
Long-Term Functional Efficacy and Safety of Viltolarsen in Patients with Duchenne Muscular Dystrophy
Abstract
Background:
Duchenne muscular dystrophy (DMD) is a rare, genetic disease caused by mutations in the DMD gene resulting in an absence of functional dystrophin protein. Viltolarsen, an exon 53 skipping therapy, has been shown to increase endogenous dystrophin levels. Herein, long-term (>2 years) functional outcomes in viltolarsen treated patients were compared to a matched historical control group.
Objective:
To evaluate long-term efficacy and safety of the anti-sense oligonucleotide viltolarsen in the treatment of patients with DMD amenable to exon 53 skipping therapy.
Methods:
This trial (NCT03167255) is the extension of a previously published 24-week trial in North America (NCT02740972) that examined dystrophin levels, timed function tests compared to a matched historical control group (Cooperative International Neuromuscular Research Group Duchenne Natural History Study, CINRG DNHS), and safety in boys 4 to < 10 years (N = 16) with DMD amenable to exon 53 skipping who were treated with viltolarsen. Both groups were treated with glucocorticoids. All 16 participants elected to enroll in this long-term trial (up to 192 weeks) to continue evaluation of motor function and safety.
Results:
Time to stand from supine and time to run/walk 10 meters showed stabilization from baseline through week 109 for viltolarsen-treated participants whereas the historical control group showed decline (statistically significant differences for multiple timepoints). Safety was similar to that observed in the previous 24-week trial, which was predominantly mild. There have been no treatment-related serious adverse events and no discontinuations.
Conclusions:
Based on these results at over 2 years, viltolarsen can be a new treatment option for patients with DMD amenable to exon 53 skipping.
INTRODUCTION
Duchenne muscular dystrophy (DMD), an X-linked recessive disorder, affects approximately 1 : 3600 to 1 : 9300 male newborns worldwide [1]. In patients with DMD, loss-of-function variants in the DMD gene result in an absence of functional dystrophin protein, leading to progressive weakness and degeneration of skeletal muscle [2, 3], cardiac dysfunction, respiratory failure, and, ultimately, premature death [2, 4]. However, individuals with DMD mutations who have low levels of normal dystrophin protein from birth [5, 6] and those with in-frame deletions that result in the production of truncated dystrophin protein [7] may exhibit a milder, slower-progressing, and less severe disease [5, 7].
Exon skipping therapies developed for the treatment of DMD have the potential to increase levels of functional dystrophin. The exon skipping approach aims to shift the reading frame to convert a DMD out-of-frame variant to an in-frame, Becker muscular dystrophy-like deletion, leading to an endogenously produced, internally shortened version of the dystrophin protein that retains essential functional portions [7–9]. Viltolarsen is a phosphorodiamidate morpholino oligomer/antisense oligonucleotide designed to treat DMD in patients with a confirmed mutation of the DMD gene that is amenable to exon 53 skipping [9, 10]. The exon 53 skipping approach could be therapeutic in approximately 8% to 10% of patients with DMD caused by an out-of-frame deletion, including, but not limited to, those with deletions such as exons 43-52, 45-52, 47-52, 48-52, 49-52, 50-52, or 52 alone [3, 11, 12].
An initial Phase 2, two period, randomized, dose-finding trial consisted of a 4-week double-blinded, placebo-controlled period for safety of viltolarsen followed by a 20-week open-label period to evaluate the efficacy, safety, and tolerability of viltolarsen (40 or 80 mg/kg/week) in boys 4 to < 10 years of age with DMD (N = 16) amenable to exon 53 skipping [13]. Participants were ambulatory and could complete motor function assessments at screening. Participants had been treated with a stable dose of glucocorticoids for at least 3 months prior to enrollment and were expected to remain on the stable dose for the duration of the study. Treatment with viltolarsen resulted in significant increases in dystrophin production, as assessed by Western blot after 20 to 24 weeks of treatment (80 mg/kg/wk: 5.9% of normal at week 25 vs 0.6% at baseline) [13]. This initial Phase 2 study also included timed function tests, designed to provide evidence of treatment-related improvements in clinical functioning when compared with DMD natural history controls provided by the Cooperative International Neuromuscular Research Group (CINRG) Duchenne Natural History Study (DNHS), who were matched for age, geographic location, ambulatory ability at baseline, and glucocorticoid treatment. The viltolarsen cohort demonstrated significant improvements from baseline on timed function tests, including time to stand from supine (TTSTAND), time to run/walk 10 meters (TTRW), and 6-minute walk test (6MWT) [13]. Adverse events (AEs) were predominantly mild; no treatment-related serious AEs were reported, and no participants discontinued from the study. Based on these data, viltolarsen received accelerated approval for the treatment of DMD patients with mutations amenable to exon 53 skipping in the United States and Japan [14, 15].
At the conclusion of the Phase 2 study, participants were given the opportunity to enroll in the long-term extension (LTE) study (NCT03167255), the primary objective of which was to evaluate the clinical efficacy and safety of viltolarsen over a longer period of time (up to an additional 192 weeks) [16]. All participants elected to continue in the LTE study [17]. Here, we present results at 109 weeks (approximately 2 years) from the open-label LTE study of the original 16 participants enrolled in the North American study [17, 18].
MATERIALS AND METHODS
As previously described, participants for the initial Phase 2 study (NCT02740972) were recruited from 6 sites in North America (UC Davis (Sacramento, California); Lurie Children’s Hospital (Chicago, Illinois); Washington University in St. Louis (St. Louis, Missouri); Duke University Medical Center (Durham, North Carolina); Children’s Hospital of Richmond at VCU (Richmond, Virginia); Alberta Children’s Hospital (Calgary, Alberta, Canada)) [13]. Following completion of this 24-week study, all 16 patients, who were 4 to < 10 years old when enrolled in the initial Phase 2 study, elected to enroll and were followed in the LTE study for up to 192 weeks (NCT03167255). Inclusion criteria again included the requirement for participants to be taking a stable dose of glucocorticoid and to remain on the stable dose for the duration of the study. Treatment for the purpose of dystrophin protein induction or treatment with other investigational drugs after completion of the initial Phase 2 trial were prohibited [16].
Viltolarsen was administered as an IV infusion at a dosage of 40 mg/kg or 80 mg/kg once weekly. All patients received viltolarsen by peripheral venous access during the initial Phase 2 study, and five patients requested placement of a central port at some point during the LTE study.
Efficacy assessments were conducted every 12 weeks [16], and safety was assessed throughout the open-label extension study [19]. [See Fig. 1.] The primary endpoint was TTSTAND; secondary endpoints included TTRW, TTCLIMB, 6MWT, and North Star Ambulatory Assessment (NSAA). Results for the endpoints TTSTAND, TTRW, and TTCLIMB were also converted to velocity, which provides a better and often more reliable accounting of those participants who can no longer perform a test [20]. Clinical assessments were compared to historical controls (CINRG DNHS), who were group-matched for age, geographic location, ambulatory ability (defined as all participants being able to perform TTRW, TTSTAND, and TTCLIMB at baseline), and glucocorticoid use. Analysis at 2 years (109 weeks) was selected because the timepoint aligned with sufficient numbers of patients in the CINRG DNHS group, who had functional assessments less frequently than the viltolarsen-treated patients.
Fig. 1
Design of the Phase 2 and LTE studies. mg, milligrams; kg, kilograms; wk, week.
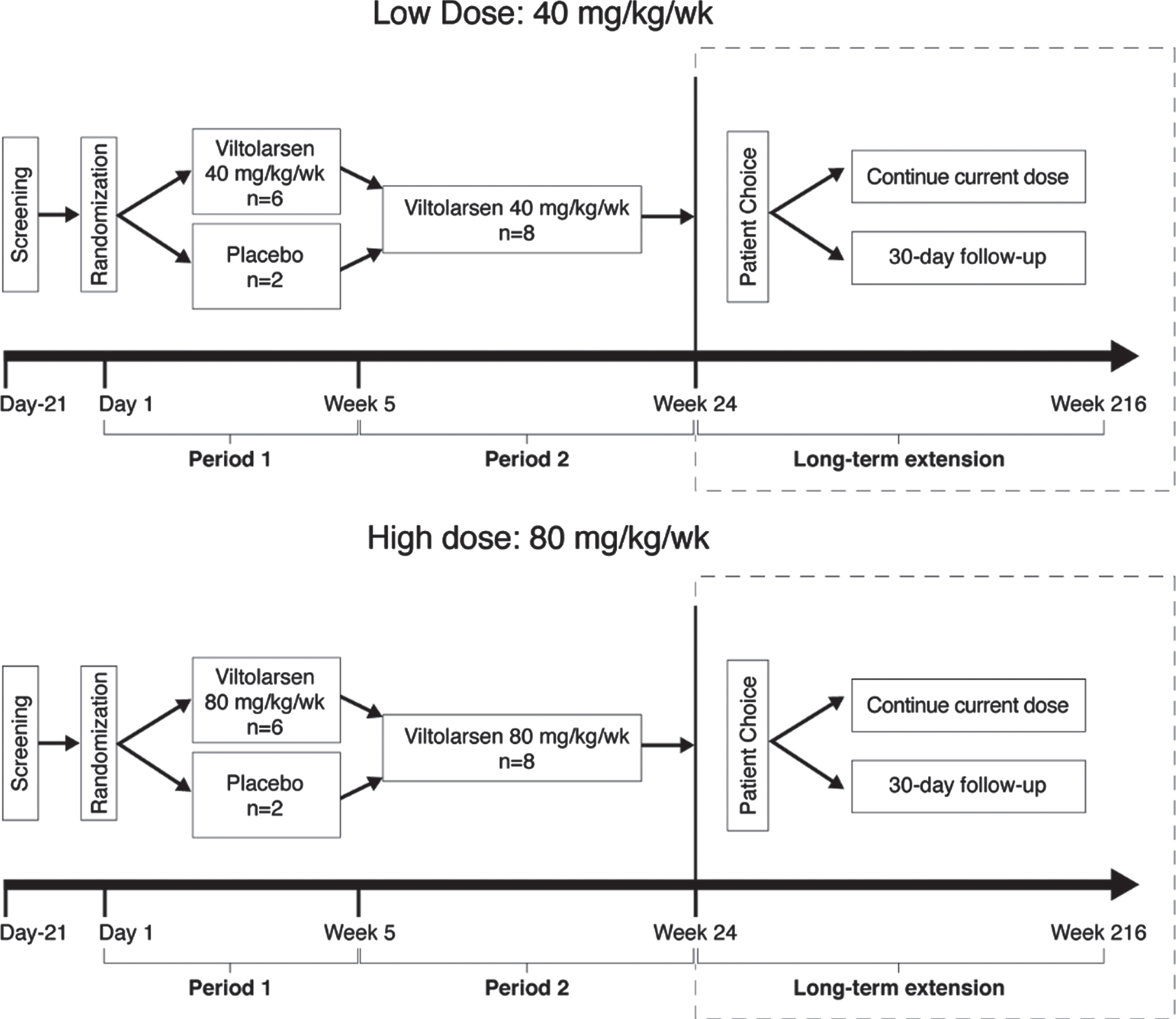
The trial protocol was approved by ethics review panels of each participating recruitment center. Prior to any study-related procedures, participants, who were minors, provided written or verbal informed assent appropriate for age and developmental status, and the parent or legal guardian of participants provided written informed consent and/or HIPAA authorization. The trial was performed according to the principles of the Declaration of Helsinki and the Good Clinical Practice regulations. Activities were overseen by an independent data and safety monitoring board.
RESULTS
Participants
All participants (N = 16) who completed the Phase 2 study continued participation in the LTE study throughout the period of this report. Most participants (15/16) were white; one was Asian. Their mean age (range) of enrollment in the initial Phase 2 study was 7.4 (4.3, 9.8) years. For this LTE study, participants continued therapy according to their previously assigned viltolarsen dose cohort (low-dose [40 mg/kg per week, n = 8] or high-dose [80 mg/kg per week, n = 8>]). Overall, baseline characteristics between participants in the 2 dose cohorts were balanced. Participants in the external comparator group, whose data were drawn from the CINRG DNHS, were group-matched to the viltolarsen-treated participants (see Methods and Table 1). Nine patients in the CINRG DNHS comparator group had DMD amenable to exon 53 skipping and 56 had DMD with non–exon 53 skipping mutations, excluding patients with deletion of exons 3–7 or gene deletions amenable to exon skipping of exon 44, as these mutation groups have a milder disease progression [13, 19].
Table 1
Baseline Demographic Characteristics of the Participants Enrolled in the Long-term Extension Study of Viltolarsen and the CINRG DNHS Control Cohort
| Characteristics | Viltolarsen cohort, mean (range) | CINRG DNHS control cohort, mean (range) | ||||
| 40 mg/kg/week (n = 8) | 80 mg/kg/week (n = 8) | Total (N = 16) | Exon 53 amenable controls (n = 9) | Non-exon 53 amenable controls (n = 56) | Total (N = 65) | |
| Age, y | 7.5 | 7.2 | 7.4 | 6.3 | 7.2 | 7.1 |
| (4.3, 9.8) | (4.8, 9.8) | (4.8, 9.8) | (4.5, 7.8) | (4.2, 9.6) | (4.2, 9.6) | |
| Weight, kg | 23.7 | 22.3 | 23.0 | 21.6 | 24.4 | 24.0 |
| (14.9, 30.4) | (15.5, 35.4) | (14.9, 35.4) | (16.6, 28.1) | (14.8, 38.7) | (14.8, 38.7) | |
| Height, cm | 114.6 | 112.2 | 113.4 | 111.3 | 117.0 | 116.2 |
| (102.5, 123.4) | (99.4, 127.1) | (99.4, 127.1) | (102.2, 122.2) | (96.1, 135.9) | (96.1, 135.9) | |
| BMI, mg/kg2 | 17.9 | 17.4 | 17.7 | 17.3 | 17.6 | 17.5 |
| (14.2, 20.0) | (15.4, 21.9) | (14.2, 21.9) | (15.5, 21.6) | (13.9, 22.9) | (13.9, 22.9) | |
CINRG DNHS, Cooperative International Neuromuscular Research Group Duchenne Natural History Study; y, years.
Efficacy outcomes
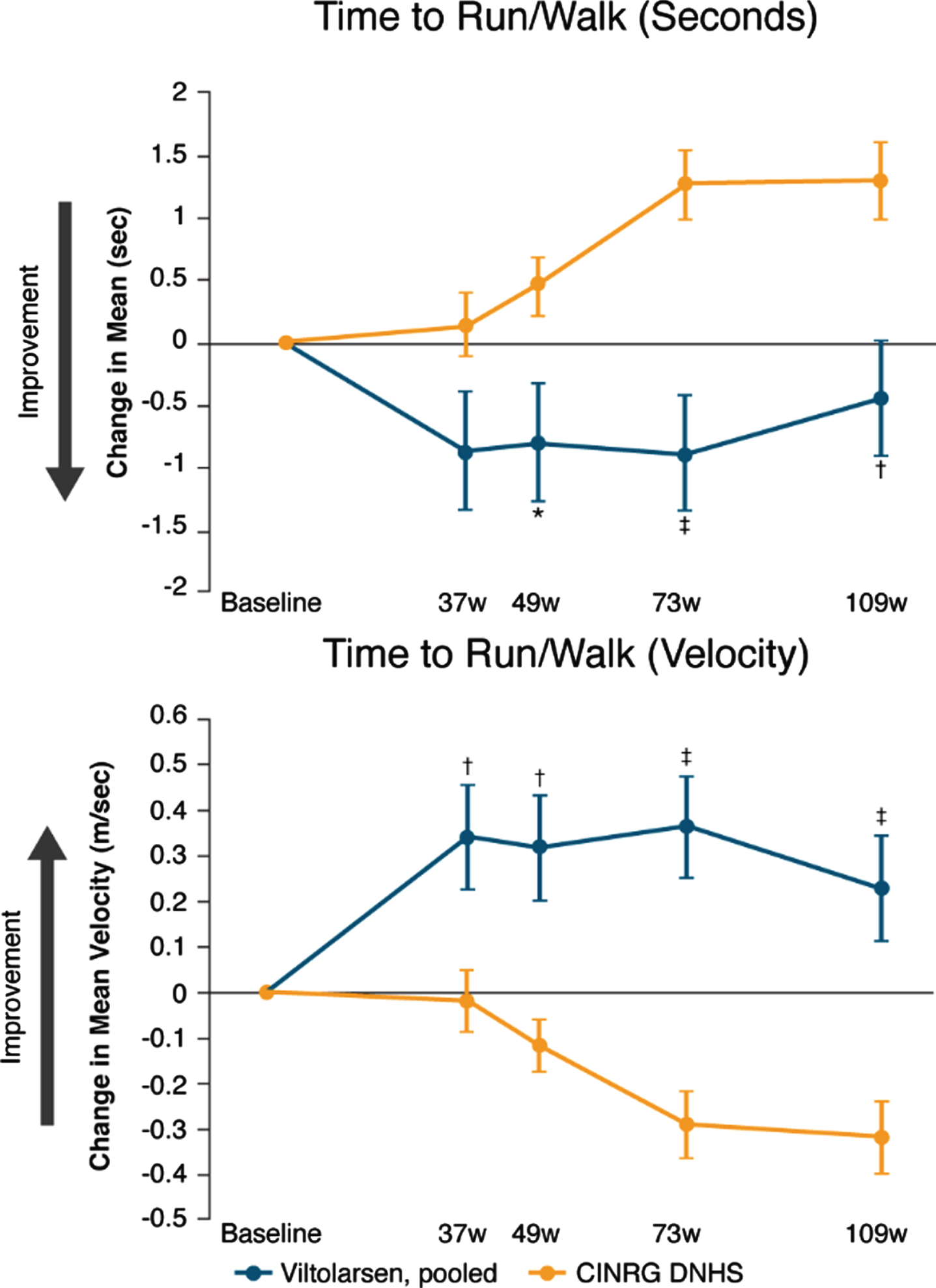
Timed function tests and muscle strength assessments were used to measure disease progression. Data for the primary efficacy endpoint of time to stand from supine (TTSTAND) indicate that function was maintained over 109 weeks in participants treated with viltolarsen, whereas the CINRG DNHS group experienced functional decline (Fig. 2 and Table 2). Improvements were significant at timepoints 73 and 109 weeks for TTSTAND (seconds) and at all timepoints for TTSTAND (velocity). Change from baseline in the secondary efficacy endpoint of TTRW likewise indicated maintenance of function through week 109 in participants treated with viltolarsen versus functional decline in the historical control group (Fig. 3 and Table 2), with significant differences shown at all timepoints after 37 weeks for TTRW (seconds) and at all timepoints for TTRW (velocity). Although TTCLIMB (seconds) and TTCLIMB (velocity) showed numerical improvement at all timepoints, TTCLIMB (seconds) was not significantly different from the control group, and TTCLIMB (velocity) was significantly different from the control group only at 73 weeks (viltolarsen change from baseline [CFB]: 0.05 task/s [SE 0.03], DNHS CFB: –0.05 task/s [SE 0.02]; P = 0.0082).
Fig. 2
Change from baseline in time to stand from supine. CINRG DNHS, Cooperative International Neuromuscular Research Group Duchenne Natural History Study; TTSTAND, time to stand from supine; w, weeks. Calculation of TTSTAND velocity: rise/second. *P<0.05; †P<0.01; ‡P≤0.0001. Observed sample size for viltolarsen at baseline and for each of the subsequent timepoints on viltolarsen were: n = 16, n = 16, n = 15, n = 14, and n = 14. Observed sample size for DNHS cohort at baseline and for each of the subsequent timepoints for this cohort were: n = 65, n = 31, n = 57, n = 24, n = 24 for TTSTAND (seconds); n = 65, n = 31, n = 58, n = 28, n = 28 for TTSTAND (velocity).
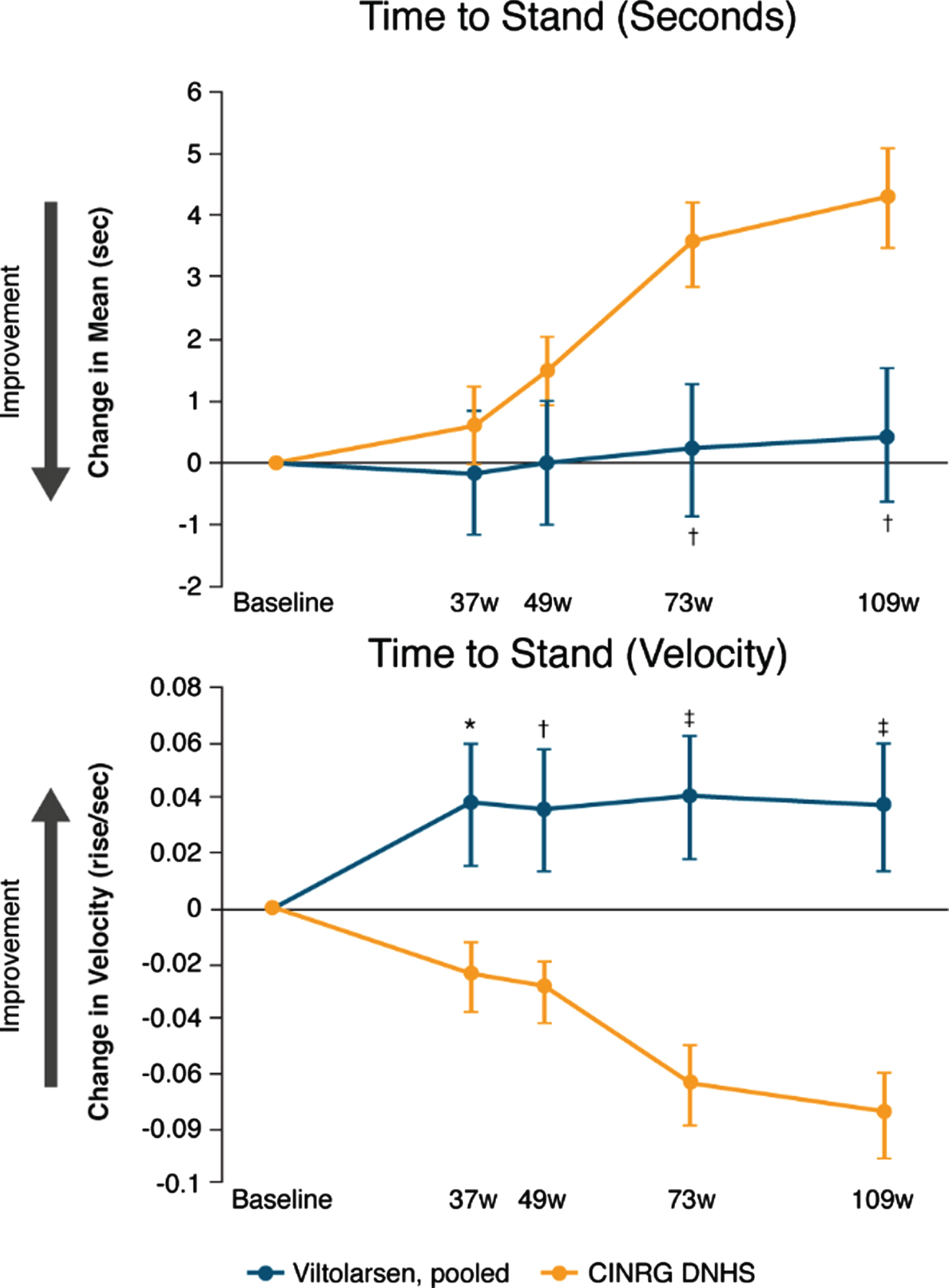
Table 2
Efficacy at Week 109
| Efficacy at week 109, change from baseline comparison | |||
| Parameter | Viltolarsen LSM (SE) | DNHS LSM (SE) | Between-group comparison, P-value |
| TTSTAND (seconds) | 0.43 (1.077) | 4.31 (0.786) | 0.0044 |
| TTSTAND (velocity) | 0.04 (0.023) | –0.08 (0.015) | <0.0001 |
| TTRW (seconds) | –0.44 (0.464) | 1.29 (0.309) | 0.0024 |
| TTRW (velocity) | 0.23 (0.113) | –0.32 (0.080) | 0.0001 |
| TTCLIMB (seconds) | 1.02 (0.972) | 3.00 (0.690) | 0.0992 |
| TTCLIMB (velocity) | 0.004 (0.029) | –0.05 (0.020) | 0.1578 |
DNHS, Duchenne Natural History Study; LSM, least-squares mean; SE, standard error; TTCLIMB, time to climb 4 stairs; TTRW, time to run/walk 10 meters; TTSTAND, time to stand from supine. Change from baseline was modeled using a residual maximum likelihood (REML)-based, mixed-model repeated measures analysis with treatment group, week of the visit, and treatment-by-week interaction as factors, and age at study entry for viltolarsen/baseline for DNHS as one covariate, and baseline response as a second covariate.
Fig. 3
Change from baseline in time to run/walk 10 meters. CINRG DNHS, Cooperative International Neuromuscular Research Group Duchenne Natural History Study; TTRW, time to run/walk 10 meters; w, weeks. Calculation of TTRW velocity: meters/second. *P<0.05; †P<0.01; ‡P≤0.0001. Observed sample size for viltolarsen at baseline and for each of the subsequent timepoints on viltolarsen were: n = 16, n = 16, n = 15, n = 16, n = 16 for TTRW. Observed sample size for DNHS cohort at baseline and for each of the subsequent timepoints for this cohort were: n = 65, n = 32, n = 58, n = 28, n = 29 for TTRW (seconds); n = 65, n = 32, n = 59, n = 29, n = 29 for TTRW (velocity).
The secondary efficacy endpoints of 6MWT and NSAA were added late in the study to the CINRG DNHS protocol, and thus the CINRG DNHS comparator group did not have sufficient data on NSAA and 6MWT to adequately compare with the viltolarsen-treated group.
Safety
The overall safety profile of viltolarsen over the LTE study was similar to that observed in the previous 24-week Phase 2 study and was predominantly mild in nature. All 16 participants experienced treatment-emergent adverse events (TEAE) (Table 3). Only one TEAE, an IV infiltration that was considered related to both the study drug and procedure, and graded as mild, occurred in the 80 mg/kg/week viltolarsen group. The most common TEAEs (Table 4), which occurred in≥25% of patients, were cough, nasopharyngitis, insect bite, and rash. There were two serious TEAEs, a lower limb fracture and a case of rhabdomyolysis; both were considered unrelated to viltolarsen treatment. There were no discontinuations due to AEs, and no deaths occurred during the study.
Table 3
Safety Profile of Viltolarsen During the Long-term Extension Study
| Viltolarsen Treatment | |||
| 40 mg/kg/wk (n = 8) n (%) | 80 mg/kg/wk (n = 8) n (%) | Total (N = 16) n (%) | |
| Any TEAE* | 8 (100) | 8 (100) | 16 (100) |
| Any drug-related TEAE | 0 | 1 (12.5) | 1 (6.3) |
| Discontinuation due to TEAEs | 0 | 0 | 0 |
| Any serious treatment- related TEAE | 0 | 0 | 0 |
| Death | 0 | 0 | 0 |
*TEAE, treatment-emergent adverse event. Note: 80 mg/kg/wk is the FDA-approved dosing regimen for viltolarsen.
Table 4
Patients with Treatment-emergent Adverse Events≥25% Total in Long-term Extension Study
| Adverse Events | Viltolarsen Treatment | |||
| 40 mg/kg/wk (n = 8) n (%) | 80 mg/kg/wk (n = 8) n (%) | Total (N = 16) n (%) | ||
| Cough | 5 (62.5) | 4 (50.0) | 9 (56.3) | |
| Nasopharyngitis | 4 (50.0) | 4 (50.0) | 8 (50.0) | |
| Insect bite | 4 (50.0) | 2 (25.0) | 6 (37.5) | |
| Rash | 2 (25.0) | 4 (50.0) | 6 (37.5) | |
| Fever | 2 (25.0) | 3 (37.5) | 5 (31.3) | |
| Vomiting | 3 (37.5) | 2 (25.0) | 5 (31.3) | |
| Fall | 4 (50.0) | 0 | 4 (25.0) | |
| Headache | 2 (25.0) | 2 (25.0) | 4 (25.0) | |
| Influenza | 3 (37.5) | 1 (12.5) | 4 (25.0) | |
| Nasal congestion | 3 (37.5) | 1 (12.5) | 4 (25.0) | |
Note: 80 mg/kg/wk is the FDA-approved dosing regimen for viltolarsen.
Discussion
We report here that the effects of viltolarsen on timed function tests were maintained over 2 years in this analysis of data from the long-term extension study, compared to a decline in the DNHS historical control group that was matched on key factors such as age, treatment with corticosteroids, ambulatory ability (all participants were required to perform TTRW, TTSTAND, and TTCLIMB at baseline), and geographic location. The present findings represent the first published long-term functional data for viltolarsen therapy used in the treatment of patients with DMD who were 4 to < 10 years of age, ambulant, and taking glucocorticoids at baseline [13]. The safety profile of viltolarsen over 2 years was also similar to that observed in the 24-week Phase 2 trial. This analysis at the 2-year mark suggests clinically relevant motor function was maintained and supports the safety profile of viltolarsen.
Long-term data on the use of FDA-approved DMD therapies are beginning to emerge. A recent paper by Servais and colleagues found that golodirsen treatment in DMD patients (n = 25, mean age 8.4 years) amenable to exon 53 skipping did not significantly reduce loss of ambulation, 6MWT, or forced vital capacity evaluations at 3 years relative to external controls [21]. Viltolarsen (presented here) and golodirsen target the same DMD gene exon 53. For a different exon-specific drug, two studies evaluated eteplirsen over a period of 3 to 4 years. Mendell and colleagues assessed 6MWT and pulmonary function in exon 51 skip-amenable patients compared with patient-level control data from the Italian DMD Registry and found that eteplirsen (n = 13, mean age 9.3 years) treatment provided a significant benefit compared with the historical controls, and pulmonary function remained stable [22]. Similarly, Kinane and colleagues found that eteplirsen treatment attenuated respiratory decline compared with United Dystrophinopathy Project controls over 216 weeks [23].
Limitations
DMD is a rare disease, and only 8% –10% of DMD patients have a variant amenable to exon 53 skipping [3]. While the small sample size (N = 16) represents a limitation of this study, both the study design and sample size are consistent with other studies investigating potential approaches to treatment for this patient population [21–24]. An additional limitation is the use of an historical control group (group-level matched) in this study, rather than a placebo arm. The use of an historical control group, although less rigorous than a randomized, placebo-controlled study design, is appropriate for a Phase 2 trial in a rare disease with a surrogate primary outcome. However, an analysis of the consistency of change in 6-minute walk distance in DMD using group-matched DMD natural history data from 5 separate natural history datasets found that external controls were not different from placebo controls drawn from placebo datasets included in 6 randomized, blinded DMD treatment studies encompassing 4 sets of eligibility criteria [25]. While this study did not use patient-level matching due to the small patient population size in the DMD historical control group; it used group-level matching, which included age, geographic location, ambulatory ability, and glucocorticoid use. To confirm the clinical findings, a Phase 3 randomized, double-blind, placebo-controlled, multi-center study (NCT04060199; RACER53) to assess the efficacy and safety of viltolarsen in boys with DMD who are able to walk independently without assistive devices is being conducted [26].
Conclusions
Using timed function tests as markers of clinical efficacy, the effects of viltolarsen were maintained in this long-term study over 2 years, compared to a decline in the DMD historical control group that was matched on key factors such as age, ambulatory ability, and glucocorticoid treatment. Further, the safety profile of viltolarsen over this long-term study was mild and similar to that observed in the initial 24-week Phase 2 study. Based on the efficacy and safety results of this Phase 2 long-term extension study, we consider that treatment with viltolarsen offers an important option for patients with DMD with mutations that are amenable to exon 53 skipping.
ACKNOWLEDGMENTS
The authors thank all study participants and their families, as well as the study team members, at participating Cooperative International Neuromuscular Research Group (CINRG) sites for assistance in collecting and managing the data. The authors are grateful to Robert Crozier, PhD, and Leslie Magnus, MD, of NS Pharma, Inc., for discussions and a critical reading of the manuscript.
The CINRG Duchenne Natural History Study (DNHS) was supported by grants H133B031118 and H133B090001 from US Department of Education and National Institute on Disability, Independent Living, and Rehabilitation Research; grant W81XWH-12-1-0417 from the US Department of Defense; grant R01AR061875 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases; and grants from the Parent Project Muscular Dystrophy. The viltolarsen treatment study was funded by NS Pharma.
Writing and editorial support were provided by Marilyn G. Stearns, MD; and Steven D. Stockton, Jr., MS, PhD; from PRI Healthcare Solutions, and were funded by NS Pharma.
Author Contributions
1. Contributed to study conception and design: P.C., T.N., E.H.
2. Analyzed and interpreted data: P.C., V.R., A.C., A.H., J.M., C.M., E.S., C.Z., T.N., E.H.
3. Drafted and reviewed the final publication: P.C., V.R., A.C., A.H., J.M., C.M., E.S., C.Z., T.N., E.H.
The CINRG DNHS Investigators contributed to CINRG DNHS data collection. Please see Supplementary Table for the investigator group’s members and affiliations.
Conflicts of Interest
a. Dr Clemens has received grants from NS Pharma during the conduct of the study as well as grants from Spark Therapeutics, Amicus Therapeutics, Sanofi Genzyme, ReveraGen BioPharma, NIH, MDA, and TRiNDS and personal fees from Epirium, outside the submitted work.
b. Dr Rao has received grants and personal fees from NS Pharma during the conduct of the study and nonfinancial support from Ann and Robert H. Lurie Children’s Hospital of Chicago during the conduct of the study as well as personal fees from Biogen, Avexis/Novartis, Capricor, Regenxbio, Genentech-Roche, Scholar Rock, PTC Therapeutics, Sarepta Therapeutics, France Foundation, and MDA outside the submitted work.
c. Dr Connolly has received grants from the Washington University School of Medicine and TRiNDS during the conduct of the study as well as grants from Sarepta Therapeutics, AveXis, and Fibrogen and personal fees from Sarepta Therapeutics, Scholar Rock, Genentech-Roche, Dyne Therapeutics, and Edgewise Therapeutics outside the submitted work.
d. Dr Harper has received grants from NS Pharma, Italfarmaco, ReveraGen BioPharma, Catabasis Pharmaceuticals, Astellas Pharmaceuticals, MLBio, AveXis, CSL Behring, Teva Pharmaceutical Industries, Novartis, National Institutes of Health, and US Centers for Disease Control and Prevention.
e. Dr Mah has received grants from NS Pharma during the conduct of the study as well as personal fees from PTC Therapeutics, Biogen and Roche and grants from PTC Therapeutics, Pfizer, Roche, Sarepta Therapeutics, Italfarmaco, Novartis, Biogen, ReveraGen BioPharma, Catabasis Pharmaceuticals, Sanofi-Genzyme, and Alberta Children’s Hospital Foundation outside the submitted work.
f. Dr McDonald has received grants from NS Pharma during the conduct of the study as well as personal fees from Avidity Biosciences, Astellas, Capricor Therapeutics, Catabasis Pharmaceuticals, Edgewise Therapeutics, Entrada Therapeutics, Epirium Bio, FibroGen, Italfarmaco, Pfizer, PTC Therapeutics, Roche, Santhera Pharmaceuticals, Solid Biosciences, and Sarepta Therapeutics outside the submitted work.
g. Dr Smith has received personal fees from NS Pharma during the conduct of the study as well as personal fees from Pfizer and Sarepta Therapeutics outside the submitted work.
h. Dr Zaidman has received grant support from Biogen and Novartis outside the submitted work.
i. Mr Nakagawa is an employee of NS Pharma, Inc.
j. Dr Hoffman has received fees from NS Pharma, holds stock and held oversight roles in AGADA Biosciences and TRiNDS during the conduct of the study, and holds stock and has management roles in ReveraGen BioPharma outside the submitted work.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-220811.
REFERENCES
[1] | Mah JK , Korngut L , Dykeman J , Day L , Pringsheim T , Jette N , A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord (2014) ;24: (6):482–91. |
[2] | Hoffman EP , Brown RH Jr , Kunkel LM , Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell (1987) ;51: (6):919–28. |
[3] | Bladen CL , Salgado D , Monges S , Foncuberta ME , Kekou K , Kosma K , et al., The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat (2015) ;36: (4):395–402. |
[4] | Bushby K , Finkel R , Birnkrant DJ , Case LE , Clemens PR , Cripe L , et al., Diagnosis and management of Duchenne muscular dystrophy, part diagnosis, and pharmacological and psychosocial management. Lancet Neurol. (2010) ;9: (1):77–93. |
[5] | de Feraudy Y , Ben Yaou R , Wahbi K , Stalens C , Stantzou A , Laugel V , et al., Very low residual dystrophin quantity Is associated with milder dystrophinopathy. Ann Neurol (2021) ;89: (2):280–92. |
[6] | Hoffman EP , Clemens PR , Concerns regarding therapeutic implications of very low-level dystrophin. Ann Neurol (2021) ;90: (1):176. |
[7] | Beggs AH , Hoffman EP , Snyder JR , Arahata K , Specht L , Shapiro F , et al., Exploring the molecular basis for variability among patients with Becker muscular dystrophy: dystrophin gene and protein studies. Am J Hum Genet (1991) ;49: (1):54–67. |
[8] | Verhaart IEC , Aartsma-Rus A , Therapeutic developments for Duchenne muscular dystrophy. Nat Rev Neurol (2019) ;15: (7):373–86. |
[9] | Komaki H , Nagata T , Saito T , Masuda S , Takeshita E , Sasaki M , et al., Systemic administration of the antisense oligonucleotide NS-065/NCNP-01 for skipping of exon 53 in patients with Duchenne muscular dystrophy. Sci Transl Med (2018) ;10: (437). |
[10] | VILTEPSO® (viltolarsen) injection. Prescribing information. NS Pharma, Inc. |
[11] | Watanabe N , Nagata T , Satou Y , Masuda S , Saito T , Kitagawa H , et al., NS-065/NCNP- an antisense oligonucleotide for potential treatment of exon 53 skipping in Duchenne muscular dystrophy. Mol Ther Nucleic Acids (2018) ;13: :442–9. |
[12] | Komaki H , Takeshima Y , Matsumura T , Ozasa S , Funato M , Takeshita E , et al., Viltolarsen in Japanese Duchenne muscular dystrophy patients: A phase 1/2 study. Ann Clin Transl Neurol (2020) ;7: (12):2393–408. |
[13] | Clemens PR , Rao VK , Connolly AM , Harper AD , Mah JK , Smith EC , et al., Safety, tolerability, and efficacy of viltolarsen in boys with Duchenne muscular dystrophy amenable to exon 53 skipping: a phase 2 randomized clinical trial. JAMA Neurol (2020) ;77: (8):982–91. |
[14] | US Food and Drug Administration (FDA). FDA approves targeted treatment for rare Duchenne muscular dystrophy mutation. Accessed December 16, 2021. https://www.fda.gov/news-events/press-announcements/fda-approves-es-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation |
[15] | NS Pharma announces marketing authorization in Japan of VILTEPSO® for the treatment of Duchenne. Accessed December 16, 2021. https://www.parentprojectmd.org/nspharma-announces-marketing-authorization-in-japan-of-viltepso-for-the-treatment-of-duchenne/. |
[16] | ClinicalTrials.gov. Extension study of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD) 2021. Accessed December 16, 2021. https://clinicaltrials.gov/ct2/show/record/NCT03167255 |
[17] | Clemens PR , Rao VK , Connolly AM , Harper AD , Mah JK , Smith EC , et al., Efficacy and safety analysis of viltolarsen in DMD: open-label, long-term extension study. Presented at: World Muscle Society 2021 Virtual Congress; September 20-24, 2021; London, UK. |
[18] | Rao VK , Clemens PR , Connolly AM , Harper AD , Mah JK , Smith EC , et al., Efficacy and safety analysis of viltolarsen in DMD: open-label, long-term extension study. Presented at: Child Neurology Society 50th Annual Meeting; September 29–October 2, 2021; Boston, Massachusetts. |
[19] | NS Pharma. Data on file. 2021. |
[20] | Bushby K , Connor E , Clinical outcome measures for trials in Duchenne muscular dystrophy: report from International Working Group meetings. Clin Investig (Lond) (2011) ;1: (9):1217–35. |
[21] | Servais L , Mercuri E , Straub V , Guglieri M , Seferian AM , Scoto M , et al., SKIP-NMD Study GrouLong-term safety and efficacy data of golodirsen in ambulatory patients with Duchenne muscular dystrophy amenable to exon 53 skipping: a first-in-human, multicenter, two-part, open-label, Phase 1/2 trial. Nucleic Acid Ther (2022) ;32: (1):29–39. |
[22] | Mendell JR , Goemans N , Lowes LP , Alfano LN , Berry K , Shao J , et al., Eteplirsen Study Group and Telethon Foundation DMD Italian Network Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol (2016) ;79: (2):257–71. |
[23] | Kinane TB , Mayer OH , Duda PW , Lowes LP , Moody SL , Mendell JR , Long-term pulmonary function in Duchenne muscular dystrophy: comparison of eteplirsen-treated patients to natural history J Neuromuscul Dis (2018) ;5: (1):47–58. |
[24] | Mendell JR , Khan N , Sha N , Eliopoulos H , McDonald CM , Goemans N , et al., Comparison of long-term ambulatory function in patients with Duchenne muscular dystrophy treated with eteplirsen and matched natural history controls. J Neuromuscul Dis (2021) ;8: (4):469–79. |
[25] | Goemans N , Signorovitch J , Sajeev G , Yao Z , Gordish-Dressman H , McDonald CM , et al., Suitability of external controls for drug evaluation in Duchenne muscular dystrophy. Neurology (2020) ;95: (10):e1381–e1391 |
[26] | ClinicalTrials.gov. Study to assess the efficacy and safety of viltolarsen in ambulant boys with DMD (RACER53), 2021. Accessed December 16, 2021. https://clinicaltrials.gov/ct2/show/NCT04060199. |


