
Open-Label Evaluation of Eteplirsen in Patients with Duchenne Muscular Dystrophy Amenable to Exon 51 Skipping: PROMOVI Trial
Abstract
Background
Eteplirsen received accelerated FDA approval for treatment of Duchenne muscular dystrophy (DMD) with mutations amenable to exon 51 skipping, based on demonstrated dystrophin production.
Objective
To report results from PROMOVI, a phase 3, multicenter, open-label study evaluating efficacy and safety of eteplirsen in a larger cohort.
Methods
Ambulatory patients aged 7–16 years, with confirmed mutations amenable to exon 51 skipping, received eteplirsen 30 mg/kg/week intravenously for 96 weeks. An untreated cohort with DMD not amenable to exon 51 skipping was also enrolled.
Results
78/79 eteplirsen-treated patients completed 96 weeks of treatment. 15/30 untreated patients completed the study; this cohort was considered an inappropriate control group because of genotype-driven differences in clinical trajectory. At Week 96, eteplirsen-treated patients showed increased exon skipping (18.7-fold) and dystrophin protein (7-fold) versus baseline. Post-hoc comparisons with patients from eteplirsen phase 2 studies (4658-201/202) and mutation-matched external natural history controls confirmed previous results, suggesting clinically notable attenuation of decline on the 6-minute walk test over 96 weeks (PROMOVI: –68.9 m; phase 2 studies: –67.3 m; external controls: –133.8 m) and significant attenuation of percent predicted forced vital capacity annual decline (PROMOVI: –3.3%, phase 2 studies: –2.2%, external controls: –6.0%; p < 0.001). Adverse events were generally mild to moderate and unrelated to eteplirsen. Most frequent treatment-related adverse events were headache and vomiting; none led to treatment discontinuation.
Conclusions
This large, multicenter study contributes to the growing body of evidence for eteplirsen, confirming a positive treatment effect, favorable safety profile, and slowing of disease progression versus natural history.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a fatal, X-linked disease caused by mutations that disrupt production of a functional dystrophin protein [1, 2]. With an estimated incidence of 1 in 3500–5000 newborn boys worldwide, DMD leads to loss of muscle function and premature death. Its debilitating effects diminish quality of life for patients, their families, and caregivers. In the absence of dystrophin, muscle cells degenerate, leading to progressive muscle weakness [3], loss of ambulation by age 8–14 years, and ultimately life-threatening complications including cardiomyopathy and respiratory insufficiency [4, 5]. Exon skipping to restore the dystrophin open reading frame has been shown to be an effective treatment approach for patients with DMD [6–9]. Exon skipping enables translation of an internally truncated, yet functional dystrophin protein, thereby reducing the effects of the mutations underlying pathogenesis of the disease [10].
The majority of dystrophin pathogenic variants cluster between exons 45–55. Many of these deletions are amenable to exon skipping, with the most common being amenable to exon 51 skipping, representing approximately 13% of all DMD patients [10]. Clear differences in clinical trajectories have been linked to genotype differences in DMD [11–13]. For example, patients with mutations amenable to exon 51 or 53 skipping have steeper declines in walking ability, with loss of ambulation at earlier ages, and have worse pulmonary outcomes, compared with patients amenable to exon 44 skipping [11, 12, 14]. These observations have made it clear that studies of exon-skipping therapies must include genotype-matched controls as comparators, and that genotype-mismatched groups are inappropriate controls.
Eteplirsen is a phosphorodiamidate morpholino oligomer (PMO) designed to skip dystrophin exon 51 [1, 15, 16]. In phase 2 studies (studies 4658-201/202; NCT01396239), eteplirsen demonstrated a significant increase in dystrophin protein expression in patient muscle biopsy samples over 48 weeks [16]. Furthermore, studies 201/202 provided evidence that eteplirsen may slow muscle deterioration, prolong ambulation, and preserve pulmonary function [15–19]. Eteplirsen was granted accelerated approval by the United States Food and Drug Administration (FDA) in 2016 [7, 8] based on increased dystrophin protein expression in study 201 (n = 12). Further evaluation of efficacy was needed to determine whether these results could be replicated in a larger patient population across multiple study sites, and to determine whether exon skipping and dystrophin production increased over time. PROMOVI was a large, multicenter, phase 3 trial evaluating the efficacy and safety of eteplirsen over 96 weeks in ambulatory patients with DMD amenable to exon 51 skipping.
MATERIALS AND METHODS
Study design
PROMOVI (ClinicalTrials.org identifier: NCT02255552) was a 96-week, multicenter, open-label, non-randomized trial evaluating the efficacy and safety of eteplirsen in patients with DMD and genetic deletions amenable to exon 51 skipping, with an intended control group of patients with DMD and genetic deletions not amenable to exon 51 skipping. The trial was conducted at 40 sites in the United States between November 2017 and June 2019.
Independent ethics committees or institutional review boards at each site approved the protocol. The trial was conducted in accordance with the ethical principles of Good Clinical Practice, according to the International Council for Harmonisation Tripartite Guideline [20]. Written informed consent was obtained from each patient’s parent(s) or legal guardian(s), and informed assent was obtained from patients where applicable.
Patients
Eligible patients were male, aged 7–16 years, with DMD and genotypically confirmed mutations amenable to exon 51 skipping (eteplirsen-treated group) or with genotypically confirmed mutations not amenable to exon 51 skipping (untreated control group). Patients were required to have stable pulmonary function (percent predicted forced vital capacity [FVC%p]≥50%, not requiring nocturnal ventilation, and unlikely to decompensate over the duration of the study), be on a stable dose of oral corticosteroids for at least 24 weeks before study entry and be able to walk≥300 m on the 6-minute walk test (6MWT). Patients may or may not have been genotyped prior to the screening period. For those who were not previously genotyped, screening procedures (other than informed consent) and treatment group assignment were not conducted until after genotype results were received.
Exclusion criteria included use of any pharmacologic treatment, other than corticosteroids, that may have had an effect on muscle strength or function within 12 weeks of study entry; previous treatment with ezutromid at any time and treatment with drisapersen within the last 3 months; previous treatment with any other RNA antisense agent or any gene therapy within the last 6 months, or participation in any other DMD interventional clinical study within 12 weeks of study entry. Also excluded were patients with left ventricular ejection fraction of < 50%, based on the screening echocardiogram (ECHO) or QT interval corrected by Fridericia’s formula (QTcF) ≥450 ms based on the screening electrocardiogram (ECG).
Treatment
Patients in the treated group received a weekly intravenous (IV) infusion of 30 mg/kg eteplirsen, administered over approximately 35–60 minutes, from baseline (Week 1) until the end of the study (Week 96).
Assessments
The effect on ambulation, endurance, and muscle function was assessed using the 6MWT; the 6MWT was performed by standardized procedures, in which patients were asked to walk a set course of 25 m for 6 minutes and the distance walked was recorded, as previously described [21]. The North Star Ambulatory Assessment (NSAA) scale, performed on 2 consecutive days, was used to assess patient performance on 17 different functional activities [22]. Patients were graded as follows: 2 = normal, no obvious modification of activity; 1 = modified method but achieves goal independent of physical assistance from another; and 0 = unable to achieve goal independently. Ability to rise independently from the floor (without external support) was defined as an NSAA subscore of 2 or 1. Loss of ambulation (LOA) was defined as: i) an NSAA walk subscore of 0; ii) or NSAA was not done due to reason related to nonambulation; or iii) if 6MWT was not done with any reason related to permanent nonambulation. It was also required that no later data showed the patient was still ambulatory. Respiratory muscle function was evaluated as FVC%p using standard spirometry procedures. FVC%p was calculated using the Hankinson formula [23], and calculations were based on the use of both standing height (measured with shoes off) and ulnar-calculated height (the ulna was measured with an anthropometer or calipers and height was calculated as: height [cm] = 4.605×ulnar length [cm] + 1.308×patient age [years] + 28.003). Patients in the eteplirsen-treated group underwent muscle biopsy at baseline and were randomized to a muscle biopsy at either Week 24, Week 48, Week 72, or Week 96 in a 1:2:1:1 ratio. Muscle biopsies were analyzed for quantity of dystrophin protein expression using Western blot, intensity of dystrophin expression and percentage of dystrophin-positive fibers using immunofluorescence histochemistry (IHC), and exon 51 skipping using quantitative digital droplet polymerase chain reaction (ddPCR) assay. All expression analyses were performed as previously described [8].
Study endpoints
The primary efficacy endpoint was the change from baseline to Week 96 in 6MWT distance. Secondary efficacy endpoints included change from baseline to Week 96 for the ability to rise independently from the floor, LOA, NSAA total score, and FVC%p. In the original study protocol, comparisons were to be made with the untreated group for the primary and secondary endpoints; however, due to inadequate recruitment and retention of untreated patients with mutations not amenable to exon 51 skipping a revised statistical analysis plan was finalized prior to database lock, which specified that only descriptive summaries would be presented for each group. During the time of trial design and initiation, natural history data began to emerge demonstrating disparate disease trajectories for patients with different mutations [11–14]. As these data were confirmed, it became clear that the untreated control group, which consisted entirely of patients with mutations not amenable to exon 51 skipping, did not provide an adequate comparator group, and nonmutation-matched comparisons may be inappropriate.
Biological endpoints included the change from baseline to Week 96 in exon 51 skipping by quantitative digital droplet polymerase chain reaction (ddPCR) and dystrophin protein expression by Western blot. Safety assessments included all adverse events (AEs), vital signs, weight, height and ulnar length, physical examination, ECG, ECHO, and routine clinical laboratory evaluations.
Post-hoc analyses with matched comparators
Post-hoc analyses were performed with genotype- and baseline characteristic-matched comparisons of a subset of PROMOVI data with external natural history control cohorts and data from previous eteplirsen phase 2 studies, 4658-201 [16]/202 [15]. For the 6MWT analysis, a subgroup of PROMOVI patients that matched predefined criteria was compared with patients from studies 201/202 [24]. An external natural history cohort (exon 51 skipping amenable patients from the Italian DMD Telethon Registry [12, 25, 26], and the Leuven NMRC Registry [27]) is also shown to give context to the study results. These registries are based in expert neuromuscular centers that use a standard of care consistent with that used in the United States, where PROMOVI was conducted. The matched subgroup of PROMOVI patients was defined as those in the trial who had baseline 6MWT distance of ≥300–≤450 m, a baseline NSAA total score of ≥17–≤31 and were aged 7–13 years. For the FVC%p analysis, a matched subgroup of PROMOVI patients was compared with data from matched untreated patients (aged 10–18 years) in the Cooperative International Neuromuscular Research Group (CINRG) Duchenne Natural History Study (DNHS) exon 51 cohort [11]; data from studies 201/202 were also used for comparisons with previous reports of eteplirsen treatment effect [28].
Statistical analyses
The efficacy set consisted of all patients in the eteplirsen-treated and untreated control groups who had at least 1 post-baseline functional assessment. The primary efficacy set consisted of all patients in the efficacy set who had a baseline 6MWT distance of≥300–≤450 m. The safety set consisted of all patients who were enrolled in the study and either received at least 1 dose of eteplirsen in the eteplirsen-treated group or had at least 1 post-baseline safety assessment in the untreated group. For patients in the untreated group, any safety assessment on or after the Week 1 visit was considered a post-baseline safety assessment. Changes in 6MWT, ability to rise, LOA, NSAA total score, and FVC%p from baseline were summarized by treatment group using descriptive statistics. Changes in dystrophin protein expression and exon 51 skipping were analyzed using a 1-sample permutation t-test; for all hypothesis testing, significance level was 0.05. Correlation between exon 51 skipping and dystrophin protein expression was analyzed using Pearson’s coefficient and Spearman’s r correlation coefficient. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) Version 17.1 or higher, and treatment-emergent AEs (TEAEs) were summarized using descriptive statistics.
For the post-hoc analyses: FVC%p was compared between PROMOVI and the CINRG DNHS exon 51 cohort using mixed model repeated measures (MMRM) model with age, treatment, and age by treatment interaction as fixed effects and patient as a random effect; the slope of each treatment group is estimated from least square means in the MMRM.
RESULTS
Untreated arm (not amenable to exon 51 skipping)
Study enrollment occurred over a 2.5-year period starting October 1, 2014. A total of 30 patients entered the untreated arm; 15 (50%) completed the study (Fig. 1 and Table 1). As their mutations were not amenable to exon 51 skipping, they could not cross over into the treatment group. This factor contributed to poor retention, which precluded statistically and clinically meaningful comparisons. Because the untreated control group was not mutation-matched, it was also an inappropriate comparator group and was thus not analyzed for efficacy comparisons. Clinical outcomes in the untreated group are shown in Table 2. TEAEs were reported in 25 (83.3%) of these untreated patients and were serious in 2 (6.7%) patients. Of 78 TEAEs, 58 were mild and 20 were moderate.
Fig. 1
Participant flow diagram and analysis subsets. Abbreviations: CINRG = Cooperative International Neuromuscular Research Group; DNHS = Duchenne Natural History Study (DNHS); FVC%p = percent predicted forced vital capacity; LOA = loss of ambulation; 6MWT = 6-minute walk test; NSAA = North Star Ambulatory Assessment. aEteplirsen studies 201 (NCT01396239) [24] and 202 (NCT01540409) [28]. bUntreated patients in the CINRG DNHS exon 51 cohort (age 10–18 years) [11]. cItalian DMD Telethon Registry [12, 25, 26] and the Leuven NMRC Registry [27].
![Participant flow diagram and analysis subsets. Abbreviations: CINRG = Cooperative International Neuromuscular Research Group; DNHS = Duchenne Natural History Study (DNHS); FVC%p = percent predicted forced vital capacity; LOA = loss of ambulation; 6MWT = 6-minute walk test; NSAA = North Star Ambulatory Assessment. aEteplirsen studies 201 (NCT01396239) [24] and 202 (NCT01540409) [28]. bUntreated patients in the CINRG DNHS exon 51 cohort (age 10–18 years) [11]. cItalian DMD Telethon Registry [12, 25, 26] and the Leuven NMRC Registry [27].](https://content.iospress.com:443/media/jnd/2021/8-6/jnd-8-6-jnd210643/jnd-8-jnd210643-g001.jpg)
Table 1
Untreated patients: Baseline characteristics and demographics
| Characteristic | Untreated groupa (N = 30) |
| Age, y | |
| Mean±SD | 8.8±1.8 |
| Min, max | 7.0, 13.0 |
| Weight, kg | |
| Mean (SD) | 30.6±9.9 |
| Min, max | 17.9, 53.2 |
| Standing height, cm | |
| Mean (SD) | 122.8±9.0 |
| Min, max | 108.0, 142.5 |
| Time since DMD diagnosis at baseline, mo | |
| Mean (SD) | 54.7±31.6 |
| Min, max | 12.7, 114.9 |
| Corticosteroid treatment, n (%) | |
| Deflazacort | 21 (70.0) |
| Prednisone | 9 (30.0) |
| Corticosteroid schedule, n (%) | |
| Continuous | 24 (80.0) |
| Intermittent | 2 (20.0) |
Abbreviations: DMD = Duchenne Muscular Dystrophy; SD =standard deviation. aIncluded patients with the following mutations: exon 12–44 (n = 1); exon 42–45 (n = 1); exon 43 (n = 1); exon 44 (n = 1); exon 45 (n = 2); exon 45–52 (n = 4); exon 46–47 (n = 3); exon 46–48 (n = 5); exon 46–50 (n = 1); exon 48–52 (n = 2); exon 49–52 (n = 2); exon 51 (n = 2); exon 51–53 (n = 2); exon 51–55 (n = 3).
Table 2
Untreated patients: Clinical outcomes
| Untreated group | ||
| Endpoints | Baseline (n = 20) | Week 96 (n = 9) |
| 6MWT distance, m | ||
| Mean±SD | 382.6±45.7 | 252.2±133.1 |
| Range | 301.5, 448.0 | 0.0, 453.5 |
| Ability to rise, n (%) | 18 (90.0) | 3 (33.3) |
| Patients with LOA, n (%) | - | 1 (5.0)a |
| NSAA total score | ||
| Mean±SD | 22.3±7.3 | 12.0±8.6 |
| Range | 12.0, 32.0 | 1.0, 27.5 |
| FVC%p, % | ||
| Mean±SD | 96.9±17.7 | 91.9±14.2 |
| Range | 67.5, 125.8 | 70.5, 113.8 |
Abbreviations: 6MWT = 6-minute walk test; FVC%p = percent predicted forced vital capacity; LOA = loss of ambulation; NSAA = North Star Ambulatory Assessment; SD = standard deviation. an = 20.
Eteplirsen-treated patients
A total of 79 patients were enrolled into the eteplirsen-treated arm, 78 (98.7%) of whom completed 96 weeks of treatment. Mean age at baseline was 9.1 years, and mean time since DMD diagnosis was 53.3 months (Table 3). Patients were ambulatory at baseline.
Table 3
Eteplirsen-treated patients: Baseline characteristics and demographics
| Characteristic | Eteplirsen-treated group (N = 79) |
| Age, y | |
| Mean±SD | 9.1±2.0 |
| Min, max | 7.0, 16.0 |
| Weight, kg | |
| Mean (SD) | 34.2±11.3 |
| Min, max | 18.4, 68.9 |
| Standing height, cm | |
| Mean (SD) | 125.5±9.0 |
| Min, max | 106.0, 148.5 |
| Time since DMD diagnosis at baseline, mo | |
| Mean (SD) | 53.3±33.3 |
| Min, max | 5.5, 147.1 |
| Corticosteroid treatment, n (%) | |
| Deflazacort | 22 (27.8) |
| Prednisone | 57 (72.2) |
| Corticosteroid schedule, n (%) | |
| Continuous | 65 (82.3) |
| Intermittent | 14 (17.7) |
Abbreviations: DMD = Duchenne Muscular Dystrophy; SD =standard deviation.
Biological assessments
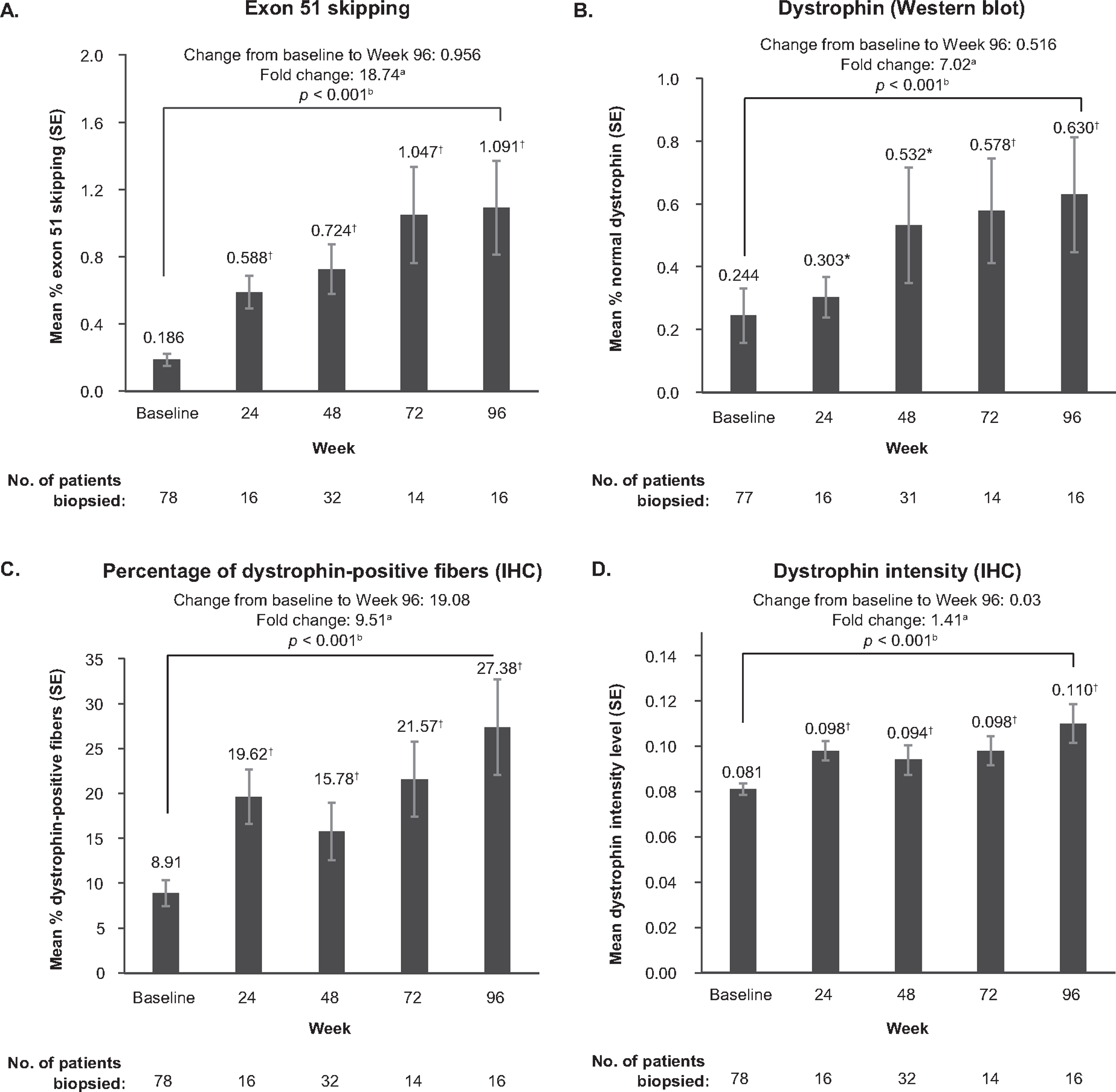
With eteplirsen treatment, exon 51 skipping in muscle fibers significantly increased over baseline at the earliest time point assessed (Week 24), continued to increase until Week 72, and was 18.7-fold higher at Week 96 versus baseline (p < 0.001; Fig. 2A). Similarly, dystrophin protein expression increased over time, increasing 7.0-fold over baseline by Week 96 (p < 0.001; Fig. 2B). Positive correlation was observed between exon 51 skipping and protein expression (Pearson coefficient = 0.710 [p < 0.001]; Spearman coefficient = 0.692 [p < 0.001]). IHC analyses indicated that weekly treatment with eteplirsen resulted in a significant mean increase in percentage of dystrophin-positive fibers at Week 96 (p < 0.001) and dystrophin intensity levels (p < 0.001) (Fig. 2C & 2D, respectively).
Fig. 2
Exon 51 skipping by quantitative ddPCR (A), dystrophin protein quantification by western blot (B), percentage of dystrophin-positive fibers by IHC (C) and dystrophin intensity by IHC (D) from Baseline to Week 96 in eteplirsen-treated patients. Abbreviations: SE = standard error. *p < 0.05. †p < 0.001. aMean of individual patient fold changes. bp value is based on one-sample permutation t-test.
Efficacy assessments with eteplirsen treatment
Primary and secondary efficacy outcomes for eteplirsen-treated patients are shown in Table 4. Mean 6MWT distance decreased from 374.6 m at baseline to 256.2 m at Week 96. At Week 96, 54.1%of eteplirsen-treated patients were able to rise from the floor independently compared with 86.6% at baseline. The Kaplan-Meier estimate for remaining ambulatory at Week 96 was 81.9%; LOA occurred in 17.9% of eteplirsen-treated patients. The NSAA total score for eteplirsen-treated patients was 14.9 at Week 96 compared with 21.4 at baseline. Mean FVC%p decreased from 90.4% at baseline to 87.3% at Week 96.
Table 4
Eteplirsen-treated patients: Primary and secondary efficacy measures at Baseline and Week 96
| Eteplirsen-treated group | ||
| Endpoints | Baseline (n = 67) | Week 96 (n = 66) |
| 6MWT distance, m | ||
| Mean±SD | 374.6±44.1 | 256.2±148.7a |
| Range | 303.0, 449.5 | 0.0, 496.0 |
| Ability to rise independently, n (%) | 58 (86.6) | 33 (54.1)b |
| Patients with LOA, n (%) | - | 12 (17.9)c |
| NSAA total score | ||
| Mean±SD | 21.4±6.9 | 14.9±8.8b |
| Range | 4.5, 34.0 | 0.0, 34.0 |
| FVC%p, % | ||
| Mean±SD | 90.4±16.0 | 87.3±16.3 |
| Range | 50.0, 126.0 | 56.0, 128.4 |
Abbreviations: FVC%p = percent predicted forced vital capacity; LOA = loss of ambulation; 6MWT = 6-minute walk test; NSAA = North Star Ambulatory Assessment; SD = standard deviation. an = 65. bn = 61. cn = 67.
Post-hoc analyses
Baseline characteristics of the PROMOVI and studies 201/202 subgroup populations for the 6MWT-matched comparator analysis are shown in Table S1. At Week 96, the 6MWT comparator analysis showed that PROMOVI results were consistent with those of studies 201/202. Mean change from baseline in 6MWT in eteplirsen-treated patients was –68.9 m in PROMOVI compared with –67.3 m in patients from studies 201/202; mean change in external natural history controls (n = 11; Italian DMD Telethon Registry, Leuven NMRC Registry), who were matched to the study 201/202 population, was –133.8 m (Fig. 3A).
Fig. 3
Post-hoc analysis: Mean change from baseline to Week 96 in 6MWT (A), and FVC%p (B) in eteplirsen-treated patients and matched comparisons. Abbreviations: FVC%p = percent predicted forced vital capacity; 6MWT = 6-minute walk test; SE = standard error. aAt Weeks 12, 72, and 96 (n = 41). One patient did not have a value at Week 12, but had at later visits. Another patient withdrew after Week 48. bEteplirsen studies 201 (NCT01396239) [24] and 202 (NCT01540409) [28]. cItalian DMD Telethon Registry [12, 25, 26] and the Leuven NMRC Registry [27]. dUntreated patients in the CINRG DNHS exon 51 cohort (age 10–18 years) [11].
![Post-hoc analysis: Mean change from baseline to Week 96 in 6MWT (A), and FVC%p (B) in eteplirsen-treated patients and matched comparisons. Abbreviations: FVC%p = percent predicted forced vital capacity; 6MWT = 6-minute walk test; SE = standard error. aAt Weeks 12, 72, and 96 (n = 41). One patient did not have a value at Week 12, but had at later visits. Another patient withdrew after Week 48. bEteplirsen studies 201 (NCT01396239) [24] and 202 (NCT01540409) [28]. cItalian DMD Telethon Registry [12, 25, 26] and the Leuven NMRC Registry [27]. dUntreated patients in the CINRG DNHS exon 51 cohort (age 10–18 years) [11].](https://content.iospress.com:443/media/jnd/2021/8-6/jnd-8-6-jnd210643/jnd-8-jnd210643-g003.jpg)
Baseline characteristics of the PROMOVI, studies 201/202, and CINRG DNHS subgroup populations for the matched FVC%p comparator analysis are shown in Table S2. Eteplirsen-treated patients experienced a significant, clinically meaningful attenuation in pulmonary function decline (p < 0.001) compared with the untreated CINRG DNHS exon 51 cohort (Fig. 3B). In PROMOVI, the annual rate of decline in FVC%p was 3.3% based on ulnar length calculated height and 3.1% based on standing height, approximately half of the natural history decline in the matched CINRG DNHS group (–6.0%).
The proportion of PROMOVI patients with LOA at Week 96 (17.9%) was comparable to results from the phase 2 studies 201/202 at that timepoint (year 2, 17%) [24].
Safety assessment
The majority of the TEAEs reported were mild or moderate in severity, and there were no treatment-related discontinuations due to TEAEs (Table 5). Infusion-related reactions occurred in 45 of 79 (57.0%) patients; of these, headache (15/79; 19.0%) and vomiting (13/79; 16.5%) were the most common infusion-related reactions. Most infusion-related reactions were mild and all resolved. One treatment-related serious AE of urticaria was observed approximately 15–20 minutes after infusion and resolved approximately 1 hour after an IV steroid and antihistamine were administered; although the patient continued on eteplirsen without subsequent events and without corticosteroid pretreatment, the event was considered related to eteplirsen by the investigator and may reflect drug hypersensitivity. Overall, 8 eteplirsen-treated patients (10.1%) experienced renal TEAEs; each as proteinuria, which resolved by end of study in all but one individual. Proteinuria did not result in eteplirsen interruption or require treatment for any patient. Eteplirsen did not appear to adversely affect clinical laboratory values, vital signs, ECGs, or ECHOs. Percentages of patients with low, normal, or high serum chemistry, hematology, and urinalysis values were generally comparable at baseline and the last post-baseline measurement. No clinically significant mean changes in ECHOs were observed, and medical review found no clinically important changes in ECG parameters. One infected venous port serious AE was reported as severe and unrelated to treatment, and the patient recovered after removal of the port and antibiotic treatment.
Table 5
Treatment-emergent adverse events
| Eteplirsen-treated group (N = 79) | |
| Treatment-emergent AEs, n (%) | 78 (98.7) |
| Any treatment-emergent AE related to study drug and reported in≥2 patients, n (%) | 28 (35.4) |
| Vomiting | 7 (8.9) |
| Headache | 5 (6.3) |
| Diarrhea | 4 (5.1) |
| Nausea | 4 (5.1) |
| Dizziness | 3 (3.8) |
| Proteinuria | 3 (3.8) |
| Flushing | 3 (3.8) |
| Rash | 2 (2.5) |
| Urticaria | 2 (2.5) |
| Catheter site pain | 2 (2.5) |
| Treatment-emergent serious AEs, n (%) | 11 (13.9) |
| Treatment-emergent AE leading to study discontinuation, n (%) | 0 |
| Severity of treatment-emergent AEs, n | |
| Mild | 1610 |
| Moderate | 170 |
| Severe | 19 |
Abbreviations: AE = adverse event.
DISCUSSION
PROMOVI is the largest trial to date using a PMO for treating DMD. The findings from this study contribute to the growing body of evidence for eteplirsen and confirm the treatment effect and the safety profile observed in studies 201/202. Together, the available data support that eteplirsen slows disease progression in patients with DMD.
The randomized, double-blind, placebo-controlled phase 2 study (study 201) demonstrated that eteplirsen increased dystrophin protein expression in muscle biopsies after 24 and 48 weeks of treatment, and improved ambulation outcomes compared with placebo [16]. Animal [29] and clinical studies [30–32] have shown that increasing functional dystrophin protein levels is key to improving functional outcomes in DMD, with even low expression levels of dystrophin being associated with milder dystrophinopathy. In one study, a milder clinical phenotype and a delay to LOA was observed with dystrophin quantities as low as < 0.5% of normal [31]. Hence, dystrophin increase is likely to predict clinical benefit. In PROMOVI, exon skipping increased post-eteplirsen treatment, demonstrating target engagement, and a positive correlation was demonstrated between exon 51 skipping and protein expression. Dystrophin protein accumulated over time, with dystrophin levels significantly increased over baseline at each time point.
Functional benefits of increased dystrophin in PROMOVI were evident in post-hoc analyses. Matched comparisons of PROMOVI patients with patients from previous eteplirsen studies (201/202) and natural history data (Italian DMD Telethon Registry, Leuven NMRC Registry, CINRG DNHS) suggested that eteplirsen treatment slows disease progression, as demonstrated by 6MWT distance and annual change in FVC%p. The magnitude of the mean decrease in 6MWT distance after 96 weeks’ treatment with eteplirsen in PROMOVI was consistent with that seen in eteplirsen-treated patients in the studies 201/202 (69 m vs. 67 m, respectively). A matched comparison of studies 201/202 to an untreated natural history cohort amenable to exon 51 skipping (Italian/Leuven Registries) found that eteplirsen-treated patients had a smaller decline in 6MWT distance than controls (67 m vs. 134 m), indicating a clinical benefit of eteplirsen treatment. Previous work comparing long-term (4-year) follow-up data from the studies 201/202 and the matched Italian/Leuven controls showed an increasing separation of the 6MWT distance curves after the 2-year timepoint [24]. The 96-week duration of the PROMOVI trial may therefore not have been sufficiently long to show notable separation in 6MWT. The LOA findings of the PROMOVI trial were also comparable to results from the studies 201/202 [24]. Significant, clinically meaningful attenuation in pulmonary function decline was seen in eteplirsen-treated PROMOVI patients compared with the untreated CINRG DNHS exon 51 cohort (annual rate of decline in FVC%p, 3.3% vs. 6.0%, respectively).
Long-term eteplirsen treatment had a favorable safety profile. There were no discontinuations due to safety, and AEs were generally mild or moderate. AEs observed among patients who received eteplirsen were generally consistent with AEs observed in a younger population with DMD and in patients with DMD receiving chronic corticosteroid treatment. Cases of proteinuria were generally transient and self-resolved. The incidence of proteinuria during PROMOVI (10%) was consistent with the incidence of mild proteinuria found in pediatric patients with DMD [33].
A limitation of the study was the open-label design with lack of a placebo-control group amenable to exon 51 skipping. This study design was employed due to ethical concerns associated with 96 weeks of placebo IV therapy in a population with predicted disease progression. PROMOVI was initiated with a flawed comparison of eteplirsen-treated patients to a mismatched control arm (patients with mutations not amenable to exon 51 skipping). The inadequacy of the untreated control group with mutations not amenable to exon 51 skipping became clear only after the PROMOVI study was initiated. Emerging research by several groups using natural history data demonstrated that DMD patients with different mutations have different disease trajectories [11–14], and therefore comparisons between non-mutation-matched groups are inappropriate. The lack of a prospective, mutation-matched untreated control arm was a major limitation of the study. Another factor that may limit the efficacy findings is that the study population consisted of more disease-progressed patients at an age of declining ambulation; potential treatment effects in patients earlier in the disease course were not addressed.
Data from PROMOVI confirmed the positive treatment effect and favorable safety profile of eteplirsen in a large population of patients with DMD mutations amenable to exon 51 skipping treated at multiple centers.
PROMOVI trial principal clinical investigators
Hoda Z. Abdel-Hamid (UPMC Children’s Hospital of Pittsburgh, Pittsburgh, PA, USA); Gyula Acsadi (Connecticut Children’s Medical Center, Hartford, CT, USA); Susan Apkon (Seattle Children’s Hospital, Seattle, WA, USA); Ibrahim Binalsheikh (Carolinas Medical Center, Charlotte, NC, USA); William B. Burnette (Vanderbilt University Medical Center, Nashville, TN, USA); Russell Butterfield (University of Utah, Salt Lake City, UT, USA); Barry Byrne (University of Florida Health, Shands Hospital, Gainesville, FL, USA); Emma Ciafaloni (University of Rochester Clinical Research Center, Rochester, NY, USA); Anne M. Connolly (Washington University in St Louis, St Louis, MO, US); Basil Darras (Harvard Medical School, Boston Children’s Hospital, Boston, MA, USA); John Day (Stanford University School of Medicine, Stanford, CA, USA); Darryl C. De Vivo (Columbia University Irving Medical Center, New York, NY, USA); Erika Finanger (Shriners Hospitals for Children, Portland, OR, USA); Richard Finkel (Nemours Children’s Hospital, Orlando, FL, USA); Carla Grosmann (Rady Children’s Hospital, University of California at San Diego, San Diego, CA, USA); Susan Iannaccone (Children’s Medical Center of Dallas, Dallas, TX, USA); Huiyuan Jiang (Wayne State University, Detroit, MI, USA); Ashutosh Kumar (Pennsylvania State University, Hershey, PA, USA); Nancy Kuntz (Ann and Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL, USA); Tim Lotze (Texas Children’s Hospital, Houston, TX, USA); Katherine Mathews (University of Iowa Children’s Hospital, Iowa City, IA, USA); Craig M. McDonald (University of California Davis Health System and School of Medicine, Sacramento, CA, USA); Jerry R. Mendell (Nationwide Children’s Hospital, Columbus, OH, USA); Julie Parsons (Children’s Hospital Colorado, Aurora, CO, USA); Han C. Phan (Rare Disease Research, Atlanta, GA, USA); Gerald Raymond (University of Minnesota, Minneapolis, MN, USA); Ben Renfroe (Northwest Florida Clinical Research Group, Gulf Breeze, FL, USA); Perry B. Shieh (David Geffen School of Medicine at UCLA, Los Angeles, CA, USA); Kumarswamy Sivakumar (Neuromuscular Research Center, Phoenix, AZ, USA); Cuixia Tian (Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA); Mathula Thangarajh (Children’s National Health System, Washington D.C., USA); Kathryn R. Wagner (Kennedy Krieger Institute, Baltimore, MD, USA); David Wolf (Emory University, Atlanta, GA, USA).
Italian DMD telethon registry study group investigators
Antonella Pini (IRCCS Istituto delle Scienze Neurologiche di Bologna, Bologna, Italy); Maria A. Donati, Michele Sacchini (Ospedale Pediatrico Meyer, Florence, Italy); Maria P. Sormani (University of Genoa, Genoa, Italy); Claudio Bruno (IRCCS ‘Giannina Gaslini’, Genoa, Italy); Sonia Messina, Gianluca Vita (University of Messina, Messina, Italy); Giovanni Baranello, Maria T. Arnoldi (IRCCS Istituto Neurologico ‘Carlo Besta’, Milan, Italy); Giacomo P. Comi, Francesca Magri, Yvan Torrente (University of Milan and IRCSS Fondazione Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy); Luisa Politano (Seconda Università di Napoli, Naples, Italy); Stefano C. Previtali (University of Campania Luigi Vanvitelli, Naples, Italy); Elena Pegoraro (University of Padua, Padua, Italy); Angela Berardinelli (IRCCS ‘C. Mondino’ Fondazione, Pavia, Italy); Roberta Battini, Silvia Frosini (Stella Maris Institute, Pisa, Italy); Enrico Bertini, Adele D’Amico (Bambino Gesù Children’s Hospital, Rome, Italy); Claudia Brogna, Giorgia Coratti, Lavinia Fanelli, Elena S. Mazzone, Giulia Norcia, Concetta Palermo, Marika Pane (Catholic University of Sacred Heart, Rome, Italy); Tiziana Mongini, Enrica Rolle, Francesca Rossi (University of Turin, Turin, Italy).
Leuven NMRC registry investigator
Marleen van den Hauwe (University Hospitals Leuven, Leuven, Belgium).
Cooperative International Neuromuscular Research Group (CINRG) investigators
Alberto Dubrovsky (Instituto de Neurosciencias Fundacion Favaloro, Buenos Aires, Argentina); Andrew Kornberg, Monique Ryan (Royal Children’s Hospital, Melbourne, Victoria, Australia); Richard Webster (Children’s Hospital at Westmead, Sydney, New South Wales, Australia); Jean K. Mah (Alberta Children’s Hospital, Calgary, Alberta, Canada); Hanna Kolski (University of Alberta, Edmonton, Alberta, Canada); W. Douglas Biggar, Laura C. McAdam (Holland Bloorview Kids Rehab Hospital, Toronto, Ontario, Canada); S. Chidambaranathan, V. Viswanathan (Sundaram Medical Foundation and Apollo Children’s Hospital, Chennai, India); Yoram Nevo (Hadassah Hebrew University Hospital, Jerusalem, Israel); Ksenija Gorni (University of Pavia and Niguarda Ca’ Granda Hospital, Milan, Italy); Jose Carlo (University of Puerto Rico, San Juan, Puerto Rico); Mar Tulinius (Queen Silvia Children’s Hospital, Gothenburg, Sweden); Richard T. Abresch, Erik K. Henricson, Nanette C. Joyce, Craig M. McDonald (University of California Davis Health System and School of Medicine, Sacramento, CA, USA); Avital Cnaan, Tina Duong, Robert Leshner, Lauren P. Morgenroth, Carolina Tesi-Rocha, Mathula Thangarajh (Children’s National Medical Center, Washington, DC, USA); John W. Day, Peter Karachunski (University of Minnesota, Minneapolis, MN, USA); Sherilyn Driscoll, Nancy Kuntz (Mayo Clinic, Rochester, MN, USA); Anne M. Connolly, Alan Pestronk (Washington University in St Louis, St Louis, MO, USA); Hoda Z. Abdel-Hamid, Paula R. Clemens (UPMC Children’s Hospital of Pittsburgh, Pittsburgh, PA, USA); Tulio E. Bertorini (University of Tennessee, Memphis, TN, USA); Timothy Lotze (Texas Children’s Hospital Houston, TX, USA); Amy D. Harper, Jean Teasley (Children’s Hospital of Virginia, Richmond, VA, USA).
ACKNOWLEDGMENTS
The authors and Sarepta Therapeutics, Inc. thank the patients and their families for participating in the trial. This study was funded by Sarepta Therapeutics, Inc. Medical writing support was provided by Paraskevi Briassouli, PhD, and Valerie Zediak, PhD of Eloquent Scientific Solutions, and funded by Sarepta.
CONFLICTS OF INTEREST
C. M. McDonald reports consulting (Astellas/Mitobridge, Bristol Myers Squibb, Capricor, Catabasis Pharmaceuticals, Edgewise Therapeutics, Eli Lilly, Epirium Bio [formerly Cardero Therapeutics], Gilead, Halo Therapeutics, Italfarmaco, Novartis, Pfizer, Prosensa, PTC Pharmaceuticals, Santhera Pharmaceuticals, and Sarepta Therapeutics, Inc.), and research funding, principal investigator, and speaking fees (Sarepta Therapeutics, Inc.). P. B. Shieh reports consulting fees for ad hoc advisory boards (AveXis, Biogen, PTC Therapeutics, and Sarepta Therapeutics, Inc.) and speakers’ bureau participation (Alexion, Biogen, and Grifols). H. Z. Abdel-Hamid reports advisory board participation (Audentes, AveXis, Biogen, and Sarepta Therapeutics, Inc.). A. M. Connolly reports advisory board participation (Acceleron, AveXis, Genentech, NS Pharma, Sarepta Therapeutics, Inc. and Scholar Rock) and DMSB participation (Catabasis). E. Ciafaloni reports advisory board participation (AveXis, Biogen, and Sarepta Therapeutics, Inc.). K. R. Wagner reports consulting fees (AskBio, Dyne, Vita, Dynacure, PTC Therapeutics, Roche, and Sarepta Therapeutics, Inc). N. Goemans reports advisory board participations and/or speaking fees (Italfarmaco, Biogen, Roche, AveXis, Pfizer, PTC Pharmaceuticals, Santhera Pharmaceuticals, and Sarepta Therapeutics, Inc.). E. Mercuri reports paid consulting (Sarepta Therapeutics, Inc.). J. R. Mendell reports receiving grants (Parent Project Muscular Dystrophy) and consulting fees (Sarepta Therapeutics, Inc. and Nationwide Children’s Hospital). N. Khan, E. Koenig, J. Malhotra, W. Zhang, and B. Han are employees of Sarepta Therapeutics, Inc. and own stock/options in the company.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-210643.
REFERENCES
[1] | Birnkrant DJ , Bushby K , Bann CM , Apkon SD , Blackwell A , Brumbaugh D , et al. Diagnosis and management of Duchenne muscular dystrophy, part diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. (2018) ;17: (3):251–67. doi: 10.1016/S1474-4422(18)30024-3 |
[2] | Crisafulli S , Sultana J , Fontana A , Salvo F , Messina S , Trifiro G . Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta-analysis. Orphanet J Rare Dis. (2020) ;15: (1):141. doi: 10.1186/s13023-020-01430-8 |
[3] | Peverelli L , Testolin S , Villa L , D’Amico A , Petrini S , Favero C , et al. Histologic muscular history in steroid-treated and untreated patients with Duchenne dystrophy. Neurology. (2015) ;85: (21):1886–93. doi: 10.1212/wnl.0000000000002147 |
[4] | Bushby K , Finkel R , Birnkrant DJ , Case LE , Clemens PR , Cripe L , et al. Diagnosis and management of Duchenne muscular dystrophy, part implementation of multidisciplinary care. Lancet Neurol. (2010) ;9: (2):177–89. doi: 10.1016/s1474-4422(09)70272-8 |
[5] | Bushby K , Finkel R , Birnkrant DJ , Case LE , Clemens PR , Cripe L , et al. Diagnosis and management of Duchenne muscular dystrophy, part diagnosis, and pharmacological and psychosocial management. Lancet Neurol. (2010) ;9: (1):77–93. doi: 10.1016/s1474-4422(09)70271-6 |
[6] | Popplewell LJ , Trollet C , Dickson G , Graham IR . Design of phosphorodiamidate morpholino oligomers (PMOs) for the induction of exon skipping of the human DMD gene. Mol Ther. (2009) ;17: (3):554–61. doi: 10.1038/mt.2008.287 |
[7] | Sarepta Therapeutics. EXONDYS 51 Prescribing Information. 2020. |
[8] | Cirak S , Arechavala-Gomeza V , Guglieri M , Feng L , Torelli S , Anthony K , et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. (2011) ;378: (9791):595–605. doi: 10.1016/s0140-6736(11)60756-3 |
[9] | Lim KR , Maruyama R , Yokota T . Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des Devel Ther. (2017) ;11: :533–45. doi: 10.2147/dddt.S97635 |
[10] | Aartsma-Rus A , Fokkema I , Verschuuren J , Ginjaar I , van Deutekom J , van Ommen GJ , et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat. (2009) ;30: (3):293–9. doi: 10.1002/humu.20918 |
[11] | Bello L , Morgenroth LP , Gordish-Dressman H , Hoffman EP , McDonald CM , Cirak S . DMD genotypes and loss of ambulation in the CINRG Duchenne Natural History Study. Neurology. (2016) ;87: (4):401–9. doi: 10.1212/wnl.0000000000002891 |
[12] | Brogna C , Coratti G , Pane M , Ricotti V , Messina S , D’Amico A , et al. Long-term natural history data in Duchenne muscular dystrophy ambulant patients with mutations amenable to skip exons 44, 45, 51 and 53. PLoS One. (2019) ;14: (6):e0218683. doi: 10.1371/journal.pone.0218683 |
[13] | Ricotti V , Mandy WP , Scoto M , Pane M , Deconinck N , Messina S , et al. Neurodevelopmental, emotional, and behavioural problems in Duchenne muscular dystrophy in relation to underlying dystrophin gene mutations. Dev Med Child Neurol. (2016) ;58: (1):77–84. doi: 10.1111/dmcn.12922 |
[14] | Bello L , D’Angelo G , Villa M , Fusto A , Vianello S , Merlo B , et al. Genetic modifiers of respiratory function in Duchenne muscular dystrophy. Ann Clin Transl Neurol. (2020) ;7: (5):786–98. doi: 10.1002/acn3.51046 |
[15] | Mendell JR , Goemans N , Lowes LP , Alfano LN , Berry K , Shao J , et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol. (2016) ;79: (2):257–71. doi: 10.1002/ana.24555 |
[16] | Mendell JR , Rodino-Klapac LR , Sahenk Z , Roush K , Bird L , Lowes LP , et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. (2013) ;74: (5):637–47. doi: 10.1002/ana.23982 |
[17] | Kinane TB , Mayer OH , Duda PW , Lowes LP , Moody SL , Mendell JR . Long-term pulmonary function in Duchenne muscular dystrophy: comparison of eteplirsen-treated patients to natural history. J Neuromuscul Dis. (2018) ;5: (1):47–58. doi: 10.3233/jnd-170272 |
[18] | Alfano LN , Charleston JS , Connolly AM , Cripe L , Donoghue C , Dracker R , et al. Long-term treatment with eteplirsen in nonambulatory patients with Duchenne muscular dystrophy. Medicine (Baltimore). (2019) ;98: (26):e15858. doi: 10.1097/md.0000000000015858 |
[19] | Mendell JR , Khan N , Sha N , Eliopoulos H , McDonald CM , Goemans N , et al. Comparison of long-term ambulatory function in patients with Duchenne muscular dystrophy treated with eteplirsen and matched natural history controls. J Neuromuscul Dis. 2021. doi: 10.3233/jnd-200548 |
[20] | International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonized Tripartite Guideline: Guideline for Good Clinical Practice. J Postgrad Med. (2001) ;47: (1):45–50. |
[21] | McDonald CM , Henricson EK , Han JJ , Abresch RT , Nicorici A , Elfring GL , et al. The 6-minute walk test as a new outcome measure in Duchenne muscular dystrophy. Muscle Nerve. (2010) ;41: (4):500–10. doi: 10.1002/mus.21544 |
[22] | Scott E , Eagle M , Mayhew A , Freeman J , Main M , Sheehan J , et al. Development of a functional assessment scale for ambulatory boys with Duchenne muscular dystrophy. Physiother Res Int. (2012) ;17: (2):101–9. doi: 10.1002/pri.520 |
[23] | Hankinson JL , Odencrantz JR , Fedan KB . Spirometric reference values from a sample of the general U.S. population. . Am J Respir Crit Care Med. (1999) ;159: (1):179–87. doi: 10.1164/ajrccm.159.1.9712108 |
[24] | Shieh BP . Poster at the 2nd Symposium of the Latin American Society of Neuromuscular Diseases (SOLANE). June 7–9, 2018. Rio de Janeiro, Brazil. 2018. |
[25] | Ambrosini A , Calabrese D , Avato FM , Catania F , Cavaletti G , Pera MC , et al. The Italian neuromuscular registry: a coordinated platform where patient organizations and clinicians collaborate for data collection and multiple usage. Orphanet J Rare Dis. (2018) ;13: (1):176. doi: 10.1186/s13023-018-0918-z |
[26] | Pane M , Mazzone ES , Sivo S , Sormani MP , Messina S , D’Amico A , et al. Long term natural history data in ambulant boys with Duchenne muscular dystrophy: 36-month changes. PLoS One. (1082) ;9: (10):e108205. doi: 10.1371/journal.pone.0108205 |
[27] | Roy AJ , Van den Bergh P , Van Damme P , Doggen K , Van Casteren V , BNMDR Scientific Committee. Early stages of building a rare disease registry, methods and data from the Belgian Neuromuscular Disease Registry (BNMDR). Acta Neurol Belg. (2015) ;115: (2):97–104. doi: 10.1007/s13760-014-0320-0 |
[28] | Khan N , Eliopoulos H , Han L , Kinane TB , Lowes LP , Mendell JR , et al. Eteplirsen treatment attenuates respiratory decline in ambulatory and non-ambulatory patients with Duchenne muscular dystrophy. J Neuromuscul Dis. (2019) ;6: (2):213–25. doi: 10.3233/JND-180351 |
[29] | van Putten M , Hulsker M , Young C , Nadarajah VD , Heemskerk H , van der Weerd L , et al. Low dystrophin levels increase survival and improve muscle pathology and function in dystrophin/utrophin double-knockout mice. FASEB J. (2013) ;27: (6):2484–95. doi: 10.1096/fj.12-224170 |
[30] | Anthony K , Arechavala-Gomeza V , Ricotti V , Torelli S , Feng L , Janghra N , et al. Biochemical characterization of patients with in-frame or out-of-frame DMD deletions pertinent to exon 44 or 45 skipping. JAMA Neurol. (2014) ;71: (1):32–40. doi: 10.1001/jamaneurol.2013.4908 |
[31] | de Feraudy Y , Ben Yaou R , Wahbi K , Stalens C , Stantzou A , Laugel V , et al. Very low residual dystrophin quantity is associated with milder dystrophinopathy. Ann Neurol. 2020. doi:10.1002/ana.25951 |
[32] | Waldrop MA , Gumienny F , El Husayni S , Frank DE , Weiss RB , Flanigan KM . Low-level dystrophin expression attenuating the dystrophinopathy phenotype. Neuromuscul Disord. (2018) ;28: (2):116–21. doi: 10.1016/j.nmd.2017.11.007 |
[33] | Kutluk MG , Doğan Ç S . Kidney involvement and associated risk factors in children with Duchenne muscular dystrophy. Pediatr Nephrol. (2020) ;35: (10):1953–8. doi: 10.1007/s00467-020-04587-3 |


