
Phase II/III Study of Aceneuramic Acid Administration for GNE Myopathy in Japan
Abstract
Background:
GNE myopathy is an ultra-rare muscle disease characterized by a reduction in the synthesis of sialic acid derived from pathogenic variants in the GNE gene. No treatment has been established so far.
Objective:
We evaluated the safety and efficacy of oral supplementation of aceneuramic acid in patients with GNE myopathy.
Methods:
This multicenter, placebo-controlled, double-blind study comprised genetically confirmed GNE myopathy patients in Japan who were randomly assigned into treatment groups of sialic acid-extended release (SA-ER) tablets (6 g/day for 48 weeks) or placebo groups (4:1). The primary endpoint of effectiveness was set as the change in total upper limb muscle strength (upper extremity composite [UEC] score) from the start of administration to the final evaluation time point.
Results:
Among the 20 enrolled patients (SA-ER group, 16; placebo group, 4), 19 completed this 48-week study. The mean value of change in UEC score (95% confidence interval [CI]) at 48 weeks was –0.1 kg (–2.1 to 2.0) in the SA-ER group and –5.1 kg (–10.4 to 0.3) in the placebo group. The least squares mean difference (95% CI) between the groups in the covariance analysis was 4.8 kg (–0.3 to 9.9; P = 0.0635). The change in UEC score at 48 weeks was significantly higher in the SA-ER group compared with the placebo group (P = 0.0013) in the generalized estimating equation test repeated measurement analysis. In one patient in the SA-ER group, who was found to be pregnant 2 weeks after drug administration fetal death with tangled umbilical cord occurred at 13 weeks after the discontinuation of treatment. No other serious adverse effects were observed.
Conclusions:
The present study indicates that oral administration of SA-ER tablets is effective and safe in patients with GNE myopathy in Japan.
Trial registration number: UMIN000020683 (UMIN Clinical Trials Registry) (https://center6.umin.ac.jp/cgi-open-bin/ctr_e/ctr_view.cgi?recptno=R000023425)
INTRODUCTION
GNE myopathy (also known as distal myopathy with rimmed vacuoles, hereditary inclusion body myopathy, or Nonaka disease) is a hereditary muscle disorder characterized by a decrease in muscle strength, especially in distal muscles [1–6]. Particularly, tibialis anterior muscles are affected early at the onset of this disease, resulting in a gradually progressive decrease in muscle strength in the four limbs. However, the quadriceps femoris muscle is relatively spared even after considerable progression of the disease. The onset of GNE myopathy is usually from the late teens to the latter half of the thirties. According to the information in the Registry of Muscular Dystrophy (REMUDY), the estimated mean period from onset to loss of ambulation is 21 years [7]. It is an ultrarare disease affecting about 200 to 300 people in Japan.
Patients with GNE myopathy present with pathogenic variants in the GNE gene, which encodes uridine diphosphate-N-acetylglucosamine 2-epimerase/N-acetyl mannosamine kinase (GNE/MNK) that is a rate limiting enzyme for biosynthesizing of sialic acid [8]. Lack of sialic acid supply in vivo is suspected to be involved in the pathomechanism of GNE myopathy.
The effectiveness of replacement therapy of the most abundant sialic acid in vivo, aceneuramic acid, was reported in a mouse model of GNE myopathy [9, 10]. Continuous administration of aceneuramic acid before the onset of the disease to model mice restored the motor ability, contractile forces of the skeletal muscles, biochemical parameters, and muscular pathology of mice to the normal level [9, 10]. Thus, aceneuramic acid was considered an effective therapeutic agent for the amelioration or control of the progression of GNE myopathy.
The development of effective treatment modalities for GNE myopathy proved to be challenging because of the limited number of patients, slow progression, and lack of quantitative clinical outcomes of the disease. We conducted an investigator-initiated Phase I clinical trial for GNE myopathy patients from November 2010 using aceneuramic acid for the first time in Japan [11]. Phase I studies conducted in the US using sialic acid-extended release (SA-ER) tablets up to 6 g daily (2 g each time; three times daily; oral dose) demonstrated a dose-dependent increase in the serum free concentration of aceneuramic acid, with no major concerns about safety [12]. Subsequently, a placebo-controlled phase II study was conducted, wherein administration of SA-ER (6 g daily) was found to be effective and safe [13]. Meanwhile, we conducted an additional investigator-initiated Phase I study using SA-ER tablets (6 g daily for up to 7 days), and found that the concentrations of serum free aceneuramic acid were two to three times higher than those observed before administration, and the safety for the patients [11] was similar to the international phase II study [13]. However, international Phase III study [14] ended in negative results.
On the basis of these findings, we conducted a double-blind, placebo-controlled Phase II/III study for up to 48 weeks, and evaluated the efficacy and safety of a daily 6 g of SA-ER provided to patients with GNE myopathy in Japan.
METHODS
Study design
This investigator-initiated multicenter collaborative double-blind trial was conducted from March 2016 to July 2017, as cooperative research by the Department of Neurology at Tohoku University Hospital, National Center Hospital, National Center of Neurology and Psychiatry (NCNP), Nagoya University Hospital, Osaka University Hospital, and Kumamoto University Hospital in Japan. A procedure manual for each item was prepared for the evaluation of the effectiveness of this trial in order to avoid differences among evaluators. In order to maintain consistency in the measurement methods used, a video showing the standard measurement procedure in accordance with the procedure manual was created and used to explain to the examiners at the start-up meeting. GNE myopathy patients were recruited with the cooperation of Patients Association for Distal Myopathies (PADM) and REMUDY in Japan [7]. Qualified individuals were registered using Electronic Data Capture (EDC). Investigational drugs were randomly assigned to participants by an independent third party.
As GNE myopathy is an ultra-rare disease, we decided to assign the number of patients in the SA-ER group to four times that of the placebo group so as to gather as much information as possible from the SA-ER group. These numbers are based on statistical calculation and achieved required power. The subject registration center made dynamic assignments, taking into consideration a balance of factors between groups. The doctor started the administration of the drug and monitored the patient every 8 weeks for 48 weeks. As SA-ER (6 g daily) was found to be effective and safe in a placebo-controlled phase II study [13], and decline of muscle power was observed during 48 weeks in placebo group, we conducted our study in the period of 48 weeks. The assignment table and the emergency key created based on the table were strictly preserved by the investigational drug assignment managers until the data were fixed after the trial.
Standard protocol approvals, registrations, and patient consents
The study protocol was approved by the institutional review board of each medical institution. All participants in this study provided informed consent prior to any study-specific procedures. The study was conducted in accordance with the ministerial ordinance on clinical trial standards for pharmaceuticals (Good Clinical Practice [GCP], the Declaration of Helsinki). This study was registered in UMIN-CTR (UMIN000020683).
Inclusion and exclusion criteria
Patients aged 20 to 50 years, irrespective of gender, with confirmed pathogenic variants of the GNE/MNK enzyme along with a written diagnosis of GNE myopathy were included in this study. As our target population was mild to moderately affected patients with selected muscles to be in active degeneration, we decided to register more than 15 patients who could walk ≥200 m in the 6-minute walking test (6 MWT) and reproduce the muscle strength of the elbow extensor, as measured by a dynamometer at the time of screening with the difference between two measured values of the dominant hand being less than 15%. The use of ankle foot orthoses was allowed. If the patient or partner was capable of fertile, we agreed to provide effective contraceptive measures from the time of providing consent to 3 months after the trial. Female patients underwent pregnancy tests during the trial.
The main exclusion criteria included the following: those who had ingested sialic acid, N-acetyl-D-mannosamine (ManNAc), intravenous immunoglobulin (IVIG), or similar drug, sports supplements or whey protein, which are metabolized to produce sialic acid within 60 days before screening; a treatment history of sialic acid for ≥30 days; hypersensitive to sialic acid; and those (or their partners) who were pregnant or lactating at the time of screening or planning to become pregnant during the trial.
Investigational drugs and dosage regimen
The investigational drugs included a SA-ER tablet containing 500 mg of aceneuramic acid and a placebo. Both tablets were white, film-coated, and indistinguishable from each other. The patients were orally given four SA-ER (2 g/each time) or placebo tablets, three times daily (in the morning, early evening, and at bedtime) (SA-ER 6 g/day) after meals (within 30 min after eating meals or snacks) for a period of 48 weeks.
Evaluation and survey contents
The evaluation schedule used in this study is shown in Supplementary Table 1. Details regarding age, sex, height, weight, present and past medical history, complications, pretreatment drugs, concomitant medications, and combination therapy were collected. Blood and urine sampling were performed before the start of administration and before administration at Week 8, 16, 24, 32, 40, and 48 (or at the early termination). Serum-free aceneuramic acid (trough value), urinary total, free and bound aceneuramic acid concentrations (creatinine correction; trough values) as well as urinary ManNAc concentration (to determine the presence or absence of ingested ManNAc) were measured by triple-stage quadrupole mass spectrometry (LC-API/MS/MS), which was performed by Intertek Pharmaceutical Services. Efficacy measurements included muscle strength composite scores (UEC and LEC scores) [13, 14], the GNE myopathy functional activity scale (GNEM-FAS) [15], sit-to-stand test [16, 17], weighted arm lift test [16], and 6 MWT [18]. Muscle strength was measured using hand held dynamometer. UEC is the sum of muscle strength in upper limbs. The GNEM-FAS is a GNE myopathy-specific measure developed to assess the functional impact of changes in muscle strength. The scale consists of 3 domains; upper extremity, mobility, and self-care, and scores for each domain and a total score will be obtained. The GNEM-FAS was administered at baseline and Week 8, 16, 24, 32, 40, and 48 (or at early termination) to evaluate physical functioning. Muscle strength and walking distance in the 6 MWT were determined based upon the predicted standard values for healthy adults, which were ascertained by gender, age, weight, and height (depending on the parameters); the percentages of these measurements were also calculated [19–23]. Efficacy parameters were measured every 8 weeks from the start of administration until the 48th week [24, 25]. Furthermore, the Individual Neuromuscular Quality of Life Questionnaire (INQoL) [26] was used along with measurements of clinical global impression [27, 28] and serum creatinine kinase (CK) levels. The Columbia Suicide Severity Rating Scale (C-SSRS) [29] regarding suicide ideation/attempt, vital signs, ECG, clinical examination, interval-clinical history, and adverse events were monitored as safety measures. In addition, the medication status, presence or absence of overdose, pregnancy tests, and partner’s pregnancy were recorded.
Evaluation criteria
The primary endpoint was the change in the total score of the UEC score from the start of administration to the final evaluation time point, which was also employed as the primary endpoint in the international phase II and III studies [13, 14]. As the effect of the drug was expected to be small in each muscle from the results of the international phase II clinical trials that preceded the current study, UEC using the dynamometer was considered the appropriate primary endpoint. Secondary endpoints were changes in GNEM-FAS scores in the upper extremity domain and mobility domain, lower extremity composite (LEC) score, sit-to-stand test score (30-second), Number of lifts in the weighted arm lift test (30-second), muscle strength and its percentage relative to the predicted standard value of knee extensors, and 6 MWT at the time of final evaluation. The tertiary endpoints were changes in the percentages of the number of UEC and LEC scores relative to their predicted standard values, muscle strength and the percentages relative to the predicted standard values of individual muscle groups in the upper and lower extremities, self-care domain and total GNEM-FAS scores, clinical global impression (CGI) score, total score of the nine health-related items in the INQoL score, CK concentration, serum free aceneuramic acid concentration (trough value), and urinary aceneuramic acid concentration (free, total, and bound: creatinine correction; trough value) at the time of final evaluation.
The severity of adverse events was evaluated using or according to the National Cancer Institute Common Terminology Criteria for Adverse Events ver. 4.03 (NCI CTCAE).
Statistical analysis
Regarding demographic and baseline characteristics the frequency distribution (%) for categorized items and summary statistics for continuous data were calculated. The full analysis set (FAS) was used for the efficacy analysis excluding ineligible cases, those untreated with investigational drugs, and those from which efficacy information after the start of the study was not obtained.
For primary, secondary, and tertiary endpoints, the summary statistics and their 95% confidence intervals (CI) of the mean changes from the start of administration to the final evaluation time point in each dose group were calculated. Last Observation Carried Forward (LOCF) was used as the final evaluation point. Covariance analysis was performed with the changes at the time of the final evaluation as response variables, the pre-administration values as covariates, and administration group, gender, and the total distance walked (meters) in 6 MWT (<200 m, ≥200 m) as explanatory variables.
Generalized estimating equation (GEE) [13, 14] repeated measures analysis with the change from the start of administration as response variable, as a covariate, and with administration group, time point, gender, and 6 MWT (<200 m, ≥200 m) as fixed effect was employed. Missing data in individual patients was treated as missing.
In the safety assessment, all adverse events were encoded using the Medical Dictionary for Regulatory Activities (MedDRA/J Ver. 20.0) drug regulation glossary. The frequency of adverse events as well as their rates were summarized.
Sample size and statistical power
To collect as much information as possible from the SA-ER group, the ratio of subjects receiving SA-ER to placebo was set to be 4:1. The planned total number of subjects was 15 (SA-ER 12, placebo 3). The power that can evaluate the similarity to the results of the international phase III study was calculated as follows.
1) The change in UEC score across the SA-ER groups of both the present study and the international phase III study [14] was assumed to be 2 kg, the SD to be 6 kg, and the number of subjects to be 12 and 40 in the two studies, respectively. The probability of the change in UEC score in the present study being below the lower limit of 95% CI of the change in UEC score in the international phase III study was calculated to be 85%. 2) In the present study, the change in UEC score was assumed to be 2 kg in the SA-ER group and –3 kg in the placebo group, and the SD to be 6 kg. The probability of the point estimate of the change in UEC score in the SA-ER group being over that in the placebo group was calculated to be 91%.
Data availability
All participant data, study protocol, and statistical analysis plan will be provided upon request.
RESULTS
Figure 1 presents CONSORT flow diagram of this clinical trial. Among the 21 patients who were provisionally registered during the start of the clinical trial, 20 were finally selected and randomly assigned to two groups (16 in the SA-ER group and 4 in the placebo group). One patient in the SA-ER group discontinued because they turned out to be pregnant at 2 weeks after the start of drug administration. The nineteen patients who completed the trial formed the FAS.
Fig. 1
CONSORT flow diagram. Twenty patients were finally selected and randomly assigned to two treatment groups (16 in the SA-ER group and 4 in the placebo group).
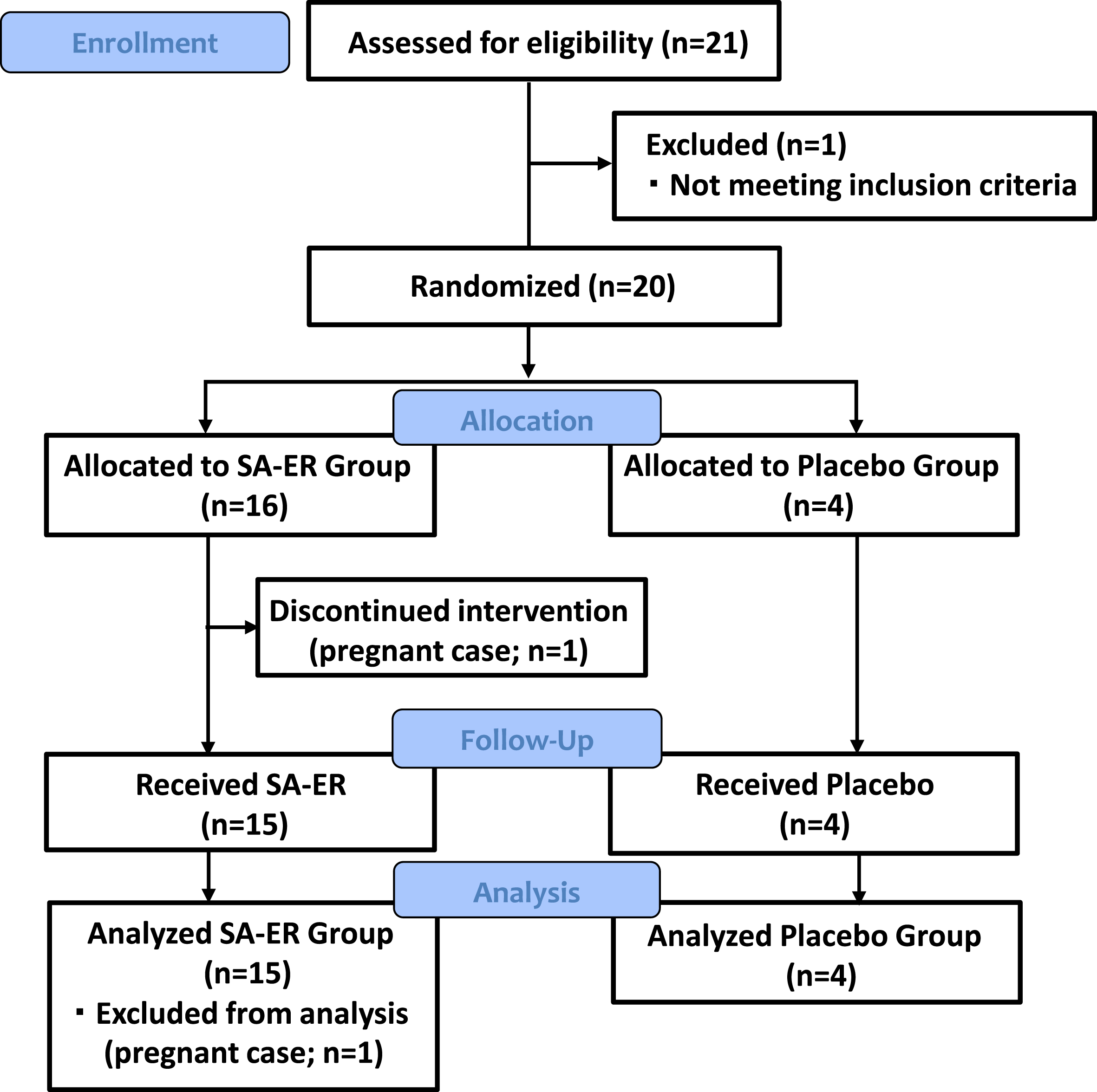
Table 1 and Supplementary Table 2 show the demographic characteristics and other baseline characteristics at the start of administration in the FAS. The pre-UEC value was 31.5±14.6 kg in the SA-ER group and 46.0±22.7 kg in the placebo group (P = 0.131; t-test). No differences (t-test or χ2 test; P≥0.15) in the other items were noted between the two groups. The ratio of males to females was 5:10 in the SA-ER group and 2:2 in the placebo group, respectively. The number of females in the SA-ER group was higher but with no significance (P = 0.539; χ2 test). Six males and 12 females (SA-ER, 14; placebo, 4) were able to walk ≥200 m in 6 minutes in the current study (Table 1). Any significant co-morbidities were not reported.
Table 1
Demographic and baseline characteristics (selected: Full analysis set is in supplementary table 2)
| Characteristic | SA-ER 6 g/day | Placebo | Total | t-test or | ||
| n = 15 | n = 4 | n = 19 | χ2 –test | |||
| Age(years) | mean±SD | 41±7 | 37±1 | 40±6 | 0.238 | |
| median(min,max) | 42(26, 48) | 37(35, 38) | 42(26, 48) | |||
| Sex | male | n(%) | 5(33.3) | 2(50.0) | 7(36.8) | 0.539 |
| Female | 10(66.7) | 2(50.0) | 12(63.2) | |||
| Height(cm) | mean±SD | 164.5±8.2 | 169.7±11.3 | 165.6±8.8 | 0.307 | |
| median(min,max) | 164.2(153.4, 177.6) | 168.7(158.7, 182.8) | 164.2(153.4, 182.8) | |||
| Weight(kg) | mean±SD | 60.6±15.8 | 63.0±10.5 | 61.1±14.6 | 0.781 | |
| median(min,max) | 60.0(39.7, 94.1) | 65.1(48.5, 73.4) | 63.8(39.7, 94.1) | |||
| Age at symptom onset(years) | mean±SD | 30±7 | 27±1 | 29±6 | 0.450 | |
| median(min,max) | 30(13, 40) | 27(26, 27) | 27(13,40) | |||
| Disease duration(years) | mean±SD | 11±5 | 10±1 | 11±5 | 0.618 | |
| median(min,max) | 10(5, 22) | 11(8, 11) | 10(5, 22) | |||
| 6-minutes walking test | mean±SD | 323.4±104.1 | 281.0±65.0 | 314.5±97.2 | 0.454 | |
| median(min,max) | 324.0(140.0, 507.4) | 273.5(210.0, 367.0) | 296.3(140.0,507.4) | |||
| <200 m | n(%) | 1 (6.7) | 0 (0.0) | 1 (5.3) | ||
|
| 14 (93.3) | 4 (100.0) | 18 (94.7) | 0.596 | ||
| 6-minutes walking test(% predicted) | mean±SD | 46.8±15.6 | 38.7±7.1 | 45.1±14.5 | 0.339 | |
| median(min,max) | 45.3(19.3,70.0) | 38.1(30.7, 48.0) | 41.0(19.3, 70.0) | |||
| Sit-to-stand test | mean±SD | 11.0±6.6 | 12.8±3.9 | 11.4±6.1 | 0.623 | |
| median(min,max) | 12(0, 19) | 12(9, 18) | 12(0, 19) | |||
| Weighted arm lift test | mean±SD | 24.8±9.9* | 23.8±18.5 | 24.5±11.8 | 0.890 | |
| median(min,max) | 23.5(10, 46.5) | 25(0, 45) | 24.8(0.0, 46.5) | |||
| Serum free aceneuramic acid (μg/mL) | mean±SD | 0.150±0.058 | 0.149±0.029 | 0.150±0.052 | 0.972 | |
| median(min,max) | 0.140(0.106, 0.335) | 0.152(0.118, 0.175) | 0.140(0.106, 0.335) | |||
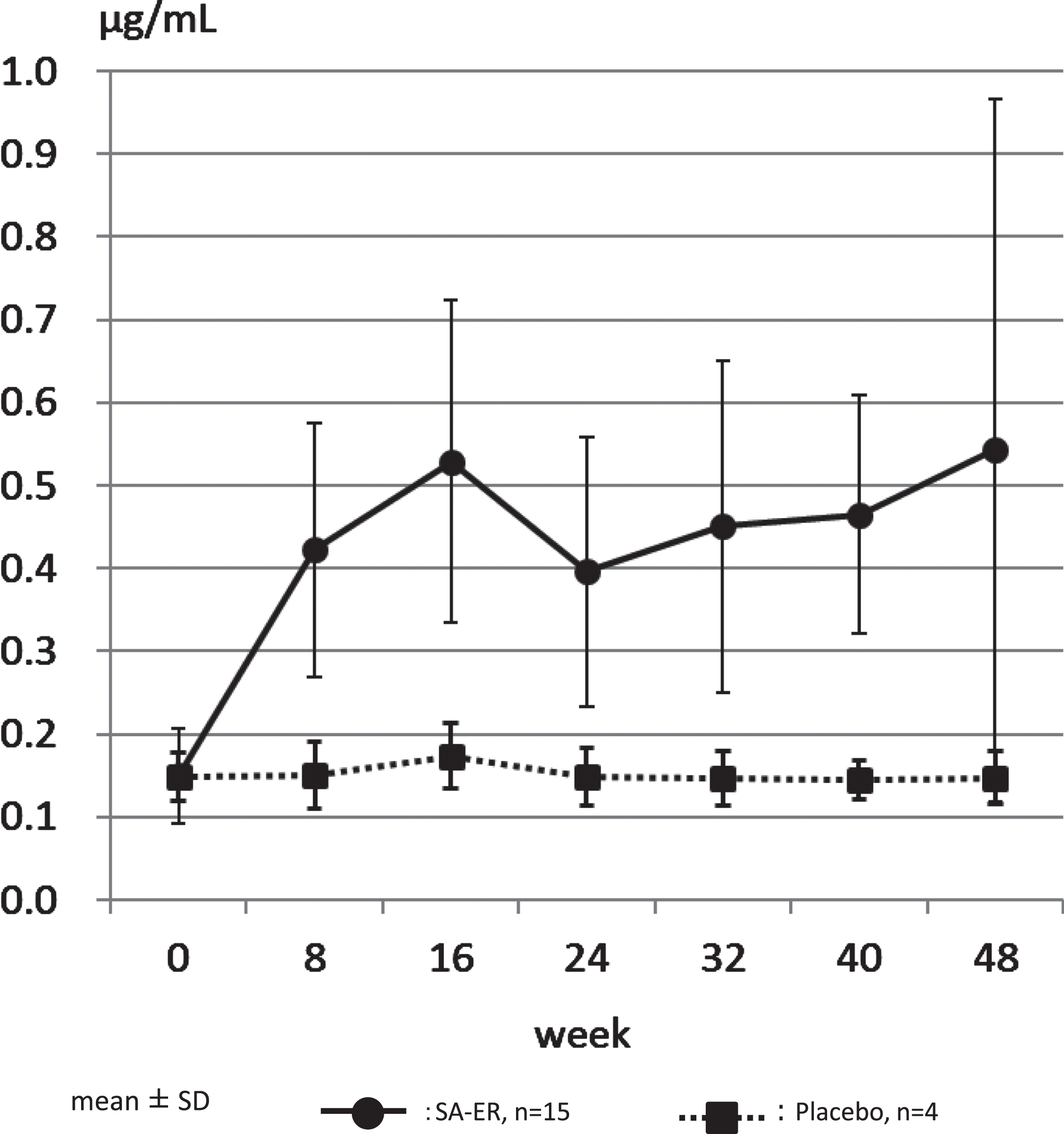
The medication compliance rate was high, with all cases presenting with >70%, which was the highest category of evaluation. Confirming this, the concentration of serum free aceneuramic acid measured was 0.150±0.058μg/mL (mean±SD) before administration and 0.543±0.425μg/mL after 48 weeks of administration in the SA-ER group (n = 15), compared to 0.149±0.029 and 0.147±0.031μg/mL before and after 48 weeks of administration, respectively, in the placebo group (n = 4) (Table 1, Fig. 2, Supplementary Table 2). The levels of serum free aceneuramic acid in the 15 patients of the SA-ER group after 48 weeks were more than three times those in the non-administration period.
Fig. 2
Serum free aceneuramic concentration. The concentration of serum free aceneuramic acid measured was 0.150±0.058μg/mL (mean±standard deviation) before administration and 0.543±0.424μg/mL after 48 weeks of administration in the SA-ER group (solid line; n = 15). In comparison, 0.149±0.029 and 0.147±0.031μg/mL before and after 48 weeks of administration, respectively, in the placebo group (dotted line; n = 4).
Efficacy evaluation
Primary endpoint
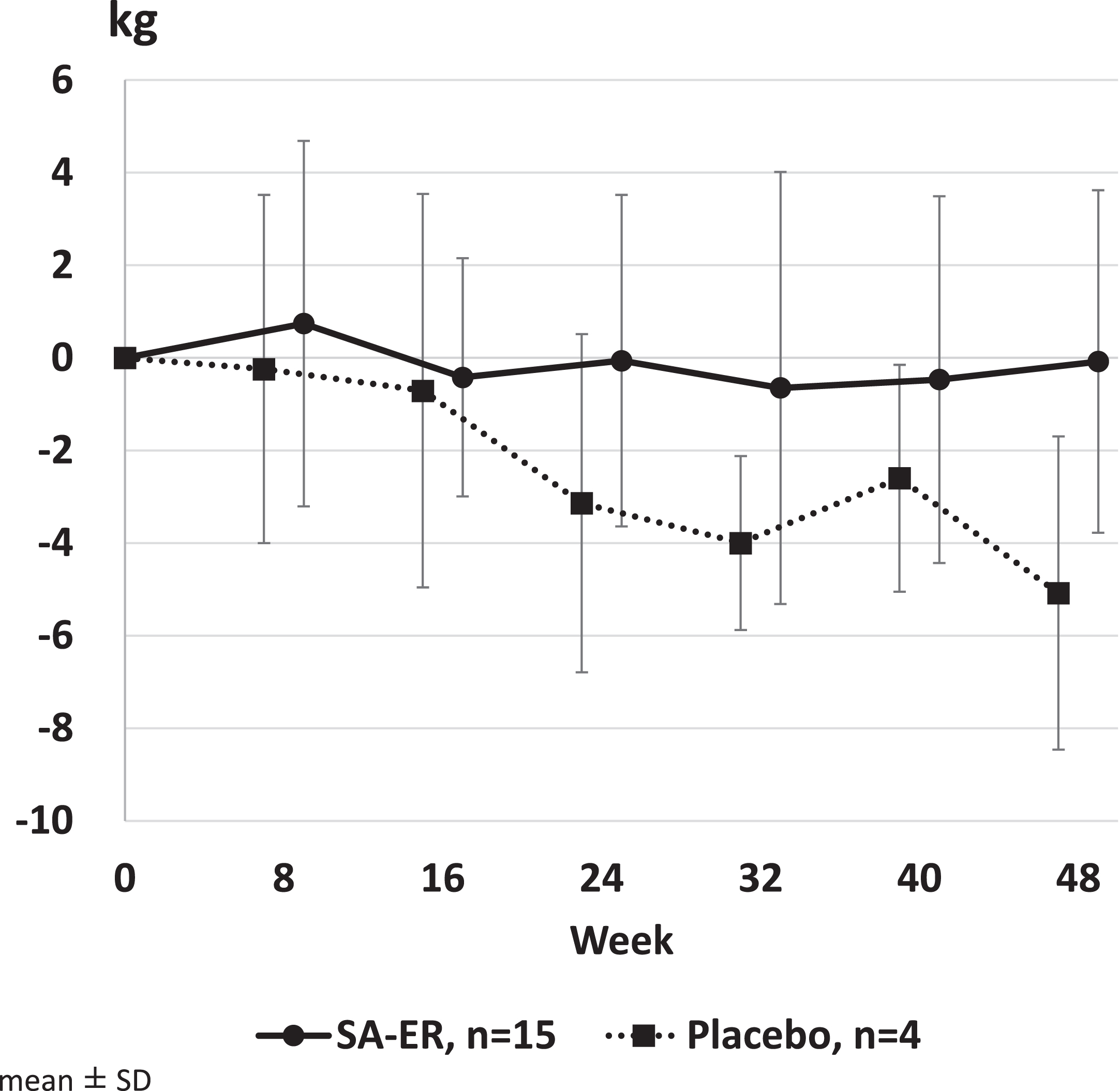
The mean value (95% CI) of UEC score change was –0.1 kg (–2.1 to 2.0) in the SA-ER group and –5.1 kg (–10.4 to 0.3) in the placebo group (Table 2). Covariance analysis revealed that the least squares mean (95% CI) of score change after 48 weeks of treatment was –1.4 kg (–5.9 to 3.1) in the SA-ER group and –6.2 kg (–12.4 to 0.1) in the placebo group, with a difference of 4.8 kg (–0.3 to 9.9; P = 0.0635) between the two groups. Figure 3 shows the transition of the change in UEC scores before and after administration in each group. GEE repeated measures analysis showed that the estimated values of change (95% CI) at 48 weeks were –1.0 kg (–2.5 to 0.5) in the SA-ER group and –5.7 kg (–8.0 to –3.4) in the placebo group, with a significant difference of 4.7 kg (1.8 to 7.5; P = 0.0013) between the two groups. Thus, the UEC scores decreased more in the placebo group compared with the SA-ER group during the study.
Table 2
Primary and secondary endpoints
| SA-ER | Placebo | |||||
| mean±SD (n) | 95% confidence interval of mean | median(min,max) | mean±SD (n) | 95% confidence interval of mean | median(min,max) | |
| Primary endpoint | ||||||
| UEC score | –0.1±3.7(15) | (–2.1, 2.0) | –0.4(–5.2, 7.6) | –5.1±3.4(4) | (–10.4, 0.3) | –4.8(–9.3, –1.5) |
| Secondary endpoints | ||||||
| GNEM-FAS mobility domain | –0.6±2.8(15) | (–2.2, 1.0) | 0.0(–7, 5) | 0.5±1.7(4) | (–2.3, 3.3) | 1.0(–2, 2) |
| GNEM-FAS upper extremity domain | 0.9±2.8(15) | (–0.7, 2.4) | 0.0(–2, 8) | –0.8±1.7(4) | (–3.5, 2.0) | –0.5(–3, 1) |
| LEC score | 2.1±9.2(14) | (–3.2, 7.4) | 3.8(–15.7, 16.9) | –3.8±4.5(4) | (–10.9, 3.3) | –1.8(–10.5, –1.1) |
| Sit-to-stand test | 1.9±2.0(15) | (0.7, 3.0) | 2.0(–1, 5) | 1.5±1.9(4) | (–1.5, 4.5) | 2.0(–1, 3) |
| Weighted arm lift test | 2.1±6.2(12) | (–1.8, 6.1) | 1.5(–6.5, 14.0) | 1.5±5.8(3) | (–12.8, 15.8) | –0.5(–3.0, 8.0) |
| Knee extensors | 1.2±4.3(14) | (–1.3, 3.7) | 1.3(–6.5, 10.4) | –1.9±4.8(4) | (–9.5, 5.7) | –2.7(–6.0, 3.9) |
| Knee extensors (% predicted) | 2.5±12.8 (14) | (–4.9, 9.9) | 3.5(–25.4, 27.4) | –4.9±11.1(4) | (–22.5, 12.7) | –5.1(–17.1, 7.6) |
| Six-minutes walk test | –19.3±30.3(15) | (–36.1, –2.5) | –20.8(–96.0, 27.2) | 4.5±23.5(4) | (–32.9, 42.0) | 1.4(–20.8, 36.0) |
| Six-minutes walk test(% predicted) | –2.7±4.1(15) | (–4.9, –0.4) | –2.8(–12.1, 3.8) | 0.5±3.2(4) | (–4.6,5.6) | 0.2(–3.0, 4.7) |
*1) Use baseline data as covariate. Also treatment group, sex and 6MWT as explanatory variables. *2) Use baseline data as covariate. Also treatment group, time point, sex and 6MWT as fixed effects.
Fig. 3
Change in upper extremity composite (UEC) score. GEE repeated measures analysis showed that the estimated values of change (95% CI) at 48 weeks were –1.0 kg (–2.5 to 0.5) in the SA-ER group and –5.7 kg (–8.0 to –3.4) in the placebo group, with a difference of 4.7 kg (1.8 to 7.5; P = 0.0013) between the two groups.
Secondary and tertiary endpoints
The mean values of changes at the final evaluation time point and the 95% CI for each dosage group with regards to secondary and tertiary endpoints are shown in Table 2 and Supplementary Table 3. The covariance analysis was performed in the same way as that for the primary endpoint. In most of the secondary and tertiary endpoints, the SA-ER group showed a better tendency than the placebo group; moreover, significant differences in the muscle strength of shoulder abductors and the percentage of the predicted standard value thereof were noted.
Changes over time in GNEM-FAS scores, lower extremity composite score, muscle strength of knee extensors, sit-to-stand test, weighted arm lift test, INQoL, and CK before administration to after drug administration between the groups are shown in Supplementary Figures 1–5.
Safety evaluation
A total of 67 incidences of adverse events were observed in 14 out of 16 cases (87.5%) in the SA-ER group and 10 incidences in 3 out of 4 cases (75.0%) in the placebo group (Supplementary Table 4). On the basis of System Organ Class (SOC), infections and infestations were the most common in both groups at 75.0%, with gastrointestinal disorder at 31.3% and 50.0% in the SA-ER group and placebo group, respectively. Although these events were considered mild or moderate, the number of cases was higher in the SA-ER group compared with the placebo group. Adverse reactions (events judged to have a causal relationship with the investigational drug, probably and possibly related) in the SA-ER group included angular cheilitis, headache, constipation, rash, pain in extremity, and fetal death, while in the placebo group, stomatitis was observed (Table 3).
Table 3
Adverse reactions during the period of clinical trial
| SA-ER n = 16 | Placebo n = 4 | |||||
| no. of events | no. of subjects | % | no. of events | no. of subjects | % | |
| Total | 6 | 4 | 25 | 1 | 1 | 25 |
| Infections and infestations | 1 | 1 | 6.3 | 0 | 0 | 0 |
| Angular cheilitis | 1 | 1 | 6.3 | 0 | 0 | 0 |
| Nervous system disorder | 1 | 1 | 6.3 | 0 | 0 | 0 |
| Headache | 1 | 1 | 6.3 | 0 | 0 | 0 |
| Gastrointestinal disorder | 1 | 1 | 6.3 | 1 | 1 | 25 |
| Constipation | 1 | 1 | 6.3 | 0 | 0 | 0 |
| Stomatitis | 0 | 0 | 0 | 1 | 1 | 25 |
| Skin and subcutaneous tissue disorders | 1 | 1 | 6.3 | 0 | 0 | 0 |
| Rash | 1 | 1 | 6.3 | 0 | 0 | 0 |
| Musculoskeletal and connective tissue disorders | 1 | 1 | 6.3 | 0 | 0 | 0 |
| Pain in extremity | 1 | 1 | 6.3 | 0 | 0 | 0 |
| Pregnancy, puerperium and perinatal conditions | 1 | 1 | 6.3 | 0 | 0 | 0 |
| Fetal death | 1 | 1 | 6.3 | 0 | 0 | 0 |
The trial was initiated in one pregnant patient because a urine pregnancy test was negative at the start of administration; the drug was terminated when she was found to be pregnant about 2 weeks later. It is likely that the patient was pregnant at the start of administration. Fetal growth was good until 13 weeks of pregnancy, but fetal death was confirmed 112 days after the start of administration. The umbilical cord was wrapped around the fetus’ neck. The fetus was removed shortly afterward, and the patient recovered without any problems. This event occurred in one patient from the SA-ER group, and the causality was thought to be low.
DISCUSSION
This study met its primary efficacy outcome measure and showed a significant improvement in the change from baseline to 48-week observational period between the SA-ER and placebo groups. GEE repeated measures analysis, which was used to compare the effect of SA-ER and placebo in the previous studies [13, 14], revealed rather preserved muscle strength in the SA-ER group than in the placebo group at Week 48 with a significant difference of 4.7 kg (1.8 to 7.5; P = 0.0013) between the two groups. A covariance analysis showed a numerically smaller change in UEC at Week 48 in the SA-ER group compared with the placebo group, although the difference did not reach the significant level. This may be due to insufficient statistical power from the small sample size in this study. Evaluation of the secondary and tertiary endpoints supported the favorable effect of SA-ER in the primary endpoint, with the numerical values of most items higher in the SA-ER group compared with the placebo group.
Although the results of this study indicated an efficacy similar to that in the international Phase II study [13], the findings are different from those in the international Phase III study [14], which ended in negative results. The international Phase III SA-ER study enrolled 89 adults with GNE myopathy able to walk ≥200 meters in the 6 MWT. Patients were randomized 1:1 to SA-ER at a dose of 6 g/day or a placebo for 48 weeks. The study did not meet the primary endpoint of demonstrating a statistically significant improvement in UEC score (+0.74 kg, P = 0.5387) for SA-ER treated patients (n = 45, –2.25 kg) compared to the placebo (n = 43, –2.99 kg) patients for the change from baseline to 48 weeks [14]. A comparative analysis of detailed data between the studies is difficult. However, a possible reason for the discrepancy could be difference in patient background variation. The international Phase II trial was conducted only in two countries (the United States and Israel) and the current trial only in Japan, while the international Phase III trial was conducted widely by the joint faculties of United States, Israel, Bulgaria, France, Canada, Italy, and the UK. Therefore, variation in ethnicity and possibly in genetic background in the international Phase III trial was larger compared to that in the phase II study and the present study. Interestingly, a natural history study suggests ethnic differences in disease severity and progression, with Bulgarians having more severe symptoms and faster progression [30]. It appears likely that a relatively homogenous genetic background may have contributed to revealing a benefit in the present study.
One of the problems of multi-center trials is inter-institution and inter-investigator variation in measurement and evaluation of outcomes. In this regard, a limited number of well-trained institutions and investigators participating in a multi-center trial may have the advantage of minimizing such variations. It may be particularly important for a disease presenting with substantial individual differences in pathology and progression such as GNE myopathy. Overall, it is considered that minimizing dispersion in UEC score will result in an increase in statistical power to detect a significant difference between the SA-ER and placebo groups.
Further, the decrease in UEC score to Week 48 (about 3 kg) in the placebo group in the international phase III study was clearly smaller than that in the present study (about 5 kg) and below the originally assumed difference (5 kg) between the SA-ER and placebo groups. The unexpectedly small decrease in UEC score in the placebo group probably diminished the power to detect a difference between treatment groups. In addition, serum free aceneuramic acid concentration at Week 48 was lower in the international phase III study for unknown reasons, which might be associated with the lower effect of SA-ER observed in the study.
Overall, aceneuramic acid was well tolerated without any serious adverse effects considered to have an apparent causal relationship. Fetal death was observed as a serious adverse event in one patient belonging to the SA-ER group. Although a relationship with the investigational drug could not be denied, it was thought that the relevance was low because reproductive and developmental toxicity were not observed in the preclinical tests of aceneuramic acid. Likewise, findings suggestive of mutagenicity and chromosomal aberration induction were not observed in the genotoxicity test of aceneuramic acid. Moreover, no abnormalities were detected in a chromosomal examination of the dead fetus. The investigational drug administration period was 2 weeks, and the growth of the fetus was good at 9 weeks after discontinuation of the drug. According to the obstetrician and gynecologist at the time of fetal removal, the umbilical cord was tangled around the neck of the fetus. Owing to the absence of any other serious adverse events, we believe that this drug is safe for use in a clinical setting.
This study has some limitations. First, the number of patients is limited (15 SA-ER and 4 placebo). The current investigator-initiated Phase II/III trial targeting GNE myopathy invited these patients with the cooperation of PADM and REMUDY. At the time of registration, a total of 121 cases in Japan have been recorded in the REMUDY database. To avoid the variance of evaluation among countries or ethnicity, we designed the trial independently from the global clinical trial with the tradeoff of the limitation of the size of the study. However, the small size may have reduced the statistical power to detect the significant efficacy of SA-ER in the covariance analysis. In addition, in order to collect as much information as possible from the SA-ER group and based on ethical consideration, the assigned number of patients in the SA-ER group was four times that of the placebo group. The unequal randomization ratio (4:1) may have also reduced the statistical power.
Second, to evaluate walking ability as well as UECs we decided to include as many patients as possible who could walk >200 m in the 6 MWT. As a result, 20 of the 21 registered cases qualified for participation, 19 of whom were able to walk >200 m at the start of administration. Comparatively mild patients were evaluated in this study. The effect in patients at an advanced stage (e.g. wheelchair bound) cannot be warranted. Finally, as GNE myopathy progresses slowly over a long period of time with large individual differences, the long-term effect of SA-ER also needs to be elucidated. A subsequent extension and confirmation study is warranted to ascertain whether there is comprehensive effectiveness.
CONCLUSION
In the present study, SA-ER tablets were orally administered (6 g/day for 48 weeks) to patients with GNE myopathy in Japan. Disease progression in the total value of upper limb muscle strength was suppressed. As side effects of the drug did not appear to be of major concern, SA-ER administration could be beneficial for GNE myopathy.
ACKNOWLEDGMENTS
We thank all the patients and their families who participated in this clinical trial, Registry of Muscular Dystrophy (REMUDY), and Patients Association for Distal Myopathies (PADM). We thank Clinical Research Innovation and Education Center Tohoku University (CRIETO) for their help in scheduling during the study. We also sincerely appreciate the support from Dr. Posner of Columbia University for permitting our us free use of the Colombian Suicide Severity Rating Scale in this trial. Medical writing of this paper was supported by Tetsuyoshi Inoue, Ph.D (SunFlare Co., Ltd., Tokyo, Japan) and the support was funded by Nobelpharma Inc., Ltd.
This study was supported by a grant for Research on Rare and Intractable Diseases (H27,28,29-intractable disease) from the Japan Agency for Medical Research and Development (AMED; 16ek0109085h0002, 17ek0109085h0003). The investigational drug used for the trials was were provided by Nobelpharma Inc., Ltd., and subsidized by the Japan Agency for Medical Research and Development as a Project Promoting Support for Drug Discovery “Support Program for Orphan drug prior to the Designation.”
CONFLICT OF INTEREST
Ichizo Nishino is an Editorial Board Member of this journal, but was not involved in the peer-review process nor had access to any information regarding its peer-review.
REFERENCES
[1] | Argov Z , Yarom R . “Rimmed vacuole myopathy” sparing the quadriceps. A unique disorder in Iranian Jews. J Neurol Sci. (1984) ;64: (1):33–43. |
[2] | Mizusawa H , Inoue K , Toyokura Y , Nakanishi T . Distribution of skeletal muscle involvement in distal myopathy with rimmed vacuoles: A clinical and computed tomographic study. Rinsho Shinkeigaku. (1986) ;26: (11):1174–81. |
[3] | Mizusawa H , Nakano I , Inoue K , Takagi A , Mannen T . Distal myopathy. A variety characterized by prominent vacuolar degeneration of muscle. Shinkeinaika. (1980) ;12: :40–7. |
[4] | Nonaka I , Sunohara N , Ishiura S , Satoyoshi E . Familial distal myopathy with rimmed vacuole and lamellar (myeloid) body formation. J Neurol Sci. (1981) ;51: (1):141–55. |
[5] | Nonaka I , Sunohara N , Satoyoshi E , Terasawa K , Yonemoto K . Autosomal recessive distal muscular dystrophy: A comparative study with distal myopathy with rimmed vacuole formation. Ann Neurol. (1985) ;17: (1):51–9. |
[6] | Sunohara N , Nonaka I , Kamei N , Satoyoshi E . Distal myopathy with rimmed vacuole formation. A follow-up study. Brain. (1989) ;112: :65–83. |
[7] | Mori-Yoshimura M , Hayashi YK , Yonemoto N , Nakamura H , Murata M , Takeda S , et al. Nationwide patient registry for GNE myopathy in Japan. Orphanet J Rare Dis. (2014) ;9: :150. |
[8] | Eisenberg I , Avidan N , Potikha T , Hochner H , Chen M , Olender T , et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. (2001) ;29: (1):83–7. |
[9] | Malicdan MC , Noguchi S , Hayashi YK , Nonaka I , Nishino I . Prophylactic treatment with sialic acid metabolites precludes the development of the myopathic phenotype in the DMRV-hIBM mouse model. Nat Med. (2009) ;15: (6):690–5. |
[10] | Malicdan MC , Noguchi S , Nonaka I , Hayashi YK , Nishino I . A Gne knockout mouse expressing human GNE D176V mutation develops features similar to distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Hum Mol Genet. (2007) ;16: (22):2669–82. |
[11] | Suzuki N , Kato M , Warita H , Izumi R , Tateyama M , Kuroda H , Asada R , Suzuki A , Yamaguchi T , Nishino I and Aoki M . Phase I clinical trial results of aceneuramic acid for GNE myopathy in Japan. Translational Medicine Communications. (2018) ;3: (7):7. |
[12] | Kakkis E MM , Shah P , Donikyan M , Ahmed R . UX001-CL101 - A Phase 1 Study to Evaluate the Safety and Pharmacokinetics of Single and Repeat Doses of Sialic Acid Extended-Release (SA-ER) Tablets in Patients with Hereditary Inclusion Body Myopathy (HIBM). World Muscle Society; Perth, Australia, 2012. |
[13] | Argov Z , Caraco Y , Lau H , Pestronk A , Shieh PB , Skrinar A , et al. Aceneuramic acid extended release administration maintains upper limb muscle strength in a 48-week study of subjects with GNE myopathy: Results from a phase 2, randomized, controlled study. J Neuromuscul Dis. (2016) ;3: (1):49–66. |
[14] | Lochmuller H , Behin A , Caraco Y , Lau H , Mirabella M , Tournev I , et al. A phase 3 randomized study evaluating sialic acid extended-release for GNE myopathy. Neurology. (2109) ;92: (18):e2109. |
[15] | Mayhew J , Bonner N , Arbuckle R , Turnbull A , Bowden A , Skrinar A . Development and preliminary evidence of the psychometric properties of the GNE myopathy functional activity scale. J Comp Eff Res. (2018) ;7: (4):381–95. |
[16] | Agarwal S , Kiely PD . Two simple, reliable and valid tests of proximal muscle function, and their application to the management of idiopathic inflammatory myositis. Rheumatology (Oxford). (2006) ;45: (7):874–9. |
[17] | Ozalevli S , Ozden A , Itil O , Akkoclu A . Comparison of the Sit-to-Stand Test with 6min walk test in patients with chronic obstructive pulmonary disease. Respir Med. (2007) ;101: (2):286–93. |
[18] | Laboratories ATSCoPSfCPF. ATS statement: Guidelines for the six-minute walk test. Am J Respir Crit Care Med. (2002) ;166: (1):111–7. |
[19] | Muscular weakness assessment: Use of normal isometric strength data. The National Isometric Muscle Strength (NIMS) Database Consortium. Arch Phys Med Rehabil. (1996) ;77: (12):1251–5. |
[20] | Bohannon RW . Reference values for extremity muscle strength obtained by hand-held dynamometry from adults aged 20 to 79 years. Arch Phys Med Rehabil. (1997) ;78: (1):26–32. |
[21] | Gibbons WJ , Fruchter N , Sloan S , Levy RD . Reference values for a multiple repetition 6-minute walk test in healthy adults older than 20 years. J Cardiopulm Rehabil. (2001) ;21: (2):87–93. |
[22] | Peters MJ , van Nes SI , Vanhoutte EK , Bakkers M , van Doorn PA , Merkies IS , et al. Revised normative values for grip strength with the Jamar dynamometer. J Peripher Nerv Syst. (2011) ;16: (1):47–50. |
[23] | Stoll T , Huber E , Seifert B , Michel BA , Stucki G . Maximal isometric muscle strength: Normative values and gender-specific relation to age. Clin Rheumatol. (2000) ;19: (2):105–13. |
[24] | Aitkens S , Lord J , Bernauer E , Fowler WM Jr, Lieberman JS , Berck P . Relationship of manual muscle testing to objective strength measurements. Muscle Nerve. (1989) ;12: (3):173–7. |
[25] | Sisto SA , Dyson-Hudson T . Dynamometry testing in spinal cord injury. J Rehabil Res Dev. (2007) ;44: (1):123–36. |
[26] | Vincent KA , Carr AJ , Walburn J , Scott DL , Rose MR . Construction and validation of a quality of life questionnaire for neuromuscular disease (INQoL). Neurology. (2007) ;68: (13):1051–7. |
[27] | Busner J , Targum SD . The clinical global impressions scale: Applying a research tool in clinical practice. Psychiatry (Edgmont). (2007) ;4: (7):28–37. |
[28] | W G. The Clinical Global Impression Scale. ECDEU Assessment Manual for Psychopharmacology-Revised. Rockville, MD: US Dept. of Health, Education and Welfare, ADAMHA, MIMH Psychopharmacology Research Branch; 1976, pp. 218-22. |
[29] | Posner K , Brown GK , Stanley B , Brent DA , Yershova KV , Oquendo MA , et al. The Columbia-Suicide Severity Rating Scale: Initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. (2011) ;168: (12):1266–77. |
[30] | Lochmuller H , Behin A , Tournev I , Tarnopolsky M , Horvath R , Pogoryelova O , et al. Results from a 3-year Non-interventional, observational disease monitoring program in adults with GNE myopathy. J Neuromuscul Dis. (2021) ;8: (2):225–34. |


