
Tracking Parkinson’s: Study Design and Baseline Patient Data
Abstract
Background:
There is wide variation in the phenotypic expression of Parkinson’s disease (PD), which is driven by both genetic and epidemiological influences.
Objectives:
To define and explain variation in the clinical phenotype of PD, in relation to genotypic variation.
Methods:
Tracking Parkinson’s is a multicentre prospective longitudinal epidemiologic and biomarker study of PD. Patients attending specialist clinics in the United Kingdom with recent onset (<3.5 years) and young onset (diagnosed <50 years of age) PD were enrolled. Motor, non-motor and quality of life assessments were performed using validated scales. Cases are followed up 6 monthly up to 4.5 years for recent onset PD, and up to 1 year for young onset PD. We present here baseline clinical data from this large and demographically representative cohort.
Results:
2247 PD cases were recruited (1987 recent onset, 260 young onset). Recent onset cases had a mean (standard deviation, SD) age of 67.6 years (9.3) at study entry, 65.7% males, with disease duration 1.3 years (0.9), MDS-UPDRS 3 scores 22.9 (12.3), LEDD 295 mg/day (211) and PDQ-8 score 5.9 (4.8). Young onset cases were 53.5 years old (7.8) at study entry, 66.9% male, with disease duration 10.2 years (6.7), MDS-UPDRS 3 scores 27.4 (15.3), LEDD 926 mg/day (567) and PDQ-8 score 11.6 (6.1).
Conclusions:
We have established a large clinical PD cohort, consisting of young onset and recent onset cases, which is designed to evaluate variation in clinical expression, in relation to genetic influences, and which offers a platform for future imaging and biomarker research.
BACKGROUND
Parkinson’s disease (PD) is the second most common neurodegenerative disease affecting the elderly, the prevalence of which is projected to double by 2030, which will have significant implications on future healthcare delivery and economics [1, 2].
Our understanding of the pathogenesis of PD changed significantly with the discovery of alpha-synuclein aggregation in Lewy bodies and Lewy neurites, the neuropathological hallmarks of the disease, as a central mechanism in the underlying disease process [3]. From the initial genetic study linking a specific mutation in the gene coding for alpha-synuclein, SNCA, to a familial form of PD [4], a variety of rare genetic mutations including LRRK2, PARK2, and PINK-1 were subsequently linked to PD. Collectively, however, these Mendelian genes account for less than 10% of all PD cases in the general population [5]. More recently, large genome-wide association studies have collectively identified susceptibility variants at over 18 loci that increase risk for ‘idiopathic’ PD [5–11]. However, in common with other complex traits, the pathogenesis in the large majority of cases of PD is expected to be multifactorial, involving a combination of genetic and environmental risk factors [12].
Differences in the clinical phenotype between patients with PD linked to Mendelian genes, compared to sporadic cases, have been recently reviewed [13]. Detailed genotyping will be performed in the current study, while also examining the role of environmental influences.
Finding a serum biomarker for PD would be a major clinical advance, given known diagnostic error rates, but would have even greater research implications for early diagnosis and recording of disease progression. Our study is collecting data in a large cohort of PD patients to facilitate detailed genetic studies and as a resource for linked biomarker research. Here we present the study protocol and descriptive baseline data, as a background to subsequent analytical reports emerging from this study.
METHODS
General outline
The study is carried out in accordance with the Declaration of Helsinki [14] and is supported by research nurses from the dementia and neurodegenerative research network (DeNDRON), a division of the National Health Service National Institute of Health Research in the United Kingdom (UK). Grant funding is from Parkinson’s UK, the national patient care and research organization. The primary objective is to define and explain the variation in the clinicalphenotype of Parkinson’s disease. Secondary objectives are: (a) to relate the variation in the clinical phenotype of PD to genetic influences; (b) to support additional studies exploring genetic, serum and imaging biomarkers for the diagnosis, stratification and progression of PD.
Tracking Parkinson’s is a large prospective, observational, multicentre project. Patients were recruited with a clinical diagnosis of PD, corroborated by Queen Square Brain Bank criteria [15] and supported by neuroimaging performed when the diagnosis was not firmly established clinically. Both drug-naïve and treated patients, aged 18 to 90 years were eligible. Young onset cases were diagnosed at or below age 50 years, and recent onset cases were diagnosed within the preceding 3.5 years. Baseline recruitment is complete and patients are currently engaged in 6 monthly follow up. Recruitment of first degree relatives, to a target of 840 unaffected siblings, is underway. All participants have LRRK2 and GBA mutation carrier status assessed with young onset cases also screened for PARK2 and PINK1 mutations. Exclusion criteria were severe comorbid illness e.g severe COPD or symptomatic heart failure that would not allow patient participation in clinic visits, other degenerative forms of parkinsonism e.g. progressive supranuclear palsy, or, parkinsonism attributable to significant cerebrovascular disease eg. lower body parkinsonism with prominent vascular history (patients with ‘incidental’ small vessel disease on brain imaging were not excluded). Patients with drug-induced parkinsonism were excluded, but drug-unmasked PD was allowed if justified by abnormal functional dopaminergic imaging with dopamine transporter (DaT) single photon emission computed tomography (SPECT) or fluorodopa (18F) positron emission tomography (F-DOPA PET).
72 sites in the UK providing secondary care treatment for PD patients as part of the UK National Health Service (NHS) (and in selected sites, their linked academic institutions) are participating, with multicentre ethics committee and local research and development department approvals.
Data handling
Data capture by the local clinical and research team allowed direct entry to a secure anonymized web-based electronic data capture system, backed by a paper case record form. Data collection for clinical assessments followed the standards of the Clinical Data Interchange Standards Consortium (CDISC) PD user guide, which incorporates common data elements developed by the US National Institute of Neurological Disorders and Stroke. Missing and erroneous data points were identified and pursued by central study coordinators. Statistical analysis was performed in Bristol. All genetic data are generated, analyzed and stored at the central laboratory in Cardiff.
Clinical assessments
Clinical assessments were made in out-patient clinics, using standardized and validated scales, to document the motor and non-motor features, quality of life and drug responsiveness of the enrolled subjects. Home visits were performed in a few remote settings. Study follow up visits were 6-monthly with more detailed observations at 0, 18, 36 and 54 months (Fig. 1). Levodopa equivalent daily dose (LEDD) was calculated using established formulas for dose equivalence [16, 17]. Orthostatic hypotension was defined as a systolic drop ≥20 mmHg or diastolic drop ≥10 mmHg. Montreal cognitive assessment (MoCA) was adjusted for education years and mild cognitive impairment defined as a score 21–25, and dementia defined as a score <21) [18]. Non-motor symptom severity (NMSS) was graded according to predefined severity categories: mild 1–20, mod 21–40, severe 41–70 and >70 very severe [19]. Depression and anxiety were identified from scores above 6 in the Leeds hospital anxiety and depression scale (LADS) [20]. Epworth sleep scale (ESS) was used to define excessive daytime sleep when the score exceeded 9 [21]. Rapid eye movement (REM) sleep behaviour disorder (RBD) was defined from a score above 4 [22]. Constipation was defined as laxative use or less than one bowel motion per day. Impaired olfaction was defined as an UPSIT (University of Pennsylvania smellidentification test) <24/40 for age for age 60 years and older, and <29/40 for under 60 years old [23].
Genetic analysis
Blood samples were collected at all sites at study entry: an ethylene diamine tetraacetic acid (EDTA) sample for DNA extraction and an acid citrate dextrose (ACD) sample for cryopreservation of peripheral blood lymphocytes at the European Centre for Cell Cultures (ECACC) in Wiltshire, England to generate a long term backup resource. All DNA samples are stored for analysis and distribution at the study’s centralized laboratory at Cardiff University, Wales.
In all PD patients the G2019S mutation at LRRK2 is genotyped using a Kompetitive Allele Specific Polymerase (KASP) assay (LGC Genomic solutions) and the GBA mutation carrier status established by DNA sequencing of all coding exons. The genes PARK2 and PINK1 are screened for mutations using DNA sequencing and multiplex ligation-dependent probe amplification (MLPA) (MRC Holland) in all young onset PD patients. All DNA samples will also be genotyped using the Illumina Human Core Exome array, which has been supplemented with custom content. This will allow for the analysis of approximately 250,000 common single nucleotide polymorphisms (SNPs) and 250,000 rare variants, plus over 27,000 custom variants selected due to their previous implication in a range of neurodegenerative, neurological, and psychiatric disorders.
Proteomic analysis
Serum samples are stored in 6 aliquots at study entry only in young onset cases; every 18 months in recent onset patients; and every 3 years in siblings of patients. Storage is at –80° centigrade. A proteomic biomarker research program is coordinated in Oxford, England, involving samples from the current study and other ongoing UK PD cohort studies.
Statistical analysis
Sample sizes were calculated pragmatically using known UK incidence rates and NHS clinic activity levels, but sufficient to allow prognostic modelling involving random splits of the samples into training and validation cohorts. Standard statistical methods (survival curves and Cox proportional hazard models) and more complex multivariable models such as multi-level, latent class and/or growth curve models will be used to examine for heterogeneity in the presenting features and natural history of PD. Collaboration with other linked cohort studies, such as the Oxford Parkinson Disease Centre (OPDC) Discovery cohort, has been established and will be used for replication of findings for external validation [18].
RESULTS
Demographic characteristics
The average age of recent onset cases at diagnosis was 66.3 years, with mean disease duration from diagnosis of 1.3 years at the time of recruitment. 43.9% of these were recruited within 1 year of diagnosis, 28.5% between 1 and 2 years and 27.6% between 2 and 3.5 years.
The average age of young onset cases at diagnosis was 43.3 years, with a mean disease duration from diagnosis of 10.2 years at the time of recruitment. In both groups, males outnumbered females by approximately 2:1. Young onset cases, as expected, had a longer disease duration from symptom onset to diagnosis (2.2 years, SD 3.4) compared to 1.8 years (SD 2.9) in recent onset patients. Additional demographic data are in Table 1 and Supplementary Table 1.
Comorbid disease
Recent onset cases had more than double the comorbid cardiovascular disease, cerebrovascular disease and vascular risk factors compared to young onset cases (Supplementary Table 2). With the exception of lung cancer, cancer diagnoses overall were also more prevalent in the recent onset group. The rates of breast cancer though were similar between the groups.
Family history
In young onset cases, 25.5% had a family history of PD, which was more frequent than in recent onset cases (19.9% ) (p = 0.038). A family history of dementia did not differ significantly between recent and young onset cases (p = 0.076), and a family history of stroke was similar across groups (Table 2).
Neuroimaging
Data relating to the mode and results of neuroimaging undertaken as part of the patient’s routine clinical care prior to recruitment were extracted. The proportion of patients with brain computed tomography (CT), magnetic resonance imaging (MRI), FP-CIT SPECT and F-DOPA PET imaging are detailed in Supplementary Table 3.
Clinical observations
Baseline lying/standing pulse, and blood pressure in cases are detailed in Supplementary Table 4. Orthostatic hypotension (Table 4) was more frequent at baseline in recent onset cases (17.2 vs. 11.3% ). BMI (Table 1) in both groups was also slightly above the normal range (defined as 18.5–24.9) [24].
Taking into account the longer disease duration of young onset cases, the following features are noted. Young onset cases had higher MDS-UPDRS Part 3 scores (Table 3), as well as total MDS-UPDRS scores at study entry (mean 63.6) compared to recent onset cases (mean 42.7). Despite these differences both groups had a median Hoehn and Yahr stage of 2. In recent onset cases, 36.8% had mild cognitive impairment (MCI) and 9.8% dementia at study entry. Young onset cases had a similar frequency of MCI (36.2% ) to the recent onset cases but a lower frequency of dementia (8.3% ). The spectrum and severity of both motor features and motor complications, disability andcognitive impairment in recent and young onset cases, according to the clinical impression of severity index for Parkinson’s disease, is shown in Fig. 2.
Results of patient scored questionnaires at study entry assessing non-motor symptoms are in Tables 4 and 5. Overall non-motor symptoms were common in both groups, but young onset patients reported a greater frequency and/or severity of these at study entry, including sleep disturbance, depression, anxiety, autonomic symptoms (except constipation and laxative use) and impulsive-compulsive behaviours, with perceived poorer quality of life compared to recent onset patients, while again noting their longer disease duration.
Medication use
Over 90% of patients were prescribed antiparkinson medication at study entry (Fig. 3). The mean LEDD was 295 mg/day in recent onset cases and 926 mg/day in young onset cases, reflecting disease duration.
Withdrawal rates
Study retention has been successful to date (Table 6). Fourteen cases were excluded from the recent onset cohort following diagnostic revision: 4 were later diagnosed with PSP, one with multiple sclerosis, one with MSA, one with essential or dystonic tremor, one with corticobasal degeneration, one had a normal FP-CIT SPECT scan, and 5 without a clear diagnosis. One case was excluded from the young onset cohort following a subsequent normal FP-CIT SPECT scan.
DISCUSSION
Young onset cases
Impulsivity data from such a large cohort of young onset cases has not been reported. The presence of one or more impulsive/compulsive behaviours in two-thirds of our young onset cases considerably exceeds the prevalence rate found in our recent onset cases, and prior reports [28]. This may relate to the known greater risk of impulse control disorders (ICD) in young onset PD [29] and frequent use of dopamine agonists. The multiplicity of involved domains from the ICD questionnaire was striking in these young onset cases.
Family history of PD was more likely in young onset than recent onset cases, consistent with known higher rates of genetic mutations in young onset disease [30] but the difference between groups was not striking. One in 4 young onset patients had at least one other family member with PD, compared to less than 1 in 5 recent onset cases. This familial association of PD also applied when considering cases where more than 1 family member was affected by PD, matching prior reports [31].
We did not identify an increased likelihood of a dementia diagnosis in relatives of young onset PD cases, compared to our recent onset cases, which differs from earlier reports [32, 33]. Our definition of dementia in relatives depended on patient reporting, without corroboration from their medical records, matching the methods of one of those studies [33]. The age cut-off for defining young onset disease in those studies was however pragmatically set at a rather older age (60 or 66 years) representing the youngest quintile [32] or tertile [33] of their series, which may have influenced their findings.
RBD is proposed as a sensitive pre-motor marker of PD [34] and can be screened for by using the RBD screening questionnaire (RBDSQ) [22]. RBDSQ scores in our young onset cases were slightly worse than recent onset cases, which is contrary to another study that found a higher prevalence of RBD in older patients [35]. Further, the Oxford Discovery study noted that patients with RBD (47.2% of cases) had a greater prevalence of non-motor features [36]. RBD at baseline significantly correlated with an increase in total UPDRS scores over time in another study [37]. RBD therefore appears to be a marker of more advanced neurodegeneration, being described in patients with more motor complications, a higher rate of falls, the emergence of cognitive problems and psychotic symptoms, and it may therefore be a predictor of entering a more advanced stage of disease [38]. The longer disease duration in our young onset cases may therefore account for our worse RBDSQ scores.
While our young onset patients reported greater diagnostic delay (around 4 months longer than recent onset cases), recall bias affected by their longer disease duration may have contributed. Somewhat shorter delays are reported, being 15 months in young onset patients (aged <46) [39], and under 2 years in most studies [40, 41]. A timely diagnosis of PD is important to initiate necessary treatment and relay prognostic information, but has greater significance for the early implementation (in clinical trials) of potential disease modifying treatments.
Recent onset cases
There is a potential two-way association between PD and stroke, in both of which oxidative stress may be pathogenic [42]. In a prospective study, a 1.5 to 2 fold increased risk of developing PD was associated with previous stroke, with a similarly increased relativerisk for a first time diagnosis of ischemic stroke in patients diagnosed with PD [43]. A high prevalence of vascular disease, and risk factors for this, were present particularly in our recent onset cases where combining high cholesterol, hypertension, and diabetes gave a ‘vascular risk’ rate of around half of cases. Vascular risk factors are of interest as several are potentially modifiable. The presence of diabetes as a risk factor for developing PD is controversial, with one case-control study showing no increased risk [44], but a cohort study reporting almost doubling of the risk [45]. The presence of vascular risk factors may modify disease expression in PD; elevated cardiovascular risk scores were an independent predictor of higher axial motor impairments in PD [46]. Hypertension, which was present in one-third of our recent onset cases, correlated with impaired cognitive performance (executive function and verbal memory) in PD in another study [47]. Continued observation of the relationship between vascular and parkinsonian features is planned in the follow-up phase.
Out of four cancer types recorded, the highest rates were for breast and prostate cancer in our recent onset cases, with no cases of prostate cancer in our young onset cases. A recent meta-analysis found no overall increased risk of breast or prostate cancer in PD patients compared to the general population [48]. The overall higher cancer prevalence in recent onset cases therefore most likely reflects the known increased incidence rates of most cancers with age [49].
The findings that 13% of recent onset cases had a mixed motor phenotype is very similar to prior observations [50]. While tremor was recorded at onset in approximately three quarters of cases in each group, PIGD was more likely in young onset cases, probably due to evolution of motor subtypes [51], which is important when evaluating the association of early clinical features and genetic and biomarker traits. For example, dementia is more likely in cases presenting with PIGD, compared to tremor dominant or indeterminate subtypes, and is also more likely in cases evolving to a PIGD subtype [52, 53]. Patients with a PIGD motor subtype also have faster disease progression [54] and worse health related quality of life [55].
The proportion of our recent onset cases with mild cognitive impairment (MCI, 36.8% ) and dementia (9.8% ) is similar to the Oxford Discovery cohort, at a similar disease duration (MCI 40.5% , dementia 11.7% ) using similar methods [18]. These findings are consistent with the ICICLE-PD study which found MCI in 42.5% at baseline, in an incident cohort, using Movement Disorder Society level II criteria [56] that recommend cut-off scores of 1.5 SD below normal values [57]. Both results are higher than the Parkinson’s progression marker initiative (PPMI) study (average disease duration 6.5 months, untreated) [58] where 21.5% had MCI at baseline, 34.2% at 1 year, and 35.5% at 2 years, and may reflect different selection criteria.
Scores of daytime somnolence were similar to those reported in cohorts of a similar age (but generally with a longer disease duration), compared to our recent onset cases. Worse ESS scores correlate with more depression, higher disease severity, and higher doses of dopaminergic agents [59]. A correlation between daytime somnolence and cognitive impairment is also reported [60]. Our PD sleep scale (PDSS) scores in young onset cases (mean 91.8, SD 28.7) were slightly worse than those previously reported (mean 120.9, SD 20.0) [61]. While reports of a positive correlation between severity of sleep disturbance and motor severity are not uniform [62] some report such an association [26, 63]. The reported prevalence of RLS in PD is variable, ranging between 5.5 and 27% in European cohort studies [64], against which our rate was 28.3% in the young onset cases and 23.6% in recent onset cases, is at the higher end of this range.
Our SCOPA-AUT results lie between results found in newly diagnosed, drug naive patients [65] and those with a longer disease duration in cross-sectional studies [66, 67]. As well as disease duration, the prevalence and severity of autonomic symptoms have been associated with more severe impairments in motor symptoms, cognition, depression, sleep disturbance, psychiatric complications, and the prescription of dopaminergic drugs [68]. These factors may account for the higher scores seen in our young onset patients in all individual components of the SCOPA-AUT, except sexual function where recent onset patients reported a greater prevalence of such symptoms.
In keeping with previous reports, a proportion of our recent onset patients exhibited orthostatic hypotension (OH) early in the course of disease, as a marker of autonomic instability. Baseline autonomic dysfunction may serve as a predictor of cognitive impairment, with a systolic drop of >10 mmHg at baseline being associated with 7-fold increased risk of dementia at 4.4 years in a prospective study [69].
Constipation is of particular interest in PD, as one marker of autonomic involvement that may predate motor diagnosis [70, 71]. However around a quarter of our recent onset cases had 2 or more bowel motions per day (without laxative use), indicating that constipation is not a universal feature, even after the diagnosis of PD. Our results are consistent with the finding that the majority of PD patients have only mild colorectal symptoms [72].
Non-motor symptoms such as constipation, depression, restless legs, particularly when they cluster together are now well recognised as early clinical markers of PD but given that the majority of our patients reported motor symptoms as a presenting feature, the inclusion of motor assessments in studies designed to identify ‘at risk’ subjects would be appropriate.
Limitations
One limitation of the study design is that comparison of recent onset and young onset groups will inevitably show differences because of different disease duration. We included a young onset population primarily to enrich the proportion with known genetic mutations, given the involvement of siblings and the genetic focus of the study. While statistically correcting for age, gender and disease duration would aid some comparisons, it has limited capacity to correct for evolution of disease characteristics over time.
CONCLUSION
In conclusion, we present the baseline data of a large clinical research network which is actively following cases in a combined clinical-laboratory program, evaluating variation in the clinical expression of PD which will be studied in relation to genetic influences. This offers a platform for serum and imaging biomarker research. We hope that the scale and linkage of this research program will help to understand the pathogenesis of PD, and identify new pathways leading towards preventive treatments. The longitudinal follow-up of our PD cases and siblings is a key component, and will be the subject of further reports.
CONFLICTS OF INTEREST
N Malek, DMA Swallow, KA Grosset, MA Lawton, AC Lehn, Y Ben-Shlomo: No conflicts of interest.
SL Marrinan has received an honorarium from Britannia and research grant support from Parkinson’s UK, GSK, and the Michael J Fox Foundation.
N Bajaj has received payment for advisory board attendance from UCB, Teva Lundbeck, Britannia, GSK, Boehringer, and honoraria from UCB Pharma, GE Healthcare, Lily Pharma, Medtronic. He has received research grant support from GE Healthcare.
RA Barker has received grants from Parkinson’s UK, NIHR, Cure Parkinson’s Trust, Evelyn Trust, Rosetrees Trust, MRC and EU along with payment for advisory board attendance from Oxford Biomedica and LCT, and honoraria from Wiley and Springer.
DJ Burn has received grants from NIHR, Wellcome Trust, GlaxoSmithKline Ltd, Parkinson’s UK, and Michael J Fox Foundation. He has acted as consultant for GSK.
T Foltynie has received payment for advisory board meetings for Abbvie and Oxford Biomedica, and honoraria for presentations at meetings sponsored by Medtronic, St Jude Medical, Britannia and Teva pharmaceuticals.
J Hardy has received honoraria from Eisai, and grant support from MRC/Wellcome, Parkinson’s UK, and the Michael J Fox Foundation.
DG Grosset has received payment for advisory board attendance from AbbVie, and honoraria from UCB Pharma, GE Healthcare, and Civitas Inc.
AUTHORS’ CONTRIBUTIONS
NM assisted in data collection, statistical analysis, and interpreted the data and drafted the manuscript, DMAS interpreted the data and drafted the manuscript, KAG participated in study design, coordinating data collection, interpretation, and critical appraisal of manuscript, MAL performed data coordination and interpretation and statistical analysis, CB coordinated and performed DNA sample handling and testing, SLM, ACL, NB, RAB, DJB, TF, HRM, and NW participated in the design of the study, data collection, interpretation and critical appraisal of the manuscript. YBS participated in the design of the study, data interpretation, and critical appraisal of the manuscript. JH participated in the design of the study and critical appraisal of the manuscript. NMW participated in design of the study and DNA sample testing. DGG participated in study design, data collection and interpretation, manuscript preparation and editing.
ACKNOWLEDGMENTS
The study group was (principal investigators):Grosset D, Bajaj N, Sugathapala L, Burn D, GrahamA, Bathgate D, Bland R, Worth P, Mamutse G, AmarK, Walker R, Raw J, Carroll C, Clarke CE, HemsleyZ, Fackrell R, Roberts H, Guptha S, Nath U, BarkerR, Counsell C, Sheridan R, Silverdale M, Sharma J,Piccini P, Hindle J, Arianayagam S, Ellis S, Ward T,Lennox G, Carson M, Sveinbjornsdottir S, Boothman B, Paviour D, Misbahuddin A, Schrag A, Athey R, Sarda P, Steiger M, Dhakam Z, Kock N, Molloy S, O’Neill M, Stern J, Capps E, Critchley P, Foltynie T, George J, Bandmann O, Harper G, Andrews T,Woodward W, Whone A, Borland C, Wilson M,Adenwala Y, Tidswell P, Chaudhuri R, Watt A, Church A, Morris H, Hu M, Kamath S, Adler B, Barber S, De Pablo-Fernandez E, Sophia R; and (research nurses and healthcare professionals): Agarwal V, Alderton L, Amor K, Andrew A, Arif S, Bennett J, Birchall K, Birt J, Blachford K, Brooke J, Brown A, Brown P, Brugaletta C, Bryden N, Burrows M, Butler S, Cable S, Callaghan R, Canovas L, Carey G, Cattarall L, Clipsham K, Colwell W, Cowen Z, Cox C, Craw S, Creaser-Smith A, Croucher Y, Daniel S, De Pietro A, Dellafera D, Dodds S, Donaldson A, Donaldson D, Dougherty A, Downes C, Dube S, Dwornik W, Edwards C, Ekins E, Fernandes R, Foale C, Forbes H, Ford S, Frost J, Fuller T, Gallagher L, Gentle R,Gethin L, Gilford J, Gray C, Gunter E, Hall S,Hamilton C, Hare M, Henderson A, Hetherington V, Higgins R, Higham A, Hill L, Hodgson K, Humphries R, Hurlstone S, Hursey A, Inniss R, James R, JohnsonE, Joyce R, Kefalopoulou Z, Kelly M, Korley M,Lehn A, Levy S, Lithgo K, Long C, Lyle A, Lynn H, MacKinnon L, Makahamadze C, Mahan T, Marks N, Marrinan S, Marshall M, Martin-Forbes P, Massey I,McBrearty C, McEntee J, McNichol A, Mills D,Morgan S, Mullan D, Murphy T, Newman J,O’Connell H, O’Donnell A, O’Donnell D, O’ReillyC, Olanrewaju O, Oughton E, Owen C, Painter S,Palfreeman S, Paterson P, Perkins L, Pilcher A,Powell K, Price C, Rachman P, Renton L, Rickett J, Rizos A, Roberts T, Roche M, Roopun R, Roussakis A, Rowland R, Saunders G, Sequeira C, Shields S,Simmons D, Snape C, Stickley J, Strong L, SunderlandC, Sutherland S, Temple N, Thomson E, Trimmer M, Tuazon J, Tyrrell E, Visentin E, Vandor C, Vernon N, Verstraelen N, Visick M, Walsh H, Walsh S, Ward K, Watson A, Watt A, Whelan E, Williams J, Williams M, Williams S, Wilson B, Witherington K, Woodcock R, Wyatt L.
The research was funded by Parkinson’s UK and supported by the National Institute for Health Research (NIHR) DeNDRoN network, the NIHR Newcastle Biomedical Research Unit based at Newcastle upon Tyne Hospitals NHS Foundation Trust and Newcastle University, and the NIHR funded Biomedical Research Centre in Cambridge. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Appendices
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-150662.
REFERENCES
1 | Nussbaum RL, Ellis CE (2003) Alzheimer’s disease and Parkinson’s disease N Engl J Med 348: 1356 1364 |
2 | Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderowf A, Tanner CM (2007) Projected number of people with Parkinsondisease in the most populous nations, 2005 through 2030 Neurology 68: 384 386 |
3 | Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewybodies Nature 388: 839 840 |
4 | Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease Science 276: 2045 2047 |
5 | Lesage S, Brice A (2012) Role of mendelian genes in “sporadic” Parkinson’s disease Parkinsonism Relat Disord 18: Suppl 1 S66 S70 |
6 | Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, Saad M, Simon-Sanchez J, Schulte C, Lesage S, Sveinbjornsdottir S, Stefansson K, Martinez M, Hardy J, Heutink P, Brice A, Gasser T, Singleton AB, Wood NW (2011) Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: Ameta-analysis of genome-wide association studies Lancet 377: 641 649 |
7 | Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M, Schulte C, Keller MF, Arepalli S, Letson C, Edsall C, Stefansson H, Liu X, Pliner H, LeeJH , Cheng R, Ikram MA, Ioannidis JP, Hadjigeorgiou GM, Bis JC, Martinez M, Perlmutter JS, Goate A, Marder K, Fiske B, Sutherland M, Xiromerisiou G, Myers RH, Clark LN, Stefansson K, Hardy JA, Heutink P, Chen H, Wood NW, Houlden H, Payami H, Brice A, Scott WK, Gasser T, Bertram L, Eriksson N, Foroud T, Singleton AB (2014) Large-scale meta-analysis of genome-wide association dataidentifies six new risk loci for Parkinson’s disease NatGenet 46: 989 993 |
8 | Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Kruger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T (2009) Genome-wide association study reveals genetic risk underlying Parkinson’s disease Nat Genet 41: 1308 1312 |
9 | Liu X, Cheng R, Verbitsky M, Kisselev S, Browne A, Mejia-Sanatana H, Louis ED, Cote LJ, Andrews H, Waters C, Ford B, Frucht S, Fahn S, Marder K, Clark LN, Lee JH (2011) Genome-wide association study identifies candidategenes for Parkinson’s disease in an Ashkenazi Jewish population BMC Med Genet 12: 104 |
10 | Pankratz N, Nichols WC, Uniacke SK, Halter C, Murrell J, Rudolph A, Shults CW, Conneally PM, Foroud T (2003) Genome-wide linkage analysis and evidence of gene-by-gene interactions in a sample of 362 multiplex Parkinsondisease families Hum Mol Genet 12: 2599 2608 |
11 | Pankratz N, Beecham GW, DeStefano AL, Dawson TM, Doheny KF, Factor SA, Hamza TH, Hung AY, Hyman BT, Ivinson AJ, Krainc D, Latourelle JC, Clark LN, Marder K, Martin ER, Mayeux R, Ross OA, Scherzer CR, Simon DK, Tanner C, Vance JM, Wszolek ZK, Zabetian CP, Myers RH, Payami H, Scott WK, Foroud T (2012) Meta-analysis of Parkinson’s disease: Identification of a novel locus, RIT2 Ann Neurol 71: 370 384 |
12 | Riess O, Kruger R (1999) Parkinson’s disease–a multifactorial neurodegenerative disorder J Neural Transm Suppl 56: 113 125 |
13 | Puschmann A (2013) Monogenic Parkinson’s disease and parkinsonism: Clinical phenotypes and frequencies of known mutations Parkinsonism Relat Disord 19: 407 415 |
14 | Declaration of Helsinki (1967) Recommendations guiding doctors in clinical research. Adopted by the World MedicalAssociation in 1964 Wis Med J 66: 25 26 |
15 | Hughes AJ, Daniel SE, Lees AJ (2001) Improved accuracy of clinical diagnosis of Lewy body Parkinson’s disease Neurology 57: 1497 1499 |
16 | Tomlinson CL, Stowe R, Patel S, Rick C, Gray R, Clarke CE (2010) Systematic review of levodopa dose equivalency reporting in Parkinson’s disease Mov Disord 25: 2649 2653 |
17 | Brennan KA, Genever RW (2010) Managing Parkinson’s disease during surgery BMJ 341: c5718 |
18 | Hu MT, Szewczyk-Królikowski K, Tomlinson P, Nithi K, Rolinski M, Murray C, Talbot K, Ebmeier KP, Mackay CE, Ben-Shlomo Y (2014) Predictors of cognitive impairment in an early stage Parkinson’s disease cohort Mov Disord 29: 3 351 359 |
19 | Ray Chaudhuri K, Rojo JM, Schapira AH, Brooks DJ, Stocchi F, Odin P, Antonini A, Brown RJ, Martinez-Martin P (2013) Aproposal for a comprehensive grading of Parkinson’s disease severity combining motor and non-motor assessments: Meeting anunmet need PLoS One 8: e57221 |
20 | Snaith RP, Bridge GW, Hamilton M (1976) The Leeds scales for the self-assessment of anxiety and depression Br J Psychiatry 128: 156 165 |
21 | Johns MW (1991) A new method for measuring daytime sleepiness: The Epworth sleepiness scale Sleep 14: 540 545 |
22 | Stiasny-Kolster K, Mayer G, Schafer S, Moller JC, Heinzel-Gutenbrunner M, Oertel WH (2007) The REM sleep behavior disorder screening questionnaire–a new diagnostic instrument Mov Disord 22: 2386 2393 |
23 | Silveira-Moriyama L, Petrie A, Williams DR, Evans A, Katzenschlager R, Barbosa ER, Lees AJ (2009) The use of a color coded probability scale to interpret smell tests in suspected parkinsonism Mov Disord 24: 1144 1153 |
24 | WHOGlobal Database on Body Mass Index. http://apps.who.int/bmi/index.jsp?introPage=intro_3.html. Accessed on 04/05/2015 |
25 | Weintraub D, Papay K, Siderowf A (2013) Screening for impulse control symptoms in patients with de novo Parkinson disease: A case-control study Neurology 80: 176 180 |
26 | Chaudhuri KR, Pal S, DiMarco A, Whately-Smith C, Bridgman K, Mathew R, Pezzela FR, Forbes A, Hogl B, Trenkwalder C (2002) The Parkinson’s disease sleep scale: A new instrument forassessing sleep and nocturnal disability in Parkinson’s disease J Neurol Neurosurg Psychiatry 73: 629 635 |
27 | Visser M, Marinus J, Stiggelbout AM, Van Hilten JJ (2004) Assessment of autonomic dysfunction in Parkinson’s disease: The SCOPA-AUT Mov Disord 19: 1306 1312 |
28 | Callesen MB, Weintraub D, Damholdt MF, Moller A (2014) Impulsive and compulsive behaviors among Danish patients with Parkinson’s disease: Prevalence, depression, and personality Parkinsonism Relat Disord 20: 22 26 |
29 | Weintraub D, David AS, Evans AH, Grant JE, Stacy M (2015) Clinical spectrum of impulse control disorders in Parkinson’s disease Mov Disord 30: 121 127 |
30 | Alcalay RN, Caccappolo E, Mejia-Santana H, Tang MX, Rosado L, Ross BM, Verbitsky M, Kisselev S, Louis ED, Comella C, Colcher A, Jennings D, Nance MA, Bressman SB, Scott WK, Tanner C, Mickel S, Andrews H, Waters C, Fahn S, Cote L, Frucht S, Ford B, Rezak M, Novak K, Friedman JH, Pfeiffer R, Marsh L, Hiner B, Siderowf A, Ottman R, Marder K, Clark LN (2010) Frequency ofknown mutations in early-onset Parkinson disease: Implication forgenetic counseling: The consortium on risk for early onset Parkinson disease study Arch Neurol 67: 1116 1122 |
31 | Elbaz A, Grigoletto F, Baldereschi M, Breteler MM, Manubens-Bertran JM, Lopez-Pousa S, Dartigues JF, Alperovitch A, Tzourio C, Rocca WA (1999) Familial aggregationof Parkinson’s disease: A population-based case-control study in Europe. EUROPARKINSON Study Group Neurology 52: 1876 1882 |
32 | Rocca WA, Bower JH, Ahlskog JE, Elbaz A, Grossardt BR, McDonnell SK, Schaid DJ, Maraganore DM (2007) Risk of cognitive impairment or dementia in relatives of patients with Parkinson disease Arch Neurol 64: 1458 1464 |
33 | Costello S, Bordelon Y, Bronstein J, Ritz B (2010) Familial associations of Alzheimer disease and essential tremor with Parkinson disease Eur J Neurol 17: 871 878 |
34 | Schenck CH, Boeve BF, Mahowald MW (2013) Delayed emergence of a parkinsonian disorder or dementia in 81% of older men initially diagnosed with idiopathic rapid eye movement sleep behavior disorder: A 16-year update on a previously reported series Sleep Med 14: 744 748 |
35 | Mahale R, Yadav R, Pal PK (2014) Rapid eye movement sleep behaviour disorder in young- and older-onset Parkinson disease: A questionnaire-based study Sleep Med 15: 642 646 |
36 | Rolinski M, Szewczyk-Krolikowski K, Tomlinson PR, Nithi K, Talbot K, Ben-Shlomo Y, Hu MT (2014) REM sleep behaviour disorder isassociated with worse quality of life and other non-motor featuresin early Parkinson’s disease J Neurol Neurosurg Psychiatry 85: 560 566 |
37 | Bugalho P, Viana-Baptista M (2013) REM sleep behavior disorder and motor dysfunction in Parkinson’s disease–a longitudinal study Parkinsonism Relat Disord 19: 1084 1087 |
38 | Sixel-Doring F, Trautmann E, Mollenhauer B, Trenkwalder C (2011) Associated factors for REM sleep behavior disorder in Parkinson disease Neurology 77: 1048 1054 |
39 | Rana AQ, Siddiqui I, Yousuf MS (2012) Challenges in diagnosis of young onset Parkinson’s disease J Neurol Sci 323: 113 116 |
40 | Breen DP, Evans JR, Farrell K, Brayne C, Barker RA (2013) Determinants of delayed diagnosis in Parkinson’s disease J Neurol 260: 1978 1981 |
41 | Saunders-Pullman R, Wang C, Stanley K, Bressman SB (2011) Diagnosis and referral delay in women with Parkinson’s disease Gend Med 8: 209 217 |
42 | Ryu H, Lee J, Olofsson BA, Mwidau A, Dedeoglu A, Escudero M, Flemington E, Azizkhan-Clifford J, Ferrante RJ, Ratan RR (2003) Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway Proc Natl Acad Sci U S A 100: 4281 4286 |
43 | Becker C, Jick SS, Meier CR (2010) Risk of stroke in patients with idiopathic Parkinson disease Parkinsonism Relat Disord 16: 31 35 |
44 | Lu L, Fu DL, Li HQ, Liu AJ, Li JH, Zheng GQ (2014) Diabetes and risk of Parkinson’s disease: An updated meta-analysis of case-control studies PLoS One 9: e85781 |
45 | Hu G, Jousilahti P, Bidel S, Antikainen R, Tuomilehto J (2007) Type 2 diabetes and the risk of Parkinson’s disease Diabetes Care 30: 842 847 |
46 | Kotagal V, Albin RL, Muller ML, Koeppe RA, Frey KA, Bohnen NI (2014) Modifiable cardiovascular risk factors and axial motor impairments in Parkinson disease Neurology 82: 1514 1520 |
47 | Jones JD, Jacobson C, Murphy M, Price C, Okun MS, Bowers D (2014) Influence of hypertension on neurocognitive domains in nondemented Parkinson’s disease patients Parkinsons Dis 2014: 507529 |
48 | Wang T (2014) The link between Parkinson’s disease and breast and prostate cancers: A meta-analysis. Int J Neurosci, 2014 Dec 18. [Epub ahead of print] |
49 | Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics CA Cancer J Clin 61: 69 90 |
50 | Jankovic J, McDermott M, Carter J, Gauthier S, Goetz C, Golbe L, Huber S, Koller W, Olanow C, Shoulson I (1990) Variable expression of Parkinson’s disease: A base-line analysis of the DATATOP cohort. The Parkinson Study Group Neurology 40: 1529 1534 |
51 | Williams-Gray CH, Mason SL, Evans JR, Foltynie T, Brayne C, Robbins TW, Barker RA (2013) The CamPaIGN study of Parkinson’s disease: 10-year outlook in an incident population-based cohort J Neurol Neurosurg Psychiatry 84: 1258 1264 |
52 | Alves G, Larsen JP, Emre M, Wentzel-Larsen T, Aarsland D (2006) Changes in motor subtype and risk for incident dementiain Parkinson’s disease Mov Disord 21: 1123 1130 |
53 | Burn DJ, Rowan EN, Allan LM, Molloy S, O’Brien JT, McKeith IG (2006) Motor subtype and cognitive decline in Parkinson’s disease, Parkinson’s disease with dementia, and dementia with Lewy bodies J Neurol Neurosurg Psychiatry 77: 585 589 |
54 | Baumann CR, Held U, Valko PO, Wienecke M, Waldvogel D (2014) Body side and predominant motor features at the onset of Parkinson’s disease are linked to motor and nonmotor progression Mov Disord 29: 207 213 |
55 | Duncan GW, Khoo TK, Yarnall AJ, O’Brien JT, Coleman SY, Brooks DJ, Barker RA, Burn DJ (2014) Health-related quality of life in early Parkinson’s disease: The impact of nonmotor symptoms Mov Disord 29: 195 202 |
56 | Litvan I, Goldman JG, Troster AI, Schmand BA, Weintraub D, Petersen RC, Mollenhauer B, Adler CH, Marder K, Williams-Gray CH, Aarsland D, Kulisevsky J, Rodriguez-Oroz MC, Burn DJ, Barker RA, Emre M (2012) Diagnostic criteria for mild cognitive impairmentin Parkinson’s disease: Movement Disorder Society Task Force guidelines Mov Disord 27: 349 356 |
57 | Yarnall AJ, Breen DP, Duncan GW, Khoo TK, Coleman SY, Firbank MJ, Nombela C, Winder-Rhodes S, Evans JR, Rowe JB, Mollenhauer B, Kruse N, Hudson G, Chinnery PF, O’Brien JT, Robbins TW, Wesnes K, Brooks DJ, Barker RA, Burn DJ (2014) Characterizing mild cognitive impairment in incident Parkinson disease: The ICICLE-PD study Neurology 82: 308 316 |
58 | de la Riva P, Smith K, Xie SX, Weintraub D (2014) Course of psychiatric symptoms and global cognition in early Parkinson disease Neurology 83: 1096 1103 |
59 | Suzuki K, Miyamoto T, Miyamoto M, Okuma Y, Hattori N, Kamei S, Yoshii F, Utsumi H, Iwasaki Y, Iijima M, Hirata K (2008) Excessive daytime sleepiness and sleep episodes in Japanese patients with Parkinson’s disease J Neurol Sci 271: 47 52 |
60 | Goldman JG, Ghode RA, Ouyang B, Bernard B, Goetz CG, Stebbins GT (2013) Dissociations among daytime sleepiness, nighttime sleep, and cognitive status in Parkinson’s disease Parkinsonism Relat Disord 19: 806 811 |
61 | Mahale R, Yadav R, Pal PK (2015) Quality of sleep in young onset Parkinson’s disease: Any difference fromolder onset Parkinson’s disease Parkinsonism Relat Disord 21: 461 464 |
62 | Svensson E, Beiske AG, Loge JH, Beiske KK, Sivertsen B (2012) Sleep problems in Parkinson’s disease: A community-based study inNorway BMC Neurol 12: 71 |
63 | Pellecchia MT, Antonini A, Bonuccelli U, Fabbrini G, Ferini Strambi L, Stocchi F, Battaglia A, Barone P (2012) Observational study of sleep-related disorders in Italian patients with Parkinson’s disease: Usefulness of the Italian version of Parkinson’s disease sleep scale Neurol Sci 33: 689 694 |
64 | Rijsman RM, Schoolderman LF, Rundervoort RS, Louter M (2014) Restless legs syndrome in Parkinson’s disease Parkinsonism Relat Disord 20: Suppl 1 S5 S9 |
65 | Mollenhauer B, Trautmann E, Sixel-Doring F, Wicke T, Ebentheuer J, Schaumburg M, Lang E, Focke NK, Kumar KR, Lohmann K, Klein C, Schlossmacher MG, Kohnen R, Friede T, Trenkwalder C (2013) Nonmotor and diagnostic findings in subjects with de novo Parkinson disease of the DeNoPa cohort Neurology 81: 1226 1234 |
66 | Kurtis MM, Rodriguez-Blazquez C, Martinez-Martin P (2013) Relationship between sleep disorders and other non-motor symptoms in Parkinson’s disease Parkinsonism Relat Disord 19: 1152 1155 |
67 | Rodriguez-Blazquez C, Forjaz MJ, Frades-Payo B, de Pedro-Cuesta J, Martinez-Martin P (2010) Independent validation of the scales for outcomes in Parkinson’s disease-autonomic (SCOPA-AUT) Eur J Neurol 17: 194 201 |
68 | Verbaan D, Marinus J, Visser M, van Rooden SM, Stiggelbout AM, van Hilten JJ (2007) Patient-reported autonomic symptoms inParkinson disease Neurology 69: 333 341 |
69 | Anang JB, Gagnon JF, Bertrand JA, Romenets SR, Latreille V, Panisset M, Montplaisir J, Postuma RB (2014) Predictors of dementia in Parkinson disease: A prospective cohort study Neurology 83: 1253 1260 |
70 | Noyce AJ, Bestwick JP, Silveira-Moriyama L, Hawkes CH, Giovannoni G, Lees AJ, Schrag A (2012) Meta-analysis of early nonmotor features and risk factors for Parkinson disease Ann Neurol 72: 893 901 |
71 | Siderowf A, Jennings D, Eberly S, Oakes D, Hawkins KA, Ascherio A, Stern MB, Marek K (2012) Impairedolfaction and other prodromal features in the Parkinson At-Risk Syndrome Study Mov Disord 27: 406 412 |
72 | Krogh K, Ostergaard K, Sabroe S, Laurberg S (2008) Clinical aspects of bowel symptoms in Parkinson’s disease Acta Neurol Scand 117: 60 64 |
Figures and Tables
Fig.1
Assessments and timeline for recent onset patients. Visits occur every 6 months, with repeated observations and blood sampling every 18 months.
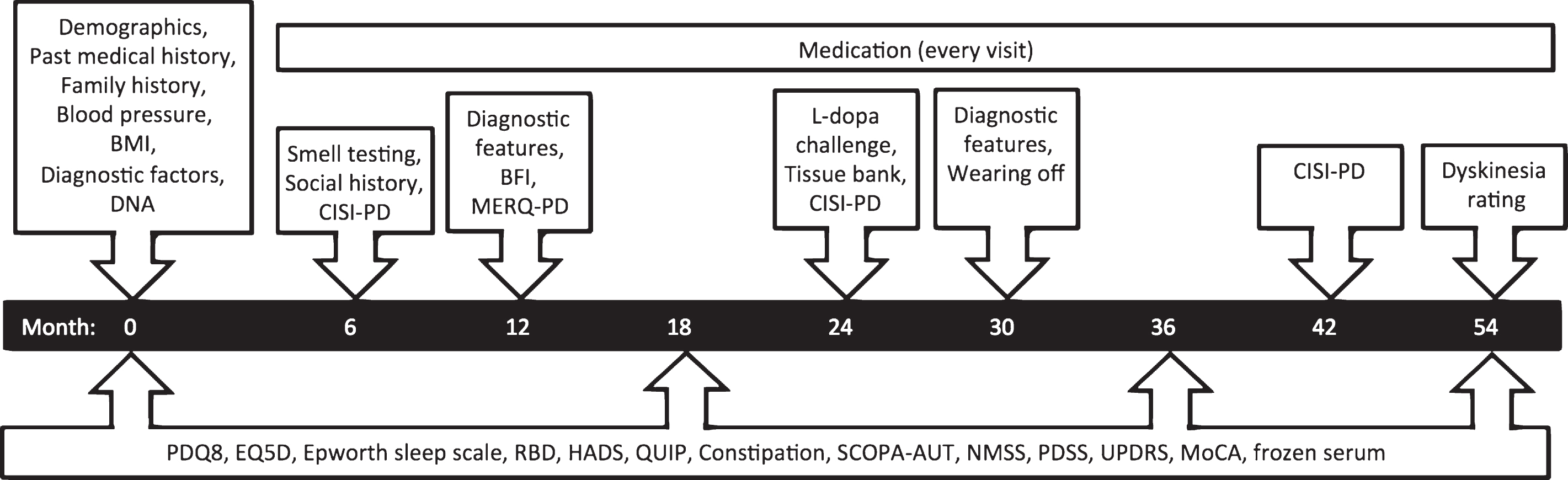
Fig.2
Clinical impression of severity index scores in 2247 PD patients. Recent onset cases had a significantly shorter disease duration than young onset cases, explaining their milder motor features and disability, while the cognitive pattern was more equal, given the greater risk of cognitive impairment with age.
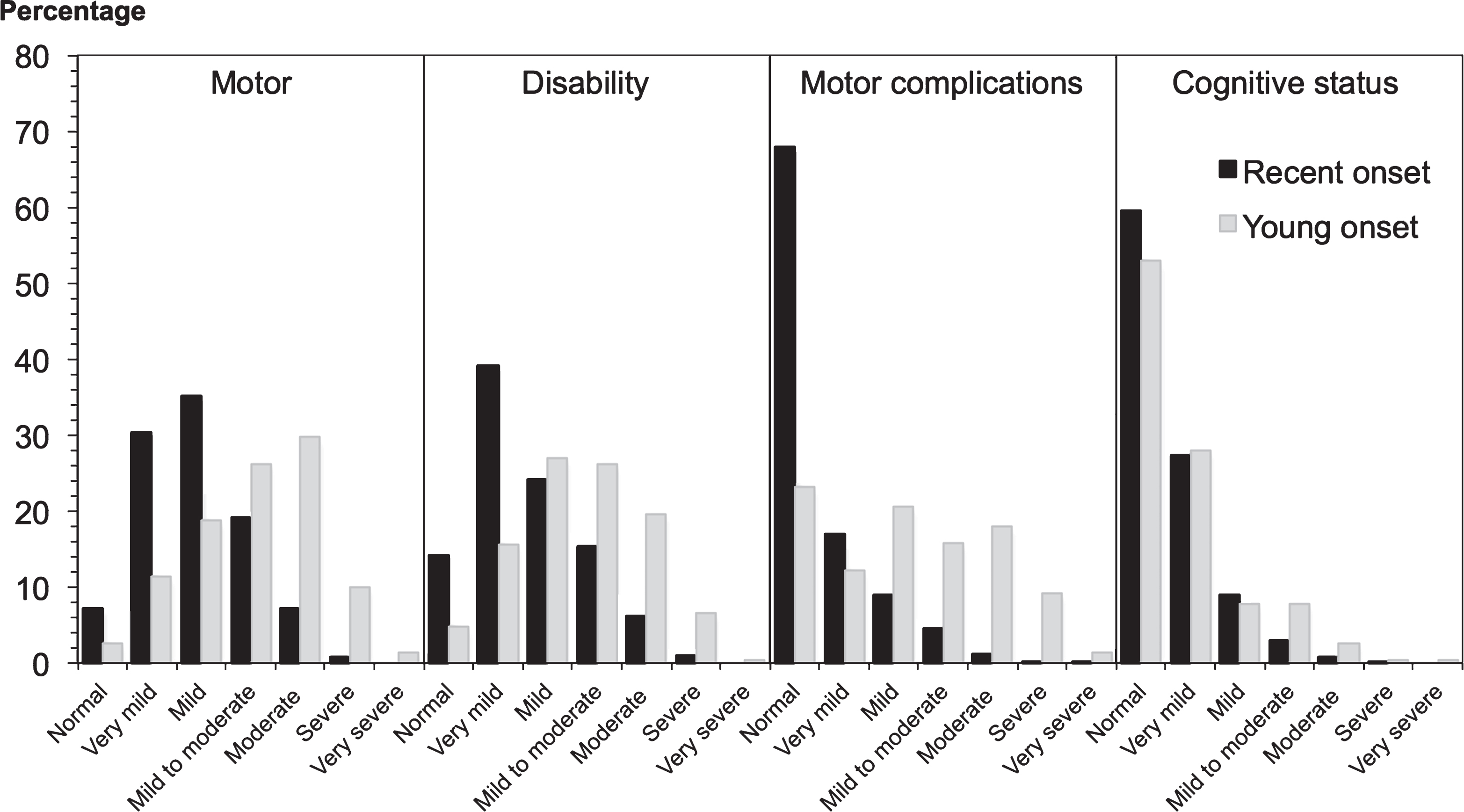
Fig.3
Antiparkinson medication at baseline in 2247 PD patients. Most patients were already on antiparkinson medication at recruitment. Young onset cases had a longer disease duration than recent onset cases, which will affect usage rates and the proportions on more than one drug class. MAOB-I = monoamine oxidase B inhibitor; COMT-I = catechol-O-methyl transferase inhibitor.
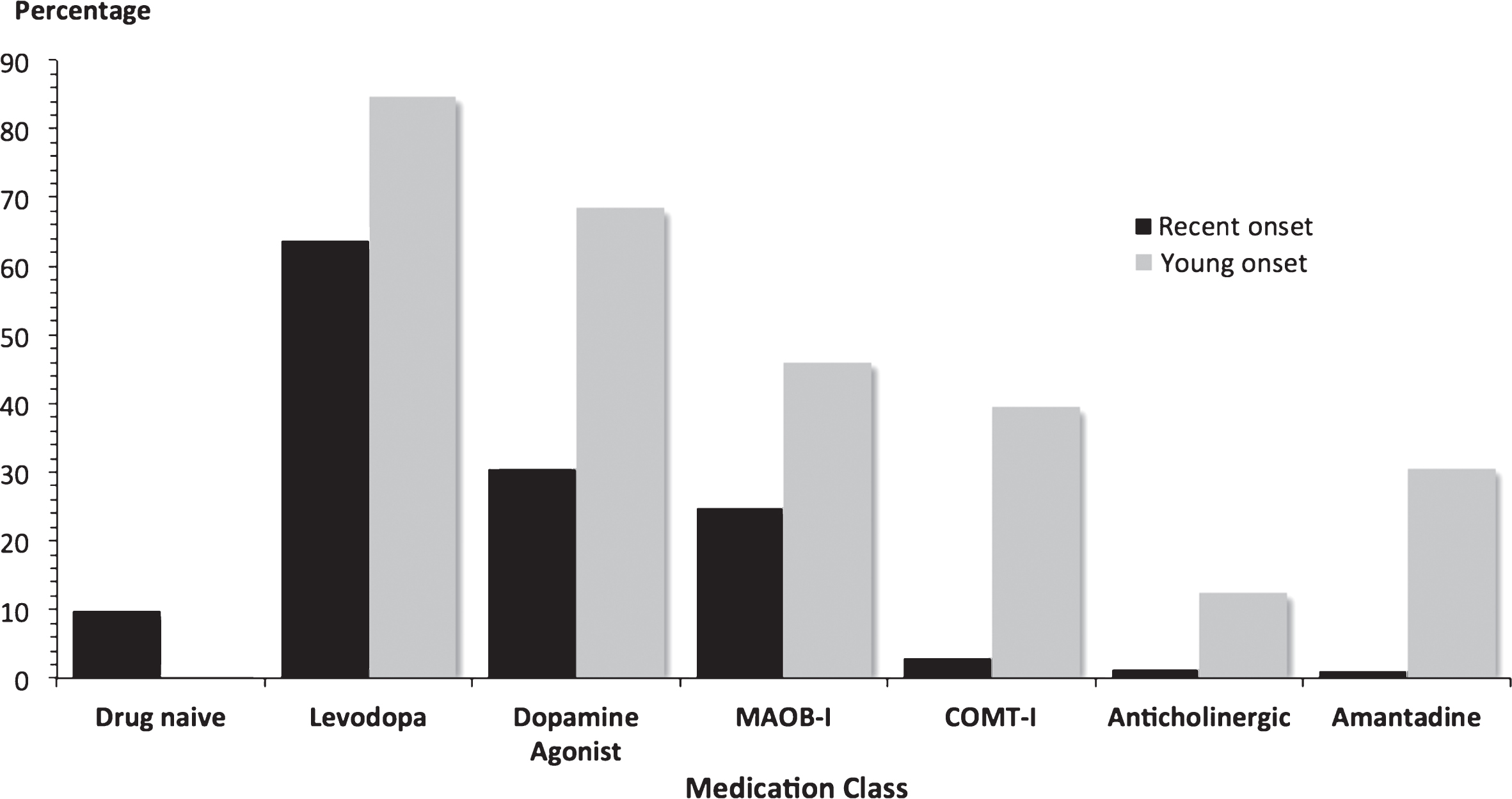
Table 1
Baseline demographics in 2247 PD patients
| Variable | Recent onset N = 1987 | Young onset N = 260 |
| Age in years | ||
| At study entry | 67.6 (9.3) | 53.5 (7.8) |
| At diagnosis | 66.3 (9.3) | 43.3 (5.7) |
| At symptom onset | 64.4 (9.8) | 41.6 (6.8) |
| Years since diagnosis | 1.3 (0.9) | 10.2 (6.7) |
| Male sex | 65.7% | 66.9% |
| Handedness (right/left/mixed) | 85.6/9.6/4.8% | 87.5/9.0/3.5% |
| Ethnicity | ||
| White | 1920 (98.0%) | 244 (95.3%) |
| Asian or Asian British | 19 (1.0%) | 10 (3.9%) |
| Black or Black British | 14 (0.7%) | 2 (0.8%) |
| Mixed | 4 (0.2%) | 0 |
| Other | 3 (0.2%) | 0 |
| BMI (kg/m2) | 27.0 (4.7) | 27.5 (5.7) |
Data are mean (standard deviation) or percentage, BMI = body mass index.
Table 2
Family history of Parkinson’s disease, dementia, and stroke in 2247 PD patients
| Positive family history | Recent onset n = 1987 | Young onset n = 260 |
| Parkinson’s disease | ||
| Only one affected | 311 (15.8%) | 51 (19.7%) |
| More than 1 affected | 82 (4.2%) | 15 (5.8%) |
| Recessive PD historya | 38 (1.9%) | 4 (1.5%) |
| Dominant PD historyb | 355 (18.0%) | 62 (23.9%) |
| Dementia or Alzheimer’s disease | ||
| Mother only | 159 (8.1%) | 13 (5.0%) |
| Maternalc | 28 (1.4%) | 2 (0.8%) |
| Father only | 46 (2.3%) | 6 (2.3%) |
| Paternald | 10 (0.5%) | 1 (0.4%) |
| Stroke | ||
| Mother only | 68 (3.5%) | 12 (4.6%) |
| Maternalc | 12 (0.6%) | 1 (0.4%) |
| Father only | 74 (3.8%) | 8 (3.1%) |
| Paternald | 4 (0.2%) | 1 (0.4%) |
Data are number (percentage), aOnly siblings affected, bAny other relative affected (could also include a sibling affected), cMother and another maternal family member affected, dFather and another paternal family member affected.
Table 3
Baseline clinician scored items in 2247 PD patients
| Variable | Recent onset N = 1987 | Young onset N = 260 |
| UPDRS | ||
| Part 1 | 9.3 (5.4) | 13.8 (7.3) |
| Part 2 | 9.8 (6.6) | 17.6 (9.5) |
| Part 3 | 22.9 (12.3) | 27.4 (15.3) |
| Part 4 | 0.8 (1.8) | 5.7 (5.0) |
| Total | 42.7 (19.8) | 63.6 (29.6) |
| Motor subtype | ||
| Tremor dominant | 46% | 27.5% |
| Postural instability gait difficulty | 41% | 63% |
| Indeterminate | 13% | 9.6% |
| Hoehn and Yahr stage, median (IQR) | 2 (1-2) | 2 (1.5–2.5) |
| LEDD | 295 (211.3) | 926 (566.6) |
| Montreal cognitive assessment | ||
| Normal | 53.4% | 55.5% |
| Mild cognitive impairment | 36.8% | 36.2% |
| Dementia | 9.8% | 8.3% |
Data are mean (standard deviation) or percentage unless otherwise stated. UPDRS = Movement Disorder Society unified Parkinson’s disease rating scale; IQR = Interquartile range; LEDD = levodopa equivalent daily dose.
Table 4
Non-motor features in 2247 PD patients
| Variable | Recent onset n = 1987 | Young onset n = 260 |
| Non-motor symptom severity | ||
| Mild | 37.1% | 19.7% |
| Moderate | 34.5% | 19.1% |
| Severe | 19.6% | 27.3% |
| Very severe | 8.8% | 33.9% |
| Restless legs | 23.6% | 28.3% |
| Depression | 23.6% | 43.0% |
| Anxiety | 24.0% | 50.0% |
| Excessive daytime sleepiness | 24.5% | 49.0% |
| REM sleep behaviour disorder | 43.7% | 60.0% |
| Constipation | 33.6% | 33.7% |
| Hyposmia | 75.3% | 75.9% |
| Orthostatic hypotension | 17.2% | 11.3% |
REM = Rapid Eye Movement.
Table 5
Impulsivity and autonomic features in 2247 PD patients
| Variable | Recent onset n = 1987 | Young onset n = 260 | Controlsa |
| Impulsivity | |||
| Gambling | 1.6% | 23.2% | 0.7% |
| Sex | 5.3% | 25.0% | 3.5% |
| Buying | 3.7% | 25.7% | 2.1% |
| Eating | 5.4% | 21.7% | 10.5% |
| Medication | 1.5% | 8.4% | NA |
| Hobbyism | 9.7% | 37.6% | 11.9% |
| Punding | 5.5% | 17.1% | 2.1% |
| Walkabout | 1.1% | 6.4% | 0.7% |
| One or more | 22.7% | 67.6% | 20.3% |
| PDSS | 109 (23.5) | 91.8 (28.7) | 120.7 (21.0) |
| SCOPA – AUT | |||
| Gastrointestinal | 3.0 (2.5) | 4.2 (3.2) | 1.4 (1.6) |
| Urinary | 4.7 (3.3) | 5.3 (3.6) | 3.9 (2.4) |
| Cardiovascular | 0.7 (1.1) | 1.1 (1.3) | 0.3 (0.6) |
| Thermoregulatory | 1.6 (1.8) | 3.1 (2.5) | 1.8 (2.0) |
| Pupillomotor | 0.4 (0.7) | 0.6 (0.8) | 0.4 (0.7) |
| Sexual male | 1.9 (2.0) | 1.7 (1.8) | 1.3 (1.7) |
| Sexual female | 1.6 (1.7) | 1.4 (1.4) | 1.4 (1.5) |
| Total autonomic score | 11.8 (7.1) | 15.6 (9.2) | 8.8 (5.4) |
Table 6
Rates and reasons for study withdrawal in 2247 PD patients
| Variable | Recent onset n = 1987 | Young onset n = 260 |
| Withdrawn | 156 (7.9%) | 11 (4.2%) |
| Time to withdrawal years | 1.4 (0.7) | 0.7 (0.6) |
| Reasona | ||
| Intercurrent illness | 14.7% | 9.1% |
| Patient died | 18.0% | 27.3% |
| Patient choice | 43.6% | 27.3% |
| Other | 23.7% | 36.4% |
Data are number (percentage). aPercentage of those withdrawn.


