
Clinical and Genetic Heterogeneity of Nuclear Envelopathy Related Muscular Dystrophies in an Indian Cohort
Abstract
Introduction:
Nuclear envelopathies occur due to structural and/or functional defects in various nuclear envelope proteins such as lamin A/C and lamin related proteins. This study is the first report on the phenotype-genotype patterns of nuclear envelopathy-related muscular dystrophies from India.
Methods:
In this retrospective study, we have described patients with genetically confirmed muscular dystrophy associated with nuclear envelopathy. Data on clinical, laboratory findings and muscle MRI were collected.
Results:
Sixteen patients were included with median age at onset of 3 years (range: 1 month – 17 years). Three genes were involved: LMNA (11, 68.75%), EMD (4, 25%) and SYNE1 (1, 6.25%). The 11 patients with LMNA variants were Congenital muscular dystrophy (MDCL)=4, Limb Girdle Muscular Dystrophy (LGMD1B)=4 and Emery-Dreifuss Muscular Dystrophy (EDMD2)=3. On muscle biopsy, one patient from each laminopathy phenotype (n = 3) revealed focal perivascular inflammatory infiltrate. Other notable features were ophthalmoparesis in one and facial weakness in one. None had cardiac involvement. Patients with EDMD1 had both upper (UL) and lower limb (LL) proximo-distal weakness. Cardiac rhythm disturbances such as sick sinus syndrome and atrial arrhythmias were noted in two patients with EDMD1. Only one patient with variant c.654_658dup (EMD) lost ambulation in the 3rd decade, 18 years after disease onset. Two had finger contractures with EMD and SYNE1 variants respectively. All patients with LMNA and SYNE1 variants were ambulant at the time of evaluation. Mean duration of illness (years) was 11.6±13 (MDCL), 3.2±1.0 (EDMD2), 10.4±12.8 (LGMD1B), 11.8±8.4 (EDMD1) and 3 (EDMD4). One patient had a novel SYNE1 mutation (c.22472dupA, exon 123) and presented with UL phenotype and prominent finger and wrist contractures.
Conclusion:
The salient features included ophthalmoparesis and facial weakness in LMNA, prominent finger contractures in EMD and SYNE1 and upper limb phenotype with the novel pathogenic variant in SYNE1.
INTRODUCTION
Nuclear envelopathies are a heterogenous group of disorders due to defects in the protein coding genes of nuclear envelope which most commonly include Lamin A/C and Emerin [1]. There are varied neuromuscular manifestations such as muscular dystrophy, cardiomyopathy and inherited neuropathies. The skeletal muscle involvement has a wide spectrum of clinical manifestations ranging from congenital muscular dystrophies to adult onset phenotypes. Emery-Dreifuss Muscular Dystrophy (EDMD) is a genetically heterogenous group of hereditary muscular dystrophies. It is a rare disorder with an estimated incidence of 3 in 100,0000 population [2]. The classical triad of EDMD includes joint contractures, progressive muscle weakness with wasting and cardiac involvement. The most common pattern of weakness is the humero-peroneal form involving biceps, triceps and peroneal muscles with scapular winging and sparing of facial muscles [3]. EDMD is further classified as Emerin (EDMD1 – OMIM 310300), Lamin A/C (EDMD2 – OMIM 181350/EDMD3 – OMIM 616516), Nesprin-1 (EDMD4 – OMIM 612998) [4]. Other phenotypes described in laminopathies include congenital muscular dystrophy (MDCL – OMIM 613205) and proximal limb-girdle pattern (LGMD1B – OMIM 159001) [5]. There are no previous studies on the clinical and genetic findings of nuclear envelopathies related muscular dystrophies from India. Thus, this study aims to describe the phenotype-genotype heterogeneity of an Indian cohort.
METHODS
This is a retrospective descriptive study done on genetically confirmed nuclear envelopathy patients with muscular dystrophies from a quaternary neurology referral centre in southern India. Detailed clinical features including onset of symptoms, pattern of muscle involvement, progression, family history, laboratory features such as serum creatine kinase (CK) levels; muscle magnetic resonance imaging (MRI) graded for fatty infiltration using modified Mercuri grading [6] and muscle biopsy findings on light microscopy histopathology (eosin and hematoxylin) and myosin ATPase staining was obtained. Genetic analysis was done by next generation sequencing in all patients as previously described [7] (Supplementary Table 1). Identified variants were classified as pathogenic/likely pathogenic as per American College of Medical Genetics (ACMG) criteria using Franklin variant classification tool (https://franklin.genoox.com/clinical-db/home) [8]. Institutional ethics committee approval was obtained [IEC no: NIMH/DO/(BS&NS) 2022]. Informed consent was obtained from all the patients for publication of clinical data and images. Descriptive statistics such as mean, median and range were used to describe the data.
RESULTS
Sixteen patients were included with a median age at onset of 3 years (range: 1month – 17 years). The median duration of illness was 4 years (range: 1–31years). The male to female ratio was 4.6:1. Three genes were involved including LMNA (11, 68.8%), EMD (4, 25%) and SYNE1 (1, 6.3%). Other genes associated with EDMD such as SYNE2, TMEM43 and FHL1 were not detected in our cohort.
(Phenotype and genotype details of cohort in Table 1)
Table 1
Clinico-genetic details of the patients
| Patient number | Sex | Age at onset (months) | Age at presen-tation (years) | Upper limb weakness (Proximal /Distal) | Lower limb weakness (Proximal /Distal) | Truncal weakness | Bulbar weakness | Joint contractures (Involved joint) | Cardiac involvement | Clinical phenotype | Gene involved | Exon/intron | Variant | Reported / Novel | Zygosity | Inheri-tance pattern | ACMG classi-fication | Serum Creatine kinase (IU/L) |
| 1 | F | 12 | 32 | +/- | +/- | – | – | – | No | MDCL | LMNA | Exon 1 | c.305T>C (p.Leu102Pro) | ClinvarID: 520647 | He | AD | LP | 835 |
| 2 | M | 1 | 8 | +/- | +/+ | – | – | – | No | MDCL | LMNA | Exon 1 | c.115A>C (p.Asn39His) | Ishiyama et al 2018 [32] | He | AD | LP | 2603 |
| 3 | F | 24 | 4 | +/- | +/+ | Scapular winging | – | +(ankle,knee, hip) | No | EDMD2 | LMNA | Exon 7 | c.1357 C>T (p.Arg453Trp) | Bonne et al 1999 [18] | He | AD | P | 1666 |
| 4 | M | 30 | 10 | -/- | -/+ | – | – | – | No | LGMD1B | LMNA | Exon 6 | c.1129 C>T (p.Arg377Cys) | Astejada et al 2007 [14] | He | AD | LP | 476 |
| 5 | M | 36 | 7 | +/- | +/+ | Scapular winging, kyphoscoliosis | Facial weakness | +(elbow,neck) | No | EDMD2 | LMNA | Exon 7 | c.1357 C>T (p.Arg453Trp) | Bonne et al 1999 [18] | He | AD | P | 673 |
| 6 | M | 12 | 6 | +/- | +/- | Hyperlordosis | – | – | No | MDCL | LMNA | Exon 4 | c.746 G>A (p.Arg249Gln) | Di Barletta et al 2000 [19] | He | AD | LP | 444 |
| 7 | M | 12 | 4 | +/- | +/- | – | Ophthal-moparesis | – | No | MDCL | LMNA | Exon 3 | c.590T>C (p.Leu197Pro) | Nishiuchi et al 2017 [33] | He | AD | LP | 947 |
| 8 | M | 42 | 7 | -/- | +/+ | – | – | +(elbow, ankle, knee, hip) | No | EDMD2 | LMNA | Exon 6 | c.1072 G>A (p.Glu358Lys) | Bonne et al 2000 [3] | He | AD | P | 1050 |
| 9 | M | 108 | 12 | +/- | +/+ | +, Scapular winging | – | +(ankle) | No | LGMD1B | LMNA | Exon 9 | c.1583 C>A (p.Thr528Lys) | Di Barletta et al 2000 [19] | He | AD | LP | 885 |
| 10 | M | 36 | 4 | +/- | +/+ | Hyperlordosis | – | +(ankle) | No | LGMD1B | LMNA | Exon 1 | c.94A>G (p.Lys32Glu) | Sframeli et al 2017 [34] | He | AD | LP | 1319 |
| 11 | F | 204 | 46 | -/- | +/- | Hyperlordosis | – | – | No | LGMD1B | LMNA | Exon 10 | c.1527dup (p.Thr510TyrfsTer42) | Scharner et al 2011 [35] | He | AD | LP | 134 |
| 12 | M | 96 | 28 | +/- | +/+ | + | – | +(ankle) | Sick sinus syndrome and left ventricular hypertrophy with implanted pacemaker | EDMD1 | EMD | Exon 6 | c.654_658dup (p.Asp220AlafsTer19) | Novel | Hemi | XLR | LP | 1371 |
| 13 | M | 12 | 19 | +/+ | +/- | – | – | +(elbow, ankle, hip, fingers) | Atrial arrthymias | EDMD1 | EMD | Exon 6 | c.599 G>A (p.Trp200Ter) | Brown et al 2011 [17] | Hemi | XLR | LP | 180 |
| 14 | M | 36 | 7 | +/- | +/+ | – | – | +(ankle, neck, spine) | No | EDMD1 | EMD | Exon 6 | c.618del (p.Arg207GlyfsTer30) | Astejada et al 2007 [14] | Hemi | XLR | LP | 1180 |
| 15 | M | 36 | 8 | +/- | +/+ | Hyperlordosis | – | +(ankle) | No | EDMD1 | EMD | Exon 4 | c.350del(p.Val117AlafsTer5) | Novel | Hemi | XLR | LP | 608 |
| 16 | M | 180 | 18 | +/+ | -/- | – | – | +(wrist, fingers) | No | EDMD4 | SYNE1 | Exon 123 | c.22472dup (p.Leu7491PhefsTer27) | Novel | Ho | AR | LP | 945 |
Footnotes: AD – Autosomal Dominant, AR – Autosomal Recessive, EMD, Emerin, EDMD – Emery Dreifuss Muscular dystrophy, F- Female, He – Heterozygous, Ho – Homozygous, Hemi – Hemizygous, LMNA – Lamin A/C, LP – Likely Pathogenic, LGMD1B – Limb girdle muscular dystrophy 1B, M- male, MDCL – Congenital muscular dystrophy – Lamin related, P – Pathogenic, SYNE1 – Spectrin Repeat Containing Nuclear Envelope Protein-1/Nesprin-1, XLR – X-linked Recessive.
EMERIN (EMD)
There were 4 patients with EMD pathogenic variants with phenotypic presentation of EDMD1. The median age at onset of symptoms was 3 years (range: 1 – 8 years) with varying duration of presentation ranging from 4 to 20 years. All patients were males. Consanguinity and positive family history of sudden cardiac death in first cousin was noted in P14. Both upper (UL) and lower limb (LL) weakness were noted in all patients. Truncal weakness was noted in only one. Only one patient (P12) lost ambulation and required wheel chair assistance at 26 years of age (18 years after disease onset). Cardiac involvement was noted in two patients– P12: sick sinus syndrome and left ventricular hypertrophy with implanted pacemaker and P13: atrial arrythmias. Talipes equino-varus deformity was noted in two patients (P12, P13) and lumbar lordosis in one (P15). All had ankle contractures with additional elbow, hip and finger contractures in P13 and posterior cervical contractures in P14. However, patients P12 (onset at 8 years) and P15 (onset at 3years) had atypical pattern with only ankle contractures at presentation. The median CK level was 894 U/L (range: 180–1371 U/L). Muscle biopsy was done in only one patient (P13) showing neurogenic changes with fibre type II grouping (Fig. 1F). All four males had X-linked recessive disorders with truncating hemizygous variations identified in EMD gene with exon 6 involved in three out of four. While two (P13, P14) have previously reported nonsense and frameshift variations respectively, P12 and P15 have frameshift variations identified for the first time in this study (Table 1). Notably both had novel pathogenic variants of c.654_658dup (P12) and c.350del(P15).
Fig. 1
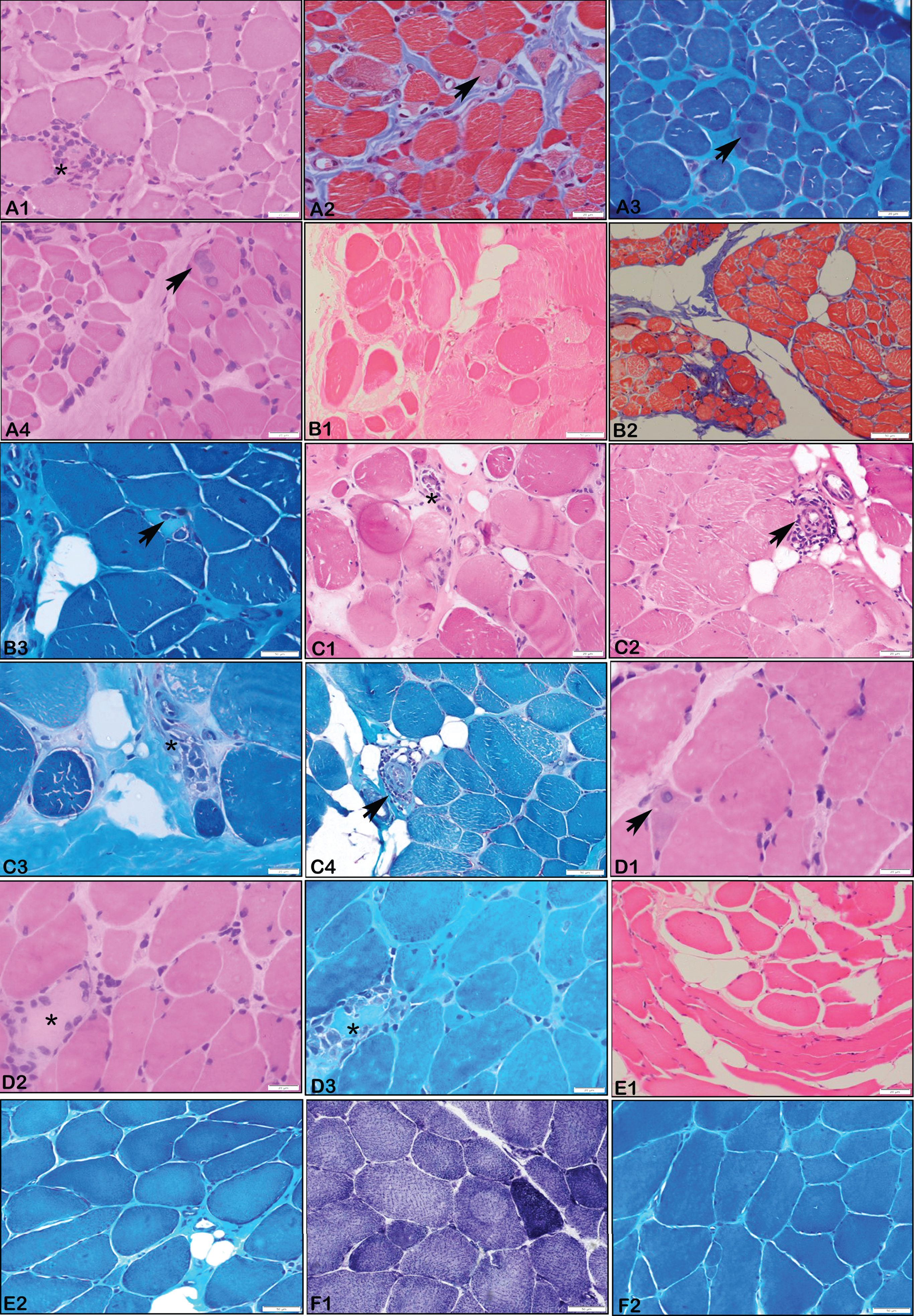
Muscle Biopsy Histopathology. A1 – A4 (P7 – LMNA, 4 years): Microphotograph showing muscle tissue with myopathic changes with scattered regenerating fibres (Arrows), endomyseal fibrosis and focal perivascular inflammation (asterix). (X 200 A1, A4 – H&E; A2 – Masson’s Trichrome stain, A3 – MGT stain). B1 – B3 (P8 – LMNA, 7 years): Microphotograph showing muscle tissue with myopathic changes with myophagocytosis (asterix), endomyseal fibrosis and perivascular inflammation (arrow). Note the myonecrosis in B 3 (asterix) (X 200 B1 – H&E;, B2 – Masson’s Trichrome stain; B3 – MGT stain). C1 – C4 (P9 – LMNA, 12 years): Microphotograph showing muscle tissue with myopathic changes with myophagocytosis (asterix), endomyseal fibrosis and perivascular inflammation (arrow). Variation in fibre size, nuclear clumps and adipocytic infiltration. (X 200 C1, C2 – H&E; C3, C4 – MGT stain). D1, D2, D3 (P3 – LMNA, 4 years): Microphotograph showing muscle tissue with myopathic changes with myonecrosis (asterix), endomyseal fibrosis and regenerating fibres (arrow). Variation in fibre size, nuclear clumps and adipocytic infiltration. (X 200 D1,D2 – H&E; D3 – MGT stain). E1, E2 (P6 – LMNA, 6 years): Microphotograph showing Muscle tissue with variation in fibre size, endomyseal fibrosis and adipocytic infiltration. (X200 E1, E2 – H&E; E2 – MGT stain). F1, F2 (P13 – EMD, 19 years): Microphotograph showing muscle tissue with neurogenic changes comprising of angulated atrophic fibres and type II fibre grouping (based on SDH stain, F1; X200, F2 – MGT stain).
LAMIN-A/C (LMNA)
Of the 11 patients with LMNA pathogenic variants, 4 (P1, P2, P6, P7) had MDCL, 4 (P4, P9, P10, P11) had LGMD1B and 3 (P3, P5, P8) had EDMD2 phenotypes. Positive family history was noted in one (P11) and consanguinity in one (P4).
All patients with MDCL phenotype had onset of weakness at 1 year of age except P2 who had at 1 month of age. Developmental delay followed by limb girdle weakness was noted in all MDCL patients with associated distal LL weakness in P2. None of these patients had contractures at presentation. Other notable features were ophthalmoparesis in P7 and hyperlordosis in P6. Muscle biopsy done in P6 and P7 showed myopathic pattern with focal perivascular inflammation in P7 (Fig. 1).
Patients with LGMD1B phenotype had a median age at onset of 6 years (range: 2.5–17years). The pattern of weakness was proximal UL and LL (P9, P10), proximal LL (P11) and distal LL (P4). Other features such as hyperlordosis (P10, P11) and scapular winging (P9) were also noted. Contractures at ankles was seen in only two subjects (P9, P10). Muscle biopsy was done in P9 and showed myopathic changes with perivascular inflammation (Fig. 1).
The age at onset in EDMD2 ranged from 2 – 3.5 years. Except P8, all had proximal UL with proximal and distal LL weakness. Facial weakness (P5), scapular winging (P3, P5) and kyphoscoliosis (P5) were other features. Muscle biopsy done in two cases (P3, P8) showed myopathic features with myonecrosis and perivascular inflammation in P8.
Mean duration of illness (years) are 11.6±13 (MDCL), 3.2±1.0 (EDMD2), 10.4±12.8 (LGMD1B). All patients were ambulant at the time of evaluation. Median serum CK level was 860 U/L (range: 134–2603). None had cardiac symptoms and electrocardiogram and 2D echocardiography were normal in all probands. Muscle MRI done in two showed global fatty infiltration of thigh (P1, P11) and leg muscles (P11). All patients except P11 were sporadic and were found to have heterozygous variations in LMNA, with the most common variations being missense involving exon 1 (n = 3). One duplication variant at exon 10 causing frameshift (c.1527dup, p.Thr510TyrfsTer42) was noted in P11 (Table 1).
NESPRIN-1/SPECTRIN REPEAT CONTAINING NUCLEAR ENVELOPE PROTEIN-1/(SYNE1)
Patient P16 had onset of symptoms at 15 years of age and presented at 18 years. The proband had atypical phenotype with proximal and distal UL weakness without LL weakness. Prominent finger and wrist flexion contractures were noted. There were no cardiac symptoms, truncal or bulbar weakness. Elevated serum CK of 945 U/L was present. P16 had a novel homozygous frameshift variation affecting exon 123 of SYNE1 (Table 1).
DISCUSSION
Here we have described in detail the phenotype – genotype characteristics of 16 patients with envelopathies which result from structural and/or functional defects in the various nuclear envelope proteins.
Emery-Dreifuss muscular dystrophy
EDMD due to emerin and lamin A/C defects contribute to 40% of EDMD cases. Lamin A/C encoded by LMNA gene is the major component of nuclear membrane localised to the inner nuclear membrane and nucleoplasm [9]. Pathogenic variants in LMNA have a broad spectrum of heterogenous manifestations including congenital muscular dystrophy, autosomal dominant dilated cardiomyopathy with conduction defect, familial partial lipodystrophy and autosomal recessive Charcot Marie Tooth disease and progeria [10]. Lamin A/C binds with other proteins such as emerin (EMD), lamin- A-associated polypeptide (LAP) and MAN1 [11]. It plays a major role in chromatin organisation, replication, cell cycle regulation and structural stability to mechanical stress especially in tissues such as skeletal and cardiac muscles [12]. The estimated prevalence of all types of EDMD is about 1.3:100,000 – 2:100,000 [13]. There is significant inter- and intrafamilial variability in age of onset, progression and severity of muscle and cardiac manifestations. The variability in clinical features ranges from severe childhood onset to late onset slowly progressive disease. Astejada et al., have described in a large series of EDMD patients with a mean age at onset being 10.1 and 3.3 years in pathogenic variants in EMD and LMNA respectively [14]. However, in the present study the median age at onset was much earlier (early 1st decade) in patients with variants in both EMD and LMNA genes. Generally joint contractures occur during the first decade followed by muscle weakness which is typically described in EDMD1. However, in EDMD2 contractures may appear after onset of muscle weakness [13]. The most common sites include elbow, ankle and posterior cervical muscles [3]. In the present study all patients with EMD had ankle contracture, while in LMNA, the ankle, elbow, knee and hip joints were equally involved. Neck contractures occurred in only one patient each with LMNA and EMD pathogenic variants. In a study done on LMNA related muscular dystrophies in Chinese cohort by Fan et al, contractures were noted in 75% of EDMD2 with ankle being the first joint involved followed by elbow joint [15]. Prominent finger contractures similar to collagenopathies was noted in two patients, one each with EMD and SYNE1 variants, which has not been reported previously. Serum CK level ranges from normal to moderately elevated [3]. A similar trend was noted in our patients.
EDMD1: Typically, symptoms begin in the first decade with ankle followed by elbow contractures. The major cardiac manifestations include rhythm disturbances such as sinus bradycardia, supraventricular extrasystolic beats, atrioventricular blocks (AVB), paroxysmal atrial fibrillation or flutter with gradual development of cardiomyopathy [2]. Skeletal muscle weakness usually precedes cardiac symptoms with onset in the second decade with predominant lower limb involvement and rarely affecting ambulation [2]. Similar cardiac manifestations and muscle weakness patterns were noted in our cohort. One patient lost independent ambulation in the 3rd decade of life. Muscle biopsy was done in one patient with (P13) which showed neurogenic changes. This might be possibly due to the site of muscle sampling in a particular muscle (near NMJ), evolving disease or nonspecific secondary changes [16]. The majority of cases are known to occur due to nonsense variations and frame shifting small insertion/deletions. Exons 1 and 2 bear recurrent variations especially in codons 1 or 34 [17]. In contrast, in the present cohort exon 6 was most commonly involved with deletions.
EDMD2: The major LMNA related muscular dystrophy phenotypes are autosomal dominant EDMD, LGMD1B and MDCL. Lamin A/C related EDMD are EDMD2 (autosomal dominant) and EDMD3 (autosomal recessive) [18, 19]. Most of the patients present with typical scapula-peroneal weakness along with pelvic girdle weakness. In contrast to previous studies [5, 15], all patients had contractures most commonly in elbow, proximal and distal LL joints with posterior cervical region in one patient. Fan Y et al., reported cardiac arrthymias in 43.8% of EDMD2 patients in their cohort [15]. However, in the current study none had cardiac manifestations. Except two patients, all our patients with LMNA variants presented in their first decade or early second decade after the onset of skeletal muscle symptoms. Cardiac involvement in EDMD2 is often reported later, usually after the second decade following skeletal muscle involvement [3, 15]. Hence, our patients might require further follow up to ascertain cardiac involvement. None had loss of ambulation, thus reflecting an overall milder phenotype. EDMD2 is most commonly caused by missense heterozygous variations with dominant negative effect followed by deletions/duplications/nonsense variations resulting in loss of function variation [20]. Similarly, in the present study all patients had missense variations with autosomal dominant inheritance.
EDMD4: Nesprins (encoded by SYNE1 and SYNE2) and Transmembrane protein 43 (TMEM43) and are other nuclear envelop proteins which co-localise to lamins [21]. Nesprins are expressed mainly in skeletal muscle and cerebellum and plays a key role in anchorage of nuclei [22]. There is a wide spectrum of phenotypes associated with SYNE1 such as EDMD4, spinocerebellar ataxia 8, myogenic type of arthrogryposis congenita with EDMD features and spastic paraplegia with intellectual disability and axonal neuropathy [23–25]. There are only few cases of EDMD4 described in literature characterized by slowly progressive muscle atrophy with contractures without cardiac abnormalities [24, 26–28]. Attali et al., reported two patients with congenital hypotonia with foot deformities and loss of ambulation in the early second decade [24]. Fanin et al., and Chen et al., reported three cases and one case with childhood onset of lower limb weakness with deformities respectively [27, 28]. Patient P16 in the present study had novel features of onset of symptoms in the middle of second decade with predominant proximal and distal upper limb weakness with severe finger and wrist contractures which has not been reported previously. The insertion variation c.22472dupA is also a novel variant. The previously reported EDMD4 variants were mostly heterozygous single nucleotide variants [26, 28].
Comparison with previous studies is summarized in Table 2.
Table 2
Comparison with previous studies on EDMD
| Author /year/ ethnicity | Age at onset (years) | Pattern of weakness | Cardiac features | Respiratory insufficiency | Inheritance | Variants |
| Emerinopathy | ||||||
| Astejada et al, 2007, Japan (n = 20) [14] | 10.1±9.5 (mean±SD) | Humeroperoneal and limb-girdle with joint contractures | Conduction defects and DCM (90%) | – | XL | c.31delG, p.(Glu11SerfsTer2); c.82 + 5 G>C, c.83-2A>G, c.123 C>G, p.(Tyr41Ter); c.144dupC, p.(Ser49LeufsTer12); c.251-55delTCTAC; c.359-62delCAGT, c.400-2A>G; c.677 G>A, p.(Trp226Ter); c.619delC; c.650_50dup |
| Ellis et al, 2000, UK (n = 2) [36] | 2 | Limb girdle weakness with neck stiffness | – | – | XL | Mutations in residue 236 and starting at residue 247 in the C-terminal tail |
| Our study (n = 4) | 3 [range: 1–8] (median) | Limb-girdle and humeroperoneal with joint contractures, prominent finger contractures in one patient, | Sick sinus syndrome (1), atrial arrthymias (1). | – | XL | c.654_658dup (p.Asp220AlafsTer19), c.599 G>A (p.Trp200Ter), c.618del (p.Arg207GlyfsTer30), c.350del(p.Val117AlafsTer5). |
| Laminopathy | ||||||
| Astejada et al, 2007, Japan (n = 27) [14] | 3.3±2.9 | Proximal LL weakness with contractures | Conduction defects (63%) | – | AD | c.73 G>C (p.Glu25Gln), c.306 C>A (p.Ala102=), c.374 G>C (p.Gly125Ala), c.746 G>A (p.Arg249Gln), c.931A>G (p.Lys311Glu), c.1058A>G (p.Gln353Arg), c.1063 C>T (p.Gln355Ter), c.1129 C>T (p.Arg377Cys), c.1357 C>T (p.Arg453Trp), c.1366A>C(p.Asn456His), c.1412 G>A (p.Arg471His), c.1540T>C(p.Trp514Arg), c.1580 G>C (p.Arg527Pro), c.1583 C>A(p.Thr528Lys), c.1622 G>A (p.Arg541His). |
| Bendetti et al, 2007, Italy [20] | 2.4 to 30.5 | Limb girdle syndrome with contractures | – | – | AD | c.992 G>A (p.Arg331Gln), c.992 G>C (p.Arg331Pro), c.1039 G>A (p.Glu347Lys), c.1130 G>A(p.Arg377His), c.1072 G>A(p.Glu358Lys) |
| Our study (n = 11) MDCL (4), LGMD1B (4), EDMD2 (3). | MDCL – 1, LGMD1B – 6 EDMD2 – 2. –3.5 | MDCL – developmental delay and limb-girdle weakness, ophthaloparesis in one.LGMD1B – limb-girdle weakness with only ankle contractures in two. EDMD2 – humeroperoneal weakness with multiple joint contractures. | – | – | AD | c.305T>C (p.Leu102Pro), c.115A>C (p.Asn39His), c.1357 C>T (p.Arg453Trp), c.1129 C>T (p.Arg377Cys), c.1357 C>T (p.Arg453Trp), c.746 G>A (p.Arg249Gln), c.590T>C (p.Leu197Pro), c.1072 G>A (p.Glu358Lys), c.94A>G (p.Lys32Glu), c.1527dup (p.Thr510TyrfsTer42). |
| Nesprinopathy | ||||||
| Chen Z et al, 2017, China [28] | 24 | LL proximal with neck and elbow contractures | – | – | AD | c.6910A>G (p.Ala2304Pro) |
| Fanin M et al, 2015, n = 3 Italy [27] | I – 6–7II – childhoodIII – 3 | Proximal weakness with foot and elbow joint contracturesIII – additional spine contractures. | – | – | AD | c.323 C>T p.(Ser108Leu) |
| Our study (n = 1) | 15 | Proximal and distal UL weakness with prominent finger and wrist contractures | – | – | AR | c.22472dup (p.Leu7491PhefsTer27) |
Footnotes: AD – Autosomal Dominant, AR – Autosomal Recessive, EDMD – Emery Dreifuss Muscular dystrophy, LGMD1B – Limb girdle muscular dystrophy 1B, MDCL – Congenital muscular dystrophy – Lamin related, XLR – X-linked Recessive.
OTHER LAMINOPATHY RELATED MUSCULAR DYSTROPHIES
MDCL: The most common initial manifestation in MDCL is decreased fetal movements followed by delayed motor milestones [29]. MDCL were classified into two groups by Quijano-Roy et al., as severe congenital muscular dystrophy and dropped head syndrome respectively [29]. All MDCL patients in the current study presented with developmental delay as described previously followed by proximal limb weakness without cervico-axial involvement. In-contrast to studies in the Italian [5] and Chinese cohorts [15] which showed contractures in 88.9% and 51.2% of MDCL patients, none of our patients had contractures at presentation depicting a milder phenotype of our cohort. Ophthalmoparesis in MDCL and facial weakness in EDMD2 noted were similar to the cohort reported by Maggi et al., [5]. MDCL has only minimal cardiac involvement as noted in previous studies [15, 29]. A recent study by Ben Yaou R et al., on 151 MDCL patients showed cardiac involvement in 48.3% with median age at first cardiac abnormality being 9.3 years (range: 0.2 – 34 years) [30]. The lack of cardiac involvement in the current study may be because of the young age of the patients and hence continued follow-up for cardiac dysfunction is required.
LGMD1B: Previous studies have shown mild pelvic girdle weakness in LGMD1B with rare cardiac involvement [5, 15]. Our study also showed pelvic and shoulder girdle weakness without cardiac involvement. Comparable to the Italian cohort, contractures were noted in 50% of LGMD1B at ankle joint only [5].
Muscle biopsy done in EDMD2, MDCL and LGMD1B showed most frequently the features of myopathy followed by focal perivascular infiltrates in all these three phenotypes. Studies by Komaki et al., [31] and Quijano-Roy et al., [29] have shown that inflammatory infiltrates were noted commonly in MDCL especially in the perimysial connective tissue. However, Fan et al., [15] have shown that 50% of MDCL and 33.3% of EDMD2 patients in their cohort showed inflammatory infiltrates in the entire muscle biopsy specimen including necrotic/non-necrotic fibres, endomysium and perivascular region of the perimysium. To ascertain the diagnostic importance in laminopathies related muscular dystrophies of this finding similar to other conditions such as facioscapulohumeral dystrophy and dysferlinopathy requires further dedicated studies. All pathogenic variants in both MDCL and LGMD1B are missense heterozygous variants except for one patient with LGMD1B.
CONCLUSION
This study on muscular dystrophies associated with nuclear envelopathies shows phenotype and genotype variability. The salient features include ophthalmoparesis and facial weakness in LMNA (in one patient each) and upper limb phenotype with novel variation in SYNE1.
The occurrence of finger contractures in EMD and SYNE1 is a novel finding which may aid in specific genetic diagnosis.
ACKNOWLEDGMENTS
None.
FUNDING
K.P. holds a Canadian Institutes of Health Research postdoctoral fellowship award under grant no. MFE-491707.
DATA AVAILABILITY
Data will be available on request from the corresponding author.
SUPPLEMENTARY MATERIALS
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-230172.
REFERENCES
[1] | Bonne G , Quijano-Roy S . Emery-Dreifuss muscular dystrophy, laminopathies, and other nuclear envelopathies. Handb Clin Neurol. (2013) ;113: :1367–76. |
[2] | Madej-Pilarczyk A . Clinical aspects of Emery-Dreifuss muscular dystrophy. Nucleus. (2018) ;9: (1):268–274. |
[3] | Bonne G , Mercuri E , Muchir A , et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann Neurol. (2000) ;48: (2):170–180. |
[4] | Dauer WT , Worman HJ . The nuclear envelope as a signaling node in development and disease. Dev Cell. (2009) ;17: :626–38. |
[5] | Maggi L , D’Amico A , Pini A , Sivo S , Pane M , Ricci G , et al. LMNA-associated myopathies: the Italian experience in a large cohort of patients. Neurology. (2014) ;83: (18):1634–44. |
[6] | Stramare R , Beltrame V , Dal Borgo R , Gallimberti L , Frigo AC , Pegoraro E , et al. MRI in the assessment of muscular pathology: a comparison between limb-girdle muscular dystrophies, hyaline body myopathies and myotonic dystrophies. Radiol Med (Torino). (2010) ;115: (4):585–99. |
[7] | Polavarapu K , Mathur A , Joshi A , Nashi S , Preethish-Kumar V , Bardhan M , et al. A founder mutation in the GMPPB gene [c.1000G >A (Asp334Asn)] causes a mild form of limb-girdle muscular dystrophy/congenital myasthenic syndrome (LGMD/CMS) in South Indian patients. Neurogenetics. (2021) ;22: (4):271–285. |
[8] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) ;17: (5):405–24. |
[9] | Shimi T1 , Pfleghaar K , Kojima S , et al. The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev. (2008) ;22: :3409–21. |
[10] | Heller SA , Shih R , Kalra R , Kang PB . Emery-Dreifuss muscular dystrophy. Muscle Nerve. (2020) ;61: (4):436–448. |
[11] | Lin F , Blake DL , Callebaut I , et al. MAN1, an inner nuclear membrane protein that shares the LEM domain with lamina-associated polypeptide 2 and emerin. J Biol Chem. (2000) ;275: :4840–7. |
[12] | Gruenbaum Y , Foisner R . Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu Rev Biochem. (2015) ;84: :131–64. |
[13] | Bonne G , Lturcq F , Ben Yaou R . Emery-Dreifuss Muscular Dystrophy. GeneReviews. Seattle (WA): University of Washington, Seattle. 2004 Sep 29 [Updated 2019Aug15];1993-2021. |
[14] | Astejada MN , Goto K , Nagano A , Ura S , Noguchi S , Nonaka I , Nishino I , Hayashi YK . Emerinopathy and laminopathy clinical, pathological and molecular features of muscular dystrophy with nuclear envelopathy in Japan. Acta Myol. (2007) ;26: (3):159–64. |
[15] | Fan Y , Tan D , Song D , Zhang X , Chang X , Wang Z , et al. Clinical spectrum and genetic variations of LMNA-related muscular dystrophies in a large cohort of Chinese patients. J Med Genet. (2021) ;58: (5):326–333. |
[16] | Dastur DK , Razzak ZA . Possible neurogenic factor in muscular dystrophy: its similarity to denervation atrophy. J Neurol Neurosurg Psychiatry. (1973) ;36: (3):399–410. |
[17] | Brown CA , Scharner J , Felice K , et al. Novel and recurrent EMD mutations in patients with Emery-Dreifuss muscular dystrophy, identify exon 2 as a mutation hotspot. J Hum Genet. (2011) ;56: :589–94. |
[18] | Bonne G , Di Barletta MR , Varnous S , et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. (1999) ;21: :285–8. |
[19] | Raffaele Di Barletta M , Ricci E , Galluzzi G , Tonali P , Mora M , Morandi L , et al. Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. Am J Hum Genet. (2000) ;66: (4):1407–12. |
[20] | Benedetti S , Menditto I , Degano M , et al. Phenotypic clustering of lamin A/C mutations in neuromuscular patients. Neurology. (2007) ;69: :1285–92. |
[21] | Zhang Q , Ragnauth CD , Skepper JN , et al. Nesprin-2 is a multi-isomeric protein that binds lamin and emerin at the nuclear envelope and forms a subcellular network in skeletal muscle. J Cell Sci. (2005) ;118: :673–87. |
[22] | Zhang X , Xu R , Zhu B , et al. Syne-1 and Syne-2 play crucial roles in myonuclear anchorage and motor neuron innervation. Development. (2007) ;134: :901–8. |
[23] | Gros-Louis F , Dupre N , Dion P , Fox MA , Laurent S , Verreault S , et al. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat Genet. (2007) ;39: :80–85. |
[24] | Attali R , Warwar N , Israel A , Gurt I , McNally E , Puckelwartz M , Glick B , Nevo Y , Ben-Neriah Z , Melki J . Mutation of SYNE-1, encoding an essential component of the nuclear lamina, is responsible for autosomal recessive arthrogryposis. Hum Mol Genet. (2009) ;18: :3462–9. |
[25] | Schuurs-Hoeijmakers JH , Vulto-van Silfhout AT , Vissers LE , van de Vondervoort II , van Bon BW , de Ligt J , et al. Identification of pathogenic gene variants in small families with intellectually disabled siblings by exome sequencing. J Med Genet. (2013) ;50: (12):802–11. |
[26] | Zhang Q , Bethmann C , Worth NF , Davies JD , Wasner C , Feuer A , Ragnauth CD , Yi Q , Mellad JA , Warren DT , et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet. (2007) ;16: :2816–33. |
[27] | Fanin M , Savarese M , Nascimbeni AC , et al. Dominant muscular dystrophy with a novel SYNE1 gene mutation. Muscle Nerve. (2015) ;51: (1):145–7. |
[28] | Chen Z , Ren Z , Mei W , et al. A novel SYNE1 gene mutation in a Chinese family of Emery-Dreifuss muscular dystrophy-like. BMC Med Genet. (2017) ;18: :63. |
[29] | Quijano-Roy S , Mbieleu B , Bönnemann CG , Jeannet PY , Colomer J , Clarke NF , et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann Neurol. (2008) ;64: (2):177–86. |
[30] | Ben Yaou R , Yun P , Dabaj I , Norato G , Donkervoort S , Xiong H , et al. International retrospective natural history study of LMNA-related congenital muscular dystrophy. Brain Commun. (2021) ;3: (3):fcab075. |
[31] | Komaki H , et al. Inflammatory changes in infantile-onset LMNA-associated myopathy. Neuromuscul Disord. (2011) ;21: :563–568. |
[32] | Ishiyama A , Iida A , Hayashi S , Komaki H , Sasaki M , Nonaka I , et al. A novel LMNAmutation identified in a Japanese patient with LMNA-associated congenital muscular dystrophy. Hum Genome Var. (2018) ;5: :19. |
[33] | Nishiuchi S , Makiyama T , Aiba T , Nakajima K , Hirose S , Kohjitani H , et al. Gene-Based Risk Stratification for Cardiac Disorders in LMNA Mutation Carriers. Circ Cardiovasc Genet. (2017) ;10: (6):e001603. |
[34] | Sframeli M , Sarkozy A , Bertoli M , Astrea G , Hudson J , Scoto M , et al. Congenital muscular dystrophies in the UK population: Clinical and molecular spectrum of a large cohort diagnosed over a 12-year period. Neuromuscul Disord. (2017) ;27: (9):793–803. |
[35] | Scharner J , Brown CA , Bower M , Iannaccone ST , Khatri IA , Escolar D , et al. Novel LMNA mutations in patients with Emery-Dreifuss muscular dystrophy and functional characterization of four LMNA mutations. Hum Mutat. (2011) ;32: (2):152–67. |
[36] | Ellis JA , Brown CA , Tilley LD , Kendrick-Jones J , Spence JE , Yates JR . Two distal mutations in the gene encoding emerin have profoundly different effects on emerin protein expression. Neuromuscul Disord. (2000) ;10: (1):24–30. |


