuclein aggregation in the DMNV following intragastric administration of PSI [185]. Furthermore, intra-cerebral administration models provide further evidence of non-catecholaminergic degeneration following proteasome inhibition, such as the loss of cholinergic neurons in the PPN after intranigral lactacystin administration (Table 3), while injection of lactacystin into the NBM leads to loss of cholinergic neurons within this structure and formation of α-synuclein positive inclusion bodies in surviving cholinergic neurons [186]. The lack of specificity of proteasome inhibitors towards DA neurons might in fact favor the use of this model to study extra-nigral pathology and its impact on motor and non-motor symptoms in PD. In a recent study employing the intranigral lactacystin model to study PPN cholinergic degeneration in PD, Pienaar and collaborators found that selective stimulation of PPN cholinergic neurons using the expression of an excitatory DREADD reversed the lesion-induced motor deficits [187], which might offer additional insights into the clinical benefits observed after deep brain stimulation of this structure in patients withPD [188].
INTERACTIONS BETWEEN PROTEASOME INHIBITION AND PARKINSON’S DISEASE-RELATED GENES
Gene-environment interactions play a key role in the pathogenesis of PD [189]. Various studies have investigated the interactions between proteasomal dysfunction and genetic background, utilizing PD-related mutations. Notably, a synergistic effect could be observed between expression of PD-related α-synuclein mutants A30P and A53T and proteasomal inhibition in cultured cells. Expression of A53T α-synuclein in SH-SY5Y cells has been found to increase the formation of α-synuclein aggregates after lactacystin administration compared to cells expressing wild-type α-synuclein [63]. Furthermore, expression of A30P α-synuclein increased the sensitivity of PC12 cells to lactacystin-induced apoptotic cell death when compared to PC12 cell expressing wild-type α-synuclein [34]. Consistent with these findings, expression of either A53T or A30P α-synuclein in NT-2, SK-N-MC or M17 cells was found to increase susceptibility to proteasome inhibition-induced cell death compared to expression of the wild-type protein [170, 190]. Besides α-synuclein, expression of the G2019S LRRK2 mutation in mice has been found to increase striatal DA loss following intra-mfb administration of lactacystin [89] (Fig. 3). This is particularly interesting as the penetrance of the G2019S LRRK2 in PD patients is incomplete, suggesting that environmental factors might play a role in the pathogenic expression of thismutation.
INTERACTIONS BETWEEN PROTEASOME INHIBITION AND PARKINSON’S DISEASE-RELATED TOXINS
PD is a multifactorial disorder and the various pathogenic pathways interact in a complex manner to produce death of DA neurons [191]. Although proteasomal dysfunction can lead on its own to DA cell death, a critical question remains on how it interacts with other pathogenic pathways occurring in PD. In order to address this question, studies were performed investigating the reaction of DA cells to simultaneous application of proteasome inhibitors and PD related neurotoxins, such as 6-OHDA, MPP+ or rotenone.
Hoglinger and collaborators reported that treatment of primary mesencephalic neurons with epoxomicin sensitized DA neurons to death induced by either rotenone, MPP+ or 6-OHDA [192]. Consistent with these findings, exposure of SH-SY5Y cells to PSI or lactacystin was found to increase cell death induced by 6-OHDA [62] or rotenone [60]. In contrast, the use of sub-toxic levels of proteasome inhibitors has been associated with neuroprotective effects (Fig. 2, lower panel). Using primary cortical cultures, Lee and collaborators reported that low concentrations of proteasome inhibitors MG132 and clasto-lactacystin-β-lactone increased the resistance of cells to oxidative injury associated with paraquat and H2O2 treatment [193]. In line with these findings, treatment of PC12 cells [194] or primary mesencephalic neurons [195] with low concentration of lactacystin was protective against 6-OHDA. Comparable findings were observed in vivo. Sub-lethal concentrations of lactacystin and MG132, when co-administered intranigrally with 6-OHDA [196] or MPP+ [197], were found to significantly protect nigral DA neurons against degeneration. Similarly, systemic delivery of PSI at a sub-toxic dose protects against MPTP-induced loss of nigral TH-positive neurons [198]. The mechanisms underlying proteasome inhibition-induced neuroprotection might be related to activation of the Nrf2-ARE pathway and induction of antioxidant defenses [194, 195, 199]. Indeed, Nrf2 is degraded via the proteasome, and its accumulation following proteasome impairment can lead to activation of ARE-dependent transcription of antioxidant genes [199]. In addition, Sawada and collaborators reported that sub-lethal concentrations of lactacystin, PSI and MG132 protect DA neurons in primary mesencephalic cultures against MPP+-induced neurotoxicity and simultaneously led to the generation of synuclein- and ubiquitin- positive intracellular inclusions which are not observed with either toxic treatment alone [197]. Similarly, Inden and collaborators observed the presence of α-synuclein-positive inclusion bodies after intranigral co-administration of 6-OHDA and lactacystin or MG132, that were not observed with 6-OHDA treatment alone [196]. This suggests that an additional neuroprotective mechanism of proteasome inhibitors might be to stimulate the formation of larger α-synuclein aggregates, which are possibly less toxic than smaller oligomericspecies [196].
In conclusion, the level of inhibition of proteasome activity seems to be critical in dictating neuroprotection vs. enhanced cell death following toxic stimuli: while partial inhibition is neuroprotective, a more widespread inhibition enhances the sensitivity to subsequent toxic stimuli.
THE VALIDITY OF THE RODENT PROTEASOME INHIBITION MODEL OF PARKINSON’S DISEASE
The proteasome inhibition model has emerged as a new model of PD that might provide novel clues in our attempts to decipher the complex pathogenesis of PD as well as represent a platform to investigate neuroprotective therapies. Its implementation in pre-clinical research is conditioned, however, by its ability to fulfill the validity criteria for modeling PD. These include requirements of construct validity (i.e. whether the model is induced in a manner that replicates pathogenic pathways relevant in the human disease), face validity (i.e. how closely does the model replicates key biochemical, pathological and behavioral changes as identified in the human disease) and predictive validity (i.e. the fidelity with which the model can positively identify clinically effective therapies) (Table 5).
Construct validity
Post-mortem data obtained from sporadic PD indicate structural and functional defects in the 26/20S proteasome (Table 1). In PD patients, the proteasome seems to be specifically inhibited in the SNc, with sparing of proteasome function in other areas such as the striatum or cortex [17, 19, 161]. As such, animal models based on inhibition of proteasome function fulfill construct validity, and might be especially relevant when administered intranigrally.
At the same time, it should be acknowledged that the causes of proteasome dysfunction in patients with PD are currently unknown. These might include, among others, brain ageing and factors related to underlying pathogenic processes, such as mitochondrial dysfunction, oxidative stress or α-synuclein aggregation. In addition, the decreased expression of structural 20S α-subunits [15, 16] can impede the assembly of the 26S/20S proteasome and, subsequently limit degradation of targeted substrates. Although proteasome inhibitors such as lactacystin are present in the environment as Streptomyces natural products and Streptomyces spp. (especially when isolated from agricultural soils) can cause loss of DA-ergic neurons in C. elegans, no epidemiological links exist between exposure to proteasome inhibitors and risk of developing PD. As such, the construct validity of the proteasome inhibition model of PD is limited to reproducing the same functional outcome as in patients with the disorder (i.e. decreased catalytic activity of the 20S proteasome), and not necessarily the same cause of proteasome inhibition as it occurs in PD.
Face validity
Administration of proteasome inhibitors to the nigrostriatal pathway has been found to consistently replicate key pathological hallmarks of PD, including nigral DA neurodegeneration and aberrant α-synuclein accumulation and/or aggregation [50, 79, 81, 82, 85, 87, 88, 90]. The effect on α-synuclein is notable, as accumulation and/or aggregation of endogenous α-synuclein is a feature difficult to reproduce in other toxin-based models of sporadic PD. Similar as in PD, nigral cell loss following proteasome inhibition is accompanied by a persistent neuroinflammatory reaction [82, 88]. Furthermore, the intranigral lactacystin model replicates cholinergic degeneration in the PPN as observed in the human condition [82], while volumetric changes described after intranigral [81] or intra-mfb [87] administration of lactacystin mirror morphological changes (such as reductions in cortical and striatal volume and hypertrophy of the lateral ventricles) occurring in PD. With regards to behavioral changes, proteasome inhibition models induce impairment in spontaneous motor activity, decreased motor coordination, and impaired skilled motor function [78, 85, 87, 88].
In contrast to motor symptoms, the evaluation of PD-relevant non-motor symptoms in proteasome inhibition models has been much more limited. We have recently characterized changes in non-motor behavior in mice intranigrally infused with lactacystin [85]. Our results indicate that nigral proteasome inhibition leads to the manifestation of a complex phenotype, characterized by anxiety-like behavior, somatosensory dysfunction and perseverative motor behavior suggestive of loss of cognitive flexibility. In addition, lactacystin-lesioned mice demonstrate hyperactivity/restlessness and develop spontaneous contralateral circling [85]. Although it is regarded that unilateral damage to the nigrostriatal DA-ergic pathway leads to spontaneous ipsilateral turning [201], intranigral epoxomicin administration in mice was similarly found to induce contralateral turning asymmetry, possibly linked with a hyperactive phenotype of the surviving DA neurons [84]. Furthermore, Konieczny and collaborators recently described an increase in DA release in the ipsilateral striatum after intranigral administration of lactacystin to rats, leading to development of spontaneous contralateral rotations [202]. Interestingly, this spontaneous rotational behavior could be blocked by combined administration of DA D1 and D2 receptor antagonists and represented an early event following toxin administration [202]. The appearance of a hyperkinetic behavior in mice intranigrally lesioned with lactacystin biased our evaluation of depressive-like behavior using the tail suspension test [85], and might require the use of depression-based paradigms less reliant on motor function and/or investigation of lesioned mice at a later time point post-lesion. In a separate study, Lu and collaborators described excessive somnolence in rats intra-nigrally lesioned with MG132, which might reproduce the nocturnal sleep disturbance and excessive daytime somnolence observed in PD patients [203]. Future studies evaluating non-motor behavior following application of proteasome inhibitors are critical in order to better define the spectrum of PD-related symptoms present in this model.
Predictive validity
With regards to PD-related symptoms, motor impairment in proteasome inhibition induced models of PD has been found to be successfully reversed using clinically effective compounds including L-DOPA [76], apomorphine [79] and pramipexole [88]. In addition, the proteasome inhibition model has been employed as platform to identify (or confirm) promising targets for achieving nigral DA neuroprotection. Genetic or pharmacological studies identified various strategies that can interfere with proteasome-inhibition induced nigral DA cell death, including increasing the expression of glial cell line-derived neurotrophic factor [178, 204–206], genetic [122] or chemical [123] iron chelation, and administration of a wide range of compounds such as MAO-B inhibitors selegiline and rasagiline [207], DA receptor agonists pramipexole [208] and D-264 [209], sodium valproate [210], and rapamycin [211] (Fig. 3). Interestingly, while most of these strategies were found to demonstrate similar effects in classical toxin-based models such as 6-OHDA and MPTP, a recent study failed to identify neuroprotective properties of celastrol (a compound with strong anti-oxidant and anti-inflammatory effects) in the lactacystin model, although being effective in the MPTP model [212]. This suggests that proteasome inhibition models induce DA neurodegeneration by distinct pathways when compared to classical models and might be useful in cross-validation of therapeutic agents.
CONCLUDING REMARKS
Local application of proteasome inhibitors to various regions of the nigrostriatal DA-ergic pathway has been found to consistently lead to nigral DA neuron degeneration and aberrant protein accumulation. This suggests a connection between proteasomal impairment and pathological processes occurring in human PD, including formation of LBs and chronic degeneration of nigral DA neurons.
The proteasome inhibition model is particular by inducing nigral DA neurodegeneration via a distinct, but highly relevant mechanism of action. Notably, the model emulates the accumulation and aggregation of endogenous α-synuclein, a feature which has been difficult to reproduce in non-genetic models of PD. With regards to behavior, rodents treated with proteasome inhibitors show motor impairment which is responsive to DA-ergic drugs. The non-motor phenotype of treated mice is, however, incompletely understood and should be more comprehensively evaluated in future studies. Similar to intracerebral injection of other PD-related toxins, the local proteasome inhibition model is characterized by an acute reaction which, depending on the site of injection, may not be progressive. Systemic proteasome inhibition models might offer a better alternative to model the chronic and progressive nature of neurodegeneration and protein accumulation as it occurs in the human disease. While systemic administration of PSI has been found to lead to inconsistent results, additional studies are warranted to better establish this approach, employing distinct proteasome inhibitors, with favorable pharmacokinetics and blood-brain barrier permeability. Critically, such studies should be controlled for successful engagement of the peripherally delivered proteasome inhibitor with its target in the brain.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
The authors are supported by grants of the Fund for Scientific Research Flanders (FWO Belgium, grant G.0384.12N), the Queen Elisabeth Medical Foundation (G.S.K.E.) and the Vrije Universiteit Brussel (SRP40). Selected icons used in the figures and tables were supplied by Brad Ashburn, Maurizio Fusillo, Felipe Perucho, Vivian Ziereisen, Anbileru Adaleru, Creative Stall, Meaghan Hendricks, Alex Tai and Boudewijn Mijnlieff from The Noun Project.
REFERENCES
[1] | Kalia LV , & Lang AE ((2015) ) Parkinson’s disease. Lancet, 386: , 896–912. |
[2] | Shulman JM , De Jager PL , & Feany MB ((2011) ) Parkinson’s disease: Genetics and pathogenesis. Annu Rev Pathol, 6: , 193–222. |
[3] | Spillantini MG , Schmidt ML , Lee VM , Trojanowski JQ , Jakes R , & Goedert M ((1997) ) Alpha-synuclein in Lewy bodies. Nature, 388: , 839–840. |
[4] | Jellinger KA ((2003) ) Neuropathological spectrum of synucleinopathies. S. Mov Disord, 18: (Suppl 6), 2–12. |
[5] | Dexter DT , & Jenner P ((2013) ) Parkinson disease: From pathology to molecular disease mechanisms. Free Radic Biol Med, 62: , 132–144. |
[6] | Ciechanover A , Orian A , & Schwartz AL ((2000) ) Ubiquitin-mediated proteolysis: Biological regulation via destruction. Bioessays, 22: , 442–451. |
[7] | Goldberg AL ((2003) ) Protein degradation and protection against misfolded or damaged proteins. Nature, 426: , 895–899. |
[8] | Ben-Nissan G , & Sharon M ((2014) ) Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules, 4: , 862–884. |
[9] | Pickering AM , & Davies KJ ((2012) ) Degradation of damaged proteins: The main function of the 20S proteasome. Prog Mol Biol Transl Sci, 109: , 227–248. |
[10] | Wang X , Yen J , Kaiser P , & Huang L ((2010) ) Regulation of the 26S proteasome complex during oxidative stress. Sci Signal, 3: , ra88. |
[11] | Haj-Yahya M , Fauvet B , Herman-Bachinsky Y , Hejjaoui M , Bavikar SN , Karthikeyan SV , Ciechanover A , Lashuel HA , & Brik A ((2013) ) Synthetic polyubiquitinated alpha-Synuclein reveals important insights into the roles of the ubiquitin chain in regulating its pathophysiology. Proc Natl Acad Sci U S A, 110: , 17726–17731. |
[12] | Rott R , Szargel R , Haskin J , Bandopadhyay R , Lees AJ , Shani V , & Engelender S ((2011) ) alpha-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc Natl Acad Sci U S A, 108: , 18666–18671. |
[13] | Liu CW , Corboy MJ , DeMartino GN , & Thomas PJ ((2003) ) Endoproteolytic activity of the proteasome. Science, 299: , 408–411. |
[14] | Tofaris GK , Layfield R , & Spillantini MG ((2001) ) alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett, 509: , 22–26. |
[15] | Bukhatwa S , Zeng BY , Rose S , & Jenner P ((2010) ) A comparison of changes in proteasomal subunit expression in the substantia nigra in Parkinson’s disease, multiple system atrophy and progressive supranuclear palsy. Brain Res, 1326: , 174–183. |
[16] | McNaught KS , Belizaire R , Jenner P , Olanow CW , & Isacson O ((2002) ) Selective loss of 20S proteasome alpha-subunits in the substantia nigra pars compacta in Parkinson’s disease. Neurosci Lett, 326: , 155–158. |
[17] | McNaught KS , Belizaire R , Isacson O , Jenner P , & Olanow CW ((2003) ) Altered proteasomal function in sporadic Parkinson’s disease. Exp Neurol, 179: , 38–46. |
[18] | McNaught KS , & Jenner P ((2001) ) Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci Lett, 297: , 191–194. |
[19] | Tofaris GK , Razzaq A , Ghetti B , Lilley KS , & Spillantini MG ((2003) ) Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J Biol Chem, 278: , 44405–44411. |
[20] | McNaught KS , Jackson T , JnoBaptiste R , Kapustin A , & Olanow CW ((2006) ) Proteasomal dysfunction in sporadic Parkinson’s disease. Neurology, 66: , S37–S49. |
[21] | Alexopoulou Z , Lang J , Perrett RM , Elschami M , Hurry ME , Kim HT , Mazaraki D , Szabo A , Kessler BM , Goldberg AL , Ansorge O , Fulga TA , & Tofaris GK ((2016) ) Deubiquitinase Usp8 regulates alpha-synuclein clearance and modifies its toxicity in Lewy body disease. Proc Natl Acad Sci U S A, 113: , E4688–E4697. |
[22] | Collier TJ , Kanaan NM , & Kordower JH ((2011) ) Ageing as a primary risk factor for Parkinson’s disease: Evidence from studies of non-human primates. Nat Rev Neurosci, 12: , 359–366. |
[23] | Keller JN , Huang FF , & Markesbery WR ((2000) ) Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neuroscience, 98: , 149–156. |
[24] | Saez I , & Vilchez D ((2014) ) The Mechanistic Links Between Proteasome Activity, Aging and Age-related Diseases. Curr Genomics, 15: , 38–51. |
[25] | Zeng BY , Medhurst AD , Jackson M , Rose S , & Jenner P ((2005) ) Proteasomal activity in brain differs between species and brain regions and changes with age. Mech Ageing Dev, 126: , 760–766. |
[26] | Betarbet R , Canet-Aviles RM , Sherer TB , Mastroberardino PG , McLendon C , Kim JH , Lund S , Na HM , Taylor G , Bence NF , Kopito R , Seo BB , Yagi T , Yagi A , Klinefelter G , Cookson MR , & Greenamyre JT ((2006) ) Intersecting pathways to neurodegeneration in Parkinson’s disease: Effects of the pesticide rotenone on DJ-1, alpha-synuclein, and the ubiquitin-proteasome system. Neurobiol Dis, 22: , 404–420. |
[27] | Sun F , Kanthasamy A , Anantharam V , & Kanthasamy AG ((2007) ) Environmental neurotoxic chemicals-induced ubiquitin proteasome system dysfunction in the pathogenesis and progression of Parkinson’s disease. Pharmacol Ther, 114: , 327–344. |
[28] | Wang XF , Li S , Chou AP , & Bronstein JM ((2006) ) Inhibitory effects of pesticides on proteasome activity: Implication in Parkinson’s disease. Neurobiol Dis, 23: , 198–205. |
[29] | Zhou Y , Shie FS , Piccardo P , Montine TJ , & Zhang J ((2004) ) Proteasomal inhibition induced by manganese ethylene-bis-dithiocarbamate: Relevance to Parkinson’s disease. Neuroscience, 128: , 281–291. |
[30] | Zeng BY , Iravani MM , Lin ST , Irifune M , Kuoppamaki M , Al-Barghouthy G , Smith L , Jackson MJ , Rose S , Medhurst AD , & Jenner P ((2006) ) MPTP treatment of common marmosets impairs proteasomal enzyme activity and decreases expression of structural and regulatory elements of the 26S proteasome. Eur J Neurosci, 23: , 1766–1774. |
[31] | Song L , & Cortopassi G ((2015) ) Mitochondrial complex I defects increase ubiquitin in substantia nigra. Brain Res, 1594: , 82–91. |
[32] | Martins-Branco D , Esteves AR , Santos D , Arduino DM , Swerdlow RH , Oliveira CR , Januario C , & Cardoso SM ((2012) ) Ubiquitin proteasome system in Parkinson’s disease: A keeper or a witness? Exp Neurol, 238: , 89–99. |
[33] | Stefanis L , Larsen KE , Rideout HJ , Sulzer D , & Greene LA ((2001) ) Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci, 21: , 9549–9560. |
[34] | Tanaka Y , Engelender S , Igarashi S , Rao RK , Wanner T , Tanzi RE , Sawa A , V LD , Dawson TM , & Ross CA ((2001) ) Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet, 10: , 919–926. |
[35] | Emmanouilidou E , Stefanis L , & Vekrellis K ((2010) ) Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol Aging, 31: , 953–968. |
[36] | Snyder H , Mensah K , Theisler C , Lee J , Matouschek A , & Wolozin B ((2003) ) Aggregated and monomeric alpha-synuclein bind to the S6’ proteasomal protein and inhibit proteasomal function. J Biol Chem, 278: , 11753–11759. |
[37] | Keller JN , Huang FF , Dimayuga ER , & Maragos WF ((2000) ) Dopamine induces proteasome inhibition in neural PC12 cell line. Free Radic Biol Med, 29: , 1037–1042. |
[38] | Zafar KS , Inayat-Hussain SH , & Ross D ((2007) ) A comparative study of proteasomal inhibition and apoptosis induced in N27 mesencephalic cells by dopamine and MG132. J Neurochem, 102: , 913–921. |
[39] | Shamoto-Nagai M , Maruyama W , Akao Y , Osawa T , Tribl F , Gerlach M , Zucca FA , Zecca L , Riederer P , & Naoi M ((2004) ) Neuromelanin inhibits enzymatic activity of 26S proteasome in human dopaminergic SH-SY5Y cells. J Neural Transm (Vienna), 111: , 1253–1265. |
[40] | Zhou ZD , & Lim TM ((2009) ) Dopamine (DA) induced irreversible proteasome inhibition via DA derived quinones. Free Radic Res, 43: , 417–430. |
[41] | Lee DH , & Goldberg AL ((1998) ) Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell Biol, 8: , 397–403. |
[42] | Bogyo M , & Wang EW ((2002) ) Proteasome inhibitors: Complex tools for a complex enzyme. Curr Top Microbiol Immunol, 268: , 185–208. |
[43] | Dick LR , Cruikshank AA , Grenier L , Melandri FD , Nunes SL , & Stein RL ((1996) ) Mechanistic studies on the inactivation of the proteasome by lactacystin: A central role for clasto-lcystin beta-lactone. J Biol Chem, 271: , 7273–7276. |
[44] | Dick LR , Cruikshank AA , Destree AT , Grenier L , McCormack TA , Melandri FD , Nunes SL , Palombella VJ , Parent LA , Plamondon L , & Stein RL ((1997) ) Mechanistic studies on the inactivation of the proteasome by lactacystin in cultured cells. J Biol Chem, 272: , 182–188. |
[45] | Fenteany G , Standaert RF , Lane WS , Choi S , Corey EJ , & Schreiber SL ((1995) ) Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science, 268: , 726–731. |
[46] | Craiu A , Gaczynska M , Akopian T , Gramm CF , Fenteany G , Goldberg AL , & Rock KL ((1997) ) Lactacystin and clasto-lactacystin beta-lactone modify multiple proteasome beta-subunits and inhibit intracellular protein degradation and major histocompatibility complex class I antigen presentation. J Biol Chem, 272: , 13437–13445. |
[47] | Ostrowska H , Wojcik C , Wilk S , Omura S , Kozlowski L , Stoklosa T , Worowski K , & Radziwon P ((2000) ) Separation of cathepsin A-like enzyme and the proteasome: Evidence that lactacystin/beta-lactone is not a specific inhibitor of the proteasome. Int J Biochem Cell Biol, 32: , 747–757. |
[48] | Geier E , Pfeifer G , Wilm M , Lucchiari-Hartz M , Baumeister W , Eichmann K , & Niedermann G ((1999) ) A giant protease with potential to substitute for some functions of the proteasome. Science, 283: , 978–981. |
[49] | Shafer TJ , & Atchison WD ((1991) ) Transmitter, ion channel and receptor properties of pheochromocytoma (PC12) cells: A model for neurotoxicological studies. Neurotoxicology, 12: , 473–492. |
[50] | Fornai F , Lenzi P , Gesi M , Ferrucci M , Lazzeri G , Busceti CL , Ruffoli R , Soldani P , Ruggieri S , Alessandri MG , & Paparelli A ((2003) ) Fine structure and biochemical mechanisms underlying nigrostriatal inclusions and cell death after proteasome inhibition. J Neurosci, 23: , 8955–8966. |
[51] | Lee CS , Han ES , Park ES , & Bang H ((2005) ) Inhibition of MG132-induced mitochondrial dysfunction and cell death in PC12 cells by 3-morpholinosydnonimine. Brain Res, 1036: , 18–26. |
[52] | Lopes UG , Erhardt P , Yao R , & Cooper GM ((1997) ) p53-dependent induction of apoptosis by proteasome inhibitors. J Biol Chem, 272: , 12893–12896. |
[53] | Nair VD , McNaught KS , Gonzalez-Maeso J , Sealfon SC , & Olanow CW ((2006) ) p53 mediates nontranscriptional cell death in dopaminergic cells in response to proteasome inhibition. J Biol Chem, 281: , 39550–39560. |
[54] | Rideout HJ , Larsen KE , Sulzer D , & Stefanis L ((2001) ) Proteasomal inhibition leads to formation of ubiquitin/alpha-synuclein-immunoreactive inclusions in PC12 cells. J Neurochem, 78: , 899–908. |
[55] | Li X , Du Y , Fan X , Yang D , Luo G , & Le W ((2008) ) c-Jun N-terminal kinase mediates lactacystin-induced dopamine neuron degeneration. J Neuropathol Exp Neurol, 67: , 933–944. |
[56] | Wang R , Zhao J , Zhang J , Liu W , Zhao M , Li J , Lv J , & Li Y ((2015) ) Effect of lysosomal and ubiquitin-proteasome system dysfunction on the abnormal aggregation of alpha-synuclein in PC12 cells. Exp Ther Med, 9: , 2088–2094. |
[57] | Yoshimoto Y , Nakaso K , & Nakashima K ((2005) ) L-dopa and dopamine enhance the formation of aggregates under proteasome inhibition in PC12 cells. FEBS Lett, 579: , 1197–1202. |
[58] | Sun F , Anantharam V , Zhang D , Latchoumycandane C , Kanthasamy A , & Kanthasamy AG ((2006) ) Proteasome inhibitor MG-132 induces dopaminergic degeneration in cell culture and animal models. Neurotoxicology, 27: , 807–815. |
[59] | Sun F , Kanthasamy A , Song C , Yang Y , Anantharam V , & Kanthasamy AG ((2008) ) Proteasome inhibitor-induced apoptosis is mediated by positive feedback amplification of PKCdelta proteolytic activation and mitochondrial translocation. J Cell Mol Med, 12: , 2467–2481. |
[60] | Yong-Kee CJ , Warre R , Monnier PP , Lozano AM , & Nash JE ((2012) ) Evidence for synergism between cell death mechanisms in a cellular model of neurodegeneration in Parkinson’s disease. Neurotox Res, 22: , 355–364. |
[61] | Bir A , Sen O , Anand S , Khemka VK , Banerjee P , Cappai R , Sahoo A , & Chakrabarti S ((2014) ) alpha-Synuclein-induced mitochondrial dysfunction in isolated preparation and intact cells: Implications in the pathogenesis of Parkinson’s disease. J Neurochem, 131: , 868–877. |
[62] | Perez-Alvarez S , Solesio ME , Manzanares J , Jordan J , & Galindo MF ((2009) ) Lactacystin requires reactive oxygen species and Bax redistribution to induce mitochondria-mediated cell death. Br J Pharmacol, 158: , 1121–1130. |
[63] | Lev N , Melamed E , & Offen D ((2006) ) Proteasomal inhibition hypersensitizes differentiated neuroblastoma cells to oxidative damage. Neurosci Lett, 399: , 27–32. |
[64] | McNaught KS , Mytilineou C , Jnobaptiste R , Yabut J , Shashidharan P , Jennert P , & Olanow CW ((2002) ) Impairment of the ubiquitin-proteasome system causes dopaminergic cell death and inclusion body formation in ventral mesencephalic cultures. J Neurochem, 81: , 301–306. |
[65] | Mytilineou C , McNaught KS , Shashidharan P , Yabut J , Baptiste RJ , Parnandi A , & Olanow CW ((2004) ) Inhibition of proteasome activity sensitizes dopamine neurons to protein alterations and oxidative stress. J Neural Transm (Vienna), 111: , 1237–1251. |
[66] | Reaney SH , Johnston LC , Langston WJ , & Di Monte DA ((2006) ) Comparison of the neurotoxic effects of proteasomal inhibitors in primary mesencephalic cultures. Exp Neurol, 202: , 434–440. |
[67] | Rideout HJ , Lang-Rollin IC , Savalle M , & Stefanis L ((2005) ) Dopaminergic neurons in rat ventral midbrain cultures undergo selective apoptosis and form inclusions, but do not up-regulate iHSP70, following proteasomal inhibition. J Neurochem, 93: , 1304–1313. |
[68] | Du Y , Wang F , Zou J , Le W , Dong Q , Wang Z , Shen F , Yu L , & Li Y ((2014) ) Histone deacetylase 6 regulates cytotoxic alpha-synuclein accumulation through induction of the heat shock response. Neurobiol Aging, 35: , 2316–2328. |
[69] | Caldwell KA , Tucci ML , Armagost J , Hodges TW , Chen J , Memon SB , Blalock JE , DeLeon SM , Findlay RH , Ruan Q , Webber PJ , Standaert DG , Olson JB , & Caldwell GA ((2009) ) Investigating bacterial sources of toxicity as an environmental contributor to dopaminergic neurodegeneration. PLoS One, 4: , e7227. |
[70] | Martinez BA , Kim H , Ray A , Caldwell GA , & Caldwell KA ((2015) ) A bacterial metabolite induces glutathione-tractable proteostatic damage, proteasomal disturbances, and PINK1-dependent autophagy in C. elegans. Cell Death Dis, 6: , e1908. |
[71] | Matsui H , Ito H , Taniguchi Y , Inoue H , Takeda S , & Takahashi R ((2010) ) Proteasome inhibition in medaka brain induces the features of Parkinson’s disease. J Neurochem, 115: , 178–187. |
[72] | Matsui H , Gavinio R , & Takahashi R ((2012) ) Medaka fish Parkinson’s disease model. Exp Neurobiol, 21: , 94–100. |
[73] | Ahn TB , & Jeon BS ((2006) ) Protective role of heat shock and heat shock protein 70 in lactacystin-induced cell death both in the rat substantia nigra and PC12 cells. Brain Res, 1087: , 159–167. |
[74] | Harrison IF , Anis HK , & Dexter DT ((2016) ) Associated degeneration of ventral tegmental area dopaminergic neurons in the rat nigrostriatal lactacystin model of parkinsonism and their neuroprotection by valproate. Neurosci Lett, 614: , 16–23. |
[75] | Konieczny J , Czarnecka A , Kaminska K , Lenda T , & Nowak P ((2015) ) Decreased behavioral response to intranigrally administered GABAA agonist muscimol in the lactacystin model of Parkinson’s disease may result from partial lesion of nigral non-dopamine neurons: Comparison to the classical neurotoxin 6-OHDA. Behav Brain Res, 283: , 203–214. |
[76] | Konieczny J , Czarnecka A , Lenda T , Kaminska K , & Lorenc-Koci E ((2014) ) Chronic L-DOPA treatment attenuates behavioral and biochemical deficits induced by unilateral lactacystin administration into the rat substantia nigra. Behav Brain Res, 261: , 79–88. |
[77] | Lorenc-Koci E , Lenda T , Antkiewicz-Michaluk L , Wardas J , Domin H , Smialowska M , & Konieczny J ((2011) ) Different effects of intranigral and intrastriatal administration of the proteasome inhibitor lactacystin on typical neurochemical and histological markers of Parkinson’s disease in rats. Neurochem Int, 58: , 839–849. |
[78] | Mackey S , Jing Y , Flores J , Dinelle K , & Doudet DJ ((2013) ) Direct intranigral administration of an ubiquitin proteasome system inhibitor in rat: Behavior, positron emission tomography, immunohistochemistry. Exp Neurol, 247: , 19–24. |
[79] | McNaught KS , Bjorklund LM , Belizaire R , Isacson O , Jenner P , & Olanow CW ((2002) ) Proteasome inhibition causes nigral degeneration with inclusion bodies in rats. Neuroreport, 13: , 1437–1441. |
[80] | Niu C , Mei J , Pan Q , & Fu X ((2009) ) Nigral degeneration with inclusion body formation and behavioral changes in rats after proteasomal inhibition. Stereotact Funct Neurosurg, 87: , 69–81. |
[81] | Vernon AC , Johansson SM , & Modo MM ((2010) ) Non-invasive evaluation of nigrostriatal neuropathology in a proteasome inhibitor rodent model of Parkinson’s disease. BMC Neurosci, 11: , 1. |
[82] | Pienaar IS , Harrison IF , Elson JL , Bury A , Woll P , Simon AK , & Dexter DT ((2015) ) An animal model mimicking pedunculopontine nucleus cholinergic degeneration in Parkinson’s disease. Brain Struct Funct, 220: , 479–500. |
[83] | Elson JL , Yates A , & Pienaar IS ((2016) ) Pedunculopontine cell loss and protein aggregation direct microglia activation in parkinsonian rats. Brain Struct Funct, 221: , 2319–2341. |
[84] | Subramaniam M , Kern B , Vogel S , Klose V , Schneider G , & Roeper J ((2014) ) Selective increase of in vivo firing frequencies in DA SN neurons after proteasome inhibition in the ventral midbrain. Eur J Neurosci, 40: , 2898–2909. |
[85] | Bentea E , Van der Perren A , Van Liefferinge J , El Arfani A , Albertini G , Demuyser T , Merckx E , Michotte Y , Smolders I , Baekelandt V , & Massie A ((2015) ) Nigral proteasome inhibition in mice leads to motor and non-motor deficits and increased expression of Ser129 phosphorylated alpha-synuclein. Front Behav Neurosci, 9: , 68. |
[86] | Fujiwara H , Hasegawa M , Dohmae N , Kawashima A , Masliah E , Goldberg MS , Shen J , Takio K , & Iwatsubo T ((2002) ) alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol, 4: , 160–164. |
[87] | Vernon AC , Crum WR , Johansson SM , & Modo M ((2011) ) Evolution of extra-nigral damage predicts behavioural deficits in a rat proteasome inhibitor model of Parkinson’s disease. PLoS One, 6: , e17269. |
[88] | Xie W , Li X , Li C , Zhu W , Jankovic J , & Le W ((2010) ) Proteasome inhibition modeling nigral neuron degeneration in Parkinson’s disease. J Neurochem, 115: , 188–199. |
[89] | Xiao Q , Yang S , & Le W ((2015) ) G2019S LRRK2 and aging confer susceptibility to proteasome inhibitor-induced neurotoxicity in nigrostriatal dopaminergic system. J Neural Transm (Vienna), 122: , 1645–1657. |
[90] | Miwa H , Kubo T , Suzuki A , Nishi K , & Kondo T ((2005) ) Retrograde dopaminergic neuron degeneration following intrastriatal proteasome inhibition. Neurosci Lett, 380: , 93–98. |
[91] | Laser H , Mack TG , Wagner D , & Coleman MP ((2003) ) Proteasome inhibition arrests neurite outgrowth and causes dying-back degeneration in primary culture. J Neurosci Res, 74: , 906–916. |
[92] | McNaught KS , Perl DP , Brownell AL , & Olanow CW ((2004) ) Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease. Ann Neurol, 56: , 149–162. |
[93] | Bukhatwa S , Iravani MM , Zeng BY , Cooper JD , Rose S , & Jenner P ((2009) ) An immunohistochemical and stereological analysis of PSI-induced nigral neuronal degeneration in the rat. J Neurochem, 109: , 52–59. |
[94] | Bukhatwa S , Zeng BY , Rose S , & Jenner P ((2010) ) The effects of dose and route of administration of PSI on behavioural and biochemical indices of neuronal degeneration in the rat brain. Brain Res, 1354: , 236–242. |
[95] | Fitzgerald P , Mandel A , Bolton AE , Sullivan AM , & Nolan Y ((2008) ) Treatment with phosphotidylglycerol-based nanoparticles prevents motor deficits induced by proteasome inhibition: Implications for Parkinson’s disease. Behav Brain Res, 195: , 271–274. |
[96] | Schapira AH , Cleeter MW , Muddle JR , Workman JM , Cooper JM , & King RH ((2006) ) Proteasomal inhibition causes loss of nigral tyrosine hydroxylase neurons. Ann Neurol, 60: , 253–255. |
[97] | Zeng BY , Bukhatwa S , Hikima A , Rose S , & Jenner P ((2006) ) Reproducible nigral cell loss after systemic proteasomal inhibitor administration to rats. Ann Neurol, 60: , 248–252. |
[98] | Bove J , Zhou C , Jackson-Lewis V , Taylor J , Chu Y , Rideout HJ , Wu DC , Kordower JH , Petrucelli L , & Przedborski S ((2006) ) Proteasome inhibition and Parkinson’s disease modeling. Ann Neurol, 60: , 260–264. |
[99] | Hawlitschka A , Haas SJ , Schmitt O , Weiss DG , & Wree A ((2007) ) Effects of systemic PSI administration on catecholaminergic cells in the brain, adrenal medulla and carotid body in Wistar rats. Brain Res, 1173: , 137–144. |
[100] | Kadoguchi N , Kimoto H , Yano R , Kato H , & Araki T ((2008) ) Failure of acute administration with proteasome inhibitor to provide a model of Parkinson’s disease in mice. Metab Brain Dis, 23: , 147–154. |
[101] | Kordower JH , Kanaan NM , Chu Y , Suresh Babu R , Stansell J 3rd , Terpstra BT , Sortwell CE , Steece-Collier K , & Collier TJ ((2006) ) Failure of proteasome inhibitor administration to provide a model of Parkinson’s disease in rats and monkeys. Ann Neurol, 60: , 264–268. |
[102] | Manning-Bog AB , Reaney SH , Chou VP , Johnston LC , McCormack AL , Johnston J , Langston JW , & Di Monte DA ((2006) ) Lack of nigrostriatal pathology in a rat model of proteasome inhibition. Ann Neurol, 60: , 256–260. |
[103] | Mathur BN , Neely MD , Dyllick-Brenzinger M , Tandon A , & Deutch AY ((2007) ) Systemic administration of a proteasome inhibitor does not cause nigrostriatal dopamine degeneration. Brain Res, 1168: , 83–89. |
[104] | Landau AM , Kouassi E , Siegrist-Johnstone R , & Desbarats J ((2007) ) Proteasome inhibitor model of Parkinson’s disease in mice is confounded by neurotoxicity of the ethanol vehicle. Mov Disord, 22: , 403–407. |
[105] | Hirst SJ , & Ferger B ((2008) ) Systemic proteasomal inhibitor exposure enhances dopamine turnover and decreases dopamine levels but does not affect MPTP-induced striatal dopamine depletion in mice. Synapse, 62: , 85–90. |
[106] | McNaught KS , & Olanow CW ((2006) ) Proteasome inhibitor-induced model of Parkinson’s disease. Ann Neurol, 60: , 243–247. |
[107] | Shin M , Jan C , Jacquard C , Jarraya B , Callebert J , Launay JM , Hantraye P , Remy P , Palfi S , & Brouillet E ((2011) ) Chronic systemic treatment with a high-dose proteasome inhibitor in mice produces akinesia unrelated to nigrostriatal degeneration. Neurobiol Aging, 32: , 2100–2102. |
[108] | Beal F , & Lang A ((2006) ) The proteasomal inhibition model of Parkinson’s disease: Boon or bust? Ann Neurol, 60: , 158–161. |
[109] | Bedford L , Hay D , Devoy A , Paine S , Powe DG , Seth R , Gray T , Topham I , Fone K , Rezvani N , Mee M , Soane T , Layfield R , Sheppard PW , Ebendal T , Usoskin D , Lowe J , & Mayer RJ ((2008) ) Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J Neurosci, 28: , 8189–8198. |
[110] | Oren M ((1999) ) Regulation of the p53 tumor suppressor protein. J Biol Chem, 274: , 36031–36034. |
[111] | Dietrich P , Rideout HJ , Wang Q , & Stefanis L ((2003) ) Lack of p53 delays apoptosis, but increases ubiquitinated inclusions, in proteasomal inhibitor-treated cultured cortical neurons. Mol Cell Neurosci, 24: , 430–441. |
[112] | Sullivan PG , Dragicevic NB , Deng JH , Bai Y , Dimayuga E , Ding Q , Chen Q , Bruce-Keller AJ , & Keller JN ((2004) ) Proteasome inhibition alters neural mitochondrial homeostasis and mitochondria turnover. J Biol Chem, 279: , 20699–20707. |
[113] | Paine SM , Anderson G , Bedford K , Lawler K , Mayer RJ , Lowe J , & Bedford L ((2013) ) Pale body-like inclusion formation and neurodegeneration following depletion of 26S proteasomes in mouse brain neurones are independent of alpha-synuclein. PLoS One, 8: , e54711. |
[114] | Ding Q , Dimayuga E , Markesbery WR , & Keller JN ((2004) ) Proteasome inhibition increases DNA and RNA oxidation in astrocyte and neuron cultures. J Neurochem, 91: , 1211–1218. |
[115] | Kikuchi S , Shinpo K , Tsuji S , Takeuchi M , Yamagishi S , Makita Z , Niino M , Yabe I , & Tashiro K ((2003) ) Effect of proteasome inhibitor on cultured mesencephalic dopaminergic neurons. Brain Res, 964: , 228–236. |
[116] | Lee MH , Hyun DH , Jenner P , & Halliwell B ((2001) ) Effect of proteasome inhibition on cellular oxidative damage, antioxidant defences and nitric oxide production. J Neurochem, 78: , 32–41. |
[117] | Elkharaz J , Ugun-Klusek A , Constantin-Teodosiu D , Lawler K , Mayer RJ , Billett E , Lowe J , & Bedford L ((2013) ) Implications for oxidative stress and astrocytes following 26S proteasomal depletion in mouse forebrain neurones. Biochim Biophys Acta, 1832: , 1930–1938. |
[118] | Reinheckel T , Sitte N , Ullrich O , Kuckelkorn U , Davies KJ , & Grune T ((1998) ) Comparative resistance of the 20S and 26S proteasome to oxidative stress. Biochem J, 335: (Pt 3), 637–642. |
[119] | Hyun DH , Lee MH , Halliwell B , & Jenner P ((2002) ) Proteasomal dysfunction induced by 4-hydroxy-2,3-trans-nonenal, an end-product of lipid peroxidation: A mechanism contributing to neurodegeneration? J Neurochem, 83: , 360–370. |
[120] | Le W ((2014) ) Role of iron in UPS impairment model of Parkinson’s disease. Parkinsonism Relat Disord, 20: (Suppl 1), S158–S161. |
[121] | Li XP , Xie WJ , Zhang Z , Kansara S , Jankovic J , & Le WD ((2012) ) A mechanistic study of proteasome inhibition-induced iron misregulation in dopamine neuron degeneration. Neurosignals, 20: , 223–236. |
[122] | Zhu W , Li X , Xie W , Luo F , Kaur D , Andersen JK , Jankovic J , & Le W ((2010) ) Genetic iron chelation protects against proteasome inhibition-induced dopamine neuron degeneration. Neurobiol Dis, 37: , 307–313. |
[123] | Zhu W , Xie W , Pan T , Xu P , Fridkin M , Zheng H , Jankovic J , Youdim MB , & Le W ((2007) ) Prevention and restoration of lactacystin-induced nigrostriatal dopamine neuron degeneration by novel brain-permeable iron chelators. FASEB J, 21: , 3835–3844. |
[124] | Zecca L , Youdim MB , Riederer P , Connor JR , & Crichton RR ((2004) ) Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci, 5: , 863–873. |
[125] | Eriksen JL , Przedborski S , & Petrucelli L ((2005) ) Gene dosage and pathogenesis of Parkinson’s disease. Trends Mol Med, 11: , 91–96. |
[126] | Oliveras-Salva M , Van der Perren A , Casadei N , Stroobants S , Nuber S , D’Hooge R , Van den Haute C , & Baekelandt V ((2013) ) rAAV2/7 vector-mediated overexpression of alpha-synuclein in mouse substantia nigra induces protein aggregation and progressive dose-dependent neurodegeneration. Mol Neurodegener, 8: , 44. |
[127] | Van der Perren A , Toelen J , Casteels C , Macchi F , Van Rompuy AS , Sarre S , Casadei N , Nuber S , Himmelreich U , Osorio Garcia MI , Michotte Y , D’Hooge R , Bormans G , Van Laere K , Gijsbers R , Van den Haute C , Debyser Z , & Baekelandt V ((2015) ) Longitudinal follow-up and characterization of a robust rat model for Parkinson’s disease based on overexpression of alpha-synuclein with adeno-associated viral vectors. Neurobiol Aging, 36: , 1543–1558. |
[128] | Taschenberger G , Garrido M , Tereshchenko Y , Bahr M , Zweckstetter M , & Kugler S ((2012) ) Aggregation of alphaSynuclein promotes progressive in vivo neurotoxicity in adult rat dopaminergic neurons. Acta Neuropathol, 123: , 671–683. |
[129] | Winner B , Jappelli R , Maji SK , Desplats PA , Boyer L , Aigner S , Hetzer C , Loher T , Vilar M , Campioni S , Tzitzilonis C , Soragni A , Jessberger S , Mira H , Consiglio A , Pham E , Masliah E , Gage FH , & Riek R ((2011) ) In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A, 108: , 4194–4199. |
[130] | Ebrahimi-Fakhari D , Wahlster L , & McLean PJ ((2012) ) Protein degradation pathways in Parkinson’s disease: Curse or blessing. Acta Neuropathol, 124: , 153–172. |
[131] | Alvarez-Castelao B , Goethals M , Vandekerckhove J , & Castano JG ((2014) ) Mechanism of cleavage of alpha-synuclein by the 20S proteasome and modulation of its degradation by the RedOx state of the N-terminal methionines. Biochim Biophys Acta, 1843: , 352–365. |
[132] | Dyllick-Brenzinger M , D’Souza CA , Dahlmann B , Kloetzel PM , & Tandon A ((2010) ) Reciprocal effects of alpha-synuclein overexpression and proteasome inhibition in neuronal cells and tissue. Neurotox Res, 17: , 215–227. |
[133] | Liu CW , Giasson BI , Lewis KA , Lee VM , Demartino GN , & Thomas PJ ((2005) ) A precipitating role for truncated alpha-synuclein and the proteasome in alpha-synuclein aggregation: Implications for pathogenesis of Parkinson disease. J Biol Chem, 280: , 22670–22678. |
[134] | Bennett MC , Bishop JF , Leng Y , Chock PB , Chase TN , & Mouradian MM ((1999) ) Degradation of alpha-synuclein by proteasome. J Biol Chem, 274: , 33855–33858. |
[135] | Rott R , Szargel R , Haskin J , Shani V , Shainskaya A , Manov I , Liani E , Avraham E , & Engelender S ((2008) ) Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J Biol Chem, 283: , 3316–3328. |
[136] | Webb JL , Ravikumar B , Atkins J , Skepper JN , & Rubinsztein DC ((2003) ) Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem, 278: , 25009–25013. |
[137] | Yang F , Yang YP , Mao CJ , Liu L , Zheng HF , Hu LF , & Liu CF ((2013) ) Crosstalk between the proteasome system and autophagy in the clearance of alpha-synuclein. Acta Pharmacol Sin, 34: , 674–680. |
[138] | Ancolio K , Alves da Costa C , Ueda K , & Checler F ((2000) ) Alpha-synuclein and the Parkinson’s disease-related mutant Ala53Thr-alpha-synuclein do not undergo proteasomal degradation in HEK293 and neuronal cells. Neurosci Lett, 285: , 79–82. |
[139] | Batelli S , Peverelli E , Rodilossi S , Forloni G , & Albani D ((2011) ) Macroautophagy and the proteasome are differently involved in the degradation of alpha-synuclein wild type and mutated A30P in an in vitro inducible model (PC12/TetOn). Neuroscience, 195: , 128–137. |
[140] | Biasini E , Fioriti L , Ceglia I , Invernizzi R , Bertoli A , Chiesa R , & Forloni G ((2004) ) Proteasome inhibition and aggregation in Parkinson’s disease: A comparative study in untransfected and transfected cells. J Neurochem, 88: , 545–553. |
[141] | Paxinou E , Chen Q , Weisse M , Giasson BI , Norris EH , Rueter SM , Trojanowski JQ , Lee VM , & Ischiropoulos H ((2001) ) Induction of alpha-synuclein aggregation by intracellular nitrative insult. J Neurosci, 21: , 8053–8061. |
[142] | Vogiatzi T , Xilouri M , Vekrellis K , & Stefanis L ((2008) ) Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem, 283: , 23542–23556. |
[143] | Cuervo AM , Stefanis L , Fredenburg R , Lansbury PT , & Sulzer D ((2004) ) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science, 305: , 1292–1295. |
[144] | Lan D , Wang W , Zhuang J , & Zhao Z ((2015) ) Proteasome inhibitor-induced autophagy in PC12 cells overexpressing A53T mutant alpha-synuclein. Mol Med Rep, 11: , 1655–1660. |
[145] | Rideout HJ , Lang-Rollin I , & Stefanis L ((2004) ) Involvement of macroautophagy in the dissolution of neuronal inclusions. Int J Biochem Cell Biol, 36: , 2551–2562. |
[146] | Shen YF , Tang Y , Zhang XJ , Huang KX , & Le WD ((2013) ) Adaptive changes in autophagy after UPS impairment in Parkinson’s disease. Acta Pharmacol Sin, 34: , 667–673. |
[147] | Ebrahimi-Fakhari D , Cantuti-Castelvetri I , Fan Z , Rockenstein E , Masliah E , Hyman BT , McLean PJ , & Unni VK ((2011) ) Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J Neurosci, 31: , 14508–14520. |
[148] | Ebrahimi-Fakhari D , McLean PJ , & Unni VK ((2012) ) Alpha-synuclein’s degradation in vivo: Opening a new (cranial) window on the roles of degradation pathways in Parkinson disease. Autophagy, 8: , 281–283. |
[149] | Lashuel HA , Overk CR , Oueslati A , & Masliah E ((2013) ) The many faces of alpha-synuclein: From structure and toxicity to theraeutic target. Nat Rev Neurosci, 14: , 38–48. |
[150] | Oueslati A , Fournier M , & Lashuel HA ((2010) ) Role of post-translational modifications in modulating the structure, function and toxicity of alpha-synuclein:L Imlications for Parkinson’s disease pathogenesis and therapies. Prog Brain Res, 183: , 115–145. |
[151] | Schildknecht S , Gerding HR , Karreman C , Drescher M , Lashuel HA , Outeiro TF , Di Monte DA , & Leist M ((2013) ) Oxidative and nitrative alpha-synuclein modifications and proteostatic stress: Implications for disease mechanisms and interventions in synucleinopathies. J Neurochem, 125: , 491–511. |
[152] | Anderson JP , Walker DE , Goldstein JM , de Laat R , Banducci K , Caccavello RJ , Barbour R , Huang J , Kling K , Lee M , Diep L , Keim PS , Shen X , Chataway T , Schlossmacher MG , Seubert P , Schenk D , Sinha S , Gai WP , & Chilcote TJ ((2006) ) Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem, 281: , 29739–29752. |
[153] | Tenreiro S , Eckermann K , Outeiro TF ((2014) ) Protein phosphorylation in neurodegeneration: Friend or foe? Front Mol Neurosci, 7: , 42. |
[154] | Chen L , & Feany MB ((2005) ) Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci, 8: , 657–663. |
[155] | Sato H , Arawaka S , Hara S , Fukushima S , Koga K , Koyama S , & Kato T ((2011) ) Authentically phosphorylated alpha-synuclein at Ser129 accelerates neurodegeneration in a rat model of familial Parkinson’s disease. J Neurosci, 31: , 16884–16894. |
[156] | Machiya Y , Hara S , Arawaka S , Fukushima S , Sato H , Sakamoto M , Koyama S , & Kato T ((2010) ) Phosphorylated alpha-synuclein at Ser-129 is targeted to the proteasome pathway in a ubiquitin-independent manner. J Biol Chem, 285: , 40732–40744. |
[157] | Zhang NY , Tang Z , & Liu CW ((2008) ) alpha-Synuclein protofibrils inhibit 26S proteasome-mediated protein degradation: Understanding the cytotoxicity of protein protofibrils in neurodegenerative disease pathogenesis. J Biol Chem, 283: , 20288–20298. |
[158] | Martinez-Vicente M , & Vila M ((2013) ) Alpha-synuclein and protein degradation pathways in Parkinson’s disease: A pathological feed-back loop. Exp Neurol, 247: , 308–313. |
[159] | Xilouri M , Brekk OR , & Stefanis L ((2013) ) alpha-Synuclein and protein degradation systems: A reciprocal relationship. Mol Neurobiol, 47: , 537–551. |
[160] | Chu Y , Dodiya H , Aebischer P , Olanow CW , & Kordower JH ((2009) ) Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol Dis, 35: , 385–398. |
[161] | Furukawa Y , Vigouroux S , Wong H , Guttman M , Rajput AH , Ang L , Briand M , Kish SJ , & Briand Y ((2002) ) Brain proteasomal function in sporadic Parkinson’s disease and related disorders. Ann Neurol, 51: , 779–782. |
[162] | Braak H , & Del Tredici K ((2008) ) Invited Article: Nervous system pathology in sporadic Parkinson disease. Neurology, 70: , 1916–1925. |
[163] | Choy MS , Chen MJ , Manikandan J , Peng ZF , Jenner AM , Melendez AJ , & Cheung NS ((2011) ) Up-regulation of endoplasmic reticulum stress-related genes during the early phase of treatment of cultured cortical neurons by the proteasomal inhibitor lactacystin. J Cell Physiol, 226: , 494–510. |
[164] | Yew EH , Cheung NS , Choy MS , Qi RZ , Lee AY , Peng ZF , Melendez AJ , Manikandan J , Koay ES , Chiu LL , Ng WL , Whiteman M , Kandiah J , & Halliwell B ((2005) ) Proteasome inhibition by lactacystin in primary neuronal cells induces both potentially neuroprotective and pro-apoptotic transcriptional responses: A microarray analysis. J Neurochem, 94: , 943–956. |
[165] | Zhang L , Chang M , Li H , Hou S , Zhang Y , Hu Y , Han W , & Hu L ((2007) ) Proteomic changes of PC12 cells treated with proteasomal inhibitor PSI. Brain Res, 1153: , 196–203. |
[166] | Hu X , Zhang H , Zhang Y , Bai L , Chen Q , Wu J , & Zhang L ((2014) ) Differential protein profile of PC12 cells exposed to proteasomal inhibitor lactacystin. Neurosci Lett, 575: , 25–30. |
[167] | Keightley JA , Shang L , & Kinter M ((2004) ) Proteomic analysis of oxidative stress-resistant cells: A specific role for aldose reductase overexpression in cytoprotection. Mol Cell Proteomics, 3: , 167–175. |
[168] | Miura T , Ohnishi Y , Kurushima H , Horie H , Kadoya T , & Nakabeppu Y ((2005) ) Regulation of the neuronal fate by DeltaFosB and its downstream target, galectin-1. Curr Drug Targets, 6: , 437–444. |
[169] | Ding Q , Bruce-Keller AJ , Chen Q , & Keller JN ((2004) ) Analysis of gene expression in neural cells subject to chronic proteasome inhibition. Free Radic Biol Med, 36: , 445–455. |
[170] | Petrucelli L , O’Farrell C , Lockhart PJ , Baptista M , Kehoe K , Vink L , Choi P , Wolozin B , Farrer M , Hardy J , & Cookson MR ((2002) ) Parkin protects against the toxicity associated with mutant alpha-synuclein: roteasome dysfunction selectively affects catecholaminergic neurons. Neuron, 36: , 1007–1019. |
[171] | Casarejos MJ , Solano RM , Rodriguez-Navarro JA , Gomez A , Perucho J , Castano JG , Garcia de Yebenes J , & Mena MA ((2009) ) Parkin deficiency increases the resistance of midbrain neurons and glia to mild proteasome inhibition: The role of autophagy and glutathione homeostasis. J Neurochem, 110: , 1523–1537. |
[172] | Jansen AH , Reits EA , & Hol EM ((2014) ) The ubiquitin proteasome system in glia and its role in neurodegenerative diseases. Front Mol Neurosci, 7: , 73. |
[173] | Dasuri K , Ebenezer PJ , Zhang L , Fernandez-Kim SO , Uranga RM , Gavilan E , Di Blasio A , & Keller JN ((2010) ) Selective vulnerability of neurons to acute toxicity after proteasome inhibitor treatment: Implications for oxidative stress and insolubility of newly synthesized proteins. Free Radic Biol Med, 49: , 1290–1297. |
[174] | Goldbaum O , Vollmer G , & Richter-Landsberg C ((2006) ) Proteasome inhibition by MG-132 induces apoptotic cell death and mitochondrial dysfunction in cultured rat brain oligodendrocytes but not in astrocytes. Glia, 53: , 891–901. |
[175] | Snider BJ , Tee LY , Canzoniero LM , Babcock DJ , & Choi DW ((2002) ) NMDA antagonists exacerbate neuronal death caused by proteasome inhibition in cultured cortical and striatal neurons. Eur J Neurosci, 15: , 419–428. |
[176] | Goldbaum O , Riedel M , Stahnke T , & Richter-Landsberg C ((2009) ) The small heat shock protein HSP25 protects astrocytes against stress induced by proteasomal inhibition. Glia, 57: , 1566–1577. |
[177] | Tydlacka S , Wang CE , Wang X , Li S , & Li XJ ((2008) ) Differential activities of the ubiquitin-proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J Neurosci, 28: , 13285–13295. |
[178] | Kumar A , Kopra J , Varendi K , Porokuokka LL , Panhelainen A , Kuure S , Marshall P , Karalija N , Harma MA , Vilenius C , Lillevali K , Tekko T , Mijatovic J , Pulkkinen N , Jakobson M , Ola R , Palm E , Lindahl M , Stromberg I , Voikar V , Piepponen TP , Saarma M , & Andressoo JO ((2015) ) GDNF overexpression from the native locus reveals its role in the nigrostriatal dopaminergic system function. PLoS Genet, 11: , e1005710. |
[179] | Fronczek R , Overeem S , Lee SY , Hegeman IM , van Pelt J , van Duinen SG , Lammers GJ , & Swaab DF ((2007) ) Hypocretin (orexin) loss in Parkinson’s disease. Brain, 130: , 1577–1585. |
[180] | Gai WP , Blumbergs PC , Geffen LB , & Blessing WW ((1992) ) Age-related loss of dorsal vagal neurons in Parkinson’s disease. Neurology, 42: , 2106–2111. |
[181] | Halliday GM , Blumbergs PC , Cotton RG , Blessing WW , & Geffen LB ((1990) ) Loss of brainstem serotonin- and substance P-containing neurons in Parkinson’s disease. Brain Res, 510: , 104–107. |
[182] | Hirsch EC , Graybiel AM , Duyckaerts C , & Javoy-Agid F ((1987) ) Neuronal loss in the pedunculopontine tegmental nucleus in Parkinson disease and in progressive supranuclear palsy. Proc Natl Acad Sci U S A, 84: , 5976–5980. |
[183] | Thannickal TC , Lai YY , & Siegel JM ((2007) ) Hypocretin (orexin) cell loss in Parkinson’s disease. Brain, 130: , 1586–1595. |
[184] | Zarow C , Lyness SA , Mortimer JA , & Chui HC ((2003) ) Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol, 60: , 337–341. |
[185] | Miwa H , Kubo T , Suzuki A , & Kondo T ((2006) ) Intragastric proteasome inhibition induces alpha-synuclein-immunopositive aggregations in neurons in the dorsal motor nucleus of the vagus in rats. Neurosci Lett, 401: , 146–149. |
[186] | MacInnes N , Iravani MM , Perry E , Piggott M , Perry R , Jenner P , & Ballard C ((2008) ) Proteasomal abnormalities in cortical Lewy body disease and the impact of proteasomal inhibition within cortical and cholinergic systems. J Neural Transm (Vienna), 115: , 869–878. |
[187] | Pienaar IS , Gartside SE , Sharma P , De Paola V , Gretenkord S , Withers D , Elson JL , & Dexter DT ((2015) ) Pharmacogenetic stimulation of cholinergic pedunculopontine neurons reverses motor deficits in a rat model of Parkinson’s disease. Mol Neurodegener, 10: , 47. |
[188] | Stefani A , Lozano AM , Peppe A , Stanzione P , Galati S , Tropepi D , Pierantozzi M , Brusa L , Scarnati E , & Mazzone P ((2007) ) Bilateral deep brain stimulation of the pedunculopontine and subthalamic nuclei in severe Parkinson’s disease. Brain, 130: , 1596–1607. |
[189] | Horowitz MP , & Greenamyre JT ((2010) ) Gene-environment interactions in Parkinson’s disease: The importance of animal modeling. Clin Pharmacol Ther, 88: , 467–474. |
[190] | Lee M , Hyun D , Halliwell B , & Jenner P ((2001) ) Effect of the overexpression of wild-type or mutant alpha-synuclein on cell susceptibility to insult. J Neurochem, 76: , 998–1009. |
[191] | Lim KL , & Zhang CW ((2013) ) Molecular events underlying Parkinson’s disease - an interwoven tapestry. Front Neurol, 4: , 33. |
[192] | Hoglinger GU , Carrard G , Michel PP , Medja F , Lombes A , Ruberg M , Friguet B , & Hirsch EC ((2003) ) Dysfunction of mitochondrial complex I and the proteasome: Interactions between two biochemical deficits in a cellular model of Parkinson’s disease. J Neurochem, 86: , 1297–1307. |
[193] | Lee CS , Tee LY , Warmke T , Vinjamoori A , Cai A , Fagan AM , & Snider BJ ((2004) ) A proteasomal stress response: Pre-treatment with proteasome inhibitors increases proteasome activity and reduces neuronal vulnerability to oxidative injury. J Neurochem, 91: , 996–1006. |
[194] | Yamamoto N , Sawada H , Izumi Y , Kume T , Katsuki H , Shimohama S , & Akaike A ((2007) ) Proteasome inhibition induces glutathione synthesis and protects cells from oxidative stress: Relevance to Parkinson disease. J Biol Chem, 282: , 4364–4372. |
[195] | Yamamoto N , Izumi Y , Matsuo T , Wakita S , Kume T , Takada-Takatori Y , Sawada H , & Akaike A ((2010) ) Elevation of heme oxygenase-1 by proteasome inhibition affords dopaminergic neuroprotection. J Neurosci Res, 88: , 1934–1942. |
[196] | Inden M , Kondo J , Kitamura Y , Takata K , Nishimura K , Taniguchi T , Sawada H , & Shimohama S ((2005) ) Proteasome inhibitors protect against degeneration of nigral dopaminergic neurons in hemiparkinsonian rats. J Pharmacol Sci, 97: , 203–211. |
[197] | Sawada H , Kohno R , Kihara T , Izumi Y , Sakka N , Ibi M , Nakanishi M , Nakamizo T , Yamakawa K , Shibasaki H , Yamamoto N , Akaike A , Inden M , Kitamura Y , Taniguchi T , & Shimohama S ((2004) ) Proteasome mediates dopaminergic neuronal degeneration, and its inhibition causes alpha-synuclein inclusions. J Biol Chem, 279: , 10710–10719. |
[198] | Oshikawa T , Kuroiwa H , Yano R , Yokoyama H , Kadoguchi N , Kato H , & Araki T ((2009) ) Systemic administration of proteasome inhibitor protects against MPTP neurotoxicity in mice. Cell Mol Neurobiol, 29: , 769–777. |
[199] | Izumi Y , Yamamoto N , Matsushima S , Yamamoto T , Takada-Takatori Y , Akaike A , & Kume T ((2015) ) Compensatory role of the Nrf2-ARE pathway against paraquat toxicity: Relevance of 26S proteasome activity. J Pharmacol Sci, 129: , 150–159. |
[200] | Watkins AL , Ray A , L RR , Caldwell KA , & Olson JB ((2016) ) The Prevalence and Distribution of Neurodegenerative Compound-Producing Soil Streptomyces spp. Sci Rep, 6: , 22566. |
[201] | Ungerstedt U ((1976) ) 6-hydroxydopamine-induced degeneration of the nigrostriatal dopamine pathway: The turning syndrome. Pharmacol Ther B, 2: , 37–40. |
[202] | Konieczny J , Lenda T , & Czarnecka A ((2016) ) Early increase in dopamine release in the ipsilateral striatum after unilateral intranigral administration of lactacystin produces spontaneous contralateral rotations in rats. Neuroscience, 324: , 92–106. |
[203] | Lu CY , Yi PL , Tsai CH , Cheng CH , Chang HH , Hsiao YT , & Chang FC ((2010) ) TNF-NF-kappaB signaling mediates excessive somnolence in hemiparkinsonian rats. Behav Brain Res, 208: , 484–496. |
[204] | Du Y , Li X , Yang D , Zhang X , Chen S , Huang K , & Le W ((2008) ) Multiple molecular pathways are involved in the neuroprotection of GDNF against proteasome inhibitor induced dopamine neuron degeneration in vivo. Exp Biol Med (Maywood), 233: , 881–890. |
[205] | Du Y , Zhang X , Tao Q , Chen S , & Le W ((2013) ) Adeno-associated virus type 2 vector-mediated glial cell line-derived neurotrophic factor gene transfer induces neuroprotection and neuroregeneration in a ubiquitin-proteasome system impairment animal model of Parkinson’s disease. Neurodegener Dis, 11: , 113–128. |
[206] | Wu J , Yu W , Chen Y , Su Y , Ding Z , Ren H , Jiang Y , & Wang J ((2010) ) Intrastriatal transplantation of GDNF-engineered BMSCs and its neuroprotection in lactacystin-induced Parkinsonian rat model. Neurochem Res, 35: , 495–502. |
[207] | Zhu W , Xie W , Pan T , Jankovic J , Li J , Youdim MB , & Le W ((2008) ) Comparison of neuroprotective and neurorestorative capabilities of rasagiline and selegiline against lactacystin-induced nigrostriatal dopaminergic degeneration. J Neurochem, 105: , 1970–1978. |
[208] | Li C , Guo Y , Xie W , Li X , Janokovic J , & Le W ((2010) ) Neuroprotection of pramipexole in UPS impairment induced animal model of Parkinson’s disease. Neurochem Res, 35: , 1546–1556. |
[209] | Li C , Biswas S , Li X , Dutta AK , & Le W ((2010) ) Novel D3 dopamine receptor-preferring agonist D-264: Evidence of neuroprotective property in Parkinson’s disease animal models induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and lactacystin. J Neurosci Res, 88: , 2513–2523. |
[210] | Harrison IF , Crum WR , Vernon AC , & Dexter DT ((2015) ) Neurorestoration induced by the HDAC inhibitor sodium valproate in the lactacystin model of Parkinson’s is associated with histone acetylation and up-regulation of neurotrophic factors. Br J Pharmacol, 172: , 4200–4215. |
[211] | Pan T , Kondo S , Zhu W , Xie W , Jankovic J , & Le W ((2008) ) Neuroprotection of rapamycin in lactacystin-induced neurodegeneration via autophagy enhancement. Neurobiol Dis, 32: , 16–25. |
[212] | Konieczny J , Jantas D , Lenda T , Domin H , Czarnecka A , Kuter K , Smialowska M , Lason W , & Lorenc-Koci E ((2014) ) Lack of neuroprotective effect of celastrol under conditions of proteasome inhibition by lactacystin in in vitro and in vivo studies: Implications for Parkinson’s disease. Neurotox Res, 26: , 255–273. |
[213] | Chang KH , Lee-Chen GJ , Wu YR , Chen YJ , Lin JL , Li M , Chen IC , Lo YS , Wu HC , & Chen CM ((2016) ) Impairment of proteasome and anti-oxidative pathways in the induced pluripotent stem cell model for sporadic Parkinson’s disease. Parkinsonism Relat Disord, 24: , 81–88. |
[214] | Wojcik S , Spodnik JH , Spodnik E , Dziewiatkowski J , & Morys J ((2014) ) Nigrostriatal pathway degeneration in rats after intraperitoneal administration of proteasome inhibitor MG-132. Folia Neuropathol, 52: , 41–55. |
Figures and Tables
Fig.1
The 26S/20S proteasome structure and function. Top: Ubiquitin- and ATP-dependent protein degradation of substrates via the 26S proteasome. The 26S proteasome can also degrade proteins in an ubiquitin-independent manner (not shown). Bottom: Ubiquitin- and ATP-independent protein degradation of natively unfolded or damaged and partly unfolded substrates via the 20S proteasome. Inset shows the structural composition of the 26S proteasome. ATP adenosine triphosphate.
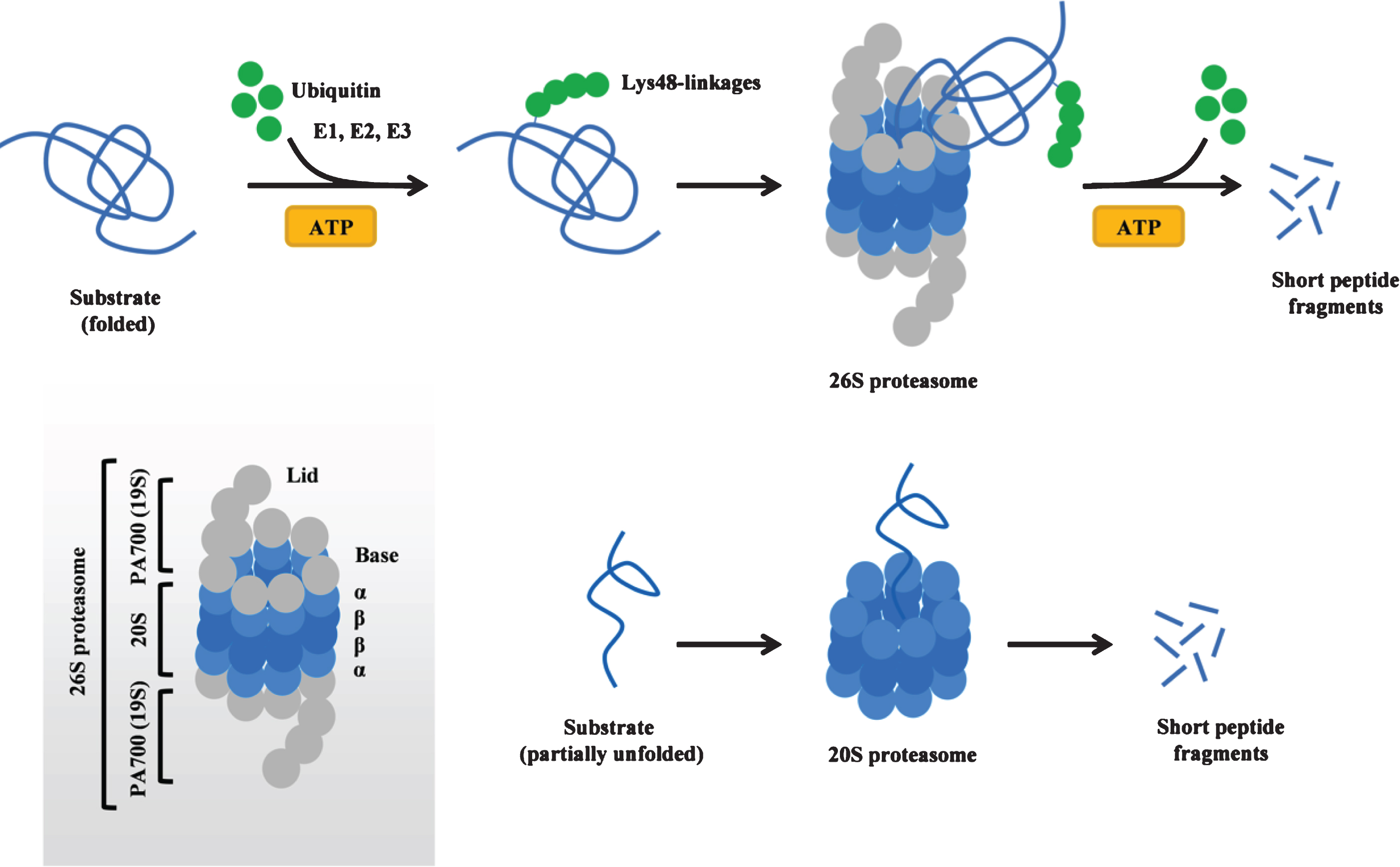
Fig.2
Molecular pathways recruited following proteasome inhibition. At high, lethal doses (upper panel), proteasome inhibitors activate multiple downstream toxic pathways that together converge in inducing neuronal death. At lower, sub-lethal doses (lower panel), proteasome inhibitors can lead to a neuroprotective response, characterized by activation of anti-oxidant pathways and clearance of α-synuclein oligomers into larger, potentially less toxic fibrils. Lactacystin is exemplified as a typical proteasome inhibitor, although similar effects are observed with structurally distinct inhibitors. See text for details. ER endoplasmic reticulum, ROS reactive oxygen species.
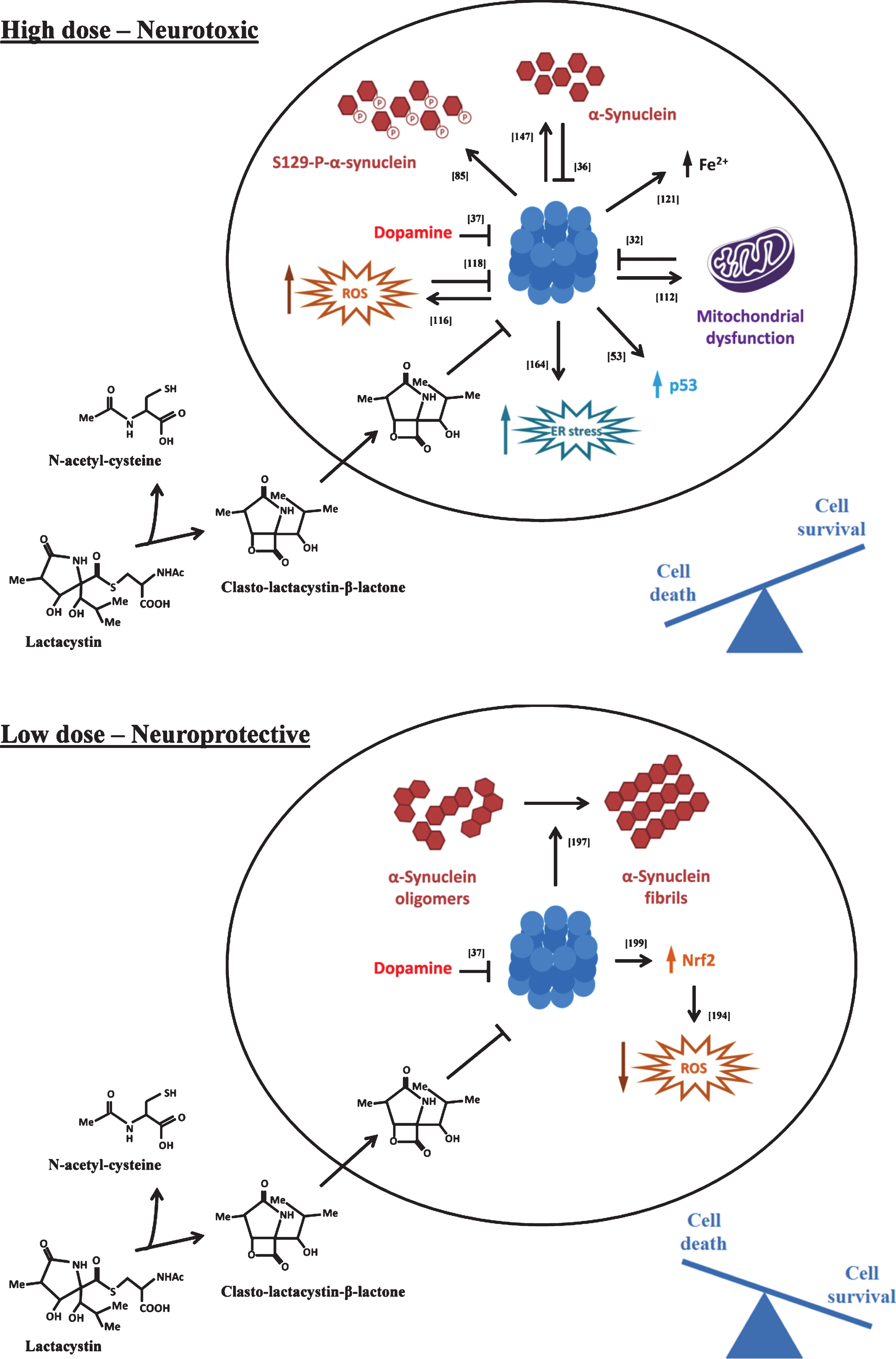
Fig.3
Factors influencing cell death following application of proteasome inhibitors. See text for details.
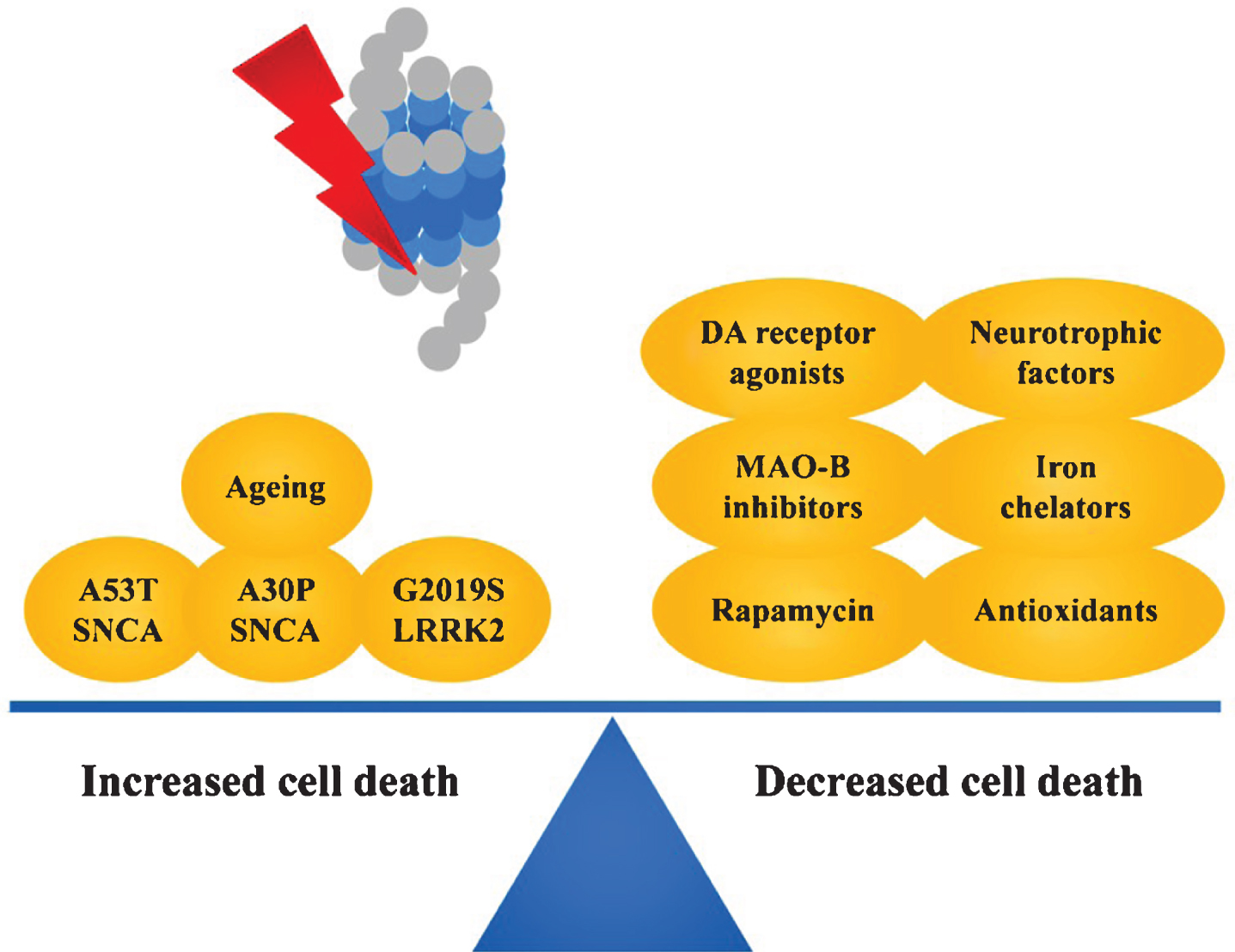
Table 1
Functional and structural defects in the proteasome system in sporadic PD
| Ref | Origin | Structural and/or functional changes |
| [15] | Post-mortem SN | Decreased immunoreactivity for 20S α-subunits in nigral neurons. No change in the expression of 20S β-subunits. |
| [213] | PD iPSCs | Decreased 20S chymotrypsin-like activity. |
| [160] | Post-mortem SN | Decreased immunoreactivity for 20S proteasomes in nigral neurons containing α-synuclein inclusions. |
| [32] | PD cybrids | Decreased 20S trypsin-like and caspase-like activities. |
| [18] | Post-mortem SN | Decreased 20S chymotrypsin-like, trypsin-like, and caspase-like activities. |
| [16] | Post-mortem SN | Decreased expression of 20S α-subunits. |
| [17] | Post-mortem SN | Decreased expression of 20S α-subunits. No change in the expression of 20S β-subunits. Decreased expression of PA700. Decreased 20S chymotrypsin-like, trypsin-like, and caspase-like activities. |
| [19] | Post-mortem SN | Decreased 20S chymotrypsin-like activity. |
iPSC induced pluripotent stem cells, SN substantia nigra, PD Parkinson’s disease.
Table 2
Key findings obtained using proteasome inhibitors to model PD in vitro and in vivo
| Cell cultures | |
 | Dose-dependent apoptotic death of cultured DA-ergic cells and DA-ergic neurons in primary ventral mesencephalic cultures. Formation of aggregates containing α-synuclein in surviving cells |
| Medaka fish | |
 | Loss of DA-ergic neurons with protein accumulation / aggregation. Motor impairment. |
| Rodents: intra-nigral administration | |
 | Loss of nigral DA-ergic neurons with α-synuclein accumulation / aggregation in surviving neurons. Motor impairment. Comparable findings in mice and rats. Extensively replicated as model for PD. |
| Rodents: intra-mfb administration | |
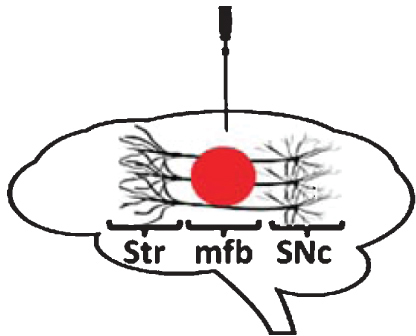 | Loss of nigral DA-ergic neurons with α-synuclein accumulation / aggregation in surviving neurons. Motor impairment. Comparable findings in mice and rats. Replicated as model for PD. |
| Rodents: intra-striatal administration | |
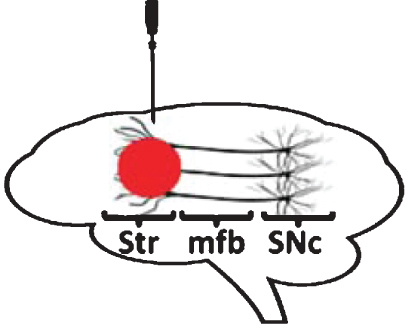 | Loss of nigral DA-ergic neurons with α-synuclein accumulation / aggregation in surviving neurons, however not replicated in one study. Approach not attempted in mice. Reproducibility of the model unclear. |
| Rodents: systemic administration | |
 | Controversy regarding the effects of systemic administration of proteasome inhibitors in both rats and mice. Not a reliable PD model with the current approach. |
| Non-human primates: systemic administration | |
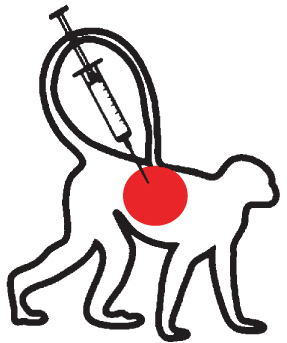 | Approach not successful in generating a PD model. Possibly linked with the failure of the systemic model in rodents. |
DA dopamine, mfb medial forebrain bundle, PD Parkinson’s disease.
Table 3
Overview of the local proteasome inhibition model of PD
| Site | Species | Inhibitor | Effects on SNc DA-ergic neurons | Effects on Str DA-ergic fibers | Additional effects on the brain | Effects on α-syn | Effects on motor function | Ref |
| SNc | Rats | LAC 3 μg | 2d: ↓ TH+ cells (∼20%) | NR | NR | NR | NR | [73] |
| 1w: ↓ TH+ cells (∼65%) | ||||||||
| SNc | Rats | LAC 10 μg | 1w: ↓ TH+ cells (∼45%) | NR | 5w: ↓ TH+ cells in VTA (∼30%) | NR | NR | [74] |
| 5w: ↓ TH+ cells (∼50%) | ||||||||
| SNc | Rats | LAC 2.5 μg | 6w: ↓ TH+ cells (∼90%) | 6w: ↓ DA levels (∼90%) | 6w: ↓ Volume of SN (∼20%). ↓ Non-TH+ cells in SN (∼25%). ↓ GABA levels in SN (∼15%) | NR | 4w: ↓ Spontaneous motor activity (open-field test) | [75] |
| SNc | Rats | LAC 2.5 μg | NR | 6w: ↓ DA levels (∼90%) | 6w: No effect on 5HT levels in Str | NR | 4w: ↓ Initiation of movement / akinesia (catalepsy test). ↓ Use of contralateral forelimb (cylinder test) | [76] |
| SNc | Rats | LAC 0.5 μg, 1 μg, 2.5 μg, 5 μg | 1w: ↓ TH+ cells (∼80% at 1 μg, other doses NR) | 1w: ↓ DA levels (∼25% at 0.5 μg, ∼70% at 1 μg, ∼75% at 2.5 μg and ∼85% at 5 μg) | NR | 1w: ↓ α-Syn expression in SN (∼50% at 1 μg, other doses NR) | No changes in motor behavior (qualitative) | [77] |
| SNc | Rats | LAC 0.5 μg, 1 μg, 2 μg, 10 μg, 20 μg | 2-3 w or 8-10w: ↓ TH+ cells (∼40% at 1 μg, ∼70% at 2 μg, ∼80% at 10 μg and ∼80% at 20 μg) | 2-3 w or 8-10w: ↓ [11C] DTBZ binding to VMAT2 (∼55% at 1 μg, ∼75% at 2 μg, ∼80% at 10 μg and ∼75% at 20 μg) (PET) | 2-3 w or 8-10w: ↓ TH+ cells in VTA (∼20% at 1 μg, ∼25% at 2 μg, ∼50% at 10 μg and ∼50% at 20 μg) | NR | 2-3 w or 8-10w: ↓ Motor function of contralateral hindlimb (tapered ledged beam task) (at all doses, except 2 μg) | [78] |
| The effects of the lesion spread partially to the contralateral SNc | The effects of the lesion spread partially to the contralateral VTA | |||||||
| SNc | Rats | LAC 0.2 μg, 2 μg, 20 μg | 3w: ↓ TH+ cells (∼75% at 2 μg and ∼95% at 20 μg) | NR | NR | 3w: Accumulation / aggregation of α-syn in TH+ cells in SNc (reported at 2 μg) | 3w: ↓ Motor function (qualitative) | [79] |
| SNc | Rats | LAC 10 μg | 2w: ↓ TH+ cells (∼40%) | ↓ TH fiber density | NR | Accumulation / aggregation of α-syn in SNc | 4w: ↓ Spontaneous motor activity (open-field test) | [80] |
| 3w: ↓ TH+ cells (∼85%) | ||||||||
| SNc | Rats | LAC 10 μg | 3w: ↓ TH+ cells (∼80%) | 3w: ↓ TH fiber density (∼70%) | 3w: ↓ Volume of the Str and midbrain (MRI). ↓ TH+ and NeuN+ cells in the VTA, SNr and extra-nigral nuclei close to injection (e.g. red nucleus). No effect on NeuN+ or DARPP-32 + cells in the Str | 3w: Accumulation / aggregation of α-syn in SNc | 3w: ↑ Median neurological score. ↓ Grip strength of the ipsilateral forelimb (grip strength test) | [81] |
| SNc | Rats | LAC 10 μg | 5w: ↓ TH+ cells (∼50%) | NR | 5w: ↓ ChAT+ cells in PPN (∼35%) | 5w: Accumulation / aggregation of α-syn in TH+ cells in the SNc and ChAT+ cells in PPN | 5w: ↓ Use of contralateral forelimb (cylinder test) | [82] |
| SNc | Rats | LAC 10 μg | 5w: ↓ TH+ cells (∼50%) | NR | 5w: ↓ ChAT+ cells (∼45%) and ↓ non-ChAT+ cells (∼25%) in PPN | 5w: Accumulation / aggregation of α-syn in ChAT+ and non-ChAT+ cells in PPN | NR | [83] |
| SNc | Rats | MG 20 μg | 8d: ↓ TH+ cells (∼40%) | NR | NR | NR | 7d: No effect on spontaneous motor behavior (cage activity monitoring) | [203] |
| SNc | Mice | MG 0.4 μg | 12d: ↓ TH+ cells (∼40%) | 12d: ↓ DA levels (∼55%) | NR | NR | NR | [58] |
| Bilateral | ||||||||
| SNc | Mice | EPO 0.02 μg | 2w: ↓ TH+ cells (∼60%) | NR | 2w: No effect on TH+ cells in VTA | NR | 2w: No effect on spontaneous motor behavior (open-field test) | [84] |
| SNc | Mice | LAC 3 μg | 1 w and 3w: ↓ TH+ cells (∼40%) | 1 w and 3w: ↓ DA levels (∼40%) | 1 w and 3w: ↓ TH+ cells in the lateral VTA (PBP) at the injection site (∼30%). No effect on TH+ cells in the entire VTA. ↓ Volume of the SN (∼20%). No effect on PV+ cells in the SNr at the injection site | 1 w and 3w: Accumulation of α-syn and Ser129-P α-syn in SN | 1 w and 3w: ↓ Motor coordination and balance (rotarod test). ↓ Use of contralateral forelimb (cylinder test). ↓ Sensorimotor function (adhesive removal test) | [85] |
| Mfb | Rats | LAC 3 μg | 3w: ↓ TH+ cells (∼20%) | 3w: ↓ DA levels (∼35%) | NR | NR | NR | [55] |
| 4w: ↓ TH+ cells (∼30%) | 4w: ↓ DA levels (∼40%) | |||||||
| Mfb | Rats | LAC 10 μg | 5w: ↓ TH+ cells (∼85%) | 5w: ↓ TH fiber density (∼70%) | 5w: ↓ Volume of Str, ventral midbrain and cortex (M1, S1BF) (MRI). No effect on NeuN+ cells in Str or M1/S1BF cortex. Loss of NeuN+ cells in thalamic and ventral midbrain nuclei. Loss of NeuN+ cells in the VTA and SNr | 5w: Accumulation / aggregation of α-syn in TH+ cells in SNc | 5w: ↑ Median neurological score. ↓ Grip strength of the ipsilateral forelimb (grip strength test). ↓ Motor coordination and balance (rotarod test) | [87] |
| Mfb | Mice | LAC 1.25 μg | 3w: ↓ TH+ cells (∼35%) | 3w: ↓ TH fiber density (∼55%) | NR | 3w: Accumulation of α-syn oligomers in SN | NR | [68] |
| Mfb | Mice | LAC 1.25 μg, 5 μg | 4w: ↓ TH+ cells (∼45% at 1.25 μg and ∼60% at 5 μg) | NR | 4w: No effect on TH+ cells in VTA | 2w: Accumulation of α-syn in TH+ cells of SNc | NR | [88] |
| PSI 0.4 μg | 4w: ↓ TH+ cells (∼25%) | 4w: ↓ DA levels (∼45%) | 4w: No effect on 5HT levels in Str | NR | NR | [88] | ||
| MG 0.4 μg | 4w: ↓ TH+ cells (∼25%) | 4w: ↓ DA levels (∼45%) | 4w: No effect on 5HT levels in Str | NR | NR | [88] | ||
| Mfb | Mice | LAC 5 μg | 1w: ↓ TH+ cells (∼25%) | 1w: ↓ DA levels (∼20%) | NR | 2w: Accumulation / aggregation of α-syn in TH+ cells of SNc | 4w: ↓ Spontaneous motor activity (open-field test). ↓ Forelimb use and skilled motor function (dowel test, suspended thread test) | [88] |
| Bilateral | 3w: ↓ TH+ cells (∼30%) | 2w: ↓ DA levels (∼40%) | ||||||
| 4w: ↓ TH+ cells (∼45%) | 4w: ↓ DA levels (∼50%) | |||||||
| 12w: ↓ DA levels (∼50%) | ||||||||
| Mfb | Mice | LAC 1 μg | 4w: ↓ TH+ cells (∼25% in 5 month old mice and ∼50% in 12 month old mice) | 4w: ↓ DA levels (∼55% in 5 month old mice and ∼65% in 12 month old mice) | No effect on TH+ cells in VTA | Accumulation / aggregation of α-syn in TH+ cells in SNc (in 12 month old mice) | NR | [89] |
| Str | Rats | LAC 0.04 μg | 1w: ↓ TH+ cells (∼40%) | 1w: ↓ DA levels (∼30%) | 1w: No effect on GAD-67 + cells in Str. No effect on TH+ cells in VTA. No effect on 5HT levels in Str | 1w: Accumulation / aggregation of α-syn in SNc | NR | [50] |
| EPO 0.05 μg | 1w: ↓ TH+ cells (∼40%) | 1w: ↓ DA levels (∼40%) | NR | 1w: Accumulation / aggregation of α-syn in SNc | NR | [50] | ||
| Str | Rats | LAC 5 μg, 10 μg | 2w: No effect on TH expression in SN (either dose) | 2w: No effect on DA levels (either dose) | NR | 2w: No effect on α-syn expression in SN (either dose) | NR | [77] |
| Str | Rats | LAC 1 μg, 10 μg | 2w: ↓ TH+ cells (∼20% at 1 μg and ∼75% at 10 μg) | NR | Non-specific toxic effects on striatal cells (CV; observed only at 10 μg) | Accumulation /aggregation of α-syn in SNc (reported at 10 μg) | NR | [90] |
All injections are unilateral unless stated otherwise. 5HT 5-hydroxytryptamine, α-syn α-synuclein, ChAT choline acetyl transferase, CV cresyl violet, DA dopamine, DARPP-32 α-dopamine and cAMP-regulated phosphoprotein-32, DTBZ dihydrotetrabenazine, EPO epoxomicin, GABA gamma-aminobutyric acid, GAD-67 glutamic acid decarboxylase-67, LAC lactacystin, Mfb medial forebrain bundle, MG MG132, MRI magnetic resonance imaging, NR not reported, PBP parabrachial pigmented nucleus, PET positron emission tomography, PPN pedunculopontine nucleus, PV parvalbumin, SN substantia nigra, SNc substantia nigra pars compacta, SNr substantia nigra pars reticulata, Str striatum, TH tyrosine hydroxylase, VMAT2 vesicular monoamine transporter 2, VTA ventral tegmental area.
Table 4
Overview of the systemic proteasome inhibition model of PD
| Species | Protocol | UPS activity | Effects on SNc DA-ergic neurons | Effects on Str DA-ergic fibers | Additional effects on the brain | Effects on α-syn | Effects on motor function | Ref |
| Rats | 3 mg/kg PSI s.c.: 6x over 2w | 1w: ↑ Activity in ventral midbrain (∼50%) | 2w: ↓ TH+ cells (∼55%) | 2 w and 6w: ↓ TH+ fiber density | 2 w and 6w: VTA TH+ cells relatively spared. SNr neurons spared (H&E) | 6w: Accumulation / aggregation of α-syn in SNc, LC and DMN | 2 w and 6w: Motor dysfunction (semiquantitative behavioral rating scale). ↓ Spontaneous motor activity (open-field test) | [92] |
| 2w: ↓ Activity in ventral midbrain (∼50%) | 6w: ↓ TH+ cells (∼70%) | 17-19w: ↓ [11C] CFT binding to DAT (PET) (∼40%) | 6w: ↓ TH+ cells in LC. ↓ Cells in DMNV (H&E). ↓ ChAT+ cells in NMB. No effect on DARPP-32 + and ChAT+ cells in Str | |||||
| 1.5 mg/kg EPO i.p.: 6x over 2w | 4w: ↓ Activity in ventral midbrain (∼50%) | 2w: ↓ TH+ cells (∼60%) | 2w: ↓ DA levels (∼70%) | NR | 2w: Accumulation / aggregation of α-syn in SNc | 2w: Motor dysfunction (semiquantitative behavioral rating scale) | [92] | |
| Rats | 8 mg/kg PSI s.c.: 6x over 2w | NR | 5w: ↓ FG+ cells (total counts) (∼20%). ↓ FG+ cells (morphologically normal) (∼40%) | NR | NR | NR | 5w: ↓ Spontaneous motor activity (cage activity monitoring) | [93] |
| Rats | 8 mg/kg PSI s.c.: 6x over 2w | NR | 15 w and 22w: ↓ TH+ cells (∼60%) | NR | 15 w and 22w: ↓ 5HT+ cells in RN. ↓ DBH+ cells in LC.↓ ChAT+ cells in DMNV. No effect on ChAT+ cells in NBM | NR | 15 w and 22w: ↓ Spontaneous motor activity (open-field test) | [94] |
| 8 mg/kg PSI p.o.: 6x over 2w | NR | 22w: ↓ TH+ cells (∼30%) | NR | 22w: No effect on 5HT+ cells in RN or DBH+ cells in LC.↓ ChAT+ cells in DMNV and NBM | NR | 22w: ↓ Spontaneous motor activity (open-field test) | [94] | |
| 8 mg/kg PSI i.p.: 6x over 2w | NR | 22w: No effect on TH+ cells | NR | 22w: No effect on 5HT+ cells in RN, DBH+ cells in LC, ChAT+ cells in DMNV or ChAT+ cells in NBM | NR | 22w: No effect on spontaneous motor activity (open-field test) | [94] | |
| 12 mg/kg PSI s.c.: 6x over 2w | NR | 15w: ↓ TH+ cells (∼30%) | NR | 15w: No effect on 5HT+ cells in RN or DBH+ cells in LC.↓ ChAT+ cells in DMNV and NBM | NR | 15w: ↓ Spontaneous motor activity (open-field test) | [94] | |
| Rats | 16 mg/kg PSI s.c.: 6x over 2w | NR | 15w: ↓ TH+ cells (∼30%) | NR | 15w: No effect on 5HT+ cells in RN or DBH+ cells in LC.↓ ChAT+ cells in DMNV. No effect on ChAT+ cells in NBM | NR | 15: No effect on spontaneous motor activity (open-field test) | [94] |
| Rats | 3 mg/kg PSI s.c.: 6x over 2w | NR | NR | 6w: ↓ DA levels (∼25%) | NR | NR | 6w: ↓ Spontaneous motor activity (open-field test) | [95] |
| 10w: No effect on DA levels | 6 w and 10w: ↓ Motor coordination and balance (rotarod test) | |||||||
| Rats | 3 mg/kg PSI s.c.: 6x over 2w | After last injection: ↓ Activity in midbrain | 6w: ↓ TH+ cells (∼40%) | NR | NR | 6w: No evidence of α-syn aggregation in SNc | 6w: No effect on motor function (SHIRPA scale) | [96] |
| 2w: No effect on activity in midbrain | ||||||||
| Rats | 5-8 mg/kg PSI s.c.: 6x over 2 w | NR | 8w: ↓ TH+ cells (∼50%) | NR | 20 w and 40w: ↓ 5HT+ neurons in RP. ↓ DBH+ cells in LC. ↓ ChAT+ cells in DMNV | 8 w, 20 w and 40w: Accumulation / aggregation of α-syn in TH+ cells in SNc | 8 w, 20 w and 40w: ↓ Spontaneous motor activity (hole-board apparatus) | [97] |
| 20w: ↓ TH+ cells (∼50%) | ||||||||
| 40w: ↓ TH+ cells (∼45%) | ||||||||
| Rats | 3 mg/kg or 6 mg/kg PSI s.c.: 6x over 2w | NR | 12w: No effect on TH+ cells (either dose) | 12w: No effect on TH+ fiber density (either dose) | NR | 12w: No evidence of α-syn aggregation in SNc (either dose) | 12w: No effect on spontaneous motor activity (either dose) | [98] |
| GFPu mice | 6 mg/kg or 9 mg/kg PSI s.c.: 6x over 2w | Lack of GFPu accumulation in the brain (either dose) | NR | NR | NR | NR | NR | [98] |
| Rats | 3 mg/kg PSI s.c.: 6x over 2w | NR | 8w: No effect on TH expression in SN | 8w: No effect on TH expression | 8w: ↑ TH expression in the glomerular layer of olfactory bulb | 8w: No evidence of α-syn aggregation in SNc | 8w: No effect on motor function (footprint test) | [99] |
| Rats | 3 mg/kg PSI s.c.: 6x over 2w | NR | 8w: No effect on TH+ cells | 8w: No effect on TH+ fiber density | 8w: No cell loss in LC (CV). No effect on ChAT+ cells in NBM | 8w: No evidence of α-syn aggregation in SNc | 8w: No effect on spontaneous motor activity (cage activity monitoring) | [101] |
| Rats | 3 mg/kg PSI s.c.: 6x over 2w | NR | 6w: No effect on TH+ cells | 6w: No effect on DA levels. No effect on TH or DAT expression | NR | 6w: No evidence of α-syn aggregation in SNc | 6w: No effect on spontaneous motor activity (open-field test). No effect on forelimb stride length (gait analysis) | [102] |
| Rats | 3 mg/kg PSI s.c.: 6x over 2w | 2w: No effect on SN activity, despite ↓ brain activity | 12w: No effect on TH+ cells | 12w: No effect on DA levels. No effect on TH+ fiber density | 12w: No effect on norepinephrine or 5HT levels in Str | NR | 12w: No effect on spontaneous motor activity (cage activity monitoring) | [103] |
| 12w: No effect on SN or brain activity | ||||||||
| Rats | 0.5 mg/kg MG i.p.: 7x over 2w | NR | 6w: ↓ TH+ cells (∼20%) | 6w: ↓ TH+ fiber density in dorsolateral Str. No effect on TH fiber density in dorsomedial or ventral Str | NR | NR | 1 w and 6w: No effect on spontaneous motor activity (open-field test). No effect on motor coordination and balance (rotarod test) | [214] |
| Mice | 3 mg/kg PSI s.c.: 6x over 2w | NR | 6w: ↓ TH+ cells (∼45%) | NR | NR | NR | NR | [53] |
| Mice | 3 mg/kg PSI s.c.: 3x over 6 h | NR | NR | 5d: No effect on DA levels. No effect on TH expression | NR | NR | NR | [100] |
| Mice | 6 mg/kg PSI s.c.: 6x over 2w | NR | 8w: No effect on TH+ cells (vs. sham (70% ethanol)-injected mice), although ↓ 35% TH+ cells compared to saline-injected mice | 8w: No effect on DA levels (vs. sham (70% ethanol)-injected mice), although ↓ 80% loss of DA compared to saline-injected mice | NR | NR | 8w: No effect on spontaneous motor activity (cage activity monitoring), although transient decrease at 2w | [104] |
| Mice | 10 mg/kg/ day PSI: osmotic pump 3w | NR | 3w: No effect on TH+ cells | 3w: No effect on DA levels | NR | 3w: No evidence of ubiquitin accumulation / aggregation in SNc | 3w: ↓ Spontaneous motor activity (cage activity monitoring) | [107] |
All time points are counted from cessation of treatment. 5HT 5HT 5-hydroxytryptamine, ChAT choline acetyl transferase, CV cresyl violet, DA dopamine, DAT dopamine transporter, CFT 2β-carbomethoxy-3β-(4-fluorophenyl)tropane, DARPP-32 α-dopamine and cAMP-regulated phosphoprotein-32, DBH DA β-hydroxylase, DMNV dorsal motor nucleus of the vagus, EPO epoxomicin, FG fluorogoldTM, H&E hematoxylin and eosin, i.p. intraperitoneal, LC locus coeruleus, NMB nucleus basalis of Meynert, NR not reported, PET positron emission tomography, p.o. oral, PSI carbobenzoxy-L-isoleucyl-L-gamma-t-butyl-L-glutamyl-L-alanyl-L-leucinal, RP raphe nuclei, s.c. subcutaneous, SN substantia nigra, SNc substantia nigra pars compacta, SNr substantia nigra pars reticulata, Str striatum, TH tyrosine hydroxylase, UPS ubiquitin-proteasome system, VTA ventral tegmental area.
Table 5
Validity criteria of the proteasome inhibition model of PD
| Construct validity |
| Catalytic activity of the proteasome decreased in the SN of PD patients. Good construct validity, especially for intra-nigral administration of proteasome inhibitors. |
| Face validity |
| Pattern of pathology similar to PD, including vulnerable degeneration of nigral DA-ergic neurons (>VTA DA-ergic neurons), α-synuclein accumulation / aggregation, neuroinflammation and extra-nigral pathology (e.g. cell loss in the PPN). Motor impairment consistently observed. |
| Predictive validity |
| Motor dysfunction improved with L-DOPA, apomorphine, and pramipexole. |
| DA dopamine, SN substantia nigra, PD Parkinson’s disease, PPN pedunculopontine nucleus, VTA ventral tegmental area. |


