
Intrahepatic Cholestasis Is a Clinically Significant Feature Associated with Natural History of X-Linked Myotubular Myopathy (XLMTM): A Case Series and Biopsy Report
Abstract
X-linked myotubular myopathy (XLMTM) is a rare, life-threatening congenital myopathy characterized by profound skeletal muscle weakness, respiratory distress, and motor dysfunction. However, pathology is not limited to muscle and can be associated with life-threatening hepatic peliosis. Hepatobiliary disease has been reported in up to 17% of XLMTM patients but has not been extensively characterized. We report on five XLMTM patients who experienced intrahepatic cholestasis in their disease natural history, illustrating the need to further investigate these manifestations. These patients shared presentations that included pruritus, hypertransaminemia, and hyperbilirubinemia with normal gamma-glutamyl transferase, following infection or vaccination. Three patients who had genetic testing showed no evidence of genetic mutations associated with familial cholestasis. In one patient, progression to cirrhotic, decompensated liver disease occurred. Further investigations into the molecular pathomechanism underpinning these clinical observations in XLMTM patients will be important for informing patient care.
INTRODUCTION
X-linked myotubular myopathy (XLMTM) is a rare, life-threatening congenital myopathy caused by mutations in MTM1, leading to absent or insufficient functional myotubularin [1], a protein required for normal development, maturation, and maintenance of muscle cells [2]. XLMTM affects 1 in 40,000–50,000 newborn males [3]. While XLMTM is most notably characterized by profound muscle weakness, resulting in severe respiratory distress at birth in approximately 80% of patients [3–6], myotubularin is ubiquitously expressed and its functions outside of skeletal muscle are unclear.
Hepatic peliosis, a life-threatening vascular lesion characterized by blood-filled, cystic cavities, has been the primary liver abnormality reported in XLMTM, affecting 5–10% of patients [5, 7]. Hepatobiliary disease has been reported in 7–17% of XLMTM patients [4, 5, 7, 8] and can manifest as jaundice, cholelithiasis, pruritus, hepatomegaly, and elevated transaminases, but has not been fully characterized or associated with clinically significant morbidity or mortality in these patients.
In 2020, three study participants with XLMTM treated with AT132 (Astellas Gene Therapies, formerly Audentes Therapeutics), an investigational AAV8 gene replacement therapy in the ASPIRO clinical study (NCT03199469) died following serious adverse events related to decompensated liver function associated with cholestasis. They were among the heavier participants in the study, receiving among the highest total doses, and experienced rapidly worsening liver dysfunction within 3-4 weeks following treatment and ultimately died [9]. Given that these patients, and more than half of ASPIRO participants, had evidence of pre-existing hepatobiliary disease [9], characterizing disease phenotypes related to myotubularin deficiency in the liver is a compelling area of interest.
CASE REPORTS
We present the cases of five patients with genetically confirmed XLMTM who were not treated with any investigational agent and demonstrated clinically significant intrahepatic cholestasis with elevated serum bile acids, significant hyperbilirubinemia with normal gamma-glutamyl transferase (GGT), fluctuating hypertransaminemia, presenting initially with pruritus and jaundice (Table 1). Muscle biopsies performed on Patients 1–4 (muscle biopsy was not performed on Patient 5) were consistent with XLMTM muscle pathology, including marked myofiber smallness, organelle accumulation in the center of fibers, and the presence of centrally nucleated fibers in the absence of inflammation or myofiber degeneration [10].
Table 1
Summary of clinical presentations
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | |
| MTM1 mutation | c.1262G > A, p.R421Q | c.1420C > T, p.R474* | 79 kb deletion (chrX:149761067-149840078, exons 3–14) | c.1644 + 1G > A | c.1088_1089del p.K363Sfs*14 |
| Age at initial presentation of cholestasis | 7.5 months | 8 months | 14 months | 16 months | 5 months |
| Laboratory values at initial presentation (BL) and/or maximum level (max) | |||||
| Maximum serum bile acids | 310.6μmol/L | 144.9μmol/L | 345.5μmol/L | 56.7μmol/L | 180.0μmol/L |
| 30×ULN | 18×ULN | 35×ULN | 6×ULN | 18×ULN | |
| Total bilirubin | BL and max: 5.6 mg/dL | BL: 11.9 mg/dL | BL and max: 2.8 mg/dL | BL and max: 4.0 mg/dL | BL: 2.7 mg/dL |
| Max (1 wk): 12.9 mg/dL | Max (21.5 wks): 15.3 mg/dL | ||||
| Direct bilirubin | BL and max: 4.2 mg/dL | BL and max: 11.2 mg/dL | BL and max: 2.1 mg/dL | BL and max: 3.9 mg/dL | BL: 1.5 mg/dL |
| Max (61 wks): 9.5 mg/dL | |||||
| Aspartate aminotransferase (AST) | Normal range | BL: normal range | BL: normal range | BL and max: 1.2×ULN | BL: 2×ULN |
| Max (18.4 wks): 28×ULN | Max (10 wks): 2×ULN | Max (21.5 wks): 26×ULN | |||
| Alanine aminotransferase (ALT) | BL and max: 1.3×ULN | BL: normal range | BL: 2×ULN | BL and max: 1.2×ULN | BL: 2×ULN |
| Max (18.4 wks): 39×ULN | Max (10 wks): 4×ULN | Max (21.5 wks): 24×ULN | |||
| Gamma-glutamyl transferase (GGT) | Normal range | BL: 2×ULN Max (18.4 wks): 7×ULN | BL: 1.4×ULN Max (0.5 wk): 1.7×ULN | BL and max: 1.1×ULN | Normal range |
| Max (at symptom resolution): 1.2×ULN | |||||
| Alpha-1-antitrypsin | Normal range | Normal range | Normal range | Normal range | Normal range |
| Status | Resolution of cholestasis at age 10 months; death, age 12 months | Resolution of cholestasis | Resolution of cholestasis | Resolution of cholestasis | Liver failure due to fibrosis |
ULN: upper limit of normal; wk: week.
Patient 1
Patient 1 had a pathologic MTM1 missense mutation (c.1262G > A, p.R421Q) [11]. He required nighttime noninvasive ventilation starting at the age of 10 months due to pulmonary atelectasis. He presented with neonatal hyperbilirubinemia, treated with phototherapy for 12 hours daily but not exchange transfusion, and resolved at day 6. Maximum total bilirubin was 16 mg/dl (direct, 0.95 mg/dl). ABO/Rh incompatibility was ruled out. He presented at age 7.5 months with jaundice, intense pruritus, choluria and transient acholic stools without hepatomegaly or splenomegaly following a respiratory infection in the preceding days. Concurrent with onset of cholestasis, platelets decreased to 96,000–120,000/μL possibly related to infection as the platelet count increased after the infection resolved. PCR testing for adenovirus, enterovirus and echovirus and urine culture were all negative.
At presentation, bilirubin and total serum bile acids were elevated, with near-normal transaminase, and normal GGT and prothrombin time and international normalized ratio (PT/INR) values. Abdominal ultrasonography showed normal liver size and echogenicity, no dilation in the biliary tree, and gallbladder distension without overt sludge or cholelithiasis. The hepatic arterial resistance index was slightly elevated (0.82) without evidence of portal hypertension. Alpha-1-antitrypsin deficiency and inborn errors of bile acid synthesis were ruled out, but genetic testing for known familial cholestatic disease was not performed.
The patient responded to treatment with ursodeoxycholic acid (UDCA) 17 mg/kg/day, fat-soluble vitamins, and antihistamines, resulting in symptomatic and laboratory resolution at age 10 months. He died at age 12 months due to cardiopulmonary failure.
Patient 2
Patient 2 had a pathogenic MTM1 nonsense mutation (c.1420C > T, p.R474*) [11]. He required 24-hour mechanical ventilation support and had a gastrostomy at 3 months old. He did not present with neonatal hyperbilirubinemia and initially presented with cholestasis at age 8 months and experienced recurrence four months later (Fig. 1A). He had a longstanding history of intense pruritus and presented initially with jaundice and choluria without acholic stools, which started days after respiratory congestion that was treated symptomatically without antibiotics or antipyretics. Bilirubin and total serum bile acids were elevated (Table 1), with unremarkable transaminase, GGT, and PT/INR values. Usual causes of cholestasis were excluded (alpha-1-antitrypsin deficiency, hepatotropic virus, inborn metabolic disorders, thyroid dysfunction). There were no mutations in genes related to familial cholestatic disorders (ABCB11, ABCB4, ATP8B1, CLDN1, DCDC2, MYO5B, NR1H4, TJP2, VPS33B), including progressive familial intrahepatic cholestasis (PFIC).
Fig. 1
Time course of laboratory findings and clinical events in select patients. Summary of liver function tests and select events in the clinical courses starting with initial symptomatic presentation (denoted as Week 0) in (A) Patient 2, (B) Patient 3, and (C) Patient 5. Total and direct bilirubin levels are shown as mg/dl on the left vertical axis. ALT, AST, and GGT levels are shown as multiples of their respective ULN on the right vertical axis. ALT: alanine aminotransferase; AST: aspartate aminotransferase; GGT: gamma-glutamyl transferase; INR: international normalized ratio; PT: prothrombin time; UDCA: ursodeoxycholic acid; ULN: upper limit of normal.
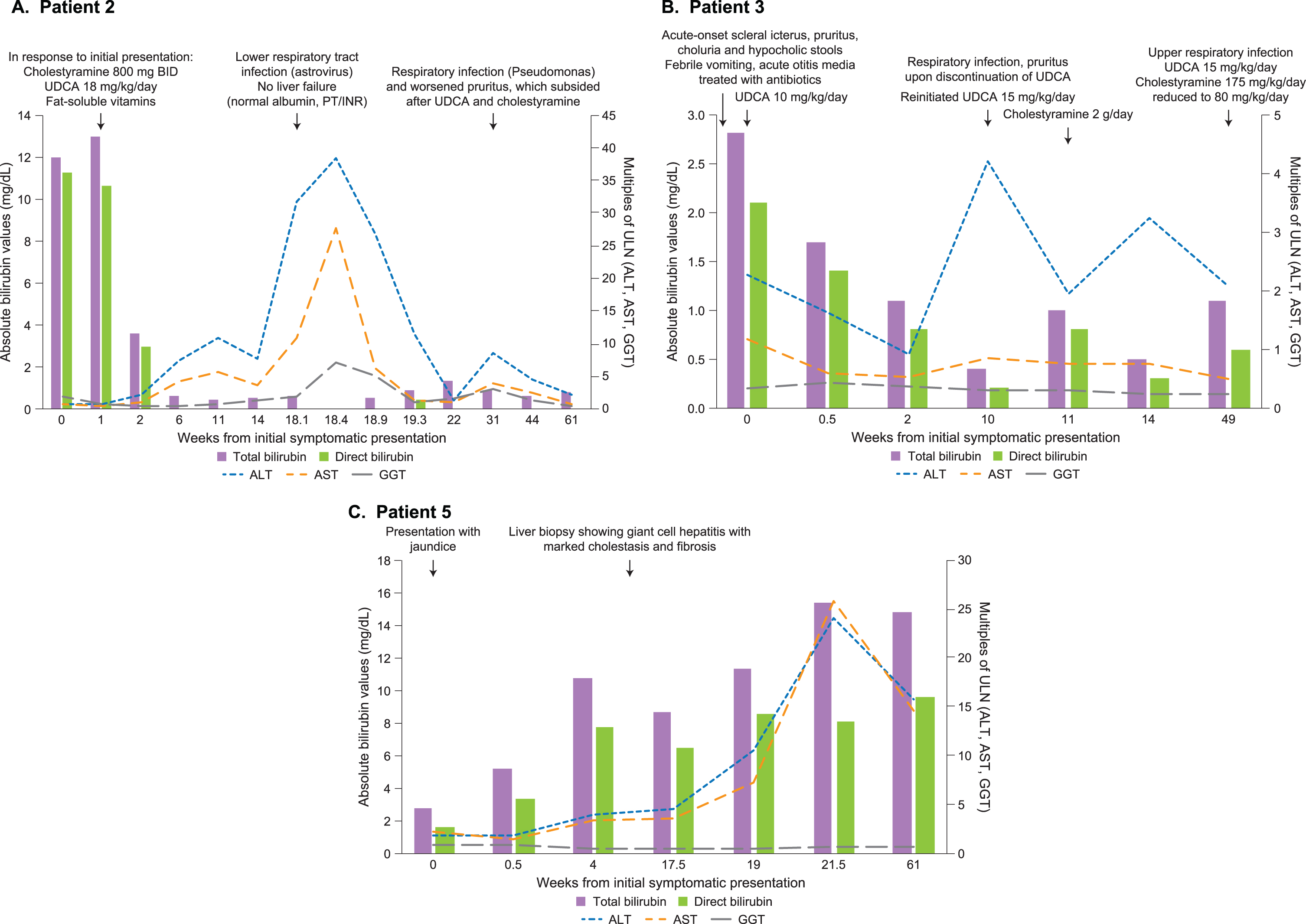
Abdominal ultrasonography showed moderate hepatomegaly with normal echogenicity, moderate dilation of the distal intrahepatic bile duct (1.8 mm), normal extrahepatic duct, adenopathy in the hepatic hilum, and moderate signs of periportal edema. Magnetic resonance cholangiogram revealed moderate signs of periportal edema, apparently normal hepatic parenchyma, distended, empty gallbladder, and discrete, non-pathologic prominence of the intrahepatic bile duct. His symptoms and laboratory abnormalities improved with cholestyramine 800 mg every 12 hours, UDCA 18 mg/kg/day, and fat-soluble vitamins; he was discharged 16 days after initial ultrasound following symptomatic and laboratory resolution.
Four months later, he presented with febrile lower respiratory tract infection and was treated with intravenous gentamicin. Laboratory evaluation at admission showed elevated transaminases (ALT > 30×ULN, AST > 10×ULN) with normal bilirubin, albumin, and PT/INR values. Over the next 30 days, transaminase levels spontaneously decreased to normal ranges.
Three months later, he presented with a Pseudomonas respiratory infection (treated with ciprofloxacin and colistin) and elevated transaminases (ALT > 8×ULN, AST > 3×ULN), normal GGT and bilirubin, and worsened pruritus. Following treatment with UDCA and cholestyramine, pruritus subsided and transaminases normalized. The positive response to UDCA and cholestyramine suggests the pruritus was likely due to hepatic causes. One month after presenting with respiratory infection, the patient remained on cholestyramine 2.4 g/day and was stable.
Patient 3
Patient 3 was a genetically confirmed XLMTM patient with a pathogenic 79 kb deletion in MTM1, spanning exons 3–14 [11]. He required 24-hour mechanical ventilation support and gastrostomy. He did not present with neonatal hyperbilirubinemia. At 14 months of age, he presented with acute-onset scleral icterus, pruritus, choluria, and hypocholic stools. Two days prior to onset, he experienced febrile vomiting and acute otitis media, which was treated with oral amoxicillin. At admission, total serum bile acids and bilirubin levels were elevated with clinically unremarkable transaminase and GGT values (Fig. 1B and Table 1). Four days after initiating UDCA 10 mg/kg/day, total/direct bilirubin levels decreased. Viral serology was negative for hepatitis A (IgM/IgG), hepatitis B surface antigen, hepatitis C (IgG), cytomegalovirus (IgG), Epstein-Barr virus (IgG/IgM), adenovirus, parvovirus B19 and enterovirus (PCR). Alpha-1-antitrypsin was within normal range and normal genotype. There were no mutations in genes related to familial cholestatic disorders including PFIC (ABCB11, ABCB4, ATP8B1, CLDN1, DCDC2, MYO5B, NR1H4, TJP2, VPS33B).
Abdominal ultrasonography with elastography showed diffuse hyperechogenicity of liver parenchyma without hepatomegaly, hypertrophic intrahepatic branches of the hepatic artery, mild dilation of intrahepatic bile duct without dilation of extrahepatic duct, and increased stiffness (mean [SD] 14.5 [1.3] KPa; mean healthy value, 2.1 kPa [12]). Treatment was continued with UDCA 8 mg/kg/day until laboratory and symptomatic resolution of cholestasis.
Two months later, he presented again with pruritus following discontinuation of UDCA and respiratory infection treated with oral amoxicillin. Upon admission, laboratory tests showed mild hypertransaminemia and elevated bile acids (33×ULN) with normal bilirubin and GGT. UDCA 15 mg/kg/day was reinitiated with cholestyramine 2 g/day due to persistent pruritus (cholestyramine later discontinued due to normochloremic metabolic acidosis).
Ten months later, he presented with severe pruritus following an upper respiratory infection and hypocholic stools. He was treated with UDCA 15 mg/kg/day and cholestyramine 175 mg/kg/day (reduced two weeks later to 80 mg/kg/day due to normochloremic metabolic acidosis) until laboratory and symptomatic resolution of cholestasis.
Patient 4
Patient 4 had an MTM1 missense mutation (c.1644 + 1G > A) and required mechanical ventilation support 20–24 hours daily and gastrostomy. He did not present with neonatal hyperbilirubinemia. He presented at age 16 months with jaundice, pruritus and choluria, without acholic stools or hepatomegaly after pneumococcal (two weeks prior) and measles, mumps, rubella (four weeks prior) vaccinations. Bilirubin and total serum bile acids were significantly elevated with unremarkable ALT, AST, and GGT values (Table 1). Alpha-1 antitrypsin was within normal range and serological testing was negative for hepatitis A/B/C viruses, cytomegalovirus, Epstein-Barr virus and HSV-1/2.
Ultrasonography revealed a lesion in the right hepatic lobe, which could correspond to a hemangioma given the patient’s age and sonographic characteristics, which would not be consistent with peliosis. Genetic evaluation for familial cholestasis was not conducted.
The patient was treated with UDCA 15 mg/kg/day and hydroxyzine 1 mg/kg/day until laboratory and symptomatic resolution.
Patient 5
Patient 5 had a pathogenic frameshift deletion in MTM1 (c.1088_1089del, p.K363Sfs*14) [11]. He required invasive mechanical ventilation since birth. He presented with jaundice at 5 months of age but did not present with neonatal hyperbilirubinemia. There was no known infectious trigger or history of medications associated with cholestatic liver injury.
Total serum bile acids and bilirubin were elevated while transaminases were modestly elevated (Table 1, Fig. 1C). Laboratory findings over the following 14 months showed consistently elevated direct bilirubin and transaminases, hypoalbuminemia, and normal GGT, alpha-1-antitrypsin and PT/INR (PT/INR data not available after 12 months of age). Whole exome sequencing excluded genetic mutations associated with cholestatic or bile acid synthesis disorders. The patient remains hospitalized with end-stage liver failure since the age of 7 months.
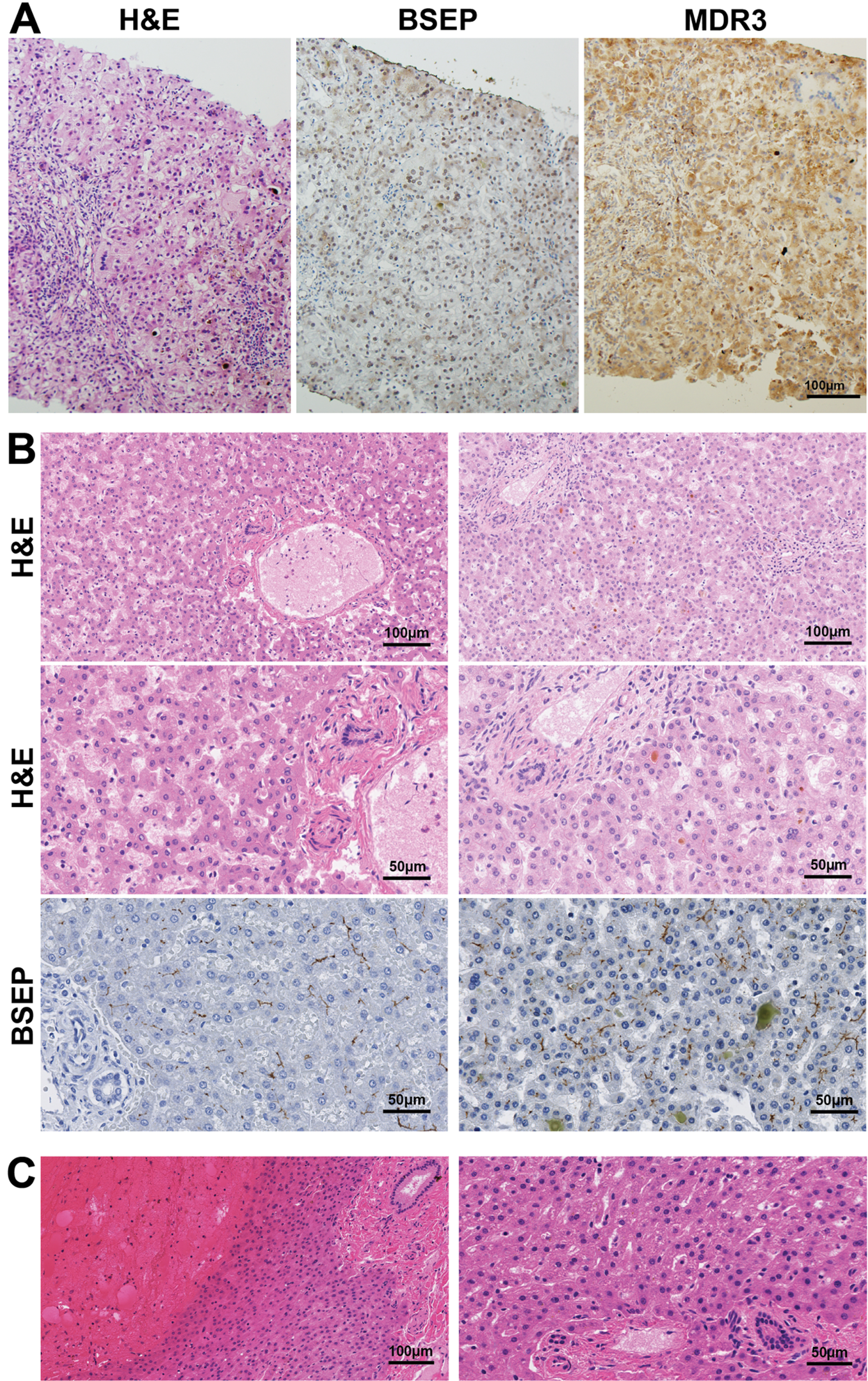
Liver biopsy with hematoxylin and eosin (H&E) staining revealed giant cell hepatitis and cirrhosis consistent with severe intrahepatic cholestasis including canalicular bile plugs, accumulation of bile material within hepatocytes, portal field fibrosis, and focal inflammation (Fig. 2A). Cytokeratin 7 staining showed unremarkable bile ducts without inflammation. There was no evidence of hepatic peliosis or other vascular abnormalities. Staining for bile salt export pump (BSEP) and multi-drug resistant protein 3 (MDR3) showed loss of canalicular expression of both (Fig. 2A), likely due to a loss of canalicular architecture from cirrhosis.
Fig. 2
Liver histopathology in XLMTM. (A) H&E-stained liver tissue from Patient 5 in this series revealed findings consistent with severe intrahepatic cholestasis including canalicular bile plugs, accumulation of bile material within hepatocytes, giant cell transformation, portal field fibrosis and focal inflammation. Staining for bile salt export pump (BSEP) and multi-drug resistant protein 3 (MDR3) showed loss of canalicular expression of both, likely due to a loss of canalicular architecture from cirrhosis. (B) Evaluation of autopsy livers from XLMTM patients (available through the Congenital Muscle Disease Tissue Repository) illustrates variable histopathology in the livers of XLMTM patients. Hematoxylin and eosin (H&E) and bile salt export protein (BSEP) staining of samples shows normal liver histopathology (left column), compared with one patient who showed intracanalicular bile plugs consistent with cholestasis (right column). (C) H&E-stained slides from an XLMTM patient’s liver at autopsy that displayed acute liver hemorrhage, presumably as a complication of hepatic peliosis. The high-magnification image (right) illustrates appropriate liver structure and no cholestatic changes in areas that are not involved in the hemorrhage. BSEP and MDR3 stains on this specimen were uninterpretable due to tissue quality.
Histopathological review of liver tissue from untreated XLMTM patients
To better establish the histological features of untreated XLMTM patients’ livers, we evaluated five autopsy liver samples from XLMTM patients in the Congenital Muscle Disease Tissue Repository (CMDTR, a tissue bank comprised of samples from patients with congenital myopathies), unrelated to Patients 1–5 described in detail above. These samples reveal normal liver histology (accounting for variable autolytic changes) in four patients (Fig. 2B) and hepatic parenchymal hemorrhage consistent with peliosis in one patient (Fig. 2C).
In one additional untreated XLMTM patient with cholestasis, histologic studies showed bile plugs and increased bile within and outside of hepatocytes, providing evidence of mild and potentially reversible intrahepatic cholestasis without evidence of more severe or irreversible changes, such as giant cell transformation, bile ductular proliferation, or fibrosis (Fig. 2B). This patient’s death was associated with intracranial hemorrhage in the context of vitamin K deficiency likely related to otherwise subclinical intrahepatic cholestasis that predated his acute hemorrhage [13]. Immunostaining of liver samples from five XLMTM non-cholestatic patients, including one with intraparenchymal liver hemorrhage, revealed appropriate canalicular staining of BSEP and MDR3 (Fig. 2B), mutations of which are responsible for PFIC types 2 and 3, respectively [14]. The remaining sample could not be evaluated for BSEP or MDR3 due to poor tissue quality.
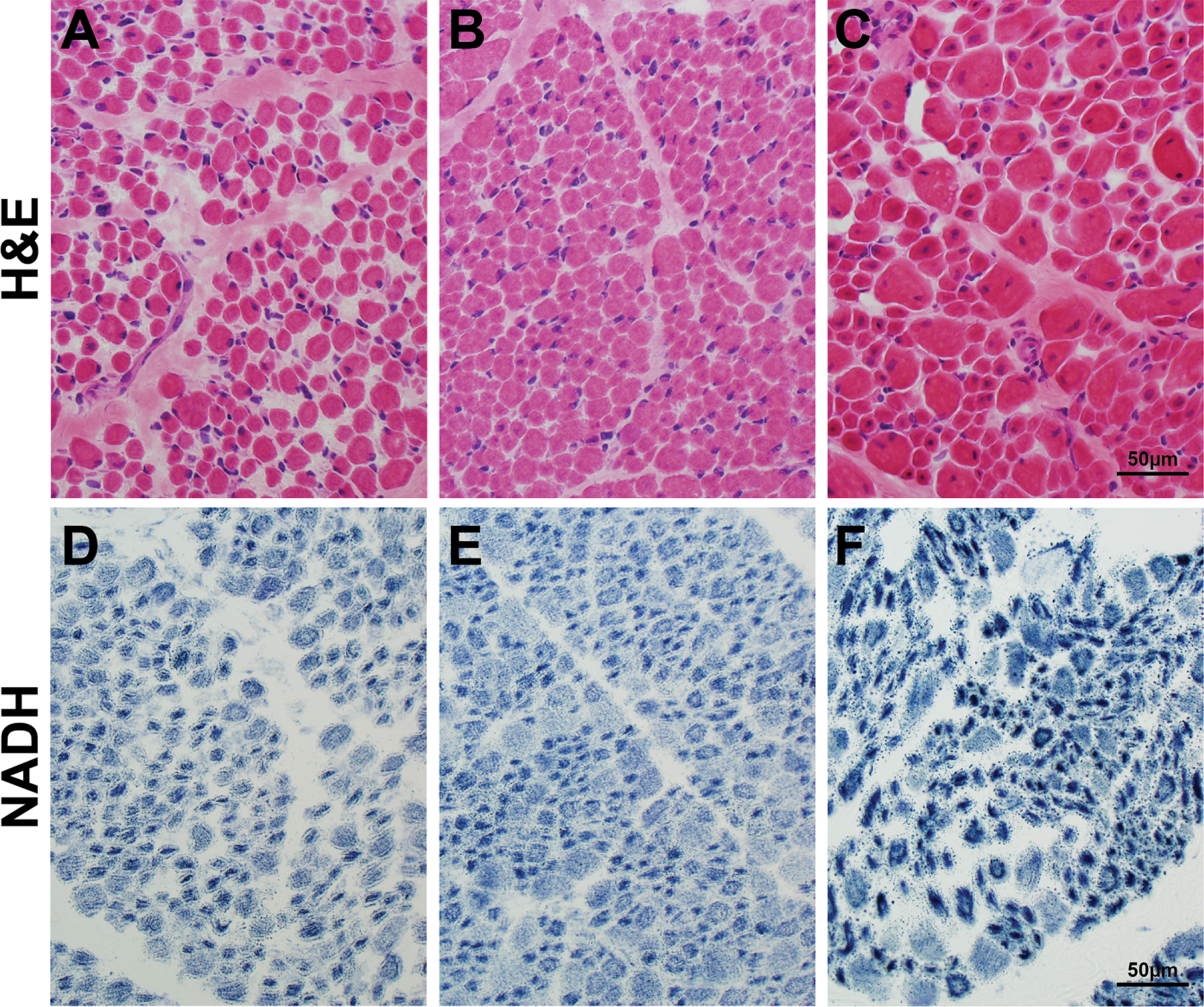
Muscle samples from the CMDTR from XLMTM patients without cholestasis (Fig. 3A, 3D), with cholestasis (Fig. 3B, 3E), and with peliosis (Fig. 3C, 3F) all showed characteristic features of XLMTM muscle pathology, and no additional pathology in patients with liver dysfunction was observed in the muscle tissue.
Fig. 3
Muscle histopathology in autopsy tissue from patients with liver disease. Skeletal muscle tissue taken at autopsy from patients in the Congenital Muscle Disease Tissue Repository who showed variable degrees of liver pathology. H&E staining (A-C) reveals similar pathology that is characteristic of XLMTM, including myofiber smallness and increased numbers of fibers with internal nucleation, in the absence of inflammation or active myofiber degeneration. NADH staining (D-F) shows a pattern of organelle mislocalization that is characteristic of XLMTM in humans, with central aggregation of mitochondria and sarcotubular elements surrounded by an area of absent staining in the subsarcolemmal region. The muscle pathology findings in these three cases are extremely similar, despite differential levels of liver disease in these patients. One patient (panels A, D) had no histological evidence of liver disease, one patient (panels B, E) showed intrahepatic cholestasis at autopsy, and one patient (panels C, F) showed hepatic hemorrhage due to presumed hepatic peliosis.
DISCUSSION
Hepatobiliary disease has not been extensively characterized in XLMTM patients, thus comprehensive laboratory studies typically are not collected, and histopathology is not routinely performed due to risk of hemorrhage from peliosis [15]. Among the five patients in this case series with genetically confirmed XLMTM who developed significant cholestasis, two had missense mutations (Patients 1 and 4), two had nonsense mutations (Patients 2 and 5) and one had a large deletion (Patient 3). Generally, these patients presented at early ages with signs of cholestasis, including jaundice, pruritus, and choluria, and cholestatic episodes followed infection or vaccination in four of the five patients. While pruritus is a common symptom of cholestasis, this mechanism is not entirely understood; serum or tissue concentrations of bile salts do not correlate with the degree of pruritus, though patients with pruritus related to liver disease typically have significant elevations of serum bile salts [16]. The recently described untreated XLMTM patient with cholestasis who died following intracranial hemorrhage had also had an upper respiratory infection shortly prior to hospitalization [13]. Infections are common in chronically ventilated XLMTM patients, with 45% of patients in the RECENSUS study requiring hospitalization due to infection [5].
All five patients showed significant serum bile acid elevation and hyperbilirubinemia with fluctuating bilirubin levels over time. Variations in hypertransaminemia were noted, with some patients showing moderate elevations and others showing severe increases with substantial fluctuations (e.g., Patient 2). Relatively unremarkable GGT levels were noted across all patients over time. Patients 1–4 responded well to choleretic treatment with UDCA and cholestyramine. Recommendations for monitoring the liver function of patients with XLMTM include a hepatic panel—transaminases, GGT, total and direct bilirubin, and serum bile acids—and abdominal ultrasonography. In the case of intercurrent illness and/or presentation with the signs of cholestasis mentioned above, we recommend repeating these analyses until resolution.
The disease courses of the five patients described here are apparently similar to benign recurrent intrahepatic cholestasis (BRIC), which is associated with mutations in the ATP8B1 and ABCB11 genes. These genes encode for ATP-binding hepatocanalicular transporter proteins involved in bile formation and export across the canalicular membrane. BRIC is characterized by recurring episodes of cholestasis with severe acute pruritus usually lasting for weeks, normal GGT, which is also observed in PFIC types 1 and 2 [14], and spontaneous resolution with a recurrent course that remains asymptomatic between episodes and typically does not progress to cirrhosis. However, caution must be used before concluding that cholestasis of XLMTM is truly benign, as the short life expectancy of XLMTM [17] may obfuscate our understanding of the long-term hepatobiliary natural history. A recent publication of fatal intracranial hemorrhage in a 6-month-old boy with XLMTM implicates intrahepatic cholestasis as a key driver of his vitamin K-deficient coagulopathy [13]. Within our cohort, Patient 5 has progressed to decompensated liver failure due to intrahepatic cholestasis, and with no other apparent genetic or environmental explanation beyond his underlying XLMTM. Conjugated hyperbilirubinemia originating with or without systemic infection and progression to severe cholestatic liver injury has been associated with altered expression, localization, and function of BSEP, the product of ABCB11 in the canalicular membrane, in PFIC type 2 [18, 19]. Patients 2, 3, and 5 had extensive genetic testing that excluded mutations associated with familial cholestasis despite having disease courses consistent with familial cholestasis, indicating that there may be another pathomechanism involved.
Myotubularin, the lipid phosphatase protein absent or mutated in XLMTM, primarily dephosphorylates the cell membrane phospholipid phosphatidylinositol 3-phosphate (PI3P), which is essential for normal excitation and contraction in muscle fibers (causing a myopathic effect when it is absent) [20] and in the regulation of bile acid transport [21]. Muscle biopsies conducted in four of the five patients in our cohort were consistent with XLMTM muscle pathology, including marked myofiber smallness, central aggregates of organelles, and centrally nucleated fibers in the absence of inflammation or myofiber degeneration. In addition, muscle tissue collected at autopsy from three patients in the CMDTR with either normal liver histology, liver hemorrhage, or intrahepatic cholestasis were indistinguishable despite their differences in liver pathology, suggesting that significant liver disease in XLMTM does not induce additional pathological changes in muscle tissue.
In hepatocytes, autophagosome formation and endosome recycling to the cell surface requires PI3P hydrolysis by myotubularin phosphatases (e.g., MTM1) to enable exocytosis [22]. The accumulation of PI3P may also perturb the activity of phosphatidylinositol 3-kinase (PI3K) [23], which is essential for the translocation of BSEP to the canalicular membrane [24]. In Patient 5, urine bile acid analysis showed increased glycine and taurine conjugates of hydroxy- and dihydroxy-oxo-cholenoic acids, the synthesis of which are regulated by PI3K pathways in hepatocytes [25]. Thus, insufficient hepatocellular myotubularin activity in XLMTM patients and its associated loss of PI3P homeostasis [2] may impair bile acid transport across the canalicular membrane [26].
There are currently no approved disease-modifying therapies for XLMTM. Greater understanding of the mechanism of liver disease in patients with XLMTM will be important to the development and study of potential therapies for patients with XLMTM.
AUTHOR CONTRIBUTIONS
• CM, TS, AN, IR, GMB, SFC, JVF, BMC actively managed the patients discussed in the report and gathered and analyzed data.
• CM, WM, BS, BMK, MWL drafted the manuscript with the support of Laurie LaRusso, MS, ELS with funding from Astellas Gene Therapies.
• RJG assisted in patient identification and coordinated and participated in discussions related to clinical care related to these patients.
• HM and MWL gathered and analyzed pathology data.
• All authors reviewed and approved manuscript content.
DECLARATION OF INTERESTS
• CM, AN, RJG: advisors for Astellas Gene Therapies outside of the current work
• TS, IR, GMB, SFC, JVF, BMC, HM: no disclosures
• WM, BS: employees of Astellas Gene Therapies
• BMK: advisor for Astellas Gene Therapies outside of the current work, Consultant for Mirum, Consultant for Albireo; Unrestricted Educational Grants from Mirum and Albireo
• MWL: grants and/or personal fees from Astellas Gene Therapies outside of the current work as well as Solid, Encoded, Modis, Ichorion, Kate Therapeutics, Taysha Therapeutics, Prothelia, Lacerta, AGADA, BioMarin, Dynacure, Affinia
ACKNOWLEDGMENTS
This work and the ASPIRO clinical trial were supported by Astellas Gene Therapies. We thank Laurie LaRusso, MS, ELS, of Chestnut Medical Communications for medical writing support paid for by Astellas Gene Therapies. We would like to thank Stacy Cossette for her work with the Congenital Muscle Disease Tissue Repository and Ms. Margaret Beatka for her work in figure design and preparation. We would like to thank Naciba Zetchi from Astellas Gene Therapies for her support in connecting the authors of this manuscript.
REFERENCES
[1] | Dowling JJ , Lawlor MW , Das S . X-Linked Myotubular Myopathy. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. Gene-Reviews((R))1993-2019 [updated 2018 Aug 23]. Seattle (WA) 2002. |
[2] | Hnia K , Vaccari I , Bolino A , Laporte J . Myotubularin phosphoinositide phosphatases: Cellular functions and disease pathophysiology. Trends Mol Med. (2012) ;18: (6):317–27. |
[3] | Vandersmissen I , Biancalana V , Servais L , Dowling JJ , Vander Stichele G , Van Rooijen S , et al. An integrated modelling methodology for estimating the prevalence of centronuclear myopathy. Neuromuscul Disord. (2018) ;28: (9):766–77. |
[4] | Annoussamy M , Lilien C , Gidaro T , Gargaun E , Che V , Schara U , et al. X-linked myotubular myopathy: A prospective international natural history study. Neurology. (2019) ;92: (16):e1852–e67. |
[5] | Beggs AH , Byrne BJ , De Chastonay S , Haselkorn T , Hughes I , James ES , et al. A multicenter, retrospective medical record review of X-linked myotubular myopathy: The RECENSUS study. Muscle Nerve. (2018) ;57: (4):550–60. |
[6] | Graham RJ , Muntoni F , Hughes I , Yum SW , Kuntz NL , Yang ML , et al. Mortality and respiratory support in X-linked myotubular myopathy: A RECENSUS retrospective analysis. Arch Dis Child. 2019. |
[7] | Herman GE , Finegold M , Zhao W , de Gouyon B , Metzenberg A . Medical complications in long-term survivors with X-linked myotubular myopathy. J Pediatr. (1999) ;134: (2):206–14. |
[8] | Amburgey K , Tsuchiya E , de Chastonay S , Glueck M , Alverez R , Nguyen CT , et al. A natural history study of X-linked myotubular myopathy. Neurology. (2017) ;89: (13):1355–64. |
[9] | Shieh PB , Bonnemann CG , Muller-Felber W , Blaschek A , Dowling JJ , Kuntz NL , et al. Re: “Moving Forward After Two Deaths in a Gene Therapy Trial of Myotubular Myopathy” by Wilson and Flotte. Hum Gene Ther. (2020) ;31: (15-16):787. |
[10] | Lawlor MW , Beggs AH , Buj-Bello A , Childers MK , Dowling JJ , James ES , et al. Skeletal muscle pathology in X-linked myotubular myopathy: Review with cross-species comparisons. J Neuropathol Exp Neurol. (2016) ;75: (2):102–10. |
[11] | Oliveira J , Oliveira ME , Kress W , Taipa R , Pires MM , Hilbert P , et al. Expanding the MTM1 mutational spectrum: Novel variants including the first multi-exonic duplication and development of a locus-specific database. Eur J Hum Genet. (2013) ;21: (5):540–9. |
[12] | Trout AT , Anupindi SA , Gee MS , Khanna G , Xanthakos SA , Serai SD , et al. Normal Liver Stiffness Measured with MR Elastography in Children. Radiology. (2020) ;297: (3):663–9. |
[13] | Neese JM , Yum S , Matesanz S , Raffini LJ , Whitworth HB , Loomes KM , et al. Intracranial Hemorrhage Secondary to Vitamin K Deficiency in X-linked Myotubular Myopathy. Neuromuscul Disord. 2021;31(7):651-55. |
[14] | Srivastava A . Progressive familial intrahepatic cholestasis. J Clin Exp Hepatol. (2014) ;4: (1):25–36. |
[15] | Biswas S , Gogna S , Patel P . A Fatal Case of Intra-Abdominal Hemorrhage Following Diagnostic Blind Percutaneous Liver Biopsy in a Patient With Peliosis Hepatis. Gastroenterology Res. (2017) ;10: (5):318–21. |
[16] | Sanjel B , Shim W-S . Recent advances in understanding the molecular mechanisms of cholestatic pruritus: A review. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. (2020) ;1866: (12):165958. |
[17] | McEntagart M , Parsons G , Buj-Bello A , Biancalana V , Fenton I , Little M , et al. Genotype-phenotype correlations in X-linked myotubular myopathy. Neuromuscul Disord. (2002) ;12: (10):939–46. |
[18] | Minemura M , Tajiri K , Shimizu Y . Liver involvement in systemic infection. World J Hepatol. (2014) ;6: (9):632–42. |
[19] | Kubitz R , Dröge C , Stindt J , Weissenberger K , Häussinger D . The bile salt export pump (BSEP) in health and disease. Clin Res Hepatol Gastroenterol. (2012) ;36: (6):536–53. |
[20] | Al-Qusairi L , Weiss N , Toussaint A , Berbey C , Messaddeq N , Kretz C , et al. T-tubule disorganization and defective excitation-contraction coupling in muscle fibers lacking myotubularin lipid phosphatase. Proceedings of the National Academy of Sciences. (2009) ;106: (44):18763–8. |
[21] | Misra S , Ujházy P , Varticovski L , Arias IM . Phosphoinositide 3-kinase lipid products regulate ATP-dependent transport by sister of P-glycoprotein and multidrug resistance associated protein 2 in bile canalicular membrane vesicles. Proc Natl Acad Sci U S A. (1999) ;96: (10):5814–9. |
[22] | Ketel K , Krauss M , Nicot AS , Puchkov D , Wieffer M , Müller R , et al. A phosphoinositide conversion mechanism for exit from endosomes. Nature. (2016) ;529: (7586):408–12. |
[23] | Cao C , Backer JM , Laporte J , Bedrick EJ , Wandinger-Ness A . Sequential actions of myotubularin lipid phosphatases regulate endosomal PI(3)P and growth factor receptor trafficking. Mol Biol Cell. (2008) ;19: (8):3334–46. |
[24] | Misra S , Ujhazy P , Gatmaitan Z , Varticovski L , Arias IM . The role of phosphoinositide 3-kinase in taurocholate-induced trafficking of ATP-dependent canalicular transporters in rat liver. J Biol Chem. (1998) ;273: (41):26638–44. |
[25] | Hohenester S , Gates A , Wimmer R , Beuers U , Anwer MS , Rust C , et al. Phosphatidylinositol-3-kinase p110γ contributes to bile salt-induced apoptosis in primary rat hepatocytes and human hepatoma cells. J Hepatol. (2010) ;53: (5):918–26. |
[26] | Trauner M , Boyer JL . Bile salt transporters: Molecular characterization, function, and regulation. Physiol Rev. (2003) ;83: (2):633–71. |


