
Decoding Parkinson’s Disease Pathogenesis: The Role of Deregulated mRNA Translation
Abstract
Mutations in a number of genes cause rare familial forms of Parkinson’s disease and provide profound insight into potential mechanisms governing disease pathogenesis. Recently, a role for translation and metabolism of mRNA has emerged in the development of various neurodegenerative disorders including Parkinson’s disease (PD). In PD, preliminary evidence supports a role for aberrant translation in the disease process stemming from mutations in several genes. Translation control is central to maintaining organism homeostasis under variable environmental conditions and deregulation of this may predispose to certain stressors. Hypothetically, deregulated translation may be detrimental to neuronal viability in PD through the misexpression of a subset of transcripts or through the impact of excessive bulk translation on energy consumption and burden on protein homeostatic mechanisms. While compelling preliminary evidence exists to support a role for translation in PD, much more work is required to identify specific mechanisms linking altered translation to the disease process.
INTRODUCTION
Protein homeostasis (hereafter referred to as proteostasis) is fundamental to cell viability and promotes favorable conditions for organismal development and somatic maintenance [1]. At its core, protein homeostasis is achieved through balancing the synthesis, folding and effective trafficking of proteins with their degradation through proteasomal or autophagy-mediated pathways [2]. Each of these processes is tightly controlled based on nutrient availability and cellular requirements while their deregulation has been hypothesized as a major driver of aging and disease. For neurodegenerative diseases, there is substantial evidence supporting a central role for the misfolding and aggregation of proteins in disease development [3]. Given that a breakdown of proteostasis is observed in these diseases, it comes as no surprise that inappropriate mRNA translation and metabolism can contribute to the establishment of disease. Loss of translation control has previously been implicated in a number of diseases including cancer, obesity, diabetes and growth disorders [4]. Considering the high energetic cost of protein synthesis to the cell, its coordinated regulation is key to organismal development and homeostasis in response to rapidly changing environmental conditions [4]. Under nutrient abundance, anabolic processes such as protein synthesis are favored while low nutrient conditions oppose global translation to conserve resources and favors expression of proteins important in stress response and organism survival. Hence, tightly regulated translation is crucial for cellular function and survival in response to adverse conditions and loss of this control may predispose to disease. A growing body of evidence now also highlights an important role for loss of translation control in neurodegenerative diseases including Parkinson’s disease. The purpose of this review is to present evidence from the recent literature describing the contribution of mRNA translation to the pathogenesis of Parkinson’s disease. For introductions to eukaryotic translation [5, 6] and genes linked to Parkinson’s disease [7], the reader is directed to a number of excellent reviews on these topics.
ALTERED mRNA TRANSLATION IN NEUROLOGICAL DISEASE
A number of neurodegenerative diseases have roots in genetic mutations or cellular pathology that affects mRNA translation. Large poly-glutamine expansions in Ataxin-2 cause dominantly-inherited spinocerebellar ataxia type 2 while intermediate-length polyQ expansions have been associated with an increased risk for developing amyotrophic lateral sclerosis (ALS) [8]. Ataxin-2 interacts with multiple RNA-binding proteins with roles in RNA processing and also to poly(A)-binding protein PABPC1, mediating its association with polyribosomes [9–12]. Ataxin-2, PABPC1 and ribosomes are localized to stress granules under cell stress conditions, suggesting that Ataxin-2 may play a role in the adaptation of mRNA processing and translation to stress in a manner that could be perturbed via Ataxin-2 mutations [13]. Dominant mutations in five transfer RNA (tRNA) synthetases, which catalyze the aminoacylation of tRNAs with appropriate amino acids, result in axonal Charcot-Marie-Tooth (CMT) neuropathy. In a Drosophila model of CMT, disease-associated mutations in glycyl-tRNA synthetase (GARS) or tyrosyl-tRNA synthetase (YARS) cause a decrease in protein synthesis rates in motor and sensory neurons [14], although this does not appear to be linked to altered aminoacylation activity and mechanisms underlying this effect are unknown. Neurodegeneration can also result from a tRNA synthetase mutation that impairs its proofreading ability to incorporate the correct amino acid. This mutation was shown in a mouse model to result in cerebellar Purkinje cell death and ataxia highlighting the consequences of impaired translation fidelity on neuronal viability [15]. A role for globally reduced protein synthesis in the etiology of prion disease has been proposed based on mouse models of disease [16, 17]. The accumulation of misfolded prion protein in mouse brain triggers ER stress, prolonged activation of PERK and PERK-mediated eIF2α phosphorylation in the unfolded protein response. The resulting repression of protein synthesis has been suggested to cause neurodegeneration through loss of synaptic proteins leading to eventual synaptic failure [17]. Finally, a growing body of literature indicates that microsatellite expansions in a number of genes can lead to repeat-associated non-ATG (RAN) translation, wherein cap-independent translation of protein fragments is initiated downstream of these expansions [18, 19]. It is currently unclear how expanded repeats cause translation initiation although there is evidence that triplet expansions known to express RAN proteins form hairpins or G-quadruplexes, providing possible secondary structure to recruit ribosomes in a process that may be analogous to cap-independent translation [18, 19]. RAN proteins have been reported in an increasing number of diseases, including spinocerebellar ataxia type 8 (SCA8) [20], myotonic dystrophy type 1 (DM1) [20], Fragile-X tremor ataxia syndrome (FXTAS) [21], and C9ORF72 amyotrophic lateral sclerosis (ALS)/frontotemporal dementia (FTD) [22]. While there is some evidence that RAN proteins accumulate in degenerating neurons (e.g. SCA8 polyAla RAN protein in Purkinje cells), more work is needed to determine their contribution to neurodegeneration.
DEREGULATED TRANSLATION IN PARKINSON’S DISEASE
Accumulating evidence indicates an important role for mRNA translation in Parkinson’s disease pathogenesis resulting from disease-linked mutations in genes including LRRK2, pink1, parkin and DJ-1. This evidence is presented below followed by a discussion on current hypotheses addressing the contribution of translation to the disease process.
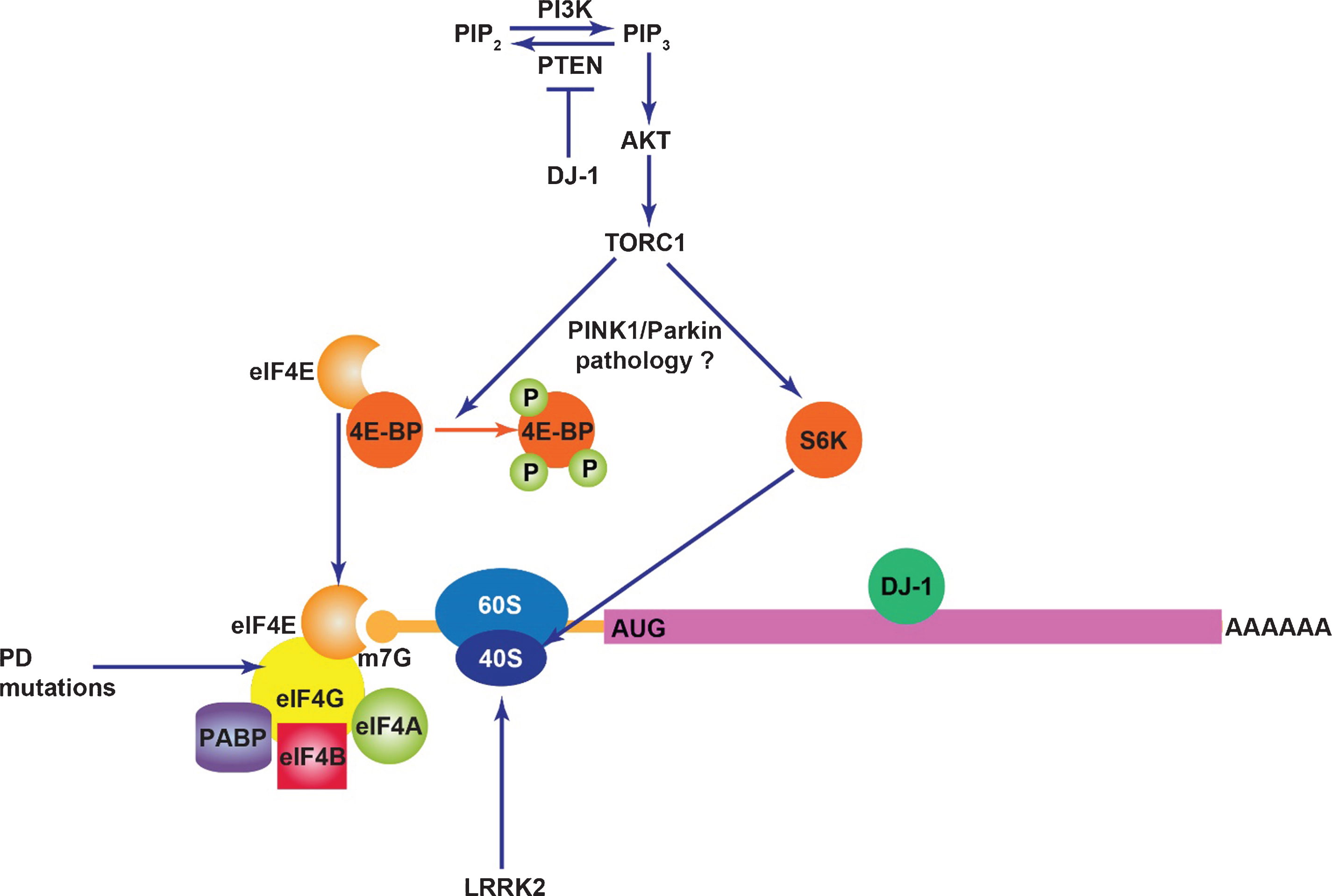
LRRK2 (leucine-rich repeat kinase 2) is a large multi-domain protein with enzymatic RAS-like GTPase and kinase domains in which numerous disease-linked mutations are clustered [23]. Mutations in LRRK2 segregate with familial PD with age-related penetrance [24, 25] and are also found in sporadic PD [26–28], indicating a broad role for LRRK2 dysfunction in PD neurodegeneration. Mutations in LRRK2 results in clinical phenotypes very similar to late-onset PD although Lewy body pathology is not always present [29, 30]. It is now well established that the common G2019S mutation in LRRK2 results in enhanced kinase activity (autophosphorylation and exogenous substrate phosphorylation) that is instrumental in neuronal toxicity, as observed in several in vitro and in vivo models [31–49]. There is currently debate about how other disease-segregating mutations affect kinase activity, although fairly good consensus exists that the I2020T mutation also increases kinase activity [38, 42, 45, 49–53]. It is less clear how ROC-COR domain mutations affect LRRK2 kinase activity, as the number of studies reporting an increase, decrease or no change in kinase activity is much more evenly spread [32–34, 39, 42, 45, 47, 48, 54]. These inconsistencies may reflect variable methodology used across these studies and results from in vitro kinase assays may not necessarily accurately represent the effects of these mutations on in vivo kinase activity which awaits clarification. A number of studies on Drosophila expressing kinase-enhancing mutations in human LRRK2 or its fly ortholog (dLRRK) support a functional interaction of LRRK2 with the translation machinery that may be altered by these mutations. Early evidence indicated that LRRK2 genetically interacts with the TOR (target of rapamycin) pathway in flies [38]. Loss of dLRRK worsens phenotypes associated with downregulated TOR signaling caused by overexpression of a constitutively-active form of 4E-BP1 (eIF4E-binding protein 1) or co-overexpression of TSC1 and TSC2. The stimulatory effects of TOR signaling on general translation are mediated in part via phosphorylation of 4E-BP1 which disrupts its eIF4E binding activity, thereby promoting cap complex formation through binding of eIF4G to free eIF4E (Figure). Both human LRRK2 and dLRRK were reported to phosphorylate 4E-BP1 and hence kinase-enhancing mutations that hyper-phosphorylate 4E-BP1 were suggested to promote PD-related phenotypes via eIF4E-dependent translation initiation [38]. Contrastingly, in LRRK2-transfected HEK cells or mammalian brain, 4E-BP1 does not appear to be a direct LRRK2 substrate [40, 55] and increased phosphorylation of 4E-BP1 is not found in G2019S LRRK2 knock-in mice but, conversely has been reported in LRRK2 knock-out mice and LRRK2 kinase-dead transgenic mice [56]. Another study sought to identify specific transcripts with altered translation in the presence of LRRK2. While numerous targets were identified, the authors concentrated on e2f1 and dp1 upregulation and report that pathogenic LRRK2 represses the activity of let-7 and miR-184, respectively to result in these effects [57]. The inhibitory effects of mutant LRRK2 on miRNA function are proposed to occur through two independent mechanisms. First, pathogenic LRRK2 associates with Drosophila Argonaute-1, a key component of the RNA-induced silencing complex, leading to decreased Argonaute-1 levels by an unknown mechanism. Second, mutant LRRK2 stimulates the association of phospho-4E-BP1 with human Argonaute-2 thereby impairing its function [57].
More recently, LRRK2 was found to interact with and directly phosphorylate a number of ribosomal proteins through interactor and kinase screening in series [42]. Phosphorylation of ribosomal protein s15 is enhanced by G2019S and I2020T mutations in the kinase domain of LRRK2, and this was identified as a key mediator of mutant LRRK2 toxicity in primary neuron and Drosophila models of LRRK2 toxicity. Reporter assays revealed that mutant LRRK2 stimulates both cap-dependent and cap-independent mRNA translation in an s15-dependent manner. Additionally, G2019S LRRK2 transgenic Drosophila exhibit elevated bulk protein synthesis which was shown to be important in age-related neurodegenerative phenotypes through rescue experiments with the protein synthesis inhibitor anisomycin. These data corroborate previous findings implicating that LRRK2 has a stimulatory effect on mRNA translation. Currently unresolved are how s15 phosphorylation affects mRNA translation and whether there are specific translational targets that mediate LRRK2 toxicity. Interestingly, G2019S LRRK2 was also found to positively regulate levels of numerous ribosomal protein transcripts in mouse brain when compared to the brains of LRRK2 KO mice, raising the possibility that altered levels of ribosomal proteins and potentially whole ribosomes could be another mechanism whereby LRRK2 impacts mRNA translation [58]. Considering the clinical and pathological overlap between LRRK2-linked PD with sporadic cases, it will be informative to determine if excess mRNA translation as a route to PD pathogenesis is also supported in sporadic PD, for example by examining if s15 or 4E-BP1 phosphorylation are elevated in postmortem brain tissue from these patients.
A role for the initiation step of mRNA translation in PD is supported by the identification of rare missense mutations in eIF4G1 proposed to cause autosomal-dominant disease [59, 60]. While PD-causing mutations in a translation initiation factor such as eIF4G1 would represent a strong link between aberrant translation and disease, it should be noted that several recent studies have suggested that these mutations may rarely cause disease or not at all [61–63]. eIF4G1 is a large protein which acts as a scaffold for the binding of eIF4A and eIF4E to form the mRNA cap complex, required for initiating cap-dependent translation [5]. eIF4G additionally binds PABP at the poly A tail to circularize mRNA which stabilizes it and activates it for translation [5]. While eIF4G1 has an important role in translation initiation, it is yet to be described whether the mutations associated with PD cause a gain, loss or toxic gain of function to the individual protein or cap-complex and how this affects translation initiation. There is, however, evidence for a genetic interaction between eIF4G1 and another PD gene, VPS35. The VPS35 protein is a retromer complex component that mediates retrograde protein transport from endosomes to the trans-Golgi network [64, 65]. eIF4G1 overexpression is reportedly toxic in yeast lacking VPS35 or expressing mutant D620N VPS35 associated with PD, suggesting that this toxicity may be due to loss of retromer function [64]. Additional experiments suggest that toxicity is further related to proteotoxic stress, ER stress and the unfolded protein response [64]. This study highlights the interconnectedness of protein synthesis and transport in preventing protein misfolding and also provides clues to how gain-of-function mutations in eIF4G1 (consistent with an autosomal dominant mode of inheritance) could potentially impact neuronal function. Similarly, a link between protein trafficking and LRRK2 in the context of PD is suggested by other studies. LRRK2 was found to interact with RAB7L1 in a co-complex that additionally includes BCL2-associated athanogene 5 and Cyclin-G-associated kinase that may promote the autophagic clearance of trans-Golgi-derived vesicles [66]. RAB7L1 is a candidate sporadic PD risk factor identified by genome-wide association studies [27, 28]. Expression of constitutively active RAB7L1 was found to rescue mutant LRRK2-induced loss of neurites in neuronal cultures and loss of dopamine neurons in flies while knock-down of RAB7L1 in dopamine neurons causes neurodegeneration [67]. RAB7L1 localizes heavily to the Golgi and a link to defective retromer function in these phenotypes is supported by evidence that both VPS35 and VPS29 levels are decreased by expression of mutant LRRK2 or knock-down of RAB7L1 while VPS35 overexpression rescues dopamine neuron viability in both of these conditions [67].
Translational suppression may also be neuroprotective in the context of other PD mutations. Genetic interaction studies in flies showed that overexpression of 4E-BP is protective against loss of dopaminergic neurons in pink1 and parkin mutants [68]. 4E-BP overexpression is expected to enhance eIF4E sequestration thereby inhibiting binding to eIF4G and mRNA cap complex formation. An upregulation of un-phosphorylated 4E-BP can also be achieved via the mTOR kinase inhibitor rapamycin, and indeed, rapamycin treatment phenocopies the neuroprotective effect of 4E-BP upregulation in flies [68]. Consistent with this, another study showed that knock-down or overexpression of S6K ameliorated or exacerbated muscle degeneration in pink1 mutant flies, respectively, and that S6K overexpression also worsened dopamine neuron loss [69]. Interestingly, in both of these studies there was evidence of downregulated Akt/TORC1 signaling (levels of phospho-4E-BP, phospho-Akt and phospho-S6K) in pink1 and parkin mutants. Collectively, these results suggest that the Akt/TORC1 pathway is suppressed in these mutants typical of a protective stress response and that augmenting this blockade of TOR signaling may be an effective strategy to prevent degenerative phenotypes [68, 69]. The TORC1 pathway in which 4E-BP1 and S6K reside regulates numerous intracellular processes including autophagy, transcription, metabolism, cell survival and translation [70]. Given that specific manipulations of 4E-BP1 and S6K expression modulate pink1 and parkin phenotypes without need for whole TORC1 pathway inhibition, it is reasonable to postulate that protection occurs via a reduction in translation. Downregulation of protein synthesis typically occurs in response to cellular stress and in the case of pink1/parkin mutants may be important in lowering energy consumption under conditions predisposing to loss of mitochondrial quality control and mitochondrial dysfunction.
In contrast with LRRK2, there is no direct evidence to suggest that pink1 or parkin regulate global mRNA translation in a manner that goes awry in Parkinson’s disease. Screening for parkin interactors in mammalian cells revealed many ribosomal proteins and translation initiation/elongation factors [71]. Despite this, validation of these interactions and evidence to support a functional role of parkin on mRNA translation is currently lacking. Similarly, the p38/AIMP2 scaffolding protein required for tRNA synthetase complex assembly is a substrate of parkin’s E3 ubiquitin ligase activity [72]. AIMP2 levels accumulate in the brain of PD patients with Parkin mutations and ventral midbrain of Parkin knockout mice, yet studies in AIMP2 overexpressing mice suggest that global translation is normal [73]. Studies on Drosophila, however, implicate a role for pink1 and parkin in the local translation of nuclear-encoded respiratory chain complex transcripts on the mitochondrial outer membrane [74]. It has been suggested that pink1 and parkin may recruit targeting of these transcripts to the mitochondrial outer membrane and also displace translational suppressors such as Pumilio and Glorund/hnRNP-F to enable translation. Impairment of this process in pink1/parkin mutants could lead to defects in oxidative phosphorylation and underlie mitochondrial pathology in PD. There is, however, data to suggest that pink1 may be important in the upregulation of cap-dependent and cap-independent translation under hypoxia conditions in vitro [75]. Loss of pink1 had a greater impact on cap-independent translation and specifically, the production of HIF-1a and hypoxia-responsive gene upregulation, possibly through loss of hypoxia-induced 4E-BP1 dephosphorylation.
While DJ-1 is best characterized to have a role protecting against oxidative stress and mitochondrial dysfunction, it may also impact translation. The PI3K/Akt pathway that activates mTORC1 signaling which in turn promotes mRNA translation is negatively regulated by the PTEN tumor suppressor [76]. DJ-1 was originally identified as a putative oncogene that transforms NIH3T3 cells cooperatively with ras [77]. It was subsequently revealed that DJ-1 acts as a suppressor of PTEN function in a Drosophila screen and its expression was found to correlate with the extent of PKB/Akt phosphorylation in mammalian cells and primary breast cancer samples [78]. Hence, it is possible that through its stimulatory effects on Akt activation, DJ-1 could be indirectly involved in regulating mRNA translation in a manner impaired by loss-of-function mutations. Although DJ-1 lacks canonical RNA binding motifs, it has been reported to bind to several mRNA targets including glutathione peroxidases, the selenoproteins necessary to produce glutathione peroxidase and members of the PTEN/PI3K pathway by a poorly understood mechanism [79]. Pathogenic variants of DJ-1 exhibit deficits in this ability to bind mRNA. Interestingly, DJ-1 has been proposed to partially inhibit mRNA translation upon mRNA binding and dissociate under conditions of oxidative stress [79], suggesting that it could in theory act to regulate specific antioxidant defenses in response to intracellular oxidative stress cues. It would be interesting to further investigate whether DJ-1 acts as both redox sensor and regulator of antioxidant defenses in cells through regulating translation of antioxidants such as glutathione peroxidase.
Collectively, the evidence presented above indicates a significant role for mRNA translation in the pathogenesis of PD linked to several genes (Table). It is important to note, however, that the field is in the very early stages of describing this relationship and many of the recently-published studies presented here await independent confirmation. Further investigation into the role of mRNA translation in PD warranted by these studies will also be necessary to go beyond initial observations and pinpoint specific mechanisms by which deregulated mRNA translation might play a role in dopamine neuron degeneration. A common theme for several of these genes (LRRK2, pink1 and parkin) is that translational suppression can be neuroprotective against pathology. Downregulation of global protein synthesis prevents mutant LRRK2-induced neurodegeneration while reducing TOR-mediated translation can block pink1 and parkin pathology. These findings give rise to a number of possible hypotheses that may be investigated further. For example, the increase in global protein synthesis rates observed in LRRK2 transgenic Drosophila are directly linked to neuronal toxicity as multiple independent methods to reduce translation are neuroprotective. Possible mechanisms linking an increase in bulk translation to neurodegeneration may be generally categorized into (i) an upregulation of pathogenic translational targets or downregulation of neuroprotective proteins resulting from loss of translational control and (ii) an overall loss of protein homeostasis leading to neuronal dysfunction and death. Of course, these mechanisms may act in combination to promote neurodegeneration and may also be interdependent. Identifying translational targets of LRRK2 in dopamine neurons and then determining the impact of their elevated expression on mutant LRRK2 neurotoxicity, e.g. through knock-down screening assays, will test the hypothesis that upregulation of key translational targets mediates LRRK2 toxicity. On the loss of protein homeostasis, an overall excess in synthesized proteins could conceivably place an overwhelming burden on protein chaperone, trafficking and turnover capacity in a manner that promotes ER stress, protein misfolding and aggregation. LRRK2 has previously been linked to protein turnover via autophagy (reviewed in [80]). PD-causing mutations result in complex effects on the autophagy/lysosomal pathway which are poorly understood and may represent either increased autophagic flux or conversely, an impairment of autophagosome fusion with lysosomes resulting in autophagy arrest. While these effects may be mediated at least in part through direct impact of LRRK2 on the function of autophagosomes [81–83] or lysosomes [67, 82, 84–86] it is also possible that LRRK2’s stimulatory effect on protein synthesis could have downstream consequences on protein degradation pathways. For mutant LRRK2, an overall increase in protein synthesis could conceivably result in depletion of the pool of free amino acids in cells that simulates nutrient starvation conditions. One anticipated outcome of depleted amino acids might be an upregulation of autophagy in order to restore amino acid homeostasis. Increased global protein production may also require a counterbalancing increase in protein turnover through autophagy, especially if the increased burden on protein homeostatic mechanisms predisposes to an accumulation of misfolded or damaged protein. Along these lines, it will be informative to determine if the autophagy phenotypes observed via mutant LRRK2 expression can be blocked through suppression of protein synthesis.
As an accumulation of misfolded protein is an established hallmark of PD pathology, deficient proteostasis is an attractive hypothesis to link excess protein synthesis with neurotoxicity. More broadly, if insufficient protein trafficking, chaperone capacity or protein turnover are important contributors to pathology, then it may be possible to ameliorate neurodegeneration through bolstering these processes in an attempt to restore proteostasis as suggested by some studies. It may be hard, however, to reconcile this with the specific vulnerability of dopamine neurons in PD, unless the excess burden on proteostatic mechanisms does not directly result in toxicity but predisposes to one or more insults that dopamine neurons are particularly vulnerable to, such as oxidative stress. Hence, a slightly modified hypothesis from the one stated above is that translational reprogramming necessary forcellular adaptation under conditions of stress is impaired by LRRK2 mutations that predisposes dopamine neurons to dysfunction and death. An accumulation of oxidative damage to intracellular macromolecules and a decline in mechanisms to handle and eradicate reactive oxygen species are observed in aging organisms, including in brain. This may represent a major driving force for the decline in cognitive function with advanced age. Dopamine neurons are reported to be particularly susceptible to oxidative stress caused by dopamine metabolism and their autonomous pacemaking activity which promotes mitochondrial dysfunction [87, 88]. An age-related decline in antioxidant defenses coupled to an impaired ability to downregulate anabolic processes such as translation in the face of oxidative stress might predispose neurons in aged organisms to LRRK2 mutations. This hypothesis could conceivably account for a role of aging in Parkinson’s disease linked to these mutations. Pervasively high levels of mRNA translation may also be detrimental through consumption of energy, given that protein synthesis is one of the major energy-consuming process. Neurons consume high levels of energy during active processes such as intraneuronal transport, neurotransmitter release and synaptic plasticity. Depleted energy reserves may also affect the ability of neurons to perform these activities. Neurons with mutations in pink1 or parkin that lead to mitochondrial dysfunction and reduced energy output may be especially vulnerable to robust levels of protein synthesis, and this may account for the observation that lowering translation through altered TOR signaling can prevent age-related neurodegeneration.
CONCLUSIONS
A number of studies implicating translation in the pathogenesis of PD have recently emerged in conjunction with an increased recognition for the role of deregulated mRNA translation and metabolism in neurodegenerative diseases. There is a compelling need for more detailed investigation into the relationship between translation and PD in order to better understand the role of proteostasis decline in disease and with the ultimate goal of providing significant advances in the development of diagnostic tools and therapeutics.
CONFLICT OF INTEREST
The author has no conflict of interest to report.
ACKNOWLEDGMENTS
I thank the Martin lab for stimulating discussions on the literature reviewed here. This work was supported by NIH/NIA K01AG050718.
REFERENCES
[1] | Taylor RC , & Dillin A ((2011) ) Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol, 3: , pii: a004440. |
[2] | Chen B , Retzlaff M , Roos T , & Frydman J ((2011) ) Cellular strategies of protein quality control. Cold Spring Harb Perspect Biol, 3: , a004374. |
[3] | Brettschneider J , Del Tredici K , Lee VM , & Trojanowski JQ ((2015) ) Spreading of pathology in neurodegenerative diseases: A focus on human studies. Nat Rev Neurosci, 16: , 109–120. |
[4] | Kapahi P , Chen D , Rogers AN , Katewa SD , Li PW , Thomas EL , & Kockel L ((2010) ) With TOR, less is more: A key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab, 11: , 453–465. |
[5] | Sonenberg N , & Hinnebusch AG ((2009) ) Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell, 136: , 731–745. |
[6] | Hershey JW , Sonenberg N , & Mathews MB ((2012) ) Principles of translational control: An overview. Cold Spring Harb Perspect Biol, 4: , pii: a011528. |
[7] | Dawson TM , Ko HS , & Dawson VL ((2010) ) Genetic animal models of Parkinson’s disease. Neuron, 66: , 646–661. |
[8] | Elden AC , Kim HJ , Hart MP , Chen-Plotkin AS , Johnson BS , Fang X , Armakola M , Geser F , Greene R , Lu MM , Padmanabhan A , Clay-Falcone D , McCluskey L , Elman L , Juhr D , Gruber PJ , Rub U , Auburger G , Trojanowski JQ , Lee VM , Van Deerlin VM , Bonini NM , & Gitler AD ((2010) ) Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature, 466: , 1069–1075. |
[9] | Albrecht M , & Lengauer T ((2004) ) Survey on the PABC recognition motif PAM2. Biochem Biophys Res Commun, 316: , 129–138. |
[10] | Nonhoff U , Ralser M , Welzel F , Piccini I , Balzereit D , Yaspo ML , Lehrach H , & Krobitsch S ((2007) ) Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol Biol Cell, 18: , 1385–1396. |
[11] | Satterfield TF , & Pallanck LJ ((2006) ) Ataxin-2 and its Drosophila homolog, ATX2, physically assemble with polyribosomes. Hum Mol Genet, 15: , 2523–2532. |
[12] | Shibata H , Huynh DP , & Pulst SM ((2000) ) A novel protein with RNA-binding motifs interacts with ataxin-2. Hum Mol Genet, 9: , 1303–1313. |
[13] | Fittschen M , Lastres-Becker I , Halbach MV , Damrath E , Gispert S , Azizov M , Walter M , Muller S , & Auburger G ((2015) ) Genetic ablation of ataxin-2 increases several global translation factors in their transcript abundance but decreases translation rate. Neurogenetics, 16: , 181–192. |
[14] | Niehues S , Bussmann J , Steffes G , Erdmann I , Kohrer C , Sun L , Wagner M , Schafer K , Wang G , Koerdt SN , Stum M , RajBhandary UL , Thomas U , Aberle H , Burgess RW , Yang XL , Dieterich D , & Storkebaum E ((2015) ) Impaired protein translation in Drosophila models for Charcot-Marie-Tooth neuropathy caused by mutant tRNA synthetases. Nat Commun, 6: , 7520. |
[15] | Lee JW , Beebe K , Nangle LA , Jang J , Longo-Guess CM , Cook SA , Davisson MT , Sundberg JP , Schimmel P , & Ackerman SL ((2006) ) Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature, 443: , 50–55. |
[16] | Moreno JA , Halliday M , Molloy C , Radford H , Verity N , Axten JM , Ortori CA , Willis AE , Fischer PM , Barrett DA , & Mallucci GR ((2013) ) Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med, 5: , 206ra138. |
[17] | Moreno JA , Radford H , Peretti D , Steinert JR , Verity N , Martin MG , Halliday M , Morgan J , Dinsdale D , Ortori CA , Barrett DA , Tsaytler P , Bertolotti A , Willis AE , Bushell M , & Mallucci GR ((2012) ) Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature, 485: , 507–511. |
[18] | Belzil VV , Gendron TF , & Petrucelli L ((2013) ) RNA-mediated toxicity in neurodegenerative disease. Mol Cell Neurosci, 56: , 406–419. |
[19] | Cleary JD , & Ranum LP ((2014) ) Repeat associated non-ATG (RAN) translation: New starts in microsatellite expansion disorders. Curr Opin Genet Dev, 26: , 6–15. |
[20] | Zu T , Gibbens B , Doty NS , Gomes-Pereira M , Huguet A , Stone MD , Margolis J , Peterson M , Markowski TW , Ingram MA , Nan Z , Forster C , Low WC , Schoser B , Somia NV , Clark HB , Schmechel S , Bitterman PB , Gourdon G , Swanson MS , Moseley M , & Ranum LP ((2011) ) Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A, 108: , 260–265. |
[21] | Todd PK , Oh SY , Krans A , He F , Sellier C , Frazer M , Renoux AJ , Chen KC , Scaglione KM , Basrur V , Elenitoba-Johnson K , Vonsattel JP , Louis ED , Sutton MA , Taylor JP , Mills RE , Charlet-Berguerand N , & Paulson HL ((2013) ) CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron, 78: , 440–455. |
[22] | Ash PE , Bieniek KF , Gendron TF , Caulfield T , Lin WL , Dejesus-Hernandez M , van Blitterswijk MM , Jansen-West K , Paul JW , Rademakers R , Boylan KB , Dickson DW , & Petrucelli L ((2013) ) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron, 77: , 639–646. |
[23] | Greggio E , & Cookson MR ((2009) ) Leucine-rich repeat kinase 2 mutations and Parkinson’s disease: Three questions. ASN Neuro, 1: , pii: e00002. |
[24] | Paisan-Ruiz C , Jain S , Evans EW , Gilks WP , Simon J , van der Brug M , Lopez de Munain A , Aparicio S , Gil AM , Khan N , Johnson J , Martinez JR , Nicholl D , Carrera IM , Pena AS , de Silva R , Lees A , Marti-Masso JF , Perez-Tur J , Wood NW , & Singleton AB ((2004) ) Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron, 44: , 595–600. |
[25] | Zimprich A , Biskup S , Leitner P , Lichtner P , Farrer M , Lincoln S , Kachergus J , Hulihan M , Uitti RJ , Calne DB , Stoessl AJ , Pfeiffer RF , Patenge N , Carbajal IC , Vieregge P , Asmus F , Muller-Myhsok B , Dickson DW , Meitinger T , Strom TM , Wszolek ZK , & Gasser T ((2004) ) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron, 44: , 601–607. |
[26] | Healy DG , Falchi M , O’Sullivan SS , Bonifati V , Durr A , Bressman S , Brice A , Aasly J , Zabetian CP , Goldwurm S , Ferreira JJ , Tolosa E , Kay DM , Klein C , Williams DR , Marras C , Lang AE , Wszolek ZK , Berciano J , Schapira AH , Lynch T , Bhatia KP , Gasser T , Lees AJ , Wood NW , & International LC ((2008) ) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol, 7: , 583–590. |
[27] | Satake W , Nakabayashi Y , Mizuta I , Hirota Y , Ito C , Kubo M , Kawaguchi T , Tsunoda T , Watanabe M , Takeda A , Tomiyama H , Nakashima K , Hasegawa K , Obata F , Yoshikawa T , Kawakami H , Sakoda S , Yamamoto M , Hattori N , Murata M , Nakamura Y , & Toda T ((2009) ) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet, 41: , 1303–1307. |
[28] | Simon-Sanchez J , Schulte C , Bras JM , Sharma M , Gibbs JR , Berg D , Paisan-Ruiz C , Lichtner P , Scholz SW , Hernandez DG , Kruger R , Federoff M , Klein C , Goate A , Perlmutter J , Bonin M , Nalls MA , Illig T , Gieger C , Houlden H , Steffens M , Okun MS , Racette BA , Cookson MR , Foote KD , Fernandez HH , Traynor BJ , Schreiber S , Arepalli S , Zonozi R , Gwinn K , van der Brug M , Lopez G , Chanock SJ , Schatzkin A , Park Y , Hollenbeck A , Gao J , Huang X , Wood NW , Lorenz D , Deuschl G , Chen H , Riess O , Hardy JA , Singleton AB , & Gasser T ((2009) ) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet, 41: , 1308–1312. |
[29] | Gaig C , Marti MJ , Ezquerra M , Rey MJ , Cardozo A , & Tolosa E ((2007) ) G2019S LRRK2 mutation causing Parkinson’s disease without Lewy bodies. J Neurol Neurosurg Psychiatry, 78: , 626–628. |
[30] | Marti-Masso JF , Ruiz-Martinez J , Bolano MJ , Ruiz I , Gorostidi A , Moreno F , Ferrer I , & de Munain AL ((2009) ) Neuropathology of Parkinson’s disease with the R1441G mutation in LRRK2. Mov Disord, 24: , 1998–2001. |
[31] | Lee BD , Shin JH , VanKampen J , Petrucelli L , West AB , Ko HS , Lee YI , Maguire-Zeiss KA , Bowers WJ , Federoff HJ , Dawson VL , & Dawson TM ((2010) ) Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat Med, 16: , 998–1000. |
[32] | Smith WW , Pei Z , Jiang H , Dawson VL , Dawson TM , & Ross CA ((2006) ) Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci, 9: , 1231–1233. |
[33] | Greggio E , Jain S , Kingsbury A , Bandopadhyay R , Lewis P , Kaganovich A , van der Brug MP , Beilina A , Blackinton J , Thomas KJ , Ahmad R , Miller DW , Kesavapany S , Singleton A , Lees A , Harvey RJ , Harvey K , & Cookson MR ((2006) ) Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis, 23: , 329–341. |
[34] | Anand VS , Reichling LJ , Lipinski K , Stochaj W , Duan W , Kelleher K , Pungaliya P , Brown EL , Reinhart PH , Somberg R , Hirst WD , Riddle SM , & Braithwaite SP ((2009) ) Investigation of leucine-rich repeat kinase 2: Enzymological properties and novel assays. FEBS J, 276: , 466–478. |
[35] | Angeles DC , Gan BH , Onstead L , Zhao Y , Lim KL , Dachsel J , Melrose H , Farrer M , Wszolek ZK , Dickson DW , & Tan EK ((2011) ) Mutations in LRRK2 increase phosphorylation of peroxiredoxin 3 exacerbating oxidative stress-induced neuronal death. Hum Mutat, 32: , 1390–1397. |
[36] | Biosa A , Trancikova A , Civiero L , Glauser L , Bubacco L , Greggio E , & Moore DJ ((2013) ) GTPase activity regulates kinase activity and cellular phenotypes of Parkinson’s disease-associated LRRK2. Hum Mol Genet, 22: , 1140–1156. |
[37] | Covy JP , & Giasson BI ((2009) ) Identification of compounds that inhibit the kinase activity of leucine-rich repeat kinase 2. Biochem Biophys Res Commun, 378: , 473–477. |
[38] | Imai Y , Gehrke S , Wang HQ , Takahashi R , Hasegawa K , Oota E , & Lu B ((2008) ) Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J, 27: , 2432–2443. |
[39] | Jaleel M , Nichols RJ , Deak M , Campbell DG , Gillardon F , Knebel A , & Alessi DR ((2007) ) LRRK2 phosphorylates moesin at threonine-558: Characterization of how Parkinson’s disease mutants affect kinase activity. Biochem J, 405: , 307–317. |
[40] | Kumar A , Greggio E , Beilina A , Kaganovich A , Chan D , Taymans JM , Wolozin B , & Cookson MR ((2010) ) The Parkinson’s disease associated LRRK2 exhibits weaker in vitro phosphorylation of 4E-BP compared to autophosphorylation. PLoS One, 5: , e8730. |
[41] | Luzon-Toro B , Rubio de la Torre E , Delgado A , Perez-Tur J , & Hilfiker S ((2007) ) Mechanistic insight into the dominant mode of the Parkinson’s disease-associated G2019S LRRK2 mutation. Hum Mol Genet, 16: , 2031–2039. |
[42] | Martin I , Kim JW , Lee BD , Kang HC , Xu JC , Jia H , Stankowski J , Kim MS , Zhong J , Kumar M , Andrabi SA , Xiong Y , Dickson DW , Wszolek ZK , Pandey A , Dawson TM , & Dawson VL ((2014) ) Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson’s disease. Cell, 157: , 472–485. |
[43] | Matta S , Van Kolen K , da Cunha R , van den Bogaart G , Mandemakers W , Miskiewicz K , De Bock PJ , Morais VA , Vilain S , Haddad D , Delbroek L , Swerts J , Chavez-Gutierrez L , Esposito G , Daneels G , Karran E , Holt M , Gevaert K , Moechars DW , De Strooper B , & Verstreken P ((2012) ) LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron, 75: , 1008–1021. |
[44] | Reynolds A , Doggett EA , Riddle SM , Lebakken CS , & Nichols RJ ((2014) ) LRRK2 kinase activity and biology are not uniformly predicted by its autophosphorylation and cellular phosphorylation site status. Front Mol Neurosci, 7: , 54. |
[45] | Sheng Z , Zhang S , Bustos D , Kleinheinz T , Le Pichon CE , Dominguez SL , Solanoy HO , Drummond J , Zhang X , Ding X , Cai F , Song Q , Li X , Yue Z , van der Brug MP , Burdick DJ , Gunzner-Toste J , Chen H , Liu X , Estrada AA , Sweeney ZK , Scearce-Levie K , Moffat JG , Kirkpatrick DS , & Zhu H ((2012) ) Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci Transl Med, 4: , 164ra161. |
[46] | Silva RG , Geoghegan KF , Qiu X , & Aulabaugh A ((2014) ) A continuous and direct assay to monitor leucine-rich repeat kinase 2 activity. Anal Biochem, 450: , 63–69. |
[47] | Webber PJ , Smith AD , Sen S , Renfrow MB , Mobley JA , & West AB ((2011) ) Autophosphorylation in the leucine-rich repeat kinase 2 (LRRK2) GTPase domain modifies kinase and GTP-binding activities. J Mol Biol, 412: , 94–110. |
[48] | West AB , Moore DJ , Biskup S , Bugayenko A , Smith WW , Ross CA , Dawson VL , & Dawson TM ((2005) ) Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A, 102: , 16842–16847. |
[49] | West AB , Moore DJ , Choi C , Andrabi SA , Li X , Dikeman D , Biskup S , Zhang Z , Lim KL , Dawson VL , & Dawson TM ((2007) ) Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet, 16: , 223–232. |
[50] | Martin I , Kim JW , Dawson VL , & Dawson TM ((2014) ) LRRK2 pathobiology in Parkinson’s disease. J Neurochem, 131: , 554–565. |
[51] | Gloeckner CJ , Kinkl N , Schumacher A , Braun RJ , O’Neill E , Meitinger T , Kolch W , Prokisch H , & Ueffing M ((2006) ) The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet, 15: , 223–232. |
[52] | Kamikawaji S , Ito G , Sano T , & Iwatsubo T ((2013) ) Differential effects of familial parkinson mutations in LRRK2 revealed by a systematic analysis of autophosphorylation. Biochemistry, 52: , 6052–6062. |
[53] | Ray S , Bender S , Kang S , Lin R , Glicksman MA , & Liu M ((2014) ) The Parkinson disease-linked LRRK2 protein mutation I2020T stabilizes an active state conformation leading to increased kinase activity. J Biol Chem, 289: , 13042–13053. |
[54] | Delbroek L , Van Kolen K , Steegmans L , da Cunha R , Mandemakers W , Daneels G , De Bock PJ , Zhang J , Gevaert K , De Strooper B , Alessi DR , Verstreken P , & Moechars DW ((2013) ) Development of an enzyme-linked immunosorbent assay for detection of cellular and in vivo LRRK2 S935 phosphorylation. J Pharm Biomed Anal, 76: , 49–58. |
[55] | Trancikova A , Mamais A , Webber PJ , Stafa K , Tsika E , Glauser L , West AB , Bandopadhyay R , & Moore DJ ((2012) ) Phosphorylation of 4E-BP1 in the mammalian brain is not altered by LRRK2 expression or pathogenic mutations. PLoS One, 7: , e47784. |
[56] | Herzig MC , Kolly C , Persohn E , Theil D , Schweizer T , Hafner T , Stemmelen C , Troxler TJ , Schmid P , Danner S , Schnell CR , Mueller M , Kinzel B , Grevot A , Bolognani F , Stirn M , Kuhn RR , Kaupmann K , van der Putten PH , Rovelli G , & Shimshek DR ((2011) ) LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum Mol Genet, 20: , 4209–4223. |
[57] | Gehrke S , Imai Y , Sokol N , & Lu B ((2010) ) Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature, 466: , 637–641. |
[58] | Nikonova EV , Xiong Y , Tanis KQ , Dawson VL , Vogel RL , Finney EM , Stone DJ , Reynolds IJ , Kern JT , & Dawson TM ((2012) ) Transcriptional responses to loss or gain of function of the leucine-rich repeat kinase 2 (LRRK2) gene uncover biological processes modulated by LRRK2 activity. Hum Mol Genet, 21: , 163–174. |
[59] | Chartier-Harlin MC , Dachsel JC , Vilarino-Guell C , Lincoln SJ , Lepretre F , Hulihan MM , Kachergus J , Milnerwood AJ , Tapia L , Song MS , Le Rhun E , Mutez E , Larvor L , Duflot A , Vanbesien-Mailliot C , Kreisler A , Ross OA , Nishioka K , Soto-Ortolaza AI , Cobb SA , Melrose HL , Behrouz B , Keeling BH , Bacon JA , Hentati E , Williams L , Yanagiya A , Sonenberg N , Lockhart PJ , Zubair AC , Uitti RJ , Aasly JO , Krygowska-Wajs A , Opala G , Wszolek ZK , Frigerio R , Maraganore DM , Gosal D , Lynch T , Hutchinson M , Bentivoglio AR , Valente EM , Nichols WC , Pankratz N , Foroud T , Gibson RA , Hentati F , Dickson DW , Destee A , & Farrer MJ ((2011) ) Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet, 89: , 398–406. |
[60] | Nuytemans K , Bademci G , Inchausti V , Dressen A , Kinnamon DD , Mehta A , Wang L , Zuchner S , Beecham GW , Martin ER , Scott WK , & Vance JM ((2013) ) Whole exome sequencing of rare variants in EIF4G1 and VPS35 in Parkinson disease. Neurology, 80: , 982–989. |
[61] | Huttenlocher J , Kruger R , Capetian P , Lohmann K , Brockmann K , Csoti I , Klein C , Berg D , Gasser T , Bonin M , Riess O , & Bauer P ((2015) ) EIF4G1 is neither a strong nor a common risk factor for Parkinson’s disease: Evidence from large European cohorts. J Med Genet, 52: , 37–41. |
[62] | Nichols N , Bras JM , Hernandez DG , Jansen IE , Lesage S , Lubbe S , & Singleton AB , International Parkinson’s Disease Genomics C ((2015) ) EIF4G1 mutations do not cause Parkinson’s disease. Neurobiol Aging, 36: , 2444 e2441–2444 |
[63] | Siitonen A , Majounie E , Federoff M , Ding J , Majamaa K , & Singleton AB ((2013) ) Mutations in EIF4G1 are not a common cause of Parkinson’s disease. Eur J Neurol, 20: , e59. |
[64] | Dhungel N , Eleuteri S , Li LB , Kramer NJ , Chartron JW , Spencer B , Kosberg K , Fields JA , Stafa K , Adame A , Lashuel H , Frydman J , Shen K , Masliah E , & Gitler AD ((2015) ) Parkinson’s disease genes VPS35 and EIF4G1 interact genetically and converge on alpha-synuclein. Neuron, 85: , 76–87. |
[65] | Ross OA , Cook C , & Petrucelli L ((2015) ) Linking the VPS35 and EIF4G1 pathways in Parkinson’s disease. Neuron, 85: , 1–3. |
[66] | Beilina A , Rudenko IN , Kaganovich A , Civiero L , Chau H , Kalia SK , Kalia LV , Lobbestael E , Chia R , Ndukwe K , Ding J , Nalls MA , International Parkinson’s Disease Genomics C, North American Brain Expression C, Olszewski M , Hauser DN , Kumaran R , Lozano AM , Baekelandt V , Greene LE , Taymans JM , Greggio E , & Cookson MR ((2014) ) Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc Natl Acad Sci U S A, 111: , 2626–2631. |
[67] | MacLeod DA , Rhinn H , Kuwahara T , Zolin A , Di Paolo G , McCabe BD , Marder KS , Honig LS , Clark LN , Small SA , & Abeliovich A ((2013) ) RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron, 77: , 425–439. |
[68] | Tain LS , Mortiboys H , Tao RN , Ziviani E , Bandmann O , & Whitworth AJ ((2009) ) Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat Neurosci, 12: , 1129–1135. |
[69] | Liu S , & Lu B ((2010) ) Reduction of protein translation and activation of autophagy protect against PINK1 pathogenesis in Drosophila melanogaster. PLoS Genet, 6: , e1001237. |
[70] | Lipton JO , & Sahin M ((2014) ) The neurology of mTOR. Neuron, 84: , 275–291. |
[71] | Zanon A , Rakovic A , Blankenburg H , Doncheva NT , Schwienbacher C , Serafin A , Alexa A , Weichenberger CX , Albrecht M , Klein C , Hicks AA , Pramstaller PP , Domingues FS , & Pichler I ((2013) ) Profiling of Parkin-binding partners using tandem affinity purification. PLoS One, 8: , e78648. |
[72] | Corti O , Hampe C , Koutnikova H , Darios F , Jacquier S , Prigent A , Robinson JC , Pradier L , Ruberg M , Mirande M , Hirsch E , Rooney T , Fournier A , & Brice A ((2003) ) The p38 subunit of the aminoacyl-tRNA synthetase complex is a Parkin substrate: Linking protein biosynthesis and neurodegeneration. Hum Mol Genet, 12: , 1427–1437. |
[73] | Lee Y , Karuppagounder SS , Shin JH , Lee YI , Ko HS , Swing D , Jiang H , Kang SU , Lee BD , Kang HC , Kim D , Tessarollo L , Dawson VL , & Dawson TM ((2013) ) Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat Neurosci, 16: , 1392–1400. |
[74] | Gehrke S , Wu Z , Klinkenberg M , Sun Y , Auburger G , Guo S , & Lu B ((2015) ) PINK1 and Parkin control localized translation of respiratory chain component mRNAs on mitochondria outer membrane. Cell Metab, 21: , 95–108. |
[75] | Lin W , Wadlington NL , Chen L , Zhuang X , Brorson JR , & Kang UJ ((2014) ) Loss of PINK1 attenuates HIF-1alpha induction by preventing 4E-BP1-dependent switch in protein translation under hypoxia. J Neurosci, 34: , 3079–3089. |
[76] | Mayer IA , & Arteaga CL ((2015) ) The PI3K/AKT pathway as a target for cancer treatment. Annu Rev Med, doi: 10.1146/annurev-med-062913-051343 |
[77] | Nagakubo D , Taira T , Kitaura H , Ikeda M , Tamai K , Iguchi-Ariga SM , & Ariga H ((1997) ) DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem Biophys Res Commun, 231: , 509–513. |
[78] | Kim RH , Peters M , Jang Y , Shi W , Pintilie M , Fletcher GC , DeLuca C , Liepa J , Zhou L , Snow B , Binari RC , Manoukian AS , Bray MR , Liu FF , Tsao MS , & Mak TW ((2005) ) DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell, 7: , 263–273. |
[79] | van der Brug MP , Blackinton J , Chandran J , Hao LY , Lal A , Mazan-Mamczarz K , Martindale J , Xie C , Ahmad R , Thomas KJ , Beilina A , Gibbs JR , Ding J , Myers AJ , Zhan M , Cai H , Bonini NM , Gorospe M , & Cookson MR ((2008) ) RNA binding activity of the recessive parkinsonism protein DJ-1 supports involvement in multiple cellular pathways. Proc Natl Acad Sci U S A, 105: , 10244–10249. |
[80] | Gan-Or Z , Dion PA , & Rouleau GA ((2015) ) Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy, 11: , 1443–1457. |
[81] | Alegre-Abarrategui J , Christian H , Lufino MM , Mutihac R , Venda LL , Ansorge O , & Wade-Martins R ((2009) ) LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum Mol Genet, 18: , 4022–4034. |
[82] | Gomez-Suaga P , Luzon-Toro B , Churamani D , Zhang L , Bloor-Young D , Patel S , Woodman PG , Churchill GC , & Hilfiker S ((2012) ) Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum Mol Genet, 21: , 511–525. |
[83] | Schapansky J , Nardozzi JD , Felizia F , & LaVoie MJ ((2014) ) Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum Mol Genet, 23: , 4201–4214. |
[84] | Dodson MW , Zhang T , Jiang C , Chen S , & Guo M ((2012) ) Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum Mol Genet, 21: , 1350–1363. |
[85] | Higashi S , Moore DJ , Yamamoto R , Minegishi M , Sato K , Togo T , Katsuse O , Uchikado H , Furukawa Y , Hino H , Kosaka K , Emson PC , Wada K , Dawson VL , Dawson TM , Arai H , & Iseki E ((2009) ) Abnormal localization of leucine-rich repeat kinase 2 to the endosomal-lysosomal compartment in lewy body disease. J Neuropathol Exp Neurol, 68: , 994–1005. |
[86] | Orenstein SJ , Kuo SH , Tasset I , Arias E , Koga H , Fernandez-Carasa I , Cortes E , Honig LS , Dauer W , Consiglio A , Raya A , Sulzer D , & Cuervo AM ((2013) ) Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci, 16: , 394–406. |
[87] | Chan CS , Guzman JN , Ilijic E , Mercer JN , Rick C , Tkatch T , Meredith GE , & Surmeier DJ ((2007) ) ’Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature, 447: , 1081–1086. |
[88] | Guzman JN , Sanchez-Padilla J , Wokosin D , Kondapalli J , Ilijic E , Schumacker PT , & Surmeier DJ ((2010) ) Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature, 468: , 696–700. |
Figures and Tables
Figure
The impact of PD proteins on mRNA translation. DJ-1 inhibits PTEN, a negative regulator of PI3K/Akt signaling and also binds to certain mRNA transcripts in a manner that inhibits their translation. TORC1 activity may contribute to PINK1/Parkin pathology potentially through mRNA translation. Mutations in the translation initiation factor eIF4G1 likely cause PD. LRRK2 stimulates mRNA translation through interaction with the ribosome and phosphorylation of s15.
Table
Summary of PD genes linked to neuronal toxicity through mRNA translation
| Gene | PD locus | Mode of Inheritance | Relationship to mRNA Translation |
| LRRK2 | PARK8 | AD | Kinase-enhancing mutations stimulate bulk translation linked to toxicity |
| eIF4G1 | PARK18 | AD | Implicated by eIF4G1 being constituent of mRNA cap complex, effect of mutations unknown |
| PINK1 | PARK6 | AR | Translation suppression prevents pathology, pink1 may regulate translation of nuclear-encoded respiratory chain mRNAs |
| Parkin | PARK2 | AR | Translation suppression prevents pathology, pink1 may regulate translation of nuclear-encoded respiratory chain mRNAs |
| VPS35 | PARK17 | AD | Genetically interacts with eIF4G1, important in protein trafficking |
| RAB7L1 | PARK16 | ND | Genetically interacts with LRRK2, important in protein trafficking |
| DJ-1 | PARK7 | AR | Interacts with certain mRNAs, potentially to suppress their translation |
AD, autosomal dominant; AR, autosomal recessive; ND, not determined.


