
Gene Therapy for Duchenne Muscular Dystrophy
Abstract
Duchenne muscular dystrophy (DMD) is an X-linked, muscle wasting disease that affects 1 in 5000 males. Affected individuals become wheelchair bound by the age of twelve and eventually die in their third decade due to respiratory and cardiac complications. The disease is caused by mutations in the DMD gene that codes for dystrophin. Dystrophin is a structural protein that maintains the integrity of muscle fibres and protects them from contraction-induced damage. The absence of dystrophin compromises the stability and function of the muscle fibres, eventually leading to muscle degeneration. So far, there is no effective treatment for deteriorating muscle function in DMD patients. A promising approach for treating this life-threatening disease is gene transfer to restore dystrophin expression using a safe, non-pathogenic viral vector called adeno-associated viral (AAV) vector. Whilst microdystrophin gene transfer using AAV vectors shows extremely impressive therapeutic success so far in large animal models of DMD, translating this advanced therapy medicinal product from bench to bedside still offers scope for many optimization steps. In this paper, the authors review the current progress of AAV-microdystrophin gene therapy for DMD and other treatment strategies that may apply to a subset of DMD patients depending on the mutations they carry.
DISCOVERY AND CHARACTERISATION OF DMD: HISTORICAL PERSPECTIVES
Identification of muscular dystrophy as a primary disease of muscles happened in the 19th century and this distinguished muscular dystrophies from diseases where muscle weakness was secondary to disease of motor neurons and their roots [1]. DMD is one of this family of conditions and is a lethal, muscle wasting disease that affects 1 in 5000 male births [2]. The disease was originally described by Edward Meryon (1807–1880) and was noted by Emery as a muscle disease that caused the integrity of the sarcolemma to be compromised [3]. Meryon followed cases of nine boys in three different families and noted some of the clinical descriptions of the progressive muscle wasting such as enlargement of calf muscles [1]. The disease was later further characterised by Guillaume B.A. Duchenne de Boulogne (1806–1875) as he performed comprehensive evaluation of the first patient believed to be affected by “progressive muscular atrophy with degeneration” (which was later named Duchenne muscular dystrophy) [1]. As Duchenne de Boulogne conducted further pathological studies, he changed the initial description of the disease to “pseudo-hypertrophic muscular paralysis” or “myo-sclerotic paralysis” [1]. Sir William Richard Gowers later added to the clinical picture of DMD by reporting the inherited nature of the disease, its preferred bias towards males and the characteristic way in which patients rise from the floor to the standing position (called The Gower’s Sign) [4].
In one of the first applications of the new molecular genetics of positional cloning, Koenig et al. discovered the DMD gene to be associated with the disease phenotype [5]. The protein product of the DMD gene was called dystrophin [6]. The progressive X-linked disease is degenerative in nature and patients become wheelchair bound in their teens. In addition to respiratory insufficiency and lung infection, cardiomyopathy also contributes in a major way to the early death of DMD patients [7]. In previous decades, DMD patients used to die in their teens, but the use of respiratory and cardiac support have however contributed to extended lifespan and survival of patients often even into the 30 s [8]. A combination of physiotherapy and corticosteroids have also helped to improve the quality of life of patients; nevertheless these palliative treatments are not a cure. Survival of patients into their fourth decade is rare [9, 10].
FROM GENOTYPE TO PHENOTYPE: THE DMD GENE AND DYSTROPHIN
The DMD gene is one of the largest protein-coding gene in the human genome, covering over 2.6 million base pairs with 79 exons that code for a family of dystrophin protein isoforms [11]. The large size of the gene makes it prone to mutations such as deletions (about 60%), duplications (about 6%), translocations and point mutations which disrupts the reading frame and eventually causes abnormal truncated dystrophin fragments to be synthesised [11]. The large 427 kD skeletal and cardiac muscle isoform of dystrophin is a cytoplasmic sarcolemmal protein that links the extracellular matrix (ECM) and cortical cytoskeleton [12]. Dystrophin is widely expressed in skeletal, cardiac and smooth muscle, and a small amount at specific locations in the CNS [13, 14]. Dystrophin is localised at the sarcolemma via the dystrophin-associated protein complex (DAPC) (Fig. 1) [12].
Fig. 1
Full length dystrophin and dystrophin-associated protein complex (DAPC). The dystrophin protein contains an actin binding domain (which is also the N-terminal domain), a rod domain made of 24 spectrin repeats (labelled R), four hinges (labelled H1-H4), a cysteine-rich domain and a C-terminal domain. Dystrophin is attached to the DAPC via the cysteine-rich domain (which bind dystroglycan) and C terminus (which bind syntrophins and dystrobrevin), linking the internal cytoskeleton and extracellular matrix (12) (13). R16 and R17 contains nNOS binding site that is important for localisation of nNOS at the sarcolemma (19).
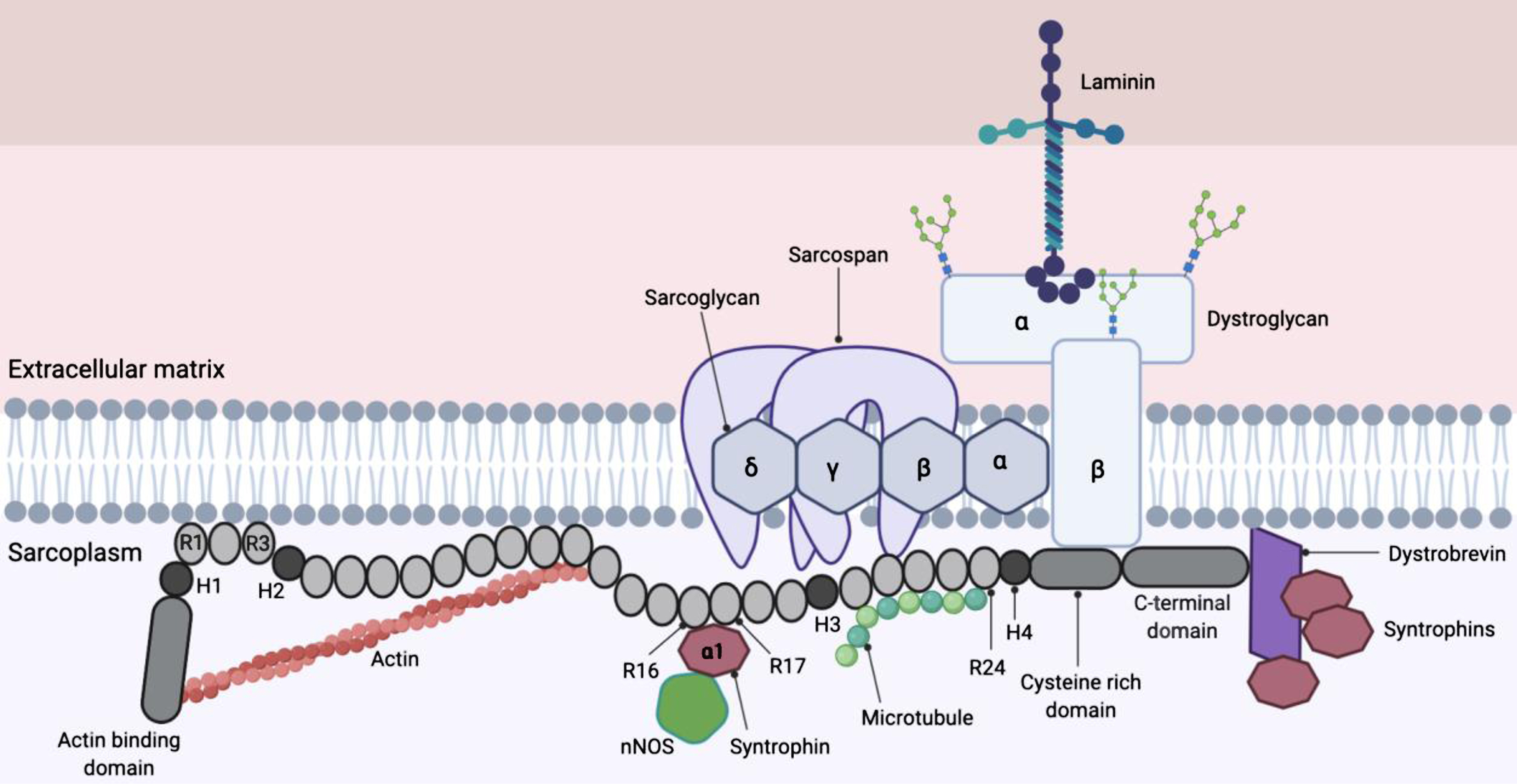
The structural role that dystrophin plays by linking the cytoskeleton to the ECM is thought to allow the transmission of force from the contractile elements inside the cell to extracellular matrix structures, while at the same time maintaining the integrity of the muscle fibers [15]. Syntrophins recruits multiple proteins to the DAPC (shown in Fig. 1) as well as neuronal nitric oxide synthase (nNOS) to the sarcolemma [16]. The rod domain of dystrophin also contains a nNOS binding domain (R16 and R17) that is necessary to localise nNOS at the sarcolemma [17, 18]. This phenomenon is believed to enable immediate diffusion of nitric oxide into the blood vessels to promote vasodilation in contracting muscles, allowing sufficient blood perfusion [19]. The delocalisation of nNOS due to absence of functional dystrophin has been proposed to cause (i) functional ischaemia and (ii) nitrosative stress that damages cells, compromising force production in the muscle [19].
The DAPC consists of multiple transmembrane, cytoplasmic and extracellular proteins and in DMD patients, the absence of functional dystrophin destabilises the DAPC [20, 21]. Cell signalling mediated by the dystrophin-glycoprotein complex are also disrupted due to the loss of dystrophin [22]. Many proteins found within the DAPC including alpha-dystrobrevin, sarcoglycan, dystroglycan, and sarcospan are essential components in cellular signalling that maintain healthy muscle function and development [10], and when mutated are associated with distinct muscular dystrophies in their own right.
In the absence of functional dystrophin in skeletal and cardiac muscle, the integrity of muscle cell membrane (also known as sarcolemma) is proposed to be affected, making the muscle fibers more prone to damage induced by contractile activity [23]. The absence of functional dystrophin protein due to mutations in the DMD gene leads to progressive muscle wasting in patients, with loss of myofibers and replacement with fatty and fibrotic tissue [24].
A less severe form of dystrophinopathy is called Becker muscular dystrophy (BMD) [8]. Like DMD, BMD is also caused by mutations in the same X-linked gene, however the disease is much milder than DMD and the age of onset of BMD can be as late as 30 or 40 years [25]. The transcript reading frame flanking mutations is not disturbed in BMD but the aberrant transcripts code for shorter dystrophins forms that are partially and variably functional, and often with reduced expression and/or stability [11]. In patients with deletion mutations, the ‘reading frame’ hypothesis suggests that semi-functional, internally deleted dystrophin proteins are produced in BMD patients giving rise to a less severe form of dystrophinopathy [26]. One striking case study has revealed a 61-year old BMD patient who was still ambulant even though 46% of the coding information was lost in the deletion in his DMD gene [25]. On the other hand, the ‘reading frame’ hypothesis suggests that in DMD patients, a truncated protein that is unstable is produced, causing the more severe form of dystrophinopathy [26]. An analysis of 258 independent deletion mutations at the DMD/BMD locus revealed that there was a correlation between phenotype and type of deletion mutations in 92% of cases; and in fact, many “in-frame” deletions of the DMD gene are not detected as the individuals with such mutations are either asymptomatic or exhibit non-DMD/non-BMD clinical features [26]. This finding was not only intriguing suggesting size of deletions in the DMD gene are not necessarily related to severity of the disease, but also revealed the possibility of manipulating and engineering the large DMD transcript in several gene therapy and antisense strategies to restore muscle function in affected patients.
ADENO-ASSOCIATED VIRUS (AAV) AS A GENE THERAPY VECTOR
Originally discovered by accident in a preparation of simian adenovirus [27], AAV was soon found to be present in human tissues [28]. A nursery population (infants six months to three years of age) at Junior Village, Washington D.C. that experienced frequent infection with various adenovirus serotypes provided the opportunity to isolate and characterise AAV strains of human origin [28]. The journey of understanding the basic biology of AAV was driven purely by scientific curiosity, at a time when the potential of this virus as a gene therapy vector was unknown [29]. Nineteen years later in 1984, AAV was vectorised for the purpose of gene transfer [30]. Eleven years following this, the viral vector was first used in a cystic fibrosis human patient in 1995 [31]. Fast forwarding the timeline, the EMA approved the first AAV-based gene therapy drug Glybera for the treatment of lipoprotein lipase deficiency in 2012 [32], followed by the approval of Luxturna by the FDA in 2017 for the treatment of an inherited retinal disease [33] and another FDA approval of Zolgensma in 2019 for the treatment of spinal muscular atrophy [34].
A further breakthrough in the field came when Samulski and colleagues described a method for the production of recombinant AAV (rAAV) using a so-called two-plasmid system, where the final virus stock contained no wild type AAV (wtAAV) [35]. In this system the rep and cap genes on the wtAAV genome are replaced with genes of interest to produce rAAV [36]. The virus inverted terminal repeats (ITRs), are however required in cis for replication and packaging of rAAV [37] and as such, the ITRs are the only sequences of wtAAV origin that are incorporated into rAAV [29]. The rep, cap and helper virus genes are provided in trans [37]. Importantly, and unlike the wtAAV, recombinant AAVs do not undergo site-specific integration into the human chromosome AAVS1 locus, and the bulk of rAAV genomes exist as extra-chromosomal episomes in transduced cells [38]. This very robust and popular method of rAAV production is via plasmid transfection of adherent human embryonic kidney 293 (HEK293) cells, where the transgene in between ITRs is delivered in cis and the rep, cap and helper genes are delivered in trans [29]. A modified version of this method is suspension culture of HEK293 that allows rapid production of rAAV and scaling-up the yield which are essential in clinical applications [39]. Following production, rAAVs are purified via either column chromatography or gradient centrifugation; and it is worth mentioning that the purity differs based on the method used which can impact the outcome of preclinical and clinical studies [40].
The diversity and occurrence of multiple AAV serotypes in human and non-human primate tissues was first elucidated by studies performed by Gao et al. [41]. Epidemiological studies show that 40–80% of the human population contain antibodies against AAV indicating previous exposure to the virus, and some AAV serotypes, AAV1, AAV2, AAV3, AAV5, AAV6, AAV7, AAV8 and AAV9, are endemic to humans [29].
Variations between studies pose some difficulties in precisely establishing AAV serotype tissue tropism [42]. The tropism of the viral capsids are largely due to presence of respective viral receptors on target cells, which can vary between species [43, 44]. The difference in receptor expression between species causes variations in potency of different AAV capsids, a fact that is sometimes overlooked in translational studies [44].
MICRO-DYSTROPHIN GENE THERAPY: SHRINKING THE DMD GENE
The mRNA ORF coding sequence of the DMD gene is some 11.5 kb long and this large size poses a huge challenge in devising gene transfer therapies [19]. The observations made in BMD patients have however suggested that transfer of full-length dystrophin ORF may not be obligatory in developing a gene therapy and the disease phenotype may be alleviated with a smaller gene construct [25]. Whilst AAVs possess many desirable qualities for use in muscle gene therapy, the small virus is only capable of packaging genomes and transgenes of limited size (respectively ∼4.7 kb and ∼4.5 kb) [45–47].
The availability of the full-length sequence of dystrophin and the knowledge of corresponding protein domains have allowed scientists to design the careful removal of some coding sequences within the gene, while aiming to maximise retention of protein function. As a result, recombinant genes encoding multiple variants of mini-/ micro-dystrophins with clinical potential have been generated [9, 48, 49, 50] (Fig. 2). Large animal models of DMD have shown significant patho-physiological improvements following systemic delivery of engineered micro-dystrophins using AAV, leading to human clinical trials [19]. The successful single-dose gene-replacement therapy for Spinal Muscular Atrophy (SMA) using AAV with dosage up to 2.0×1014 vg per kilogram, has provided proof-of-principle for systemic gene transfer using AAV in humans [19, 51]. Three clinical trials are currently ongoing in the United States by Sarepta Therapeutics, Pfizer and Solid Biosciences, each with a slightly different mini-/ micro-dystrophin constructs delivered using AAVs of differing serotype (rh74 and AAV9 respectively, Fig. 2). Another clinical trial in Europe using an AAV8-microdystrophin by Genethon in collaboration with Sarepta Therapeutics began in April 2021, where the first patient was dosed with the investigational gene therapy drug GNT 0004 [52–54]. It is also worth noting that AAV microdystrophin gene therapy treatment strategy is applicable to all DMD patients regardless of the mutations they carry in their DMD gene.
Fig. 2
Clinically relevant miniaturised dystrophin constructs. (Top) The promoters utilised in the therapeutic constructs are shown by yellow arrows. These promoters are shown to be muscle and heart specific and will preferentially express the transgene in those tissues. The transgene of all the constructs contain the coding sequence for protein domains that are clinically relevant and functional. The AAV serotype of choice is also based on tissue tropism, where they are muscle and heart tropic. Clinical constructs for Sarepta Therapeutics (in partnership with Roche), Pfizer, Solid Biosciences and Genethon adapted from (48) (49) (50) and (9) respectively. ABD: Actin-binding domain; H: hinge; R: rod; CRD: cysteine-rich domain; CTD: C-terminal domain. (a) The localisation of microdystrophin-1 (MD1) protein (utilised by Sarepta Therapeutics and Genethon) to the DAPC. (b) The localisation of mini-dystrophin protein utilised by Pfizer to the DAPC. There remains a possibility of the recruitment of nNOS to the sarcolemma by the mini-dystrophin via an unknown mechanism (114) (115). (c) The localisation of microdystrophin protein utilised by Solid Biosciences to the DAPC.
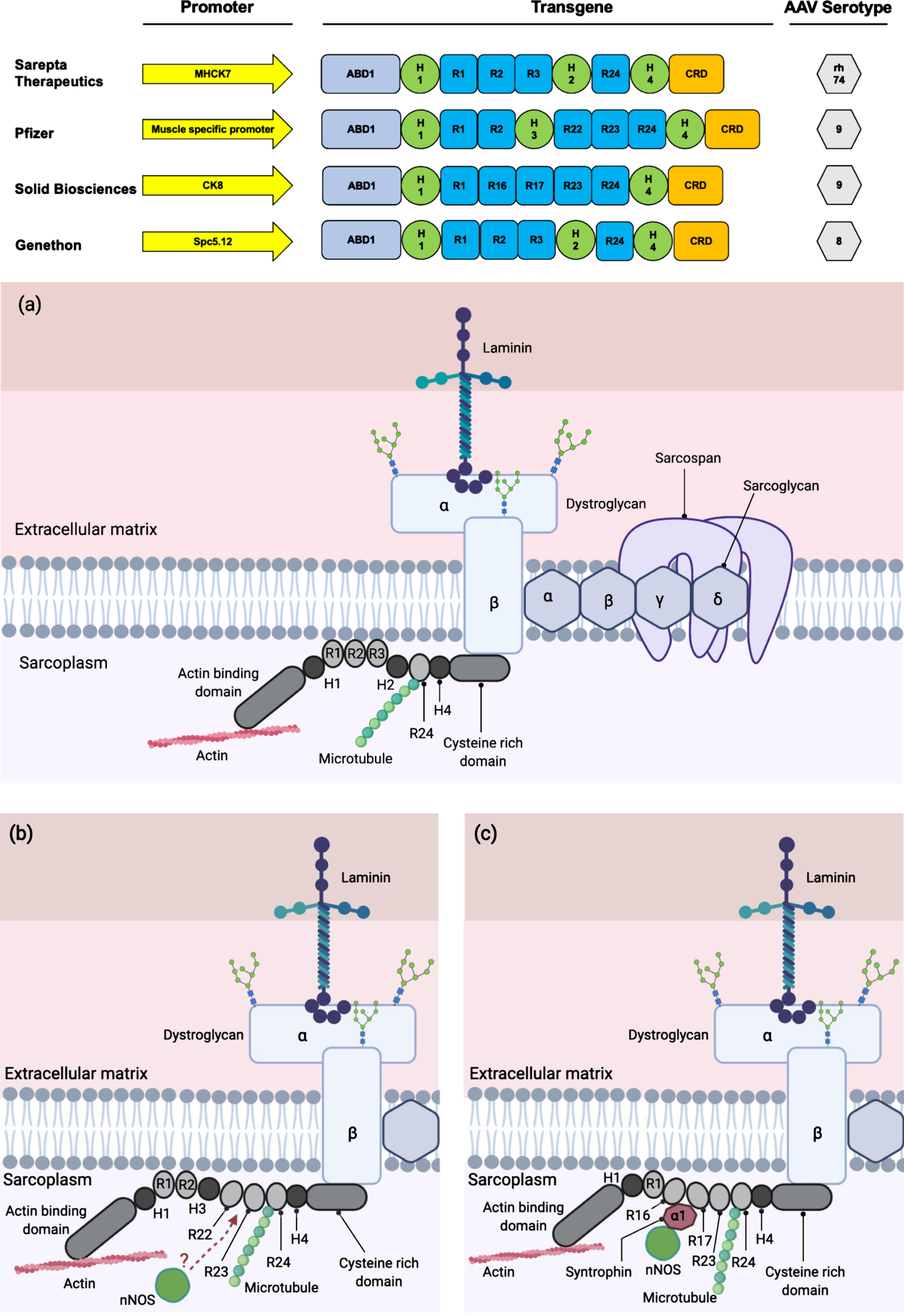
Some encouraging data have emerged from the clinical trials, notably from Sarepta Therapeutics and Pfizer (Table 1). These data provide proof-of-principle that AAV gene transfer to target the human muscle is possible from a clinical and manufacturing perspective [55]. Whilst translating these advanced therapy medicinal products (ATMPs) from bench to bedside has been possible and successful in many settings [32–34], a range of issues still remain to be tackled to fully optimise dosage, immune control and manufacture and supply challenges in AAV gene therapy for DMD.
Table 1
Clinical trial history and early data on outcomes of the AAV-microdystrophin trials
| Intervention | Clinical trial history | Outcome evaluation | Ref |
| SGT-001 (Solid Biosciences) | •Low dose of 5E13 vg/kg was administered in the first cohort of phase 1/2 IGNITE-DMD trial. First patient suffered serious adverse effect (SAE) which was resolved. •Age matched dose escalation study has been initiated with a higher dose of 2E14 vg/kg. Patient in the treatment group suffered SAE which was resolved with an increase in oral glucocorticoids. •Patients are currently being enrolled. | •Variable amounts of microdystrophin (5% to 50% of normal dystrophin) was present in muscle biopsies. •After an FDA clinical halt was released, patient dosing continued with modified manufacturing and trial protocols. No subsequent SAEs reported. •Trends in clinical benefit reported in news releases. | (55) (104), (105) (106) |
| NCT03368742 | |||
| PF06939926 (Pfizer) NCT03362502 | •1E14 vg/kg was administered in the first cohort of Phase 1b study. •Patients in the second cohort of the dose escalation study received 3E14 vg/kg. •Two patients suffered SAE but was resolved. Immune responses to AAV of variable magnitudes were observed in all patients. •99-patient phase 3 trial launched. | •Immunohistochemistry data showed 69% of dystrophin-positive fibers in the high dose group. •Two patients that have reached one-year time point showed improvements in North Star Ambulatory Assessment (NSAA) score. •Trends in clinical benefit reported in news releases. | (94) (95) (107) |
| AAVrh74.MHCK. Microdystrophin [also known as SRP-9001] (Nationwide Children’s/ Sarepta Therapeutics) | •Phase 1/2 open-label study enrolled 4 DMD subjects with no placebo group dosed with 2E14 vg/kg. •Whilst three patients had elevated liver enzyme gamma-glutamyl transpeptidase, steroid treatment resolved this. •Patients are being recruited for placebo-controlled phase 2 trial. •Interim findings from phase 2 trial reported four patients suffered SAE but was resolved. | •An average of 3.3 copies of the therapeutic gene present per nucleus. Western blot data showed mean microdystrophin expression of 96% after adjusting for fat and fibrotic tissue. •Immunohistochemistry data showed 81% of dystrophin- positive fibers. •Improvements in North Star Ambulatory Assessment (NSAA) score seen in all 4 patients (mean of 5.5) in Phase 1/2 trial. •No functional improvement seen from NSAA in Phase 2 trial. | (108), (109) (110), (111) (112), (113) |
| NCT03375164 NCT03769116 | |||
| GNT 0004 (Genethon in collaboration with Sarepta Therapeutics) | First patient was dosed. | Clinical trial is ongoing. | (53), (54) |
| 2020-002093-27 |
EXON SKIPPING GENE THERAPIES: ANTISENSE AND GENOME EDITING TECHNOLOGIES
In DMD patients with mutations in the DMD gene, non-functional dystrophin protein is produced; however, in some patients, the reading frame can be restored to produce a partially functional dystrophin protein [11]. It is important to note that this treatment strategy is mutation dependant and the individual exon-skipping strategies are only applicable to a small subset of the DMD patients. Nevertheless, collectively, with multiple medicinal products, the exon skipping strategy could potentially treat ∼60% of the DMD patients. So far, four antisense oligonucleotide therapies using a specific morpholino (PMO) chemistry have been approved by the Food and Drug Administration (FDA) for DMD treatment, targeting exon 51 (ExonDys-51), exon 53 (VyonDys-53 and Viltolarsen) and exon 45 (AmonDys-45); however, these drugs have very modest treatment outcome (variably 0.4%–5% increase in dystrophin level after many weeks of treatment) [55]. Another AON with different chemistry targeting exon 51 (Drisapersen) was stopped from development as it failed to secure FDA authorisation and demonstrated very low clinical benefits and significant toxicities [56]. Whilst the exact mechanism behind why the drug failed is not known, high doses of the drug with 2′-OMePS AON chemistry and the route of delivery subcutaneously might have contributed to the life-threatening toxicity [57, 58]. Another antisense drug, Spinraza, with 2′-OMOEPS chemistry delivered intrathecally for treatment of spinal muscular atrophy (SMA) had been effective and approved by the FDA. The delivery method intrathecally may have resulted in better delivery efficiency of the drug to the central nervous system [59].
DMD exon 2 is one of the most commonly duplicated exons resulting in formation of incorrect dystrophin protein [60]. For this duplication mutation of the DMD gene, exon skipping has a potential to restore the reading frame for production of full-length dystrophin. This strategy is currently being investigated by Audentes Therapeutics, where an AAV vector is used to deliver four copies of a modified U7 small nuclear RNA (snRNA) that contains antisense sequences targeting the splice donor (2 copies) and splice acceptor (2 copies) of the DMD exon 2 [60, 61]. Three-month post-infusion data of this ongoing study in the first 2 subjects treated with a vector dose of 3.0 × 1013 vg/kg showed expression of apparently full-length dystrophin protein and stable or improved functional outcome [60].
Genome editing enables mutations to be corrected at the DNA level for the production of functional dystrophin protein [62]. The clustered regularly interspaced palindromic repeats (CRISPR)-associated protein 9 [CRISPR/Cas9] nuclease is used to make double-strand break (DSB) at specific DNA sequences [63]. The cellular DNA repair mechanism is then activated to repair the DBS either via nonhomologous end joining (NHEJ) or homology directed repair (HDR) [64]. NHEJ pathway is more error prone as it introduces small insertions or deletions as ligation of the DNA break happens; which can be used to (i) disrupt genomic sequences or (ii) induce targeted deletions where DSBs are introduced at each end of the target sequence which is deleted as NHEJ mediates joining of the ends [64]. HDR on the other hand is a more precise pathway, where a homologous donor template is used to repair the lesion to restore the original sequence; however, this approach is not widely used in DMD as this is an uncommon event especially in post-mitotic cells [65]. In the context of DMD, the genome editing technology aims to produce the full-length dystrophin protein; however, if larger deletions of the gene are involved, then truncated, functional dystrophin protein will be produced [62]. Whilst many proof-of-principles studies in vitro and in mdx mouse and DMD dog models have been successful in restoring functional, truncated dystrophin; the gene editing approach has multiple disadvantages such as: (i) very low efficiency in deleting multiple exons, (ii) lack of efficiency in delivering the CRISPR-Cas9 system in vivo, (iii) possible need for repeated treatments, and (iv) safety concerns regarding off-target effects of Cas9 activity [55]. There are no clinical trials so far using genome editing approaches for DMD. Nevertheless, it should be noted that since CRISPR therapy can efficiently edit muscle stem cells, it is possible that repeated therapy will not be needed [66, 67]. However, Cas9 immunity is a major concern for CRISPR therapy [68].
EFFECTIVE LONG-TERM AAV GENE THERAPY: THE INTERPLAY BETWEEN AAV VECTOR DOSE AND IMMUNE RESPONSE
Whilst mouse models have been extremely useful in proof-of-principle studies, they fail to capture the magnitude of potential immune response caused by AAV delivery, often times raising no safety concerns in the studies [19]. The first barrier posed by the immune system is against rAAV capsids, similar to wtAAV, by producing neutralising antibodies (NAbs) that will eventually prevent gene delivery by rAAV [29]. Approximately 80% of the human population are estimated to be seropositive for AAV due to natural exposure to the virus [69], which will enable them to develop NAbs following such exposure [70]. Vector immunogenicity could thus arise from such pre-existing neutralising anti-capsid antibodies and also from cells that have been successfully transduced by rAAV, where the rAAV capsid antigens are displayed on the MHC-I complex, eliciting cytotoxic CD8+ T cell response [40]. While studies involving mouse models have shown promising results, the AAV capsid and transgene product however seem to elicit cytotoxic T cell response in larger animal models such as dogs [71], which are better and more sensitive models to predict potential adverse immune responses against AAV gene therapy [62].
Systemic AAV gene delivery of high doses of ∼1013 to 1014 vector genomes/kg have also been shown to elicit the activation of the innate immune response in large animal models [19]. In dystrophin-deficient neonatal dog model, in one study, two pups treated at 4-day-old had to be euthanised at 16 weeks of age as toxicity related to innate immune response were observed [72]. Another alert came from studies in juvenile non-human primates (NHPs) and piglets, where all three piglets and one NHP were euthanised due to observed toxicity and the remaining two NHPs demonstrated elevated level of transaminase (liver enzyme) and thrombocytopenia (platelet deficiency in blood) [73]. It is also important to note that none of these studies have clearly demonstrated strong evidence supporting a cellular immune response [19].
The determinants of immunogenicity of AAV vectors are still unclear, but the likelihood of AAV causing an immune response is high and should not be underestimated [74]. If AAV delivery elicits cytotoxic T lymphocyte (CTL)-mediated immune responses, this would cause short-lived gene expression [74, 75]. This CTL-mediated immune response following AAV transduction leading to clearance of transduced cells would pose a problem in the clinical setting as efficient gene therapy requires stable, long-term expression of the therapeutic gene [29, 74]. In the case of AAVs targeted to the skeletal muscles, the effects of CTL-mediated immune responses may not be as detrimental as previously thought due to the role played by regulatory T (Treg) cells in suppressing the effect of CTLs [76]. A study by Mueller et al., 2013 [76] also reported persistent, long-term transgene expression for up to 12 months and as such, immunomodulation of T cell populations may not be needed for muscle-directed AAV gene delivery. In addition, healthy skeletal muscle cells have low expression of MHC I and this could also contribute to the reduced T-lymphocyte mediated immune response [74]. In another clinical case, skeletal muscle samples from a patient with severe haemophilia B, that was injected with human factor IX-encoding AAV tested positive for factor IX 10 years post-treatment [77].
Following AAV administration, the innate immune system can also provoke a deleterious adaptive immune response via the toll-like receptor 9 (TLR9)-MyD88 pathway [78]. The activation of the TLR9-MyD88 pathway can result in a cytokine storm that induces the production of type I interferons [79]. TLR9 is a pathogen recognition receptor (PRR) that binds to unmethylated CpG sequence clusters, and CG content is significantly raised in codon optimised AAV vector genomes [80]. Self-complimentary genomes are shown to cause a stronger TLR9-dependent immune response in a dose-dependent manner [81]. Such dose dependent innate immune response eventually initiated adaptive immune response against the viral capsid and against the transgene [81]. CpG depletion from rAAV vector genomes [82] and improvements in vector production strategy by artificially increasing CpG methylation could alleviate these issues around the innate immune response against AAV [80].
The barrier posed by NAbs can potentially be overcome by some of the strategies currently being developed such as plasmapheresis, capsid engineering and using empty capsids as decoys [29]. Nevertheless, excluding seropositive patients from the trials is currently the only practical strategy in overcoming issues related to NAbs [19]. In the case of DMD, glucocorticosteroids (GCs) that are widely used as standard of care [83] also help in managing some of the immune system complications through the immunomodulatory activities of these drugs [84].
High vector doses in AAV gene therapy could potentially elicit undesirable immune responses, however, such high doses are also effective in rescuing the disease phenotype [72]. AAV gene therapy in a canine model of X-linked myotubular myopathy (MTM) showed great improvements in muscle strength and survival of the dogs in a dose-dependent manner, where the highest survival was observed in the group receiving the highest dose of rAAV expressing the therapeutic gene myotubularin (MTM1) [85]. However, high capsid dose could also trigger unwanted immune response; therefore, a balance needs to be achieved between the capsid dose and therapeutic efficacy for a good clinical outcome [74]. Recent human clinical trial of MTM gene therapy utilising AAV8 reported three deaths in the high dose group further reiterating the need for carefully managing vector dose for safety and therapeutic efficacy [86]. It is also a reminder that both in positive and sadly negative senses the most crucial data in the field of gene therapy are indeed derived from human studies, as pre-clinical studies pose some limitations in capturing the full effects of a treatment.
The choice of appropriate AAV serotype in targeting the respective tissues is also crucial in clinical outcome as they may directly influence the dosage needed in treatment. Care should also be taken as some AAV serotypes that efficiently transduce tissues in one mouse strain may not show similar transduction profile in another mouse strain, non-human primates or humans potentially due to molecular differences between the strains/ species [87]. In the case of muscle gene transfer, previously, the most commonly utilized route to administer AAV vectors is via direct intramuscular (IM) injection [71]. Multiple studies however have shown that large doses of vectors injected at one site such as the muscle can potentially trigger immune response against the transgene [75]. The route of delivery is changing over the years as human clinical trials are currently being planned and conducted using systemic intravascular (IV) administration to deliver the gene therapy products [9, 62, 72, 88, 89]. The intravenous (IV) route of vector delivery enables in principle body wide transduction that is essential in any comprehensive gene therapy of muscle diseases affecting all or many of the 700 muscles in the human. However a large vector dose delivered systemically could potentially trigger severe innate and adaptive immune responses towards the viral capsid [75]. Careful planning, selection and safety trial escalation of the vector dose is indicated. However, conventional dose escalation is problematic since patients treated with sub-clinical doses may become immunised and thus ineligible or refractive to subsequent clinical dosing. This is a difficult ethical conundrum and careful selection of early phase clinical trial dosing based on preclinical data is necessary to ensure patients are safe but may yet benefit.
Concerns regarding genotoxicity have emerged as in certain circumstances there seems to be a dose-dependent correlation between AAV gene delivery and hepatocellular carcinoma (HCC), indicating that increasing the AAV dosage could increase genotoxicity [90]. Theoretically, both the wtAAV and rAAV containing transgene could integrate part or all of their genome into the host genomic DNA, perhaps disrupting or activating oncogenic genes and thus predisposing patients to the formation of cancer [91]. This genotoxic phenomenon with AAV vectors however seems more prominent in rodent models and was not very much evident in larger animal models such as dogs and nonhuman primates [40]. Patients in human clinical trials for haemophilia-B that have received AAV gene therapy (>9 years post gene transfer) have not developed any tumors so far [92]. Studies investigating location of integration events that originated from rAAV genome have also reported no genotoxicity of systemically administered rAAV in non-human primates and humans [93]. Although no genotoxic safety concerns have been raised so far in humans, this is a risk that should remain on the radar in AAV gene therapy studies and extended periods of follow-up are still recommended [40].
Stable, long-term transgene expression is a key in achieving therapeutic efficacy following AAV gene therapy and this is even more important in chronic diseases such as DMD that requires continuous expression of modified dystrophin [19]. The biggest challenges in enabling persistent transgene expression are the muscle regeneration characteristic of the illness and immune responses to AAV capsid and transgene. Immunosuppression may partially resolve the immune responses, and other approaches to evade the immune responses such as tolerance induction may also be beneficial [44]. In post-mitotic and slowly replicating adult tissues, persistence may be achieved and when translating observations from studies in animal models to humans, it is essential to note the difference in turnover rates of different tissues in different species [40]. It has been shown that in healthy muscle AAV-mediated transgene delivery may persist for over a decade. However, in DMD, if transgene expression is below the threshold necessary for disease prevention then myofibre degeneration and regeneration will result in gradual elimination of episomal transgenes and micro-dystrophin expression. In addition to these considerations with DMD, there is the requirement for near body-wide muscle and heart transduction, and myoblast-mediated growth of patients treated at an early age. It is thus very likely that repeat administration of any DMD gene therapy will be needed. However, the required interval between treatments and the mechanism of managing the presence of circulating AAV NAbs induced by primary treatment remain as yet to be determined [19].
FUTURE PERSPECTIVES
DMD is a lethal paediatric genetic disease and so far, there is no cure for this disease. Many therapeutic strategies are being tested in clinical trials but AAV gene therapy shows the most promising success so far [94, 95]. AAV gene therapy also has the potential of treating the majority of the DMD patient population regardless of genotype. As microdystrophin gene transfer using AAV has been proven to be widely effective in preclinical models and to be relatively safe in early clinical trials [55], the focus of the research now is geared towards successfully translating this ATMP from bench to bedside, looking at issues around dose levels, repeat treatment, immune modulation and pharmaceutical scale manufacture and supply.
Of these several issues still remaining to be tackled in AAV gene therapy and one is potency and efficacy [40]. The systemic nature of DMD that requires body-wide dystrophin restoration also means a high AAV dosage may be needed to treat this disease. Whilst AAV is a safe virus, it can still provoke adverse immune responses at a high dosage [74]. This would then reduce the efficacy of the treatment. A more potent gene expression cassette would mean in theory that less virus will be needed to achieve therapeutic effect. One way of enhancing gene expression is via addition of gene regulatory elements such as novel promoters and introns [96] into the gene expression cassette. Apart from making the therapy safer due to reduced chances of provoking the immune system, a more potent gene expression cassette would also contribute to solving the challenge in AAV manufacturing at a large scale, as less virus will be needed to treat each patient. This in turn could also bring the cost of therapy down, as currently even the ‘cheapest’ AAV gene therapy drug carries a six-digit price tag.
Since rAAVs first were mooted to have clinical gene therapy promise more than 20 years ago, there has been a constant pursuit to generate and isolate novel AAV capsids with new properties [29]. As AAV gene therapy is actively moving into human clinical trials, there is a constant need and desire to improve the virus properties in terms of transduction efficiency, tissue tropism and immunogenicity [97]. In the case of DMD, an AAV serotype that is muscle-tropic and preferentially transfers gene into muscle cells would be ideal [98], especially because currently, a high dosage to achieve body-wide transduction is needed to treat DMD. Delivering therapeutic constructs using muscle-specific AAVs will improve the clinical outcome of the treatments. In addition, a lower dosage will be required for treatment, reducing potential immune response and also inevitably reducing the cost of treatment.
It should be noted that, poorly understood and even perhaps as yet unidentified; age, body-mass, or sex-related differences may exist in the efficiency of systemic AAV transduction. For example, in the context of SMA gene therapy, the effectiveness seen in neonates seems to be reduced when later-onset forms are treated [99]. In the context of DMD gene therapy, the main target organ, skeletal musculature, is undergoing progressive wasting, diminution and fibrotic remodelling as patients age [100]. Furthermore, the impact of relevant immune reactions may also be evolving with age in patient cohorts. Currently, GCs are routinely used prophylactically or reactively to control elevated serum levels of liver marker enzymes commonly considered to result from autoimmune reactivity to AAV transduced hepatocytes and appears to be a successful strategy in young patients. Several alternative immunomodulatory drug regimens are also being evaluated to either suppress existing or dose-reactive AAV immunity which may allow primary dosing in AAV seropositive individuals or re-dosing of gene therapies [101, 102]. The prevention, circumvention or control of adverse immunity is an area of ongoing research both clinically and in animal model systems which is crucial to the future development of the AAV gene therapy field in general. In addition, despite the fact that the current microdystrophins in clinical trial have been shown to be highly effective in rodent and canine models of DMD [9, 50, 103], and with indications of safety and some clinical benefit in humans (Table 1), it remains a formal possibility that they will prove less effective in the context of human clinical trials. Only time will tell, but it seems unlikely that current AAV-microdystrophins and current dose administration regimens with lead to a complete disease remission.
The AAV gene therapy field is expanding exponentially, and we are just at the beginning of an important era [29]. Other therapies such as exon skipping, stop codon readthrough, novel steroids, anti-inflammatories and anti-fibrotics may also prove beneficial, although sometimes only to a proportion of DMD patients that carry specific mutations, but there is nevertheless the realistic potential of combinations of therapies to achieve greater clinical efficacy.
This article was written as part of a special issue published on the occasion of the 80th birthday of Professor Terry Partridge and to celebrate his ongoing contributions to science. We would like to acknowledge the support, enthusiasm, inspiration and friendship of Professor Partridge, and his important contributions to the neuromuscular biology, Duchenne muscular dystrophy and the gene and cell therapy field, the community and literature over many decades.
ACKNOWLEDGMENTS
Figures were created using Biorender.
CONFLICTS OF INTEREST
The authors have no conflict to report. GD has acted as a consultant for SynPromics (AskBio), Sarepta and RegeneX. GD is a named inventor on patents related to DMD gene therapy and exon skipping.
REFERENCES
[1] | Angelini C . Muscular dystrophy. Handbook of clinical neurology: Elsevier; (2009) . p. 477–488. |
[2] | Duan D , Goemans N , Takeda S , Mercuri E , Aartsma-Rus A . Duchenne muscular dystrophy. Nature Reviews Disease Primers. (2021) ;7: (1):1–19. |
[3] | Emery AE . The muscular dystrophies. The Lancet. (2002) ;359: (9307):687–95. |
[4] | Gowers WR . Pseudo-hypertrophic muscular paralysis: a clinical lecture. : J. & A. Churchill; 1879. |
[5] | Koenig M , Hoffman EP , Bertelson CJ , Monaco AP , Feener C , Kunkel LM . Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. (1987) ;50: (3):509–17. |
[6] | Koenig M , Monaco AP , Kunkel LM . The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. (1988) ;53: (2):219–28. |
[7] | Kamdar F , Garry DJ . Dystrophin-deficient cardiomyopathy. J Am Coll Cardiol. (2016) ;67: (21):2533–46. |
[8] | Mercuri E , Bönnemann CG , Muntoni F . Muscular dystrophies. The Lancet. 3943). (2019) ;394: (10213):2025–38. |
[9] | Le Guiner C , Servais L , Montus M , Larcher T , Fraysse B , Moullec S , et al. Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nature Communications. (2017) ;8: (1):1–15. |
[10] | Abdul-Razak H , Malerba A , Dickson G . Advances in gene therapy for muscular dystrophies. F1000Research. (2016) ;5: . |
[11] | Van Deutekom JC , Van Ommen GB . Advances in Duchenne muscular dystrophy gene therapy. Nature Reviews Genetics. (2003) ;4: (10):774–83. |
[12] | Davies KE , Nowak KJ . Molecular mechanisms of muscular dystrophies: old and new players. Nature Reviews Molecular Cell Biology. (2006) ;7: (10):762–73. |
[13] | Fairclough RJ , Wood MJ , Davies KE . Therapy for Duchenne muscular dystrophy: renewed optimism from genetic approaches. Nature Reviews Genetics. (2013) ;14: (6):373–8. |
[14] | Lionarons JM , Hoogland G , Hendriksen RG , Faber CG , Hellebrekers DM , Van Koeveringe GA , et al. Dystrophin is expressed in smooth muscle and afferent nerve fibers in the rat urinary bladder. Muscle Nerve. (2019) ;60: (2):202–10. |
[15] | Douglas AG , Wood MJ . Splicing therapy for neuromuscular disease. Molecular and Cellular Neuroscience. (2013) ;56: :169–85. |
[16] | Allen DG , Whitehead NP , Froehner SC . Absence of dystrophin disrupts skeletal muscle signaling: roles of Ca2, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol Rev. (2016) ;96: (1):253–305. |
[17] | Lai Y , Thomas GD , Yue Y , Yang HT , Li D , Long C , et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest. (2009) ;119: (3):624–35. |
[18] | Lai Y , Zhao J , Yue Y , Duan D . α2 and α3 helices of dystrophin R16 and R17 frame a microdomain in the α1 helix of dystrophin R17 for neuronal NOS binding. Proceedings of the National Academy of Sciences. (2013) ;110: (2):525–30. |
[19] | Duan D . Micro-dystrophin gene therapy goes systemic in Duchenne muscular dystrophy patients. Hum Gene Ther. (2018) ;29: (7):733–6. |
[20] | Ervasti JM , Ohlendieck K , Kahl SD , Gaver MG , Campbell KP . Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. (1990) ;345: (6273):315–9. |
[21] | Yoshida M , Ozawa E . Glycoprotein complex anchoring dystrophin to sarcolemma. The Journal of Biochemistry. (1990) ;345: (6273):315–9. |
[22] | Rando TA . The dystrophin–glycoprotein complex, cellular signaling, and the regulation of cell survival in the muscular dystrophies. Muscle Nerve. (2001) ;24: (12):1575–94. |
[23] | Foster H , Popplewell L , Dickson G . Genetic therapeutic approaches for Duchenne muscular dystrophy. Hum Gene Ther. (2012) ;23: (7):676–87. |
[24] | Ennen JP , Verma M , Asakura A . Vascular-targeted therapies for Duchenne muscular dystrophy. Skeletal Muscle. (2013) ;3: (1):1–12. |
[25] | England SB , Nicholson L , Johnson MA , Forrest SM , Love DR , Zubrzycka-Gaarn EE , et al. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature. (1990) ;343: (6254):180–2. |
[26] | Koenig M , Beggs AH , Moyer M , Scherpf S , Heindrich K , Bettecken T , et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet. (1989) ;45: (4):498. |
[27] | Atchison RW , Casto BC , Hammon WM . Adenovirus-associated defective virus particles. Science. (1965) 149: (3685):754–5. |
[28] | Blacklow NR , Hoggan MD , Rowe WP . Isolation of adenovirus-associated viruses from man. Proc Natl Acad Sci U S A. (1967) ;58: (4):1410. |
[29] | Wang D , Tai PW , Gao G . Adeno-associated virus vector as a platform for gene therapy delivery. Nature Reviews Drug Discovery. (2019) ;18: (5):358–78. |
[30] | Hermonat PL , Muzyczka N . Use of adeno-associated virus as a mammalian DNA cloning vector: transduction of neomycin resistance into mammalian tissue culture cells. Proceedings of the National Academy of Sciences. (1984) ;81: (20):6466–70. |
[31] | Flotte T , Carter B , Conrad C , Guggino W , Reynolds T , Rosenstein B , et al. A phase i study of an adeno-associated virus-cftr gene vector in adult cf patients with mild lung disease. Johns hopkins children’s center, baltimore, maryland. Hum Gene Ther. (1996) ;7: (9):1145–59. |
[32] | Ylä-Herttuala S . Endgame: glybera finally recommended for approval as the first gene therapy drug in the European union. Molecular Therapy. (2012) ;20: (10):1831–2. |
[33] | Miraldi Utz V , Coussa RG , Antaki F , Traboulsi EI . Gene therapy for RPE65-related retinal disease. Ophthalmic Genet. (2018) ;39: (6):671–7. |
[34] | Hoy SM . Onasemnogene abeparvovec: first global approval. Drugs. (2019) ;79: (11):1255–62. |
[35] | Samulski RJ , Chang L , Shenk T . Helper-free stocks of recombinant adeno-associated viruses: normal integration does not require viral gene expression. J Virol. (1989) ;63: (9):3822–8. |
[36] | Kotterman MA , Schaffer DV . Engineering adeno-associated viruses for clinical gene therapy. Nature Reviews Genetics. (2014) ;15: (7):445–51. |
[37] | Flotte TR . Gene therapy progress and prospects: recombinant adeno-associated virus (rAAV) vectors. Gene Ther. (2004) ;11: (10):805–10. |
[38] | Duan D , Sharma P , Yang J , Yue Y , Dudus L , Zhang Y , et al. Circular intermediates of recombinant adeno-associated virus have defined structural characteristics responsible for long-term episomal persistence in muscle tissue. J Virol. (1998) ;72: (11):8568. |
[39] | Grieger JC , Soltys SM , Samulski RJ . Production of recombinant adeno-associated virus vectors using suspension HEK293 cells and continuous harvest of vector from the culture media for GMP FIX and FLT1 clinical vector. Molecular Therapy. (2016) ;24: (2):287–97. |
[40] | Colella P , Ronzitti G , Mingozzi F . Emerging issues in AAV-mediated in vivo gene therapy. Molecular Therapy-Methods & Clinical Development. (2018) ;8: :87–104. |
[41] | Gao G , Vandenberghe LH , Alvira MR , Lu Y , Calcedo R , Zhou X , et al. Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol. (2004) ;78: (12):6381–8. |
[42] | Wu Z , Asokan A , Samulski RJ . Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol Ther. (2006) ;14: :316–27. |
[43] | Duan D , Yue Y , Yan Z , Yang J , Engelhardt JF . Endosomal processing limits gene transfer to polarized airway epithelia by adeno-associated virus. J Clin Invest. (2000) ;105: (11):1573–87. |
[44] | Keeler AM , Flotte TR . Recombinant adeno-associated virus gene therapy in light of Luxturna (and Zolgensma and Glybera): where are we, and how did we get here? Annual Review of Virology. (2019) ;6: :601–21. |
[45] | Lai Y , Yue Y , Duan D . Evidence for the failure of adeno-associated virus serotype 5 to package a viral genome≥8.2kb. Molecular Therapy. (2010) ;18: (1):75–9. |
[46] | Wu Z , Yang H , Colosi P . Effect of genome size on AAV vector packaging. Molecular Therapy. (2010) ;18: (1):80–6. |
[47] | Dong B , Nakai H , Xiao W . Characterization of genome integrity for oversized recombinant AAV vector. Molecular Therapy. (2010) ;18: (1):87–92. |
[48] | Wang B , Li J , Xiao X . Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proceedings of the National Academy of Sciences. (2000) ;97: (25):13714–9. |
[49] | Harper SQ , Hauser MA , DelloRusso C , Duan D , Crawford RW , Phelps SF , et al. Modular flexibility of dystroph: implications for gene therapy of Duchenne muscular dystrophy. Nat Med. (2002) ;8: (3):253–61. |
[50] | Hakim CH , Wasala NB , Pan X , Kodippili K , Yue Y , Zhang K , et al. A five-repeat micro-dystrophin gene ameliorated dystrophic phenotype in the severe DBA/2J-mdx model of Duchenne muscular dystrophy. Molecular Therapy-Methods & Clinical Development. (2017) ;6: :216–30. |
[51] | Mendell JR , Al-Zaidy S , Shell R , Arnold WD , Rodino-Klapac LR , Prior TW , et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. (2017) ;377: (18):1713–22. |
[52] | Genethon. Genethon gets the green light from the ANSM to start an innovative gene therapy trial for Duchenne muscular dystrophy. 2020; Available at: https://www.genethon.fr/en/2020/12/02/genethon-gets-the-green-light-from-the-ansm-to-start-an-innovative-gene-therapy-trial-for-duchenne-muscular-dystrophy/. Accessed 1 March, 2021. |
[53] | Genethon. Genethon announces First Patient dosed in Clinical Trial of Investigational Gene therapy GNT 0004 for Duchenne Muscular Dystrophy. 2021; Available at: https://www.genethon.fr/en//04/20/genethon-announces-first-patient-dosed-in-clinical-trial-of-investigational-gene-therapy-gnt–for-duchenne-muscular-dystrophy/. Accessed 20 June, 2021. |
[54] | EUClinicalTrials Register. Clinical trial 2020-002093-27. 2020;. Accessed 20 June, 2021. |
[55] | Verhaart IE , Aartsma-Rus A . Therapeutic developments for Duchenne muscular dystrophy. Nature Reviews Neurology. (2019) ;15: (7):373–86. |
[56] | Aartsma-Rus A , Krieg AM . FDA approves eteplirsen for Duchenne muscular dystrophy: the next chapter in the eteplirsen saga. Nucleic Acid Therapeutics. (2017) ;27: (1):1–3. |
[57] | Le BT , Adams AM , Fletcher S , Wilton SD , Veedu RN . Rational design of short locked nucleic acid-modified 2′-O-methyl antisense oligonucleotides for efficient exon-skipping in vitro. Molecular Therapy-Nucleic Acids. (2017) ;9: :155–61. |
[58] | Hilhorst N , Spanoudi-Kitrimi I , Goemans N , Morren M . Injection site reactions after long-term subcutaneous delivery of drisapersen: a retrospective study. Eur J Pediatr. (2019) ;178: (2):253–8. |
[59] | van der Bent ML , da Silva Filho OP , van Luijk J , Brock R , Wansink DG . Assisted delivery of antisense therapeutics in animal models of heritable neurodegenerative and neuromuscular disorders: a systematic review and meta-analysis. Scientific Reports. (2018) ;8: (1):1–12. |
[60] | Waldrop M , Lawlor M , Vetter TM , Frair E , Beatka M , Meng H , et al. LATE BREAKING NEWS ORAL PRESENTATION: LBO 3 Expression of apparent full-length dystrophin in skeletal muscle in a first-in-human gene therapy trial using the scAAV9. U7-ACCA vector. Neuromuscular Disorders. (2020) ;30: :S166–S167. |
[61] | NCT04240314. AAV9 U7snRNA Gene Therapy to Treat Boys With DMD Exon 2 Duplications. 2020; Available at: https://clinicaltrials.gov/ct2/show/NCT04240314. Accessed 20/03/, 2021. |
[62] | Chamberlain JR , Chamberlain JS . Progress toward gene therapy for Duchenne muscular dystrophy. Molecular Therapy. (2017) ;25: (5):1125–31. |
[63] | Popplewell L , Koo T , Leclerc X , Duclert A , Mamchaoui K , Gouble A , et al. Gene correction of a duchenne muscular dystrophy mutation by meganuclease-enhanced exon knock-in. Hum Gene Ther. (2013) ;24: (7):692–701. |
[64] | Prakash V , Moore M , Yáñez-Muñoz RJ . Current progress in therapeutic gene editing for monogenic diseases. Molecular Therapy. (2016) ;24: (3):465–74. |
[65] | Lim KRQ , Yoon C , Yokota T . Applications of CRISPR/Cas9 for the treatment of Duchenne muscular dystrophy. Journal of Personalized Medicine. (2018) ;8: (4):38. |
[66] | Nance ME , Shi R , Hakim CH , Wasala NB , Yue Y , Pan X , et al. AAV9 edits muscle stem cells in normal and dystrophic adult mice. Molecular Therapy. (2019) ;27: (9):1568–85. |
[67] | Kwon JB , Ettyreddy AR , Vankara A , Bohning JD , Devlin G , Hauschka SD , et al. In vivo gene editing of muscle stem cells with adeno-associated viral vectors in a mouse model of Duchenne muscular dystrophy. Molecular Therapy-Methods & Clinical Development. (2020) ;19: :320–9. |
[68] | Crudele JM , Chamberlain JS . Cas9 immunity creates challenges for CRISPR gene editing therapies. Nature Communications. (2018) ;9: (1):1–3. |
[69] | Calcedo R , Vandenberghe LH , Gao G , Lin J , Wilson JM . Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis. (2009) ;199: (3):381–90. |
[70] | Arruda VR , Xiao W . It’s all about the clothing: capsid domination in the adeno-associated viral vector world. Journal of Thrombosis and Haemostasis. (2007) ;5: (1):12–5. |
[71] | Wang Z , Tapscott SJ , Chamberlain JS , Storb R . Immunity and AAV-mediated gene therapy for muscular dystrophies in large animal models and human trials. Frontiers in Microbiology. (2011) ;2: :201. |
[72] | Kornegay JN , Li J , Bogan JR , Bogan DJ , Chen C , Zheng H , et al. Widespread muscle expression of an AAV9 human mini-dystrophin vector after intravenous injection in neonatal dystrophin-deficient dogs. Molecular Therapy. (2010) ;18: (8):1501–8. |
[73] | Hinderer C , Katz N , Buza EL , Dyer C , Goode T , Bell P , et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum Gene Ther. (2018) ;29: (3):285–98. |
[74] | Mingozzi F , High KA . Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood, The Journal of the American Society of Hematology. (2013) ;122: (1):23–36. |
[75] | Boisgerault F , Mingozzi F . The skeletal muscle environment and its role in immunity and tolerance to AAV vector-mediated gene transfer. Current Gene Therapy. (2015) ;15: (4):381–94. |
[76] | Mueller C , Chulay JD , Trapnell BC , Humphries M , Carey B , Sandhaus RA , et al. Human Treg responses allow sustained recombinant adeno-associated virus–mediated transgene expression. J Clin Invest. (2013) ;123: (12):5310–8. |
[77] | Buchlis G , Podsakoff GM , Radu A , Hawk SM , Flake AW , Mingozzi F , et al. Factor IX expression in skeletal muscle of a severe hemophilia B patient 10 years after AAV-mediated gene transfer. Blood, The Journal of the American Society of Hematology. (2012) ;119: (13):3038–41. |
[78] | Rogers GL , Martino AT , Aslanidi GV , Jayandharan GR , Srivastava A , Herzog RW . Innate immune responses to AAV vectors. Frontiers in Microbiology. (2011) ;2: :194. |
[79] | Uematsu S , Akira S . Toll-like receptors and Type I interferons. J Biol Chem. (2007) ;282: (21):15319–23. |
[80] | Wright JF . Codon Modification and PAMPs in Clinical AAV Vectors: The Tortoise or the Hare? Molecular Therapy. (2020) ;28: (3):701–3. |
[81] | Martino AT , Suzuki M , Markusic DM , Zolotukhin I , Ryals RC , Moghimi B , et al. The genome of self-complementary adeno-associated viral vectors increases Toll-like receptor 9–dependent innate immune responses in the liver. Blood. The Journal of the American Society of Hematology. (2011) ;117: (24):6459–68. |
[82] | Faust SM , Bell P , Cutler BJ , Ashley SN , Zhu Y , Rabinowitz JE , et al. CpG-depleted adeno-associated virus vectors evade immune detection. J Clin Invest. (2013) ;123: (7):2994–3001. |
[83] | Matthews E , Brassington R , Kuntzer T , Jichi F , Manzur AY . Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database of Systematic Reviews. 2016: (5). |
[84] | Rosenberg AS , Puig M , Nagaraju K , Hoffman EP , Villalta SA , Rao VA , et al. Immune-mediated pathology in Duchenne muscular dystrophy. Science Translational Medicine. (2015) ;7: (299):299rv4. |
[85] | Mack DL , Poulard K , Goddard MA , Latournerie V , Snyder JM , Grange RW , et al. Systemic AAV8-mediated gene therapy drives whole-body correction of myotubular myopathy in dogs. Molecular Therapy. (2017) ;25: (4):839–54. |
[86] | Keown A . Third Patient Dies In Audentes’ Gene Therapy Study For Neuromuscular Disease | Biospace. 2020; Available at: https://www.biospace.com/article/third-pediatric-patient-dies-in-audentes-gene-therapy-study-for-rare-neuromuscular-disease/. Accessed 13 October, 2020. |
[87] | Hordeaux J , Wang Q , Katz N , Buza EL , Bell P , Wilson JM . The neurotropic properties of AAV-PHP. B are limited to C57BL/6J mice. Molecular Therapy. (2018) ;26: (3):664–8. |
[88] | Yue Y , Pan X , Hakim CH , Kodippili K , Zhang K , Shin J , et al. Safe and bodywide muscle transduction in young adult Duchenne muscular dystrophy dogs with adeno-associated virus. Hum Mol Genet. (2015) ;24: (20):5880–90. |
[89] | Duan D . Systemic delivery of adeno-associated viral vectors. Current Opinion in Virology. (2016) ;21: :16–25. |
[90] | Chandler RJ , LaFave MC , Varshney GK , Trivedi NS , Carrillo-Carrasco N , Senac JS , et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J Clin Invest. (2015) ;125: (2):870–80. |
[91] | Russell DW , Grompe M . Adeno-associated virus finds its disease. Nat Genet. (2015) ;47: (10):1104–5. |
[92] | Nathwani AC , Reiss UM , Tuddenham EG , Rosales C , Chowdary P , McIntosh J , et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. (2014) ;371: (21):1994–2004. |
[93] | Gil-Farina I , Fronza R , Kaeppel C , Lopez-Franco E , Ferreira V , D’avola D , et al. Recombinant AAV integration is not associated with hepatic genotoxicity in nonhuman primates and patients. Molecular Therapy. (2016) ;24: (6):1100–5. |
[94] | Pfizer. A Study to Evaluate the Safety and Tolerability of PF-06939926 Gene Therapy in Duchenne Muscular Dystrophy.. 2018; Available at: https://clinicaltrials.gov/ct2/show/record/NCT (ClinicalTrials.gov Identifier: NCT03362502). |
[95] | Gene Therapy Trials: Pfizer. PPMD 2019 Annual Conference; 28 June 2019; Orlando Florida. |
[96] | Powell SK , Rivera-Soto R , Gray SJ . Viral expression cassette elements to enhance transgene target specificity and expression in gene therapy. Discovery Medicine. (2015) ;19: (102):49. |
[97] | Zinn E , Pacouret S , Khaychuk V , Turunen HT , Carvalho LS , Andres-Mateos E , et al. In silico reconstruction of the viral evolutionary lineage yields a potent gene therapy vector. Cell Reports. (2015) ;12: (6):1056–68. |
[98] | Nance ME , Duan D . Perspective on adeno-associated virus capsid modification for Duchenne muscular dystrophy gene therapy. Hum Gene Ther. (2015) ;26: (12):786–800. |
[99] | Dangouloff T , Servais L . Clinical evidence supporting early treatment of patients with spinal muscular atrophy: current perspectives. Therapeutics and Clinical Risk Management. (2019) ;15: :1153. |
[100] | Wilson K , Faelan C , Patterson-Kane JC , Rudmann DG , Moore SA , Frank D , et al. Duchenne and Becker muscular dystrophies: a review of animal models, clinical end points, and biomarker quantification. Toxicol Pathol. (2017) ;45: (7):961–76. |
[101] | Conklin LS , Damsker JM , Hoffman EP , Jusko WJ , Mavroudis PD , Schwartz BD , et al. Phase IIa trial in Duchenne muscular dystrophy shows vamorolone is a first-in-class dissociative steroidal anti-inflammatory drug. Pharmacological Research. (2018) ;136: :140–50. |
[102] | Meliani A , Boisgerault F , Hardet R , Marmier S , Collaud F , Ronzitti G , et al. Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration. Nature Communications. (2018) ;9: (1):1–13. |
[103] | Duan D . Systemic AAV micro-dystrophin gene therapy for Duchenne muscular dystrophy. Molecular Therapy. (2018) ;26: (10):2337–56. |
[104] | Solid Biosciences. Microdystrophin Gene Transfer Study in Adolescents and Children With DMD (IGNITE DMD).. 2017; Available at: https://clinicaltrials.gov/ct2/show/record/NCT (ClinicalTrials.gov Identifier: NCT03368742). |
[105] | Gene Therapy Trials: Solid Biosciences.. PPMD 2019 Annual Conference; 28 June (2019) ; Orlando Florida. |
[106] | Solid Biosciences. Solid Biosciences Reports Efficacy and Safety Data from the Ongoing IGNITE DMD Clinical Trial and Resumption of Patient Dosing in the 2E14vg/kg Cohort. 2021;. Accessed 3 June, 2021. |
[107] | CureDuchenne. Summary of Pfizer Phase 1 Microdystrophin Gene Therapy Trial. 2019; Available at: https://www.cureduchenne.org/blog/summary-of-pfizer-phase-1-microdystrophin-gene-therapy-trial/. Accessed 20 June, 2021. |
[108] | Sarepta Therapeutics I . Systemic Gene Delivery Clinical Trial for Duchenne Muscular Dystrophy. 2017; Available at: https://clinicaltrials.gov/ct2/show/NCT03375164. |
[109] | Sarepta Therapeutics I . A Randomized, Double-blind, Placebo-controlled Study of SRP-9001 for Duchenne Muscular Dystrophy (DMD). 2018; Available at: https://clinicaltrials.gov/ct2/show/NCT03769116. |
[110] | Gene Therapy Trials: Sarepta.. PPMD 2019 Annual Conference; 28 June (2019) ; Orlando Florida. |
[111] | Mendell J , Sahenk Z , Lehman K , Nease C , Lowes L , Miller N , et al. DMD–THERAPY: P. 280 Systemic gene transfer with rAAVrh74. MHCK7. micro-dystrophin in patients with Duchenne muscular dystrophy. Neuromuscular Disorders. (2020) ;30: :S128. |
[112] | Mullard A . Sarepta’s DMD gene therapy falls flat. Nature Reviews Drug Discovery. (2021) ;20: (91). |
[113] | GlobalData Healthcare. AAN 2021: Sarepta’s SRP-9001 set to redeem its prospects in DMD. 2021; Available at: https://www.clinicaltrialsarena.com/comment/aan-2021-sarepta-srp-9001-dmd/Accessed 20 June, 2021. |
[114] | Chu X , Li J , Qiao C , Wang J , Wang Y , Jiang X , et al. Long-term effect of human mini-dystrophin in transgenic mdx mice improves muscle physiological function. The FASEB Journal. (2021) ;35: (6):e21628. |
[115] | Adams ME , Odom GL , Kim MJ , Chamberlain JS , Froehner SC . Syntrophin binds directly to multiple spectrin-like repeats in dystrophin and mediates binding of nNOS to repeats 16–17. Hum Mol Genet. (2018) ;27: (17):2978–85. |


