
Pathophysiological Concepts and Treatment of Camptocormia
Abstract
Camptocormia is a disabling pathological, non-fixed, forward bending of the trunk. The clinical definition using only the bending angle is insufficient; it should include the subjectively perceived inability to stand upright, occurrence of back pain, typical individual complaints, and need for walking aids and compensatory signs (e.g. back-swept wing sign). Due to the heterogeneous etiologies of camptocormia a broad diagnostic approach is necessary. Camptocormia is most frequently encountered in movement disorders (PD and dystonia) and muscles diseases (myositis and myopathy, mainly facio-scapulo-humeral muscular dystrophy (FSHD)). The main diagnostic aim is to discover the etiology by looking for signs of the underlying disease in the neurological examination, EMG, muscle MRI and possibly biopsy. PD and probably myositic camptocormia can be divided into an acute and a chronic stage according to the duration of camptocormia and the findings in the short time inversion recovery (STIR) and T1 sequences of paravertebral muscle MRI. There is no established treatment of camptocormia resulting from any etiology. Case series suggest that deep brain stimulation (DBS) of the subthalamic nucleus (STN-DBS) is effective in the acute but not the chronic stage of PD camptocormia. In chronic stages with degenerated muscles, treatment options are limited to orthoses, walking aids, physiotherapy and pain therapy. In acute myositic camptocormia an escalation strategy with different immunosuppressive drugs is recommended. In dystonic camptocormia, as in dystonia in general, case reports have shown botulinum toxin and DBS of the globus pallidus internus (GPi-DBS) to be effective. Camptocormia in connection with primary myopathies should be treated according to the underlying illness.
DEFINITION OF CAMPTOCORMIA
Camptocormia (from the Greek “kamptein” = to bend and “kormos” = trunk) is an involuntary flexion of the thoracolumbar spine when standing, walking or sitting, which disappears completely in the supine position. The syndrome is also known as “bent spine syndrome”, was first described by Henry Earle in 1815 [1] and reported by James Parkinson in some of his cases in 1817 [2]. The term was coined by the French neurologist A. Souques in 1915 to describe an “incurvation du tronc” in soldiers of World War I indicating a “cyphose hystérique” [3]. Until the 1980 s, camptocormia was considered to be a psychiatric condition. Kiuru & Iivanainen [4] and Laroche et al. [5] were the first to describe camptocormia in association with organic diseases.
The criteria for defining camptocormia are a matter of debate. Most studies use a forward bending angle of between 15° to 45° as the main criterion [6–17]. A large number of studies use only a descriptive term without a bending angle, indicating the difficulties in defining camptocormia [18–28]. Based on a control group of patients with Parkinson’s disease (PD) who disclaimed suffering from camptocormia, a recent study demonstrated that the stooped posture of advanced PD does not exceed a forward bending angle of 25°. Oeda et al. found a similar forward bending angle distribution in PD patients without camptocormia [29]. Furthermore, analysis of the group of photo-documented PD camptocormia patients (n = 145) showed that the bending angle as the sole criterion is insufficient to define camptocormia [Margraf et al., 2016 under review] because a third of the patients who suffered subjectively from camptocormia had an angle of less than 30°.
Others have defined camptocormia by a score of≥2 of item 28 (posture) of the Unified Parkinson Disease Rating Scale part III (UPDRS III) [30]. This definition does not differentiate between the stooped posture of advanced PD and camptocormia. Even the revised MDS-UPDRS item “posture” (3.13) cannot differentiate between stooped posture and camptocormia. The ability of a patient to straighten up temporarily does not rule out camptocormia and forward bending by orthopedic diseases of the spine must be excluded.
As camptocormia is a very disabling syndrome that frequently causes social isolation of patients [13], the diagnosis of the syndrome may be supported by typical individual complaints resulting from the loss of function of paraspinal muscles [31]. Characteristic complaints are the inability to drive a car (because of the inability to turn the body backwards), inability to look people in the eyes, inability to carry something in front of the body, inability to pick up things that lay higher than the table or the shoulder position and problems in eating and swallowing or dyspnoea due to body position (Margraf et al., 2016 under review).
In summary, camptocormia is an involuntary pathological flexion of the thoracolumbar spine that is passively reversible (recumbent position) and that causes relevant impairment in daily life as well as back pain in most cases. The definition and the clinical diagnostic criteria should focus on the following aspects: Involuntary flexion of the thoracolumbar spine, reversibility in recumbent position, forward bending angle, individual complaints, back pain, hardening of the paraspinal muscle and the course during the day.
Camptocormia is unlikely to be confused with the dropped-head syndrome, even when these two occur together in rare instances [32]. A relevant number of PD camptocormia patients present with additional laterodeviation [8, 10, 31], but there are no accepted criteria of how to differentiate the combination of Pisa syndrome and camptocormia. Seen pragmatically, the predominant abnormality gives the name but the underlying abnormality may be the same.
OCCURRENCE OF CAMPTOCORMIA
Camptocormia occurs in association with primary and secondary myopathies, inflammation, dystonia, as a pharmacological side effect or as a functionaldisorder (Fig. 1). Primary muscle diseases with involvement of axial muscles, focal myositis and secondary myopathies associated with neurodegenerative diseases seem to be the most frequent causes of camptocormia. The overall incidence of camptocormia is unknown. Data on the incidence have only been published for movement disorders. The relative frequency of primary or secondary myopathies or focal myositis varies with the specialty of the reporting departments. In a cohort of camptocormia patients from a rheumatology center, 65% of the patients suffered from a dystrophy, 17% from PD and 13% from a myositis [33]. In a cohort from a neuromuscular center, 25% suffered from a facio-scapulo-humeral muscular dystrophy (FSHD) and 18% from sporadic inclusion body myositis [32]. There is some evidence that FSHD is underdiagnosed in camptocormia [34].
Camptocormia occurs in a broad range of primary muscle diseases. It can be due to dystrophies, especially FSHD, limb-girdle dystrophies and myotonic dystrophy, structural myopathies and metabolic myopathies including defects in energy metabolism and hypothyroidism. It may occur in all forms of inflammatory muscle diseases. Camptocormia may occur in diseases of the neuromuscular junction, especially myasthenia gravis and as radiation-induced secondary myopathy. In some patients, degenerative changes of the spinal column may cause camptocormia [35]. A list of underlying diseases is given in Table 1.
Among neurodegenerative diseases, camptocormia occurs most frequently in PD [6]. The published prevalence rates for camptocormia in PD differ between 3% and 17.6% [10, 13, 22, 75], and large studies reported a prevalence of around 10% [76]. As camptocormia is frequently observed in advanced PD, the mean disease duration of the observed patients influences the prevalence rate found in the studies [77, 78].
Besides PD, camptocormia also occurs in other α-synuclein aggregation disorders, such as multisystem atrophy (MSA) [79, 80] and dementia with Lewy bodies [81]. Camptocormia is also observed in amyotrophic lateral sclerosis. Paraspinal muscles are involved quite frequently in ALS [82], and camptocormia may occur, especially in patients in whom the onset of ALS was in the respiratory muscles [83–85]. With regard to other neurodegenerative diseases, there have been individual reports of camptocormia in Alzheimer’s disease [86]. Focal lenticular lesions have also been associated with camptocormia [87].
Patients with dystonia and involvement of axial muscles are another group in which camptocormia is common. DYT 1 mutations have been described in individual patients [88]. Camptocormia can occur in isolated as well as in combined dystonia [34, 89] and some authors categorize camptocormia in PD as combined (formerly secondary) dystonia [90]. Recent work points out that hereditary muscle diseases are not infrequently misdiagnosed as dystonia [34].
Camptocormia can also be a rare pharmacological side effect. The substances that may induce camptocormia are mainly dopaminergic drugs, anti-psychotics, anti-epileptics and cholinesterase inhibitors/anti-cholinergics. The Pisa syndrome, which has a clinical overlap with camptocormia, was initially assumed to be induced by anti-psychotic drugs. The drug list includes: L-dopa/carbidopa, L-dopa/benzaseride [91], L-dopa/carbidopa/entacapone [92], pramipexole [93], ropinirole [94], olanzapine [95–97], valproate [4], atomoxetine (selective norepinephrine reuptake inhibitor) [98] donepezil and rivastigmine [99].
Camptocormia can be a functional disorder. It can develop following a blast injury [100] or as a conversion disorder to escape anxiety-provoking situations and conflicts [101, 102]. It may occur in adolescents, as, for example, in a 13-year old boy with concomitant anorexic behavior who refused to accept the cultural change associated with immigration [103]. When psychotherapy is able to uncover the purposes of the symptom, the symptoms may no longer serve any purpose, and the camptocormia may disappear [101].
DIAGNOSIS OF CAMPTOCORMIA
Clinical examination
The main diagnostic approach is to search for the etiology. The clinical examination should focus primarily on signs of myopathies and movement disorders. Beside typical aspects of a Parkinson syndrome this includes a search for indicators of dystonia, such as sensory tricks (e.g. in these cases backward walking, pressing on the thighs, stepping with one foot on a chair, carrying a back pack) or painful contractions of the abdominal muscles while standing with a feeling of being pulled forward. In contrast, in camptocormia associated with PD a hardening of the lumbar paravertebral muscles can regularly be palpated. These patients sometimes hold their arms in a backward position to stabilize their body position (back-swept wing sign). In the absence of a movement disorder, a careful search for a paresis somewhere else is important, and the pattern of extra-axial paretic muscles could be the key to the diagnosis. In myositis there could be asymmetrical painful paravertebral muscles with erythema and elevated temperature.
The neurological examination should describe the extent of the forward bending, record the individual peculiarities of the posture (e.g. additional laterodeviation or other axial disorders) and verify that the bending is not fixed. At present, there is no generally accepted method for assessing the angle. Assessment of the angle is not trivial because the presentation may change [104]. We suggest measuring the bending angle after having had the patient stand free for 3 minutes. Photographs or videos can be very useful for this purpose.
The course and duration of the camptocormia may give a hint as to its etiology. While inflammatory camptocormia usually shows a more rapid course over weeks to months, there are basically two patterns of camptocormia in PD: A more rapid one and one with symptoms worsening even after more than one year [8, 11, 12, 22, 105]. Afterwards, symptoms may remain on a plateau in some patients, while others might experience a second acceleration of symptoms later on. A more stepwise and slow progression of camptocormia could indicate a primary myopathic process. A slight to moderate CK elevation is not conclusive for myositis but is also seen in PD camptocormia [11, 17].
In documenting the disease severity, it is helpful to record the need for technical support (like orthoses and walking aids), the main individual complaints due to camptocormia, the severity of pain, if present, on a numeric pain rating scale (0–10) as well as its frequency and localization, and the subjective burden of camptocormia on a Likert scale. Aspects such as back disorders in the medical history and the current and former medication (side effects, neuroleptics) are of interest as are the family history.
In the literature, camptocormia in PD is much more accurately described than camptocormia of other etiologies. Frequently, only the forward bending aspect is reported, the existence of pain is occasionally noted but further details are lacking.
EMG
EMG is a helpful diagnostic tool in camptocormia. With it, one can identify myopathic, myositic or neurogenic changes. The choice of muscles to beexamined should be determined by the neurological examination. These might comprise dorsal and ventral trunk muscles (mainly paravertebral and rectus abdominis muscles) in patients with movement disorders. Performing EMG in muscles of the abdominal wall can be challenging, especially with the patient in a standing position, which is necessary when searching for dystonic EMG-activity. When myopathy is suspected a distal and proximal limb muscle should be added. While the EMG of the paravertebral muscles can show spontaneous activity in a relaxed patient, the assessment of neurogenic and motor unit pattern may be more difficult, as the patients have to partially activate the paravertebrals. In older patients, neurogenic pattern and spontaneous activity are not obligatory pathological [106, 107]. Motor units of the paravertebral muscles differ from limb muscles in that they have a smaller amplitude and a shorter duration [108].
Muscle imaging
Muscle MRI is an important diagnostic tool in camptocormia as it shows typical muscle abnormalities and the extent of the muscle affection. In addition, imaging is necessary to exclude orthopedic diseases. It is essential to use special MRI sequences that show acute and chronic processes. Acute changes in the muscle can be identified using contrast-enhanced MRI sequences or the short time inversion recovery (STIR) sequence. The STIR sequence is sensitive for edema and is suppressing fat tissue. However, the findings are unspecific and pathological STIR findings can be seen e.g. in myositis, trauma, denervation and a wide range of muscle diseases [109–115].
Ideally, the STIR sequence is combined with a T1 sequence that best covers the chronic changes of camptocormia: These are atrophy, often asymmetrical, and fatty degeneration shown by hyperintense signals in T1. The frequently used T2 sequence is not a substitute for the T1 sequence because hyperintensities in T2 can resemble both edema and fat and cannot support the classification of camptocormia into acute or chronic stages.
In PD camptocormia the spectrum of muscle MRI findings ranges from swelling and edema of the paravertebral and the quadratus lumborum muscles [65] to findings primarily described as atrophy and fatty degeneration [9, 13, 20, 22]. Previously, we suggested that the duration of camptocormia in PD corresponds with radiological muscle findings [116]. Edema and swelling in the paravertebral, quadratus lumborum and psoas muscles as acute changes were found in camptocormia duration of up to 31 months. Swelling as a hallmark of severe edema was only found in the camptocormia patients and not in the matched PD control group. The occurrence of fatty degeneration in the paravertebral muscles characterized a chronic phase and was found in camptocormia duration of 37 months and longer. This model of categorizing can also be possibly found in camptocormia of other etiologies [7, 61, 65] as shown in Table 2. A disadvantage of the muscle MRI is the lack of specificity. In particular, if muscle MRI shows exclusively acute changes, a muscle biopsy is indicated, because STIR hyperintensities cannot prove myositis. The muscle MRI can be used to determine the most appropriate biopsy site [117].
Muscle biopsy: Methodological aspects
Selection of biopsy site
The most frequent indication for a muscle biopsy in camptocormia is to identify or exclude myositis involving paraspinal muscles. The biopsy site should be a muscle that is involved in the disease but does not yet show end-stage changes only. In camptocormia, the biopsy of a paraspinal muscle cannot be replaced by a limb muscle biopsy. Inflammatory diseases can show a very focal involvement even within a single muscle and can easily be missed. The clinical examinations and imaging by MRI or ultrasound may be helpful in identifying the best biopsy site. Prior EMG investigations exclude the muscle from biopsy, because needle tracks may mimic inflammatoryprocesses.
The histology of paraspinal muscles differs from that of limb muscles
When comparing paraspinal muscles of elderly patients with their limb muscles, a higher number of ragged red fibers is seen in paraspinal muscles [118, 119]. Incidental focal chronic endomysial inflammation has been reported for paraspinal necropsy muscles [120]. From these investigations of control patients, we conclude that some ragged-red fibers, rimmed vacuoles, COX-deficient fibers and sparse infiltrates of lymphocytes may be found randomly in paraspinal muscles and are not necessarily pathological [118].
Myopathological findings in camptocormia
In inflammatory diseases associated with camptocormia, the myopathological findings in paraspinal muscles are the same as in other muscles involved in these diseases. In myopathies that present as axial myopathy, histology of paraspinal muscles may show pathology even if limb muscle biopsies were normal [121] but the experience with paraspinal muscle biopsies in these diseases is limited [122]. The spectrum of diseases that should be considered is listed in Fig. 1 and Table 1.
Camptocormia in PD, however, shows a specific and consistent lesion pattern as was recently shown [118]. It is composed of myopathic changes with type-1 fiber hypertrophy, loss of type-2 fibers, loss of oxidative enzyme activity and a fine granular acid phosphatase reactivity of lesions. Ultrastructurally, myofibrillar disorganization and Z-band streaming up to electron-dense patches/plaques were seen in the lesions. Endomyseal fibrosis and fatty degeneration were observed to a variable extent and seem to correlate with the duration of camptocormia [118]. In principle, these changes have been described in some case series before [12, 20, 123], but without the knowledge of the physiological spectrum of paraspinal muscle morphology, the interpretation was inconsistent.
PATHOPHYSIOLOGICAL CONCEPTS
The pathophysiological concepts for camptocormia are as heterogenous as the causes of the syndrome. Primary muscle diseases, secondary myopathies, inflammation and dystonia seem to be the main associated diseases in camptocormia, and different concepts must be assumed for each group. Diseases may directly involve paraspinal muscles, as with inflammatory diseases, specific muscle diseases such as FSHD, limb-girdle muscular dystrophies (LGMD), myotonia and metabolic myopathies. In these diseases, camptocormia seem to be associated with a loss of function of paravertebral muscles. Fibrosis may be the reason for radiation-induced myopathies [58].
Some neurodegenerative diseases may cause secondary myopathy. In ALS the mechanism leading to camptocormia is obviously the degeneration of the secondary or primary motor neurons of the respective muscles. In PD the pathophysiological mechanism of camptocormia is not as apparent. Frequently, camptocormia in PD was interpreted as dystonia [90] but several clinical aspects (EMG results, negative sensory trick testing, lack of positive effects of botulinum toxin treatment) as well as the myopathological lesion profile in biopsies argues against this assumption.
Explanation of camptocormia in PD by a proprioceptive dysregulation
The pathophysiology of camptocormia is a matter of debate. The lesion patterns that were seen in biopsies of paraspinal muscles in camptocormia give a first hint on the pathophysiological mechanisms [118]. The myopathological lesions of paraspinal muscles in camptocormia show remarkable similarities to lesions found in the soleus muscle after experimental tenotomy of the Achilles tendon [124]. The lesions in experimental tenotomy were generated only when tendons were cut but the motor nerve and the spinal tracts remained intact. By transsecting the motor nerve or the spinal tracts of the polysynaptic reflex arch in addition to the tendon, the muscle lesions as well as the motor unit activity of the muscle are abolished. This led to the conclusion that the muscle lesions are the consequence of a dysregulation of the functionally intact polysynaptic reflex arch: After tenotomy the input information from the Golgi tendon organ is “muscle tension is too low”, and the consequence is a hypercontraction of the muscle, as observed, for example, as “Popeye sign” after biceps tendon tears [125], and result in myopathy. In camptocormia, the same muscle lesion pattern is observed [118], but the tendons seem to be intact. Clinically, paraspinal muscles in camptocormia show a painful hardening like muscles after tendon tear [125]. As opposed to tenotomy, where the myopathological changes are due to a dysregulation of the polysynaptic reflex arch at the level of tendons, the myopathological changes in PD-related camptocormia may be due to a dysregulation of proprioception at the level of the central nervous system. Such an impairment of proprioception has been described in PD patients [126]. This pathophysiological concept is supported by the observation that lenticular lesions [87] can cause camptocormia and that neurostimulation of the subthalamic nucleus can relieve PD-related camptocormia [31] as it (partially) reverts the proprioceptive dysregulation [127]. Later in the course of camptocormia, intrafascicular fibrosis and fatty replacement of muscle fibers occur as secondary changes [118]. In this stage, camptocormia cannot be treated by procedures that act on muscle fibers. Consequently, in this state neurostimulation of the subthalamic nucleus has no effect on the bending angle [31].
Pathophysiology of camptocormia in dystonia and as side effect of medication
In isolated and combined dystonia, camptocormia can be addressed as an imbalance in the activity of agonistic and antagonistic muscles, which also may be the reason for camptocormia as a side effect of drugs. Most of these drugs interfere with neurotransmitters such as L-dopa. Among these are dopamine agonists, anti-cholinergics, anti-psychotics, and anti-epileptics.
TREATMENT OPTIONS FOR CAMPTOCORMIA
Treatment of camptocormia depends mainly on the etiology and may therefore be difficult. At present there is an unfortunate lack of controlled studies for many of the different etiologies. Figure 2 illustrates a proposed work-up and therapy algorithm for common etiologies and diseases with which camptocormia can occur.
Treatment of PD-associated camptocormia
Drug therapy
In the majority of patients, treatment with L-dopa gives only minimal or no improvement of camptocormia in PD [6, 8, 19]. The two exceptions are the rare cases of L-dopa responsive camptocormia, possibly an off-dystonia [128], and camptocormia in patients with end of dose fluctuations.
Optimizing the PD therapy may improve camptocormia as was described with continuous subcutaneous infusions of apomorphine in a case series of five PD patients with a follow-up of up to three years [129].
Botulinum toxin
In five studies in PD patients with camptocormia, botulinum toxin (BTX) was injected under EMG, ultrasound or CT guidance [6, 130–133]. BTX was injected into the rectus abdominis, abdominal external oblique and ilio-psoas muscles either singly or as a combination of several. Different types of botulinum toxin A were used with different dosages per muscle [134]. Of a total of 23 PD patients with camptocormia treated with BTX, four (17%) improved to some extent while 19 showed no effect. In the studies with negative results, the injection guidance and dosage were sufficient. However, several studies [6, 73, 130] reported that BTX injections were partially successful in PD camptocormia patients with signs of dystonia of the rectus abdominis muscle.
In conclusion, some patients with PD camptocormia may be suitable for treatment with BTX, although according to the data, BTX treatment would not be effective in the vast majority of them.
Lidocaine
Lidocaine was used in two non-blinded, open-label studies [135, 136]. In these studies the authors defined an upper and a lower subtype of PD camptocormia. They performed repeated injections of lidocaine into the external oblique muscle for upper camptocormia and into the psoas major muscle for lower camptocormia. The average improvement of the bending angle was approximately 12°. However, almost none of the patients showed a bending angle below 30° after treatment. Treatment with lidocaine shows only limited improvement, so far, but further investigation is needed [137].
Trans-spinal magnetic stimulation (TSMS)
Arii et al. compared repetitive TSMS with sham stimulation in PD patients with camptocormia in a randomized, crossover study [138]. In the standing position the angle improved immediately after the stimulation by a mean of 10.9° and in the sitting position by 8.1° while there were no significant changes in the sham group. The observed effects remain ambiguous as they are too limited to be clinically relevant and a proposed influence of the stimulation on ascending and descending pathways in the spine is conceivable but were not proven [139].
Deep brain stimulation (DBS)
Although bilateral neurostimulation of the subthalamic nuclei has been shown to improve core symptoms of PD [140], the effect on PD-associated camptocormia is heterogeneous. Of about 80 studied patients, only about 60% showed improvement [16, 31, 141]. Multifactorial analysis showed a correlation between an improvement of the bending angle and the time between onset of camptocormia and start of neurostimulation [31]. All patients with camptocormia of up to 1.5 years duration showed a beneficial effect, while patients with camptocormia persisting between 1.5 and ca. 3 years showed mixed results. No patient with a duration of more than 40 months showed any improvement except for a single patient whose camptocormia was L-dopa-responsive. The bending angle was not a prognostic factor. Similar results were seen in an independent study [16].
Surgical treatment of PD camptocormia
Each of the so far reported 10 PD patients who were treated with spinal surgery due to camptocormia [142–146] had acute or delayed complications of various degrees after surgical intervention. Regularly these patients had at least one revision [147]. The high revision rate may more likely to be connected with the diagnosis of PD than camptocormia, as Babat et al. [148] reported for PD patients with spine surgery to various reasons other than camptocormia also a revision rate of 86%. A possible explanation might be the susceptibility of PD patients for osteoporosis [142, 146, 148, 149]. PD patients are a high-risk population in adult spinal surgery [150]. Surgical spine procedures increases the risk of a later formation of camptocormia [17] and minor surgical interventions in PD patients can lead to instability of the spine [146]. Specifically in PD camptocormia an imminent limitation is the adjacent segment instability after surgical correction of camptocormia leading again to an abnormal posture. However, in 10 surgically treated PD camptocormia patients 5 reported even in the face of relevant complications and a prolonged postoperative course for recovery a benefit in their posture and a reduction in back pain, 3 had an unfavorable outcome and in 2 cases it was not reported.
Treatment of dystonic camptocormia
Drug therapy
There are some case reports of patients with dopamine-responsive dystonia presenting with camptocormia who benefitted greatly from long-term L-dopa therapy in low [151, 152] or sometimes high doses [88]. In an earlier case series of patients with adult-onset axial dystonia a third of these patients improved under treatment with either a combination of tetrabenazine, pimozide, and an anticholinergic drug, or with high dose anticholinergic drugs alone [153].
Botulinum toxin
Overall the effects of BTX in patients with dystonic camptocormia are more homogenous than in those with PD-associated camptocormia. However, experience is still limited by small datasets and the lack of controlled studies. Beneficial effects have been reported in patients with segmental or abdominal dystonia with dystonic findings in the EMG after treatment with BTX injections in the upper abdominal muscles or the rectus abdominis [89, 154]. However, BTX injections in abdominal dystonia were ineffective in two cases [6], or had only partial effects [155].
Deep brain stimulation
To date, 17 dystonic camptocormia patients were reported to have been treated by deep brain stimulation of the GPi [26, 27, 155–160]. Fifteen of these were diagnosed primary dystonia, and two probably had secondary dystonia. The Burke Fahn Marsden Dystonia Rating Scale (BFMDRS) sub-item “trunk” showed a reduction by 50% to 100% on the last follow up at between six to sixty months after DBS.
As in other types of dystonia, a possible treatment algorithm for dystonic camptocormia would start with medication followed by botulinum toxin as secondary therapy option, and if this is unsuccessful by DBS of the GPi.
Backpack treatment
One PD patient with camptocormia who did not respond to BTX injections in the rectus abdominis muscle showed complete resolution of camptocormia when wearing a low-slung backpack weighting approximately 6 kg [161]. This benefit was prolonged and only ended on removal of the backpack. The backpack possibly stimulates a sensory trick indicating a dystonic symptom.
Treatment of inflammatory camptocormia
Camptocormia in inflammatory myopathies and chronic inflammatory demyelinating polyradiculoneuropathy can be successfully treated with immunosuppressive therapy but therapy response depends on the presence of inflammation and absence of secondary changes such as atrophy and fatty degeneration. A MRI of the paravertebral muscles (e.g. T1, STIR) and camptocormia duration might predict the treatment outcome with good results in acute changes and very limited results depending on the degree of chronic muscle fatty degeneration. The choice of immunosuppressive drug and its dosage should be adjusted to the disease activity (cf. Tab. 2). An exception is inclusion body myositis (IBM) in which immunosuppressive drugs and immunoglobulins are ineffective [162]. The best therapeutic approach in IBM to slow progression is regular physiotherapy [163].
Treatment of camptocormia in primary myopathies and myasthenia gravis
Primary myopathies and myasthenia gravis should be treated according to the underlying disease. This may resolve the axial syndrome as was shown for riboflavin treatment of late-onset multiple acyl-CoA dehydrogenase deficiency (MADD), a lipid storage myopathy [165], and thyroid hormone replacement therapy in hypothyroid myopathy [55]. Treatment effects in myasthenia gravis are possibly favorable in case of acute muscle changes and limited if chronic muscles changes have already occurred (please compare Table 2).
Camptocormia as side effect of several medications
Discontinuing medications reported to induce camptocormia as a side effect may improve the posture. This has been shown in a study in which pramipexole was discontinued in 34 PD patients after which camptocormia improved [76], as well as in a case report [93].
Camptocormia as a dose-dependent side effect of valproate monotherapy resolved within 1-2 weeks after dose reduction [4]. Olanzapine-induced camptocormia normalized within weeks to months after olanzapine was discontinued [95–97].
Supportive therapy independent of camptocormia etiology
Pardessus et al. showed the efficiency of a leather orthosis that straightened the patients and reduced back pain [166]. About 68% wore the leather orthosis at least 9 hours a day. In a prospective study, de Séze et al. evaluated a new orthosis based on the principle of thoraco-pelvic anterior distraction in camptocormia combined with a physiotherapy program [167]. Camptocormia was significantly improved as shown by clinical and radiological data in a follow-up after 90 days. Back pain was reduced by 70%. In a case series with 3 PD patients with camptocormia a high-frame walker with forearm support was shown to be an effective support material [168]. Back pain was reduced and the patients’ ability to walk for a longer distance in an upright position was enhanced. PD patients with anterior trunk bending (UPDRS item posture ≥ 2) benefit from muscle stretching, posture training and proprioceptive stimulation, as was shown in a randomized control study with a 2-month follow-up [169]. The median effects on the degree of bending were minimal, although statistically significant for the treatment group but not for the control group.
Orthoses, technical support material and physiotherapy show a relevant potential in the treatment of camptocormia. Considering the large number of chronic PD camptocormia patients with fewer therapeutic options, more studies of this type are warranted.
Pain therapy
Severe back pain is a frequent complaint in camptocormia and is a relevant therapy target. Its importance is also shown by its significant correlation with the subjective impairment due to camptocormia in everyday life (Margraf et al., 2016 under review). The most effective treatment of this pain is to reduce the forward bending [66, 142, 143, 166, 167]. This includes the use of orthoses indicating that there are options for pain therapy even in chronic camptocormia. It is sometimes possible to obtain pain relief without any change in the posture [31]. A possible explanation could be a reduced rigidity of the paravertebral muscles. Classical analgetics such as non-steroidal anti-inflammatory drugs, opioids or muscle relaxants frequently have only limited effects [6].
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Earle H ((1815) ) Reply to the review of Mr. Bayrton’s essay on the cure of crooked spine. Edinburgh Med Surg J, 11: , 35–51. |
[2] | Parkinson J ((2002) ) An essay on the shaking palsy. 1817. J Neuropsychiatry Clin Neurosci, 14: , 223–236. |
[3] | Souques A ((1916) ) Reformes, incapacites, gratifications dans la camptocormie. Rev Neurol, 23: , 757–758. |
[4] | Kiuru S , & Iivanainen M ((1987) ) Camptocormia, a new side effect of sodium valproate. Epilepsy Res, 1: , 254–257. |
[5] | Laroche M , Rousseau H , Mazieres B , Bonafe A , Joffre F , & Arlet J ((1989) ) [Value of x-ray computed tomography in muscular pathology. Personal cases and review of the literature]. Rev Rhum Mal Osteoartic, 56: , 433–439. |
[6] | Azher SN , & Jankovic J ((2005) ) Camptocormia: Pathogenesis, classification, and response to therapy. Neurology, 65: , 355–359. |
[7] | Diederich NJ , Goebel HH , Dooms G , Bumb A , Huber F , Kompoliti K , & Meinck HM ((2006) ) Camptocormia associated with focal myositis in multiple-system atrophy. Mov Disord, 21: , 390–394. |
[8] | Bloch F , Houeto JL , Tezenas du MS , Bonneville F , Etchepare F , Welter ML , Rivaud-Pechoux S , Hahn-Barma V , Maisonobe T , Behar C , Lazennec JY , Kurys E , Arnulf I , Bonnet AM , & Agid Y ((2006) ) Parkinson’s disease with camptocormia. J Neurol Neurosurg Psychiatry, 77: , 1223–1228. |
[9] | Bonneville F , Bloch F , Kurys E , du Montcel ST , Welter ML , Bonnet AM , Agid Y , Dormont D , & Houeto JL ((2008) ) Camptocormia and Parkinson’s disease: MR imaging. Eur Radiol, 18: , 1710–1719. |
[10] | Tiple D , Fabbrini G , Colosimo C , Ottaviani D , Camerota F , Defazio G , & Berardelli A ((2009) ) Camptocormia in Parkinson disease: An epidemiological and clinical study. J Neurol Neurosurg Psychiatry, 80: , 145–148. |
[11] | Margraf NG , Wrede A , Rohr A , Schulz-Schaeffer WJ , Raethjen J , Eymess A , Volkmann J , Mehdorn MH , Jansen O , & Deuschl G ((2010) ) Camptocormia in idiopathic Parkinson’s disease: A focal myopathy of the paravertebral muscles. Mov Disord, 25: , 542–551. |
[12] | Spuler S , Krug H , Klein C , Medialdea IC , Jakob W , Ebersbach G , Gruber D , Hoffmann KT , Trottenberg T , & Kupsch A ((2010) ) Myopathy causing camptocormia in idiopathic Parkinson’s disease: A multidisciplinary approach. Mov Disord, 25: , 552–559. |
[13] | Abe K , Uchida Y , & Notani M ((2010) ) Camptocormia in Parkinson’s disease. Parkinsons Dis, pii: 267640. |
[14] | Doherty KM , van de Warrenburg BP , Peralta MC , Silveira-Moriyama L , Azulay JP , Gershanik OS , & Bloem BR ((2011) ) Postural deformities in Parkinson’s disease. Lancet Neurol, 10: , 538–549. |
[15] | Lyons M , Boucher O , Patel N , Birch B , & Evidente V ((2012) ) Long-term benefit of bilateral subthalamic deep brain stimulation on camptocormia in Parkinson’s disease. Turk Neurosurg, 22: , 489–492. |
[16] | Yamada K , Shinojima N , Hamasaki T , & Kuratsu JI ((2015) ) Subthalamic nucleus stimulation improves Parkinson’s disease-associated camptocormia in parallel to its preoperative levodopa responsiveness. J Neurol Neurosurg Psychiatry. doi: 10.1136/jnnp-2015-310926 |
[17] | Nakane S , Yoshioka M , Oda N , Tani T , Chida K , Suzuki M , Funakawa I , Inukai A , Hasegawa K , Kuroda K , Mizoguchi K , Shioya K , Sonoda Y , & Matsuo H ((2015) ) The characteristics of camptocormia in patients with Parkinson’s disease: A large cross-sectional multicenter study in Japan. J Neurol Sci, 358: , 299–303. |
[18] | Laroche M , Delisle MB , Aziza R , Lagarrigue J , & Mazieres B ((1995) ) Is camptocormia a primary muscular disease? Spine (Phila Pa 1976.), 20: , 1011–1016. |
[19] | Djaldetti R , Mosberg-Galili R , Sroka H , Merims D , & Melamed E ((1999) ) Camptocormia (bent spine) in patients with Parkinson’s disease–characterization and possible pathogenesis of an unusual phenomenon. Mov Disord, 14: , 443–447. |
[20] | Schäbitz WR , Glatz K , Schuhan C , Sommer C , Berger C , Schwaninger M , Hartmann M , Hilmar GH , & Meinck HM ((2003) ) Severe forward flexion of the trunk in Parkinson’s disease: Focal myopathy of the paraspinal muscles mimicking camptocormia. Mov Disord, 18: , 408–414. |
[21] | Micheli F , Cersosimo MG , & Piedimonte F ((2005) ) Camptocormia in a patient with Parkinson disease: Beneficial effects of pallidal deep brain stimulation. Case report. J Neurosurg, 103: , 1081–1083. |
[22] | Lepoutre AC , Devos D , Blanchard-Dauphin A , Pardessus V , Maurage CA , Ferriby D , Hurtevent JF , Cotten A , Destee A , & Defebvre L ((2006) ) A specific clinical pattern of camptocormia in Parkinson’s disease. J Neurol Neurosurg Psychiatry, 77: , 1229–1234. |
[23] | Yamada K , Goto S , Matsuzaki K , Tamura T , Murase N , Shimazu H , Nagahiro S , Kuratsu J , & Kaji R ((2006) ) Alleviation of camptocormia by bilateral subthalamic nucleus stimulation in a patient with Parkinson’s disease. Parkinsonism Relat Disord, 12: , 372–375. |
[24] | Hellmann MA , Djaldetti R , Israel Z , & Melamed E ((2006) ) Effect of deep brain subthalamic stimulation on camptocormia and postural abnormalities in idiopathic Parkinson’s disease. Mov Disord, 21: , 2008–2010. |
[25] | Gdynia HJ , Sperfeld AD , Unrath A , Ludolph AC , Sabolek M , Storch A , & Kassubek J ((2009) ) Histopathological analysis of skeletal muscle in patients with Parkinson’s disease and ‘dropped head’/’bent spine’ syndrome. Parkinsonism Relat Disord, 15: , 633–639. |
[26] | O’Riordan S , Paluzzi A , Liu X , Aziz TZ , Nandi D , & Bain PG ((2009) ) Camptocormia - Response to bilateral globus pallidus interna stimulation in three patients. Mov Disord, 24: (Suppl 1), 489. |
[27] | Capelle HH , Schrader C , Blahak C , Fogel W , Kinfe TM , Baezner H , & Krauss JK ((2011) ) Deep brain stimulation for camptocormia in dystonia and Parkinson’s disease. J Neurol, 258: , 96–103. |
[28] | Asahi T , Taguchi Y , Hayashi N , Hamada H , Dougu N , Takashima S , Tanaka K , & Endo S ((2011) ) Bilateral subthalamic deep brain stimulation for camptocormia associated with Parkinson’s disease. Stereotact Funct Neurosurg, 89: , 173–177. |
[29] | Oeda T , Umemura A , Tomita S , Hayashi R , Kohsaka M , & Sawada H ((2013) ) Clinical factors associated with abnormal postures in Parkinson’s disease. PLoS One, 8: , e73547. |
[30] | Umemura A , Oka Y , Ohkita K , Yamawaki T , & Yamada K ((2010) ) Effect of subthalamic deep brain stimulation on postural abnormality in Parkinson disease. J Neurosurg, 112: , 1283–1288. |
[31] | Schulz-Schaeffer WJ , Margraf NG , Munser S , Wrede A , Buhmann C , Deuschl G , & Oehlwein C ((2015) ) Effect of neurostimulation on camptocormia in Parkinson’s disease depends on symptom duration. Mov Disord, 30: , 368–372. |
[32] | Ghosh PS , & Milone M ((2015) ) Camptocormia as presenting manifestation of a spectrum of myopathic disorders. Muscle Nerve, 52: , 1008–1012. |
[33] | Laroche M , & Cintas P ((2010) ) Bent spine syndrome (camptocormia): A retrospective study of 63 patients. Joint Bone Spine, 77: , 593–596. |
[34] | Doherty KM , Noyce AJ , Silveira-Moriyama L , Nisbet A , Quinn N , & Lees AJ ((2012) ) Familial camptocormia: From dystonia to myopathy. J Neurol Neurosurg Psychiatry, 83: , 350–351. |
[35] | Duman I , Baklaci K , Tan AK , & Kalyon TA ((2008) ) Unusual case of camptocormia triggered by lumbar-disc herniation. Clin Rheumatol, 27: , 525–527. |
[36] | Wood-Allum C , Brennan P , Hewitt M , Lowe J , Tyfield L , & Wills A ((2004) ) Clinical and histopathological heterogeneity in patients with 4q35 facioscapulohumeral muscular dystrophy (FSHD). Neuropathol Appl Neurobiol, 30: , 188–191. |
[37] | Jordan B , Eger K , Koesling S , & Zierz S ((2011) ) Camptocormia phenotype of FSHD: A clinical and MRI study on six patients. J Neurol, 258: , 866–873. |
[38] | Kottlors M , Kress W , Meng G , & Glocker FX ((2010) ) Facioscapulohumeral muscular dystrophy presenting with isolated axial myopathy and bent spine syndrome. Muscle Nerve, 42: , 273–275. |
[39] | Papadopoulos C , Papadimas GK , Spengos K , & Manta P ((2011) ) Bent spine syndrome in facioscapulohumeral muscular dystrophy. Muscle Nerve, 43: , 615–616. |
[40] | Doherty KM , Silveira-Moriyama L , Giladi N , Bhatia KP , Parton M , & Lees AJ ((2012) ) Camptocormia: Don’t forget muscle disease in the movement disorder clinic. J Neurol, 259: , 1752–1754. |
[41] | Umapathi T , Chaudhry V , Cornblath D , Drachman D , Griffin J , & Kuncl R ((2002) ) Head drop and camptocormia. J Neurol Neurosurg Psychiatry, 73: , 1–7. |
[42] | Liewluck T , & Goodman BP ((2012) ) Late-onset axial myopathy and camptocormia in a calpainopathy carrier. J Clin Neuromuscul Dis, 13: , 209–213. |
[43] | Dupeyron A , Stober N , Gelis A , Castelnovo G , Labauge P , & Pelissier J ((2010) ) Painful camptocormia: The relevance of shaking your patient’s hand. Eur Spine J, 19: (Suppl 2), S87–S90. |
[44] | Lawson VH , King WM , & Arnold WD ((2013) ) Bent spine syndrome as an early manifestation of myotonic dystrophy type 1. J Clin Neuromuscul Dis, 15: , 58–62. |
[45] | Kocaaga Z , Bal S , Turan Y , Gurgan A , & Esmeli F ((2008) ) Camptocormia and dropped head syndrome as a clinic picture of myotonic myopathy. Joint Bone Spine, 75: , 730–733. |
[46] | Serratrice J , Weiller PJ , Pouget J , Serratrice G ((2000) ) [An unrecognized cause of camptocormia: Proximal myotonic myopathy]. Presse Med, 29: , 1121–1123. |
[47] | Findlay AR , Lewis S , Sahenk Z , & Flanigan KM ((2013) ) Camptocormia as a late presentation in a manifesting carrier of duchenne muscular dystrophy. Muscle Nerve, 47: , 124–127. |
[48] | Loseth S , Voermans NC , Torbergsen T , Lillis S , Jonsrud C , Lindal S , Kamsteeg EJ , Lammens M , Broman M , Dekomien G , Maddison P , Muntoni F , Sewry C , Radunovic A , de VM , Straub V , van EB , & Jungbluth H ((2013) ) A novel late-onset axial myopathy associated with mutations in the skeletal muscle ryanodine receptor (RYR1) gene. J Neurol, 260: , 1504–1510. |
[49] | Windpassinger C , Schoser B , Straub V , Hochmeister S , Noor A , Lohberger B , Farra N , Petek E , Schwarzbraun T , Ofner L , Loscher WN , Wagner K , Lochmuller H , Vincent JB , & Quasthoff S ((2008) ) An X-linked myopathy with postural muscle atrophy and generalized hypertrophy, termed XMPMA, is caused by mutations in FHL1. Am J Hum Genet, 82: , 88–99. |
[50] | Renard D , Castelnovo G , Fernandez C , De Paula AM , Penttila S , Suominen T , & Udd B ((2012) ) Camptocormia as presenting sign in myofibrillar myopathy. Neuromuscul Disord, 22: , 987–989. |
[51] | Kemta LF , Chevalier X , Dubourg O , & Dimitri D ((2013) ) Isolated camptocormia revealing sporadic late onset nemaline myopathy. Presse Med, 42: , 1142–1144. |
[52] | Delcey V , Hachulla E , Michon-Pasturel U , Queyrel V , Hatron PY , Boutry N , Lemaitre V , Vanhille P , Serratrice J , Disdier P , Juhan V , Devulder B , & Thevenon A ((2002) ) [Camptocormia: A sign of axial myopathy. Report of 7 cases]. Rev Med Interne, 23: , 144–154. |
[53] | Gomez-Puerta JA , Peris P , Grau JM , Martinez MA , & Guanabens N ((2007) ) Camptocormia as a clinical manifestation of mitochondrial myopathy. Clin Rheumatol, 26: , 1017–1019. |
[54] | Sakiyama Y , Okamoto Y , Higuchi I , Inamori Y , Sangatsuda Y , Michizono K , Watanabe O , Hatakeyama H , Goto Y , Arimura K , & Takashima H ((2011) ) A new phenotype of mitochondrial disease characterized by familial late-onset predominant axial myopathy and encephalopathy. Acta Neuropathol, 121: , 775–783. |
[55] | Kim JM , Song EJ , Seo JS , Nam EJ , & Kang YM ((2009) ) Polymyositis-like syndrome caused by hypothyroidism, presenting as camptocormia. Rheumatol Int, 29: , 339–342. |
[56] | Kelly L , Perju-Dumbrava LD , Thyagarajan D , & Lee YC ((2013) ) Delayed postirradiation camptocormia. BMJ Case Rep, pii: bcr2013200083. |
[57] | Psimaras D , Maisonobe T , Delanian S , Leclercq D , Lenglet T , Feuvret L , Ricard D , Hoang-Xuan K , & Pradat PF ((2011) ) Late onset radiation-induced camptocormia. J Neurol, 258: , 1723–1725. |
[58] | Seidel C , Kuhnt T , Kortmann RD , & Hering K ((2015) ) Radiation-induced camptocormia and dropped head syndrome: Review and case report of radiation-induced movement disorders. Strahlenther Onkol, 191: , 765–770. |
[59] | Rezania K , Pytel P , Smit LJ , Mastrianni J , Dina MA , Highsmith WE , & Dogan A ((2013) ) Systemic transthyretin amyloidosis in a patient with bent spine syndrome. Amyloid, 20: , 131–134. |
[60] | Friedman Y , Paul JT , Turley J , Hazrati LN , & Munoz D ((2007) ) Axial myopathy due to primary amyloidosis. Muscle Nerve, 36: , 542–546. |
[61] | Mattar MA , Gordo JM , Halpern AS , & Shinjo SK ((2013) ) Camptocormia secondary to polymyositis. Rev Bras Reumatol, 53: , 368–370. |
[62] | Kuo SH , Vullaganti M , Jimenez-Shahed J , & Kwan JY ((2009) ) Camptocormia as a presentation of generalized inflammatory myopathy. Muscle Nerve, 40: , 1059–1063. |
[63] | Goodman BP , Liewluck T , Crum BA , & Spinner RJ ((2012) ) Camptocormia due to inclusion body myositis. J Clin Neuromuscul Dis, 14: , 78–81. |
[64] | Ma H , McEvoy KM , & Milone M ((2013) ) Sporadic inclusion body myositis presenting with severe camptocormia. J Clin Neurosci, 20: , 1628–1629. |
[65] | Wunderlich S , Csoti I , Reiners K , Gunthner-Lengsfeld T , Schneider C , Becker G , & Naumann M ((2002) ) Camptocormia in Parkinson’s disease mimicked by focal myositis of the paraspinal muscles. Mov Disord, 17: , 598–600. |
[66] | Charpentier P , Dauphin A , Stojkovic T , Cotten A , Hurtevent JF , Maurage CA , Thevenon A , Destee A , & Defebvre L ((2005) ) [Parkinson’s disease, progressive lumbar kyphosis and focal paraspinal myositis]. Rev Neurol (Paris), 161: , 459–463. |
[67] | Dominick J , Sheean G , Schleimer J , & Wixom C ((2006) ) Response of the dropped head/bent spine syndrome to treatment with intravenous immunoglobulin. Muscle Nerve, 33: , 824–826. |
[68] | Rojana-Udomsart A , Fabian V , Hollingsworth PN , Walters SE , Zilko PJ , & Mastaglia FL ((2010) ) Paraspinal and scapular myopathy associated with scleroderma. J Clin Neuromuscul Dis, 11: , 213–222. |
[69] | Zenone T , Streichenberger N , & Puget M ((2013) ) Camptocormia as a clinical manifestation of polymyositis/systemic sclerosis overlap myositis associated with anti-Ku. Rheumatol Int, 33: , 2411–2415. |
[70] | Zwecker M , Iancu I , Zeilig G , & Ohry A ((1998) ) Camptocormia: A case of possible paraneoplastic aetiology. Clin Rehabil, 12: , 157–160. |
[71] | Terashima M , Kataoka H , Sugie K , Horikawa H , & Ueno S ((2009) ) Coexistence of chronic inflammatory demyelinating polyneuropathy and camptocormia. J Neurol Neurosurg Psychiatry, 80: , 1296–1297. |
[72] | Devic P , Choumert A , Vukusic S , Confavreux C , & Petiot P ((2013) ) Myopathic camptocormia associated with myasthenia gravis. Clin Neurol Neurosurg, 115: , 1488–1489. |
[73] | Kataoka H , Tonomura Y , Eura N , Terashima M , Kawahara M , & Ueno S ((2012) ) Painful abdominal contractions in patients with Parkinson disease. J Clin Neurosci, 19: , 624–627. |
[74] | Abboud H , Sivaraman I , Ontaneda D , & Tavee J ((2015) ) Camptocormia and Pisa syndrome as manifestations of acute myasthenia gravis exacerbation. J Neurol Sci, 359: , 8–10. |
[75] | Ashour R , & Jankovic J ((2006) ) Joint and skeletal deformities in Parkinson’s disease, multiple system atrophy, and progressive supranuclear palsy. Mov Disord, 21: , 1856–1863. |
[76] | Yoritaka A , Shimo Y , Takanashi M , Fukae J , Hatano T , Nakahara T , Miyamato N , Urabe T , Mori H , & Hattori N ((2013) ) Motor and non-motor symptoms of 1453 patients with Parkinson’s disease: Prevalence and risks. Parkinsonism Relat Disord, 19: , 725–731. |
[77] | Seki M , Takahashi K , Koto A , Mihara B , Morita Y , Isozumi K , Ohta K , Muramatsu K , Gotoh J , Yamaguchi K , Tomita Y , Sato H , Nihei Y , Iwasawa S , & Suzuki N ((2011) ) Camptocormia in Japanese patients with Parkinson’s disease: A multicenter study. Mov Disord, 26: , 2567–2571. |
[78] | Song W , Guo X , Chen K , Huang R , Zhao B , Cao B , Chen Y , & Shang HF ((2014) ) Camptocormia in Chinese patients with Parkinson’s disease. J Neurol Sci, 337: , 173–175. |
[79] | Skidmore F , Mikolenko I , Weiss H , & Weiner W ((2005) ) Camptocormia in a patient with multiple system atrophy. Mov Disord, 20: , 1063–1064. |
[80] | Slawek J , Derejko M , Lass P , & Dubaniewicz M ((2006) ) Camptocormia or Pisa syndrome in multiple system atrophy. Clin Neurol Neurosurg, 108: , 699–704. |
[81] | Gavrylova N , Limousin N , Belin J , de TB , Praline J ((2013) ) Camptocormia as presenting sign in dementia with Lewy bodies. Clin Neurol Neurosurg, 115: , 2397–2398. |
[82] | Kuncl RW , Cornblath DR , & Griffin JW ((1988) ) Assessment of thoracic paraspinal muscles in the diagnosis of ALS. Muscle Nerve, 11: , 484–492. |
[83] | Van Gerpen JA ((2001) ) Camptocormia secondary to early amyotrophic lateral sclerosis. Mov Disord, 16: , 358–360. |
[84] | Gautier G , Verschueren A , Monnier A , Attarian S , Salort-Campana E , & Pouget J ((2010) ) ALS with respiratory onset: Clinical features and effects of non-invasive ventilation on the prognosis. Amyotroph Lateral Scler, 11: , 379–382. |
[85] | Castrillo SA , Rodriguez VJ , Hernandez BM , & Duarte Garcia-Luis J ((2013) ) Early appearance of camptocormia in motor neuron disease: An association? J Clin Neuromuscul Dis, 15: , 43–44. |
[86] | Brucki S , & Nitrini R ((2008) ) Camptocormia in Alzheimer’s disease: An association? Mov Disord, 23: , 156–157. |
[87] | Nieves AV , Miyasaki JM , & Lang AE ((2001) ) Acute onset dystonic camptocormia caused by lenticular lesions. Mov Disord, 16: , 177–180. |
[88] | Oravivattanakul S , Abboud H , Fernandez H , & Itin I ((2014) ) Unusual case of levodopa-responsive camptocormia in a patient with negative dopamine transporter scan and normal DYT 5 gene. Clin Neuropharmacol, 37: , 63–64. |
[89] | Reichel G , Kirchhofer U , & Stenner A ((2001) ) [Camptocormia–segmental dystonia. Proposal of a new definition for an old disease]. Nervenarzt, 72: , 281–285. |
[90] | Jankovic J , & Tintner R ((2001) ) Dystonia and parkinsonism. Parkinsonism Relat Disord, 8: , 109–121. |
[91] | Cannas A , Solla P , Floris G , Tacconi P , Serra A , Piga M , Marrosu F , & Marrosu MG ((2009) ) Reversible Pisa syndrome in patients with Parkinson’s disease on dopaminergic therapy. J Neurol, 256: , 390–395. |
[92] | Solla P , Cannas A , Congia S , Floris G , Aste R , Tacconi P , & Marrosu MG ((2008) ) Levodopa/carbidopa/entacapone-induced acute Pisa syndrome in a Parkinson’s disease patient. J Neurol Sci, 275: , 154–156. |
[93] | Nakayama Y , & Miwa H ((2012) ) Drug-induced camptocormia: A lesson regarding vascular Parkinsonism. Intern Med, 51: , 2843–2844. |
[94] | Galati S , Moller JC , & Stadler C ((2014) ) Ropinirole-induced Pisa syndrome in Parkinson disease. Clin Neuropharmacol, 37: , 58–59. |
[95] | Robert F , Koenig M , Robert A , Boyer S , Cathebras P , & Camdessanche JP ((2010) ) Acute camptocormia induced by olanzapine: A case report. J Med Case Rep, 4: , 192. |
[96] | Vela L , Jimenez MD , Sanchez C , Pareja JA , & Baron M ((2006) ) Camptocormia induced by atypical antipsychotics and resolved by electroconvulsive therapy. Mov Disord, 21: , 1977–1980. |
[97] | Gonzalez-Pablos E , Valles-de la Calle JM , Iglesias-Santa PF , & Camara BS ((2016) ) A case report of camptocormia coinciding with olanzapine use. J Clin Psychopharmacol, 36: , 183–184. |
[98] | Bhattacharya A , Praharaj SK , & Sinha VK ((2014) ) Persistent camptocormia associated with atomoxetine in a child with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol, 24: , 596–597. |
[99] | Kwak YT , Han IW , Baik J , & Koo MS ((2000) ) Relation between cholinesterase inhibitor and Pisa syndrome. Lancet, 355: , 2222. |
[100] | Perez-Sales P ((1990) ) Camptocormia. Br J Psychiatry, 157: , 765–767. |
[101] | Rosen JC , & Frymoyer JW ((1985) ) A review of camptocormia and an unusual case in the female. Spine (Phila Pa 1976.), 10: , 325–327. |
[102] | Skidmore F , Anderson K , Fram D , & Weiner W ((2007) ) Psychogenic camptocormia. Mov Disord, 22: , 1974–1975. |
[103] | Pfeiffer E , von MA ((2000) ) Camptocormia in an adolescent. J Am Acad Child Adolesc Psychiatry, 39: , 944–945. |
[104] | Melamed E , & Djaldetti R ((2006) ) Camptocormia in Parkinson’s disease. J Neurol, 253: (Suppl 7), VII16–VII14. |
[105] | Lenoir T , Guedj N , Boulu P , Guigui P , & Benoist M ((2010) ) Camptocormia: The bent spine syndrome, an update. Eur Spine J, 19: , 1229–1237. |
[106] | Date ES , Mar EY , Bugola MR , & Teraoka JK ((1996) ) The prevalence of lumbar paraspinal spontaneous activity in asymptomatic subjects. Muscle Nerve, 19: , 350–354. |
[107] | Barkhaus PE , Periquet MI , & Nandedkar SD ((1997) ) Quantitative motor unit action potential analysis in paraspinal muscles. Muscle Nerve, 20: , 373–375. |
[108] | Serratrice G , Pouget J , & Pellissier JF ((1996) ) Bent spine syndrome. J Neurol Neurosurg Psychiatry, 60: , 51–54. |
[109] | West GA , Haynor DR , Goodkin R , Tsuruda JS , Bronstein AD , Kraft G , Winter T , & Kliot M ((1994) ) Magnetic resonance imaging signal changes in denervated muscles after peripheral nerve injury. Neurosurgery, 35: , 1077–1085. |
[110] | McDonald CM , Carter GT , Fritz RC , Anderson MW , Abresch RT , & Kilmer DD ((2000) ) Magnetic resonance imaging of denervated muscle: Comparison to electromyography. Muscle Nerve, 23: , 1431–1434. |
[111] | Tasca G , Monforte M , Iannaccone E , Laschena F , Ottaviani P , Leoncini E , Boccia S , Galluzzi G , Pelliccioni M , Masciullo M , Frusciante R , Mercuri E , & Ricci E ((2014) ) Upper girdle imaging in facioscapulohumeral muscular dystrophy. PLoS One, 9: , e100292. |
[112] | Diekman EF , van der Pol WL , Nievelstein RA , Houten SM , Wijburg FA , & Visser G ((2014) ) Muscle MRI in patients with long-chain fatty acid oxidation disorders. J Inherit Metab Dis, 37: , 405–413. |
[113] | Subhawong TK , Wang X , Machado AJ , Mammen AL , Christopher-Stine L , Barker PB , Carrino JA , & Fayad LM ((2013) ) 1H Magnetic resonance spectroscopy findings in idiopathic inflammatory myopathies at 3 T: Feasibility and first results. Invest Radiol, 48: , 509–516. |
[114] | Hayashi D , Hamilton B , Guermazi A , de VR , Crema MD , & Roemer FW ((2012) ) Traumatic injuries of thigh and calf muscles in athletes: Role and clinical relevance of MR imaging and ultrasound. Insights Imaging, 3: , 591–601. |
[115] | Tomasova SJ , Charvat F , Jarosova K , & Vencovsky J ((2007) ) The role of MRI in the assessment of polymyositis and dermatomyositis. Rheumatology (Oxford), 46: , 1174–1179. |
[116] | Margraf NG , Rohr A , Granert O , Hampel J , Drews A , & Deuschl G ((2015) ) MRI of lumbar trunk muscles in patients with Parkinson’s disease and camptocormia. J Neurol, 262: , 1655–1664. |
[117] | Ohana M , Durand MC , Marty C , Lazareth JP , Maisonobe T , Mompoint D , & Carlier RY ((2014) ) Whole-body muscle MRI to detect myopathies in non-extrapyramidal bent spine syndrome. Skeletal Radiol, 43: , 1113–1122. |
[118] | Wrede A , Margraf NG , Goebel HH , Deuschl G , & Schulz-Schaeffer WJ ((2012) ) Myofibrillar disorganization characterizes myopathy of camptocormia in Parkinson’s disease. Acta Neuropathol, 123: , 419–432. |
[119] | Askmark H , Eeg-Olofsson K , Johansson A , Nilsson P , Olsson Y , & Aquilonius S ((2001) ) Parkinsonism and neck extensor myopathy: A new syndrome or coincidental findings? Arch Neurol, 58: , 232–237. |
[120] | Wharton SB , Chan KK , Pickard JD , & Anderson JR ((1996) ) Paravertebral muscles in disease of the cervical spine. J Neurol Neurosurg Psychiatry, 61: , 461–465. |
[121] | Narayanaswami P , Bertorini TE , & Halford H , III((2000) ) Selective paraspinal muscle amyotrophy. J Neurol Sci, 176: , 70–74. |
[122] | Witting N , Andersen LK , & Vissing J ((2016) ) Axial myopathy: An overlooked feature of muscle diseases. Brain, 139: , 13–22. |
[123] | Lava NS , & Factor SA ((2001) ) Focal myopathy as a cause of anterocollis in Parkinsonism. Mov Disord, 16: , 754–756. |
[124] | Karpati G , Carpenter S , & Eisen AA ((1972) ) Experimental core-like lesions and nemaline rods. A correlative morphological and physiological study. Arch Neurol, 27: , 237–251. |
[125] | Delle Rose G , Borroni M , Silvestro A , Garofalo R , Conti M , De NP , & Castagna A ((2012) ) The long head of biceps as a source of pain in active population: Tenotomy or tenodesis? A comparison of 2 case series with isolated lesions. Musculoskelet Surg, 96: (Suppl 1), S47–S52. |
[126] | Zia S , Cody F , & O’Boyle D ((2000) ) Joint position sense is impaired by Parkinson’s disease. Ann Neurol, 47: , 218–228. |
[127] | Maschke M , Tuite PJ , Krawczewski K , Pickett K , & Konczak J ((2006) ) Perception of heaviness in Parkinson’s disease. Mov Disord, 21: , 1013–1018. |
[128] | Ho B , Prakash R , Morgan JC , & Sethi KD ((2007) ) A case of levodopa-responsive camptocormia associated with advanced Parkinson’s disease. Nat Clin Pract Neurol, 3: , 526–530. |
[129] | Mensikova K , Kaiserova M , Vastik M , Kurcova S , & Kanovsky P ((2015) ) Treatment of camptocormia with continuous subcutaneous infusions of apomorphine: 1-year prospective pilot study. J Neural Transm (Vienna), 122: , 835–839. |
[130] | Wijemanne S , & Jimenez-Shahed J ((2014) ) Improvement in dystonic camptocormia following botulinum toxin injection to the external oblique muscle. Parkinsonism Relat Disord, 20: , 1106–1107. |
[131] | Fietzek UM , Schroeteler FE , & Ceballos-Baumann AO ((2009) ) Goal attainment after treatment of parkinsonian camptocormia with botulinum toxin. Mov Disord, 24: , 2027–2028. |
[132] | Colosimo C , & Salvatori FM ((2009) ) Injection of the iliopsoas muscle with botulinum toxin in camptocormia. Mov Disord, 24: , 316–317. |
[133] | von Coelln R , Raible A , Gasser T , & Asmus F ((2008) ) Ultrasound-guided injection of the iliopsoas muscle with botulinum toxin in camptocormia. Mov Disord, 23: , 889–892. |
[134] | Bertram KL , Stirpe P , & Colosimo C ((2015) ) Treatment of camptocormia with botulinum toxin. Toxicon, 107: , 148–153. |
[135] | Furusawa Y , Mukai Y , Kobayashi Y , Sakamoto T , & Murata M ((2012) ) Role of the external oblique muscle in upper camptocormia for patients with Parkinson’s disease. Mov Disord, 27: , 802–803. |
[136] | Furusawa Y , Mukai Y , Kawazoe T , Sano T , Nakamura H , Sakamoto C , Iwata Y , Wakita M , Nakata Y , Kamiya K , Kobayashi Y , Sakamoto T , Takiyama Y , & Murata M ((2013) ) Long-term effect of repeated lidocaine injections into the external oblique for upper camptocormia in Parkinson’s disease. Parkinsonism Relat Disord, 19: , 350–354. |
[137] | Furusawa Y , Hanakawa T , Mukai Y , Aihara Y , Taminato T , Iawata Y , Takei T , Sakamoto T , & Murata M ((2015) ) Mechanism of camptocormia in Parkinson’s disease analyzed by tilt table-EMG recording. Parkinsonism Relat Disord, 21: , 765–770. |
[138] | Arii Y , Sawada Y , Kawamura K , Miyake S , Taichi Y , Izumi Y , Kuroda Y , Inui T , Kaji R , & Mitsui T ((2014) ) Immediate effect of spinal magnetic stimulation on camptocormia in Parkinson’s disease. J Neurol Neurosurg Psychiatry, 85: , 1221–1226. |
[139] | Thevathasan W , Mazzone P , Jha A , Djamshidian A , Dileone M , Di Lazzaro V , & Brown P ((2010) ) Spinal cord stimulation failed to relieve akinesia or restore locomotion in Parkinson disease. Neurology, 74: , 1325–1327. |
[140] | Deuschl G , Schade-Brittinger C , Krack P , Volkmann J , Schafer H , Botzel K , Daniels C , Deutschlander A , Dillmann U , Eisner W , Gruber D , Hamel W , Herzog J , Hilker R , Klebe S , Kloss M , Koy J , Krause M , Kupsch A , Lorenz D , Lorenzl S , Mehdorn HM , Moringlane JR , Oertel W , Pinsker MO , Reichmann H , Reuss A , Schneider GH , Schnitzler A , Steude U , Sturm V , Timmermann L , Tronnier V , Trottenberg T , Wojtecki L , Wolf E , Poewe W , & Voges J ((2006) ) A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med, 355: , 896–908. |
[141] | Srivanitchapoom P , & Hallett M ((2016) ) Camptocormia in Parkinson’s disease: Definition, epidemiology, pathogenesis and treatment modalities. J Neurol Neurosurg Psychiatry, 87: , 75–85. |
[142] | Peek AC , Quinn N , Casey AT , & Etherington G ((2009) ) Thoracolumbar spinal fixation for camptocormia in Parkinson’s disease. J Neurol Neurosurg Psychiatry, 80: , 1275–1278. |
[143] | Upadhyaya CD , Starr PA , & Mummaneni PV ((2010) ) Spinal deformity and Parkinson disease: A treatment algorithm. Neurosurg Focus, 28: , E5. |
[144] | Sutter P , Forster T , & Kulling F ((2013) ) [Spinal deformity in Parkinson’s disease: A treatment proposal]. Unfallchirurg, 116: , 955–959. |
[145] | Wadia PM , Tan G , Munhoz RP , Fox SH , Lewis SJ , & Lang AE ((2011) ) Surgical correction of kyphosis in patients with camptocormia due to Parkinson’s disease: A retrospective evaluation. J Neurol Neurosurg Psychiatry, 82: , 364–368. |
[146] | Siewe J , Zarghooni K , Rollinghoff M , Herren C , Koy T , Eysel P , & Sobottke R ((2013) ) [Complication analysis of spinal interventions in adult central movement disorders and scoliosis]. Z Orthop Unfall, 151: , 454–462. |
[147] | Ha Y , Oh JK , Smith JS , Ailon T , Fehlings MG , Shaffrey CI , & Ames CP ((2015) ) Impact of movement disorders on management of spinal deformity in the elderly. Neurosurgery, 77: (Suppl 4), S173–S185. |
[148] | Babat LB , McLain RF , Bingaman W , Kalfas I , Young P , & Rufo-Smith C ((2004) ) Spinal surgery in patients with Parkinson’s disease: Construct failure and progressive deformity. Spine (Phila Pa 1976.), 29: , 2006–2012. |
[149] | Invernizzi M , Carda S , Viscontini GS , & Cisari C ((2009) ) Osteoporosis in Parkinson’s disease. Parkinsonism Relat Disord, 15: , 339–346. |
[150] | Koller H , Acosta F , Zenner J , Ferraris L , Hitzl W , Meier O , Ondra S , Koski T , & Schmidt R ((2010) ) Spinal surgery in patients with Parkinson’s disease: Experiences with the challenges posed by sagittal imbalance and the Parkinson’s spine. Eur Spine J, 19: , 1785–1794. |
[151] | Van Gerpen JA ((2006) ) Dopa-responsive dystonic camptocormia. Neurology, 66: , 1779. |
[152] | Micheli F , & Pardal MM ((2007) ) Dopa-responsive dystonic camptocormia. Neurology, 68: , 1543. |
[153] | Bhatia KP , Quinn NP , & Marsden CD ((1997) ) Clinical features and natural history of axial predominant adult onset primary dystonia. J Neurol Neurosurg Psychiatry, 63: , 788–791. |
[154] | Mahjneh I , Edstrom B , & Sandstrom G ((2009) ) [Bent spine straightens up–a case of camptocormia]. Duodecim, 125: , 1889–1893. |
[155] | Yadav R , Ansari AZ , Surathi P , Srinivas D , Somanna S , & Pal P ((2015) ) Bilateral pallidal deep brain stimulation in idiopathic dystonic camptocormia. Neurol India, 63: , 911–914. |
[156] | Nandi D , Parkin S , Scott R , Winter JL , Joint C , Gregory R , Stein J , & Aziz TZ ((2002) ) Camptocormia treated with bilateral pallidal stimulation: Case report. ECP. Neurosurg Focus, 12: , 2. |
[157] | Fukaya C , Otaka T , Obuchi T , Kano T , Nagaoka T , Kobayashi K , Oshima H , Yamamoto T , & Katayama Y ((2006) ) Pallidal high-frequency deep brain stimulation for camptocormia: An experience of three cases. Acta Neurochir Suppl, 99: , 25–28. |
[158] | Sakas DE , Panourias IG , Stavrinou LC , Boviatsis EJ , Themistocleous M , Stathis P , Tagaris G , Angelopoulos E , & Gatzonis S ((2010) ) Restoration of erect posture in idiopathic camptocormia by electrical stimulation of the globus pallidus internus. J Neurosurg, 113: , 1246–1250. |
[159] | Hagenacker T , Gerwig M , Gasser T , Miller D , Kastrup O , Jokisch D , Sure U , & Frings M ((2013) ) Pallidal deep brain stimulation relieves camptocormia in primary dystonia. J Neurol, 260: , 1833–1837. |
[160] | Reese R , Knudsen K , Falk D , Mehdorn HM , Deuschl G , & Volkmann J ((2014) ) Motor outcome of dystonic camptocormia treated with pallidal neurostimulation. Parkinsonism Relat Disord, 20: , 176–179. |
[161] | Gerton BK , Theeler B , & Samii A ((2010) ) Backpack treatment for camptocormia. Mov Disord, 25: , 247–248. |
[162] | Benveniste O ((2014) ) [Inclusion-body myositis]. Rev Med Interne, 35: , 472–479. |
[163] | Spector SA , Lemmer JT , Koffman BM , Fleisher TA , Feuerstein IM , Hurley BF , & Dalakas MC ((1997) ) Safety and efficacy of strength training in patients with sporadic inclusion body myositis. Muscle Nerve, 20: , 1242–1248. |
[164] | Hund E , Heckl R , Goebel HH , & Meinck HM ((1995) ) Inclusion body myositis presenting with isolated erector spinae paresis. Neurology, 45: , 993–994. |
[165] | Peng Y , Zhu M , Zheng J , Zhu Y , Li X , Wei C , & Hong D ((2015) ) Bent spine syndrome as an initial manifestation of late-onset multiple acyl-CoA dehydrogenase deficiency: A case report and literature review. BMC Neurol, 15: , 114. |
[166] | Pardessus V , Compere S , Tiffreau V , Blanchard A , & Thevenon A ((2005) ) [Leather corset for the treatment of camptocormia: 31 cases]. Ann Readapt Med Phys, 48: , 603–609. |
[167] | de Seze MP , Creuze A , de SM , & Mazaux JM ((2008) ) An orthosis and physiotherapy programme for camptocormia: A prospective case study. J Rehabil Med, 40: , 761–765. |
[168] | Schroeteler FE , Fietzek UM , Ziegler K , & Ceballos-Baumann AO ((2011) ) Upright posture in parkinsonian camptocormia using a high-frame walker with forearm support. Mov Disord, 26: , 1560–1561. |
[169] | Capecci M , Serpicelli C , Fiorentini L , Censi G , Ferretti M , Orni C , Renzi R , Provinciali L , & Ceravolo MG ((2014) ) Postural rehabilitation and Kinesio taping for axial postural disorders in Parkinson’s disease. Arch Phys Med Rehabil, 95: , 1067–1075. |
Figures and Tables
Fig.1
Different disease principles can cause camptocormia, and camptocormia may be symptom of a variety of diseases. The graph does not show the diseases in proportion to their occurrence. Abbreviations: ALS = amyotrophic lateral sclerosis; CIDP = chronic inflammatory demyelinating polyradiculoneuropathy; dermatomyos. = dermatomyositis; DLB = dementia with Lewy bodies; FHL1 = four and a half LIM domain 1 gene; IBM = inclusion body myositis; MSA = multiple-system atrophy; NMJ = neuromuscular junction; periph. = peripheral; polysyn. = polysynaptic; striat. = striatal.
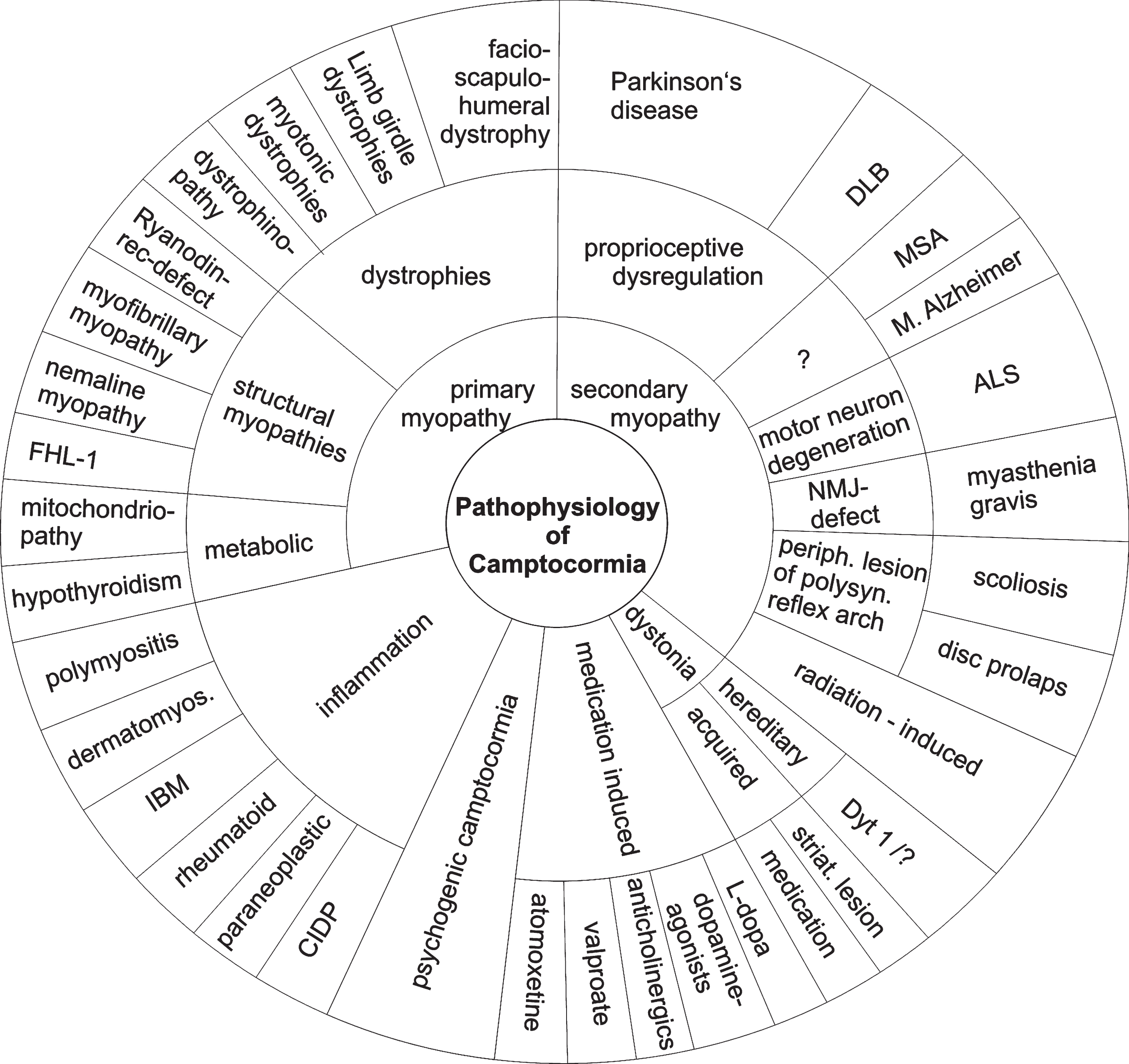
Fig.2
Proposal for a work-up and treatment algorithm for camptocormia. Abbreviations: DBS = deep brain stimulation; GPi = globus pallidus internus; STN = subthalamic nucleus.
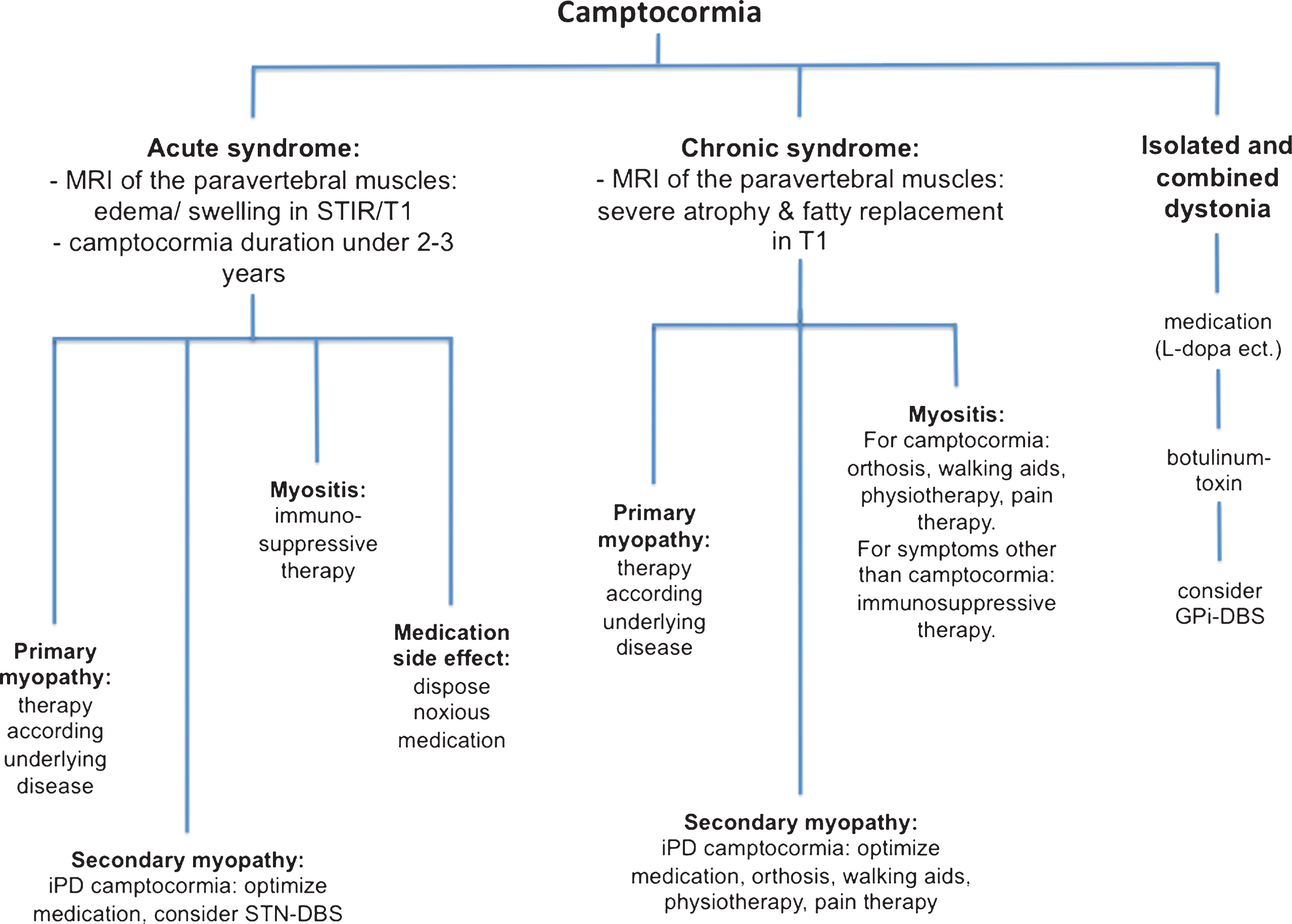
Table 1
Muscle diseases that may present with camptocormia
| Dystrophies: | Facio-scapulo-humeral muscular dystrophy (FSHD) [34, 36–41] |
| Limb girdle dystrophies: Calpainopathy [42] | |
| Myotonic dystrophies: DM1 [43, 44], DM2 [45, 46] | |
| Duchenne muscular dystrophy [47] | |
| Structural myopathies: | Ryanodine receptor (RYR1)-defect [48] |
| Four and a half LIM domain (FHL1)-mutation [49] | |
| Myofibrillar myopathy [50] | |
| Nemaline myopathy /late onset [51] | |
| Congenital myopathy [52] | |
| Metabolic myopathies: | Mitochondriopathies [52–54] |
| Hypothyroidism [55] | |
| Varia: | Radiation-induced myopathy [56–58] |
| Amyloidosis [52, 59, 60] | |
| Inflammation/autoimmune: | Polymyositis [52, 61, 62] |
| Dermatomyositis [52] | |
| Inclusion body myositis (IBM) [52, 63, 64] | |
| Unspecific focal myositis [7, 65–69] | |
| Paraneoplastic myositis [70] | |
| Chronic inflammatory polyneuropathy [71] | |
| Myasthenia gravis [72–74] |
Table 2
Treatment of inflammatory camptocormia
| Study | Diagnosis | n | Treatment | Effect on | Camptocormia | Findings in the |
| camptocormia | duration/existence of pain | paravertebral muscles | ||||
| Wunderlich et al., 2002 [65] | focal myositis, PD | 1 | methylprednisolone i.v. and oral | marked improvement | 2 months (also pain) | EMG: spontaneous activity, myopathic |
| MRI: severe unilateral edema | ||||||
| Charpentier et al., 2005 [66] | focal myositis, PD | 1 | IVIG, prednisolone i.v. | partial improvement | 21 months (also pain) | EMG: myositic |
| MRI: unilateral accentuated edema | ||||||
| Diederich et al., 2006 [7] | focal myositis, MSA | 2 | only in one case: methylprednisolone i.v. | good response | 1 year | EMG: myositicMRI: bilateral focal edema |
| Dominick et al., 2006 [67] | focal myositis | 1 | IVIG | very good response | 1 year | EMG: myositic |
| Delcey et al., 2002 [52] | poly- and dermatomyositis | 4 | methylprednisolone i.v. and oral, IVIG, cyclosporine | 3 improved | n.r. (back pain: n = 4) | EMG: myositicMRI: in 3 pat. fatty atrophic changes, in 1 acute changes |
| Kuo et al., 2009 [62] | polymyositis | 1 | methylprednisolone i.v. | modest improvement | 1.5 years | back pain |
| MRI: diffuse atrophy with fatty replacement | ||||||
| Mattar et al., 2013 [61] | polymyositis | 1 | oral prednisolone, azathioprine, methotrexate, cyclosporine | none | for several years | MRI: total fatty replacement |
| Rojana-Udomsart et al., 2010 [68] | myositis associated with scleroderma | 2 | in one case prednisolone, methotrexate, azathioprine | none | n.r. | EMG: in one case spontaneous activityCT: severe fatty atrophy |
| Zenone et al., 2013 [69] | polymyositis/systemic sclerosis overlap myositis | 1 | oral prednisolone, methotrexate, IVIG | modest improvement | 3 months (also pain) | EMG (limbs): myogenicSpine MRI: normal |
| Hund et al., 1995 [164] | inclusion body myositis | 1 | dexamethasone oral | mild &transient | 4 years | EMG: neurogenic-myopathic pattern |
| Goodman et al., 2012 [63] | inclusion body myositis | 1 | n.r. | n.r. | 18 months | EMG: axial myopathyMRI: atrophy and fatty replacement |
| Ma et al., 2013 [64] | inclusion body myositis | 2 | n.r. | n.r. | 18 years, respectively 7 years | EMG: myopathic-neurogenic pattern, respectively myopathic |
| MRI: no structural abnormalities in one case; n.r. in the other case | ||||||
| Terashima et al., 2009 [71] | CIDP | 1 | IVIG | markedly reduction | 6 months | EMG (during follow-up): chronic neurogenic, spontaneous activity |
| MRI (during follow-up): No abnormal signals | ||||||
| Kataoka et al., 2012 [73] | myasthenia gravis | 1 | i.v. edrophonium, oral prednisolone | markedly improved | 14 months | EMG: no myopathyMRI: mild atrophy |
| Devic et al., 2013 [72] | myasthenia gravis | 1 | oral pyridostigmine, i.v. edrophonium, IVIG, mycophenolate mofetil | none | n.r. | EMG: no myopathy, no spontaneous activity MRI: severe atrophy with major fatty replacement |
| Abboud &Sivaraman, 2015 [74] | myasthenia gravis | 1 | without effect: oral pyridostigmine, oral steroids; with effect: IVIG plus oral steroids | significantly improvement | 5 weeks | MRI: scoliosis, nothing else reported |
Abbreviations: CIDP = chronic inflammatory demyelinating polyradiculoneuropathy; i.v. = intravenous; IVIG = intravenous immunoglobulins; MSA = multiple-system atrophy; n.r. = not reported; PD = Parkinson’s disease.


