
Immunotherapy in Parkinson’s Disease: Micromanaging Alpha-Synuclein Aggregation
Abstract
Currently, several α-synuclein immunotherapies are being tested in experimental Parkinson’s disease models and in clinical trials. Recent research has revealed that α-synuclein is not just an intracellular synaptic protein but also exists extracellularly. Moreover, the transfer of misfolded α-synuclein between cells might be a crucial step in the process leading to a progressive increase in deposition of α-synuclein aggregates throughout the Parkinson’s disease brain. The revelation that α-synuclein is present outside cells has increased the interest in antibody-based therapies and opens up for the notion that microglia might play a key role in retarding Parkinson’s disease progression. The objectives of this review are to describe and contrast the use of active and passive immunotherapy in treating α-synucleinopathies and highlight the likely important role of microglia in clearing misfolded α-synuclein from the extracellular space.
INTRODUCTION: ALPHA-SYNUCLEIN and PARKINSON’S DISEASE
In Parkinson’s disease (PD), misfolded α-synuclein (α-syn) is the main constituent of Lewy bodies and neurites, which are the pathological hallmarks of the disorder. Several lines of evidence indicate that α-syn plays a key role in PD pathogenesis, both in rare genetic forms as well as the common idiopathic forms [1]. Missense mutations (A53T, A30P, E46K, H50Q) in the α-syn gene are associated with autosomal dominant PD [2–6]. Duplications and triplications in the α-syn gene lead to a syndrome that includes parkinsonian features [7, 8]. Importantly, certain single-nucleotide polymorphisms in the α-syn gene are associated with increased PD risk [9]. It is not clear which molecular form of α-syn is toxic to neurons. Current thinking suggests that oligomeric species of α-syn cause neuronal death, but larger fibrillar assemblies, visible in the microscope as Lewy pathology, might also be the culprit. Earlier studies emphasized the importance of intracellular α-syn in the pathogenesis of PD. Importantly, just over a decade ago, it was recognized that α-syn is also present in extracellular fluid [10–12]. In 2008, it was proposed that misfolded α-syn that has been released from neurons into the extracellular space can be taken up by adjacent cells where it seeds further aggregation of endogenous protein [13]. This process was suggested to underpin the spreading of Lewy neuropathology from one brain region to another [14–19]. Thus, this hypothesis might explain the progressive, stereotypical spreading of intraneuronal α-syn aggregates in PD in accordance with the six neuropathological stages proposed by Braak[20, 21].
IMMUNOTHERAPY TRIALS IN ALZHEIMER’S DISEASE TAKE A POSITIVE TURN
Recent developments in experimental immunotherapy for Alzheimer’s disease (AD) have been positive, and have also contributed to the surge of interest in antibody-based approaches to PD therapy. Therefore, we briefly describe some of these recent developments in this section.
Extracellular aggregates of amyloid β (Aβ) protein, along with intracellular tangles of the neurofilament protein tau, form the pathological hallmarks of AD. Importantly in relation to immunotherapies, Aβ is a secreted molecule that is found in blood and can be detected by antibodies. Similarly, tau (normally cytoplasmic in neurons) can also be found extracellularly and targeted by antibodies [22]. The first immunotherapy trials in AD targeted Aβ and were preceded by several positive preclinical studies [23–25]. In contrast to the encouraging preclinical experiments, the outcomes of these clinical trials were disappointing with no significant benefits in cognition despite a reduction in Aβ plaques on brain scans in some patients [26]. In addition, adverse events, such as meningoencephalitis, vasogenic oedema and intracerebral microhaemorrhages have been observed in a significant number of patients in the early trials [27]. In hindsight, it has been suggested that these unfavorable outcomes were primarily due to poor patient selection with several of the recruited cognitively impaired individuals probably not suffering from AD. Furthermore, it was not clear if the antibodies were directed to the optimal Aβ epitopes and second generation antibodies target specific Aβ epitopes to avoid off target effects [28]. Recent preliminary reports from clinical trials have been more positive and have stimulated enthusiasm for immunotherapies against Aβ. Specifically, preliminary results released recently indicated that the antibody BIIB37 (aducanumab) was relatively safe and reduced cognitive deficits in a large Phase 1b trial in early AD (Jeffrey Sevigny ADPD meeting, 2015 [29]). Different factors might have contributed to the tentative success of this trial. First of all, only patients with clear Aβ pathology on brain scans were included. Secondly, the BIIB37 antibody has a long half-life and recognizes the N-terminus of Aβ in aggregates, not monomers (Jeffrey Sevigny ADPD meeting, 2015 [29]). Thirdly, the antibody was originally derived from healthy, aged donors who were cognitively normal. There are also hints of treatment benefits in a recent conferencereport on a pre-specified secondary analysis of the Phase 3 EXPEDITION trial testing a monoclonal antibody to soluble Aβ called solanezumab. Although the primary outcome was negative [30], this secondary analysis suggested that early immunotherapy in patients with mild AD might delay decline in cognitive function [31]. With these positive developments, immunotherapy for AD and other neurodegenerative diseases is attracting renewed interest. Further trials with antibody therapies against Aβ are planned, as well as therapies targeting misfolded tau in AD, as immunotherapy against tau can modulate Aβ pathology [32–34]. As mentioned above, interest into immunotherapy in PD has also received a boost as a consequence of the outcome of these recentAD trials.
BLOOD BRAIN BARRIER AND ANTIBODIES
While the blood brain barrier (BBB) is potentially an obstacle to effective immunotherapy in neurodegenerative disorders, in this section we briefly discuss that it is not an absolute hurdle and that novel approaches can promote the entry of antibodies, across the BBB, into the brain. The BBB clearly limits the transport of substances from the peripheral circulation into the central nervous system [35]. The BBB concept comprises a monolayer of brain endothelial cells that exhibit interconnected transmembrane tight junctions, resulting in a low permeability of molecules, including antibodies, from the blood to the brain [36–40]. Transporters and enzymes present on both sides of the BBB regulate the movement of substances between the periphery and the extracellular space of the central nervous system [41]. Astrocytes and pericytes surrounding the endothelial cell layer are believed to contribute to BBB integrity [38]. Given this tight regulation surrounding the central nervous system, the permeability of antibodies is low with approximately 0.1–0.2 % of peripherally administered antibody crossing the BBB and gaining access to the extracellular space of the central nervous system [36]. Evidence from AD immunotherapy studies demonstrate that anti-Aβ antibodies cross the BBB, albeit at a low rate [42–44] and Aβ-directed IgG antibodies cross the BBB using extracellular pathways [40]. A brain-to-blood efflux system may exist at the BBB for IgG molecules [45]. A promising approach for targeting antibody therapeutics to the brain is through receptor-mediated transcytosis. The specificity of this mechanism for antibody delivery takes advantage of endogenous receptors expressed on the luminal side of the BBB that normally function to deliver macromolecule nutrients to the brain [46–48]. Future trials might be able to take advantage of receptor-mediated transcytosis to improve the efficacy of immunotherapies for neurodegenerative diseases.
NEUROINFLAMMATION AND MICROGLIA ACTIVATION IN PARKINSON’S DISEASE
A recent ground breaking paper, that discovered lymphatic vessels in the brain, highlights the role for the immune system in the brain [49]. Microglia and astrocytes are the resident immune cells of the brain, and in this capacity they are likely to play crucial roles in the success of immunotherapies forneurodegenerative diseases. In PD, these cells are activated, as part of a neuroinflammatory response, implicating that the immune system plays a role in the pathogenesis of PD. However, observations of activated microglia and astroglia do not clarify whether neuroinflammation is the primary or secondary event in PD pathogenesis (for review, see [50]). Studies in PD patient brains have revealed neuroinflammation particularly in the basal ganglia and neocortex [51]. The microglia in these regions exhibit an activated morphology, and in the substantia nigra they surround the degenerating dopaminergic neurons [52]. They may consist of a mixed population of invading peripheral monocytes who act in concert with resident microglia [50]. The adaptive immune response also plays a role with the substantia nigra of PD patients exhibiting a 10-fold increase in numbers of CD4+ and CD8+ T-lymphocytes compared to controls [50]. Further support for inflammation being important in PD pathogenesis comes from studies showing that PD is associated with certain HLA-variants [53, 54] and that intake of some non-steroidal anti-inflammatory drugs reduces the risk of developing PD [50].
The trigger for the inflammatory response in PD is not clear. While it might just be a reaction to ongoing neuronal cell death, it has also been suggested that misfolded α-syn itself might play a direct role in this regard. Extracellular, misfolded α-syn activates microglia [55], and this, in turn, is linked to neuronal death. The extracellular α-syn is primarily believed to derive from non-classical exocytic or endocytic pathways, but degenerating neurons are another potential source [56]. Oligomeric α-syn derived from neurons can activate microglia through stimulation of Toll like receptor (TLR) 2 [57]. Further evidence linking α-syn to microglia stimulation was observed in transgenic mice overexpressing α-syn, which exhibit microglia activation, as assessed by levels of pro-inflammatory cytokines, nitric acid and reactive oxygen species [58–60]. This microglia activation in α-syn PD mouse models is related spatially and temporally to α-syn expression [58]. The interplay between α-syn and immune cells of the brain, however, is not limited to α-syn-induced activation of the microglia. In a neural transplant model of α-syn cell-to-cell transfer, we have observed that microglia take up α-syn released from nigrostriatal terminals in mice overexpressing human wild type α-syn in the nigrostriatal dopamine neurons (Fig. 1). Cell culture studies have also shown that microglia can phagocytose different molecular species of α-syn and this process is enhanced in the presence of monomeric but not aggregated α-syn [61]. Additionally, it has been suggested that aggregated α-syn (even when present in the extracellular space) is particularly damaging to dopamine neurons when activated microglia are present [62]. Interestingly, lipopolysaccharide-induced activation of microglia in vitro slows α-syn degradation, leading to its accumulation in the cytoplasm [63]. This suggests that when the microglia participate in an inflammatory response, they might be less efficient at clearing extracellular α-syn, which, in turn, could exacerbate the development of α-syn pathology. Therefore, in the context of immunotherapies for PD, it is of paramount importance to understand how the activation of microglia by the underlying disease process impacts their ability to clear α-syn that has been bound by therapeutic antibodies.
IMMUNOTHERAPY TARGETING α-SYNUCLEIN IN PARKINSON’S DISEASE
As mentioned above, mounting evidence points to the existence of a significant extracellular pool ofα-syn. α-Syn has been identified in the cerebrospinal fluid [11] and plasma [12] of patients with PD. Clinical immunotherapy trials in PD are currently targeting the “prion-like” cell-to-cell transfer of α-syn [64]. Indeed, for α-syn to transfer from neuron-to-neuron, it most likely has to transcend the extracellular space. Consequently, the development of immunotherapies against α-syn is a fast growing area of interest. Several studies have tested immunotherapies targeting α-syn in PD animal models in attempts to remove α-syn from the extracellular space and thereby reduce the progressive deposition of α-syn aggregates throughout the brain. Two immunotherapeutic strategies, inspired by numerous studies in the AD field, have been explored in PD models: active immunization, using the animal’s own immune system to generate antibodies against α-syn or passive immunization with the direct administration of antibodies against different domains of α-syn. It is thought that α-syn antibodies will stimulate microglial cells to scavenge the extracellular α-syn and prevent its transfer from one neuron to another. As current immunotherapy studies targeting α-syn rely on microglia for its effective degradation, in each section we briefly discuss the role that microglia might play in immunotherapy for PD and how they might assist in reducing α-syn pathology.
ACTIVE IMMUNIZATION THERAPY
Masliah and colleagues pioneered experimental immunotherapy targeting α-syn [65] (Table 1A). Over a decade ago, their first study utilized a transgenic mouse overexpressing human wild type α-syn under control of the platelet-derived growth factor-β promoter [66]. These mice exhibit α-syn accumulation in neurons and glia of the neocortex, hippocampus and substantia nigra [66]. Mice were immunized with recombinant human α-syn resulting in the production of high affinity α-syn antibodies. The antibodies that had the highest affinity were directed to the C-terminus of α-syn. The treated mice exhibited a decreased accumulation of α-syn in neuronal cell bodies and synapses in temporal cortex, and preservation of a higher number of synaptophysin-positive nerve terminals, as well as reduced neurodegeneration [65]. It is believed that α-syn was targeted to the lysosome for degradation [65]. This study presented experimental evidencesuggesting that antibodies can act inside neurons by reducing intraneuronal inclusions. Interestingly, only mild microglial activation was observed followingvaccination (immunohistochemical analysis using antibodies against the microglia marker, Iba1) suggesting, somewhat surprisingly, that microglia might not have been major players in clearing the α-syn from the extracellular space once it had bound to the therapeutic antibodies.
A more recent study utilized a rat model of PD involving virus-mediated delivery of human α-syn into the nigrostriatal pathway to test the effects of active immunization against α-syn [67]. Vaccination with human recombinant α-syn combined with complete or incomplete Freund adjuvant via systemic injections on two occasions, 6–10 weeks before the intracerebral injection of the α-syn viral vector, reduced α-syn inclusions in the substantia nigra. This vaccination regime generated an adaptive memory response, as the immunogen was not present at the time of human α-syn overexpression. Subsequently, memory lymphocytes (CD 4 + T cells) and microglial activation were induced. The microglia response was associated with increased cell numbers, morphologic microglial changes (increased numbers exhibiting a “protective” phenotype, as opposed to “neurotoxic”, phenotype); major histocompatibility complex (MHC) class II and CD 4 expression [68]. The resulting increase in protective microglia was coupled to changes in surveillance and reaction to α-syn, suggesting that activated microglia can reduce α-syn pathology [67].
To test active immunization against PD, the biotechnology company AFFIRiS AG, has developed a therapeutic peptide vaccine. Their team has performed active immunization studies in animal models with their AFFITOPE® vaccine AFF1 in two mouse models of PD [69]. This vaccine contains short peptide fragments that mimic the C-terminal of human α-syn, conjugated to a carrier and administered with aluminum hydroxide as an immunological adjuvant [69]. The use of a short peptide in the conjugate is to avoid T cell autoimmunity, as it occurred in early AD clinical trials, a possible side effect with immunotherapy treatment [70]. The results demonstrated that AFF1 vaccine reduces neuropathology in the mice and improves motor function (body suspension test) and long term memory (Morris water maze) [69]. Aggregated α-syn was reduced in the synaptic terminals of treated mice and associated with microglia activation as well as increases in anti-inflammatory cytokines [69]. In a mouse model of multiple system atrophy (MSA), where α-syn is expressed under control of an oligodendrocyte-specific promoter [71], treatment with AFF1 led to a reduced transfer of α-syn from oligodendrocytes to astrocytes and an increase of α-syn that had been taken up by microglia [71]. AFFITOPE® vaccines have already undergone safety testing in normal subjects and are in a Phase 1 trial for PD and a Phase 2 trial for MSA.
Despite the promising pre-clinical data, active immunization is also associated with some potential disadvantages. Neuroinflammation is frequently reported as a side effect in human trials [72]. A significant variability in the antibody response across treated subjects has also been observed. This might be due to a variable age-related reduction in the immune competency of elderly subjects [27]. In response to vaccination, the elderly immune system is less likely to generate adequate antibody titers and might develop autoimmune side effects instead [27]. Additionally, life-long adverse effects can occur after active vaccination, due to the polyclonal antibodies potentially binding non-desired epitopes on other proteins [27]. Notwithstanding these risks, active vaccination has theoretical advantages in that its long-lasting immune responses might also be advantageous and it might lead to higher antibody levels than passive immunization strategies [72].
PASSIVE IMMUNIZATION
The first study to move from active α-syn immunization to passive, in the hope to circumvent the potential side effects associated with active immunization, was conducted by Masliah and coworkers [73] (Table 1B). A major advantage of passive immunization is the opportunity to reduce the dose or potentially stop treatment if adverse events appear. In order to be effective, treatment most likely needs to be chronic, with repeated administration of antibodies a few weeks apart. The immunogen used to generate the therapeutic antibody in the first published passive immunization study was a monoclonal α-syn antibody against the C-terminus of α-syn (9E4). The C-terminus of α-syn probably plays an important role in the pathogenic process in PD. Thus, C-terminus truncation of α-syn results in formation of toxic fragments and facilitates α-syn oligomerization and propagation [74–77]. In a transgenic mouse model of PD (expressing human wild type α-syn under control of the platelet derived growth factor-β promoter) [66] a weekly injection of 9E4 antibody resulted in a decrease in the accumulation of calpain-cleaved α-syn fragments in axons and synaptic terminals. Despite the reduction in α-syn fragments, no reduction in overall α-syn levels (i.e. including non-truncated forms) was observed [73]. The antibody 9E4 binds an epitope in the C-terminal site of α-syn that includes a calpain cleavage site. Therefore this antibody is believed to decrease enzymatic cleavage of α-syn and instead promote lysosomal clearance ofα-syn. Notably, motor skills (rotarod test and pole test) and learning and memory (Morris water maze) significantly improved in the 9E4 antibody-treated mice.
In a related study by Bae and coworkers (2012) [16], using the same mouse model as in the study described immediately above [73], a different anti-body against the C-terminal region of α-syn was utilized. This antibody promotes the clearance of extracellular α-syn primarily by microglia. In different experiments included in this study, antibodies were directly injected into the hippocampus of mice or were given as intraperitoneal injections once a week for four weeks. Internalization of α-syn aggregates was reported to occur via the Fcγ receptors on the surface of microglial cells, as evidenced by in vitro culture experiments using the murine BV-2 microglia cell line [16]. The passive immunotherapy resulted in decreased motor deficits, attenuated α-syn accumulation in glia and neurons and less severe neurodegeneration. Interestingly, the antibodies used in both the study by Bae et al. [16] and that of Masliah and coworkers [73] although different, recognize a similar epitope on α-syn on the C-terminal region. The antibody utilized in [73] promoted the degradation of α-syn by lysosome/autophagosomes within neurons. The authors did not monitor what happened to the microglia following the antibody injections, so it is unknown whether microglia participated in removing extracellular α-syn in their paradigm [73]. In Bae et al., [16] degradation of α-syn was primarily by microglia and the antibody used is more likely to cause inflammation (and this was evidenced by an increase in Iba1 staining of the microglia in the brain of treated mice) due to the involvement of glial cells in the phagocytosis of α-syn.
Recently, Prothena Biosciences Inc. in collaboration with Roche, completed a Phase 1 safety trial in normal subjects, using a humanized form of the antibody 9E4 (called PRX002) which was based on the one tested in the study published by Masliah et al. [73] (Table 1C). The study design was a phase 1, randomized, double-blind, placebo-controlled, single ascending-dose paradigm. The results indicate that the antibody was well tolerated, and it was possible to detect target engagement in peripheral tissue. The antibody treatment reduced unbound α-syn in serum by 96% , indicating that most of the α-syn is bound by the antibody. The reductions and pharmacodynamic responses of total α-syn (free and bound) were dose-dependent. The average half-life of serum PRX002 exposure was around 18 days (Schenk et al., 2015 http://www.mdscongress2015.org/Congress-2015-Files/pdfs/2015LateBreakingAbstractsPubDoc.pdf).
In June 2014, Prothena initiated a multicenter, multiple-ascending-dose trial comparing a six-month course of PRX002 to placebo in patients with idiopathic PD to assess safety and pharmacokinetic parameters and this study is ongoing and will conclude in April 2016. It remains to be demonstrated that this antibody affects α-syn levels in the central nervous system, and this will be a most crucial issue as the program develops.
Another approach has been the use of antibodies targeting the N-terminal or central region of α-syn. In a model of nigrostriatal degeneration in rats (due to adenoviral overexpression of α-syn) intraperitoneal injections of α-syn antibodies over the course of three months was neuroprotective, with the N-terminal antibody being the more effective of the two [77]. Antibodies were delivered via intraperitoneal injections every two weeks for three months. Both the antibodies directed against N-terminal and the central region of α-syn reduced α-syn-induced nigral cell death, and decreased the number of activated microglia in the substantia nigra that received the adenoviral injection.
To avoid using antibodies that are either directed to the C-terminus or N-terminus of monomeric α-syn and thereby potentially interfering with the normal function of the protein, the biotechnology company BioArctic Neuroscience AB has generated monoclonal antibodies selective for oligomeric and protofibril forms of α-syn. As mentioned above, different oligomeric species of α-syn have been suggested to be neurotoxic and play an important role in the pathogenesis of PD and related synucleinopathies. A recent paper describing a new technique, α-syn proximity ligation assay, revealed previously unrecognized diffuse pathology of α-syn oligomeric deposits frequently observed in the absence of Lewy bodies [78]. Importantly, the α-syn proximity ligation assay preferentially detects early-stage, loosely compacted lesions, and not Lewy bodies which have been the focus of most prior neuropathology studies [78]. These novel findings highlight that oligomeric α-syn might represent early stage PD pathology, and that targeting α-syn oligomers might be a fruitful immunotherapy approach to halting the disease process before too much neuronal damage is done. The oligomer-specific anti-human α-syn antibodies developed by BioArctic Neuroscience AB have been suggested to reduce intracellular oligomerization of α-syn in vitro [79]. Their oligomer-specific antibodies detect early brain pathology in post-mortem human PD and dementia with Lewy bodies brains, as well as in A53T α-syn transgenic mice [80]. Chronic (14 wks) antibody administration to mice expressing human A30P α-syn under control of the Thy-1 promoter reduced the levels of α-syn protofibrils in the spinal cord [81]. In treated and untreated A30P α-syn transgenic mice, the levels of protofibrillar α-syn correlated with the levels of activated microglia suggesting that protofibrillar α-syn drives inflammation [81]. The antibody treatment did not further enhance the microglial activation [81]. The team at BioArctic Neuroscience AB is testing the safety and efficacy of a humanized version of their antibody directed against α-syn protofibrils in a first-in-man clinical trial (http://www.bioarctic.se/news/-2nd-us-patent-granted-on-therapeutic-antibodies-for-parkinsons-disease).
Another research group has used a conceptually different approach to immunotherapy against α-syn, by using a recombinant fusion protein consisting of the variable regions of the light and heavy chains of immunoglobulin, called single-chain fragment variable [82]. To facilitate cellular clearance of α-syn and passage through the BBB, they linked a recombinant single-chain fragment variable targeting α-syn oligomers to the low-density lipoprotein receptor-binding domain of apolipoprotein B. The hybrid protein crossed the BBB effectively, leading to cellular uptake and degradation of the complex via the lysosome through the endosomal sorting complex required for transport (ESCRT) pathway [82]. When administered intraperitoneally to mice overexpressing human wild type α-syn under the promoter for platelet-derived growth factor-β, the protein conjugate decreased α-syn neuropathology, neuronal loss, astrogliosis, and behavioral deficits in the water maze [82]. In this study, microgliosis was not assessed so the effect of the single-chain fragment variable on inflammation is not possible to discern.
As mentioned earlier, the notion that α-syn can transfer between cells, and that this is a key element in the pathogenic PD process, has gained increasingtraction in recent years [83]. The passive immunization strategy has been utilized in a model of α-syn cell-to-cell transfer, where antibodies disrupt the spread of α-syn pathology [84]. In the paradigm where mice receive intrastriatal injection of preformed α-syn fibrils, long-term systemic administration of antibodies against misfolded α-syn (targeting N-terminus, Syn303) reduced the spread of aggregates. In this model, inflammation was assessed by immunohistochemistry and there was no difference in microgliosis or astrogliosis between α-syn antibody-treated mice and untreated controls. The authors tentatively suggest that this is due to the lack of neurodegeneration in this particular mouse model [84].
Overall, the preclinical studies evaluating both active and passive immunization strategies for α-synucleinopathies have been positive. The experimental therapies have been tested in various models of α-synucleinopathies. Antibodies against theC-terminus of α-syn have been evaluated using transgenic mice overexpressing human α-syn using the PDGF-β promoter [16, 73] and under the Thy-1 promoter [85]. Passive immunization in mice overexpressing α-syn under the Thy1 promoter led to reduced brain levels of truncated α-syn in axons and was reported to protect both synapses and dopaminergic neurons, and improve motor and cognitive functions [85]. In the same treated mice, astrogliosis and microgliosis were significantly reduced [85]. Both active and passive immunization strategies reduce α-syn pathology and improve motor and cognitive behavior. The effectiveness of antibodies that can recognize different conformations of α-syn has also been evaluated in α-syn transgenic mice using the PDGF-β promoter. Antibodies recognizing monomeric and oligomeric species of α-syn were proven effective [75]. Further investigations are needed into whether antibodies that are specific to certain α-syn conformations are more effective. It also remains unknown whether immunotherapy would primarily work as a “preventive” treatment or also might retard further progression in moderate to severe disease [28]. Studies in animal models that define the effects of immunotherapy at different stages of synucleinopathy are going to be crucial when the design of future clinical trials is optimized.
CONCLUDING REMARKS
Targeting the immune system and using immunotherapy are promising strategies for treating PD [86]. Immunotherapy holds the key to be the one of the disease-modifying strategies against PD, as immunizing patients in their early stages and prior to the accumulation of α-syn deposits would be the most effective strategy in preventing the progression of the disease [28]. Although there is evidence from preclinical trials for the efficacy of immunotherapy to reduce α-syn pathology and improve behavioral outcomes in several animal models, the details of how immunotherapy achieves this, remains unclear. Current immunotherapy studies targeting α-syn most likely require microglia to effectively degrade extracellular α-syn [62]. Microglia can be activated in many different states characterized by morphology and cytokine profile [87]. Microglia have been traditionally characterized as pro-inflammatory or anti-inflammatory in their phenotype but this does not account for the myriad of cells that comprise of diverse phenotypes; beneficial and detrimental to the central nervous system [87]. In terms of immunotherapy, this leads us to ask questions regarding how microglia activation affect the efficacy of microglia in clearing α-syn that has been bound by antibody? Once phagocytosed, is the degradation of ingested α-syn affected by the activation state of the microglia? In addition, it is unclear how the various activation states of microglia differentially affect the capacity of the cells to phagocytose antibody bound α-syn. Understanding the role that microglia play will allow us to determine which type of functional microglia most effectively reduce the spread of the pathogenic protein.
As α-syn aggregates are the target of immunotherapy trials in PD, an important consideration and possible side effect is the induction of inflammatory autoimmunity mediated by Th17 cells (subset of T helper cells) as α-syn is an endogenous self-protein and Th17 cells have been shown to be involved in the neurodegenerative neuroinflammatory process [86, 88]. No study using antibodies to induce protection in PD models have studied the T cell response [86]. Therefore for immunotherapy against α-syn to be an effective treatment, it must consider the adaptive immune response to regulate the response against α-syn and immunotherapy must be initiated prior to the formation of α-syn aggregates. Using imaging ligands specific for aggregated α-syn will also likely prove instructive as when used as a surrogate endpoint and to assess target engagement [89]. In conclusion, lessons learnt from the AD field have greatly informed the PD immunotherapy field and have lead us to promising results from both preclinical and clinical trials.
ACKNOWLEDGMENTS
Dr. Brundin reports grants from Michael J Fox Foundation USA, National Institutes of Health, Cure Parkinson’s Trust, KiMe Foundation, Campbell Foundation outside but relevant to the submitted work. Dr. Brundin has received commercial support as a consultant from Renovo Neural, Inc., Roche, and Teva/Lundbeck, ClearView Healthcare Partners and Capital Technologies, Inc. He has received travel support from Prothena Corporation. Additionally he has received commercial support for grants/research from Renovo, Bioarctic Neuroscience AB and Teva/Lundbeck. Dr. Brundin has ownership interests in Acousort AB and Parkcell AB.
REFERENCES
1 | Wales P, Pinho R, Lázaro DF, Outeiro TF (2013) Limelight on alpha-synuclein: Pathological and mechanistic implications in neurodegeneration J Parkinsons Dis 3: 415 459 |
2 | Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease Nat Genet 18: 106 108 |
3 | Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease Science 276: 2045 2047 |
4 | Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E, del Ser T, Muñoz DG, de & Yebenes JG (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia Ann Neurol 55: 164 173 |
5 | Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, ShermanH , Yu I, Shah B, Weir D, Thompson C, Szu-Tu C, Trinh J, Aasly JO, Rajput A, Rajput AH, Jon Stoessl A, Farrer MJ (2013) Alpha-synuclein p. H50Q, a novel pathogenic mutation forParkinson’s disease Mov Disord 28: 811 813 |
6 | Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL (2013) A novel α-synuclein missense mutation in Parkinson disease Neurology 80: 11 1062 1064 |
7 | Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-& Hardy K (2003) α -Synuclein Locus Triplication Causes Parkinson’s Disease Science 302: 5646 841 841 |
8 | Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S (2004) Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease The Lancet 364: 9440 1167 1169 |
9 | Maraganore DM, de Andrade M, Lesnick TG, Strain KJ, Farrer MJ, Rocca WA (2005) High-resolution whole-genome association study of Parkinson disease Am J Hum Genet 77: 5 685 693 |
10 | El-Agnaf OM, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ (2003) Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma FASEB J 17: 13 1945 1947 |
11 | Tokuda T, Qureshi MM, Ardah MT, Varghese S (2010) Detection of elevated levels of α-synuclein oligomers in CSF from patients with Parkinson disease Neurology 75: 20 1766 1772 |
12 | El-Agnaf OMA, Salem SA, Paleologou KE, Curran MD, Gibson MJ, Court JA (2006) Detection of oligomeric forms of α-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease FASEB J 20: 3 419 425 |
13 | Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ (2008) Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation Nat Med 14: 5 501 503 |
14 | George S, Rey NL, Reichenbach N, Steiner JA, Brundin P (2013) α-Synuclein: The long distance runner Brain Pathol 23: 3 350 357 |
15 | Aguzzi A, Rajendran L (2009) The Transcellular Spread of Cytosolic Amyloids, Prions, and Prionoids Neuron 64: 6 783 790 |
16 | Bae E-J, Lee H-J, Rockenstein E, Ho D-H, Park E-B, Yang N-Y (2012) Antibody-aided clearance of extracellularα-synuclein prevents cell-to-cell aggregate transmission J Neurosci 32: 39 13454 13469 |
17 | Danzer KM, Krebs SK, Wolff M, Birk G, Hengerer B (2009) Seeding induced by α-synuclein oligomers provides evidence for spreading of α-synuclein pathology J Neurochem 111: 1 192 203 |
18 | Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein Proc Nat Acad Sci 106: 31 13010 13015 |
19 | Frost B, Diamond MI (2010) Prion-like mechanisms inneurodegenerative diseases Nat Rev Neurosci 11: 3 155 159 |
20 | Braak H, R b U, Gai WP, Del Tredici K (2003) Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen J Neural Transm 110: 5 517 536 |
21 | Braak H, Ghebremedhin E, R b U, Bratzke HR, Tredici K (2004) Stages in the development of Parkinson’s disease-related pathology Cell Tissue Res 318: 1 121 134 |
22 | Yamada K, Holth JK, Liao F, Stewart FR, Mahan TE, Jiang H (2014) Neuronal activity regulates extracellular tau} J Exp Med 211: 3 387 393 |
23 | Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD (2000) A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease Nature 408: 6815 979 982 |
24 | Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J (2000) A|[beta]| peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease Nature 408: 6815 982 985 |
25 | McLaurin J, Cecal R, Kierstead ME, Tian X, Phinney AL, Manea M (2002) Therapeutically effective antibodies against amyloid-beta peptide target amyloid-beta residues 4-10 and inhibit cytotoxicity and fibrillogenesis Nat Med 8: 11 1263 1269 |
26 | Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A (2008) Long-term effects of Abeta42 immunisation in Alzheimer’s disease: Follow-up of a randomised, placebo-controlled phase I trial The Lancet 372: 9634 216 223 |
27 | Lannfelt L, Relkin NR, Siemers ER (2014) Amyloid-ß-directed immunotherapy for Alzheimer’s disease J Intern Med 275: 3 284 295 |
28 | Lemere CA, Masliah E (2010) Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol 6: 2 108 119 |
29 | Mechanisms, Clinical Strategies (2015) Promising Treatments ofNeurodegenerative Diseases. 12th International Conference AD/PDNice, France, March 18-22, 2015: Abstracts. Neuro-degenerativediseases 15 Suppl 1: 1 1969 |
30 | Doody R, Thomas RG, Farlow M, Iwatsubo T (2014) Phase 3 Trials of Solanezumab for Mild-to-Moderate Alzheimer’s Disease NEJM 370: 311 321 |
31 | Liu-Seifert H, Siemers E, Holdrige K, Andersen S, et al. Delayed-Start of up to 3.5 Years in the Phase 3 SolanezumabExpedition Program in Mild Alzheimer’s Disease. Presented atAlzheimer’s Association International Conference 2015, July18–23, 2015. |
32 | Spencer B, Masliah E (2014) Immunotherapy for Alzheimer’sdisease: Past, present and future Front Aging Neurosci 6: 114 |
33 | Pedersen JT, Sigurdsson EM (2015) Tau immuno-therapy forAlzheimer’s disease Trends Mol Med 21: 6 394 402 |
34 | Castillo-Carranza DL, Guerrero-Munoz MJ, Sengupta U, Hernandez C, Barrett ADT, Dineley K (2015) Tau Immunotherapy Modulates Both Pathological Tau and Upstream Amyloid Pathology in an Alzheimer’s Disease Mouse Model J Neurosci 35: 12 4857 4868 |
35 | Neuwelt EA, Bauer B, Fahlke C, Fricker G, Iadecola C, Janigro D (2011) Engaging neuroscience to advance translational research in brain barrier biology Nat RevNeurosci 12: 3 169 182 |
36 | Brightman MW, Reese TS (1969) Junctions between intimately apposed cell membranes in the vertebrate brain J Cell Biol 40: 3 648 677 |
37 | Reese TS, Karnovsky MJ (1967) Fine structural localization of a blood-brain barrier to exogenous peroxidase J Cell Biol 34: 1 207 217 |
38 | Yu YJ, Watts RJ (2013) Developing Therapeutic Antibodies for Neurodegenerative Disease Neurotherapeutics 10: 3 459 472 |
39 | Rubin LL, Staddon JM (1999) The Cell Biology of the blood brain barrier Annu Rev Neurosci 22: 1 11 28 |
40 | Tam SJ, Watts RJ (2010) Connecting vascular and nervous system development: Angiogenesis and the blood-brain barrier Ann Rev Neurosci 33: 1 379 |
41 | Lee G, Dallas S, Hong M, Bendayan R (2001) Drug transporters inthe central nervous system: Brain barriers and brain parenchymaconsiderations Pharmacol Rev 53: 4 569 596 |
42 | Banks WA, Terrell B, Farr SA, Robinson SM, Nonaka N, Morley JE (2002) Passage of amyloid beta protein antibody across the blood-brain barrier in a mouse model of Alzheimer’s disease Peptides 23: 12 2223 2226 |
43 | Banks WA, Farr SA, Morley JE, Wolf KM, Geylis V, Steinitz M (2007) Anti-amyloid beta protein antibody passage across theblood-brain barrier in the SAMP8 mouse model of Alzheimer’sdisease: An age-related selective uptake with reversal of learningimpairment Exp Neurol 206: 2 248 256 |
44 | Wang A, Das P, Switzer RC, Golde TE, Jankowsky JL (2011) Robust amyloid clearance in a mouse model of Alzheimer’s disease provides novel insights into the mechanism of amyloid-beta immunotherapy J Neurosci 31: 11 4124 4136 |
45 | Schlachetzki F, Zhu C, Pardridge WM (2002) Expression of the neonatal Fc receptor (FcRn) at the blood-brain barrier J Neurochem 81: 1 203 206 |
46 | Jefferies WA, Brandon MR, Hunt SV, Williams AF, Gatter KC, Mason DY (1984) Transferrin receptor on endothelium of brain capillaries Nature 312: 5990 162 163 |
47 | Duffy KR, Pardridge WM (1987) Blood-brain barrier transcytosis of insulin in developing rabbits Brain Res 420: 1 32 38 |
48 | Golden PL, Maccagnan TJ, Pardridge WM (1997) Human blood-brain barrier leptin receptor. Binding and endocytosis in isolated human brain microvessels J Clin Invest 99: 1 14 18 |
49 | Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD (2015) Structural and functional features of centralnervous system lymphatic vessels Nature 523: 337 341 |
50 | Allen Reish HE, Standaert DG (2015) Role of α-Synuclein in Inducing Innate and Adaptive Immunity in ParkinsonDisease J Parkinsons Dis 5: 1 |
51 | Hirsch EC, Hunot S (2009) Neuroinflammation inParkinson’s disease: A target for neuroprotection? Lancet Neurol 8: 4 382 397 |
52 | Blandini F (2013) Neural and Immune Mechanisms in the Pathogenesisof Parkinson’s Disease J NeuroimmunePharmacol 8: 1 189 201 |
53 | Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D (2010) Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease Nat Genet 42: 9 781 785 |
54 | Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease Nat Genet 1 7 |
55 | Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W (2012) Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia Glia 61: 3 349 360 |
56 | Marques O, Outeiro TF (2012) Alpha-synuclein: From secretion to dysfunction and death Cell Death Dis 3: 7 e350 |
57 | Kim C, Ho D-H, Suk J-E, You S, Michael S, Kang J (2013) Neuron-released oligomeric α-synuclein is an endogenousagonist of TLR2 for paracrine activation of microglia NatComms 1: 4 1562 1572 |
58 | Watson MB, Richter F, Lee SK, Gabby L, Wu J, Masliah E (2012) Regionally-specific microglial activation in young mice over-expressing human wildtype alpha-synuclein Exp Neurol 237: 2 318 334 |
59 | Gardai SJ, Mao W, Schüle B, Babcock M, Schoebel S, Lorenzana C (2013) Elevated Alpha-Synuclein Impairs Innate Immune Cell Function and Provides a Potential Peripheral Biomarker for Parkinson’s Disease. Vekrellis K, editor. PLoS ONE 8: 8 e71634 |
60 | Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, &Federoff HJ (2008) Synuclein activates microglia in a model ofParkinson’s disease Neurobiol Aging 29: 11 1690 1701 |
61 | Park J-Y, Paik SR, Jou I, Park SM (2008) Microglial phagocytosis is enhanced by monomeric α-synuclein, not aggregated α-synuclein: Imlications for Parkinson’s disease Glia 56: 11 1215 1223 |
62 | Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML (2005) Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease FASEB J 19: 6 533 542 |
63 | Lee H-J, Suk J-E, Bae E-J, Lee S-J (2008) Clearance and deposition of extracellular α-synuclein aggregates in microglia Biochem Biophys Res Commun 372: 3 423 428 |
64 | Lindström V, Ihse E, Fagerqvist T, Bergström J, Nordström E, Möller C (2014) Immunotherapy targeting α-synuclein, with relevance for future treatment of Parkinson’s disease and other Lewy body disorders Immunotherapy 6: 2 141 153 |
65 | Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M (2005) Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease Neuron 46: 6 857 868 |
66 | Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I (2002) Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters J Neurosci Res 68: 5 568 578 |
67 | Sanchez-Guajardo V, Annibali A, Jensen PH, Romero-Ramos M (2013) α-Synuclein vaccination prevents the accumulationof parkinson disease-like pathologic inclusions in striatum inassociation with regulatory T cell recruitment in a rat model J Neuropathol Exp Neurol 72: 7 624 645 |
68 | Perry VH, Nicoll JAR, Holmes C (2010) Microglia in neurodegenerative disease Nat Rev Neurol 6: 4 193 201 |
69 | Mandler M, Valera E, Rockenstein E, Weninger H, Patrick C, Adame A (2014) Next-generation active immunization approach for synucleinopathies: Implications for Parkinson’s disease clinical trials Acta Neuropathol 127: 6 861 879 |
70 | Monsonego A, Zota V, Karni A, Krieger JI, Bar-Or A, Bitan G (2003) Increased T cell reactivity to amyloid β protein in older humans and patients with Alzheimer disease J Clin Invest 112: 3 415 422 |
71 | Mandler M, Valera E, Rockenstein E, Mante M, Weninger H, Patrick C (2015) Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy Mol Neurodegen 10: 1 721 |
72 | Valera E, Masliah E (2013) Immunotherapy for neurodegenerative diseases: Focus on α-synucleinopathies PharmacolTherap 138: 3 311 322 |
73 | Masliah E, Rockenstein E, Mante M, Crews L, Spencer B, Adame A (2011) Passive Immunization Reduces Behavioral and Neuropathological Deficits in an Alpha-Synuclein Transgenic Model of Lewy Body Disease. McAlonan GM, editor PLoS ONE 6: 4 e19338 |
74 | Mishizen-Eberz AJ, Guttmann RP, Giasson BI, Day GA, Hodara R, Ischiropoulos H (2003) Distinct cleavage patterns of normal and pathologic forms of alpha-synuclein by calpain I in vitro J Neurochem 86: 4 836 847 |
75 | Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L (2005) Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations Proc Natl Acad Sci USA 102: 6 2162 2167 |
76 | Dufty BM, Warner LR, Hou ST, Jiang SX, Gomez-Isla T, Leenhouts KM (2007) Calpain-Cleavage of α-Synuclein The American Journal of Pathology 170: 5 1725 1738 |
77 | Shahaduzzaman M, Nash K, Hudson C, Sharif M, GrimmigB , Lin X (2015) Anti-human α-synuclein N-terminal peptide antibody protects against dopaminergic celldeath and ameliorates behavioral deficits in anAAV-α-synuclein rat model of Parkinson’s disease PLoS ONE 10: 2 e0116841 |
78 | Roberts RF, Wade-Martins R, Alegre-Abarrategui J (2015) Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinson’s disease brain Brain 138: Pt 6 1642 1657 |
79 | Näsström T, Goncalves S, Sahlin C, Nordström E, Screpanti Sundquist V, Lannfelt L (2011) Antibodies against Alpha-Synuclein Reduce Oligomerization in Living Cells. e PLoS ONE 6: 10 27230 |
80 | Fagerqvist T, Lindström V, Nordström E, Lord A, TuckerSME , Su X (2013) Monoclonal antibodies selective forα-synuclein oligomers/protofibrils recognize brainpathology in Lewy body disorders andα-synuclein transgenic mice withthe disease-causing A30P mutation J Neurochem 126: 1 131 144 |
81 | Lindström V, Fagerqvist T, Nordström E, Eriksson F, Lord A, Tucker S (2014) Immunotherapy targetingα-synuclein protofibrils reduced pathology in (Thy-1)-h[A30P] α-synuclein mice Neurobiol Dis 69: 134 143 |
82 | Spencer B, Emadi S, Desplats P, Eleuteri S, Michael S, Kosberg K (2014) ESCRT-mediated Uptake andDegradation of Brain-targeted α-synuclein Single Chain Antibody Attenuates Neuronal Degeneration Mol Ther 22: 10 1753 1767 |
83 | Chu Y, Kordower JH (2015) The prion hypothesis ofParkinson’s disease Curr Neurol Neurosci Rep 15: 5 28 |
84 | Tran HT, Chung CH-Y, Iba M, Bin Zhang, Trojanowski JQ, Luk KC (2014) a-Synuclein Immunotherapy Blocks Uptakeand Templated Propagation of Misfolded a-Synuclein and Neurodegeneration Cell Rep 7: 6 2054 2065 |
85 | Games D, Valera E, Spencer B, Rockenstein E, Mante M, Adame A (2014) Reducing C-Terminal-Truncated Alpha-Synuclein by Immunotherapy Attenuates Neurodegeneration and Propagation in Parkinson’s Disease-Like Models J Neurosci 34: 28 9441 9454 |
86 | Romero-Ramos M, Euler Chelpin von M, Sanchez-Guajardo V (2014) Vaccination strategies for Parkinson disease Hum Vaccin Immunother 10: 4 852 867 |
87 | Schwartz M, Butovsky O, Brück W (2006) Hanisch U-K. Microglial phenotype: Is the commitment reversible? Trends Neurosci 29: 2 68 74 |
88 | Reynolds AD, Stone DK, Hutter JAL, Benner EJ, Mosley RL, Gendelman HE (2010) Regulatory T cells attenuate Th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of parkinson’s disease J Immunol 184: 5 2261 2271 |
89 | Eberling JL, Dave KD, Frasier MA (2013) α-synuclein imaging: A critical need for Parkinson’s disease research.(4) J Parkinsons Dis 3: 565 567 |
Figures and Tables
Fig.1
Microglia take up human α-syn in mouse striatum in an in vivo model of α-syn cell-to-cell transfer. A. Confocal three-dimensional reconstruction of microglia (Iba1 positive, green) containing human α-syn (red). B. Reconstructed confocal orthogonal projections are presented as viewed in the x-z (bottom) and y-z(right) planes displays a perinuclear localisation of human α-syn within-microglia (nucleus stainedblue, DAPI).
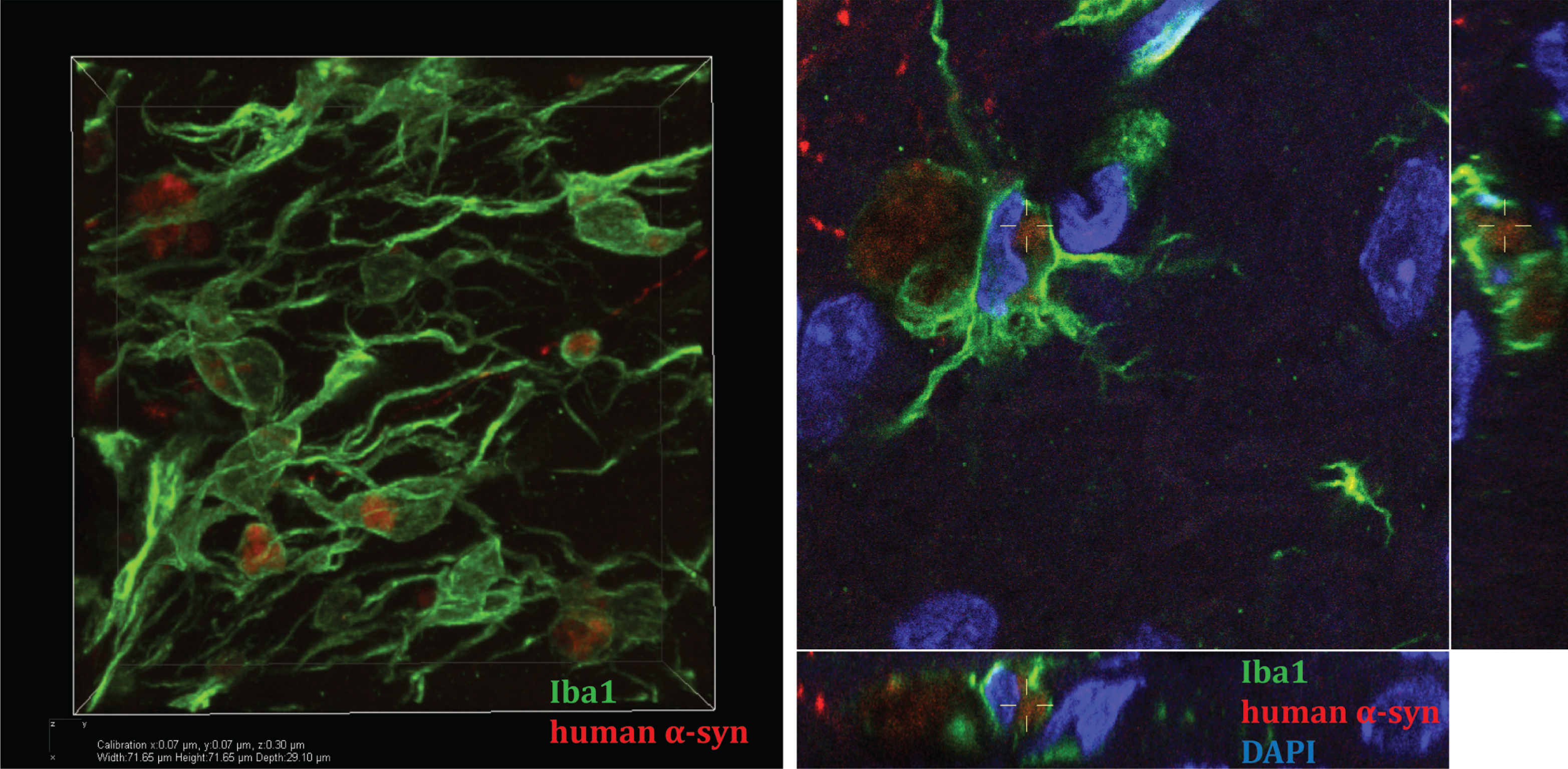
Table 1
α-Synuclein active immunization studies to date
| Animal Model | Antigen/antibody | Outcome | Reference |
| Transgenic mice expressing human α-syn under the platelet-derived growth factor β (PDGF-β) promoter, D-line | Recombinant human α-syn | Reduction of accumulated α-syn in neurons and higher number of synaptophysin-positive nerve terminals ameliorating neuronal damage. Mild microglia activation. | [65] |
| Sprague-Dawley rats injected with recombinant adeno-associated virus α-syn into substantia nigra | Recombinant human α-syn | Reduction of α-syn inclusions in substantia nigra. Induction of regulated T cells and activated microglia. | [67] |
| Two models: PDGF-α-syn mice expressing human α-syn under the PDGF-β promoter and mThy1-α-syn mice expressing human α-syn under the murine Thy1 promoter | C-terminus of α-syn (aa 110–130), also able to bind to full-length and N-terminal-truncated forms of α- syn, such as α-syn 96–140 | Reduced α-syn oligomers in axons and synapses. Reduced degeneration of striatal tyrosine hydroxylase-immunoreactive fibers. Clearance of α-syn involved microglia. Improved motor (body suspension test) and memory (Morris water maze) deficits in both models. | [69] |
| Mice expressing human α-syn under the control of the myelin basic protein promoter | Antigen mimicking the C-terminus of α-syn or the original C-terminus peptide (aa 110–130) | Decreased accumulation of α-syn, reduced demyelination in neocortex, striatum and corpus callosum. Reduced neurodegeneration. Activation and clearance of α-syn by microglia. Reduced spreading of α-syn to astrocytes. | [71] |
Table 2
α-Synuclein passive immunization studies to date
| Animal model | Antigen/antibody | Outcome | Reference |
| Transgenic mice expressing human α-syn under the PDGF-β promoter, D-line | α-syn C-terminus antibody 9E4 (IgG1), epitope 118-126 | Reduction of calpain-cleaved α-syn in neurons. No difference in microglia activation between control and antibody-treated mice. Less motor (rotarod) and cognitive impairment (Morris water maze). | [73] |
| Transgenic mice expressing human α-syn under the PDGF-β promoter, M-line | α-syn C-terminus antibody 274 (IgG2a), epitope 120-140 | Reduced accumulation of α-syn in neurons and astroglia. Increased presence of α-syn in microglia. Improved function in open field and pole tests. | [16] |
| Transgenic mice expressing human α-syn under the Thy-1 promoter, line 61 | C-Terminus α-syn antibodies: 1H7, 9E4, 5C1, and 5D12 | Attenuated synaptic and axonal pathology in cortex, hippocampus and striatum. Reduced accumulation of C-terminus-truncated α-syn in striatal axons and mitigated loss of tyrosine hydroxylase fibers. Reduced astrogliosis and microgliosis. Improved motor (round beam test) and memory deficits (Morris water maze). | [85] |
| Transgenic mice expressing human A30P α-syn under the Thy-1 promoter | α-syn protofibril-selective monoclonal antibody (mAb47) | Reductions of soluble and membrane-associated α-syn protofibrils in spinal cord. No increase in astrocytic or microglial activation. | [80] |
| Mice overexpressing human α-syn under the PDGF-β promoter (line D) | Single-chain fragment variables against oligomeric α-syn fused to the low-density lipoprotein receptor-binding domain of apolipoprotein B | Decreased α-syn accumulation in neocortex and hippocampus. Oligomeric and phosphorylated α-syn was reduced. Increased neuron numbers and synapses, as well as reduced levels of astrocytes. Improved memory function (Morris water maze). | [82] |
| Intrastriatal stereotaxic injections of α-syn preformed fibrils in wild type C57Bl6/C3H-mice | Monoclonal antibody: Syn303 (binds pathological conformations of human and mouse α-syn) targeting N-terminus) | Reduction of Lewy-like pathology, amelioration of nigral dopamine neuron loss. No differences in astrogliosis and microgliosis. Improved motor behavior (rotarod, wire hang test and open field activity). | [84] |
| Fisher 344 male rats injected into substantia nigra with recombinant adeno-associated viral vector expressing human wild type α-syn | Antibodies against the N-terminal or central region of α-syn | Lowered levels of α-syn, reduced α-syn-induced dopaminergic cell loss. Decreased number of activated microglia. Partial improvement of behavioral deficits (cylinder test). | [77] |
Table 3
α-Synuclein human immunization studies reported at conferences or in media to date
| Trial design/ Subjects | Antigen/antibody | Outcome | Reference |
| Passive immunization, Phase 1/ Parkinson’s disease patients | α-syn conjugate (AFFITOPE® PD01; Affiris, Vienna, Austria) | Safe and tolerated. Induction of α-syn antibodies | https://clinicaltrials.gov/ct2/show/NCT01568099 |
| Passive immunization, Phase 1/ Healthy subjects | Monoclonal antibody targeting α-syn (PRX002; Prothena, Dublin, Ireland) | Safe and tolerated. Reduced free serum α-syn levels of up to 96% | http://www.prothena.com/pipeline/prx002-for-parkinsons-disease/ |


