
Pre Clinical Assessment of AAVrh74.MCK.GNE Viral Vector Therapeutic Potential: Robust Activity Despite Lack of Consistent Animal Model for GNE Myopathy
Abstract
Background:
GNE myopathy is a unique adult onset rare neuromuscular disease caused by recessive mutations in the GNE gene. The pathophysiological mechanism of this disorder is not well understood and to date, there is no available therapy for this debilitating disease. We have previously established proof of concept that AAV based gene therapy can effectively deliver the wild type human GNE into cultured muscle cells from human patients and in mice, using a CMV promoter driven human wild type GNE plasmid delivered through an adeno associated virus (AAV8) based platform.
Objective:
In the present study we have generated a muscle specific GNE construct, driven by the MCK promoter and packaged with the AAVrh74 serotype for efficacy evaluation in an animal model of GNE Myopathy.
Methods:
The viral vector was systemically delivered at 2 doses to two age groups of a Gne–/– hGNED207V Tg mouse described as a preclinical model of GNE Myopathy, and treatment was monitored for long term efficacy.
Results:
In spite of the fact that the full described characteristics of the preclinical model could not be reproduced, the systemic injection of the rAAVrh74.MCK.GNE viral vector resulted in a long term presence and expression of human wt GNE in the murine muscles and in some improvements of their mild phenotype. The Gne–/– hGNED207V Tg mice are smaller from birth, but cannot be differentiated from littermates by muscle function (grip strength and Rotarod) and their muscle histology is normal, even at advanced age.
Conclusions:
The rAAVrh74.MCK.GNE vector is a robust tool for the development of GNE Myopathy therapies that supply the intact GNE. However, there is still no reliable animal model to fully assess its efficacy since the previously developed Gne–/– hGNED207V Tg mice do not present disease characteristics.
INTRODUCTION
GNE myopathy is a unique adult onset rare neuromuscular disease caused by recessive mutations in the UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine-kinase (GNE) gene, which encodes for an essential bifunctional enzyme in sialic acid biosynthesis [1–4]. To date, no therapy is available for this debilitating disease. GNE mutations only partially diminish sialic acid synthesis and it remains controversial whether decreased sialic acid production directly results in the characteristic muscle dysfunction and atrophy of this disorder [5–11]. Recently, a Phase III sialic acid supplementation clinical trial has failed [12]. Accumulating data point to other postulated functions of GNE that may be impaired, leading to the human myopathy [13–22]. Therefore, a comprehensive approach for treatment, such as gene therapy, could result in amelioration of this myopathy. This approach intends to address the notion that GNE myopathy is not driven by the presence of the mutated GNE protein, but because of the lack of normal GNE protein. Therefore, a supply of normal GNE protein into patients’ muscles should overcome this deficiency and allow the muscle cell to function normally, while providing higher levels of sialic acid but also the additional, still unknown, functions essential for normal muscle physiology. We have established proof of concept that AAV based gene therapy can effectively deliver the wild type human GNE into cultured muscle cells from human patients and into mouse muscle cells and tissues [23]. CMV promoter-driven human wild type GNE delivery, through an adeno associated virus (AAV8)-based platform, was shown in vitro to infect normal mouse muscle cells as well as muscle cells from GNE myopathy patients with efficient expression. In vivo, we have demonstrated that this AAVCMVGNE viral vector when administered intravenously, incorporates GNE in muscle cells with mRNA expression detected for at least 12 months [23].
In the present studies we have generated an MCK promoter-driven GNE construct packaged with the AAVrh74 serotype. This viral vector was systemically delivered to two age groups of the Gne–/– h GNED207V Tg mice, described as a preclinical mouse model of GNE Myopathy [24]. To test the efficacy of the vector to prevent disease symptoms, a young asymptomatic mice cohort was injected at 10 weeks of age, and an expected symptomatic mice cohort was treated at 50 weeks of age. GNE expression was monitored and treatment efficacy was followed up to one year post injection for the first cohort and up to 85 weeks of age for the second one, by functional and histopathological assays. In spite of the fact that the fully described characteristics of the preclinical model could not be reproduced, systemic injection of the viral vector resulted in the long term presence and expression of wt human GNE in the murine muscles and some improvements of their mild phenotype.
MATERIALS AND METHODS
Mice maintenance
Breeding pairs of the Gne(–/–)hGNED207V-Tg mice, carrying a transgene encoding a frequent mutation in the Japanese cluster of GNE Myopathy patients, on a mouse endogeneous Gne KO background were provided by Dr Nishino’s laboratory [24] and were used to establish a colony for our studies. All animals, including C57Bl mice, were maintained at the facilities of the Authority for Biological and Biomedical Models at the Faculty of Medicine of the Hebrew University of Jerusalem.
All animal procedures were performed in accordance with institutional guidelines under protocols approved by the Institutional Committee for Animal Care of the Hebrew University-Hadassah Medical Center.
Vector generation
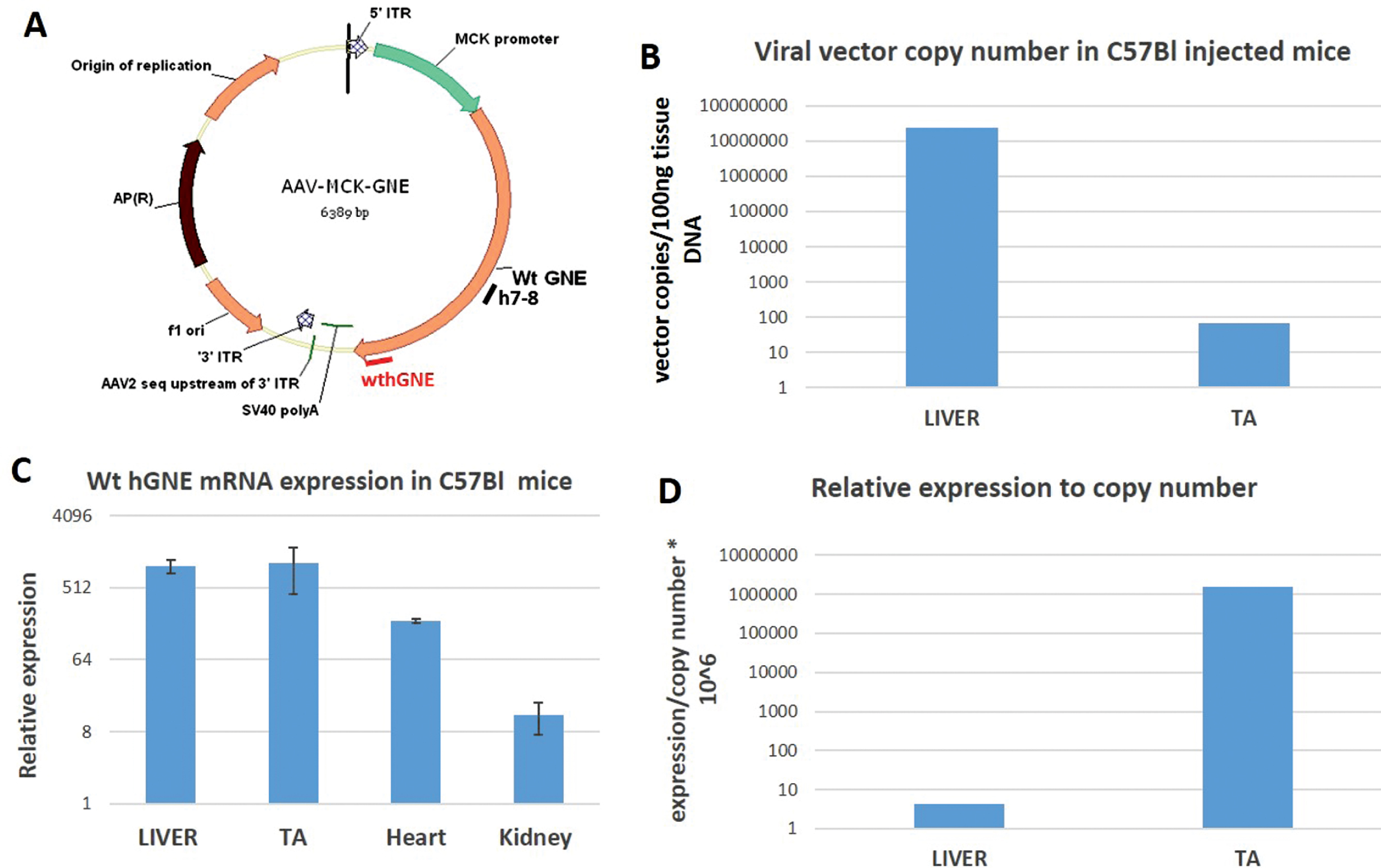
We have constructed an AAV based vector by cloning the MCK promoter instead of the CMV promoter of our previous used construct ITR AAV2.CMV.GNE vector using the full-length human GNE cDNA (NM_005476) ATG to TAG, (2169bp) [23]. In our final construct (AAV.MCK.GNE), we replaced the entire sequences between the 5’ AAV2 ITR and the SV40 polyA signal with a cassette including the MCK promoter+a consensus Kozak sequence (CCACC)+ an SV40 chimeric intron -that was derived from the pAAVmck human microdystrophin plasmid (a kind gift of Dr Mendell, Nationwide Children’s Hospital, Columbus, Ohio), and GNE cDNA. Packaged viral vector particles with the AAVrh74 serotype, that was isolated at Nationwide Children’s Hospital, were generated at large scale at the Viral Vector Core laboratory of the Research Institute at Nationwide Children’s Hospital, Columbus Ohio (Fig. 1A).
Fig. 1
rAAVrh74MCKGNE plasmid functionality in C57Bl mice. A. Schematic representation of the generated AAVMCKGNE plasmid showing the human wt GNE cDNA driven from the MCK muscle specific promoter, the SV40 polyadenylation signal (polyA) and the 5′ and 3′ AAV2 inverted terminal repeats (ITR); B. rAAVrh74MCKGNE viral copy number in liver and tibialis anterior (TA) of C57Bl mice 45 days after systemic delivery of rAAVrh74MCKGNE viral vector at 5.1013 vg/kg. C. Relative expression of wt human GNE mRNA in liver, tibialis anterior (TA), heart and kidney in C57Bl mice 45 days after systemic delivery of 5.1013 vg/kg. D. Expression of wt human GNE mRNA relative to copy number in liver and tibialis anterior of injected mice.
Mice injections
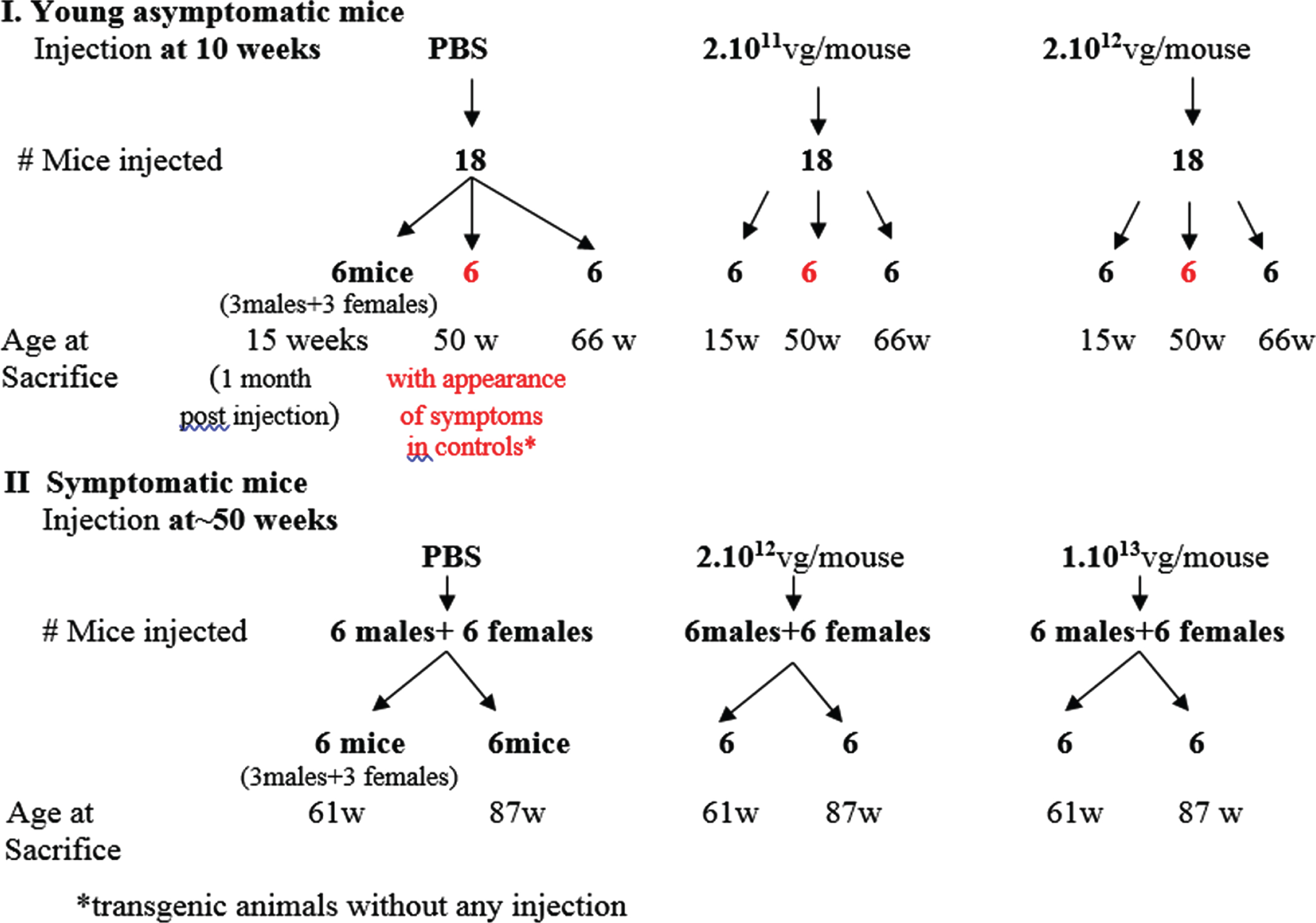
C57Bl mice 5–6 weeks old were injected at a dose of 1.1012 viral vector genomes/mouse (equivalent of 5. 1013 vg/kg body weight). Experiments with the Gne(–/–) hGNED207V-Tg mice were designed as follows (see schema, Fig. 2): a first set of experiments was designed to examine whether human GNE wt injection can prevent the appearance of muscle pathology in mice. Three groups of 18 mice each (9 males and 9 females), aged 10 weeks, were injected with a total of either 2.1011 (low dose-LD- 1.1013 vg/kg), 2.1012 (high dose-HD-1.1014 vg/kg) viral vector genomes, or PBS respectively. A second set of experiments was designed to inject mice at 50 weeks (after the phenotype will be established) to examine whether the gene therapy approach can stop or even ameliorate muscle damage progression. Three groups of 18 mice each (9 males and 9 females), aged 50 weeks, were injected with a total of either 2.1012 (high dose-HD-1.1014 vg/kg), 1.1013 (higher dose-HHD-5.1014 vg/kg) viral vector genomes, or PBS respectively. Viral vectors were diluted in PBS. All mice were injected in the tail vein with a 250 microliters solution volume.
Fig. 2
Schematic representation of the experimental design for AAVrh74MCKGNE systemic delivery to the Gne(–/–) hGNED207V-Tg young and old mice.
Mice follow up
The mice were followed up for diverse parameters: weight (once a week); hindlimb grip peak force (in grams) using an Electronic Grip Strength Meter (Columbus Instruments, Columbus, OH, USA). Five measurements were made for each animal, and the three highest measurements were averaged to give the strength score. Measurements were not normalized relative to weight. Rotarod performance measurements (time in seconds till fall) were performed in a USB Rota-Rod treadmill. Grip strength and Rotarod measurements were performed every 2 weeks alternately. All strength tests were performed at a similar time of day in identical environmental conditions. After sacrifice, mice were evaluated by histology examination and analysis of expression of the introduced human wt GNE. Three timepoints were determined for sacrifice of the young injected mice: 1 month post injection- at 15 weeks of age- when no muscle defects are expected; the second one -at 50 weeks of age- with expected presence of symptoms; the third at 66 weeks of age, after the phenotype would have been well established. The older mice, injected at 50 weeks of age, were euthanized at 2 time points: 11 weeks and 37 weeks post injection. At each time point 6 mice of each group (3 males and 3 females) were euthanized and tissues specimens were immediately collected, fixed in paraformaldehyde (4 %) and paraffin embedded for histological examination. Tissue sections (5μm) were stained with H&E following standard procedures. Different muscles (gastrocnemius, quadriceps, tibialis anterior) were snap frozen in liquid nitrogen cooled in isopentane and stored at –80°C till processing for tissue sections (8μm). For RNA and DNA analyses, the different organs and muscle specimens were snap frozen in liquid nitrogen and stored at –80°C till further processing.
Relative human wt GNE expression determination
Total RNA from tissues stored in liquid nitrogen was extracted with Tri-Reagent (Sigma, St. Louis, MO, USA) according to the manufacturer’s protocol. The Tri-Reagent containing the non RNA sample fractions were stored at –80°C for further DNA processing. After DNAse (Invitrogen) treatment, RNA was reverse transcribed using random hexamer primers (Roche, Germany) by the SuperscriptIII reverse transcriptase enzyme (Invitrogen) following the protocol supplied by the manufacturer. The cDNA products were amplified by PCR. To detect wild type human GNE cDNA transgene expression in C57Bl mice tissues, we used quantitative real time PCR with a TaqMan set (h7-8) consisting of primers and probe specifically designed for human GNE cDNA detection solely (human GNE exons 7–8): hF7-5′ TCTTGGCGGGACGAACCTCCGA 3′; hR8-5′ ACACACATCTGTAGGATTAAAT 3′; hGNE probe-FAM-TTGCAATAGTCAGCATGAAG-BQ. Endogenous mouse GNE expression was simultaneously measured in the same samples with a TaqMan set consisting of primers and probe specifically designed for mouse endogenous GNE cDNA detection, in the very same region (mouse GNE exons 7–8):
1 mF7-5′TCTTGGCGGGACAAACCTGAGG 3′;
2 mR8-5′CACACATCTGCAGGATTAAAC 3′;
3 mGNE probe FAM- TGGCAATAGTTAGCATGAAG-BQ.
All measurements were done in duplicates and normalized relative to the endogenous mouse Gne expression of the corresponding tissues and mouse. In addition, as an internal control, the mouse Hprt gene was also quantitated with the Hprt, Mm00446968_m1 probe assay (Thermo-Fisher Scientific USA). Expression was calculated relative to the corresponding tissue of the PBS injected mice. Indeed, running the reaction for 50 cycles did allow faint detection of amplification with the human primers in the mouse control.
For detection of wild type hGNE expression driven by the injected viral vector in the Gne(–/–)hGNED207V-Tg mice, we designed a primers/probe set (wthGNE) detecting specifically sequences expressed only from the rAAVrh74.MCK.GNE vector at the 3′ end of GNE: a forward primer F: 5′ GGTTTCGGATTTGGTTGACC 3′; a reverse primer specific to the plasmid AAV.MCK.GNE, AAVR: 5′ TGGTTTGTCCAAACTCATCAA 3′; a Taqman probe specific for the viral vector which does not detect the transgenic construct, CGGCCGCTTCGAGCAGACATGAT. Transgenic expression of the mutant GNE in the Gne(–/–)hGNED207V-Tg mice was also measured, with the same forward primer F: 5′ GGTTTCGGATTTGGTTGACC 3′; a reverse primer specific for the transgenic construct: 5′CCAGGAGCTCTGGAGAGAAG 3′; a Taqman probe specific for the transgenic construct which does not detect the viral vector: ACCTCCAGGAACAGACATGGACC.
Quantification of human wt GNE (hGNE) expression was relative to the lowest value detected among the low dose injected mice, since even when running the reaction for 50 cycles, no amplification could be detected for the human wt injected GNE in the control samples, injected with PBS, thus confirming the high specificity of the primers/probe set used. All measurements were done in duplicates and normalized relative to mouse Gne exons 7–8 expression (present also in endogenous Gne KO- the missing exon is exon 3) of the corresponding tissues andmouse.
Copy number determination
Vector copy number was evaluated by quantitative PCR of the tissue DNA (in duplicate) versus a standard curve established from the plasmid AAV.MCK.GNE DNA with the same primers/probe set designed to detect sequences expressed from hGNE (h7-8) for evaluation in C57Bl mice. For evaluation in the transgenic mice, the primer/probe set specifically detecting sequences from the rAAVrh74.MCK.GNE vector, as above, wthGNE, was used. All copy number and expression experiments were run in a StepOnePlus Real time PCR system (Applied Biosystems, UK).
Statistical analysis
Statistical analysis for the comparison between groups was performed by the Student’s two tailed unpaired t test.
RESULTS
rAAVrh74.MCK.GNE vector functionality and experimental design
The AAV.MCK.GNE plasmid was generated by replacing the CMV promoter of our original plasmid (23) by the MCK promoter, and packaged in the AAVrh74 serotype capsid, as detailed in the Material and Methods section (Fig. 1A). To assess vector functionality, rAAvRh74.MCK.GNE viral vectors were injected at the tail vein of C57Bl mice at a dose of 1.1012 viral vector genomes/mouse (equivalent of 5. 1013 vg/kg body weight). Mice were euthanized 45 days post injection. Vector copy number (Fig. 1B) and human wt GNE mRNA expression (Fig. 1C) were evaluated by quantitative PCR. As expected, most viral vectors accumulated in the liver, but also reached muscle. However, the expression of hGNE relatively to copy number was much higher in muscle than in liver -up to 1.106fold, as the MCK promoter highly enriches muscle expression (Fig. 1D). Notably, no abnormal phenotype could be detected in the treated mice.
The next step was to assess the efficacy of this vector in a GNE Myopathy mouse model.
The Gne–/– hGNED207V Tg mice, carrying a transgene encoding a frequent mutation in the Japanese cluster of GNE Myopathy patients, on a mouse endogeneous Gne KO background, were described as presenting muscle disease symptoms from week 40 [24], including reduction in motor performance and appearance of muscle pathological findings recapitulating muscle histology from GNE Myopathy patients. We obtained few breeding pairs and established a colony of these mice in our laboratory, to evaluate the effect of the human wt GNE to either prevent disease onset by injecting the vector at early age, well before the appearance of symptoms, or/and to stop or even ameliorate muscle pathology and function by treating the mice after the appearance of symptoms. The experimental design is illustrated in Fig. 2.
rAAVrh74.MCK.GNE injections to 10 week old Gne–/– hGNED207V Tg mice
First, young mice aged 10 weeks (before the appearance of any symptom is expected) were infected by systemic injection of the viral vector at either low dose (LD-1.1013 vg/kg) or high dose (HD-1.1014 vg/kg). A third group of mice was injected with PBS. The mice were followed up for diverse parameters, weight, grip force and rotarod performance measurements, histology examination and expression of the injected human wt GNE, up to 70 weeks of age.
Follow up of Gne(–/–) hGNED207V-Tg young mice injected with AAVrh74.MCK.GNE at 10 weeks
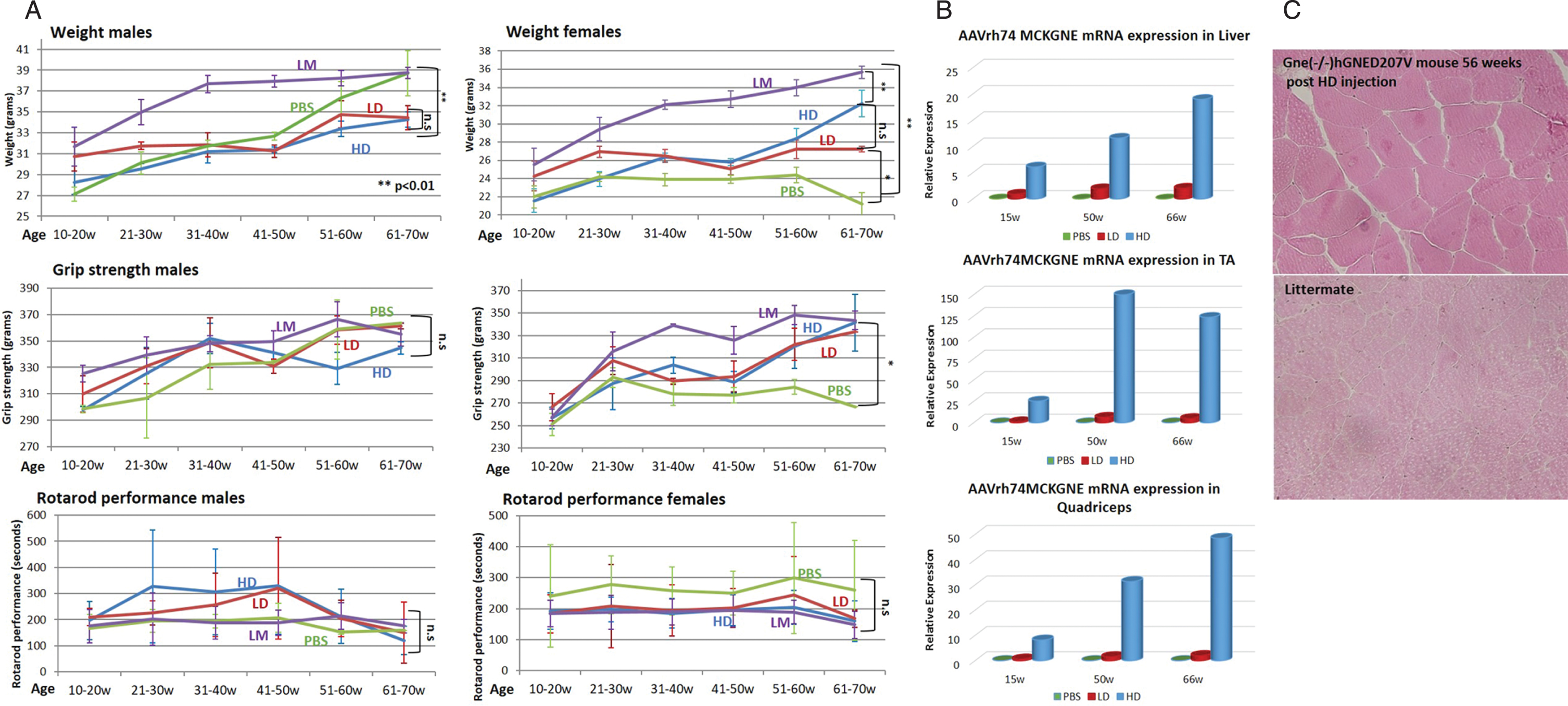
Once the presence of the rAAVrh74.MCK.GNE vector was assessed in the tissues of the injected mice (data not shown), all mice were observed for general behaviour, weight and muscle function up to 70 weeks of age (Fig. 3). As described, the transgenic mice are smaller and weigh less than their littermates (Fig. 3A, higher panel). However, in males we noted that from the age of 50 weeks, the male transgenic untreated mice (PBS) decreased the weight gap progressively and reached the weight of their littermates (LM) at 60–70 weeks. The weight of the treated male mice did not change significantly, while in female mice, the GNE viral vector improved weight with age, compared to the PBS injected ones, which stayed smaller than littermates during the entire follow up period. There was no significant change in grip strength of the injected male mice with age, also both untreated (PBS) and littermate (LM) mice displayed similar values (Fig. 3A, middle panel). In contrast grip strength of the GNE vector injected female mice improved progressively with age: the treated and littermate groups begin to differentiate from the untreated PBS injected model mice after 20 weeks of age, and this difference increased at least till age 70 weeks. Beginning at age 60 weeks, the GNE viral vector injected mice reached a level of grip strength similar to their littermate healthy mice. In the rotarod assay (Fig. 3A, lower panel), we see a strong improvement of muscle motor performance in male mice injected with both LD and HD viral vectors, during the 30 to 50 weeks period, and then a sharp decrease reaching the values of both untreated (PBS) and littermates (LM) mice (which could not be differentiated along the entire 20 weeks to 70 weeks of age period). In female, there is a difference between untreated and littermate mice, but inverse to the expected behaviour (untreated mice performed better than the healthy littermates). The GNE viral vector injected female mice performed similarly to their untreated littermates during the entire examined period.
Fig. 3
Functional efficacy of gene therapy and wild type hGNE expression in Gne(–/–) hGNED207V-Tg 10 week old treated mice. A. Weight, grip strength and rotarod performance of Gne(–/–) hGNED207V-Tg mice were monitored at different time points after systemic delivery of low dose (LD-1.1013 vg/kg) and high dose (HD-2.1014 vg/kg) rAAVrh74MCKGNE viral vector to 10 week old mice; * = p value < 0.05; ** = p value < 0.01; ns = not significant. LM, non transgenic littermate mice; PBS, Gne(–/–) hGNED207V-Tg mice injected with PBS. B. human wt GNE mRNA relative expression in muscle of the LD and HD treated 10 week old Gne(–/–) hGNED207V-Tg mice in liver, tibialis anterior (TA) and quadriceps. C. Representative H&E gastrocnemius histological section of a 10 week old treated Gne(–/–) hGNED207V-Tg mouse, 56 weeks after injection of HD rAAVrh74MCKGNE viral vector. Littermate: untreated wt mice.
Human wt GNE expression in the Gne(–/–)hGNED207V-Tg mice injected with rAAVrh74.MCK.GNE at 10 weeks of age.
To assess expression of the injected human wt GNE, mice in each group were sacrificed simultaneously at each time point, at 15, 50 and 66 weeks of age. Human wt GNE expression was measured in various tissues by real time PCR, with a Taqman probe specifically designed to differentiate between the mutated human GNE transgene present in the mice and the wt human GNE injected with the viral vector. For each tissue, the level of human wt GNE mRNA present in the low dose (LD) injected animals examined at 15 weeks was used as the reference (1fold) to which the level of expression of human wt GNE mRNA in all samples analyzed was compared (Fig. 3B). There is a clear difference between the expression level of human wt GNE between the low dose and the high dose injected animals at the 3 time points, in both liver and muscle tissue, but the difference is more pronounced in muscle tissue. As expected, in the mice injected with PBS, human wt GNE mRNA is undetectable. All measurements have been normalized to the level of expression of the corresponding endogenous mouse GNE mRNA. Injected GNE is expressed in liver and muscles. The level of expression correlates with the dose injected. The expression level remains stable for the period analyzed, from injection at 10 weeks of age, up to 66 weeks of age, thus for more than one year after injection.
Histology of Gne(–/–) hGNED207V-Tg mice injected with rAAVrh74MCKGNE at 10 weeks
Different organs of sacrificed mice were processed for histological examination at the different time points. Microscope examination of the different sections did not detect any consistent pathological change in the untreated model mice compared to littermates, in any of the organs examined, including muscle sections of tibialis anterior, gastrocnemius, quadriceps or diaphragm, nor at age 15 weeks, as expected, but neither at week 50 or 66, where severe pathological changes in the untreated Gne(–/–) hGNED207V-Tg mice should have been present. Similarly, no pathological changes were observed in the human wt GNE viral vector injected mice as illustrated for gastrocnemius in Fig. 3C.
AAVrh74.MCK.GNE injection to 50 week old Gne(–/–) hGNED207V-Tg mice
Concomitantly with the first experiment, we injected the rAAVrh74.MCK.GNE viral vector in mice aged 50 weeks. At this age, the transgenic mice should have shown clear myopathy symptoms. We expected to evaluate whether the treatment would be able to stop or even ameliorate the muscle pathology of the diseased mice. Since the low dose injected young mice showed low GNE expression, we decided to increase the dose injected in mice aged 50 weeks to a higher dose (5.1014 vg/kg-HHD), in addition to the 1.1014 vg/kg high dose (HD). Low dose (1.1013 vg/kg-LD) was not injected. These mice were evaluated for body weight, grip strength and Rotarod performance up to the age of 84–85 weeks (Fig. 4A). Male body weight was shown to slightly improve upon treatment with the high dose (HD) viral vector, but the higher dose (HHD) did not show the same pattern. In females no improvement was observed with either dose. Grip force strength and Rotarod tests did not show any significant difference between the different groups of animals. To note, no significant differences were observed between the untreated model mice (PBS) and their wt littermates (LM). Mice were sacrificed at 2 time points, 11 and 37 weeks after injection, thus at age 61 and 87 weeks, to assess GNE expression and histology, particularly in muscle.
Fig. 4
Functional efficacy of gene therapy and GNE expression in Gne(–/–) hGNED207V-Tg 50 week old treated mice. Weight, grip force and rotarod performance follow up of Gne(–/–) hGNED207V-Tg mice after systemic delivery of high dose (HD-1.1014 vg/kg) and higher dose (HHD-5.1014 vg/kg) of rAAVrh74MCKGNE viral vector to 50 week old mice; LM, littermate; PBS, Gne(–/–) hGNED207V-Tg mice injected with PBS. ** = p value < 0.01; ns = not significant. B. human wt GNE mRNA relative expression of the HD and HHD treated 50 week old Gne(–/–) hGNED207V-Tg mice (right panel), 11 and 37 weeks post injection, compared to the 10 week old injected mice (left panel); C. Representative H&E gastrocnemius histological section of a 50 week old treated Gne(–/–) hGNED207V-Tg mouse, 37 weeks after injection of an HHD dose of rAAVrh74MC.GNE viral vector.
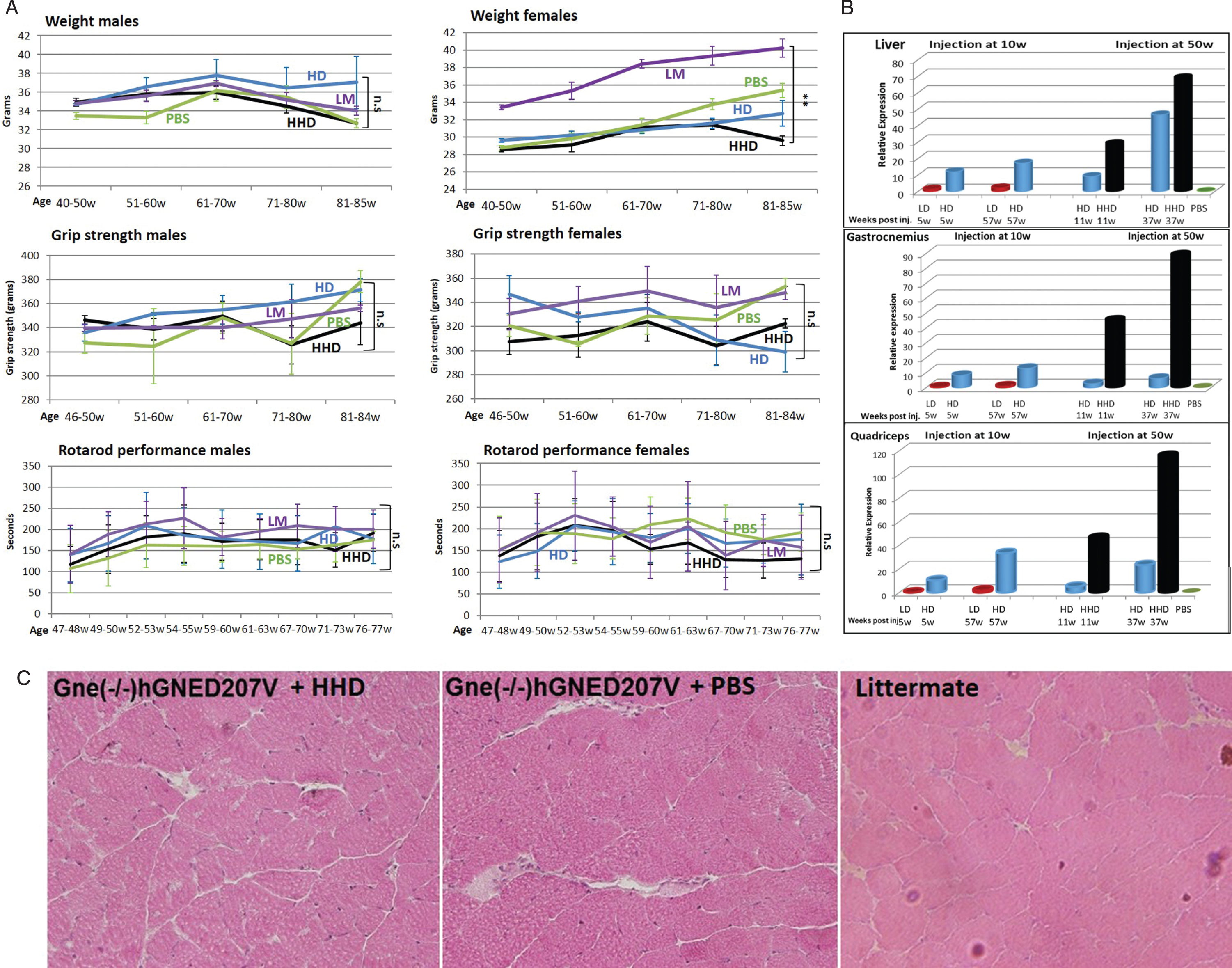
Human wt GNE expression was measured in various tissues by real time PCR, with the same Taqman differential probe (Fig. 4B). As before, all measurements were normalized to the level of expression of the corresponding endogenous mouse Gne mRNA. The level of human wt GNE mRNA present in the low dose (LD) injected animals examined at 15 weeks was used as the reference (1fold) to which the level of expression of human wt GNE mRNA in all samples analyzed was compared. There was a very significant difference in the level of expression of the human wt GNE between the 1.1014 vg/kg (HD) and the 5.1014 vg/kg (HHD) dose in liver and even larger differences in the various muscles examined. More importantly, the level of expression increased with time and was highest at the last time point examined, 37 weeks after injection. To note, in liver, at long term (37 weeks pi), the high dose injection (HD) results in higher expression of human wt GNE than injection at young age. However in gastrocnemius and quadriceps, the high dose injection results in higher expression when performed at youngage.
Histology. Microscopic examination of the various processed tissues could not detect any consistent pathological changes in any organ examined, including muscle, in any of the 3 mice groups. More importantly no muscle changes could be detected between the untreated model mice and the wt littermates (Fig. 4C).
Follow up of the Gne(–/–) hGNED207V-Tg colony
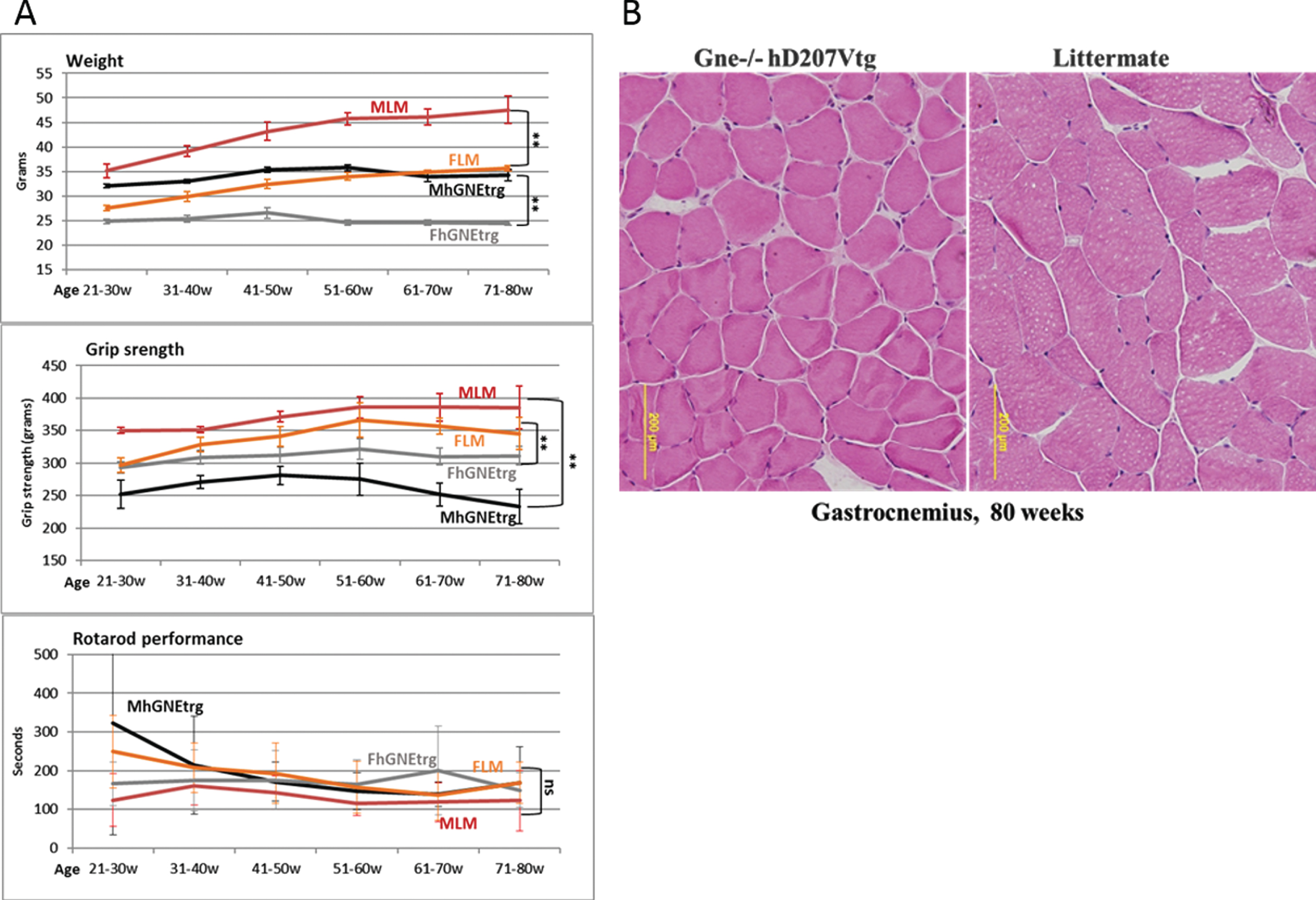
In parallel with the viral vector injections experiments, we have thoroughly observed the Gne (–/–) hGNED207V-Tg mouse model colony from birth, compared to their littermates (either Gne+/- or Gne+/+with or without hGNED207V transgene). As described in the original publication [24], we observed that these mice were much smaller than their littermates and there was some decrease in their grip strength (Fig. 5A), but no significant differences in the Rotarod performance. However, unlike the findings reported in the original report, we were unable to replicate the histological changes. This colony of GNE transgenic mice showed no consistent changes in muscle pathology and no histological findings of atrophy or rimmed vacuoles for up to 80 weeks of age (Fig. 5B).
Fig. 5
Muscle performance follow up of Gne(–/–) hGNED207V-Tg mice. A. Weight, grip force and rotarod performance follow up of Gne(–/–) hGNED207V-Tg mice; MLM, male littermate; FLM, female littermate; MhGNEtrg, male Gne(–/–) hGNED207V-Tg mice; FhGNEtrg, female male Gne(–/–) hGNED207V-Tg mice. B. Representative H&E gastrocnemius histological section of an 80 week old Gne(–/–) hGNED207V-Tg mouse and an 80 week old wt littermate.
DISCUSSION
The objective of these studies was to develop a viral vector that could efficiently target expression of wild type human GNE in diseased muscles and restore normal GNE related functions in a previously described GNE Myopathy preclinical model, Gne (–/–) hGNED207V-Tg. Our previous studies [23] had assessed gene delivery and shown long term expression of wt human GNE transcripts in healthy mice: taking advantage of the relative small size of GNE cDNA driven by the CMV promoter, to be accommodated in the AAV8 serotype, we have shown that AAV8 delivery of GNE leads to robust expression in muscle of the intact transcript by both intramuscular and vascular routes for a long period of time. Here we expanded this approach to deliver the same GNE cDNA driven by a striated muscle specific promoter -the MCK promoter- that was packaged into the capsid of the AAVrh.74 serotype. This serotype transduces muscle very efficiently when delivered via the vascular system (IV injection) and has been used in various preclinical studies [25–30] and even clinical trials in patients with DMD [31] and with limb-girdle muscular dystrophy types2E/R4, 2D/R3, and 2B/R2 (NCT02376816, NCT03769116, NCT03652259, NCT01976091, NCT02710500). This serotype has also the advantage to have a relatively low prevalence in the general population, thus allowing its potential delivery to more patients [32]. Since we intended to use this vector for preclinical studies prior to requesting regulatory agencies approval for human use, we did not include a tag that would have facilitated the monitoring of the production of the wt hGNE protein but could affect approval for human usage.
Currently, there is no reliable antibody against GNE that can be used for muscle immunohistological or Western blot testing. Therefore to evaluate GNE expression we relied on the presence of human wt GNE mRNA transcripts, as a marker for gene delivery and expression success. To measure the expression of the injected human wt GNE in the Gne–/– hGNED207V Tg mice, which carry the human mutated cDNA GNE transgene, we had to develop a Taqman probe system that differentiatesbetween this “endogenous” human GNED207V cDNA and the wt human GNE introduced via the viral infection. We took advantage of a sequence difference existing at the 3′ end of the GNE gene between the human transgene in the mouse model and the injected AAVrh74.MCK.GNE viral vector. Both primers and probe were designed to detect sequences expressed specifically and solely from the rAAVrh74.MCK.GNE vector (and not detecting the human mutated GNE expressed from the transgene present in the mice). Since we wanted to assess the expression level of the injected human wt GNE in every tissue, and particularly in muscle- and not compare its expression between tissues- even if mouse Gne is expressed at different levels at different tissues, we decided to normalize the measurements relative to mouse Gne expression of the corresponding tissues and mouse, and not to a housekeeping gene such as Hprt. Once we assessed the presence of the injected vector in muscle of the Gne–/– hGNED207V Tg at the first time point, we did not determine viral copy number in each organ at each time point since it has been shown that there is no clear correlation between this number and the expression level of the AAV packaged transgene mRNA [33].
In our mouse studies, the MCK muscle specific promoter does not absolutely restrict GNE expression solely to muscle tissue: the expression of GNE in other tissues is not completely shut off as we can still see a certain level of expression in the liver, although very low relatively to the high number of viral vectors accumulating in this organ. In contrast, in muscles, the expression of GNE is much higher relative to the number of viral vectors reaching the tissue. Although it is difficult to compare the absolute numbers with the ones obtained in C57Bl mice injected with rAAV8.CMV.GNE in our previous studies(a different AAV serotype- AAV8- was used at a different dose- 4.1013 vg/kg body weight), the expression of human wt GNE relatively to copy number was then up to 1.103 fold higher in muscle than in liver [23], and it is up to 1.106 fold in our present studies. Thus, the use of a muscle specific promoter in this set up seems to be justified to maximize gene expression in muscle tissue and minimize it in other organs, in particular the liver since systemic delivery targets mostly this organ. In the present studies, the rAAVrh74.MCK.GNE viral vector has proved as a robust tool since it infects muscle cells when injected to Gne (–/–) hGNED207V-Tg mice and allows constant expression of the wild type human GNE gene for a long period of time (at least one year) in this tissue, with no obvious safety concerns, even at high doses (5.1014 vg/kg). This is in line with other reports of such high doses of the same backbone viral vector that have been injected in various mice models and found to be safe [34]. However, dosing of patients must be carefully evaluated for each disease, to avoid possible adverse effects as recently two fatal cases resulting from liver disease were reported in a gene therapy trial for myotubular myopathy in a patients’ cohort receiving high dose AAV8 based vectors (3. 1014 vg/kg), in spite of the lack of safety concerns in a canine model injected with 5. 1014 vg/kg [35, 36]. We have also observed that injection at younger age allows higher mRNA expression of human GNE in muscles compared with injection at older age. These findings could have important implications regarding the time window for treatment of patients. Among the parameters examined, we could see, mostly at high dose, some slight functional improvement in the young injected Gne (–/–)hGNED207V-Tg mice for weight and grip force in the female mice group only. Indeed, better motor performance of females has been observed recently in a study on calpain 3 deficient mice [30]. This theoretically could be attributed to the different muscle fiber type distribution observed in humans between males and females [37], males presenting a more glycolytic phenotype and females a more oxidative phenotype, as also seen in transcriptome analyses [38]. In contrast, for several parameters measuring muscle function or muscle histology, the determination of any effect of human wt GNE delivery is very difficult to assess: indeed, except for weight, we could not see significant differences between the Gne (–/–) hGNED207V-Tg mice and their healthy littermates, making difficult to truly evaluate the effect of the treatment on muscle function. In the animals injected at 50 weeks, no significant changes could be detected in either parameter, even at the higher dose (5.1014 vg/kg). In parallel to our experimental injections of the rAAVrh74.MCK.GNE viral vector, we followed up the Gne(–/–) hGNED207V-Tg and wt littermates colony for as long as 80 weeks. We could not detect significant differences between the transgenic model and the wt littermates. The first publication describing this model [24] reported that mice had progressive muscle pathology appearing from 30–40 weeks of age. We received few breeding pairs of these mice from the original lab, expanded the colony and experimented with them. We decided not to wait and see the development of the described muscle phenotype, after a 40 weeks roughly, but rather inject young mice at age 10 weeks to examine whether the rAAVrh74.MCK.GNE could prevent the appearance of those symptoms. Surprisingly, at around 50 weeks of age, we could not see a muscle phenotype in the model mice. Assuming it could appear slightly later, we decided to continue the experiment and injected 50 week old mice with the viral vector. However, compared to the original description, the Gne(–/–) hGNED207V-Tg mice did only show a minimal phenotype (lower weight from birth), but no muscle phenotype at any time point during their lifespan, thus not allowing a reliable evaluation of any treatment effect for GNE Myopathy. Later publications from the laboratory that originally developed the model have shown ‘dilution’ of the phenotype: mice showed significant weight changes only from week 50 and became weak even later, at 70–80 weeks, as their more recent study shows [39]. Other labs also had difficulties in establishing a consistent model based on the original breeding pairs (Dr P. Martin, 2019, personal communication; [40]). The disappearance of most of the disease features in these mice could be due to changes in the transgene copy number, as it may occur in transgenic models, however in the Gne(–/–) hGNED207V-Tg model, since Gne KO is lethal and the transgene is supposedly rescuing the mice from death, we would expect that a lower expression of the transgene would at least shorten the lifespan of the mice. Although the lack of a clear, muscle related phenotype in the transgenic mice could be interpreted as a suggestion that mutated GNE is not the cause of the disease in humans, it should be pointed out that the GNE D207V mutation is considered to be very mild in humans too [41]. Thus it seems more likely that the cause of the lack of phenotype in the transgenic mice is due to a different response between humans and mice to the mutations found in GNE Myopathy patients.
Therefore, not surprisingly, no significant clinical and histological differences were observed before and after treatment with the viral vector. The functional parameters for muscle activity and their pathology were not consistently different either between the different injected groups, nor the controls Gne(–/–)hGNED207V-Tg mice and wt littermates. The present studies determine that the viral vector rAAVrh74.MCK.GNE allows the long term expression of wt human GNE in muscles. However, the efficiency of this vector for the treatment of GNE Myopathy could not be evaluated since the mouse model studied is not a reliable model for GNE Myopathy. To date, there is no animal model consistently recapitulating even some of the features of human GNE Myopathy. Gne KO mice are lethal in utero [42] and die from brain hemorrhages [43]. Knock in mice models have been generated to introduce common mutations prevalent in the human disease, by homologous recombination. Both the knocked in M743T [44,45] and V603L [46] mice died few days or weeks from severe renal failure. A surviving subcolony of the M743T mice present no renal or muscle pathology during their entire healthy normal lifespan (>1.5 years). Their “protective” transcriptomic profile includes expression enrichment of muscle related genes [47]. Therefore it seems that the response of mice to the mutations found in GNE Myopathy patients is totally different than in humans. Some attempts for novel animal models in mice and in zebrafish are in progress (Mitrani-Rosenbaum et al., unpublished results). Development of different mouse models may be one avenue to solve the current model deficiency in GNE myopathy. Any successful model would be relevant also for evaluating therapeutic approaches other than gene therapy. Indeed, currently an additional clinical trial is underway with substrate replacement, using ManNAc supplementation (NCT02346461; [48]). However such model may take a long time to develop and, it is possible that it will still be unsuccessful in mimicking the human disease. The issue is whether regulatory agencies will allow human trial based on safety of this vector in larger animals and experience gained by very similar vectors in other human myopathy trials. Recently, gene editing based techniques have gain popularity as they show a strong potential for mutation correction in several disorders, including neuromuscular diseases [49–51]. This approach could be much harder to implement in the clinic, specifically for GNE Myopathy where hundreds of pathologic mutations have been detected spanning the entire gene sequence, since an experimental system would have to be developed and more importantly fully validated for each mutation separately. Also it will take a long time till the off target issue as well as potential collateral damage extent will be reliably evaluated [52]. Alternatives to animal models could be considered to allow implementation of this rAAVrh74.MCK.GNE viral vector or any other treatment to clinical trials for GNE Myopathy patients. However human muscle cell lineages derived from GNE patients have not been very informative in terms of biomarkers for assessing GNE Myopathy treatments [53–55], certainly not for functional improvement assessment, where at least an entire organ is necessary. It is very likely that iPSCs cells derived from patients would have the same non informativepatterns.
Our present long-term follow-up studies one year post rAAVrh74.MCK.GNE treatment in GNE mutated mice demonstrate some functional improvement that can be attributed to sustained wt GNE expression. Thus, there is a solid basis for the development of GNE Myopathy therapies that supply the intact GNE, but still no reliable animal model is available to fully assess its efficacy.
ACKNOWLEDGMENTS
This work was supported by the Neuromuscular Disease Foundation (NDF), Los Angeles, California.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Argov Z , Mitrani-Rosenbaum S , Hereditary Inclusion Body Myopathies. In: KarpatiG, Hilton-JonesD, BushbyK, GriggsRC, editors. Disorders of Voluntary Muscle. 8th edition, Cambridge, (2010) p. 492–498. |
[2] | Eisenberg I , AvidanN, PotikhaT, HochnerH, ChenM, OlenderT, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nature Genetics. (2001) ;29: :83–87. |
[3] | Hinderlich S , Stasche R , Zeitler R , Reutter W . A bifunctional enzyme catalyzes the first two steps inN-acetylneuraminic acid biosynthesis of rat liver. Purification and characterization of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase. J Biol Chem. (1997) ;272: :24313–24318. |
[4] | Keppler OT , Hinderlich S , Langner J , Schwartz-Albiez R , Reutter W , Pawlita M (1999). UDP-GlcNAc 2-epimerase: A regulator of cell surface sialylation. Science. (1999) ;284: :1372–1376. |
[5] | Noguchi S , Keira Y , Murayama K , Ogawa M , Fujita M , Kawahara G , et al. Reduction of UDP-N acetylglucosamine 2-epimerase/N-acetylmannosamine kinase activity and sialylation in distal myopathy with rimmed vacuoles. J. Biol. Chem. (2004) ;279: :11402–11407. |
[6] | Saito F , Tomimitsu H , Arai K , Nakai S , Kanda T , Shimizu T , et al. A Japanese patient with distal myopathy with rimmed vacuoles: missense mutations in the epimerase domain of the UDP-N-acetylglucosamine 2-epimerase N-acetylmannosaminekinase (GNE) gene accompanied by hyposialylation of skeletal muscle glycoproteins. Neuromuscul. Disord. (2004) ;14: :158–161. |
[7] | Hinderlich S , Salama I , Eisenberg I , Potikha T , Mantey L , Yarema K , et al. The homozygous M712T mutation of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase results in reduced enzyme activities but not in altered cellular sialylation in hereditary inclusion body myopathy. FEBS letters. (2004) ;566: :105–109. |
[8] | Salama I , Hinderlich S , Shlomai Z , Eisenberg I , Krause S , Yarema K , et al. No overall hyposialylation in hereditary inclusion body myopathy myoblasts carrying the homozygous M712T GNE mutation. Biochem. Biophys. Res. Commun. (2005) ;328: :221–226. |
[9] | Broccolini A , Gliubizzi C , Pavoni E , Gidaro T , Morosetti R , Sciandra F , et al. Alpha-Dystroglycan does not play a major pathogenic role in autosomal recessive hereditary inclusion body myopathy. Neuromuscul. Disord. (2005) ;15: :177–184. |
[10] | Ricci E , Broccolini A , Gidaro T , Morosetti R , Gliubizzi C , Frusciante R , et al. NCAM is hyposialylated in hereditary inclusion body myopathy due to GNE mutations. Neurology. (2006) ;66: :755–758. |
[11] | Sela I , Goss V , Becker-Cohen M , Dell A , Haslam SM , Mitrani-Rosenbaum S . The glycomic sialylation profile of GNE Myopathy muscle cells does not point to consistent hyposialylation of individual glycoconjugates. Neuromuscul. Disord. (2020) ;30: :621–630. |
[12] | Lochmuller H , Behin A , Caraco Y , Lau H , Mirabella M , Tournev I , et al. A Phase 3 randomized study evaluating sialic acid extended-release for GNE myopathy. Neurology. (2019) ;92: :1–9. |
[13] | Wang Z , Li AV , Yarema KJ . Roles for UDP-GlcNAc 2-epimerase/ManNAc 6-kinase outside of sialic acid biosynthesis: modulation of sialyltransferase and BiP expression, GM3 and GD3 biosynthesis, proliferation, and apoptosis, and ERK1/2 phosphorylation. J. Biol. Chem. (2006) ;281: :27016–27028. |
[14] | Weidemann W , Stelzl U , Lisewski U , Bork K , Wanker EE , Hinderlich S , et al. The collapsin response mediator protein 1 (CRMP-1) and the promyelocytic leukemia zinc finger protein (PLZF) bind to UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE), the key enzyme of sialic acid biosynthesis. FEBS Lett. (2006) ;580: :6649–6654. |
[15] | Amsili S , Shlomai Z , Levitzki R , Krause S , Lochmuller H , Ben-Bassat , et al. Characterization of hereditary inclusion body myopathy myoblasts: possible primary impairment of apoptotic events. Cell Death Differ. (2007) ;14: :1916–1924. |
[16] | Amsili S , Zer H , Hinderlich S , Krause S , Becker-Cohen M , MacArthur DG , et al. UDP- N -acetylglucosamine2-epimerase/ N - acetylmannosamine kinase (GNE) binds to alpha-actinin novel pathways in skeletal muscle. PLoS One. (2008) ;3: :e2477. |
[17] | Eisenberg I , Novershtern N , Itzhaki Z , Becker-Cohen M , Sadeh M , Willems P , et al. Mitochondrial processes are impaired in hereditary inclusion body myopathy. Hum. Mol. Genet. (2008) ;17: :3663–3674. |
[18] | Sela I , Milman Krentsis I , Shlomai Z , Sadeh M , Dabby R , Argov Z , et al. The proteomic profile of hereditary inclusion body myopathy. PLoS One. (2011) ;6: :e16334. |
[19] | Harazi A , Chaouat M , Shlomai Z , Levitzki R , Becker-Cohen M , Sadeh M , et al. Survival-apoptosis associated signaling in GNE myopathy-cultured myoblasts. J. Recept. Signal Transduct. Res. (2015) ;35: :249–257. |
[20] | Harazi A , Becker-Cohen M , Zer H , Moshel O , Hinderlich S , Mitrani-Rosenbaum S . The interaction of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) and alpha-actinin 2 is altered in GNE Myopathy M743T mutant. Mol. Neurobiol. (2017) ;54: :2928–2938. |
[21] | Grover S , Arya R . Role of UDP-N-acetylglucosamine2-epimerase/N-acetylmannosamine kinase (GNE) in beta1-integrin-mediated cell adhesion. Mol. Neurobiol. (2014) ;50: :257–273. |
[22] | DeviSS, YadavR, AryaR. Altered Actin Dynamics in Cell Migration of GNE Mutant Cells. Front. Cell. Dev. Biol. (2021) ;9: :603742 eCollection 2021. |
[23] | Mitrani-Rosenbaum S , Yakovlev L , Becker Cohen M , Telem M , Elbaz M , Yanay N , et al. Sustained expression and safety of human GNE in normal mice after gene transfer based on AAV8 systemic delivery. Neuromuscul. Disord. (2012) ;22: :1015–1024. |
[24] | Malicdan MC , Noguchi S , Nonaka I , Hayashi WK , Nishino I . A Gne knockout mouse expressing human GNE D176V mutation develops features similar to distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Hum. Mol. Genet. (2007) ;16: :2669–2682. |
[25] | Chicoine LG , Rodino-Klapac LR , Shao G , Xu R , Bremer WG , Camboni M , et al. Vascular delivery of rAAVrh74.MCK.GALGT2 to the gastrocnemius muscle of the rhesus macaque stimulates the expression of dystrophin and laminin a2 surrogates. Mol. Ther. (2014) ;22: :713–724. |
[26] | SondergaardP GriffinD PozsgaiE JohnsonRW GroseWE HellerKN, et al. AAV.dysferlin overlap vectors restore function in dysferlinopathy animal models. Ann. Clin. Transl. Neurol. (2015) ;2: :256–270. |
[27] | Xu R , Jia Y , Zygmunt DA , Cramer ML , Crowe KE , Shao G , Maki AE , et al. An isolated limb infusion method allows for broad distribution of rAAVrh74.MCK.GALGT2 to leg skeletal muscles in the rhesus macaque. Mol. Ther. Methods Clin. Dev. (2018) ;10: :89–104. |
[28] | Zygmunt DA , Xu R , Jia Y , Ashbrook A , Menke C , Shao G , et al. rAAVrh74.MCK.GALGT2 Demonstrates Safety and Widespread Muscle Glycosylation after Intravenous Delivery in C57BL/6J Mice. Mol Ther Methods Clin. Dev. (2021) ;15: :305–319. |
[29] | Martin PT , Zygmunt DA , Ashbrook A , Hamilton S , Packer D , Birch SM , et al. Short-term treatment of golden retriever muscular dystrophy (GRMD) dogs with rAAVrh74.MHCK7.GALGT2 induces muscle glycosylation and utrophin expression but has no significant effect on muscle strength. PLoS One. (2021) ;16: :e0248721. |
[30] | Sahenk Z , Ozes B , Murrey D , Myers M , Moss K , Yalvac ME , et al. Systemic delivery of AAVrh74.tMCK.hCAPN3 rescues the phenotype in a mouse model for LGMD2A/R1. Mol. Ther. Methods Clin.Dev. (2021) ;22: :401–414. |
[31] | Mendell JR , Sahenk Z , Lehman K , et al. Assessment of systemic delivery of rAAVrh74.MHCK7.micro-dystrophin in children with Duchenne muscular dystrophy: A nonrandomized trial. JAMA Neurol. (2020) ;77: :1122–1131. |
[32] | Zygmunt DA , Crowe KE , Flanigan KM , Martin PT . Comparison of serum rAAV serotype-specific antibodies in patients with Duchenne muscular dystrophy, Becker muscular dystrophy, inclusion body myositis, or GNE myopathy. Hum. Gene Ther. (2017) ;28: :737–746. |
[33] | Tabebordbar M , Lagerborg KA , Stanton A , King EM , Ye S , Tellez L , et al. Directed evolution of a family of AAV capsid variantsenabling potent muscle-directed gene delivery across species. Cell. (2021) ;184: :4919–4938. |
[34] | Potter RA , Griffin DA , Heller KN , Peterson EL , Clark EK , Mendell JR , et al. Dose-Escalation Study of Systemically Delivered rAAVrh74.MHCK7.micro-dystrophin in the mdx Mouse Model of Duchenne Muscular Dystrophy. Hum. Gene Ther. (2021) ;32: :375–389. |
[35] | Morales L , Gambhir Y , Bennett J , Stedman H . Broader Implications of Progressive Liver Dysfunction and Lethal Sepsis in Two Boys following Systemic High-Dose AAV. Molec. Ther. (2020) ;28: :1753–1755. |
[36] | Elverman M , Goddard MA , Mack D , Snyder JM , Lawlor MW , Meng H , et al. Long-term effects of systemic gene therapy in a canine model of myotubular myopathy. Muscle Nerve. (2017) ;56: :943–953. |
[37] | Staron RS , Hagerman FC , Hikida RS , Murray TF , Hostler DP , Crill MT , et al. Fiber type composition of the vastus lateralis muscle of young men and women. J. of Histochem. and Cytochem. (2000) ;48: :623–629. |
[38] | Lindholm ME , Huss M , Solnestam BW , Kjellqvist S , Lundeberg J , Sundberg CJ . The human skeletal muscle transcriptome: sex differences, alternative splicing, and tissue homogeneity assessed with RNA sequencing. FASEBJ. (2014) ;28: :4571–4581. |
[39] | Yonekawa T , Malicdan MC , Cho A , Hayashi YK , Nonaka I , Mine T , et al. Sialyllactose ameliorates myopathic phenotypes in symptomatic GNE myopathy model mice Brain. (2014) ;137: :2670–2679. |
[40] | Crowe KE , Zygmunt DA , Heller K , Rodino-Klapac L , Noguchi S , Nishino I , et al. Visualizing Muscle Sialic Acid Expression in the GNED207VTgGne–/– Cmah–/– Model of GNE Myopathy: A Comparison of Dietary and Gene Therapy Approaches. J. Neuromuscul. Dis. (2021) ; online ahead of print. |
[41] | Pogoryelova O , Wilson IJ , Mansbach H , Argov Z , Nishino I , Lochmüller H . GNE genotype explains 20% of phenotypic variability in GNE myopathy. Neurol. Genet. (2019) Feb 1;5: (1):e308. e308. |
[42] | Schwarzkopf M , Knobeloch KP , Rohde E , Hinderlich S , Wiechens N , Lucka L , et al. Sialylation is essential for early development in mice. Proc. Natl. Acad. Sci. USA. (2002) ;99: :5267–5270. |
[43] | Wedekind H , Kats E , Weiss AC , Thiesler H , Klaus C , Kispert A , et al. Gne deletion in mice leads to lethal intracerebral hemorrhage during embryonic development. Glycobiology. (2021) , Online ahead of print. |
[44] | Galeano B , Klootwijk R , Manoli I , Sun M , Ciccone C , Darvish D , et al. Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J. Clin. Invest. (2007) ;117: :1585–1594. |
[45] | Sela I , Yakovlev L , Becker Cohen M , Elbaz M , Yanay N , Ben Shlomo U , et al. Variable phenotypes of knockin mice carrying the M712T Gne mutation. Neuromol. Med. (2013) ;15: :180–191. |
[46] | Ito M , Sugihara K , Asaka T , Toyama T , Yoshihara T , Furuichi K , et al. Glycoprotein Hyposialylation Gives Rise to a Nephrotic-Like Syndrome That Is Prevented by Sialic Acid Administration in GNE V572L Point-Mutant Mice. PLoS One. (2012) ;7: :e29873. |
[47] | Benyamini H , Kling Y , Yakovlev L , Becker Cohen M , Nevo Y , Elgavish S , et al. Upregulation of Hallmark Muscle Genes Protects GneM743T/M743T Mutated Knock-In Mice From Kidney and Muscle Phenotype. J. Neuromuscul. Dis. (2020) ;7: :119–136. |
[48] | Carrillo N , Malicdan MC , Leoyklang P , Shrader JA , Joe G , Slota C , Perreault J , Heiss JD , Class B , Liu CY , et al. Safety and efficacy of N-acetylmannosamine (ManNAc) in patients with GNE myopathy: an open-label phase 2 study. Genet Med. (2021) ; online ahead of print. |
[49] | Gillmore JD , Gane E , Taubel J , Kao J , Fontana M , Maitland ML , et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N Engl J Med. (2021) ; online ahead of print. |
[50] | Min YL , Li H , Rodriguez-Caycedo C , Mireault AA , Huang J , Shelton JM , et al. CRISPR-Cas9 corrects Duchennemuscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. (2019) ;5: :eaav4324. |
[51] | Amoasii L , Hildyard JCW , Li H , Sanchez-Ortiz E , Mireault A , Caballero D , et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. (2018) ;362: :86–91. |
[52] | Leibowitz ML , Papathanasiou S , Doerfler PA , Blaine LJ , Sun L , Yao Y , et al. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet. (2021) ;53: :895–905. |
[53] | Amsili S , Shlomai Z , Levitzki R , Krause S , Lochmuller H , Ben-Bassat H , et al. Characterization of hereditary inclusion body myopathy myoblasts: possible primary impairment of apoptotic events. Cell Death Differ. (2007) ;14: :1916–1924. |
[54] | Sela I , Milman Krentsis I , Shlomai Z , Sadeh M , Dabby R , Argov Z , et al. The Proteomic Profile of Hereditary Inclusion Body Myopathy. PLoS ONE. (2011) ;6: (1):e16334, 2011. |
[55] | Harazi A , Chaouat M , Shlomai Z , Levitzki R , Becker-Cohen M , Sadeh M , et al. Survival-apoptosis associated signaling in GNE myopathy-cultured myoblasts. J. Recept. Signal Transduct. Res. (2015) ;35: :249–257. |


