
Retrograde Axonal Degeneration in Parkinson Disease
Abstract
In spite of tremendous research efforts we have not yet achieved two of our principal therapeutic goals in the treatment of Parkinson’s disease (PD), to prevent its onward progression and to provide restoration of systems that have already been damaged by the time of diagnosis. There are many possible reasons for our inability to make progress. One possibility is that our efforts thus far may not have been directed towards the appropriate cellular compartments. Up until now research has been largely focused on the loss of neurons in the disease. Thus, neuroprotection approaches have been largely aimed at blocking mechanisms that lead to destruction of the neuronal cell body. Attempts to provide neurorestoration have been almost entirely focused on replacement of neurons. We herein review the evidence that the axonal component of diseased neuronal systems merit more of our attention. Evidence from imaging studies, from postmortem neurochemical studies, and from genetic animal models suggests that the axons of the dopaminergic system are involved predominantly and early in PD. Since the mechanisms of axonal destruction are distinct from those of neuron cell body degeneration, a focus on axonal neurobiology will offer new opportunities for preventing their degeneration. At present these mechanisms remain largely obscure. However, defining them is likely to offer new opportunities for neuroprotection. In relation to neurorestoration, while it has been classically believed that neurons of the adult central nervous system are incapable of new axon growth, recent evidence shows that this is not true for the dopaminergic projection. In conclusion, the neurobiology of axons is likely to offer many new approaches to protective and restorative therapeutics.
INTRODUCTION: PD AND THE NEUROBIOLOGY OF AXONS
Parkinson’s disease (PD) has been estimated to affect over 4 million individuals in 2005 in the five most populous nations in Western Europe and the ten most populous nations of the rest of the world. This number is projected to more than double, to about 9 million, by 2030 [1]. In the United States the economic burden of the disease was estimated to be $14.4 billion in 2010, with a predicted 1.7-fold increase in medical costs alone by 2030 [2]. In spite of the social, economic and emotional burdens imposed by this disease we have not yet been able to provide therapies that either forestall its progression or provide restoration of damaged neural systems. This deficiency in available therapeutics persists in spite of decades of intensive studies that have shed much light on our understanding of the molecular and genetic aspects of PD and have shown that a number of cellular mechanisms, such as protein clearance, mitochondria failure, programmed cell death, autophagy and inflammation are likely to contribute to its pathogenesis. In spite of these many gains, the specific cause of the disease remains unknown, and this lack of knowledge has undoubtedly contributed to our lack of success thus far in efforts to develop better treatments [3].
In this review we will try to make the case that another possible reason for our lack of progress in developing neuroprotective and restorative therapies for PD is that we have neglected the importance of the axonal degeneration that occurs. In spite of the evidence, which we shall review, suggesting that axons are involved early and predominantly, most research discussions up until now about disease pathogenesis and experimental therapeutics have focused mainly on the loss of neurons alone. Thus, in formulating strategies for neuroprotection, the discussion almost always is about preventing neuron death. Likewise, in exploring approaches to restoration in PD, the focus has been entirely on diverse approaches to cell replacement with very little consideration being given to axon growth, pathfinding and target contact. There are a number of possible reasons why PD research has been so focused on neuronal cell bodies, perhaps the simplest being that neurons are, of course, easily observed in postmortem material, whereas the demonstration of axons requires special techniques and especially high quality preservation of the tissue samples. In addition, neuron cell bodies are much easier to study experimentally both in vitro and in vivo and consequently we have a more extensive knowledge about the molecular mechanisms of their survival and death. In the case of cell death the tremendous amount of information that we have acquired about the many pathways of programmed cell death for the most part applies to the neuron soma. The mechanisms of axon degeneration are largely distinct from those of programmed cell death and much less well known [4].
We will review the evidence that axon loss is an early and predominant feature of PD and likely to be a retrograde form of degeneration. We will in addition provide an overview of the different mechanisms of axonal degeneration which indicate that this process may occur through a local self-destruction program, distinct from the events that take place during the death of the neuronal soma. Based on these observations, we will suggest that the biology of axons is more deserving of our attention in trying to understand the pathogenesis of PD and to improve its treatment.
AXON DEGENERATION IS AN EARLY AND PREDOMINANT FEATURE OF PD
Evidence from postmortem and imaging studies in humans
There have been varying estimates in the literature of how many dopaminergic neurons are lost in the substantia nigra (SN) by the time the first motor signs of the disease appear. Estimates as high as 80% have been made [5], but more often estimates of about 60 to 70% are given [6, 7]. However, the number is likely to be considerably less. Among three independent quantitative morphologic studies there was a remarkable consistency, with each suggesting about a 30% loss of SN neurons at the time of onset of motor signs [8–10] (reviewed in Cheng et al., 2010 [11]).
In contrast, several observations suggest that at the time of disease onset there is a greater extent of loss of striatal axon terminal markers. Numerous imaging studies have examined the relationship between striatal dopaminergic marker loss and onset of motor signs. Among those that have used ligands for the dopamine transporter (DAT) or for the vesicular monoamine transporter (VMAT) (which are preferable for this purpose [12]), estimates of 50–70% loss are typical (reviewed in Cheng et al., 2010 [11]).
Kordower at al., have evaluated the integrity of the nigrostriatal dopamine system in 28 brains from PD patients at multiple time points following diagnosis [13]. Using tyrosine hydroxylase (TH) and DAT as markers for dopamine function, they showed that fibers in the dorsal striatum, which underlie motor function in PD, are only moderately affected at 1 year, more severely affected at 3 years and virtually absent by years 4–5 and thereafter. The decrease in the number of TH positive neurons in the SN was highly variable but robust even at the earlier time points with a relatively minor loss over the time and with a residual population of TH positive neurons even decades after diagnosis. In an earlier study the Kordower laboratory had reported that little difference was seen between age-matched controls and Hoehn and Yahr Stage I PD patients in the immunostaining for TH-positive neurons in the SN, but a severe reduction in staining of axonal terminal structures in the putamen was observed in the patient group, indicating an early involvement of axons [14].
These observations suggesting that about 30% of SN dopamine neurons and about 50–70% of striatal dopaminergic terminals are lost by the time of symptom onset are supported by observations in incidental Lewy body disease (ILBD). This is a condition in which many Lewy bodies are observed in postmortem brains, but the patient was asymptomatic during life. There is a good consensus that these cases are likely to represent presymptomatic PD. Among such cases the loss of SN dopamine neurons was found by Fearnley and Lees to be 27% , just beneath the estimated threshold of 30% for symptomatic patients [8]. Measurement of striatal TH levels by ELISA in such patients found a 50% reduction, in keeping with an estimate of a 50–70% reduction as symptoms appear [15].
Given the critical role that synuclein plays in the pathogenesis of PD [16], it is worthwhile to consider what is known about the site of appearance of synuclein pathology. There has been a great focus on the appearance of Lewy bodies, which are composed primarily of synuclein, and which typically are identified in the neuron soma. However, with the development of new antibodies and staining protocols it has become apparent that synuclein pathology is abundant in axons [17, 18] and in some studies it has been identified predominantly in axons. With the use of a paraffin-embedded tissue blot technique, in which tissue sections are subjected to digestion, the greatest abundance of synuclein aggregates is found in the neuropil, not in cell bodies [19] in dementia with Lewy bodies (DLB) brains. DLB is believed by many to be closely related to PD [20]. Furthermore, these studies show that the preponderance of synuclein small aggregates in DLB brains are entrapped within presynaptic terminals. Thus, there is evidence that synuclein pathology is abundant in axons and presynaptic terminals, consistent with the observation that its normal localization is predominantly in presynaptic terminals. A role for synuclein pathology in axonal dysfunction is also supported at the single fiber level by the observations of Chu and colleagues, who noted a loss of the axon transport protein kinesin in axons with positive synuclein phospho-serine 129 immunostaining [14].
While the above cited evidence indicates that axons are involved early and predominantly in PD, it does not provide information about the pattern of axonal involvement; i.e., does it start proximal to the cell soma and proceed anterograde, or does it begin distally and proceed retrograde? There have been few studies attempting to examine this question. Orimo and colleagues have used the known involvement of peripheral autonomic neurons and their axons in PD to explore the timing of events [21]. Based on patterns of synuclein pathology and TH immunostaining in cardiac sympathetic axons and ganglia in patients with ILBD, PD, and controls, they concluded that the disease process begins in the distal axon and proceeds retrograde.
Evidence from genetic animal models
Many genetic animal models of PD have been created and although none of them fulfills all the key features of the disease, particularly neurodegenerative change in the nigrostriatal dopaminergic projection, mild deficits in dopamine transmission and behavioral impairments have been identified in some, thereby providing insight into the pathophysiological changes that precede actual neurodegeneration [22]. While these models have been informative for understanding some molecular pathways that may be relevant to PD [23], because they have not recapitulated the pathological hallmarks, their implications are limited. However, more recently, some transgenic and virus-induced models using more specific gene promoters and more efficient vector constructs have been able to recreate some of the cardinal morphological features of the disease observed in the nigrostriatal dopaminergic system. A number of these models support the idea that axonopathy is an early feature of PD (Table 1).
Among the models that have used synuclein, either the wildtype or mutant forms, those using viral vector transduction of dopamine neurons of the SN have been the most helpful in delineating the sequence of pathological events. Chung and colleagues created a model of synuclein overexpression in SN dopaminergic neurons in the rat by transduction with an AAV 2 vector in which human A53T synuclein expression was regulated by the neuronal synapsin promoter [24]. At 4 and 8 weeks following AAV injection they observed no SN dopamine neuron loss, but at both time points they identified by TH immunohistochemistry striatal axonal pathology characterized by dystrophic, swollen neurites. In addition, before neuron loss occurred there were specific reductions in axonal anterograde transport proteins. In association with these alterations in axon morphology and proteins, an inflammatory response was identified in the striatum, but not SN, by the demonstration of activated microglia.
Using an AAV vector construct containing the neuron-specific synapsin-1 promoter and a WPRE enhancer element to drive human synuclein expression, Decressac and co-investigators were able to detect important structural and functional impairments in the dopaminergic system [25]. Three weeks after virus injection they observed loss of striatal TH-positive axon terminals, the appearance of synuclein-positive axonal swellings and dystrophic neurites. These abnormalities occurred when the mice showed limited dopamine neuron cell loss and before they displayed any motor deficits. The authors concluded that the progressive degenerative changes seen in the transduced dopaminergic neurons over time support the idea that the synuclein pathology affects the axons and terminals first and later progresses to involve the neuronal cell bodies.
Observations in a BAC transgenic mouse model expressing the human disease-causing LRRK2 (R1441G) mutant indicate that the earliest pathology in the dopaminergic system occurs in the axons. Between 2 to 4 months of age, these mice demonstrate an axonopathy that is characterized by the presence of “giant”, polymorphic axonal spheroids (>90μm3) observed in the medial forebrain bundle (MFB) and striatum [26]. These spheroids are packed with autophagic vacuoles. In addition, these mice show autophagy in the axoplasm of axons in the MFB and intra-axonal myelin invagination. At 2 to 4 months, when these axonal abnormalities appear, there is a normal number and morphology of dopamine neuronal cell bodies and dendrites in the SN by light microscopy and ultrastructural analysis. As they age, the mutant animals develop progressive motor deficits and robust signs of axonopathy with the presence of dystrophic neurites, expression of phosphorylated tau, and axonal spheroids in dopaminergic fibers [27]. Even as they age, these animals show no decrease in the number of dopamine neurons in the SN pars compacta (SNpc). The observation that axonal pathology is the initial abnormality in the SN dopaminergic system in these transgenic mice suggests that the role of LRRK2 in the biology of axons may be of primary importance in the pathogenesis of PD. Further evidence indicates that LRRK2 may play in role in the development and maintenance of neurites. Several mutant forms of LRRK2 induced degeneration of neurites in primary cortical neuron cultures [28]. Plowey and co-investigators observed in differentiated SH-SY5Y cells that the G2019S mutant induced shortening of neurites accompanied by the activation of autophagy [29].
The gene engrailed1 (En1), encodes a protein that is expressed in developing and mature dopamine neurons [30, 31] and it has been postulated to be a risk factor for sporadic PD [32, 33]. Heterozygous null En1 transgenic mice show progressive degeneration of the dopaminergic nigrostriatal system at the level of the axonal terminals several weeks before the loss of nigral dopaminergic neurons is evident [34]. These mice show VMAT2 and DAT positive spheroidal dystrophic terminals in the striatum as early as postnatal day 8 and an increase in their number through 4 and 24 weeks. However, the number of TH positive neurons in the SN was normal at 4 weeks and a decrease of 18% was not observed until 16 weeks. In the striatum, swollen axons contained autophagic vacuoles. In association with these changes, increased phosphorylation of the kinase mTor and one of its downstream targets, pS6, were identified. Since mTor activation negatively regulates autophagy, this combination of findings suggests that either mTor is being upregulated as a compensatory response to attempt to maintain axonal integrity, or that the increased number of autophagic vacuoles is due to diminished ‘throughput’. The latter possibility is suggested by the diminished amount of LC3, an autophagic marker.
The transcription factor Nurr1 is critical for the development of dopamine neurons of the midbrain [35]. In addition, it may play a role as a genetic risk factor for PD. Mutations in exon 1 of Nurr1 were reported to be associated with PD [36]. While other investigators were unable to confirm these early observations [37, 38], more recently a mutation in the 5’ UTR of the Nurr1 gene has been identified and shown to reduce Nurr1 mRNA expression in neuronal cell lines and human brain tissue [39]. Functionally, the mutation abrogated the ability of Nurr1 to protect against apoptotic stress. Kadkhodaei and colleagues have shown that expression of Nurr1 is essential in the adult brain for the maintenance of the viability of the SN dopaminergic system [40]. Following conditional deletion of Nurr1 in SN dopamine neurons in adult mice, there is a slow, progressive degeneration of the SN dopaminergic system that predominantly affects the axons [41]. At 2 months following Nurr1 ablation there was no loss of SN dopamine neurons, but as early as 1 and 4 weeks there was diminished expression of TH, VMAT2 and the DAT in the axons of the striatum. At 4 months following ablation, there was a diminished number of axons in the MFB. Thus, as in the models described above, Nurr1 ablation in the adult leads to an early and predominant degeneration of axons.
Another pair of transcription factors that are essential for the differentiation of dopamine neurons are Lmx1a and b. Compelling evidence for a role of these factors in the genetics of PD is lacking [42], but diminished levels of LMX1B protein expression have been identified in neuromelanin-containing neurons in PD brains [43]. Following conditional deletion of Lmx1a and b in adult mice results in the early appearance of striking abnormalities in dopaminergic terminals; they become enlarged and filled with autophagic vacuoles. At this time there is no loss of dopaminergic neurons. Laguna et al conclude that following Lmx1a and b ablation in adulthood, there is an early disturbance in the autophagic-lysosomal pathway, associated with axon terminal pathology long before cell body loss.
Thus, there is evidence from both PD patient studies and genetic animal models to support the concept that the degeneration of the dopaminergic system in PD starts in the axon and synaptic terminals and only later involves the neuronal cell bodies in the SNpc. It is important to emphasize that although this evidence suggests that axons are the first site of degenerative change, this does not necessarily mean that the primary disturbance at the molecular level first occurs in the axons. For example, a primary disturbance at the nuclear level may result in transcriptional, translational or trafficking abnormalities that first manifest as axonal dysfunction and degeneration. The critical need to focus on axons for both neuroprotective and restorative therapeutics derives not only from their early involvement, but also their essential role in dopamine release in the striatum. A particularly striking example of the dependence of functional preservation on the axonal projection is the demonstration that intrastriatal GDNF protects axons as well as cell bodies and preserves behavioral performance, but intranigral injection preserves only cell bodies and does not preserve function [44].
DISTURBANCES OF AXON TRANSPORT IN PD AND MODELS
Axonal transport is an essential process in the maintenance of neuronal viability. Anterograde axonal transport keeps axons and nerve terminals supplied with proteins, membrane bound-organelles, lipids and mitochondria, while retrograde axonal transport mediates the clearance of recycled, misfolded proteins and damaged mitochondria, and the transport of trophic signaling molecules (see reviews by Maday et al. [45] and Millecamps and Julien [46]). Therefore, it is not surprising that impairment of axonal transport has emerged as a possible common factor in several neurodegenerative disorders. The unique architecture of dopamine neurons in the SN, with long, thin and arborized axons, makes them especially vulnerable to axonal transport disruption, as discussed in greater detail below. In PD, synuclein has been proposed to alter axon transport. Transfection of cultured neurons with PD-associated A30P and A53T mutations, as well as mutation of serine-129 to mimic permanent phosphorylation, reduces axonal transport of the protein [47]. Such an effect may increase the propensity of the protein to self-aggregate. As described above, Chung and collaborators used a recombinant AAV2 to deliver mutant (A53T) human synuclein under the synapsin promoter into the rat SN. Four weeks after injection, and before neuronal death, they observed in the striatum altered levels of motor proteins involved in axonal transport. While the levels of anterograde transport motor proteins were diminished, the levels of retrograde transport motor proteins were increased. They also observed an imbalance of cytoskeletal proteins with high levels of actin and reduced levels of α/γ tubulin. Taken together, these results suggest early impairment of axonal transport before dopamine neurons loss occurs in the SN [24]. In a human study using postmortem tissue from 16 patients with sporadic PD (10 patients with early PD and 6 with late PD with no difference in age at the time of death) and 9 age-matched controls Chu and co-authors found altered levels of both anterograde and retrograde motor proteins. Diminished levels of anterograde motor proteins were detected during early stages of the disease whereas a reduction in the levels of retrograde motor proteins was observed only at late PD stages. These data were recapitulated in a rat model based on viral overexpression of mutant (A30P) human synuclein [14].
Abnormalities of axonal transport have also been identified in neurotoxin models of PD. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and its metabolite 1-methyl-4-phenylpyridinium (MPP+) cause mitochondrial impairment at complex I. Morfini and coinvestigators demonstrated that the injection of MPP+ into isolated squid axoplasm induced an increase in dynein-dependent fast retrograde axonal transport and a decrease in kinesin-mediated fast anterograde axonal transport of vesicles [48].
Although direct evidence of impairment of axonal transport associated with LRRK2 mutations is lacking, BAC-transgenic mice carrying the human LRRK2(R1441G) mutation exhibit early signs of axonopathy with giant axonal spheroids with disrupted cytoskeleton and accumulation of autophagic vacuoles and mitochondria. Moreover, the axons containing these giant axonal spheroids displayed fewer mitochondria than normal axons. This abnormality could be due to a defect at the level of the axon in mitochondria trafficking, but more study is needed [26].
UNIQUE MORPHOLOGIC FEATURES OF THE NIGROSTRIATAL AXONAL PROJECTION AND SELECTIVE VULNERABILITY OF SNPC DOPAMINERGIC NEURONS
It has been suggested that select types of neurons are more vulnerable in neurodegenerative disorders based on their axonal morphology. Projection neurons with long, thin, unmyelinated or poorly myelinated axons have been proposed to be the most vulnerable [49]. Dopaminergic neurons of the SNpc are of this type. Myelination is postulated to diminish metabolic demands by facilitating transmission of nerve impulses. Another unique feature of dopaminergic neurons of the SNpc is that they give rise to a massive striatal terminal arborization with a vast number of axon terminals providing a dense innervation to the striatum [50]. Matsuda and colleagues demonstrated that, in the rat brain, the axonal arbor of a single SNpc dopamine neuron covered more than 5% of the total volume of the striatum [50]. Bolam and Pissadaki have estimated that a single SN dopamine neuron in the rat gives rise to as many as 245,000 synapses, whereas, in contrast, a typical globus pallidus pars externa neuron will give rise to about 2,000 and striatal GABA neurons about 5,000 [51]. Thus, an enormous synapse to cell body ratio seems to be a unique feature of dopamine neurons in the SNpc, distinguishing these neurons even from adjacent dopamine neurons in the ventral tegmental area, which establish fewer synapses and have much smaller axonal arbors [51]. Bolam and Pissadaki propose that the maintenance of such a large axonal architecture may put dopamine neuron under a phenomenal energy demand such that any disturbance inducing a negative energy balance might lead to functional failure and, eventually, cell death [51].
MECHANISMS OF AXON DEGENERATION
The distinct nature of pathways of neuron soma and axon destruction
If we hope to utilize the concept that axons are involved early in PD, and are therefore a prime target for early neuroprotective intervention, then it is critical to recognize that the molecular mechanisms of axon degeneration are separate and distinct from the canonical pathways of programmed cell death which mediate destruction of the neuron soma. This distinction was made years ago by Raff and colleagues [52] based on their observations that activation of caspase 3 did not occur in a variety of models of axon destruction [4]. While activation of programmed cell death pathways can occur in axon destruction in some settings, particularly in the developmental context [53, 54], typically it does not, especially in models in adult animals [55]. Striking evidence for the distinct nature of the pathways of cell and axonal destruction derives from observations in the slow Wallerian degeneration (Wlds) mutant mouse [56]. This mutation arose spontaneously in C57Bl/6 mice, and it was demonstrated to cause delayed Wallerian degeneration in peripheral nerve after axotomy [57]. The mutation was identified as an 85-kb tandem triplication that results in a novel chimeric mRNA that encodes for the N-terminal 70 amino acids of ubiquitination factor E4B (Ube4B), followed by the complete coding sequence for the nicotinamide adenine dinucleotide (NAD) synthesizing enzyme nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1) [58, 59]. These mRNA sequences splice to encode a fusion protein that does not occur in wildtype mice [60]. Strikingly, the Wlds mutant was shown by Deckwerth and Johnson to provide protection of axons of sympathetic ganglion neurons following withdrawal of nerve growth factor in spite of induction of apoptosis in the cell soma [61], thus dramatically demonstrating the distinct nature of the pathways of programmed cell death and those of axon destruction.
Two modes of axon degeneration: Anterograde and retrograde
In the setting of neurological injury or disease there are two forms of axon degeneration that can occur: Anterograde and retrograde [52, 62]. During development, degenerative processes of synapse elimination and axon pruning occur, but they will not be considered here. Anterograde (or Wallerian) degeneration occurs after injury to the proximal axon, followed by degeneration of the axon distal to the site of injury. The degenerative process is orderly; there is a breakdown of cytoskeletal elements, swelling of the mitochondria and fragmentation of the axon [63]. Retrograde degeneration (or ‘dying back’) is characterized by degeneration proceeding from the distal axon terminals to the neuron cell body [52, 62]. It has been identified in neurodegenerative diseases, peripheral neuropathies and neuropathies due to neurotoxin exposure. In the late stages of retrograde axon degeneration, the axonal fragmentation looks similar to that observed in anterograde degeneration, but the molecular basis of the two forms is likely to be distinct, as discussed below.
Anterograde axon degeneration
A great deal of what we now know about the molecular basis of axon degeneration derives from studies of the WldS mutation [60], and most of that work has been performed in models of anterograde degeneration in peripheral nerves. As stated earlier, the mutation results in the production of a fusion protein that contains the N-terminal 70 amino acids of Ube4B, followed by the complete coding sequence of NMNAT1. Studies based on the selective expression of transgenic Wlds in Drosophila have shown that it acts cell-autonomously within neurons to mediate axon protection; when expression is restricted to glia, the axon protection phenotype is not observed [64]. Analysis of the effect of expression of selected regions of the Wlds protein have revealed that NMNAT1 is capable of recapitulating the Wlds axon protection phenotype [65, 66]. Wlds appears to act within the cytoplasm to mediate axon protection, as elimination of an intrinsic nuclear localization signal within the NMNAT1 domain enhances it [67]. Babetto and colleagues have further shown that a mutant form of NMNAT1 with disrupted nuclear targeting and the addition of an axon-targeting peptide sequence is more potent than Wlds in providing axon protection [68]. At a subcellular level, the NMNAT isoform NMNAT3 localizes to mitochondria [69]. The potential significance of this observation is that Wlds has been shown to suppress the loss of mitochondrial motility after axotomy lesion [69].
While a great deal of this experimental work has been performed in models of anterograde degeneration (i.e., classic Wallerian degeneration), some studies have been performed in models of retrograde degeneration. There have been mixed results for the ability of Wlds to protect from degeneration in this context. Wlds inhibits axon loss and synapse degeneration in progressive motor neuronopathy mice (pmn/pmn), a model of motor neuron disease in which a ‘dying back’ axonal degeneration occurs [70]. In gracile axonal dystrophy mice (gad), in which dying back axon degeneration also occurs, Wlds inhibits the appearance of axon spheroid pathology [71], which is often associated with axon degeneration. Nevertheless, Wlds did not provide protection in other models of motor neuron disease or primary spinal atrophy [60] in which ‘dying back’ is likely to occur.
In relation to PD in particular, Wlds does provide protection for axons of the SN dopaminergic projection in models of anterograde degeneration, but it does not in models of retrograde degeneration. Sajadi and colleagues reported that Wlds provided protection in a model of injection of 6-hydroxydopamine (6OHDA) directly into the MFB. This lesion is made near the SN and it results in anterograde degeneration of the nigrostriatal projection. However, when 6OHDA was injected into the striatum, a lesion that produces retrograde degeneration, no protection was observed [72]. Our laboratory subsequently confirmed their observations made with 6OHDA; Wlds was protective in the 6OHDA MFB (anterograde) model, but not the intra-striatal (retrograde) model [73]. To exclude the possibility that the difference was due to some other aspect of the two lesion techniques, rather than the anterograde/retrograde difference, we also examined the ability of Wlds to protect in two different axotomy models; proximal and distal MFB axotomies to induce anterograde and retrograde degeneration, respectively. In these models as well, Wlds protected from anterograde, but not retrograde degeneration [73]. We conclude that Wlds does not have the capacity to inhibit retrograde degeneration in the dopaminergic nigrostriatal projection and other approaches must be sought to develop axonal neuroprotection in PD, in which ‘dying back’ is likely to occur.
Although we have learned a tremendous amount about the neurobiology of axon degeneration from studies of the Wlds mutation, the fusion protein, as a product of a novel chimeric mRNA, is not normally expressed in mice, so it does not play a role as an endogenous regulator of axon integrity. By identification of relevant domains in the Wlds protein and by characterization of the pathways with which they interact, it will be possible to identify endogenous regulators. Some progress has been made in the identification of endogenous regulators of axon integrity independently of Wlds. Genetic screening for loss of function mutations that delay axonal degeneration in D. melanogaster after antennal ablation revealed that three loss-of-function alleles of dsarm (a Toll-like receptor adaptor) induced a level of axonal protection comparable to that of Wlds. Neuroprotection in D. melanogaster extended to the synapses since synaptobrevin expression was detected in synaptic terminals even 30 days after axotomy [74]. The same group showed robust axonal protection both in vitro and in vivo in null mutants for Sarm1, the mouse ortholog of dsarm. Cultured Sarm1–/– neurons from the superior cervical ganglia, cortex and dorsal root ganglia (DRG) exhibited robust axonal protection up to 72 hours after lesion. Interestingly, DRG explants were not protected from nerve growth factor deprivation. Studies in vivo showed that Sarm1–/– mutation strongly preserved axons after sciatic nerve lesion for at least 14 days before cytoskeleton breakdown takes place. Most synaptic terminals were also preserved at six days after injury. Gerdts and colleagues independently identified SARM in an RNAi-based screen of mouse DRG neurons [75] as a regulator of axon destruction and demonstrated that the SAM and TIR domains are necessary and sufficient for destruction. Thus, these motifs offer targets for therapeutic intervention. Importantly, most of these investigations were performed in models of peripheral nerve injury, so the role of Sarm in axon degeneration in the central nervous system requires further investigation.
Another endogenous mediator of axon degeneration is dual leucine kinase (DLK), a mitogen activated protein kinase kinase kinase which has, as one of its downstream targets, c-jun N-terminal kinase (JNK). JNK has previously been shown to be activated following axonal injury [76]. The absence of DLK both in vitro and in vivo has been shown to delay Wallerian degeneration, although its effect is very modest compared to that of Wlds and Sarm1 [77]. Yang and colleagues have demonstrated a relationship between Sarm1 and the DLK/JNK signaling pathway for axon degeneration. Activation of the DLK/JNK signaling pathway is abolished in Sarm1 null mice. Furthermore, there is no additivity of phenotype when components of the DLK/JNK cascade are knocked down in the Sarm1 null axons, indicating that these mediators are on the same genetic pathway [78].
Loss of the Phr1 (PAM-Highwire-Rpm-1) E3 ubiquitin ligase in mice has been shown to dramatically delay Wallerian degeneration after injury in several neuronal types including myelinated and unmyelinated axons, central and peripheral nerves and sensory and motor terminals [79]. The extent of axonal protection provided by the absence of PHR1 is similar to that of the loss of Sarm1 and the expression of Wlds. Interestingly, Babetto and colleagues find that Phr1 depletion increases the axonal level of the NMNAT2 isoform, and it is necessary for Phr1-dependent axon stability.
Retrograde axon degeneration
Despite the importance of axonal retrograde degeneration in neurodegenerative diseases, such as PD, little is known about the underlying mechanisms. As discussed above, much of what we know about the process of axonal degeneration comes from studies of anterograde degeneration and the ability of Wlds to protect axons during this process. However, there are many examples of Wlds failing to provide protective effects in models of retrograde degeneration, particularly in models of motor neuron disease, including spinal muscular atrophy [80, 81], and amyotrophic lateral sclerosis [82]. One might raise the possibility that the failure of Wlds in these instances might be due to the unique biology of motor neurons; perhaps they simply do not express essential mediators of the Wlds effect. However, as outlined previously, we have shown that although the dopaminergic nigrostriatal projection manifests Wlds– mediated protection in two models of anterograde degeneration (proximal axotomy and lesion of the MFB with 6OHDA), it does not demonstrate Wlds– mediated protection in two models of retrograde degeneration (distal axotomy and lesion of striatal dopaminergic terminals with 6OHDA), Therefore, although this system is capable of responding to Wlds in anterograde models, it provides no protection for retrograde degeneration, suggesting that different mechanisms are involved [73].
A number of studies have suggested that retrograde axonal degeneration can be suppressed by inhibition of macroautophagy. We have observed that AAV2/1 mediated transduction of SN neurons to induce expression of a constitutively active form of the survival-signaling kinase myristoylated Akt (Myr-Akt) suppresses retrograde degeneration of dopaminergic axons in vivo in two different models of nigrostriatal axon injury, the intrastriatal injection of 6OHDA and axotomy of the MFB distal to the SN [83]. Myr-Akt preserved the number of dopaminergic axons in the MFB and their structural integrity, since axonal fragmentation and the appearance of axonal spheroids were suppressed. In addition, Myr-Akt also protected nerve terminals, indicated by preserved striatal DAT immunostaining. This ability of Myr-Akt to protect axons is likely to be mediated by mTor signaling, because it is recapitulated by a constitutively active form of Rheb, a GTPase that mediates downstream effects of Akt by direct activation of mTor. Transduction of SN neurons with hRheb(S16H), a mutant resistant to negative regulation by TSC1/2, resulted in a robust increase in the number of neurons positive for p-4E-BP1, an mTor substrate [83, 84]. The ability of Myr-Akt and hRheb(S16H) to suppress retrograde axon degeneration by enhanced mTor signaling is likely to be due to the ability of mTor to suppress macroautophagy. Induction of retrograde axonal degeneration by either the intrastriatal injection of 6OHDA or axotomy of the MFB distal to the SN results in the abundant appearance of autophagic vacuoles in the neuron soma and in axons. Induced expression of Myr-Akt or hRheb(S16H) suppresses their appearance [83]. Furthermore, direct abrogation of macroautophagy by selective deletion of Atg7 in SN dopamine neurons results in a striking resistance of dopamine axons to retrograde degeneration (Fig. 1). Thus, these studies suggest that the ability of Myr-Akt to protect axons from retrograde degeneration is likely to be due, at least in part, to its ability to suppress autophagy through mTor signaling [83].
Other studies, based on in vitro models of injury and both in vitro and in vivo genetic models of PD, have also implicated macroautophagy as a mediator of retrograde axonal degeneration. Yang and colleagues have shown that both pharmacologic and genetic disruption of autophagy signaling protects from retrograde axon degeneration induced by NGF withdrawal or vinblastine treatment [85]. In a genetic model of neurite retraction induced by the expression of the LRRK2(G2019S) mutant in SH-SY5Y cells, Plowey et al observed an association with activation of macroautophagy since RNA interference-mediated knockdown of Atg7 expression prevented the neurite degeneration [29].
Studies of spinal cord injury in vivo in rats have identified increases in autophagy mediators and in number of autophagic vacuoles in both retrograde and anterograde axon degeneration following the injury [86]. Interestingly, expression of unc-51-like kinase (UNK1), an early mediator of autophagy that is negatively regulated by mTor, was predominantly within axon retraction bulbs early after injury. A mechanistic role for autophagy in mediating axon degeneration following traumatic injury is supported by observations in an optic nerve crush model in which inhibition of autophagy by 3-methyladenine results in a slower time course for axon degeneration [87].
It is also noteworthy that macroautophagy frequently accompanies early axonal degeneration observed in a number of genetic models of PD in vivo. We have identified accumulation of autophagic vacuoles in giant axonal spheroids in the MFB in BAC transgenic mice carrying the LRRK2(R1441G) mutation but not in their littermate controls (Fig. 2) [26]. This abnormality is the first pathology to appear in these mice. In another LRRK2 transgenic mouse model expressing the G2019S mutant, Ramonet and collaborators identified vacuoles with multiple membranes resembling autophagosomes within regions enriched in axons and synapses and clusters of mitochondria, reminiscent of mitochondria undergoing autophagocytosis, within neuronal soma [88]. In the engrailed heterozygous mouse, previously described, autophagic vacuoles are identified within dystrophic axons [34]. Thus, macroautophagy frequently accompanies axonal dystrophy in these models, but its functional role remains unknown.
CONCLUDING REMARKS
In spite of recent tremendous strides in our understanding of PD, particularly its genetic basis, and of the cellular basis of neurodegeneration, we remain unable to offer patients truly disease modifying neuroprotective therapy. In addition, although we know that PD patients have already lost about 60% of their dopamine projection at the time of diagnosis [11], we remain unable to offer restorative therapies. In our efforts in the past to develop therapies for these needs we have largely focused on the loss of neurons in the SN. For example, in efforts to provide neuroprotection, a number of approaches, including some clinical trials, have focused on blockade of the programmed cell death pathways that mediate destruction of the cell body. In experimental efforts to provide restoration, the focus has been almost entirely on cell replacement approaches to correct the loss of neurons.
However, there is now an emerging consensus that it is the axons of the dopamine neurons that are involved initially and predominantly in this disease. One of the implications of this new concept is that in the development of neuroprotective therapies, where it is critical to target early events, it would make more sense to target mechanisms of axonal destruction. These mechanisms are now recognized to be different from those involved in canonical programmed cell death and the destruction of the cell body. The ‘bad news’ is that today we know very little about the patterns of axonal degeneration in the disease. Furthermore, we know very little about the molecular mechanisms of both anterograde and retrograde axon degeneration, giving us very little basis for the development of new therapeutics. However the ‘good news’ is that as our understanding of these mechanisms grows we will almost certainly be provided with many potential therapeutic opportunities.
In relation to neurorestoration, we need to consider the observation that at the time of diagnosis patients have lost only about 30% of their neurons. Therefore, rather than replacing cells, it may be more efficient and effective to restore axons instead. In the past, the classic notion was that adult neurons in the brain are unable to regrow their axons, but we now know that this is not always true. Efforts to induce new axon growth in mature neurons by rejuvenating the pathways that are responsible for axon growth during development have had substantial success [84, 89]. Thus with a focus on axons we will have available to us an entirely new approach to restoration in PD, one that will avoid the many challenges of cell replacement therapies.
In conclusion the biology of axons in the mature central nervous system, both their mechanisms of degeneration and their potential for regrowth, offer many new opportunities for the development of critical neuroprotective and restorative therapies in PD. This conclusion is not meant to suggest that neuronal cell bodies are not important. The cell bodies are required of course to host new axon growth. Furthermore they provide essential cellular functions related to nuclear genomic DNA, to support of dendrites and their arborizations and many others. Our conclusion is meant only to encourage a better balance – axons, as well as the cell bodies, are critically important.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGMENTS
This work was supported by DOD W81XVVH-12-1-0051.
REFERENCES
[1] | Dorsey ER , Constantinescu R , Thompson JP , Biglan KM , Holloway RG , Kieburtz K , Marshall FJ , Ravina BM , Schifitto G , Siderowf A , & Tanner CM ((2007) ) Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology, 68: , 384–386. |
[2] | Kowal SL , Dall TM , Chakrabarti R , Storm MV , & Jain A ((2013) ) The current and projected economic burden of Parkinson’s disease in the United States. Mov Disord, 28: , 311–318. |
[3] | Olanow CW , Kieburtz K , & Schapira AH ((2008) ) Why have we failed to achieve neuroprotection in Parkinson’s disease? Ann Neurol, 64: (Suppl 2), S101–S110. |
[4] | Finn JT , Weil M , Archer F , Siman R , Srinivasan A , & Raff MC ((2000) ) Evidence that Wallerian degeneration and localized axon degeneration induced by local neurotrophin deprivation do not involve caspases. J Neurosci, 20: , 1333–1341. |
[5] | Le WD , Chen S , & Jankovic J ((2009) ) Etiopathogenesis of Parkinson disease: A new beginning? Neuroscientist, 15: , 28–35. |
[6] | Dauer W , & Przedborski S ((2003) ) Parkinson’s disease: Mechanisms and models. Neuron, 39: , 889–909. |
[7] | Lang AE , & Lozano AM ((1998) ) Parkinson’s disease. Second of two parts. N Engl J Med, 339: , 1130–1143. |
[8] | Fearnley JM , & Lees AJ ((1991) ) Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain, 114: , 2283–2301. |
[9] | Ma SY , Roytta M , Rinne JO , Collan Y , & Rinne UK ((1997) ) Correlation between neuromorphometry in the substantia nigra and clinical features in Parkinson’s disease using disector counts. J Neurol Sci, 151: , 83–87. |
[10] | Greffard S , Verny M , Bonnet AM , Beinis JY , Gallinari C , Meaume S , Piette F , Hauw JJ , & Duyckaerts C ((2006) ) Motor score of the Unified Parkinson Disease Rating Scale as a good predictor of Lewy body-associated neuronal loss in the substantia nigra. Arch Neurol, 63: , 584–588. |
[11] | Cheng HC , Ulane CM , & Burke RE ((2010) ) Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol, 67: , 715–725. |
[12] | Nandhagopal R , McKeown MJ , & Stoessl AJ ((2008) ) Functional imaging in Parkinson disease. Neurology, 70: , 1478–1488. |
[13] | Kordower JH , Olanow CW , Dodiya HB , Chu Y , Beach TG , Adler CH , Halliday GM , & Bartus RT ((2013) ) Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain, 136: , 2419–2431. |
[14] | Chu Y , Morfini GA , Langhamer LB , He Y , Brady ST , & Kordower JH ((2012) ) Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain, 135: , 2058–2073. |
[15] | Beach TG , Adler CH , Sue LI , Peirce JB , Bachalakuri J , Dalsing-Hernandez JE , Lue LF , Caviness JN , Connor DJ , Sabbagh MN , & Walker DG ((2008) ) Reduced striatal tyrosine hydroxylase in incidental Lewy body disease. Acta Neuropathol, 115: , 445–451. |
[16] | Devine MJ , Gwinn K , Singleton A , & Hardy J ((2011) ) Parkinson’s disease and alpha-synuclein expression. Mov Disord, 26: , 2160–2168. |
[17] | Galvin JE , Uryu K , Lee VM , & Trojanowski JQ ((1999) ) Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc Natl Acad Sci U S A, 96: , 13450–13455. |
[18] | Duda JE , Giasson BI , Mabon ME , Lee VM , & Trojanowski JQ ((2002) ) Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann Neurol, 52: , 205–210. |
[19] | Kramer ML , & Schulz-Schaeffer WJ ((2007) ) Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci, 27: , 1405–1410. |
[20] | McKeith I ((2007) ) Dementia with Lewy bodies and Parkinson’s disease with dementia: Where two worlds collide. Pract Neurol, 7: , 374–382. |
[21] | Orimo S , Uchihara T , Nakamura A , Mori F , Kakita A , Wakabayashi K , & Takahashi H ((2008) ) Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain, 131: , 642–650. |
[22] | Lee Y , Dawson VL , & Dawson TM ((2012) ) Animal models of Parkinson’s disease: Vertebrate genetics. Cold Spring Harb Perspect Med, 2: , pii–a009324. |
[23] | Dawson TM , Ko HS , & Dawson VL ((2010) ) Genetic animal models of Parkinson’s disease. Neuron, 66: , 646–661. |
[24] | Chung CY , Koprich JB , Siddiqi H , & Isacson O ((2009) ) Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J Neurosci, 29: , 3365–3373. |
[25] | Decressac M , Mattsson B , Lundblad M , Weikop P , & Bjorklund A ((2012) ) Progressive neurodegenerative and behavioural changes induced by AAV-mediated overexpression of alpha-synuclein in midbrain dopamine neurons. Neurobiol Dis, 45: , 939–953. |
[26] | Tagliaferro P , Kareva T , Oo TF , Yarygina O , Kholodilov N , & Burke RE ((2015) ) An early axonopathy in a hLRRK2(R1441G) transgenic model of Parkinson disease. Neurobiol Dis, 82: , 359–371. |
[27] | Li Y , Liu W , Oo TF , Wang L , Tang Y , Jackson-Lewis V , Zhou C , Geghman K , Bogdanov M , Przedborski S , Beal MF , Burke RE , & Li C ((2009) ) Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat Neurosci, 12: , 826–828. |
[28] | MacLeod D , Dowman J , Hammond R , Leete T , Inoue K , & Abeliovich A ((2006) ) The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron, 52: , 587–593. |
[29] | Plowey ED , Cherra SJ III , Liu YJ , & Chu CT ((2008) ) Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem, 105: , 1048–1056. |
[30] | Le Pen G , Sonnier L , Hartmann A , Bizot JC , Trovero F , Krebs MO , & Prochiantz A ((2008) ) Progressive loss of dopaminergic neurons in the ventral midbrain of adult mice heterozygote for Engrailed1: A new genetic model for Parkinson’s disease? Parkinsonism Relat Disord, 14: Suppl 2, S107–S111. |
[31] | Sonnier L , Le Pen G , Hartmann A , Bizot JC , Trovero F , Krebs MO , & Prochiantz A ((2007) ) Progressive loss of dopaminergic neurons in the ventral midbrain of adult mice heterozygote for Engrailed1. J Neurosci, 27: , 1063–1071. |
[32] | Fuchs J , Mueller JC , Lichtner P , Schulte C , Munz M , Berg D , Wullner U , Illig T , Sharma M , & Gasser T ((2009) ) The transcription factor PITX3 is associated with sporadic Parkinson’s disease. Neurobiol Aging, 30: , 731–738. |
[33] | Haubenberger D , Reinthaler E , Mueller JC , Pirker W , Katzenschlager R , Froehlich R , Bruecke T , Daniel G , Auff E , & Zimprich A ((2011) ) Association of transcription factor polymorphisms PITX3 and EN1 with Parkinson’s disease. Neurobiol Aging, 32: , 302–307. |
[34] | Nordstrom U , Beauvais G , Ghosh A , Pulikkaparambil Sasidharan BC , Lundblad M , Fuchs J , Joshi RL , Lipton JW , Roholt A , Medicetty S , Feinstein TN , Steiner JA , Escobar Galvis ML , Prochiantz A , & Brundin P ((2015) ) Progressive nigrostriatal terminal dysfunction and degeneration in the engrailed1 heterozygous mouse model of Parkinson’s disease. Neurobiol Dis, 73: , 70–82. |
[35] | Zetterstrom RH , Solomin L , Jansson L , Hoffer BJ , Olson L , & Perlmann T ((1997) ) Dopamine neuron agenesis in Nurr1-deficient mice. Science, 276: , 248–250. |
[36] | Le WD , Xu P , Jankovic J , Jiang H , Appel SH , Smith RG , & Vassilatis DK ((2003) ) Mutations in NR4A2 associated with familial Parkinson disease. Nat Genet, 33: , 85–89. |
[37] | Nichols WC , Uniacke SK , Pankratz N , Reed T , Simon DK , Halter C , Rudolph A , Shults CW , Conneally PM , Foroud T , & Parkinson Study G ((2004) ) Evaluation of the role of Nurr1 in a large sample of familial Parkinson’s disease. Mov Disord, 19: , 649–655. |
[38] | Levecque C , Destee A , Mouroux V , Amouyel P , & Chartier-Harlin MC ((2004) ) Assessment of Nurr1 nucleotide variations in familial Parkinson’s disease. Neurosci Lett, 366: , 135–138. |
[39] | Sleiman PM , Healy DG , Muqit MM , Yang YX , Van Der Brug M , Holton JL , Revesz T , Quinn NP , Bhatia K , Diss JK , Lees AJ , Cookson MR , Latchman DS , & Wood NW ((2009) ) Characterisation of a novel NR4A2 mutation in Parkinson’s disease brain. Neurosci Lett, 457: , 75–79. |
[40] | Kadkhodaei B , Ito T , Joodmardi E , Mattsson B , Rouillard C , Carta M , Muramatsu S , Sumi-Ichinose C , Nomura T , Metzger D , Chambon P , Lindqvist E , Larsson NG , Olson L , Bjorklund A , Ichinose H , & Perlmann T ((2009) ) Nurr1 is required for maintenance of maturing and adult midbrain dopamine neurons. J Neurosci, 29: , 15923–15932. |
[41] | Kadkhodaei B , Alvarsson A , Schintu N , Ramskold D , Volakakis N , Joodmardi E , Yoshitake T , Kehr J , Decressac M , Bjorklund A , Sandberg R , Svenningsson P , & Perlmann T ((2013) ) Transcription factor Nurr1 maintains fiber integrity and nuclear-encoded mitochondrial gene expression in dopamine neurons. Proc Natl Acad Sci U S A, 110: , 2360–2365. |
[42] | Bergman O , Hakansson A , Westberg L , Belin AC , Sydow O , Olson L , Holmberg B , Fratiglioni L , Backman L , Eriksson E , & Nissbrandt H ((2009) ) Do polymorphisms in transcription factors LMX1A and LMX1B influence the risk for Parkinson’s disease? J Neural Transm (Vienna), 116: , 333–338. |
[43] | Laguna A , Schintu N , Nobre A , Alvarsson A , Volakakis N , Jacobsen JK , Gomez-Galan M , Sopova E , Joodmardi E , Yoshitake T , Deng Q , Kehr J , Ericson J , Svenningsson P , Shupliakov O , & Perlmann T ((2015) ) Dopaminergic control of autophagic-lysosomal function implicates Lmx1b in Parkinson’s disease. Nat Neurosci, 18: , 826–835. |
[44] | Kirik D , Rosenblad C , & Bjorklund A ((2000) ) Preservation of a functional nigrostriatal dopamine pathway by GDNF in the intrastriatal 6-OHDA lesion model depends on the site of administration of the trophic factor. Eur J Neurosci, 12: , 3871–3882. |
[45] | Maday S , Twelvetrees AE , Moughamian AJ , & Holzbaur EL ((2014) ) Axonal transport: Cargo-specific mechanisms of motility and regulation. Neuron, 84: , 292–309. |
[46] | Millecamps S , & Julien JP ((2013) ) Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci, 14: , 161–176. |
[47] | Saha AR , Hill J , Utton MA , Asuni AA , Ackerley S , Grierson AJ , Miller CC , Davies AM , Buchman VL , Anderton BH , & Hanger DP ((2004) ) Parkinson’s disease alpha-synuclein mutations exhibit defective axonal transport in cultured neurons. J Cell Sci, 117: , 1017–1024. |
[48] | Morfini G , Pigino G , Opalach K , Serulle Y , Moreira JE , Sugimori M , Llinas RR , & Brady ST ((2007) ) 1-Methyl-4-phenylpyridinium affects fast axonal transport by activation of caspase and protein kinase C. Proc Natl Acad Sci U S A, 104: , 2442–2447. |
[49] | Braak H , & Del Tredici K ((2004) ) Poor and protracted myelination as a contributory factor to neurodegenerative disorders. Neurobiol Aging, 25: , 19–23. |
[50] | Matsuda W , Furuta T , Nakamura KC , Hioki H , Fujiyama F , Arai R , & Kaneko T ((2009) ) Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci, 29: , 444–453. |
[51] | Bolam JP , & Pissadaki EK ((2012) ) Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov Disord, 27: , 1478–1483. |
[52] | Raff MC , Whitmore AV , & Finn JT ((2002) ) Axonal self-destruction and neurodegeneration. Science, 296: , 868–871. |
[53] | El-Khodor BF , & Burke RE ((2002) ) Medial forebrain bundle axotomy during development induces apoptosis in dopamine neurons of the substantia nigra and activation of caspases in their degenerating axons. J Comp Neurol, 452: , 65–79. |
[54] | Nikolaev A , McLaughlin T , O’Leary DD , & Tessier-Lavigne M ((2009) ) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature, 457: , 981–989. |
[55] | Ries V , Silva RM , Oo TF , Cheng HC , Rzhetskaya M , Kholodilov N , Flavell RA , Kuan CY , Rakic P , & Burke RE ((2008) ) JNK2 and JNK3 combined are essential for apoptosis in dopamine neurons of the substantia nigra, but are not required for axon degeneration. J Neurochem, 107: , 1578–1588. |
[56] | Coleman MP , & Perry VH ((2002) ) Axon pathology in neurological disease: A neglected therapeutic target. Trends Neurosci, 25: , 532–537. |
[57] | Lunn ER , Perry VH , Brown MC , Rosen H , & Gordon S ((1989) ) Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci, 1: , 27–33. |
[58] | Conforti L , Tarlton A , Mack TG , Mi W , Buckmaster EA , Wagner D , Perry VH , & Coleman MP ((2000) ) A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse. Proc Natl Acad Sci U S A, 97: , 11377–11382. |
[59] | Mack TG , Reiner M , Beirowski B , Mi W , Emanuelli M , Wagner D , Thomson D , Gillingwater T , Court F , Conforti L , Fernando FS , Tarlton A , Andressen C , Addicks K , Magni G , Ribchester RR , Perry VH , & Coleman MP ((2001) ) Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci, 4: , 1199–1206. |
[60] | Coleman MP , & Freeman MR ((2010) ) Wallerian degeneration, wld(s), and nmnat. Annu Rev Neurosci, 33: , 245–267. |
[61] | Deckwerth TL , & Johnson EM Jr ((1994) ) Neurites can remain viable after destruction of the neuronal soma by programmed cell death (apoptosis). Dev Biol, 165: , 63–72. |
[62] | Luo L , & O’Leary DD ((2005) ) Axon retraction and degeneration in development and disease. Annu Rev Neurosci, 28: , 127–156. |
[63] | Griffin JW , George EB , & Chaudhry V ((1996) ) Wallerian degeneration in peripheral nerve disease. Baillieres Clin Neurol, 5: , 65–75. |
[64] | Hoopfer ED , McLaughlin T , Watts RJ , Schuldiner O , O’Leary DD , & Luo L ((2006) ) Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron, 50: , 883–895. |
[65] | Araki T , Sasaki Y , & Milbrandt J ((2004) ) Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science, 305: , 1010–1013. |
[66] | Avery MA , Sheehan AE , Kerr KS , Wang J , & Freeman MR ((2009) ) Wlds requires Nmnat1 enzymatic activity and N16-VCP interactions to suppress Wallerian degeneration. J Cell Biol, 184: , 501–513. |
[67] | Beirowski B , Babetto E , Gilley J , Mazzola F , Conforti L , Janeckova L , Magni G , Ribchester RR , & Coleman MP ((2009) ) Non-nuclear Wld(S) determines its neuroprotective efficacy for axons and synapses in vivo. J Neurosci, 29: , 653–668. |
[68] | Babetto E , Beirowski B , Janeckova L , Brown R , Gilley J , Thomson D , Ribchester RR , & Coleman MP ((2010) ) Targeting NMNAT1 to axons and synapses transforms its neuroprotective potency in vivo. J Neurosci, 30: , 13291–13304. |
[69] | Avery MA , Rooney TM , Pandya JD , Wishart TM , Gillingwater TH , Geddes JW , Sullivan PG , & Freeman MR ((2012) ) WldS prevents axon degeneration through increased mitochondrial flux and enhanced mitochondrial Ca2+ buffering. Curr Biol, 22: , 596–600. |
[70] | Ferri A , Sanes JR , Coleman MP , Cunningham JM , & Kato AC ((2003) ) Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Curr Biol, 13: , 669–673. |
[71] | Mi W , Beirowski B , Gillingwater TH , Adalbert R , Wagner D , Grumme D , Osaka H , Conforti L , Arnhold S , Addicks K , Wada K , Ribchester RR , & Coleman MP ((2005) ) The slow Wallerian degeneration gene, WldS, inhibits al spheroid pathology in gracile axonal dystrophy mice. Brain, 128: , 405–416. |
[72] | Sajadi A , Schneider BL , & Aebischer P ((2004) ) Wlds-mediated protection of dopaminergic fibers in an animal model of Parkinson disease. Curr Biol, 14: , 326–330. |
[73] | Cheng HC , & Burke RE ((2010) ) The Wld(S) mutation delays anterograde, but not retrograde, axonal degeneration of the dopaminergic nigro-striatal pathway in vivo . J Neurochem, 113: , 683–691. |
[74] | Osterloh JM , Yang J , Rooney TM , Fox AN , Adalbert R , Powell EH , Sheehan AE , Avery MA , Hackett R , Logan MA , MacDonald JM , Ziegenfuss JS , Milde S , Hou YJ , Nathan C , Ding A , Brown RH Jr , Conforti L , Coleman M , Tessier-Lavigne M , Zuchner S , & Freeman MR ((2012) ) dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science, 337: , 481–484. |
[75] | Gerdts J , Summers DW , Sasaki Y , DiAntonio A , & Milbrandt J ((2013) ) Sarm1-mediated axon degeneration requires both SAM and TIR interactions. J Neurosci, 33: , 13569–13580. |
[76] | Cavalli V , Kujala P , Klumperman J , & Goldstein LS ((2005) ) Sunday Driver links axonal transport to damage signaling. J Cell Biol, 168: , 775–787. |
[77] | Miller BR , Press C , Daniels RW , Sasaki Y , Milbrandt J , & DiAntonio A ((2009) ) A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat Neurosci, 12: , 387–389. |
[78] | Yang J , Wu Z , Renier N , Simon DJ , Uryu K , Park DS , Greer PA , Tournier C , Davis RJ , & Tessier-Lavigne M ((2015) ) Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell, 160: , 161–176. |
[79] | Babetto E , Beirowski B , Russler EV , Milbrandt J , & DiAntonio A ((2013) ) The Phr1 ubiquitin ligase promotes injury-induced axon self-destruction. Cell Rep, 3: , 1422–1429. |
[80] | Kariya S , Mauricio R , Dai Y , & Monani UR ((2009) ) The neuroprotective factor Wld(s) fails to mitigate distal axonal and neuromuscular junction (NMJ) defects in mouse models of spinal muscular atrophy. Neurosci Lett, 449: , 246–251. |
[81] | Rose FF Jr , Meehan PW , Coady TH , Garcia VB , Garcia ML , & Lorson CL ((2008) ) The Wallerian degeneration slow (Wld(s)) gene does not attenuate disease in a mouse model of spinal muscular atrophy. Biochem Biophys Res Commun, 375: , 119–123. |
[82] | Vande Velde C , Garcia ML , Yin X , Trapp BD , & Cleveland DW ((2004) ) The neuroprotective factor Wlds does not attenuate mutant SOD1-mediated motor neuron disease. Neuromolecular Med, 5: , 193–203. |
[83] | Cheng HC , Kim SR , Oo TF , Kareva T , Yarygina O , Rzhetskaya M , Wang C , During M , Talloczy Z , Tanaka K , Komatsu M , Kobayashi K , Okano H , Kholodilov N , & Burke RE ((2011) ) Akt suppresses retrograde degeneration of dopaminergic axons by inhibition of macroautophagy. J Neurosci, 31: , 2125–2135. |
[84] | Kim SR , Kareva T , Yarygina O , Kholodilov N , & Burke RE ((2012) ) AAV transduction of dopamine neurons with constitutively active Rheb protects from neurodegeneration and mediates axon regrowth. Mol Ther, 20: , 275–286. |
[85] | Yang Y , Fukui K , Koike T , & Zheng X ((2007) ) Induction of autophagy in neurite degeneration of mouse superior cervical ganglion neurons. Eur J Neurosci, 26: , 2979–2988. |
[86] | Ribas VT , Schnepf B , Challagundla M , Koch JC , Bahr M , & Lingor P ((2015) ) Early and sustained activation of autophagy in degenerating axons after spinal cord injury. Brain Pathol, 25: , 157–170. |
[87] | Knoferle J , Koch JC , Ostendorf T , Michel U , Planchamp V , Vutova P , Tonges L , Stadelmann C , Bruck W , Bahr M , & Lingor P ((2010) ) Mechanisms of acute axonal degeneration in the optic nerve in vivo. Proc Natl Acad Sci U S A, 107: , 6064–6069. |
[88] | Ramonet D , Daher JP , Lin BM , Stafa K , Kim J , Banerjee R , Westerlund M , Pletnikova O , Glauser L , Yang L , Liu Y , Swing DA , Beal MF , Troncoso JC , McCaffery JM , Jenkins NA , Copeland NG , Galter D , Thomas B , Lee MK , Dawson TM , Dawson VL , & Moore DJ ((2011) ) Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One, 6: , e18568 |
[89] | Kim SR , Chen X , Oo TF , Kareva T , Yarygina O , Wang C , During MJ , Kholodilov N , & Burke RE ((2011) ) Dopaminergic pathway reconstruction by Akt/Rheb-induced axon regeneration. Ann Neurol, 70: , 110–120. |
Figures and Tables
Fig.1
Each panel shows nigrostriatal axons in the MFB visualized by confocal microscopy in TH-GFP mice. Following deletion of Atg7, axons of SNpc dopamine neurons are resistant to retrograde axon degeneration. In the absence of AAV Cre injection, Atg7fl/fl:TH-GFP mice show a loss of MFB dopaminergic axons, and the appearance of axonal spheroid pathology (red arrows) following unilateral 6OHDA injection. Atg7wt/wt:TH-GFP mice injected with AAV Cre show a similar axon loss and pathology. However, following injection of AAV Cre, Atg7fl/fl:TH-GFP mice show minimal axon loss and pathology following 6OHDA injection. Atg7fl/fl:TH-GFP mice without AAV Cre (n = 5) and Atg7wt/wt:TH-GFP mice given AAV Cre (n = 6) show a mean loss of 31 (or 32% ) and 32 (or 34% ) MFB axons respectively, whereas Atg7fl/fl:TH-GFP mice treated with AAV Cre (n = 6) show a mean loss of only 5 (or 5% ) (p < 0.001, ANOVA).
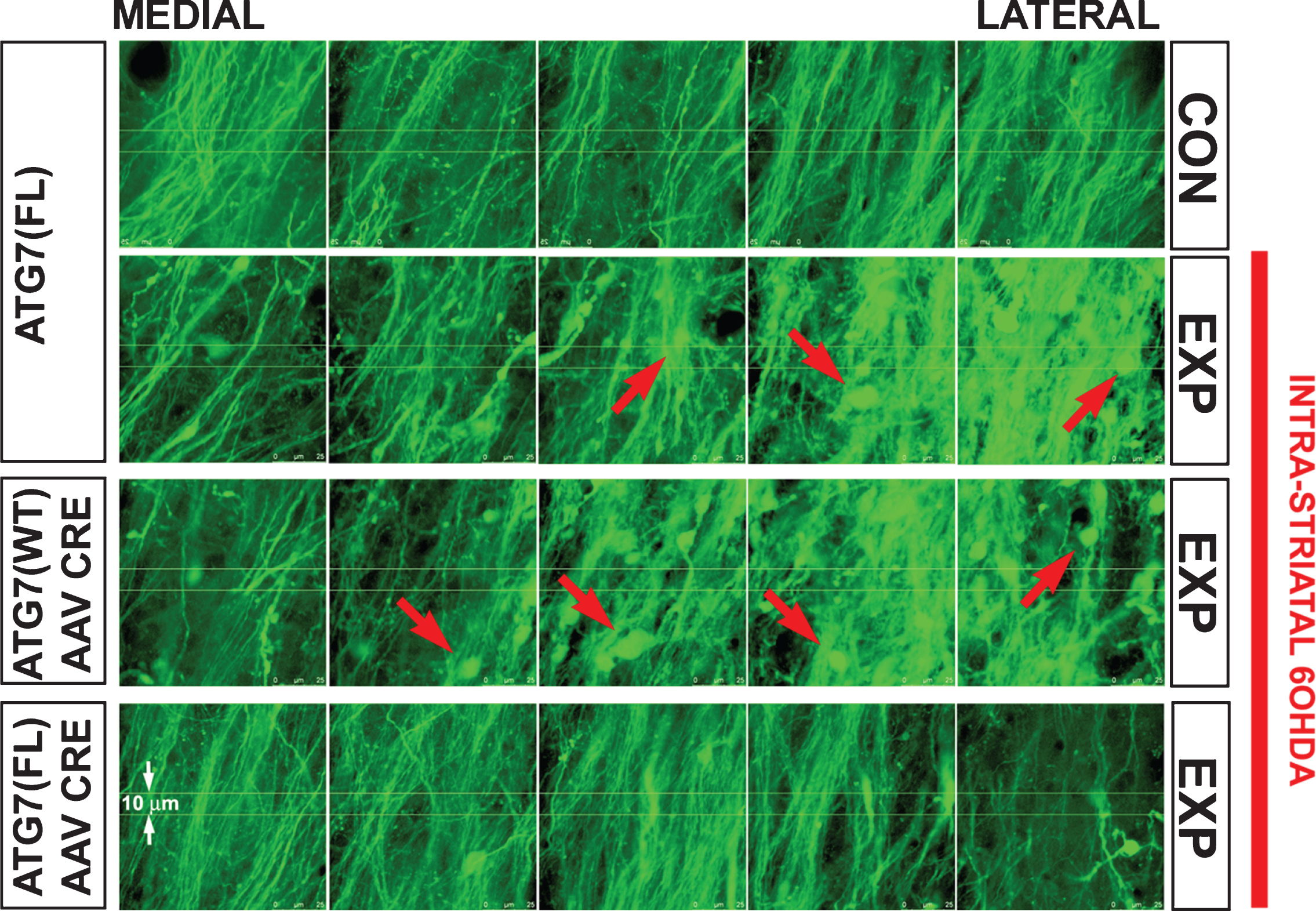
Fig.2
Transmission electron microscopic analysis of axonal spheroids in the medial forebrain bundle in the hLRRK2(R1441G) BAC transgenic mice revealed the presence of giant axonal spheroids containing large numbers of autophagic vacuoles. In A, a thinly myelinated axon (arrowheads) with well-organized cytoskeletal elements leads into a large spheroid that lacks myelin, contains disorganized cytoskeletal structures and is packed with autophagic vacuoles. The region in the white box is shown at higher magnification in B. Numerous vacuoles with double membranes, characteristic of autophagic vacuoles (AV), are observed. Interspersed among them are numerous multilamellar bodies (MLB).
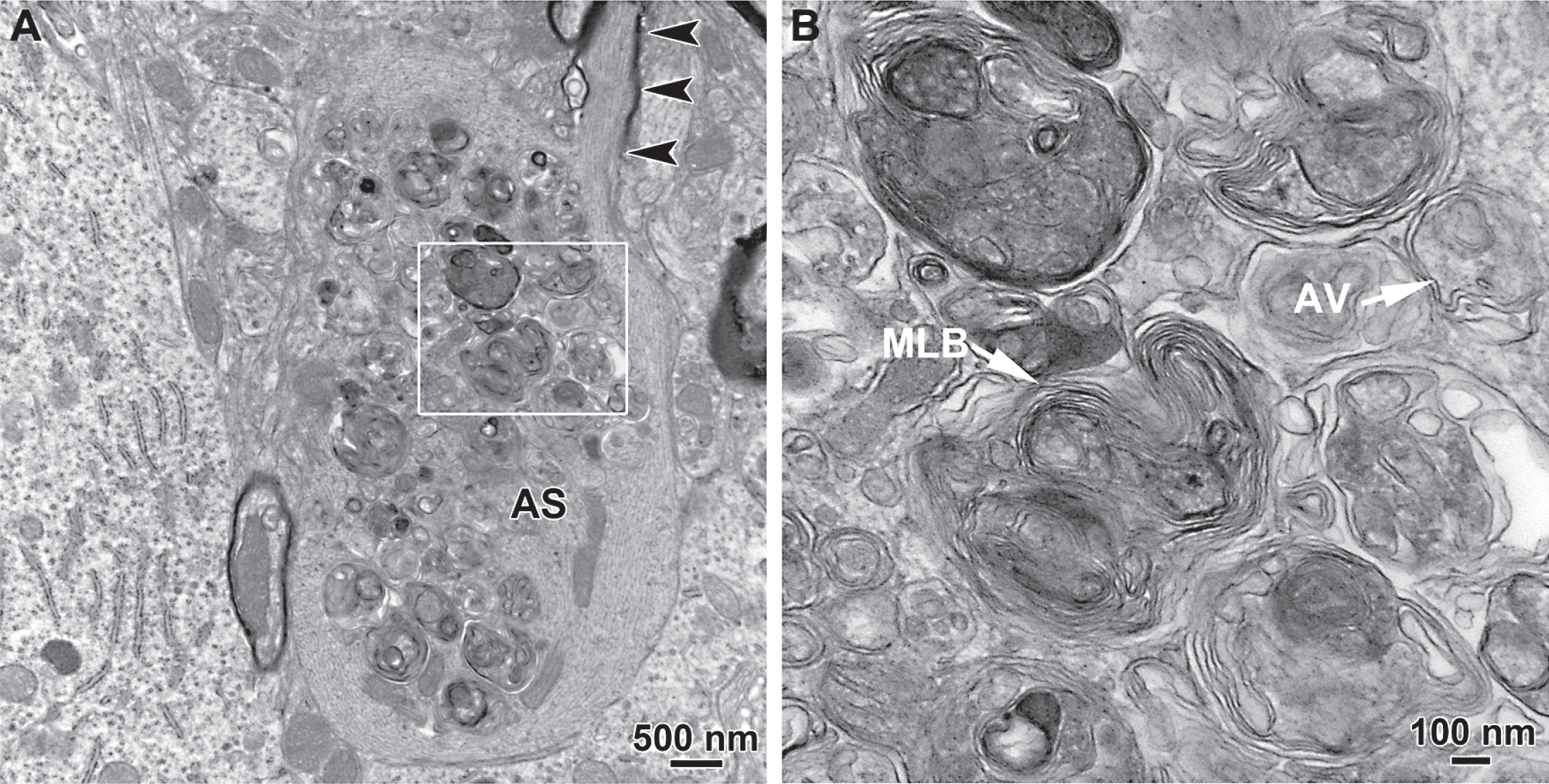
Table 1
Animal models of PD revealing an axonopathy
| Animal model (transgene) (species) | Promoter | Time course | Axonopathy | Striatal DA markers | SN Changes in DA structures | Motor deficits/DA dysfunction | Other | References |
| AAV/2 h α-syn-A53T Rat | Synapsin | 4 to 24 weeks | Dystrophic/swollen α-syn+ neurites in STR at 4&8 weeks. | N/A | ↓TH+ cells at 16 weeks. | STR ↑DOPAC, STR ↑DOPAC/DA | Changes in levels of axonal transport proteins. Inflammatory response in STR | [24] |
| AAV/6 h α-syn-WT Rat | Synapsin1 | 10 days to 16 weeks | Dystrophic/swollen α-syn+ neurites in STR at 10 days. | ↓TH+ fibers in STR at 3 weeks | ↓TH+ cells at 3 weeks. | Motor deficits starting at 5 weeks. | [25] | |
| hBAC-LRRK2(R1441G) Mouse | hLRRK2 in BAC | 3–12 mo 2–4 mo [ref] | Giant axonal spheroids at 2–4 and 9 months; AT8 axonal swellings at 9 months. Dystrophic neurites at 9 months. | No change STR TH IHC | ↓number of TH+ dendrites in SNpr at 9 mo ↓TH+ cells size at 9 mo | Motor deficits and ↓DA levels in STR at 9 months. | AVs in axonal spheroids at 2–4 months. | [27]; [26] |
| mBAC-Nurr1 (fl/fl) X mBACDAT-CreERT2) Mouse | mDAT in BAC | Up to 12 mo after tamoxifen | ↓ Number of TH+ axons in MFB at 4 months. TH+ fragmented axons in GP at 11 months. | ↓TH, VMAT2, DAT in STR at 1 and 4 weeks | No ↓in TH+ cells after at 2 months | Motor deficits at 4–7 months ↓DA, DOPAC, HVA in STR at 11 months | [41] | |
| Engrailed1(+/–) mouse | Endogenous engrailed | 4 to 24 weeks. | Swollen TH+ axon terminals at 8 days, 4 and 24 weeks. Fragmented axons in MFB at 8 weeks. | ↓TH and DAT protein by Western in STR at 4, 16 and 24 weeks | ↓TH+ cells at 16 and 24weeks. | ↓DA in STR at 4 &24 weeks | AVs in axons at 8 weeks. Abnormal mitochondria. ↓LC3B staining in SN. Upregulation of pmTor and PS6 in SN. | [34] |
A summary of the characterization of animal models of PD that display axon pathology. See text for further detail. Abbreviations: AV, autophagic vacuoles; BAC, bacterial artificial chromosome; DA, dopamine; DAT, dopamine transporter; DOPAC, 3, 4-dihydroxyphenylacetic acid; GP, globus pallidus; h, human; HVA, homovanillic acid; IHC, immunohistochemistry; LC3B, microtubule associated protein 1A/1B light chain 3B; MFB, medial forebrain bundle; mo, month; N/A, not available; pmTor, phosphorylated-mechanistic target of rapamycin; PS6, phospho-S6 ribosomal protein; SN, substantia nigra; SNpr, substantia nigra pars reticulata; STR, striatum; syn, synuclein; TH, tyrosine hydroxylase; VMAT2, vesicular monoamine transporter 2; WPRE, woodchuck posttranscriptional regulatory element; WT, wildtype.


