
Development of Assays to Measure GNE Gene Potency and Gene Replacement in Skeletal Muscle
Abstract
Background:
GNE myopathy (GNEM) is a severe muscle disease caused by mutations in the UDP-GlcNAc-2-epimerase/ManNAc-6-kinase (GNE) gene, which encodes a bifunctional enzyme required for sialic acid (Sia) biosynthesis.
Objective:
To develop assays to demonstrate the potency of AAV gene therapy vectors in making Sia and to define the dose required for replacement of endogenous mouse Gne gene expression with human GNE in skeletal muscles.
Methods:
A MyoD-inducible Gne-deficient cell line, Lec3MyoDI, and a GNE-deficient human muscle cell line, were made and tested to define the potency of various AAV vectors to increase binding of Sia-specific lectins, including MAA and SNA. qPCR and qRT-PCR methods were used to quantify AAV biodistribution and GNE gene expression after intravenous delivery of AAV vectors designed with different promoters in wild-type mice.
Results:
Lec3 cells showed a strong deficit in MAA binding, while GNE–/–MB135 cells did not. Overexpressing GNE in Lec3 and Lec3MyoDI cells by AAV infection stimulated MAA binding in a dose-dependent manner. Use of a constitutive promoter, CMV, showed higher induction of MAA binding than use of muscle-specific promoters (MCK, MHCK7). rAAVrh74.CMV.GNE stimulated human GNE expression in muscles at levels equivalent to endogenous mouse Gne at a dose of 1×1013vg/kg, while AAVs with muscle-specific promoters required higher doses. AAV biodistribution in skeletal muscles trended higher when CMV was used as the promoter, and this correlated with increased sialylation of its viral capsid.
Conclusions:
Lec3 and Lec3MyoDI cells work well to assay the potency of AAV vectors in making Sia. Systemic delivery of rAAVrh74.CMV.GNE can deliver GNE gene replacement to skeletal muscles at doses that do not overwhelm non-muscle tissues, suggesting that AAV vectors that drive constitutive organ expression could be used to treat GNEM.
INTRODUCTION
GNE myopathy (GNEM) is a severe muscle disease characterized by progressive muscle weakness and muscle atrophy [1]. Patients with GNEM typically begin to have muscle weakness in the third or fourth decade of life, often rendering patients non-ambulatory due to extensive muscle wasting by the second decade after diagnosis [1]. Mitrani-Rosenbaum and colleagues first showed that GNEM is a recessive disease caused by mutations in the GNE gene [2]. GNE encodes the uridine diphosphate-N-acetylglucosamine(UDP-GlcNAc)-2-epimerase/N-acetylmannosamine(ManNAc)-6-kinase, a bifunctional enzyme required for synthesis of sialic acid (Sia) [3]. Common founder missense mutations in GNE exist in GNEM patients of Japanese and Middle Eastern descent [1, 4], including D207V and M743T respectively, though there are many other disease-causing mutations as well [5, 6]. GNE mutations that cause GNEM typically reduce GNE enzyme activity and are thought to cause disease by lowering Sia expression [1, 7], though there is considerable variability in enzyme activity as it relates to disease presentation [5].
Proof-of-concept studies to support clinical GNEM studies have largely involved oral glycan supplementation therapies, but these studies are complicated by the phenotypic variability of GNEM mouse models, where disease phenotypes can range from absent to severe within the same genetic strain [8–12]. Such studies have largely relied on testing in the GNED207VTgGne–/– mouse model [10], where human GNE bearing the D207V [4] mutation is constitutively expressed in the absence of endogenous mouse Gne, and GneM743T mice [11, 12], where the M743T disease-causing mutation has been placed in the Gne locus. Two oral glycan therapies have been tested in GNEM clinical trials, extended-release Sia (ACE-ER) and N-acetylmannosamine (ManNAc) [12–16]. ACE-ER therapy showed pre-clinical therapeutic effects in the GNED207VTgGne–/– mouse model [9, 15] but met no clinical milestones when tested in a placebo-controlled phase 3 clinical trial [17]. A more recent phase II/III clinical trial with fewer subjects, however, showed improvement in upper extremity composite strength score [18]. ManNAc supplementation showed pre-clinical effects with continuous administration over a one year period in GNED207VTgGne–/– mice [9, 14] and rescued perinatal lethality in GneM743T mice [12, 13]. ManNAc has been tested in one open label phase 2 clinical trial of 12 subjects, where it had no impact on 6-minute walk test results but showed some changes in other strength measures [19]. ManNAc is currently being tested in a placebo-controlled phase 2 clinical trial (NCT04231266).
The results of clinical studies on oral glycan therapy have highlighted the importance of developing a gene therapy for GNEM patients that may promise greater therapeutic efficacy. GNE gene delivery has been tested in a single patient using lipoplex DNA transfection [20]. This subject displayed evidence of GNE gene expression, some potential stabilization of muscle function, and modest improvement in muscle sialylation. Several studies have tested systemic delivery of GNE gene replacement using recombinant Adeno Associated Virus (rAAV) vectors in wild-type and Gne mutant mice [8, 21, 22]. To date, however, there are no assays developed to demonstrate the potency of AAV gene therapy vectors in making Sia, the ultimate product of GNE activity, and no study to determine the dose of human GNE gene therapy required to replace endogenous mouse muscle Gne gene expression. An AAV potency assay to demonstrate bioactivity of various production lots of AAV and their relative stability over time would be required for the clinical development of any AAV-based GNE gene therapy. Here, we have undertaken studies aimed at accomplishing these goals.
MATERIALS AND METHODS
Plasmid construction and AAV production
The full-length cDNA for the human GNE cDNA (GenBank: NM_005476.6) was synthesized to include a 5’ consensus Kozak sequence (GCCGCCACC) using Invitrogen GeneArt Gene Synthesis (Thermo Fisher Scientific; Waltham, MA). GNE was then cloned in the pAAV plasmid under control of a Cytomegalovirus (CMV), muscle creatine kinase (MCK), or MHCK7 promoter [8, 23–25]. Each vector also contained polyA signal following the transgene. Sanger sequencing was performed to confirm the sequence of the plasmids and the integrity of the inverted terminal repeats (ITRs) (Genewiz; South Plainfield, NJ). Packaged rAAV, rhesus 74 (rh74) serotype, was produced using the triple transfection method in HEK293 cells [26], followed by purification by iodixanol ultracentrifugation and anion exchange chromatography (Andelyn Biosciences; Columbus, OH) [27]. AAV titers were quantified by quantitative polymerase chain reaction (qPCR) against a linear standard.
Generation of GNE-deficient MB135 cells
Human immortalized myoblast MB135 cells [28] were grown in F-10 growth media (Gibco N6908; Grand Island, NY) with 20% FBS (R&D Systems S11150; Minneapolis, MN), 1μM dexamethasone (Sigma-Aldrich D4902; St. Louis, MO), 10 ng/mL basic fibroblast growth factor (Gibco PHG0026; Grand Island, NY), and 1X Antibiotic/Antimycotic Solution (Gibco A5955; Grand Island, NY). GNE-deleted (GNE–/–) MB135 cells were generated via CRISPR/Cas9 RNP nucleofection via the CRISPRevolution sgRNA EZ Kit (Synthego; Redwood City, CA) as per manufacturers’ instructions, utilizing a sgRNA targeting exon 3 of GNE (NM_001128227.3, sgRNA: 5’-AGGCUACACACAAUUGUGAG-3’). Nucleofection was performed using the Lonza 4D nucleofector system, using the SE cell line 4D-nucleofector kit (Lonza V4XC-1032; Basel, Switzerland), pulse code CA-137, 80k cells per nucleocuvette. Following nucleofection, single colonies were isolated, and gDNA was extracted using QuickExtract (LGC Biosearch Technologies QE09050; Middlesex, UK) as per manufacturers’ instructions. Successful insertion/deletion (indel) generation was confirmed through PCR amplification of the target region, followed by Sanger sequencing of the PCR product, confirming a homozygous insertion of A at hg38 genomic coordinates chr9:36,246,420 A >AA corresponding to NM_001128227.3:c.[320_321insT] and NP_001121699.1:p.(Arg108GlufsTer5).
Lectin binding assays in MB135 and GNE–/–MB135 cells
MB135 cells and GNE–/–MB135 cells were plated in a 96-well dish at 50,000 cells/well. After 24 hours in F-10 growth media, media was removed and replaced with Skeletal Muscle Differentiation Media (PromoCell C-23061; Heidelberg, Germany) with 1X Antibiotic/Antimycotic Solution (Gibco A5955; Grand Island, NY). Cells remained in Skeletal Muscle Differentiation Media for 5, 7, or 13 days, and cells were fed with new media every 3 days. Cells were rinsed once with PBS and were fixed with 4% paraformaldehyde (PFA) in PBS for 15 minutes at room temperature. Cells were blocked for an hour at room temperature with 2% fish gelatin in PBS. HRP-conjugated Maackia amurensis agglutinin (MAA, a combined mixture of types I and II), HRP-conjugated Sambucus nigra (SNA-1) agglutinin, HRP-conjugated Erythrina cristagalli agglutinin (ECA), or HRP-conjugated Arachis hypogaea agglutinin (also peanut agglutinin, PNA) (all from EY Laboratories; San Mateo, CA) were diluted at 1:500 in 2% fish gelatin in PBS, and cells were incubated for one hour with the appropriate lectin. Binding was developed by addition of 3,3’,5,5’-tetramethylbenzidine (TMB) solution (R&D Systems; Minneapolis, MN) for 15–20 minutes. Sulfuric acid (H2SO4) was added to stop the reaction, and plates were read on a microplate reader at 450 nm. Each assay condition was tested in triplicate for all experiments.
Generation of Lec3MyoDI cell lines
Pro-Lec3.4B cells (hereafter called Lec3 cells) were grown in Minimal Essential Media alpha (MEMα, Thermo Fisher Scientific; Grand Island, NY) supplemented with 10% fetal calf serum, penicillin (100U/mL), and streptomycin (100μg/mL) (Thermo Fisher Scientific; Grand Island, NY). 1.75×106 Lec3 cells were plated in a 10 cm dish, and 15μg of the pSINTREMyoD_hPGKrtTA2S_M221306_sv40_hygro plasmid was transfected using Mirus TransIT-X2 transfection reagent according to manufacturer’s instructions. After 72 hours, transfection media was removed and fresh growth media was added with 300μg/mL hygromycin B (Gold Biotechnology; St. Louis, MO). Cells were allowed to grow in growth media with 300μg/mL hygromycin B (hereafter called selection media) until death of all non-transfected cells had occurred and the remaining stably transfected cells became confluent. These cells are called Lec3MyoDI (MyoD-inducible) cells.
To subclone Lec3MyoDI cells, 100μL of selection media was added to each well of a 96-well plate. 200μL of Lec3MyoDI cell suspension was added at 2×104 cells/ml to each A1 well, and then 1:2 serial dilutions were made down the column from A1 to H1, with the volume in each well brought to 200μL. 1:2 serial dilutions were then made across each row, then the final volume of all wells was brought to 200μL using selection media. Wells with a single colony of cells were selected for expansion.
Immunofluorescence staining
Lec3, Lec3MyoDI, and CHO cells were plated at 2×104 cells/well on a gelatin-coated glass coverslips in a 24-well dish. For pAAV.CMV.GNE plasmid transfection, 2×104 cells were plated in each well of a 24-well dish, and 500 ng of DNA was transfected using Mirus TransIT-X2 transfection reagent (Mirus Bio; Madison, WI) according to manufacturer’s instructions. After 48 hours, cells were fixed with 4% PFA for 10 minutes. For staining with anti-GNE antibody, cells were blocked with 2% fish gelatin in PBS for 1 hour, then incubated with anti-GNE antibody (HPAA027258, Sigma Aldrich; St. Louis, MO) at room temperature for 2 hours. Coverslips were washed three times with PBS, incubated with TRITC-conjugated MAA (EY Laboratories; San Mateo, CA) and Cy2 AffiniPure Goat anti-rabbit IgG (H+L) (Jackson ImmunoResearch; West Grove, PA). Coverslips were then mounted on slides using ProLong Gold antifade mountant with DAPI (Invitrogen; Waltham, MA). For MyoD staining, Lec3MyoDI were tested with and without doxycycline, which was added at 2μg/mL to the appropriate wells for 72 hours. Cells were blocked with 5% donkey serum in PBS and then incubated with anti-MyoD antibody (NBP1-54153, Novus Biologicals; Littleton, CO). Coverslips were washed three times with PBS, incubated with Alexa Fluor Plus 555 donkey anti-rabbit antibody (Invitrogen; Waltham, MA), and then mounted with ProLong Gold antifade mountant with DAPI.
Western blot
Cells were harvested and protein extracted using RIPA Lysis and Extraction buffer (Thermo Scientific; Waltham, MA) with EDTA and cOmplete Protease Inhibitors (Millipore Sigma; St. Louis, MO) following manufacturer’s instructions. Protein amounts were quantified using the MicroBCA Protein assay (Thermo Scientific; Waltham, MA). 40μg of protein was incubated with NuPage LDS sample buffer (Invitrogen; Carlsbad, CA) with β-mercaptoethanol, boiled for 10 minutes, and separated on a Bolt 4–12% Bis-Tris Plus gel (Invitrogen; Carlsbad, CA). Proteins were transferred to a nitrocellulose membrane and probed with anti-MyoD antibody (Novus Biologicals; Littleton, CO). After washing, horseradish peroxidase-coupled secondary antibody was added and blots were developed using the ECL chemiluminescence method (Lumigen; Southfield, MI) after further washing.
GNE potency assay
One day before plating cells, growth media was removed from Lec3 cells (MEMα supplemented with 10% fetal bovine serum and penicillin/streptomycin) and CHO cells (Ham’s F12 supplemented with 10% fetal bovine serum and penicillin/streptomycin). Cells were rinsed once with PBS and placed in Opti-MEM reduced-serum media (Thermo Fisher Scientific; Waltham, MA). Plates were incubated at 37°C with 5% CO2. After 24 hours, cells were trypsinized, plated at 25,000 cells/well (in 200μL Opti-MEM) in 96-well plates, and incubated for another 24 hours. Cells were then infected with AAV at doses ranging from 104 to 107 multiplicity of infection (MOI) for 72 hours. After removal of media, cells were washed once with PBS and fixed with 4% PFA in PBS for 15 minutes at room temperature. Cells were blocked for an hour at room temperature with 2% fish gelatin in PBS. HRP-conjugated Maackia amurensis agglutinin (MAA, a combined mixture of types I and II, EY Laboratories; San Mateo, CA) or HRP-conjugated Concanavalin A (ConA) (EY Laboratories; San Mateo, CA) was diluted at 1:500 in 2% fish gelatin in PBS, and the cells were incubated with lectin for one hour. 3,3’,5,5’-tetramethylbenzidine (TMB) solution (R&D Systems; Minneapolis, MN) was added for 15–20 minutes to develop lectin binding, and sulfuric acid (H2SO4) was added to stop the reaction. Absorbance was read on a microplate reader at 450 nm. Each assay condition was tested in triplicate for all experiments.
Potency assays using Lec3MyoDI cells followed the same protocol with the following modifications. Growth media for Lec3MyoDI cells was MEMα supplemented with 10% fetal bovine serum, penicillin/streptomycin and 300μg/mL hygromycin B. AAVs were tested with and without doxycycline, which was added at 2μg/mL to the appropriate wells with or without AAV for 72 hours.
Lectin binding to HEK293 cells
HEK293 cells were grown in MEMα supplemented with 10% fetal bovine serum and penicillin/streptomycin. Cells were plated in a 96-well dish at 50,000 cells/well. After 24 hours, serum-free media was added to cells. Cells remained in serum-free media for a total of 4 days and were then assayed for binding of HRP-conjugated lectins with the same protocol used for MB135 cells and GNE–/–MB135 cells.
Lectin binding to AAV capsid protein
A 96-well ELISA plate was coated with 0.2M bicarbonate buffer, pH 9.4, overnight at 4°C, with some wells containing rAAVrh74.CMV.GNE, rAAVrh74.MCK.GNE, or rAAVrh74.MHCK7.GNE at 3×1010 particles/well. Wells were blocked for an hour at room temperature with 2% fish gelatin in PBS. HRP-conjugated Maackia amurensis agglutinin (MAA, a combined mixture of types I and II), HRP-conjugated Sambucus nigra agglutinin (SNA-1), HRP-conjugated Erythrina cristagalli agglutinin (ECA), or HRP-conjugated Arachis hypogaea agglutinin (PNA) (all from EY Laboratories; San Mateo, CA) were diluted at 1:500 in 2% fish gelatin in PBS and added to the appropriate wells for one hour. Lectin binding was developed by addition of 3,3’,5,5’-tetramethylbenzidine (TMB) solution (R&D Systems; Minneapolis, MN) for 25 minutes, and sulfuric acid (H2SO4) was added to stop the reaction. Absorbance was read on a microplate reader at 450 nm. Each assay condition was tested in triplicate for all experiments.
Mice
All animal experiments were conducted with approval from the Institutional Animal Use and Care Committee (IACUC) at The Research Institute at Nationwide Children’s Hospital. Mice were fed food and water ad libitum (Cat # 2919, Teklad Global Rodent diet, Harlan, USA). 2 month-old C57Bl/6J mice (000664, Jackson Laboratories; Bar Harbor, ME) were weighed and then injected via the tail vein with rAAVrh74.CMV.GNE, rAAVrh74.MCK.GNE, or rAAVrh74.MHCK7.GNE at doses of 1×1014 vector genomes per kilogram body weight (vg/kg), 6.6×1013 vg/kg, 3.3×1013 vg/kg, 2×1013 vg/kg, or 1×1013 vg/kg. After a 2 month incubation period, mice were euthanized and muscles dissected and analyzed for AAV biodistribution and GNE gene expression. An additional cohort of untreated 4 month-old C57Bl/6J mice were analyzed for expression of endogenous mouse Gne gene expression.
qPCR measures of AAV biodistribution
Qiagen DNeasy Blood and Tissue kit (Qiagen; Germantown, MD) was used to extract DNA from snap-frozen tissues following manufacturer’s protocol. 100 ng of DNA was assayed on an Applied Biosystems 7500 Real-Time PCR System using a primer designed in the lab and produced by Integrated DNA Technologies (Coralville, IA): Forward 5’-GCC TGT ACG GAA GTG TTA CT-3’, Probe 5’-/56-FAM/TAG CCG CCA TGG AGA ATG /36-TAMSp/-3’, and Reverse 5’-CCG CAG CTT TCG GTT ATT TC-3’. The pAAV.CMV.GNE plasmid was linearized with SwaI (New England Biolabs; Ipswich, MA) and purified with Qiagen QIAquick Gel Extraction Kit for use as standard DNA. The linearized standard was logarithmically measured from 50 to 50 million copies for use as a linear calibration, with correlation coefficients always exceeding 0.98.
qRT-PCR measures of GNE/Gne mRNA tissue ratios
RNA was isolated from snap-frozen tissues using Trizol reagent (Invitrogen; Carlsbad, CA) and purified with Zymo Directzol RNA kit (Zymo Research; Irvine, CA). RNA concentration was measured using a nano-drop spectrophotometer (Thermo Fisher Scientific; Waltham, MA) and 600 ng of RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription kit from Applied Biosystems (Applied Biosystems; Waltham, MA). Primer/Probe sets for human GNE (Hs01103400_m1) and mouse Gne (Mm00450174_m1) were purchased from Applied Biosystems. All samples were measured on a Applied Biosystems 7500 Real-Time PCR System, with results analyzed using the delta-delta CT method [29] with 18S ribosomal (r)RNA (4319413E, Applied Biosystems) as an internal reference. Delta CT values for human GNE (vs. 18S rRNA) from AAV-infected tissues and mouse Gne (vs. 18S rRNA) from uninfected wild type tissues were compared to identify doses that yielded equivalent delta CT values (GNE/Gne = 1).
Statistics
Determinations of significance when comparing 3 or more groups were done using one- or two-way ANOVA with Tukey’s post-hoc test. Comparisons between only 2 groups were done using an unpaired two-tailed Student’s t test. All statistics were calculated using Prism software version 9.3.0 (GraphPad; San Diego, CA).
RESULTS
Development of an AAV potency assay for GNE gene therapy
Plant lectins that bind sialic acid, including Maackia amurensis agglutinin (MAA) and Sambucus nigra agglutinin (SNA), have commonly been used to assess sialic acid (Sia) expression in cells and tissues. MAA preferentially binds α2,3 linked Sia, while SNA preferentially binds α2,6-linked Sia. In addition, increased binding of lectins such as peanut agglutinin (PNA) and Erythrina cristagalli agglutinin (ECA), which bind Galβ1,3GalNAc or Galβ1,4GlcNAc respectively, can also reflect reduced Sia, as Sia often caps these structures in a manner that prevents lectin binding. We used these lectins to assess the extent of reduced Sia expression in GNE-deficient human MB135 muscle cells and Gne-deficient Lec3 Chinese Hamster Ovary (CHO) cells (Fig. 1).
Fig. 1
Lectin binding to GNE-deficient muscle and CHO cells. A. Lectin binding comparing human MB135 cells and GNE–/–MB135 cells at 5, 7, or 13 days after serum starvation. B. Percent reduction in Sia staining by MAA and SNA in GNE–/–MB135 cells compared to MB135 control. C. Lectin binding comparing Chinese hamster ovary (CHO) cells and Lec3 cells, a Gne-deficient CHO cell line. D. Percent reduction in Sia staining by MAA and SNA in Lec3 cells compared to normal CHO control. Errors are SD for n = 3–5/group (A–D). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 Maackia amurensis agglutinin, MAA; Sambucus nigra agglutinin, SNA, Peanut agglutinin, PNA; Erythrina cristagalli agglutinin, ECA
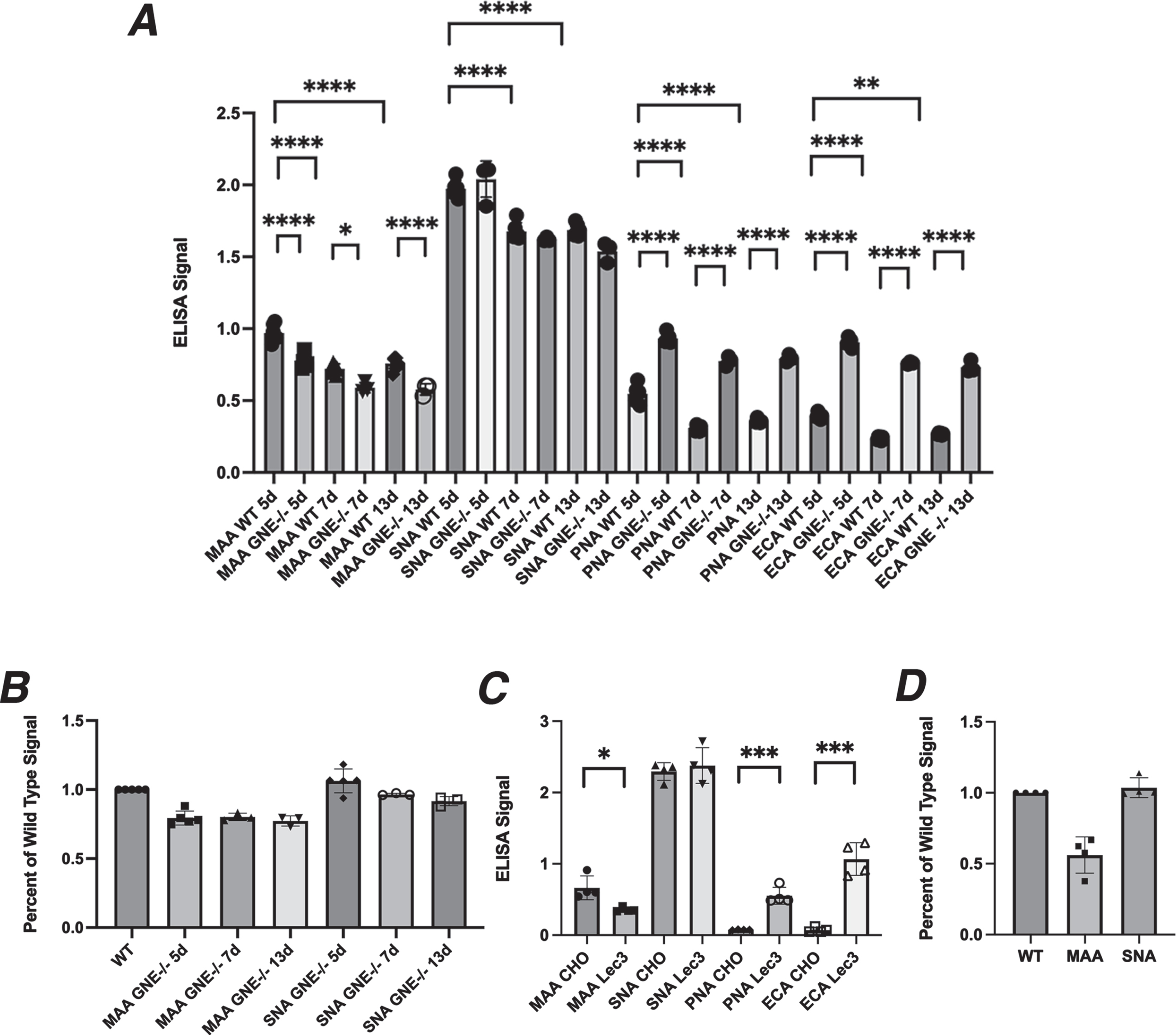
MB135 cells are an immortalized myoblast cell line made by Tapscott and colleagues [28]. Cas9-CRISPR methods were used to induce deletion of the GNE gene in these cells, selecting a GNE–/– MB135 cell variant. We grew normal MB135 cells and GNE–/– MB135 cells in the presence of serum on ELISA plates for several days, after which we induced differentiation into myotubes by placement in serum-free media for 5, 7, or 13 days. GNE–/–MB135 cells formed myotubes normally and on the same schedule as MB135 cells(ns). GNE–/–MB135 cells showed only a very modest reduction in MAA binding compared to control MB135 cells, with a maximal reduction of 24% at 13 days (Fig. 1A, B). SNA binding was reduced even less, with a maximal reduction of 9% at 13 days (Fig. 1A, B). MAA and SNA binding in control MB135 cells was reduced by 24–25% from day 5 to day 13 day, a level equivalent to the greatest differences seen between GNE–/–MB135 and normal MB135 cells on any given day. PNA and ECA binding were increased significantly at all three time points in GNE–/– MB135 cells relative to control (Fig. 1A). These data suggest that serum starvation over time in GNE–/–MB135 cells failed to create a large deficit in binding of Sia-specific lectins, though a trend toward reduced binding was evident.
We next compared CHO cells, which express endogenous Gne activity, to the Lec3 CHO cell variant, which lacks such activity [30]. Unlike GNE-deficient muscle cells, there was a 50% reduction, on average, in MAA binding in Lec3 cells compared to normal CHO cells (Fig. 1C, D), while SNA binding was unchanged. PNA and ECA both showed increased binding in Lec3 cells relative to normal CHO cells (Fig. 1C).
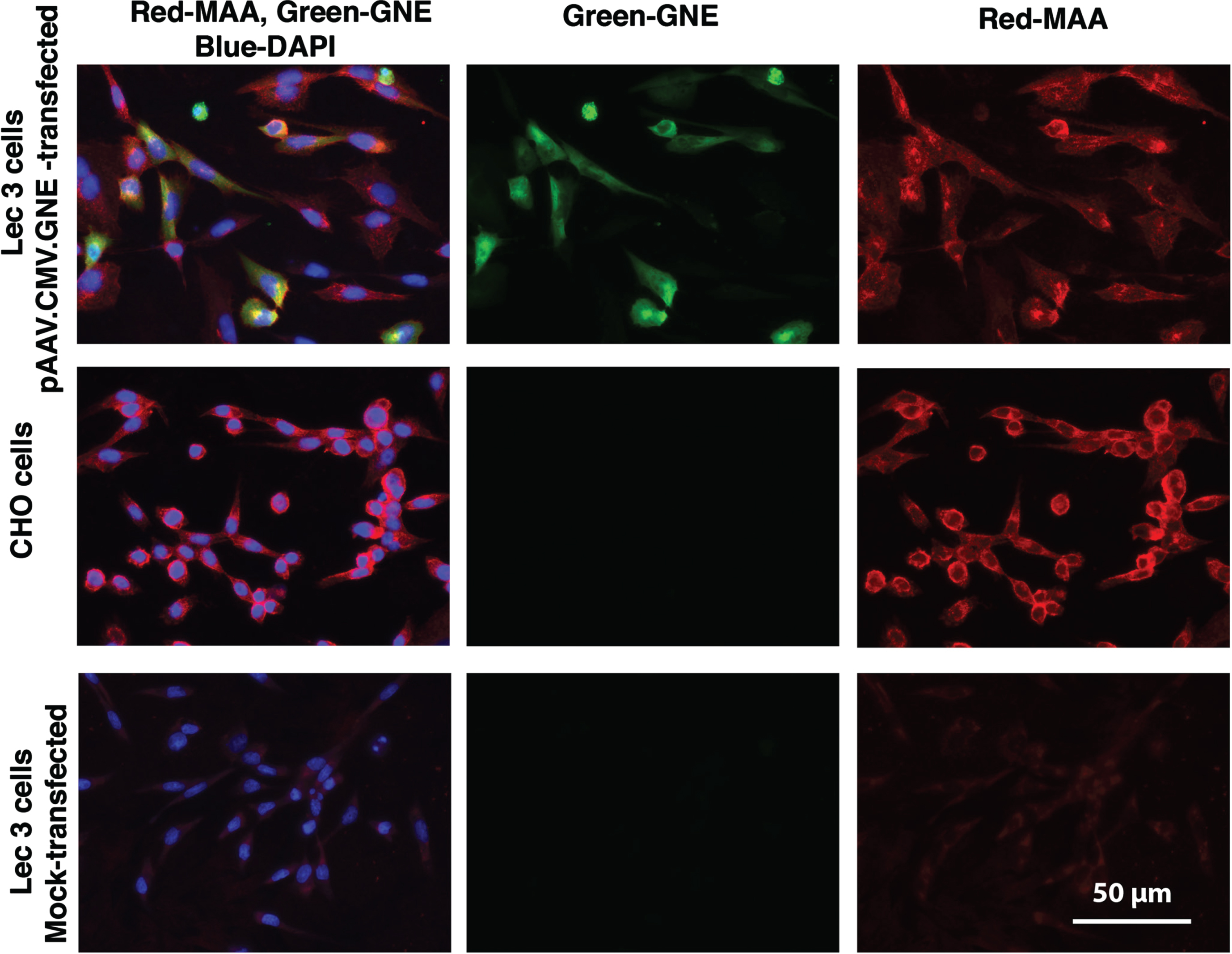
Because Lec3 cells showed a more robust reduction in MAA binding, we next tested if reintroduction of wild type GNE in Lec3 cells would recover Sia expression in these cells. Lec3 cells showed minimal to no MAA staining, while CHO cells were brightly stained by MAA. Transfection of Lec3 cells with pAAV.CMV.GNE plasmid led to human GNE protein expression and increased MAA staining (Fig. 2). MAA also stained some untransfected Lec3 cells in the transfection experiment, perhaps reflecting binding of secreted sialylated proteins from transfected cells to their untransfected neighbors.
Fig. 2
Increased MAA staining after pAAV.CMV.GNE plasmid transfection of Lec3 cells. MAA, a sialic acid binding lectin (red), anti-GNE antibody (green), and DAPI (blue) were used to stain Chinese Hamster Ovary (CHO) cells and Lec3 cells, a Gne-deficient CHO cell line. Lec3 cells were either mock-transfected (no DNA) or transfected with the pAAV.CMV.GNE plasmid to express the human GNE gene. Bar is 50μm.
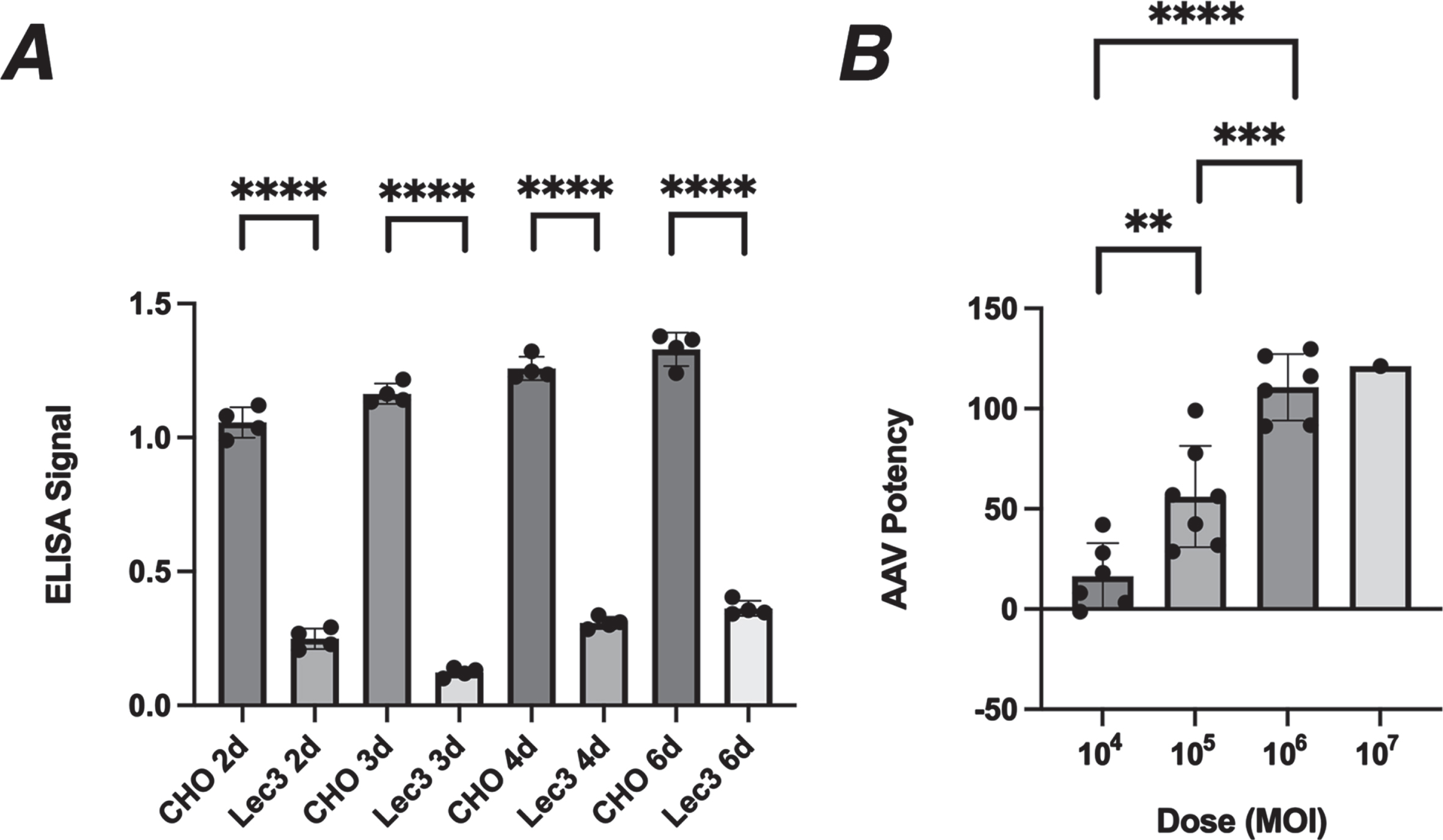
Reduced MAA binding was relatively constant in Lec3 cells between 2 to 6 days of serum starvation, in this experiment showing a reduction of 74–90% from normal (Fig. 3A). We therefore decided to use MAA binding in serum-starved Lec3 cells to assay changed Sia expression resulting from GNE expression in AAV gene therapy vectors. We infected serum-starved Lec3 cells with of rAAVrh74.CMV.GNE using doses from 104 MOI (multiplicities of infection) to 107 (Fig. 3B). Plating of even numbers of cells was confirmed by Concanavalin A (ConA) binding, which binds mannose sugars [31, 32] and shows unchanged binding in Lec3 cells relative to CHO cells (ns). Potency was defined as the percentage MAA binding in AAV-infected Lec3 cells relative to control CHO cells. Background signal from uninfected Lec3 cells was subtracted from both conditions prior to performing the potency calculation. While there was some experiment to experiment variability, infection with rAAVrh74.CMV.GNE caused a significant increase in MAA binding with increasing dose; A dose of 104 MOI, on average, replaced 20% of MAA binding, a dose of 105 MOI replaced 60%, and a dose of 106 replaced 100%. Thus, AAV infection of Lec3 cells can be used to assess AAV vector potency.
Fig. 3
Potency assay for rAAVrh74.CMV.GNE in Lec3 cells. A. MAA binding to CHO and Lec3 cells was compared after 2, 3, 4, or 6 days of serum starvation. B. Lec3 cells starved of serum for 4 days were infected with rAAVrh74.CMV.GNE for the final 3 days and then assayed for MAA binding. Percentage of normal CHO cell signal is reported for different doses. Errors are SD for n = 4/group in A and n = 6-7/group in B (but with only one experiment for the 107 dose). **p < 0.01, ***p < 0.001, ****p < 0.0001. MOI, multiplicities of infection
CHO cells are derived from an ovary and not from muscle, so muscle-specific promoters such as MCK and MHCK7 have little to no activity in these cells. MyoD is a master regulator of skeletal muscle gene expression [33, 34], and both MCK and MHCK7 have MyoD binding sites on their promoters. To increase signal from muscle promoters, we stably transfected Lec3 cells with a doxycycline (Dox)-inducible MyoD expression plasmid (Lec3MyoDI). Transfected Lec3 cells that had been batch-selected were stained (Fig. 4A) and immunoblotted (Fig. 4B) for MyoD either before or after addition of Dox. No MyoD protein was seen in the absence of Dox, while addition of Dox led to strong MyoD expression in most, but not all, cells by 3 days after addition. Immunoblotting showed strong Dox induction of MyoD protein at 1, 2 or 3 days post-addition.
Fig. 4
MyoD immunostaining and immunoblotting of Lec3MyoDI cells. (A) MyoD immunostaining (magenta) and DAPI staining (blue) of batch-selected Lec3MyoDI cells is shown. Cells were grown in the presence or absence of Dox for 3 days. (B) MyoD immunoblot of CHO, Lec3, and Lec3MyoDI cell lysates at different times after Dox addition. (C) Clonally selected Lec3MyoDI variant DZ2.G3 was immunostained for MyoD (magenta) and stained with DAPI (blue). (D) Western blot of MyoD in CHO and Lec3MyoDI clonal variant cell lysates, before or after addition of Dox for 3 days. Expected MW of native MyoD protein is 45kDa. Bar is 200μm for all panels in A and C.
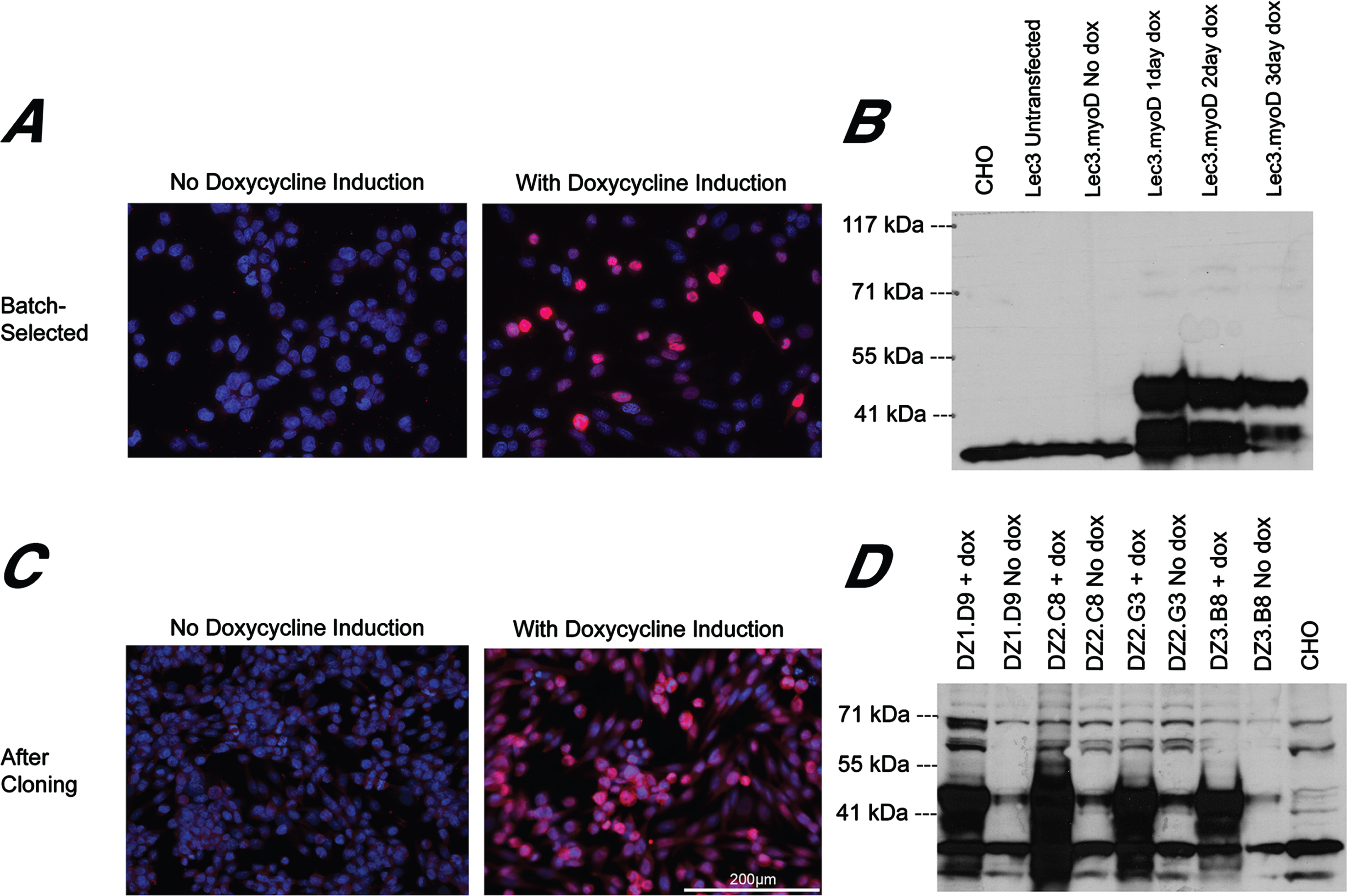
To improve uniformity of MyoD expression, we isolated and expanded Lec3MyoDI single cell clones. In clonal cultures, most cells showed consistent MyoD expression by immunostaining (Fig. 4C) and many clonal variants showed strong MyoD expression by immunoblotting (Fig. 4D). Some MyoD was evident in these clonal cell cultures by western blotting even in the absence of Dox addition, and this may reflect leakiness in the tet-ON regulatory element used to drive MyoD.
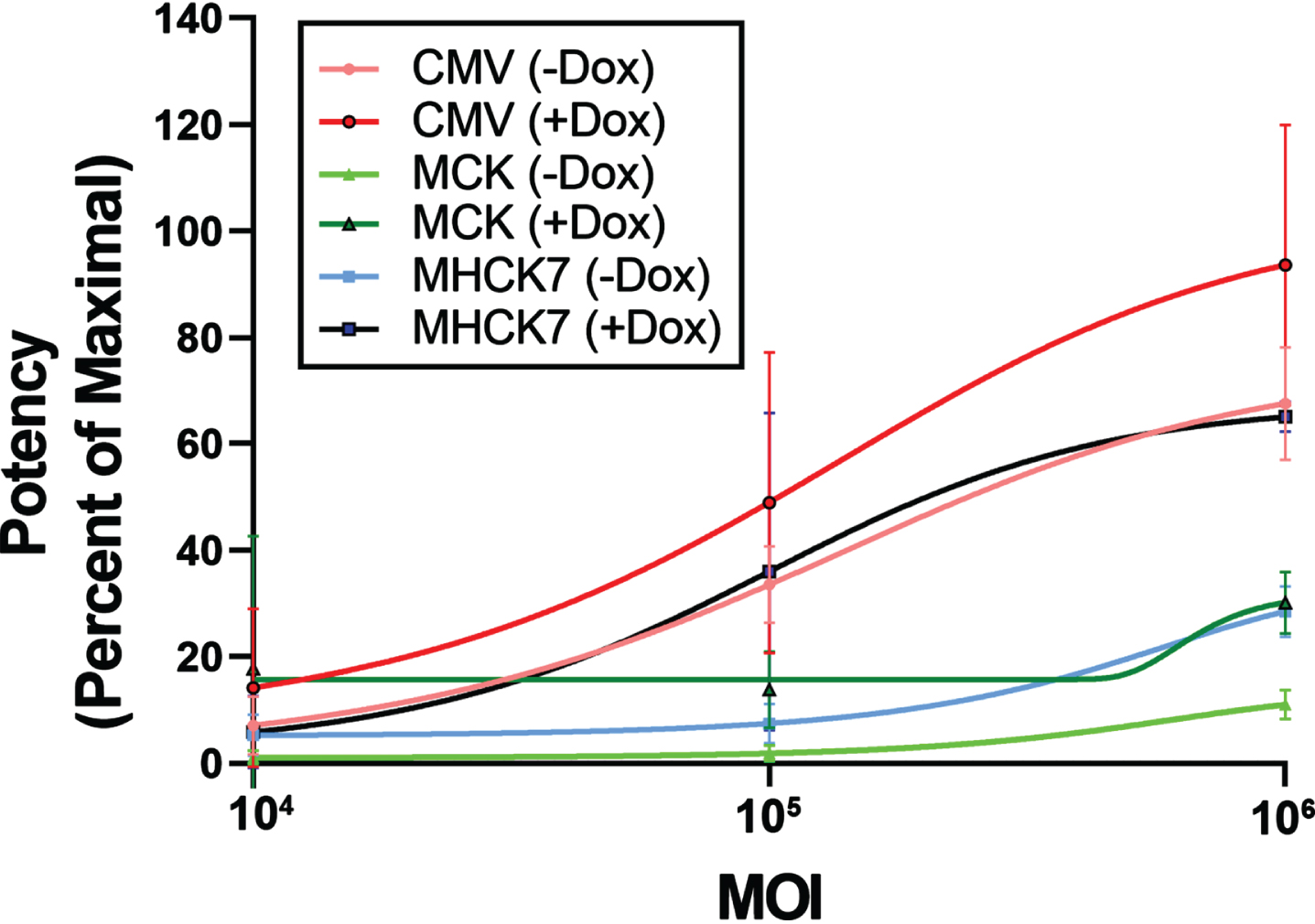
We expanded the DZ2.G3 Lec3MyoDI cell clone and tested three different AAV vectors bearing the human GNE gene: rAAVrh74.CMV.GNE, rAAVrh74.MCK.GNE, and rAAVrh74.MHCK7.GNE (Fig. 5). In the presence of Dox, 106 MOI rAAVrh74.MCK.GNE showed a 30% recovery of MAA binding and rAAVrh74.MHCK7.GNE showed a 65% recovery. rAAVrh74.CMV.GNE was more potent than AAVs using the MCK or MHCK7 promoter at all doses tested, even in the presence of MyoD.
Fig. 5
Potency assay of AAV vectors in Lec3MyoDI cells. AAV potency assays were done to compare promoters (CMV, MCK, or MHCK7) driving GNE expression for rAAVrh74 vectors in Lec3MyoDI cells with or without doxycycline (Dox). Errors are SD for n = 4/group.
Development of an assay to measure GNE gene replacement in vivo
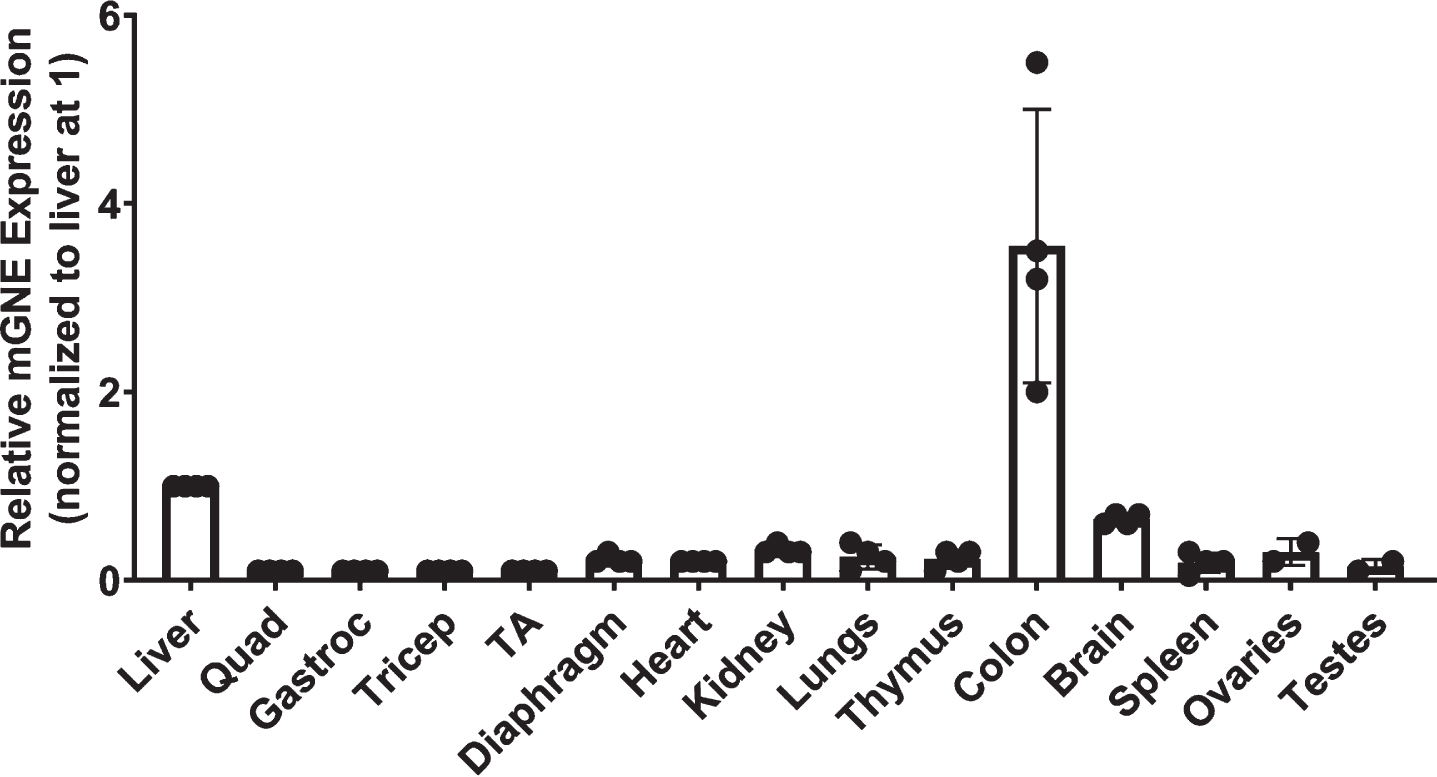
While the Lec3 cell potency assay allowed us to demonstrate the activity of AAV vectors in increasing Sia at various doses, we also wanted to determine the dose at which AAV vectors would be able to replace endogenous mouse Gne gene expression in skeletal muscles in vivo with human GNE. We first tested human-specific and mouse-specific oligonucleotide primer/probe sets to see if they would give the same output signal on their respective target transgenes (Fig. 6). These primer/probe sets showed no signal against the transgene from the other species (ns) and showed the same linear response to their host species transgene. We also used the mouse-specific Gne primer/probe set to measure the amount of endogenous gene expression in non-AAV-treated wild type mouse muscles and in non-muscle organs (Fig. 7). When comparing such tissues to each other (and normalizing to liver at 1), we found that the highest level of mouse Gne gene expression occurred in the colon and liver, much as in human studies (https://www.proteinatlas.org/ENSG00000159921-GNE/tissue). Skeletal muscles, including the quadriceps femoris (quad), gastrocnemius (gastroc), triceps brachii (triceps), and tibialis anterior (TA) had the lowest levels of gene expression. Gne expression in diaphragm and heart muscle was higher than in limb muscles, and this was also the case for kidney, lung, thymus, spleen, ovary, and testes.
Fig. 6
Comparison of human and mouse primer probe sets by qPCR. qPCR was performed on human GNE cDNA using human-specific (hGNE) primer-probe set or on mouse Gne cDNA using a mouse-specific (mGne) primer-probe set. Cq is quantification cycle. Errors are SD for n = 2/group.
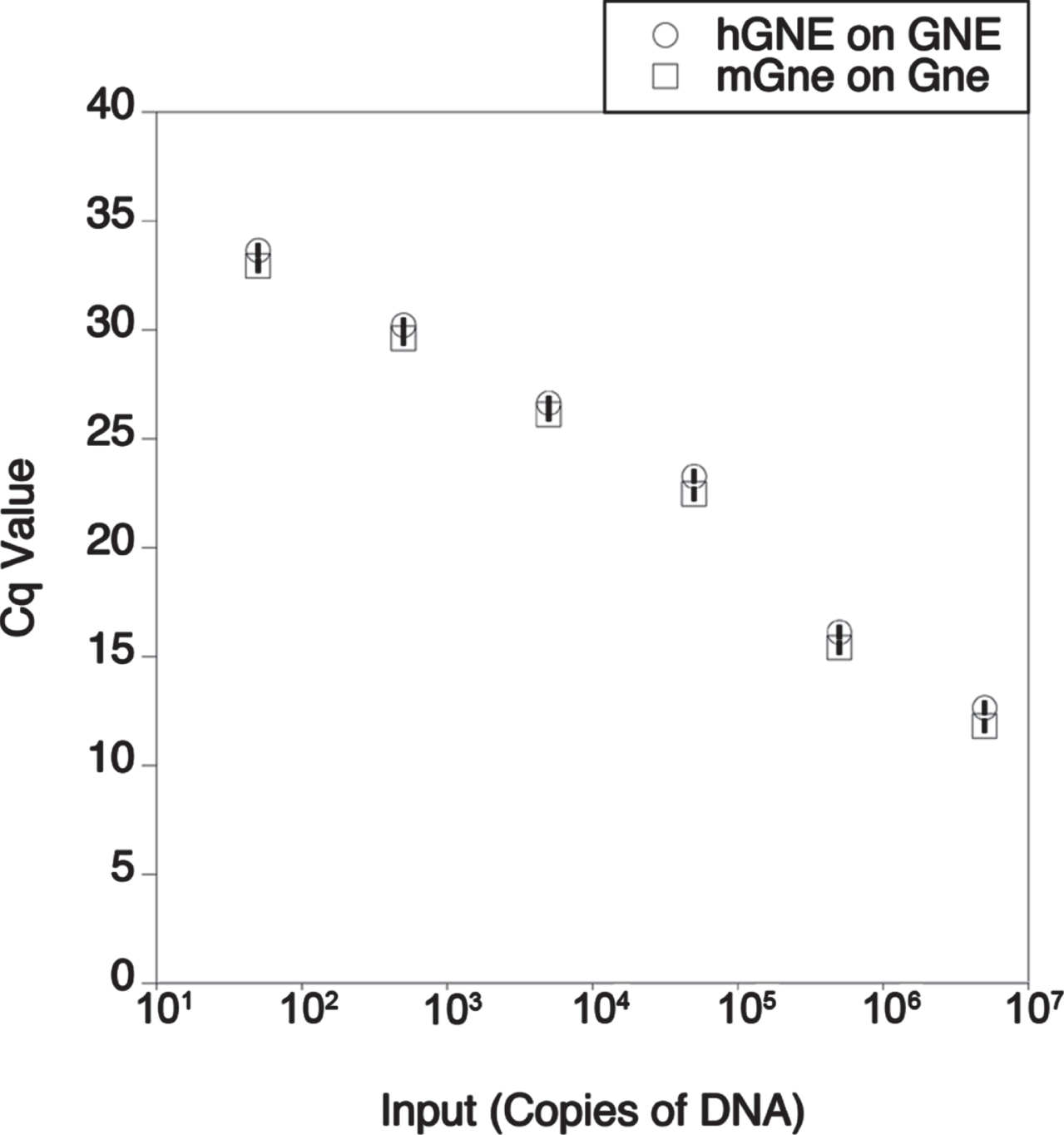
Fig. 7
Relative expression of Gne in wild type mouse organs. mRNA levels for mouse Gne were measured by qRT-PCR in various organs of wild-type (C57Bl/6J) mice. Levels of expression were normalized to liver, set at 1. Errors are SD for n = 4/group.
To measure the dose of human GNE required to replace endogenous mouse Gne expression, we treated wild type (C57Bl/6J) mice with high doses (1×1014vg/kg, 6.6×1013vg/kg, and 3.3×1013vg/kg) of rAAVrh74.CMV.GNE, rAAVrh74.MCK.GNE, or rAAVrh74.MHCK7.GNE. 2-month-old mice were treated via intravenous injection of the tail vein, and muscle expression was analyzed at 2-months post-injection. No phenotypes were apparent as the result of any AAV injected, and muscles appeared normal in H&E stains (ns). We assessed the biodistribution of AAV vector genomes (vgs) in each tissue and the mRNA ratio of human GNE in AAV-infected mice to mouse Gne in uninfected wild type mice (GNE/Gne), where human GNE and mouse Gne levels were each independently referenced to 18S rRNA expression (Table 1). While the rAAVrh74 serotype is known to target heart and skeletal muscles, it still is preferentially taken up by liver [35]; vgs/nucleus were highest in the liver for all three AAV vectors tested, ranging from 80 vg/nucleus at the highest dose to 12 vg/nucleus at the lowest dose. Biodistribution in the heart ranged from 11 to 3 vg/nucleus, while biodistribution in the gastrocnemius muscle ranged from 3.6 to 0.6 vg/nucleus. For gastrocnemius and heart muscle at the 3.3×1013 vg/kg dose, CMV and MHCK7 promoters showed a GNE/Gne ratio of 8.5 or 4.1, respectively, despite having only 1.3 or 0.6 vgs/nucleus present, while MCK showed a GNE/Gne ratio of 0.6 with 0.7 vgs/nucleus present. GNE/Gne ratios in the heart were higher for the 3.3×1013 vg/kg dose of CMV and MHCK7 than they were for MCK, while the GNE/Gne ratio in the liver never exceeded 0.4 for any promoter.
Table 1
Screening of gene expression and biodistribution of AAA.GNE gene therapy vectors at high AAV doses in gastrocnemius muscle, heart, and liver
| GNE Expression Normalized to Wild-Type Endogenous Mouse Gne Expression | |||||||||
| Gastrocnemius | Heart | Liver | |||||||
| 3.3E13vg/kg | 6.6E13vg/kg | 1E14vg/kg | 3.3E13vg.kg | 6.6E13vg/kg | 1E14vg/kg | 3.3E13vg.kg | 6.6E13vg/kg | 1E14vg/kg | |
| CMV | 15.8±1.5 | 54±9.8 | 50.5±11.3 | 32.4±2 | 93.7±13.4 | 100.1±29.2 | 0.6±0.03 | 1.0±0.3 | 3.9±0.9 |
| MCK | 1.4±0.1 | 5.2±0.8 | 7.7±5.4 | 1.1±0.2 | 3.3±1.1 | 2.3±0.8 | 0.7±0.3 | 0.7±0.2 | 1.6±0.6 |
| MHCK7 | 8.7±1.7 | 40.4±15.3 | 46±20.6 | 24.4±3.8 | 57.9±9.5 | 71.1±28.1 | 1.1±0.6 | 1.3±0.3 | 3.7±1.0 |
| Biodistribution of AAV (vg/nucleus) | |||||||||
| Gastrocnemius | Heart | Liver | |||||||
| 3.3E13vg/kg | 6.6E13vg/kg | 1E14vg/kg | 3.3E13vg.kg | 6.6E13vg/kg | 1E14vg/kg | 3.3E13vg.kg | 6.6E13vg/kg | 1E14vg/kg | |
| CMV | 1.3±0.3 | 2.6±1 | 3.6±1.3 | 4.1±0.5 | 8.2±0.4 | 8.2±1.9 | 12.5±3.9 | 56.8±31.7 | 83.2±15.2 |
| MCK | 0.7±0.1 | 1.3±0.3 | 1.5±0.5 | 2.7±0.7 | 6.1±2 | 4.9±1.1 | 16.1±7.2 | 30.3±5.6 | 99.6±58.4 |
| MHCK7 | 0.6±0.3 | 3.4±1 | 3.3±1.2 | 3.6±1.6 | 9±2.7 | 11.3±2.6 | 39±20.3 | 35.8±5.6 | 41.5±3.2 |
2 month-old wild type (C57Bl/6J) mice were dosed IV with rAAVrh74.GNE gene therapy vectors using different promoters (CMV, MCK, or MHCK7). Human GNE transgene expression delivered by AAV gene therapy is normalized to endogenous mouse Gne gene expression found in wild type tissue. AAV biodistribution of vector genomes (vg) per host nucleus is reported below. Errors are SD for n = 4 per condition.
We next compared AAV doses of 3.3×1013 vg/kg, 2×1013 vg/kg, and 1×1013 vg/kg for all three vectors, with additional skeletal muscles (quadriceps femoris, triceps brachii, tibialis anterior, and diaphragm) and non-muscle organs (kidney, lung, thymus, colon, brain, spleen, ovary, and testes) tested (Table 2). AAV biodistribution in skeletal muscles at the 3.3×1013 vg/kg dose was, by and large, above 1vg/nucleus for all three vectors, while this dropped to 1 or below for the two lower doses. AAV biodistribution in the diaphragm was about twice that of all limb muscles. At the 3.3×1013 vg/kg dose, the GNE/Gne mRNA ratio for all five skeletal muscles, averaged together, was 12 for CMV, 1.5 for MCK, and 4.5 for MHCK7 (Table 2). These were all above the targeted 1:1 ratio needed for complete replacement of mouse Gne by human GNE. At the 2×1013 vg/kg dose, the muscle GNE/Gne average measure was 2.6 and 2 for CMV and MHCK7, respectively, and 0.3 for MCK. At the 1×1013vg/kg dose, this same measure was 1 for CMV, 0.8 for MHCK7, and 0.1 for MCK. Thus, rAAVrh74.CMV.GNE, given IV at a dose of 1×1013 vg/kg, would be sufficient to provide Gne gene replacement in skeletal muscles. MHCK7 would reach this measure at 2×1013vg/kg and MCK at 3.3×1013vg/kg.
Table 2
Gene expression and biodistribution of AAA.GNE gene therapy vectors at lower AAV doses
| GNE Expression Normalized to Wild-Type Endogenous Mouse Gne Expression | ||||||||||||||||
| Promoter & Dose | Quad | Gastroc | Tricep | TA | Diaphragm | Average of All Muscles | Heart | Liver | Kidney | Lungs | Thymus | Colon | Brain | Spleen | Ovaries | Testes |
| CMV @3.3E13 vg/kg | 18.1±3.4 | 15.8±1.5 | 6.6±2.1 | 11.3±1.2 | 8.1±1.6 | 12±1.8 | 32.4±2 | 0.6±0.03 | 0.03±0.01 | 0.4±0.1 | 1.5±0.9 | 0.003±0.001 | 0.004±0.001 | 0.04±0.01 | 0.9±0 | 0.2±0.1 |
| MCK @3.3E13 vg/kg | 2.5±0.3 | 1.4±0.1 | 1.6±0.4 | 1.4±0.5 | 0.6±0.2 | 1.5±0.3 | 1.1±0.2 | 0.7±0.3 | 0.02±0.01 | 0.03±0.01 | 0.1±0.02 | 0.001±0.0002 | 0.004±0.002 | 0.02±0.01 | 0.3±0.01 | 0.01±0.002 |
| MHCK7 @3.3E13 vg/kg | 6.3±1.4 | 8.7±1.7 | 1.9±0.4 | 4.1±0.9 | 1.6±0.4 | 4.5±1.1 | 24.4±3.8 | 1.1±0.6 | 0.01±0.003 | 0.03±0.01 | 0.1±0.03 | 0.0003±0.0001 | 0.002±0.0003 | 0.01±0.004 | 0.2±0.01 | 0.01±0.0002 |
| CMV @2E13 vg/kg | 4.9±2.6 | 3.0±1 | 0.9±0.2 | 1.7±0.7 | 2.5±0.7 | 2.6±0.6 | 19.8±4.8 | 0.5±0.1 | 0.04±0.01 | 0.1±0.02 | 4.7±1.9 | 0.001±0.0004 | 0.003±0.001 | 0.05±0.01 | 0.8±0.1 | 0.1±0.03 |
| MCK @2E13 vg/kg | 0.5±0.4 | 0.7±0.4 | 0.1±0.04 | 0.2±0.1 | 0.2±0.1 | 0.3±0.1 | 0.7±0.2 | 0.2±0.02 | 0.04±0.02 | 0.02±0.005 | 0.02±0.01 | 0.0002±0.0001 | 0.001±0.0001 | 0.01±0.001 | 0.1±0.1 | 0.003±0.001 |
| MHCK7 @2E13vg/kg | 2.6±1.8 | 4.4±2.4 | 0.8±0.5 | 1.0±0.5 | 1.1±0.4 | 2±0.7 | 9.1±1.2 | 0.8±0.1 | 0.1±.02 | 0.01±0.02 | 0.7±0.4 | 0.0004±0.0002 | 0.002±0.0005 | 0.1±0.03 | 0.9±0.3 | 0.01±0.003 |
| CMV @1E13 vg/kg | 1.2±0.7 | 1.3±0.5 | 0.4±0.2 | 1.1±0.6 | 1±0.3 | 1.0±0.2 | 5.6±1.8 | 0.2±0.03 | 0.02±0.004 | 0.04±0.01 | 0.2±0.1 | 0.0003±0.0001 | 0.002±0.0003 | 0.05±0.02 | 0.02±0.1 | 0.03±0.001 |
| MCK @1E13 vg/kg | 0.2±0.1 | 0.2±0.1 | 0.1±0.03 | 0.2±0.2 | 0.1±0.04 | 0.1±0.04 | 0.3±0.1 | 0.1±0.03 | 0.01±0.01 | 0.01±0.001 | 0.02±0.01 | 0.0002±0.00001 | 0.001±0.0003 | 0.01±0.01 | 0.1±0.1 | 0.003±0.00005 |
| MHCK7 @1E13vg/kg | 1.5±1.2 | 1.0±0.4 | 0.4±0.2 | 0.6±0.4 | 0.8±0.3 | 0.8±0.2 | 2.7±0.5 | 0.4±0.1 | 0.01±0.002 | 0.02±0.01 | 0.1±0.04 | 0.0002±0.00003 | 0.001±0.0002 | 0.01±0.01 | 0.6±0.2 | 0.01±0.003 |
| Biodistribution of AAV (vg/nucleus) | ||||||||||||||||
| Promoter & Dose | Quad | Gastroc | Tricep | TA | Diaphragm | Average of All Muscles | Heart | Liver | Kidney | Lungs | Thymus | Colon | Brain | Spleen | Ovaries | Testes |
| CMV @3.3E13 vg/kg | 4.7±0.6 | 1.3±0.2 | 2.9±0.4 | 4.9±0.8 | 6.5±2 | 4.1±0.5 | 4.1±0.5 | 12.5±3.9 | 1.6±.5 | 1.9±0.4 | 1.4±0.7 | 1.4±0.4 | 0.2±0.1 | 0.4±0.2 | 2.2±1.4 | 0.1±0.0003 |
| MCK @3.3E13 vg/kg | 1.6±0.5 | 0.7±0.1 | 1.3±0.2 | 0.9±0.2 | 3.0±1.2 | 1.5±0.3 | 2.7±0.7 | 16.1±7.2 | 1±0.2 | 0.9±0.3 | 0.6±0.1 | 0.4±0.3 | 0.04±0.01 | 0.4±0.1 | 1.6±0.4 | 0.02±0.002 |
| MHCK7 @3.3E13 vg/kg | 2.7±0.7 | 0.6±0.2 | 2.5±1.1 | 2.5±0.3 | 4.6±1.3 | 2.6±0.4 | 3.6±1.6 | 39±20.3 | 1.5±0.4 | 2.9±1.3 | 1.4±0.8 | 0.3±0.1 | 0.1±0.03 | 0.5±0.2 | 4.8±0.02 | 0.04±0.01 |
| CMV @2E13 vg/kg | 0.4±0.2 | 0.6±0.2 | 0.3±0.1 | 0.5±0.1 | 2.2±1.6 | 0.8±0.3 | 3.7±0.5 | 79±41.1 | 0.8±0.1 | 1.8±0.2 | 3±1.5 | 0.3±0.3 | 0.05±0.01 | 0.4±0.1 | 2.7±1.1 | 0.1±0.02 |
| MCK @2E13 vg/kg | 0.3±0.05 | 0.6±0.1 | 0.3±0.02 | 0.5±0.1 | 1.2±0.6 | 0.6±0.1 | 1.7±0.4 | 59.2±9.4 | 0.7±0.3 | 0.7±0.1 | 0.8±0.5 | 0.3±0.1 | 0.03±0.02 | 0.4±0.1 | 5.9±4.9 | 0.1±0.04 |
| MHCK7 @2E13 vg/kg | 0.7±0.1 | 1.0±0.2 | 0.8±0.1 | 1.0±0.1 | 1.7±0.7 | 1.1±0.2 | 3.1±0.3 | 82.6±14 | 1.1±0.3 | 0.8±0.2 | 1.1±0.4 | 0.4±0.1 | 0.03±0.01 | 0.3±0.1 | 2.6±0.1 | 0.05±0.02 |
| CMV @1E13 vg/kg | 0.4±0.2 | 0.4±0.1 | 0.4±0.1 | 0.6±0.2 | 2.9±1.6 | 0.9±0.3 | 0.8±0.1 | 19±2.7 | 0.2±0.04 | 0.3±0.03 | 0.1±0.1 | 0.1±0.1 | 0.003±0.001 | 0.1±0.04 | 0.4±0.2 | 0.02±0.01 |
| MCK @1E13 vg/kg | 0.4±0.1 | 0.4±0.1 | 0.2±0.1 | 0.4±0.2 | 0.9±0.5 | 0.5±0.1 | 0.4±0.2 | 14.8±3.8 | 0.2±0.1 | 0.2±0.02 | 0.2±0.1 | 0.1±0.05 | 0.01±0.005 | 0.04±0.01 | 1.2±0.5 | 0.004±0.004 |
| MHCK7 @1E13vg/kg | 0.5±0.3 | 0.4±0.1 | 0.4±0.05 | 0.4±0.1 | 0.8±0.3 | 0.5±0.1 | 0.9±0.5 | 15.3±5.3 | 0.2±0.1 | 0.04±0.02 | 0.2±0.1 | 0.1±0.03 | 0.02±0.004 | 0.2±0.1 | 0.2±0.1 | 0.01±0.01 |
2 month-old wild type (C57Bl/6J) mice were dosed IV with rAAVrh74.GNE gene therapy vectors using different promoters (CMV, MCK, or MHCK7). Human GNE transgene expression delivered by AAV gene therapy is normalized to endogenous mouse Gne gene expression found in wild type tissue, with AAV biodistribution of vector genomes (vg) per host nucleus reported below. Errors are SD for n = 4 per condition (2 males and 2 females), or n = 2 per condition for gonads. Quad, quadriceps femoris; Gastroc, gastrocnemius; tricep, triceps brachii; TA, tibialis anterior.
At the 1×1013 vg/kg dose, the GNE/Gne ratio in the heart was greater than that seen in skeletal muscle, with CMV at 5.6, MCK at 0.3, and MHCK7 at 2.7. Despite this, GNE/Gne values were only as high as 0.4 in the liver (for MHCK7), 0.2 in the thymus (for CMV), and 0.6 in the ovaries (for MHCK7). GNE/Gne values for kidney, lung, colon, brain, spleen, and testes were all below 0.1 for all three promoters at this dose. Thus, an AAV dose of 1×1013 vg/kg using CMV could induce GNE expression to normal levels in skeletal muscles without greatly increasing relative expression in non-muscle tissues.
One oddity of the in vivo study was that the transduction of the AAV vectors into skeletal muscle appeared to be different for various promoters even though all three AAVs were produced using the same rh74 serotype. For example, at the 3×1013vg/kg dose, biodistribution in skeletal muscles of AAV containing the CMV promoter averaged 4.1 vg/nucleus, while it was 1.5 for MCK and 2.6 for MHCK7 (Fig. 8A). All of the AAVs we produced were made by the triple transfection method in HEK293 cells, where the expression plasmid to be packaged into AAV was co-transfected along with a rep-cap plasmid, to make the capsid protein, and a third plasmid to make additional necessary proteins. As each GNE expression plasmid that was to be packaged into AAV contained a unique promoter (CMV, MCK, or MHCK7), transfection of these plasmids might also lead to differential expression of GNE in HEK293 cells while the AAVs were being produced. We surmised that the rAAVrh74 capsid protein itself might therefore be differentially sialylated as the result of transfection of these plasmids into HEK293 cells.
Fig. 8
Altered biodistribution and glycosylation of AAV vectors. (A) AAV biodistribution of rAAVrh74 vectors in five skeletal muscles (TA, gastroc, quad, triceps, diaphragm) after IV dosing at 3.3×1013vg/kg. (B) Lectin binding to human embryonic kidney 293 (HEK293) cells grown with or without serum for 4 days. (C) Lectin binding to purified rAAVrh74 containing the CMV, MCK, or MHCK7 promoter with GNE. All AAV vectors were produced in serum-starved HEK293 cells. Errors are SD for n = 5/group (A), 12/group (B), or 3/group (C). *p < 0.05, ***p < 0.001, ****p < 0.0001
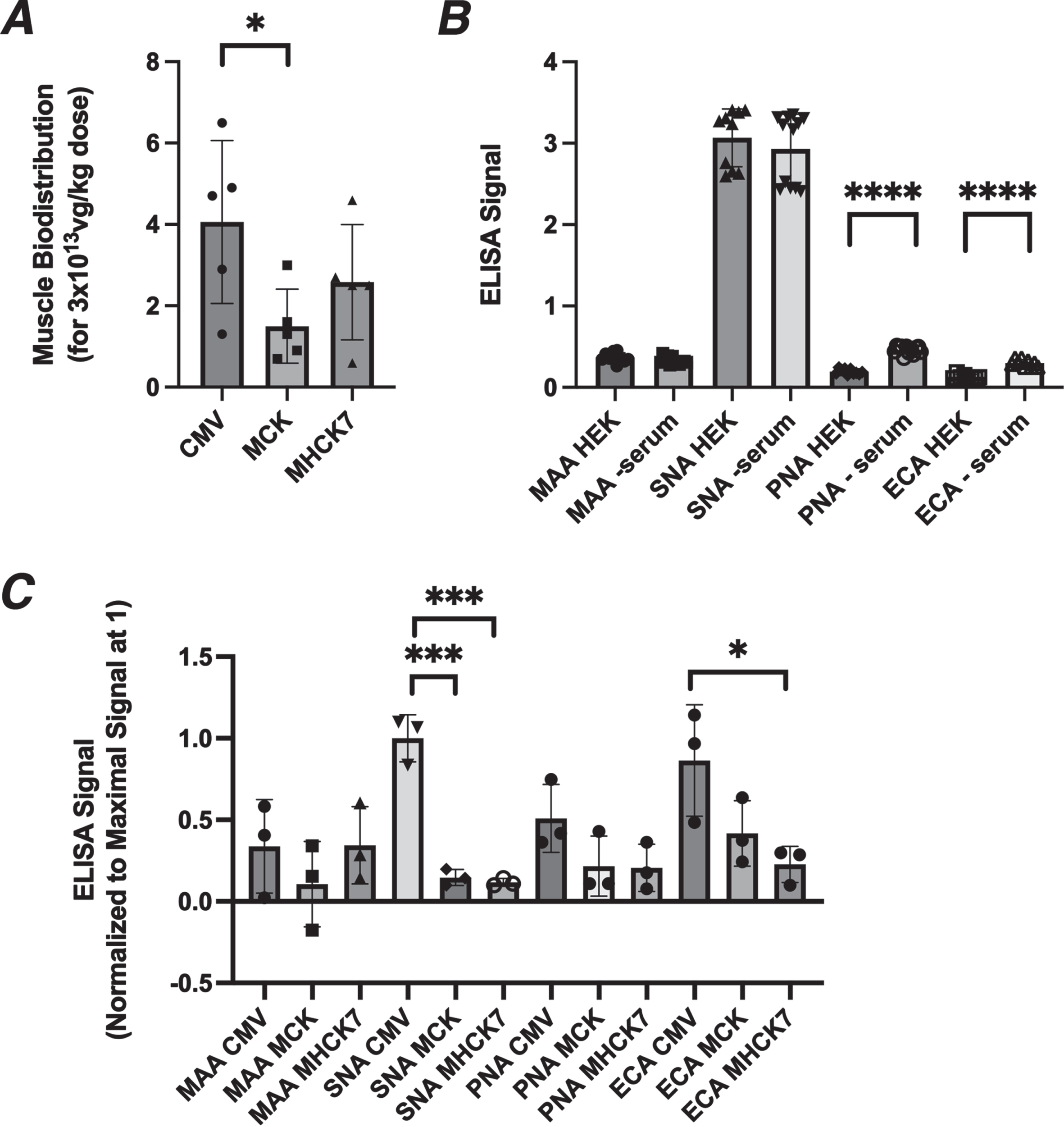
HEK293 cells are starved for serum for four days after they are transfected with the three plasmids used to make an AAV. Like GNE-deficient muscle cells and Lec3 cells (Fig. 1), we found that serum starvation of HEK cells for 4 days led to reductions in MAA and SNA binding and to increased PNA and ECA binding (Fig. 8B). Transfection of the pAAV.CMV.GNE plasmid in serum-starved HEK293 cells for four days significantly increased MAA and SNA binding (MAA by 30±2% (p < 0.0001, n = 6/grp) and SNA by 7±0.2% (p < 0.01, n = 6/grp)), while transfection of pAAV.MCK.GNE showed no increase for either lectin.
We next assayed MAA and SNA binding to the purified rAAV vectors used in this study (Fig. 8C). MAA binding to rAAVrh74.CMV.GNE and rAAVrh74.MHCK7.GNE was higher than to rAAVrh74.MCK.GNE, and SNA binding was 10 times higher to rAAVrh74.CMV.GNE than to rAAVrh74.MCK.GNE or rAAVrh74.MHCK7.GNE. ECA and PNA binding were also elevated for rAAVrh74.CMV.GNE relative to rAAVrh74.MCK.GNE or rAAVrh74.MHCK7.GNE. Thus, rAAVrh74.CMV.GNE production in HEK293 cells led to increased sialylation of its rAAVrh74 capsid protein relative to rAAVrh74.MCK.GNE and rAAVrh74.MHCK7.GNE. While not proven here, such glycosylation changes may help to explain the altered transduction properties of the three rAAVrh74 vectors.
DISCUSSION
The recent findings in ACE-ER sialic acid [17, 18] and ManNAc [19] clinical trials suggest that gene therapy merits serious consideration as an alternative to glycan treatment for patients with GNE myopathy. Gene replacement using AAV has recently been shown to be successful in a number of monogenic genetic diseases [36–39], but there are a number of obstacles to its development for GNEM. We have addressed two of those obstacles in this study: development of an assay to demonstrate AAV potency in producing sialic acid (Sia) and identification of doses where AAV treatment can replace endogenous Gne expression with human GNE in vivo.
An AAV potency assay is necessary to describe the function and stability of clinical production lots of AAV. The FDA requires that AAV potency be reported for each clinical AAV lot in each year that it is used. Our AAV potency assay takes advantage of a CHO mutant cell line, Lec3 [30], that is deficient in Gne enzyme activity. When placed in serum-free media, Lec3 cells show reduced, though not absent, binding of the Sia-binding lectin MAA. Without serum starvation, Lec3 cells show no deficit in Sia, as serum contains large amounts of Sia that cultured cells can consume and utilize. Reduced MAA binding was also seen in GNE-deficient human muscle cells (GNE–/–MB135) placed in serum-free media, but the extent of reduced binding was much greater for Lec3 cells than for GNE–/–MB135 cells. The difficulty in working with muscle cell lines to develop a potency assay include the fact that myoblasts differentiate in the absence of serum, meaning only myotube or differentiated myoblast cultures can be assayed after days in the serum-free media. Myotube cell cultures also secrete extracellular matrix proteins that can stick on the plate and remain there for long periods of time after serum is removed. Thus, myotube cultures likely have increased the expression of stably sialylated proteins relative to non-muscle cell lines, and this lessens the window for seeing lower Sia levels after serum removal. MAA, which binds α2,3-linked sialic acids, showed reduced binding in both Lec3 and GNE–/–MB135 cells, but, SNA, which binds α2,6-linked sialic acids, showed less change. While both Lec3 and GNE–/– human muscle cells showed elevations in PNA and ECA binding, two lectins that are unmasked when Sia is removed from subterminal glycan structures, these lectins do not directly bind Sia. Therefore, MAA, which does bind Sia, provides the best direct measure of changed Sia expression.
Lec3 cells provided a robust cell line to test potency of AAV vectors. CHO cells are not of a myogenic origin, so Lec3 cells cannot be used to assay some muscle-specific promoters such as MCK. To address this, we stably transfected an inducible Tet-ON MyoD expression plasmid into Lec3 cells to stimulate increased responsiveness to muscle-specific promoters after doxycycline (Dox) addition. The MCK and MHCK7 promoters both responded better in Lec3 potency assays when MyoD was expressed. CMV, however, yielded higher potency than MCK or MHCK7, even in the presence of MyoD.
One of the most important facets of developing a gene therapy for clinical use is understanding the dose at which gene replacement has therapeutic value. Because of the variable disease outcomes of current GNEM disease mouse models [9–12, 22, 40], we bypassed their study here and instead asked what AAV dose would be required to induce human GNE expression in wild type mouse muscles at a level that was equivalent to the endogenous mouse Gne gene. While such studies do not demonstrate prevention of disease, and while they also do not consider possible differences between gene and protein expression, they provide an important foundation for dosing of AAV vectors in GNEM patients. Intravenous dosing of AAV vectors that either used a strong constitutive promoter (rAAVrh74.CMV.GNE) or a strong “muscle-specific” promoter (rAAVrh74.MHCK7.GNE) allowed 1:1 GNE:Gne gene replacement at a dose of 1×1013vg/kg or 2×1013vg/kg, respectively, averaged over five different skeletal muscles. This dose is about 10 times lower than the dose of rAAVrh74.MHCK7.micro-dystrophin (DMD) or rAAV9.CBA.SMN gene therapy currently being used to treat Duchenne muscular dystrophy or spinal muscular atrophy type 1 [36, 37]. The lowered dose requirement likely reflects the fact that endogenous Gne gene expression is quite low in skeletal muscles, making the amount of human GNE needed to replace it equivalently lower.
Use of CMV to express GNE also altered sialylation in serum-starved HEK293 cells that are used to make AAV vectors. This led to increased glycosylation of the AAV capsid for rAAVrh74.CMV.GNE relative to rAAVrh74.MCK.GNE and rAAVrh74.MHCK7GNE. Such a difference in glycosylation for rAAVrh74.CMV.GNE may help improve muscle transduction for this vector. While glycosylation in serum-starved HEK293 cells, which express GNE, was less changed than was the case in Lec3 cells (which do not express functional Gne), the large amount of capsid protein produced with the triple transfection method in HEK293 cells may stress the glycosylation system, leading to more significant changes for this protein. Future work is needed to prove a direct relationship between altered capsid glycosylation and altered AAV transduction, but these data suggest that altered Sia on AAV capsids may change their therapeutic potential.
Because of the low dose needed to achieve gene replacement in skeletal muscles, use of the constitutive CMV promoter led to only modest levels of GNE overexpression in non-muscle tissues. rAAVrh74.CMV.GNE yielded 1:1 gene replacement, on average, at 1×1013 vg/kg, in skeletal muscles, but this dose only elevated liver GNE expression 0.2-fold despite transduction with 19 vector genomes per liver cell nucleus. The only non-muscle organ of concern here was the heart, where GNE expression was 5-6 times normal at this dose. GNE enzyme activity is negatively regulated by CMP-Sia [41], the last step in the Sia biosynthetic pathway, so increased GNE expression in heart may not necessarily result in excessive Sia production. Regardless, these data suggest that use of constitutive promoters may be a viable option to deliver gene replacement to muscles in GNE myopathy. Because certain glycoproteins containing Sia are delivered to muscle from other organs, particularly the liver [8], and because GNE is normally expressed in all tissues and its mutation may have disease impacts outside muscle, for example thrombocytopenia [42–46] and motor neuron disease [47–49], it may be that use of a constitutive promoter for AAV gene therapy would facilitate a more complete recovery of function for GNEM patients than would use of muscle-specific promoters.
CONCLUSION
In the present study, an assay was developed to assess the potency of AAV vectors expressing the GNE gene to make sialic acid. Such an assay will allow different lots of AAV vectors expressing GNE to be compared for potency and will allow an assessment of AAV stability over time. This study also demonstrated that rAAVrh74.CMV.GNE can be used to replace endogenous mouse Gne with human GNE at a relatively low dose, and that this dose caused little GNE overexpression in non-muscle tissues. Therefore, AAV vectors that utilize constitutive promoters to express GNE could be used to treat GNE myopathy in a way that might not significantly impact non-muscle organs.
ACKNOWLEDGMENTS
We would like to thank Pamela Stanley (Albert Einstein University, New York NY) for the gift of Lec3 cells, Stephen Tapscott (U. Washington) for the gift of MB135 cells, and GENETHON (Evry, France) for the gift of the pSINTREMyoD_hPGKrtTA2S_M221306_sv40_hygro plasmid. Funding was received from the Neuromuscular Disease Foundation (Beverly Hills, CA) and from the Technology Development Fund at Nationwide Children’s Hospital (Columbus, OH).
CONFLICTS OF INTEREST
PTM and NCH receive licensing fees from Sarepta Therapeutics. PTM is also CSO and co-founder of Genosera, Inc. None of the other authors have any conflicts to declare.
REFERENCES
[1] | Nishino I , Carrillo-Carrasco N , Argov Z . GNE myopathy: Current update and future therapy. J Neurol Neurosurg Psychiatry. (2015) ;86: (4):385–92. |
[2] | Eisenberg I , Avidan N , Potikha T , Hochner H , Chen M , Olender T , et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. (2001) ;29: (1):83–7. |
[3] | Stasche R , Hinderlich S , Weise C , Effertz K , Lucka L , Moormann P , et al. A bifunctional enzyme catalyzes the first two steps in N-acetylneuraminic acid biosynthesis of rat liver. Molecular cloning and functional expression of UDP-N-acetyl-glucosamine 2-epimerase/N-acetylmannosamine kinase. J Biol Chem. (1997) ;272: (39):24319–24. |
[4] | Huizing M , Carrillo-Carrasco N , Malicdan MC , Noguchi S , Gahl WA , Mitrani-Rosenbaum S , et al. GNE myopathy: New name and new mutation nomenclature. Neuromuscul Disord. (2014) ;24: (5):387–9. |
[5] | Pogoryelova O , Wilson IJ , Mansbach H , Argov Z , Nishino I , Lochmuller H . GNE genotype explains 20% of phenotypic variability in GNE myopathy. Neurol Genet. (2019) ;5: (1):e308. |
[6] | Celeste FV , Vilboux T , Ciccone C , de Dios JK , Malicdan MC , Leoyklang P , et al. Mutation update for GNE gene variants associated with GNE myopathy. Hum Mutat. (2014) ;35: (8):915–26. |
[7] | Chan YM , Lee P , Jungles S , Morris G , Cadaoas J , Skrinar A , et al. Substantial deficiency of free sialic acid in muscles of patients with GNE myopathy and in a mouse model. PLoS One. (2017) ;12: (3):e0173261. |
[8] | Crowe KE , Zygmunt DA , Heller K , Rodino-Klapac L , Noguchi S , Nishino I , et al .Visualizing muscle sialic acid expression in the GNED207VTgGne-/- Cmah-/- model of GNE myopathy: A comparison of dietary and gene therapy approaches. J Neuromuscul Dis. 2021. |
[9] | Malicdan MC , Noguchi S , Hayashi YK , Nonaka I , Nishino I . Prophylactic treatment with sialic acid metabolites precludes the development of the myopathic phenotype in the DMRV-hIBM mouse model. Nat Med. (2009) ;15: (6):690–5. |
[10] | Malicdan MC , Noguchi S , Nonaka I , Hayashi YK , Nishino I . A Gne knockout mouse expressing human GNE D176V mutation develops features similar to distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Hum Mol Genet. (2007) ;16: (22):2669–82. |
[11] | Sela I , Yakovlev L , Becker Cohen M , Elbaz M , Yanay N , Ben Shlomo U , et al. Variable phenotypes of knockin mice carrying the M712T Gne mutation. Neuromolecular Med. (2013) ;15: (1):180–91. |
[12] | Galeano B , Klootwijk R , Manoli I , Sun M , Ciccone C , Darvish D , et al. Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J Clin Invest. (2007) ;117: (6):1585–94. |
[13] | Niethamer TK , Yardeni T , Leoyklang P , Ciccone C , Astiz-Martinez A , Jacobs K , et al. Oral monosaccharide therapies to reverse renal and muscle hyposialylation in a mouse model of GNE myopathy. Mol Genet Metab. (2012) ;107: (4):748–55. |
[14] | Malicdan MC , Noguchi S , Tokutomi T , Goto Y , Nonaka I , Hayashi YK , et al. Peracetylated N-acetylmannosamine, a synthetic sugar molecule, efficiently rescues muscle phenotype and biochemical defects in mouse model of sialic acid-deficient myopathy. J Biol Chem. (2012) ;287: (4):2689–705. |
[15] | Malicdan MC , Noguchi S , Nishino I . A preclinical trial of sialic acid metabolites on distal myopathy with rimmed vacuoles/hereditary inclusion body myopathy, a sugar-deficient myopathy: A review. Ther Adv Neurol Disord. (2010) ;3: (2):127–35. |
[16] | Xu X , Wang AQ , Latham LL , Celeste F , Ciccone C , Malicdan MC , et al. Safety, pharmacokinetics and sialic acid production after oral administration of N-acetylmannosamine (ManNAc) to subjects with GNE myopathy. Mol Genet Metab. (2017) ;122: (1-2):126–34. |
[17] | Lochmuller H , Behin A , Caraco Y , Lau H , Mirabella M , Tournev I , et al. A phase 3 randomized study evaluating sialic acid extended-release for GNE myopathy. Neurology. (2109) ;92: (18):e2109–e17. |
[18] | Suzuki N , Mori-Yoshimura M , Katsuno M , Takahashi MP , Yamashita S , Oya Y , et al. Phase II/III study of aceneuramic acid administration for GNE myopathy in Japan. J Neuromuscul Dis. 2023. |
[19] | Carrillo N , Malicdan MC , Leoyklang P , Shrader JA , Joe G , Slota C , et al. Safety and efficacy of N-acetylmannosamine (ManNAc) in patients with GNE myopathy: An open-label phase 2 study. Genet Med. (2021) ;23: (11):2067–75. |
[20] | Nemunaitis G , Jay CM , Maples PB , Gahl WA , Huizing M , Yardeni T , et al. Hereditary inclusion body myopathy: Single patient response to intravenous dosing of GNE gene lipoplex. Hum Gene Ther. (2011) ;22: (11):1331–41. |
[21] | Mitrani-Rosenbaum S , Yakovlev L , Becker Cohen M , Telem M , Elbaz M , Yanay N , et al. Sustained expression and safety of human GNE in normal mice after gene transfer based on AAV8 systemic delivery. Neuromuscul Disord. (2012) ;22: (11):1015–24. |
[22] | Mitrani-Rosenbaum S , Yakovlev L , Becker Cohen M , Argov Z , Fellig Y , Harazi A . Pre clinical assessment of AAVrh74. MCK.GNE viral vector therapeutic potential: Robust activity despite lack of consistent animal model for GNE myopathy. J Neuromuscul Dis. (2022) ;9: (1):179–92. |
[23] | Salva MZ , Himeda CL , Tai PW , Nishiuchi E , Gregorevic P , Allen JM , et al. Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol Ther. (2007) ;15: (2):320–9. |
[24] | Levitt N , Briggs D , Gil A , Proudfoot NJ . Definition of an efficient synthetic poly(A) site. Genes Dev. (1989) ;3: (7):1019–25. |
[25] | Xu R , Jia Y , Zygmunt DA , Cramer ML , Crowe KE , Shao G , et al. An isolated limb infusion method allows for broad distribution of rAAVrh74. MCK.GALGT2 to leg skeletal muscles in the rhesus macaque. Mol Ther Methods Clin Dev. (2018) ;10: , 89–104. |
[26] | Xiao X , Li J , Samulski RJ . Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. (1998) ;72: (3):2224–32. |
[27] | Clark KR , Liu X , McGrath JP , Johnson PR . Highly purified recombinant adeno-associated virus vectors are biologically active and free of detectable helper and wild-type viruses. Human Gene Therapy. (1999) ;10: (6):1031–9. |
[28] | Jagannathan S , Shadle SC , Resnick R , Snider L , Tawil RN , van der Maarel SM , et al. Model systems of DUX4 expression recapitulate the transcriptional profile of FSHD cells. Hum Mol Genet. (2016) ;25: (20):4419–31. |
[29] | Livak KJ , Schmittgen TD . Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. (2001) ;25: (4):402–8. |
[30] | Hong Y , Stanley P . Lec3 Chinese hamster ovary mutants lack UDP-N-acetylglucosamine 2-epimerase activity because of mutations in the epimerase domain of the Gne gene. J Biol Chem. (2003) ;278: (52):53045–54. |
[31] | Tsaneva M , Van Damme EJM . 130 years of plant lectin research. Glycoconj J. (2020) ;37: (5):533–51. |
[32] | Kim J , Albarghouthi M . Rapid monitoring of high-mannose glycans during cell culture process of therapeutic monoclonal antibodies using lectin affinity chromatography. J Sep Sci. (2022) ;45: (12):1975–83. |
[33] | Esteves de Lima J , Relaix F . Master regulators of skeletal muscle lineage development and pluripotent stem cells differentiation. Cell Regen. (2021) ;10: (1):31. |
[34] | Weintraub H , Tapscott SJ , Davis RL , Thayer MJ , Adam MA , Lassar AB , et al. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc Natl Acad Sci U S A. (1989) ;86: (14):5434–8. |
[35] | Zygmunt DA , Xu R , Jia Y , Ashbrook A , Menke C , Shao G , et al. rAAVrh74. MCK.GALGT2 demonstrates safety and widespread muscle glycosylation after intravenous delivery in C57BL/6J Mice. Mol Ther Methods Clin Dev. (2019) ;15: , 305–19. |
[36] | Mendell JR , Sahenk Z , Lehman K , Nease C , Lowes LP , Miller NF , et al. Assessment of systemic delivery of rAAVrh74.MHCK7.micro-dystrophin in children with duchenne muscular dystrophy:Anonrandomized controlled trial. JAMA Neurol. 2020. |
[37] | Mendell JR , Al-Zaidy S , Shell R , Arnold WD , Rodino-Klapac LR , Prior TW , et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. (2017) ;377: (18):1713–22. |
[38] | High KA , Anguela XM . Adeno-associated viral vectors for the treatment of hemophilia. Hum Mol Genet. (2016) ;25: (R1):R36–41. |
[39] | Michalakis S , Gerhardt M , Rudolph G , Priglinger S , Priglinger C . Gene therapy for inherited retinal disorders: Update on clinical trials. Klin Monbl Augenheilkd. (2021) ;238: (3):272–81. |
[40] | Crowe KE , Zygmunt DA , Heller K , Rodino-Klapac L , Noguchi S , Nishino I , et al. Visualizing muscle sialic acid expression in the GNED207VTgGne-/- Cmah-/- model of GNE myopathy: A comparison of dietary and gene therapy approaches. J Neuromuscul Dis. (2022) ;9: (1):53–71. |
[41] | Hinderlich S , Weidemann W , Yardeni T , Horstkorte R , Huizing M . UDP-GlcNAc 2-Epimerase/ManNAc Kinase (GNE): A master regulator of sialic acid synthesis. Top Curr Chem. 2013. |
[42] | Beecher G , Liewluck T . GNE myopathy: Don’t sleep on the platelets. Muscle Nerve. (2022) ;65: (3):263–5. |
[43] | Yoshioka W , Shimizu R , Takahashi Y , Oda Y , Yoshida S , Ishihara N , et al. Extra-muscular manifestations in GNE myopathy patients: A nationwide repository questionnaire survey in Japan. Clin Neurol Neurosurg. (2022) ;212: , 107057. |
[44] | Xu Z , Xiang J , Luan X , Geng Z , Cao L . Novel compound heterozygous mutations in a GNE myopathy with congenital thrombocytopenia: A case report and literature review. Clin Case Rep. (2022) ;10: (4):e05659. |
[45] | Paul P , Liewluck T . Distal myopathy and thrombocytopenia due to a novel GNE mutation. J Neurol Sci. (2020) ;415: , 116954. |
[46] | Izumi R , Niihori T , Suzuki N , Sasahara Y , Rikiishi T , Nishiyama A , et al. GNE myopathy associated with congenital thrombocytopenia: A report of two siblings. Neuromuscul Disord. (2014) ;24: (12):1068–72. |
[47] | Grecu N , Villa L , Cavalli M , Ristaino A , Choumert A , Butori C , et al. Motor axonal neuropathy associated with GNE mutations. Muscle Nerve. (2021) ;63: (3):396–401. |
[48] | Koroglu C , Yilmaz R , Sorgun MH , Solakoglu S , Sener O . GNE missense mutation in recessive familial amyotrophic lateral sclerosis. Neurogenetics. (2017) ;18: (4):237–43. |
[49] | Huang YN , Chuang HJ , Hsueh HW , Huang HC , Lee NC , Chao CC , et al. A case of GNE myopathy mimicking hereditary motor neuropathy. Eur J Neurol. (2020) ;27: (11):2389–91. |


