
Progression to Loss of Ambulation Among Patients with Autosomal Recessive Limb-girdle Muscular Dystrophy: A Systematic Review
Abstract
Background
The impact of age at autosomal recessive limb girdle muscular dystrophy (LGMDR) onset on progression to loss of ambulation (LOA) has not been well established, particularly by subtype.
Objectives:
To describe the characteristics of patients with adult-, late childhood-, and early childhood-onset LGMDR by subtype and characterize the frequency and timing of LOA.
Methods:
A systematic review was conducted in MEDLINE, Embase and the Cochrane library. Frequency and timing of LOA in patients with LGMDR1, LGMDR2/Miyoshi myopathy (MM), LGMDR3-6, LGMDR9, and LGMDR12 were synthesized from published data.
Results:
In 195 studies, 695 (43.4%) patients had adult-, 532 (33.2%) had late childhood-, and 376 (23.5%) had early childhood-onset of disease across subtypes among those with a reported age at onset (n = 1,603); distribution of age at onset varied between subtypes. Among patients with LOA (n = 228), adult-onset disease was uncommon in LGMDR3-6 (14%) and frequent in LGMDR2/MM (42%); LGMDR3-6 cases with LOA primarily had early childhood-onset (74%). Mean (standard deviation [SD]) time to LOA varied between subtypes and was shortest for patients with early childhood-onset LGMDR9 (12.0 [4.9] years, n = 19) and LGMDR3-6 (12.3 [10.7], n = 56) and longest for those with late childhood-onset LGMDR2/MM (21.4 [11.5], n = 36).
Conclusions:
This review illustrated that patients with early childhood-onset disease tend to have faster progression to LOA than those with late childhood- or adult-onset disease, particularly in LGMDR9. These findings provide a greater understanding of progression to LOA by LGMDR subtype, which may help inform clinical trial design and provide a basis for natural history studies.
INTRODUCTION
Limb girdle muscular dystrophy (LGMD) comprises a group of rare muscular dystrophies caused by mutations in genes encoding proteins involved with muscle maintenance, function, and repair [1]. Over 30 subtypes of LGMD have been identified, of which 90% are autosomal recessive (LGMDR) [2, 3]. The most common of these (along with their European NeuroMuscular Centre [ENMC] nomenclature/original nomenclature, and affected protein), are calpainopathy (LGMDR1/LGMD2A, calpain-3), dysferlinopathy (LGMDR2/LGMD2B and Miyoshi myopathy [MM], dysferlin), sarcoglycanopathies (LGMDR3/LGMD2D, α-sarcoglycan; LGMDR4/LGMD2E, β-sarcoglycan; LGMDR5/LGMD2 C, γ-sarcoglycan; LGMDR6/LGMD2F, δ-sarcoglycan), fukutin-related protein (FKRP)-related dystroglycanopathy (LGMDR9/LGMD2I, FKRP), and anoctaminopathy (LGMDR12/LGMD2 L, anoctamin-5).
Data on the natural history of LGMDR are few and studies describing larger clinical cohorts followed over long periods of time are lacking. Nonetheless, understanding the clinical course and likely disease prognosis is important. Patients with LGMD, including LGMDR, primarily present with proximal skeletal muscle weakness [4], but the clinical course of different LGMDR subtypes appears highly variable and the age at disease onset can range from childhood through adulthood. Disease severity also varies from mild forms in which patients have relatively normal function, to severe forms with rapid onset and progression [1, 4]. Patients with LGMD may experience signs and symptoms of muscle weakness at any age and these typically worsen over time [3, 5]. At an early stage, patients may present with an abnormal walking gait, difficulties in walking, and/or difficulties in running. As the disease progresses, ambulatory function may deteriorate, patients may require walking assistance, and ultimately progress to loss of ambulation (LOA) and wheelchair dependence.
Progression to LOA has been described across LGMDR subtypes [6, 7]; however, it is unclear how the frequency and timing of LOA compares between patients with different subtypes. Previous studies have suggested that the clinical trajectories of patients with LGMD onset before 10 years of age (i.e. early childhood onset) may differ from those with onset after 10 years of age [8]. Although there has been some indication that patients with pediatric-onset LGMDR may experience a more severe progression than those with adult-onset disease [9], the impact of age at symptomatic disease onset on progression to LOA has not been well established. Understanding the natural history by subtype and age at onset is important to characterize disease burden and understand likely disease trajectories among those with LGMDR. In the absence of data from large clinical studies, a systematic review can help fill this gap. As such, through a systematic review and synthesis of the published literature, this study sought to describe the characteristics of patients with pediatric- and adult-onset LGMDR according to subtype and to characterize the frequency and timing of LOA among patients with pediatric- and adult-onset LGMDR, by subtype.
METHODS
Search strategy and study identification
A systematic literature review was conducted in MEDLINE, Embase and the Cochrane library to identify all published data from database inception to September 3, 2019, on the frequency and timing of LOA in patients with LGMDR1, LGMDR2/MM, LGMDR3-6, LGMDR9, and LGMDR12. The search strategy was developed to include terms related to the Population, Exposure/Comparator, Outcomes, Study design (PECOS) criteria specified to be of interest for the study (Supplementary Table 1). Two reviewers worked independently to screen all abstracts identified by the search strategy against the PECOS criteria, and then reviewed the full text of all potentially relevant abstracts. If any discrepancies occurred between the studies selected for inclusion by the two reviewers, a third reviewer provided arbitration. Patient-level data from epidemiologic studies (retrospective and prospective cohort studies), clinical trials, as well as case series and reports were included.
Outcomes and data synthesis
Pediatric-onset disease was characterized based on two categories: onset before age 10 (early childhood-onset) and onset from age 10 to 18 years (late childhood-onset). Adult-onset was defined as disease onset at 18 years or older. Patients whose age at LGMD onset was reported descriptively rather than numerically were analyzed according to the category corresponding to the appropriate age at onset definition. For example, those whose age at onset was reported as occurring “in their third decade” were categorized as having adult-onset disease. Baseline demographics and clinical characteristics of the patients included in the studies were characterized, using counts and proportions, and means and standard deviations (SD), as appropriate. These were stratified by age at onset and LGMDR subtype. Patients were considered to have LOA if they were described as being wheelchair dependent or non-ambulant. Those who were described as ambulatory at their last assessment were considered to not have LOA.
The total number of patients with patient-level data in the included studies and the number of patients with age at LGMD onset reported (and the subset among whom an ambulatory status was reported) were determined by subtype and among the overall sample in order to characterize data availability.
Outcomes of interest (overall and for each LGMDR subtype)
The following outcomes were extracted from the overall sample and for each LGMDR subtype. Among patients with age at LGMD onset reported, the n (%) with adult- (>18 years), late childhood- (10-18 years), and early childhood-onset (<10 years) LGMDR was tabulated. This was determined for the overall cohort as well as separately for male and female patients. Among patients with adult-, late childhood-, and early childhood-onset of disease, the n (%) with ambulatory status reported was assessed; patients could be either ambulatory or non-ambulatory at the time of each assessment. Among those with LOA, mean (SD) time from LGMD onset to LOA, stratified by age of onset (adult-, late childhood-, and early childhood-onset LGMDR, as per the above) was tabulated. Time to LOA was calculated for patients whose age at disease onset was reported numerically, rather than descriptively (e.g., in the third decade). Patients were analyzed based on the subgroup reported by study authors, except for LGMDR2 and MM which were combined into an overall dysferlinopathies subgroup for analysis (LGMDR2/MM).
As all data used in the study were from previously published sources, participant consent or ethical review and approval was not necessary.
RESULTS
Study characteristics
The systematic search identified 2,929 abstracts which after screening resulted in the inclusion of 298 studies, of which 195 studies provided data informing this analysis (Fig. 1). Data were primarily derived from case reports and case series (n = 123) [10–132], although patient-level data were also available from 31 retrospective [133–163], 21 prospective [164–184], and 4 cross-sectional studies [185–188], as well as 2 randomized controlled trials [189, 190] and 14 reports based on other designs (Supplementary Table 2) [98, 191–203]. Of studies reporting geographic location, over 35 countries were represented, with the most studies conducted in the United States (n = 28) and Italy (n = 23).
Fig. 1
PRISMA diagram. aRemoval of duplicates was built into the search strategy.
Data availability
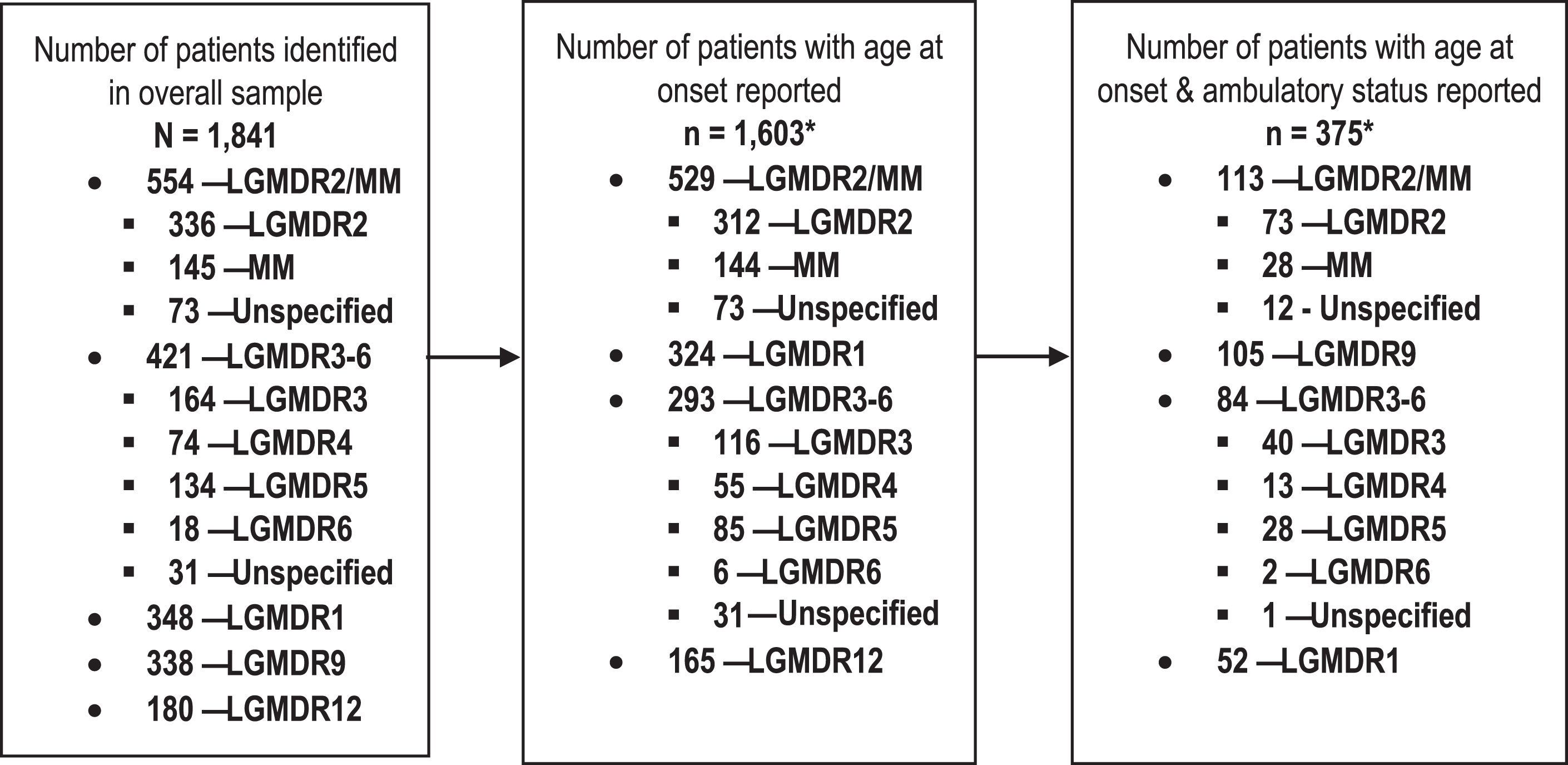
In total, data from 1,841 patients were identified to comprise the overall sample (Fig. 2); of these, 1,603 (87.1%) patients had a reported age at onset. LGMDR2/MM and LGMDR1 were the most common subtypes among patients with a reported age at onset (n = 529, 33.0%; n = 324, 20.2%, respectively), while LGMDR6 was uncommon (n = 6, 0.4%). Among those with age at onset reported, 375 patients also had ambulatory status reported (23.4%), and the percentages with ambulatory status reported ranged from 0.5% (n = 2; LGMDR6) to 30.1% (n = 113; LGMDR2/MM) when considered by subtype. Data on ambulatory status were generally cross-sectional in nature; ambulatory function was typically reported at one assessment only.
Fig. 2
Summary of data availability across LGMDR subtypes. *Includes patients whose age at onset was reported descriptively rather than numerically, with sufficient information to be categorized as having either adult-, late childhood-, or early childhood-onset disease (e.g. onset in “first decade”). Abbreviations: LGMDR, autosomal recessive limb girdle muscular dystrophy; MM, Miyoshi myopathy.
Patient characteristics
Patient characteristics by age at onset category
Of the 1,603 patients with a reported age at onset, 695 (43.4%) had adult-, 532 (33.2%) had late childhood-, and 376 (23.5%) had early childhood-onset disease overall (Fig. 3A). The distribution of patients with pediatric- compared to adult-onset disease was highly variable between subtypes. Patients with LGMDR12 had onset predominantly in adulthood (95.2%), while adult-onset disease was infrequent among patients with LGMDR3-6 (7.5%). When the distribution of pediatric- compared to adult-onset disease was evaluated by sex, there were no real differences by sex among included patients with age at onset data, although the number of female patients with LGMDR12 (n = 39) was comparatively lower than the number of male patients with LGMDR12 (n = 124) identified for this analysis (data not shown). Among patients with adult-onset (n = 695) and late childhood-onset LGMDR (n = 532), the most common subtype was LGMDR2/MM (n = 308, 44.3% adult-onset; n = 211, 39.7% late childhood-onset; Table 1, Fig. 3B). Among those with early childhood-onset disease (n = 376), LGMDR3-6 was the most common subtype (n = 211; 56.1%). There were no patients with early childhood-onset LGMDR12.
Fig. 3
A) Distribution of early childhood-, late childhood- and adult- onset LGMD, by subtype; B) distribution of LGMD subtypes by age at onset category. Note: Percentages are rounded to the nearest whole number. Abbreviations: LGMDR, autosomal recessive limb girdle muscular dystrophy; MM, Miyoshi myopathy.
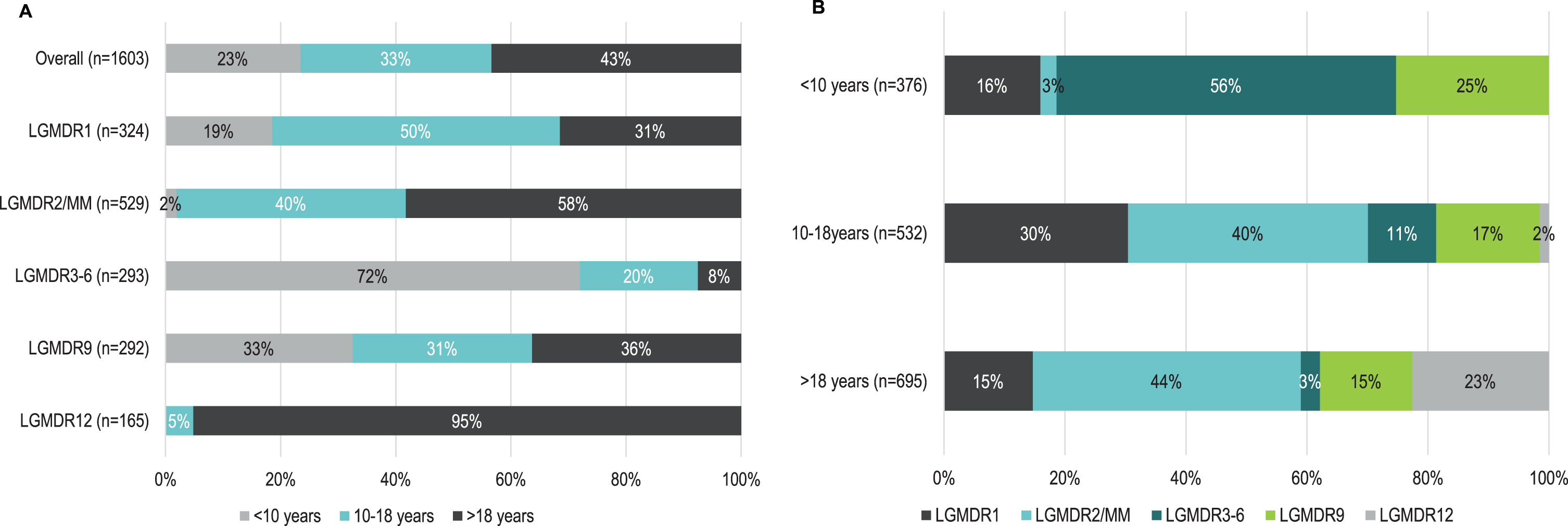
Table 1
Characteristics of patients contributing data to the analysis, stratified by age at onset and subtype (n = 1,603)
| Subtype | N with age at onset reported | Mean (SD) age at onset | n with sex reported | n male (%) |
| Adult-onset (>18 years) | ||||
| Overall* | 695 | 29.2 (10.1) | 672 | 359 (51.7) |
| LGMDR1 | 102 | 30.4 (10.4) | 100 | 39 (38.2) |
| LGMDR2/MM | 308 | 26.3 (7.9) | 290 | 140 (45.5) |
| LGMDR3-6 | 22 | 29.9 (8.2) | 21 | 9 (40.9) |
| LGMDR5 | 7 | 23 | 7 | 3 (42.9) |
| LGMDR3 | 12 | 32.1 (9.5) | 11 | 4 (33.3) |
| LGMDR4 | 0 | N/A | 0 | 0 |
| LGMDR6 | 0 | N/A | 0 | 0 |
| LGMDR9 | 106 | 29.6 (8.2) | 106 | 54 (50.9) |
| LGMDR12 | 157 | 35.8 (13.1) | 155 | 117 (74.5) |
| Late childhood-onset (10-18 years) | ||||
| Overall* | 532 | 14.0 (2.7) | 516 | 242 (45.5) |
| LGMDR1 | 162 | 13.1 (2.5) | 160 | 62 (38.3) |
| LGMDR2/MM | 211 | 15.5 (2.1) | 197 | 97 (46.0) |
| LGMDR3-6 | 60 | 11.8 (2.1) | 60 | 31 (51.7) |
| LGMDR5 | 10 | 12.3 (2.8) | 10 | 5 (50.0) |
| LGMDR3 | 32 | 11.6 (1.7) | 32 | 14 (43.8) |
| LGMDR4 | 11 | 12.8 (2.4) | 11 | 8 (72.7) |
| LGMDR6 | 0 | N/A | 0 | 0 |
| LGMDR9 | 91 | 13.7 (2.7) | 91 | 45 (49.5) |
| LGMDR12 | 8 | 15.2 (2.3) | 8 | 7 (87.5) |
| Early childhood-onset (<10 years) | ||||
| Overall* | 376 | 5.1 (2.5) | 374 | 201 (53.5) |
| LGMDR1 | 60 | 5.9 (2.4) | 60 | 24 (40.0) |
| LGMDR2/MM | 10 | 4.3 (2.6) | 8 | 8 (80.0) |
| LGMDR3-6 | 211 | 5.1 (2.3) | 211 | 112 (53.1) |
| LGMDR5 | 68 | 5.5 (2.3) | 68 | 39 (57.4) |
| LGMDR3 | 72 | 5.1 (2.4) | 72 | 35 (48.6) |
| LGMDR4 | 44 | 4.4 (2.4) | 44 | 23 (52.3) |
| LGMDR6 | 6 | 3.8 (2.3) | 6 | 4 (66.7) |
| LGMDR9 | 95 | 4.8 (2.8) | 95 | 57 (60.0) |
| LGMDR12 | 0 | N/A | 0 | 0 |
*Within each age at onset category, “Overall” refers to unique patients in that category. As patients with LGMDR3, LGMDR4, LGMDR5, and LGMDR6 may be counted in both the individual subtype and combined LGMDR3-6 groups, the number of patients added across subtypes exceed the counts reflected under “Overall.” Abbreviations: LGMDR, autosomal recessive limb girdle muscular dystrophy; MM, Miyoshi myopathy; N/A, not applicable; SD, standard deviation.
Age at onset and ambulatory status
The frequency of ambulation assessment varied across age at onset categories and LGMDR subtypes (Table 2). Patients with early childhood-onset disease had ambulation assessed most often (33.2%, 121/376), while ambulation was infrequently assessed in patients with adult-onset disease (17.6%, 122/695). Among the different subtypes, ambulation assessments were generally common in LGMDR3-6 and LGMDR9 regardless of age at onset. The frequency of ambulation assessment was lowest in LGMDR1 (11.8% of adult-onset, 18.5% of late childhood-onset, and 16.7% of early childhood-onset disease).
Table 2
Ambulatory status and age at ambulation assessment, stratified by age at onset and subtype
| Subtype | n (%) with ambulation assessment | n (%) LOA | Mean (SD) age at LOA | Mean (SD) age at last assessment if ambulatory |
| Adult-onset (>18 years) | ||||
| Overall (n = 695) | 122 (17.6) | 64 (52.5) | 44.7 (13.6) | 43.4 (12.0) |
| LGMDR1 (n = 102) | 12 (11.8) | 11 (91.7) | 48.4 (14.2) | 43.0 |
| LGMDR2/MM (n = 308) | 56 (18.2) | 32 (57.1) | 45.2 (13.1) | 37.5 (10.6) |
| LGMDR3-6 (n = 22) | 11 (50.0) | 11 (100.0) | 32.5 (6.9) | N/A |
| LGMDR9 (n = 106) | 23 (21.7) | 5 (21.7) | 49.4 (14.4) | 43.2 (12.4) |
| LGMDR12 (n = 157) | 20 (12.7) | 5 (25.0) | 55.0 (12.5) | 53.2 (7.3) |
| Late childhood-onset (10-18 years) | ||||
| Overall (n = 532) | 132 (24.8) | 78 (59.1) | 32.8 (11.4) | 26.5 (11.4) |
| LGMDR1 (n = 162) | 30 (18.5) | 17 (56.7) | 30.6 (12.1) | 20.9 (7.8) |
| LGMDR2/MM (n = 211) | 54 (25.6) | 42 (77.8) | 36.2 (11.1) | 24.0 (6.6) |
| LGMDR3-6 (n = 60) | 13 (21.7) | 9 (69.2) | 21.7 (9.4) | 28.3 (14.3) |
| LGMDR9 (n = 91) | 34 (37.4) | 10 (29.4) | 32.1 (8.6) | 30.2 (13.4) |
| LGMDR12 (n = 8) | 1 (12.5) | 0 (0.0) | N/A | 35.0 |
| Early childhood-onset (<10 years) | ||||
| Overall (n = 376) | 121 (32.2) | 86 (71.1) | 17.7 (10.6) | 13.1 (10.1) |
| LGMDR1 (n = 60) | 10 (16.7) | 7 (70.0) | 21.6 (10.6) | 17.2 (7.1) |
| LGMDR2/MM (n = 10) | 3 (30.0) | 3 (100.0) | 36.0 (12.1) | N/A |
| LGMDR3-6 (n = 211) | 60 (28.4) | 57 (95.0) | 16.9 (11.1) | 8.0 (1.0) |
| LGMDR9 (n = 95) | 48 (50.5) | 19 (39.6) | 15.5 (5.9) | 13.2 (10.8) |
| LGMDR12 (n = 0) | N/A | N/A | N/A | N/A |
Abbreviations: LGMDR, autosomal recessive limb girdle muscular dystrophy; LOA, loss of ambulation; MM, Miyoshi myopathy; N/A, not applicable; SD, standard deviation.
LOA was observed in 60.8% (228/375) of patients with ambulatory function reported. Among patients with adult-onset disease, LOA was reported in 52.5%, and at a mean (SD) age of 44.7 (13.6) years (Table 2). LOA was most frequently reported among patients with LGMDR3-6, who also experienced the earliest LOA (mean [SD]: 32.5 [6.9] years) in this age at onset group. Among patients with late childhood-onset disease, 59.1% experienced LOA overall, at a mean (SD) age of 32.8 (11.4) years (Table 2). Compared to patients with adult-onset disease (52.5%), the percentage of patients with late childhood-onset disease who had LOA was slightly higher. Among patients with early childhood-onset disease, 71.1% experienced LOA overall, at a mean (SD) age of 17.7 (10.6) years (Table 2). LOA was most common in this age at onset group. This trend was generally consistent across subtypes, except in cases where low numbers of patients with ambulatory status were available. The number and percentage of patients with LOA and adult- or late childhood-onset disease was highest for LGMDR2/MM (n = 32, 57.1%; n = 42, 77.8%, respectively); while n (%) with LOA and early childhood-onset was highest for LGMDR3-6 (n = 57, 95.0%). All 3 patients with early childhood-onset LGMDR2/MM with an ambulatory assessment had LOA and their mean age at LOA was comparable to the mean age of LOA for the late-childhood onset patients.
Time to LOA
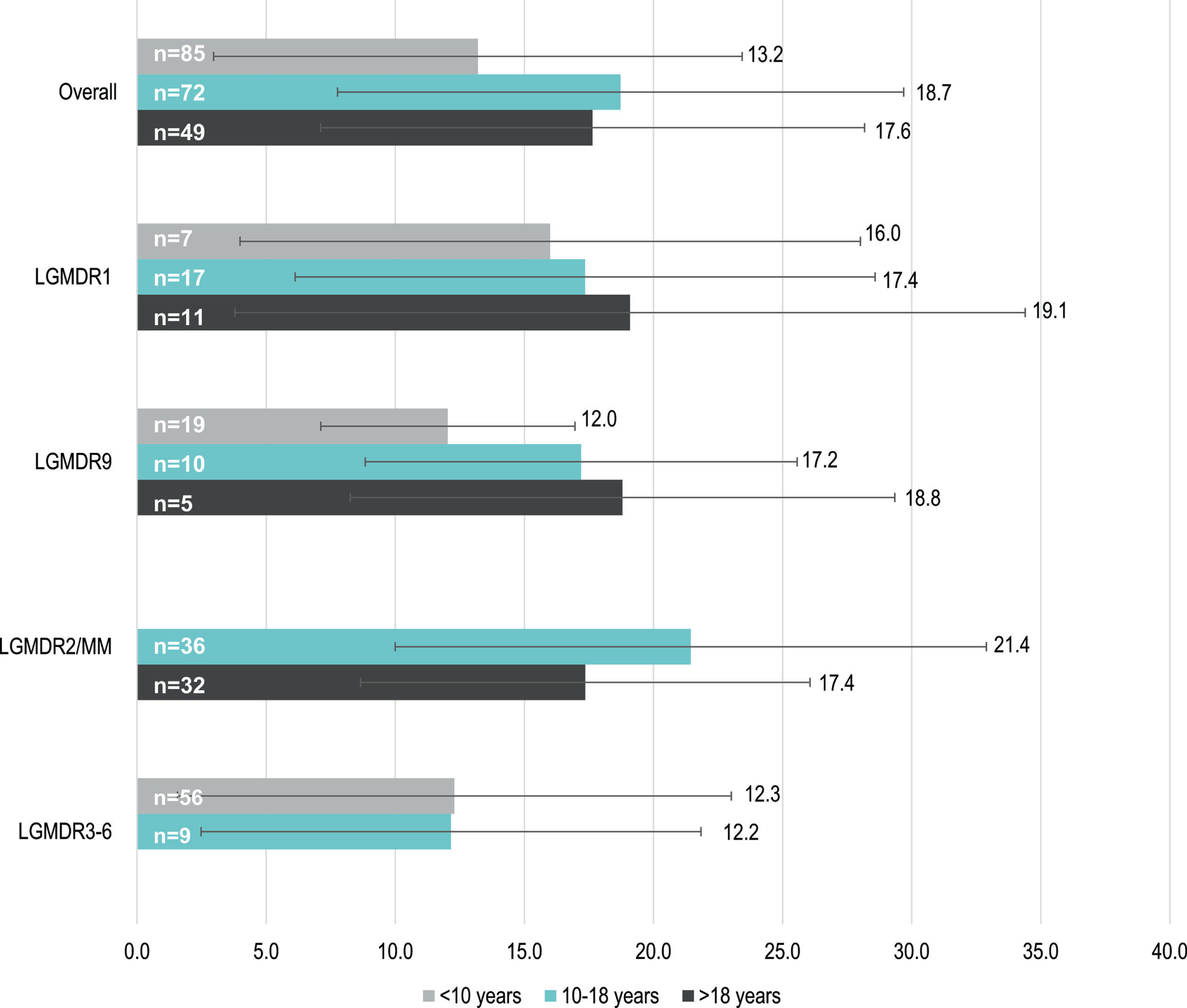
Data to assess time from disease onset to LOA according to age at onset category were limited. Mean (SD) time to LOA varied between subtypes and was shortest for patients with early childhood-onset LGMDR9 (12.0 [4.9] years, n = 19) and LGMDR3-6 (12.3 [10.7] years, n = 56) and longest for those with late childhood-onset LGMDR2/MM (21.4 [11.5] years, n = 36) (Fig. 4). In LGMDR1 and LGMDR9, patients with earlier disease onset appeared to have a faster progression to LOA than those with later disease onset.
Fig. 4
Mean (SD) time (years) to LOA of early childhood-, late childhood- and adult- onset LGMDR, by subtype. **Note: time to LOA was only determined for patients whose age at onset was reported numerically; e.g. patients who were reported as having onset “in adulthood” are not reflected. In addition, subtypes and age groups with n<5 are not reflected (e.g. LGMDR12). Abbreviations: LGMDR, autosomal recessive limb girdle muscular dystrophy; LOA, loss of ambulation; MM, Miyoshi myopathy; SD, standard deviation.
DISCUSSION
This systematic review described the frequency and timing of LOA in LGMDR according to age at disease onset and subtype. LOA represents a severe manifestation of LGMDR, in which muscle weakness and contractures have progressed and worsened over time [204]. Given the heterogeneity of clinical progression among different patients [6–8], the occurrence of LOA was characterized separately for each LGMDR subtype, and for patients with adult-, late childhood-, and early childhood-onset disease.
Understanding the natural history of the progression to LOA and intervening prior to its occurrence would be critical to reduce the burden of disability in LGMDR. To date, there have been few large observational studies conducted that follow patients longitudinally to understand the natural history of LGMDR. However, the Global FKRP Registry has been established to support research in LGMDR9 [205]; as long-term data become available, this registry would offer important insights into the clinical trajectories for patients with this LGMD subtype.
The findings from this systematic review confirm that there is variability in age at LGMD onset across subtypes and provide novel details about LOA. Of note, the frequencies of individual subtypes within adult-, late childhood-, and early childhood-onset categories may be driven by data availability rather than true incidence, as some subtypes had considerably more patients with age at onset reported (e.g. LGMDR2/MM). Nevertheless, patients with LGMDR12 had onset predominantly in adulthood, which corresponds with previous reports that LGMD12 is characterized by late-onset lower limb weakness [206]. In comparison, patients with LGMDR3-6 frequently had early childhood-onset disease, similar to what was observed in a large cohort of patients with sarcoglycanopathy [8]. Variability in the occurrence of LOA was observed across LGMDR subtypes and across age at onset categories. LOA was more common among patients with early childhood- (71%) compared to late childhood- (59%) and adult-onset disease (52%). As ambulatory assessments were more frequent among patients with early childhood-onset disease, data on the occurrence and age at LOA may be more robust in this group than for those with adult-onset disease.
Regardless of age at disease onset, patients with LGMDR3-6 generally had the earliest LOA among the subtypes (at 32 years among adult-onset, 22 years among late childhood-onset, 17 years among early childhood-onset disease). Patients with early childhood-onset disease tended to have faster progression to LOA than those with late childhood- or adult-onset disease, particularly in LGMDR9. This observation is consistent with the findings for LGMDR3-5 from the European Sarcoglycanopathy Consortium, which reported that patients with disease onset before age 10 experienced significantly faster progression to ambulation loss than those with onset after 10 years of age [8].
Given the inclusion of case report data in this systematic review, limitations to study findings include that case report data are prone to bias as investigators may have different motivations for their analyses. In particular, there may be a bias of reporting more severe patients as case studies of the disease. For example, patients who had ambulatory function assessments likely had issues with ambulation in the first place that would prompt such an assessment. Moreover, given the cross-sectional nature of data included in this review, the reported absence of a clinical outcome such as LOA in a patient may not necessarily mean that this patient would not experience LOA subsequently. The small numbers of patients with LGMDR12 identified with LOA may be a consequence of such limitations, as these patients have a later onset of weakness and ambulation loss. In addition, as LGMDR12 was not characterized as a separate entity until recently [44], data availability for these patients may have been particularly affected. Large registries or observational studies that follow patients over time would be valuable to better characterize the natural history of LGMDR, as well as the prevalence of the different subtypes. Large observational studies would also be important for understanding other factors that may influence disease progression, such as socioeconomic characteristics and access to medical care, which are difficult to capture in a systematic review. For the present analyses, data availability was limited as patients were considered eligible only if both their age at LGMD onset and the age at which ambulation was assessed was reported. Therefore, the small numbers of patients meeting these criteria for some subtypes may limit the generalizability of these findings. However, given the rarity of some subtypes such as LGMDR6, small sample sizes were expected. Among included patients, it was often unclear whether they presented symptomatically, were asymptomatic but diagnosed as a result of early recognition through genetic screening, or were asymptomatic and diagnosed when they become symptomatic. As a result, the reported age at disease onset, and consequently the calculated time from onset to LOA, may reflect different experiences for different patients, even for the same LGMDR subtype. Selection and reporting bias may also have resulted in ambulatory status being preferentially investigated among those who were experiencing severe mobility difficulties or who eventually progressed to LOA, which would further impact generalizability. The frequency of LOA among patients with adult-onset disease may particularly be impacted by such biases given the challenge of capturing data for patients with mild disease. Despite the potential for overestimating the frequency of LOA for LGMDR from this review, the differences in frequency and age at LOA observed between subtypes may still hold true, e.g. that patients with sarcoglycanopathies experience faster progression to LOA than other subtypes. Although overall estimates of mean age at onset and age at LOA were determined for patients with adult-, late childhood-, and early childhood-onset LGMDR, data according to subtype may be more meaningful within these categories given the clinical heterogeneity across subtypes.
CONCLUSIONS
Using currently available data, this review illustrated that LOA occurred most commonly among LGMDR patients with early childhood-onset disease, and least commonly among those with adult-onset disease. Moreover, patients with early childhood-onset disease tended to have faster progression to LOA than those with late childhood- or adult-onset disease, suggesting that age at disease onset may be an important risk factor for disease progression. While this systematic review was informed by patient-level data including case report data, which has its caveats such as reporting bias, it provides an in-depth assessment of patients with LGMDR across multiple countries and is a valuable addition to the literature for this rare disease. To complement and strengthen these findings, large registries such as the Global FKRP Registry or observational studies that follow patients over time would be important for better characterizing the natural history of LGMDR. Despite limited data, these findings provide a greater understanding of progression to LOA by subtype in LGMDR, which may help inform clinical trial design and provide a basis for natural history studies.
ACKNOWLEDGMENTS
Funding: This study was funded by Sarepta Therapeutics, Inc.
Authors’ contributions: All authors contributed to the study conception and design. Data analysis was performed by AC, EF and SMS. All authors contributed to the writing of the manuscript. All authors read and approved the final manuscript.
CONFLICTS OF INTEREST
IFA and KLG are employees of Sarepta Therapeutics, Inc. AC, EF and SMS are employees of Broadstreet HEOR which was contracted by Sarepta Therapeutics, Inc for the conduct of this study. CCW received consulting fees from Sarepta Therapeutics, Inc.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-210771.
REFERENCES
[1] | Lo HP , Cooper ST , Evesson FJ , Seto JT , Chiotis M , Tay V , et al. Limb-girdle muscular dystrophy: Diagnostic evaluation, frequency and clues to pathogenesis. Neuromuscular Disorders. (2008) ;18: (1):34–44. |
[2] | Chu ML , Moran E The Limb-Girdle Muscular Dystrophies: Is Treatment on the Horizon? Neurotherapeutics. (2018) ;15: (4):849–62. |
[3] | Taghizadeh E , Rezaee M , Barreto GE , Sahebkar A Prevalence, pathological mechanisms, and genetic basis of limb-girdle muscular dystrophies: A review. J Cell Physiol. (2019) ;234: (6):7874–84. |
[4] | Nigro V , Savarese M Genetic basis of limb-girdle muscular dystrophies: The update. Acta Myol. (2014) ;33: (1):1–12. |
[5] | Iyadurai SJ , Kissel JT The Limb-Girdle Muscular Dystrophies and the Dystrophinopathies. Continuum (Minneap Minn). (2016) ;22: (6, Muscle and Neuromuscular Junction Disorders):1954–77. |
[6] | Mahmood OA , Jiang XM Limb-girdle muscular dystrophies: Where next after six decades from the first proposal (Review). Molecular Medicine Reports. (2014) ;9: (5):1515–32. |
[7] | Siciliano G , Simoncini C , Giannotti S , Zampa V , Angelini C , Ricci G Muscle exercise in limb girdle muscular dystrophies: Pitfall and advantages. Acta Myologica. (2015) ;34: (1):3–8. |
[8] | Alonso-Pérez J , González-Quereda L , Bello L , Guglieri M , Straub V , Gallano P , et al. New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy. Brain. (2020) ;143: (9):2696–708. |
[9] | Angelini C LGMD. Identification, description and classification. Acta myologica: Myopathies and cardiomyopathies: Official Journal of the Mediterranean Society of Myology. (2020) ;39: (4):207–17. |
[10] | Aharoni S , Sadeh M , Silver EL , Straussberg R Dysferlinopathy and very-long-chain acyl coenzyme a dehydrogenase deficiency segregating in the same family. Israel Medical Association Journal. (2011) ;13: (10):632–4. |
[11] | Akarsu EO , Kotan D , Alemdar M , Tunc A Myopathy and epilepsy: A rare phenotype in limb-girdle muscular dystrophy 2l associated with ANO5 mutation. Acta Myologica. (2018) ;37: (2):161. |
[12] | Algahtani H , Shirah B , Alassiri AH , Habib BA , Almuhanna R , Ahamed MF Limb-girdle muscular dystrophy type 2B: An unusual cause ofproximal muscular weakness in Saudi Arabia. Journal of Back andMusculoskeletal Rehabilitation. (2018) ;31: (5):999–1004. |
[13] | Al-Harbi KM , Abdallah AM LGMD2D syndrome: The importance of clinical and molecular genetics in patient and family management. Neuroendocrinology Letters. (2016) ;37: (4):277–81. |
[14] | Angelini C , Fanin M , Menegazzo E , Freda MP , Duggan DJ , Hoffman EP Homozygous alpha-sarcoglycan mutation in two siblings: One asymptomatic and one steroid-responsive mild limb-girdle muscular dystrophy patient. Muscle and Nerve. (1998) ;21: (6):769–75. |
[15] | Angelini C , Peterle E , Gaiani A , Bortolussi L , Borsato C Dysferlinopathy course and sportive activity: Clues for possible treatment. Acta Myologica. (2011) ;30: (OCTOBER):127–32. |
[16] | Bandyopadhyay S A novel phenotype of miyoshi myopathy or LGMD2B muscular dystrophy with early severe contractures and normal CK, simulating atypical bethlem myopathy. Muscle and Nerve. (2018) ;58: (Supplement 2):S67. |
[17] | Barresi R , Morandi L , Mora M , Di Blasi C , Negri T , Brugnoni R , et al. Disruption of heart sarcoglycan complex and severe cardiomyopathy caused by beta sarcoglycan mutations. Journal of Medical Genetics. (2000) ;37: (2):102–7. |
[18] | Baumeister SK , Todorovic S , Milic-Rasic V , Dekomien G , Lochmuller H , Walter MC Eosinophilic myositis as presenting symptom in gamma-sarcoglycanopathy. Neuromuscular Disorders. (2009) ;19: (2):167–71. |
[19] | Bhatt AD , Shah K , Puvar A , Joshi CG , Joshi M A case of limb girdle muscular dystrophy type 2A from India: Copy number variation analysis using targeted amplicon sequencing. Journal of Clinical and Diagnostic Research. (2019) ;13: (4):GD01–GD2. |
[20] | Bird TD , Lipe HP , Steinbart EJ Geriatric neurogenetics: Oxymoron or reality? Archives of Neurology. (2008) ;65: (4):537–9. |
[21] | Blackburn PR , Selcen D , Jackson JL , Guthrie KJ , Cousin MA , Boczek NJ , et al. Early-onset limb-girdle muscular dystrophy-2L in a female athlete. Muscle and Nerve. (2017) ;55: (5):E19–E21. |
[22] | Bouquet F , Cossee M , Behin A , Deburgrave N , Romero N , Leturcq F , et al. Miyoshi-like distal myopathy with mutations in anoctamin 5 gene. Revue Neurologique (2012) . |
[23] | Burke G , Hillier C , Cole J , Sampson M , Bridges L , Bushby K , et al. Calpainopathy presenting as foot drop in a 41 year old. Neuromuscular Disorders. (2010) ;20: (6):407–10. |
[24] | Cagliani R , Comi GP , Tancredi L , Sironi M , Fortunate F , Giorda R , et al. Primary beta-sarcoglycanopathy manifesting as recurrent exercise-induced myoglobinuria. Neuromuscular Disorders. (2001) ;11: (4):389–94. |
[25] | Carman KB , Yimenicioglu S , Yarar C Limb-girdle muscular distrophy type 2B: A case presented with abdominal and ankle pain. Acta Myologica. (2018) ;37: (2):160. |
[26] | Catteruccia M , Primiano G , Fattori F , Bertini E , Servidei S , D’Amico A Exercise intolerance and myoglobinuria as presenting symptom of alpha-sarcoglycanopathy. Acta Myologica. 37. (2018) ;37: (1):58. |
[27] | Chourasia N , Biliciler S EDB hypertrophy associated with FKRP mutation in LGMD 21. Journal of Clinical Neuromuscular Disease. (2017) ;18: (Supplement 1):S11. |
[28] | Connolly AM , Pestronk A , Mehta S , Al-Lozi M Primary alpha-sarcoglycan deficiency responsive to immunosuppression over three years. Muscle and Nerve. (1998) ;21: (11):1549–53. |
[29] | D’Amico A , Petrini S , Parisi F , Tessa A , Francalanci P , Grutter G , et al. Heart transplantation in a child with LGMD2I presenting as isolated dilated cardiomyopathy. Neuromuscular Disorders. (2008) ;18: (2):153–5. |
[30] | Darin N , Kroksmark AK , Ahlander AC , Moslemi AR , Oldfors A , Tulinius M Inflammation and response to steroid treatment in limb-girdle muscular dystrophy 2I. European Journal of Paediatric Neurology. (2007) ;11: (6):353–7. |
[31] | Dicapua D , Patwa H Puerto rican founder mutation G787A in the SGCG Gene: A case report of 2 siblings with LGMD 2C. Journal of Clinical Neuromuscular Disease. (2014) ;15: (3):105–7. |
[32] | Diers A , Carl M , Stoltenburg-Didinger G , Vorgerd M , Spuler S Painful enlargement of the calf muscles in limb girdle muscular dystrophy type 2B (LGMD2B) with a novel compound heterozygous mutation in DYSF. Neuromuscular Disorders. (2007) ;17: (2):157–62. |
[33] | Diniz G , Tekgul H , Hazan F , Yararbas K , Tukun A Sarcolemmal deficiency of sarcoglycan complex in an eighteen months old Turkish boy with a huge deletion in the beta sarcoglycan gene. Neuromuscular Disorders. (2014) ;24: (9–10):884–5. |
[34] | Diniz G , Tekgul H , Hazan F , Yararbas K , Tukun A Sarcolemmal deficiency of sarcoglycan complex in an 18-month-old Turkish boy with a large deletion in the beta sarcoglycan gene. Balkan Journal of Medical Genetics. (2015) ;18: (2):71–6. |
[35] | Dirik E , Aydin A , Kurul S , Sahin B Limb girdle muscular dystrophy type 2A presenting with cardiac arrest. Pediatric Neurology. (2001) ;24: (3):235–7. |
[36] | Fanin M , Melacini P , Boito C , Pegoraro E , Angelini C LGMD2E patients risk developing dilated cardiomyopathy. Neuromuscular Disorders. (2003) ;13: (4):303–9. |
[37] | Frosk P , Del Bigio MR , Wrogemann K , Greenberg CR Hutterite brothersboth affected with two forms of limb girdle muscular dystrophy:LGMD2H and LGMD2I. European Journal of Human Genetics. (2005) ;13: (8):978–82. |
[38] | Fuschillo S , Torrente Y , Balzano G Severe respiratory and skeletal muscles involvement in a carrier of dysferlinopathy with chronic obstructive pulmonary disease. Respiratory Care. (2010) ;55: (8):1091–3. |
[39] | Gemelli C , Fiorillo C , Fabbri S , Cabona C , Zara F , Madia F , et al. A case of limb-girdle muscular dystrophy type 2L mimicking Dermatomyositis. Acta Myologica. (2017) ;36: (2):94. |
[40] | Ghafouri-Fard S , Hashemi Gorji F , Fardaei M , Miryounesi M Limb girdle muscular dystrophy type 2E due to a novel large deletion in SGCB gene. Iranian Journal of Child Neurology. (2017) ;11: (3):57–60. |
[41] | Gulati S , Leekha S , Sharma MC , Kalra V Gamma-Sarcoglycanopathy. Indian Pediatrics. (2003) ;40: (11):1077–81. |
[42] | Habig K , Wurzer B , Gunther B , Schanzer A , Nolte D , Kramer HH A slight change with a major impact-a report on a patient with limb girdle muscular dystrophy. Clinical Neurophysiology. (2017) ;128: (10):e354. |
[43] | Heikali D , Bokhoor P New onset cardiomyopathy and cardiogenic shock leading to a diagnosis of limbgirdle muscular dystrophy 2I. Journal of the American College of Cardiology. (2017) ;69: (11 Supplement 1):2352. |
[44] | Hicks D , Sarkozy A , Muelas N , Koehler K , Huebner A , Hudson G , et al. A founder mutation in Anoctamin 5 is a major cause of limb-girdle muscular dystrophy. Brain. (2011) ;134: (1):171–82. |
[45] | Hong D , Zhang W , Wang W , Wang Z , Yuan Y Asian patients with limb girdle muscular dystrophy 2I (LGMD2I). Journal of Clinical Neuroscience. (2011) ;18: (4):494–9. |
[46] | Illa I , De Luna N , Dominguez-Perles R , Rojas-Garcia R , Paradas C , Palmer J , et al. Symptomatic dysferlin gene mutation carriers: Characterization of two cases. Neurology. (2007) ;68: (16):1284–9. |
[47] | Izzedine H , Brocheriou I , Eymard B , Le Charpentier M , Romero NB , LeNaour G , et al. Loss of Podocyte Dysferlin Expression Is Associated With Minimal Change Nephropathy. American Journal of Kidney Diseases. (2006) ;48: (1):143–50. |
[48] | Jenne DE , Kley RA , Vorgerd M , Schroder JM , Weis J , Reimann H , et al. Limb girdle muscular dystrophy in a sibling pair with a homozygous Ser606Leu mutation in the alternatively spliced IS2 region of calpain 3. Biological Chemistry. (2005) ;386: (1):61–7. |
[49] | Jethwa H , Jacques TS , Gunny R , Wedderburn LR , Pilkington C , Manzur AY Limb girdle muscular dystrophy type 2B masquerading as inflammatory myopathy: Case report. Pediatric Rheumatology. (2013) ;11: (1) (no pagination)(19). |
[50] | Jomaa R , Star MH , Mhenni SY , Jenzri S , Jerbi S , Zantour B , et al. Isolated neurosarcoidosis revealed by diabetes insipidus, visual loss and diplopia in a child patient: A diagnostic problem. Clinical Pediatric Endocrinology. (2009) ;18: (1):51–4. |
[51] | Kapoor S , Tatke M , Aggarwal S , Gupta A Beta-sarcoglycanopathy. Indian Journal of Pediatrics. (2005) ;72: (1):71–4. |
[52] | Kefi M , Amouri R , Chabrak S , Mechmeche R , Hentati F Variable cardiac in involvement in Tunisian siblings harboring FKRP gene mutations. Neuropediatrics. (2008) ;39: (2):113–5. |
[53] | Khadilkar SV , Singh RK , Agarwal P , Krahn M , Levy N Twenty-two year follow-up of an Indian family with dysferlinopathy-clinical, immunocytochemical, western blotting and genetic features. Neurology India. (2008) ;56: (3):388–90. |
[54] | Khaiboullina SF , Martynova EV , Bardakov SN , Mavlikeev MO , Yakovlev IA , Isaev AA , et al. Serum Cytokine Profile in a Patient Diagnosed with Dysferlinopathy. Case Reports in Medicine. (no pagination) (3615354). |
[55] | Kida H , Sano K , Yorita A , Miura S , Ayabe M , Hayashi Y , et al. First Japanese case of muscular dystrophy caused by a mutation in the anoctamin 5 gene. Neurology and Clinical Neuroscience. (2015) ;3: (4):150–2. |
[56] | Klinge L , Dean AF , Kress W , Dixon P , Charlton R , Muller JS , et al. Late onset in dysferlinopathy widens the clinical spectrum. Neuromuscular Disorders. (2008) ;18: (4):288–90. |
[57] | Kocak Eker H , Balasar O Limb-girdle muscular dystrophy: A family with three cases. Gazi Medical Journal. (2019) ;30: (1):P93. |
[58] | Koken OY , Kayilioglu H , Sel CG , Oztoprak U , Talim B , Yuksel D A case of limb-girdle muscular dystrophy 2I due to FKRP mutation presenting as acute myositis. Acta Myologica. (2018) ;37: (2):161–2. |
[59] | Krishnaiah B , Lee JJ , Wicklund MP , Kaur D Young girl presenting with exercise-induced myoglobinuria. Muscle and Nerve. (2016) ;54: (1):161–4. |
[60] | Lanzillo R , Aurino S , Fanin M , Aguennoz M , Vitale F , Fiorillo C , et al. Early onset calpainopathy with normal non-functional calpain 3 level. Developmental Medicine and Child Neurology. (2006) ;48: (4):304–6. |
[61] | Lerario A , Cogiamanian F , Marchesi C , Belicchi M , Bresolin N , Porretti L , et al. Effects of rituximab in two patients with dysferlin-deficient muscular dystrophy. BMC Musculoskeletal Disorders. (2010) ;11: , 157. |
[62] | Li F , Yin G , Xie Q , Shi G Late-onset dysferlinopathy presented as“liver Enzyme” abnormalities: A technical note. Journal ofClinical Rheumatology. (2014) ;20: (5):275–7. |
[63] | Liewluck T , Goodman BP Late-onset axial myopathy and camptocormia in a calpainopathy carrier. Journal of Clinical Neuromuscular Disease. (2012) ;13: (4):209–13. |
[64] | Lin YC , Murakami T , Hayashi YK , Nishino I , Nonaka I , Yuo CY , et al. A novel FKRP gene mutation in a Taiwanese patient with limb-girdle muscular dystrophy 2I. Brain and Development. (2007) ;29: (4):234–8. |
[65] | Lindberg C , Sixt C , Oldfors A Episodes of exercise-induced dark urine and myalgia in LGMD 2I. Acta Neurologica Scandinavica (2011) . |
[66] | Little AA , McKeever PE , Gruis KL Novel mutations in the anoctamin 5 gene (ANO5) associated with limb-girdle muscular dystrophy 2L. Muscle and Nerve. (2013) ;47: (2):287–91. |
[67] | Lomma C , Ransom D Chemotherapy dosing and toxicity in a patient with muscular dystrophy. Cancer Reports. (2018) ;1: (2) (no pagination)(e1106). |
[68] | Lu Y , Song X , Ji G , Wu H , Li D , Sun S Identification of a novel SGCA missense mutation in a case of limb-girdle muscular dystrophy 2D with the absence of four sarcoglycan proteins. Neuropathology. (2019) ;39: (3):207–11. |
[69] | Margeta M , Connolly AM , Winder TL , Pestronk A , Moore SA Cardiac pathology exceeds skeletal muscle pathology in two cases of limb-girdle muscular dystrophy type 2I. Muscle and Nerve. (2009) ;40: (5):883–9. |
[70] | Martinez-Thompson JM , Moore SA , Liewluck T A novel CAPN3 mutation in late-onset limb-girdle muscular dystrophy with early respiratory insufficiency. Journal of Clinical Neuroscience. (2018) ;53: :229–31. |
[71] | Mashiah J , Harel A , Bitterman O , Sagi L , Gat A , Fellig Y , et al. Isotretinoin treatment of autosomal recessive congenital ichthyosis complicated by coexisting dysferlinopathy. Clin Exp Dermatol. (2016) ;41: (4):390–3. |
[72] | Matsubara E , Tsuchiya A , Minami N , Nishino I , Pappolla MA , Shoji M , et al. A unique case of limb-girdle muscular dystrophy type 2A carrying novel compound heterozygous mutations in the human CAPN3 gene. European Journal of Neurology. (2007) ;14: (7):819–22. |
[73] | McMillan HJ , Carter MT , Jacob PJ , Laffan EE , O’Connor MD , Boycott KM Homozygous contiguous gene deletion of 13q12 causing LGMD2C and ARSACS in the same patient. Muscle and Nerve. (2009) ;39: (3):396–9. |
[74] | McNally EM , Passos-Bueno MR , Bonnemann CG , Vainzof M , De Moreira ES , Lidov HGW , et al. Mild and severe muscular dystrophy caused by a single gamma-sarcoglycan mutation. American Journal of Human Genetics. (1996) ;59: (5):1040–7. |
[75] | Meznaric-Petrusa M , Kralj E , Angelini C , Fanin M , Trinkaus D Cardiomyopathy in a patient with limb-girdle muscular dystrophy type 2D: Pathomorphological aspects. Forensic Science International Supplement Series. (2009) ;1: (1):58–62. |
[76] | Miskew Nichols B , Nikhanj A , Wang F , Freed DH , Oudit GY Advanced Dilated Cardiomyopathy in a Patient with Hutterite Limb-Girdle Muscular Dystrophy: Use of a Left Ventricular Assist Device. Circulation: Heart Failure. (2018) ;11: (4) (no pagination) (e004960). |
[77] | Mojbafan M , Nilipour Y , Tonekaboni SH , Bagheri SD , Bagherian H , Sharifi Z , et al. A rare form of limb girdle muscular dystrophy (type 2E) seen in an Iranian family detected by autozygosity mapping. Journal of Neurogenetics. (2016) ;30: (1):1–4. |
[78] | Moody S , Mancias P Dysferlinopathy presenting as rhabdomyolysis and acute renal failure. Journal of Child Neurology. (2013) ;28: (4):502–5. |
[79] | Muller T , Krasnianski M , Witthaut R , Deschauer M , Zierz S Dilated cardiomyopathy may be an early sign of the C826A Fukutin-related protein mutation. Neuromuscular Disorders. (2005) ;15: (5):372–6. |
[80] | Mustafa E , Khandaker MAH , Rashid MM , Ghose SK , Chowdhury MK Sarcoglycanopathy - A rare case report and literature review. Journal of Medicine (Bangladesh). (2014) ;15: (1):77–9. |
[81] | Nagashima T , Chuma T , Mano Y , Goto YI , Hayashi YK , Minami N , et al. Dysferlinopathy associated with rigid spine syndrome. Neuropathology. (2004) ;24: (4):341–6. |
[82] | Negrao L , Geraldo A , Matos A , Rebelo O , Santos R , Marques C The first portuguese families with limb-girdle muscular dystrophy 2L. Sinapse. (2012) ;12: (1):39–44. |
[83] | Nemes A , Dezsi L , Domsik P , Kalapos A , Forster T , Vecsei L Left ventricular deformation abnormalities in a patient with calpainopathy-a case from the three-dimensional speckle-tracking echocardiographic magyar-path study. Quantitative Imaging in Medicine and Surgery. (2017) ;7: (6):685–90. |
[84] | Neto ARC , Leoni TB , De Lima FD , Martinez ARM , Nucci A , Franca MC EMG findings in patients with limb-girdle muscular dystrophy due to heterozygous CAPN3 mutations. Clinical Neurophysiology. (2018) ;129: (Supplement 1):e82. |
[85] | Nowak KJ , Walsh P , Jacob RL , Johnsen RD , Peverall J , McNally EM , et al. Severe gamma-sarcoglycanopathy caused by a novel missense mutation and a large deletion. Neuromuscular Disorders. (2000) ;10: (2):100–7. |
[86] | Oexle K , Herrmann R , Dode C , Leturcq F , Hubner C , Kaplan JC , et al. Neurosensory hearing loss in secondary adhalinopathy. Neuropediatrics. (1996) ;27: (1):32–6. |
[87] | Oflazer PS , Gundesli H , Zorludemir S , Sabuncu T , Dincer P Eosinophilic myositis in calpainopathy: Could immunosuppression of the eosinophilic myositis alter the early natural course of the dystrophic disease? Neuromuscular Disorders. (2009) ;19: (4):261–3. |
[88] | Okahashi S , Ogawa G , Suzuki M , Ogata K , Nishino I , Kawai M Asymptomatic sporadic dysferlinopathy presenting with elevation of serum creatine kinase. Typical distribution of muscle involvement shown by MRI but not by CT. Internal Medicine. (2008) ;47: (4):305–7. |
[89] | Okere A , Reddy SS , Gupta S , Shinnar M A cardiomyopathy in a patient with limb girdle muscular dystrophy type 2A. Circulation: Heart Failure. (2013) ;6: (1):e12–3. |
[90] | Oliveira Santos M , Ninitas P , Conceicao I Severe limb-girdle muscular dystrophy 2A in two young siblings from Guinea-Bissau associated with a novel null homozygous mutation in CAPN3 gene. Neuromuscular Disorders. (2018) ;28: (12):1003–5. |
[91] | Patel NJ , Van Dyke KW , Espinoza LR Limb-Girdle Muscular Dystrophy 2B and Miyoshi Presentations of Dysferlinopathy. American Journal of the Medical Sciences. (2017) ;353: (5):484–91. |
[92] | Peddareddygari LR , Oberoi K , Baisre-DeLeon A , Grewal RP Novel mutation in anoctamin 5 gene causing limb-girdle muscular dystrophy 2L. Journal of Clinical Neuromuscular Disease. (2018) ;19: (4):228–31. |
[93] | Pena L , Kim K , Charrow J Episodic myoglobinuria in a primary gamma-sarcoglycanopathy. Neuromuscular Disorders. (2010) ;20: (5):337–9. |
[94] | Penisson-Besnier I , Richard I , Dubas F , Beckmann JS , Fardeau M Pseudometalic variability of and phenotypic variability of calpain deficiency in two siblings. Muscle and Nerve. (1998) ;21: (8):1078–80. |
[95] | Pimentel LHC , Alcantara RNM , Fontenele SMDA , Costa CMDC , Gondim FDAA Limb-girdle muscular dystrophy type 2B mimicking polymyositis. Arquivos de Neuro-Psiquiatria. (2008) ;66: (1):80–2. |
[96] | Pizzanelli C , Mancuso M , Galli R , Choub A , Fanin M , Nascimbeni AC , et al. Epilepsy and limb girdle muscular dystrophy type 2A: Double trouble, serendipitous finding or new phenotype? Neurological Sciences. (2006) ;27: (2):134–6. |
[97] | Prahm KP , Feldt-Rasmussen U , Vissing J Human growth hormone stabilizes walking and improves strength in a patient with dominantly inherited calpainopathy. Neuromuscular Disorders. (2017) ;27: (4):358–62. |
[98] | Prelle A , Comi GP , Tancredi L , Rigoletto C , Ciscato P , Fortunato F , et al. Sarcoglycan deficiency in a large Italian population of myopathic patients. Acta Neuropathologica. (1998) ;96: (5):509–14. |
[99] | Quinlivan RM , Robb SA , Sewry C , Dubowitz V , Piccolo F , Kaplan JC Absence of alpha-sarcoglycan and novel missense mutations in thealpha-sarcoglycan gene in a young British girl with musculardystrophy. Developmental Medicine and Child Neurology. (1997) ;39: (11):770–4. |
[100] | Rekik S , Sakka S , Ben Romdhan S , Farhat N , Baba Amer Y , Lehkim L , et al. Novel Missense CAPN3 Mutation Responsible for Adult-Onset Limb Girdle Muscular Dystrophy with Calves Hypertrophy. Journal of Molecular Neuroscience (2019) . |
[101] | Renard D , Fernandez C , Bouchet-Seraphin C , Labauge P Cortical heterotopia in LGMD2I. Neuromuscular Disorders. (2012) ;22: (5):443–4. |
[102] | Saenz A , Ono Y , Sorimachi H , Goicoechea M , Leturcq F , Blazquez L , et al. Does the severity of the LGMD2A phenotype in compound heterozygotes depend on the combination of mutations? Muscle and Nerve. (2011) ;44: (5):710–4. |
[103] | Sahai S , Khanlou N , Shieh P A case of bilateral scapular winging. Journal of Clinical Neuromuscular Disease. (2019) ;20: (3):143. |
[104] | Schessl J , Kress W , Schoser B Novel ANO5 mutations causing hyper-CK-emia, limb girdle muscular weakness and miyoshi type of muscular dystrophy. Muscle and Nerve. (2012) ;45: (5):740–2. |
[105] | Schneider I , Stoltenburg G , Deschauer M , Winterholler M , Hanisch F Limb girdle muscular dystrophy type 2L presenting as necrotizing myopathy. Acta Myologica: Myopathies and Cardiomyopathies: Official Journal of the Mediterranean Society of Myology / Edited by the Gaetano Conte Academy for the Study of Striated Muscle Diseases. (2014) ;33: (3):19–21. |
[106] | Schutz PW , Scalco RS , Barresi R , Houlden H , Parton M , Holton JL Calpainopathy with macrophage-rich, regional inflammatory infiltrates. Neuromuscular Disorders. (2017) ;27: (8):738–41. |
[107] | Sczesny-Kaiser M , Kowalewski R , Schildhauer TA , Aach M , Jansen O , Grasmucke D , et al. Treadmill training with HAL exoskeleton-A novel approach for symptomatic therapy in patients with limb-girdle muscular dystrophy-preliminary study. Frontiers in Neuroscience. (2017) ;11: (AUG) (no pagination)(449). |
[108] | Selva-O’Callaghan A , Labrador-Horrillo M , Gallardo E , Herruzo A , Grau-Junyent JM , Vilardell-Tarres M Muscle inflammation, autoimmune Addison’s disease and sarcoidosis in a patient with dysferlin deficiency. Neuromuscular Disorders. (2006) ;16: (3):208–9. |
[109] | Shouman K , Morren J , Soltanzadeh P Phenotypic heterogeneity in limbgirdle muscular dystrophy type 2l (anoctaminopathy). Neurology Conference: 70th Annual Meeting of the American Academy of Neurology, AAN. (2018) ;90: (15 Supplement 1). |
[110] | Strafella C , Campoli G , Galota RM , Caputo V , Pagliaroli G , Carboni S , et al. Limb-Girdle Muscular Dystrophies (LGMDs): The clinical application of NGS analysis, a family case report. Frontiers in Neurology. (2019) ;10: (JUN) (no pagination)(619). |
[111] | Svahn J , Streichenberger N , Benveniste O , Menassa R , Michel L , Fayolle H , et al. Significant response to immune therapies in a case of subacute necrotizing myopathy and FKRP mutations. Neuromuscular Disorders. (2015) ;25: (11):865–8. |
[112] | Szymanska S , Rokicki D , Karkucinska-Wieckowska A , Szymanska-Debinska T , Ciara E , Ploski R , et al. Case report of an adolescent girl with limb-girdle muscular dystrophy type 2B - The usefulness of muscle protein immunostaining in the diagnosis of dysferlinopathies. Folia Neuropathologica. (2014) ;52: (4):452–6. |
[113] | Takahashi K Effects of prednisone on a patient with dysferlinopathy assessed by maximal voluntary isometric contraction: Alternate-day low-dose administration for a 17-year period. Case Reports in Neurology. (2019) :10–6. |
[114] | Takahashi T , Aoki M , Imai T , Yoshioka M , Konno H , Higano S , et al. A case of dysferlinopathy presenting choreic movements. Movement Disorders. (2006) ;21: (9):1513–5. |
[115] | Takano A , Bonnemann CG , Honda H , Sakai M , Feener CA , Kunkel LM , et al. Intrafamilial phenotypic variation in limb-girdle muscular dystrophy type 2C with compound heterozygous mutations. Muscle and Nerve. (2000) ;23: (5):807–10. |
[116] | Tarnopolsky M , Hoffman E , Giri M , Shoffner J , Brady L Alpha-sarcoglycanopathy presenting as exercise intolerance and rhabdomyolysis in two adults. Neuromuscular Disorders. (2015) ;25: (12):952–4. |
[117] | Tasca G , Evila A , Pane M , Monforte M , Graziano A , Hackman P , et al. Isolated semitendinosus involvement in the initial stages oflimb-girdle muscular dystrophy 2L. Neuromuscular Disorders. (2014) ;24: (12):1118–9. |
[118] | Tsuburaya R , Suzuki T , Saiki K , Nonaka I , Sugita H , Hayashi YK , et al. Lobulated fibers in a patient with 46-year history of limb-girdle muscle weakness. Neuropathology. (2011) ;31: (4):455–7. |
[119] | Van der Kooi AJ , De Visser M , Van Meegen M , Ginjaar HB , Van Essen AJ , Jennekens FGI , et al. A novel gamma-sarcoglycan mutation causing childhood onset, slowly progressive limb girdle muscular dystrophy. Neuromuscular Disorders. (1998) ;8: (5):305–8. |
[120] | Viloria-Alebesque A , Bellosta-Diago E , Santos-Lasaosa S , Mauri-Llerda JA Familial association of genetic generalised epilepsy with limb-girdle muscular dystrophy through a mutation in CAPN3. Epilepsy and Behavior Case Reports. (2019) ;11: :122–4. |
[121] | Von Der Hagen M , Kaindl AM , Koehler K , Mitzscherling P , Hausler HJ , Stoltenburg-Didinger G , et al. Limb girdle muscular dystrophy type 2I caused by a novel missense mutation in the FKRP gene presenting as acute virus-associated myositis in infancy. European Journal of Pediatrics. (2006) ;165: (1):62–3. |
[122] | Von Guionneau A , Burford C , Stone S Limb-girdle muscular dystrophy type 2i (LGMD2I) and dilated cardiomyopathy: Management during Pregna. International Journal of Gynecology and Obstetrics. (2018) ;143: (Supplement 3):583–4. |
[123] | Vorgerd M , Gencik M , Mortier J , Epplen JT , Malin JP , Mortier W Isolated loss of gamma-sarcoglycan: Diagnostic implications in autosomal recessive limb-girdle muscular dystrophies. Muscle and Nerve. (2001) ;24: (3):421–4. |
[124] | Walsh R , Hill F , Breslin N , Connolly S , Brett FM , Charlton R , et al. Progressive dysphagia in limb-girdle muscular dystrophy type 2B. Muscle and Nerve. (2011) ;43: (5):761–4. |
[125] | Walter MC , Braun C , Vorgerd M , Poppe M , Thirion C , Schmidt C , et al. Variable reduction of caveolin-3 in patients with LGMD2B/MM. Journal of Neurology. (2003) ;250: (12):1431–8. |
[126] | Walter MC , Dekomien G , Schlotter-Weigel B , Reilich P , Pongratz D , Muller-Felber W , et al. Respiratory insufficiency as a presenting symptom of LGMD2D in adulthood. Acta Myologica. (2004) ;23: (1):1–5. |
[127] | Wang CH , Liang WC , Minami N , Nishino I , Jong YJ Limb-girdle muscular dystrophy type 2A with mutation in CAPN The first report in Taiwan. Pediatrics and Neonatology. (2015) ;56: (1):62–5. |
[128] | Wang DN , Wang ZQ , Chen YQ , Xu GR , Lin MT , Wang N Limb-girdle muscular dystrophy type 2I: Two Chinese families and a review in Asian patients. International Journal of Neuroscience. (2018) ;128: (3):199–207. |
[129] | Wong-Kisiel LC , Kuntz NL Two siblings with limb-girdle muscular dystrophy type 2E responsive to deflazacort. Neuromuscular Disorders. (2010) ;20: (2):122–4. |
[130] | Xu C , Chen J , Zhang Y , Degree M , Li J Limb-girdle muscular dystrophy type 2B misdiagnosed as polymyositis at the early stage Case report and literature review. Medicine (United States). (2018) ;97: (21) (no pagination)(e10539). |
[131] | Yilmaz A , Gdynia HJ , Mahrholdt H , Sechtem U Cardiovascular magnetic resonance reveals similar damage to the heart of patients with Becker and limb-girdle muscular dystrophy but no cardiac symptoms. Journal of Magnetic Resonance Imaging. (2009) ;30: (4):876–7. |
[132] | Yuksel D , Dedeoglu O , Ozbay F , Talim B A rare form of limb-girdle muscular dystrophy (LGMD type 2E) in trisomy 21. Acta Myologica. (2018) ;37: (2):161. |
[133] | Alavi A , Esmaeili S , Nilipour Y , Nafissi S , Tonekaboni SH , Zamani G , et al. LGMD2E is the most common type of sarcoglycanopathies in the Iranian population. Journal of Neurogenetics. (2017) ;31: (3):161–9. |
[134] | Alhamidi M , Brox V , Stensland E , Liset M , Lindal S , Nilssen O Limb girdle muscular dystrophy type 2I: No correlation between clinical severity, histopathology and glycosylated alpha-dystroglycan levels in patients homozygous for common FKRP mutation. Neuromuscular Disorders. (2017) ;27: (7):619–26. |
[135] | Angelini C , Nardetto L , Borsato C , Padoan R , Fanin M , Nascimbeni AC , et al. The clinical course of calpainopathy (LGMD2A) and dysferlinopathy (LGMD2B). Neurological Research. (2010) ;32: (1):41–6. |
[136] | Aoki M , Liu J , Richard I , Bashir R , Britton S , Keers SM , et al. Genomic organization of the dysferlin gene and novel mutations in Miyoshi myopathy. Neurology. (2001) ;57: (2):271–8. |
[137] | Argov Z , Sadeh M , Mazor K , Soffer D , Kahana E , Eisenberg I , et al. Muscular dystrophy due to dysferlin deficiency in Libyan Jews. Clinical and genetic features. Brain. (2000) ;123: (6):1229–37. |
[138] | Boito CA , Melacini P , Vianello A , Prandini P , Gavassini BF , Bagattin A , et al. Clinical and molecular characterization of patients with limb-girdle muscular dystrophy type 2I. Archives of Neurology. (2005) ;62: (12):1894–9. |
[139] | Cagliani R , Magri F , Toscano A , Merlini L , Fortunato F , Lamperti C , et al. Mutation finding in patients with dysferlin deficiency and role of the dysferlin interacting proteins annexin A1 and A2 in muscular dystrophies. Human mutation. (2005) ;26: (3):283. |
[140] | Cai S , Gao M , Xi J , Liu Z , Yue D , Wu H , et al. Clinical spectrum and gene mutations in a Chinese cohort with anoctaminopathy. Neuromuscular Disorders (2019) . |
[141] | de Albuquerque MAV , Abath Neto O , da Silva FMA , Zanoteli E , Reed UC Limb-girdle muscular dystrophy type 2A in Brazilian children. Arquivos de Neuro-Psiquiatria. (2015) ;73: (12):993–7. |
[142] | Fanin M , Fulizio L , Nascimbeni AC , Spinazzi M , Piluso G , Ventriglia VM , et al. Molecular diagnosis in LGMD2A: Mutation analysis of protein testing? Human Mutation. (2004) ;24: (1):52–62. |
[143] | Fayssoil A , Nguyen LS , Ogna A , Meng P , Nardi O , Laforet P , et al. Effects of Home Mechanical Ventilation on Left Ventricular Function in Sarcoglycanopathies (Limb Girdle Muscular Dystrophies). American Journal of Cardiology. (2018) ;122: (2):353–5. |
[144] | Figarella-Branger D , El-Dassouki M , Saenz A , Cobo AM , Malzac P , Tong S , et al. Myopathy with lobulated muscle fibers: Evidence for heterogeneous etiology and clinical presentation. Neuromuscular Disorders. (2002) ;12: (1):4–12. |
[145] | Guglieri M , Magri F , D’Angelo MG , Prelle A , Morandi L , Rodolico C , et al. Clinical, molecular, and protein correlations in a large sample of genetically diagnosed Italian limb girdle muscular dystrophy patients. Human Mutation. (2008) ;29: (2):258–66. |
[146] | Hanisch F , Muller CR , Grimm D , Xue L , Traufeller K , Merkenschlager A , et al. Frequency of calpain-3 c. 550delA mutation in limb girdle muscular dystrophy type 2 and isolated hyperCKemia in German patients. Clinical Neuropathology. (2007) ;26: (4):157–63. |
[147] | Liewluck T , Winder TL , Dimberg EL , Crum BA , Heppelmann CJ , Wang Y , et al. ANO5-muscular dystrophy: Clinical, pathological and molecular findings. European Journal of Neurology. (2013) ;20: (10):1383–9. |
[148] | Paradas C , Llauger J , Diaz-Manera J , Rojas-Garcia R , De Luna N , Iturriaga C , et al. Redefining dysferlinopathy phenotypes based on clinical findings and muscle imaging studies. Neurology. (2010) ;75: (4):316–23. |
[149] | Park HJ , Hong JM , Suh GI , Shin HY , Kim SM , Sunwoo IN , et al. Heterogeneous characteristics of Korean patients with dysferlinopathy. Journal of Korean Medical Science. (2012) ;27: (4):423–9. |
[150] | Passos-Bueno MR , Moreira ES , Marie SK , Bashir R , Vasquez L , Love DR , et al. Main clinical features of the three mapped autosomal recessive limb-girdle muscular dystrophies and estimated proportion of each form in 13 Brazilian families. Journal of Medical Genetics. (1996) ;33: (2):97–102. |
[151] | Penttila S , Palmio J , Suominen T , Raheem O , Evila A , Gomez NM , et al. Eight new mutations and the expanding phenotype variability in muscular dystrophy caused by ANO5. Neurology. (2012) ;78: (12):897–903. |
[152] | Petersen JA , Kuntzer T , Fischer D , von der Hagen M , Huebner A , Kana V , et al. Dysferlinopathy in Switzerland: Clinical phenotypes and potential founder effects. BMC Neurology. (2015) ;15: (1) (no pagination)(182). |
[153] | Pollitt C , Anderson LVB , Pogue R , Davison K , Pyle A , Bushby KMD The phenotype of calpainopathy: Diagnosis based on a multidisciplinary approach. Neuromuscular Disorders. (2001) ;11: (3):287–96. |
[154] | Poppe M , Cree L , Bourke J , Eagle M , Anderson LVB , Birchall D , et al. The phenotype of limb-girdle muscular dystrophy type 2I. Neurology. (2003) ;60: (8):1246–51. |
[155] | Rasmussen M , Scheie D , Breivik N , Mork M , Lindal S Clinical and muscle biopsy findings in Norwegian paediatric patients with limb girdle muscular dystrophy 2I. Acta Paediatrica, International Journal of Paediatrics. (2014) ;103: (5):553–8. |
[156] | Rosales XQ , Gastier-Foster JM , Lewis S , Vinod M , Thrush DL , Astbury C , et al. Novel diagnostic features of dysferlinopathies. Muscle and Nerve. (2010) ;42: (1):14–21. |
[157] | Sarkozy A , Hicks D , Hudson J , Laval SH , Barresi R , Hilton-Jones D , et al. ANO5 gene analysis in a large cohort of patients with anoctaminopathy: Confirmation of male prevalence and high occurrence of the common exon 5 gene mutation. Human Mutation. (2013) ;34: (8):1111–8. |
[158] | Takahashi T , Aoki M , Tateyama M , Kondo E , Mizuno T , Onodera Y , et al. Dysferlin mutations in Japanese Miyoshi myopathy: Relationship to phenotype. Neurology. (2003) ;60: (11):1799–804. |
[159] | Tetreault M , Srour M , Allyson J , Thiffault I , Loisel L , Robitaille Y ,et al. Founder mutation for alpha-sarcoglycan-LGMD2D in a Magdalen Islands Acadian cluster. The Canadian journal of neurological sciences. 2011;Le journal canadien des sciences neurologiques. 38: (5):747–52. |
[160] | Todorova A , Georgieva B , Tournev I , Todorov T , Bogdanova N , Mitev V , et al. A large deletion and novel point mutations in the calpain 3 gene (CAPN3) in Bulgarian LGMD2A patients. Neurogenetics. (2007) ;8: (3):225–9. |
[161] | Van der Kooi AJ , Ten Dam L , Frankhuizen WS , Straathof CSM , Van Doorn PA , De Visser M ,et al. ANO5 mutations in the Dutch limb girdle muscular dystrophy population. Neuromuscular Disorders. (2013) ;23: (6):456–60. |
[162] | Weiler T , Greenberg CR , Nylen E , Halliday W , Morgan K , Eggertson D , et al. Limb-girdle muscular dystrophy and Miyoshi myopathy in an aboriginal Canadian kindred map to LGMD2B and segregate with the same haplotype. American Journal of Human Genetics. (1996) ;59: (4):872–8. |
[163] | Ylikallio E , Auranen M , Mahjneh I , Lamminen A , Kousi M , Traskelin AL , et al. Decreased aerobic capacity in ANO5-muscular dystrophy. Journal of Neuromuscular Diseases. (2016) ;3: (4):475–85. |
[164] | Arrigoni F , De Luca A , Velardo D , Magri F , Gandossini S , Russo A , et al. Multiparametric quantitative MRI assessment of thigh muscles in limb-girdle muscular dystrophy 2A and 2B. Muscle and Nerve. (2018) ;58: (4):550–8. |
[165] | Fatehi F , Nafissi S , Urtizberea JA , Blanck-Labelle V , Levy N , Krahn M , et al. Dysferlinopathy in Iran: Clinical and genetic report. Journal of the Neurological Sciences. (2015) ;359: (1-2):256–9. |
[166] | Fu X , Yang H , Wei C , Jiao H , Wang S , Yang Y , et al. FKRP mutations, including a founder mutation, cause phenotype variability in Chinese patients with dystroglycanopathies. Journal of Human Genetics. (2016) ;61: (12):1013–20. |
[167] | Gaul C , Deschauer M , Tempelmann C , Vielhaber S , Klein HU , Heinze HJ , et al. Cardiac involvement in limb-girdle muscular dystrophy 2I: Conventional cardiac diagnostic and cardiovascular magnetic resonance. Journal of Neurology. (2006) ;253: (10):1317–22. |
[168] | Ginjaar HB , Van Der Kooi AJ , Ceelie H , Kneppers ALJ , Van Meegen M , Barth PG , et al. Sarcoglycanopathies in Dutch patients with autosomal recessive limb girdle muscular dystrophy. Journal of Neurology. (2000) ;247: (7):524–9. |
[169] | Hollingsworth KG , Willis TA , Bates MGD , Dixon BJ , Lochmuller H , Bushby K , et al. Subepicardial dysfunction leads to global left ventricular systolic impairment in patients with limb girdle muscular dystrophy 2I. European Journal of Heart Failure. (2013) ;15: (9):986–94. |
[170] | Kang PB , Feener CA , Estrella E , Thorne M , White AJ , Darras BT , et al. LGMD2I in a North American population. BMC Musculoskeletal Disorders. (2007) ;8: (no pagination)(115). |
[171] | Mercuri E , Brockington M , Straub V , Quijano-Roy S , Yuva Y , Herrmann R , et al. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Annals of Neurology. (2003) ;53: (4):537–42. |
[172] | Ro LS , Lee-Chen GJ , Lin TC , Wu YR , Chen CM , Lin CY , et al. Phenotypic features and genetic findings in 2 Chinese families with miyoshi distal myopathy. Archives of Neurology. (2004) ;61: (10):1594–9. |
[173] | Rosales XQ , Moser SJ , Tran T , McCarthy B , Dunn N , Habib P , et al. Cardiovascular magnetic resonance of cardiomyopathy in limb girdle muscular dystrophy 2B and 2I. Journal of Cardiovascular Magnetic Resonance. (2011) ;13: (1) (no pagination)(39). |
[174] | Seong MW , Cho A , Park HW , Seo SH , Lim BC , Seol D , et al. Clinical applications of next-generation sequencing-based gene panel in patients with muscular dystrophy: Korean experience. Clinical Genetics. (2016) ;89: (4):484–8. |
[175] | Tasca G , Monforte M , Diaz-Manera J , Brisca G , Semplicini C , D’Amico A , et al. MRI in sarcoglycanopathies: A large international cohort study. Journal of Neurology, Neurosurgery and Psychiatry. (2018) ;89: (1) (no pagination)(jnnp–2017–316736). |
[176] | Topaloglu H , Dincer P , Richard I , Akcoren Z , Alehan D , Ozme S , et al. Calpain-3 deficiency causes a mild muscular dystrophy in childhood. Neuropediatrics. (1997) ;28: (4):212–6. |
[177] | Umakhanova ZR , Bardakov SN , Mavlikeev MO , Chernova ON , Magomedova RM , Akhmedova PG , et al. Twenty-year clinical progression of dysferlinopathy in patients from Dagestan. Frontiers in Neurology. (2017) ;8: (MAR) (no pagination)(77). |
[178] | Vissing CR , Preisler N , Husu E , Prahm KP , Vissing J Aerobic training in patients with anoctamin 5 myopathy and hyperckemia. Muscle and Nerve. (2014) ;50: (1):119–23. |
[179] | Wahbi K , Meune C , Hamouda EH , Stojkovic T , Laforet P , Becane HM , et al. Cardiac assessment of limb-girdle muscular dystrophy 2I patients: An echography, Holter ECG and magnetic resonance imaging study. Neuromuscular Disorders. (2008) ;18: (8):650–5. |
[180] | Woudt L , Di Capua GA , Krahn M , Castiglioni C , Hughes R , Campero M , et al. Toward an objective measure of functional disability in dysferlinopathy. Muscle Nerve. (2016) ;53: (1):49–57. |
[181] | Xi J , Blandin G , Lu J , Luo S , Zhu W , Beroud C , et al. Clinical heterogeneity and a high proportion of novel mutations in a chinese cohort of patients with dysferlinopathy. Neurology India. (2014) ;62: (6):635–9. |
[182] | Xie Z , Hou Y , Yu M , Liu Y , Fan Y , Zhang W , et al. Clinical and genetic spectrum of sarcoglycanopathies in a large cohort of Chinese patients. Orphanet Journal of Rare Diseases. (2019) ;14: (1) (no pagination)(43). |
[183] | Xie Z , Xiao J , Zheng Y , Wang Z , Yuan Y Magnetic resonance imaging findings in the muscle tissue of patients with limb girdle muscular dystrophy type 2I harboring the founder mutation c545A>G in the FKRP gene. BioMed Research International. (2018) ;2018: (no pagination)(3710814). |
[184] | Yu M , Zheng Y , Jin S , Gang Q , Wang Q , Yu PL,H , et al. Mutational spectrum of Chinese LGMD patients by targeted next-generation sequencing. PLoS ONE. (2017) ;12: (4)(no pagination)(e0175343). |
[185] | Dincer P , Bonnemann CG , Erdir Aker O , Akcoren Z , Nigro V , Kunkel LM , et al. A homozygous nonsense mutation in delta-sarcoglycan exon 3 in a case of LGMD2F. Neuromuscular Disorders. (2000) ;10: (4-5):247–50. |
[186] | Feng X , Luo S , Li J , Yue D , Xi J , Zhu W , et al. Fatty infiltration evaluation and selective pattern characterization of lower limbs in limb-girdle muscular dystrophy type 2A by muscle magnetic resonance imaging. Muscle and Nerve. (2018) ;58: (4):536–41. |
[187] | Lokken N , Hedermann G , Thomsen C , Vissing J Contractile properties are disrupted in Becker muscular dystrophy, but not in limb girdle type 2I. Annals of Neurology. (2016) ;80: (3):466–71. |
[188] | Willis TA , Hollingsworth KG , Coombs A , Sveen ML , Andersen S , Stojkovic T , et al. Quantitative magnetic resonance imaging in limb-girdle muscular dystrophy 2i: A multinational cross-sectional study. PLoS ONE. (2014) ;9: (2) (no pagination)(e90377). |
[189] | Mendell JR , Rodino-Klapac LR , Rosales XQ , Coley BD , Galloway G , Lewis S , et al. Sustained alpha-sarcoglycan gene expression after gene transfer in limb-girdle muscular dystrophy, type 2D. Annals of Neurology. (2010) ;68: (5):629–38. |
[190] | Walter MC , Reilich P , Thiele S , Schessl J , Schreiber H , Reiners K , et al. Treatment of dysferlinopathy with deflazacort: A double-blind, placebo-controlled clinical trial. Orphanet Journal of Rare Diseases. (2013) ;68: (1) (no pagination)(26). |
[191] | Bonnemann CG , Passos-Bueno MR , McNally EM , Vainzof M , Moreira EDS , Marie SK , et al. Genomic screening for beta-sarcoglycan gene mutations: Missense mutations may cause severe limb-girdle muscular dystrophy type 2E (LGMD 2E). Human Molecular Genetics. (1996) ;5: (12):1953–61. |
[192] | Cagliani R , Fortunato F , Giorda R , Rodolico C , Bonaglia MC , Sironi M , et al. Molecular analysis of LGMD-2B and MM patients: Identification of novel DYSF mutations and possible founder effect in the Italian population. Neuromuscular Disorders. (2003) ;13: (10):788–95. |
[193] | Calvo F , Teijeira S , Fernandez JM , Teijeiro A , Fernandez-Hojas R , Fernandez-Lopez XA , et al. Evaluation of heart involvement in gamma-sarcoglycanopathy (LGMD2C). A study of ten patients. Neuromuscular Disorders. (2000) ;10: (8):560–6. |
[194] | de Paula F , Vieira N , Starling A , Yamamoto LU , Lima B , de Cassia Pavanello R , et al. Asymptomatic carriers for homozygous novel mutations in the FKRP gene: The other end of the spectrum. European Journal of Human Genetics. (2003) ;11: (12):923–30. |
[195] | Driss A , Amouri R , Ben Hamida C , Souilem S , Gouider-Khouja N , Ben Hamida M , et al. A new locus for autosomal recessive limb-girdle muscular dystrophy in a large consanguineous Tunisian family maps to chromosome 19q13.3. Neuromuscular Disorders. (2000) ;10: (4-5):240–6. |
[196] | Fischer D , Walter MC , Kesper K , Petersen JA , Aurino S , Nigro V , et al. Diagnostic value of muscle MRI in differentiating LGMD2I from other LGMDs. Journal of Neurology. (2005) ;252: (5):538–47. |
[197] | Gomez-Andres D , Diaz J , Munell F , Sanchez-Montanez A , Pulido-Valdeolivas I , Suazo L , et al. Disease duration and disability in dysfeRlinopathy can be described by muscle imaging using heatmaps and random forests. Muscle Nerve. (2019) ;59: (4):436–44. |
[198] | Peric S , Stevanovic J , Johnson K , Kosac A , Peric M , Brankovic M , et al. Phenotypic and genetic spectrum of patients with limb-girdle muscular dystrophy type 2A from Serbia. Acta Myologica: Myopathies and Cardiomyopathies: Official Journal of the Mediterranean Society of Myology. (2019) ;38: (3):163–71. |
[199] | Quick S , Schaefer J , Waessnig N , Schultheiss T , Reuner U , Schoen S , et al. Evaluation of heart involvement in calpainopathy (LGMD2A) using cardiovascular magnetic resonance. Muscle and Nerve. (2015) ;52: (4):661–3. |
[200] | Sarkozy A , Deschauer M , Carlier RY , Schrank B , Seeger J , Walter MC , et al. Muscle MRI findings in limb girdle muscular dystrophy type 2L. Neuromuscular Disorders. (2012) ;22: (SUPPL. 2):S122–S9. |
[201] | Sharma MC , Mannan R , Singh NG , Gulati S , Kalra V , Sarkar C Sarcoglycanopathies: a clinicopathological study of 13 cases [corrected]. Neurology India. (2004) ;52: (4):446–9. |
[202] | Ueyama H , Kumamoto T , Nagao SI , Masuda T , Horinouchi H , Fujimoto S , et al. A new dysferlin gene mutation in two Japanese families with limb-girdle muscular dystrophy 2B and Miyoshi myopathy. Neuromuscular Disorders. (2001) ;11: (2):139–45. |
[203] | Vainzof M , Moreira ES , Canovas M , Anderson LVB , Pavanello RCM , Passos-Bueno MR , et al. Partial alpha-sarcoglycan deficiency with retention of the dystrophin- glycoprotein complex in a LGMD2D family. Muscle and Nerve. (2000) ;23: (6):984–8. |
[204] | Mohassel P , Bönnemann CG . Chapter 34 - Limb-girdle Muscular Dystrophies. In: Darras BT, Jones HR, Ryan MM, De Vivo DC, editors. Neuromuscular Disorders of Infancy, Childhood, and Adolescence (Second Edition). San Diego: Academic Press; (2015) . p. 635–66 |
[205] | Murphy LB , Schreiber-Katz O , Rafferty K , Robertson A , Topf A , Willis TA , et al. Global FKRP Registry: Observations in more than 300 patients with Limb Girdle Muscular Dystrophy R9. Ann Clin Transl Neurol. (2020) ;7: (5):757–66. |
[206] | Penttilä S , Vihola A , Palmio J , Bjarne U . ANO5 Muscle Disease. Seattle, WA: University of Washington, Seattle; 1993-2021 2012 Nov 29 [Updated 2019 Aug 22]. |

