
A 20-year Clinical and Genetic Neuromuscular Cohort Analysis in Lebanon: An International Effort
Abstract
Background:
Clinical and molecular data on the occurrence and frequency of inherited neuromuscular disorders (NMD) in the Lebanese population is scarce.
Objective:
This study aims to provide a retrospective overview of hereditary NMDs based on our clinical consultations in Lebanon.
Methods:
Clinical and molecular data of patients referred to a multi-disciplinary consultation for neuromuscular disorders over a 20-year period (1999–2019) was reviewed.
Results:
A total of 506 patients were diagnosed with 62 different disorders encompassing 10 classes of NMDs. 103 variants in 49 genes were identified. In this cohort, 81.4% of patients were diagnosed with motor neuron diseases and muscular dystrophies, with almost half of these described with spinal muscular atrophy (SMA) (40.3% of patients). We estimate a high SMA incidence of 1 in 7,500 births in Lebanon. Duchenne and Becker muscular dystrophy were the second most frequently diagnosed NMDs (17% of patients). These disorders were associated with the highest number of variants (39) identified in this study. A highly heterogeneous presentation of Limb Girdle Muscular Dystrophy and Charcot-Marie-Tooth disease was notably identified. The least common disorders (5.5% of patients) involved congenital, metabolic, and mitochondrial myopathies, congenital myasthenic syndromes, and myotonic dystrophies. A review of the literature for selected NMDs in Lebanon is provided.
Conclusions:
Our study indicates a high prevalence and underreporting of heterogeneous forms of NMDs in Lebanon- a major challenge with many novel NMD treatments in the pipeline. This report calls for a regional NMD patient registry.
INTRODUCTION
Neuromuscular disorders (NMDs) constitute a group of heterogeneous conditions that affect different components of the neuromuscular system, involved in controlling body movements. NMDs are characterized by dysfunction or damage of muscle or peripheral nerves, which results in partial or total loss of voluntary movement, and more seldom, leads to fatal complications such as cardiac or respiratory insufficiency. Clinical characteristics observed in NMD patients develop due to the dysfunction of motor units - composed of the lower motor neurons, the neuromuscular junction, and the skeletal muscle itself. The underlying cause for this dysfunction can be inherited or acquired [1]. To date, 1079 NMDs, classified into 16 categories, have been associated with 608 nuclear and mitochondrial genes (http://musclegenetable.fr/) [2].
The overlapping and often non-specific symptoms observed in NMD patients have made the process of differential diagnosis rather complex [1]. Moreover, the large number of genes and wide range of genetic alterations linked to these diseases have further complicated the diagnosis. Advances in the molecular biology field and the emergence of high-throughput approaches such as microarray analysis and next generation sequencing (NGS) have significantly enhanced the identification of novel NMD genes and improved the diagnostic process [3]. A systematic approach towards the diagnosis and management of hereditary NMDs is needed, especially in the Middle East, where the prevalence of these diseases is thought to be high and underestimated [4, 5]. The lack of NMDs-related data in Lebanon makes it difficult to understand and quantify their socio-economic impact, concurrent with retrospective studies calling for a much needed infrastructure for data collection and sharing in the Middle East [5, 6]. Establishing a patient registry would address this challenge and enable clinicians, researchers, policy makers, and industry experts to unify their efforts in introducing new treatment methods and providing better patient care [7]. In an effort towards starting a regional patient registry, we report herein a 20-year retrospective overview of a cohort of NMD patients from Lebanon.
METHODS
This study was conducted over a period of twenty years, from 1999 to 2019, by a multidisciplinary team from Lebanon and France that included clinicians, mainly neurologists, orthopedists, pediatricians, genetic counsellors, biologists, psychologists, and psychomotor therapists. It was further supported by two patient advocacy groups, AFM-Telethon (France) and Sesobel (Lebanon). Patients were mainly referred by neuropediatricians, adult neurologists, pathologists, and orthopedic surgeons from various Lebanese hospitals and clinics. Clinical and genetic reports of patients have been compiled from available medical records. Female and male patients of any age presenting with neuromuscular phenotypes, confirmed only clinically or coupled with molecular diagnosis, were included in this study. Ataxias, spastic paraplegias, primary cardiomyopathies, or mitochondrial diseases without neuromuscular expression were excluded. Genetic testing was carried out in different Lebanese private laboratories specialized in molecular genetic diagnosis, as well as in public or private laboratories abroad, whenever the required test was not available in Lebanon. This study was approved by the institutional ethics review board. Blood samples for DNA extraction were obtained upon informed consent. All procedures performed in studies involving the human participants were in accordance with the ethical standards of the 1975 Helsinki declaration and molecular testing was performed in distinct laboratories by different biologists specialized in molecular and human genetics. All patients underwent thorough clinical and medical investigations, before being recommended suitable molecular testing. WES analysis was performed for cases with unclear clinical diagnosis and for patients with a suspected genetically heterogeneous disease. Up until 2014, tools used for molecular diagnostics included Multiplex PCR, Southern blot, Multiplex Ligation-Dependent Probe Amplification (MLPA), as well as Restriction Fragment Length Polymorphism PCR mainly for the diagnosis of Spinal Muscular Atrophy (SMA) and Duchenne/Becker Muscular Dystrophy (DMD; BMD). Sanger sequencing was applied for all other monogenic diseases. The use of Next Generation Sequencing (NGS) techniques has gradually become routine practice since then. A panel including 378 genes associated with NMDs was sequenced by NGS in patients suspected to present a clinically or genetically heterogeneous disease. Genomic DNA obtained from the tested sample was enriched for targeted regions using a hybridization-based protocol, and sequenced. All targeted regions were sequenced with ≥50X depth. When the genetic panel failed to identify the molecular diagnosis, Whole Exome Sequencing (WES) was performed [8]. Approximately 37 Mb (214,405 exons) of the Consensus Coding Sequences (CCS) were enriched from fragmented genomic DNA by more than 340,000 probes designed against the human genome (Nextera Rapid Capture Exome, Illumina) and the generated library sequenced on an Illumina NextSeq or HiSeq 4,000 platform to an average coverage depth 70–100X. An end to end bioinformatics pipeline including base calling, primary filtering of low quality reads and probable artefacts, and annotation of variants was applied. To select the deleterious variants, variants with a frequency greater than 1% in public databases (including gnomAD and 1000 Genomes) were filtered out in order to only select rare variants that might be relevant. Furthermore, due to the challenging interpretation of variants found in non-coding areas such as UTRs and intronic regions, these were discarded from our analysis unless their implication in the disease was previously reported. Relevant genetic variants were only selected if their depth is ≥50X. All identified variants were evaluated with respect to their pathogenicity and causality and categorized based on the guidelines of the American College of Medical Genetics and Genomics (ACMG).
mtDNA sequencing was performed by Centogene AG, Germany. For select patients who underwent mitochondrial analysis, the entire mitochondrial genome was amplified using two overlapping long range PCRs. Amplicons were examined for commonly reported deletions within the mitochondrial genome using Bioanalyzer. Libraries generated using Illumina compatible adapters were sequenced on Illumina platforms to an average sequencing depth of > 1000X.
All variants identified by means of NGS analysis were subsequently confirmed by Sanger sequencing. Segregation studies were performed for all families wishing to complete the genetic study for their genealogy as a part of the offered genetic counselling.
RESULTS
The NMD cohort we present here consisted of 506 patients, including 291 males (57.5%) and 215 females (42.5%) from referrals across clinics in Lebanon. Patients were diagnosed between the ages of 0 and 60, and mainly exhibited a pediatric disease presentation (median = 2yrs; mean = 8yrs). 62 genetic disorders and clinical presentations classified into 10 groups were identified in this study (Fig. 1). Motor Neuron Diseases (MND), Muscular Dystrophies (MD), and Charcot-Marie-Tooth disease (CMT), represented the bulk of disorders identified with 227 (44.9%), 170 (36.6%), and 66 (13%) patients respectively. The remaining 28 (5.5%) patients exhibited congenital, metabolic, and mitochondrial myopathies, congenital myasthenic syndromes, and myotonic dystrophy. Disorders identified in this study encompassed monogenic inherited disorders, mostly involving autosomal recessive (n = 33) and dominant (n = 10) NMDs; X-linked (n = 4) and mitochondrial (n = 1) disorders constituted the remaining disorders identified (Table 1). Expectedly, SMA, DMD, and BMD were diagnosed in over half of the patients (290 patients; 57.3%). 95 patients of our cohort were diagnosed clinically without molecular testing. The remaining 411 patients underwent molecular diagnosis; variant data was available for 324 of those patients, thus generating a success rate of 79% for the diagnostic strategy followed. 103 distinct variants in 49 genes were identified. Based on zygosity, patients exhibited 35 homozygous, 14 heterozygous, 7 compounds heterozygous, 41 hemizygous variants, and 1 mitochondrial variant (Table 2). 39 distinct variants in DMD were notably identified (Fig. 2). A heterogeneous presentation of Limb-Girdle MD (LGMD) and congenital MD (CMD), involving 15 subtypes (Fig. 3), as well as CMT, involving 12 subtypes, was prominent. All families with no exceptions underwent genetic counselling and segregation studies.
Fig. 1
Sunburst representing the spectrum of hereditary NMDs in the patient cohort. Arranged by number of patients clockwise, the inner ring represents NMD groups as classified by The 2021 version of the gene table of neuromuscular disorders [2]; the outer ring represents short names of NMD subtypes separated by number of patients. Full names and OMIM numbers for each disorder are included in Table 2. *Marked disorders indicate either an undetermined NMD subtype with all patients having been only clinically diagnosed, or disease-gene correlations not currently recognized as a subtype on OMIM.
![Sunburst representing the spectrum of hereditary NMDs in the patient cohort. Arranged by number of patients clockwise, the inner ring represents NMD groups as classified by The 2021 version of the gene table of neuromuscular disorders [2]; the outer ring represents short names of NMD subtypes separated by number of patients. Full names and OMIM numbers for each disorder are included in Table 2. *Marked disorders indicate either an undetermined NMD subtype with all patients having been only clinically diagnosed, or disease-gene correlations not currently recognized as a subtype on OMIM.](https://content.iospress.com:443/media/jnd/2022/9-1/jnd-9-1-jnd210652/jnd-9-jnd210652-g001.jpg)
Table 1
List of hereditary NMDs identified in this study, distributed by modes of inheritance
| Autosomal Recessive | |
| BVVL2 | LGMDR5 |
| CMS25 | LGMDR6 |
| CMT2S | LGMDR10 |
| CMT2T | LGMDR23 |
| CMT4A | MDCL |
| CMT4B3 | MDDGB6 |
| CMT4C | MDDGC1 |
| CMT4F | MDDGC5 |
| CMT4H | MTPD |
| CMT6C | NEM2 |
| CMTRIC | RSMD1 |
| DSMA1 | SMA1 |
| GSD2 | SMA2 |
| LGMDR1 | SMA3 |
| LGMDR2 | UCMD1 |
| LGMDR3 | NBIA2A |
| LGMDR4 | |
| Autosomal Dominant | |
| FSHD1 | CMTDIE |
| BTHLM1 | CCD |
| FTDALS4* | TAM1 |
| ALS1* | MD1 |
| CMT1A | EDMD4* |
| X-Linked Recessive | |
| BMD | |
| CNMX | |
| DMD | |
| X-Linked Dominant | |
| CMTX1 | |
| Mitochondrial | |
| MM (MTRNR2) | |
Full names of the disorders and respective OMIM numbers are included in Table 2. *Sporadic manifestations of genetic NMDs were identified in patients with EDMD4, ALS1, and FTDALS4, associated with de novo mutations in SYNE1, SOD1, and TBK1 respectively.
Table 2
List of hereditary NMDs with associated genes and variants identified in the patient cohort
| NMD group | Disorder (Short name; MIM#) | Patients (#); Families (#) | Ages at Diagnosis (years or range in years with avgerage)† | Associated Gene or Genomic Region (MIM#) | Mutations identified (homozygous; heterozygous; compound heterozygous; hemizygous)† | References† |
| Motor Neuron Diseases | Amyotrophic Lateral Sclerosis 1 (ALS1; 105400) | 8; 6 | 50-51 | SOD1 (147450) | c.352C > G p.Leu118Val | – |
| Brown-Vialetto-Van Laere syndrome 2 (BVVL2; 614707) | 6; 2 | 12–19 (avg. 16) | SLC52A2 (607882) | c.916G4A p.Gly306Arg | [28, 29] | |
| Distal Spinal Muscular Atrophy 1 (DSMA1; 604320) | 6; 3 | < 1 | IGHMBP2 (600502) | c.1540G > A p.Glu514Lys | [14] | |
| Distal Spinal Muscular Atrophy (DSMA; Unknown subtype) | 1; 1 | – | – | – | ||
| Frontotemporal Dementia and/or Amyotrophic Lateral Sclerosis 4 (FTDALS4; 616439) | 2; 2 | 35; 39 | TBK1 (604834) | c.2079_2082del p.Glu695Argfs*16 | [8] | |
| c.2079_2082del p.Glu695Argfs*16 | ||||||
| Spinal Muscular Atrophy 1 (SMA1; 253300) | 190; – | < 1 | SMN1a (600354) | exon 7 &8 deletion | – | |
| Spinal Muscular Atrophy 2 (SMA2; 253550) | 7; 7 | 2–15 (avg. 6) | exon 8 deletion | |||
| Spinal Muscular Atrophy 3 (SMA3; 253550) | 7; 4 | 7–33 (avg. 20) | c.549del; p.Lys184Serfs* and exon 7 & 8 deletion | |||
| Hereditary Motor and Sensory Neuropathies | Charcot-Marie-Tooth Disease, Axonal, Type 2S (CMT2S; 616155) | 4b; 2 | 32–53 | IGHMBP2 (600502) | c.62G > T p.Arg21Ileb | [8] |
| Charcot-Marie-Tooth Disease, Axonal, Type 2T (CMT2T; 617017) | 2; 2 | – | MME (120520) | Under publication (Delague V. et al.) | ||
| Charcot-Marie-Tooth Disease, Demyelinating, Type 1A (CMT1A; 118220) | 4; 4 | 6 | PMP22 (601097) | 1.5-Mb duplication at 17p11.2 | – | |
| Charcot-Marie-Tooth Disease, Type 4A (CMT4A; 214400) | 5; 4 | 7–26 (avg. 13) | GDAP1 (606598) | c.668T > A p.Leu223* | [78] | |
| Charcot-Marie-Tooth Disease, Type 4B3 (CMT4B3; 615284) | 2; 1 | 7; 12 | SBF1 (615284) | c.1004T > C p.Lys335Pro | [65] | |
| Charcot-Marie-Tooth Disease, Type 4C (CMT4C; 601596) | 1; 1 | – | SH3TC2 (608206) | Unpublished data (Delague V. et al.) | ||
| Charcot-Marie-Tooth Disease, Demyelinating, Type 4F (CMT4F; 614895) | 10; 3 | 0–19 (avg. 6) | PRX (605725) | c.586C > T p.Arg196* | [67] | |
| Charcot-Marie-Tooth Disease, Demyelinating, Type 4H (CMT4H; 609311) | 2; 2 | 17 | FGD4 (611104) | c.1698G > A p.Met566Ile | [79] | |
| Charcot-Marie-Tooth Disease, Type 6C (CMT6C; 618511) | 4; 2 | – | PDXK (179020) | c.628G > A p.Ala228Thr | Under publication (Delague V. et al.) | |
| Charcot-Marie-Tooth Disease, Dominant Intermediate E (CMTDIE; 614455) | 1; 1 | – | INF2 (610982) | c.312C > G p.Cys104Trp | – | |
| Charcot-Marie-Tooth Disease, Recessive Intermediate C (CMTRIC; 615376) | 5; 4 | 7– 15 (avg. 12) | PLEKHG5 (611101) | c.909C > A p.Tyr303* | [14] Under publication | |
| c.1452_1453del p.His485Profs*169 | (Delague V. et al.) | |||||
| Charcot-Marie-Tooth Disease Type I (CMT1; Unknown subtype) | 3; 2 | 13; 28; 37 | – | – | ||
| Charcot-Marie-Tooth Disease Type I (CMT1; Associated with MTMR4 mutation) | 1; 1 | 0 | MTMR4 (603559) | Unpublished data (Delague V. et al.) | ||
| Charcot-Marie-Tooth Disease Type 2 (CMT2; Unknown subtype) | 3; 3 | 23; 44 | – | – | ||
| Charcot-Marie-Tooth Disease Type 2 (CMT2; Associated with BAG3 mutation) | 1; 1 | 25 | BAG3 (603883) | Under publication (Delague V. et al.) | ||
| Charcot-Marie-Tooth Disease, X-Linked Dominant, 1 (CMTX1; 302800) | 2; 2 | 7; 34 | GJB1 (304040) | c.164_184dup p.Thr55_Asn61dup | [8] | |
| c.G139A p.Glu47Lys | ||||||
| Charcot-Marie-Tooth Disease (CMT; Associated with DNAJB2 mutation) | 1; 1 | – | DNAJB2 (604139) | Under publication (Delague V. et al.) | ||
| Charcot-Marie-Tooth Disease (CMT; Associated with VRK1 mutation) | 1; 1 | – | VRK1 (602168) | |||
| Charcot-Marie-Tooth Disease (CMT; Unknown subtype) | 14; 14 | 14– 50 | – | – | ||
| Muscular Dystrophies | Becker Muscular Dystrophy (BMD; 300376) | 13c; 5 | 6– 30 (avg. 15) | DMD (300377) | See Fig. 2 for exonal deletions | – |
| Bethlem Myopathy 1 (BTHLM1; 158810) | 3; 2 | 12; 15; 43 | COL6A1 (120220) | c.928_930delAAG p.Lys310del | – | |
| c.868G > A p.Gly290Arg | ||||||
| Duchenne Muscular Dystrophy (DMD; 310200) | 73c; 62 | 0– 41 (avg. 10) | DMD (300377) | See Fig. 2 for exonal deletions and exonal positions of truncating variants | [8] | |
| exon 53 duplication | ||||||
| t(X;1)(p21;q23) | ||||||
| c.94-1G > T | ||||||
| c.4071 + 1G > A | ||||||
| Emery-Dreifuss Muscular Dystrophy 4 (EDMD4; 612998) | 1; 1 | 7 | SYNE1 (608441) | c.18445C > A p.Arg6149Ser | [14] | |
| Facioscapulohumeral Muscular Dystrophy 1 (FSHD1; 158900) | 21d; 13 | 12– 52 (avg. 27) | Chr.4q35 [DUX4] (606009) | 4qA haplotype with 3– 6 D4Z4 microsatellite repeats | – | |
| Limb Girdle Muscular Dystrophy Recessive 1 (LGMDR1; 253600) | 11; 5 | 7– 22 (avg. 19) | CAPN3 (114240) | c.257C > T p.Ser86Phe | – | |
| c.[257C > T];[956C > T] p.[Ser86Phe];[Pro319Leu] | ||||||
| c.[956C > T; 310-4_310-1delACAG] p.[Pro319Leu];[?] | ||||||
| Limb Girdle Muscular Dystrophy Recessive 2 (LGMDR2; 253601) | 2; 2 | 21; 42 | DYSF (603009) | c.5438T > C p.Leu1813Pro | – | |
| Limb Girdle Muscular Dystrophy Recessive 3 (LGMDR3; 608099) | 6; 4 | 3– 15 (avg. 11) | SGCA (600119) | c.[157G > A];[c.574C > T] p.[Ala53Thr];[Arg192*] | [8] | |
| Limb Girdle Muscular Dystrophy Recessive 4 (LGMDR4; 604286) | 3; 3 | 10 | SGCB (600900) | c.-10_22dup p.Ala8fs | – | |
| Limb Girdle Muscular Dystrophy Recessive 5 (LGMDR5; 253700) | 6; 4 | 2– 24 (avg. 13) | SGCG (608896) | exon 7 deletion | [8] | |
| Limb Girdle Muscular Dystrophy Recessive 6 (LGMDR6; 601287) | 1; 1 | 8 | SGCD (601411) | c.784G > A p.Glu262Lys | – | |
| Limb Girdle Muscular Dystrophy Recessive 10 (LGMDR10; 608807) | 1; 1 | 6 | TTN (188840) | c.36040A > T; p.Lys12014* | [8] | |
| Limb Girdle Muscular Dystrophy Recessive 23 (LGMDR23; 618138) | 8; 7 | 4– 30 (avg. 11) | LAMA2 (156225) | c.8244 + 3_8244 + 6del | [8] | |
| c.8244 + 2dupT | ||||||
| c.[3829C > T];[1300C > T] p.[Arg1277*];[Arg434*] | ||||||
| Limb Girdle Muscular Dystrophy Recessive (LGMDR; Unknown Subtype) | 12; 7 | 10– 45 (avg. 22) | – | – | ||
| Muscular Dystrophy-Dystroglycanopathy, Limb-Girdle, Type C, 1 (MDDGC1; 609308) | 6; 3 | 4– 22 (avg. 12) | POMT1 (607423) | c.[1858C > T];[221C > T] p.[Arg620*];[Ala74Val] | [8, 14] | |
| c.[200C > A];[2005G > A] p.[Pro67Gln];[Ala669Thr] | ||||||
| Muscular Dystrophy-Dystroglycanopathy, Limb-Girdle, Type C, 5 (MDDGC5; 607155) | 3; 3 | 1; 6; 15 | FKRP (606596) | c.823C > T p.Arg275Cys | [8] | |
| Congenital Muscular Dystrophies | Muscular Dystrophy-Dystroglycanopathy, Congenital with Impaired Intellectual Development, Type B, 6 (MDDGB6; 608840) | 2; 1 | 4; 8 | LARGE1 (603590) | Intron 10 42.9-KB insertion/4.1-KB deletion | [80] |
| Muscular Dystrophy, Congenital, LMNA-Related (MDCL; 613205) | 1; 1 | 6 | LMNA (150330) | c.1867A > G p.Thr623Ala | [14] | |
| Muscular Dystrophy, Congenital (CMD; Unknown subtype) | 4; 4 | 8 | – | – | ||
| Rigid Spine Muscular Dystrophy 1 (RSMD1; 602771) | 2; 2 | 14 | SELENON (606210) | c.1405C > T p.Arg469Trp | – | |
| Ullrich Congenital Muscular Dystrophy 1 (UCMD1; 254090) | 6e; 3 | 0– 3 | COL6A1 (120220) | COL6A1: c.2923G > C p.Gly975Arg | [14] | |
| COL6A2 (120240) | COL6A2: c.2611G > A p.Asp871Asne | |||||
| COL6A3 (120250) | COL6A3: c.8136del p.Arg2713Glyfs*3 | |||||
| Congenital Myopathies | Central Core Disease of Muscle (CCD; 117000) | 1; 1 | 25 | RYR1 (180901) | c.8758C > T p.Arg2920* | [8] |
| Myopathy, Centronuclear, X-Linked (CNMX; 310400) | 2; 2 | 0 | MTM1 (300415) | c.969dupA p.Val324Serfs*8 | – | |
| c.1190A > G; p.Tyr397Cys | ||||||
| Nemaline Myopathy 2 (NEM2; 256030) | 1; 1 | 5 | NEB (161650) | c.5452-1G > A | – | |
| Congenital Myopathy (CM; Unknown subtype) | 3; 3 | 1; 7; 9 | – | – | ||
| Metabolic Myopathies | Glycogen Storage Disease II (GSD2; 232300) | 2; 2 | 7 | GAA (606800) | c.266G > A p.Arg89His | [8] |
| c.1927G > A p.Gly643Arg | ||||||
| Neurodegeneration with Brain Iron Accumulation 2A (NBIA2A; 256600) | 3; 3 | 3; 4; 5 | PLA2G6 (603604) | c.2257G > T p.Val753Phe | [8] | |
| c.2370T > G p.Tyr790* | ||||||
| Other Myopathies | Tubular Aggregate Myopathy 1 (TAM1; 160565) | 1; 1 | 12 | STIM1 (605921) | c.57G > C p.Gln19His | – |
| Myotonic Syndromes | Myotonic Dystrophy 1 (MD1; 160900) | 3; 2 | 30; 33; 60 | DMPK (605377) | 3’ CTG triplet repeat expansion (> 400 repeats) | – |
| Other Neuromuscular Disorders | Mitochondrial DNA depletion syndrome (MTDPS; Unknown subtype) | 2; 1 | 15 | – | – | |
| Congenital Myasthenic Syndromes | Myasthenic Syndrome, Congenital, 25, Presynaptic (CMS25; 618323) | 1; 1 | 14 | VAMP1 (185880) | c.97C > T p.Arg33* | [8] |
| Unspecified | Mitochondrial Myopathy (MM; Associated with MTRNR2 mutation) | 1; 1 | 11 | MTRNR2 (561010) | m.2119T > Cf | – |
| Mitochondrial Trifunctional Protein Deficiency (MTPD; 609015) | 1; 1 | 28 | HADHA (600890) | c.703C > T p.Arg235Trpg | – | |
| Unclassified NMDs | 7; 6 | 3– 52 (avg. 27) | – | – | ||
†Patient data on age, variants, and relevant published studies are available only for independent subsets of patients. a: Spinal Muscular Atrophy patients presented with varying copy numbers of SMN2 b: One patient presented with uncharacteristic syndromic features including delayed motor language development, intellectual disability, microcephaly, and dysmorphic facial features; c: All female patients were manifesting carriers of DMD mutations; d: One patient presented with typical FSHD1 symptoms and a negative molecular test; e: One patient presented with uncharacteristic cognitive features in the form of psychomotor delay; f: Heteroplasmic MTRNR2 variant in a patient exhibiting features of mitochondrial myopathy; g: Variant position is based on the precursor from of the mitochondrial protein.
Fig. 2
Exonal dystrophin (DMD) deletion and truncating mutation patterns in patients with DMD and BMD in this study. Values over each bar represent the number of patients for each of the 36 variants. Remaining DMD mutations including duplication, translocation, and splice site variants are described in Table 2.
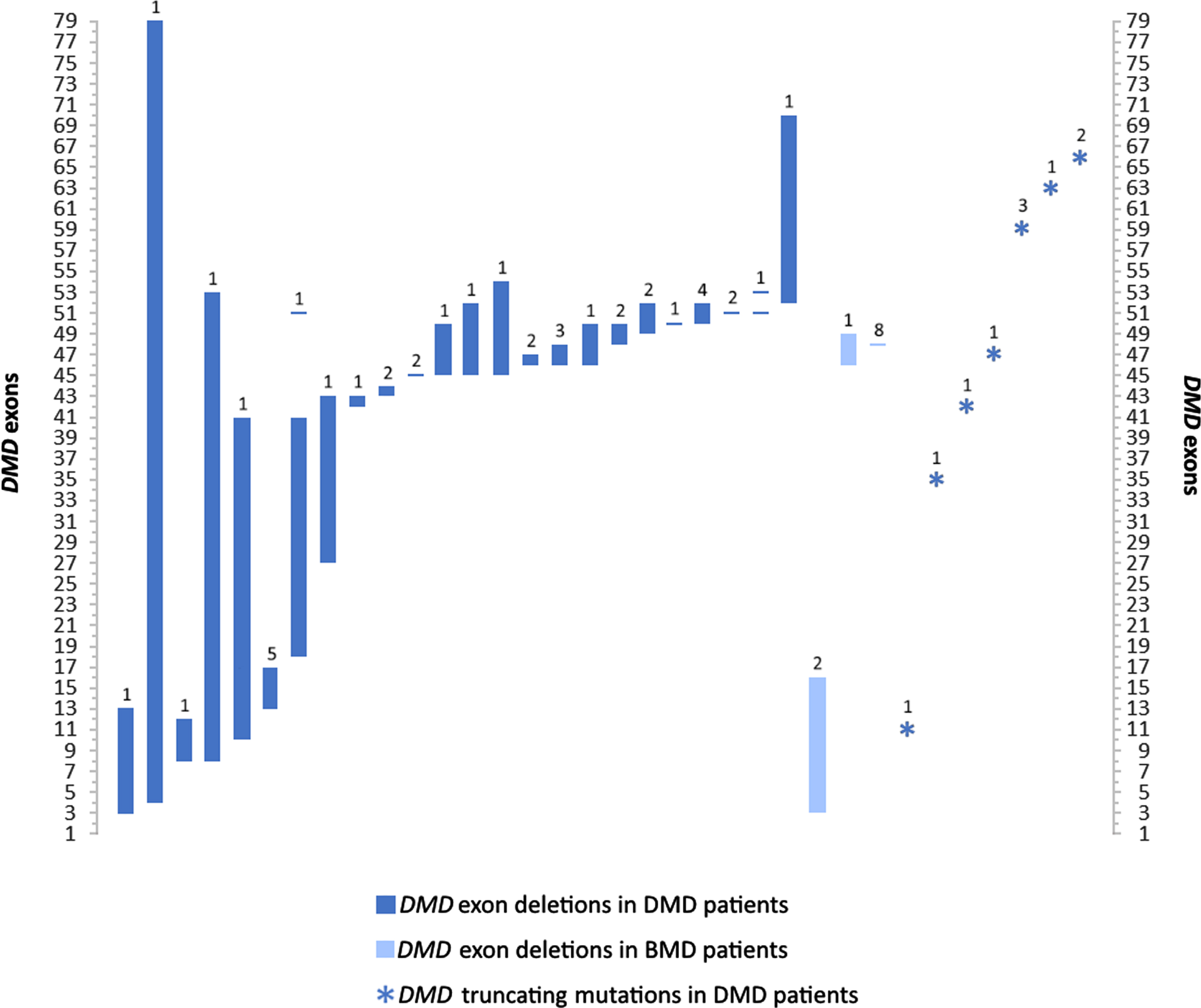
Fig. 3
Distribution of Limb Girdle Muscular Dystrophies and Congenital Muscular Dystrophies in this cohort. MD disorders listed are based on the current revised classification of LGMD [81]; known aliases are included in a dotted box to the left of the official OMIM disease name. Disease subgroups are based on associated genes, derived from Liewluck and Milone [82].
![Distribution of Limb Girdle Muscular Dystrophies and Congenital Muscular Dystrophies in this cohort. MD disorders listed are based on the current revised classification of LGMD [81]; known aliases are included in a dotted box to the left of the official OMIM disease name. Disease subgroups are based on associated genes, derived from Liewluck and Milone [82].](https://content.iospress.com:443/media/jnd/2022/9-1/jnd-9-1-jnd210652/jnd-9-jnd210652-g003.jpg)
DISCUSSION
5q-Spinal Muscular Atrophy
The most prevalent NMD in this study cohort was proximal SMA linked to chromosome 5q (40.3% of patients). SMA is an autosomal recessive lower alpha motor neuron disease globally estimated to affect ∼1 in 4,000 to 20,000 births [9]. Patients in this study presented with characteristic features of acute infantile and intermediate forms of (SMA1-3). Assessment of SMN1 variants in 185 patients (90.7% of SMA patients) from this cohort revealed homozygous deletions of exons 7 and 8 to be the most common molecular diagnosis. Assessment of SMN2 gene copy number, which typically correlates inversely with disease severity [10], was not performed for most patients as it was not standard protocol at the time of the patients’ diagnoses; access to second-tier investigations remains challenging for many Lebanese families. One of the patients in our cohort, presenting with SMA1, exhibited compound heterozygous mutations (c.549del; p.Lys184Serfs* and exon 7 and 8 deletion). Genotypes with deleterious point variants in addition to exon deletions are uncommon and homozygosity for such point variants are extremely rare. Interestingly, the same variant from our study (c.549del) was observed in an Iranian cohort where only one patient presenting with SMA1 exhibited it in homozygous state, whilst the remaining patients exhibited exon 7 and 8 deletions [11].
Published genetic studies on SMN1-associated SMA in Lebanon are limited to a few reports on patients with chronic forms of the disorder [12, 13]. These include a report on a Lebanese family with SMA3, described amongst 24 families in a linkage analysis study associating chronic proximal SMA with chromosome 5q preceding the discovery of the gene in 1994 [12]. Another study highlighted a homozygous variant (c.859G > C) in a Lebanese individual, which was also reported to be a protective marker against severe SMA [14, 15].
While the reported carrier frequency of SMA in the US, Australia, Europe, and the UK is 1:40–80, it has been reported to be much higher (∼1:20) in many parts of the Middle East and North Africa regions [16–21]. Based on a calculated average number of births per year for the 20-year period covered by this study using population (World Bank) and birth rate (UNICEF) data, we estimate a high annual incidence of ∼1 in 7,500 individuals with SMA in Lebanon. This is comparable to rates in Saudi Arabia (∼1 in 7000) [22], Egypt [17], and Libya (∼1 in 12500 for SMA1) [23]. Early treatment and intervention can improve clinical outcomes in SMA patients with the advent of new therapies [24]. Although Lebanon is far away from adopting SMA into neonatal screening programs, increased awareness about these disorders was noticed throughout the duration of this study. Additionally, the importance of pre-implantation and prenatal genetic screening in families with a history of SMA has become more highlighted in the general community.
Non-5q Spinal Muscular Atrophy
Other MNDs in this study included Amyotrophic Lateral Sclerosis (ALS), which has a reported prevalence and annual incidence of 3–5:100,000 and 1.5–2.7:100,000 individuals respectively [25]. In our study, six families were described with ALS1 and two families with FTDALS4 (Frontotemporal dementia and/or amyotrophic lateral sclerosis 4). Notably, a novel homozygous TBK1 mutation (c.2079_2082del; p.Glu695Argfs*16) was identified in a 35 year old patient who presented with an abnormal gait and walking difficulty. Another 39 year old patient, heterozygous for the same variant presented with an abnormal gait, juvenile arthritis, dysphagia, dysarthria, and respiratory symptoms.
Literature from Lebanon is lacking, however the country has joined other regions in clinical trials for ALS modifying treatments [26]. A recent clinical study of ALS, which described 140 patients from Lebanon, revealed 113 patients with ALS and a lower mean age of onset relative to global reports, with the remaining patients presenting with six subtypes of atypical ALS [27].
Other non-5q MND disorders identified in this cohort included Brown-Vialetto-Van Laere syndrome 2 (BVVLS2), diagnosed in six patients from two families in our cohort [28]. All patients exhibited the same homozygous mutation (c.916G > A) in the riboflavin transporter gene SLC52A2. Interestingly, six other Lebanese patients from three families were described with BVVLS2 attributed to the same mutation [29]; haplotype and functional analysis revealed a shared ancestral allele highlighting the c.916G > A as a founder mutation in the Lebanese population. A subsequent study identified the same mutation in another large consanguineous Lebanese family [30].
Distal Spinal Muscular Atrophy 1 (DSMA1), another non-5q SMA associated with mutations in the IGHMBP2 gene, was diagnosed in six patients from three families in this cohort. In the literature, a Lebanese family was among the first three families presenting with SMA features and normal SMN1, with haplotype analysis revealing linkage at 11q13-q21 [31]. Subsequent studies have identified homozygous mutations in IGHMBP2 in the same [32]. Interestingly, a homozygous mutation in IGHMBP2 (c.449 + 1G > T) was more recently associated with CMT2S in two Lebanese siblings [33].
Duchenne and Becker Muscular Dystrophies
MDs constituted the second most prevalent group of disorders among patients in this study, with a predominantly male cohort of DMD and BMD patients (78 males and 8 females; 17% of patients). DMD is an X-linked progressive muscle wasting disorder with a global prevalence of 1 in 4000–6000 live male births [34]. The average age of confirmatory diagnosis for DMD in our cohort was 10 years, with half of the patients diagnosed at the age of eight or below. This is above the general global average and median diagnosis age in Western countries [35] and in the Middle East [5]. The average age of diagnosis for BMD was also higher than the general mean age of 12 [35]. DMD molecular data in our study was available for 61 patients, encompassing single and multiple exon deletions (71.1%), truncating mutations (18.4%), one duplication (exon 53), two splice site mutations, as well as a balanced translocation (Table 2; Fig. 2). Multi-exon deletions in our study encompassed most of the dystrophin gene, with the most common regions (exons 45–52 and 3–17) falling within previously characterized hotspot regions identified in large databases [36]. Interestingly, despite the rarity of DMD diagnoses in females, we report a manifesting female carrier with an 8–53 exon deletion. BMD-associated molecular data involved multi-exon deletions (exons 3–16 and 46–49) identified in three patients from two unrelated families (Fig. 2). Eight patients, including four males and four female carriers, harboring a deletion of exon 48 exhibited a very mild BMD phenotype, consistent with previous reports [37].
Comparable to our molecular findings, a recent study described a male cohort of 52 unrelated Lebanese DMD patients with a relatively lower average age of diagnosis (7 years) [38]. They described similar hotspot regions of multi-exon and single-exon deletions (in addition to exons 19, 52, and 53).
With Lebanese patients representing the typical DMD cohort, there is potential benefit from recent disease-modifying treatments, especially those targeting exon skipping [39]. Truncating point variants were more prominent than expected in our cohort relative to the previous Lebanese study [38]. These mutations are also targets of approved drug therapies [40]. An early diagnosis of DMD however is better and sometimes necessary to ensure drug efficacy. The adoption of promising therapies is contingent on the systematic overhaul of the DMD diagnostic process in Lebanon. To date, only a handful of male DMD patients have had access to these costly therapies.
Facioscapulohumeral Muscular Dystrophy
Facioscapulohumeral Muscular Dystrophy Type 1 (FSHD1) was the third most diagnosed MD and NMD in this study (4.2% of patients). Globally, FSHD has an estimated prevalence range of 1 in 8000 to 20000 individuals [41]. We report on 21 patients, 17 of whom had a confirmed genetic diagnosis with distal subtelomeric contractions of polymorphic repeats in chromosome 4. All patients exhibited the FSHD1 4qA allele with D4Z4 microsatellite deletions resulting in a range of 3 to 6 repeats. One of the patients in our cohort exhibited typical FSHD features, however no structural anomalies were detected on chromosome 4, suggesting that he may have FSHD2 or a mimicking muscle disorder.
Published data on FSHD in Lebanon is scarce. In a recent in-depth genetic analysis study on a large cohort of FSHD patients, a Lebanese family with five affected individuals was discussed, four of whom were diagnosed with distal 4qA repeat deletions [42]. Literature on FSHD in the Middle East is limited to a few studies from Iran, Egypt and Libya reporting relatively low numbers of patients in NMD cohorts [43–46]. The molecular diagnosis of FSHD1 remains technically challenging even with NGS, thus explaining in part the low rate of detection of this condition in Middle Eastern populations.
Limb Girdle Muscular Dystrophy
LGMD affects approximately 1 in 61,000 (35,000–106,000) individuals globally [47], with 34 subtypes currently associated with 34 genes (OMIM 2020). 11 subtypes of LGMD were identified in this study. LGMDR1 was the most frequently diagnosed subtype, associated with homozygous mutations in CAPN3. Another notable subtype from our study was LGMDR5, identified in four families, exhibited a recurring exon 7 deletion in SGCG. Published literature on hereditary LGMD in Lebanon is limited, mainly involving familial case reports of LGMDR1 with variants described in this study (c.257C > T and c.956C > T). Additionally, c.717ΔT and c.2242C > G variants were reported in Lebanese families presenting with mild and severe features respectively [48, 49]. Novel polymorphisms in LAMA2 and SGCG, potentially influencing gene expression profiles, were identified in 35 healthy Lebanese individuals among other ethnicities [50].
The cohort described in our study is reflective of global reports, with LGMDR1 being the most frequently observed subtype [51, 52]. In the Middle East, a study on 60 Iranian families with LGMD showed a broadly similar pattern of LGMD relative to our cohort, involving –in order of frequency –LGMDR1, LGMDR4, followed by other sarcoglycanopathies and dysferlinopathy [53]. Clinics throughout the MENA region including Algeria, Tunisia, and Egypt have notably reported other trends [54]. More recently, a review of muscle biopsies from NMD patients in Saudi Arabia revealed mainly dysferlinopathies, sarcoglycanopathies, and congenital MDs including merosin-deficient CMD and Walker Warburg Syndrome [6].
Congenital Muscular Dystrophy
CMDs involve heterogeneous autosomally inherited disorders with severe features (relative to LGMD) typically manifesting at birth or infancy. Limited epidemiological data suggest a low prevalence of 0.68–2.5:100,000 [55]. We report on four forms of CMD including Rigid Spine MD (associated with SEPN1 or SELENON), LMNA-associated CMD, Ullrich Congenital Muscular Dystrophy 1 (UCMD1; associated with COL6), and LARGE1-related CMD. In addition, other dystroglycanopathies considered to be CMDs, such as FKRP and POMT1 related MDs, were included among the LGMD group in this cohort due to prominent features of slowly progressive or late-onset limb-girdle muscle weakness (Fig. 3). Notably, a novel homozygous variant in COL6A3 (c.8136del) was identified in association with UCMD1; another variant in COL6A1, which we report for the first time at the homozygous state (c.2923G > C), was also associated with UCMD1. Literature on CMD in Lebanon is scarce involving multiple families with distinct genotype-phenotype correlations mainly involving dystroglycanopathies. Variants in POMT1 were reported in Lebanese patients with MDDGB1 (MIM# 613155) and Walker-Warburg Syndrome [56, 57]. A Lebanese family was reported with MDDGA12 (MIM# 615249), associated with homozygous mutations in the POMK gene [58]. Additionally, heterozygous LMNA variants were associated with cardiomyopathy and with Emery-Dreifuss Muscular Dystrophy 2 [59, 60].
Charcot-Marie-Tooth disease
The remaining major group of NMDs identified in our study involved CMT disease. CMT is the most common hereditary neuropathy estimated to affect ∼1 in 1,200 to 10,000 individuals [61]; it involves a large group of genetically and clinically heterogeneous HMSN disorders. Before the emergence of NGS, molecular diagnostic rates for CMT were relatively poor. The adoption of whole exome sequencing has rapidly increased gene discovery, with multigene-panels set to cover future diagnoses [3, 62]. There are currently 75 CMT subtypes associated with 58 genes (OMIM 2020). We report 12 subtypes including recessive demyelinating forms of CMT (CMT4A, CMT4B3, CMT4F, CMT4H, CMT4C, and CMT6C); followed by axonal (CMT2S and CMT2T); recessive and dominant intermediate forms (CMTRIC and CMTDIE); dominant demyelinating CMT1A; as well as X-linked axonal and demyelinating form - CMTX1. A recently characterized subtype, CMT6C, was identified in two families from our cohort, associated with homozygous mutations in PDXK (under publication; Dr. Delague). Additionally, CMT patients in this study were observed with mutations in BAG3, DNAJB2, MTMR4, and VRK1 (under publication; Dr. Delague); CMT genotype-phenotype correlations with variants in BAG3 and DNAJB2 genes were recently described [63, 64]. Our cohort also included CMT4B3 patients presenting with unique syndromic features. These patients –from a single family –lacked sensory symptoms and exhibited a fork and bracket MRI sign related to brainstem abnormalities [65]. A recent study in the Lebanese population involving 116 sporadic and familial cases with CMT revealed CMT1A (associated with a commonly described duplication variant in the PMP22 gene) to be the most prevalent type, followed by CMT4H and CMT4F [66]. Loci associated with the latter two subtypes (FGD4 and PRX respectively) were first identified in Lebanese patients [67, 68]. Novel CMT genes continue to be identified in the Lebanese population, including the second reported study on MCM3AP-associated CMT in a Lebanese family [69].
Comparison with other NMD reports
Global reports have shown markedly different spectrums of NMDs. A study on hereditary NMDs in North England identified 31 disorders in 1,100 patients, most commonly involving myotonic dystrophy followed by dystrophinopathies and FSHD. In contrast to our cohort, the study reported fewer cases of SMA [51]. A review of NMD registry data in the Netherlands revealed ALS and myotonic dystrophy to be most common, whereas SMA was diagnosed as frequently as CMT in their study [70].
Clinically focused community surveys and hospital studies have offered brief insight into the spectrum of NMDs in the Middle East, including studies in Egypt, Saudi Arabia, and Libya, with sparse reporting on a multitude of NMD subtypes [22, 44–46, 71]. While large, in-depth, and locus-specific mutational studies are gaining prominence in the region [72, 73], extensive studies are needed to address the broad spectrum of genetic NMDs that are rapidly being identified and incorporated into globally recognized patient registries. A few reports have revealed the prevalence and profiles of NMDs in the region: in a 43-year study with 823 NMD patients from Cairo, DMD and autosomal MDs were reported as most frequent followed by lower rates of SMA and congenital myopathies. CMT disorders were less frequent, along with myotonic dystrophy identified in relatively few patients [74]. In Kuwait, muscular dystrophies are the most frequently diagnosed NMDs; this is followed by SMA and less frequently by myotonic syndromes, congenital and mitochondrial myopathies, CMT, and congenital myasthenic syndrome (unpublished data; personal communication Dr. Laila Bastaki). Another study analyzing 337 adult Iranian patients with NMDs revealed a similar constellation to our cohort of muscular dystrophies and mild proximal SMA [43]. Most recently, a 30-year overview of muscle biopsy analysis from 461 NMD patients in Saudi Arabia delineated multiple forms of MDs, as well as congenital and metabolic myopathies, with the study calling for a national NMD registry [6].
CONCLUSION
The efforts throughout the 20 years have successfully identified and helped NMD patients in Lebanon primarily because of the multidisciplinary approach that our team and collaborators have followed. In addition, our partner patient support organizations have been a tremendous help in supporting patients and their families, advocating for patients’ social rights and lobbying for more support and treatment coverage by the government. With significant efforts made towards streamlining and standardizing the collection of patient data over the last two decades [75], the retrospective study presented here could form the backbone of a future Lebanese neuromuscular registry. This is a contribution to a better understanding of the epidemiology of neuromuscular diseases in the region, which is necessary with many novel NMD therapies in the pipeline.
Limitations
The cohort described here consists predominantly of genetic NMD cases, mostly due to recruitment bias, as our team rarely receives referrals of patients with non-genetic disorders. With the exception of SMA, for which we have been the main diagnostic centre in Lebanon to the best of our knowledge, we were unable to determine an accurate prevalence of NMDs in general or that of particular disorders within the Lebanese population. This study also overlooks patients diagnosed by other genetic centres. Although we do not report on parental consanguinity, inherited NMDs in this study constituted a majority of autosomal recessive disorders; this may be attributed to high rates of consanguinity, estimated to be an average between 28.4% and 35.5% in Lebanon [76, 77]. The limited access to genetic testing, mainly due to the associated costs given the lack of coverage by insurance companies and the government, is the reason why some of the patients described here received just a clinical diagnosis without a molecular confirmation. Additionally, seven patients exhibited presentations compatible with primary NMDs, however a specific diagnosis could not be reached. With the advent of NGS and its gradual affordability, the number of such patients has slowly decreased during the period of this study. It is important to note that even with genetic sequencing, some NMDs such as mitochondrial disorders remain challenging to diagnose due to difficulties in variants’ interpretation or due to phenotypic complexity. The literature review above does not cover uncommonly described NMDs in Lebanon; more details can be found on the Catalogue of Transmission Genetics in Arabs database: https://www.cags.org.ae/en/ctga-overview.
ACKNOWLEDGEMENTS
We are thankful to Dr Laila Bastaki (Kuwait Medical Genetics Centre, Kuwait) for providing unpublished data. This work was partially funded by the Lebanese CNRS.
AUTHOR CONTRIBUTIONS
All authors have made substantial contributions to conception and design and for important intellectual content. SB, AD, EC, SEH, JAU, GL, AM have made substantial contributions in acquisition of data, analysis and interpretation of data. SB, AD, CM, EC, SEH, VD, AU, AM, have been involved in drafting the manuscript and revising it critically. All authors have given final approval of the version to be published.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
Annex 1: Neuromuscular Diseases panel genes
AAAS, AARS1, ABCC9, ABHD12, ABHD5, ACAD9, ACADL, ACADM, ACADS, ACADVL, ACTA1, ACTN2, ADCY6, ADGRG6, ADSS1, AGL, AGRN, AIFM1, ALDOA, ALG14, ALG2, ALG3, AMPD1, ANO5, ARHGEF10, ASAH1, ASCC1, ATL1, ATL3, ATP1A1, ATP1A2, ATP2A1, ATP7A, B3GALNT2, B4GAT1, BAG3, BICD2, BIN1, BSCL2, BVES, C12orf65, C1orf194, CACNA1E, CACNA1S, CAPN3, CASQ1, CAV3, CAVIN1, CCDC78, CCT5, CFL2, CHAT, CHCHD10, CHKB, CHRNA1, CHRNB1, CHRND, CHRNE, CHRNG, CHST14, CLCN1, CNTN1, CNTNAP1, COA7, COL12A1, COL13A1, COL6A1, COL6A2, COL6A3, COLQ, COX6A1, COX6A2, CPT2, CRYAB, CTDP1, DAG1, DCAF8, DCTN1, DCTN2, DES, DGAT2, DHTKD1, DMD, DNA2, DNAJB2, DNAJB5, DNAJB6, DNM2, DNMT1, DOK7, DPAGT1, DPM1, DPM2, DPM3, DRP2, DST, DYNC1H1, DYSF, ECEL1, EGR2, ELP1, EMD, EMILIN1, ENO3, ERBB3, ERGIC1, ETFA, ETFB, ETFDH, EXOSC3, EXOSC8, FBLN5, FBN2, FBXO38, FGD4, FHL1, FIG4, FKBP10, FKBP14, FKRP, FKTN, FLAD1, FLNB, FLNC, FXR1, G6PC, GAA, GAN, GARS1, GBE1, GDAP1, GFPT1, GJB1, GJB3, GLA, GLDN, GLE1, GMPPB, GNB4, GNE, GOLGA2, GSN, GYG1, GYS1, HACD1, HADH, HADHA, HADHB, HARS1, HEXA, HINT1, HK1, HNRNPDL, HOXD10, HSPB1, HSPB3, HSPB8, IGHMBP2, INF2, INPP5K, ISCU, ISPD, ITGA7, KARS1, KBTBD13, KCNE3, KCNJ2, KCNJ5, KIF1A, KIF1B, KIF5A, KLHL40, KLHL41, KLHL9, KY, LAMA2, LAMA5, LAMP2, LARGE1, LAS1L, LDB3, LDHA, LGI4, LIMS2, LITAF, LMNA, LMOD3, LPIN1, LRP12, LRP4, LRSAM1, MAP3K20, MARS1, MATR3, MB, MCM3AP, MED25, MEGF10, MET, MFN2, MICU1, MME, MORC2, MPV17, MPZ, MSTO1, MTM1, MTMR14, MTMR2, MUSK, MYBPC1, MYH14, MYH2, MYH3, MYH7, MYH8, MYL1, MYO9A, MYOD1, MYOF, MYOT, MYPN, NAGLU, NALCN, NDRG1, NEB, NEFH, NEFL, NEK9, NGF, NPL, NTRK1, NUP88, OPA1, ORAI1, PABPN1, PAX7, PDHA1, PDK3, PDXK, PFKM, PGAM2, PGK1, PGM1, PHKA1, PHKB, PHKG2, PIEZO2, PIP5K1C, PLEC, PLEKHG5, PLOD2, PMP2, PMP22, PNPLA2, POGLUT1, POLG, POLG2, POMGNT1, POMGNT2, POMK, POMT1, POMT2, POPDC3, PPP3CA, PRDM12, PREPL, PRKAG2, PRPS1, PRX, PUS1, PYGM, PYROXD1, RAB7A, RAPSN, RBCK1, RBM7, REEP1, RETREG1, RRM2B, RXYLT1, RYR1, RYR3, SBF1, SBF2, SCARF2, SCN10A, SCN11A, SCN4A, SCN9A, SCO2, SCYL2, SELENON, SEPTIN9, SETX, SGCA, SGCB, SGCD, SGCG, SGPL1, SH3TC2, SIGMAR1, SIL1, SLC12A6, SLC16A1, SLC18A3, SLC22A5, SLC25A1, SLC25A20, SLC25A21, SLC25A42, SLC25A46, SLC5A7, SLC9A3R1, SMCHD1, SMPD4, SNAP25, SOD1, SORD, SOX10, SPEG, SPG11, SPTAN1, SPTBN4, SPTLC1, SPTLC2, STAC3, STIM1, SUCLA2, SURF1, SYNE1, SYNE2, SYT2, TAZ, TCAP, TECPR2, TFG, TIA1, TIMM22, TK2, TMEM43, TMEM65, TNNI2, TNNT1, TNNT3, TNPO3, TOR1A, TOR1AIP1, TPM2, TPM3, TRAPPC11, TRIM2, TRIM32, TRIM54, TRIM63, TRIP4, TRPA1, TRPV4, TTN, TTR, TWNK, TYMP, UBA1, UBA5, UNC50, VAMP1, VAPB, VCP, VIPAS39, VMA21, VPS33B, VRK1, WARS1, WNK1, YARS1, YARS2, ZC4H2, ZFHX2.
REFERENCES
[1] | Laing NG . Genetics of neuromuscular disorders. Critical reviews in clinical laboratory sciences. (2012) ;49: (2):33–48. 10.3109/10408363.2012.658906 |
[2] | Benarroch L , Bonne G , Rivier F , Hamroun D . The version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscular disorders: NMD. (2020) ;30: (12):1008–48. 10.1016/j.nmd.2020.11.009 |
[3] | Efthymiou S , Manole A , Houlden H . Next-generation sequencing in neuromuscular diseases. Current opinion inneurology. (2016) ;29: (5):527–36. 10.1097/wco.0000000000000374 |
[4] | Salih MA , editor Muscular Dystrophies and Myopathies in Arab Populations 2010. |
[5] | Jumah MA , Muhaizea MA , Rumayyan AA , Saman AA , Shehri AA , Cupler E , et al. Current management of Duchenne muscular dystrophy in the Middle East: expert report. Neurodegenerative disease management. (2019) ;9: (3):123–33. 10.2217/nmt-2019-0002 |
[6] | Alassiri AH , Alyami AA , Alshabibi MI , Alhusain AM , Nasradeen MH , Barri AT , et al. The spectrum of muscle pathologies: Three decades of experience from a reference laboratory in Saudi Arabia. Annals of Diagnostic Pathology. (2020) ;47: :151532. 10.1016/j.anndiagpath.2020.151532 |
[7] | Leary R , Oyewole AO , Bushby K , Aartsma-Rus A . Translational Research in Europe for the Assessment and Treatment for Neuromuscular Disorders (TREAT-NMD). Neuropediatrics. (2017) ;48: (4):211–20. 10.1055/s-0037-1604110 |
[8] | Nair P , Sabbagh S , Mansour H , Fawaz A , Hmaimess G , Noun P , et al. Contribution of next generation sequencing in pediatric practice in Lebanon. A Study on 213 cases. Molecular Genetics & Genomic Medicine. (2018) ;6: (6):1041–52. 10.1002/mgg3.480 |
[9] | Verhaart IEC , Robertson A , Wilson IJ , Aartsma-Rus A , Cameron S , Jones CC , et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet journal of rare diseases. (2017) ;12: (1):124. 10.1186/s13023-017-0671-8 |
[10] | Prior TW , Nagan N . Spinal Muscular Atrophy: Overview of Molecular Diagnostic Approaches. Current protocols in human genetics. (2016) ;88: :9 27 1–9 13. 10.1002/0471142905.hg0927s88 |
[11] | Bagheri M , Abdirad M , Ghazavi A . Deletion and point mutation analysis of the SMN1 gene in patients with spinal muscular atrophy (SMA) in west Azerbaijan province of Iran. Studies in Medical Sciences. (2018) ;29: (7):474–80. |
[12] | Melki J , Abdelhak S , Sheth P , Bachelot MF , Burlet P , Marcadet A , et al. Gene for chronic proximal spinal muscular atrophies maps to chromosome 5q. Nature. (1990) ;344: (6268):767–8. 10.1038/344767a0 |
[13] | Lakkis B , El Chediak A , Hashash JG , Koubar SH . Severe ketoacidosis in a patient with spinal muscular atrophy. CENcase reports. (2018) ;7: (2):292–5. 10.1007/s13730-018-0345-y |
[14] | Jalkh N , Corbani S , Haidar Z , Hamdan N , Farah E , Abou Ghoch J , et al. The added value of WES reanalysis in the field of genetic diagnosis: lessons learned from 200 exomes in the Lebanese population. BMC Med Genomics. (2019) ;12: (1):11. 10.1186/s12920-019-0474-y |
[15] | Bernal S , Alías L , Barceló MJ , Also-Rallo E , Martínez-Hernández R , Gámez J , et al. The c. 859G > C variant in the SMN2 gene is associated with types II and III SMA and originates from a common ancestor. Journal of Medical Genetics. (2010) ;47: (9):640–2. 10.1136/jmg.2010.079004 |
[16] | Jumah MA , Majumdar R , Rehana Z , Al Rajeh S , Eyaid W . A pilot study of spinal muscular atrophy carrier screening in Saudi Arabia. Journal of Pediatric Neurology. (2007) ;05: (03):221–4. 10.1055/s-0035-1557384694 |
[17] | Shawky RM , El-Sayed NS . Clinico-epidemiologic characteristics of spinal muscular atrophy among Egyptians. Egyptian Journal of Medical Human Genetics. (2011) ;12: (1):25–30. https://doi.org/10.1016/j.ejmhg.2011.02.015. |
[18] | Lyahyai J , Sbiti A , Barkat A , Ratbi I , Sefiani A . Spinal muscular atrophy carrier frequency and estimated prevalence of the disease in Moroccan newborns. Genetic testing and molecular biomarkers. (2012) ;16: (3):215–8. 10.1089/gtmb.2011.0149 |
[19] | Hasanzad M , Azad M , Kahrizi K , Saffar BS , Nafisi S , Keyhanidoust Z , et al. Carrier frequency of SMA by quantitative analysis of the SMN1 deletion in the Iranian population. European journal of neurology. (2010) ;17: (1):160–2. 10.1111/j.1468-1331.2009.02693.x |
[20] | Basel-Vanagaite L , Taub E , Drasinover V , Magal N , Brudner A , Zlotogora J , et al. Genetic carrier screening for spinal muscular atrophy and spinal muscular atrophy with respiratory distress 1 in an isolated population in Israel. Genetic testing. (2007) ;12: (1):53–6. 10.1089/gte.2007.0030 |
[21] | Fried K , Mundel G . High incidence of spinal muscular atrophy type I (Werdnig - Hoffmann disease) in the Karaite community in Israel. Clinical genetics. (1977) ;12: (4):250–1. 10.1111/j.1399-0004.1977.tb00934.x |
[22] | al Rajeh S , Bademosi O , Ismail H , Awada A , Dawodu A , al-Freihi H , et al. A community survey of neurological disorders in Saudi Arabia: the Thugbah study. Neuroepidemiology. (1993) ;12: (3):164–78. 10.1159/000110316 |
[23] | Radhakrishnan K , Thacker AK , Maloo JC , Gerryo SE , Mousa ME . Descriptive epidemiology of some rare neurological diseases in Benghazi, Libya. Neuroepidemiology. (1988) ;7: (3):159–64. 10.1159/000110150 |
[24] | Gibbons M , Stratton A , Parsons J . Spinal Muscular Atrophy (SMA) in the Therapeutic Era. Current Genetic Medicine Reports. (2019) ;7: (3):162–7. 10.1007/s40142-019-00172-9 |
[25] | Al-Chalabi A , Hardiman O . The epidemiology of ALS: a conspiracy of genes, environment and time. Nature Reviews Neurology. (2013) ;9: (11):617–28. 10.1038/nrneurol.2013.203 |
[26] | Chiò A , Mazzini L , Mora G . Disease-modifying therapies in amyotrophic lateral sclerosis. Neuropharmacology. (2020) ;167: :107986. 10.1016/j.neuropharm.2020.107986 |
[27] | Malek E , Ismail H , Doumiati H , Salameh J . Characteristics of amyotrophic lateral sclerosis in Lebanon-a chart review. Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration. 2020:1-6. 10.1080/21678421.2020.1788095 |
[28] | Mégarbané A , Desguerres I , Rizkallah E , Delague V , Nabbout R , Barois A , et al. Brown-Vialetto-Van Laere syndrome in a large inbred Lebanese family: confirmation of autosomal recessive inheritance? American Journal of Medical Genetics. (2000) ;92: (2):117–21. 10.1002/(sici)1096-8628(20000515)92:2<117::aid-ajmg7 > 3.0.co;2-c |
[29] | Foley AR , Menezes MP , Pandraud A , Gonzalez MA , Al-Odaib A , Abrams AJ , et al. Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain: a Journal of Neurology. (2014) ;137: (Pt 1):44–56. 10.1093/brain/awt315 |
[30] | Srour M , Putorti ML , Schwartzentruber J , Bolduc V , Shevell MI , Poulin C , et al. Mutations in riboflavintransporter present with severe sensory loss and deafness in childhood. Muscle & Nerve. (2014) ;50: (5):775–9. 10.1002/mus.24224 |
[31] | Grohmann K , Wienker TF , Saar K , Rudnik-Schöneborn S , Stoltenburg-Didinger G , Rossi R , et al. Diaphragmatic spinal muscular atrophy with respiratory distress is heterogeneous, and one form Is linked to chromosome 11q13-q21. American Journal of Human Genetics. (1999) ;65: (5):1459–62. 10.1086/302636 |
[32] | Grohmann K , Schuelke M , Diers A , Hoffmann K , Lucke B , Adams C , et al. Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nature Genetics. (2001) ;29: (1):75–7. 10.1038/ng703 |
[33] | Schottmann G , Jungbluth H , Schara U , Knierim E , Morales Gonzalez S , Gill E , et al. Recessive truncating IGHMBP2 mutations presenting as axonal sensorimotor neuropathy. Neurology. (2015) ;84: (5):523–31. 10.1212/wnl.0000000000001220 |
[34] | Crisafulli S , Sultana J , Fontana A , Salvo F , Messina S , Trifirò G . Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta-analysis. Orphanet Journal of Rare Diseases. (2020) ;15: (1):141. 10.1186/s13023-020-01430-8 |
[35] | Wilson K , Faelan C , Patterson-Kane JC , Rudmann DG , Moore SA , Frank D , et al. Duchenne and Becker Muscular Dystrophies: A Review of Animal Models, Clinical End Points, and Biomarker Quantification. Toxicologic Pathology. (2017) ;45: (7):961–76. 10.1177/0192623317734823 |
[36] | Bladen CL , Salgado D , Monges S , Foncuberta ME , Kekou K , Kosma K , et al. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Human Mutation. (1002) ;36: (4):395–402. 10.1002/humu.22758 |
[37] | Taglia A , Petillo R , D’Ambrosio P , Picillo E , Torella A , Orsini C , et al. Clinical features of patients with dystrophinopathy sharing the 45-55 exon deletion of DMD gene. Acta Myologica: Myopathies and Cardiomyopathies: Official Journal of the Mediterranean Society of Myology. (2015) ;34: (1):9–13. |
[38] | El-Khoury R , Ahdab-Barmada M , Souaid M , Farra C . Molecular Characteristics of Duchenne Muscular Dystrophy in a Lebanese Cohort. Journal of Molecular and Genetic Medicine. 2018;12. |
[39] | Echigoya Y , Lim KRQ , Nakamura A , Yokota T . Multiple Exon Skipping in the Duchenne Muscular Dystrophy Hot Spots: Prospects and Challenges. Journal of Personalized Medicine. (2018) ;8: (4). 10.3390/jpm8040041 |
[40] | Salmaninejad A , Valilou SF , Bayat H , Ebadi N , Daraei A , Yousefi M , et al. Duchenne muscular dystrophy: an updated review of common available therapies. The International Journal of Neuroscience. (2018) ;128: (9):854–64. 10.1080/00207454.2018.1430694 |
[41] | Deenen JC , Arnts H , van der Maarel SM , Padberg GW , Verschuuren JJ , Bakker E , et al. Population-based incidenceand prevalence of facioscapulohumeral dystrophy. Neurology. (2014) ;83: (12):1056–9. 10.1212/wnl.0000000000000797 |
[42] | Nguyen K , Broucqsault N , Chaix C , Roche S , Robin JD , Vovan C , et al. Deciphering the complexity of the 4q and 10q subtelomeres by molecular combing in healthy individuals and patients with facioscapulohumeral dystrophy. Journal of Medical Genetics. (2019) ;56: (9):590–601. 10.1136/jmedgenet-2018-105949 |
[43] | Tehrani KHN , Hajiloo M , Asadollahi E , Lagini FP . Prevalence of muscular dystrophy in patients with muscular disorders in Tehran, Iran. Eur J Transl Myol [Internet]. 2018 2018/04//; 28: (2):[7380 p.]. Available from: http://europepmc.org/abstract/MED/29991987https://doi.org/10.4081/ejtm.2018.7385;https://europepmc.org/articles/PMC6036313; https://europepmc.org/articles/PMC6036313?pdf=render. |
[44] | Radhakrishnan K , el-Mangoush MA , Gerryo SE . Descriptive epidemiology of selected neuromuscular disorders in Benghazi, Libya. Acta neurologica Scandinavica. (1987) ;75: (2):95–100. 10.1111/j.1600-0404.1987.tb07901.x827 |
[45] | El-Tallawy HN , Khedr EM , Qayed MH , Helliwell TR , Kamel NF . Epidemiological study of muscular disorders in Assiut, Egypt. Neuroepidemiology. (2005) ;25: (4):205–11. 10.1159/000088674 |
[46] | Khedr EM , Fawi G , Abbas MA , Abo El-Fetoh N , Zaki AF , Gamea A , et al. Prevalence of neuromuscular disorders in Qena governorate/Egypt: population-based survey. Neurological Research. (2016) ;38: (12):1056–63. 10.1080/01616412.2016.1243640 |
[47] | Mah JK , Korngut L , Fiest KM , Dykeman J , Day LJ , Pringsheim T , et al. A Systematic Review and Meta-analysis on the Epidemiology of the Muscular Dystrophies. Canadian Journal of Neurological Sciences / Journal Canadien des Sciences Neurologiques. (2015) ;43: (1):163–77. 10.1017/cjn.2015.311 |
[48] | Richard I , Brenguier L , Dinçer P , Roudaut C , Bady B , Burgunder JM , et al. Multiple independent molecular etiology for limb-girdle muscular dystrophy type 2A patients from various geographical origins. American Journal of Human Genetics. (1997) ;60: (5):1128–38. |
[49] | El-Khoury R , Traboulsi S , Hamad T , Lamaa M , Sawaya R , Ahdab-Barmada M . Divergent Features of Mitochondrial Deficiencies in LGMD2A Associated With Novel Calpain-3 Mutations. Journal of Neuropathology & Experimental Neurology. (2018) ;78: (1):88–98. 10.1093/jnen/nly113 |
[50] | Siala O , Salem IH , Tlili A , Ammar I , Belguith H , Fakhfakh F . Novel sequence variations in LAMA2 andSGCG genes modulating cis-acting regulatory elements and RNA secondary structure. Genetics and Molecular Biology. (2010) ;33: (1):190–7. 10.1590/s1415-47572010005000008 |
[51] | Norwood FL , Harling C , Chinnery PF , Eagle M , Bushby K , Straub V . Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain: A Journal of Neurology. (2009) ;132: (Pt 11):3175–86. 10.1093/brain/awp236 |
[52] | Magri F , Nigro V , Angelini C , Mongini T , Mora M , Moroni I , et al. The italian limb girdle muscular dystrophy registry: Relative frequency, clinical features, and differential diagnosis. Muscle & Nerve. (1002) ;55: (1):55–68. 10.1002/mus.25192 |
[53] | Mojbafan M , Bahmani R , Bagheri SD , Sharifi Z , Zeinali S . Mutational spectrum of autosomal recessive limb-girdlemuscular dystrophies in a cohort of 112 Iranian patients and reporting of a possible founder effect. OrphanetJournal of Rare Diseases. (2020) ;15: (1):14. 10.1186/s13023-020-1296-x |
[54] | Topaloglu H . Epidemiology of muscular dystrophies in the Mediterranean area. Acta Myologica: Myopathies and Cardiomyopathies: Official Journal of the Mediterranean Society of Myology. (2013) ;32: (3):138–41. |
[55] | Bertini E , D’Amico A , Gualandi F , Petrini S . Congenital muscular dystrophies: a brief review. Seminars in Pediatric Neurology. (2011) ;18: (4):277–88. 10.1016/j.spen.2011.10.010 |
[56] | von der Hagen M , Becker L-L , Wienker TF , Smitka M , Musante L , Ropers H-H , et al. Just Expect It: Compound Heterozygous Variants of POMT1 in a Consanguineous Family—The Role of Next Generation Sequencing in Neuromuscular Disorders. Neuropediatrics. (2020) ;51: (01):072–5.10.1055/s-0039-1695787 |
[57] | van Reeuwijk J , Maugenre S , van den Elzen C , Verrips A , Bertini E , Muntoni F , et al. The expanding phenotype of POMT1 mutations: from Walker-Warburg syndrome to congenital muscular dystrophy, microcephaly, and mental retardation. Human Mutation. 10./humu. (1002) ;27: (5):453–9. 10.1002/humu.20313 |
[58] | von Renesse A , Petkova MV , Lützkendorf S , Heinemeyer J , Gill E , Hübner C , et al. POMK mutation in a family with congenital muscular dystrophy with merosin deficiency, hypomyelination, mild hearing deficit and intellectual disability. Journal of Medical Genetics. (1136) ;51: (4):275–82.10.1136/jmedgenet-2013-102236 |
[59] | de Andrade NXS , Adiyaman SC , Yuksel BD , Ferrari CT , Eldin AJ , Saydam BO , et al. Unusual presentations of lmna-associated lipodystrophy with complex phenotypes and generalized fat loss: when the genetic diagnosis uncovers novel features. AACE Clinical Case Reports. (2020) ;6: (2):e79–e85.10.4158/accr-2019-0366 |
[60] | Refaat MM , Hassanieh S , Ballout JA , Zakka P , Hotait M , Khalil A , et al. Non-familial cardiomyopathies in Lebanon: exome sequencing results for five idiopathic cases. BMC Medical Genomics. (2019) ;12: (1):33. 10.1186/s12920-019-0478-7 |
[61] | Barreto LCLS , Oliveira FS , Nunes PS , de França Costa IMP , Garcez CA , Goes GM , et al. Epidemiologic Study of Charcot-Marie-Tooth Disease: A Systematic Review. Neuroepidemiology. (1159) ;46: (3):157–65. 10.1159/000443706 |
[62] | Hoyle JC , Isfort MC , Roggenbuck J , Arnold WD . The genetics of Charcot-Marie-Tooth disease: current trends and future implications for diagnosis and management. The Application of Clinical Genetics. (2147) ;8: :235–43. 10.2147/tacg.s69969 |
[63] | Shy M , Rebelo AP , Feely SM , Abreu LA , Tao F , Swenson A , et al. Mutations in BAG3 cause adult-onset Charcot-Marie-Tooth disease. Journal of Neurology, Neurosurgery, and Psychiatry. (1136) ;89: (3):313–5. 10.1136/jnnp-2017-315929 |
[64] | Martin PB , Hicks AN , Holbrook SE , Cox GA . Overlapping spectrums: The clinicogenetic commonalities between Charcot-Marie-Tooth and other neurodegenerative diseases. Brain Research. (2020) ;1727: :146532. https://doi.org/10.1016/j.brainres.2019.146532. |
[65] | Romani M , Mehawej C , Mazza T , Mégarbané A , Valente EM . “Fork and bracket” syndrome expands the spectrum of SBF1-related sensory motor polyneuropathies. Neurology Genetics. (2016) ;2: (2):e61. 10.1212/nxg.0000000000000061. |
[66] | Jabbour R , Koussa S , Delague V , Megarbane A , Atweh S . Epidemiology of Charcot-Marie- Tooth in Lebanon: Clinical, genetic and electrophysiological correlation_Pilot Study. (P4.154). Neurology. (2015) ;84: (14 Supplement):P4.154. |
[67] | Guilbot A , Williams A , Ravisé N , Verny C , Brice A , Sherman DL , et al. A mutation in periaxin is responsible for CMT4F, an autosomal recessive form of Charcot-Marie-Tooth disease. Human Molecular Genetics. (2001) ;10: (4):415–21. 10.1093/hmg/10.4.415. |
[68] | Delague V , Jacquier A , Hamadouche T , Poitelon Y , Baudot C , Boccaccio I , et al. Mutations in FGD4 encoding the Rho GDP/GTP exchange factor FRABIN cause autosomal recessive Charcot-Marie-Tooth type 4H. American Journal of Human Genetics. (2007) ;81: (1):1–16. 10.1086/518428 |
[69] | Kennerson ML , Corbett AC , Ellis M , Perez-Siles G , Nicholson GA . A novel MCM3AP mutation in a Lebanese family with recessive Charcot-Marie-Tooth neuropathy. Brain: A Journal of Neurology. (2018) ;141: (9):e66. 10.1093/brain/awy184 |
[70] | Deenen JC , van Doorn PA , Faber CG , van der Kooi AJ , Kuks JB , Notermans NC , et al. The epidemiology of neuromuscular disorders: Age at onset and gender in the Netherlands. Neuromuscular disorders: NMD. (2016) ;26: (7):447–52. 10.1016/j.nmd.2016.04.011 |
[71] | Salih MA , Mahdi AH , al-Jarallah AA , al Jarallah AS , al-Saadi M , Hafeez MA , et al. Childhood neuromuscular disorders: a decade’s experience in Saudi Arabia. Annals of Tropical Paediatrics. (1996) ;16: (4):271–80. 10.1080/02724936.1996.11747838 |
[72] | Mohammed F , Elshafey A , Al-Balool H , Alaboud H , Al Ben Ali M , Baqer A , et al. Mutation spectrum analysis of Duchenne/Becker muscular dystrophy in 68 families in Kuwait: The era of personalized medicine. PloS One. (2018) ;13: (5):e0197205. 10.1371/journal.pone.0197205 |
[73] | Elhawary NA , Jiffri EH , Jambi S , Mufti AH , Dannoun A , Kordi H , et al. Molecular characterization of exonic rearrangements and frame shifts in the dystrophin gene in Duchenne muscular dystrophy patients in a Saudi community. Human Genomics. (2018) ;12: (1):18. 10.1186/s40246-018-0152-8 |
[74] | Shawky R , El-Sayed NS , Ibrahim D , Seifeldin N . Profile of genetic disorders prevalent in northeast region of Cairo, Egypt. Egyptian Journal of Medical Human Genetics. (2012) ;13: :45–62. |
[75] | Thompson R , Robertson A , Lochmüller H . Natural History, Trial Readiness and Gene Discovery: Advances in Patient Registries for Neuromuscular Disease. Advances in Experimental Medicine and Biology. (2017) ;1031: :97–124. 10.1007/978-3-319-67144-45 |
[76] | Barbour B , Salameh P . Consanguinity in Lebanon: prevalence, distribution and determinants. Journal of Biosocial Science. (1017) ;41: (4):505–17. 10.1017/s0021932009003290 |
[77] | El-Kheshen G , Saadat M . Prevalence of consanguineous marriages among shi’a populations of Lebanon. Journal of Biosocial Science. (2013) ;45: (5):675–82. 10.1017/s0021932012000843 |
[78] | De Sandre-Giovannoli A , Chaouch M , Boccaccio I , Bernard R , Delague V , Grid D , et al. Phenotypic and genetic exploration of severe demyelinating and secondary axonal neuropathies resulting from GDAP1 nonsense and splicing mutations. Journal of Medical Genetics. (2003) ;40: (7):e87. 10.1136/jmg.40.7.e87 |
[79] | Baudot C , Esteve C , Castro C , Poitelon Y , Mas C , Hamadouche T , et al. Two novel missense mutations in FGD4/FRABIN cause Charcot-Marie-Tooth type 4H (CMT4H). Journal of the Peripheral Nervous System: JPNS. (2012) ;17: (2):141–6. 10.1111/j.1529-8027.2012.00405.x |
[80] | Clarke NF , Maugenre S , Vandebrouck A , Urtizberea JA , Willer T , Peat RA , et al. Congenital muscular dystrophy type 1D (MDC1D) due to a large intragenic insertion/deletion, involving intron 10 of the LARGE gene. European Journal of Human Genetics: EJHG. (2010) ;19: (4):452–7. 10.1038/ejhg.2010.212 |
[81] | Straub V , Murphy A , Udd B . 229th ENMC international workshop: Limb girdle muscular dystrophies - Nomenclature and reformed classification Naarden, the Netherlands, 17–19 March 2017. Neuromuscular disorders: NMD. (2018) ;28: (8):702–10. 10.1016/j.nmd.2018.05.007 |
[82] | Liewluck T , Milone M . Untangling the complexity of limb-girdle muscular dystrophies. Muscle & Nerve. (1002) ;58: (2):167–77. 10.1002/mus.26077 |

