
Real-World Outcomes in Patients with Spinal Muscular Atrophy Treated with Onasemnogene Abeparvovec Monotherapy: Findings from the RESTORE Registry
Abstract
Background:
Long-term, real-world effectiveness and safety data of disease-modifying treatments for spinal muscular atrophy (SMA) are important for assessing outcomes and providing information for a larger number and broader range of SMA patients than included in clinical trials.
Objective:
We sought to describe patients with SMA treated with onasemnogene abeparvovec monotherapy in the real-world setting.
Methods:
RESTORE is a prospective, multicenter, multinational, observational registry that captures data from a variety of sources.
Results:
Recruitment started in September 2018. As of May 23, 2022, data were available for 168 patients treated with onasemnogene abeparvovec monotherapy. Median (IQR) age at initial SMA diagnosis was 1 (0–6) month and at onasemnogene abeparvovec infusion was 3 (1–10) months. Eighty patients (47.6%) had two and 70 (41.7%) had three copies of SMN2, and 98 (58.3%) were identified by newborn screening. Infants identified by newborn screening had a lower age at final assessment (mean age 11.5 months) and greater mean final (SD) CHOP INTEND score (57.0 [10.0] points) compared with clinically diagnosed patients (23.1 months; 52.1 [8.0] points). All patients maintained/achieved motor milestones. 48.5% (n = 81/167) experienced at least one treatment-emergent adverse event (AE), and 31/167 patients (18.6%) experienced at least one serious AE, of which 8/31 were considered treatment-related.
Conclusion:
These real-world outcomes support findings from the interventional trial program and demonstrate effectiveness of onasemnogene abeparvovec over a large patient population, which was consistent with initial clinical data and published 5-year follow-up data. Observed AEs were consistent with the established safety profile of onasemnogene abeparvovec.
INTRODUCTION
Spinal muscular atrophy (SMA) is a rare, debilitating neuromuscular disease characterized by loss of motor neurons, leading to progressive weakness and atrophy of skeletal and bulbar muscles [1–8]. Although almost all cases of SMA have the same underlying genetic cause —a biallelic deletion or mutation in the survival motor neuron 1 (SMN1) gene —clinical severity is heterogeneous, with varying copy number of the SMN2 “backup” gene being strongly correlated with disease onset and severity as an important phenotypic modifier of SMA [2–11]. Historically, SMA phenotype has been classified by five clinical types based on the age at onset and maximum motor function ranging from type 0 (most severe with prenatal onset) to type 4 (less severe with later onset) [3, 4, 6, 12–14]. SMA type 1 (severe with onset during the first 6 months of life) is the most common phenotype, accounting for approximately 60% of SMA cases [1, 6, 8, 9, 15, 16]. Because SMA type is based on maximum motor function achieved by untreated patients, it does not take into account the more recent changes in disease phenotype caused by administration of disease-modifying treatments (DMTs) [14].
SMA has long been cited as the leading genetic cause of infant mortality, with natural history studies of SMA type 1 reporting nearly all patients (90%) dying or requiring permanent ventilation by 2 years of age with supportive treatment only [9, 15, 17–20]. However, prognoses have improved markedly in recent years, mainly owing to the advent of DMTs [13, 14, 21–38]. The first of these, nusinersen, became available for the treatment of SMA in 2016. Nusinersen is an intrathecally administered antisense oligonucleotide that alters the splicing of SMN2 to increase the amount of functional SMN protein produced [22, 23]. An oral SMN2 splicing modifier, risdiplam, became available in 2020 [24, 25]. In 2019, the US Food and Drug Administration approved onasemnogene abeparvovec, a one-time, intravenous, adeno-associated virus 9 (AAV9) vector-based gene replacement therapy that delivers a fully functional copy of the human SMN1 cDNA into target cells [26––28]. Since then, onasemnogene abeparvovec has been approved for the treatment of SMA by the European Medicines Agency and in many other countries.
Knowledge of these new treatments for SMA has been largely based on interventional trial data, which is limited most notably by narrow eligibility criteria and limited follow-up duration. In onasemnogene abeparvovec clinical studies, which included patients with SMA type 1 who had two or three copies of SMN2, almost all of whom were younger than 6 months of age at infusion (mean age range, 0.5–7.9 months), and presymptomatic infants with either two or three copies of SMN2 at risk for developing SMA type 1 or SMA types 2 or 3, respectively, gene therapy administration resulted in longer survival (free from permanent ventilation), achievement of motor milestones, and improved motor function, especially for patients treated earlier, or in the prodromal phase of the disease [27, 31–34]. The inclusion criteria for these onasemnogene abeparvovec clinical trials were defined based on the need to produce robust evidence of clinical efficacy within the duration of the clinical trial setting; however, the real-world use of onasemnogene abeparvovec also targets patients with different criteria from those of the clinical trials [27, 31–34]. Because the approved indication for onasemnogene abeparvovec is broader than the study population upon which initial approval was based [26, 27], real-world evidence became especially important shortly after the approval of onasemnogene abeparvovec. In addition, long-term safety and durability data could not be obtained in the course of the onasemnogene abeparvovec clinical studies [27, 31–34], and long-term extension studies will fill this data gap [38, 39].
Published real-world evidence to date, which mostly consists of individual case reports, small case series, and single-center experiences, has contributed a limited amount of clinical information to the body of knowledge that assists clinicians and caregivers with treatment decisions and guides therapeutic expectations for SMA patients receiving DMTs [40–46]. Therefore, additional long-term, real-world data that are more reflective of the broader SMA patient population are needed to fill this knowledge gap and characterize the effectiveness and safety of these DMTs and to guide treatment decisions [47–49].
RESTORE is an ongoing, prospective, multinational, multicenter, observational disease registry assessing real-world treatment patterns and outcomes for patients with SMA, with the goal of informing treatment decisions and improving patient outcomes [47]. The RESTORE registry was designed to augment data from existing SMA registries [38, 50–54] by providing important information about the clinical course of SMA for patients receiving DMTs that were not available as treatment options when earlier SMA patient registries were created. To enhance data from long-term extensions of completed and ongoing clinical trials and existing real-world study data, we describe patients with SMA treated with onasemnogene abeparvovec monotherapy from the RESTORE registry, including patient demographics and clinical characteristics and real-world effectiveness and safety outcomes.
METHODS
The RESTORE registry is governed by a steering committee of international SMA experts committed to ensuring data quality and the sharing of RESTORE registry data [47]. RESTORE is sponsored by Novartis Gene Therapies, Inc., the manufacturer of onasemnogene abeparvovec [47], and was designed to capture SMA history and treatment, effectiveness, and safety data for patients receiving DMTs [47]. Detailed study methodology that encompasses study design, ethical considerations, patient eligibility, data acquisition, and variables assessed has been published (Supplementary Figure 1) [47].
Patient care in RESTORE follows usual SMA treatment practices in each country and participating clinical site and depends upon local resources. Patients are recruited worldwide to provide sufficient data to document differences between patients who receive a variety of treatments as they evolve during the 15-year duration of this registry [47]. Patients are enrolled consecutively at each site to minimize selection bias [47]. Data for patients with SMA meeting RESTORE eligibility criteria were collected from individual de novo (i.e., new, independent) clinical study sites [47]. Data cutoff for the current analysis was May 23, 2022.
Clinical care in RESTORE is not dictated by a research protocol [47], and no additional support is provided by Novartis. RESTORE participation does not require any mandatory visits, tests, or assessments [47]. Participating centers contributing data are not required to have experience or expertise in performing rigorous clinical trials. RESTORE enrollment is rolling (i.e., continuous, allowing consecutive, ongoing patient enrollment without waiting or deadlines) over a 5-year period, with a follow-up period of 15 years, until death, or until the patient is withdrawn from the registry, which may occur at the discretion of the patient or the patient’s parent/legal representative or physician [47].
Patients
In the current study, real-world effectiveness and safety outcomes were assessed for patients following onasemnogene abeparvovec monotherapy (i.e., patients who received only onasemnogene abeparvovec gene therapy and have not received any dose of another DMT [i.e., nusinersen and/or risdiplam [55]. This population was selected because it represents a large percentage of the patients enrolled in RESTORE, and follow-up evidence in the first years of treatment with onasemnogene abeparvovec is critical to more fully characterize patient impacts in larger populations, given the often smaller populations associated with most rare disease clinical studies, and including current advanced treatments that most frequently center on niche populations. Patient variables collected in RESTORE include sociodemographics (i.e., age, year of birth, sex, race), medical history of SMA (i.e., date of and age at diagnosis, genetic status, SMN2 copy number, weight and height at diagnosis, newborn screening [NBS] status), date of onasemnogene abeparvovec administration, and prednisolone treatment [47].
Effectiveness
Patient assessments in RESTORE included event-free survival, motor function, motor milestones, and bulbar function [47].
Event-free survival
Event-free survival was assessed by SMN2 copy number using Kaplan-Meier estimates [47] and was defined as the avoidance of death or the requirement of permanent ventilatory support. Permanent ventilatory support was defined as requiring either a tracheostomy or respiratory support for 16 or more hours per day (including non-invasive ventilatory support) continuously for 14 or more days in the absence of an acute reversible illness, excluding perioperative ventilation. Patients requiring permanent ventilatory support prior to treatment were excluded.
Motor function
Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND), Hammersmith Infant Neurological Examination –Section 2 (HINE-2), and Hammersmith Functional Motor Scale –Expanded (HFMSE) were collected in RESTORE (Supplementary Table 1) [47]. The interval for each collection (e.g., at enrollment or at every follow-up) was dependent on the routine practice of follow-up visits at each participating SMA clinic and the family’s ability to bring the patient into the SMA clinic for a routine follow-up visit. Motor function was assessed for patients in RESTORE treated with onasemnogene abeparvovec monotherapy having two or more assessments (one or more occurring post-treatment) with at least 6 months between first and last assessments. Scores were not required to have been collected pretreatment; therefore, two scores available for analysis may both reflect post-treatment changes.
Motor milestones
The evaluation of motor milestones was assessed using performance criteria from the World Health Organization (WHO) [56, 57] and Bayley Scales of Infant Development, Third Edition (BSID) [58]. Ten select performance criteria were used to define the achievement of developmental milestones on the case report form, specifically: holds head erect 3 seconds; rolls from back to sides; sits without support for 10 seconds (WHO) and 30 seconds (BSID); stands with assistance; crawls; pulls to stand; walks with assistance; stands alone; and walks alone.
Bulbar function
Measures of bulbar function in RESTORE included total oral nutrition, ability to swallow, and aspiration pneumonia/pneumonia.
Safety
Safety data, including pulmonary assessments (i.e., was it performed; normal/abnormal; if abnormal, details), ventilatory support (i.e., cough assist details, noninvasive details, invasive details), use of non-oral feeding support, safety laboratories including liver function tests, and start and stop dates of serious adverse events (SAEs) and adverse events of special interest (AESIs), including dates and primary causes of death, were collected in RESTORE [47]. An SAE was defined as being an AE with at least one of the following outcomes: death, is life-threatening, requires inpatient hospitalization or causes prolongation of existing hospitalization, results in persistent or significant disability/incapacity, may have caused a congenital anomaly/birth defect, or requires intervention to prevent permanent impairment or damage. Treatment-emergent was defined as an AE with start date on or after onasemnogene abeparvovec administration or an AE with a severity that worsened on or after onasemnogene abeparvovec administration. AEs were coded using MedDRA®, Version 25.0, with patients counted only once at each level of summarization.
AESIs included hepatotoxicity, transient thrombocytopenia, cardiac AEs, and thrombotic microangiopathy (TMA). In the RESTORE registry, physicians reported AEs at their discretion, with any reported AESIs matching the predefinitions being counted as an AESI. Hepatotoxicity was predefined as having clinically significant laboratory values (i.e., alanine aminotransferase [ALT] or aspartate aminotransferase [AST] > 3×upper limit of normal [ULN] with associated bilirubin > 2×ULN); hepatic failure, fibrosis, cirrhosis and other liver damage-related conditions, or hepatic disorders. Thrombocytopenia was predefined as having an AE identified as hematopoietic thrombocytopenia, hemorrhages, or platelet disorders. Cardiac AESIs were predefined as having ischemic heart disease, cardiomyopathy, cardiac arrhythmias, myocardial infarction, or embolic and thrombotic events.
Descriptive analyses
Demographic, clinical, and treatment characteristics were analyzed using descriptive analyses with means (±standard deviation [SD]), median and range, or percentages, as applicable. The relationships of the endpoints were investigated qualitatively [47]. Missing observations were not imputed in the analysis of individual questions or items [47]. Time-to-event outcomes, such as event-free survival, were assessed using Kaplan-Meier analysis. Analyses were performed using SAS Version 9.4 (Cary, NC). Mean change in first observation to last, mean monthly change, and time between assessments were assessed for CHOP INTEND, HINE-2, and HFMSE for overall population, infants identified by NBS, and patients with any SMN2 copy number. Minimal clinically important differences (MCID) were defined for CHOP INTEND (≥4-point change), HFMSE, (≥3-point change), and HINE-2 (≥2-point change). Patients were classified into three categories: improved (patients who achieved a higher milestone at the last assessment compared with the first assessment); maintained (patients remained stable and were at the same milestone at the first and the last assessments); and other (patients who achieved a milestone that was not recorded at a subsequent evaluation).
RESULTS
Patients
Recruitment started in September 2018, and, as of the May 23, 2022, data cutoff, data were available for 385 patients globally. There were 168 patients with SMA treated with onasemnogene abeparvovec monotherapy from seven countries worldwide (United States n = 138; Japan n = 19; Taiwan n = 4; Portugal n = 3; Israel n = 2; Greece n = 1; and Russia n = 1). The median (IQR) age at initial diagnosis of SMA was 1 (0–6) month (Table 1). The median (IQR) age at onasemnogene abeparvovec infusion was 3 (1–10) months. The mean (SD) weight was 5.05 (2.31) kg at initial diagnosis and 5.92 (2.34) kg at onasemnogene abeparvovec infusion. Approximately half of the patients (n = 80/168; 47.6%) had two copies of SMN2, 82 patients (48.8%) were asymptomatic at diagnosis, and 98 infants (58.3%) were identified by NBS (see Table 1). Nine of the 168 (5.4%) patients treated with onasemnogene abeparvovec monotherapy had gestational age ≤35 weeks at birth, with eight of these nine patients (88.9%) receiving treatment before 6 months of age (median age 2.0 [range 1–7] months). Mean (SD) time from onasemnogene abeparvovec infusion to last known visit was 13.68 (8.94) months, with patients who received onasemnogene abeparvovec monotherapy having a range of post-treatment follow-up of 0.03 to 37.06 months.
Table 1
Demographics and baseline clinical characteristics for all patients identified by newborn screening, clinical diagnosis, and in the overall cohort
| Characteristics | Newborn screening | Clinical diagnosis | All patients |
| (n = 98) | (n = 70) | (N = 168) | |
| Age, months | |||
| At initial SMA diagnosis | |||
| Mean (SD) | 1.34 (7.12) | 9.49 (6.54) | 4.73 (7.96) |
| Median (IQR) | 0 (0–1) | 8.5 (4–14) | 1 (0–6) |
| Min, Max | 0, 70 | 0, 27 | 0, 70 |
| At onasemnogene abeparvovec infusion | |||
| Mean (SD) | 3.30 (7.99) | 10.70 (6.66) | 6.38 (8.29) |
| Median (IQR) | 1 (1–2) | 9.5 (5–15) | 3 (1–10) |
| Min, Max | 0, 72 | 0, 28 | 0, 72 |
| Weight, mean (SD), kg | |||
| At initial SMA diagnosis | 3.53 (0.86) | 7.20 (1.98) | 5.05 (2.31) |
| At onasemnogene abeparvovec infusion | 4.65 (1.74) | 7.56 (1.96) | 5.92 (2.34) |
| Weight range (kg) at onasemnogene abeparvovec infusion | 1.6–10.5 | 3.3–12.3 | 1.6–12.3 |
| Weight category at onasemnogene abeparvovec infusion | |||
| < 8.5 kg | 76 (77.5) | 45 (64.3) | 121 (72.0) |
| ≥8.5 kg | 4 (4.1) | 17 (24.3) | 21 (12.5) |
| Missing | 18 (18.4) | 8 (11.4) | 26 (15.5) |
| Duration, months | |||
| Time from diagnosis to treatment | |||
| Mean (SD) | 1.96 (3.61) | 1.23 (1.17) | 1.65 (2.87) |
| Median (IQR) | 1 (1–1) | 1 (1–2) | 1 (1–2) |
| Min, Max | 0, 23 | 0, 6 | 0, 23 |
| Time from first treatment to last known visit | |||
| Mean (SD) | 12.06 (8.18) | 15.95 (9.5) | 13.68 (8.94) |
| Median (IQR) | 11.15 (5.26–17.81) | 13.44 (9.17–24.02) | 12.24 (6.03–19.22) |
| Min, Max | 0.03, 30.62 | 0.03, 37.06 | 0.03, 37.06 |
| Sex | |||
| Female, n (%) | 52 (53.1) | 35 (50.0) | 87 (51.8) |
| Male, n (%) | 46 (46.9) | 35 (50.0) | 81 (48.2) |
| SMA type, n (%) | |||
| 1 | 27 (27.6) | 43 (61.4) | 70 (41.7) |
| 2 | 3 (3.1) | 23 (32.9) | 26 (15.5) |
| 3 | 2 (2.0) | 0 (0.0) | 2 (1.2) |
| Missing/not specified where not applicable | 66 (67.4) | 4 (5.7) | 70 (41. 7) |
| Asymptomatic at SMA diagnosis, n (%) | |||
| Yes (asymptomatic) | 79 (80.6) | 3 (4.3) | 82 (48.8) |
| No (symptomatic) | 19 (19.4) | 67 (95.7) | 86 (51.2) |
| SMN2 copy number, n (%) | |||
| One | 3 (3.1) | 0 (0.0) | 3 (1.8) |
| Two | 42 (42.9) | 38 (54.3) | 80 (47.6) |
| Three | 39 (39.8) | 31 (44.3) | 70 (41.7) |
| Four or more than four | 14 (14.3) | 0 (0.0) | 14 (8.3) |
| Unknown | 0 (0.0) | 1 (1.4) | 1 (0.6) |
| Country, n (%) | |||
| United States | 92 (93.9) | 46 (65.7) | 138 (82.1) |
| Japan | 1 (1.0) | 18 (25.7) | 19 (11.3) |
| Greece | 0 (0.0) | 1 (1.4) | 1 (0.6) |
| Israel | 1 (1.0) | 1 (1.4) | 2 (1.2) |
| Portugal | 0 (0.0) | 3 (4.3) | 3 (1.8) |
| Russia | 0 (0.0) | 1 (1.4) | 1 (0.6) |
| Taiwan | 4 (4.1) | 0 (0.0) | 4 (2.4) |
| Race, n (%) | |||
| White | 61 (62.2) | 38 (54.3) | 99 (58.9) |
| Asian | 6 (6.1) | 20 (28.6) | 26 (15.5) |
| Black | 6 (6.1) | 2 (2.9) | 8 (4.8) |
| Multiracial | 6 (6.1) | 0 (0.0) | 6 (3.6) |
| Native Hawaiian or other Pacific Islander | 0 (0.0) | 1 (1.4) | 1 (0.6) |
| Not reported | 13 (13.3) | 5 (7.1) | 18 (10.7) |
| Missing | 6 (6.1) | 4 (5.7) | 10 (6.0) |
IQR, interquartile range; SD, standard deviation; SMA, spinal muscular atrophy; SMN2, survival motor neuron 2 gene.
Effectiveness
Event-free survival
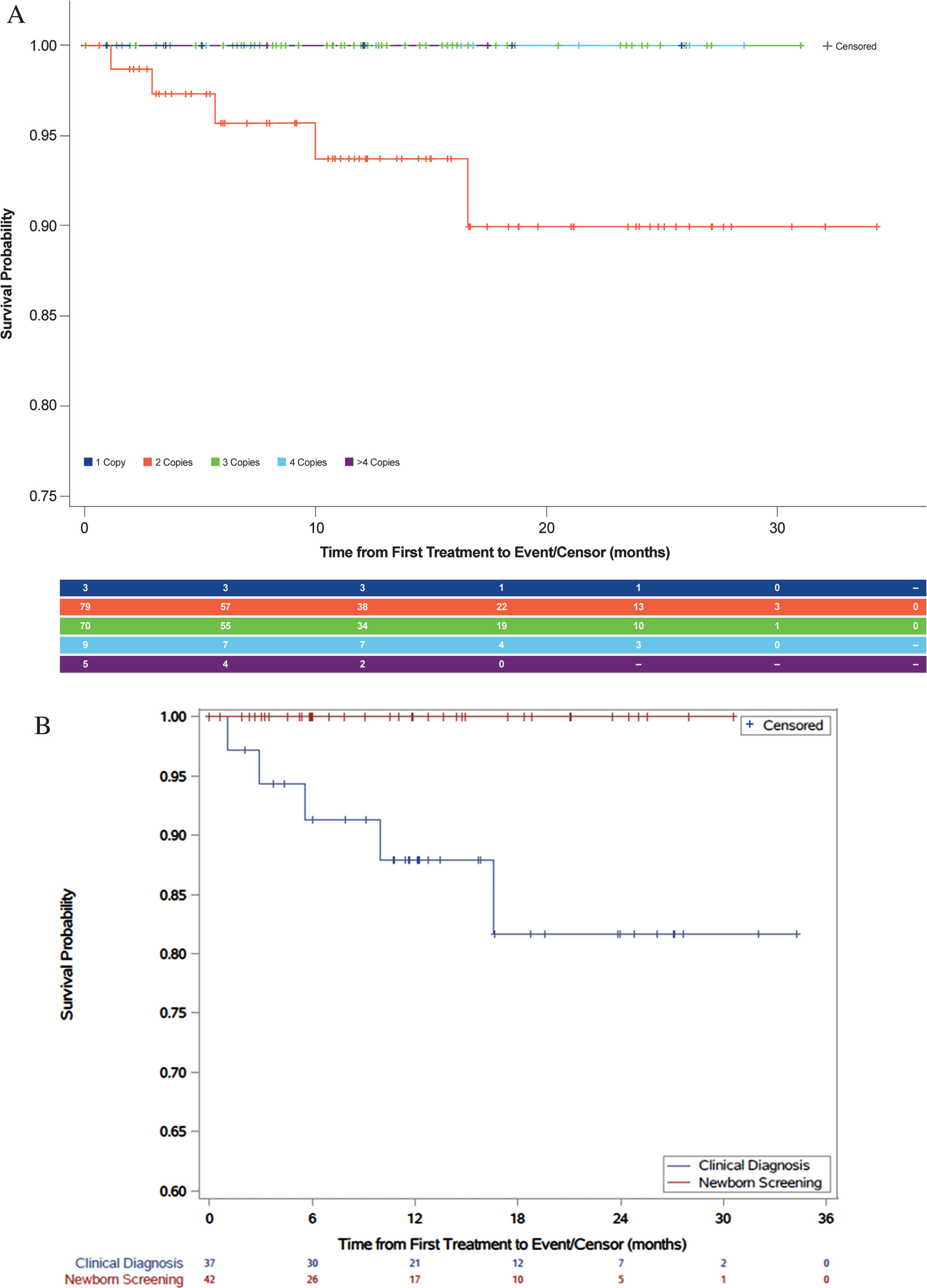
Event-free survival for patients with two copies of SMN2 was dramatically improved compared with natural history (Fig. 1a). Event-free survival of 93.7% was observed for 79 patients with two copies of SMN2 post-treatment at 1 year, and 90% event-free survival was observed for these patients at 2 years (three two-copy patients had a tracheostomy). Event-free survival of 100% was observed for 70 patients with three copies of SMN2 post-treatment at 1 year. Although one patient required ventilatory support, there were insufficient data to estimate event-free survival for patients with one copy (n = 3) or four or more copies (n = 14) of SMN2. Treatment benefits were observed for patients with two copies of SMN2 identified by NBS who were treated with onasemnogene abeparvovec (Fig. 1b). For the 42 infants with two copies of SMN2 identified by NBS, 100% event-free survival was observed at 1 year and 2 years post-treatment. For the 37 patients with two copies of SMN2 diagnosed clinically, 87.9% event-free survival was observed at 1 year and 81.7% at 2 years post-treatment.
Fig. 1
Event-free survival: patients treated with onasemnogene abeparvovec (A) by SMN2 copy number and (B) patients with two copies of SMN2 gene identified by newborn screening or clinical diagnosis.
Motor function
Of the 66 patients treated with onasemnogene abeparvovec monotherapy who had CHOP INTEND scores, 41 (62.1%) had two or more CHOP INTEND assessments, with ≥6 months between first and last assessments. It must be noted that in nine patients, the first CHOP INTEND was performed after onasemnogene abeparvovec infusion. CHOP INTEND scores increased overall, with treatment benefits observed for the 41 patients having two or more assessments (n = 20 identified by NBS and n = 21 diagnosed clinically) (Table 2). A total of 13 of 20 (65.0%) patients identified by NBS and 13 of 21 (61.9%) clinically diagnosed patients having two or more CHOP INTEND assessments had two SMN2 gene copies (Supplementary Table 2). For patients having two or more assessments, the mean (range) age at onasemnogene abeparvovec infusion was 2.15 (1–11) months for NBS patients and 8.2 (0–19) months for clinically diagnosed patients. For patients with two SMN2 copies, the mean (SD) age at onasemnogene abeparvovec infusion was 1.41 (0.9) months for NBS patients and 4.50 (3.56) months for clinically diagnosed patients. The range of CHOP INTEND total scores for patients with two copies of SMN2 was much greater than the range for patients with three copies of SMN2, for both patients identified by NBS and those clinically diagnosed (Fig. 2). It should be noted that, unlike the other CHOP INTEND evaluations, this analysis included all patients with at least two assessments, regardless of time between assessments (i.e., not necessarily 6 months between first and last assessments). An increase in scores of ≥4 points between first and last assessment was realized in the infants identified by NBS and the patients who were clinically diagnosed (85.0% [n = 17/20] and 95.2% [n = 20/21], respectively) (see Table 2). Three patients identified by NBS did not achieve a ≥4-point increase between first and last assessment. Infants identified by NBS were identified and assessed at the mean age of 1.7 months and had initial mean (SD) CHOP INTEND scores of 44.9 (13.0) points. Patients who were clinically diagnosed were identified and assessed at the mean age of 11.0 months and had initial mean (SD) CHOP INTEND scores of 37.3 (11.3) points. The difference in mean age and initial mean CHOP INTEND scores for patients identified by NBS and clinically diagnosed patients demonstrate relatively lower motor neuron loss. For infants identified by NBS, mean age at final assessment was 11.5 months and final mean (SD) CHOP INTEND scores were 57.0 (10.0) points. For clinically diagnosed patients, mean age at final assessment was 23.1 months and final mean (SD) CHOP INTEND scores were 52.1 (8.0) points.
Table 2
Changes in CHOP INTEND total score: patients identified by newborn screening or clinical diagnosis
| Newborn screening (n = 20) | Clinical diagnosis (n = 21) | |
| Score at first assessment, mean (SD) | 44.9 (13.0) | 37.3 (11.3) |
| Score at last assessment, mean (SD) | 57.0 (10.0) | 52.1 (8.0) |
| Difference between scores, mean (SD) | 12.1 (7.8) | 14.1 (9.8) |
| Months between assessments, mean (SD) | 9.7 (4.6) | 12.1 (4.9) |
| Age at first assessment, months, mean (SD) | 1.7 (1.5) | 11.0 (5.7) |
| Age at last assessment, months, mean (SD) | 11.5 (94.7) | 23.1 (7.2) |
| Achieved ≥4-point increase between first and last assessment, n (%)a | 17 (85.0) | 20 (95.2) |
CHOP INTEND, Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders; SD, standard deviation. aThree patients identified by newborn screening did not achieve a ≥4-point increase between first and last assessment. This was most likely related to the ceiling effect.
Fig. 2
CHOP INTEND scores: newborn screening versus clinical diagnosis.a CHOP INTEND, Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders. aCHOP INTEND scores were not required to have been collected pretreatment; therefore, patients with two scores available for analysis may both reflect post-treatment changes. Unlike the other CHOP INTEND evaluations, the data in this figure include all patients with at least two assessments, regardless of time between assessments.
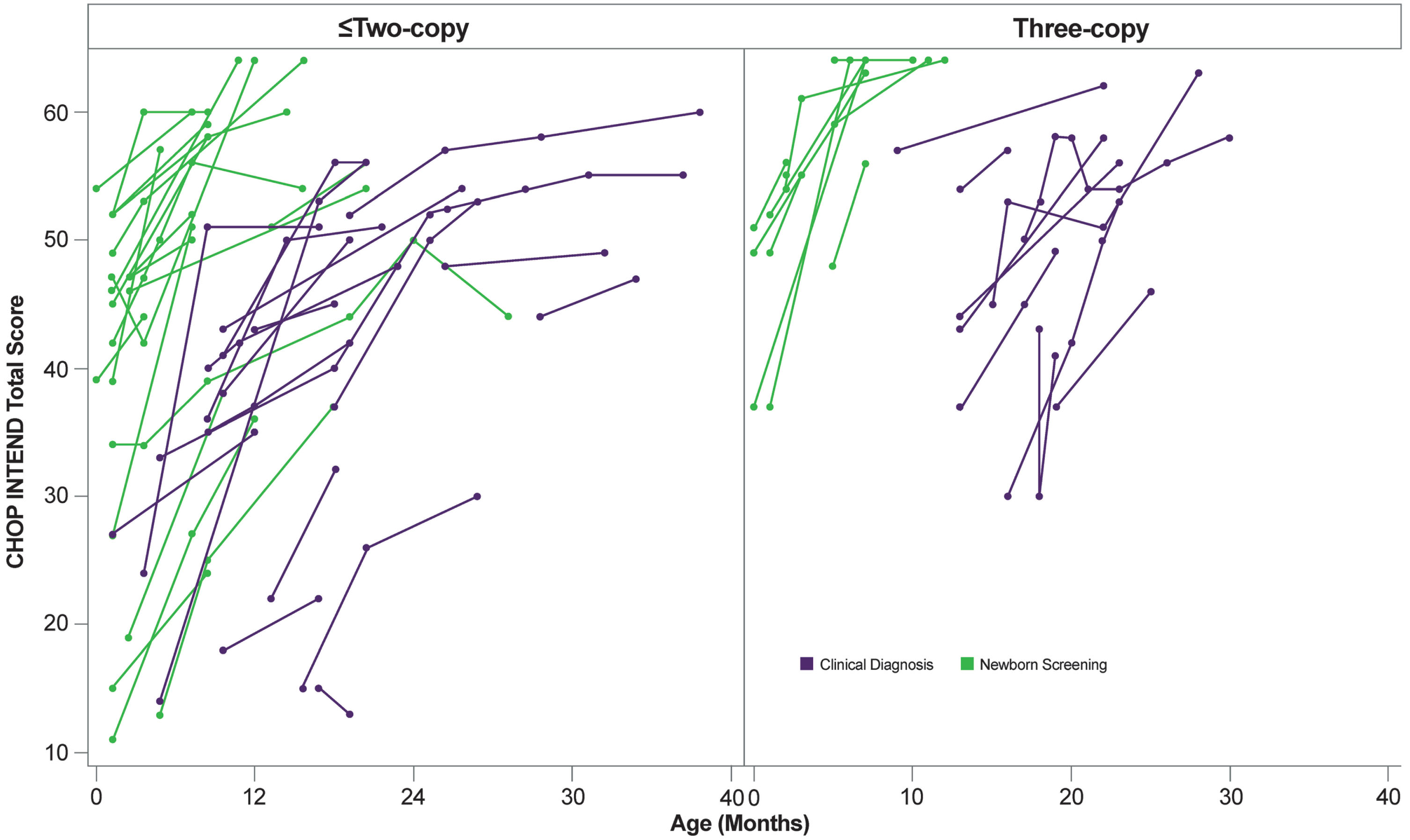
Of infants identified by NBS, 85.0% (n = 17/20) either achieved or maintained CHOP INTEND scores ≥40 points (Supplementary Figure 2). The three patients (15.0%) who had scores that remained < 40 points indicated symptoms at diagnosis but experienced an increase in CHOP INTEND scores (see Supplementary Figure 2). Of patients identified by clinical diagnosis, 90.5% (n = 19/21) either achieved or maintained CHOP INTEND scores≥40 points, with the two patients who remained with scores below 40 points also demonstrating increased scores (see Supplementary Figure 2).
Overall, there was a mean (SD) increase in the HFMSE score of 10.5 (8.73) points for patients (n = 20) treated with onasemnogene abeparvovec monotherapy (Supplementary Table 3). Mean (SD) age at time of assessment was 20.5 (10.3) (range 6.9–45.0) months. Mean (SD) change in score/month was 0.9 (0.69) points, with a mean (SD) time between assessments of 11.0 (5.57) months. The mean (SD) overall increase in HFMSE score for NBS infants (n = 6) was 11.3 (9.63) points and the mean (SD) age at time of assessment was 23.6 (16.5) months. The mean (SD) overall increase for clinically diagnosed patients (n = 14) was 10.1 (8.68) points.
Mean (SD) increase in the HINE-2 score for all patients treated with onasemnogene abeparvovec monotherapy (n = 22) was 7.3 (4.43) points (see Supplementary Table 3). Mean (SD) age at time of assessment was 6.4 (4.9) (range 0.2–16.6) months. Mean (SD) change in score/month was 0.9 (0.56) points, with a mean (SD) time between assessments of 8.4 (4.34) months. The mean (SD) overall increase in the HINE-2 score for NBS infants (n = 10) was 8.6 (3.95) points, and the mean (SD) age at time of assessment was 2.7 (2.7) months. The mean (SD) overall increase for clinically diagnosed patients (n = 12) was 6.2 (4.67) points.
Overall, 95.5% of patients (n = 21/22) had a > 1-point change in HINE-2 scores, and therefore achieved the MCID (HINE-2 ≥2-point change) (Supplementary Table 4). One hundred percent (n = 10/10) of NBS infants achieved MCID in HINE-2. For clinically diagnosed patients, 91.7% (n = 11/12) achieved MCID in HINE-2. Overall, 80.0% of patients (n = 16/20) achieved MCID in HFMSE (see Supplementary Table 4). For NBS infants, 83.3% (n = 5/6) achieved MCID in HFMSE. For clinically diagnosed patients, 78.6% (n = 11/14) achieved MCID in HFMSE.
Overall, 95.0% of patients improved or maintained HINE-2 and HFMSE scores (see Supplementary Table 4). All NBS infants (100%) improved or maintained HINE-2 and HFMSE scores. Of clinically diagnosed patients, 91.7% improved HINE-2 scores, and 92.9% improved or maintained HFMSE scores.
Motor milestones
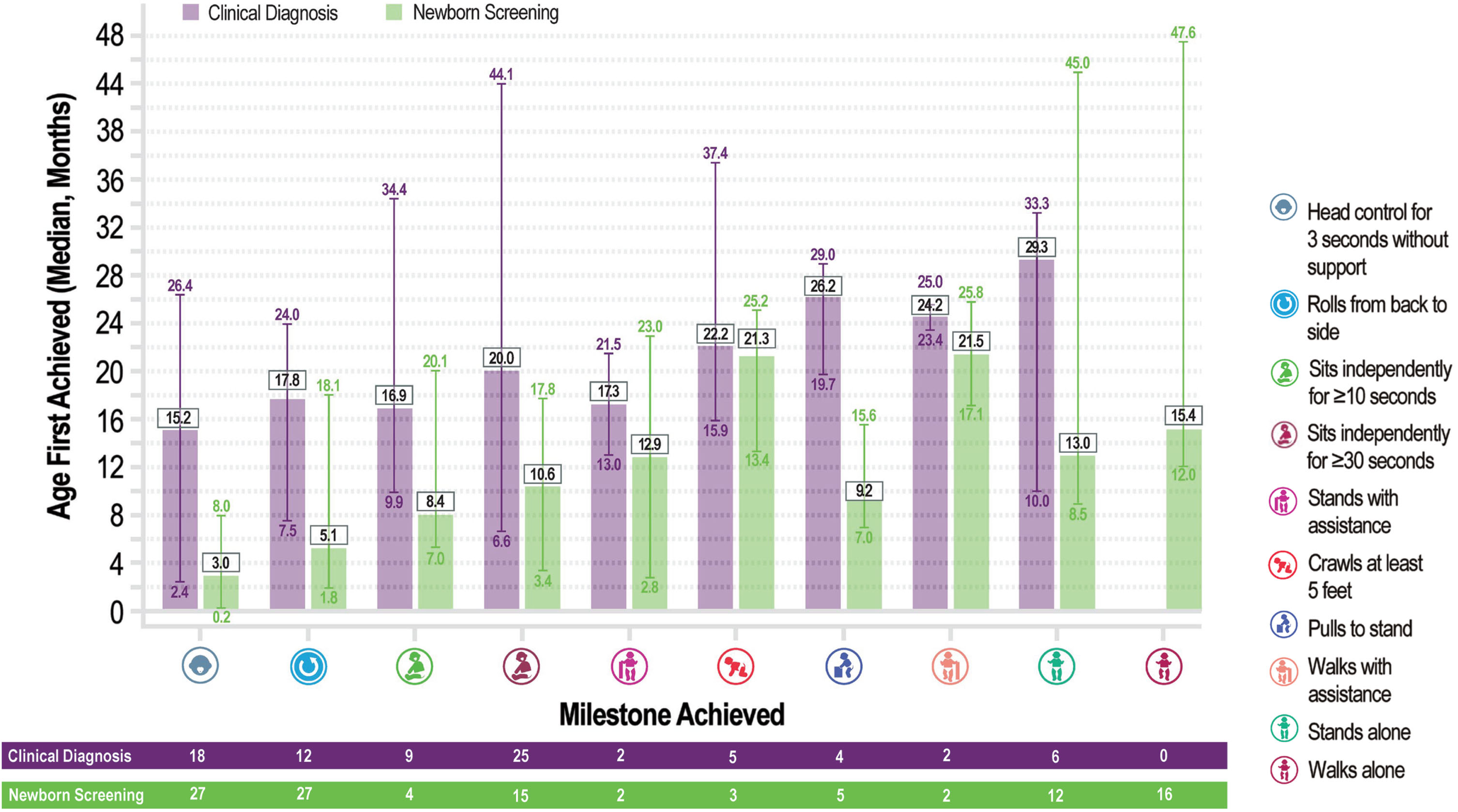
Median age of all first motor milestone achievements was lower for infants diagnosed through NBS (range, 3.03–21.46 months) than clinically diagnosed patients (range, 15.24–29.32 months) (Fig. 3). The median age that NBS infants first reported the milestone of independent sitting for ≥30 seconds was 10.63 months. The median age that clinically diagnosed patients first reported the milestone of independent sitting for ≥30 seconds was 20.03 months. No clinically diagnosed patients reached the milestone of walking independently (at a median age 18.0 months). A total of 16 of 32 evaluable NBS infants reached the milestone of walking independently (median age 15.4 months). Six of the 16 patients (37.5%) were > 18 months of age at the time when the milestone of independent walking was recorded. Although 16 NBS infants did not achieve standing by 11 months of age (median [SD] age 18 [5.77] months), four of these patients did achieve walking with assistance (at a median age of 16.1 months). Of the three two-copy patients with tracheostomy, two patients achieved new motor milestones, and one patient maintained achieved milestones during the observation period.
Fig. 3
Median age of milestone achievements: newborn screening versus clinical diagnosis.
Bulbar function
Twelve months post-onasemnogene abeparvovec infusion, 162/168 patients (96.6%) were fed exclusively by mouth. Most patients with recorded data on calorie intake during the last 4 weeks before the data cutoff (35/39, 89.7%) reported sufficient intake by oral feeding alone. Twenty-two patients reported dysphagia/swallowing and/or feeding difficulty. Eight patients experienced pneumonia/aspiration pneumonia.
Safety
After enrollment, AE data were available for 99.4% (n = 167/168) of patients with SMA treated with onasemnogene abeparvovec monotherapy (Table 3). Of these patients, 48.5% (n = 81/167) experienced at least one treatment-emergent AE, 18.6% (n = 31/167) experienced an SAE, and 4.8% (n = 8/167) experienced AEs that were determined by the investigator to be serious and treatment-related.
Table 3
Treatment-emergent adverse events
| Patients (n = 167) | |
| Any grade TEAE, n (%) | 81 (48.5) |
| ≥ Grade 3 TEAE, n (%) | 40 (24.0) |
| Any serious AE, n (%) | 31 (18.6) |
| Related AE, n (%) | 54 (32.3) |
| Serious related AE, n (%) | 8 (4.8) |
| AESIs | |
| Hepatotoxicity, n (%) | 49 (29.3) |
| Transient thrombocytopenia, n (%) | 23 (13.8) |
| Cardiac AEs, n (%) | 22 (13.2) |
| Thrombotic microangiopathy, n (%) | 1 (0.6) |
AE, adverse event; AESI, adverse event of special interest; TEAE, treatment-emergent adverse event.
AEs often occurred during the short-term (commonly within three months) post-infusion period. No malignancies were reported. These data are consistent with the overall safety profile for onasemnogene abeparvovec.
AESIs included hepatotoxicity, transient thrombocytopenia, cardiac AEs, and TMA (see Table 3). Hepatotoxicity events were reported in 29.3% (n = 49/167) of patients. Hepatotoxicity pertained to elevated aminotransferases. No cases of acute liver failure or acute serious liver injury were reported or identified in this patient cohort. Transient thrombocytopenia was reported for 13.8% (n = 23/167) of patients. Thrombocytopenic events comprised isolated decreases in platelet counts without clinical significance or sequelae. Cardiac AEs (13.2%, n = 22/167) were mainly elevated troponin without clinical significance or sequelae. No events suggestive of dorsal root ganglion toxicity were reported.
TMA was reported in one case (0.6%). The patient was a 15-month-old female, and SMA was suspected at the age of 13 months. Increased leukocytes were observed within 2 weeks of onasemnogene abeparvovec infusion, and after treatment, leukocytes continued to rise. One week after receiving onasemnogene abeparvovec, TMA was diagnosed based on the clinical evidence of thrombocytopenia, hemolytic anemia, and acute kidney injury. Aspiration pneumonia was also suspected at the time of the TMA diagnosis. After 1 month of treatments including plasmapheresis, peritoneal dialysis, hemofiltration, and blood pressure control with a calcium channel blocker, the patient completely recovered from TMA.
Deaths were reported for two patients. Neither case was assessed to be related to onasemnogene abeparvovec monotherapy. One was found prone and dead on an adult bed after being placed supine with his head and torso elevated by pillows, suggesting accidental obstruction of the airway. The other patient experienced aspiration pneumonia, followed by respiratory insufficiency, followed by death.
At least one treatment emergent AE was experienced by 36.1% (n = 35/97) of infants identified by NBS, and 9.3% (n = 9/97) experienced any SAE (Supplementary Table 5). For patients who were clinically diagnosed, 65.7% (n = 46/70) experienced at least one treatment-emergent AE, and 31.4% (n = 22/70) experienced any SAE (see Supplementary Table 5). Hepatotoxicity events were reported in 19.6% (n = 19/97) of NBS patients; transient thrombocytopenia was reported for 5.2% (n = 5/97); and cardiac AEs were reported for 8.2% (n = 8/97) (see Supplementary Table 5). Hepatotoxicity events were reported in 42.9% (n = 30/70) of clinically diagnosed patients; transient thrombocytopenia was reported for 25.7% (n = 18/70); and cardiac AEs were reported for 20.0% (n = 14/70) (see Supplementary Table 5).
Any treatment-emergent AE was observed in 49.2% (n = 59/120) of patients weighing < 8.5 kg, and 21.7% (n = 26/120) experienced any SAE (Supplementary Table 6). For heavier patients weighing ≥8.5 kg, 61.9% (n = 13/21) experienced any treatment-emergent AE, and 4.8% (n = 1/21) had any SAE (see Supplementary Table 6). Similar incidences were observed for ≥Grade 3 treatment-emergent AEs and transient thrombocytopenia for patients weighing < 8.5 kg and ≥8.5 kg (see Supplementary Table 6). No heavier patients weighing ≥13.5 kg experienced an AE, and 5.8% (n = 7/120) of patients weighing < 8.5 kg experienced AEs that were determined by the investigator to be serious and treatment-related (see Supplementary Table 6).
Any treatment-emergent AE was observed in 80% (n = 4/5) of older patients ≥24 months of age, and 20% (n = 1/5) experienced a treatment-related SAE (Supplementary Table 7). For patients ≥12 months and < 24 months of age, 69.0% (n = 20/29) reported a treatment-emergent AE, and 10.3% (n = 3/29) had a treatment-related SAE (see Supplementary Table 7). Any treatment-emergent AE was experienced by 57.7% (n = 15/26) of patients ≥6 months and < 12 months of age, and 3.8% (n = 1/26) had a treatment-related SAE (see Supplementary Table 7). For patients < 6 months of age, any treatment-emergent AE was observed in 39.3% (n = 42/107) of patients, and 2.8% (n = 3/107) had a treatment-related SAE (see Supplementary Table 7). A greater percentage of older patients ≥24 months of age experienced AESIs compared with younger patients (see Supplementary Table 7).
DISCUSSION
These findings from the RESTORE registry provide extended assessments (up to 37 months of post-treatment follow-up) of real-world outcomes from routine clinical practice for patients with SMA who received onasemnogene abeparvovec monotherapy. Patients receiving onasemnogene abeparvovec demonstrated improved survival and achieved motor milestones, and AE and SAE profiles were consistent with those described during clinical trials of onasemnogene abeparvovec [27, 29–38]. These findings further validate substantial improvement and outcome gains versus the natural history of SMA [9, 15–17, 27, 29, 31–34, 38]. In the real world, SMA presentation is more heterogeneous than for patients included in clinical trials [59], and the current enrollment in the RESTORE registry for patients treated with onasemnogene abeparvovec monotherapy reflects a range of SMA phenotypes and SMN2 genotypes. Real-world data, including experience from RESTORE, has been crucial for filling knowledge gaps and demonstrating through clinical experience that onasemnogene abeparvovec is associated with improvements in motor function, bulbar function, and pulmonary function in a varied SMA patient population and over an extended period of observation [37, 40–43, 59–61]. Ongoing patient follow-up in RESTORE will further extend long-term data on the effectiveness and safety of gene replacement therapy for SMA.
Given the pathophysiology of SMA, characterized by rapid death of motor neurons, early intervention is necessary to optimize outcomes [32, 33, 62–64]. RESTORE provides information on early intervention for the broad population of patients with SMA, including those identified by NBS, patients with two or three copies of SMN2, and patients who were asymptomatic or symptomatic at diagnosis. Our observations underscore that initial diagnosis by NBS does not directly equate with presymptomatic diagnosis or treatment, which may explain low initial CHOP INTEND scores observed for some NBS patients in RESTORE, as well as real-world results that only partially reproduced the outcomes observed in the presymptomatic patients in SPR1NT. The SPR1NT trial of onasemnogene abeparvovec demonstrated that neonates treated presymptomatically achieved greater and earlier developmental milestones than both untreated patients and patients treated after symptom onset [32, 33]. All patients in SPR1NT achieved their primary efficacy endpoint (independent sitting ≥30 seconds for two-copy patients at a median age of 265 [range, 172–354] days; independent standing ≥3 seconds for three-copy patients at a median age of 377 [range, 284–549] days) [32, 33]. Moreover, nine of 14 two-copy patients (64.3%) walked independently ≥5 steps at a median age of 526 (range, 367–564) days [32]. All but one of the three-copy patients (14/15; 93.3%) achieved independent walking at a median age of 422 (range, 362–563) days [33]. To better clarify the “true” presymptomatic state for patients with SMA, a new classification has been proposed that includes clinically silent disease (no symptoms with normal motor examination), prodromal disease (subtle symptoms and/or findings consistent with SMA but not definitive), and symptomatic SMA (definite clinical findings typical of SMA) [65]. Indeed, infants identified by NBS who present with CHOP INTEND scores < 40 may not be truly presymptomatic and may be considered as exhibiting very early features of SMA, warranting prompt treatment [66]. An important finding of the current study is that patients identified by NBS with CHOP INTEND scores < 40 demonstrated substantial improvements after treatment with onasemnogene abeparvovec monotherapy, as exemplified by CHOP INTEND increases and motor milestone acquisition. Additional follow-up is needed to evaluate the long-term evolution of these patients.
Our data confirm other real-world and clinical study data that infants identified by NBS are diagnosed and assessed earlier, have greater initial CHOP INTEND scores, and overall greater improvements [32, 33, 63, 67, 68]. Mean overall increase in HFMSE and HINE-2 scores were greater for NBS infants compared with clinically diagnosed patients, and a greater percentage of NBS infants achieved MCID in HINE-2 and HFMSE. In addition, all NBS infants improved or maintained HINE-2 and HFMSE scores, which was not the case for the clinically diagnosed patients. Median age of all first milestone achievements was lower for infants diagnosed though NBS compared with clinically diagnosed patients. These results support the benefits of early identification and treatment of patients with SMA, and, as more data become available, the RESTORE registry could allow for further evaluation of NBS and early intervention.
Evaluating the safety and effectiveness of treatment with onasemnogene abeparvovec in older patients ≥6 months of age and heavier patients weighing ≥8.5 kg remains critical. These real-world data from RESTORE indicate clear clinical benefit for older, heavier patients. Since onasemnogene abeparvovec dosing is weight-based and immune-mediated reactions may be in direct relation to vector load [61, 69], further investigation of onasemnogene abeparvovec is warranted and ongoing in clinical trials for SMA patients ≤24 months of age and weighing ≤17 kg (OFELIA [NCT05073133]) [70] and for patients weighing ≥8.5 kg to ≤21 kg (SMART [NCT04851873]) [69]. RESTORE could allow for further evaluation of SMA treatment for older and heavier patients as additional data become available [71, 72].
The observed AEs in RESTORE were consistent with the established safety profile of onasemnogene abeparvovec in clinical trials [27, 31–34] and in limited case reports (e.g., TMA has been reported only outside published clinical trials) [37]. These data are also supported by other studies in the real-world setting [37, 40–43, 60]. A key finding from this study was the improved safety profile that was demonstrated for infants identified by NBS. However, current data are insufficient to draw a firm conclusion regarding any potential differences in safety profile based on patient weight or age for the following reasons: (1) greater or similar percentage of serious AEs, ≥ Grade 3 AEs, and thrombocytopenia were observed in patients weighing < 8.5 kg, (2) no serious and related AEs were reported in the heavier group (weighing ≥8.5 kg), and (3) body weight was not available for all patients, especially for older patients, therefore greater incidence of AEs in older patients may not be fully explained. In addition, the lower AE incidence observed for younger patients was likely driven by the infants diagnosed via NBS. Although the current safety findings and those reported for 102 patients weighing ≥8.5 kg administered onasemnogene abeparvovec from a global managed care access program were consistent with AEs described for patients weighing < 8.5 kg from clinical trials, overall safety data for heavier patients weighing > 8.5 kg are limited and analyses may be underpowered [73]. Factors driving AAV-related toxicity are not completely understood, and although ostensibly related AEs are detected in the real-world setting (with older and heavier patients associated with greater risk), age or weight may not be the only risk factors [74–78]. Additional studies and real-world data are needed to better understand why some patients experience these AEs and to assess the potential impact of patient weight and age.
There are methodologic difficulties with assessing motor milestone evolution for patients with SMA. Standardized assessment during real-world studies lacks the precision obtained in controlled study methodology. Therefore, evaluators did not have the same degree of training and familiarity with the different scales, and the assessments were conducted at different time points, which makes descriptive analysis more complex. Motor milestones could constitute a valuable alternative, but it should be noted that the order in which children acquire and/or exhibit motor milestones is variable (i.e., attainment of milestones is not an ordinal progression), and children with SMA may not acquire milestones in the anticipated pattern of a typically developing child [79]. The 10 milestones originally selected for RESTORE were initially ranked from motor milestones that were supposed to be achieved earliest to milestones that were supposed to be achieved later, but the motor milestone results clearly demonstrate that the order can be different than initially anticipated, as displayed in Fig. 3. In this context, the original design of the case report form could lead an evaluator to interpret that a patient who acquired a new milestone outside of the anticipated order of milestone acquisition was actually losing a milestone, even though the patient had truly achieved a new milestone. The case report form has since been adapted to capture the dynamics of motor milestone acquisition and any potential milestone loss more precisely. Further analysis of RESTORE using the revised case report form will provide additional data regarding achievement of motor milestones.
Our study presents several limitations. Because of the descriptive nature of the study, statistical testing was not performed on the available real-world data for patients treated with onasemnogene abeparvovec monotherapy. Future studies may incorporate statistical modeling as data permit. The duration of follow-up is variable and necessarily limited. Because patients’ clinical care is not dictated by the RESTORE protocol, the nature of data generated across study sites and the completeness of these data at the time of analysis are variable. Several patients, likely identified through NBS, had greater baseline motor function, making it more difficult to observe improvements for these patients when using the CHOP INTEND. Other motor function measures will be needed as these patients, and patients with SMA types 2 or 3, mature and grow. From a safety perspective, AEs occurring before enrollment may not be captured. In addition, the RESTORE registry did not capture specific SAEs for each patient. This potentially biases the safety signals, as it is likely that most treatment-related AEs occur soon after administration, and these AEs would not be recorded for patients with a substantial gap in time between treatment and enrollment.
Regional differences impact SMA treatment, including disparities between countries regarding access to care, NBS and diagnosis, specialty care, and ventilation support, as well as differences in regulatory approvals of DMTs [80]. The majority of patients in the current data cut were from the United States (n = 138; 82.1%) largely because RESTORE enrollment initiated in the United States before initiation in other countries. A greater representation of patients from countries outside of the United States is necessary to enhance these real-world data. As more countries approve onasemnogene abeparvovec for the treatment of patients with SMA, more information is needed regarding its use in clinical practice that is not addressed in the prescribing information, clinical trial experience, or real-world assessments [81–83].
Although RESTORE enrolls patients with SMA regardless of treatment(s) received, those treated with onasemnogene abeparvovec alone represent the largest percentage of patients in the registry at the time of this analysis. These findings add further support for gene therapy as a treatment modality that can deliver durable transformative effects for patient populations with profound unmet needs [84, 85]. Although some patients in RESTORE received onasemnogene abeparvovec in sequence or combination with other DMTs, these patients were not included in the current report. This may have induced a positive bias by including only patients who did not receive treatment with another DMT in a potential effort to maximize benefit or because of a perceived regression. The definitions for sequential/combination treatment with onasemnogene abeparvovec have been addressed in a separate analysis [55].
CONCLUSIONS
The RESTORE registry represents a successful collaboration between industry and academia and is a robust real-world instrument providing a wealth of long-term data on patients receiving DMTs for SMA. RESTORE continues to enroll new patients and engage new study sites around the world. The findings in this report reflect a range of SMA patient types treated with onasemnogene abeparvovec monotherapy. Generally, patients with SMA who received onasemnogene abeparvovec derived benefit from treatment, with most evaluable patients achieving improvements in CHOP INTEND scores during the follow-up period. The observed AEs in RESTORE were consistent with the safety profile of onasemnogene abeparvovec in clinical trials. An improved safety profile was observed for infants diagnosed via NBS compared with those who were clinically diagnosed. Treatment with onasemnogene abeparvovec monotherapy was transformative for this vulnerable patient population. Further analyses of the RESTORE real-world data will be conducted for various patient populations to increase our understanding of SMA in the era of DMTs.
TRANSPARENCY
Acknowledgments
Writing and editing assistance, including preparation of a draft manuscript under the direction and guidance of the authors, incorporating author feedback, and manuscript submission, was provided by Wynne Dillon, MS (Kay Square Scientific, Newtown Square, PA, USA) and David Wolff, MS (Novartis Gene Therapies, Inc., Bannockburn, IL, USA). This support was funded by Novartis Gene Therapies, Inc. Nicole Gusset of SMA Schweiz, Heimberg, Switzerland, and SMA Europe, Frieberg, Germany, was a member of the RESTORE steering committee and was part of the discussions that contributed to this analysis. The authors also wish to thank the investigators and site coordinators and, most importantly, all the patients, families, and caregivers for their willingness to participate in RESTORE, which is sponsored by Novartis Gene Therapies, Inc.
Declaration of funding
All financial and material support for this research was provided by Novartis Gene Therapies, Inc.
Declaration of financial/other relationships
LS has received personal compensation from Novartis Gene Therapies, Inc., Biogen, Inc., Biophytis, Cytokinetics, Dynacure, Roche, Santhera, and Sarepta Therapeutics, and research funding from Novartis Gene Therapies, Inc., Biogen, Dynacure, and Roche. JWD has received consulting fees from Novartis Gene Therapies, Inc., Biogen, Cytokinetics, Ionis Pharmaceuticals, Pfizer, Roche, and Sarepta Therapeutics; license fees or royalty payments from Athena Diagnostics; and research funding from Novartis Gene Therapies, Inc., Biogen, Cytokinetics, Roche, Sanofi-Genzyme, and Sarepta Therapeutics. He is an Editorial Board Member of this journal, but was not involved in the peer-review process nor had access to any information regarding its peer-review. DCD has received personal compensation for consulting for advisory board participation from Novartis Gene Therapies, Inc., Biogen, Cytokinetics, Ionis Pharmaceuticals, Inc., Mallinckrodt, Metafora, PTC Therapeutics, Roche, Sanofi, Sarepta Therapeutics, and Ultragenyx, and the n-lorem Foundation and SMA Foundation. He has no financial interests in these companies. He has received compensation as a consultant from Novartis Gene Therapies, Inc., Biogen, and Sarepta Therapeutics. He has received research grants from the Hope for Children Research Foundation, National Institutes of Health, SMA Foundation, Cure SMA, GliaPharm, Rocket Pharma, Glut1 Deficiency Foundation, and the US Department of Defense; he also has received clinical trial funding from Biogen, Mallinckrodt, PTC Therapeutics, Sarepta Therapeutics, Scholar Rock, and Ultragenyx; and serves as a member of the DSMB for Aspa Therapeutics. JK has received honoraria for clinical research and/or consultancy activities from Biogen, Novartis Gene Therapies, Inc., Roche, and Scholar Rock; and personal fees from PTC Therapeutics, Pfizer, Roche, and Sarepta Therapeutics. He is an Editorial Board Member of this journal, but was not involved in the peer-review process nor had access to any information regarding its peer-review. EM has received personal compensation for clinical trial consulting, serving on scientific advisory boards, and research funding from Novartis Gene Therapies, Inc. FM has received honoraria for scientific advisory board participation from Novartis Gene Therapies, Inc., Biogen, Novartis, PTC, Roche, and Sarepta, and grants and personal fees from Novartis Gene Therapies, Inc., Biogen, and Roche. CMP is a site principal investigator for Astellas, Biogen, Catabasis, CSL Behring, Novartis Gene Therapies, Inc., Pfizer, PTC, Sarepta, and Scholar Rock clinical trials, and has received honoraria for advisory board participation from Biogen, Novartis Gene Therapies, Inc., Novartis, Roche, and Sarepta; and speaker’s fees from Biogen and Novartis Gene Therapies, Inc. PBS has received personal compensation from Novartis Gene Therapies, Inc., Alexion, argenx, Biogen, Catalyst, Grifols, CSL Behring, Genentech, Pfizer, PTC Therapeutics, and Sarepta Therapeutics; research funding from Astellas Gene Therapies, Biogen, Catalyst, Pfizer, PTC, Sanofi-Genzyme, Sarepta, Solid Biosciences, and Zogenix. EFT has received personal compensation for consultancy from Novartis Gene Therapies, Inc., Biogen, Biologiz, Cytokinetics, Novartis, and Roche, and research funding from Biogen/Ionis and Roche. SQ-R is a site principal investigator for clinical trials of Biogen and Novartis Gene Therapies, Inc.; has served as a consultant and has participated on advisory boards for Novartis Gene Therapies, Inc., Biogen, and Roche; and has received travel and speaker honoraria from Biogen, Novartis, and Roche. ID has received personal compensation for lectures and/or scientific board meetings from Novartis Gene Therapies, Inc., Biogen, BioMarin, PTC Therapeutics, and Sarepta. KS is a site principal investigator for Biogen, Novartis Gene Therapies, Inc., and Chugai/Roche clinical trials; has served on advisory boards for Novartis Gene Therapies, Inc./Novartis, Biogen, and Chugai/Roche; and has received speaker’s fees from Biogen, Novartis and Chugai/Roche. EF, KB, DR, and RS are employees of Novartis Gene Therapies, Inc., and own Novartis stock or other equities. NL was an employee of Novartis Gene Therapies, Inc., at the time of the study. FAA has received personal compensation from United Biosciences Corporation and research funding from Portola. RSF has received personal compensation for consulting and for advisory board participation from Novartis Gene Therapies, Inc., Biogen, Novartis, Roche, and Scholar Rock; editorial fees from Elsevier for co editing a neurology textbook; license fees from the Children’s Hospital of Philadelphia; and research funding from Novartis Gene Therapies, Inc., Biogen, Roche/Genentech, and Scholar Rock. He is an Editorial Board Member of this journal, but was not involved in the peer-review process nor had access to any information regarding its peer-review.
Author contributions
As stipulated in the RESTORE bylaws, all publication topics were authored and approved by the members of the steering committee, and analyses were performed by statisticians employed by the CRO that manages the study data. All authors had access to and analyzed and interpreted the data, participated in the development and critical review of the manuscript, approved the final version of the manuscript submission for publication, and are accountable for the accuracy and integrity of the work.
Data-sharing statement
The data sets generated and analyzed during the study are available from the RESTORE registry. These data sets are not publicly available, but are available from the corresponding author/RESTORE Steering Committee on reasonable request.
Supplementary material
The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-230122.
REFERENCES
[1] | Sugarman EA , Nagan N , Zhu H , Akmaev VR , Zhou Z , Rohlfs EM , et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet. (2012) ;20: (1):27–32. |
[2] | Kolb SJ , Kissel JT . Spinal muscular atrophy. Neurol Clin. (2015) ;33: (4):831–46. |
[3] | Sheng-Yuan Z , Xiong F , Chen YJ , et al. Molecular characterization of SMN copy number derived from carrier screening and from core families with SMA in a Chinese population. Eur J Hum Genet. (2010) ;18: (9):978–84. |
[4] | Messina S , Sframeli M . New treatments in spinal muscular atrophy: positive results and new challenges. J Clin Med. (2020) ;9: (7):2222. |
[5] | Lefebvre S , Bürglen L , Reboullet S , Clermont O , Burlet P , Viollet L , et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. (1995) ;80: (1):155–65. |
[6] | Awano T , Kim JK , Monani UR . Spinal muscular atrophy: journeying from bench to bedside. Neurotherapeutics. (2014) ;11: (4):786–95. |
[7] | Lorson CL , Hahnen E , Androphy EJ , Wirth B . A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. (1999) ;96: (11):6307–11. |
[8] | Arnold WD , Kassar D , Kissel JT . Spinal muscular atrophy: diagnosis and management in a new therapeutic era. Muscle Nerve. (2015) ;51: (2):157–67. |
[9] | Kolb SJ , Coffey CS , Yankey JW , Krosschell K , Arnold WD , Rutkove SB , et al. Natural history of infantile-onset spinal muscular atrophy. Ann Neurol. (2017) ;82: (6):883–91. |
[10] | Mailman MD , Heinz JW , Papp AC , Snyder PJ , Sedra MS , Wirth B , et al. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med. (2002) ;4: :20–6. |
[11] | Calucho M , Bernal S , Alías L , March F , Venceslá A , Rodríguez-Álvarez FJ , et al. Correlation between SMA type and SMN2 copy number revisited: an analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul Disord.. (2018) ;28: (3):208–15. |
[12] | Finkel RS , Mercuri E , Meyer OH , Simonds AK , Schroth MK , Graham RJ , et al. Care group. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. (2018) ;28: (3):197–207. |
[13] | Mercuri E , Finkel RS , Muntoni F , Wirth B , Montes J , Main M , et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. (2018) ;28: (2):103–15. |
[14] | Mercuri E , Sumner CJ , Muntoni F , Darras BT , Finkel RS . Spinal muscular atrophy. Nat Rev Dis Primers. (2022) ;8: :52. https://doi.org/10.1038/s41572-022-00380-8. |
[15] | Ogino S , Wilson RB , Gold B . New insights on the evolution of the SMN1 and SMN2 region: simulation and meta-analysis for allele and haplotype frequency calculations. Eur J Hum Genet. (2004) ;12: (12):1015–23. |
[16] | Finkel RS , McDermott MP , Kaufmann P , Darras BT , Chung WK , Sproule DM , et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. (2014) ;83: :810–7. |
[17] | Farrar MA , Vucic S , Johnston HM , du Sart D , Kiernan MC . Pathophysiological insights derived by natural history and motor function of spinal muscular atrophy. J Pediatr. (2013) ;162: (1):155–9. |
[18] | Lomonte P , Baklouti F , Binda O . The biochemistry of survival motor neuron protein is paving the way to novel therapies for spinal muscle atrophy. Biochemistry. (2020) ;59: (14):1391–97. |
[19] | Lin Y , Lin CH , Yin X , Zhu L , Yang J , Shen Y , et al. Newborn screening for spinal muscular atrophy in China using DNA mass spectrometry. Front Genet. (2019) ;10: :1–8. |
[20] | Miller N , Shi H , Zelikovich AS , Ma YC . Motor neuron mitochondrial dysfunction in spinal muscular atrophy. Hum Mol Genet. (2016) ;25: (16):3395–3406. |
[21] | Wang CH , Finkel RS , Bertini ES , Schroth M , Simonds A , Wong B , et al. Participants of the International Conference on SMA Standard of Care. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. (2007) ;22: (8):1027–49. |
[22] | SPINRAZA® Prescribing Information. 2023 [updated Feb 2023; cited 2023 Apr 1]. Available from: https://www.spinraza.com/content/dam/commercial/specialty/spinraza/caregiver/enus/pdf/spinraza-prescribing-information.pdf. |
[23] | Hoy SM . Nusinersen: a review in 5q spinal muscular atrophy. CNS Drugs. (2021) ;35: (12):1317–28. |
[24] | EVRYSDI® Prescribing Information. 2023 [updated Feb 2023; cited 2023 Apr 1]. Available from: https://www.gene.com/download/pdf/evrysdiprescribing.pdf. |
[25] | Paik J . Risdiplam: a review in spinal muscular atrophy. CNS Drugs. (2022) ;36: :401–10. |
[26] | ZOLGENSMA® Prescribing Information. 2023 [updated Feb 2023; cited 2023 Apr 1] Available from: https://www.novartis.com/us-en/sites/novartisus/files/zolgensma.pdf. |
[27] | Mendell JR , Al-Zaidy S , Shell R , Arnold WD , Rodino-Klapac LR , Prior TW , et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. (2017) ;377: (18):1713–22. |
[28] | Darras BT , De Vivo DC . Precious SMA natural history data: a benchmark to measure future treatment successes. Neurology. (2018) ;91: (8):337–339. |
[29] | Al-Zaidy SA , Kolb SJ , Lowes L , Alfano LN , Shell R , Church KR , et al. AVXS-101 (onasemnogene abeparvovec) for SMA comparative study with a prospective natural history cohort. J Neuromuscul Dis. (2019) ;6: (3):307–17. |
[30] | Naveed A , Calderon H . Onasemnogene abeparvovec (AVXS-101) for the treatment of spinal muscular atrophy. J Pediatr Pharmacol Ther. (2021) ;26: (5):437–44. |
[31] | Day JD , Finkel RS , Chiriboga CA , Connolly AM , Crawford TO , Darras BT , et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. (2021) ;120: (4):284–93. |
[32] | Strauss KA , Farrar MA , Muntoni F , Saito K , Mendell JR , Servais L , et al. Onasemnogene abeparvovec for presymptomatic infants with two copies of SMN2 at risk for spinal muscular atrophy type 1: the Phase III SPR1NT trial. Nat Med. (2022) ;28: :1381–9. |
[33] | Strauss KA , Farrar MA , Muntoni F , Saito K , Mendell JR , Servais L , et al. Onasemnogene abeparvovec for presymptomatic infants with three copies of SMN2 at risk for spinal muscular atrophy: the Phase III SPR1NT trial. Nat Med. (2022) ;28: :1390–7. |
[34] | Mercuri E , Muntoni F , Baranello G , Masson R , Boespflug-Tanguy O , Bruno C , et al. STR1VE-EU study group. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy type 1 (STR1VEEU): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. (2021) ;20: (10):832–41. |
[35] | Finkel RS , Day JW , Darras BT , Kuntz NL , Zaidman C , Connolly AM , et al. Intrathecal (IT) administration of AVXS-101 IT gene therapy for spinal muscular atrophy: Phase 1 Study (STRONG) interim data analysis. Neurology. (2020) ;94: (15s). |
[36] | Lowes L , Al-Zaidy S , Shell R , Arnold W , Rodino-Klapac L , Prior T , et al. P.374 - AVXS-101 phase 1 gene therapy clinical trial in SMA type 1: patients treated early with the proposed therapeutic dose were able to sit unassisted at a younger age. Neuromuscul Disord. (2017) ;27: :S208–9. |
[37] | Day JW , Mendell JR , Mercuri E , Finkel RS , Strauss KA , Kleyn A , et al. Clinical trial and postmarketing safety of onasemnogene abeparvovec therapy. Drug Saf. (2021) ;44: (10):1109–19. |
[38] | Mendell JR , Al-Zaidy SA , Lehman KJ , McColly M , Lowes LP , Alfano LN , et al. Five-year extension results of the phase 1 START trial of onasemnogene abeparvovec in spinal muscular atrophy. JAMA Neurol. (2021) ;78: :834–41. |
[39] | Darras BT , Mercuri E , Strauss KA , Day JW , Chien YH , Masson R , et al. Intravenous and intrathecal onasemnogene abeparvovec gene therapy in symptomatic and presymptomatic spinal muscular atrophy: long-term follow-up study. Abstract presented at: Muscular Dystrophy Association Clinical & Scientific Conference; Dallas, TX; March 19-22, 2023. |
[40] | D’Silva AM , Holland S , Kariyawasam D , Herbert K , Barclay P , Cairns A , et al. Onasemnogene abeparvovec in spinal muscular atrophy: an Australian experience of safety and efficacy. Ann Clin Transl Neurol.. (2022) ;9: (3):339–50. |
[41] | Erdos J , Wild C . Mid- and long-term (at least 12 months) follow-up of patients with spinal muscular atrophy (SMA) treated with nusinersen, onasemnogene abeparvovec, risdiplam or combination therapies: a systematic review of real-world study data. Eur J Paediatr Neurol. (2022) ;39: :1–10. |
[42] | Weiß C , Ziegler A , Becker LL , Johannsen J , Brennenstuhl H , Schreiber G , et al. Gene replacement therapy with onasemnogene abeparvovec in children with spinal muscular atrophy aged 24 months or younger and bodyweight up to 15kg: an observational cohort study. Lancet Child Adolesc Health. (2022) ;6: (1):17–27. |
[43] | Bitetti I , Lanzara V , Margiotta G , Varone A . Onasemnogene abeparvovec gene replacement therapy for the treatment of spinal muscular atrophy: a real-world observational study. Gene Ther. (2023) ;30: :592–7 doi: 10.1038/s41434-022-00341-6.. |
[44] | Hahn A , Günther R , Ludolph A , Schwartz O , Trollmann R , Weydt P , et al. Risdiplam Compassionate Use Program Group. Short-term safety results from compassionate use of risdiplam in patients with spinal muscular atrophy in Germany. Orphanet J Rare Dis. (2022) ;17: (1):276. |
[45] | Berglund A , Berkö S , Lampa E , Sejersen T . Survival in patients diagnosed with SMA at less than 24 months of age in a population-based setting before, during and after introduction of nusinersen therapy. Experience from Sweden. Eur J Paediatr Neurol. (2022) ;40: :57–60. |
[46] | Pane M , Coratti G , Pera MC , Sansone VA , Messina S , d’Amico A , et al. Nusinersen efficacy data for 24-month in type 2 and 3 spinal muscular atrophy. Ann Clin Transl Neurol. (2022) ;9: (3):404–9. |
[47] | Finkel RS , Day JW , De Vivo DC , Kirschner J , Mercuri E , Muntoni F , et al. RESTORE: a prospective multinational registry of patients with genetically confirmed spinal muscular atrophy - rationale and study design. J Neuromuscul Dis. (2020) ;7: :145–52. |
[48] | Harada Y , Rao VK , Arya K , Kuntz NL , DiDonato CJ , Napchan-Pomerantz G , et al. Combination molecular therapies for type 1 spinal muscular atrophy. Muscle Nerve. (2020) ;62: (4):550–4. |
[49] | Matesanz SE , Curry C , Gross B , Rubin AI , Linn R , Yum SW , et al. Clinical course in a patient with spinal muscular atrophy type 0 treated with nusinersen and onasemnogene abeparvovec. J Child Neurol. (2020) ;35: (11):717–23. |
[50] | Mercuri E , Finkel R , Scoto M , Hall S , Eaton S , Rashid A , et al. Development of an academic disease registry for spinal muscular atrophy. Neuromuscul Disord. (2019) ;29: (10):794–99. |
[51] | Lemoine M , Gomez M , Grimaldi L , Andoni Urtizberea J Quijano-Roy. Le registre national SMA France: des résultats déjà encourageants [The SMA France national registry: already encouraging results].MedSci (Paris). 2021; 37 Hors série n°. 1:25-29. doi: 10.1051/medsci/2021187. |
[52] | Cure SMA Cure SMA Care Center Network. 2023. [updated 2023 Mar 31; cited 2023 Apr 1]. Available from: https://www.curesma.org/sma-care-center-network/. |
[53] | Pechmann A , Konig K , Bernert G , Schachtrup K , Schara U , Schorling D , et al. SMArtCARE: a platform to collect real life outcome data of patients with spinal muscular atrophy. Orphanet J Rare Dis. (2019) ;14: (1):18. |
[54] | Segovia-Simon S , Nungo C , Vazquez Costa J , Pitarch I , Caballero J , et al. P.42 CuidAME: Registry for longitudinal data collection of Spanish SMA patients.. Neuromuscul Disord. (2022) ;32: (1):60. |
[55] | Proud CM , Mercuri E , Finkel RS , Kirschner J , De Vivo DC , Muntoni F , et al. Combination disease-modifying treatment in spinal muscular atrophy: a proposed classification. Ann Clin Transl Neurol. (2023) ;10: (11):2155–60. doi: 10.1002/acn3.51889. |
[56] | de Onis M , Garza C , Victora CG , Onyango AW , Frongillo EA , Martines J . The WHO Multicentre Growth Reference Study: planning, study design, and methodology. Food Nutr Bull. (2004) ;25: :S15–26. |
[57] | WHO Multicentre Growth Reference Study Group WHO Motor Development Study: windows of achievement for six gross motor development milestones. Acta Paediatr Suppl. (2006) ;450: :86–95. |
[58] | Albers CA , Grieve AJ . Test Reviews. Bayley, N. Bayley Scales of Infant and Toddler Development - Third Edition. San Antonio, TX: Harcourt Assessment. J Psychoeduc Assess. (2006) ;25: (2):180–90. doi: 10.1177/0734282906297199 |
[59] | Swoboda KJ , Kissel JT , Crawford TO , Bromberg MB , Acsadi G , D’Anjou G , et al. Perspectives on clinical trials in spinal muscular atrophy. J Child Neurol. (2007) ;22: (8):957–66. |
[60] | Friese J , Geitmann S , Holzwarth D , Müller N , Sassen R , Baur U , et al. Safety monitoring of gene therapy for spinal muscular atrophy with onasemnogene abeparvovec-a single centre experience. J Neuromuscul Dis.. (2021) ;8: (2):209–16. |
[61] | Kirschner J , Butoianu N , Goemans N , Haberlova J , Kostera-Pruszczyk A , Mercuri E , et al. European ad-hoc consensus statement on gene replacement therapy for spinal muscular atrophy. Eur J Paediatr Neurol. (2020) ;28: :38–43. |
[62] | Govoni A , Gagliardi D , Comi GP , Corti S . Time is motor neuron: therapeutic window and its correlation with pathogenetic mechanisms in spinal muscular atrophy. Mol Neurobiol. (2018) ;55: :6307–18. |
[63] | Ramdas S , Servais L . New treatments in spinal muscular atrophy: an overview of currently available data. Expert Opin Pharmacother. (2020) ;21: (3):307–15. |
[64] | Dangouloff T , Hiligsmann M , Deconinck N , D’Amico A , Seferian AM , Boemer F , et al. Financial cost and quality of life of patients with spinal muscular atrophy identified by symptoms or newborn screening. Dev Med Child Neurol. (2023) ;65: (1):67–77. |
[65] | Finkel RS , Benatar M . Pre-symptomatic spinal muscular atrophy: a proposed nosology. Brain. (2022) . 29;145: (7):2247–49. |
[66] | Tizzano EF , Zafeiriou D . Prenatal aspects in spinal muscular atrophy: From early detection to early presymptomatic intervention. Eur J Paediatr Neurol. (2018) ;22: (6):944–50. |
[67] | Pane M , Berti B , Capasso A , Coratti G , Varone A , D’Amico A , et al. ;ITASMAc grouOnasemnogene abeparvovec in spinal muscular atrophy:predictors of efficacy and safety in naïve patients with spinalmuscular atrophy and following switch from other therapies. EClinicalMedicine. (2023) ;59: :101997. |
[68] | Kariyawasam DS , D’Silva AM , Sampaio H , Briggs N , Herbert K , Wiley V , et al. Newborn screening for spinal muscular atrophy in Australia: a non-randomised cohort study. Lancet Child Adolesc Health. (2023) ;7: (3):159–70. |
[69] | ClinicalTrials.gov. Safety and Efficacy of Intravenous OAV101 (AVXS-101) in Pediatric Patients with Spinal Muscular Atrophy (SMA) (SMART). 2021 [updated 2023 Aug 18; cited 2023 Nov 21]. Available from: https://clinicaltrials.gov/ct2/show/NCT04851873. |
[70] | ClinicalTrials.gov. Safety and Efficacy of Intravenous OAV101 (AVXS-101) in Pediatric Patients With Spinal Muscular Atrophy (SMA) (OFELIA). 2021 [updated 2023 Nov 14; cited 2023 Nov 21]. Available from: https://clinicaltrials.gov/ct2/show/NCT05073133. |
[71] | Servais L , De Vivo DC , Kirschner J , Mercuri E , Muntoni F , Tizzano EF , et al. P.79 Effectiveness and safety of onasemnogene abeparvovec in older patients with spinal muscular atrophy: Real-world outcomes from the RESTORE registry. Poster presented at: Muscular Dystrophy Association Clinical and Scientific Congress. 2022. [updated 2022; cited 2023 Apr 1]. Available from: https://www.mdaconference.org/abstractlibrary/effectiveness-and-safety-of-onasemnogeneabeparvovec-in-older-patients-with-spinal-muscularatrophy-real-world-outcomes-from-the-restore-registry/. |
[72] | Servais L , Benguerba K , De Vivo D , Kirschner J , Mercuri E , Muntoni F , et al. P.103 Onasemnogene abeparvovec (OA) treatment outcomes by patient weight at infusion: Initial findings from the RESTORE registry. Neuromuscul Disord. (2022) ;32: (1):S87. |
[73] | Chand DH , Mitchell S , Sun R , LaMarca N , Reyna SP , Sutter T . Safety of onasemnogene abeparvovec for patients with spinal muscular atrophy 8.5kg or heavier in a global managed access program. Pediatr Neurol. (2022) ;132: :27–32. |
[74] | Ertl HCJ . Immunogenicity and toxicity of AAV gene therapy. Front Immunol. (2022) ;13: :975803. |
[75] | Kishimoto TK , Samulski RJ . Addressing high dose AAV toxicity - ‘one and done’ or ‘slower and lower’? Expert Opin Biol Ther. (2022) ;22: (9):1067–71. |
[76] | Chand D , Mohr F , McMillan H , Tukov FF , Montgomery K , Kleyn A , et al. Hepatotoxicity following administration of onasemnogene abeparvovec (AVXS-101) for the treatment of spinal muscular atrophy. J Hepatol. (2021) ;74: (3):560–6. |
[77] | Wijngaarde CA , Blank AC , Stam M , Wadman RI , van den Berg LH , van der Pol WL . Cardiac pathology in spinal muscular atrophy: a systematic review. Orphanet J Rare Dis. (2017) ;12: (1):67. |
[78] | Chand DH , Zaidman C , Arya K , Millner R , Farrar MA , Mackie FE , et al. Thrombotic microangiopathy following onasemnogene abeparvovec for spinal muscular atrophy: a case series. J Pediatr. (2021) ;231: :265–68. |
[79] | De Sanctis R , Coratti G , Pasternak A , Montes J , Pane M , Mazzone ES , et al. Developmental milestones in type I spinal muscular atrophy. Neuromuscul Disord. (2016) ;26: (11):754–9. |
[80] | Dangouloff T , Vrsčaj E , Servais L , Osredkar D , SMA NBS World Study GrouNewborn screening programs for spinal muscular atrophy worldwide: Where we stand and where to go Neuromuscul Disord. (2021) ;31: (6):574–82. |
[81] | McMillan HJ , Proud CM , Farrar MA , Alexander IE , Muntoni F , Servais L . Onasemnogene abeparvovec for the treatment of spinal muscular atrophy. Expert Opin Biol Ther. (2022) ;22: (9):1075–90. |
[82] | Al-Zaidy SA , Mendell JR . From clinical trials to clinical practice:practical considerations for gene replacement therapy in SMA type 1. Pediatr Neurol. (2019) ;100: :3–11. |
[83] | Kichula EA , Proud CM , Farrar MA , Kwon JM , Saito K , Desguerre I , McMillan HJ . Expert recommendations and clinical considerations in the use of onasemnogene abeparvovec gene therapy for spinal muscular atrophy. Muscle Nerve. (2021) ;64: (4):413–27. |
[84] | Faulkner E , Spinner DS , Ringo M , Carroll M . Are global health systems ready for transformative therapies? Value Health. (2019) ;22: (6):627–41. |
[85] | Faulkner E , Ringo M , Spinner D , Carroll , Latif E , Miller TM Guiding principles of rare disease care and patient access. Global Genes and The Child Neurology Foundation. September 2020. [updated 2022; cited 2023 Apr 1]. Available at: https://globalgenes.org/white-paper/accessto-critical-therapies-guiding-principles-of-rare-diseasecare-and-patient-access/. |


