Role of Different Alpha-Synuclein Strains in Synucleinopathies, Similarities with other Neurodegenerative Diseases
Abstract
Misfolded protein aggregates are the hallmark of several neurodegenerative diseases in humans. The main protein constituent of these aggregates and the regions within the brain that are affected differ from one neurodegenerative disorder to another. A plethora of reports suggest that distinct diseases have in common the ability of protein aggregates to spread and amplify within the central nervous system. This review summarizes briefly what is known about the nature of the protein aggregates that are infectious and the reason they are toxic to cells. The chameleon property of polypeptides which aggregation into distinct high-molecular weight assemblies is associated to different diseases, in particular, that of alpha-synuclein which aggregation is the hallmark of distinct synucleinopathies, is discussed. Finally, strategies targeting the formation and propagation of structurally distinct alpha-synuclein assemblies associated to different synucleinopathies are presented and their therapeutic and diagnostic potential is discussed.
INTRODUCTION
Age-related dementias constitute a major health problem associated to increased life expectancy. These progressive diseases affect increasing number of people (presently 45 million people world wide, expected to reach 76 millions in 2030). These devastating diseases have high societal costs and lead, directly or indirectly, to a dramatic loss in life expectancy and quality of life. They represent therefore priorities for medical research and a threat to ageing societies. These diseases have in common the aggregation after misfolding and/or incorrect elimination of distinct polypeptides. The misfolded or incorrectly cleared proteins form aggregates that accumulate with time. The latter damage the cell, disrupt important neuronal structures and are therefore toxic. Most important however is their ability to amplify by recruiting the soluble form of homologous polypeptides and to propagate from one cell to another. Thus, through their ‘template-driven misfolding’ of similar proteins over time, e.g. amplification, spread from one cell from the central nervous system to another, e.g. transmissibility and accumulation within the central nervous system, misfolded protein aggregates create a self-sustaining, exponential vicious circle leading to disease.
PROTEIN MISFOLDING AND DISEASE
Within our cells, nascent proteins fold spontaneously or following interaction with molecular chaperones in order to be functional [1]. At any given time a fraction of newly synthesized polypeptides populate non-native conformers [2–4], with a sub-fraction corresponding to mis-processed peptides (for example Aβ after APP cleavage by β and γ secretases instead of the α and γ secretase product p3) or polypeptides where translation errors occurred. A fraction of these conformers is given another chance to fold correctly after full/partial unfolding by molecular chaperones [1] while another, never reaches the native state and is cleared or exported outside the cell. Thus, by opposing to protein misfolding, unfolding and aggregation, molecular chaperones, play a central role in the maintenance of normal cellular proteostasis [5]. Upon cellular stress the amounts of misfolded/unfolded proteins and/or mis-processed and mutant polypeptides increase [6]. This triggers heat shock response via the activation of the heat shock transcription factor 1 leading to partial correction of misfolding/unfolding and incorrect processing [5, 7, 8]. As sustained activation of the heat shock response is detrimental [9], normal cellular proteostasis is imbalanced with saturation of proteostatic networks [1, 5, 6]. As a consequence, misfolded proteins aggregate into intra- or extracellular protein deposits, depending on whether they are within the cytosol of in the extracellular milieu. It is widely accepted that the Ubiquitin Proteasome System degrades efficiently misfolded/unfolded or incorrectly processed proteins as long as the monomeric and aggregated forms are in dynamic equilibrium [10]. In contrast, high molecular weight irreversible assemblies are cleared by autophagy [11]. When the latter process fails either because of defects in autophagosome formation, transport, fusion with lysosomes or recognition of the substrate, protein aggregates accumulate with deleterious consequences [11].
Misfolded proteins aggregation can cause both loss of function and gain of pathologic function [12–14]. This is the consequence of either i - misfolded protein aggregates-mediated permeabilization of plasma membrane and/or membranous compartments, ii - perturbation in membrane protein dynamics and distribution they may cause, iii - the formation of novel pathogenic signaling platforms they may mediate, iv - the trapping of significant amounts of molecular chaperones and other partner proteins within the aggregates and v - the ability of the misfolded protein aggregates to recruit the soluble form of the proteins they are formed of and amplify.
Protein aggregates made of fibrillar material, in some cases straight as needles, in others curved and twisted, are the hallmarks of a variety of diseases affecting the central nervous system, in particular, Alzheimer’s (AD), Parkinson’s (PD), Huntington’s (HD) and Creutzfeldt-Jacob (CJD) diseases. The number of deposits within the affected tissues correlates most often with disease severity [15–17]. As the probability of occurrence of events such as i - misfolding and aggregation events of newly synthesized polypeptide chains, ii - unfolding of native polypeptide chains following a variety of stresses, iii - errors during translation and iv - incorrect processing and elimination of proteins targeted to degradation, is dependent on time, misfolding diseases are associated to ageing [18].
SPREAD OF MISFOLDED PROTEIN AGGREGATES
Until recently, the spread and transmission of disease via misfolded protein aggregates was thought to be restricted to the prion protein (PrP) [19]. A wide range of recent experimental evidence suggest that other protein aggregates that are the hallmarks of major neurodegenerative diseases spread in a prion-like manner [20, 21]. Thus, misfolded protein aggregates propagation and amplification appears to be a widespread phenomenon in neurodegenerative diseases as opposed to a curiosity limited to a unique protein: PrP. Indications for such a process originated from the work of Heiko Braak and co-workers who described distinctive patterns of pathological changes over time throughout the brain for the major neurodegenerative diseases in man. Indeed, based on autopsy cases, Braak and co-workers noticed that protein aggregates associated to pathology initiate in circumscribed areas of the brain specific to each disease and progress in a topographically predictable manner following anatomical connections [15–17]. These observations led them to establish a disease progression scale and to hypothesize that neurotropic pathogens were spreading through defined pathways to interconnected regions within the central nervous system. As for PrP, these pathogens were first thought to be of viral nature.
The first evidence for the transmission of misfolded protein aggregates not involving PrP in man came from the observation that Lewy bodies in PD brains contaminate grafted fetal mesencephalic progenitor neurons decades after transplantation [22, 23]. Additional evidence came from in vivo and in vitro experiments. Injection of brain extracts containing amyloid plaques, which main constituent is the aggregated form of the amyloid β-peptide (Aβ), neurofibrillary tangles, that are mainly made of the microtubule associated proteinTau, and Lewy bodies, which main constituent is the protein alpha-synuclein (α-syn), into the brain of model animal induced lesions characteristic of AD and PD [24–34]. No such lesions where observed when control brain extracts lacking protein aggregates were injected. The induced lesions, that were initially confined to the injected brain region, propagated to neighboring and/or axonally connected areas over time, suggesting directed spreading and amplification through neuronal transport processes [32–35]. The intraperitoneal injection of brain extracts containing amyloid plaques and neurofibrillary tangles also induced cerebral lesions characteristic of AD (Aβ and Tau deposits) [36, 37]. Similarly, the injection of brain extracts and Lewy bodies enriched fractions from patients developing PD into the brain of non-human primates, wild-type and model mice, induced several months after injection the formation of lesions characteristic of PD [31–35]. Finally, Lewy bodies of human nature appeared in rodents neural grafts implanted in the brains of mice expressing human α-syn that develop Lewy bodies over time [30] and aggregated huntingtin (Htt) with a polyQ stretch length associated to HD and huntingtin N-terminal moiety (Htt-Exon1) were shown to move between cultured cells, in organotypic slice cultures of a Htt mouse model and between host and grafted tissues [38–40].
Luckily, we can reproduce in test tubes the aggregation of Aβ, Tau, α-syn, Htt-Exon1 with a polyQ stretch length associated to HD, and Cu/Zn superoxide dismutase 1 (SOD1) point mutants associated to motor neuron death in familial cases of amyotrophic lateral sclerosis (ALS). Pathologic Htt-Exon1, only in its fibrillar form, binds to cultured cells, is internalized through yet an unknown mechanism, and induces the aggregation of sub-pathologic Htt-Exon1 within the cytosol of the recipient cells [38, 41]. α-syn mega-Dalton protein assemblies were also shown to be taken up [30] and to trigger the aggregation of endogenous cytosolic α-syn in cultured primary cortical neurons and in neuronal cell lines [42–45]. They were shown to be transported anterogradely and retrogradely following their uptake in primary neurons and to be transmitted to secondary neurons [46]. In vitro generated Tau assemblies were also shown to bind and enter cells and to transmit a misfolded state to intracellular Tau [44, 47–49]. Finally, mutant SOD1 aggregates made in vitro were shown to penetrate inside neurons by macropinocytosis and induce the aggregation of soluble mutant SOD1 in the cytoplasm [50].
Tau and α-syn assemblies made in vitro were also inoculated to transgenic model mice. They were taken up by neurons and glial cells and shown to transfer from neurons to oligodendrocytes [51]. Fibrillar assemblies induced several months after injection the formation of lesions characteristic of PD and AD [32, 37].
THE NATURE OF THE TOXIC PROTEIN ASSEMBLIES
Under physiological pH, salt and temperature conditions, Aβ, Tau, α-syn, Htt-Exon1 with a pathologic polyQ stretch and mutant SOD1, self-associate into assemblies that can elongate in a cooperative manner by incorporation of monomers. Growth is unlimited as the incorporation of a monomer creates a new binding site for another monomer (Fig. 1). Metastable, prefibrillar oligomeric intermediates, considered as the precursors of mature fibrils, are populated prior or concomitant to fibrils formation [52, 53]. In some cases, it is the binding of ligands to aggregation prone polypeptides or their modifications that yield oligomeric species that are off-assembly pathway as for example upon the interaction of α-syn with dopamine [54]. The nature of the assemblies that exhibit the highest toxicity and/or seeding capacity is subject to intense debate. Fibrillar assemblies made under physiological conditions have been shown to be the only entities capable of recruiting the soluble form of homolog proteins in vitro, in cell cultures and in vivo, i.e. with seeding capacities, and a body of evidence demonstrates their cytotoxic potential [30, 33, 38, 41, 43–45, 48–50, 55–58]. Nonetheless, they are considered as storage, inert form of misfolded proteins by those who consider the oligomers that form during the early stages of assembly as toxic [59–63]. Toxicity in the latter case is thought to be the consequence of impairment of membrane integrity and permeability [59, 64, 65]. The conflict is certainly due in some cases to non-physiological assembly conditions such as assembly after polypeptide dehydration, under high or low pH, at very low ionic strength (e.g. distilled water), extreme salt conditions and temperatures that are suitable for extremophiles. It is also and mostly due to i - comparison of the properties of protein particles at different concentrations, ii - difficulties in defining what is precisely an oligomer and iii- oligomers - fibrils inter-conversion. At a given concentration of monomeric Aβ, Tau, α-syn, Htt-Exon1 with a pathologic polyQ stretch or mutant SOD1 the concentrations of on-assembly pathway oligomers and fibrils diverge by several orders of magnitude as the molecular masses, number of constituting molecules and sizes of oligomeric and fibrillar species differ very significantly. Indeed, oligomeric species are made of less than 10 to 50 monomeric proteins at most while fibrils that are over 0.1 μm in length are made of thousands of polypeptide chains. Thus, it is critical to compare the toxicity of the different assemblies at identical particle concentrations as cells interact with the particles as a whole, not the precursor monomeric protein within the assemblies. Measurement of the molecular weight of defined on-assembly pathway oligomeric assemblies and fibrils with homogeneous length allowed calculating the particle concentration of assemblies at any given concentration of precursor monomeric polypeptide. When the relative toxicities of monomeric, oligomeric, and fibrillar assemblies were compared, the fibrillar assemblies, with homolog polypeptide seeding and spreading propensity, were found most toxic [66]. As fibrils and oligomers have distinct molecular weights they are frequently separated by centrifugation. However as the solutions are often viscous, a significant amount of short fibrils may remain in the supernatant. It is therefore important to bear in mind that one cannot exclude that observations derived from such fractionation procedures are due to the presence of short fibrils. Finally as fibrils and precursor oligomeric species are in equilibrium [67] one needs to keep in mind that oligomers can form fibrils in the time course of the experiment either in solution or when associated to the cell membranes as their concentrations increase when moving from a three- (test tube) to a bi- (cell membrane) dimensional space. Similarly, while fibrils most often exhibit some resistance to detergents [68], one cannot exclude their possible disassembly by cellular factors [69].
WHY PROTEIN ASSEMBLIES ARE TOXIC?
The toxicity of fibrillar and oligomeric assemblies is in part the consequence of their binding to and permeabilization of the cell membrane [70]. The binding of assemblies, besides inducing lipid rafts and the monosialoganglioside GM1 clustering [71], certainly impinge membrane curvature [72], thus further affecting cell survival. Furthermore, the lateral diffusion of assemblies in the plane of the plasma membrane together or independently of membranous components such as receptors, channels, adhesion molecules, integrins certainly contributes to the redistribution and/or coalescence of protein receptors at the surface of the cell [73]. Misfolded protein assemblies may even selectively recruit membranous proteins thus creating novel signaling platform with undesirable properties, thus contributing to cell apoptosis. Once internalized, they appear to leak out of the endosomal-lysosomal compartments as fibrils in particular reach the cytosol and seed the aggregation of their soluble cytosolic counterparts [38, 42, 45, 47]. By crossing these membranous compartments, through yet an unknown mechanism, they may cause leakage of the lysosomal content or rupture of lysozomes, with cathepsin B-dependent increase in reactive oxygen species within the cytosol, thus triggering pathogenesis [74]. The latter may account for the mitochondrial dysfunction and inflammation described in synucleinopathies [75]. Upon reaching the cytosol, exogenous misfolded protein assemblies certainly imbalance proteostasis following the titration of the cytosolic molecular chaperones and/or saturation of the clearance machinery of the cell leading to additional deleterious effects [5, 76]. Finally, toxicity may also be the consequence of the seeding capacity of the protein assemblies, e.g. the recruitment of fibrils soluble counterpart and exhaustion of the functional form of the protein.
As exogenous Aβ, Tau, α-syn etc ... assemblies bind and penetrate cells, amplify by recruiting their endogenous soluble counterparts through a seeding process and propagate to other cells they can be considered as infectious [77] in accord with the modifications that have been proposed to Koch’s postulates [78].
THE NOTION OF INFECTIOUS PROTEINS STRAINS
The observation that the aggregation of non-mutant PrP give rise to distinct diseases (Creutzfeldt Jakob’s disease (CJD), new variant CJD (vCJD) and Kuru in man, classical and Nor98 scrapie in sheep, classical and atypical (H-BSE and L-BSE) bovine spongiform encephalopathies (BSE)) that are distinguishable by the disease incubation time, lesions type in the brain and proteolytic cleavage patterns has led to the notion that PrP can adopt different conformations into the mega-Dalton assemblies associated to disease. Indeed, each conformation an amino acid stretch adopts can establish a defined set of inter-molecular interactions. As a consequence alternative packing is possible yielding structurally unlike assemblies that expose distinct surfaces (Fig. 1). The lateral surfaces of these different assemblies rule their ability to interact with receptors and lipids at the surface of the cells and protein partners within the cell. They dictate functional properties such as toxicity. The surfaces at the tips of structurallydistinct assemblies determine their growth rate. These distinct conformations are indeed imprinted to the newly incorporated PrP upon assemblies growth either through a templating process or the selective recruitment of defined PrP conformers that can establish intermolecular interactions with the well defined assemblies tips thus leading to the faithful amplification of distinct prion strains.
In a manner similar to PrP, the deposition of α-syn within the central nervous system is not only tightly associated to PD but also to distinct pathological phenotypes: multiple system atrophy (MSA) and dementia with Lewy bodies (DLB). These diseases are grouped under the banner “synucleinopathies”. Besides being intimately linked to AD, the aggregation of Tau also leads to distinct tauopathies: argyrophilic grain disease, tangle-only dementia, Pick disease, progressive supra-nuclear palsy and cortico-basal degeneration. The involvement of the deposits made of the same protein in diseases exhibiting different phenotypic traits can be accounted for either by the existence of distinct i - α-syn and Tau strains, ii - areas within the central nervous system affected by neurodegeneration or iii - neuroinflammatory responses, given that neuroinflammation has been repeatedly linked to neurodegeneration [76]. Indeed, mounting evidence from epidemiological, postmortem, brain imaging and animal studies indicate that neuroinflammatoryprocesses are tightly linked to neurodegeneration [79–82]. Nonetheless, it is still unclear whether the inflammatory process is involved in disease etiology or a secondary consequence of neurodegeneration and whether neuroinflammation affects to different extents distinct brain regions.
STRAINS OF α-SYN, Aβ AND TAU
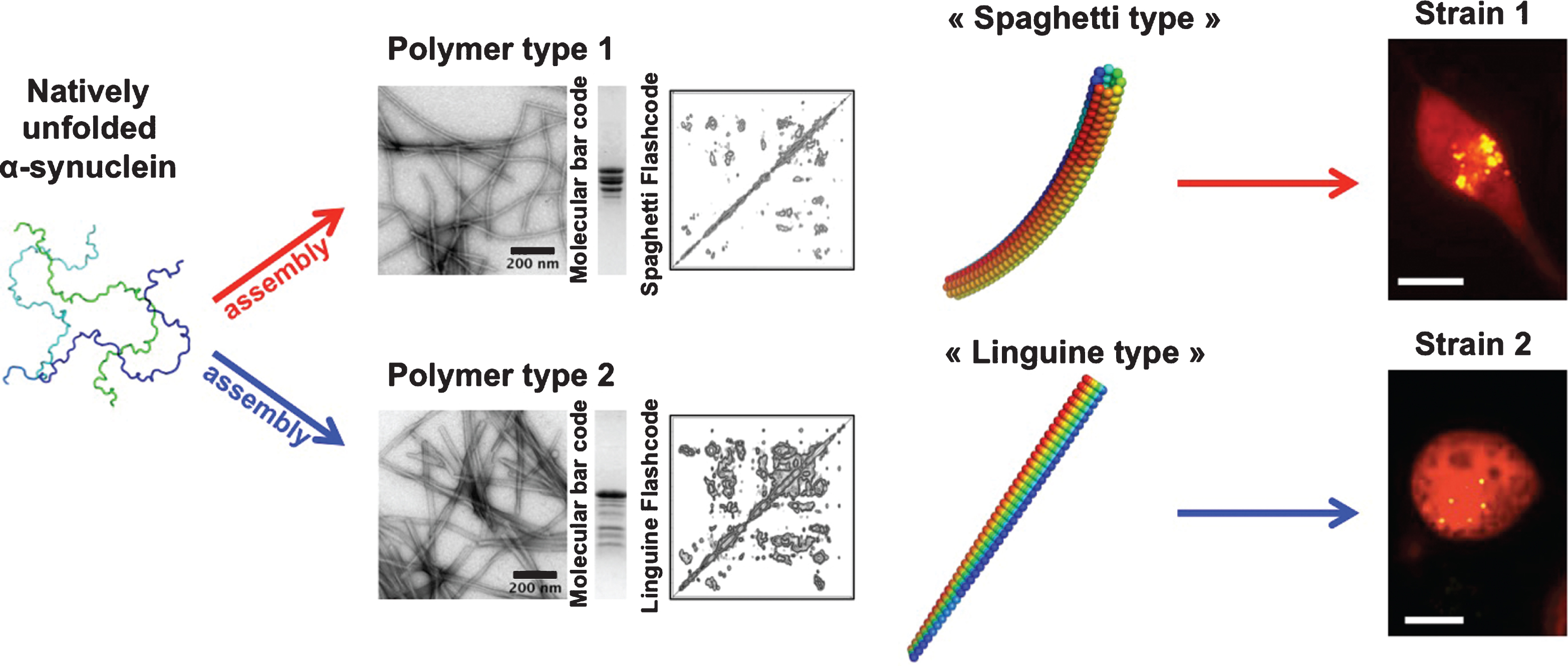
Differences in the biochemical properties of aggregated α-syn in different genetic backgrounds in humans have been reported suggesting that α-syn aggregates may be unlike [83]. The existence of α-syn strains was established upon the generation of pure α-syn fibrillar polymorphs under stringent and tighly controlled assembly conditions in vitro. Distinct fibrillar polymorphs made of wild-type, full-length α-syn, exhibited different shapes in the electron microscope, limited proteolytic and X-ray fiber diffraction patterns and were distinguishable by conformational antibodies. Assessment of their intrinsic structures by solid-state nuclear magnetic resonance (NMR) revealed that although they have high beta-sheet content, α-syn molecules within the fibrillar polymorphs are packed in distinct manner. As a consequence they recruit soluble α-syn with different efficacy, elongate at distinguishable rates and must have different surface properties. Functional studies revealed that the distinct polymorphs bind and permeabilize cells to different extents; they exhibit different toxicities, efficacy of recruitment of cellular α-syn and persistence/propagation propensity. They were also shown to imprint their distinct intrinsic structures to the recruited cellular α-syn [45] (Fig. 2).
The finding that distinct α-syn strains bind to cells and are taken up with different efficiencies suggest that they may exhibit different tropism for cells from the central nervous system, in other words, bind with different affinities to neurons, glial cells or astrocytes. By targeting different cells from the central nervous system or circuits, they may be at the origin of distinct synucleinopathies. As distinct strains differ by their surface properties they also may trigger the inflammatory response to different extents. Finally, the progression of distinct synucleinopathies may also reflect the different seeding propensity, resistance to clearance and persistence in cell cultures of distinct α-syn strains.
Baffling evidence for the existence of α-syn strains in vivo were brought by the groups of S. Prusiner, E. Bezard and M. Vila who demonstrated that semi-purified aggregated α-syn from patients developing MSA or PD faithfully led to MSA or PD-like neuropathological disorders in recipient animals [34, 84].
Additional evidence for the existence of putative α-syn strains came from the lesion profiles observed after injection of either newly formed α-syn fibrils orrepeatedly seeded α-syn fibrils and the differential induction of Tau aggregation by the two kinds of assemblies [85]. It also came from comparing the proteinase K degradation profiles of sarkosyl-insoluble protein fractions isolated from patients diagnosed PD or PD and AD because of abundant tau neurofibrillary tangles and senile plaque [85]. While one cannot rule out that the differences in the degradation profile of aggregated α-syn are due to the abundance of tau neurofibrillary tangles and A-beta these findings remain striking [85].
Recently, amyloid plaques extracted from the brain of two patients developing AD, with distinct neuropathology and clinical history, were used to seed the aggregation of Aβ in vitro. The resulting fibrils exhibited somewhat different shapes as judged by electron microscopy observations. They also yielded distinct solid-state NMR spectra [86]. Although Aβ aggregation in control brain extracts from healthy aged individuals and patients developing unrelated neurodegenerative diseases was not assessed, these observations suggest that Aβ within the initial seeds recruit soluble Aβ in vitro and imprint their structures to the synthetic, isotopically labeled, Aβ. Further evidence for the existence of Aβ strains came from the use of tightly controlled assembly conditions in vitro of synthetic Aβ1–40 and 1–42 [87]. Starting from the observation that synthetic fibrillar Aβ1–40 recruits when injected into the brain of recipient animals both Aβ1–40 and Aβ1–42 while synthetic fibrillar Aβ1–42 induce the aggregation of Aβ1–42, it was shown that slightly different assembly conditions lead synthetic fibrillar Aβ1–42 to stably acquire the seeding properties characteristic of fibrillar Aβ1–40.
Evidence for the existence of Tau strains have been brought through the use of truncated Tau. When exposed to the fibrillar form of truncated Tau, cells expressing the same truncated Tau fused to a reporter fluorescent protein develop truncated Tau clusters of different shapes. The pattern of these clusters is stably inherited over generations and transmissible to naïve cells. When lysates from cells exhibiting distinct clusters were injected into the brain of recipient model animals, they induced distinguishable neuropathological patterns that could be stably propagated in mice. Finally, brain homogenate from patients developing different tauopathies induced the clustering of reporter Tau in cultured cell lines. Reporter Tau inclusion morphology and limited degradation patterns revealed homogeneous for most pathological cases with exceptions suggesting that a number of patients may suffer from more than one Taustrain [49].
PROPAGATION OF Aβ, TAU AND α-SYN STRAINS AND DISEASE MODIFYING STRATEGIES
Symptomatic treatments are the only therapeutic tools available for patients developing AD and PD. In PD, for example drugs aiming at restoring dopamine levels, such as L-DOPA, dopamine agonists, MAO-B and COMT inhibitors, although powerful, lose efficiency and occasion side effects with time [88–90]. Invasive interventions, such as deep-brain stimulation, are implemented when drugs fail to relieve the patients. They are used when neurodegeneration has become sufficiently severe to induce a clinical phenotype. The discovery that Aβ, Tau and α-syn fibrillar assemblies transit between cells within the central nervous system and amplify by recruitment of their soluble counterparts in recipient cells, thus contributing to disease progression, suggests that pathogenesis starts many years before the first symptoms become apparent. Targeting early enough protein assemblies involved in disease formation and propagation may therefore represent a novel and effective preventive therapeutic avenue. Thus, in compliment to the classical approaches aimed at either inhibiting the initial aggregation of proteins or favoring their clearance by boosting the level of molecular chaperones within the cells and/or the ubiquitin-proteasome clearance machinery, or both, therapeutic strategies targeting disease-associated protein assemblies propagation and seeding capacities become highly relevant.
Small molecules such as the polyphenols rottlerin, curcumin or Epigallocatechin-3-gallate have been shown to either interfere with the aggregation of α-syn, stimulate proteostasis, activate the ubiquitin-proteasome clearance machinery or counteract the neuro-inflammatory response associated to α-syn aggregation [91–93]. A novel class of small molecules that repair proteostasis networks specifically through HSF-1, FOXO and Nrf-2 and restore proteome balance has been identified [94]. These molecules have a promising therapeutic potential in a variety of protein conformational diseases as they enhance cell stress pathways and chaperone activity [95]. They protect therefore the cells in a generic manner against misfolding and/or aggregation and the resulting pleiotropic damages. In addition, strategies aimed at protecting and/or stimulating neuronal cells through the use of neurotrophic factors such as brain-derived neurotrophic factor [96] or clearing the polypeptides associated to neurodegeneration prior to their aggregation/deposition through immunotherapy are explored [97, 98]. Finally, neural stem cell-based treatments for neurodegenerative diseases are considered [99].
In the case of PD, innovative strategies aimed at interfering specifically with α-syn assemblies propagation and seeding capacities have not yet been developed to a stage where they can be used for therapeutic purposes. The key step in this process is the docking of α-syn assemblies to the cell. One can interfere with this process either by i - perturbing the binding capacity of extrinsic membrane proteins that interact with α-syn assemblies or ii - changing the surface properties of α-syn assemblies so that they become incapable of binding to their receptors. As no specific receptors of α-syn assemblies have been identified so far, the strategy consisting of rendering those receptors unavailable for binding α-syn assemblies is impossible. Furthermore changing the binding capacities of these receptors may cause undesirable pleiotropic effects. Designing α-syn assemblies binders that change their surface properties in such a way that they either become unable to bind the cell surface or incapable of elongating by recruitment of the endogenous protein because their ends are capped appears more promising. Two classes of binders, molecular chaperones and antibodies, have been shown to interfere with α-syn assemblies uptake by cells and/or toxicity [100, 101]. These molecules cross the blood-brain barrier with difficulty because of their size. Thus, identifying within these molecules what is necessary and sufficient for binding is critical for the design of smaller molecules with therapeutic potential. The interaction interfaces between the molecular chaperone Hsc70 client protein binding site and α-syn have been identified through the use of chemical cross-linkers and mass spectrometry [102, 103]. The same strategy can be employed to identify paratopes within antibodies exhibiting high affinity for α-syn. Peptides reproducing Hsc70 or anti-α-syn paratopes that bind with the highest affinity α-syn assemblies represent potential therapeuticcandidates.
ACKNOWLEDGMENTS
Research carried out in RM laboratory is sponsored by the Centre National de la Recherche Scientifique and grants from the Agence Nationale de la Recherche Scientifique, The European Commission FP7 Marie Curie Actions grant no. 264508, The region Ile-de-France, The Fondation Bettencourt Schueller and the charity France Parkinson.
REFERENCES
1 | Hartl FU, Bracher A, Hayer-Hartl M (2011) Molecular chaperones in protein folding and proteostasis Nature 475: 324 332 |
2 | Anfinsen CB (1973) Principles that govern folding of protein chains Science 181: 223 230 |
3 | Dill KA, Chan HS (1997) From Levinthal to pathways to funnels Nature Struct Biol 4: 10 19 |
4 | Onuchic JN, Luthey-Schulten Z, Wolynes PG (1997) Theory of protein folding: The energy landscape perspective Annu Rev Phys Chem 48: 545 600 |
5 | Morimoto RI (2011) The heat shock response: Systems biology of proteotoxic stress in aging and disease Cold Spring Harb Symp Quant Biol 76: 91 99 |
6 | Balch WE, Morimoto RI, Dillin A, Kelly JW (2008) Adapting proteostasis for disease intervention Science 319: 916 919 |
7 | Anckar J, Sistonen L (2011) Regulation of HSF1 function in the heat stress response: Implications in aging and disease Annu Rev Biochem 80: 1089 1115 |
8 | Westerheide SD, Morimoto RI (2005) Heat shock response modulators as therapeutic tools for diseases of protein conformation J Biol Chem 280: 33097 33100 |
9 | Roth DM, Hutt DM, Tong J, Bouchecareilh M, Wang N, Seeley T, Dekkers JF, Beekman JM, Garza D, Drew L, Masliah E, Morimoto RI, Balch WE (2014) Modulation of the maladaptive stress response to manage diseases of protein folding PLoS Biol 12: e1001998 |
10 | Amm I, Sommer T, Wolf DH (2014) Protein quality control and elimination of protein waste: The role of the ubiquitin-proteasome system Biochim Biophys Acta 1843: 182 196 |
11 | Mariño G, Madeo F, Kroemer G (2011) Autophagy for tissue homeostasis and neuroprotection Curr Opin Cell Biol 23: 198 206 |
12 | Ross CA, Poirier MA (2014) Protein aggregation and neurodegenerative disease. S10-S Nat Med 10: (Suppl) 17 |
13 | Park SH, Kukushkin Y, Gupta R, Chen T, Konagai A, Hipp MS, Hayer-Hartl M, Hartl FU (2013) PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone Cell 154: 134 145 |
14 | Yu A, Shibata Y, Shah B, Calamini B, Lo DC, Morimoto RI (2014) Protein aggregation can inhibit clathrin-mediated endocytosis by chaperone competition Proc Natl Acad Sci U S A 111: E1481 E1490 |
15 | Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease Neurobiol Aging 24: 197 211 |
16 | Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes Acta Neuropathol 82: 239 259 |
17 | Thal DR, Rüb U, Orantes M, Braak H (2002) Phases of A beta-deposition in the human brain and its relevance for the development of AD Neurology 58: 1791 1800 |
18 | Labbadia J, Morimoto RI (2014) Proteostasis and longevity: When does aging really begin? F1000Prime Rep 6: 7 |
19 | Prusiner SB (2012) Cell biology. A unifying role for prions in neurodegenerative diseases Science 336: 1511 1513 |
20 | Brundin P, Melki R, Kopito R (2010) Prion-like transmission of protein aggregates in neurodegenerative diseases Nat Rev Mol Cell Biol 11: 301 307 |
21 | Jucker M, Walker LC (2013) Self-propagation of pathogenic protein aggregates in neurodegenerative diseases Nature 501: 45 51 |
22 | Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Björklund A, Widner H, Revesz T, Lindvall O, Brundin P (2008) Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation Nat Med 14: 501 503 |
23 | Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW (2008) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease Nat Med 14: 504 506 |
24 | Kane MD, Lipinski WJ, Callahan MJ, Bian F, Durham RA, Schwarz RD, Roher AE, Walker LC (2000) Evidence for seeding of beta-amyloid by intracerebral infusion of Alzheimer brain extracts in beta-amyloid precursor protein-transgenic mice J Neurosci 20: 3606 3611 |
25 | Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M (2006) Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host Science 313: 1781 1784 |
26 | Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Guerrero-Munoz MJ, Kiritoshi T, Neugebauer V, Jackson GR, Kayed R (2012) Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau Sci Rep 2: 700 |
27 | Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, Jucker M, Goedert M, Tolnay M (2009) Transmission and spreading of tauopathy in transgenic mouse brain Nat Cell Biol 11: 909 913 |
28 | Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, Probst A, Winkler DT, Reichwald J, Staufenbiel M, Ghetti B, Goedert M, Tolnay M (2013) Brain homogenates from human tauopathies induce tau inclusions in mouse brain Proc Natl Acad Sci U S A 110: 9535 9540 |
29 | Kordower JH, Dodiya HB, Kordower AM, Terpstra B, Paumier K, Madhavan L, Sortwell C, Steece-Collier K, Collier TJ (2011) Transfer of host-derived α synuclein to grafted dopaminergic neurons in rat Neurobiol Dis 43: 552 557 |
30 | Hansen C, Angot E, Bergström AL, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li JY, Brundin P (2011) α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells J Clin Invest 121: 715 725 |
31 | Mougenot AL, Nicot S, Bencsik A, Morignat E, Verchère J, Lakhdar L, Legastelois S, Baron T (2012) Prion-like acceleration of a synucleinopathy in a transgenic mouse model Neurobiol Aging 33: 2225 2228 |
32 | Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM (2012) Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice Science 338: 949 953 |
33 | Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM (2012) Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice J Exp Med 209: 975 986 |
34 | Recasens A, Dehay B, Bové J, Carballo-Carbajal I, Dovero S, Pérez-Villalba A, Fernagut PO, Blesa J, Parent A, Perier C, Fariñas I, Obeso JA, Bezard E, Vila M (2014) Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys Ann Neurol 75: 351 362 |
35 | Bétemps D, Verchère J, Brot S, Morignat E, Bousset L, Gaillard D, Lakhdar L, Melki R, Baron T (2014) Alpha-synuclein spreading in M83 mice brain revealed by detection of pathological α-synuclein by enhanced ELISA Commun 2: 29 |
36 | Eisele YS, Obermüller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, Walker LC, Staufenbiel M, Heikenwalder M, Jucker M (2010) Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis Science 330: 980 982 |
37 | Clavaguera F, Hench J, Lavenir I, Schweighauser G, Frank S, Goedert M, Tolnay M (2014) Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice Acta Neuropathol 127: 299 301 |
38 | Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR (2009) Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates Nat. Cell Biol 11: 219 225 |
39 | Pecho-Vrieseling E, Rieker C, Fuchs S, Bleckmann D, Esposito MS, Botta P, Goldstein C, Bernhard M, Galimberti I, Müller M, Lüthi A, Arber S, Bouwmeester T, van der Putten H, Di Giorgio FP (2014) Transneuronal propagation of mutant huntingtin contributes to non-cell autonomous pathology in neurons Nat Neurosci 17: 1064 1072 |
40 | Kordower JH, Halliday G (2014) Misfolded proteins in Huntington disease fetal grafts: Further evidence of cell-to-cell transfer? Ann Neurol 76: 20 21 |
41 | Trevino RS, Lauckner JE, Sourigues Y, Pearce MM, Bousset L, Melki R, Kopito RR (2012) Fibrillar structure and charge determine the interaction of polyglutamine protein aggregates with the cell surface J Biol Chem 287: 29722 29728 |
42 | Danzer KM, Krebs SK, Wolff M, Birk G, Hengerer B (2009) Seeding induced by alpha-synuclein oligomers provides evidence for spreading of alpha-synuclein pathology J Neurochem 111: 192 203 |
43 | Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM (2009) Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells Proc Natl Acad Sci U S A 106: 20051 20056 |
44 | Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M (2010) Seeded aggregation and toxicity of alpha-synuclein and tau: Cellular models of neurodegenerative diseases J Biol Chem 285: 34885 34898 |
45 | Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Böckmann A, Meier BH, Melki R (2013) Structural and functional characterization of two alpha-synuclein strains Nat Commun 4: 2575 |
46 | Freundt EC, Maynard N, Clancy EK, Roy S, Bousset L, Sourigues Y, Covert M, Melki R, Kirkegaard K, Brahic M (2012) Neuron-to-neuron transmission of α-synuclein fibrils through axonal transport Ann Neurol 72: 517 524 |
47 | Frost B, Jacks RL, Diamond MI (2009) Propagation of tau misfolding from the outside to the inside of a cell J Biol Chem 284: 12845 12852 |
48 | Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI (2012) Trans-cellular propagation of Tau aggregation by fibrillar species J Biol Chem 287: 19440 19451 |
49 | Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC, Thorpe JR, Serpell LC, Miller TM, Grinberg LT, Seeley WW, Diamond MI (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies Neuron 82: 1271 1288 |
50 | Münch C, O’Brien J, Bertolotti A (2011) Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells Proc Natl Acad Sci U S A 108: 3548 3553 |
51 | Reyes JF, Rey NL, Bousset L, Melki R, Brundin P, Angot E (2014) Alpha-synuclein transfers from neurons to oligodendrocytes Glia 62: 387 398 |
52 | Goldberg MS, Lansbury PTJr (2000) Is there a cause-and-effect relationship between alpha-synuclein fibrilization and Parkinson’s disease? Nat Cell Biol 2: E115 E119 |
53 | Poirier MA, Li H, Macosko J, Cai S, Amzel M, Ross CA (2002) Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization J Biol Chem 277: 41032 41037 |
54 | Cappai R, Leck SL, Tew DJ, Williamson NA, Smith DP, Galatis D, Sharples RA, Curtain CC, Ali FE, Cherny RA, Culvenor JG, Bottomley SP, Masters CL, Barnham KJ, Hill AF (2005) Dopamine promotes alpha-synuclein aggregation into SDS-resistant soluble oligomers via a distinct folding pathway FASEB J 19: 1377 1379 |
55 | El-Agnaf OM, Jakes R, Curran MD, Middleton D, Ingenito R, Bianchi E, Pessi A, Neill D, Wallace A (1998) Aggregates from mutant and wild-type alpha-synuclein proteins and NAC peptide induce apoptotic cell death in human neuroblastoma cells by formation of beta-sheet and amyloid-like filaments FEBS Lett 440: 71 75 |
56 | Yang W, Dunlap JR, Andrews RB, Wetzel R (2002) Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells Hum Mol Genet 11: 2905 2917 |
57 | Nekooki-Machida Y, Kurosawa M, Nukina N, Ito K, Oda T, Tanaka M (2009) Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity Proc Natl Acad Sci U S A 106: 9679 9684 |
58 | Novitskaya V, Bocharova OV, Bronstein I, Baskakov IV (2006) Amyloid fibrils of mammalian prion protein are highly toxic to cultured cells and primary neurons J Biol Chem 281: 13828 13836 |
59 | Caughey B, Lansbury PTJr (2003) Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders Annu Rev Neurosci 26: 267 298 |
60 | Winner BR, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, Tzitzilonis C, Soragni A, Jessberger S, Mira H, Consiglio A, Pham E, Masliah E, Gage FH, Riek R (2011) In vivo demonstration that alpha-synuclein oligomers are toxic Proc Natl Acad Sci U S A 108: 4194 4199 |
61 | Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death Nature 431: 805 810 |
62 | Takahashi T, Kikuchi S, Katada S, Nagai Y, Nishizawa M, Onodera O (2008) Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic Hum Mol Genet 17: 345 356 |
63 | Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases Nature 416: 507 511 |
64 | Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M (2007) Different species of asynuclein oligomers induce calcium influx and seeding J Neurosci 27: 9220 9232 |
65 | Kagan BL, Hirakura Y, Azimov R, Azimova R (2001) The channel hypothesis of Huntington’s disease Brain Res Bull 56: 281 284 |
66 | Pieri L, Madiona K, Bousset L, Melki R (2012) Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells Biophys J 102: 2894 2905 |
67 | Schmit JD, Ghosh K, Dill K (2011) What drives amyloid molecules to assemble into oligomers and fibrils? Biophys J 100: 450 458 |
68 | Wanker EE, Scherzinger E, Heiser V, Sittler A, Eickhoff H, Lehrach H (1999) Membrane filter assay for detection of amyloid-like polyglutamine-containing protein aggregates Methods Enzymol 309: 375 386 |
69 | Torrente MP, Shorter J (2013) The metazoan protein disaggregase and amyloid depolymerase system: Hsp110, Hsp70, Hsp40, and small heat shock proteins Prion 7: 457 463 |
70 | Cecchi C, Stefani M (2013) The amyloid-cell membrane system. The interplay between the biophysical features of oligomers/fibrils and cell membrane defines amyloid toxicity Biophys Chem 182: 30 43 |
71 | Bucciantini M, Nosi D, Forzan M, Russo E, Calamai M, Pieri L, Formigli L, Quercioli F, Soria S, Pavone F, Savistchenko J, Melki R, Stefani M (2012) Toxic effects of amyloid fibrils on cell membranes: The importance of ganglioside GM1 FASEB J 26: 818 831 |
72 | Smith PE, Brender JR, Ramamoorthy A (2009) Induction of negative curvature as a mechanism of cell toxicity by amyloidogenic peptides: The case of islet amyloid polypeptide J Am Chem Soc 131: 4470 4478 |
73 | Rossy J, Ma Y, Gaus K (2014) The organisation of the cell membrane: Do proteins rule lipids? Curr Opin Chem Biol 20: 54 59 |
74 | Freeman D, Cedillos R, Choyke S, Lukic Z, McGuire K, Marvin S, Burrage AM, Sudholt S, Rana A, O’Connor C, Wiethoff CM, Campbell EM (2013) Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis PLoS One 8: e62143 |
75 | Dias V, Junn E, Mouradian MM (2013) The role of oxidative stress in Parkinson’s disease J Parkinsons Dis 3: 461 491 |
76 | Wolff S, Weissman JS, Dillin A (2014) Differential scales of protein quality control Cell 157: 52 64 |
77 | Fernández-Borges N1, Eraña H, Elezgarai SR, Harrathi C, Gayosso M, Castilla J (2013) Infectivity versus seeding in neurodegenerative diseases sharing a prion-like mechanism Int J Cell Bio 2013: 583498 |
78 | Walker L, Levine H, Jucker M (2006) Koch’s postulates and infectious proteins Acta Neuropathol 12: 1 4 |
79 | Hirsch EC, Hunot S (2009) Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol 8: 382 397 |
80 | Heneka MT, Rodriguez JJ, Verkhratsky A (2010) Neuroglia in neurodegeneration Brain Research Reviews 63: 189 211 |
81 | Braak H, Sastre M, Del Tredici K (2007) Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease Acta Neuropathol 114: 231 241 |
82 | Tansey MG, Goldberg MS (2010) Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention Neurobiol Dis 37: 510 518 |
83 | Mamais A, Raja M, Manzoni C, Dihanich S, Lees A, Moore D, Lewis PA, Bandopadhyay R (2013) Divergent α-synuclein solubility and aggregation properties in G2019S LRRK2 Parkinson’s disease brains with Lewy Body pathology compared to idiopathic cases Neurobiol Dis 58: 183 190 |
84 | Watts JC, Giles K, Oehler A, Middleton L, Dexter DT, Gentleman SM, DeArmond SJ, Prusiner SB (2013) Transmission of multiple system atrophy prions to transgenic mice Proc Natl Acad Sci U S A 110: 19555 19560 |
85 | Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, Lee VM (2013) Distinct α-synuclein strains differentially promote tau inclusions in neurons Cell 154: 103 117 |
86 | Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R (2013) Molecular structure of β-amyloid fibrils in Alzheimer’s disease brain tissue Cell 154: 1257 1268 |
87 | Stöhr J, Condello C, Watts JC, Bloch L, Oehler A, Nick M, DeArmond SJ, Giles K, DeGrado WF, Prusiner SB (2014) Distinct synthetic Aβ prion strains producing different amyloid deposits in bigenic mice Proc Natl Acad Sci U S A 111: 10329 10334 |
88 | Connolly BS, Lang AE (2014) Pharmacological treatment of Parkinson disease: A review JAMA 311: 1670 1683 |
89 | Olanow CW, Agid Y, Mizuno Y, Albanese A, Bonuccelli U, Damier P, De Yebenes J, Gershanik O, Guttman M, Grandas F, Hallett M, Hornykiewicz O, Jenner P, Katzenschlager R, Langston WJ, LeWitt P, Melamed E, Mena MA, Michel PP, Mytilineou C, Obeso JA, Poewe W, Quinn N, Raisman-Vozari R, Rajput AH, Rascol O, Sampaio C, Stocchi F (2004) Levodopa in the treatment of Parkinson’s disease: Current controversies Mov Disord 19: 997 1005 |
90 | Fahn S, Oakes D, Shoulson I, Kieburtz K, Rudolph A, Lang A, Olanow CW, Tanner C, Marek KParkinson Study Group (2004) Levodopa and the progression of Parkinson’s disease N Engl J Med 351: 2498 2508 |
91 | Masuda M, Suzuki N, Taniguchi S, Oikawa T, Nonaka T, Iwatsubo T, Hisanaga S, Goedert M, Hasegawa M (2006) Small molecule inhibitors of alpha-synuclein filament assembly Biochemistry 45: 6085 6094 |
92 | Maioli E, Torricelli C, Valacchi G (2012) Rottlerin and curcumin: A comarative analysis Ann N Y Acad Sci 1259: 65 76 |
93 | Mähler A, Mandel S, Lorenz M, Ruegg U, Wanker EE, Boschmann M, Paul F (2013) Epigallocatechin-3-gallate: A useful, effective and safe clinical approach for targeted prevention and individualised treatment of neurological diseases? EPMA J 5: |
94 | Calamini B, Silva MC, Madoux F, Hutt DM, Khanna S, Chalfant MA, Saldanha SA, Hodder P, Tait BD, Garza D, Balch WE, Morimoto RI (2011) Small-molecule proteostasis regulators for protein conformational diseases Nat Chem Biol 8: 185 196 |
95 | Calamini B, Morimoto RI (2012) Protein homeostasis as a Therapeutic target for diseases of protein conformation Curr Top Med Chem 12: 2623 2640 |
96 | Nagahara AH, Tuszynski MH (2011) Potential therapeutic uses of BDNF in neurological and psychiatric disorders Nat Rev Drug Discov 10: 209 219 |
97 | Broadstock M, Ballard C, Corbett A (2014) Latest treatment options for Alzheimer’s disease, Parkinson’s disease dementia and dementia with Lewy bodies Expert Opin Pharmacother 15: 1797 1810 |
98 | Hampel H, Schneider LS, Giacobini E, Kivipelto M, Sindi S, Dubois B, Broich K, Nisticò R, Aisen PS, Lista S (2015) Advances in the therapy of Alzheimer’s disease: Targeting amyloid beta and tau and perspectives for the future Expert Rev Neurother 15: 83 105 |
99 | Kim SU, Lee HJ, Kim YB (2013) Neural stem cell-based treatment for neurodegenerative diseases Neuropathology 33: 491 504 |
100 | Pemberton S, Madiona K, Pieri L, Kabani M, Bousset L, Melki R (2011) Hsc70 protein interaction with soluble and fibrillar alpha-synuclein J Biol Chem 286: 34690 34699 |
101 | Tran HT, Chung CH, Iba M, Zhang B, Trojanowski JQ, Luk KC, Lee VM (2014) α-synuclein immunotherapy blocks uptake and templated propagation of misfolded α-synuclein and neurodegeneration Cell Rep 7: 2054 2065 |
102 | Redeker V, Pemberton S, Bienvenut W, Bousset L, Melki R (2012) Identification of protein interfaces between α-synuclein, the principal component of Lewy bodies in Parkinson disease, and the molecular chaperones human Hsc70 and the yeast Ssa1p J Biol Chem 287: 32630 32639 |
103 | Nury C, Redeker V, Dautrey S, Romieu A, van der Rest G, Renard PY, Melki R, Chamot-Rooke J (2015) A novel bio-orthogonal cross-linker for improved protein/protein interaction analysis Anal Chem 87: 1853 1860 |
Figures and Tables
Fig.1
Protein misfolding and aggregation into infectious fibrillar assemblies. Native or natively unfolded polypeptide chain (sphere) undergo conformational changes that lead to distinct abnormal (cube or cylinder) forms. The rates depend on the propensity of the polypeptide to populate any given (cube or cylinder) conformation. The abnormal forms are short lived either because they are unstable or sensitive to clearance. According to the “template assistance” model, the abnormal folding intermediates interact with the native form of the infectious protein and convert it into abnormal forms. The distinct abnormal forms of the polypeptide have the ability to interact transiently with like conformers and establish longitudinal or lateral interactions following the “seeded polymerization” model. The oligomeric species are unstable and dissociate because the inter-molecular interactions do not outweigh the entropic cost of binding. Once longitudinal and lateral interactions have been established between abnormal forms of the polypeptide, distinct stable seeds are formed (in brackets). These seeds grow indefinitely from one or both ends by incorporation of like molecules, yielding fibrils of distinct physical properties. The different fibrils can break into smaller fragments because, amongst other things, of Brownian movement. The rate of breakage depends on the number of bonds established between the molecules. Each resulting fragment acts as seed.
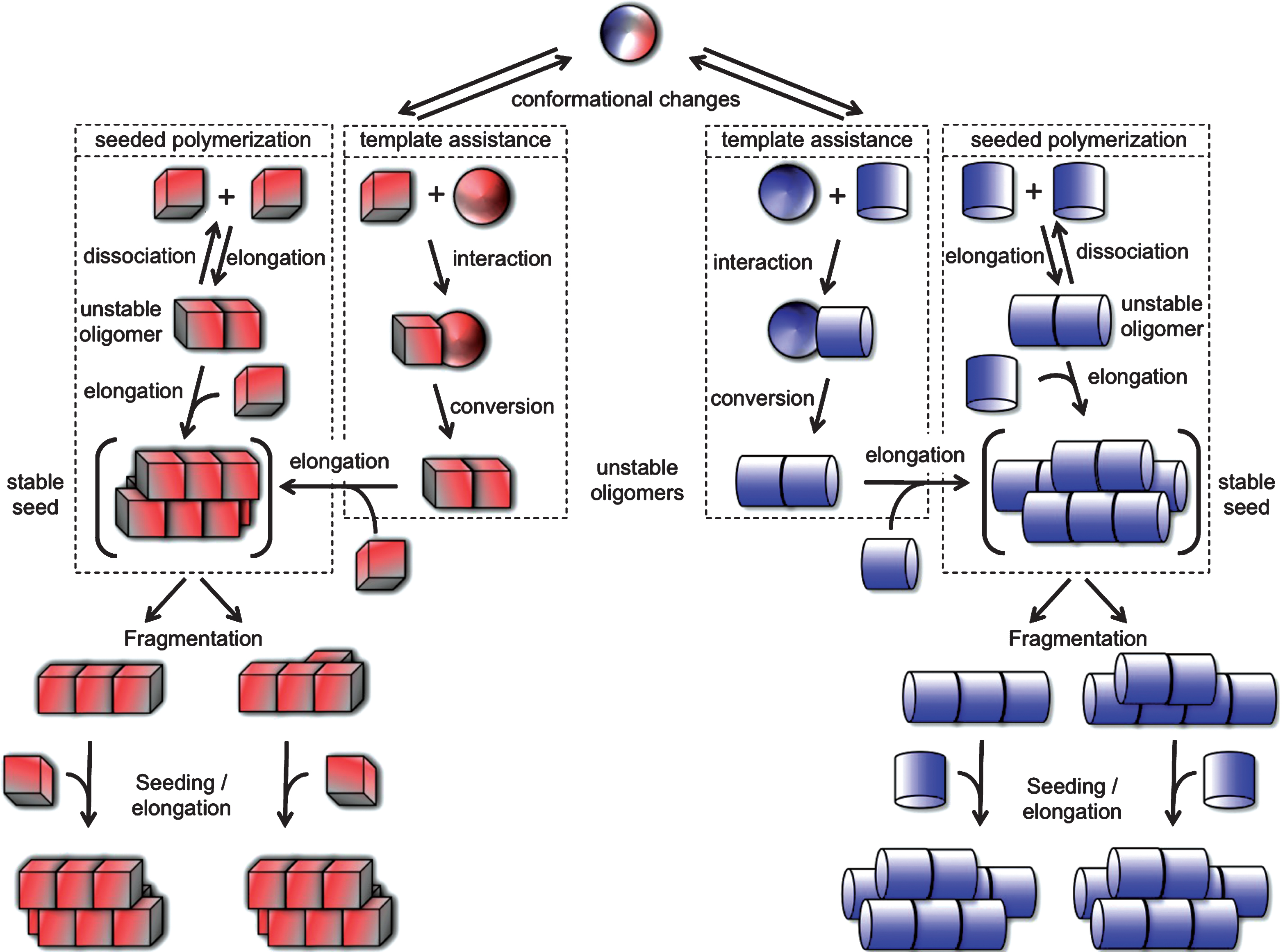
Fig.2
Alpha-synuclein strains. Natively unfolded α-syn (3 different conformations are presented), assemble into polymers that look unlike in the electron microscope. One type of polymers has the shape of spaghetti, while another type has the shape of a flat linguine. The two assemblies have distinct limited proteolysis degradation patterns (molecular bar codes) and solid-state NMR spectra (structural fingerprint). The two kinds of polymers made of the same protein have different functional properties (toxicity, binding propensity to cells, seeding capacities and persistence). They imprint their intrinsic structure to the soluble α-syn they recruit in cells. They behave therefore as two distinct strains.