
Exon-Skipping in Duchenne Muscular Dystrophy
Abstract
Duchenne muscular dystrophy (DMD) is a devastating, rare disease. While clinically described in the 19th century, the genetic foundation of DMD was not discovered until more than 100 years later. This genetic understanding opened the door to the development of genetic treatments for DMD. Over the course of the last 30 years, the research that supports this development has moved into the realm of clinical trials and regulatory drug approvals. Exon skipping to therapeutically restore the frame of an out-of-frame dystrophin mutation has taken center stage in drug development for DMD. The research reviewed here focuses on the clinical development of exon skipping for the treatment of DMD. In addition to the generation of clinical treatments that are being used for patient care, this research sets the stage for future therapeutic development with a focus on increasing efficacy while providing safety and addressing the multi-systemic aspects of DMD.
THE DMD GENE AND DYSTROPHIN PROTEIN STRUCTURE AND FUNCTION
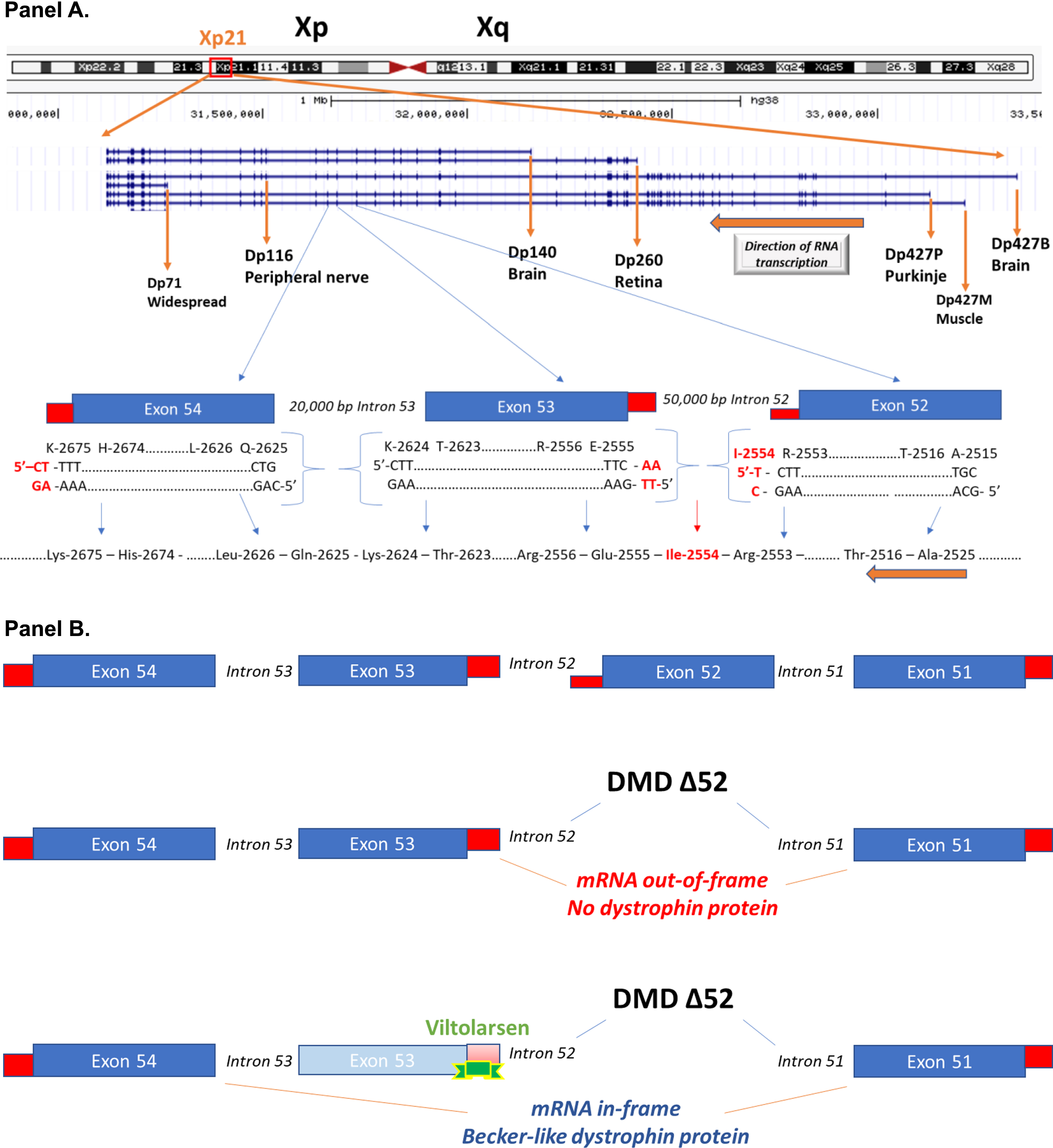
Gene mutations in the 2.24 million base pair DMD gene on the X chromosome result in biochemical loss or abnormalities of the dystrophin protein. The DMD gene has multiple gene promoters driving expression of different mRNA (and encoded protein) isoforms (Fig. 1). The ‘full-length’ 14 kb mRNA using the 3 most proximal 5’ gene promoters (Dp427B, Dp427M, Dp427P) contains 79 exons and encodes a 427 kDa membrane cytoskeletal protein (dystrophin), that is expressed in all skeletal muscles, smooth muscles (vascular and visceral), heart, peripheral nerve, and some neurons. The DMD gene also contains multiple downstream distal promoters encoding shorter mRNA and protein isoforms (Dp260, Dp140, Dp116), with the shortest Dp71 (Dystrophin protein, 71 kDa) showing relatively ubiquitous expression in non-muscle and nerve cells (Fig. 1) [1].
Fig. 1
Schematic of the DMD gene and exon skipping. Panel A: A schematic of the DMD gene from the Genome Browser (genome.ucsc.edu) at Xp21 with encoded mRNA transcripts is shown. Gene transcription is shown from left to right, with the three gene promoters driving expression of the full-length 427 kDa dystrophin (Dp427B, Dp427M, Dp427P), as well as down-stream gene promoters driving smaller molecular weight dystrophin proteins (Dp260, Dp140, Dp116, Dp71). Also shown is an expansion of exons 52, 53, and 54. The amino acids and encoding triplet codons are provided at the ends of each of these exons. Exon 52 ends in an incomplete codon for isoleucine (I-2554), requiring the last two bases from exon 53 to complete the codon. In contrast, exon 53 ends with a complete codon for lysine (K-2624), splicing to exon 54 that starts with a complete codon for glutamine (Q-2625). A gene mutation deleting exon 53 would then be out-of-frame, as an incomplete codon ending exon 52 would be fused to a complete codon on exon 54, leading to a frame shift in the resulting dystrophin mRNA. Panel B: This shows the consequence of drug-induced exon skipping by viltolarsen targeted to exon 53. A boys with DMD is shown as having a deletion mutation of exon 52, and when this patient’s dystrophin mRNA splices together the remaining exons (exon 51 to exon 52), this leads to a frame shift, mRNA out-of-frame, and no dystrophin protein. Viltolarsen binds to exon 53, and blocks its inclusion in the dystrophin mRNA. The drug-induced splicing of exon 51 to exon 54 results in an in-frame dystrophin mRNA, and Becker-like dystrophin protein.
Dystrophin is a component of the intracellular membrane cytoskeleton where it interacts with actin filaments, intermediate filaments, and transmembrane proteins that themselves interact with the extracellular basal lamina. It has been dubbed a ‘broad membrane integrator’ [2]. The primary biochemical role of dystrophin in skeletal muscle myofibers is to increase the stability of the plasma membrane, protecting it from force-related membrane disruptions. In this way, its biochemical role is similar to the structurally-related spectrin protein in red blood cells, where spectrin similarly imparts deformability and stability of plasma membranes (of erythrocytes) as they pass through small capillaries. While dystrophin does not have signaling or enzymatic roles itself, it binds directly or indirectly to multiple other proteins that do have signaling or enzymatic roles, such as neuronal nitric oxide synthase (nNOS).
The DMD gene mutations that inactivate the gene or mRNA such that little or no dystrophin is produced (e.g. null mutations) lead to dystrophin deficiency. However, the location of an inactivating (frame-shift or nonsense) mutation within the DMD gene can differentially affect different isoforms. For example, a mutation within the first 29 exons of the gene would be expected to inactivate mRNA and protein from the full-length brain, muscle, and Purkinje cell gene promoters (Dp427B, Dp427M, Dp427P), but leave the downstream Dp260 (retina), Dp140 (brain), Dp116 (peripheral nerve) and Dp71 (widespread) dystrophin proteins intact [Fig. 1]. On the other hand, mutations in the last 17 exons (3’ end of the gene) would be expected to lead to deficiency of all dystrophin isoforms in all tissues. Clinical findings are consistent with this, where loss of Dp260 (retina) leads to loss of night vision and distinctive changes in electroretinography findings, and this phenotype correlates with the location of the DMD gene mutation and predicted effect on the Dp260 isoform. The retinopathy phenotype maps to distal mutations downstream of Dp260 (retina) isoform [3], but there appears to be retinal sensitivity to ischemia that maps to the full length Dp427 isoform [4]. Likewise, mutations in the 3’ end of the gene, removing more dystrophin isoforms, are correlated with more severe cognitive involvement and developmental brain abnormalities [5– 7].
While the diagnostic term of a ‘muscular dystrophy’ is often thought of as a disorder restricted to skeletal muscle structure and function, increasing knowledge of DMD suggests that the clinical disorder has features of a multi-system disease (syndrome) with functional defects of vascular smooth muscle, visceral smooth muscle, heart, and brain/nerve. In the majority of cases, the skeletal muscle disease predominates, but abnormalities of other tissues contribute to the overall clinical picture. Use of assisted ventilation extends patient lifespan; most ventilated patients succumb to cardiac disease. This is a point to consider when developing and evaluating therapeutic approaches to DMD.
BECKER MUSCULAR DYSTROPHY AND CLINICAL VARIABILITY
The diagnosis of Becker muscular dystrophy (BMD) was originally reserved for male patients from X-linked recessive families segregating a muscular dystrophy that was clinically milder than DMD. With the cloning of the DMD gene, identification of dystrophin, and advent of molecular diagnostics, the diagnosis of BMD became synonymous with present but abnormal dystrophin protein in skeletal muscle biopsies [8, 9]. It soon became apparent in practice that a genetic characteristic of most cases of BMD, an in-frame deletion in the dystrophin gene, was not always concordant with a milder ‘Becker-like’ phenotype. This discordance impacts the design of human clinical trials focused on BMD therapeutics.
While all BMD patients show present but biochemically abnormal dystrophin in muscle, the gene mutations causing the abnormal dystrophin are highly variable, and the precise biochemical perturbations of the dystrophin protein similarly highly variable. The most common gene mutations in BMD are two in-frame deletions (Δ45– 47 [30% ]; Δ45– 48 [20% ]) [10]. The ‘reading frame rule’ defined by the out-of-frame (inactivating) DMD gene mutations in the severe DMD, and the in-frame (residual function) DMD gene mutations in BMD is correlated with protein findings in about 75% of cases; there are many exceptions to this rule. The exceptions follow some patterns. Gene mutations in the 5’ (beginning) of the gene can be out-of-frame (DMD) but show dystrophin protein on muscle biopsy and a milder clinical picture, both features that are consistent with the diagnosis of BMD. This is often due to use of alternative AUG initiator codons in mRNAs (downstream of the authentic AUG), and escape of nonsense mediated decay (cellular degradation of out-of-frame mRNAs). Out-of-frame exon 44 or 45 skippable mutations can show low levels of residual dystrophin due to endogenous (naturally occurring) alternative splicing creating low levels of in-frame transcripts, with about half of patients showing a milder phenotype [11]. Splice site mutations are ‘leaky’ in that they are often non-null (some normal dystrophin) [12]. Efforts to predict the clinical outcomes of different in-frame deletions based on systematic analyses of large DNA/phenotype databases find considerable heterogeneity in observed phenotypes [13].
Very low levels of normal dystrophin can be associated with a clinical picture that can be milder than typical DMD [15]. That said, low levels of both normal and Becker-like dystrophins can also be associated with a typical DMD phenotype. Other factors over and above gene mutation type and dystrophin protein content of muscle contribute to variation in clinical phenotypes of both DMD and BMD, including genetic modifiers [14] and socioeconomic status [15]. Natural history studies of BMD patient cohorts have shown marked clinical variability, even between those with the same in-frame mutation, ranging from just slightly milder than DMD to asymptomatic [13, 16, 17]. Importantly, muscle MRI findings of the degree of fatty replacement of skeletal muscles appear more correlated with clinical symptoms than either dystrophin protein content or gene mutation [18, 20]. The fact that MRI imaging of fatty replacement is so well-correlated with patient functional ability likely reflects the importance of the variable inflammation and fibrosis pathways downstream of the primary gene and protein defect, and the variability of different muscle groups in terms of moving into the fatty replacement phase associated with functional disability.
Overall, the expectations of therapies directed towards low levels of dystrophin should acknowledge that all approaches aim to rescue or deliver dystrophin protein that is biochemically abnormal, and, therefore, partially functional (exon-skipping, gene therapy, CRISPR DNA editing). Success at achieving any level (low or high) of partially function dystrophin protein will almost certainly be associated with marked clinical variability from patient to patient, and within a patient from muscle to muscle.
THERAPEUTIC APPROACH OF CONVERTING DUCHENNE TO BECKER MUSCULAR DYSTROPHY
Therapeutic approaches that aim to restore partially functional muscle dystrophin in patients with DMD focus on one of three approaches: 1. gene delivery using viral vectors; 2. stop codon read-through; 3. converting out-of-frame mutations to in-frame mutations (exon skipping; multiple approaches).
For gene delivery using viral vectors, the limited carrying capacity of the most suitable viral vectors based on the adeno-associated virus (AAV) requires use of a highly modified smaller molecular weight versions of dystrophin called micro-dystrophins (∼150 kDa compared to normal 427 kDa dystrophin). The micro-dystrophins are semi-functional proteins that have removed over half the normal dystrophin amino acid sequence. The biochemically abnormal dystrophin delivered with gene therapy is, in part, similar to ‘Becker-like’ dystrophins that occur naturally in patients with BMD or are induced by exon-skipping as a treatment for patients with DMD. Of note, the AAV-driven dystrophins are much smaller than those seen in Becker muscular dystrophy patients. Pre-clinical data in the mdx mouse [19– 21] and CXMD dog [22– 24] demonstrated functional benefit of AAV-delivered de novo micro-dystrophin. Preliminary results from a human clinical trial of AAV gene therapy have shown high level expression of micro-dystrophin in DMD patient muscle [25], with improvements in MRI fat fraction findings through 24 months post-treatment [26]. A key question with gene therapy in DMD is persistence of effect, as re-delivery is anticipated to be significantly limited by antibody responses against the second administration of the viral delivery vehicle, and degeneration/regeneration cycles of DMD muscle. Also, many DMD boys have pre-existing immunity to certain serotypes of AAV that are used for gene delivery, and this currently excludes these patients from receiving gene therapy [27]. Immunosuppressant strategies to address this limitation of gene therapy are being developed [28].
For stop codon read-through, about 10– 15% of boys with DMD have an amino-acid codon mutated to a premature stop codon, and enabling the ribosome to insert an amino acid at the premature stop codon, rather than terminating dystrophin protein translation, may lead to de novo dystrophin. A small molecule drug, ataluren, has been developed as a stop codon read-through drug, and has shown variable improvement in 6-minute walk times [29]. Drug-responsive increases in dystrophin in patient muscle has not yet been demonstrated, and a recent report showed 30% of boys with DMD with a stop codon to show residual dystrophin in muscle without ataluren treatment [15]. Ataluren has been approved by the EMA since 2014.
Exon skipping, the conversion of out-of-frame to in-frame deletions, can be achieved by exon skipping (with oligonucleotides or U7 snRNPs expressing an antisense sequence) or by exon deletions (with genome editing). The majority of clinical studies of drug-induced de novo dystrophin in muscle have been with exon-skipping; the removal of an additional DMD gene exon neighboring a patient’s deletion mutation, to convert an out-of-frame DMD mutation to an in-frame BMD mutation (Fig. 1, 2). For exon-skipping, there are three different experimental approaches: oligonucleotide, DNA editing (CRISPR) and U7 snRNP-mediated splice blocking. CRISPR DNA editing approaches, while not yet in clinical trials in DMD, seek to modify the myofiber genomic DNA to convert a DMD out-of-frame to a BMD-like in-frame deletion. CRISPR relies on delivery of the DNA editing machinery using viral vectors. A second approach to accomplish exon-skipping is U7 snRNP-mediated blocking of splicing, similar in mechanism of action to oligonucleotide approaches. AAV vectors have been used to deliver modified U7 snRNP genes where the normal antisense part that hybridizes to histone RNA is replaced with an antisense sequence targeting (in this case) a dystrophin exon. This does not target mRNA, but pre-mRNA (like exon skipping with oligonucleotides). A single clinical trial of AAV-mediated RNA editing (scAAV9.U7.ACCA; NCT04240314) is underway for exon 2 duplications [30]. The 3rd approach to accomplishing exon-skipping is using oligonucleotide drugs to bind to the pre-mRNA (prior to splicing) to modulate RNA splicing. These oligonucleotide approaches have used multiple chemistries for the drug, with variable success, and this is discussed further in the remainder of this text.
Fig. 2
Lead candidate selection for exon 53 exon skipping. Panel A: Shown is a schematic of the 38 oligonucleotides tested for strength in blocking exon 53 splicing, and the experimental approach leading to lead compound selection (NS-065/NCNP-01; viltolarsen). Panel B: Dose-response analyses shows NS-065/NCNP-01 (viltolarsen) to achieve ∼70% exon skipping efficiency in cell cultures. From Watanabe et al. 2018 [62].
![Lead candidate selection for exon 53 exon skipping. Panel A: Shown is a schematic of the 38 oligonucleotides tested for strength in blocking exon 53 splicing, and the experimental approach leading to lead compound selection (NS-065/NCNP-01; viltolarsen). Panel B: Dose-response analyses shows NS-065/NCNP-01 (viltolarsen) to achieve ∼70% exon skipping efficiency in cell cultures. From Watanabe et al. 2018 [62].](https://content.iospress.com:443/media/jnd/2021/8-s2/jnd-8-s2-jnd210682/jnd-8-jnd210682-g002.jpg)
To explain exon skipping in more detail, oligonucleotide drugs bind to the dystrophin pre-mRNA (prior to splicing) and block the inclusion of an exon neighboring the patient’s gene deletion (Figs. 1, 2). The 79 exons of the DMD gene often begin and end with blunt ends, where amino acids encoded by the exons are fully encoded by a triplet codon residing within the exon. For example, as shown in Fig. 1 (note gene is oriented right to left, so the bottom strand is read as encoded RNA), the end of exon 53 encodes the AAG codon for a lysine at position 2,624 (K-2624) in the dystrophin amino acid sequence and this exon 53 is spliced to the blunt end of exon 54 which itself encoded a complete codon for the following glutamic acid residue (Q-2625). However, other exons encode incomplete amino acid codons at their termini: the end of exon 52 encodes the first “C” nucleotide of isoleucine (I-2554 in red font), and requires the next 2 thymidine bases of exon 53 to complete the codon (C-TT in DNA, or CUU in RNA) (Fig. 1). Deletion mutations where remaining exons share the same reading frame are ‘in-frame’, and when spliced together lead to a relatively stable mRNA encoding partly functional dystrophin lacking amino acids corresponding to the deleted exons. On the other hand, remaining exons that do not share the same reading frame are ‘out-of-frame’, and when the remaining exons are spliced together into mRNA, a translational frame-shift is encountered by the ribosome, leading to a halt in dystrophin protein translation. Most mRNA transcripts with premature stop codons trigger a nonsense-mediated decay (NMD) mechanism that targets these out-of-frame mRNAs for degradation. With the DMD gene, it appears that premature stop codons may also lead to epigenomic changes in the gene, reducing mRNA expression as well [31].
An example for the viltolarsen drug targeting exon 53 of the dystrophin mRNA is diagrammed (Fig. 2). If the oligonucleotide drug hits its RNA target, the drug blocks the inclusion of the exon to which the drug is bound in the dystrophin RNA, bringing the transcript back into frame – effectively converting the DMD gene mutation to a BMD-like gene mutation at the level of the mRNA.
For oligonucleotide-based exon skipping, pre-clinical studies in the mdx mouse model were carried out first using intramuscular injection of 2’-O-methyl phosphorothioate (2OMePS) oligonucleotides [32], and soon after using systemic delivery of both 2OMePs and phosphorodiamidate morpholino oligomer (PMO) chemistries [33, 34]. Key to success of oligonucleotide approaches is achieving adequate drug concentrations within the myofiber, so that drug can hit its pre-mRNA target in the myofiber nucleus (prior to pre-mRNA splicing) and block the splicing of the targeted exon. Measurements of myofiber delivery of oligonucleotide drugs have been done using three different methods: in vitro cell transfections, in vivo direct intramuscular injections, and in vivo systemic delivery (intravenous or subcutaneous). Different oligonucleotide chemistries show distinct effectiveness of myofiber delivery by these three methods, and thus show different potency in driving exon skipping depending on the assay system. Morpholino chemistry (PMO) are uncharged molecules, and are difficult to transfect into cells in vitro, show little or no delivery to normal myofibers by systemic delivery, but in dystrophic muscle show high level delivery and high potency in driving exon skipping [35– 37]. The effective delivery of PMOs to dystrophic muscle seems to be mediated, at least in part, by myoblasts and macrophages as an intermediate to dystrophin-deficient myofibers [40]. On the other hand, 2OMePS chemistries are negatively charged, transfect well into cells in vitro, and can be delivered by intramuscular injection, but have not yet been shown to drive dystrophin production in patient muscle in clinical trials. Direct injection into muscle tissue destabilizes myofiber membranes near the injection site and leads to bulk delivery of any DNA or RNA payload to either normal or dystrophic muscle. Most cell types cannot recover from such overt breaches of their plasma membranes, but the enormous syncytial myofibers can recover and then retain the nucleic acids delivered by this ‘brute force’ approach.
To date, the highest levels of dystrophin rescue by shown by systemic delivery to skeletal muscle in mouse, dog, and human studies have been with the morpholino chemistry. A key advantage of the morpholino chemistry is that it has shown a good safety profile at very high systemic doses; up to 3.0 grams/kg in mice [38], 200 mg/kg in dogs [39], and 80 mg/kg in DMD boys [40, 41].
CLINICAL TRIALS OF OLIGONUCLEOTIDE-INDUCED EXON SKIPPING
Exon-skipping clinical trials in DMD have been carried out with two different chemistries; 2OMePS (drisaperson), morpholino (viltolarsen, eteplirsen, golodirsen, casimersen). In addition, a locked chirality, stereopure ASO (suvodirsen) has been tested in a clinical trial, but not yet published. Here, we focus on clinical studies using 2OmePS and morpholino (PMO) chemistries for oligonucleotide drugs.
2OMePS (drisapersen)
The earliest exon skipping clinical trials for DMD began in 2006 and were designed to test the 2OMePS chemistry targeting exon 51 skipping (alternatively named PRO-051, GSK2402968, drisapersen) as an addition to corticosteroid standard of care treatment. In an initial open label study, 0.8 mg drisapersen was injected into the tibialis anterior muscle of 4 boys with DMD. Assessment of muscle dystrophin 28 days later showed evidence of drug-related de novo dystrophin expression by muscle biopsy at the injection site [42]. This promising intramuscular injection pilot study was followed by an open label, dose-finding trial of 12 DMD boys given 5 weekly subcutaneous injections, with 3 boys at each of 4 doses (0.5, 2.0, 4.0, 6.0 mg/kg), followed by a 12-week extension with all 12 boys treated with 6.0 mg/kg/wk. There was a suggestion of improvement in 6-minute walk distance, and some evidence of dystrophin mRNA splicing and dystrophin in muscle biopsies, although both appeared to be at very low levels that were difficult to distinguish from pre-treatment samples [43]. These same 12 boys were followed in a long-term extension study of weekly drisaperson for ∼3.4 years (all participants also continued corticosteroid treatment). When compared to matched, steroid-treated, natural history controls, there was a suggestion of prolongation of ambulation compared to external controls [44].
These exploratory and dose-finding trials of drisapersen were then followed by two double-blind Phase II placebo-controlled studies. The first Phase II study (NCT01153932) enrolled 53 steroid-treated participants with DMD (7.7±1.5 yrs) into 3 arms; placebo, intermittent drispersen, and continuous (once weekly) drispersen [45] (Table 1). The treatment period was 48 wks, however the primary outcome was at study midpoint (25 wks; change in 6-minute walk test drisapersen continuous vs placebo). The continuous 6 mg/kg/wk drisapersen group showed an improvement of ∼30 meters, whereas the placebo group showed a slight decline of ∼5 meter (Δ35 meters; p = 0.014). Both groups then showed an overall decline in meters walked over the subsequent 23 wk treatment period while the 35 meter difference was maintained (p = 0.051). Dystrophin studies of muscle biopsy were carried out, however quantitative measures of dystrophin by immunoblot were not reported.
Table 1
Summary of clinical trials of systemic oligonucleotide delivery for exon-skipping
| Publication (clinicaltrials.gov) | Trial dates | Drug; dose groups | Delivery | # participants (Age±SD) | Treatment period | Dystrophin (immunoblot) | 6-minute walk change vs placebo |
| 2’-O-methyl oligonucleotide | |||||||
| Voit et al. 2014 (NCT01153932) | 2010– 2012 | drisapersen; placebo, 6.0 mg/kg/wk (weekly; intermittent) | Subcutaneous | 53 (7.7±1.5 yrs)1 | 25 wks 49 wks2 | Not quantitated | 25 wks: +35 m (p = 0.014) 49 wks: +35 m (p = 0.051) |
| McDonald et al. 2018 (NCT01462292) | 2011– 2013 | drisapersen; placebo, 3.0, 6.0 mg/kg/wk | Subcutaneous | 51 (7.6±2.7 yrs) | 24 wks | ND | +27 m (ns) |
| Goemens et al. 2017 (NCT01254019) | 2010– 2013 | drisapersen; placebo, 6.0 mg/kg/wk | Subcutaneous | 186 (8.1±2.4 yrs) | 48 wks | ND | +10 m (ns) |
| phosphorodiamidate morpholino oligonucleotide | |||||||
| Cirak et al. 2011 (NCT00844597) | 2008– 2010 | eteplirsen; 0.5, 1.0, 2.0, 4,0, 10.0, and 20.0 mg/kg/wk | Intravenous | 19 (8.7 yrs) | 12 wks | ∼4% (20.0 mg/kg; n = 4) | ns |
| Mendell et al. 2013 (NCT01396239) | 2011– 2012 | eteplirsen; placebo, 30, 50 mg/kg/wk | Intravenous | 12 (8.8±1.2 yrs) | 24 wks | 24 wks: ND33.5 yrs: 0.9% 4 | – 103 m (30 mg/kg)+25 m (50 mg/kg) |
| Frank et al. 2020 (NCT02310906) | 2015– 2019 | golodirsen; placebo, dose escalation (4, 10, 20, 30 mg/kg/wk sequentially for 2 weeks each) | Intravenous | 12 Group 1 (8.6±2.1 yrs)513 Group 2 (8.5±2.5 yrs) | 24 wks | 1% | NR |
| Komaki et al. 2018 (NCT02081625) | 2013– 2014 | viltolarsen, 1.25, 5, or 20 mg/kg/wk | Intravenous | 10 (11.0±3.0 yrs) | 12 wks | 2% (20 mg/kg)6 | NR |
| Clemens et al. 2020 (NCT02740972) | 2016– 2017 | viltolarsen, 40, 80 mg/kg/wk | Intravenous | 16 (7.4±1.8 yrs) | 24 wks | 5.7% (40 mg/kg)5.9% (80 mg/kg) | +28.9 m (+94.2 m vs. external control) |
| Komaki et al. 2020 (JAPIC CTI-163291) | 2016– 2017 | viltolarsen, 40, 80 mg/kg/wk | Intravenous | 16 (8.4±2.0 yrs) | 24 wks | 1.5% (40 mg/kg)4.8% (80 mg/kg) | – 25 m (both doses) |
1Placebo group was younger (6.9±1.2 yrs). 2The treatment period was 49 weeks, but the primary outcome was change in 6-minute walk test at 24 weeks vs. placebo. 3Immunoblot of a single post-treatment biopsy at 48 wks treatment was shown, but quantitations were not reported. 4Participants were enrolled in an extension study, and re-biopsies after 3.5 years of treatment (Charleston et al. 2018). 5Placebo group was mean 1.5 years younger. 6Of the 4 subjects in the 20 mg/kg/wk group, one showed 8% levels of dystrophin, whereas the other 3 showed undectable dystrophin.
A second placebo-controlled Phase II study (NCT01462292) of subcutaneous drisapersen administration was carried out in 51 participants with DMD randomized to 3 arms (drisapersen 3 mg/kg/wk, 6 mg/kg/wk or placebo) [46] (Table 1). For the primary outcome, 6-minute walk distance, the drisapersen 3.0 mg/kg/wk and placebo groups showed a small decline from baseline to 24 weeks, whereas the drisapersen group 6.0 mg/kg/wk showed a small improvement which did not achieve statistical significance.
The Phase 3, randomized, double-blind, placebo-controlled clinical trial, carried out from 2010– 2013 at 44 sites in 19 countries, enrolled 186 boys with DMD randomized 2:1 to drisapersen vs. placebo, with a 48-wk treatment period (NCT01254019) [47]. Recruited participants were older than 5 years at entry (mean [SD] age 8.1±2.4 yrs), and were treated with corticosteroids for over 3 months at the time of first study drug administration. At the end of the treatment period, there was no significant clinical improvement relative to placebo. A post-hoc subgroup analysis of less severe participants at entry (ability to rise from floor, and 6-minute walk 300– 400 meters), suggested a 35 meter reduction in decline in drisapersen group relative to placebo (p = 0.039) [50]. Approval from the FDA in the USA was sought, but the regulatory agency noted the lack of robust evidence of efficacy, and the safety concerns of extensive injection site reactions that continued after drug cessation [48] [https://www.fdanews.com/ext/resources/files/11-15/11-20-FDA-DMD-Briefing.pdf?1520841005], and the program was terminated.
Phosphorodiamidate morpholino (PMO) (eteplirsen, golodirsen, viltolarsen, casimersen)
Clinical trials of the phosphorodiamidate morpholino oligonucleotide (PMO) chemistries began with a study of intramuscular injection of an exon-51 oligonucleotide (AVI-4658; eteplirsen) into a small foot muscle (extensor digitorum brevis; EDB) in 7 boys with DMD [49]. Injections were 0.09 mg (n = 2) or 0.9 mg (n = 5) of the morpholino oligonucleotide, with muscle biopsy 3-4 weeks after the single injection. Strong evidence of drug-related increase in both altered mRNA (skipped exon 51) and de novo dystrophin production were seen at the higher dose. As noted above, intramuscular injections are a robust delivery method for nucleic acids to myofibers that may be a factor unique to muscle tissue.
An open-label dose-finding study of intravenously administered eteplirsen was carried out in 19 participants with DMD (mean age 8.7 yrs) [50]. Doses tested were 0.5, 1.0, 2.0, 4,0, 10.0, and 20.0 mg/kg/wk, over a 12-week treatment period, with 2 to 4 participants per dose group (Table 1). A dose-responsive increase in dystrophin in muscle biopsies was seen with the highest dose group of 4 participants showing a mean of 4% levels by Western blot (range 0 – 7.7% increase from baseline). The authors carried out motor function assessments and did not see evidence of dose-related functional improvement, although the short treatment period and small number of subjects limited interpretation of these findings. Further analysis of the biopsies from this study showed restoration of the dystrophin-associated proteins in myofibers, consistent with the degree of dystrophin restoration [51]. An extension study was not done beyond the 12-week treatment.
A placebo-controlled dose-finding study for eteplirsen was carried out in 12 participants with DMD (mean age 8.8 years), with 4 participants randomized to placebo, 30 mg/kg/wk, or 50 mg/kg/wk for a 24-week treatment period [52] (Table 1). The primary outcome was dystrophin-positive myofibers measured by immunohistochemistry (after 12 weeks treatment for 4 patients who received 50 mg/kg and 2 patients who received placebo). A drug-related increase in dystrophin-positive myofibers was seen, but this result did not appear to be dose-responsive (30 mg/kg/wk 23.0% [range 15.9 to 29.0% ]; 50 mg/kg/wk 0.8% [– 9.3 to 7.4% ]), although the time of treatment at the time of biopsy differed for the two groups (24 weeks for 30 mg/kg/wk; 12 weeks for 50 mg/kg/wk). There also appeared to be variability in measures of dystrophin positive myofibers, as the placebo biopsies showed appreciable variation from baseline to post-treatment (+4.5%, – 5.8%, – 6.5%, – 8.5%). Quantitative immunoblot data were not reported. Analysis of 6-minute walk data showed an overall decline in all groups over the 24-week period, with the greatest decline in the 30 mg/kg/day group (– 103 m vs. placebo). Subjects were enrolled in an extension study, and additional biopsies taken after 3.5 years of treatment in 11 of the participants, and this showed a mean of 0.9% post-treatment dystrophin levels [53]. This data was submitted to FDA for accelerated approval based on the surrogate biomarker of drug-related increase in dystrophin in patient muscle. The approval was granted (30 mg/kg/wk), but with considerable controversy within the FDA [54], and biomedical research community [55– 57]. There are also concerns that the eteplirsen drug was not optimized for exon skipping (e.g. not potent), with alternative morpholino sequences showing over 10-fold greater potency in driving drug-induced exon skipping [58]. Eteplirsen was not approved by the European Medicines Agency (EMA) despite two attempts [59].
Golodirsen is a PMO directed against exon 53 and was first tested in a 24-week placebo-controlled, dose-escalation study of 12 participants with DMD (4 placebo; 8 golodirsen [all escalating from 4 to 30 mg/kg/wk] over the 24-week treatment period) [60] (Table 1). Participants were then enrolled into a long-term extension study (all at 30 mg/kg/wk), and an additional 13 participants with DMD who entered the 30 mg/kg/wk golodirsen treatment arm directly. The 25 golodirsen-treated participants had skeletal muscle biopsies taken at 48-weeks post-treatment. Dystrophin immunoblot analysis showed a mean of drug-related increase of 1% dystrophin. The 6-minute walk test was assessed after 2.7 years of treatment, where FDA noted “Performance on the 6-minute walk test and pulmonary function tests, with at least 144 weeks of follow-up, showed a decline from baseline; however, these results are not interpretable in the absence of a control group” [61]. Golodirsen (30 mg/kg/wk) was approved by FDA based on the surrogate biomarker of drug-related dystrophin expression.
Viltolarsen is a PMO directed against exon 53, similar to golodirsen, and directed against a target sequence on exon 53 that partially overlaps. The approach to the optimization of exon 53 skipping potency for lead compound selection for viltolarsen, as well as the pre-clinical development program, has been fully described [62]. In the initial stage of screening, 38 overlapping 25 nucleotide 2OMePS oligonucleotides were tested for potency (dose-response) in a cell culture system (Fig. 2). This defined a region between nucleotide positions 31 and 65 on the exon 53 sequence that was most effective in blocking the inclusion of exon 53 (exon skipping). In the second stage, a series of 25 PMO oligonucleotides of varying length (15– 25 nucleotides) was designed covering the position 31– 65 region. This identified a 21 nucleotide PMO, named NS-065/NCNP-01 (viltolarsen), located between positions 36 and 56 that was most potent at blocking inclusion of exon 53. The effectiveness of viltolarsen was further confirmed using DMD patient-derived myogenic cells, with transfection of the cells facilitated by Endo-Porter transfection. Evidence for both efficient drug targeting of the pre-mRNA, as well as dose-responsive de novo dystrophin protein from the DMD patient-derived cells was seen after 3 days at a concentration of 10 mmol/L. Viltolarsen-responsive pre-mRNA exon skipping and dystrophin protein rescue was found to be dependent on drug concentration, time of drug exposure, and repeated treatment. Highest levels achieved were 80% normal levels of dystrophin rescued in patient cells.
The first human clinical study of viltolarsen was a Phase 1 dose-ranging study conducted in Japan and enrolled 10 boys with DMD (6 to 16 yrs). Study participants were treated with viltolarsen at intravenous doses of 1.25, 5, or 20 mg/kg/wk for 12 weeks [63] (Table 1). Six of the participants were treated with corticosteroids prior to and during the study, and four were not treated with corticosteroids. Seven of the 10 were non-ambulant at the initiation of treatment. Dose-responsive increases in exon skipping by RT-PCR assays of biopsy mRNA, and increased dystrophin protein production were observed. By immunoblotting, one of the four participants treated with 20 mg/kg/wk, who was also the participant with the greatest absolute dose of viltolarsen based on his body weight, showed 8% of a normal dystrophin level in the post-treatment muscle biopsy, and others showed evidence of dystrophin-positive myofibers by immunostaining.
Given the good safety profile of the PMO chemistry, the results of the Phase 1 study that had a maximum tested dose of 20 mg/kg/wk that showed a dose-dependent increase in muscle truncated dystrophin production and previous studies in mice and dogs that suggested that higher doses might be needed to drive stronger dystrophin expression, two parallel Phase II clinical trials were carried out in Japan and the US using higher doses of 40 and 80 mg/kg/wk [43, 64]. The US study was carried out by the Cooperative International Neuromuscular Research Group (CINRG), and randomized 16 steroid-treated participants with DMD to placebo [4 weeks safety only], 40 and 80 mg/kg/wk. After 4 weeks treatment period, the participants receiving placebo were randomized to viltolarsen for the remainder of the 24-week treatment period. The primary outcome was increase in dystrophin by immunoblot from baseline to 24-weeks, with secondary outcomes of dystrophin immunostaining, RT-PCR of dystrophin mRNA, and mass spectrometry quantitation of dystrophin (orthogonal approach [65]). All biopsy analyses were done blinded. Mean drug-related increase in dystrophin for the 40 mg/kg/wk group was 5.7% [range 3.2– 10.3] of normal, and for the 80 mg/kg/wk group 5.9% [range 1.1– 14.4] of normal, and the dystrophin protein rescue corresponded to observed exon-skipping in dystrophin mRNA (Table 1) (Fig. 3). The immunostaining, RT-PCR and mass spectrometry methods showed significant dose-response, with approximately a 2-fold increased rescue in the 80 mg/kg/wk group compared to the 40 mg/kg/wk group (Table 2). Functional outcome measures were studied with assessments at baseline, 12-weeks and 24-weeks (tests of 6-minute walk, 10-meter run/walk, time to climb 4 stairs, time to stand from supine). All outcome measures showed mean improvement from baseline after 24-weeks treatment, and all were significant when compared to a matched, steroid-treated natural history comparator group from the CINRG DNHS study [66, 67].
Fig. 3
Example of an RNA blot showing viltolarsen-induced exon skipping in DMD participant muscle. From Clemens et al. 2020 [40].
![Example of an RNA blot showing viltolarsen-induced exon skipping in DMD participant muscle. From Clemens et al. 2020 [40].](https://content.iospress.com:443/media/jnd/2021/8-s2/jnd-8-s2-jnd210682/jnd-8-jnd210682-g003.jpg)
Table 2
Orthogonal studies of dystrophin rescue by viltolarsen. From Clemens et al. 2020 [40]
| Samples | Dystrophin Western blot | Mass Spectrometry | Dystrophin IF Analysis | RNA RT-PCR | ||
| Participant Cohort | Normalized myosin heavy chain | Normalized to to alpha-actinin | % Dystrophin | % Dystrophin-positive fibers | % Skipped mRNA (molarity [nmol/L]) | |
| Mean±SD | Mean±SD | Mean | Mean±SD | Mean±SD | ||
| 40 mg/kg | Pre | 0.3±0.1 | 0.2±0.2 | 0.5 | 1.5±1.0 | 0.0±0.0 |
| Post | 5.7±2.4 | 5.4±2.8 | 2.1 | 14.3±7.8 | 17.4±7.2 | |
| 80 mg/kg | Pre | 0.6±0.8 | 0.4±0.7 | 0.6 | 1.8±2.4 | 0.0±0.0 |
| Post | 5.9±4.5 | 3.7±2.4 | 4.2 | 34.8±20.4 | 43.9±16.7 | |
| Overall | Pre | 0.4±0.6 | 0.3±0.5 | 0.6 | 1.7±1.8 | 0.0±0.0 |
| Post | 5.8±3.4 | 4.5±2.6 | 3.1 | 24.5±18.3 | 30.6±18.5 | |
A harmonized parallel study carried out in Japan recruited 16 participants with DMD randomized to 2 groups (40 mg/kg/wk; 80 mg/kg/wk) [64]. All subjects had a pre-treatment muscle biopsy, then 8 had a post-treatment biopsy after a 12-week treatment period, and the remaining 8 after a 24-week treatment period. A dose-related increase in dystrophin mRNA exon skipping was observed, as well as dose-related increase in dystrophin expression by both immunoblot and immunostaining (Table 1). The drug-related increase in dystrophin by immunoblot in the 80 mg/kg/wk group in the Japan trial (mean 4.8% normal) was similar to the findings with the same dose and treatment period in the US trial (mean 5.9% normal). Motor outcomes measures declined in both dose groups in the Japan study, in contrast to the US study, although the participants were older in the Japan study (mean 8.4 yrs) compared to US study (mean 7.4 yrs). Viltolarsen was granted accelerated approval based on dystrophin findings in both the US and Japan [64].
A PMO directed against exon 45 (casimersen) was approved by FDA in 2021. At the time of writing, there are no publications or FDA materials yet available to review the clinical trials that served as the basis for the approval.
LESSONS LEARNED AND NEXT STEPS
From the clinical findings to date, oligonucleotide drugs based on the 2OMePS chemistry appear to lack a sufficient therapeutic index to drive adequate levels of dystrophin without dose-limiting toxicities. Interestingly, nusinersen (Spinraza) approved for intrathecal administration for treatment of spinal muscular atrophy is based on 2OMOE, a chemistry similar to 2OMePS, but has not run into the dose-limiting toxicities that drisapersen did. This highlights the relevance of site of administration and frequency of dosing, with apparent impacts on both efficacy and safety. The intrathecal dosing of nusinersen likely provides a more targeted delivery to motor neurons, compared to the subcutaneous delivery of drispersen to skeletal muscle. The local delivery of nusinersen is done at a dose of 12 mg every 4 months, whereas drisapersen was dosed systemically at 6 mg/kg weekly; this likely leads to a much higher effective dose for motor neurons with a longer persistence of drug.
Oligonucleotide drugs for DMD based on the PMO chemistry (eteplirsen, golodirsen, viltolarsen, casimersen) show a broader therapeutic index, without the dose-limiting toxicities seen with other oligonucleotide chemistries. The morpholino backbone is not metabolized, and does not seem to trigger the innate immunity reactions triggered by the 2OMePS chemistries, which appears to contribute to greater safety of the PMO chemistry at higher doses (80 mg/kg/wk viltolarsen, compared to 6 mg/kg/wk drisapersen). That said, it is clear that only a very small fraction of the PMO drugs delivered by intravenous infusion reach the myofiber nucleus. Also, the mechanisms that have been defined for PMO delivery to skeletal muscle (intramuscular injection; systemic delivery via macrophages, myoblasts, and unstable myofiber membranes) are unlikely to provide dystrophin restoration for other cell types. Thus, the PMO technology may be a dystrophic muscle-specific chemistry. Multiple efforts are underway to achieve better delivery of oligonucleotides to muscle, including conjugates and muscle homing peptides.
In reviewing the experience with exon skipping to date, what are the lessons learned? First, it seems that the oligonucleotide delivery to myofibers in vivo remains a key variable that is relatively poorly understood. It is critical to understand this mechanism in order to optimize treatment (drug doses; frequency of dosing; mode of delivery). While studies of low levels of normal dystrophin (normal in molecular weight and biochemical composition) in some patients suggest that very low levels may have clinical benefit [15], the abnormal dystrophin (Becker-like in biochemical composition) is only partially functional. Thus, very low amounts of de novo dystrophin from therapeutic approaches (all biochemically abnormal) are expected to show less clinical benefit than the same amounts of biochemically normal dystrophin. Thus, published studies of clinical correlates of low levels of normal dystrophin are likely not relevant to similarly low levels of therapeutic dystrophin [68]. Thus, it is clear that exon skipping approaches will show more compelling evidence of clinical benefit if higher levels of drug-induced dystrophin can be obtained. Much higher levels of dystrophin have been induced by oligonucleotide approaches in mice and dog models of DMD, but these have utilized much higher doses of drug than are currently utilized in human studies (up to 10-fold higher). Efforts to optimize dosing regimens may be best studied in animal models, given the many variables to be explored [69].
Most patients with DMD show little or no dystrophin, and de novo expression of dystrophin may invoke cell-mediated immunity, as seen in murine dystrophin exon skipping studies [70], and with AAV viral gene delivery of dystrophin in the dog model of DMD [71]. Thus, efforts to monitor anti-dystrophin antibodies, prevent the onset of cell-mediated immunity, and mitigate possible clinical relevance of anti-dystrophin antibodies will be important going forward. Studies have shown that amniotic delivery of micro-dystrophin to CXMD dog fetuses may induce immune tolerance enabling later re-delivery of micro-dystrophin by AAV vectors [72].
As noted elsewhere in this review, all therapeutic efforts to restore dystrophin in DMD patient muscle involves semi-functional, biochemically abnormal types of dystrophin, not the full-length normal protein. We cannot expect the delivery of semi-functional dystrophin to ‘cure’ skeletal muscle to normal muscle tissue; success is defined as a Becker-type muscle and phenotype. As such, the dystrophic milieu of skeletal muscle will continue, even as high level production of de novo dystrophin become increasingly successful. The dystrophic milieu has been shown to lead to short-lived AAV-derived therapeutic mRNAs [73], and the pro-inflammatory state in Becker muscle leads to induction of microRNAs that bind to the dystrophin mRNA and inhibit dystrophin protein translation [74, 75]. It is likely that polypharmacy approaches will be needed to retain and stabilize Becker-like dystrophins at multiple levels.
Finally, it is increasingly clear that the skeletal muscle phenotype of a patient with DMD changes as a function of age, with early activation of innate immunity soon after birth, but later connective tissue replacement of specific muscles at different ages that drives functional disability [76, 77]. Expressing de novo dystrophin in connective tissue is unlikely to lead to any functional benefit, and some muscle groups in a boy with DMD already show connective tissue replacement even at young ages, depending on the muscle group. Thus, earlier treatment at younger ages is expected to show more clinical benefit over the disease course, assuming that the molecular pathways leading to connective tissue replacement can be slowed or stopped. Increased understanding of the transition from early successful muscle regeneration at young ages, to later unsuccessful regeneration and connective tissue replacement is critical [78].
In ending this overview, the authors wish to highlight the critical and ongoing contributions of Dr. Terence Partridge. Terry had major roles in the studies of systemic delivery to PMO drugs to the mdx mouse model [35, 36], showed that macrophages and myoblasts mediate delivery of PMO drugs to dystrophin-deficient myofibers [40], collaborated with the authors on the proof-of-concept systemic delivery studies of PMOs in the dog model [42], and noted that anti-dystrophin antibodies that may occur with drug-induced de novo dystrophin in mouse models [73]. Terry’s broad contributions and fundamental insights across the development of viltolarsen and other exon skipping drugs reflect his seminal contributions to translational muscle research, as well as his endearing and highly collaborative nature.
ACKNOWLEDGMENTS
Disclosures: S.T. has patents on sequences for exon skip by antisense nucleic acids as a member of NCNP together with Nippon Shinyaku. As principal inventor of these patents, S.T. is entitled to a share of royalties. S.T. discloses being an ad hoc consultant for Ono Pharmaceutical, Daiichisankyo, Asahikasei Pharma, Teijin Pharma, AGADA Biosciences, and Wave and being a member of the scientific advisory boards of Nippon Shinyaku, Taiho Pharma and Sarepta therapeutics. S.T. received speaker honoraria from Japan Health Science Foundation and Astellas Pharma and has also received research supports from Taiho Pharma, Daiichisankyo, Nippon Shinyaku, Takeda Pharmaceutical and the Noguchi Institute in the past 3 years.
Drs. Takeda, Clemens and Hoffman have been directly involved in the development of viltolarsen, and serve as consultants to the developer and marketer of viltolarsen, Nippon Shinyaku and NS Pharma. Drs. Clemens and Hoffman serve on the Board of TRiNDS LLC, a clinical contract research organization (CRO) that aids the design, conduct and management of clinical trials in neuromuscular disease, including viltolarsen. Dr. Hoffman is co-founder of AGADA BioSciences, a CRO that facilitates drug development in neuromuscular disease, including development of viltolarsen. Dr. Hoffman is co-founder and CEO of ReveraGen BioPharma, developer of vamorolone as a potential replacement for corticosteroids in inflammatory disease. Dr. Clemens holds contracts from ReveraGen for implementation of vamorolone trials and is on the advisory board for NS Pharma, Epirium, RegenXBio and Edgewise. Dr. Clemens has grants from the National Institutes of Health, NS Pharma, Sanofi Genzyme, Amicus and Spark.
REFERENCES
[1] | Muntoni F , Torelli S , Ferlini A . Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. (2003) ;2: (12):731–40. |
[2] | Gao QQ , McNally EM . The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr Physiol. (2015) ;5: (3):1223–39. doi: 10.1002/cphy.c140048 |
[3] | Pillers DA , Fitzgerald KM , Duncan NM , Rash SM , White RA , Dwinnell SJ , Powell BR , Schnur RE , Ray PN , Cibis GW , Weleber RG . Duchenne/Becker muscular dystrophy: Correlation of phenotype by electroretinography with sites of dystrophin mutations. Hum Genet. (1999) ;105: (1-2):2–9. doi: 10.1007/s004399900111 |
[4] | Bucher F , Friedlander MSH , Aguilar E , Kurihara T , Krohne TU , Usui Y , Friedlander M . The long dystrophin gene product Dp427 modulates retinal function and vascular morphology in response to age and retinal ischemia. Neurochem Int. (2019) ;129: :104489. doi: 10.1016/j.neuint.2019.104489 |
[5] | Taylor PJ , Betts GA , Maroulis S , Gilissen C , Pedersen RL , Mowat DR , Johnston HM , Buckley MF . Dystrophin gene mutation location and the risk of cognitive impairment in Duchenne muscular dystrophy. PLoS One. (2010) ;5: (1):e8803. doi: 10.1371/journal.pone.0008803 |
[6] | Naidoo M , Anthony K . Dystrophin Dp71 and the Neuropathophysiology of Duchenne Muscular Dystrophy. Mol Neurobiol. (2020) ;57: (3):1748–67. doi: 10.1007/s12035-019-01845-w |
[7] | Doorenweerd N , Mahfouz A , van Putten M , Kaliyaperumal R , T’ Hoen PAC , Hendriksen JGM , Aartsma-Rus AM , Verschuuren JJGM , Niks EH , Reinders MJT , Kan HE , Lelieveldt BPF . Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenne muscular dystrophy. Sci Rep. (2017) ;7: (1):12575. doi: 10.1038/s41598-017-12981-5 |
[8] | Hoffman EP , Kunkel LM , Angelini C , Clarke A , Johnson M , Harris JB . Improved diagnosis of Becker muscular dystrophy by dystrophin testing. Neurology. (1989) ;39: (8):1011–7. doi: 10.1212/wnl.39.8.1011 |
[9] | Anthony K , Cirak S , Torelli S , Tasca G , Feng L , Arechavala-Gomeza V , Armaroli A , Guglieri M , Straathof CS , Verschuuren JJ , Aartsma-Rus A , Helderman-van den Enden P , Bushby K , Straub V , Sewry C , Ferlini A , Ricci E , Morgan JE , Muntoni F . Dystrophin quantification and clinical correlations in Becker muscular dystrophy: Implications for clinical trials. Brain. (2011) ;134: (Pt 12):3547–59. doi: 10.1093/brain/awr291 |
[10] | Clemens PR , Niizawa G , Feng J , Florence J , D’Alessandro AS , Morgenroth LP , Gorni K , Guglieri M , Connolly A , Wicklund M , Bertorini T , Mah JK , Thangarajh M , Smith E , Kuntz N , McDonald CM , Henricson EK , Upadhyayula S , Byrne B , Manousakis G , Harper A , Bravver E , Iannaccone S , Spurney C , Cnaan A , Gordish-Dressman H ; CINRG BNHS Investigators. The CINRG Becker Natural History Study: Baseline characteristics. Muscle Nerve. (2020) ;62: (3):369–76. doi: 10.1002/mus.27011 |
[11] | Anthony K , Arechavala-Gomeza V , Ricotti V , Torelli S , Feng L , Janghra N , Tasca G , Guglieri M , Barresi R , Armaroli A , Ferlini A , Bushby K , Straub V , Ricci E , Sewry C , Morgan J , Muntoni F . Biochemical characterization of patients with in-frame or out-of-frame DMD deletions pertinent to exon 44 or 45 skipping. JAMA Neurol. (2014) ;71: (1):32–40. doi: 10.1001/jamaneurol.2013.4908 |
[12] | de Feraudy Y , Ben Yaou R , Wahbi K , Stalens C , Stantzou A , Laugel V , Desguerre I ; FILNEMUS Network, Servais L , Leturcq F , Amthor H . Very Low Residual Dystrophin Quantity Is Associated with Milder Dystrophinopathy. Ann Neurol. (2021) ;89: (2):280–92. doi: 10.1002/ana.25951 |
[13] | Anwar S , He M , Lim KRQ , Maruyama R , Yokota T . A Genotype-Phenotype Correlation Study of Exon Skip-Equivalent In-Frame Deletions and Exon Skip-Amenable Out-of-Frame Deletions across the DMD Gene to Simulate the Effects of Exon-Skipping Therapies: A Meta-Analysis. J Pers Med. (2021) ;11: (1):46. doi: 10.3390/jpm11010046 |
[14] | Bello L , Pegoraro E . The “Usual Suspects”: Genes for Inflammation, Fibrosis, Regeneration, and Muscle Strength Modify Duchenne Muscular Dystrophy. J Clin Med. (2019) ;8: (5):649. doi: 10.3390/jcm8050649 |
[15] | Hufton M , Roper H . Variations in Duchenne muscular dystrophy course in a multi-ethnic UK population: Potential influence of socio-economic factors. Dev Med Child Neurol. (2017) ;59: (8):837–42. doi: 10.1111/dmcn.13460 |
[16] | Bushby KM , Gardner-Medwin D . The clinical, genetic and dystrophin characteristics of Becker muscular dystrophy. I. Natural history. J Neurol. (1993) ;240: (2):98–104. doi: 10.1007/BF00858725. Erratum in: J Neurol 1993;240(7):453. |
[17] | Barp A , Bello L , Caumo L , Campadello P , Semplicini C , Lazzarotto A , Sorarú G , Calore C , Rampado A , Motta R , Stramare R , Pegoraro E . Muscle MRI and functional outcome measures in Becker muscular dystrophy. Sci Rep. (2017) ;7: (1):16060. doi: 10.1038/s41598-017-16170-2 |
[18] | van den Bergen JC , Wokke BH , Janson AA , van Duinen SG , Hulsker MA , Ginjaar HB , van Deutekom JC , Aartsma-Rus A , Kan HE , Verschuuren JJ . Dystrophin levels and clinical severity in Becker muscular dystrophy patients. J Neurol Neurosurg Psychiatry. (2014) ;85: (7):747–53. doi: 10.1136/jnnp-2013-306350 |
[19] | Wang B , Li J , Xiao X . Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc Natl Acad Sci U S A. (2000) ;97: (25):13714–9. doi: 10.1073/pnas.240335297 |
[20] | Sakamoto M , Yuasa K , Yoshimura M , Yokota T , Ikemoto T , Suzuki M , Dickson G , Miyagoe-Suzuki Y , Takeda S . Micro-dystrophin cDNA ameliorates dystrophic phenotypes when introduced into mdx mice as a transgene. Biochem Biophys Res Commun. (2002) ;293: (4):1265–72. doi: 10.1016/S0006-291X(02)00362-5 |
[21] | Yoshimura M , Sakamoto M , Ikemoto M , Mochizuki Y , Yuasa K , Miyagoe-Suzuki Y , Takeda S . AAV vector-mediated microdystrophin expression in a relatively small percentage of mdx myofibers improved the mdx phenotype. Mol Ther. (2004) ;10: (5):821–8. doi: 10.1016/j.ymthe.2004.07.025 |
[22] | Wang Z , Kuhr CS , Allen JM , Blankinship M , Gregorevic P , Chamberlain JS , Tapscott SJ , Storb R . Sustained AAV-mediated dystrophin expression in a canine model of Duchenne muscular dystrophy with a brief course of immunosuppression. Mol Ther. (2007) ;15: (6):1160–6. doi: 10.1038/sj.mt.6300161 |
[23] | Koo T , Okada T , Athanasopoulos T , Foster H , Takeda S , Dickson G . Long-term functional adeno-associated virus-microdystrophin expression in the dystrophic CXMDj dog. J Gene Med. (2011) ;13: (9):497–506. doi: 10.1002/jgm.1602 |
[24] | Yue Y , Pan X , Hakim CH , Kodippili K , Zhang K , Shin JH , Yang HT , McDonald T , Duan D . Safe and bodywide muscle transduction in young adult Duchenne muscular dystrophy dogs with adeno-associated virus. Hum Mol Genet. (2015) ;24: (20):5880–90. doi: 10.1093/hmg/ddv310 |
[25] | Mendell JR , Sahenk Z , Lehman K , Nease C , Lowes LP , Miller NF , Iammarino MA , Alfano LN , Nicholl A , Al-Zaidy S , Lewis S , Church K , Shell R , Cripe LH , Potter RA , Griffin DA , Pozsgai E , Dugar A , Hogan M , Rodino-Klapac LR . Assessment of Systemic Delivery of rAAVrh74. MHCK7.micro-dystrophin in Children With Duchenne Muscular Dystrophy: A Nonrandomized Controlled Trial. JAMA Neurol. (2020) ;77: (9):1122–1131. doi: 10.1001/jamaneurol.2020.1484 |
[26] | Willcocks RJ , Forbes SC , Walter GA , Sweeney L , Rodino-Klapac LR , Mendell JR , Vandenborne K . Assessment of rAAVrh. 74.MHCK7.micro-dystrophin Gene Therapy Using Magnetic Resonance Imaging in Children With Duchenne Muscular Dystrophy. JAMA Netw Open. (2021) ;4: (1):e2031851. doi: 10.1001/jamanetworkopen.2020.31851 |
[27] | Weber T . Anti-AAV Antibodies in AAV Gene Therapy: Current Challenges and Possible Solutions. Front Immunol. (2021) ;12: :658399. doi: 10.3389/fimmu.2021.658399 |
[28] | Salabarria SM , Nair J , Clement N , Smith BK , Raben N , Fuller DD , Byrne BJ , Corti M . Advancements in AAV-mediated Gene Therapy for Pompe Disease. J Neuromuscul Dis. (2020) ;7: (1):15–31. doi: 10.3233/JND-190426 |
[29] | Campbell C , Barohn RJ , Bertini E , Chabrol B , Comi GP , Darras BT , Finkel RS , Flanigan KM , Goemans N , Iannaccone ST , Jones KJ , Kirschner J , Mah JK , Mathews KD , McDonald CM , Mercuri E , Nevo Y , Péréon Y , Renfroe JB , Ryan MM , Sampson JB , Schara U , Sejersen T , Selby K , Tulinius M , Vílchez JJ , Voit T , Wei LJ , Wong BL , Elfring G , Souza M , McIntosh J , Trifillis P , Peltz SW , Muntoni F ; PTC124-GD-007-DMD Study Group; ACT DMD Study Group; Clinical Evaluator Training Groups. Meta-analyses of ataluren randomized controlled trials in nonsense mutation Duchenne muscular dystrophy. J Comp Eff Res. (2020) ;9: (14):973–84. doi: 10.2217/cer-2020-0095 |
[30] | Gushchina LV , Frair EC , Rohan NL , Bradley AJ , Chavan HD , Chou HJ , Eggers M , Waldrop MA , Wein N , Flanigan K . Lack of toxicity in non-human primates receiving clinically relevant doses of an AAV9.U7snRNA vector designed to induce DMD exon 2 skipping. Hum Gene Ther. 2021 Jan 6. doi: 10.1089/hum.2020.286 |
[31] | García-Rodríguez R , Hiller M , Jiménez-Gracia L , van der Pal Z , Balog J , Adamzek K , Aartsma-Rus A , Spitali P . Premature termination codons in the DMD gene cause reduced local mRNA synthesis. Proc Natl Acad Sci U S A. (2020) ;117: (28):16456–64. doi: 10.1073/pnas.1910456117 |
[32] | Mann CJ , Honeyman K , Cheng AJ , Ly T , Lloyd F , Fletcher S , Morgan JE , Partridge TA , Wilton SD . Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc Natl Acad Sci U S A. (2001) ;98: (1):42–7. doi: 10.1073/pnas.011408598 |
[33] | Lu QL , Mann CJ , Lou F , Bou-Gharios G , Morris GE , Xue SA , Fletcher S , Partridge TA , Wilton SD . Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat Med. (2003) ;9: (8):1009–14. doi: 10.1038/nm897 |
[34] | Alter J , Lou F , Rabinowitz A , Yin H , Rosenfeld J , Wilton SD , Partridge TA , Lu QL . Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med. (2006) ;12: (2):175–7. doi: 10.1038/nm1345 |
[35] | Hoffman EP , Bronson A , Levin AA , Takeda S , Yokota T , Baudy AR , Connor EM . Restoring dystrophin expression in duchenne muscular dystrophy muscle progress in exon skipping and stop codon read through. Am J Pathol. (2011) ;179: (1):12–22. doi: 10.1016/j.ajpath.2011.03.050 |
[36] | Heemskerk HA , de Winter CL , de Kimpe SJ , van Kuik-Romeijn P , Heuvelmans N , Platenburg GJ , van Ommen GJ , van Deutekom JC , Aartsma-Rus A . In vivo comparison of 2’-O-methyl phosphorothioate and morpholino antisense oligonucleotides for Duchenne muscular dystrophy exon skipping. J Gene Med. (2009) ;11: (3):257–66. doi: 10.1002/jgm.1288 |
[37] | Novak JS , Hogarth MW , Boehler JF , Nearing M , Vila MC , Heredia R , Fiorillo AA , Zhang A , Hathout Y , Hoffman EP , Jaiswal JK , Nagaraju K , Cirak S , Partridge TA . Myoblasts and macrophages are required for therapeutic morpholino antisense oligonucleotide delivery to dystrophic muscle. Nat Commun. (2017) ;8: (1):941. doi: 10.1038/s41467-017-00924-7 |
[38] | Wu B , Lu P , Benrashid E , Malik S , Ashar J , Doran TJ , Lu QL . Dose-dependent restoration of dystrophin expression in cardiac muscle of dystrophic mice by systemically delivered morpholino. Gene Ther. (2010) ;17: (1):132–40. doi: 10.1038/gt.2009.120 |
[39] | Yokota T , Lu QL , Partridge T , Kobayashi M , Nakamura A , Takeda S , Hoffman E . Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol. (2009) ;65: (6):667–76. doi: 10.1002/ana.21627 |
[40] | Clemens PR , Rao VK , Connolly AM , Harper AD , Mah JK , Smith EC , McDonald CM , Zaidman CM , Morgenroth LP , Osaki H , Satou Y , Yamashita T , Hoffman EP ; CINRG DNHS Investigators. Safety, Tolerability, and Efficacy of Viltolarsen in Boys With Duchenne Muscular Dystrophy Amenable to Exon 53 Skipping: A Phase 2 Randomized Clinical Trial. JAMA Neurol. (2020) ;77: (8):982–91. doi: 10.1001/jamaneurol.2020.1264 |
[41] | Komaki H , Takeshima Y , Matsumura T , Ozasa S , Funato M , Takeshita E , Iwata Y , Yajima H , Egawa Y , Toramoto T , Tajima M , Takeda S . Viltolarsen in Japanese Duchenne muscular dystrophy patients: A phase 1/2 study. Ann Clin Transl Neurol. (2020) ;7: (12):2393–408. doi: 10.1002/acn3.51235 |
[42] | van Deutekom JC , Janson AA , Ginjaar IB , Frankhuizen WS , Aartsma-Rus A , Bremmer-Bout M , den Dunnen JT , Koop K , van der Kooi AJ , Goemans NM , de Kimpe SJ , Ekhart PF , Venneker EH , Platenburg GJ , Verschuuren JJ , van Ommen GJ . Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med. (2007) ;357: (26):2677–86. doi: 10.1056/NEJMoa073108 |
[43] | Goemans NM , Tulinius M , van den Akker JT , Burm BE , Ekhart PF , Heuvelmans N , Holling T , Janson AA , Platenburg GJ , Sipkens JA , Sitsen JM , Aartsma-Rus A , van Ommen GJ , Buyse G , Darin N , Verschuuren JJ , Campion GV , de Kimpe SJ , van Deutekom JC . Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N Engl J Med. (2011) ;364: (16):1513–22. doi: 10.1056/NEJ-Moa1011367 |
[44] | Goemans N , Tulinius M , Kroksmark AK , Wilson R , van den Hauwe M , Campion G . Comparison of ambulatory capacity and disease progression of Duchenne muscular dystrophy subjects enrolled in the drisapersen DMD73 study with a matched natural history cohort of subjects on daily corticosteroids. Neuromuscul Disord. (2017) ;27: (3):203–213. doi: 10.1016/j.nmd.2016.11.013 |
[45] | Voit T , Topaloglu H , Straub V , Muntoni F , Deconinck N , Campion G , De Kimpe SJ , Eagle M , Guglieri M , Hood S , Liefaard L , Lourbakos A , Morgan A , Nakielny J , Quarcoo N , Ricotti V , Rolfe K , Servais L , Wardell C , Wilson R , Wright P , Kraus JE . Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): An exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. (2014) ;13: (10):987–96. doi: 10.1016/S1474-4422(14)70195-4 |
[46] | McDonald CM , Wong B , Flanigan KM , Wilson R , de Kimpe S , Lourbakos A , Lin Z , Campion G ; DEMAND V study group. Placebo-controlled Phase 2 Trial of Drisapersen for Duchenne Muscular Dystrophy. Ann Clin Transl Neurol. (2018) ;5: (8):913–926. doi: 10.1002/acn3.579 |
[47] | Goemans N , Mercuri E , Belousova E , Komaki H , Dubrovsky A , McDonald CM , Kraus JE , Lourbakos A , Lin Z , Campion G , Wang SX , Campbell C ; DEMAND III study group. A randomized placebo-controlled phase 3 trial of an antisense oligonucleotide drisapersen, in Duchenne muscular dystrophy. Neuromuscul Disord. (2018) ;28: (1):4–15. doi: 10.1016/j.nmd.2017.10.004 |
[48] | Hilhorst N , Spanoudi-Kitrimi I , Goemans N , Morren MA . Injection site reactions after long-term subcutaneous delivery of drisapersen: A retrospective study. Eur J Pediatr. (2019) ;178: (2):253–8. doi: 10.1007/s00431-018-3272-1 |
[49] | Kinali M , Arechavala-Gomeza V , Feng L , Cirak S , Hunt D , Adkin C , Guglieri M , Ashton E , Abbs S , Nihoyannopoulos P , Garralda ME , Rutherford M , McCulley C , Popplewell L , Graham IR , Dickson G , Wood MJ , Wells DJ , Wilton SD , Kole R , Straub V , Bushby K , Sewry C , Morgan JE , Muntoni F . Local restoration of dystrophin expression with the morpholino oligomer AVI-in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. (2009) ;8: (10):918–28. doi: 10.1016/S1474-4422(09)70211-X |
[50] | Cirak S , Arechavala-Gomeza V , Guglieri M , Feng L , Torelli S , Anthony K , Abbs S , Garralda ME , Bourke J , Wells DJ , Dickson G , Wood MJ , Wilton SD , Straub V , Kole R , Shrewsbury SB , Sewry C , Morgan JE , Bushby K , Muntoni F . Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet. (2011) ;378: (9791):595–605. doi: 10.1016/S0140-6736(11)60756-3 |
[51] | Cirak S , Feng L , Anthony K , Arechavala-Gomeza V , Torelli S , Sewry C , Morgan JE , Muntoni F . Restoration of the dystrophin-associated glycoprotein complex after exon skipping therapy in Duchenne muscular dystrophy. Mol Ther. (2012) ;20: (2):462–7. doi: 10.1038/mt.2011.248 |
[52] | Mendell JR , Rodino-Klapac LR , Sahenk Z , Roush K , Bird L , Lowes LP , Alfano L , Gomez AM , Lewis S , Kota J , Malik V , Shontz K , Walker CM , Flanigan KM , Corridore M , Kean JR , Allen HD , Shilling C , Melia KR , Sazani P , Saoud JB , Kaye EM ; Eteplirsen Study Group. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. (2013) ;74: (5):637–47. doi: 10.1002/ana.23982 |
[53] | Charleston JS , Schnell FJ , Dworzak J , Donoghue C , Lewis S , Chen L , Young GD , Milici AJ , Voss J , DeAlwis U , Wentworth B , Rodino-Klapac LR , Sahenk Z , Frank D , Mendell JR . Eteplirsen treatment for Duchenne muscular dystrophy: Exon skipping and dystrophin production. Neurology. (2146) ;90: (24):e2146–e2154. doi: 10.1212/WNL.0000000000005680 |
[54] | https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/206488_summary%20review_redacted.pdf |
[55] | Kesselheim AS , Avorn J . Approving a Problematic Muscular Dystrophy Drug: Implications for FDA Policy. JAMA. (2016) ;316: (22):2357–8. doi: 10.1001/jama.2016.16437 |
[56] | The Lancet. Patient need versus evidence: A balancing act. Lancet. (2016) ;388: (10052):1350. doi: 10.1016/S0140-6736(16)31765-2 |
[57] | Unger EF , Califf RM . Regarding “Eteplirsen for the treatment of Duchenne muscular dystrophy”. Ann Neurol. (2017) ;81: (1):162–4. doi: 10.1002/ana.24842 |
[58] | Echigoya Y , Lim KRQ , Trieu N , Bao B , Miskew Nichols B , Vila MC , Novak JS , Hara Y , Lee J , Touznik A , Mamchaoui K , Aoki Y , Takeda S , Nagaraju K , Mouly V , Maruyama R , Duddy W , Yokota T . Quantitative Antisense Screening and Optimization for Exon 51 Skipping in Duchenne Muscular Dystrophy. Mol Ther. (2017) ;25: (11):2561–72. doi: 10.1016/j.ymthe.2017.07.014 |
[59] | Aartsma-Rus A , Goemans N . A Sequel to the Eteplirsen Saga: Eteplirsen Is Approved in the United States but Was Not Approved in Europe. Nucleic Acid Ther. (2019) ;29: (1):13–15. doi: 10.1089/nat.2018.0756 |
[60] | Frank DE , Schnell FJ , Akana C , El-Husayni SH , Desjardins CA , Morgan J , Charleston JS , Sardone V , Domingos J , Dickson G , Straub V , Guglieri M , Mercuri E , Servais L , Muntoni F ; SKIP-NMD Study Group. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology. (2270) ;94: (21):e2270–e2282. doi: 10.1212/WNL.0000000000009233 |
[61] | https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/211970Orig1s000SumR.pdf |
[62] | Watanabe N , Nagata T , Satou Y , Masuda S , Saito T , Kitagawa H , Komaki H , Takagaki K , Takeda S . NS-065/NCNP- An Antisense Oligonucleotide for Potential Treatment of Exon 53 Skipping in Duchenne Muscular Dystrophy. Mol Ther Nucleic Acids. (2018) ;13: :442–449. doi: 10.1016/j.omtn.2018.09.017 |
[63] | Komaki H , Nagata T , Saito T , Masuda S , Takeshita E , Sasaki M , Tachimori H , Nakamura H , Aoki Y , Takeda S . Systemic administration of the antisense oligonucleotide NS-065/NCNP-01 for skipping of exon 53 in patients with Duchenne muscular dystrophy. Sci Transl Med. (2018) ;10: (437):eaan0713. doi: 10.1126/scitranslmed.aan0713 |
[64] | Komaki H , Takeshima Y , Matsumura T , Ozasa S , Funato M , Takeshita E , Iwata Y , Yajima H , Egawa Y , Toramoto T , Tajima M , Takeda S . Viltolarsen in Japanese Duchenne muscular dystrophy patients: A phase 1/2 study. Ann Clin Transl Neurol. (2020) ;7: (12):2393–408. doi: 10.1002/acn3.51235 |
[65] | Uaesoontrachoon K , Srinivassane S , Warford J , Mekhssian K , Montpetit H , Beauvois R , Keyhani A , Hathout Y , Yamashita T , Satou Y , Osaki H , Praest M , Moraca M , Malbasic M , Ross W , MacKinnon A , Rowsell J , Mullen A , Matyas M , Mummidivarpu S , Nagaraju K , Hoffman EP . Orthogonal analysis of dystrophin protein and mRNA as a surrogate outcome for drug development. Biomark Med. (2019) ;13: (14):1209–25. doi: 10.2217/bmm-2019-0242 |
[66] | McDonald CM , Henricson EK , Abresch RT , Han JJ , Escolar DM , Florence JM , Duong T , Arrieta A , Clemens PR , Hoffman EP , Cnaan A ; Cinrg Investigators. The cooperative international neuromuscular research group Duchenne natural history study–a longitudinal investigation in the era of glucocorticoid therapy: Design of protocol and the methods used. Muscle Nerve. (2013) ;48: (1):32–54. doi: 10.1002/mus.23807 |
[67] | McDonald CM , Henricson EK , Abresch RT , Duong T , Joyce NC , Hu F , Clemens PR , Hoffman EP , Cnaan A , Gordish-Dressman H ; CINRG Investigators. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: A prospective cohort study. Lancet. (2018) ;391: (10119):451–61. doi: 10.1016/S0140-6736(17)32160-8 |
[68] | Hoffman EP and Clemens PR . Letter to the Editor. Annals Neurology in press. |
[69] | Malerba A , Thorogood FC , Dickson G , Graham IR . Dosing regimen has a significant impact on the efficiency of morpholino oligomer-induced exon skipping in mdx mice. Hum Gene Ther. (2009) ;20: (9):955–65. doi: 10.1089/hum.2008.157 |
[70] | Vila MC , Novak JS , Benny Klimek M , Li N , Morales M , Fritz AG , Edwards K , Boehler JF , Hogarth MW , Kinder TB , Zhang A , Mazala D , Fiorillo AA , Douglas B , Chen YW , van den Anker J , Lu QL , Hathout Y , Hoffman EP , Partridge TA , Nagaraju K . Morpholino-induced exon skipping stimulates cell-mediated and humoral responses to dystrophin in mdx mice. J Pathol. (2019) ;248: (3):339–51. doi: 10.1002/path.5263 |
[71] | Yuasa K , Yoshimura M , Urasawa N , Ohshima S , Howell JM , Nakamura A , Hijikata T , Miyagoe-Suzuki Y , Takeda S . Injection of a recombinant AAV serotype 2 into canine skeletal muscles evokes strong immune responses against transgene products. Gene Ther. (2007) ;14: (17):1249–60. doi: 10.1038/sj.gt.3302984 |
[72] | Hayashita-Kinoh H , Yugeta N , Okada H , Nitahara-Kasahara Y , Chiyo T , Okada T , Takeda S . Intra-amniotic rAAV-mediated microdystrophin gene transfer improves canine X-linked muscular dystrophy and may induce immune tolerance. Mol Ther. (2015) ;23: (4):627–37. doi: 10.1038/mt.2015.5 |
[73] | Dupont JB , Tournaire B , Georger C , Marolleau B , Jeanson-Leh L , Ledevin M , Lindenbaum P , Lecomte E , Cogné B , Dubreil L , Larcher T , Gjata B , Van Wittenberghe L , Le Guiner C , Penaud-Budloo M , Snyder RO , Moullier P , Léger A . Short-lived recombinant adeno-associated virus transgene expression in dystrophic muscle is associated with oxidative damage to transgene mRNA. Mol Ther Methods Clin Dev. (2015) ;2: :15010. doi: 10.1038/mtm.2015.10 |
[74] | Fiorillo AA , Heier CR , Novak JS , Tully CB , Brown KJ , Uaesoontrachoon K , Vila MC , Ngheim PP , Bello L , Kornegay JN , Angelini C , Partridge TA , Nagaraju K , Hoffman EP . TNF-α-Induced microRNAs Control Dystrophin Expression in Becker Muscular Dystrophy. Cell Rep. (2015) ;12: (10):1678–90. doi: 10.1016/j.celrep.2015.07.066 |
[75] | Kinder TB , Heier CR , Tully CB , Van der Muelen JH , Hoffman EP , Nagaraju K , Fiorillo AA . Muscle Weakness in Myositis: MicroRNA-Mediated Dystrophin Reduction in a Myositis Mouse Model and Human Muscle Biopsies. Arthritis Rheumatol. (2020) ;72: (7):1170–1183. doi: 10.1002/art.41215 |
[76] | Chen YW , Nagaraju K , Bakay M , McIntyre O , Rawat R , Shi R , Hoffman EP . Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology. (2005) ;65: (6):826–34. doi: 10.1212/01.wnl.0000173836.09176.c4 |
[77] | Rooney WD , Berlow YA , Triplett WT , Forbes SC , Willcocks RJ , Wang DJ , Arpan I , Arora H , Senesac C , Lott DJ , Tennekoon G , Finkel R , Russman BS , Finanger EL , Chakraborty S , O’Brien E , Moloney B , Barnard A , Sweeney HL , Daniels MJ , Walter GA , Vandenborne K . Modeling disease trajectory in Duchenne muscular dystrophy. Neurology. (2020) ;94: (15):e1622–e1633. doi: 10.1212/WNL.0000000000009244 |
[78] | Uezumi A , Ito T , Morikawa D , Shimizu N , Yoneda T , Segawa M , Yamaguchi M , Ogawa R , Matev MM , Miyagoe-Suzuki Y , Takeda S , Tsujikawa K , Tsuchida K , Yamamoto H , Fukada S . Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J Cell Sci. (2011) ;124: (Pt 21):3654–64. doi: 10.1242/jcs.086629 |


