
Targeted Therapies for Leigh Syndrome: Systematic Review and Steps Towards a ‘Treatabolome’
Abstract
Background:
Leigh syndrome (LS) is the most frequent paediatric clinical presentation of mitochondrial disease. The clinical phenotype of LS is highly heterogeneous. Though historically the treatment for LS is largely supportive, new treatments are on the horizon. Due to the rarity of LS, large-scale interventional studies are scarce, limiting dissemination of information of therapeutic options to the wider scientific and clinical community.
Objective:
We conducted a systematic review of pharmacological therapies of LS following the guidelines for FAIR-compliant datasets.
Methods:
We searched for interventional studies within Clincialtrials.gov and European Clinical trials databases. Randomised controlled trials, observational studies, case reports and case series formed part of a wider MEDLINE search.
Results:
Of the 1,193 studies initially identified, 157 met our inclusion criteria, of which 104 were carried over into our final analysis. Treatments for LS included very few interventional trials using EPI-743 and cysteamine bitartrate. Wider literature searches identified case series and reports of treatments repleting glutathione stores, reduction of oxidative stress and restoration of oxidative phosphorylation.
Conclusions:
Though interventional randomised controlled trials have begun for LS, the majority of evidence remains in case reports and case series for a number of treatable genes, encoding cofactors or transporter proteins of the mitochondria. Our findings will form part of the international expert-led Solve-RD efforts to assist clinicians initiating treatments in patients with treatable variants of LS.
INTRODUCTION
Mitochondrial diseases are common inherited metabolic disorders, of which the commonest is Leigh syndrome (LS) [1, 2]. The clinical phenotype of LS is comprised of neurological symptoms with basal ganglia and/or brainstem dysfunction. The diagnostic criteria include clinical and radiological features and biochemical evidence of abnormal energy metabolism due to defects in the oxidative phosphorylation (OXPHOS) pathway or pyruvate dehydrogenase complex (PDHc). If the diagnostic criteria are not fulfilled, the condition can be classified as Leigh-like syndrome [2]. LS usually presents in early childhood. with developmental delay, encephalopathy, dystonia and symmetrical T2 hyperintensities in the basal ganglia and brainstem, but some cases with late-onset are known. Additionally, systemic issues outside of the central nervous system such as renal failure, cardiomyopathy, diabetes mellitus can be present, making LS a disease with diverse clinical features [3, 4].
Despite well-defined diagnostic criteria, the combination of clinical heterogeneity and rarity can lead to delay in diagnosis.Whilst there are both complete and ongoing clinical trials in LS [5], the lack of clearly-presented evidence and vast amount of literature, the majority of which lies in case reports and case series, prevent timely instigation of treatment by clinicians. Moreover, there is a lack of guidance regarding which of the over 75 causative genes and which biochemical pathways in LS [2] are treatable, without extensive literature searching. The combination of diagnostic delay and lack of systematic evaluation of treatments inevitably impacts the clinical course of LS and the likelihood of patient response to treatment.
The RD-Connect Genome-Phenome Analysis Platform (GPAP) is an online tool for diagnosis and gene discovery in rare disease research (https://rd-connect.eu/what-we-do/omics/gpap/). As part of the H2020 research project Solve-RD rare disease experts aim towards creating a database of treatable rare disease, the ‘Treatabolome’, at gene and variant levels [6]. Previous Treatabolomes have been created for congenital myasthenic syndromes [7], inherited neuropathies [8] and laminopathies [9]. As part of this international effort, we have systematically reviewed existing evidence of pharmacological treatments of LS.
METHODS
Systematic review protocol
We aimed to identify any gene/variant specific pharmacological treatments in LS and assessed the strength of the associated supporting evidence as part of the creation of a LS ‘Treatabolome’. The systematic review was designed following the guidelines set out by Atalaia et al. 2020 for creating a Treatabolome with FAIR-compliant datasets [6].
Literature search strategy
The conditions studied were genetically confirmed LS as previously described, including clinical, radiological criteria and known LS-associated genetic variants [2]. We searched electronic databases MEDLINE via PubMed, ClinicalTrials.gov and the European Clinical Trials Register. A full search strategy including search terms and filters used for each database is described in Supplementary File 1.
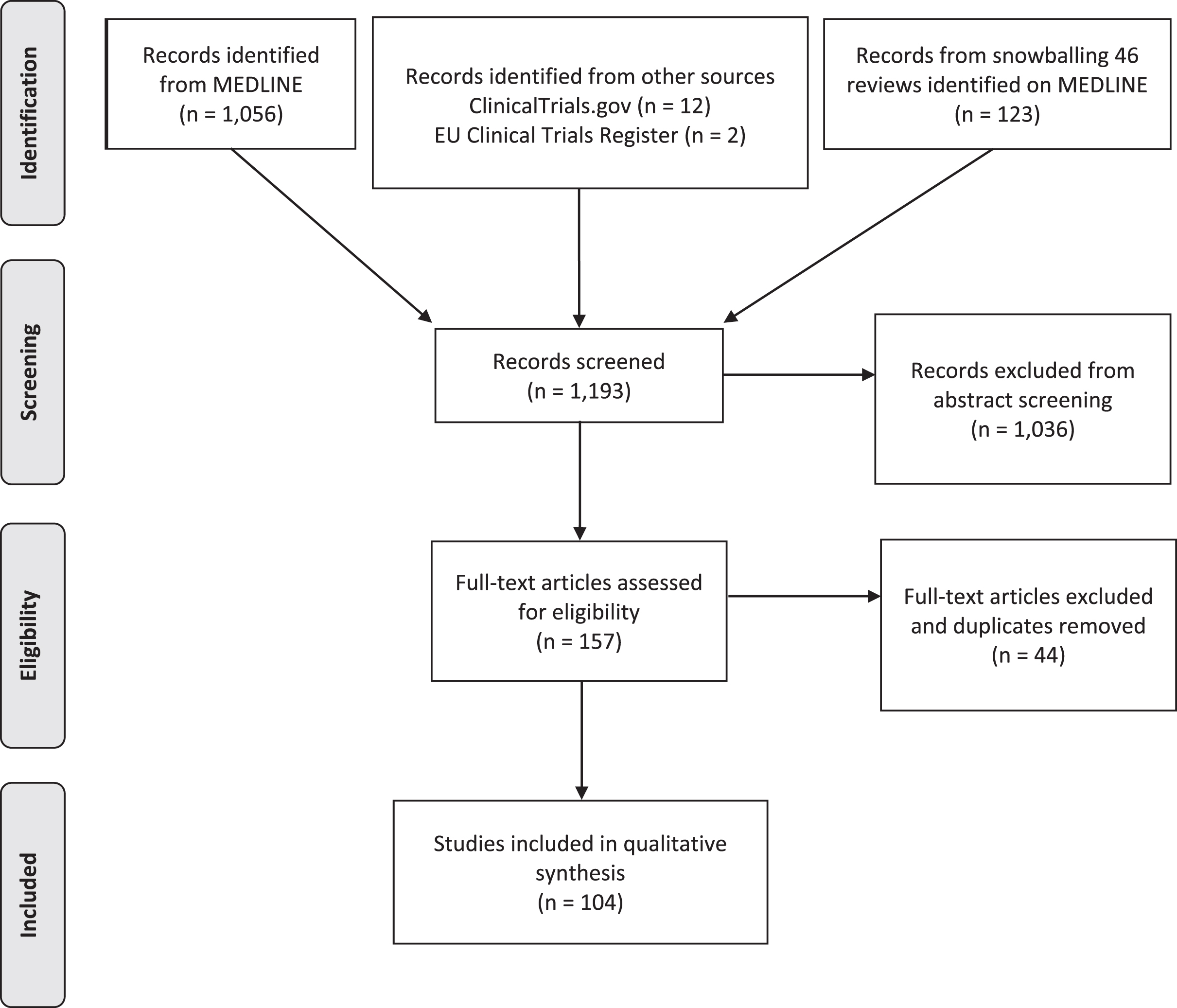
We identified 1,056 articles from PubMed, 12 clinical trials from ClinicalTrials.gov and 2 from European Clinical Trials Register (Fig. 1). Relevant reviews and other secondary literature identified from PubMed were flagged and screened for relevant primary research that met the inclusion criteria. This identified an additional 123 papers. All studies were uploaded onto the Rayyan QCRI platform for subsequent screening and removal of duplicates (Fig. 1).
Fig. 1
PRISMA flow diagram of articles screened for treatments in Leigh syndrome.
Inclusion & exclusion criteria for screening
Following compilation of all the papers identified from each database, title & abstract screening was undertaken by 2 independent blinded reviewers (ZL, MYT) and arbitrated by an expert reviewer (RH). The following inclusion and exclusion criteria were implemented for both title and abstract screening and full-text screening (Fig. 2).
Fig. 2
Inclusion and exclusion criteria for screening Leigh syndrome treatments.

Data extraction
For the 104 papers that met our inclusion criteria, all relevant information was extracted onto a standardised data form as described in the guidelines [6] (Fig. 3). In studies with multiple patients on different treatment regimes, individual gene variants and all treatments trialled for each individual patient were recorded where such information was available. Each paper was evaluated by 2 independent blinded reviewers (ZL, MYT) using both the OCEBM 2011 Levels of Evidence score [10] and the Jadad score [11]. Discrepancies in scoring were resolved by a third reviewer (RH).
Fig. 3
Data extraction from articles discussing treatment in Leigh syndrome to develop a FAIR-compliant dataset.

RESULTS
In total, 1,193 records were screened from PubMed, EU Clinical Trials Register, Clinicaltrials.gov and review articles. A total of 157 papers passed the inclusion criteria. After removing duplications, 104 studies were included in our analysis. The majority of evidence remained in case reports or case series scoring 4 (44.7%, n = 46) or 5 (50.5%, n = 52) on OCEBM and 0 (95.1%, n = 98) on Jadad scoring (Fig. 4). The highest levels of evidence were associated with completed clinical trials in only 3 compounds: Vatiquinone (EPI-743) (n = 2) KH176 (n = 1) and cysteamine bitartrate (RP103) (n = 2), but were all open-labelled studies (level 3 OCEBM, Jadad 1). Randomized multi-centre clinical trials with KH176 are ongoing, and a further trial with novel compound Nabsirolimus (ABI-009) is scheduled to begin recruitment late 2021 (Fig. 5).
Fig. 4
Level of evidence of treatment in Leigh syndrome according to treatment, number of studies and OCEBM score (4A), Jadad score (4B), combined Jadad and OCEBM score.
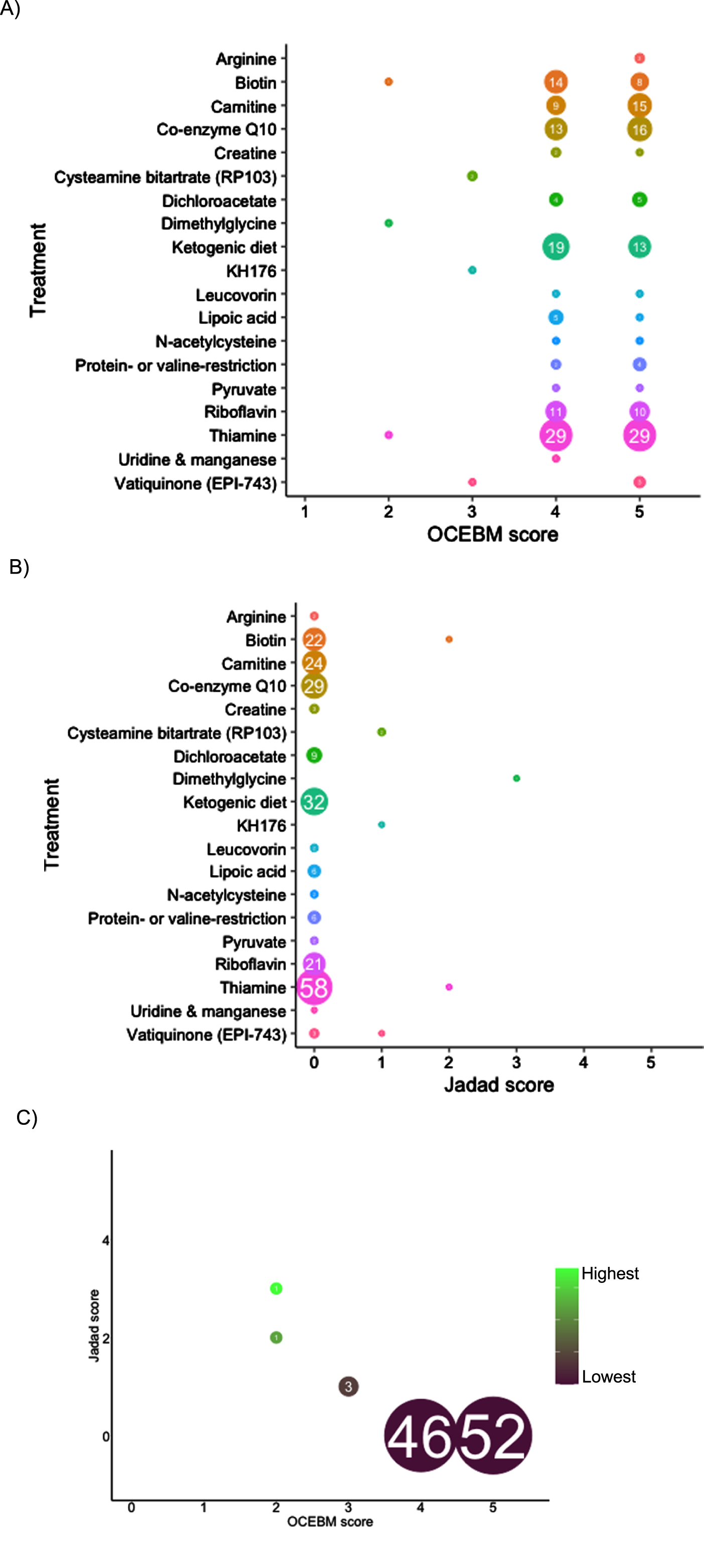
Fig. 5
Clinical trials for treatment of Leigh syndrome. LTE = long-term extension.
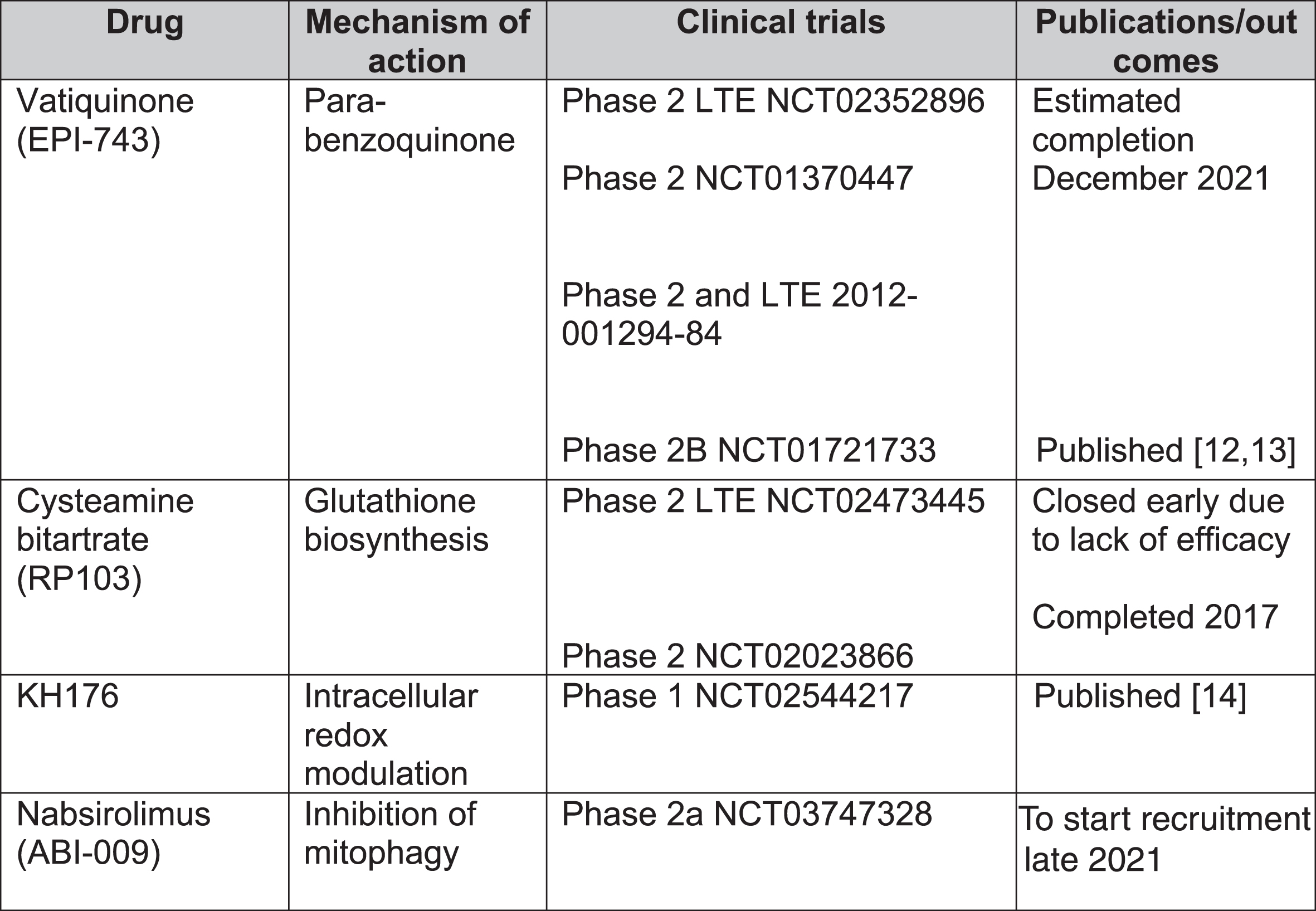
EPI-743
EPI-743, a novel para-benzoquinone that repletes intracellular gluthathione more potently than co-enzyme Q10 or idebenone, has been tested in open-label clinical trials in genetically confirmed inherited respiratory chain diseases [13]. Enns et al. reported that 11 of the 12 participants showed clinical improvement and increased regional and whole brain technetium-99m-hexamethylpropylene oxime (HMPAO) uptake on single photon emission computed tomography (SPECT) [12]. In a phase 2a single-site clinical trial, Martinelli et al. reported significant improvement of the Newcastle Paediatric Mitochondrial Disease Scale (NPMDS), Gross Motor Function Measure and PedsQL Neuromuscular Module in 10 children with genetically-confirmed Leigh syndrome [13].
Cysteamine bitartrate
Cysteamine is required for gluthathione biosynthesis to reduce oxidative stress levels and has been used for lysosomal storage disease cystinosis [15]. So far, there have been 2 open-labelled studies (NCT02473445 and NCT02023866) on various mitochondrial diseases. The phase 2 long-term extension study was terminated early due to lack of efficacy.
KH176
KH176 is a redox-modulating agent. A phase 1 study (NCT02544217) for various mitochondrial diseases, including LS, reported that the agent was well-tolerated but caused QTc prolongation and T-wave morphological changes at higher doses [14]. Randomized multi-centre phase 2 studies are currently recruiting including mitochondrial disorders such as MELAS, MIDD, LS and mitochondrial myopathies as well as mitochondrial encephalopathies. A safety and efficacy trial has been completed in m.3243A > G-associated mitochondrial disease [16].
Nabsirolimus (ABI-009)
Nabsirolimus is an albumin-bound sirolimus nanoparticle suspension which inhibits mitophagy [5]. A phase 2, open-label study for LS patients with moderate severity according to NPMDS score plans to begin recruitment of patients in August 2021 (NCT03747328).
Fig. 6
Treatments for Leigh syndrome according to causal gene. Key: + = overall positive outcome, M = mixed outcome, - = negative outcome, CoQ10 = co-enzyme Q10, EPI-743 = Vatiquinone.
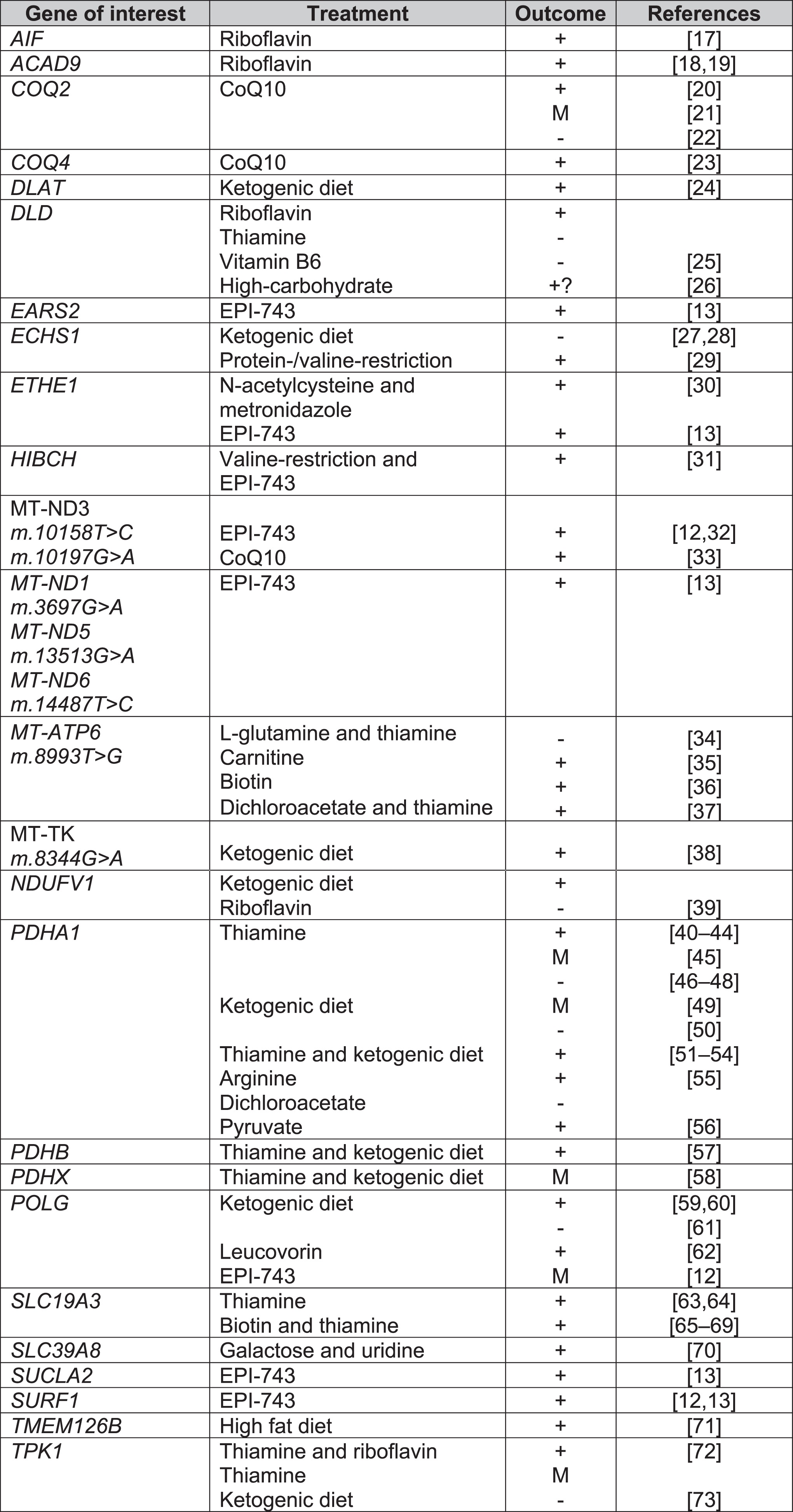
Fig. 7
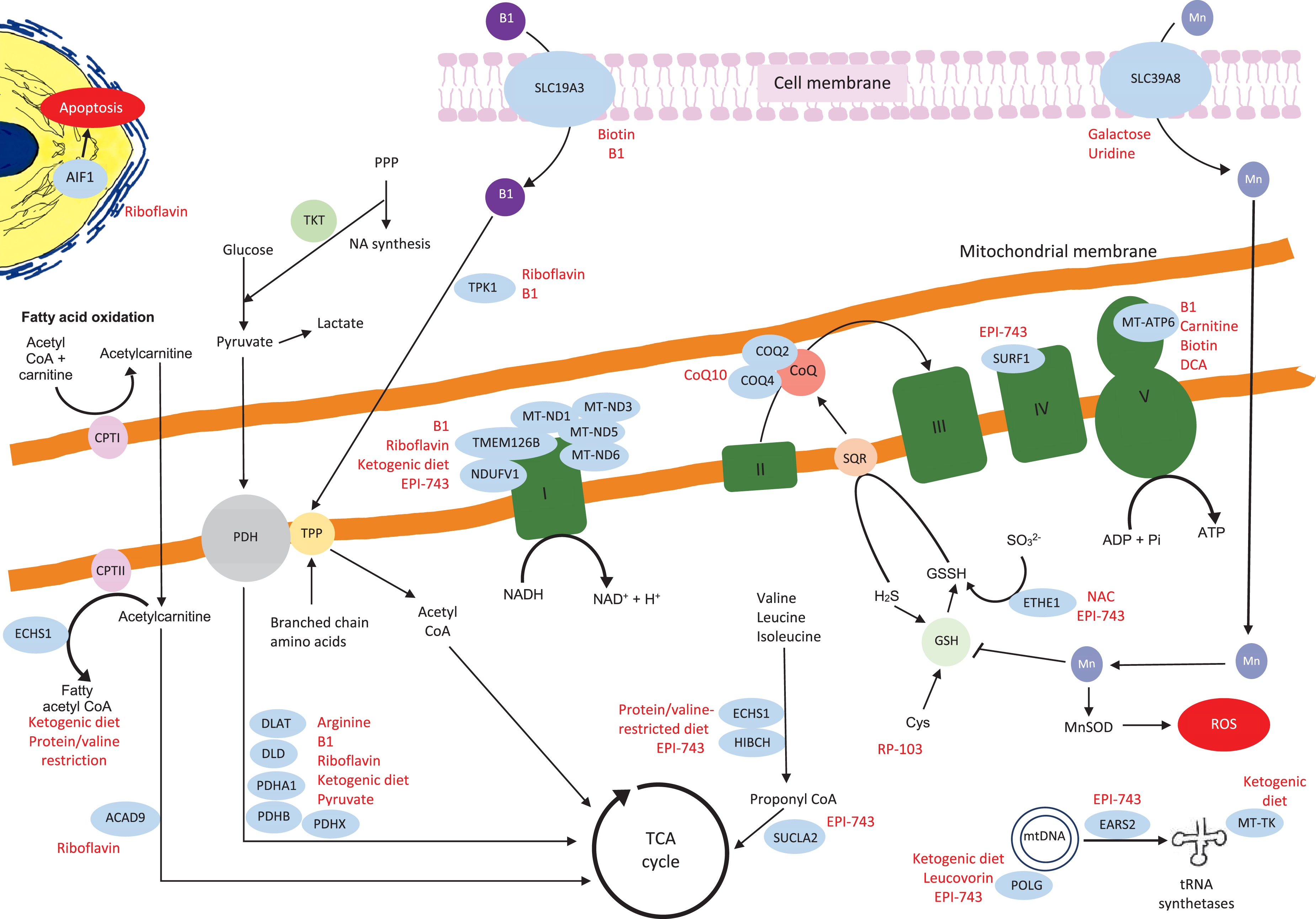
Schematic representation of the mitochondrial genes and proteins with their links to the other metabolic pathways. The genes where we identified relevant references on treatments are in blue circles, the potential treatments are highlighted in red. The details of the relevant treatments are explained above. Key:B1 = thiamine, TPP = thiamine pyrophosphate, TKT = transketolase, DCA = dichloroacetic acid, PPP = pentose phosphate pathway, NA = nucleic acid, PDH = pyruvate dehydrogenase, CPT1 = carnitine palmitoyltransferaseI, Actyl CoA = acetyl co-enzyme A, CoQ = coenzyme Q, TCA = tricarboxylic acid, H2S = hydrogen sulfide, GSH = glutathione, GSSH = glutathione persulfide, SQR = sulfide:quinone oxidoreductase, Cys = cysteine, Mn = manganese, MnSOD = manganese-superoxide dismutase, ROS = reactive oxygen species.
Biotin and thiamine
The SLC19A3 gene encodes a thiamine transporter. SLC19A3 mutations can lead to Biotin-Responsive Basal Ganglia Disease (BBGD). A total of 22 and 57 studies included biotin and thiamine supplementation respectively, most commonly in combination as a “mitochondrial cocktail” of treatments. Only 2 case series tested biotin or thiamine monotherapy versus biotin and thiamine combined treatment. Debs et al. reported clinical and radiological improvement in both biotin monotherapy and combination therapy in 2 siblings with missense mutations in SLC19A3 [74], whilst Tabarki et al. reported no significant difference in Burke-Fahn-Marsden Dystonia Rating Scale (BFMDRS) following thiamine monotherapy or combination therapy in an open-labelled prospective study in 20 children with SLC19A3 c.1264A > G (p.Thr422Ala) mutation. There was, however, significantly quicker recovery from acute crises (2 days versus 3 days) with combination therapy [65].
Delays in treatment initiation can limit meaningful clinical outcome. Yamada et al. and Kevelam et al. reported no improvement with biotin therapy in patients with advanced disease [75, 76]. Kevelam et al. initiated biotin therapy for a patient with homozygous SLC19A3. The treatment was initiated age 19 months but there was evidence of severe cortical atrophy on MRI age 14 months [75].
Co-enzyme Q10 (ubiquinone)
Co-enzyme Q10 (CoQ10) is required for oxidative phosphorylation in mitochondria, acting as an electron carrier between complex I, II and III [77]. A large number of studies were excluded for data extraction due to secondary CoQ10 deficiency or patients not fulfilling the diagnostic criteria for LS clinically, radiologically or genetically. As with biotin and thiamine, in the majority of the 28 studies which included CoQ10, CoQ10 formed part of a mitochondrial cocktail of supplements for LS patients (Supplementary Figure 2). Six studies assessed CoQ10 alone, of which 4 reported improvement in patients with mutations in m.10197G > A, COQ2, COQ4 [20, 21, 23, 33]. It was noted that clinical deterioration occurred if patients were already in late-stages of disease before treatment initiation [21]. Improvement in renal function were reported with CoQ10 initiation [20]; however, Scalais et al. reported a child who developed massive proteinuria despite CoQ10 supplementation in infancy [22].
Pyruvate
Pyruvate provides NAD+ restoring ATP production and reducing the NADH/NAD + ratio from oxidative phosphorylation disorders [78, 79]. The majority of studies included biochemical pyruvate dehydrogenase deficiencies which were not radiologically or genetically-confirmed LS. Only 2 papers passed all inclusion and exclusion criteria. The highest level of evidence was a case series by Fuji et al. including 2 genetically confirmed LS patients with the following variants m.8993T > G and m.9176 T > C, who were bedbound but had improvements in the Newcastle Paediatric Mitochondrial Disease Scale (NPMDS) after pyruvate treatment [79].
Dichloroacetic acid (DCA) inhibits pyruvate dehydrogenase kinase, which in turn inhibits pyruvate dehydrogenase [80]. Our study confirms the mixed clinical outcomes from DCA. DCA can lead to worsening peripheral neuropathy, even with prophylactic administration of thiamine [81]. Successful improvement of midbrain hyperintensities with DCA and thiamine therapy in a patient with MT-ATP6 m.8993T > C were reported by Fujii et al. [37]. Conversely, Koga et al. reported a child with a PDHE1 α mutation who deteriorated with DCA but responded to pyruvate therapy [56].
Riboflavin
Flavin adenine dinucleotide (FAD) precursor protein, riboflavin, was used in 18 studies, but the majority were part of a mitochondrial cocktail treatment. A total of 3 studies reported riboflavin monotherapy. Two studies reported improvement in patients with ACAD9 mutations [18, 19]. Gerards et al. reported restoration of complex I activity in a family with homozygous ACAD9 (c.1594C > T) mutation [19]. Ghezzi et al. described 2 children born from monozygotic twin sisters with mutations in AIF1, 1 patient received treatment with riboflavin with initial improvement but later deteriorated with further seizures and tetraplegia [17].
Carnitine
Carnitine is involved in fatty acid oxidation [82] and is used as a neutraceutical (mitochondrial cocktail) with other co-factors and antioxidants to bypass the electron transport chain [83]. Carnitine featured as a mitochondrial cocktail treatment in case series or case reports but often with no effect [28, 84, 85]. A case report by Toth et al. reported a patient with m.8993T > C mutation acutely losing ambulation with an upper respiratory tract infection. It is unclear if the clinical improvement with subsequent carnitine supplementation coincided with resolution of their respiratory tract infection. The authors reported low plasma and muscle carnitine levels prior to treatment. Further deterioration in ataxia and weakness was noted at the age of 13 and then improvement with adjusted dosing of carnitine according to weight [35].
N-acetylcysteine
N-acetylcysteine (NAC) is a precursor of antioxidant agent glutathione which binds toxic sulfide [15, 30]. We found 2 studies assessing the effectiveness of NAC treatment. Most notably, Viscomi et al. described improvement in seizure-frequency, neurological improvement, reduction of acrocyanosis and petechiae in 5 children with homozygous 505 + 1G > T splice-site mutations in ETHE1. The patients also had improvements in diarrhoea, which may be explained with the combination therapy with bactericidal and prokinetic agent Metronidazole [30]. Shayota et al. reported developmental improvement with NAC in a patient with ECSH1 mutation; however, the patient also received a valine-restricted diet [29].
Other supplements
Isolated studies have been published for arginine [55], leucovorin (folinic acid) [62], uridine and galactose [70] stating a positive effect. Manganese deficiency in SLC39A8 variants leads to the decrease in manganese superoxide dismutase and oxidative stress [86]. Treatment with uridine and galactose in SLC39A8 is reported to improve transferrin glycosylation pattern [70]. Further studies are required to fully assess the effectiveness of such treatments.
Diets
Dietary modifications are known for use of epilepsy-control. Ketogenic diets are thought to promote fatty acid β-oxidation as an alternative pathway to oxidative phosphorylation [39]. Effectiveness in epilepsy control, eye movements and mental development has been reported in ECHS1 [28], POLG [60], TMEM126B [71], PDHA1 leading to pyruvate dehydrogenase deficiency [43].
Protein and valine-restriction have been reported to be effective in patients with mutations involved in valine-degradation, such as HIBCH [31] and ECSH1 [29]. It is believed that such diets help to circumvent the accumulation of toxic metabolites such as methacrylyl-CoA and acryloyl-CoA [29]. However, ketogenic diet can induce metabolic acidosis and lead to clinical dete- rioration in LS [73]. To date, the evidence of dietary modifications is limited to case reports and small case series.
Measurable outcomes
The majority of treatments for LS are from low quality studies. Clinical improvements reported in case reports and case series are often without quantitative clinical scales. Although case series rank as low quality of evidence, we note that some studies used clinically-validated disease scores such as NPMDS [12, 13] and BFMDRS [65].
Promising biomarkers are emerging in LS, such as glutathione status as a marker of redox for EPI-743 therapy [87]. Radiological features such as basal ganglia lesions [74] and HMPAO on SPECT [12] are associated with clinical improvement in LS and could serve as potential therapeutic biomarkers.
CONCLUSIONS
We have conducted a systematic review of mutation-specific treatments in LS according to FAIR-principles, as part of an international effort to develop a rare disease Treatabolome. The majority of evidence of LS treatments relies on case reports and case series, resulting in publication bias due to the tendency towards reporting treatment-responsive cases. However, these reports should not be overlooked as a number of LS genetic variants are highly responsive to treatments. This holds promise for the development of future therapies and clinical trials. Clinicians with patients of these treatable genotypes may wish to consider experimental use of these therapies with the guidance of specialised mitochondrial services. There are a small number of clinical trials which are still actively recruiting and EPI-743 has reported clinical improvement in LS patients.
Clinical heterogeneity may affect trial outcomes. However, clinical trials may not be needed when the clinical response and disease mechanism is clear; an example would be BBGD, where prospective case series compared biotin/thiamine monotherapy or combination therapies using validated clinical outcome measures [65]. The LS Treatabolome will allow clinicians to appropriately assess the existing evidence for treatable LS genotypes. Due to the clinical heterogeneity of LS, we propose that MRI features and a genetically confirmed variant to be used as part of the selection criteria for future clinical trials. However, due to the relative low prevalence of LS, it is likely that future trials will continue to incorporate this patient group within other mitochondrial disorders.
Treatment-responsiveness, as documented in several trials and case reports, relies on early recognition and intervention. To that end, our findings will form part of the international expert-led Solve-RD efforts to assist clinicians initiating treatments in patients with treatable-variants of LS.
ACKNOWLEDGEMENTS
M.Y.T. was supported by the Addenbrookes Charitable Trust (G104114). R.H. is a Wellcome Trust Investigator (109915/Z/15/Z), who receives support from the Medical Research Council (UK) (MR/N025431/1 and MR/V009346/1), the European Research Council (309548), the Newton Fund (UK/Turkey, MR/N027302/1), the Addenbrookes Charitable Trust (G100142), the Evelyn Trust, the Stoneygate Trust, the Lily Foundation and an MRC strategic award to establish an International Centre for Genomic Medicine in Neuromuscular Diseases (ICGNMD) MR/S005021/1. The study was further supported by the Horizon 2020 research and innovation program via grant 779257 “Solve-RD”. This research was supported by the NIHR Cambridge Biomedical Research Centre (BRC-1215-20014). The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to report.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-210715.
REFERENCES
[1] | Schubert MB , Vilarinho L . Molecular basis of Leigh syndrome: A current look. Orphanet J Rare Dis. (2020) ;15: (1):1–14. doi: 10.1186/s13023-020-1297-9 |
[2] | Lake NJ , Compton AG , Rahman S , Thorburn DR . Leigh syndrome: One disorder, more than 75 monogenic causes. Ann Neurol. (2016) ;79: (2):190–203. doi: 10.1002/ana.24551 |
[3] | Rahman S , Hanna MG . Diagnosis and therapy in neuromuscular disorders: Diagnosis and new treatments in mitochondrial diseases. J Neurol Neurosurg Psychiatry. (2009) ;80: (9):943–53. doi: 10.1136/jnnp.2008.158279 |
[4] | Lake NJ , Bird MJ , Isohanni P , Paetau A . Leigh Syndrome: Neuropathology and Pathogenesis. J Neuropathol Exp Neurol. (2015) ;74: (6):482–92. |
[5] | Pitceathly RDS , Keshavan N , Rahman J , Rahman S . Moving towards clinical trials for mitochondrial diseases. J Inherit Metab Dis. (2021) ;44: (1):22–41. doi: 10.1097/NEN.0000000000000195 |
[6] | Atalaia A , Thompson R , Corvo A , Carmody L , Piscia D , Matalonga L , et al. A guide to writing systematic reviews of rare disease treatments to generate FAIR-compliant datasets: Building a Treatabolome. Orphanet J Rare Dis. (2020) ;15: (1):1–11. doi: 10.1186/s13023-020-01493-7 |
[7] | Thompson R , Bonne G , Missier P , Lochmüller H . Targeted therapies for congenital myasthenic syndromes: Systematic review and steps towards a treatabolome. Emerg Top Life Sci. (2019) ;3: (1):19–37. doi: 10.1042/ETLS20180100 |
[8] | Jennings MJ , Lochmüller A , Atalaia A , Horvath R . Targeted therapies for hereditary peripheral neuropathies: Systematic review and steps towards a ‘ treatabolome ’. J Neuromuscul Dis. (2021) ;8: (3):383–400. doi: 10.3233/JND-200546 |
[9] | Atalaia A , Ben R , Wahbi K , Sandre-giovannoli De A , Vigouroux C , Bonne G . Laminopathies ’ Treatments Systematic Review: A Contribution Towards a ‘ Treatabolome. ’ J Neuromuscul Dis. (2021) ;8: (3):419–39. doi: 10.3233/JND-200596 |
[10] | Howick J , Chalmers I , Glasziou P , Greenhalgh T , Heneghan C , Liberati A , et al. Explanation of the 2011 Oxford Centre for Evidence-Based Medicine (OCEBM) Levels of Evidence (Background Document). [Internet]. Oxford Centre for Evidence-Based Medicine. Available from: https://www.cebm.ox.ac.uk/resources/levels-of-evidence/explanation-of-the-2011-ocebm-levels-of-evidence/ |
[11] | Jadad AR , Moore RA , Carroll D , Jenkinson C , Reynolds DJM , Gavaghan DJ , et al. Assessing the quality of reports of randomized clinical trials: Is blinding necessary? Control Clin Trials. (1996) ;17: (1):1–12. doi: 10.1016/0197-2456(95)00134-4 |
[12] | Enns GM , Kinsman SL , Perlman SL , Spicer KM , Abdenur JE , Cohen BH , et al. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol Genet Metab. (2012) ;105: (1):91–102. doi: 10.1016/j.ymgme.2011.10.009 |
[13] | Martinelli D , Catteruccia M , Piemonte F , Pastore A , Tozzi G , Dionisi-Vici C , et al. EPI-743 reverses the progression of the pediatric mitochondrial disease-Genetically defined Leigh Syndrome. Mol Genet Metab. (2012) Nov; 107: (3):383–8. doi: 10.1016/j.ymgme.2012.09.007 |
[14] | Koene S , Spaans E , Van Bortel L , Van Lancker G , Delafontaine B , Badilini F , et al. KH176 under development for rare mitochondrial disease: A first in man randomized controlled clinical trial in healthy male volunteers. Orphanet J Rare Dis. (2017) ;12: (1):1–12. doi: 10.1186/s13023-017-0715-0 |
[15] | Bottani E , Lamperti C , Prigione A , Tiranti V , Persico N , Brunetti D . Therapeutic approaches to treat mitochondrial diseases: “One-size-fits-all” and “precision medicine” strategies. Pharmaceutics. (2020) ;12: (11):1–63. doi: 10.3390/pharmaceutics12111083 |
[16] | Janssen MCH , Koene S , de Laat P , Hemelaar P , Pickkers P , Spaans E , et al. The KHENERGY Study: Safety and Efficacy of KH176 in Mitochondrial m.3243A>G Spectrum Disorders. Clin Pharmacol Ther. (2019) ;105: (1):101–11. doi: 10.1002/cpt.1197 |
[17] | Ghezzi D , Sevrioukova I , Invernizzi F , Lamperti C , Mora M , D’Adamo P , et al. Severe X-Linked Mitochondrial Encephalomyopathy Associated with a Mutation in Apoptosis-Inducing Factor. Am J Hum Genet. (2010) ;86: (4):639–49. doi: 10.1016/j.ajhg.2010.03.002 |
[18] | Garone C , Donati MA , Sacchini M , Garcia-Diaz B , Bruno C , Calvo S , et al. Mitochondrial encephalomyopathy due to a novel mutation in ACAD9. JAMA Neurol. (2013) ;70: (9):1177–9. doi: 10.1001/jamaneurol.2013.3197 |
[19] | Gerards M , Van Den Bosch BJC , Danhauser K , Serre V , Van Weeghel M , Wanders RJA , et al. Riboflavin-responsive oxidative phosphorylation complex i deficiency caused by defective ACAD9: New function for an old gene. Brain. (2011) ;134: (1):210–9. doi: 10.1093/brain/awq273 |
[20] | Montini G , Malaventura C , Salviati L . Early Coenzyme Q10 Supplementation in Primary Coenzyme Q10 Deficiency. N Engl J Med. (2008) ;358: (26):2849–50. doi: 10.1056/nejmc0800582 |
[21] | Eroglu FK , Ozaltin F , Gönç N , Nalçacıoğlu H , Özçakar ZB , Yalnızoğlu D , et al. Response to Early Coenzyme Q10 Supplementation Is not Sustained in CoQ10 Deficiency Caused by CoQ2 Mutation. Pediatr Neurol. (2018) ;88: :71–4. doi: 10.1016/j.pediatrneurol.2018.07.008 |
[22] | Scalais E , Chafai R , Van Coster R , Bindl L , Nuttin C , Panagiotaraki C , et al. Early myoclonic epilepsy, hypertrophic cardiomyopathy and subsequently a nephrotic syndrome in a patient with CoQ10 deficiency caused by mutations in para-hydroxybenzoate-polyprenyl transferase (COQ2). Eur J Paediatr Neurol. (2013) ;17: (6):625–30. doi: 10.1016/j.ejpn.2013.05.013 |
[23] | Salviati L , Trevisson E , Hernandez MAR , Casarin A , Pertegato V , Doimo M , et al. Haploinsufficiency of COQ4 causes coenzyme Q10 deficiency. J Med Genet. (2012) ;49: (3):187–91. doi: 10.1136/jmedgenet-2011-100394 |
[24] | McWilliam CA , Ridout CK , Brown RM , McWilliam RC , Tolmie J , Brown GK . Pyruvate dehydrogenase E2 deficiency: A potentially treatable cause of episodic dystonia. Eur J Paediatr Neurol. (2010) ;14: (4):349–53. doi: 10.1016/j.ejpn.2009.11.001 |
[25] | Carrozzo R , Torraco A , Fiermonte G , Martinelli D , Di Nottia M , Rizza T , et al. Riboflavin responsive mitochondrial myopathy is a new phenotype of dihydrolipoamide dehydrogenase deficiency: The chaperon-like effect of vitamin B2. Mitochondrion. (2014) ;18: :49–57. doi: 10.1016/j.mito.2014.09.006 |
[26] | Quinonez SC , Leber SM , Martin DM , Thoene JG , Jirair K , Bedoyan MD . Leigh syndrome in a girl with a novel DLD mutation causing E3 deficiency. Pediatr Neurol 2013. (2013) ;48: (1):67–72. doi: 10.1016/j.pediatrneurol.2012.09.013.Leigh |
[27] | Haack TB , Jackson CB , Murayama K , Kremer LS , Schaller A , Kotzaeridou U , et al. Deficiency of ECHS1 causes mitochondrial encephalopathy with cardiac involvement. Ann Clin Transl Neurol. 2015;492-509. doi: 10.1002/acn3.189 |
[28] | Illsinger S , Korenke GC , Boesch S , Nocker M , Karall D , Nuoffer JM , et al. Paroxysmal and non-paroxysmal dystonia in 3 patients with biallelic ECHS1 variants: Expanding the neurological spectrum and therapeutic approaches. Eur J Med Genet. (2020) ;63: (11). doi: 10.1016/j.ejmg.2020.104046 |
[29] | Shayota BJ , Soler-Alfonso C , Bekheirnia MR , Mizerik E , Boyer SW , Xiao R , et al. Case report and novel treatment of an autosomal recessive Leigh syndrome caused by short-chain enoyl-CoA hydratase deficiency. Am J Med Genet Part A. (2019) ;179: (5):803–7. doi: 10.1002/ajmg.a.61074 |
[30] | Viscomi C , Burlina AB , Dweikat I , Savoiardo M , Lamperti C , Hildebrandt T , et al. Combined treatment with oral metronidazole and N-acetylcysteine is effective in ethylmalonic encephalopathy. Nat Med. (2010) ;16: (8):869–71. doi: 10.1038/nm.2188 |
[31] | Soler-Alfonso C , Enns GM , Koenig MK , Saavedra H , Bonfante-Mejia E , Northrup H . Identification of HIBCH gene mutations causing autosomal recessive Leigh syndrome: A gene involved in valine metabolism. Pediatr Neurol. (2015) ;52: (3):361–5. doi: 10.1016/j.pediatrneurol.2014.10.023 |
[32] | Kouga T , Takagi M , Miyauchi A , Shimbo H , Iai M , Yamashita S , et al. Japanese Leigh syndrome case treated with EPI-743. Brain Dev. (2018) ;40: (2):145–9. doi: 10.1016/j.braindev.2017.08.005 |
[33] | Chen Z , Zhao Z , Ye Q , Chen Y , Pan X , Sun B , et al. Mild clinical manifestation and unusual recovery upon coenzyme Q10 treatment in the first Chinese Leigh syndrome pedigree with mutation m.10197 G>A. Mol Med Rep. (2015) ;11: (3):1956–62. doi: 10.3892/mmr.2014.2911 |
[34] | Yoshinaga H , Ogino T , Ohtahara S , Sakuta R , Nonaka I , Horai S . A T-to-G Mutation at Nucleotide Pair 8993 in Mitochondrial DNA in a Patient With Leigh’s Syndrome. J Child Neurol. (1993) ;8: (2):129–33. doi: 10.1177/088307389300800204 |
[35] | Toth G, E. , Morava , Bene J , Selhorst JJ , Overmars H , Vreken P , et al. Carnitine-responsive carnitine insufficiency in a case of mtDNA 8993TbC mutation associated Leigh syndrome. J Inherit Metab Dis. (2001) ;24: (1993):421–2. doi: 10.1023/A:1010537527291 |
[36] | Balasubramaniam S , Lewis B , Mock DM , Said HM , Tarailo-Graovac M , Mattman A , et al. Leigh-Like Syndrome Due to Homoplasmic m.8993T>G Variant with Hypocitrullinemia and Unusual Biochemical Features Suggestive of Multiple Carboxylase Deficiency (MCD). JIMD Rep. (2016) ;33: :99–107. doi: 10.1007/8904 |
[37] | Fujii T , Ito M , Miyajima T , Okuno T . Dichloroacetate therapy in Leigh syndrome with a mitochondrial T8993C mutation. Pediatr Neurol. (2002) ;27: (1):58–61. doi: 10.1016/s0887-8994(02)00378-8 |
[38] | Buda P , Piekutowska-Abramczuk D , Karkuciñska-Więckowska A , Jurkiewicz E , Chełstowska S , Pajdowska M , et al. “Drop attacks” as first clinical symptoms in a child carrying MTTK m.8344A>G mutation. Folia Neuropathol. (2013) ;51: (4):347–54. doi: 10.5114/fn.2013.39726 |
[39] | Laugel V , This-Bernd V , Cormier-Daire V , Speeg-Schatz C , de Saint-Martin A , Fischbach M . Early-Onset Ophthalmoplegia in Leigh-Like Syndrome Due to NDUFV1 Mutations. Pediatr Neurol. (2007) ;36: (1):54–7. doi: 10.1016/j.pediatrneurol.2006.08.007 |
[40] | Giribaldi G , Doria-Lamba L , Biancheri R , Severino M , Rossi A , Santorelli FM , et al. Intermittent-relapsing pyruvate dehydrogenase complex deficiency: A case with clinical, biochemical, and neuroradiological reversibility. Dev Med Child Neurol. (2012) ;54: (5):472–6. doi: 10.1111/j.1469-8749.2011.04151.x |
[41] | Castiglioni C , Verrigni D , Okuma C , Diaz A , Alvarez K , Rizza T , et al. Pyruvate dehydrogenase deficiency presenting as isolated paroxysmal exercise induced dystonia successfully reversed with thiamine supplementation. Case report and mini-review. Eur J Paediatr Neurol. (2015) ;19: (5):497–503. doi: 10.1016/j.ejpn.2015.04.008 |
[42] | Lee EH , Ahn MS , Hwang JS , Ryu KH , Kim SJ , Kim SH . A Korean female patient with thiamine-responsive pyruvate dehydrogenase complex deficiency due to a novel point mutation (Y161C) in the PDHA1 gene. J Korean Med Sci. (2006) ;21: (5):800–4. doi: 10.3346/jkms.2006.21.5.800 |
[43] | Wexler ID , Hemalatha SG , McConnell J , Buist NRM , Dahl HHM , Berry SA , et al. Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diets: Studies in patients with identical mutations. Neurology. (1997) ;49: (6):1655–61. doi: 10.1212/WNL.49.6.1655 |
[44] | Satogami K , Takahashi S , Kose A , Shinosaki K . Schizophrenia-like symptoms in a patient with Leigh syndrome. Asian J Psychiatr. (2017) ;25: :249–50. doi: 10.1016/j.ajp.2016.12.012 |
[45] | van Dongen S , Brown RM , Brown GK , Thorburn DR , Boneh A . Thiamine-Responsive and Non-responsive Patients with PDHC-E1 Deficiency: A Retrospective Assessment. JIMD Rep. (2015) ;15: :13–27. doi: 10.1007/8904 |
[46] | Debray FG , Lambert M , Gagne R , Maranda B , Laframboise R , MacKay N , et al. Pyruvate dehydrogenase deficiency presenting as intermittent isolated acute ataxia. Neuropediatrics. (2008) ;39: (1):20–3. doi: 10.1055/s-2008-1077084 |
[47] | Briones P , Lopez MJ , Meirleir De L , Ribes A , Rodds M , Peris M , et al. Leigh syndrome due to pyruvate dehydrogenase Ela deficiency (point mutation R263G) in a Spanish boy. J Inher Metab Dis. (1996) ;19: :795–6. doi: 10.1007/BF01799177 |
[48] | Rubio-Gozalbo ME , Heerschap A , Trijbels JMF , Meirleir De L , Thijssen HOM , Smeitink JAM . Proton MR spectroscopy in a child with pyruvate dehydrogenase complex deficiency. Magn Reson Imaging. (1999) ;17: (6):939–44. doi: 10.1016/S0730-725X(99)00002-8 |
[49] | DeBrosse SD , Okajima K , Zhang S , Nakouzi G , Schmotzer CL , Lusk-Kopp M . Spectrum of neurological and survival outcomes in pyruvate dehydrogenase complex (PDC) deficiency: Lack of correlation with genotype. Mol Genet Metab. (2012) ;107: (3):394–402. doi: 10.1016/j.ymgme.2012.09.001 |
[50] | Steller J , Gargus JJ , Gibbs LH , Hasso AN , Kimonis VE . Mild phenotype in a male with pyruvate dehydrogenase complex deficiency associated with novel hemizygous in-frame duplication of the e1α subunit gene (PDHA1). Neuropediatrics. (2014) ;45: (1):56–60. doi: 10.1055/s-0033-1341601 |
[51] | El-Gharbawy AH , Boney A , Young SP , Kishnani PS . Follow-up of a child with pyruvate dehydrogenase deficiency on a less restrictive ketogenic diet. Mol Genet Metab. (2011) ;102: (2):214–5. doi: 10.1016/j.ymgme.2010.11.001 |
[52] | Kara B , Genç HM , Uyur-Yalçın E , Sakarya-Güneş A , Topçu U , Mülayim S , et al. Pyruvate dehydrogenase-E1α deficiency presenting as recurrent acute proximal muscle weakness of upper and lower extremities in an 8-year-old boy. Neuromuscul Disord. (2017) ;27: (1):94–7. doi: 10.1016/j.nmd.2016.11.001 |
[53] | Singhi P , Meirleir De L , Lissens W , Singhi S , Saini AG . Pyruvate Dehydrogenase-E1a Deficiency Presenting as Recurrent Demyelination: An Unusual Presentation and a Novel Mutation. JIMD Rep. (2012) ;4: :107–11. doi: 10.1007/8904 |
[54] | Marsac C , Benelli C , Desguerre I , Diry M , Fouque F , De Meirleir L , et al. Biochemical and genetic studies of four patients with pyruvate dehydrogenase E1α deficiency. Hum Genet. (1997) ;99: (6):785–92. doi: 10.1007/s004390050449 |
[55] | João Silva M , Pinheiro A , Eusébio F , Gaspar A , Tavares De Almeida I , Rivera I . Pyruvate dehydrogenase deficiency: Identification of a novel mutation in the PDHA1 gene which responds to amino acid supplementation. Eur J Pediatr. (2009) ;168: (1):17–22. doi: 10.1007/s00431-008-0700-7 |
[56] | Koga Y , Povalko N , Katayama K , Kakimoto N , Matsuishi T , Naito E , et al. Beneficial effect of pyruvate therapy on Leigh syndrome due to a novel mutation in PDH E1α gene. Brain Dev. (2012) ;34: (2):87–91. doi: 10.1016/j.braindev.2011.03.003/ |
[57] | Quintana E , Mayr JA , García Silva MT , Font A , Tortoledo MA , Moliner S , et al. PDH E1β deficiency with novel mutations in two patients with Leigh syndrome. J Inherit Metab Dis. (2009) ;32: (1):339–43. doi: 10.1007/s10545-009-1343-1 |
[58] | Schiff M , Miné M , Brivet M , Marsac C , Elmaleh-Bergés M , Evrard P , et al. Leigh’s disease due to a new mutation in the PDHX gene. Ann Neurol. (2006) ;59: (4):709–14. doi: 10.1002/ana.20818 |
[59] | Joshi CN , Greenberg CR , Mhanni AA , Salman MS . Ketogenic Diet in Alpers-Huttenlocher Syndrome. Pediatr Neurol. (2009) ;40: (4):314–6. doi: 10.1016/j.pediatrneurol.2008.10.023 |
[60] | Martikainen MH , Päivärinta M , Jääskeläinen S , Majamaa K . Successful treatment of POLG-related mitochondrial epilepsy with antiepileptic drugs and low glycaemic index diet. Vol. 14, Epileptic Disorders. Epileptic Disord; 2012. pp. 438-41. doi: 10.1684/epd.2012.0543 |
[61] | Cardenas JF , Amato RS . Compound heterozygous polymerase gamma gene mutation in a patient with alpers disease. Semin Pediatr Neurol. (2010) ;17: (1):62–4. doi: 10.1016/j.spen.2010.02.012 |
[62] | Hasselmann O , Blau N , Ramaekers VT , Quadros E V , Sequeira JM , Weissert M . Cerebral folate deficiency and CNS inflammatory markers in Alpers disease. Mol Genet Metab. (2010) ;99: (1):58–61. doi: 10.1016/j.ymgme.2009.08.005 |
[63] | Gerards M , Kamps R , Van Oevelen J , Boesten I , Jongen E , De Koning B , et al. Exome sequencing reveals a novel Moroccan founder mutation in SLC19A3 as a new cause of early-childhood fatal Leigh syndrome. Brain. (2013) ;136: (3):882–90. doi: 10.1093/brain/awt013 |
[64] | Kono S , Miyajima H , Yoshida K , Togawa A , Shirakawa K , Suzuki H . Mutations in a Thiamine-Transporter Gene and Wernicke’s-like Encephalopathy. N Engl J Med. (2009) ;360: (17):1792–24. doi: 10.1056/nejmc0809100 |
[65] | Tabarki B , Alfadhel M , Alshahwan S , Hundallah K , Alshafi S , Alhashem A . Treatment of biotin-responsive basal ganglia disease: Open comparative study between the combination of biotin plus thiamine versus thiamine alone. Eur J Paediatr Neurol. (2015) ;19: (5):547–52. doi: 10.1016/j.ejpn.2015.05.008 |
[66] | Distelmaier F , Huppke P , Pieperhoff P , Amunts K , Schaper J , Morava E , et al. Biotin-Responsive Basal Ganglia Disease: A Treatable Differential Diagnosis of Leigh Syndrome. JIMD Rep. (2013) ;13: :53–7. doi: 10.1007/8904_2013_271 |
[67] | Fassone E , Wedatilake Y , DeVile CJ , Chong WK , Carr LJ , Rahman S . Treatable Leigh-like encephalopathy presenting in adolescence. BMJ Case Rep.;2013. doi: 10.1136/bcr-2013-200838 |
[68] | Ortigoza-Escobar JD , Alfadhel M , Molero-Luis M , Darin N , Spiegel R , de Coo IF , et al. Thiamine deficiency in childhood with attention to genetic causes: Survival and outcome predictors. Ann Neurol. (2017) ;82: (3):317–30. doi: 10.1002/ana.24998 |
[69] | Lee JS , Yoo T , Lee M , Lee Y , Jeon E , Kim SY , et al. Genetic heterogeneity in Leigh syndrome: Highlighting treatable and novel genetic causes. Clin Genet. (2020) ;97: (4):586–94. doi: 10.1111/cge.13713 |
[70] | Riley LG , Cowley MJ , Gayevskiy V , Roscioli T , Thorburn DR , Prelog K , et al. A SLC39A8 variant causes manganese deficiency, and glycosylation and mitochondrial disorders. J Inherit Metab Dis. (2017) ;40: (2):261–9. doi: 10.1007/s10545-016-0010-6 |
[71] | Theunissen TEJ , Gerards M , Hellebrekers DMEI , van Tienen FH , Kamps R , Sallevelt SCEH , et al. Selection and characterization of palmitic acid responsive patients with an OXPHOS complex i defect. Front Mol Neurosci. (2017) ;10: (October):1–12. doi: 10.3389/fnmol.2017.00336 |
[72] | Invernizzi F , Panteghini C , Chiapparini L , Moroni I , Nardocci N , Garavaglia B , et al. Thiamine-responsive disease due to mutation of tpk1: Importance of avoiding misdiagnosis. Neurology. (2017) ;89: :870–1. doi: 10.1212/WNL.0000000000004270 |
[73] | Banka S , de Goede C , Yue WW , Morris AAM , von Bremen B , Chandler KE , et al. Expanding the clinical and molecular spectrum of thiamine pyrophosphokinase deficiency: A treatable neurological disorder caused by TPK1 mutations. Mol Genet Metab. (2014) ;113: (4):301–6. doi: 10.1016/j.ymgme.2014.09.010 |
[74] | Debs R , Depienne C , Rastetter A , Bellanger A , Degos B , Galanaud D , et al. Biotin-responsive basal ganglia disease in ethnic Europeans with novel SLC19A3 mutations. Arch Neurol. (2010) ;67: (1):126–30. doi: 10.1001/archneu-rol.2009.293 |
[75] | Yamada K , Miura K , Hara K , Suzuki M , Nakanishi K , Kumagai T , et al. A wide spectrum of clinical and brain MRI findings in patients with SLC19A3 mutations. BMC Med Genet. (2010) ;11: (1). doi: 10.1186/1471-2350-11-171 |
[76] | Kevelam SH , Bugiani M , Salomons GS , Feigenbaum A , Blaser S , Prasad C , et al. Exome sequencing reveals mutated SLC19A3 in patients with an early-infantile, lethal encephalopathy. Brain. (2013) ;136: (5):1534–43. doi: 10.1093/brain/awt054 |
[77] | Baertling F , Rodenburg RJ , Schaper J , Smeitink JA , Koopman WJH , Mayatepek E , et al. A guide to diagnosis and treatment of Leigh syndrome. J Neurol Neurosurg Psychiatry. (2014) ;85: (3):257–65. doi: 10.1136/jnnp-2012-304426 |
[78] | Prasad C , Rupar T , Prasad AN . Pyruvate dehydrogenase deficiency and epilepsy. Brain Dev. (2011) ;33: (10):856–65. doi: 10.1016/j.braindev.2011.08.003 |
[79] | Fujii T , Nozaki F , Saito K , Hayashi A , Nishigaki Y , Murayama K , et al. Efficacy of pyruvate therapy in patients with mitochondrial disease: A semi-quantitative clinical evaluation study. Mol Genet Metab. (2014) ;112: (2):133–8. doi: 10.1016/j.ymgme.2014.04.008 |
[80] | Bonnet S , Archer SL , Allalunis-Turner J , Haromy A , Beaulieu C , Thompson R , et al. A Mitochondria-K+Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth. Cancer Cell. (2007) ;11: (1):37–51. doi: 10.1016/j.ccr.2006.10.020 |
[81] | Spruijt L , Naviaux RK , McGowan KA , Nyhan WL , Sheean G , Haas RH , et al. Nerve conduction changes in patients with mitochondrial diseases treated with dichloroacetate. Muscle and Nerve. (2001) ;24: (7):916–24. doi: 10.1002/mus.1089 |
[82] | Gorman GS , Chinnery PF , Dimauro S , Hirano M , Koga Y , Mcfarland R , et al. Mitochondrial diseases. Nat Publ Gr. (2016) ;2: :1–23. doi: 10.1038/nrdp.2016.80 |
[83] | Tarnopolsky MA . The mitochondrial cocktail: Rationale for combined nutraceutical therapy in mitochondrial cytopathies. Adv Drug Deliv Rev. (2008) ;60: (13-14):1561–7. doi: 10.1016/j.addr.2008.05.001 |
[84] | Danis D , Brennerova K , Skopkova M , Kurdiova T , Ukropec J , Stanik J , et al. Mutations in SURF1 are important genetic causes of Leigh syndrome in Slovak patients. Endocr Regul. (2018) ;52: (2):110–8. doi: 10.2478/enr-2018-0013 |
[85] | Huang X , Bedoyan JK , Demirbas D , Harris DJ , Miron A , Edelheit S , et al. Succinyl-CoA synthetase (SUCLA2) deficiency in two siblings with impaired activity of other mitochondrial oxidative enzymes in skeletal muscle without mitochondrial DNA depletion. Mol Genet Metab. (2017) ;120: (3):213–22. doi: 10.1016/j.ymgme.2016.11.005 |
[86] | Nebert DW , Liu Z . SLC39A8 gene encoding a metal ion transporter: Discovery and bench to bedside. Hum Genomics. (2019) ;13: :1–21. doi: 10.1186/s40246-019-0233-3 |
[87] | Pastore A , Petrillo S , Tozzi G , Carrozzo R , Martinelli D , Dionisi-Vici C , et al. Glutathione: A redox signature in monitoring EPI-743 therapy in children with mitochondrial encephalomyopathies. Mol Genet Metab. (2013) ;109: (2):208–14. doi: 10.1016/j.ymgme.2013.03.011 |


