
A Combined Prospective and Retrospective Comparison of Long-Term Functional Outcomes Suggests Delayed Loss of Ambulation and Pulmonary Decline with Long-Term Eteplirsen Treatment
Abstract
Background:
Studies 4658-201/202 (201/202) evaluated treatment effects of eteplirsen over 4 years in patients with Duchenne muscular dystrophy and confirmed exon-51 amenable genetic mutations. Chart review Study 4658-405 (405) further followed these patients while receiving eteplirsen during usual clinical care.
Objective:
To compare long-term clinical outcomes of eteplirsen-treated patients from Studies 201/202/405 with those of external controls.
Methods:
Median total follow-up time was approximately 6 years of eteplirsen treatment. Outcomes included loss of ambulation (LOA) and percent-predicted forced vital capacity (FVC%p). Time to LOA was compared between eteplirsen-treated patients and standard of care (SOC) external controls and was measured from eteplirsen initiation in 201/202 or, in the SOC group, from the first study visit. Comparisons were conducted using univariate Kaplan-Meier analyses and log-rank tests, and multivariate Cox proportional hazards models with regression adjustment for baseline characteristics. Annual change in FVC%p was compared between eteplirsen-treated patients and natural history study patients using linear mixed models with repeated measures.
Results:
Data were included from all 12 patients in Studies 201/202 and the 10 patients with available data from 405. Median age at LOA was 15.16 years. Eteplirsen-treated patients experienced a statistically significant longer median time to LOA by 2.09 years (5.09 vs. 3.00 years, p < 0.01) and significantly attenuated rates of pulmonary decline vs. natural history patients (FVC%p change: –3.3 vs. –6.0 percentage points annually, p < 0.0001).
Conclusions:
Study 405 highlights the functional benefits of eteplirsen on ambulatory and pulmonary function outcomes up to 7 years of follow-up in comparison to external controls.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a severe, degenerative, X-linked neuromuscular disease which is caused by mutations in the DMD gene encoding dystrophin, a protein with a key role in upholding the structure of muscle fibers [1]. The most common mutations are deletions flanking exon 51, which account for 13% of all DMD cases and result in the failure of dystrophin synthesis due to disruptions to the transcriptional open-reading frame [2]. The incidence of DMD in the United States (US), Canada, France, and the United Kingdom is approximately one in 3,500 to 5,000 newborn boys, similar to that reported globally in a 2020 meta-analysis, of 19.8 per 100,000 newborn boys, i.e., ∼1 in 5,000 [3–6].
As patients with DMD age, they experience progressive damage and degeneration of muscle fibers due to the absence of functional dystrophin. Hallmarks of DMD include progressive loss of motor function, frequent falls and developmental delay, beginning around the age of two to three years [7]. The initial development of motor skills typically plateaus around age seven and most boys lose ambulation by their early to mid-teens [8, 9]. However, patients experience substantial heterogeneity in clinical trajectory; treatment regimens [1, 2] and the type of mutations of the DMD gene [3–8] are among the factors associated with disease progression. Respiratory failure and cardiomyopathy then develop gradually, contributing to a life expectancy of under 30 years [10].
Eteplirsen, the first disease modifying therapy for patients with DMD with confirmed exon-51 skip-amenable mutations [11] approved by the US Food and Drug Administration, is a phosphorodiamidate morpholino oligomer that hybridizes with pre-mRNA transcripts of the DMD gene [12]. This interaction allows the restoration of the mRNA open reading frame in patients with mutations amenable to exon 51 skipping, leading to the production of an internally truncated yet functional dystrophin protein [12, 13].
Patients’ functional outcomes in clinical trials are typically measured using the six-minute walk test (6MWT) [8, 14], timed function tests [15], and the North Star Ambulatory Assessment (NSAA) [16, 17]. These assessments have been used in clinical and natural history studies of DMD to monitor disease progression until loss of ambulation (LOA) [18–23]. LOA is a critical disease milestone for patients and families, and consequently is an important outcome in regulatory evaluations and health technology assessment of new DMD therapies [9, 22, 24]. An important clinical outcome for patients with DMD is forced vital capacity (FVC). The percent-predicted FVC (FVC%p) measures respiratory decline in patients with DMD. FVC%p begins to decline between the ages of 7 and 10 years in untreated patients with DMD and declines linearly at approximately 5–6% per year among those 10 to 18 years old, irrespective of corticosteroid treatment [9–12]. The steady deterioration requires increased levels of clinical intervention as the FVC%p drops below critical thresholds, impacting quality of life [13, 14].
Due to the rarity of DMD, both clinical trial and real-world data are scarce. However, understanding the long-term efficacy of eteplirsen among patients with DMD in the real world is of vital importance. To this end, the present study compared time to LOA and the annual rate of FVC%p decline between patients with DMD who have a mutation amenable to exon-51 skipping receiving eteplirsen and those receiving SOC.
MATERIALS AND METHODS
Patients
Eteplirsen-treated patients for LOA and FVC%p analyses
Eteplirsen-treated patients described in this analysis were enrolled in the pivotal trial of eteplirsen and followed into an extension study, with additional subsequent follow-up in the real world setting with eteplirsen obtained via retrospective chart review (Fig. 1).
Fig. 1
Study design and timing for studies 201, 202, and 405. Note: Patients were followed from initiation of eteplirsen treatment until the end of data availability in all studies in which they participated.

The pivotal study of eteplirsen was a single center, 24-week, double-blind, placebo-controlled trial (Study 201; ClinicalTrials.gov identifier: NCT01396239) with a 4-year, open-label extension (Study 202; NCT01540409) [24, 25]. Study 201 enrolled 12 ambulatory boys with DMD with mutations amenable to exon 51 skipping, aged 7 to 13 years, with baseline 6MWT values between 180 and 440 meters, and receiving a stable dose of steroids for≥24 weeks prior to enrollment. The patients were randomized to either eteplirsen (either 30 mg/kg [n = 4] or 50 mg/kg [n = 4]) or placebo (n = 4) [24]. In Study 202, all 12 patients were followed for a cumulative total of approximately 4 years (since the start of Study 201) while being treated with eteplirsen [26].
Study 405 was a multicenter retrospective chart review of 10 of the 12 patients who completed Studies 201/202 to assess functional outcomes in patients treated in real-world settings as they continued eteplirsen treatment. This study provides an analysis of all available follow-up data for the entire cohort of 12 patients with DMD receiving eteplirsen long-term in clinical trial and real-world settings compared to external controls, similar to previous studies comparing eteplirsen outcomes in clinical trials to those of external controls [24]. The baseline for eteplirsen-treated patients was defined at eteplirsen initiation, which occurred either at the start of Study 201 (for treatment arm patients) or at the start of Study 202 (for placebo arm patients).
Studies 201/202 were approved by the relevant institutional review boards (IRB) prior to implementation, and informed consent/assent was obtained from each subject and family before enrollment. All but one site obtained a waiver of written informed consent/assent by their IRB for Study 405, as it was a retrospective observational study of minimal risk and secondary use of de-identified clinical data, without imposing any intervention or interfering with usual patient care. One study site IRB determined the research to meet the criteria for exemption from IRB review, as it qualified as secondary research for which consent was not required.
External controls for LOA analyses
External controls were drawn from two natural history databases of patients with DMD, Italian DMD Registry (Telethon) and Leuven Neuromuscular Reference Center (NMRC) Registry (Leuven) [27], and from the placebo arm of a previously conducted clinical trial of drisapersen, DEMAND III (Fig. 2).
Fig. 2
Flowcharts for patients who completed Studies 201/202/405 (eteplirsen-treated) and patients from external control data sources.
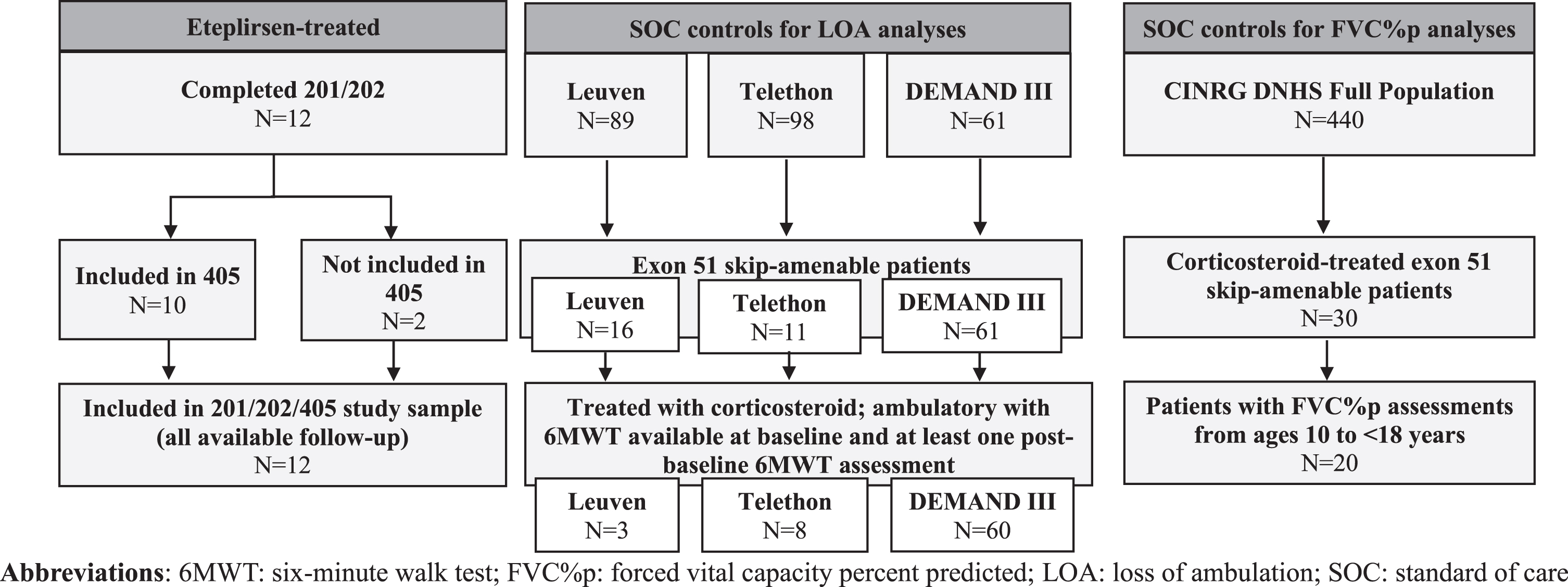
The Italian DMD Registry is a database of 13 participating tertiary care centers in Italy. Patients were eligible to participate if they had a genetically-confirmed DMD diagnosis, were aged ≥5 years, were able to walk independently at least 75 meters, and had no moderate or severe learning difficulties or behavioral problems. Data collected from routine clinical practice and curated at 12-month intervals were available from 8 patients with mutations amenable to exon 51 skipping. Most patients were treated with corticosteroids at baseline.
The Leuven NMRC Registry is a database of patients with DMD receiving care at a single pediatric neurology clinic at University Hospitals in Leuven, Belgium. This ongoing study has been approved by Ethische Commissie Onderzoek, the Ethics Committee of the University Hospitals Leuven, and was conducted in accordance with the Declaration of Helsinki. Written consent from the guardians of each participant was obtained. Patients were eligible if they had genetically-confirmed DMD, were aged 4.5 to 17.5 years, and had no severe cognitive or behavioral disorder impairing compliance. Study assessments occurred approximately every 6 months, and included the 6MWT and timed functional tests, as well as concurrent assessments of height, weight, and steroid use. Data were available from 3 patients with mutations amenable to exon 51 skipping.
DEMAND III (DMD114044; NCT01254019) was a 48-week, randomized, double-blind, placebo-controlled phase 3 trial to investigate the efficacy of drisapersen in boys with DMD [28]. Patients were aged > 5 years at baseline, had 6MWT of≥75 meters at baseline, had systemic corticosteroid treatment for≥6 months before the start of the trial, and received a stable corticosteroid dose and regimen for≥3 months prior to screening. The placebo arm of DEMAND III (n = 60 with complete data) is an important source of additional patients with mutations amenable to exon 51 skipping and was included in order to increase the number of patients in the SOC group for a more robust analysis. At baseline, the DEMAND III cohort is comparable to the eteplirsen cohort on key parameters, including age, 6MWT, ten-meter walk run test (10MWR), time to rise, and corticosteroid use.
Patients in these three sources (N = 71) who met the following criteria were included in the LOA external control group and are collectively referred to as the “SOC group” throughout the paper: 1) genetically-confirmed DMD amenable to exon 51 skipping therapy; 2) treated with corticosteroid therapy; and 3) ambulatory with 6MWT assessment available at baseline and at least 1 assessment post-baseline. The baseline was defined as the first available study visit that fulfilled the inclusion criteria.
External controls for FVC%p analyses
The Cooperative International Neuromuscular Research Group Duchenne Natural History Study (CINRG DNHS) was one of the largest prospective natural history studies of DMD conducted to date, comprising 440 patients with DMD across all ages [26]. Assessments of respiratory function (including FVC%p) were conducted every 3 months for 1 year, then at 18 and 24 months, and annually thereafter. For the analysis of FVC%p, the exon 51 CINRG DNHS cohort (n = 20) was used as a comparator, comprised of corticosteroid -treated patients between the ages of 10 and 18 years and with mutations amenable to exon 51 skipping. As for the other external control cohorts, the baseline was defined as the first available study visit that fulfilled the inclusion criteria.
Patients from additional eteplirsen studies for FVC%p analyses
Data from two additional eteplirsen studies were used to provide context for the rate of FVC%p decline among patients in this analysis. First, PROMOVI (NCT02255552) was a large, phase 3, US-based, multi-center study that compared 79 eteplirsen-treated patients for up to 96 weeks with untreated external controls [29]. This study measured exon skipping and dystrophin production and assessed functional outcomes including change from baseline 6MWT and the annual rate of change in FVC%p. Second, Study 204 (NCT02286947) was an open-label, multicenter study designed to evaluate the safety and tolerability of eteplirsen treatment in boys aged 7 to 21 years with a diagnosis of DMD and mutations amenable to exon 51 skipping. All patients were receiving a stable dose of oral corticosteroids or had not received corticosteroids for at least 24 weeks prior to study drug administration, and were minimally ambulatory or non-ambulatory. A total of 52 eteplirsen-treated patients from PROMOVI and 20 eteplirsen-treated patients from Study 204 were included as benchmarks for the eteplirsen cohort in this study.
Outcome measures
Time to LOA was measured from initiation of eteplirsen in Studies 201/202 or, in the SOC group, from the first study visit. LOA was defined as the first occurrence of the inability to complete the 6MWT as recorded in Studies 201/202, 100% wheelchair use in the retrospective chart review in Study 405, or inability to complete the 6MWT as recorded in the SOC group. The 6MWT was performed according to published methods [30] modified for use in patients with DMD in both the eteplirsen-treated and SOC group. The 6MWT is not routinely performed in real-world settings and thus a different definition was used in Study 405, consistent with previous use of continuous wheelchair used to define LOA in natural history studies [31]. Among patients who did not experience loss of ambulation during follow-up, time to loss of ambulation was censored at the end of data availability. Data end was defined as disenrollment from Study 202 for patients who did not participate in the Study 405, the date of the last visit to the medical center before the cutoff date for chart review for patients who were assessed retrospectively in Study 405, and at the date of the last available follow-up assessment in the SOC sources.
Four patients in the chart review Study 405 had uncertainty with respect to the precise date of LOA. In these cases, the date was imputed based on all the information available. For two of these patients participating in 405 who had the milestone of full-time wheelchair use recorded to the month, the exact date was imputed as the midpoint of that month. For two patients who were recorded to lose ambulation at an unspecified date during Study 202, the date of the milestone was imputed as the midpoint between the last recorded visit with 6MWT values > 0 meters and the last visit in Study 202.
In Study 405, Study 202, PROMOVI, and the CINRG DNHS, FVC%p was calculated using the NHANES III equation, derived by Hankinson et al. [32]. Standing height was available in Study 405, whereas height was calculated using ulnar lengths for patients in Study 204, PROMOVI, and the CINRG DNHS, because some non-ambulatory patients were not able to stand [33]. In Studies 201/202, FVC%p was calculated by each study site using the methodology described by Polgar and Promadhat [34], using measured standing height, as these earlier studies evaluated ambulatory patients treated with eteplirsen [16].
Sensitivity analyses of time to LOA
In the first sensitivity analysis, information from case manager polls was used for the two patients who did not participate in Study 405 and were censored for follow up at the end of Study 202 in the main analysis. One patient was assumed to lose ambulation after the study completion at the date recorded by the case manager, and a second patient was assumed to remain ambulatory until the end of Study 405, when he was censored for follow-up.
In the second sensitivity analysis, time from birth to LOA was analyzed instead of time from baseline. Although this analysis can be subject to bias, as patients were not treated with eteplirsen since birth, it is a commonly performed analysis in DMD studies when data on baseline prognostic factors are not available.
In the third sensitivity analysis, the assumption that LOA occurred in the midpoint of the interval of uncertainty for patients with imprecisely recorded date of LOA. The date of LOA was alternatively assumed to be at the earliest date of that interval (lower bound) or at the latest date of that interval (upper bound). For example, for a patient with recorded date of LOA of August 2017, the lower bound was August 1, 2017, the midpoint was August 16, 2017, and the upper bound was August 31, 2017.
In the fourth sensitivity analysis, the SOC group was expanded to include patients from the CINRG DNHS. These patients were much younger and had better ambulatory function at baseline compared to eteplirsen-treated patients analyzed here, and thus were not included in the main analyses. The CINRG DNHS patients were included in the sensitivity analysis if they had an exon-51 skip amenable mutation, were ambulatory and treated with corticosteroids, and had 6MWT values available at baseline (study entry). The definition of LOA in CINRG DNHS is identical to that used in Study 405, i.e. full-time wheelchair use. Time to LOA was compared between eteplirsen-treated patients and this expanded SOC group.
Statistical analyses
Time to LOA was described using Kaplan-Meier analyses and compared between eteplirsen-treated patients and the SOC group using a log-rank test and multivariable Cox proportional hazards models. Multivariable models adjusted for known prognostic factors (age, steroid type, and baseline measures of function) [35, 36]. A series of four adjusted analyses was conducted to elucidate the impacts of adjustment for different sets of prognostic factors. Specifically, Model 1 included an indicator for eteplirsen and adjusted for age and baseline 6MWT values, while Model 2 adjusted for these factors plus steroid type, 10MWR performance, and rise from floor performance. Models 3 and 4 further assessed whether the effect of eteplirsen was associated with baseline 6MWT values to test, in particular, the hypothesis that earlier treatment of less impaired patients might result in greater preservation of function. Model 3 included an indicator for eteplirsen and an interaction term between eteplirsen and baseline 6MWT and adjusted for age and baseline 6MWT values; Model 4 adjusted for Model 3 variables plus steroid type, 10MWR performance, and rise from floor performance. For all analyses, the proportional hazards assumption was tested using the Schoenfeld residuals test [37].
Comparisons of the rates of decline in FVC%p were conducted using segmented linear mixed models with repeated measures (MMRM). The response variable was the FVC%p calculated or reported at each visit. Covariates included fixed effects for treatment group, age at visit, treatment group by age interaction, and random effects for patient identifier. The annual rate of decline, in percentage points of FVC%p, was compared to that of the natural history cohort of exon 51 skipping–amenable untreated male patients from the CINRG DNHS. Results from other eteplirsen studies (Studies 201/202 only, Study 204, and PROMOVI) are also shown.
All analyses were conducted in R version 3.6.1 (The R Foundation, Vienna, Austria) using packages survival [17] (for time to LOA models) and lme4 [18] (for FVC%p MMRM). P-values were reported based on a two-sided test, and a type-I error level α of 0.05 was used to assess statistical significance.
RESULTS
Patients and baseline characteristics
Twelve eteplirsen-treated patients from Studies 201/202/405 and 71 external control patients from the Italian Telethon database (N = 8), Leuven database (N = 3), and DEMAND III (N = 60) were included in the analysis (Fig. 2).
The baseline characteristics of the eteplirsen-treated and external control patients with DMD are listed in Table 1. The mean ages of eteplirsen-treated (9.48 years) and external control patients (8.60 years) were similar. Additionally, the eteplirsen-treated and external control patients’ baseline functional test performance was comparable, including for the 6MWT (363.17 [SD: 42.19] versus 350.27 [89.42] meters), 10MWR (1.71 [0.44] versus 1.57 [0.55] m/s), and timed rise from floor velocity (0.18 [0.09] versus 0.19 [0.25] s–1) values. Rates of treatment with corticosteroids were identical between the two groups, with 100.0% of both the eteplirsen-treated patients and the SOC group patients treated with corticosteroids at baseline. However, the types of corticosteroids used differed between the two groups. A higher proportion of eteplirsen-treated patients (66.7% [n = 8]) than external control patients (46.8% [n = 32]) received deflazacort at baseline, while a higher proportion of external control patients than eteplirsen-treated patients (53.6% [n = 67] versus 33.3% [n = 4]) received prednisone. For 2 patients in the SOC group (from DEMAND III) baseline steroid type was unknown. The total follow-up time differed between the two groups, with eteplirsen-treated patients followed for an average of 5.72 (SD: 0.90) years, while external control patients were followed for an average of 1.34 (1.04) years.
Table 1
Baseline characteristics of patients included in the LOA analyses by treatment group
| Eteplirsen (Studies 201/202/405) N = 12 | Standard of Care N = 71 | |
| Demographics | ||
| Age (years), mean±SD | 9.48±1.18 | 8.60±2.09 |
| Median | 9.75 | 8.60 |
| IQR | [8.68, 10.57] | [7.06, 10.06] |
| Range | [7.36, 11.03] | [5.25, 15.36] |
| Function | ||
| 6MWT (meters) | 363.17±42.19 | 350.27±89.42 |
| Timed ten-meter walk/run velocity (m/s) | 1.71±0.44 | 1.57±0.55 |
| Timed rise from floor velocity (s–1) | 0.18±0.09 | 0.19±0.25 |
| Steroid type | ||
| Deflazacort | 8 (66.7%) | 32 (46.38%)a |
| Prednisone | 4 (33.3%) | 37 (53.62%)a |
| Missing | 0 / 12 (0.00%) | 2 / 71 (2.82%) |
| Total follow-up time (years) | ||
| Mean±SD | 5.72±0.90 | 1.34±1.04 |
| Median | 6.06 | 0.92 |
| IQR | [4.95, 6.29] | [0.92, 0.92] |
| Range | [4.13, 6.88] | [0.69, 4.03] |
Notes: aDue to 2 patients having missing values for the steroid type at baseline, the percentages of patients with deflazacort and prednisone are based on n = 69 patients and add up to 100%. Mean±SD shown for continuous characteristics; count (percentage) shown for categorical characteristics. Patient characteristics for the eteplirsen sample were measured at eteplirsen initiation (at the start of 201 or 202). This sample includes patients included in the Kaplan-Meier analyses (n = 83); 9 standard-of-care patients were excluded from the Cox analyses (n = 74) due to missing characteristics on baseline timed functional tests or steroid information. Abbreviations: 6MWT: six-minute walk test; IQR: interquartile range; SD: standard deviation.
Patient characteristics for the eteplirsen-treated patients and for the external control group by individual data source are shown in Supplementary Table 1. Patients were comparable at baseline across most characteristics between the different data sources; however, there were important differences in the length of follow-up. The mean (SD) follow-up was 3.16 (1.12) years in the Leuven cohort, 3.88 (0.35) years in the Italian Group cohort, and 0.92 (0.03) years in the DEMAND III cohort. All 48 weeks of data available in DEMAND III were used, without any extrapolation of data or modeling after 48 weeks.
The patient characteristics at eteplirsen initiation were similar between the group of eteplirsen-treated patients who participated in the chart review studies (Study 405; n = 10) and those who did not (n = 2) (Supplementary Table 2).
Comparison of time to LOA
Unadjusted analyses
Over the approximate 4 to 7-year follow-up, 7 of 12 patients in the eteplirsen-treated group lost ambulation and 14 of 71 external controls lost ambulation during the 1 to 4-year follow-up (Fig. 3). When times to LOA were compared between groups using the unadjusted Kaplan-Meier analyses accounting for the shorter duration of follow-up among external controls, a longer time to LOA was observed for eteplirsen patients. Specifically, the median time to LOA was 5.09 years in the eteplirsen-treated group, compared to 3.00 years for the external controls (Table 2). This difference of 2.09 years, favoring eteplirsen, was statistically significant based on the log-rank test (p < 0.01) (Fig. 3).
Fig. 3
Kaplan-Meier curves for time from baseline to LOA by treatment group. Abbreviations: CI: confidence interval; LOA: loss of ambulation.
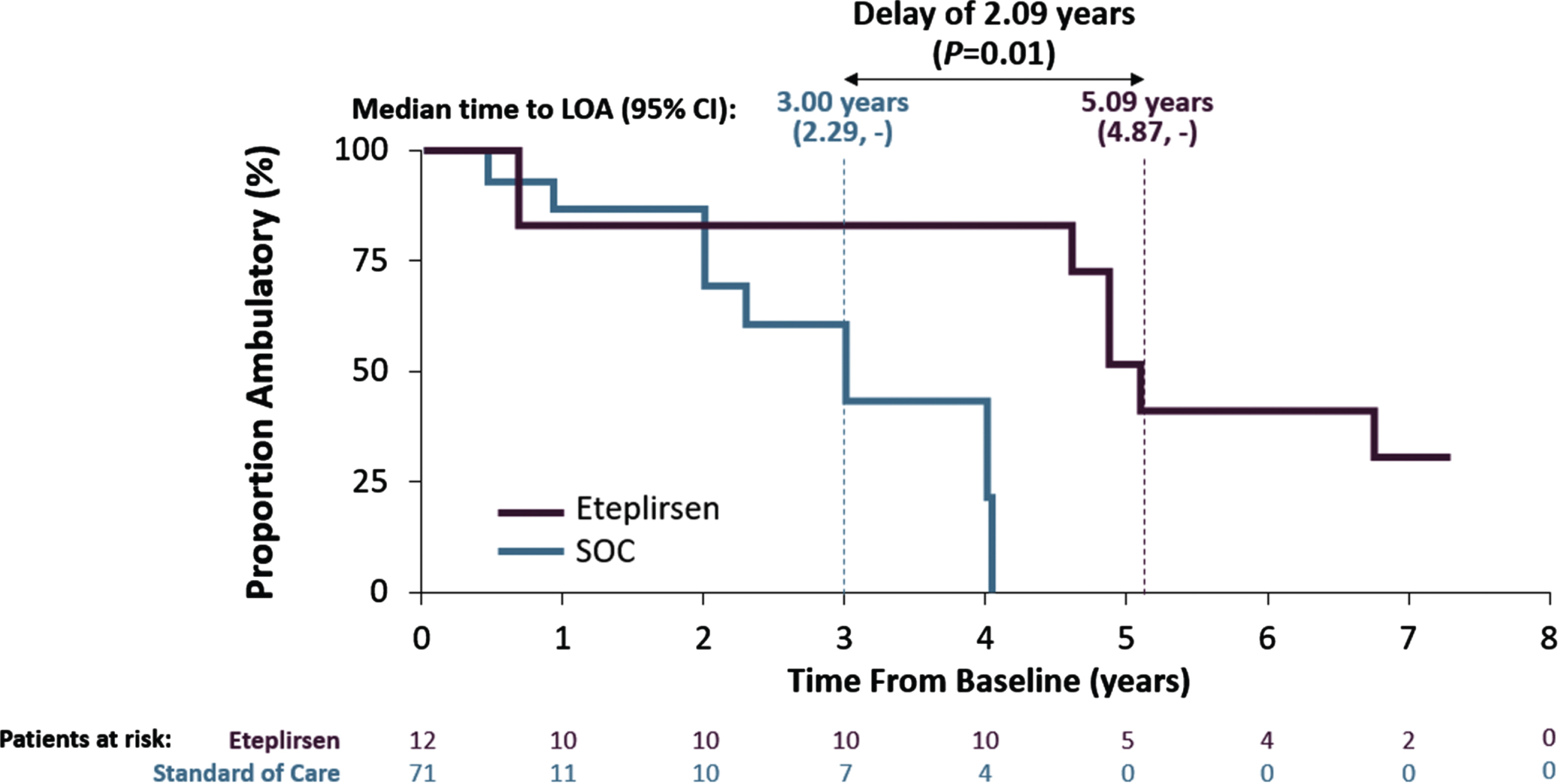
Table 2
Median time from baseline to loss of ambulation by treatment group
| N | Patients with events | Median time to LOA (years) 95% CI | |
| Eteplirsen | 12 | 7 | 5.09 (4.87, –) |
| Standard of care | 71 | 14 | 3.00 (2.29, –) |
Abbreviations: CI: confidence interval; LOA: loss of ambulation.
Adjusted analyses
In the analysis of time to LOA adjusting for baseline age and 6MWT values (Model 1), the estimates from the Cox proportional hazards model indicated an 87.5% decrease in the risk of LOA (HR [95% CI]: 0.125 [0.019, 0.829]; p < 0.05) associated with eteplirsen compared to SOC group patients (Table 3). In addition, the estimates from Model 1 indicated a significant association with baseline 6MWT values (HR: 0.985 [95% CI: 0.977, 0.993]; p < 0.001). In the analysis that adjusted for baseline age, 6MWT, 10MWR, rise from floor values, and corticosteroid type (Model 2), the estimates were generally similar to those of Model 1 and again indicated an 88.1% lower risk associated with eteplirsen compared to external controls (HR [95% CI]: 0.119 [0.016, 0.863]; p < 0.05). The results of Model 2 also indicated statistically significant associations with baseline 6MWT values (HR [95% CI]: 0.987 [0.977, 0.997]) and use of deflazacort (0.234 [0.067, 0.819]) (both p < 0.05) (Table 3). The results from a test based on Schoenfeld residuals supported the proportionality of hazards.
Table 3
Hazard ratio estimates from Cox proportional hazards models without interaction terms
| Model 1 HR Estimate [95% CI] | Model 2 HR Estimate [95% CI] | |
| Eteplirsen | 0.125* [0.019, 0.829] | 0.119* [0.016, 0.863] |
| Age (years) | 0.966 [0.701, 1.330] | 1.096 [0.720, 1.668] |
| 6MWT (meters) | 0.985*** [0.977, 0.993] | 0.987* [0.977, 0.997] |
| Timed rise from floor velocity (s–1) | 0.670 [0.001, 433.675] | |
| Timed ten-meter walk/run velocity (m/s) | 1.179 [0.210, 6.616] | |
| Steroid type: deflazacort | 0.234* [0.067, 0.819] | |
| C-statistic (SE) | 0.726 (0.110) | 0.795 (0.087) |
| Number of patients | 74 | 74 |
| Number of events | 17 | 17 |
Notes: *p < 0.05; ***p < 0.001. The sample was limited to patients with non-missing values for the covariates included in the adjusted model; 95% CIs are shown in parentheses, unless otherwise noted. Abbreviations: 6MWT: six-minute walk test; 10MWR: ten-meter walk/run; CI: confidence interval; HR: hazard ratio; SE: standard error.
The results of the Cox models with interaction terms (Models 3 and 4) indicated that patients treated with eteplirsen benefitted regardless of their baseline functional level (Model 3 HR [95% CI]: 0.059 [0.005, 0.644]; Model 4 HR: 0.054 [0.005, 0.592]; both p < 0.05) (Table 4). However, the effect of eteplirsen was stronger for eteplirsen-treated patients with higher 6MWT values at baseline, as indicated by the estimates for the eteplirsen-6MWT interaction (Model 3 HR [95% CI]: 0.946 [0.916, 0.977], p < 0.001; Model 4 HR: 0.949 [0.918, 0.982], p < 0.01).
Table 4
Hazard ratio estimates from Cox proportional hazards models with interaction terms
| Model 3 HR Estimate [95% CI] | Model 4 HR Estimate [95% CI] | |
| Eteplirsen | 0.059* [0.005, 0.644] | 0.054* [0.005, 0.592] |
| 6MWT (meters) | 0.985*** [0.976, 0.994] | 0.984** [0.973, 0.995] |
| Eteplirsen×Baseline 6MWT | 0.946*** [0.916, 0.977] | 0.949** [0.918, 0.982] |
| Age (years) | 0.860 [0.604, 1.226] | 0.961 [0.595, 1.553] |
| Timed rise from floor velocity (s–1) | 0.794 [0.001, 862.911] | |
| Timed ten-meter walk/run velocity (m/s) | 1.758 [0.316, 9.786] | |
| Steroid type: deflazacort | 0.286 [0.077, 1.062] | |
| C-statistic (SE) | 0.809 (0.099) | 0.841 (0.088) |
| Number of patients | 74 | 74 |
| Number of events | 17 | 17 |
Notes: The sample is limited to patients with non-missing values for the covariates included in the adjusted model; 95% CIs shown in parentheses, unless otherwise noted. *p < 0.05; **p < 0.01; ***p < 0.001. Abbreviations: 6MWT: six-minute walk test; 10MWR: ten-meter walk/run; CI: confidence interval; HR: hazard ratio; SE: standard error.
Sensitivity analyses
The results of the first sensitivity analysis, using assumptions about LOA for four patients informed by case manager polls, confirmed the benefit of eteplirsen and were consistent with the main analyses (Supplementary Figure 1). Specifically, in the unadjusted Kaplan-Meier analyses, the difference in median time to LOA was 2.43 years in favor of eteplirsen compared with SOC group patients (p < 0.01) (Supplementary Table 3).
Similarly, the second sensitivity analysis comparing time from birth to LOA between the eteplirsen-treated patients and external controls indicated a delay of 1.66 years in the median time to this milestone in favor of eteplirsen (p = 0.24) (Supplementary Figure 2 and Supplementary Table 4).
The results of the third sensitivity analysis using different assumptions for eteplirsen-treated patients with uncertainty in the date of LOA confirmed the delay in LOA observed in the main analysis, which assumed that LOA occurred in the midpoint of the uncertainty interval (Supplementary Figure 3 and Supplementary Table 5). When LOA was assumed to have occurred at the lower bound of the interval, the median time to LOA was virtually identical to that in the main analysis (5.05 years). Alternatively, when LOA was assumed to have occurred at the upper bound of the interval, the median time to LOA was 5.60 years.
Finally, the results of the fourth sensitivity analysis comparing eteplirsen-treated patients with an expanded SOC group including CINRG DNHS patients were consistent with the results of the main analysis. CINRG DNHS patients were on average younger and had generally better baseline function compared to eteplirsen-treated as well as Telethon, Leuven, and DEMAND III patients (Supplementary Table 6). The difference in median time from baseline to LOA between eteplirsen-treated and SOC patients in these analyses was 2.09 years (p < 0.05) (Supplementary Figure 4 and Supplementary Table 7), while the difference in median time from birth to LOA was 1.66 years (p = 0.24) (Supplementary Figure 5 and Supplementary Table 8). No LOA events occurred among CINRG DNHS patients during follow-up.
Comparison of pulmonary function (FVC%p)
In the analysis comparing pulmonary decline among eteplirsen-treated patients and external controls, the annual FVC%p decline of patients receiving eteplirsen in Studies 201, 202, and 405 was significantly attenuated compared to patients receiving SOC in the CINRG DNHS comparison cohort (p < 0.0001; Fig. 4). Over the follow-up of up to 7 years in Studies 201, 202, and 405, FVC%p declined linearly at a rate of 3.3 percentage points per year among patients treated with eteplirsen while the observed rate in CINRG DNHS external controls was 6.0 percentage points per year [33].
Fig. 4
Pulmonary function in Studies 201/202 and 405 in comparison with matched natural history controls and other eteplirsen studies. Notes: aIncludes assessments up to Week 240 for 12 patients in Studies 201/202. Post Study 202, FVC%p data include 10 patients from Study 405. Nominal P-values vs CINRG DNHS exon 51 patients. Height was measured as standing height in Studies 201/202/405 and as ulnar length in Study 204, PROMOVI, and CINRG DHNS. 1Khan N, et al. J Neuromuscul Dis 2019;6 : 213-25. doi:10.3233/JND-180351 2McDonald CM, et al. J Neuromuscul Dis. 2021; Preprint: 1–13. doi:10.3233/JND-210643.
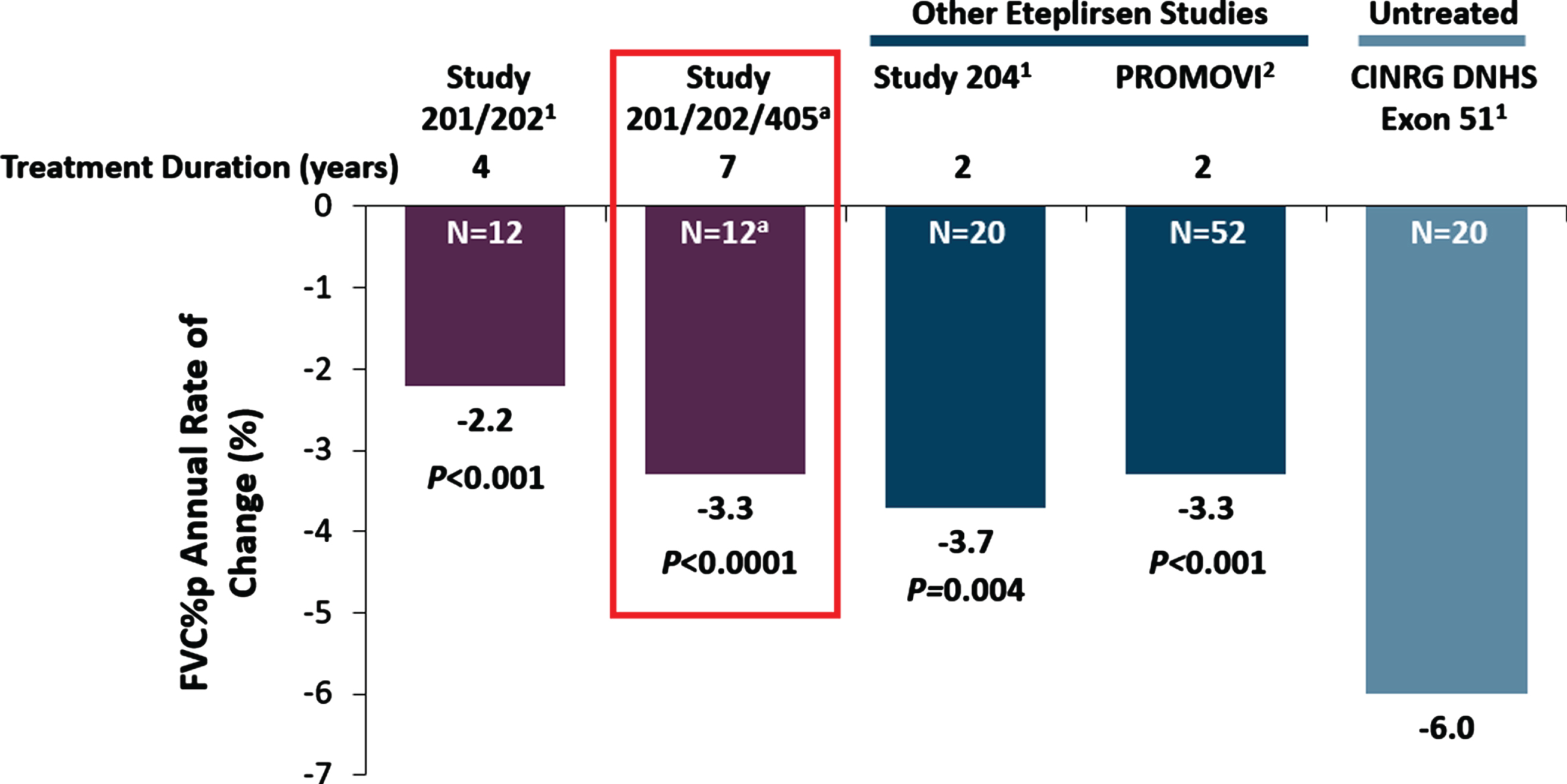
DISCUSSION
DMD is a degenerative disease for which there is currently no cure, thus, there is a great unmet need for innovative therapies that prolong patients’ functional ability and improve long-term outcomes. In the present study, based on up to 7 years of treatment, patients receiving eteplirsen had a significant delay in the time to LOA of 2.1 years (median time to LOA 5.1 vs. 3.0 years; p < 0.01). A delay in this critical disease milestone is of great importance for patients and families and also predicts a delay in reaching subsequent milestones, including the loss of motor abilities, development of scoliosis, peak FVC, need for supportive ventilation, FVC of 1L, and death [38–43]. Moreover, patients with better ambulatory function (i.e., higher 6MWT values) at the time of treatment initiation experienced an even greater delay in time to LOA compared to SOC group, consistent with a greater benefit of earlier treatment initiation.
The present LOA results are consistent with previous studies comparing patients with DMD from Studies 201/202 [24] and PROMOVI [44] to external controls, which indicated that eteplirsen preserves ambulatory function in patients with DMD and mutations amenable to exon-51 skipping. Moreover, the median age at LOA for eteplirsen-treated patients in Studies 201/202/405 (15.2 years) was higher than the previously published ages at LOA among patients in untreated natural history cohorts in the DuchenneConnect study (12.0 years) and CINRG DNHS (13.0 years) [31, 45].
A significantly attenuated decline in pulmonary function measured by FVC%p was also observed in the eteplirsen group versus comparable external controls (–3.3 vs. –6.0 percentage points annually, p < 0.0001). The rate of decline in FVC%p observed in this study, of 3.3 percentage points per year among eteplirsen-treated patients, is consistent with the findings of previous studies of patients in Studies 201/202, 204, and PROMOVI. In those studies, FVC%p declined at rates of 2.2 to 3.7 percentage points per year, highlighting the treatment benefits of eteplirsen compared with SOC. As a reduction in FVC%p decline to below 5 percentage points per year may delay critical milestones of morbidity and mortality, the benefit of eteplirsen can be considered clinically significant [44, 46]. In the current study, eteplirsen slowed disease progression compared with matched external controls, confirming the findings observed in the first 4 years of treatment during the prior studies [24].
A strength of this study is the length of follow-up time for patients with DMD. With 5.7 years on average for all patients included in Studies 201/202/405 (range: 4.13 to 6.88) and most patients having > 6 years of follow-up, this study describes the longest follow-up of eteplirsen-treated patients to date.
Previous literature has reported associations between deflazacort and better ambulatory function and longer time to LOA compared to prednisone [26, 47]. To account for the potential confounding effect of corticosteroid type, the Cox models adjusted for deflazacort versus prednisone treatment status at baseline (Model 2 in Table 3 and Model 4 in Table 4), and found associations of corticosteroid type consistent with previous studies. These analyses furthermore show that eteplirsen was associated with delayed LOA even after adjusting for the type of corticosteroid with which patients were treated at baseline.
Safety
Safety data from Studies 201/202 has been reported elsewhere [24]. During the combined 201/202 and 405 chart review period, 78% of patients used cardiac medications and 67% used pulmonary medications to treat cardiac and pulmonary complications. The most common comorbid conditions during the 405 chart review period included obesity, long bone fracture, osteoporosis, and anxiety. No serious renal abnormalities were noted.
Limitations
This study has limitations which are related to the rarity of DMD and the scarcity of both real-world and clinical trial data of this population. First, the statistical comparisons were conducted between non-randomized groups and thus may be confounded by unobserved or unadjusted baseline differences in prognostic variables between the groups. While the Cox models adjusted for known predictors of change in ambulatory function and time to loss of ambulation, identified in prior studies [36, 48], the potential for confounding bias due to unobserved differences cannot be excluded.
Second, the small sample size in the eteplirsen-treated group limits the precision of the treatment effect estimate. Despite this limitation, robust and statistically significant treatment effects were detected in both the unadjusted and adjusted time to event analyses.
Third, the outcome definitions differed between the data sources for the eteplirsen-treated patients and external controls. Full-time wheelchair use was used in conjunction with the inability to perform the 6MWT in the 201/202/405 data, while inability to perform the 6MWT was used in the SOC group data. This limitation stems from the fact that while the 6MWT has been used in clinical trials and natural history studies in DMD, this test is not frequently administered in real-world clinical practice. However, this limitation is expected to understate the effect of eteplirsen on time to LOA compared to the external controls, given that full-time wheelchair use typically occurs before 6MWT = 0. Since this definition was used only in Study 405 but not in any of the external control groups in the main analysis, eteplirsen-treated patients were recorded to lose ambulation slightly earlier than if 6MWT data were consistently available. A similarly “conservative” approach results from the fact that some uncertainty in the date of LOA is present in the external control data, particularly for the Leuven and Telethon studies, which had visits every 12 months and 6 months, respectively. Since LOA was recorded at the first visit where 6MWT = 0 but may have occurred before that, Leuven and Telethon patients may appear to have lost ambulation later than they did in reality.
Differences in outcome measurement are also a limitation for the analysis of pulmonary function, as different equations were used for calculating FVC%p in different studies, and height was either measured or calculated based on ulnar length in different studies.
Finally, there were significant differences in the available time under follow-up between the two groups, which limits the precision of estimated long-term effects. In particular, longer-term follow-up for external controls with exon-51 skip amenable mutations represented a smaller sample drawn from natural history studies, since the larger number of subjects receiving placebo in a clinical trial setting had only 48 weeks of follow-up. Therefore, due to the length of DEMAND III follow-up of 48 weeks, eteplirsen-treated patients are compared mainly to the Italian Telethon and Leuven patients in the long-term comparisons.
CONCLUSIONS
In this long-term follow-up of eteplirsen-treated patients with DMD in both a clinical trial and real-world setting, eteplirsen-treated patients experienced a significant delay in the time to LOA, by 2.1 years (p < 0.01), compared to patients receiving SOC (i.e., external controls). Additionally, eteplirsen-treated patients exhibited significantly slower rates of pulmonary decline compared to patients receiving SOC (–3.3 vs. –6.0 percentage points annually, p < 0.0001). These results were robust to multiple sensitivity analyses, and indicate that eteplirsen may help preserve ambulatory and pulmonary function longer than SOC among patients with DMD.
ACKNOWLEDGMENTS
Studies 201/202 were funded by Sarepta Therapeutics, Inc. and registered on ClinicalTrials.gov under identification numbers NCT01396239 and NCT01540409. Study 405 was funded by Sarepta Therapeutics, Inc. The authors thank the patients and their families for their participation in the studies or registries, and to Fondazione Telethon, Leuven NMRC, and CINRG network sites for making the natural history data available for the present study. The authors also thank Katherine Tsai, MPH, PhD for assistance with Study 405 as a former employee of Sarepta Therapeutics, Inc.
Editing assistance was provided by Nicolae Done and Shelley Batts (Analysis Group, Inc.), and by Zohal Ghiaszada and Aaron Novack (Sarepta Therapeutics, Inc.).
CONFLICTS OF INTEREST
Olga Mitelman, Ashish Dugar, and Sourav Santra are employees of Sarepta Therapeutics, Inc. and own Sarepta Therapeutics, Inc. stock.
Hoda Z. Abdel-Hamid served on advisory boards for AveXis, Audentes, Biogen, and Sarepta Therapeutics, Inc.
Barry J. Byrne is the co-founder of Aavanti Bio, Inc.
Anne M. Connolly served on advisory boards for Acceleron, AveXis, Genentech, Edgewise, and Sarepta Therapeutics, Inc., and on DMSB for Catabasis.
Peter Heydemann served on advisory panels for Sarepta Therapeutics, Inc. and PTC Therapeutics.
Crystal Proud served on advisory boards for AveXis, Biogen, Sarepta Therapeutics, Inc., is a speaker for AveXis, Biogen, has or is conducting research for AveXis, Astellas, Biogen, Catabasis, CSL Behring, PTC Therapeutics, Pfizer, Sarepta Therapeutics Inc., and Scholar Rock.
Perry B. Shieh is a consultant for AveXis, Biogen, PTC Therapeutics, and Sarepta Therapeutics, Inc., and serves on speaker bureaus for Alexion, Biogen, and Grifols.
Kathryn R. Wagner is a consultant for AskBio, Dynacure, PTC Therapeutics, Roche, and Sarepta Therapeutics, Inc., and serves on the DSMB for Fibrogen and on a dose escalation committee for Wave Life Sciences.
James Signorovitch is a member of the collaborative Trajectory Analysis Project and is an employee of Analysis Group, Inc., a consulting firm that received payment from Sarepta Therapeutics, Inc. to conduct this research.
Nathalie Goemans has served on clinical steering committees and/or as a consultant and received compensation from Eli Lilly, Italfarmaco SpA, PTC Therapeutics, BioMarin Pharmaceutical, Sarepta Therapeutics, Inc., Pfizer Inc., Roche, Wave Life Sciences; has served as site investigator for GlaxoSmithKline, Prosensa, BioMarin Pharmaceutical, Italfarmaco SpA, Sarepta Therapeutics, Inc., Wave Life Sciences, Roche, and Eli Lilly.
Craig M. McDonald has served as a consultant for PTC Therapeutics, BioMarin Pharmaceutical, Sarepta Therapeutics, Inc., Eli Lilly, Pfizer Inc., Santhera Pharmaceuticals, Epirium Bio (formerly Cardero Therapeutics, Inc.), Catabasis Pharmaceuticals, Capricor Therapeutics, Astellas Pharma (Mitobridge), Italfarmaco, Edgewise Therapeutics, and FibroGen, Inc.; serves on external advisory boards related to Duchenne muscular dystrophy for PTC Therapeutics, Sarepta Therapeutics, Inc., Santhera Pharmaceuticals, Avidity Biosciences, and Capricor Therapeutics; and reports grants from United States Department of Education/National Institute on Disability and Rehabilitation Research, The National Institute on Disability, Independent Living, and Rehabilitation Research, United States National Institute of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health /National Institute of Neurological Disorders and Stroke, United States Department of Defense, and Parent Project Muscular Dystrophy US, during the conduct of the study.
Eugenio Mercuri has served on clinical steering committees and/or as a consultant for Italfarmaco SpA, PTC Therapeutics, Prosensa, Sarepta Therapeutics, Santhera Pharmaceuticals, and BioMarin Pharmaceutical; has served as site investigator for GlaxoSmithKline, Prosensa, BioMarin Pharmaceutical, Italfarmaco SpA, Pfizer Inc., Sarepta Therapeutics, Inc., Santhera Pharmaceuticals, Roche, and Eli Lilly.
Jerry R. Mendell has received grants from the Parent Project Muscular Dystrophy and has received personnel fees from Sarepta Therapeutics, Inc. and National Children’s Hospital.
The LNMRC Natural History study has been supported by the Klinisch Onderzoeksfonds UZ Leuven and the Fonds voor Spierzieke Kinderen. Coinvestigator: Marleen van den Hauwe (University Hospitals Leuven, Leuven, Belgium).
The Italian DMD Registry was funded by the Fondazione Telethon (GUP 09010 and GUP 07009). Coinvestigators: Pane M, Mazzone ES, Sormani MP, Messina S, D’Amico A, Vita G, Fanelli L, Berardinelli A, Coratti G, Torrente Y, Frosini S, Norcia G, Rolle E, Magri F, Palermo C, Rossi F, Donati MA, Sacchini M, Arnoldi MT, Baranello G, Mongini T, Pini A, Battini R, Pegoraro E, Previtali S, Bruno C, Politano L, Comi GP, Bertini E, Brogna C.
The Cooperative International Neuromuscular Research Group (CINRG) Duchenne Natural History Study was funded by the United States Department of Education/National Institute on Disability and Rehabilitation Research (#H133B031118, #H133B090001); United States Department of Defense (#W81XWH-12-1-0417); National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases (#R01AR061875); Parent Project Muscular Dystrophy.
Coinvestigators: Alberto Dubrovsky (Instituto de Neurosciencias Fundacion Favaloro, Buenos Aires, Argentina); Andrew Kornberg, Monique Ryan (Royal Children’s Hospital, Melbourne, Victoria, Australia); Richard Webster (Children’s Hospital at Westmead, Sydney, New South Wales, Australia); Jean K. Mah (Alberta Children’s Hospital, Calgary, Alberta, Canada); Hanna Kolski (University of Alberta, Edmonton, Alberta, Canada); W. Douglas Biggar, Laura C. McAdam (Holland Bloorview Kids Rehab Hospital, Toronto, Ontario, Canada); S. Chidambaranathan, V. Viswanathan (Sundaram Medical Foundation and Apollo Children’s Hospital, Chennai, India); Yoram Nevo (Hadassah Hebrew University Hospital, Jerusalem, Israel); Ksenija Gorni (University of Pavia and Niguarda Ca’ Granda Hospital, Milan, Italy); Jose Carlo (University of Puerto Rico, San Juan, Puerto Rico); Mar Tulinius (Queen Silvia Children’s Hospital, Gothenburg, Sweden); Richard T. Abresch, Erik K. Henricson, Nanette C. Joyce, Craig M. McDonald (University of California Davis Health System and School of Medicine, Sacramento, CA, USA); Avital Cnaan, Tina Duong, Robert Leshner, Lauren P. Morgenroth, Carolina Tesi-Rocha, Mathula Thangarajh (Children’s National Medical Center, Washington, DC, USA); John W. Day, Peter Karachunski (University of Minnesota, Minneapolis, MN, USA); Sherilyn Driscoll, Nancy Kuntz (Mayo Clinic, Rochester, MN, USA); Anne M. Connolly, Alan Pestronk (Washington University in St Louis, St Louis, MO, USA); Hoda Z. Abdel-Hamid, Paula R. Clemens (UPMC Children’s Hospital of Pittsburgh, Pittsburgh, PA, USA); Tulio E. Bertorini (University of Tennessee, Memphis, TN, USA); Timothy Lotze (Texas Children’s Hospital Houston, TX, USA); Amy D. Harper, Jean Teasley (Children’s Hospital of Virginia, Richmond, VA, USA).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-210665.
REFERENCES
[1] | Hoffman EP , Brown RH Jr , Kunkel LM , Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell. (1987) ;51: (6):919–28. |
[2] | Aartsma-Rus A , Fokkema I , Verschuuren J , Ginjaar I , van Deutekom J , van Ommen GJ , et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat. (2009) ;30: (3):293–9. |
[3] | Emery AE . Population frequencies of inherited neuromuscular diseases–a world survey. Neuromuscul Disord. (1991) ;1: (1):19–29. |
[4] | Romitti PA , Zhu Y , Puzhankara S , James KA , Nabukera SK , Zamba GK , et al. Prevalence of Duchenne and Becker muscular dystrophies in the United States. Pediatrics. (2015) ;135: (3):513–21. |
[5] | Ryder S , Leadley RM , Armstrong N , Westwood M , de Kock S , Butt T , et al. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: An evidence review. Orphanet Journal of Rare Diseases. (2017) ;12: (1):79. |
[6] | Crisafulli S , Sultana J , Fontana A , Salvo F , Messina S , Trifirò G . Global epidemiology of Duchenne muscular dystrophy: An updated systematic review and meta-analysis. Orphanet Journal of Rare Diseases. (2020) ;15: (1):141. |
[7] | Emery AEH , Muntoni F , Quinlivan R . Duchenne muscular dystrophy. 4th ed. Oxford: Oxford University Press; (2015) . ix, pp. 308. |
[8] | McDonald CM , Henricson EK , Abresch RT , Florence J , Eagle M , Gappmaier E , et al. The 6-minute walk test and other clinical endpoints in duchenne muscular dystrophy: Reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve. (2013) ;48: (3):357–68. |
[9] | Mazzone E , Vasco G , Sormani MP , Torrente Y , Berardinelli A , Messina S , et al. Functional changes in Duchenne muscular dystrophy: A 12-month longitudinal cohort study. Neurology. (2011) ;77: (3):250–6. |
[10] | Henricson EK , Abresch RT , Cnaan A , Hu F , Duong T , Arrieta A , et al. The cooperative international neuromuscular research group Duchenne natural history study: Glucocorticoid treatment preserves clinically meaningful functional milestones and reduces rate of disease progression as measured by manual muscle testing and other commonly used clinical trial outcome measures. Muscle Nerve. (2013) ;48: (1):55–67. |
[11] | United States Food and Drug Administration. Highlights of prescribing information: EXONDYS (eteplirsen) 2020 [Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/206488lbl.pdf. |
[12] | Charleston JS , Schnell FJ , Dworzak J , Donoghue C , Lewis S , Chen L , et al. Eteplirsen treatment for Duchenne muscular dystrophy: Exon skipping and dystrophin production. Neurology. (2018) ;90: (24):e2146–e54. |
[13] | Salmaninejad A , Jafari Abarghan Y , Bozorg Qomi S , Bayat H , Yousefi M , Azhdari S , et al. Common therapeutic advances for Duchenne muscular dystrophy (DMD). Int J Neurosci. (2021) ;131: (4):370–389. |
[14] | Henricson E , Abresch R , Han JJ , Nicorici A , Goude Keller E , de Bie E , et al. The 6-Minute walk test and person reported outcomes in boys with duchenne muscular dystrophy and typically developing controls: longitudinal comparisons and clinically-meaningful changes over one year. PLoS Curr. 2013;5. |
[15] | Arora H , Willcocks RJ , Lott DJ , Harrington AT , Senesac CR , Zilke KL , et al. Longitudinal timed function tests in Duchenne muscular dystrophy: ImagingDMD cohort natural history. Muscle Nerve. (2018) ;58: (5):631–8. |
[16] | Mazzone ES , Messina S , Vasco G , Main M , Eagle M , D’Amico A , et al. Reliability of the North Star Ambulatory Assessment in a multicentric setting. Neuromuscul Disord. (2009) ;19: (7):458–61. |
[17] | Mazzone ES , Pane M , Sormani MP , Scalise R , Berardinelli A , Messina S , et al. 24 month longitudinal data in ambulant boys with Duchenne muscular dystrophy. PLoS One. (2013) ;8: (1):e52512. |
[18] | Voit T , Topaloglu H , Straub V , Muntoni F , Deconinck N , Campion G , et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): An exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. (2014) ;13: (10):987–96. |
[19] | Bushby K , Finkel R , Wong B , Barohn R , Campbell C , Comi GP , et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. (2014) ;50: (4):477–87. |
[20] | McDonald CM , Henricson EK , Abresch RT , Florence JM , Eagle M , Gappmaier E , et al. THE 6-minute walk test and other endpoints in Duchenne muscular dystrophy: Longitudinal natural history observations over 48 weeks from a multicenter study. Muscle Nerve. (2013) ;48: (3):343–56. |
[21] | Goemans N , van den Hauwe M , Wilson R , van Impe A , Klingels K , Buyse G . Ambulatory capacity and disease progression as measured by the 6-minute-walk-distance in Duchenne muscular dystrophy subjects on daily corticosteroids. Neuromuscul Disord. (2013) ;23: (8):618–23. |
[22] | Pane M , Mazzone ES , Sormani MP , Messina S , Vita GL , Fanelli L , et al. 6 Minute walk test in Duchenne MD patients with different mutations: 12 month changes. PLoS One. (2014) ;9: (1):e83400. |
[23] | Pane M , Mazzone ES , Sivo S , Sormani MP , Messina S , A DA , et al. Long term natural history data in ambulant boys with Duchenne muscular dystrophy: 36-month changes. PLoS One. (2014) ;9: (10):e108205. |
[24] | Mendell JR , Goemans N , Lowes LP , Alfano LN , Berry K , Shao J , et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol. (2016) ;79: (2):257–71. |
[25] | Mendell JR , Rodino-Klapac LR , Sahenk Z , Roush K , Bird L , Lowes LP , et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. (2013) ;74: (5):637–47. |
[26] | McDonald CM , Henricson EK , Abresch RT , Duong T , Joyce NC , Hu F , et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: A prospective cohort study. Lancet. (2018) ;391: (10119):451–61. |
[27] | Brogna C , Coratti G , Pane M , Ricotti V , Messina S , D’Amico A , et al. Long-term natural history data in Duchenne muscular dystrophy ambulant patients with mutations amenable to skip exons 44, 45, 51 and 53. PLoS One. (2019) ;14: (6):e0218683. |
[28] | Goemans N , Mercuri E , Belousova E , Komaki H , Dubrovsky A , McDonald CM , et al. A randomized placebo-controlled phase 3 trial of an antisense oligonucleotide, drisapersen, in Duchenne muscular dystrophy. Neuromuscular Disorders. (2018) ;28: (1):4–15. |
[29] | McDonald CM , Shieh PB , Abdel-Hamid HZ , Connolly AM , Ciafaloni E , Wagner KR , et al. Open-label evaluation of eteplirsen in patients with Duchenne muscular dystrophy amenable to exon 51 skipping: PROMOVI trial. Journal of Neuromuscular Diseases. (Preprint). (2021) ;(Preprint):1–13. |
[30] | ATS statement: Guidelines for the six-minute walk test. American Journal of Respiratory and Critical Care Medicine. (2002) ;166: (1):111–7. |
[31] | Bello L , Morgenroth LP , Gordish-Dressman H , Hoffman EP , McDonald CM , Cirak S , et al. DMD genotypes and loss of ambulation in the CINRG Duchenne Natural History Study. Neurology. (2016) ;87: (4):401–9. |
[32] | Hankinson JL , Odencrantz JR , Fedan KB . Spirometric reference values from a sample of the general U. S. population. American Journal of Respiratory and Critical Care Medicine. (1999) ;159: (1):179–87. |
[33] | Khan N , Eliopoulos H , Han L , Kinane TB , Lowes LP , Mendell JR , et al. Eteplirsen treatment attenuates respiratory decline in ambulatory and non-ambulatory patients with Duchenne muscular dystrophy. J Neuromuscul Dis. (2019) ;6: (2):213–25. |
[34] | Polgar G , Promadhat V . Pulmonary function testing in children: Techniques and standards: Saunders Limited.; (1971) . |
[35] | Goemans N , Signorovitch J , Sajeev G , Yao Z , Gordish-Dressman H , McDonald CM , et al. Suitability of external controls for drug evaluation in Duchenne muscular dystrophy. Neurology. (2020) ;95: (10):e1381–e1391. |
[36] | Goemans NM , Tulinius M , van den Hauwe M , Kroksmark AK , Buyse G , Wilson RJ , et al. Long-term efficacy, safety, and pharmacokinetics of drisapersen in Duchenne muscular dystrophy: results from an open-label extension study. PLoS One. (2016) ;11: (9):e0161955. |
[37] | Schoenfeld D . Partial residuals for the proportional hazards regression model. Biometrika. (1982) ;69: (1):239–41. |
[38] | Humbertclaude V , Hamroun D , Bezzou K , Bérard C , Boespflug-Tanguy O , Bommelaer C , et al. Motor and respiratory heterogeneity in Duchenne patients: Implication for clinical trials. European Journal of Paediatric Neurology. (2012) ;16: (2):149–60. |
[39] | Rall S , Grimm T . Survival in Duchenne muscular dystrophy. Acta Myol. (2012) ;31: (2):117–20. |
[40] | Kinali M , Main M , Eliahoo J , Messina S , Knight RK , Lehovsky J , et al. Predictive factors for the development of scoliosis in Duchenne muscular dystrophy. European Journal of Paediatric Neurology. (2007) ;11: (3):160–6. |
[41] | Bushby K , Bourke J , Bullock R , Eagle M , Gibson M , Quinby J . The multidisciplinary management of Duchenne muscular dystrophy. Current Paediatrics. (2005) ;15: (4):292–300. |
[42] | Birnkrant DJ , Bushby K , Bann CM , Alman BA , Apkon SD , Blackwell A , et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. The Lancet Neurology. (2018) ;17: (4):347–61. |
[43] | McDonald CM , Henricson EK , Abresch RT , Duong T , Joyce NC , Hu F , et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: A prospective cohort study. The Lancet. (2018) ;391: (10119):451–61. |
[44] | Iff J , Gerrits C , Birk E , Tuttle E , Zheng Y , Henricson E , et al. Delays in progression of Duchenne muscular dystrophy with eteplirsen treatment: attenuation of pulmonary function decline and projected freedom from continuous ventilation. Neurology. (2020) ;94: (15 Supplement):1398. |
[45] | Wang RT , Barthelemy F , Martin AS , Douine ED , Eskin A , Lucas A , et al. DMD genotype correlations from the Duchenne Registry: Endogenous exon skipping is a factor in prolonged ambulation for individuals with a defined mutation subtype. Hum Mutat. (2018) ;39: (9):1193–202. |
[46] | McDonald CM , Gordish-Dressman H , Henricson EK , Duong T , Joyce NC , Jhawar S , et al. Longitudinal pulmonary function testing outcome measures in Duchenne muscular dystrophy: Long-term natural history with and without glucocorticoids. Neuromuscular Disorders. (2018) ;28: (11):897–909. |
[47] | McDonald CM , Sajeev G , Yao Z , McDonnell E , Elfring G , Souza M , et al. Deflazacort vs prednisone treatment for Duchenne muscular dystrophy: A meta-analysis of disease progression rates in recent multicenter clinical trials. Muscle & Nerve. (2020) ;61: (1):26–35. |
[48] | Goemans N , Signorovitch J , Sajeev G , Fillbrunn M , Wong H , Ward S , et al. P. 202A composite prognostic score for time to loss of walking ability in Duchenne muscular dystrophy (DMD). Neuromus Dis. (2019) ;29: :S108. |


