
GNE Myopathy: Genotype – Phenotype Correlation and Disease Progression in an Indian Cohort
Abstract
Introduction:
GNE myopathy is a rare slowly progressive adult-onset distal myopathy with autosomal recessive inheritance. It has distinctive features of quadriceps sparing with preferential anterior tibial involvement. Most patients eventually become wheelchair bound by 10–20 years after onset. This study analyzes the phenotype-genotype characteristics and disease progression in a large cohort of GNEM patients from India.
Materials and methods:
Retrospective observational study on GNEM from a quaternary neurology referral hospital in southern India. Data was collected from clinical phenotyping, serum creatine kinase levels, muscle biopsy histopathology, genetic analysis and functional assessment scales – IBMFRS and MDFRS.
Results:
157 patients were included with mean age at onset and diagnosis: 26.5±6.2 years and 32.8±7.8 years, respectively. M:F ratio was 25 : 13. Most common presenting symptom: foot drop (46.5%) and limb girdle weakness (19.1%). Wasting and weakness of small muscles of hand and finger flexors seen in 66.2% and as an initial symptoms in 5.2%. Though tibialis anterior involvement was most common (89.2%), early quadriceps weakness was noted in 3.2% and Beevor’s sign in 59.2%. Rimmed vacuoles were present in 75% of patients with muscle biopsy. Most common variant was the Indian Founder variant identified in 129 patients (c.2179 G>A, p.Val727Met - 82.2%) and most common zygosity being compound heterozygous state (n = 115, 87.5%). Biallelic kinase domain variations predisposed to a more severe phenotype. Wheelchair bound state noted in 8.9% with a mean age and duration of 32.0±7.1 and 6.3±4.9 years respectively, earlier than previous studies on other ethnic groups.
Conclusion:
This is the largest GNEM cohort reported from South Asia. The p.Val727Met variant in compound heterozygous state is noted in majority (82.2%) of the cases. Observed relationships between genotype and clinical parameters shows that severity of the disease might be attributable to specific GNE genotype and thus could aid in predicting the disease progression.
INTRODUCTION
GNE (UDP-N-acetlyglucosamine-2 epimerase: GlcNAc/ N-acetylmannosamine: ManNAc) myopathy is a rare adult-onset distal myopathy. Following its initial descriptions by Nonaka et al., [1] and Argov et al., [2], it has been known by various names such as myopathy with rimmed vacuoles (DMRV) and hereditary inclusion body myopathy (hIBM) later unified as GNE myopathy (GNEM) [3]. The worldwide prevalence of GNEM is ≈1–9/1,000,000 [4]. In India, a first report of 23 GNEM patients were published by us in 2010 [5]. GNEM is an adult onset myopathy with appearance of symptoms in the third decade of life [6]. The most common presenting symptom is foot drop with variable progression to involve the proximal lower and upper limb muscles [7]. GNEM is an autosomal recessive disease due to biallelic mutations in the GNE gene residing in chromosome 9p13.3 with 13 exons. GNE protein has bifunctional enzymatic activity involved in biosynthetic pathway of sialic acid, 5-N-acetylneuraminic acid (Neu5Ac) [8]. Neu5Ac is involved in sialylation of glycolipids and glycoproteins of mammalian cells, reduced sialylation has been attributed to clinical features of myopathy [9].
Diagnosis of GNEM relies on the typical clinical features which can be supported by histopathology of muscle biopsy. The confirmation of diagnosis is done by next generation sequencing or targeted gene sequencing in highly prevalent regions. Most of the variations are missense and compound heterozygous involving both the epimerase and kinase domains [10]. With more than 200 mutations in GNE gene reported worldwide, many ethnic founder mutations have been described including Middle east (p.Met743Thr), Japanese (p.Asp207Val, p.Val603Leu), Roma Bulgarian (p.Ile618Thr) and Indian (p.Val727Met) [11–14].
Since GNEM is a rare myopathy, only few studies have been done on clinical correlation and disease progression. Hence, this study was done on a large cohort of GNEM in Indian population to analyse retrospectively the genotype-phenotype characteristics and milestones in disease progression.
MATERIALS AND METHODS
This is a retrospective study done on 157 GNE myopathy diagnosed by typical clinical features, histopathology and/or genetic analysis. Patients were evaluated and followed up in the neuromuscular division of department of Neurology at a quaternary neurosciences centre in India. The genetic diagnosis was confirmed either by single gene testing or next generation sequencing (targeted or exome). Heterozygous variants were noted in 14 patients who were diagnosed clinically with GNEM of whom two patients also had typical histopathological features of GNEM. Among these, 92 patients (58.6%) (other patients lost to follow-up) were followed up longitudinally during their periodic clinic visits between 2006 and 2022. The data was retrieved from patient records between the year 2006 and 2022. Patients with incomplete data were excluded from the study. The parameters recorded include demographic details, neurological examination findings, clinical / disability self-rating scales such as Inclusion body myositis functional rating scale (IBMFRS) and Muscular Dystrophy Functional Rating Scale (MDFRS), serum creatine kinase (CK) levels, muscle biopsy – histopathological features (whenever available) and details of identified GNE variations.
GNE variations in the current cohort were annotated based on RefSeq transcript NM_001128227.3 [Genome assembly: GRCh37 (hg19)]. The variant pathogenicity based on ACMG criteria was determined using Franklin variant analysis platform (https://franklin.genoox.com – Franklin by Genoox) [15].
Institutional ethics committee approved the study protocol [IEC no: NIMH/DO/(BS& NS) 2022]. All patients or their legal representatives gave informed written consent for participation and publication of the study.
Statistical analysis
Major outcome measures included age at onset/presentation, use of assistive device/ wheelchair bound state and outcome scale scores such as IBMFRS and MDFRS. These outcome measures were compared between three patient sub-groups based on type of mutation: missense, nonsense, frameshift, splicesite, intronic splice proximal and inframe mutations; location of mutations: group K (both mutations located in kinase domain); group K + E (one each mutation in epimerase and kinase); group E (both mutations in epimerase). Further, patients carrying two common founder mutations (p.Val727Met, p.Ile618Thr) were compared for disease severity and progression. The demographic data and major variables were expressed using descriptive statistics (mean±standard deviation [SD], range). The presenting clinical symptoms are represented as the number of patients with the particular symptom. Comparison of means of variables such as age at onset, age at use of assistive device / wheel chair, IBMFRS and MDFRS, of groups were done using ANOVA one factor and student t-test for unkown variance. Correlation of continuous variables and categorical variables were done using Pearson correlation and point biserial correlation respectively. A P-value of ≤0.05 was regarded as statistically significant. The statistical analysis was done using statistical package for the social sciences (SPSS Ver 25.0).
RESULTS
We studied 157 genetically confirmed GNEM patients seen over 22 years.
Demographic features: (Summarized in Table 1)
Table 1
Baseline demographic characteristics
| Characteristics (n = 157) | Patient features N (%) |
| Males | 102 (65) |
| Females | 55 (35) |
| Mean age at onset (SD), years | 26.5 (6.2) – Range: 14–53 |
| Mean age at diagnosis (SD), years | 32.8 (7.8) – Range: 19–63 |
| Initial symptoms | |
| Foot extensor weakness | 73 (46.5) |
| Difficulty in rising from floor | 32(20.4) |
| Difficulty running | 17 (11.5) |
| Gait abnormality | 11 (7) |
| Muscle cramps | 10 (6.4) |
| Upper limb distal weakness | 8 (5.1) |
| Frequent falls | 6 (3.8) |
| Positive family history | 33 (21) |
| Siblings -26 | |
| Parent – 1 | |
| Uncle and aunt – 2 | |
| Grandparents -2 | |
| Mobility status at presentation | |
| Without support | 136 (86.6) |
| With assistance | 15 (9.6) |
| Wheelchair bound | 5 (3.2) |
| Bedbound | 1 (0.6) |
The male to female ratio was 1.9 : 1. Consanguinity was noted in 27 patients. The mean duration of symptoms of 6.2±4.0 years. The reported patients were from various states of India with the highest number hailing from Karnataka (n = 46) followed by West Bengal (n = 19) and Tamil Nadu (n = 19). The p.Val727Met (Indian founder mutation) was the most common variant in all regions except Rajasthan where all patients had the p.Ile618Thr (Roma gypsy) variant.
Clinical features
The most common initial symptom was foot dorsiflexion weakness (n = 73, 46.5%) followed by difficulty rising from the floor (n = 32, 20.4%). Asymmetrical limb involvement was seen in 64 patients with a mean time to involve the opposite side being 1.8±1.5 years. Distal upper limb weakness as the presenting symptom was noted in 8 patients. Of these eight patients, 7 patients were compound heterozygous variation with variant p.Val727Met in one allele and the second allele carrying the variants such as p.Ile329Thr, p. Arg352Cys, p.Pro58Leu, p.Gln371Ter, p.Thr88Ile, p.Val336PhefsTer11, c.1727- 2A>G (intron 9). The remaining one patient was homozygous with variant p.Val727Met. Limb-girdle pattern of weakness as initial symptom was observed in 30 (19.1%). Other symptoms which were observed along with limb weakness include truncal weakness at onset (n = 68, 43.3%), myalgia (n = 72, 45.9%) and buckling of knees (n = 67, 42.7%). The following skeletal deformities were noted: pes cavus (n = 24), hammer toes (n = 28), exaggerated lumbar lordosis (n = 32) and kyphoscoliosis (n = 12). A significant number of patients had leg wasting (n = 120) and foot wasting (n = 86). Notably, upper limb wasting and weakness of forearm muscles with involvement of long flexors of hand (especially of index finger) in 91 and small muscles of hand (thenar muscles and first dorsal interossei more than hypothenar muscles) in 104 patients [Supplementary Figure 1]. Other less common features included scapular winging (n = 24) and bifacial weakness (n = 9). According to modified MRC grading the muscle power examination showed significant weakness of neck flexors [n = 51 (MRC≤3)], infraspinatus (n = 92), finger flexors and small hand muscles (n = 65), iliopsoas (n = 128), hip adductors (n = 135), hamstrings (n = 134) and tibialis anterior (n = 140). Though classically GNEM is known as quadriceps sparing myopathy, severe quadriceps weakness at onset was seen in 5 patients early in the disease course (2–3 years from onset of symptoms) with compound heterozygous variations p.Val727Met in one allele and variants p.Ala555Val, p.Pro680Arg, c.258–8 G>A (intron 2) and c.1727- 2A>G (intron 9) in the other allele respectively. One patient with early quadriceps weakness had heterozygous variation in p.Val727Met. Beevor’s sign indicating weakness of lower abdominal muscles and hip flexors was seen in 93 (59.2%) patients.
Laboratory features
The mean serum CK value was 504±392 U/L, ranging from 54 - 2445 U/L. The values were normal in 28 patients (17.8%). Electromyography done in 62 patients showed a myogenic pattern in 54 and neurogenic in 8. Thirty-six patients underwent muscle biopsy and rimmed vacuoles were detected in 27 (75%), inflammation (n = 2), ragged red fibres (n = 1) and nemaline rods(n = 1).
Genetic characteristics
Table 2
Types and allele frequency of GNE mutations
| Genotype characteristics | Results |
| Mutation type | Allele frequency - Total – 314 |
| N (%) | |
| Missense | 268 (85.4) |
| Nonsense | 20 (6.4) |
| Frameshift | 16 (5.1) |
| Splicing | 8 (2.5) |
| In-frame deletion/insertion | 2 (0.6) |
| Common variants | |
| Indian founder (p.Val727Met) | 131 (41.7) |
| Roma gypsy (p.Ile618Thr) | 15 (4.8) |
| No. of patients: Total – 157, N (%) | |
| Zygosity of mutations (n = 157) | |
| Compound heterozygous | 115 (73.2) |
| Homozygous | 28 (17.8) |
| Heterozygous | 14 (8.9) |
| Domains (n = 157) | |
| Kinase | 91 |
| Epimerase + kinase | 58 |
| Epimerase | 8 |
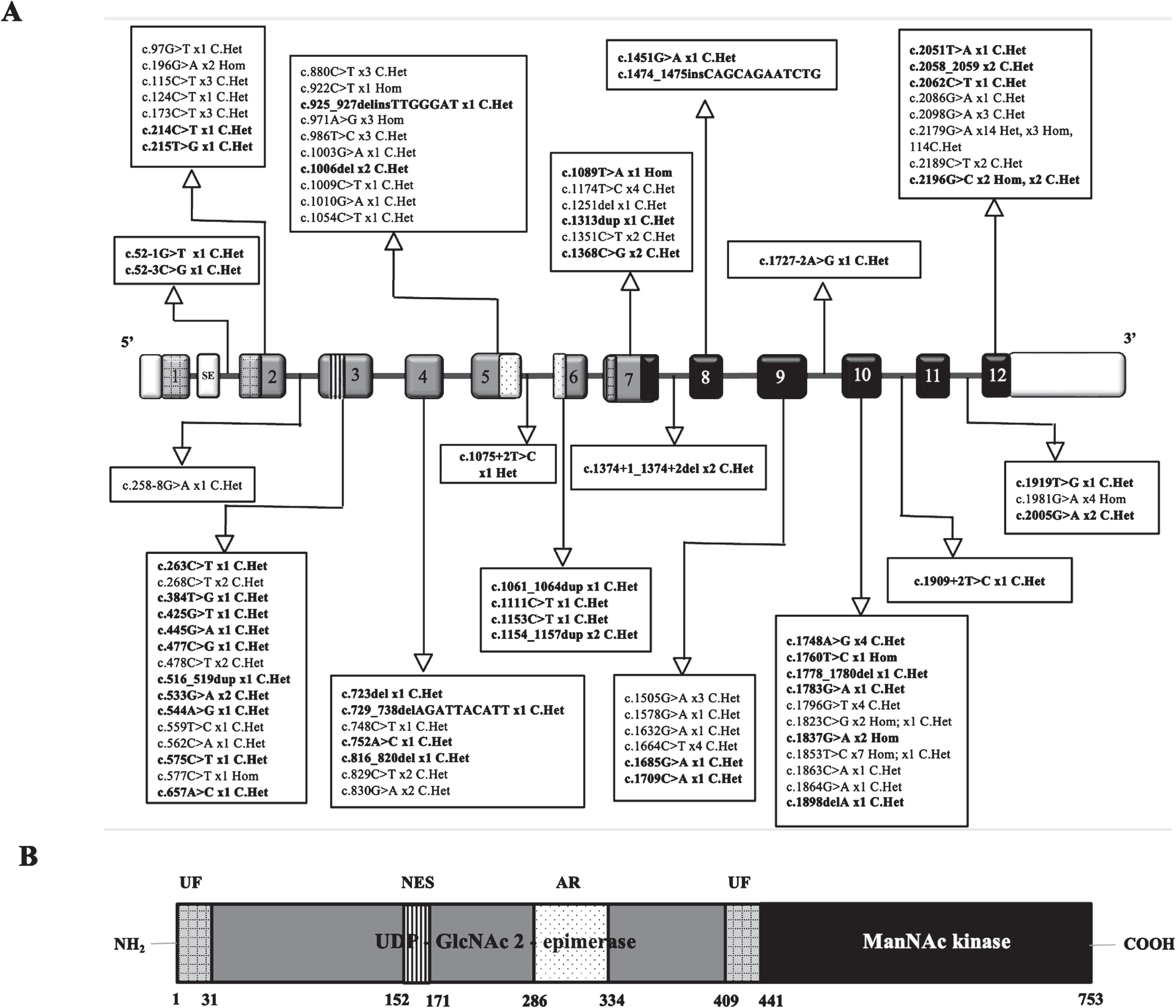
The majority of the variations were compound heterozygous (n = 115, 73.4%) with most of the alleles having missense variations. Among the compound heterozygous patients, p.Val727Met is present in one of the alleles in 129 patients (Fig. 1). The most common variant noted in homozygous state was the Roma gypsy founder mutation (p.Ile618Thr) followed by p.Ala661Thr. Homozygous variation in p.Val727Met was noted in two patients. Exon 12 was most commonly affected due to location of the Indian founder mutation (p.Val727Met) variant (Fig. 2). All patients carried missense variation in atleast one allele. Based on the type of variation in the second allele present along with Indian founder variant, early mean age of onset and lowest mean IBMFRS and mean MDFRS scores were noted with splice site variations. Novel variants are summarised in Supplementary Table 1.
Fig. 1
Types of zygosity – Pie-chart showing the various zygosities and most common pathogenic variants in each category. Parentheses next to the variant shows allele frequency. “n” represents number of patients in each zygosity.
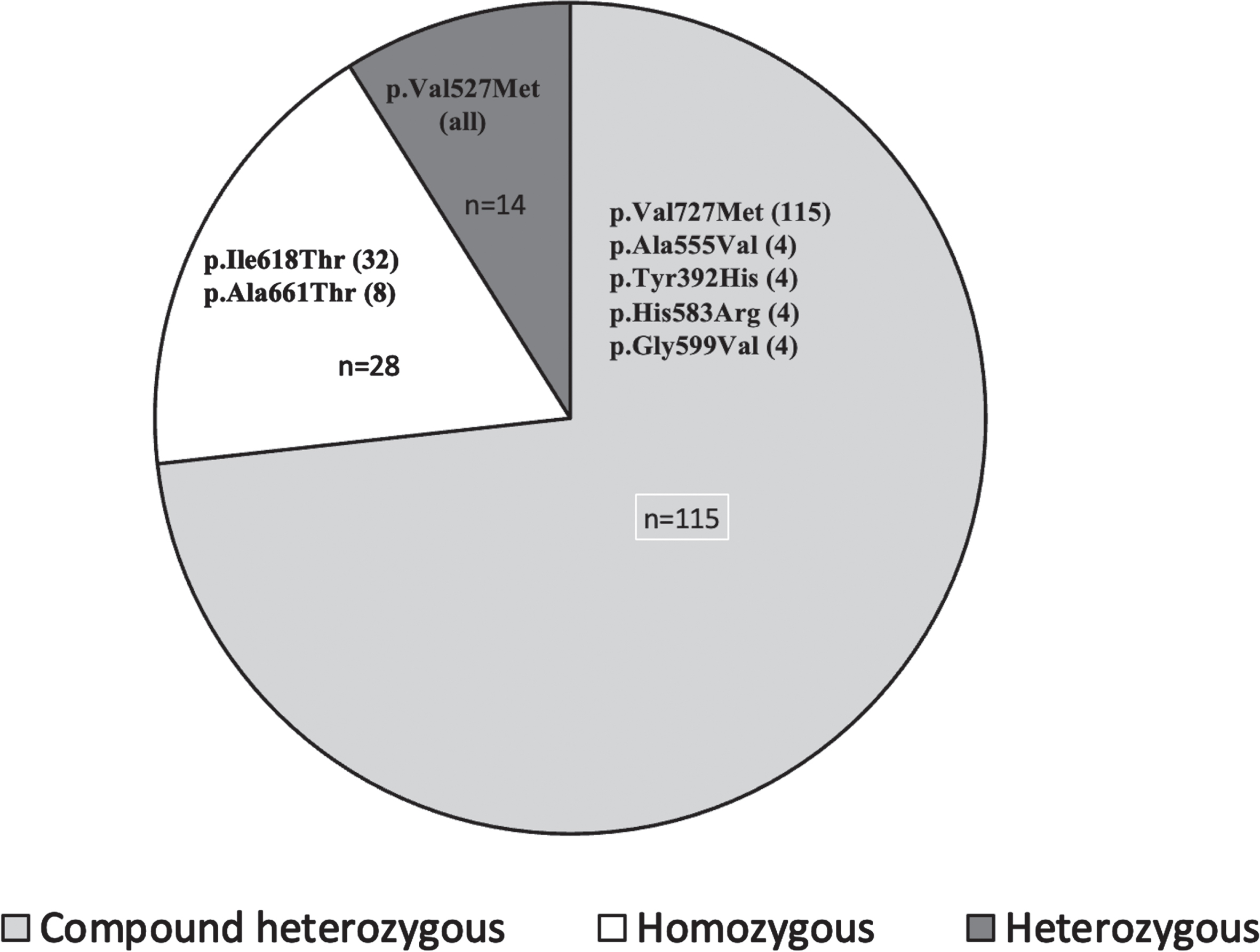
Fig. 2
Exon distribution of various pathogenic variants and mutation spectrum – A – Exons are represented as rectangular boxes with respective exonic numbers (Transcript ID: NM_001128227.3). White boxes at the ends represent untranslated regions (UTRs). The novel variants are highlighted in bold. ‘X’n represents number of individuals carrying the variant. SE – skipped exon, Het-Heterozygous, Hom-Homozygous, C.Het – Compound heterozygous. Size of exons /introns is not represented at scale. B – hGNE2 protein structure.: Gray UF - unknown function, vertical lined box denote putative nuclear export signal (NES) and dotted box denote experimental allosteric region (AR) for CMP-sialic acid binding. Light grey boxes denote UDP-GlcNAc 2-epimerase and dark grey denote ManNAc kinase domains. Amino acid numbering is indicated below the structure.
Follow-up data
Ninety-two patients were followed up with a mean duration of 2.9±2.7 years (range: 6months – 12 years). Loss of independent ambulation was seen in 29 patients with use of assistive devices (n = 20), wheelchair bound (n = 14) and bedbound state (n = 4). The mean duration of loss of ambulation from symptom onset was 11.7±3.7 years (range: 5 – 19). IBMFRS (n = 82) and MDFRS (n = 77) showed mean scores of 29±6.6 and 65.2±11 respectively at the last follow-up. Four patients developed swallowing difficulty at follow-up, with mean duration since onset of 8.3±3.5 years (range- 4.5–11.5). However, none had overt clinical cardiac or respiratory symptoms.
Genotype-Phenotype correlation (summarised in Table 3)
Table 3
Genotype – phenotype correlation for the entire cohort
| Genotype | Age at onset Mean (Range) | Age at presentation Mean (Range) | Use of Assistive device | Wheel Chair Bound | IBMFRS Mean (Range) | MDFRS Mean (Range) | ||
| N | Mean age (Range) | N | Mean age (Range) | |||||
| Domains | ||||||||
| K n = 91 | 26±5.2 (14 – 38) | 32.5±7.1 (19 – 55) | 21 | 28±6.4 (18.5 – 45) | 12 | 34.4±6.6 (27 – 49) | 28.2±6.9 (5 – 40) | 64.4±11.2 (28 – 83) |
| K+E, n = 58 | 27.1±7.2 (15 – 53) | 33.7±8.8 (21 – 63) | 8 | 33.5±7.3 (23 – 46) | 2 | 38.5±12.0 (30 – 47) | 30.4±6.0 (9 – 39) | 66.8±11.1 (22 – 80) |
| E n = 8 | 28.6±8.6 (19 – 46) | 33±7.9 (23 – 49) | 2 | 37.5±2.1 (36 – 39) | 0 | – | 28.5±8.2 (18 – 38) | 64.5±10.3 (52 – 76) |
| P-value (ANOVA – single factor) | 0.4 | 0.9 | 0.09 | 0.7 (t-test: unequal variances) | 0.4 | 0.6 | ||
| Variants | ||||||||
| c.2179 G>A (p.Val727Met) n = 129 (Indian Founder) | 32.3±6.1 (31.2-33.4) | 33.5±7.8 (32.2-34.8) | 9 | 30.9±5.3 (27.4-34.4) | 10 | 34.6±6.1 (30.8-38.4) | 29.4±6.2 (28.3-30.5) | 65.8±10.7 (64.0-67.4) |
| c.1853T>C (p.Ile618Thr) n = 8 (Roma Gypsy) | 20.4±4.1 (17.6-23.2) | 26.4±5.7 (22.5-30.4) | 1 | 31 | 1 | 20 | 27.7±11.2 (20-35.5) | 56.6±19.2 (43.3-69.9) |
| Others n = 20 | 29.9±8.3 (26.3-33.5) | 30.8±6.4 (28-33.6) | 2 | 37.5±2.1 | 3 | 27.5±3.1 | 25±6.5 (22.2-27.8) (8) | 60±11.3 (55.0-65.0) |
| Mutation Types | ||||||||
| Missense – Missense n = 97 | 25.7±6.3 [14 – 53] | 31.7±7.9 [19 – 63] | 19 | 28.9±6.5 [18.5 – 42] | 8 | 33.3±6.6 [27 – 47] | 29.1±6.9 [7 – 40] | 64±12.5 [22 – 83] |
| Missense – Nonsense n = 20 | 27.9±6.7 [16 – 41] | 35.2±7.5 [24 – 56] | 5 | 33±8.2 [23 – 46] | 0 | – | 28.5±4.3 [22 – 36] | 66.7±8.9 [55 – 77] |
| Missense – Frameshift n = 16 | 27.2±6.1 [19 – 42] | 34.3±8.4 [24 – 51] | 4 | 34±8.3 [25 – 45] | 3 | 39±10 [29 – 49] | 28.4±10.1 [5 – 37] | 67.6±8.9 [50 – 77] |
| Missense – Splice site n = 6 | 25±1.1 [24 – 26] | 30.8±6.4 [25 – 43] | 3 | 31.7±4.5 [27 – 36] | 1 | 39 | 24.2±5.9 [17 – 30] | 59.2±8.5 [47 – 66] |
| Missense – Intronic splice proximal n = 2 | 28.5±4.9 [25 – 32] | 32±1.4 [31 – 33] | 0 | – | 0 | – | – | – |
| Missense – inframe n = 2 | 25.5±3.5 [23 – 28] | 33.5±0.7 [33 – 34] | 0 | – | 1 | 28 | 31 | 71 |
| Missense (single allele) n = 14 | 29.8±5.5 [21 – 38] | 35.8±7.1 [25 – 48] | 0 | – | 0 | – | 31.8±3.6 [27 – 37] | 70.1±5.9 [63 – 80] |
| P-value (ANOVA – single factor) | 0.27 | 0.32 | 0.57 | 0.85 | 0.85 | 0.69 | ||
Footnotes: K- Kinase E – Epimerase.
Genotype-phenotype correlations were considered based on type of variations, domains and variants involved. Early age of onset and low IBMFRS and MDFRS scores were noted with missense and splice site variation combination. Similarly, age at onset and initial presentation were early in K group as compared to combination of K + E and E groups though not statistically significant. The mean age at first use of assistive device was also early in kinase group and was trending towards significance (p value-0.07). The mean IBMFRS and MDFRS scores were comparable and lower between K and E groups than K + E. Significant positive correlation [P(Z< =z) two-tail:<0.05] was noted between age at onset and age at use of assistance (r: 0.8, p-value: 0.0000002), mean IBMFRS score (r: 0.2, p-value: 0.02), mean MDFRS (r: 0.3, p-value: 0.005). Presence of variation in kinase domain resulted in early onset of symptoms (r: –0.1, p value: 0.2), early age at use of assistive device (r: –0.2, p value: 0.2), lower mean IBMFRS (r: –0.1, p value: 0.2) and lower MDFRS scores (r: –0.1, p value: 0.4) compared to presence of variation in both kinase and epimerise resulting in severe phenotype of GNEM (Supplementary Figure 2).
Among the two common founder mutations, patients with homozygous Roma gypsy mutation (p.Ile618Thr) had relatively severe phenotype compared to those with Indian founder mutation (p.Val727Met) in at least one allele. It was also noted that Indian founder mutation (p.Val727Met) in homozygous state which was noted in only two patients had later onset of disease at 30 and 25 years of age with duration of disease being 20 and 10 years, respectively. Both the patients were independently ambulant at presentation reflecting mild phenotype.
DISCUSSION
This is one of the largest study on GNEM from South Asia. The baseline demographic features of the current cohort (n = 157) such as mean age at onset and diagnosis are were comparable with previous studies [6, 16, 17]. Most of the patients had bilateral foot drop followed by proximal lower limb symptoms at the initial presentation which is typical of GNEM. Though typically upper limb involvement is noticed in about 5–10 years after the onset of symptoms [6], 8 of our patients had distal upper limb weakness as initial symptom with 3 of them having asymmetrical involvement. Previously, only a single case report by de Dios et al., have been reported with upper limb onset in GNEM [18]. Quadriceps involvement is reported to occur in upto 5% of cases. Our cohort had 3.2% of patients with this feature. About 32 patients had limb girdle pattern of weakness at presentation. This has been reported in another Indian cohort by Khadilkar et al., in which about 50% of their patients had limb girdle weakness at onset [19] and also the Korean cohort reported by Park et al., [20]. This reveals the clinical heterogeneity of GNEM with varied early presentations and diagnostic challenges. Atypical presenting features such as asymmetrical limb weakness and upper limb involvement was seen in patients with kinase domain involvement in our cohort in contrast to previous studies which showed epimerase involvement in these patients [21]. Beevor’s sign noted in > 50% of our patients has not been reported in other global cohorts. This could serve as an important clue in a given suspected case of GNEM. 17.8% of patients in the current cohort had normal serum CK levels. In GNEM patients, mild to moderate elevation of serum CK is often observed in early stages of disease. This would be expected to decrease over time with disease progression, and the CK level in non-ambulatory patients may be normal or low. Serum CK levels were more than 5 times the upper limit in 15 patients with most of them having kinase domain involvement. The most common variant noted was p.Val727Met. Due to the high prevalence of p.Val727Met variant in the Indian subcontinent and absence in other ethnic groups, it has been regarded as likely Indian founder mutation. Interestingly, the Indian founder mutation (p.Val727Met) has been most commonly reported in compound heterozygous state. This may be hypothesized that presence of this variant in homozygous state may be fatal at birth or mild manifestation requiring second allele for clinical manifestation [14]. Fifty novel GNE mutations were identified in this cohort expanding the mutational spectrum in GNEM [Supplementary Table 1]. Though classically GNEM occurs due to biallelic variations in GNE gene, 14 patients in our cohort had heterozygous mutation with all having the Indian founder mutation (p.Val727Met, c.2179 G>A). The inability to find the variation in second allele may be due to deep intronic or copy number variants which are often not detected in routine diagnostic sequencing and a follow up whole genome sequencing or RNA sequencing was not performed. On comparing previous studies on disease progression [6, 16, 22], the present cohort had earlier age at onset and varied initial presentation (Supplementary Table 2). Though only 8.9% of patients were wheelchair bound the patients in our cohort had an earlier age at wheel chair dependence of 32.0±7.1 years (mean duration – 11.7±3.7 years) compared to the above-described studies. The mean duration to wheelchair bound state in our cohort was much earlier than most of the previous studies [6, 10]. Early onset with rapid progression was noted in patients with both variations in the kinase domain compared to other two groups with either one variation each in kinase and epimerase domains or only having mutations in epimerase domain. Similarly, in a study done by Zhao et al., in the Chinese cohort [22], epimerase domain involvement had later onset than the kinase domain involvement. In contrast, a study by Pogoryelova et al., [6] showed more severe phenotype with compound heterozygous involvement of kinase and epimerase domains. On comparing the severity between the two founder mutations, p.Ile618Thr (Roma Gypsy) had earlier age at onset and lower mean IBMFRS and MDFRS score than p.Val727Met (Indian Founder mutation). Chamova et al., studied 50 patients with Roma Gypsy mutation of p.Ile618Thr with homozygous variation in 49 patients, mean age at onset of 24.4±6.31 years, mean duration from onset loss of ambulation was 10.34±4.31 years noted in 32 patients (range: 3–20 yrs) [13]. This was in contrast to the present study where Roma gypsy mutation had earlier mean age of onset (20.4±4.1 years) and only one patient had loss of ambulation. In the Japanese cohort reported by Cho A et al., the variant c.527A>T (p.Asp207Val) which was the second most common variant in the cohort was noted to be a milder variant. This was evident in both compound heterozygous and homozygous states. In contrast, the most variant of their cohort, c.1714 GC (p.Val603Leu), had severe disease phenotype in homozygous and compound heterozygous with non- c.527A>T variants [23]. Similarly, c.2179 G>A (p.Val727Met) in our cohort which was predominantly noted in compound heterozygous with only two patients in homozygous state had milder phenotype. The Roma Gypsy variant c.1853T>C (p.Ile618Thr) which occurred only in homozygous state had severe phenotype as shown by earlier age of onset and functional rating scale scores.
Thus, ethnic differences and mutation variants have an impact on disease severity and progression. Due to the ultra-rare nature of GNEM, all population-based studies tend to be underpowered with various factors affecting the link between genotype and phenotype in these patients.
CONCLUSION
This is the first study to report on the characteristics and disease progression in Indian GNEM patients and the largest genetically confirmed cohort describing the pattern on GNE mutations in India. Apart from the typical clinical features, notable characteristics of prominent and early weakness of long finger flexors of lateral fingers, positive Beevor’s sign and limb girdle pattern was seen in this study. Genotype – phenotype correlation revealed early onset with loss of ambulation noted with biallelic kinase domain and Roma gypsy founder mutations.
LIMITATIONS OF THE STUDY
The major limitations of the study include:
- Retrospective study
- Sanger sequencing/functional studies not done in heterozygous cases.
DATA AVAILABILITY
Data will be available on request from the corresponding author.
FUNDING
3Kiran Polavarapu receives support from the Canadian Institutes of Health Research (CIHR) postdoctoral fellowship award grant no: 202210MFE-491707-FPP-CECC-404816.
8Oksana Pogoryelova receives support from the Wellcome centre for mitochondrial research, Newcastle University, Newcastle upon Tyne, UK.
9Hanns Lochmuller receives support from the Canadian Institutes of Health Research (Foundation Grant FDN-167281, Team Grant ERT-174211), the Canadian Institutes of Health Research and Muscular Dystrophy Canada (Network Catalyst Grant for NMD4 C NG2-170044), the Canada Foundation for Innovation (CFI-JELF 38412), and the Canada Research Chairs program (Canada Research Chair in Neuromuscular Genomics and Health, 950-232279).
CONFLICT OF INTEREST
Hanns Lochmuller is an Editorial Board Member of this journal, but was not involved in the peer-review process nor had access to any information regarding its peer-review.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-230130.
REFERENCES
[1] | Nonaka I , Sunohara N , Ishiura S , Satoyoshi E . Familial distal myopathy with rimmed vacuole and lamellar (myeloid) body formation. J Neurol Sci. (1981) ;51: :141–55. |
[2] | Argov Z , Yarom R . “Rimmed vacuole myopathy” sparing the quadriceps. A unique disorder in Iranian Jews. J Neurol Sci. (1984) ;64: :33–43. |
[3] | Huizing M , Carrillo-Carrasco N , Malicdan MC , Noguchi S , Gahl WA , Mitrani-Rosenbaum S , Argov Z , Nishino I . GNE myopathy: new name and new mutation nomenclature. Neuromuscul Disord. (2014) ;24: :387–9. |
[4] | Carrillo N , Malicdan MC , Huizing M . GNE Myopathy: Etiology, Diagnosis, and Therapeutic Challenges. Neurotherapeutics. (2018) ;15: (4):900–914. |
[5] | Nalini A , Gayathri N , Dawn R . Distal myopathy with rimmed vacuoles: Report on clinical characteristics in 23 cases. Neurol India. (2010) ;58: :235–41. |
[6] | Pogoryelova O , Cammish P , Mansbach H , Argov Z , Nishino I , Skrinar A , et al. Phenotypic stratification and genotype-phenotype correlation in a heterogeneous, international cohort of GNE myopathy patients: first report from the GNE myopathy Disease Monitoring Program, registry portion. Neuromuscul Disord. (2018) ;28: (2):158–168. |
[7] | Nishino I , Carrillo-Carrasco N , Argov Z . GNE myopathy: current update and future therapy. J NeurolNeurosurg Psychiatry. (2015) ;86: (4):385–92. |
[8] | Schauer R . Sialic acids as regulators of molecular and cellular interactions. Curr Opin Struct Biol. (2009) ;19: (5):507–14. |
[9] | Gagiannis D , Orthmann A , Danssmann I , et al. Reduced sialylation status in UDP-N- acetylglucosamine-2-epimerase/N-acetylmannosamine kinase (GNE)-deficient mice. Glycoconj J. (2007) ;24: (2-3):125–30. |
[10] | Zhu W , Mitsuhashi S , Yonekawa T , Noguchi S , Huei JC , Nalini A , et al. Missing genetic variations in GNE myopathy: rearrangement hotspots encompassing 5′UTR and founder allele. J Hum Genet. (2017) ;62: (2):159–166. |
[11] | Argov Z , Eisenberg I , Grabov-Nardini G , et al. Hereditary inclusion body myopathy: the Middle Eastern genetic cluster. Neurology. (2003) ;60: (9):1519–23. |
[12] | Nishino I , Noguchi S , Murayama K , Driss A , Sugie K , Oya Y , Nagata T , Chida K , Takahashi T , Takusa Y , Ohi T , Nishimiya J , Sunohara N , Ciafaloni E , Kawai M , Aoki M , Nonaka I . Distal myopathy with rimmed vacuoles is allelic to hereditary inclusion body myopathy. Neurology. (2002) ;59: :1689–93. |
[13] | Chamova T , et al. GNE myopathy in Roma patients homozygous for the I618T founder mutation. NeuromusculDisord. (2015) ;25: :713–8. |
[14] | Bhattacharya S , Khadilkar SV , Nalini A , Ganapathy A , Mannan AU , Majumder PP , Bhattacharya A . Mutation Spectrum of GNE Myopathy in the Indian Sub-Continent. J Neuromuscul Dis. (2018) ;5: (1):85–92. |
[15] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , et al., ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) ;17: (5):405–24. |
[16] | Park YE , Kim DS , Choi YC , Shin JH . Progression of GNE Myopathy Based on the Patient-Reported Outcome. J Clin Neurol. (2019) ;15: (3):275–284. |
[17] | Mori-Yoshimura M , Hayashi YK , Yonemoto N , Nakamura H , Murata M , Takeda S , et al. Nationwide patient registry for GNE myopathy in Japan. Orphanet J Rare Dis. (2014) ;9: :150. |
[18] | de Dios JK , Shrader JA , Joe GO , McClean JC , Williams K , Evers R , Malicdan MC , Ciccone C , Mankodi A , Huizing M , McKew JC , Bluemke DA , Gahl WA , Carrillo-Carrasco N . Atypical presentation of GNE myopathy with asymmetric hand weakness. NeuromusculDisord. (2014) ;24: (12):1063–7. |
[19] | Khadilkar SV , Chaudhari AD , Singla MB , Dastur RS , Gaitonde PS , Bhutada AG , Hegde MR . Early and consistent pattern of proximal weakness in GNE myopathy. Muscle Nerve. (2021) ;63: (2):199–203. |
[20] | Lochmüller H , Behin A , Tournev I , Tarnopolsky M , Horváth R , Pogoryelova O , et al. Results from a 3-year Non-interventional, Observational Disease Monitoring Program in Adults with GNE Myopathy. J Neuromuscul Dis. (2021) ;8: (2):225–234. |
[21] | Park YE , Kim HS , Choi ES , et al. Limb-girdle phenotype is frequent in patients with myopathy associated with GNE mutations. J Neurol Sci. (2012) ;321: (1-2):77–81. |
[22] | Zhao J , Wang Z , Hong D , Lv H , Zhang W , Chen J , Yuan Y . Mutational spectrum and clinical features in 35 unrelated mainland Chinese patients with GNE myopathy. J Neurol Sci. (2015) ;354: (1-2):21–6. |
[23] | Cho A , Hayashi YK , Monma K , Oya Y , Noguchi S , Nonaka I , Nishino I . Mutation profile of the GNE gene in Japanese patients with distal myopathy with rimmed vacuoles (GNE myopathy). J Neurol Neurosurg Psychiatry. (2014) ;85: (8):914–7. |


