
Duchenne Muscular Dystrophy in Kazakhstan: A Journey from Diagnosis to the Treatment, the Biases and Achievements
Abstract
Background:
Neuro-muscular disorders constitutes a group of rare but heterogeneous conditions. The onset of these diseases ranges widely from birth to elderly. Many of them are life threatening and progressive. Neuromuscular science is a very specialised medical field for which specific knowledge and expertise are necessary. Such an expertise is available only partially in Kazakhstan where underdiagnosis, misdiagnosis and mismanagement of patients with muscle diseases are commonplace. Hopefully, times are changing. With the implementation of international guidelines for the diagnosis and treatment of Duchenne Muscular Dystrophy (DMD), patients are now given better care including pharmacological interventions (including steroids in DMD), respiratory and nutritional support.
Objectives:
To report on clinical data and genetic variants in a nationwide cohort of DMD patients. To describe and analyse management strategies applied in Kazakhstan in these patients.
Methods:
The medical records of 84 patients recruited by the national expert-consulting board based at the national multidisciplinary centre of reference in neuro-muscular disorders in Astana, Kazakhstan, have been ascertained for the study. The national expert committee meets monthly to decide over the prescription of disease-modifying therapies in paediatric neuromuscular disorders. Data on the age of disease onset, the age at genetic testing, spectrum of genetic variants, the stage of disease and the serum CK levels have been collected.
Results
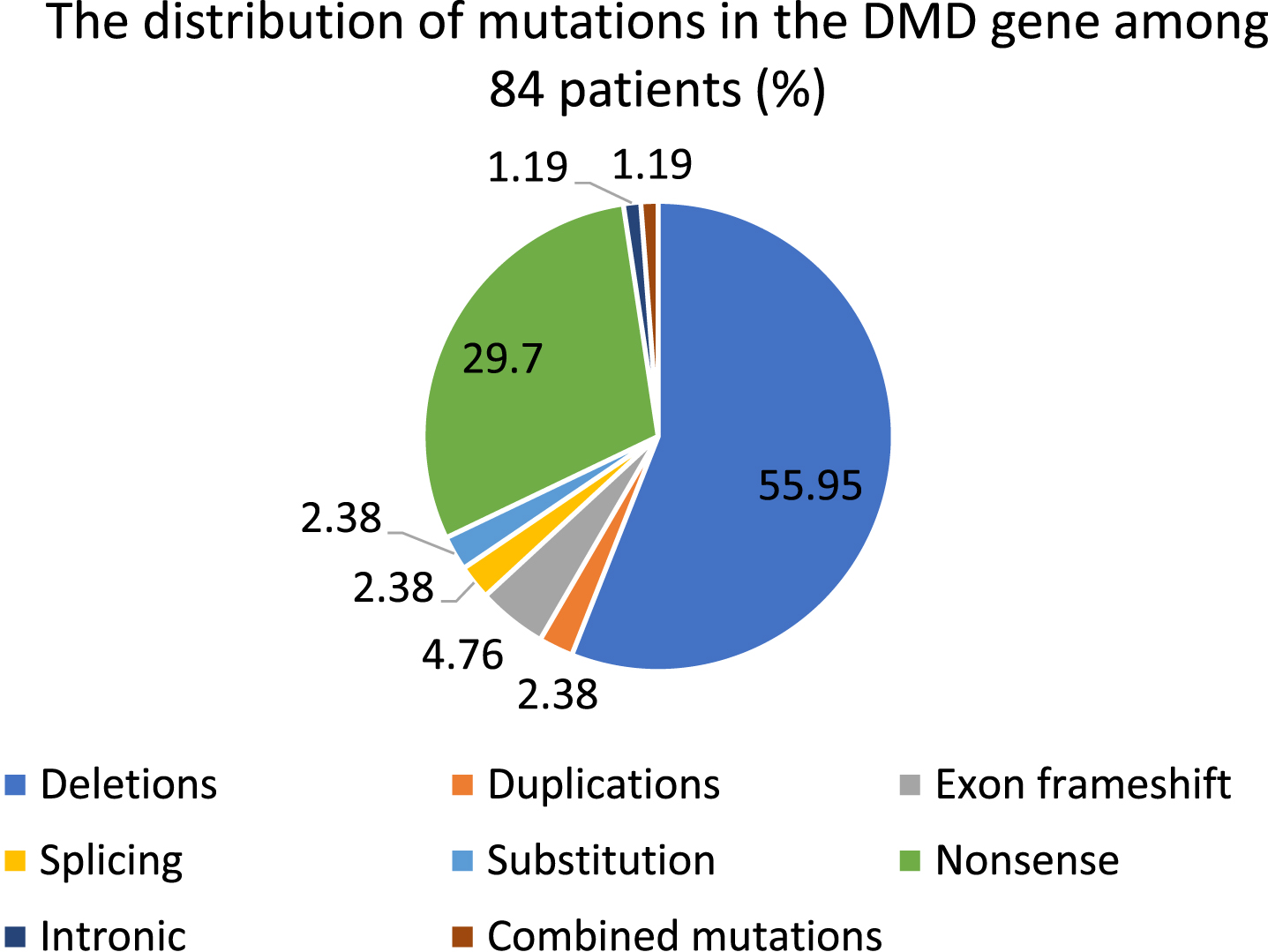
The mean age of 84 examined patients was 10 years. In Kazakhstan, the average age of disease manifestation was 3 years and 3 months. The vast majority of patients passed through genetic test due to the clinical manifestations. The average age of genetic confirmation was 7 years and 6 months. There were 58,33%of gross variations, of which 55,95%were deletions and 2,38%were duplications. Nonsense mutations were identified in 29,7%.
Conclusion:
The authors contend that strictly keeping the clinical guides in the diagnosis of DMD is essential, as the genetic variations may affect the stage and feasibility of novel therapies. The way of management of neuro-muscular diseases used in Kazakhstan is strictly recommended for implementation in developing countries.
1INTRODUCTION
Duchenne Muscular Dystrophy (DMD) is a rare neuromuscular X-linked disorder that belongs to a group of disorders known as dystrophinopathies. DMD is caused by pathogenic variations in the dystrophin gene, which is one of the largest known human genes. DMD gene variations lead to an absence or reduction of the dystrophin protein. Dystrophin is notably crucial for the stability of the membrane in skeletal muscle. Dystrophin isoforms are also expressed in the brain which accounts for cognitive impairment often seen in DMD. As such, the lack of functional dystrophin impairs the structure and function of myofibres, something critical for the physiological growth and maintenance of muscle tissue [1].
To date, more than 3,000 various disease-causing variations of the DMD have been reported. The majority of them are deletions that account for 65–78%of these variations. In most deletions, mutations may lead to alteration of the open reading frame (ORF) leading to a complete absence of dystrophin. The rest of the variations consists of duplications (6%–10%), point mutations (missense, nonsense, and splice site variations) (10–25%), or other smaller rearrangements: insertions/deletions, small inversion. Approximately 2%of mutations are caused by complex mutations and deep intronic rearrangements [3].
Since 2014 several disease-modifying medications that affect disease progression have been approved. For example, Ataluren was authorised by the European Medicine Agency in 2014 [4, 5], Eteplirsen was approved by the U.S. Food and Drug Administration in 2016, and more recently golodirsen, viltolarsen and casimersen [6]. Such medications except viltolarsen are all in the list of clinical guides for treatment of approved for DMD/BMD in Kazakhstan.
Kazakhstani doctors have started to pay closer attention to DMD diagnosis over the last 5–7 years. As this diagnosis is not very common, based on the worldwide prevalence of disease (1 : 3500 alive born boys a year by National Organization for Rare disorders, 2019) we expect that there should be around 580 children in Kazakhstan with such a diagnosis.
This retrospective study aims to describe the establishment of diagnostic approaches based on the analysis of some clinical issues such as onset, stage of disease, CK level and the spectrum of dystrophin gene variations in DMD patients in Kazakhstan.
2MATERIALS AND METHODS
In this retrospective study conducted in Kazakhstan, data has been obtained from the Multidisciplinary reference centre on neuro-muscular disorders located in Astana, Kazakhstan. 84 patients from (age 2–19 years) have been included in the study. All patients were males. Although the sample studied is not fully representative of the whole Kazakh population, it still offers a valuable snapshot of the recorded cases of DMD diagnosis and treatment in Kazakhstan. The enrolment criteria for the study was a DMD diagnosis, identified genetic mutation by Multiple Ligation Probe Amplification and/or by sequencing of DMD gene or NGS analysis. Written consent was provided by the parents, when patients proceeded with the genetic analysis. The MLPA analysis was conducted in Kazakhstan (MRC-Holland Salsa MLPA probemix P034/P035) and in Germany (Centogene variant classification (based on ACMG recommendations)). The statistical data has been processed by the Excel program.
3RESULTS
84 patient’s records were analysed. The age of patients varied between 2 years and 19 years. The median age of patients was 10 years at registration.
The onset of disease has been estimated in 58 cases (motor signs, milestones delay and transaminase elevation). The average age of onset of disease was 3 years and 4 months. The minimal age of onset was 4 months (liver transaminase elevation) and the maximal age of disease onset was 96 months (8 years). 23 records do not contain data about the time when the first clinical manifestations were noted. Three cases among 84 were diagnosed at a presymptomatic stage within the framework of the family screening.
The medical records contain data on clinical stages in almost all cases except one. On physical examination, the stage when the patient’s diagnosis was established as ambulatory –60, non-ambulatory –20 and 1 patient was on the stage of diagnosis estimation (4 months). 2 patients were discussed on Board commission twice and the clinical stage was initially ambulatory, which deteriorated to non-ambulatory later on (Table 1).
Table 1
The distribution of mobility by age
| Age | Preclinical stage | Ambulatory stage | Nonambulatory stage |
| Total | 1 | 60 | 20 |
| 1–2 years | 1 | ||
| 2–3 years | 1 | ||
| 3–4 years | 1 | ||
| 4–5 years | 4 | ||
| 5–6 years | 3 | ||
| 6–7 years | 8 | ||
| 7–8 years | 4 | ||
| 8–9 years | 12 | ||
| 9–10 years | 7 | 4 | |
| 10–11 years | 8 | 1 | |
| 11–12 years | 5 | 1 | |
| 12–13 years | 4 | ||
| 13–14 years | 3 | 3 | |
| 14–15 years | 2 | ||
| 15–16 years | 3 | ||
| 16–17 years | 1 | ||
| 17–18 years | 2 | ||
| >18 years | 3 |
The average age when the genetic diagnosis has been firmly established was 7 years and 6 months. The minimal age of genetic confirmation was 4,5 months and the maximal age when the genetic test was performed was 17 years.
There were 49 cases (58,33%) of gross rearrangements, of which 47 (55,95%) were deletions (one exon or multiple exons) and 2 (2,38%) were duplications. Nonsense mutations were identified in 25 cases (29,7%). Combined mutation was diagnosed in 1 case (1,19%). There were 4 frameshift mutations (4,76%), as well. 2 mutations (2,38%) among all pathogenic variations were substitution and splicing. 1 case (1,19%) of intronic mutation was recorded (Tables 2, 3, Diagram 1)
Table 2
Spectrum and frequency of mutations
| Type | Frequency | %of total number of mutations |
| Large mutations | 49 | 58,33% |
| Deletions | 47 | 55,95 |
| Duplications | 2 | 2,38 |
| Point and small mutations | 34 | 40, 41% |
| Exon frameshift | 4 | 4,76 |
| Splicing | 2 | 2,38 |
| Substitution | 2 | 2,38 |
| Nonsense | 25 | 29,7 |
| Intronic | 1 | 1,19 |
| Combined mutation | 1 | 1, 19% |
| Total number | 84 | 100% |
Table 3
Spectrum and frequency of deletion cases
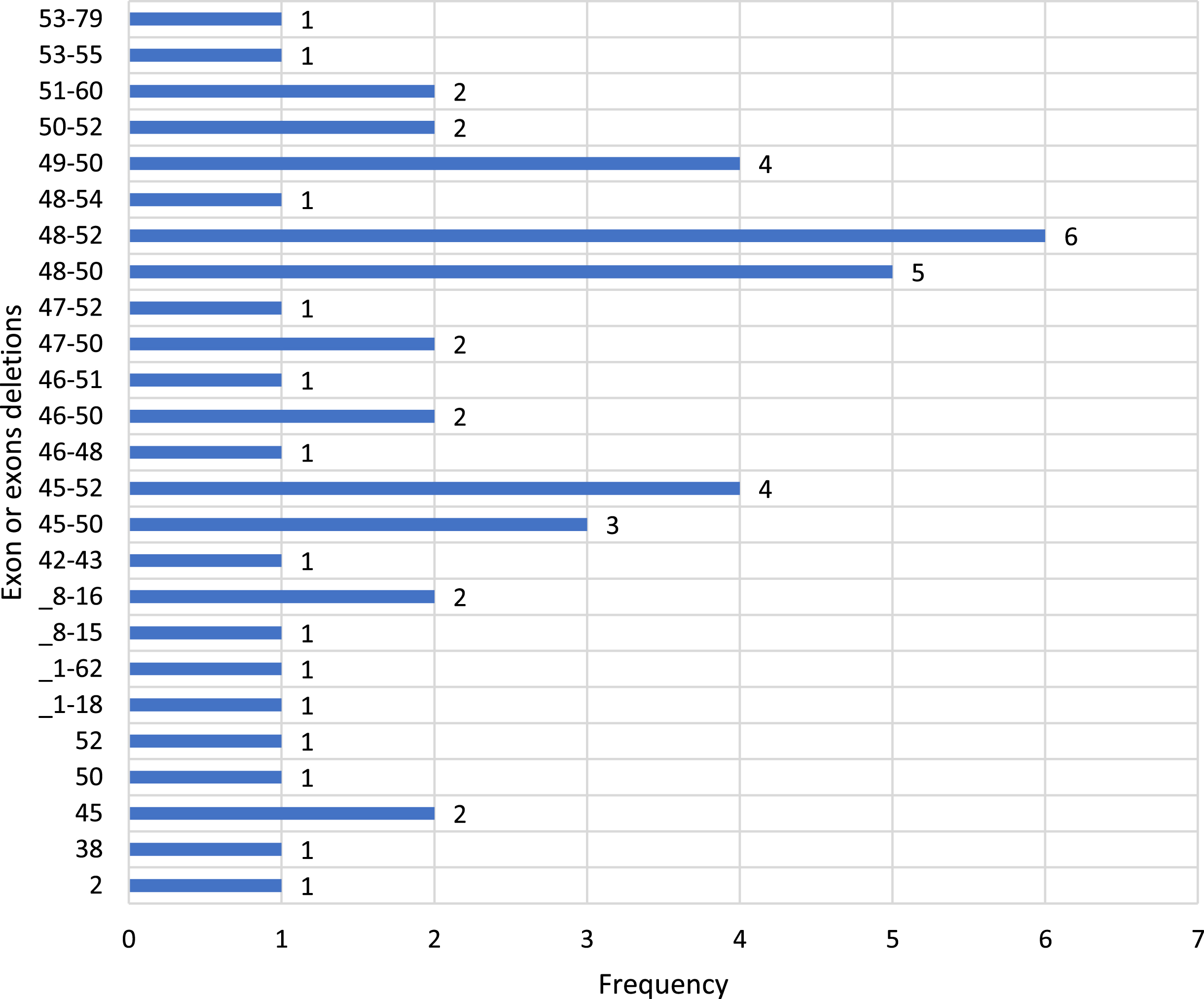
Diagram 1
The distribution of DMD pathogenic variations among patients (total number is 84 (in %)).
The level of creatine phosphokinase in primary years of disease was known in 61 patients. The average level of creatine phosphokinase is –8 516,66 IU.
4DISCUSSION
The diagnostic method in Kazakhstan remains the same as in the other countries. Initially, since 1995, the PCR analysis of 19 exons has been launched, then since 2011 the diagnostic method has been modernised and MLPA analysis has been implemented. A biochemical blood analysis (creatine phosphokinase (CK), lactate dehydrogenase (LDH), liver transaminase levels as ALT and AST), an electromyography and MLPA are conducted on patients who exhibit clinical manifestations, have specific complaints, and/or have a positive family history. Such diagnostic procedures are all available in Kazakhstani laboratories.
It should be noted, however, that while there is recognition that an electromyography is limited in the diagnosis of DMD and is not widely used internationally, its practice in Kazakhstan, as a way to narrow down the search for a diagnosis, has remained active since the Soviet era. The negative MLPA analysis in clinically manifested children gives the opportunity to proceed with NGS in order to reveal the pathological variations. The NGS method is conducted in the Centogene laboratory (Germany). Ideally, the patient with a positive genetic test should then be directed to a paediatric neurologist, who specialises in neuromuscular diseases. In reality, even with confirmed clear diagnostic algorithms many medical professionals are not familiar with neuromuscular conditions, naturally leading to wrong diagnoses and inappropriate management.
In Kazakhstan, management of patients with any neuro-muscular disorder is a burning issue now due to an increasing number of patients with muscle diseases, although the alertness of doctors is virtually lacking regarding this rare condition. Many of these patients have wrong diagnosis and they are treated more aggressively than needed due to heterogenicity of muscle diseases. In Kazakhstan, international guidelines for diagnostic and treatment approaches to Duchenne muscle dystrophy were just recently accepted, and we started to give patients more or less proper care including steroids, respiratory and nutritional support. Nevertheless, other types of muscle diseases are still underdiagnosed or even undiagnosed due to lack of knowledge and absence of a system for neuromuscular service in Kazakhstan. Proper diagnosis and management for such kinds of rare diseases are mostly dependent on the growth of scientific knowledge and the development of new therapies. Importantly, the status of some of those diseases are now moved from the untreatable to treatable side. Needless to say, it is very important to establish a system for muscle diseases in order to provide proper health care to patients through scientifically-proven approaches, such as imaging, immunohistochemistry, and molecular studies.
As a result of a lack of DMD specialists in Kazakhstan but the necessity in solving the issue at the same time, the national expert-consulting board at Multidisciplinary reference centre on neuro-muscular disorders was created in Astana, Kazakhstan in August 2020. The national expert-consulting board consists of 10 specialists, whose work focuses on neuro-muscular disorders. This Board considers the clinical, functional and genetic status of the patient, advises a diagnostic method, prescribes therapy and manages treatment.
Our study shows that the mean age of patients with DMD, when the doctors started to discuss a target treatment possibility, is 10 years. The average age for the onset of the DMD in Kazakhstani patients, according to clinical observations, is 3 years and 3 months. But the decision to implement target treatment is only possible when the doctors have a genetic confirmation of mutation at DMD gene. Our data shows that the genetic diagnosis in Kazakhstan is performed later, demonstrating a delay of diagnosis and treatment for more than 4 years. Since the discovery of the DMD gene in 1987, significant progress has been made in genetic testing using DNA analysis for DMD, so that it is now possible to identify DMD mutations from blood specimens in over 95%of patients with DMD [8]. Several clinic-based studies spanning more than a decade show a mean age for diagnosis of DMD between 4 and 5 years [9–12]. The delay of diagnosis in Kazakhstan results in lost opportunities for timely genetic counselling and initiation of steroids and target treatment. Some patients and doctors are afraid of steroids’ side effects, which is a treatment stigma. According to reports of the national expert-consulting board at Multidisciplinary reference centre on neuro-muscular disorders 4 patients among 24 non-ambulant have been prescribed Ataluren. Our data shows that the patients start to become non ambulant at the age of 9–10 years. Most of the patients can keep ambulation up to 13–14 years of age and we have not seen ambulant patients older than 14–15 years. Such findings reflect known information about the natural duration of the disease. According to the publications, the disease progresses very quickly and usually the patients are in need of a wheelchair at the age of 10 years. The life expectancy has also been significantly extended through the use of corticosteroid treatment and higher standards of medical care, such as non-invasive ventilation, although patients still tend to die from cardiac and respiratory complications [13]. DMD patients develop a severe cardiomyopathy that generally manifests at about 10 years and is prevalent in most patients by 20 years of age [14]. So, the above data demonstrated that we have to concentrate our efforts on early detection of DMD in order to optimise attempts to use a disease modification therapeutic approach.
The earlier the age of onset, the better the diagnostic opportunities and further the feasibility of specified therapy [15].
Furthermore, the lack of information on the level of CK in the initial stages or during the course of the disease on some of the patient’s records, indicates an underestimation of this laboratory feature in the cases of the DMD. Moreover, in some cases elevated CK level was misdiagnosed as hepatitis and patients were wasting time finding the correct diagnosis [16].
The number of involved patients is 84, which is not high compared with other nationwide studies. The percentage of 58,3%in large deletions by MLPA in our study is not as high as in Bladen’s paper of the TREAT-NMD DMD Global database (80%). [17] However it is close to the Italian cohort of 1162 DMD patients, where deletions were found in 57%of cases [18]. In correlation to a study in an Asian country, China (60.2%) the findings are similar [19(2)]. More often we have detected deletions in exons 48–52 in 12,7%, exons 48–50 and exons 45–52 and 49–50, respectively.
Nonsense mutations in our group of DMD males were found in 29,7%which is quite a high group of pathogenic variations. It was described that approximately 15%of DMD cases are caused by a nonsense mutation [5] and 17%of deletions are amenable for a target treatment approach. The previous studies in Kazakhstan in 2019 and 2021 demonstrated 27%among nonsense mutations in DMD gene [20, 21]. The highest level of nonsense mutations may be caused by excessive activity in seeking specified available therapy that is provided by pharmaceutical companies. So actively developed possibilities in disease course correction may lead to bias in analysis of statistical data in the country. This occurrence is more visible in low- or middle-income countries with no special programmes for disease screening due to restrictions in feasibility of free warranted genetic analysis. These statistical biases may lead to consequences within the whole medical care system, for example in terms of management (money planning for specified medicines).
Other countries do struggle with similar problems as Kazakhstani doctors: delayed diagnosis, funding deficits (22,23).
5CONCLUSION
1) The diagnostic method and connected targeted therapy are modernised. The clinical guide for DMD/BMD has been edited annually in 2020 and 2021 due the inclusion of modern medications for targeted therapy.
2) Biochemical analysis is crucial in setting up a diagnosis on time. The analysis of CK level in our patients confirms the necessity of conducting laboratory diagnosis. It is an obligatory step in the diagnostic algorithm.
3) The genotype definition is crucial for mutation-specific treatment. As there are only 10–15%of patients with point mutations among genetically confirmed cases corresponding to target therapy, we need to accelerate the alertness to DMD/BMD diagnosis. But in our group 29,7%of cases have nonsense mutation. That means that, in the case of suspicion of this diagnosis, we should perform the genetic test as early as possible. The high percentage of nonsense mutation (29,7%) may cause statistical, epidemiological bias. It is difficult to say whether our findings are representative because initially cohorts that do have modified mutations have been involved in the study (preliminary-modified). However, as the birth incidence is high, we do expect the high prevalence of pathogenic variations.
4) There is limited access to specified pathogenetic medications, so not all patients are able to get exon skipping therapies. However, even Kazakhstan, which is classified to countries with low and middle income by the World Development Bank, has broad involvement of patients among countries, and has seen improvement of health care through the implication of targeted therapy.
5) Since targeted therapy of DMD disease is a novel therapy in Kazakhstan, it is still too early to make conclusions about the effectiveness and safety of medicines.
6) The Expert-Consulting Board at the Multidisciplinary centre of excellence on neuro-muscular disorders does recommend the use of the above described management algorithm of neuro-muscular disorders in the developing countries.
CONFLICT OF INTEREST
The authors have no conflict of interest to report
REFERENCES
[1] | Birnkrant DJ , Bushby K , Bann CM , Apkon SD , Blackwell A , Brumbaugh D , et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Glob Health. (2018) ;4422: (18). https://doi.org/10.1016/S1474-4422(18)30024-3. |
[2] | Giliberto F , Radic CP , Luce L , Ferreiro V , de Brasi C , Szijan I . Symptomatic female carriers of Duchenne muscular dystrophy (DMD): Genetic and clinical characterization. J Neurol Sci. 2014: ;336: (1–2):36–41. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24135430. |
[3] | Falzarano MS , Scotton C , Passarelli C , Ferlini A . Duchenne Muscular Dystrophy: From Diagnosis to Therapy. Molecules (2015) ;20: :18168–84. doi:10.3390/molecules201018168. |
[4] | Wood CL , Cheetham T . Treatment of Duchenne muscular dystrophy: First small steps. The Lancet. (2017) ;390: (10101):1467–8. https://doi.org/10.1016/S0140-6736(17)31669-0. |
[5] | McDonald MC , Campbell C , Torricelli RE , Finkel RS , Flanigan KM , Goemans N , et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): A multicentre, randomised, double-blind, placebocontrolled, phase 3 trial. Lancet. (2017) ;390: :1489–98. |
[6] | Takeda S , Clemens PR , Hoffman EP . Exon-Skipping in Duchenne Muscular Dystrophy. Journal of Neuromuscular Diseases. (2021) ;8: :S343–58. DOI 10.3233/JND-210682. |
[7] | Jaxybayeva A . Management of neuromuscular diseases at children in Kazakhstan: Challenges and pathways for solving. Pediatria I Detskaya Chirurgiya-Pediatric and Children’s Surgery. (2020) ;2: (100):13–22. |
[8] | Flanigan KM , von Niederhausern A , Dunn DM , Alder J , Mendell JR , Weiss RB . Rapid direct sequence analysis of the dystrophin gene. Am J Hum Genet. (2003) ;72: :931–9. [PubMed: 12632325]. |
[9] | Parsons EP , Clarke AJ , Bradley DM . Developmental progress in Duchenne muscular dystrophy: Lessons for earlier detection. Eur J Paediatr Neurol. (2004) ;8: :145–53. [PubMed: 15120686]. |
[10] | Mohamed K , Appleton R , Nicolaides P . Delayed diagnosis of Duchenne muscular dystrophy. Eur J Paediatr Neurol. (2000) ;4: :219–23. [PubMed: 11030068]. |
[11] | Ciafaloni E , Fox DF , Pandya S , Westfield CR , Puzhankara S , Romitti PA , et al. Delayed Diagnosis in Duchenne Muscular Dystrophy: Data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet) /J Pediatr. (2009) ;155: (3):380–5. doi:10.1016/j.jpeds.2009.02.007. |
[12] | López-Hernández , Luz , et al., Duchenne muscular dystrophy in a developing country: Challenges in management and genetic counseling. Genetic Counseling. 2014. |
[13] | Wu B , Cloer C , Lu P , Milazi S , Shaban M , Shah SN , et al. Exon skipping restores dystrophin expression, but fails to prevent disease progression in later stage dystrophic dkomice. Gene Ther. (2014) ;21: :785–93. |
[14] | Nakamura A . X-Linked Dilated Cardiomyopathy: A Cardiospecific Phenotype of Dystrophinopathy. Pharmaceuticals (Basel). (2015) ;8: :303–20. |
[15] | Gowda VL , Fernandez M , Prasad M , Childs AM , Hughes I , Tirupathi S , et al. Prediagnosis pathway benchmarking audit in patients with Duchenne muscular dystrophy. Arch Dis Child, (2022) ;107: :160–5. doi:10.1136/archdischild-2020-32145111/. |
[16] | Zhang Y , Huang JJ , Wang ZQ , Wang N , Wu ZY . Value of muscle enzyme measurement in evaluating different neuromuscular diseases. Clinica Chimica Acta. (2012) ;413: (3–4):520–4. |
[17] | Bladen CL , Salgado D , Monges S , Foncuberta ME , Kekou K , Kosma K , et al. The TREAT-NMD DMD global database: Analysis of more than 7,000 duchenne muscular dystrophy mutations. Hum Mutat [Internet]. (2015) Apr 1 [cited 2021 Apr 25];36: (4):395–402. Available from: https://pubmed.ncbi.nlm.nih.gov/25604253/ |
[18] | Neri M , Rossi R , Trabanelli C , Mauro A , Selvatici R , Falzarano MS , et al. The Genetic Landscape of Dystrophin Mutations in Italy: A Nationwide Study. Front Genet [Internet]. (2020) Mar 3 [cited 2021 Apr 25];11: :1. Available from: /pmc/articles/PMC7063120/. |
[19] | Guo R , Zhu G , Zhu H , Ma R , Peng Y , Liang D , et al. DMD mutation spectrum analysis in 613 Chinese patients with dystrophinopathy. J Hum Genet. (2015) ;60: (8):435–42. |
[20] | Myrzaliyeva B , Lepessova M , Jaxybayeva A . Clinical and genetic characteristic of dystrophinopathy in children in Kazakhstan. Abstract book of the 13th European Paediatric Neurology Society (EPNS) Congress, 17–21 September, 2019, Athens, Greece, P. 281. |
[21] | Chunkayeva D , Jaxybayeva A . Clinical and genetic characteristics of patients with Duchenne muscular dystrophy. Proceedings XV International scientific-practical conference “Ecology. Radiation. Health” August 28, 2021, Semey city, p. 211–2. |
[22] | Alghamdi F , Al-Tawari A , Alrohaif H , Alshuaibi W , Mansour H , Aartsma-Rus A , Mégarbané A . Case Report: The Genetic Diagnosis of Duchenne Muscular Dystrophy in the Middle East. Front Pediatr. (2021) ;9: :716424. doi: 10.3389/fped.2021.716424. PMID: 34595143; PMCID: PMC8476401. |
[23] | Megarbane A , Bizzari S , Deepthi A , Sabbagh S , Mansour H , Chouery E , Hmaimess G , Jabbour R , Mehawej C , Alame S , Hani A , Hasbini D , Ghanem I , Koussa S , Al-Ali MT , Obeid M , Talea DB , Lefranc G , Lévy N , Leturcq F , El Hayek S , Delague V , Urtizberea JA . A 20-year Clinical and Genetic Neuromuscular Cohort Analysis in Lebanon: An International Effort. J Neuromuscul Dis. (2022) ;9: (1):193–210. doi: 10.3233/JND-210652. PMID: 34602496; PMCID: PMC8842757. |


