
Patients with Spinal Muscular Atrophy Type 1 Achieve and Maintain Bulbar Function Following Onasemnogene Abeparvovec Treatment1
Abstract
Background:
Improvement and maintenance of bulbar function are goals of disease-modifying treatments for spinal muscular atrophy (SMA). Lack of standardized measures and a widely accepted definition of bulbar function represents a gap in SMA care.
Objective:
A multidisciplinary team conducted post-hoc analyses of pooled data from one phase 1 (START) and two phase 3 (STR1VE-US, STR1VE-EU) studies to define and evaluate bulbar function of infants with SMA type 1 after receiving one-time gene replacement therapy, onasemnogene abeparvovec.
Methods:
We defined bulbar function as the ability to meet nutritional needs while maintaining airway protection and the ability to communicate verbally. Four endpoints represented adequate bulbar function: (1) absence of clinician-identified physiologic swallowing impairment, (2) receiving full oral nutrition, (3) absence of adverse events indicating pulmonary instability, and (4) the ability to vocalize at least two different, distinct vowel sounds. We descriptively assessed numbers/percentages of patients who achieved each endpoint and all four collectively. Patients were followed until 18 months old (STR1VE-US and STR1VE-EU) or 24 months (START) post-infusion.
Results:
Overall, 65 patients were analyzed for swallowing, nutrition intake, and adverse events, and 20 were analyzed for communication. At study end, 92% (60/65) of patients had a normal swallow, 75% (49/65) achieved full oral nutrition, 92% (60/65) had no evidence of pulmonary instability, 95% (19/20) met the communication endpoint, and 75% (15/20) achieved all four bulbar function components in the composite endpoint.
Conclusions:
In these three clinical trials, patients with SMA type 1 who received onasemnogene abeparvovec achieved and maintained the bulbar function criteria utilized within this investigation.
INTRODUCTION
Spinal muscular atrophy (SMA) is a monogenic, neurodegenerative disease related to biallelic mutations of the survival motor neuron 1 (SMN1) gene. SMN1 directs the production of the ubiquitously expressed survival motor neuron (SMN) protein, which is essential for the development and maintenance of motor neurons in the spinal cord and brain. The loss of the SMN1 gene results in the degeneration of motor neurons, leading to severe, progressive skeletal and respiratory muscle weakness and atrophy and, in severe forms, early death [1, 2]. Historically, five clinical phenotypes of SMA have been identified, with presentations ranging from profound weakness at birth (type 0) to relatively mild symptoms with adult onset (type 4) [3–6]. Type 1 is the most common phenotype of SMA. It defines those children who have a very severe presentation, and symptoms usually appear during the first 6 months of life. Without treatment, patients with SMA type 1 present with hypotonia and poor-to-absent head control. They do not sit independently and usually do not survive without permanent ventilation and nutritional support past 2 years of age [7, 8]. In addition to nutritional and respiratory deficits for patients with SMA type 1 [7–9], communication is hindered because of global muscle weakness, including components of bulbar function [10–12].
Bulbar motor neurons, located in the lower brain stem, control muscles required for mouth opening, chewing, swallowing, and speaking, and are affected by SMA. Impaired bulbar function in SMA manifests as difficulties with these activities [13] and can lead to choking, malnutrition, aspiration, and respiratory infections, potentially resulting in death [6, 13–19]. Specifically, dysfunction of bulbar motor neurons in SMA leads to tongue fasciculations, poor suck, and difficulty feeding and swallowing, and as a result, aspiration pneumonia is a risk for morbidity and mortality for patients with SMA [6, 16]. Improvement and maintenance of components of bulbar function are important goals of disease-modifying treatments for SMA, but standardized and validated measures do not exist [13], nor does a widely accepted definition of bulbar function in SMA.
Onasemnogene abeparvovec is a single-dose, intravenous gene replacement therapy that delivers a functional human SMN gene to restore expression of full-length SMN protein and is approved in multiple countries [20–22]. Clinical trials have demonstrated improved survival and motor function, as well as achievement of motor milestones such as sitting without support, standing alone, and walking alone, after administration of onasemnogene abeparvovec in infants with SMA type 1 [20–22]. The impact of onasemnogene abeparvovec on bulbar function has not yet been comprehensively evaluated.
A multidisciplinary team of academia and Novartis Gene Therapies SMA experts on deglutition, respiratory function, physical therapy, nutrition, neurology, clinical statistics, and cell and tissue engineering conducted post-hoc analyses of pooled data from one phase 1 (START NCT02122952 [20]) and two phase 3 (STR1VE-US NCT03306277 [21] and STR1VE-EU NCT03461289 [22]) studies to define and evaluate components of bulbar function in patients with symptomatic SMA type 1 who received onasemnogene abeparvovec.
MATERIALS AND METHODS
To determine the effects of onasemnogene abeparvovec on bulbar function, we first assembled a team of multidisciplinary experts in SMA to comprehensively define bulbar function. A consensus definition was reached after several roundtable discussions among the experts. We defined bulbar function as integrity within cranial nerves that enables an individual to (1) meet nutritional needs by mouth without pulmonary instability and (2) demonstrate verbal communication abilities.
Next, we retrospectively reviewed data collected during clinical trials of onasemnogene abeparvovec to identify outcomes that would reflect the definition of bulbar function, and four endpoints were selected to best represent key components of the established definition: (1) absence of clinician-identified (clinical or fluoroscopic) markers of physiologic swallowing impairment, (2) achievement of full oral nutrition (defined as not requiring a feeding tube for nutrition support and receiving 76–100% of nutrition via oral intake), (3) absence of adverse events relating to pulmonary stability (i.e., aspiration or aspiration pneumonia), and (4) achievement of item #6 or above on the Bayley Scales of Infant and Toddler Development, 3rd Edition Expressive Communication subtest, which is defined as the ability of a child to vocalize at least two different, distinct vowel sounds [23] (Table 1).
Table 1
Composite definition and assessment of bulbar function in SMA
| Bulbar function is integrity within cranial nerves that enables an individual to: | Swallow food and liquids without functional deficits | Meet nutritional needs by mouth | Maintain pulmonary stability | Establish verbal communication skills |
| Represented by: | Absence of clinician-identified (clinical or fluoroscopic) markers of swallowing impairment | Full oral nutrition | Absence of aspiration-induced morbidities because of compromised airway protection | Ability of a child to vocalize at least two different, distinct vowel sounds |
| Defined by: | Any finding of “functional swallow,” “normal swallow,” or “safe for swallow” | A lack of non-oral nutrition support and receiving 76–100% of nutrition via oral intake | No occurrence of aspiration or aspiration pneumonia | Achievement of item #6 or above on the Bayley Scales of Infant and Toddler Development 3rd Edition Expressive Communication subtest [23] |
SMA, spinal muscular atrophy.
Data from three clinical studies were pooled for these post-hoc analyses: START [20], STR1VE-US [21], and STR1VE-EU [22]. The details of the study designs and patient populations have been published [20–22]. Briefly, START was a phase 1 study of onasemnogene abeparvovec that included 15 patients, 12 of whom received a dose equivalent to the therapeutic dose of 1.1×1014 vector genomes per kilogram of body weight (vg/kg). STR1VE-US and STR1VE-EU were phase 3 studies that included 22 and 32 patients in the intention-to-treat populations, respectively. All patients in the STR1VE studies received the therapeutic dose of onasemnogene abeparvovec of 1.1×1014 vg/kg. Patients were followed until 18 months of age in STR1VE-EU and STR1VE-US and until 24 months post-infusion in START. Patients from START, STR1VE-US, and STR1VE-EU were included in this post-hoc analysis if they received the therapeutic dose (or equivalent) of onasemnogene abeparvovec and were younger than 6 months old at the time of treatment administration. We assessed numbers and percentages of patients who achieved each endpoint at predetermined intervals during the study and all four endpoints at study end as a composite endpoint.
In the three studies, swallowing assessments were performed at screening, every 6 months starting at Month 6, and at the end of the study when patients reached 18 months of age (STR1VE-US and STR1VE-EU) or 24 months after infusion (START). Swallowing was assessed by a clinical bedside examination or a fluoroscopic examination, according to standard practice and clinician decision at each study site. In START, fluoroscopy was used at each assessment. However, in STR1VE-US and STR1VE-EU, the choice of test varied between study sites and practitioners based on local standard practices, and information on the method used was not collected under the study protocols and was, therefore, not available for this analysis. Because the goal of this analysis was to evaluate bulbar function and not specific physiologic integrity, as well as the fact that “normal” fluoroscopic results are not clearly delineated in infants, any finding of “functional swallow,” “normal swallow,” or “safe for swallow” was categorized as an absence of swallowing impairment (or normal swallow) for this analysis.
The need for nutrition support was assessed via parent report or provider observation at screening and every 6 months in all three studies. The percentage of oral intake was assessed at screening and every 6 months (STR1VE-US and STR1VE-EU) and only from Month 18 (START) until the end of the study. A lack of non-oral nutrition support and receiving 76–100% of nutrition via oral intake together represented maximum oral nutrition for this analysis.
Information on adverse events was collected at every study visit beginning at screening through the end of the study. Adverse events of aspiration or aspiration pneumonia were considered evidence of respiratory compromise. A lack of these adverse events was considered evidence of pulmonary stability for this analysis.
Communication was only assessed for patients from native English-speaking families in START and STR1VE-US. Full Bayley Scales, including subtests of receptive and expressive communication, were administered at screening, every 3 (START) or 6 (STR1VE-US) months, and at the end-of-study visit. Bayley Scales were not assessed for patients in START who did not achieve a score of at least 60 out of 64 on the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND). The communication endpoint for this post-hoc analysis was considered achievement of item #6 or greater on the Bayley Expressive Communication subtest. Though true achievement of functional expressive communication varies based on patient age and is not identified by a single skill cut-point, age-based data were not available for the majority of participants in this retrospective investigation. As such, item #6 on the Bayley Scales was selected, because these data were available for most patients and it reflected early expressive communication.
Performance at the last evaluated time point was used to judge achievement of individual and composite bulbar function outcomes. Analyses were descriptive only, and no hypothesis was considered for this post-hoc analysis. Categorical variables are described as numbers and percentages.
RESULTS
Patient disposition
In all, 65 patients were included in the analyses of swallowing, nutrition intake, and adverse events: 11 patients from START, 22 from STR1VE-US, and 32 from STR1VE-EU. Twenty patients from STR1VE-US had communication assessed at baseline, and 20 patients (four from START and 16 from STR1VE-US) had communication assessed before end of study. Not all patients had data for all endpoints, and, in total, 20 were included in the evaluation of the four-component composite endpoint.
All patients received a one-time administration of onasemnogene abeparvovec. Overall, onasemnogene abeparvovec was well-tolerated, and few patients experienced serious adverse events related to the study drug (Supplementary Table). All serious adverse events resolved.
Individual endpoints
At baseline, 88% (57/65) of patients had a normal swallow. At the last evaluated time point, 92% (60/65) had evidence of a normal swallow after receiving onasemnogene abeparvovec (Table 2, Fig. 1).
Table 2
Individual and composite endpoints
| START [20] | STR1VE-US [21] | STR1VE-EU [22] | Combined | |
| (N = 11), | (N = 22), | (N = 32), | (N = 65), | |
| n (%) | n (%) | n (%) | n (%) | |
| Endpointa | ||||
| Normal swallow | ||||
| Baseline | 4 (36) | 22 (100) | 31 (97) | 57 (88) |
| End of studyb | 11 (100) | 20 (91) | 29 (91) | 60 (92) |
| Full oral nutrition | ||||
| Baseline | 7 (64) | 22 (100) | 26 (81) | 55 (85) |
| End of studyb | 6 (55) | 19 (86) | 24 (75) | 49 (75) |
| Able to maintain pulmonary stability | ||||
| Baseline | 11 (100) | 22 (100) | 32 (100) | 65 (100) |
| End of studyb | 8 (73) | 20 (91) | 32 (100) | 60 (92) |
| Able to communicatec | ||||
| Baseline | N/A | 4 (20) | N/A | 4 (20) |
| End of studyb | 4 (100) | 15 (94) | N/A | 19 (95) |
| Composite endpointd | ||||
| End of studyb | 3 (75) | 12 (75) | N/A | 15 (75) |
N/A, not assessed. aPercentages calculated as a proportion of patients with available data. b“End of study” represents the last evaluated time point during the study period. cAt baseline, communication was assessed for 20 patients in STR1VE-US; before end of study, communication was assessed for four patients in START and 16 in STR1VE-US. dFour patients from START and 16 from STR1VE-US had available data for the composite endpoint.
Fig. 1
Percentage of patients meeting the individual endpointsa representing bulbar function. aPercentages calculated as a portion of patients with available data (N = 65 for swallow, nutrition, and airway protection; N = 20 for communication). At baseline, communication was assessed for 20 patients in STR1VE-US; communication was assessed before end of study for four patients in START and 16 in STR1VE-US.
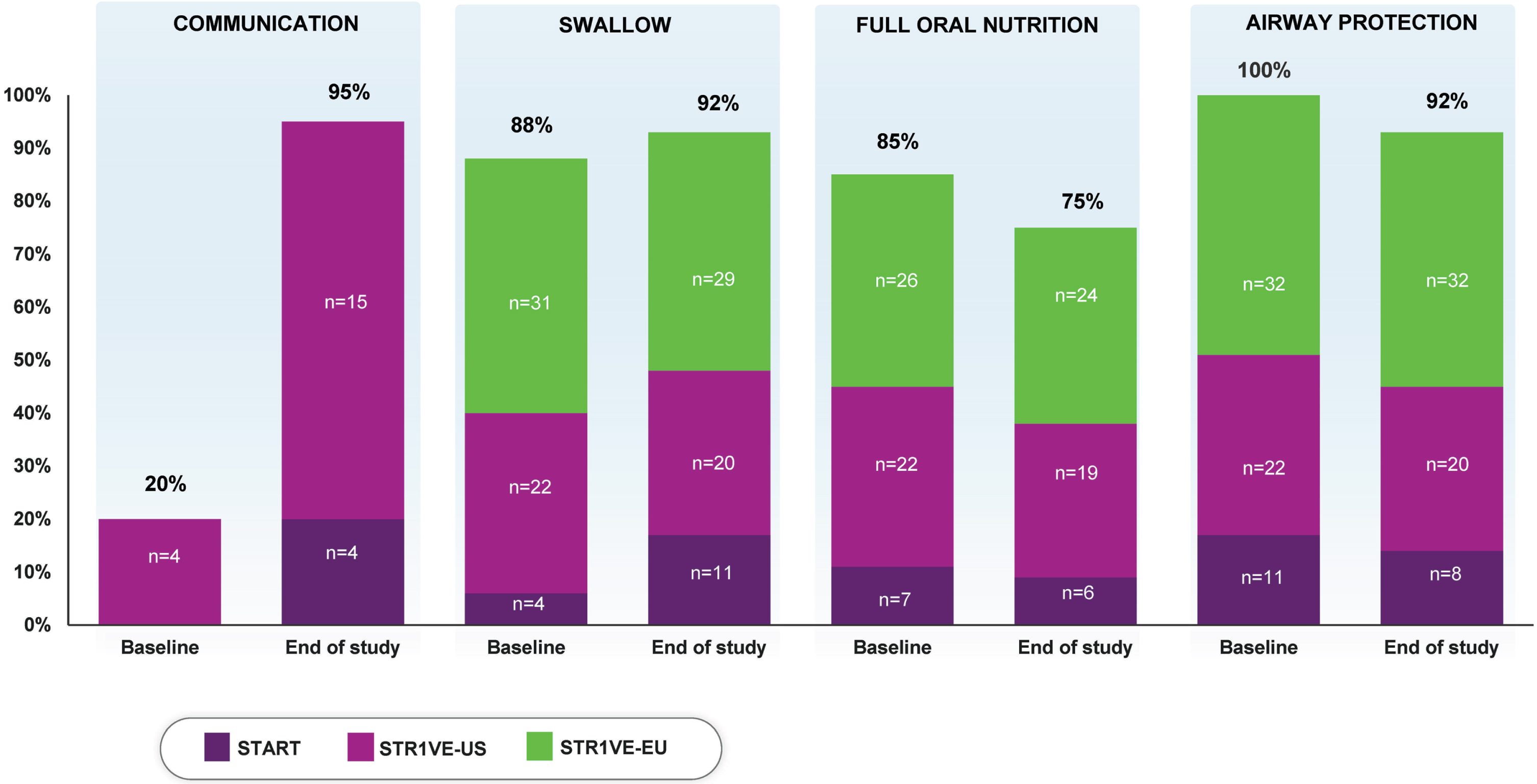
At baseline, 85% (55/65) of patients received full oral nutrition. At the last evaluated time point, 75% (49/65) of patients met this endpoint (Table 2, Fig. 1).
No patient had evidence of pneumonia or pulmonary instability at baseline. As of the last evaluated time point, 92% (60/65) of patients had no occurrence of aspiration or aspiration pneumonia reported (Table 2, Fig. 1).
At baseline, 20% (4/20) of patients met the communication endpoint. At the last evaluated time point, 95% (19/20) of patients met this criterion (Table 2, Fig. 1).
Composite endpoint
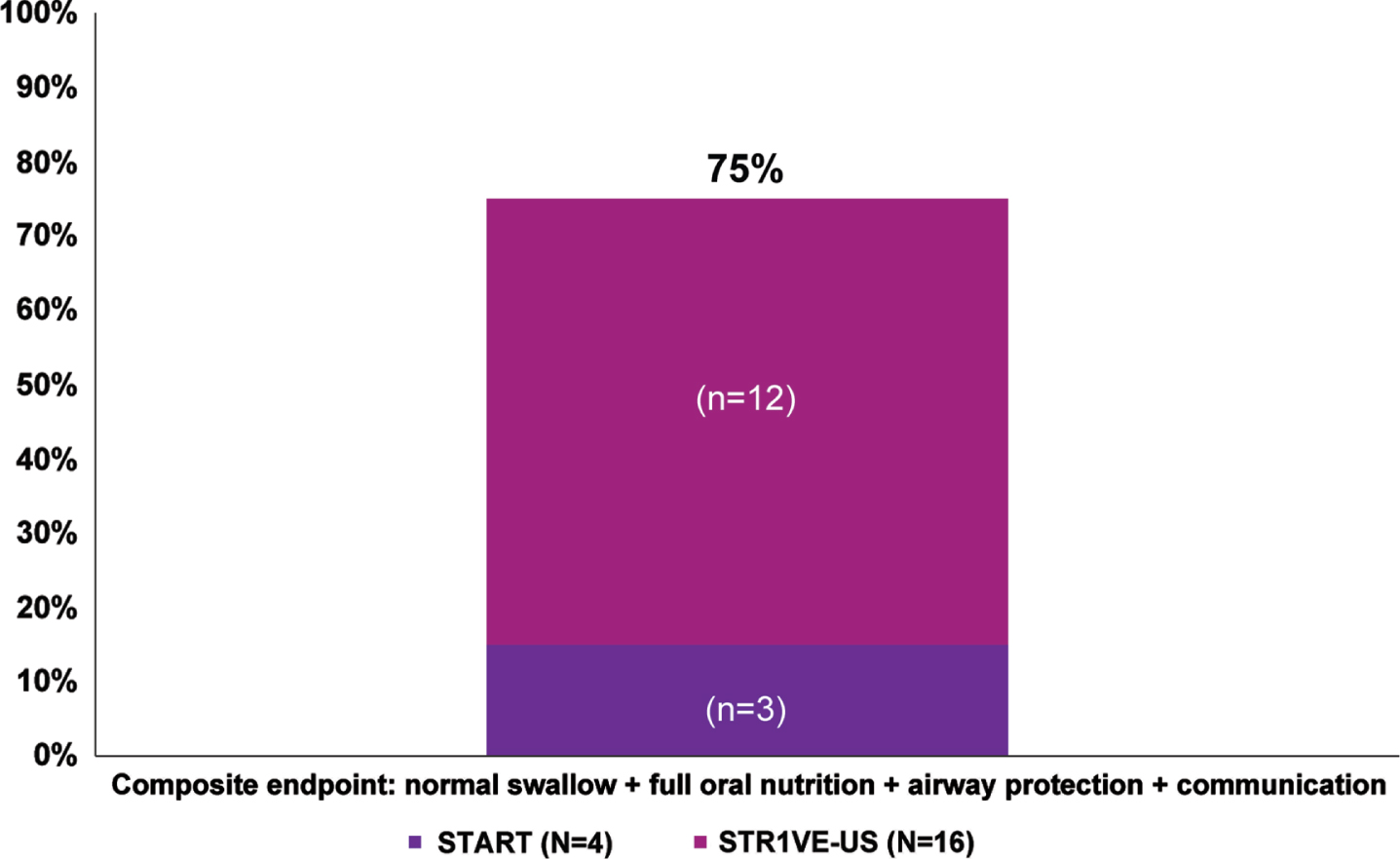
Overall, as of the last evaluated time point, 75% (15/20) of patients achieved the composite endpoint of having a normal swallow, achieving full oral nutrition, demonstrating no evidence of pulmonary instability, and demonstrating expressive communication abilities (Table 2, Fig. 2).
Fig. 2
Percentage of patients meeting the composite endpointa representing bulbar function. aPercentages were calculated as a portion of patients with available data (N = 65 for swallow, nutrition, and airway protection; N = 20 for communication; N = 20 for composite endpoint). At baseline, communication was assessed for 20 native-English speaking patients in STR1VE-US who had a CHOP INTEND score of 60 or greater; communication was assessed before end of study for four patients in START and 16 in STR1VE-US.
DISCUSSION
These post-hoc analyses indicate that patients with symptomatic SMA type 1 who were treated with onasemnogene abeparvovec as part of START, STR1VE-US, or STR1VE-EU could swallow and meet nutritional needs orally, maintain pulmonary stability, and establish early verbal communication skills. Together, these findings demonstrate promise that these patients achieved and/or maintained some integrity in bulbar function, and this rescue of bulbar function demonstrates substantial clinical benefit not observed in SMA type 1 natural history [7, 8].
Bulbar function has been rarely studied in patients with SMA. The few analyses conducted to date have mostly included patients with SMA types 2 or 3 [24–28]. Overall, patients with SMA have dysphagia and mastication problems as the tongue and pharyngeal muscles weaken, though the frequency of these manifestations varies, partly because of the inconsistent definitions used in studies and subjective measurements reported by patients.
Standardized assessment of bulbar function using reliable, objective outcome measures are lacking, and this represents a knowledge gap in the current management of SMA. Motor function and bulbar function, such as speaking, chewing, and swallowing, are assessed by different clinical evaluators and are not consistently evaluated with the tools used to assess motor function in SMA type 1, such as the CHOP INTEND, the Hammersmith Functional Motor Scale Expanded, and the Revised Upper Limb Module, and these scales do not account for impaired bulbar function [29]. To fill this gap, we established our new and stringent definition of bulbar function as a single, composite index of an individual’s ability to safely consume food or liquids via oral intake without nutritional or pulmonary compromise and to establish verbal communication skills. We then selected four distinct endpoints from available clinical trial data to best reflect this definition. Based on these endpoints, 75% of patients with SMA type 1 who were evaluated in three studies achieved and maintained bulbar function improvements. Though the retrospective nature of this investigation limited our ability to use age-based standardized assessments that may offer more information regarding true age-appropriate deglutition and communication functions, the achievement of the selected endpoints represents a tremendous success, as previous research suggests untreated patients with SMA would not achieve any of these endpoints. Specifically, the children included in START, STR1VE-US, and STR1VE-EU experienced onset of SMA symptoms prior to receiving onasemnogene abeparvovec, and, without treatment, would have been expected to require permanent ventilation and nutritional support before 2 years of age, as well as requiring alternative nonverbal communication modalities. Future investigations further elucidating the physiologic and functional swallowing parameters using standardized video-fluoroscopic and age-based communication assessments are needed.
A recently published study evaluated bulbar function in patients with SMA type 1 receiving nusinersen, an antisense oligonucleotide that increases SMN protein production. Bulbar function was assessed using the Paediatric Functional Oral Intake Scale. Twenty-four children were included in the study: three with SMA type 1a (the most severe, having a neonatal presentation), nine with SMA type 1b (symptom onset younger than 3 months of age), and 12 with SMA type 1c (symptom onset between 3 and 6 months old). Of these patients, 14 (58%) required nutrition support prior to nusinersen administration, and feeding and swallowing difficulties persisted after nusinersen treatment. In fact, the need for a feeding tube for nutrition support increased, with 20 (83%) infants requiring such support 24 months after initiation of nusinersen. Patients with the least severe phenotype (type 1c) were more likely to have maintained bulbar function after starting nusinersen treatment [30]. This evaluation only assessed swallowing and feeding and did not include pulmonary stability and communication as important components of bulbar function.
Other analyses have indicated that motor function improvements attained with nusinersen therapy did not translate to bulbar function improvements. In two recent studies of motor and bulbar function for patients with SMA type 1, CHOP INTEND scores increased after nusinersen initiation, but feeding and respiratory dysfunction persisted, and patients continued to experience dysphagia and respiratory failure that required intervention [31, 32].
Published data on bulbar function assessments following administration of risdiplam, an oral small molecule drug that increases functional SMN production, are limited. In a recent study of 41 infants with SMA type 1 (aged 1–7 months at enrollment) who received risdiplam daily for 24 months, no formal measure of bulbar function was assessed, but the abilities to swallow and feed orally were evaluated throughout the study by parent interview and subjective clinician-administered rating scales. Although most children maintained baseline function, no gains were achieved after receiving treatment with risdiplam. At baseline, 39 (95%) infants had the ability to swallow and 35 (85%) received at least some oral nutrition. By Month 24, three (7%) children had lost the ability to swallow, and no additional children were feeding orally. In addition, pneumonia was the most common adverse event, occurring in 19 (46%) infants, indicating pulmonary instability [33]. Similar to other studies, this study did not comprehensively assess all components of bulbar function and the criteria for meeting the endpoints evaluated were not rigorous nor objectively measured.
No head-to-head studies of bulbar function outcomes after disease-modifying therapy for SMA exist, and direct comparisons between previous studies and our post-hoc analyses are difficult because of differing study designs, populations, and outcomes. Still, our findings demonstrate achievement and maintenance of multiple components of bulbar function following treatment with onasemnogene abeparvovec for children with SMA type 1 who participated in these clinical trials. When considered with all improvements reported in the original START [20], STR1VE-US [21], and STR1VE-EU [22] studies, the rescue of bulbar function could be a key factor supporting the use of onasemnogene abeparvovec for SMA.
Bulbar dysfunction in neuromuscular disease, including SMA, leads to physical and emotional stress, risks of aspiration and failure to thrive, poor health-related quality of life, and dependency on caregivers and assistive devices [34, 35]. Specifically, children with SMA and their families experience social discomfort and stigma; constraints on social, academic, and employment activities; and limitations on independence [36–38]. Adequate bulbar function would allow children with SMA, as well as their families, to communicate better, socialize more, and feed orally. Simply, children who achieve and maintain bulbar function could more fully and comfortably participate in family and social interactions.
Nutrition, swallowing, airway protection, pulmonary stability, and communication are all important in the management of children with SMA. The multidisciplinary care team and patients or caregivers must monitor for the onset of or changes in signs of bulbar dysfunction, including dysphagia, aspiration, insufficient oral intake, prolonged and labored feeding time, and ineffective communication [19, 39]. Therefore, the definition of and vocabulary related to bulbar function, as well as scales and tools to measure bulbar function in SMA, should be standardized and applied consistently to guide clinical decision-making and improve outcomes for patients with SMA.
To our knowledge, these post-hoc analyses are the first to define and comprehensively evaluate bulbar function for patients with SMA, though, as with all investigations, limitations exist. Most notably, the use of pooled data from studies with different designs and study populations, including primary endpoints, timing of assessments, and ages of patients, limited the available data from which we could select endpoints to accurately reflect the established definition of bulbar function. For example, achievement of the communication component of the bulbar function definition was marked by the patient’s vocalization of at least two distinct vowel sounds. While the achievement of this milestone reflects functional early communication skills and a greater degree of integrity than untreated patients would have likely achieved, it does not reflect what would be expected for expressive communication in an older child. Likewise, because the study protocol did not include collection of data on these outcomes for patients with more severe SMA phenotypes, this limited our ability to report on their communication integrity and, therefore, our results may not fully reflect the entire breadth of SMA bulbar function. In addition, we chose not to include common, readily available measurements of growth in SMA, such as weight gain, but our endpoints serve as better representatives of bulbar function than typical metrics owing to the lack of standardized growth charts for patients with SMA [40–42]. Larger prospective studies using standardized and age-based assessments with longer follow-up periods are needed to assess the clinical stability of bulbar function for patients with SMA who receive onasemnogene abeparvovec. The authors intend to extend and validate the newly defined composite endpoint in other data sets. In addition, real-world studies and registries are currently ongoing, as are long-term follow-up studies of START, STR1VE-US, and STR1VE-EU that will help to clarify the durable and clinically significant benefits of onasemnogene abeparvovec. These post-hoc analyses represent an important advance toward consistency in the reporting of bulbar function in SMA, and in establishing a framework for its evaluation. The endpoints for our analyses were chosen based on available data from three clinical trials, providing what we believe represents an integrative basis for thorough analysis of bulbar function in this SMA type 1 population. Alternative criteria or surrogate endpoints may be used to assess bulbar function in different settings and for different patient populations.
CONCLUSIONS
Bulbar function includes the ability to meet nutritional needs orally while maintaining airway protection and demonstrating expressive communication abilities. In this post-hoc analysis, 75% of evaluable patients with SMA type 1 who received onasemnogene abeparvovec in a clinical trial setting achieved all components of the composite endpoint representing the successful rescue of bulbar function, which included no evidence of swallowing impairment, achievement of full oral nutrition, no evidence of aspiration or aspiration pneumonia, and the ability to establish verbal communication via at least two different, distinct vowel sounds. The availability of disease-modifying treatments is improving survival and motor function for patients with SMA, and care has evolved from supportive care to a proactive rehabilitative approach. However, the assessment and management of bulbar function ––for both physiology and function ––remain unclear [43]. Early intervention (perhaps even before the onset of symptoms for children identified as at-risk for SMA by pre- or post-natal screening) with onasemnogene abeparvovec may preserve and stabilize bulbar function [44].
ACKNOWLEDGMENTS
This analysis was funded by Novartis Gene Therapies, Inc. The authors would like to thank Susanne Crowe, formerly of Novartis Gene Therapies, Inc., for her contributions to the collation and analysis of the data in this manuscript. Medical writing assistance and editorial support was provided by Jennifer Gibson, PharmD, from Kay Square Scientific, Newtown Square, PA. This support was funded by Novartis Gene Therapies, Inc.
CONFLICTS OF INTERESTS
K.E.M. has received research grants from the National Institutes of Health, National Institute of Child Health and Human Development, and National Institute of Environmental Health Sciences, and consulting fees from Novartis Gene Therapies, Inc., Biogen, and Roche. R.D.S. has been a paid speaker for Novartis Gene Therapies, Inc. R.H.D. is a Cure SMA Medical Advisory Council Member. S.D.Y. is an advisory board member/consultant for Biogen, Cure SMA, Genentech/Roche, and Scholar Rock, and receives grant support from Novartis Gene Therapies, Inc., Biogen, Cure SMA, Genentech/Roche, Genzyme, Minoryx, PTC Therapeutics, and Scholar Rock. E.O. is an employee of Novartis and holds stock or other equities. A.L., S.W., N.L., and S.P.R. are employees of Novartis Gene Therapies, Inc., and hold stock or other equities. B.T.D. has served as an ad-hoc scientific advisory board member for Audentes, AveXis/Novartis Gene Therapies, Inc., Biogen, Pfizer, Sarepta, and Roche/Genentech, as steering committee chair and member for Roche FIREFISH study and Biogen ASCEND study respectively, and data and safety monitoring board member for Amicus Inc.; he has no financial interests in these companies. He has received research support from the National Institutes of Health/National Institute of Neurological Disorders and Stroke, the Slaney Family Fund for SMA, the Spinal Muscular Atrophy Foundation, Cure SMA, and the Working on Walking Fund, and has received grants from Ionis Pharmaceuticals, Inc., for the ENDEAR, CHERISH, and CS2/CS12 studies; from Biogen for CS11; and from Novartis Gene Therapies, Inc., Sarepta Pharmaceuticals, Novartis, PTC Therapeutics, Roche, Scholar Rock, and Fibrogen. He has also received royalties for books and online publications from Elsevier and UpToDate, Inc.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-221531.
REFERENCES
[1] | Lefebvre S , Bürglen L , Reboullet S , Clermont O , Burlet P , Viollet L , et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. (1995) ;80: (1):155–65. doi:10.1016/0092-8674(95)90460-3. |
[2] | Wirth B . Spinal muscular atrophy: In the challenge lies a solution. Trends Neurosci. (2021) ;44: (4):306–22. doi:10.1016/j.tins.2020.11.009. |
[3] | Bladen CL , Thompson R , Jackson JM , Garland C , Wegel C , Ambrosini A , et al. Mapping the differences in care for 5,000 spinal muscular atrophy patients, a survey of 24 national registries in North America, Australasia and Europe. J Neurol. (2014) ;261: (1):152–63. doi:10.1007/s00415-013-7154-1. |
[4] | Lally C , Jones C , Farwell W , Reyna SP , Cook SF , Flanders WD . Indirect estimation of the prevalence of spinal muscular atrophy type I, II, and III in the United States. Orphanet J Rare Dis. (2017) (1);12: :175. doi:10.1186/s13023-017-0724-z. |
[5] | Butchbach MER . Copy number variations in the Survival Motor Neuron Genes: Implications for spinal muscular atrophy and other neurodegenerative diseases. Front Mol Biosci. (2016) ;3: :7. doi:10.3389/fmolb.2016.00007. |
[6] | D’Amico A , Mercuri E , Tiziano FD , Bertini E . Spinal muscular atrophy. Orphanet J Rare Dis. (2011) ;6: :71. doi:10.1186/1750-1172-6-71. |
[7] | Finkel RS , McDermott MP , Kaufmann P , Darras BT , Chung WK , Sproule DM , et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. (2014) ;83: (9):810–7. doi:10.1212/WNL.0000000000000741. |
[8] | Kolb SJ , Coffey CS , Yankey JW , Krosschell K , Arnold WD , Rutkove SB , et al. Natural history of infantile-onset spinal muscular atrophy. Ann Neurol. (2017) ;82: (6):883–91. doi:10.1002/ana.25101. |
[9] | LoMauro A , Aliverti A , Mastella C , Arnoldi MT , Banfi P , Baranello G . Spontaneous breathing pattern as respiratory functional outcome in children with spinal muscular atrophy (SMA). PLoS One. (2016) ;11: (11):e0165818. doi:10.1371/journal.pone.0165818. |
[10] | Bach JR , Saltstein K , Sinquee D , Weaver B , Komaroff E . Long-term survival in Werdnig–Hoffmann disease. Am J Phys Med Rehabil. (2007) ;86: (5):339–45. doi:10.1097/PHM.0b013e31804a8505. |
[11] | Pane M , Palermo C , Messina S , Sansone VA , Bruno C , Catteruccia M , et al. An observational study of functional abilities in infants, children, and adults with type 1 SMA. Neurology. (2018) ;91: (8):e696–703. doi:10.1212/WNL.0000000000006050. |
[12] | Zappa G , LoMauro A , Baranello G , Cavallo E , Corti P , Mastella C , et al. Intellectual abilities, language comprehension, speech, and motor function in children with spinal muscular atrophy type 1. J Neurodev Disord. (2021) ;13: (1):9. doi:10.1186/s11689-021-09355-4. |
[13] | Brakemeier S , Stolte B , Thimm A , Kizina K , Totzeck A , Munoz-Rosales J , et al. Assessment of bulbar function in adult patients with 5q-SMA type 2 and 3 under treatment with nusinersen. Brain Sci. (2021) ;11: (9):1244. doi:10.3390/brainsci11091244. |
[14] | Tilton AH , Miller MD , Khoshoo V . Nutrition and swallowing in pediatric neuromuscular patients. Semin Pediatr Neurol. (1998) ;5: (2):106–15. doi:10.1016/s1071-9091(98)80026-0. |
[15] | Wang CH , Finkel RS , Bertini ES , Schroth M , Simonds A , Wong B , et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. (2007) ;22: (8):1027–49. doi:10.1177/0883073807305788. |
[16] | Kolb SJ , Kissel JT . Spinal muscular atrophy. Neurol Clin. (2015) ;33: (4):831–46. doi:10.1016/j.ncl.2015.07.004. |
[17] | Mongiovi P , Dilek N , Garland C , Hunter M , Kissel JT , Luebbe E , et al. Patient reported impact of symptoms in spinal muscular atrophy (PRISM-SMA). Neurology. (2018) ;91: (13):e1206–14. doi:10.1212/WNL.0000000000006241. |
[18] | Corsello A , Scatigno L , Pascuzzi MC , Calcaterra V , Dilillo D , Vizzuso S , et al. Nutritional, gastrointestinal and endo-metabolic challenges in the management of children with spinal muscular atrophy type 1. Nutrients. (2021) ;13: (7):2400. doi:10.3390/nu13072400. |
[19] | Mercuri E , Finkel RS , Muntoni F , Wirth B , Montes J , Main M , et al. Diagnosis and management of spinal muscular atrophy: Part Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. (2018) ;28: (2):103–15. doi:10.1016/j.nmd.2017.11.005. |
[20] | Mendell JR , Al-Zaidy S , Shell R , Arnold WD , Rodino-Klapac LR , Prior TW , et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. (2017) ;377: (18):1713–22. doi:10.1056/NEJMoa1706198. |
[21] | Day JW , Finkel RS , Chiriboga CA , Connolly AM , Crawford TO , Darras BT , et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): An open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. (2021) (4);20: :284–93. doi:10.1016/S1474-4422(21)00001-6. |
[22] | Mercuri E , Muntoni F , Baranello F , Masson R , Boespflug-Tanguy O , Bruno C , et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy type 1 (STR1VE-EU): An open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. (2021) ;20: (10):832–41. doi:10.1016/S1474-4422(21)00251-9. |
[23] | Bayley N . Scales of Infant and Toddler Development, 3rd ed: Administration Manual. San Antonio (TX): Harcourt Assessment; (2006) . |
[24] | van Bruggen HW , Wadman RI , Bronkhorst EM , Leeuw M , Cruegers N , Kalaykova SI , et al. Mandibular dysfunction as a reflection of bulbar involvement in SMA type 2 and 3. Neurology. (2016) ;86: (6):552–9. doi:10.1212/WNL.0000000000002348. |
[25] | van den Engel-Hoek L , Erasmus CE , van Bruggen HW , de Swart BJM , Sie LTL , Steenks MH , et al. Dysphagia in spinal muscular atrophy type II: More than a bulbar problem? Neurology. (2009) ;73: (21):1787–91. doi:10.1212/WNL.0b013e3181c34aa6. |
[26] | van der Heul AMB , Wijngaarde CA , Wadman RI , Asselman F , van den Aardweg MTA , Bartels B , et al. Bulbar problems self-reported by children and adults with spinal muscular atrophy. J Neuromuscul Dis. (2019) ;6: (3):361–8. doi:10.3233/JND-190379. |
[27] | van der Heul AMB , van Eijk RPA , Wadman RI , Asselman F , Cuppen I , Nievelstein RAJ , et al. Mastication in patients with spinal muscular atrophy types 2 and 3 is characterized by abnormal efficiency, reduced endurance, and fatigue. Dysphagia. 2022;37:715–23. doi: 10.1007/s00455-021-10351-y. [E-pub ahead of print]. |
[28] | Wadman RI , van Bruggen HW , Witkamp TD , Sparreboom-Kalaykova SI , Stam M , van den Berg LH , et al. Bulbar muscle MRI changes in patients with SMA with reduced mouth opening and dysphagia. Neurology. (2014) ;83: (12):1060–6. doi:10.1212/WNL.0000000000000796. |
[29] | Kruse T , Lehmann HC , Braumann B , Fink GR , Wunderlich G . The maximum bite force for treatment evaluation in severely affected adults SMA patients—protocol for a longitudinal study. Front Neurol. (2020) ;11: :139. doi:10.3389/fneur.2020.00139. |
[30] | Weststrate H , Stimpson G , Thomas L , Scoto M , Johnson E , Stewart A , et al. Evolution of bulbar function in spinal muscular atrophy type 1 treated with nusinersen. Dev Med Child Neurol. (2022) ;64: (7):907–14. doi:10.1111/dmcn.15171. |
[31] | Chen KA , Widget J , Teng A , Fitzgerald DA , D’Silva A , Farrar M . Real-world respiratory and bulbar comorbidities of SMA type 1 children treated with nusinersen: 2-Year single centre Australian experience. Paediatr Respir Rev. (2021) ;39: :54–60. doi:10.1016/j.prrv.2020.09.002. |
[32] | van der Heul AMB , Cuppen I , Wadman RI , Asselman F , Schoenmakers MAGC , van de Woude DR , et al. Feeding and swallowing problems in infants with spinal muscular atrophy type An observational study. J Neuromuscul Dis. (2020) ;7: (3):323–30. doi:10.3233/JND-190465. |
[33] | Masson R , Mazurkiewicz-Beldzinska M , Rose K , Servais L , Xiong H , Zanoteli E , et al. Safety and efficacy of risdiplam in patients with type 1 spinal muscular atrophy (FIREFISH part 2): Secondary analyses from an open-label trial. Lancet Neurol. 2022;21(12):1110–9. doi: 10.1016/S1474-4422(22)00339-8. |
[34] | Pena-Longobardo LM , Aranda-Reneo I , Oliva-Moreno J , Litzkendorf S , Durand-Zaleski I , Tizzano E , et al. The economic impact and health-related quality of life of spinal muscular atrophy. An analysis across Europe. Int J Environ Res Public Health. (2020) ;17: (16):5640. doi:10.3390/ijerph17165640. |
[35] | Yunasova Y , Plowman EK , Green JR , Barnett C , Bode P . Clinical measures of bulbar dysfunction in ALS. Front Neurol. (2019) ;10: :106. doi:10.3389/fneur.2019.00106. |
[36] | Aranda-Reneo I , Pena-Longobardo LM , Oliva-Moreno J , Litzkendorf S , Durand-Zaleski I , Tizzano E , et al. The burden of spinal muscular atrophy on informal caregivers. Int J Environ Res Public Health. (2020) ;17: (23):8989. doi:10.3390/ijerph17238989. |
[37] | Aksaralikitsunti M , Sanmaneechai O . Health-related quality of life in Thai children with spinal muscular atrophy. Pediatr Neonatol. (2022) ;63: (3):291–7. doi:10.1016/j.pedneo.2022.01.002. |
[38] | Qian Y , McGraw S , Henne J , Jarecki J , Hobby K , Yeh WS . Understanding the experiences and needs of individuals with spinal muscular atrophy and their parents: A qualitative study. BMC Neurol. (2015) ;15: :217. doi:10.1186/s12883-015-0473-3. |
[39] | Finkel RS , Mercuri E , Meyer OH , Simonds AK , Schroth MK , Graham RJ , et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements, and immunizations; other organ systems; and ethics. Neuromuscul Disord. (2018) ;28: (9):197–207. doi:10.1016/j.nmd.2017.11.004. |
[40] | Bertoli S , De Amicis R , Mastella C , Pieri G , Giaquinto E , Battezzati A , et al. Spinal muscular atrophy, types I and II: What are the differences in body composition and resting energy expenditure? Clin Nutr. (2017) ;36: (6):1674–80. |
[41] | Poruk KE , Davis RH , Smart AL , Chisum BS , Lasalle BA , Chan GM , et al. Observational study of caloric and nutrient intake, bone density, and body composition in infants and children with spinal muscular atrophy type I. Neuromuscul Disord. (2012) ;22: (11):966–73. |
[42] | De Amicis R , Baranello G , Foppiani A , Leone A , Battezzati A , Bedogni G , et al. Growth patterns in children with spinal muscular atrophy. Orphanet J Rare Dis. (2021) ;16: , 375. |
[43] | McGrattan KE , Graham RJ , DiDonato CJ , Darras BT . Dysphagia phenotypes in spinal muscular atrophy: The past, present, and promise for the future. Am J Soc Speech Lang Path. (2021) ;30: (3):1008–22. doi:10.1044/2021_AJSLP-20-00217. |
[44] | Tizzano EF , Finkel RS . Spinal muscular atrophy: A changing phenotype beyond the clinical trials. Neuromuscul Disord. (2017) ;27: (10):883–9. doi:10.1016/j.nmd.2017.05.011. |


