
Identifying Biomarkers of Spinal Muscular Atrophy for Further Development
Abstract
Background:
Spinal muscular atrophy (SMA) is caused by bi-allelic, recessive mutations of the survival motor neuron 1 (SMN1) gene and reduced expression levels of the survival motor neuron (SMN) protein. Degeneration of alpha motor neurons in the spinal cord causes progressive skeletal muscle weakness. The wide range of disease severities, variable rates of decline, and heterogenous clinical responses to approved disease-modifying treatment remain poorly understood and limit the ability to optimize treatment for patients. Validation of a reliable biomarker(s) with the potential to support early diagnosis, inform disease prognosis and therapeutic suitability, and/or confirm response to treatment(s) represents a significant unmet need in SMA.
Objectives:
The SMA Multidisciplinary Biomarkers Working Group, comprising 11 experts in a variety of relevant fields, sought to determine the most promising candidate biomarker currently available, determine key knowledge gaps, and recommend next steps toward validating that biomarker for SMA.
Methods:
The Working Group engaged in a modified Delphi process to answer questions about candidate SMA biomarkers. Members participated in six rounds of reiterative surveys that were designed to build upon previous discussions.
Results:
The Working Group reached a consensus that neurofilament (NF) is the candidate biomarker best poised for further development. Several important knowledge gaps were identified, and the next steps toward filling these gaps were proposed.
Conclusions:
NF is a promising SMA biomarker with the potential for prognostic, predictive, and pharmacodynamic capabilities. The Working Group has identified needed information to continue efforts toward the validation of NF as a biomarker for SMA.
INTRODUCTION
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disorder that has historically been the most common monogenic cause of death in infancy [1], affecting 1 in 11,000 live births [2, 3]. SMA is most often caused by homozygous deletions of the survival motor neuron gene (SMN1), and less often, by missense, nonsense, or frameshift mutations of SMN1 [4–6]. Both the SMN1 and SMN2 genes generate the survival motor neuron (SMN) protein, but the transcripts that arise from SMN1 are full-length and generate a functional protein, whereas the vast majority of those transcribed from SMN2 are truncated and generate a less stable version of the SMN protein that cannot interact optimally with binding partners [7]. Although the SMN2 gene is therefore unable to fully compensate for the loss of a functional SMN1 gene, SMN2 copy number is inversely correlated with disease severity [8–16].
SMA is characterized by reduced expression levels of the SMN protein and subsequent degeneration of spinal cord and brainstem motor neurons [17–19]. The disease phenotype ranges widely in severity and rate of progression. If untreated, reduced SMN protein levels can cause a range of debilitating complications such as functional motor deficits, limb and truncal muscle weakness, difficulty breathing, and eventual respiratory failure [1, 20–28]. Historically, SMA patients have been segmented into Types 0–4 depending on severity and clinical disease onset (e.g., “Type 0” refers to fetal onset and severe muscle weakness, whereas “Type 4” describes the small proportion of patients who do not display symptoms until adulthood) [8, 11, 20, 21, 29]. However, disease severity is understood to comprise a wide gradient, and this nomenclature is becoming less relevant as newborn screening and pre-symptomatic treatments are more widely utilized [30–32]. Since 2016, three disease-modifying SMA treatments have been approved by the United States Food and Drug Administration (FDA). All three therapies act by increasing SMN protein levels [33–35]. Nusinersen (SPINRAZA®) [36–38] and risdiplam (Evrysdi®) [39, 40] each modulate SMN2 mRNA splicing through different mechanisms to increase the amount of full-length, functional protein generated from the SMN2 gene. Both are indicated for use in pediatric patients, including newborns, and adults [41, 42]. Onasemnogene abeparvovec-xioi (ZOLGENSMA®) is an adeno-associated virus 9 (AAV9)-mediated, SMN1 gene replacement therapy approved for children under two years old and administered intravenously [43, 44]. Responses to nusinersen [45–54], onasemnogene abeparvovec [55–62], and risdiplam [63–67] vary widely between individuals and are affected by a number of factors including, but not limited to SMN2 copy number, age, and disease severity at the time of treatment. Because of this variability, SMA biomarkers to guide treatment decisions (prognostic biomarkers) and predict therapeutic responses (predictive biomarkers) are urgently needed. In addition, several therapeutics that do not target the SMN protein but focus on preventing or reversing motor neuron loss, improving function at the neuromuscular junction, or enhancing muscle performance are in various stages of development and clinical trials [35, 68–71]. A combination of SMN-dependent and SMN-independent therapeutics may be the most effective way to treat SMA [35, 69, 72, 73]. Furthermore, because SMA is a progressive disease that changes over the life of a patient, the specific combination of drugs used may change with time [74]. As such, new pharmacodynamic SMA biomarkers would be extremely useful as outcome measures in clinical trials and in tracking responses to evolving therapeutic regimens over time.
A great deal of research into putative biomarkers for SMA has occurred (Table 1; Fig. 1) [75–78]. Although SMN mRNA and protein are natural candidate biomarkers for SMA and are readily measurable in blood, SMN blood levels may not correlate with levels in the CNS, disease severity, or motor function gains [75–79]. For these reasons, SMN quantification has not emerged as a leading SMA biomarker thus far. However, a recent study quantified full length SMN transcript levels within the extracellular vesicles (EVs) found in blood serum of individuals with Type 2 SMA [80]. This technique was developed on the premise that EVs play a critical role in cell-to-cell communication across tissue types and cross the blood brain barrier in both directions, thereby potentially reflecting motor neuron health status. Study results indicated that full length SMN transcript levels in EVs increase with nusinersen treatment. Notably, the investigators chose to compare SMN transcript levels against levels of neurofilament-heavy chain (NF-H) in blood serum and CSF, which decreased with nusinersen treatment.
Fig. 1
SMA Pathophysiology and Candidate Biomarkers. A number of candidate biomarkers have been proposed based on the pathophysiology of SMA. Genetic factors impacting the expression of SMN protein include SMN2 copy number and polymorphisms, the presence of modifier genes that can improve downstream neuronal and motor functions associated with SMA disease state, and gene transcription factors that affect the expression of SMN2 and other genes. Muscle presence and function can be measured through a variety of techniques including imaging, action potential and electrical response following motor nerve stimulation, and quantification of molecular factors that relate to muscle metabolism or damage. The presence of neurofilament in serum and CSF has been the subject of extensive research.
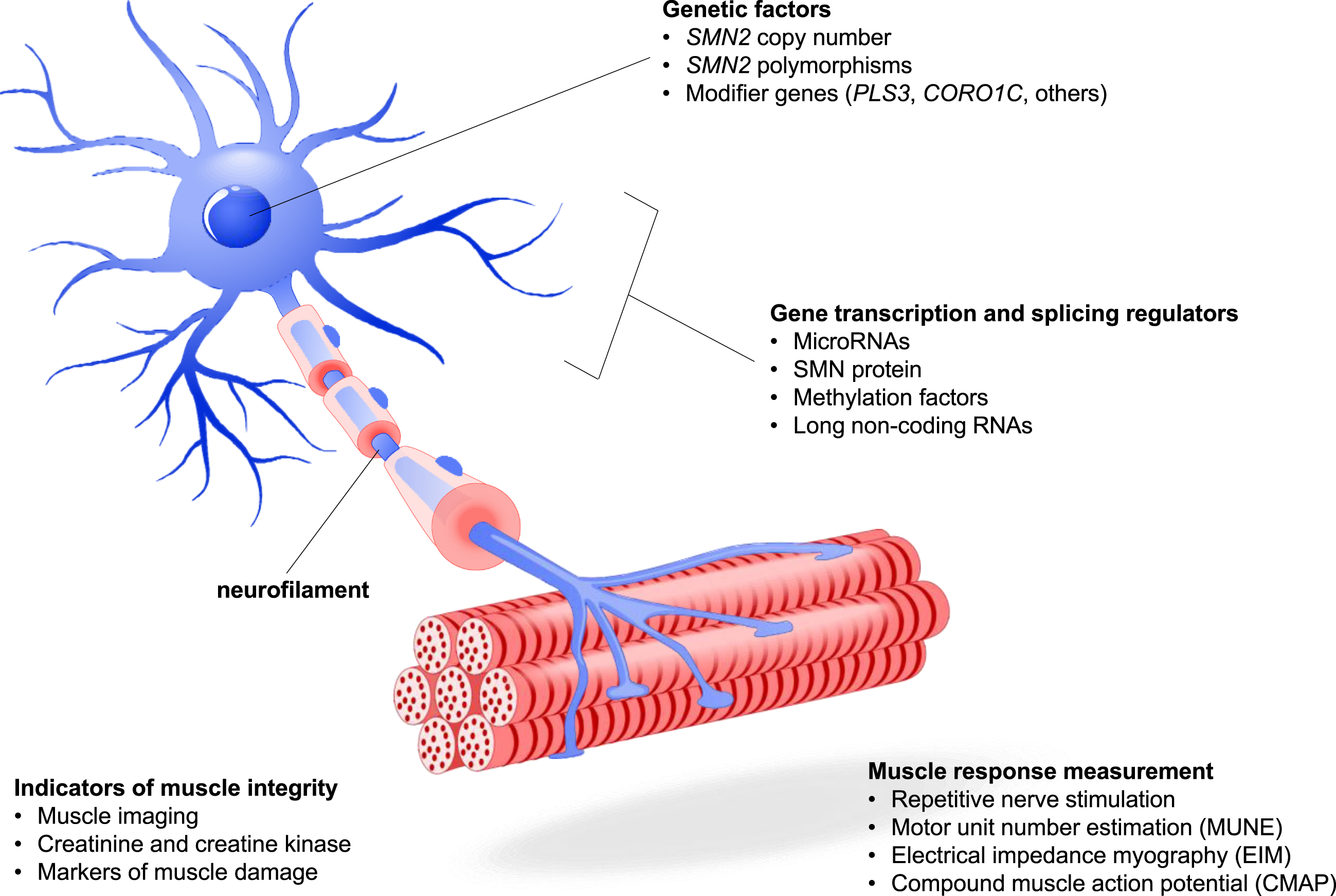
Table 1
Candidate biomarkers for SMA
| Biomarker | Notes | References |
| Biomolecular candidates | ||
| SMN protein | Lower levels of circulating SMN protein correlate with more severe/ advanced disease. | [18, 39, 48, 57, 81, 104–106] |
| Neurofilament | Higher levels of NF indicate greater axonal degradation and more severe/advanced disease. | [78, 107–118, 167] |
| Muscle indicators | Creatinine, creatine kinase, and other markers of muscle damage are elevated in more advanced SMA. | [122–124] |
| Genetic candidates | ||
| SMN2 gene copy number or polymorphisms | Fewer copies of SMN2 gene correlate with less SMN protein and more severe/advanced disease. | [10, 13–16, 81–83] |
| Modifier genes | The expression of some non-SMN genes may modify the SMA phenotype. | [73, 84–89] |
| Gene transcription and | ||
| splicing regulators | ||
| Micro RNAs | Preclinical and clinical research suggests different miRNAs may be expressed differentially according to SMA severity. | [90–94] |
| Methylation factors | Methylation of SMN2 impacts its expression and may be a way to measure SMN protein production. Genome-wide methylation patterns and methylation of genes other than SMN1 and SMN2 may correlate with disease severity. | [95–99] |
| Long non-coding RNAs | Non-coding RNAs might impact gene expression, including activation of SMN2. | [100–103] |
| Imaging candidates | ||
| Muscle imaging approaches | MRI may be useful in determining the degree of SMA severity, muscle atrophy, and response to treatment. | [128–133] |
| Electrical impedance myography (EIM) | EIM measures action potentials on the surface of muscles following stimulation and has been shown to be sensitive to nuanced advancement of disease progression. | [75, 125–127] |
| Electrophysiological parameters | ||
| Compound muscle action potential (CMAP) | CMAP measurements indicate the degree to which a muscle responds to motor nerve stimulation. CMAP responses decline with disease onset. | [45, 75, 134–138, 175] |
| Motor unit number estimation (MUNE) | MUNEs are obtained through several methods, such as applying increasing levels of stimulus to estimate the number of motor units that remain. | [134, 135, 138, 175–179] |
| Repetitive nerve stimulation (RNS) | RNS measures of function of the neuromuscular junction and is an exploratory indicator of SMA disease state. | [180–182] |
Other recent candidate biomarkers include genetic factors such as SMN2 copy number [10, 13–15, 81, 82], SMN2 polymorphisms [10, 13–16, 81–83], and modifier genes [73, 84–89]; transcription and splicing regulators, such as microRNAs [90–94], methylation factors [95–99], and long non-coding RNAs [100–103]; biomolecular factors such as SMN protein [18, 39, 48, 57, 81, 104–106], neurofilament (NF) [78, 107–118], tau protein [112, 119–121], and muscle damage indicators [122–124]; muscle imaging parameters including magnetic resonance imaging (MRI) [122–124] and electrical impedance myography (EIM) [75, 125–127]; and electrophysiological parameters including compound muscle action potential (CMAP) [128–133], motor unit number estimation (MUNE) [75, 125–127], and repetitive nerve stimulation [45, 75, 134–138].
Despite this abundance of research, there remains a need for consensus among the SMA research and clinical care community as to which prognostic, predictive, and pharmacodynamic biomarkers for SMA are the most promising candidates for widespread use [139–141]. To address this need and to advance the field of SMA biomarker research, Cure SMA convened a working group of SMA experts. The SMA Biomarkers Multidisciplinary Working Group reviewed evidence in support of lead candidate biomarkers and engaged in a modified Delphi process consisting of a series of surveys designed to build consensus and determine best next steps to advance the field of SMA biomarker research.
METHODS
The SMA Biomarkers Multidisciplinary Working Group mission
The SMA Biomarkers Multidisciplinary Working Group comprised 11 physicians, scientists, and other healthcare professionals with expertise in the treatment and research of SMA. These individuals were selected for their expertise in SMA clinical care, roles in SMA clinical trials, and their contributions to the understanding of the natural history of SMA or SMA biomarkers. For logistical simplicity, Working Group advisors were recruited from within the U.S. Therefore, discussion was held under the assumption that group members were working within the scientific and regulatory framework of the U.S. The aims were to review the existing candidate biomarkers for SMA (Table 1), select the candidate SMA biomarker best poised for further development, generate a list of data needed for the advancement of this candidate biomarker, and specify the validation studies necessary to generate those data. Cure SMA acted as a neutral moderator for this study. Employees of Cure SMA are neither invested in nor biased toward SMA biomarkers or therapies.
Achieving consensus through the Delphi technique
A modified Delphi technique was used to gain consensus and work towards selection of a biomarker for further development [142, 143]. This process consisted of surveys and phone calls to discuss the questions posed. Surveys and phone calls were reiterative in order to gain increasing consensus with each discussion [144]; initial, broad questions were designed to evoke more specific follow-up questions. Each of the six discussions was designed to build on consensus derived from the previous discussions. Surveys were distributed prior to each phone call. The questions were answered anonymously either through a multiple-choice selection or in an open-ended manner. Most multiple-choice questions also contained a comment field for working group members to utilize if they felt the choices given were insufficient, or if they wanted to add more context to their choice.
The surveys and phone calls took place between October 2020 and April of 2021. The first seven phone calls were proceeded by an online survey and occurred at a frequency of one call per month. A final survey was issued after the last working group call. Survey responses were shared with the wider Working Group prior to each round of discussions, and each voting member was given the opportunity to maintain the response they had given or to modify their response after considering the opinions of their colleagues. This approach balanced the need for voting members to provide opinions anonymously and without influence from colleagues initially while considering the group’s outlook on each question before moving to the next stage. In this manner, it was possible to determine which topics were areas of consensus and which topics necessitated more discussion. From the outset of the study, consensus was defined as a majority; however, the group achieved at least 80% consensus around most decisions. A neutral, non-voting member with expertise in the Delphi process and no conflicts of interest was present to moderate the discussion. The objectives of this modified Delphi process were presented to the Working Group as follows: “The goals of this process are 1) the selection of the biomarker most poised for further development for SMA, and 2) the recommendation of an experimental plan for the purposed validation studies. The objective for the first three calls will be to select a biomarker(s) for further development. The first three calls will be directed towards understanding which potential biomarkers exist and what data is available on those biomarkers. These calls will include guest speakers presenting relevant data, as well as data summaries from the Cure SMA moderator. The goal for the last three calls will be to determine the exact data that exists for the selected biomarker(s) and the gaps that remain for validation. The last three calls will also focus on developing recommendations for a validation study to generate missing data for the selected biomarker(s). The group will select the type of study (i.e., basic research, clinical) and map out the recommended experiments/data to be collected within the determined scope, desired cohort to study (including sample size), parameters to be assessed, assays to be utilized, etc.”
Prior to the first Working Group call, a briefing packet containing a reference list was distributed to all Working Group advisors. The reference list contained 64 peer-reviewed articles intended to provide basic background on each candidate biomarker. The articles were compiled from a literature search performed on PubMed using the following search terms: “biomarker” OR “biomarkers” AND “SMA” OR “spinal muscular atrophy.” Search limits were used to restrict the search to primary research articles and omit reviews and case studies. Working Group members were invited to suggest additions to the reference list, and an amended reference list was recirculated. The briefing packet also contained an initial/pre-call survey in which advisors were asked to rank their interest in five types of biomarkers (biomolecular, electrophysiological, genetic factors, muscle imaging, and transcription and splicing regulators) within the context of the group’s objectives (Table 2; initial/pre-call survey). Biomarker types were ranked on a scale of 1 to 5, where 1 was least interested and 5 was most interested. Scores were averaged to obtain a group score. Having ranked biomolecular and electrophysiological biomarkers of most interest, advisors were asked to indicate which specific candidate biomarkers of these types they were most interested in pursuing (Table 2; initial/pre-call survey). Working Group members could choose more than one biomarker in each category. The next three surveys and calls were directed toward the discussion of specific potential biomarkers and the data that are available about those biomarkers (Table 2). The goal for this phase of the project was to select the biomarker(s) most poised for further development. After the advisors viewed presentations by guest subject matter experts, and reviewed relevant data and data summaries from the Cure SMA moderator, three surveys followed by discussions were administered. Survey one asked questions about prognostic, predictive, and pharmacodynamic biomarkers1 [145] in infants, children and teens, and adults to narrow the discussion to a small number of candidate biomarkers (Table 2; survey one). Working Group members were asked to rank their top three candidate biomarkers in order of interest on a scale of 1 to 3, where 1 was of the most and 3 is of the least interest. Survey two asked about perceived gaps in understanding in the field of the consensus candidate biomarker (ranked 1 to 5, where 1 was least pressing and 5 was most pressing), and which data collections were of the highest priority (ranked 1 to 6, where 1 was the highest and 6 the lowest priority) (Table 2; survey two). Survey three asked Working Group members whether they favored revisiting the second and third biomarker candidates identified, as well as to priority rank the consensus biomarker candidate data that remained outstanding (Table 2; survey three).
Table 2
Interview questions and advisor responses
| Survey Question and Responses | Group Score (0–5) or Response (%) |
| Initial/Pre-call survey | |
| Please rank the following types of biomarkers in order of your interest.1 | |
| Biomolecular (i.e., SMN protein, neurofilament, muscle markers)....... | 4.7 |
| Electrophysiological (i.e., CMAP, MUNE, EIM, etc.)....... | 3.6 |
| Genetic factors (i.e., SMN2 copy number, SMN2 polymorphisms, modifier genes)....... | 3.1 |
| Transcription and splicing regulators (i.e., miRNAs, methylation factors, lncRNAs)....... | 3.0 |
| Muscle imaging (i.e., MRI, ultrasound)....... | 2.7 |
| Which biomolecular biomarker candidates are you most interested in exploring?2 | |
| Neurofilament | 100.0% |
| SMN protein | 75.0% |
| Creatinine | 50.0% |
| Creatine kinase or other markers of muscle damage | 50.0% |
| Which electrophysiological biomarker candidates are you most interested in exploring?2 | |
| CMAP | 83.3% |
| MUNE | 83.3% |
| EIM | 66.7% |
| Repetitive nerve stimulation | 66.7% |
| Electromyography | 50.0% |
| Survey One | |
| Which candidate would be the best pharmacodynamic biomarker… 3 | |
| …in infants | |
| Neurofilament | 72.7% |
| CMAP | 63.6% |
| SMN protein | 45.5% |
| …in children and teens | |
| Neurofilament | 63.6% |
| SMN protein | 54.5% |
| CMAP | 45.5% |
| …in adults | |
| CMAP | 54.5% |
| Neurofilament | 45.5% |
| MUNE | 36.4% |
| Which candidate would be the best predictive biomarker… 3 | |
| …in infants | |
| Neurofilament | 72.7% |
| CMAP | 63.6% |
| MUNE | 27.3% |
| …in children and teens | |
| Neurofilament | 63.6% |
| CMAP | 54.5% |
| MUNE | 27.3% |
| …in adults | |
| CMAP | 54.5% |
| Neurofilament | 36.4% |
| Blood markers of muscle damage | 36.4% |
| Which candidate would be the best prognostic biomarker… 3 | |
| …in infants | |
| Neurofilament | 72.7% |
| CMAP | 54.5% |
| SMN protein | 27.3% |
| …in children and teens | |
| Neurofilament | 63.6% |
| Survey Two | |
| CMAP | 54.5% |
| SMN protein | 36.4% |
| …in adults | |
| CMAP | 54.5% |
| Neurofilament | 45.5% |
| MUNE | 27.3% |
| What are the most pressing data gaps in our understanding of NF as a biomarker for SMA?4 | |
| Age-related changes in NF levels in healthy individuals compared to pathological disease changes | 4.67 |
| Identification of disease onset and tracking of disease progression in patients with ≥3 copies of SMN2 (i.e., understanding NF changes by SMA type) | 4.50 |
| Understanding if threshold values vs. change from baseline for an individual person is more pertinent in tracking disease progression/treatment response | 4.33 |
| Please rank the following data collections in order of highest priority to lowest priority according to which would be most helpful in advancing NF as a biomarker:5 | |
| Determination of the best assay to measure NF | 1 |
| Collection of control data in healthy older adults to understand NF during aging in order to create reference values | 2 |
| Comparison of both NF-H and NF-L in patients of different ages and disease status and the response to treatment | 3 |
| Collection of data in older teens/adults with SMA both on treatment and not on treatment to understand disease course and impacts of treatment on NF | 4 |
| Data from both SMA patients and controls to determine if threshold values or changes from baseline in individuals are better indicators of need for treatment/ treatment response | 5 |
| Collection of control data in healthy infants/ young children to understand developmental changes in NF in order to create reference values | 6 |
| Survey Three | |
| Two additional biomarker candidates, SMN protein and CMAP, were ranked highly by this Working Group. Do you favor revisiting these, or remaining focused on NF alone?6 | |
| Revisit SMN protein and CMAP | 71.4% |
| Continue with NF, not revisiting SMN and CMAP | 28.6% |
| The Working Group would like to delay revisiting SMN protein until further data (e.g., from Roche) are published. Should CMAP be assessed for adults (as NF may be less informative in older ages)? What approach would you suggest moving forward?6 | |
| Delay SMN protein assessment until further data become available while continuing with an assessment of CMAP for older patients | 50.0% |
| Do not continue with either assessment, focusing entire effort on NF | 33.3% |
| Continue with both an assessment of SMN protein and also with an assessment of CMAP for older patients | 16.7% |
| The group agrees that NF may not be as suitable in adult patients as in younger populations. What do you think is the most promising outcome or biomarker for adults?6 | |
| CMAP | 50.0% |
| Serum creatinine | 33.3% |
| SMN protein | 16.7% |
1Rankings of 1–5 (1 = least interested and 5 = most interested) were averaged to obtain group scores. 2Working Group advisors could choose more than one biomarker. 3Results represent the percentage of Working Group advisors who ranked these biomarkers among the top three candidates for stated age group and function. 4Rankings of 1–5 (1 = least pressing and 5 = most pressing) were averaged to obtain group scores. 5Rankings of 1–6 (1 = highest priority and 6 = lowest priority) were averaged to obtain group scores. 6Working Group advisors chose one biomarker.
The final three surveys and discussions were tailored to determine the precise data that exists for the selected biomarker(s) (Table 3). The goal of these final interviews was to identify the gaps that need to be filled to validate the candidate biomarker. Once priority knowledge gaps were agreed upon, the Working Group members tailored a list of recommendations for validation studies to generate missing data for the selected biomarker(s). Details of these experiments included 1) type of study (i.e., basic research, clinical); 2) specific experiments; 3) data to be collected within the determined size and scope; 4) desired cohort to study, including sample size; 5) parameters to be assessed; and 6) assays to be utilized.
Table 3
NF assay validation studies: Group questions and answers from surveys four-six
| Question | Common Advisor Answers |
| Would you prefer to assay NF-L or NF-H? | Both NF-L and NF-H |
| What biofluid sample should be collected? | Blood should be prioritized over CSF |
| Over what period of time should longitudinal samples be collected? | Varies depending on subject age |
| What ages should be studied? | 0–10 years old; adults; determine SMA natural history |
| How many subjects should be included? | Unknown |
| How many total studies will need to be conducted? | Approximately 10 |
| What outcomes should be measured against NF levels to support validation of the biomarker? | Motor function, CMAP, SMN protein levels, SMN2 copy number |
| Should healthy volunteers be included? | Yes, to determine thresholds and normal variation |
| What additional questions should the validation studies address? | •Can the assay be multiplexed with other assays in the future? |
| •Cost, ease of use, possibility of utilizing a commercial laboratory | |
| •Sample volume needed for readout | |
| •Reproducibility and biologic variation |
RESULTS
Building consensus around a priority candidate biomarker
When asked to rank categories of potential biomarkers for SMA, Working Group advisors favored biomolecular entities, followed by electrophysiological parameters, over genetic factors such as transcription factors and splicing regulators, as well as over muscle imaging (Table 2; initial/pre-call survey). When next asked which of several biomolecular candidates the Working Group favored, 100% of members selected NF, followed by 75% who also wanted to explore SMN protein, and 50% of members who also wished to follow up on creatinine, or creatine kinase and other markers of muscle damage. The electrophysiological biomarkers of highest interest were CMAP and MUNE.
Working Group advisors unanimously selected NF as the best biomarker to use to determine pharmacodynamic proof of concept and dose size and frequency optimization (Table 2; survey one). Second-priority candidate biomarkers differed depending on patient age; Working Group members favored CMAP for use in adult and infant patients, but SMN in children and teens. The advisors believed NF would be the best predictive biomarker to monitor disease progression and determine the timing of drug initiation in infants, children and teens, and adults. Second to NF in the predictive biomarker category were CMAP (for infants and children/teens) and blood markers of muscle damage (for adults). Furthermore, advisors felt that NF would be the best prognostic biomarker for clinical monitoring (i.e., for safety, efficacy, and stratification of phenotypic severities) for all age groups, followed by CMAP for all age groups (Table 2).
Identifying and filling gaps in knowledge about NF
The Working Group members identified several knowledge gaps in the field’s understanding of NF as a biomarker for SMA (Table 2; survey two). In healthy individuals, NF levels fluctuate during both in early childhood development and during aging [146], but it is currently unclear how age-related changes in NF levels in healthy individuals compare to changes in SMA-affected individuals [109, 111, 115, 116, 119, 121, 147–149]. It is also unknown how NF levels differ across severities of SMA and whether NF can be used to detect disease onset or track disease progression in patients with three or more copies of SMN2. Finally, it is not currently understood whether a deviation from a standardized baseline value, or a change from an individual’s own baseline, is the relevant factor when tracking NF levels alongside disease progression and treatment response [140, 151]. These unknowns were weighted by the Working Group as being of equal importance.
The advisors next ranked the additional data that should be collected to fill the identified knowledge gaps (Table 2). The information most needed to move forward with biomarker validation was deemed to be the selection of an appropriate assay, followed by data on NF in healthy control subjects, insights about differences between NF heavy (NF-H) and light (NF-L), NF natural history data in patients with SMA, and data regarding changes following treatment. The Working Group indicated that comparison of natural history data from SMA patients to normative controls could elucidate how NF changes with age and in which scenarios a threshold value (e.g., to initiate therapy) or a change from baseline (e.g., when monitoring individual disease progression) would be most relevant, as well as help establish those values. While most advisors also favored revisiting SMN protein and CMAP as additional candidate biomarkers, the group favored waiting until more information about SMN expression is made available and instead moving ahead with CMAP research in adults, for whom NF may be less useful (Table 2; survey three) (See Discussion).
Designing a pilot study to work toward validation
The Working Group concluded that the first step towards validating NF as a biomarker for SMA would be to validate an assay to measure NF in the context of SMA (i.e., to understand reference values relevant to the SMA patient population and corresponding healthy individuals). The advisors discussed the usefulness of conducting a small pilot study to this end. Several key data elements were identified as priorities for the pilot study (Table 3). Members agreed that to validate a candidate NF assay for SMA, it would be beneficial to determine the degree of biologic and measurement variability that exists and to determine the sample volume needed for accurate readout. Once these parameters of the assay were determined, the study could then outline the natural history of NF in SMA patients compared to healthy individuals to determine pathological thresholds and normal age ranges. The reproducibility of the assay (i.e., the intra-individual variation over multiple measurements taken over time) would also be an important consideration, and it would be advantageous to compare the study participants’ NF levels to their SMN2 copy numbers, levels of SMN protein, and measurements of other candidate biomarkers such as CMAP. The group discussed the availability of assay platforms for NF-L and NF-H, as well as in which populations an NF assay would be most useful. Advisors favored quantifying NF in blood rather than CSF. Potential challenges, such as performing repeated blood draws and obtaining an adequate volume of blood from infants, were also discussed. Finally, the Working Group discussed specific parameters for such a pilot study, including ideal subject age ranges and sample sizes. While no broad agreement was reached at this level of detail, the input may serve as a valuable foundation for the planning of this validation study in the future.
DISCUSSION
As of February 2023, 99% of all U.S. infants were being screened for SMA at birth [152], enabling early treatment with three FDA-approved, SMN-dependent, disease-modifying SMA therapies. Widespread SMA treatment will alter the natural history of the disease as it is currently understood, creating new sub-populations of treated SMA-affected individuals with unique responses to SMN-dependent therapies. In addition, several SMN-independent therapies are currently in the clinical trial pipeline [112, 140, 153, 154], and a combination of therapies from these two categories may be the most effective way to treat SMA [35, 69, 72, 73]. Combination treatment regimens will require new biomarkers to help predict and measure treatment responses and disease progression. This rapidly evolving SMA treatment landscape, combined with the heterogeneity of SMA phenotypes and the progressive nature of the disease, makes the need for SMA biomarkers especially urgent [140, 141, 153, 154]. It is likely that combinations of prognostic, predictive, and pharmacodynamic biomarkers will be needed [140, 153].
The SMA Multidisciplinary Biomarkers Working Group successfully reached a consensus on the suitability of NF as a candidate biomarker for SMA (Table 2), particularly at younger ages, and outlined next steps for its further development (Table 3). NFs are significant structural components of the neuronal cytoskeleton and are abundant in large, myelinated axons, such as those of motor neurons [146]. Their importance to the structural integrity of axons is not limited to their role as cellular scaffolding [155–157]. NFs also impact microtubule and actin dynamics, as well as synaptic function. When neurons degenerate or experience damage to axons, in which NFs are enriched, NFs are degraded and released into the interstitial fluid, cerebrospinal fluid (CSF), and blood in significant quantities. This process underlies the observed correlation between NF levels in blood serum or CSF and axonal injury, axonal loss, and neuronal death.
The NF protein consists of four subunits in varying states of phosphorylation, which strengthens them and confers additional resistance to protein degradation. When NF is utilized as a biomarker, researchers typically measure the NF-heavy chain (NF-H) and/or NF-light chain (NF-L) subunits, which are the most and least phosphorylated subunits, respectively [146]. NF has been considered as a biomarker in a number of neurodegenerative conditions [151, 157, 158], including multiple sclerosis [159], Parkinson’s disease [160, 161], Charcot-Marie-Tooth disease [162], peripheral neuropathy [163], and amyotrophic lateral sclerosis (ALS) [164].
NF has also been studied as a potential biomarker for SMA [110, 116]. Research has shown that NF is elevated in SMA-affected infants and young children compared to healthy controls, and it rapidly declines with treatment by nusinersen before stabilizing above normal [107, 113, 118, 165]. For example, Finkel et al. (2022) analyzed data from the CS3A and ENDEAR studies and found that in infants with SMA, lower NF-H levels in CSF were associated with increased nusinersen dosage, which in turn correlated with better treatment outcomes [165]. Studies that have evaluated the effects of nusinersen treatment on NF levels in SMA-affected adolescents and adults have yielded conflicting results, suggesting a need for further, standardized studies on these populations [109, 111, 115, 121, 147–149, 166, 167]. Factors that may have contributed to these variable results include whether NF was measured in blood plasma, serum, or CSF, and whether the NF-H and/or NF-L subunit was measured. Because NF may not be as informative a biomarker in adolescents and adults, the Working Group advisors favored CMAP as a candidate biomarker for further development for those age groups [45, 75, 134–138] (Table 2; survey one). The majority of working group members also rated SMN protein as a top biomolecular candidate, but the Working Group opted to wait for forthcoming data about SMN protein expression during risdiplam treatment. In July 2022, Mercuri et al. [168] published the results of the SUNFISH Part 1 clinical trial, a dose-finding study for risdiplam that demonstrated a dose-dependent increase in SMN protein in treated SMA-affected individuals ages 2–25 years.
The SMA Multidisciplinary Biomarkers Working Group identified additional knowledge gaps that needed to be addressed to validate NF as a biomarker for SMA (Table 2; survey two). At the time of this study, it was unclear whether NF levels change over the lifetime of healthy individuals, and how such changes vary with disease severity and disease progression [109, 111, 115, 116, 121, 147–150]. Furthermore, the working group stated that it must be determined whether the relevant metric is an increase beyond a universal NF threshold level or a deviation from each individual’s own NF baseline [140, 151, 169]. Specific parameters for NF assay validation studies were outlined, including experiments to determine biofluids volumes needed, variations in NF measurement results from within the same sample, and ages and numbers of study candidates needed to draw meaningful conclusions (Table 3). A pilot study to examine these unknown variables was determined to be of great benefit to the field and is the likely next step towards validation of NF as a biomarker for SMA.
Since the SMA Multidisciplinary Biomarkers Working Group convened in 2020, a handful of studies seeking to establish NF-L reference intervals in healthy individuals over a range of ages have been published [170–173]. These studies have added valuable information to the knowledge base about NF-L levels in individuals who are not affected by neurological disease. However, data gaps remain regarding NF-L levels in healthy infants and young children, and more studies are needed among more diverse populations. Furthermore, establishment of a standardized assay for measuring NF-L levels—as well as a consensus as to whether to measure the protein in blood plasma, serum, or CSF—would allow for better comparison of results across studies.
Limitations of the study
NF is expressed exclusively in the nervous system, and the protein is released in response to a wide range of neurologic diseases and traumatic events that result in axonal damage. Therefore, NF has been studied extensively as a biomarker, and the peer-reviewed literature on the subject is robust. This abundance of information had the potential to bias the Working Group in favor of selecting NF as the best SMA biomarker on which to focus future research. However, because the group evaluated all candidate biomarkers on the basis of multiple criteria, any bias imparted by an imbalance in published research was likely corrected. A second limitation of the study is that the Working Group consists exclusively of researchers from the U.S., which had the potential to influence the members to limit their due diligence to biomarker studies performed there. However, the group balanced this potential bias by reviewing literature published by SMA biomarker researchers from around the world. In addition, most of the Working Group members regularly participate in international collaborations on SMA research. Finally, the recent FDA approval of three disease-modifying SMA therapies, along with a dramatic increase in U.S. newborn screening, has enabled approximately 70% of SMA-affected individuals to receive treatment for the disease [174]. As the population of untreated individuals living with SMA grows smaller, it will become more difficult to establish reference values for SMA biomarkers like NF.
CONCLUSION
SMA clinical trial pipelines in the U.S. and worldwide are burgeoning with a variety of potential treatment options, many of which may be used in combination with currently approved SMN-dependent therapies [35, 68–71]. As such, future SMA treatment regimens are likely to involve the use of multiple, often concurrent therapies and evolve over the lifetime of the individual. For these reasons, the need for validated biomarkers in multiple areas of SMA clinical care and research—including predicting treatment response, monitoring disease progression, and measuring pharmacologic response—has never been more urgent.
ACKNOWLEDGMENTS
We would like to thank Biogen and the Miller McNeil Woodruff Foundation for funding this project. We also thank Hilary Scheler, PhD, for writing the early drafts of this manuscript.
CONFLICTS OF INTEREST
Thomas Crawford has served as a paid advisor, consultant, and/or speaker to commercial entities Biogen, AveXis/Novartis, Roche/Genentech, Scholar Rock Pfizer and Cytokinetics, and has received research funding/study site investigation support from Biogen, Avexis, Cytokinetics, Catalyst, and Erydel. He advises several patient organizations: Cure SMA, SMA Foundation, Muscular Dystrophy Foundation, Ataxia Telangiectasia Children’s Project.
Basil T. Darras, MD, has served as an ad hoc scientific advisory board member for AveXis/Novartis Gene Therapies, Biogen, Pfizer, Sarepta, Vertex and Roche/Genentech; Steering Committee Chair for Roche FIREFISH and MANATEE studies and DSMB member for Amicus Inc. and Lexeo Therapeutics; he has no financial interests in these companies. BTD has received research support from the National Institutes of Health/National Institute of Neurological Disorders and Stroke, the Slaney Family Fund for SMA, the Spinal Muscular Atrophy Foundation, Cure SMA, and Working on Walking Fund and has received grants from Ionis Pharmaceuticals, Inc., for the ENDEAR, CHERISH, CS2/CS12 studies; from Biogen for CS11; and from AveXis, Sarepta Pharmaceuticals, Novartis (AveXis), PTC Therapeutics, Roche, Scholar Rock, and Fibrogen. BTD has also received royalties for books and online publications from Elsevier and UpToDate, Inc.
This work was prepared while Christine J. DiDonato was employed at Ann & Robert H. Lurie Children’s Hospital and Northwestern University. The opinions expressed in this article are the author’s own and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the United States government.
Bakri Elsheikh has received compensation for consulting from Biogen, Genentech, Argenx, and Stealth Bio-therapeutics.
Wildon R. Farwell was an employee of Biogen during the period of time this manuscript was written.
Jacqueline Glascock is an employee of Cure SMA and has no financial stake in any SMA drug or company involved in developing SMA drugs.
Kelly Howell has no conflicts to disclose.
Jill Jarecki was an employee of Cure SMA at the time of writing and is currently an employee of Biomarin.
Stephen Kolb receives grant support from Novartis and has served as a paid advisor for Novartis, Biogen and Genentech.
Marco Petrillo is a former employee at Biogen Inc. All conflict statement: Marco Petrillo has received Biogen stocks during his employment period.
Charlotte J. Sumner receives or has received grant support from Roche Ltd., Ionis Pharmaceuticals, Biogen, and Argenx and has served as a paid advisor, consultant, and/or speaker to Biogen, Ionis Pharmaceuticals, PTC Therapeutics, AveXis, Novartis, Roche/Genentech, Cytokinetics and Biomarin. All conflict statement: Charlotte J. Sumner receives grant support from Roche Ltd., Biogen, Argenx, and Actio Bio and has served as a paid advisor, consultant, and/or speaker to Biogen, Ionis Pharmaceuticals, PTC Therapeutics, AveXis, Novartis, Roche/Genentech, Cytokinetics, Sarepta, Atlanta, and Biomarin.
Jessica Tingey is an employee of Cure SMA and has no financial stake in any SMA drug or company involved in developing SMA drugs.
At the time of this work, Marta Valente was a full-time employee of and held stock/stock options in Biogen.
REFERENCES
[1] | Kolb SJ , Coffey CS , Yankey JW , Krosschell K , Arnold WD , Rutkove SB , et al. Natural history of infantile-onset spinal muscular atrophy. Ann Neurol. (2017) ;82: (6):883–91. |
[2] | Sugarman EA , Nagan N , Zhu H , Akmaev VR , Zhou Z , Rohlfs EM , et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: Clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet. (2012) ;20: (1):27–32. |
[3] | Verhaart IEC , Robertson A , Wilson IJ , Aartsma-Rus A , Cameron S , Jones CC , et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet J Rare Dis. (2017) ;12: (1):124. |
[4] | Milligan JN , Blasco-Perez L , Costa-Roger M , Codina-Sola M , Tizzano EF Recommendations for interpreting and reporting silent carrier and disease-modifying variants in sma testing workflows. Genes (Basel). (2022) ; 13: (9). |
[5] | Keinath MC , Prior DE , Prior TW Spinal muscular atrophy: Mutations, testing, and clinical relevance. Appl Clin Genet. (2021) ;14: , 11–25. |
[6] | Wirth B , Karakaya M , Kye MJ , Mendoza-Ferreira N Twenty-five years of spinal muscular atrophy research: From phenotype to genotype to therapy, and what comes next. Annu Rev Genomics Hum Genet. (2020) ;21: , 231–61. |
[7] | Burnett BG , Munoz E , Tandon A , Kwon DY , Sumner CJ , Fischbeck KH Regulation of smn protein stability. Mol Cell Biol. (2009) ;29: (5):1107–15. |
[8] | Wadman RI , Stam M , Gijzen M , Lemmink HH , Snoeck IN , Wijngaarde CA , et al. Association of motor milestones, smn2 copy and outcome in spinal muscular atrophy types 0-4. J Neurol Neurosurg Psychiatry. (2017) ;88: (4):365–7. |
[9] | Wirth B , Brichta L , Schrank B , Lochmuller H , Blick S , Baasner A , et al. Mildly affected patients with spinal muscular atrophy are partially protected by an increased smn2 copy number. Hum Genet. (2006) ;119: (4):422–8. |
[10] | Calucho M , Bernal S , Alias L , March F , Vencesla A , Rodriguez-Alvarez FJ , et al. Correlation between sma type and smn2 copy number revisited: An analysis of 625 unrelated spanish patients and a compilation of reported cases. Neuromuscul Disord. (2018) ;28: (3):208–15. |
[11] | Harada Y , Sutomo R , Sadewa AH , Akutsu T , Takeshima Y , Wada H , et al. Correlation between smn2 copy number and clinical phenotype of spinal muscular atrophy: Three smn2 copies fail to rescue some patients from the disease severity. J Neurol. (2002) ;249: (9):1211–9. |
[12] | Mailman MD , Heinz JW , Papp AC , Snyder PJ , Sedra MS , Wirth B , et al. Molecular analysis of spinal muscular atrophy and modification of the phenotype by smn2. Genet Med. (2002) ;4: (1):20–6. |
[13] | Swoboda KJ , Prior TW , Scott CB , McNaught TP , Wride MC , Reyna SP , et al. Natural history of denervation in sma: Relation to age, smn2 copy number, and function. Ann Neurol. (2005) ;57: (5):704–12. |
[14] | Elsheikh B , Prior T , Zhang X , Miller R , Kolb SJ , Moore D , et al. An analysis of disease severity based on smn2 copy number in adults with spinal muscular atrophy. Muscle Nerve. (2009) ;40: (4):652–6. |
[15] | Prior TW , Krainer AR , Hua Y , Swoboda KJ , Snyder PC , Bridgeman SJ , et al. A positive modifier of spinal muscular atrophy in the smn2 gene. Am J Hum Genet. (2009) ;85: (3):408–13. |
[16] | Ruhno C , McGovern VL , Avenarius MR , Snyder PJ , Prior TW , Nery FC , et al. Complete sequencing of the smn2 gene in sma patients detects smn gene deletion junctions and variants in smn2 that modify the sma phenotype. Hum Genet. (2019) ;138: (3):241–56. |
[17] | D’Amico A , Mercuri E , Tiziano FD , Bertini E Spinal muscular atrophy. Orphanet J Rare Dis. (2011) ;6: , 71. |
[18] | Chaytow H , Huang YT , Gillingwater TH , Faller KME The role of survival motor neuron protein (smn) in protein homeostasis. Cell Mol Life Sci. (2018) ;75: (21):3877–94. |
[19] | Monani UR A deficiency in a ubiquitous protein: Spinal muscular atrophy; a motor neuron-specific disease. Neuron. (2005) ;48: (6):885–96. |
[20] | Zerres K , Rudnik-Schoneborn S Natural history in proximal spinal muscular atrophy. Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch Neurol. (1995) ;52: (5):518–23. |
[21] | Piepers S , van den Berg LH , Brugman F , Scheffer H , Ruiterkamp-Versteeg M , van Engelen BG , et al. A natural history study of late onset spinal muscular atrophy types 3b and 4. J Neurol. (2008) ;255: (9):1400–4. |
[22] | Annoussamy M , Seferian AM , Daron A , Pereon Y , Cances C , Vuillerot C , et al. Natural history of type 2 and 3 spinal muscular atrophy: 2-year nathis-sma study. Annals of Clinical and Translational Neurology. (2021) ;8: (2):359–73. |
[23] | Cances C , Vlodavets D , Comi GP , Masson R , Mazurkiewicz-Beldzinska M , Saito K , et al. Natural history of type 1 spinal muscular atrophy: A retrospective, global, multicenter study. Orphanet J Rare Dis. (2022) ;17: (1):300. |
[24] | Mercuri E , Lucibello S , Perulli M , Coratti G , de Sanctis R , Pera MC , et al. Longitudinal natural history of type i spinal muscular atrophy: A critical review. Orphanet J Rare Dis. (2020) ;15: (1):84. |
[25] | Veldhoen ES , Wijngaarde CA , Hulzebos EHJ , Wosten-van Asperen RM , Wadman RI , van Eijk RPA , et al. Natural history of respiratory muscle strength in spinal muscular atrophy: A prospective national cohort study. Orphanet J Rare Dis. (2022) ;17: (1):70. |
[26] | Finkel RS , McDermott MP , Kaufmann P , Darras BT , Chung WK , Sproule DM , et al. Observational study of spinal muscular atrophy type i and implications for clinical trials. Neurology. (2014) ;83: (9):810–7. |
[27] | Kaufmann P , McDermott MP , Darras BT , Finkel R , Kang P , Oskoui M , et al. Observational study of spinal muscular atrophy type 2 and Functional outcomes over 1 year. Arch Neurol. (2011) ;68: (6):779–86. |
[28] | Pane M , Palermo C , Messina S , Sansone VA , Bruno C , Catteruccia M , et al. An observational study of functional abilities in infants, children, and adults with type 1 sma. Neurology.e696-e. (2018) ;91: (8):703. |
[29] | Russman BS Spinal muscular atrophy: Clinical classification and disease heterogeneity. J Child Neurol. (2007) ;22: (8):946–51. |
[30] | Glascock J , Sampson J , Haidet-Phillips A , Connolly A , Darras B , Day J , et al. Treatment algorithm for infants diagnosed with spinal muscular atrophy through newborn screening. J Neuromuscul Dis. (2018) ;5: (2):145–58. |
[31] | Glascock J , Sampson J , Connolly AM , Darras BT , Day JW , Finkel R , et al. Revised recommendations for the treatment of infants diagnosed with spinal muscular atrophy via newborn screening who have 4 copies of smn2. J Neuromuscul Dis. (2020) ;7: (2):97–100. |
[32] | Schorling DC , Pechmann A , Kirschner J Advances in treatment of spinal muscular atrophy - new phenotypes, new challenges, new implications for care. J Neuromuscul Dis. (2020) ;7: (1):1–13. |
[33] | Ravi B , Chan-Cortes MH , Sumner CJ Gene-targeting therapeutics for neurological disease: Lessons learned from spinal muscular atrophy. Annu Rev Med. (2021) ;72: , 1–14. |
[34] | Aslesh T , Yokota T Restoring smn expression: An overview of the therapeutic developments for the treatment of spinal muscular atrophy. Cells. (2022) ; 11: (3). |
[35] | Mercuri E , Pera MC , Scoto M , Finkel R , Muntoni F Spinal muscular atrophy - insights and challenges in the treatment era. Nat Rev Neurol. (2020) ;16: (12):706–15. |
[36] | Hua Y , Sahashi K , Hung G , Rigo F , Passini MA , Bennett CF , et al. Antisense correction of smn2 splicing in the cns rescues necrosis in a type iii sma mouse model. Genes Dev. (2010) ;24: (15):1634–44. |
[37] | Passini MA , Bu J , Richards AM , Kinnecom C , Sardi SP , Stanek LM , et al. Antisense oligonucleotides delivered to the mouse cns ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med.72ra. (2011) ;3: (72):18. |
[38] | Passini MA , Bu J , Roskelley EM , Richards AM , Sardi SP , O’Riordan CR , et al. Cns-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J Clin Invest. (2010) ;120: (4):1253–64. |
[39] | Poirier A , Weetall M , Heinig K , Bucheli F , Schoenlein K , Alsenz J , et al. Risdiplam distributes and increases smn protein in both the central nervous system and peripheral organs. Pharmacol Res Perspect. (2018) ;6: (6):e00447. |
[40] | Sivaramakrishnan M , McCarthy KD , Campagne S , Huber S , Meier S , Augustin A , et al. Binding to smn2 pre-mrna-protein complex elicits specificity for small molecule splicing modifiers. Nature Communications. (2017) ;8: (1):1476. |
[41] | Spinraza prescribing information. Cambridge: Biogen; (2016) . |
[42] | Evrysdi prescribing information. San Francisco: Genentech; (2020) . |
[43] | Zolgensma prescribing information. Bannockburn: AveXis Inc.; (2019) . |
[44] | McMillan HJ , Proud CM , Farrar MA , Alexander IE , Muntoni F , Servais L Onasemnogene abeparvovec for the treatment of spinal muscular atrophy. Expert Opin Biol Ther. (2022) ;22: (9):1075–90. |
[45] | Darras BT , Chiriboga CA , Iannaccone ST , Swoboda KJ , Montes J , Mignon L , et al. Nusinersen in later-onset spinal muscular atrophy: Long-term results from the phase 1/2 studies. Neurology.e-e. (2492) ;92: (21):506. |
[46] | Finkel RS , Chiriboga CA , Vajsar J , Day JW , Montes J , De Vivo DC , et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet. (2016) ;388: (10063):3017–26. |
[47] | De Vivo DC , Bertini E , Swoboda KJ , Hwu WL , Crawford TO , Finkel RS , et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the phase 2 nurture study. Neuromuscul Disord. (2019) ;29: (11):842–56. |
[48] | Finkel RS , Mercuri E , Darras BT , Connolly AM , Kuntz NL , Kirschner J , et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. (2017) ;377: (18):1723–32. |
[49] | Finkel RS , Chiriboga CA , Vajsar J , Day JW , Montes J , De Vivo DC , et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: Final report of a phase 2, open-label, multicentre, dose-escalation study. The Lancet Child & Adolescent Health. (2021) ;5: (7):491–500. |
[50] | Pechmann A , Langer T , Schorling D , Stein S , Vogt S , Schara U , et al. Evaluation of children with sma type 1 under treatment with nusinersen within the expanded access program in germany. J Neuromuscul Dis. (2018) ;5: (2):135–43. |
[51] | Pechmann A , Behrens M , Dörnbrack K , Tassoni A , Wenzel F , Stein S , et al. Improved upper limb function in non-ambulant children with sma type 2 and 3 during nusinersen treatment: A prospective 3-years smartcare registry study. Orphanet J Rare Dis. (2022) ;17: (1):384. |
[52] | Walter MC , Wenninger S , Thiele S , Stauber J , Hiebeler M , Greckl E , et al. Safety and treatment effects of nusinersen in longstanding adult 5q-sma type 3 - a prospective observational study. J Neuromuscul Dis. (2019) ;6: (4):453–65. |
[53] | Hagenacker T , Wurster CD , Günther R , Schreiber-Katz O , Osmanovic A , Petri S , et al. Nusinersen in adults with 5q spinal muscular atrophy: A non-interventional, multicentre, observational cohort study. The Lancet Neurology. (2020) ;19: (4):317–25. |
[54] | Konersman CG , Ewing E , Yaszay B , Naheedy J , Murphy S , Skalsky A Nusinersen treatment of older children and adults with spinal muscular atrophy. Neuromuscul Disord. (2021) ;31: (3):183–93. |
[55] | Al-Zaidy SA , Kolb SJ , Lowes L , Alfano LN , Shell R , Church KR , et al. Avxs-101 (onasemnogene abeparvovec) for sma Comparative study with a prospective natural history cohort. J Neuromuscul Dis. (2019) ;6: (3):307–17. |
[56] | Day JW , Finkel RS , Chiriboga CA , Connolly AM , Crawford TO , Darras BT , et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of smn2 (str1ve): An open-label, single-arm, multicentre, phase 3 trial. The Lancet Neurology. (2021) ;20: (4):284–93. |
[57] | Mendell JR , Al-Zaidy S , Shell R , Arnold WD , Rodino-Klapac LR , Prior TW , et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. (2017) ;377: (18):1713–22. |
[58] | Mendell JR , Al-Zaidy SA , Lehman KJ , McColly M , Lowes LP , Alfano LN , et al. Five-year extension results of the phase 1 start trial of onasemnogene abeparvovec in spinal muscular atrophy. JAMA Neurology. (2021) ;78: (7):834–41. |
[59] | Mercuri E , Muntoni F , Baranello G , Masson R , Boespflug-Tanguy O , Bruno C , et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy type 1 (str1ve-eu): An open-label, single-arm, multicentre, phase 3 trial. The Lancet Neurology. (2021) ;20: (10):832–41. |
[60] | Pascual-Morena C , Cavero-Redondo I , Lucerón-Lucas-Torres M , Martínez-García I , Rodríguez-Gutiérrez E , Martínez-Vizcaíno V Onasemnogene abeparvovec in type 1spinal muscular atrophy: A systematic review and meta-analysis. HumGene Ther. (2022) . |
[61] | Strauss KA , Farrar MA , Muntoni F , Saito K , Mendell JR , Servais L , et al. Onasemnogene abeparvovec for presymptomatic infants with two copies of smn2 at risk for spinal muscular atrophy type The phase iii spr1nt trial. Nat Med. (2022) ;28: (7):1381–9. |
[62] | Strauss KA , Farrar MA , Muntoni F , Saito K , Mendell JR , Servais L , et al. Onasemnogene abeparvovec for presymptomatic infants with three copies of smn2 at risk for spinal muscular atrophy: The phase iii spr1nt trial. Nat Med. (2022) ;28: (7):1390–7. |
[63] | Baranello G , Darras BT , Day JW , Deconinck N , Klein A , Masson R , et al. Risdiplam in type 1 spinal muscular atrophy. N Engl J Med 2021. |
[64] | Darras BT , Masson R , Mazurkiewicz-Bełdzińska M , Rose K , Xiong H , Zanoteli E , et al. Risdiplam-treated infants with type 1 spinalmuscular atrophy versus historical controls. N Engl J Med. (2021) ;385: (5):427–35. |
[65] | Masson R , Mazurkiewicz-Bełdzińska M , Rose K , Servais L , Xiong H , Zanoteli E , et al. Safety and efficacy of risdiplam in patientswith type 1 spinal muscular atrophy (firefish part 2): Secondaryanalyses from an open-label trial. The Lancet Neurology. (2022) ;21: (12):1110–9. |
[66] | Sturm S , Gunther A , Jaber B , Jordan P , Al Kotbi N , Parkar N , et al. A phase 1 healthy male volunteer single escalating dose study of the pharmacokinetics and pharmacodynamics of risdiplam (rgro067), a smn2 splicing modifier. British Journal of Clinical Pharmacology. (2019) ;85: (1):181–93. |
[67] | Mercuri E , Deconinck N , Mazzone ES , Nascimento A , Oskoui M , Saito K , et al. Safety and efficacy of once-daily risdiplam in type 2 and non-ambulant type 3 spinal muscular atrophy (sunfish part 2): A phase 3, double-blind, randomised, placebo-controlled trial. The Lancet Neurology. (2022) ;21: (1):42–52. |
[68] | Cure SMA. Clinical trials (2022) . Available from: https://www.curesma.org/sma-care-center-network/. |
[69] | Wurster C , Petri S Progress in spinal muscular atrophy research. Curr Opin Neurol. (2022) ;35: (5):693–8. |
[70] | Day JW , Howell K , Place A , Long K , Rossello J , Kertesz N , et al. Advances and limitations for the treatment of spinal muscularatrophy. BMC Pediatr. (2022) ;22: (1):632. |
[71] | Abati E , Manini A , Comi GP , Corti S Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases. Cell Mol Life Sci. (2022) ;79: (7):374. |
[72] | Wirth B Spinal muscular atrophy: In the challenge lies a solution. Trends Neurosci. (2021) ;44: (4):306–22. |
[73] | Hosseinibarkooie S , Peters M , Torres-Benito L , Rastetter RH , Hupperich K , Hoffmann A , et al. The power of human protective modifiers: Pls3 and coro1c unravel impaired endocytosis in spinal muscular atrophy and rescue sma phenotype. Am J Hum Genet. (2016) ;99: (3):647–65. |
[74] | Harada Y , Rao VK , Arya K , Kuntz NL , DiDonato CJ , Napchan-Pomerantz G , et al. Combination molecular therapies for type 1 spinal muscular atrophy. Muscle Nerve. (2020) ;62: (4):550–4. |
[75] | Kolb SJ , Coffey CS , Yankey JW , Krosschell K , Arnold WD , Rutkove SB , et al. Baseline results of the neuronext spinal muscular atrophy infant biomarker study. Annals of Clinical and Translational Neurology. (2016) ;3: (2):132–45. |
[76] | Wadman RI , Stam M , Jansen MD , van der Weegen Y , Wijngaarde CA , Harschnitz O , et al. A comparative study of smn protein and mrna in blood and fibroblasts in patients with spinal muscular atrophy and healthy controls. PLoS One. (2016) ;11: (11):e0167087. |
[77] | Maretina M , Egorova A , Lanko K , Baranov V , Kiselev A Evaluation of mean percentage of full-length smn transcripts as a molecular biomarker of spinal muscular atrophy. Genes (Basel). (2022) ; 13: (10). |
[78] | Alves CRR , Petrillo M , Spellman R , Garner R , Zhang R , Kiefer M , et al. Implications of circulating neurofilamentsfor spinal muscular atrophytreatment early in life: A case series. Mol Ther Methods Clin Dev. (2021) ;23: , 524–38. |
[79] | Alves CRR , Zhang R , Johnstone AJ , Garner R , Eichelberger EJ , Lepez S , et al. Whole blood survival motor neuron protein levels correlate with severity of denervation in spinal muscular atrophy. Muscle Nerve. (2020) ;62: (3):351–7. |
[80] | Trifunov S , Natera-de Benito D , Carrera-García L , Codina A , Expósito-Escudero J , Ortez C , et al. Full-length smn transcript in extracellular vesicles as biomarker in individuals with spinal muscular atrophy type 2 treated with nusinersen. J Neuromuscul Dis. (2023) . |
[81] | Crawford TO , Paushkin SV , Kobayashi DT , Forrest SJ , Joyce CL , Finkel RS , et al. Evaluation of smn protein, transcript, and copy number in the biomarkers for spinal muscular atrophy (bforsma) clinical study. PLoS One. (2012) ;7: (4):e33572. |
[82] | McAndrew PE , Parsons DW , Simard LR , Rochette C , Ray PN , Mendell JR , et al. Identification of proximal spinal muscular atrophy carriers and patients by analysis of smnt and smnc gene copy number. Am J Hum Genet. (1997) ;60: (6):1411–22. |
[83] | Bernal S , Alías L , Barceló MJ , Also-Rallo E , Martínez-Hernández R , Gámez J , et al. The c. 859g>cvariant in the smn2 gene is associated with types ii and iii sma andoriginates from a common ancestor. J Med Genet. (2010) ;47: (9):640–2. |
[84] | Yanyan C , Yujin Q , Jinli B , Yuwei J , Hong W , Fang S Correlation of pls3 expression with disease severity in children with spinal muscular atrophy. Journal of Human Genetics. (2014) ;59: (1):24–7. |
[85] | Oprea GEKS , McWhorter ML , Rossoll W , Müller S , Krawczak M , Bassell GJ , Beattie CE , Wirth B Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. (2008) ;320: (5875):524–27. |
[86] | Riessland M , Kaczmarek A , Schneider S , Swoboda KJ , Löhr H , Bradler C , et al. Neurocalcin delta suppression protects against spinal muscular atrophy in humans and across species by restoring impaired endocytosis. Am J Hum Genet. (2017) ;100: (2):297–315. |
[87] | Stratigopoulos G , Lanzano P , Deng L , Guo J , Kaufmann P , Darras B , et al. Association of plastin 3 expression with disease severity in spinal muscular atrophy only in postpubertal females. Arch Neurol. (2010) ;67: (10):1252–6. |
[88] | Ackermann B , Kröber S , Torres-Benito L , Borgmann A , Peters M , Hosseini Barkooie SM , et al. Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum Mol Genet. (2013) ;22: (7):1328–47. |
[89] | McGovern VL , Massoni-Laporte A , Wang X , Le TT , Le HT , Beattie CE , et al. Plastin 3 expression does not modify spinal muscular atrophy severity in the Δ7 sma mouse. PLoS One. (2015) ;10: (7):e0132364. |
[90] | Catapano F , Zaharieva I , Scoto M , Marrosu E , Morgan J , Muntoni F , et al. Altered levels of microrna-9, -206, and -132 in spinal muscular atrophy and their response to antisense oligonucleotide therapy. Mol Ther Nucleic Acids. (2016) ;5: (7):e331. |
[91] | Bonanno S , Marcuzzo S , Malacarne C , Giagnorio E , Masson R , Zanin R , et al. Circulating myomirs as potential biomarkers to monitor response to nusinersen in pediatric sma patients. Biomedicines. (2020) ; 8: (2). |
[92] | Magri F , Vanoli F , Corti S Mirna in spinal muscular atrophy pathogenesis and therapy. Journal of Cellular and Molecular Medicine. (2018) ;22: (2):755–67. |
[93] | Magen I , Aharoni S , Yacovzada NS , Tokatly Latzer I , Alves CRR , Sagi L , et al. Muscle micrornas in the cerebrospinal fluid predict clinical response to nusinersen therapy in type ii and type iii spinal muscular atrophy patients. Eur J Neurol. (2022) ;29: (8):2420–30. |
[94] | D’Silva AM , Kariyawasam D , Venkat P , Mayoh C , Farrar MA Identification of novel csf-derived mirnas in treated paediatric onset spinal muscular atrophy: An exploratory study. Pharmaceutics. (2023) ; 15: (1). |
[95] | Hauke J , Riessland M , Lunke S , Eyüpoglu IY , Blümcke I , El-Osta A , et al. Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed by histone deacetylase inhibition. Hum Mol Genet. (2009) ;18: (2):304–17. |
[96] | Cao YY , Qu YJ , He SX , Li Y , Bai JL , Jin YW , et al. Association between smn2 methylation and disease severity in chinese children with spinal muscular atrophy. Journal of Zhejiang University Science B. (2016) ;17: (1):76–82. |
[97] | Zheleznyakova GY , Nilsson EK , Kiselev AV , Maretina MA , Tishchenko LI , Fredriksson R , et al. Methylation levels of slc23a2 and ncor2 genes correlate with spinal muscular atrophy severity. PLoS One. (2015) ;10: (3):e0121964. |
[98] | Maretina MA , Valetdinova KR , Tsyganova NA , Egorova AA , Ovechkina VS , Schioth HB , et al. Identification of specific gene methylation patterns during motor neuron differentiation from spinal muscular atrophy patient-derived ipsc. Gene. (2022) ;811: , 146109. |
[99] | Maretina M , Egorova A , Baranov V , Kiselev A Dync1h1 gene methylation correlates with severity of spinal muscular atrophy. Ann Hum Genet. (2019) ;83: (2):73–81. |
[100] | Chen KW , Chen JA Functional roles of long non-coding rnas in motor neuron development and disease. J Biomed Sci. (2020) ;27: (1):38. |
[101] | Vangoor VR , Gomes-Duarte A , Pasterkamp RJ Long non-coding rnas in motor neuron development and disease. J Neurochem. (2021) ;156: (6):777–801. |
[102] | d’Ydewalle C , Ramos DM , Pyles NJ , Ng SY , Gorz M , Pilato CM , et al. The antisense transcript smn-as1 regulates smn expression and is a novel therapeutic target for spinal muscular atrophy. Neuron. (2017) ;93: (1):66–79. |
[103] | Fenoglio C , Ridolfi E , Galimberti D , Scarpini E An emerging role for long non-coding rna dysregulation in neurological disorders. Int J Mol Sci. (2013) ;14: (10):20427–42. |
[104] | Mercuri E , Darras BT , Chiriboga CA , Day JW , Campbell C , Connolly AM , et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. (2018) ;378: (7):625–35. |
[105] | Kwon YW , Gurney ME Brain-derived neurotrophic factor transiently stabilizes silent synapses on developing neuromuscular junctions. J Neurobiol. (1996) ;29: (4):503–16. |
[106] | Tiziano FD , Lomastro R , Abiusi E , Pasanisi MB , Di Pietro L , Fiori S , et al. Longitudinal evaluation of smn levels as biomarker for spinal muscular atrophy: Results of a phase iib double-blind study of salbutamol. J Med Genet. (2019) ;56: (5):293–300. |
[107] | Darras BT , Crawford TO , Finkel RS , Mercuri E , De Vivo DC , Oskoui M , et al. Neurofilament as a potential biomarker for spinal muscular atrophy. Annals of Clinical and Translational Neurology. (2019) ;6: (5):932–44. |
[108] | Agerholm JS , Basse A Spinal muscular atrophy in calves of the red danish dairy breed. Vet Rec. (1994) ;134: (10):232–5. |
[109] | Faravelli I , Meneri M , Saccomanno D , Velardo D , Abati E , Gagliardi D , et al. Nusinersen treatment and cerebrospinal fluid neurofilaments: An explorative study on spinal muscular atrophy type 3 patients. Journal of Cellular and Molecular Medicine. (2020) ;24: (5):3034–9. |
[110] | Franco-Espin J , Gatius A , Armengol J , Arumugam S , Moradi M , Sendtner M , et al. Smn is physiologically downregulated at wild-type motor nerve terminals but aggregates together with neurofilaments in sma mouse models. Biomolecules. (2022) ; 12: (10). |
[111] | Freigang M , Steinacker P , Wurster CD , Schreiber-Katz O , Osmanovic A , Petri S , et al. Glial fibrillary acidic protein in cerebrospinal fluid of patients with spinal muscular atrophy. Annals of Clinical and Translational Neurology. (2022) ;9: (9):1437–48. |
[112] | Johannsen J , Weiss D , Daubmann A , Schmitz L , Denecke J Evaluation of putative csf biomarkers in paediatric spinal muscular atrophy (sma) patients before and during treatment with nusinersen. Journal of Cellular and Molecular Medicine. (2021) ;25: (17):8419–31. |
[113] | Nitz E , Smitka M , Schallner J , Akgun K , Ziemssen T , von der Hagen M , et al. Serum neurofilament light chain in pediatric spinal muscular atrophy patients and healthy children. Annals of Clinical and Translational Neurology. (2021) ;8: (10):2013–24. |
[114] | Orbach R , Sagi L , Sadot E , Tokatly Latzer I , Shtamler A , Zisberg T , et al. Cerebrospinal fluid characteristics of patients treated with intrathecal nusinersen for spinal muscular atrophy. Muscle Nerve 2022. |
[115] | Rich KA , Fox A , Yalvac M , Heintzman S , Tellez M , Bartlett A , et al. Neurofilament levels in csf and serum in an adult sma cohort treated with nusinersen. J Neuromuscul Dis. (2022) ;9: (1):111–9. |
[116] | Spicer C , Lu CH , Catapano F , Scoto M , Zaharieva I , Malaspina A , et al. The altered expression of neurofilament in mouse models and patients with spinal muscular atrophy. Annals of Clinical and Translational Neurology. (2021) ;8: (4):866–76. |
[117] | Winter B , Guenther R , Ludolph AC , Hermann A , Otto M , Wurster CD Neurofilaments and tau in csf in an infant with sma type 1 treated with nusinersen. J Neurol Neurosurg Psychiatry. (2019) ;90: (9):1068–9. |
[118] | Olsson B , Alberg L , Cullen NC , Michael E , Wahlgren L , Kroksmark AK , et al. Nfl is a marker of treatment response in children with sma treated with nusinersen. J Neurol. (2019) ;266: (9):2129–36. |
[119] | Šimić G , Vukić V , Babić M , Banović M , Berečić I , Španić E , et al. Total tau incerebrospinal fluid detects treatment responders among spinalmuscular atrophy types 1-3 patients treated with nusinersen. CNSNeuroscience & Therapeutics. (2022) . |
[120] | Walter MC , Wenninger S , Thiele S , Stauber J , Hiebeler M , Greckl E , et al. Safety and treatment effects of nusinersen in longstandingadult 5q-sma type 3 - a prospective observational study. JNeuromuscul Dis. (2019) ;6: (4):453–65. |
[121] | Milella G , Introna A , D’Errico E , Fraddosio A , Scaglione G , Morea A , et al. Cerebrospinal fluid and clinical profiles in adult type 2-3 spinal muscular atrophy patients treated with nusinersen: An 18-month single-centre experience. Clinical Drug Investigation. (2021) ;41: (9):775–84. |
[122] | Alves CRR , Zhang R , Johnstone AJ , Garner R , Nwe PH , Siranosian JJ , et al. Serum creatinine is a biomarker of progressive denervation in spinal muscular atrophy. Neurology.e921-e. (2020) ;94: (9):31. |
[123] | Freigang M , Wurster CD , Hagenacker T , Stolte B , Weiler M , Kamm C , et al. Serum creatine kinase and creatinine in adult spinal muscular atrophy under nusinersen treatment. Annals of Clinical and Translational Neurology. (2021) ;8: (5):1049–63. |
[124] | Lombardi V , Querin G , Ziff OJ , Zampedri L , Martinelli I , Heller C , et al. Muscle and not neuronal biomarkers correlate with severity in spinal and bulbar muscular atrophy. Neurology.e-e. (1205) ;92: (11):11. |
[125] | Rutkove SB , Aaron R , Shiffman CA Localized bioimpedance analysis in the evaluation of neuromuscular disease. Muscle Nerve. (2002) ;25: (3):390–7. |
[126] | Rutkove SB , Gregas MC , Darras BT Electrical impedance myography in spinal muscular atrophy: A longitudinal study. Muscle Nerve. (2012) ;45: (5):642–7. |
[127] | Li J , Geisbush TR , Arnold WD , Rosen GD , Zaworski PG , Rutkove SB A comparison of three electrophysiological methods for the assessment of disease status in a mild spinal muscular atrophy mouse model. PLoS One. (2014) ;9: (10):e111428. |
[128] | Sprenger-Svacina A , Haensch J , Weiss K , Grosse Hokamp N , Maintz D , Schlamann M , et al. Mri correlates of motoneuron loss in sma. J Neurol. (2022) . |
[129] | Durmus H , Yilmaz R , Gulsen-Parman Y , Oflazer-Serdaroglu P , Cuttini M , Dursun M , et al. Muscle magnetic resonance imaging in spinal muscular atrophy type Selective and progressive involvement. Muscle Nerve. (2017) ;55: (5):651–6. |
[130] | Morrow JM , Sinclair CD , Fischmann A , Machado PM , Reilly MM , Yousry TA , et al. Mri biomarker assessment of neuromuscular disease progression: A prospective observational cohort study. The Lancet Neurology. (2016) ;15: (1):65–77. |
[131] | Gallone A , Mazzi F , Bonanno S , Zanin R , Moscatelli M , Aquino D , et al. Muscle quantitative mri in adult sma patients on nusinersen treatment: A longitudinal study. Acta Myol. (2022) ;41: (2):76–83. |
[132] | Souza PVS , Pinto W , Ricarte A , Badia BML , Seneor DD , Teixeira DT , et al. Clinical and radiological profile of patients with spinal muscular atrophy type 4. Eur J Neurol. (2021) ;28: (2):609–19. |
[133] | Shimizu-Motohashi Y , Chiba E , Mizuno K , Yajima H , Ishiyama A , Takeshita E , et al. Muscle impairment in mri affect variability in treatment response to nusinersen in patients with spinal muscular atrophy type 2 and 3 A retrospective cohort study. Brain Dev. (2022) . |
[134] | Arnold WD , Porensky PN , McGovern VL , Iyer CC , Duque S , Li X , et al. Electrophysiological biomarkers in spinal muscular atrophy:Preclinical proof of concept. Annals of Clinical and TranslationalNeurology. (2014) ;1: (1):34–44. |
[135] | Kang PB , Gooch CL , McDermott MP , Darras BT , Finkel RS , Yang ML , et al. The motor neuron response to smn1 deficiency in spinal muscular atrophy. Muscle Nerve. (2014) ;49: (5):636–44. |
[136] | Lewelt A , Krosschell KJ , Scott C , Sakonju A , Kissel JT , Crawford TO , et al. Compound muscle action potential and motor function in children with spinal muscular atrophy. Muscle Nerve. (2010) ;42: (5):703–8. |
[137] | Farrar MA , Park SB , Vucic S , Carey KA , Turner BJ , Gillingwater TH , et al. Emerging therapies and challenges in spinal muscular atrophy. Ann Neurol. (2016) . |
[138] | Duque SI , Arnold WD , Odermatt P , Li X , Porensky PN , Schmelzer L , et al. A large animal model of spinal muscular atrophy and correction of phenotype. Ann Neurol. (2015) ;77: (3):399–414. |
[139] | D’Silva AM , Kariyawasam DST , Best S , Wiley V , Farrar MA , Group NSNS Integrating newborn screening for spinal muscular atrophy into health care systems: An australian pilot programme. Dev Med Child Neurol. (2022) ;64: (5):625–32. |
[140] | Pino MG , Rich KA , Kolb SJ Update on biomarkers in spinal muscular atrophy. Biomark Insights. (2021) ;16: , 11772719211035643. |
[141] | Kariyawasam DST , D’Silva A , Lin C , Ryan MM , Farrar MA Biomarkers and the development of a personalized medicine approach in spinal muscular atrophy. Frontiers in Neurology. (2019) ;10: , 898. |
[142] | Delbecq A , Ven de Van A , Gustafson D The delphi technique. In: Delbecq A, Ven de Van A, Gustafson D, editors. Group techniques for program planning: A guide to nominal group and delphi processes. Madison: Scott Foresman; (1975) , pp. 83–107. |
[143] | Rowe G , Wright G Expert opinions in forecasting. Role of the delphi technique. In: S AJ, editor. Principles of forecasting: A handbook of researchers and practitioners. Boston: Kluwer Academic Publishers. (2001) . |
[144] | Hsu CC , Sandford BA The delphi technique: Making sense of consensus. Practical Assessment, Research and Evaluation. (2007) ;12: (10):1–8. |
[145] | FDA-NIH Joint Leadership Council. Best (biomarkers, endpoints, and other tools) resource. (2016) . |
[146] | Yuan A , Rao MV , Veeranna , Nixon RA Neurofilaments and neurofilament proteins in health and disease. Cold Spring Harb Perspect Biol. (2017) ; 9: (4). |
[147] | Wurster CD , Gunther R , Steinacker P , Dreyhaupt J , Wollinsky K , Uzelac Z , et al. Neurochemical markers in csf of adolescent and adult sma patients undergoing nusinersen treatment. Ther Adv Neurol Disord. (2019) ;12: , 1756286419846058. |
[148] | Wurster CD , Steinacker P , Günther R , Koch JC , Lingor P , Uzelac Z , et al. Neurofilament light chain in serum of adolescent and adult sma patients under treatment with nusinersen. J Neurol. (2020) ;267: (1):36–44. |
[149] | Totzeck A , Stolte B , Kizina K , Bolz S , Schlag M , Thimm A , et al. Neurofilament heavy chain and tau protein are not elevated in cerebrospinal fluid of adult patients with spinal muscular atrophy during loading with nusinersen. Int J Mol Sci. (2019) ; 20: (21). |
[150] | De Wel B , De Schaepdryver M , Poesen K , Claeys KG Biochemical and clinical biomarkers in adult sma 3-4 patients treated with nusinersen for 22 months. Annals of Clinical and Translational Neurology. (2022) ;9: (8):1241–51. |
[151] | Mak G , Menon S , Lu JQ Neurofilaments in neurologic disorders and beyond. J Neurol Sci. (2022) ;441: , 120380. |
[152] | Cure SMA. Newborn screening for SMA Available from (2022) )–https://www.curesma.org/newborn-screening-for-sma/. |
[153] | Smeriglio P , Langard P , Querin G , Biferi MG The identification of novel biomarkers is required to improve adult sma patient stratification, diagnosis and treatment. J Pers Med. (2020) ; 10: (3). |
[154] | Navarrete-Opazo AGS , Waite M Molecular biomarkers for spinal muscular atrophy: A systematic review. Neurology Clinical Practice. (2021) ; 11: (4). |
[155] | Lee Y , Lee BH , Yip W , Chou P , Yip BS Neurofilament proteins as prognostic biomarkers in neurological disorders. Curr Pharm Des. (2020) ;25: (43):4560–9. |
[156] | Olsson B , Portelius E , Cullen NC , Sandelius A , Zetterberg H , Andreasson U , et al. Association of cerebrospinal fluidneurofilament light protein levels with cognition in patients withdementia, motor neuron disease, and movement disorders. JAMANeurology. (2019) ;76: (3):318–25. |
[157] | Delaby C , Bousiges O , Bouvier D , Fillée C , Fourier A , Mondésert E , et al. Neurofilaments contribution in clinic: Stateof the art. Frontiers in Aging Neuroscience. (2022) ;14: , 1034684. |
[158] | Gaetani L , Blennow K , Calabresi P , Di Filippo M , Parnetti L , Zetterberg H Neurofilament light chain as a biomarker in neurological disorders. J Neurol Neurosurg Psychiatry. (2019) ;90: (8):870–81. |
[159] | Ning L , Wang B Neurofilament light chain in blood as a diagnostic and predictive biomarker for multiple sclerosis: A systematic review and meta-analysis. PLoS One. (2022) ;17: (9):e0274565. |
[160] | Batzu L , Rota S , Hye A , Heslegrave A , Trivedi D , Gibson LL , et al. Plasma p-tau181, neurofilament light chain and association with cognition in parkinson’s disease. NPJ Parkinson’s Disease. (2022) ;8: (1):154. |
[161] | Huang Y , Huang C , Zhang Q , Shen T , Sun J Serum nfl discriminates parkinson disease from essential tremor and reflect motor and cognition severity. BMC Neurol. (2022) ;22: (1):39. |
[162] | Millere E , Rots D , Simren J , Ashton NJ , Kupats E , Micule I , et al. Plasma neurofilament light chain as a potential biomarker in charcot-marie-tooth disease. Eur J Neurol. (2021) ;28: (3):974–81. |
[163] | Sandelius A , Zetterberg H , Blennow K , Adiutori R , Malaspina A , Laura M , et al. Plasma neurofilament light chain concentration in the inherited peripheral neuropathies. Neurology.e518-e. (2018) ;90: (6):24. |
[164] | Gagliardi D , Meneri M , Saccomanno D , Bresolin N , Comi GP , Corti S Diagnostic and prognostic role of blood and cerebrospinal fluid and blood neurofilaments in amyotrophic lateral sclerosis: A review of the literature. Int J Mol Sci. (2019) ; 20: (17). |
[165] | Finkel RS , Ryan MM , Pascual Pascual SI , Day JW , Mercuri E , De Vivo DC , et al. Scientific rationale for a higher dose of nusinersen. Annals of Clinical and Translational Neurology. (2022) ;9: (6):819–29. |
[166] | Paris A , Bora P , Parolo S , MacCannell D , Monine M , van der Munnik N , et al. A pediatric quantitative systems pharmacology model of neurofilament trafficking in spinal muscular atrophy treated with the antisense oligonucleotide nusinersen. CPT: Pharmacometrics & Systems Pharmacology. (2022) . |
[167] | Li JY , Dai Y , Sun XH , Ren HT , Shen DC , Yang XZ , et al. Comparison of neurofilament light and heavy chain in spinal muscular atrophy and amyotrophic lateral sclerosis: A pilot study. Brain and Behavior. e. (2023) ), 2997. |
[168] | Mercuri E , Baranello G , Boespflug-Tanguy O , De Waele L , Goemans N , Kirschner J , et al. Risdiplam in types 2 and 3 spinal muscular atrophy: A randomised, placebo-controlled, dose-finding trial followed by 24 months of treatment. Eur J Neurol. (2022) . |
[169] | Hviid CVB , Madsen AT , Winther-Larsen A Biological variation of serum neurofilament light chain. Clin Chem Lab Med. (2022) ;60: (4):569–75. |
[170] | Bornhorst JA , Figdore D , Campbell MR , Pazdernik VK , Mielke MM , Petersen RC , et al. Plasma neurofilament light chain (nfl) reference interval determination in an age-stratified cognitively unimpaired cohort. Clin Chim Acta. (2022) ;535: , 153–6. |
[171] | Fitzgerald KC , Sotirchos ES , Smith MD , Lord HN , DuVal A , Mowry EM , et al. Contributors to serum nfl levels in people without neurologic disease. Ann Neurol. (2022) ;92: (4):688–98. |
[172] | Simrén J , Andreasson U , Gobom J , Suarez Calvet M , Borroni B , Gillberg C , et al. Establishment of reference values for plasmaneurofilament light based on healthy individuals aged 5-90 years. Brain Communications.fcac. (2022) ;4: (4):174. |
[173] | Hviid CVB , Knudsen CS , Parkner T Reference interval and preanalytical properties of serum neurofilament light chain in scandinavian adults. Scand J Clin Lab Invest. (2020) ;80: (4):291–5. |
[174] | Cure SMA. State of SMA Available from (2022) )–https://www.curesma.org/wp-content/uploads/2022/06/9042022_State-of-SMA_vweb.pdf. |
[175] | Kariyawasam D , D’Silva A , Howells J , Herbert K , Geelan-Small P , Lin CS , et al. Motor unit changes in children with symptomatic spinal muscular atrophy treated with nusinersen. J Neurol Neurosurg Psychiatry. (2020) ;92: (1):78–85. |
[176] | Galea V , Fehlings D , Kirsch S , McComas A Depletion and sizes of motor units in spinal muscular atrophy. Muscle Nerve. (2001) ;24: (9):1168–72. |
[177] | Bromberg MB , Swoboda KJ Motor unit number estimation in infants and children with spinal muscular atrophy. Muscle Nerve. (2002) ;25: (3):445–7. |
[178] | Gawel M , Kostera-Pruszczyk A , Lusakowska A , Jedrzejowska M , Ryniewicz B , Lipowska M , et al. Motor unit loss estimation by the multipoint incremental mune method in children with spinal muscular atrophy–a preliminary study. Neuromuscul Disord. (2015) ;25: (3):216–21. |
[179] | Günther R , Neuwirth C , Koch JC , Lingor P , Braun N , Untucht R , et al. Motor unit number index (munix) of hand muscles is a disease biomarker for adult spinal muscular atrophy. Clinical Neurophysiology: Official Journal of the International Federation of Clinical Neurophysiology. (2019) ;130: (2):315–9. |
[180] | Montes J , McDermott MP , Martens WB , Dunaway S , Glanzman AM , Riley S , et al. Six-minute walk test demonstrates motor fatigue in spinal muscular atrophy. Neurology. (2010) ;74: (10):833–8. |
[181] | Bertorini T Neurologic evaluation and ancillary tests. In: Bertorini T, editor. Neuromuscular case studies. Philadelphia, PA: Butterworth-Heinemann. (2008) ), 27–76. |
[182] | Wadman RI , Vrancken AF , van den Berg LH , van der Pol WL Dysfunction of the neuromuscular junction in spinal muscular atrophy types 2 and 3. Neurology. (2012) ;79: (20):2050–5. |
Notes
1 Biomarker categories are based on those outlined in the Biomarkers, Endpoints, and other Tools (BEST) Resource [145]. However, terminology has been adapted to be consistent with that used in prior peer-reviewed SMA research literature.


