
Targeted Therapies for Metabolic Myopathies Related to Glycogen Storage and Lipid Metabolism: a Systematic Review and Steps Towards a ‘Treatabolome’
Abstract
Background:
Metabolic myopathies are a heterogenous group of muscle diseases typically characterized by exercise intolerance, myalgia and progressive muscle weakness. Effective treatments for some of these diseases are available, but while our understanding of the pathogenesis of metabolic myopathies related to glycogen storage, lipid metabolism and β-oxidation is well established, evidence linking treatments with the precise causative genetic defect is lacking.
Objective:
The objective of this study was to collate all published evidence on pharmacological therapies for the aforementioned metabolic myopathies and link this to the genetic mutation in a format amenable to databasing for further computational use in line with the principles of the “treatabolome” project.
Methods:
A systematic literature review was conducted to retrieve all levels of evidence examining the therapeutic efficacy of pharmacological treatments on metabolic myopathies related to glycogen storage and lipid metabolism. A key inclusion criterion was the availability of the genetic variant of the treated patients in order to link treatment outcome with the genetic defect.
Results:
Of the 1,085 articles initially identified, 268 full-text articles were assessed for eligibility, of which 87 were carried over into the final data extraction. The most studied metabolic myopathies were Pompe disease (45 articles), multiple acyl-CoA dehydrogenase deficiency related to mutations in the ETFDH gene (15 articles) and systemic primary carnitine deficiency (8 articles). The most studied therapeutic management strategies for these diseases were enzyme replacement therapy, riboflavin, and carnitine supplementation, respectively.
Conclusions:
This systematic review provides evidence for treatments of metabolic myopathies linked with the genetic defect in a computationally accessible format suitable for databasing in the treatabolome system, which will enable clinicians to acquire evidence on appropriate therapeutic options for their patient at the time of diagnosis.
INTRODUCTION
Though individually uncommon, rare genetic disorders affect over 300 million people worldwide [1]. While gene-based therapy may not be available for many of these disorders, there are numerous rare diseases for which pharmacological therapies can ameliorate the clinical phenotype, limit disease progression, and increase life expectancy. However, as clinicians’ exposure to these disorders is limited due to the low prevalence and incidence within the population, there is often a significant lag between diagnosis and initiation of evidence-based treatment. This lag leads to an increased burden on the healthcare system and suboptimal patient care. As more patients are diagnosed genetically through high-throughput DNA sequencing, the implementation of an electronic decision-support system that links genetic diagnoses with possible treatment options through analysis tools such as the RD-Connect Genome-Phenome Analysis Platform [2] can flag treatments at the time of diagnosis, thus decreasing this delay.
The development of this readily available com-puter-aided support system, now termed the ‘treat-abolome’ [3, 4], is an ongoing international collective research effort. The process begins with individual systematic reviews to critically appraise and summarize the scientific evidence for treatments for certain rare genetic disorders at the genetic-variant level. After databasing this information and making it computationally accessible, the resulting interface will empower clinicians with the latest evidence when deciding the management plan for a patient newly diagnosed with a rare genetic disorder. Previous reviews for the treatabolome have been completed on congenital myasthenic syndromes [4] and hereditary peripheral neuropathies [5]. The present systematic review will define the treatabolome for metabolic myopathies related to glycogen and lipid storage/metabolism.
Broadly, metabolic myopathies can be classified based on the perturbed metabolism of a specific fuel source. Inborn errors that lead to abnormal glucose storage either in the muscle and/or liver are known as glycogen storage diseases (GSDs). These disorders can be the result of mutations related to glycogen synthesis, degradation, or regulation. The clinical presentation of the associated myopathy is muscle cramps, exercise intolerance, and progressive weakness in the trunk and extremities [6]. These symptoms are often accompanied by cardiomyopathy, rhabdomyolysis, and myoglobinuria. 15 types of GSDs have been identified, with five accounting for 94% of cases [7]. Four of them involve the liver: GSD I/von Gierke disease, GSD III, GSD VI/Hers disease, and GSD IX, while GSD II/Pompe disease predominantly affects the muscle [7]. Noteworthily, the gene affected in GSD III, amyloglucosidase (AGL) gene, is expressed in both skeletal muscle and the liver. The majority of cases involve both organs, known as GSD IIIa; however, 15% of patients experience liver involvement without muscle disease (GSD IIIb), [7].
The difference between GSDs and myopathies related to fatty acid metabolism defects is that the latter produces muscle symptoms, namely myalgia, after prolonged exercise [6]. Fatty acid oxidation disorders (FAODs) are inborn errors related to defects in the failure of the mitochondrial beta-oxidation pathway and/or the carnitine-based shuttle of fatty acids into the mitochondria. In addition to symptoms being triggered by prolonged exercise, infections and fasting can also trigger multisystemic symptoms such as cardiomyopathy, encephalopathy, and liver failure, in addition to the myopathy. FAODs are categorized based on the length of the carbon chain of the fatty acid that is oxidized: short-chain (two to four), medium-chain (six to 12), long-chain (14 to 18), or very-long-chain (20 or more) [7]. Myoadenylate deaminase deficiency, a deficiency related to purine metabolism, was not examined in this systematic review. Although it is the most prevalent metabolic myopathy, the majority of those with this inborn error are asymptomatic and this deficiency can co-exist with another metabolic myopathy. It would be difficult to link the response to pharmacological treatment to the genetic variant [8, 9].
Current treatment strategies for metabolic myo-pathies are limited. Most notably, a recombinant human acid alfa-glucosidase (rhGAA) enzymatic replacement therapy (ERT) was approved for the treatment of Pompe disease in 2006 [10]. This treatment has led to increased survival in patients with the severe infantile-onset form with complete amelioration of the cardiomyopathy and dysphagia, as well as partial normalization in skeletal muscle function and strength [11–13]. For other myopathic GSDs, primary management strategies are to reduce the incidence of muscle energy crises through exercise and dietary recommendations. GSD V/McArdle disease is one of the most studied myopathic GSDs, with many studies advocating for closely monitored exercise training to increase the threshold for triggering myalgias, muscle cramping, and rhabdomyolysis. Monitored exercise training can be coupled with pre-exercise simple carbohydrate, such as sucrose, supplementation to bypass glycogenolytic defects and increase available fuel. The opposite is observed in GSDs related to glycolysis defects [6, 14]. Similarly, for FAODs, education provision is instrumental in avoiding increasing susceptibility to myalgias. Avoiding strenuous exercise in fasted-states or during ongoing infections, along with dietary modifications (high carbohydrate and low fat) are the mainstays for management strategies [6]. Overall, current recommendations for metabolic myopathies advocate for supportive therapy and symptomatic management, ranging from routine cardiac monitoring to invasive ventilation [15].
In summary, there is a growing body of evidence to support the use of supplements to replenish the affected deficiency in metabolic myopathies, mostly in the form of case reports and other observational studies. While the scientific format of these publications allows for in-depth analysis of these rare diseases, they are not conducive to rapid sharing of information to guide clinical decisions. The goal of this systematic review is to summarize all forms of scientific evidence on pharmacological treatment strategies for metabolic myopathies related to glycogen storage and lipid metabolism. This novel review will be part of an on-going international research effort to create a hub, the treatabolome, that links genetic diagnosis to evidence for management strategies.
METHODS
The design of this systematic review followed the treatabolome review strategy [3], which is based on the Cochrane Collaboration methodology [16]. Evidence was assessed using the Oxford Centre for Evidence-Based Medicine (CEBM) 2011 Levels of Evidence [17], similar to previous treatabolome projects [4, 5]. The output follows the FAIR (Find-able, Accessible, Interoperable, and Reusable) guidelines to ensure expertise sharing and knowledge translation [18]. All forms of clinical and observational research examining the therapeutic efficacy of pharmacological interventions in patients with metabolic myopathies related to an inborn error with glycogen and lipid storage and metabolism were included. A key inclusion criterion was that the genetic variant of the treated patients must be provided, in order to link the outcomes to the specific genetic mutation. Systematic reviews identified in our search were considered for narrative purposes but did not undergo eligibility assessment.
Inclusion criteria
As mentioned above, we considered all forms of clinical and observational research. This included, but was not limited to, studies employing a randomized controlled trial (RCT), non-RCT or open-labelled uncontrolled trial formats or observational reports such as case reports, case series and cohort studies. The research articles’ participants were children or adults with a genetically confirmed metabolic myopathy related to defects in glycogen and lipid storage and metabolism. A list of the eligible GSDs and corresponding mutated gene involved is provided in Table 1. For the purpose of this review, GSD Type II, Pompe Disease, was considered a myopathic GSD despite the pathophysiology affecting lysosomal metabolism. A myopathic GSD was defined as a mutation in a gene related to glycogen metabolism either expressed in skeletal muscle or expressed in other tissue but with a myopathy phenotype. Mutated genes associated with myopathies related to lipid metabolism can be seen in Table 2. These metabolic myopathies were categorized based on their physiological impairment: defects in the carnitine-based shuttling, mitochondrial beta-oxidation or the storage of the lipid in the muscle cell.
Table 1
Description of the myopathic glycogen storage diseases considered in this systematic review
| Disease | Enzymatic deficiency | OMIM | Gene affected | Tissue expression | Key clinical symptoms with HPO term identifier |
| GSD 0b | Muscle glycogen synthase | 611556 | GYSA1 | Skeletal and cardiac muscle | Cardiomyopathy (0001638), Exercise intolerance (0003546) |
| GSD II/Pompe disease | Acid alpha-glucosidase deficiency | 232300 | GAA | Ubiquitous expression | Cardiomyopathy (0001638), Hypotonia (0001290), Muscle weakness (0001324), Respiratory insufficiency (0002093) |
| GSD III/Forbes-Cori disease | Glycogen debrancher deficiency | 232400 | AGL | Skeletal muscle | Cardiomyopathy (0001638), Hepatomegaly (0002240), Muscle weakness (0001324) |
| GSD IV/Andersen disease | Glycogen branching enzyme deficiency | 232500 | GBE1 | Skeletal muscle | Cardiomyopathy (0001638), Hepatomegaly (0002240), Muscle weakness (0001324) |
| GSD V/McArdle disease | Myophosphorylase deficiency | 232600 | PYGM | Skeletal muscle | Exercise intolerance (0003546), Myalgia 0003738), Muscle weakness (0001324) |
| GSD VII/Tarui disease | Muscle phosphofructokinase deficiency | 232800 | PFK | Skeletal muscle | Exercise intolerance (0003546), Hemolytic anemia (0001930), Muscle weakness (0001324) |
| GSD X | Muscle-specific Phosphoglycerate Mutase deficiency | 261670 | PGAM2 | Ubiquitous expression | Exercise intolerance (0003546), Myalgia (0003738), Muscle weakness (0001324) |
| GSD XII | Aldolase a deficiency | 611881 | ALDOA | Skeletal muscle | Hemolytic anemia (0001930), Muscle weakness (0001324) |
| GSD XIII | Enolase 3 deficiency | 612932 | ENO3 | Skeletal and cardiac muscle | Exercise intolerance (0003546), Myalgia 0003738), Muscle weakness (0001324) |
Table 2
Description of metabolic myopathies related to defects in the carnitine-based shuttling, mitochondrial beta-oxidation or the storage of the lipid in the muscle cell considered in this systematic review
| Gene | Enzymatic deficiency | OMIM | Affected Process | Key clinical symptoms with HPO term identifier |
| ACADVL | Very long-chain Acyl-CoA dehydrogenase | 609575 | Beta-oxidation | Hypoglycemia (0001985), Hepatomegaly (0002240), Cardiomyopathy (0001638), Exercise-induced rhabdomyolysis (0009045) |
| HADHA | Long-chain 3-hydroxyAcyl-CoA dehydrogenase | 609016 | Beta-oxidation | Hypoglycemia (0001985), Hepatomegaly (0002240), Cardiomyopathy (0001638), Exercise-induced rhabdomyolysis (0009045) |
| HADHA, HADHB | Either alpha or beta subunits of the mitochondrial trifunctional protein (trifunctional protein deficiency) | 609015 | Beta-oxidation | Hypoglycemia (0001985), Hepatomegaly (0002240), Cardiomyopathy (0001638), Exercise-induced rhabdomyolysis (0009045) |
| ETFA, ETFB, ETFDH | Multiple Acyl-CoA dehydrogenase | 231680 | Beta-oxidation | Hypoglycemia (0001985), Hepatomegaly (0002240), Cardiomyopathy (0001638), Muscle weakness (0001324) |
| ACADS | Short-chain Acyl-CoA dehydrogenase | 201470 | Beta-oxidation | Hypotonia (0001290), Muscle weakness (0001324), Failure to thrive (0001508) |
| SLC22A5 | ORGANIC CATION TRANSPORTER (Systemic primary carnitine deficiency) | 603377 | Carnitine-based transport | Hypoglycemia (0001985), Hepatomegaly (0002240), Cardiomyopathy (0001638), Exercise-induced rhabdomyolysis (0009045) |
| SLC25A20 | Carnitine/acylcarnitine translocase | 613698 | Carnitine-based transport | Hypoglycemia (0001985), Hepatomegaly (0002240), Cardiomyopathy (0001638) |
| CPT2 | Carnitine palmitoyltransferase 2 | 255110 | Carnitine-based transport | Hypoglycemia (0001985), Hepatomegaly (0002240), Cardiomyopathy (0001638), Exercise-induced rhabdomyolysis (0009045) |
| CPT1A | Carnitine palmitoyltransferase 1A | 600528 | Carnitine-based transport | Hepatomegaly (0002240), Cardiomyopathy (0001638), Hypoglycemia (0001985) |
| LPIN1 | Lipin-1 deficiency | 268200 | Triglyceride and phospholipid biosynthesis | Exercise-induced rhabdomyolysis (0009045), Myalgia (0003326) |
| ABHD5 | Acyltransferase (Chanarin-Dorfman syndrome/neutral lipid storage disease with ichthyosis) | 604780 | Triglyceride metabolism | Congenital ichthyosiform erythroderma (0007479), Muscle weakness (0001324), Hepatomegaly (0002240) |
| PNPLA2 | Triglyceride lipase | 610717 | Triglyceride metabolism | Hepatomegaly (0002240), Myopathy (0003198) |
We considered all appropriate outcome measures to assess the therapeutic response to pharmacological intervention in these metabolic myopathies. This included but was not limited to standardized assessment scales, objective measures of muscle strength and function, biochemical markers and reported clinical improvement in disease phenotype. Examples of improvements in disease phenotype include regaining ambulation, reducing the need for ventilation, feeding tubes or dialysis, and an increase in ability to perform both activities and instrumental activities of daily living. Studies were excluded if the response to therapy was ambiguously described in a manner that could not be extracted for the treatabolome. For example, a case report or case series explaining that a therapy was trialed after genetic diagnosis, but improvements were subjectively reported but not measured.
Literature and trial database search
Following the treatabolome methodology for systematic reviews [3], we developed an electronic search strategy of several databases to identify all the potentially relevant research articles for this treat-abolome. Search terms were generated using relevant alternative names of the aforementioned metabolic myopathies and synonyms for “genetic diagnosis” and “pharmacological management”. To be more specific with our yield we used the syntax to only search the title and abstract for our metabolic myopathies terms (see Supplementary File 1). Our first search was done on the PubMed database. Next, to ensure we included the highest forms of evidence, we searched for clinical trials and systematic reviews using the Cochrane Central Register of Controlled Trials (CENTRAL), the ClinicalTrials.gov and EU Clinical Trials registers, and the Centre for Reviews and Dissemination database, which includes the Database of Abstracts of Reviews of Effects (DARE) and NHS Economic Evaluation Database (NHS EED). When a relevant research article was identified, manually searching for the principal investigator’s previous work was undertaken to further populate our initial search. Finally, references of systematic reviews and select literature reviews were manually screened and relevant articles were included as part of the review.
After discarding duplicates and removing irrelevant research articles based on title and abstract screening, the remaining articles underwent full-text assessment. Reasons for exclusion of ineligible articles can be seen in the PRISMA flowchart (Fig. 1). Once the relevant articles were selected for the treatabolome, each article underwent data extraction. Characteristics required for a narrative summary were extracted using a standardized extraction form (see Supplementary File 2). This information included design, number of participants, genetic diagnosis, age at diagnosis and treatment initiation, treatment characteristics: type, dose, frequency, and duration of treatment, and characteristics of the outcome measures.
Fig. 1
PRISMA flow diagram showing the literature evaluation process for the systematic review.
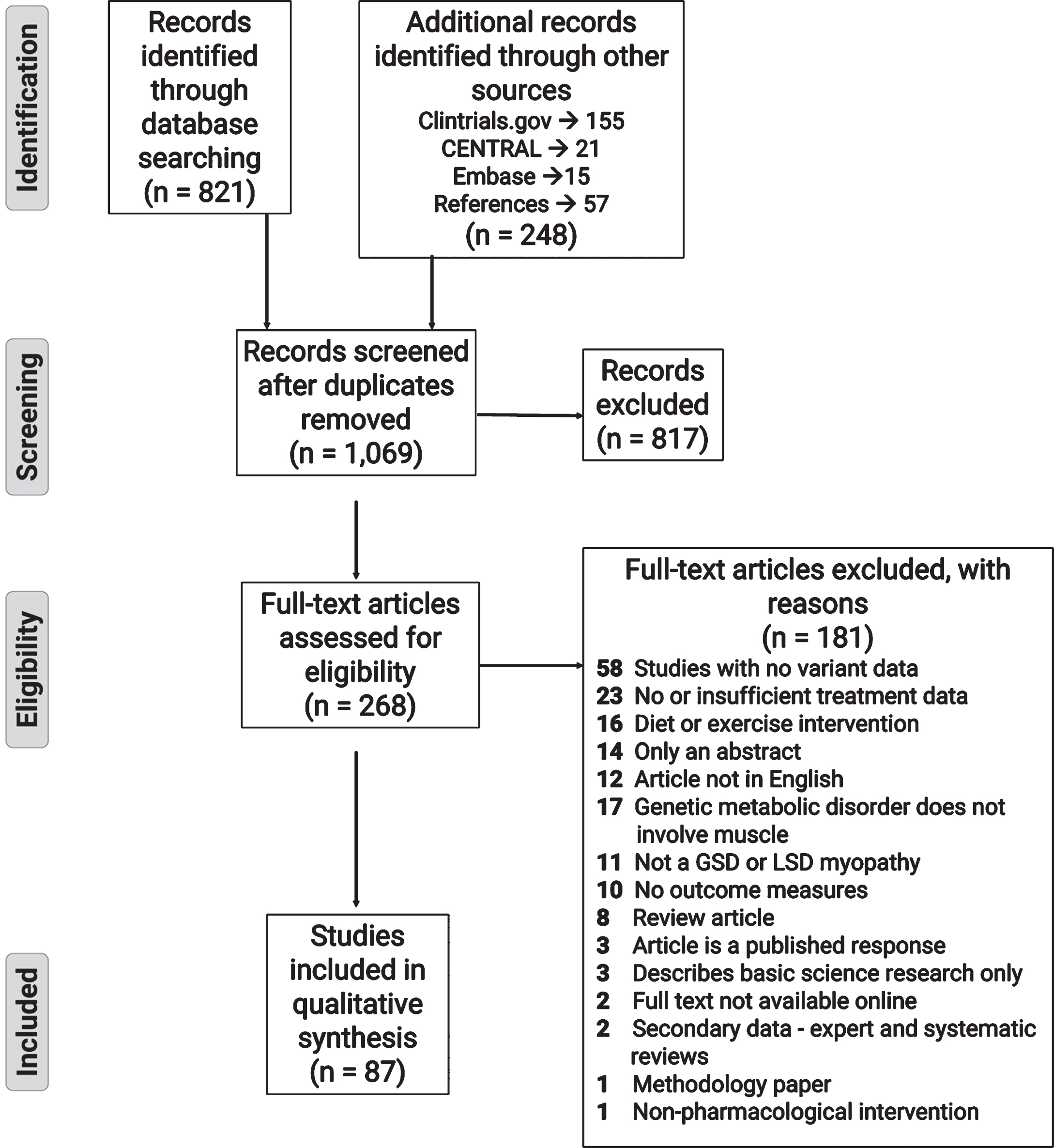
RESULTS
The initial PubMed database search retrieved 821 records for title and abstract review. An additional 191 potentially relevant articles were discovered through searching the background literature for RCTs or other clinical drug trials listed on CENTRAL, Clinicaltrials.gov and Embase. Finally, 57 articles were selected from scanning the references of included studies or relevant reviews for screening and assessment of eligibility. Following title and abstract screening, 817 articles were excluded, with the majority of these studies either being basic science publications covering muscle metabolism related to metabolic myopathies or publications pertaining to Friedreich’s ataxia. The gene responsible for Pompe disease, GAA, was frequently mistaken by the text-based search as the trinucleotide sequence (GAA) responsible for Friedreich’s ataxia, which is an unrelated (non-metabolic) neurological disorder. This similarity resulted in many research articles related to Friedreich’s ataxia being identified in our initial search. A further 181 articles were excluded following full text review. The majority of these studies were excluded because there was no confirmatory genetic diagnosis, the treatment data was insufficiently detailed for extraction, or the intervention was diet or exercise-based. The systematic review process is illustrated in the PRISMA flowchart in Fig. 1
The remaining 87 articles [19–105] underwent full data extraction. These publications included observational studies such as case reports, case series and cohort studies and clinical studies including single-arm, open label, uncontrolled and randomized, controlled trials. The most studied metabolic myopathies were Pompe disease (45 articles), multiple acyl-coenzyme a dehydrogenase deficiency (MCADD) due to electron transfer flavoprotein dehydrogenase (ETFDH) mutations (15 studies) and primary systemic carnitine deficiency (8 studies). 49.4% (43 articles) examined rhGAA for ERT in Pompe disease, followed by riboflavin (17 articles) and carnitine supplementation (11 studies) in MCADD and FAOD related to carnitine-based shuttle defects, respectively. Table 3 provides key recommendations and references from extracted data and Supplementary File 2 provides full extraction detail.
Table 3
Key recommendations for the metabolic myopathies related to defects in glycogen storage or lipid metabolism
| Gene involved | Disease | Subtype | First-line treatment recommendation | Supplemental treatment recommendation | Likely ineffective | Avoid treatment (may worsen) | Expert summary of the evidence |
| Myopathies related to glycogen storage disease | |||||||
| GAA | Pompe Disease | Infantile-onset | Myozyme© at 20– 40 mg/kg q1-2weeks | Immune tolerance induction with rituximab, methotrexate and intravenous immunoglobulin OR bortezomib | Albuterol | 18 Observational studies 7 Single-arm nonrandomized trials 2 Uncontrolled open-label study | |
| Late-onset | Myozyme© at 20– 40 mg/kg q1-2weeks | 12 Observational Studies | |||||
| 4 Single-arm nonrandomized trials 1 Uncontrolled open-label study | |||||||
| PYGM | McArdle disease | Vitamin B6 at 90 mg qd | Ramipril if two copies of ACE deletion mutation at 5 mg qd | Gentamycin | 1 Observational study | ||
| 1 Randomized Controlled Trial | |||||||
| Myopathies related to defects of beta-oxidation enzymes | |||||||
| ETFDH | Multiple acyl-coenzyme A dehydrogenase/Glutaric Aciduria II | Riboflavin at 30– 400 mg | coenzyme Q10 at 100– 500 mg qd L-Carnitine at 50– 400 mg/kg OR 1-2 g | Glucocorticoids | 15 Observational studies | ||
| ETFA | Multiple acyl-coenzyme A dehydrogenase/Glutaric acidemia IIA | sodium-D,L-3-hydroxybutyrate at 360– 2600 mg/kg qd | Riboflavin at 100 mg qd, L-Carnitine at 50 mg/kg qd, coenzyme Q10 at 100 mg qd | 2 Observational studies | |||
| SLC52A1 | Multiple acyl-coenzyme A dehydrogenase | Riboflavin at 100 mg tid | 1 Observational study | ||||
| HADHB | Trifunctional protein deficiency | Bezafibrate at 10– 20 mg/kg qd | Triheptanoin(14% of daily caloric intake) | 1 Observational study 1 Randomized controlled trial | |||
| Myopathies related to medium- and short-chain fatty acid oxidation disorders | |||||||
| ACADS | Short-chain acyl-CoA dehydrogenase deficiency | Riboflavin at 100 mg qd | L-Carnitine at 100 mg/kg qd | 1 Observational study | |||
| ACADVL | Very-long-chain acyl-CoA dehydrogenase deficiency | Bezafibrate at 100– 400 mg qd | Triheptanoin(14% of daily caloric intake) | L-Carnitine | 1 Observational study | ||
| 2 Single-arm studies | |||||||
| 1 Randomized controlled trial | |||||||
| Myopathies related to neutral lipid storage disease | |||||||
| ABDH5/CGI-58 | Chanarin-Dorfman syndrome, neutral lipid storage disease with ichthyosis | Acitretin at 10 mg bid | 2 Observational studies | ||||
| Myopathies related to carnitine cycle disorders | |||||||
| SLC25A20 | Carnitine acylcarnitine translocase deficiency | L-Carnitine at 200– 300 mg/kg qd | 1 Observational study | ||||
| SLC22A5 | Primary systemic carnitine deficiency | L-Carnitine at Carnitine 100 mg/kg qd | Triheptanoin (14% of daily caloric intake) | 8 Observational studies | |||
| 1 Randomized controlled trial | |||||||
| CPT2 | Carnitine palmitoyltransferase type 2 deficiency | L-Carnitine at 2400 mg qd | Triheptanoin (14% of daily caloric intake), Bezafibrate at 10– 20 mg/kg qd | 1 Observational study | |||
| 1 Single-arm study | |||||||
| 1 Randomized controlled trial |
While Cochrane reviews typically only consider publications with a high level of evidence (such as RCTs), this strict approach would result in very few publications in rare disease being eligible for consideration. The treatabolome approach is therefore to review all types of study returned by the search, but to clearly indicate the limitations of the evidence by providing evidence levels according to the CEBM evidence levels. The majority of the evidence included in this review is in the form of observational studies, mainly cohort and case reports or series, which are considered lower levels of evidence than randomized controlled trials. This is not surprising as cumulatively, the overall incidence of all GSD subtypes is 1:40,000 [106], while the most common lipid-metabolism related myopathy, MCADD, has an incidence of 1:9,000 [107]. The findings across these studies are generally similar and positive. For example, across all the evidence regarding ERT in both IOPD and LOPD, 42 out of 284 patients did not respond positively. This corresponded to 36 unique genetic variants, 17 of which were described to benefit from ERT in other included papers. The majority of the patients encompassed by the other 19 genetic variants did show a degree of cardiomyopathy rescue, motor improvement or prolonged life expectancy. However, later they regressed in motor milestones, became ventilator-dependent or contracted a pneumonia. Ultimately, ERT was beneficial in extending life years, however the participants’ phenotypes were frailer and at higher risk of infection and respiratory distress.
There was no similar discrepancy when summa-rizing the literature on riboflavin and carnitine supplementation for MCADD related to ETFDH mutation and FAODs related to carnitine transport deficiencies. The 3 patients with MCADD related to the ETFDH mutation across the 11 studies died of an acquired infection during the treatment observation period. Only the patient who did not have a primary carnitine deficiency did not benefit from carnitine supplementation. This is consistent with the literature, suggesting that carnitine supplementation should only be given in the case of primary deficiencies such as those related to SLC25A20 and SLC22A5 mutations but less often the case with other long-chain FAOD.
Systematic reviews
We found one systematic review and meta-analysis that evaluated the effect of ERT on long-term outcomes and survival in patients with LOPD [108]. Overall, the meta-analysis demonstrated a 5-fold reduction in mortality rate with attenuated reduction in forced vital capacity and an augmented response in ambulation gained. This study included 19 studies and trials. A confirmed genetic diagnosis was not an inclusion criterion for this review. From the 19 studies, six were included in this project [29, 33, 80, 81, 85, 98]. A further ten of the included studies had been identified by our initial search but were not included because there was no variant data for the patients. The remaining three articles were not identified by our initial search strategy because two did not have an intervention and one was a poster.
Another systematic review [109] highlighted the evidence to support newborn screening (NBS) for Pompe disease to reinforce policy change for its addi-tion to the NBS battery in healthcare systems. In this systematic review, the authors suggested it would be invaluable to study the effects of early ERT intervention following NBS confirmation of infantile-onset Pompe disease (IOPD) or pre-symptomatic detection of late-onset Pompe disease (LOPD). For IOPD, current guidelines recommend treatment initiation when diagnosis is confirmed with concurrent immune tolerance induction (ITI) if the cross-reactive immunologic material (CRIM) status is negative. CRIM-status is indicative of endogenous GAA enzyme protein, where a negative status means the patient is null for the endogenous enzyme, increasing their likelihood of developing high and sustained antibody titers to the ERT, influencing clinical outcomes and ERT effectiveness. Guidelines for LOPD management require patients to follow-up every three months during the first year of life and every three to twelve months thereafter. ERT initiation is recommended if signs or symptoms of LOPD emerge during these follow-up appointments [110]. Since the time of this systematic review, four case series [12, 111–113], two of them published on the same cohort in Taiwan [112, 113] support the recommended algorithm for IOPD and LOPD ERT initiation for both short-term and long-term survival, disease-reversal and clinical improvement. A case series [114] and open-label trial [115] examining the ERT concurrent with ITI following NBS diagnosis of CRIM-negative IOPD supports the combination therapy and early ITI therapy as a safe and efficacious treatment option. More recently, The European Pompe Consortium [116] has recommended starting ERT for LOPD when a diagnosis of Pompe is confirmed by enzyme activity testing and/or GAA variant mutation analysis and the patient has observed skeletal muscle weakness or respiratory muscle involvement. Routine follow-up should be maintained for 2 years and then its effectiveness should be re-evaluated. These guidelines are supported by a systematic review summarizing 10 years of experience in treating adults with Pompe Dis-ease with ERT. Reasons to stop ERT include the patient suffering from severe infusion-associated reactions, development of high antibody titers or no improvement nor stabilization of skeletal muscle and respiratory function in the first two years [116].
A systematic review [117] examining the pharmacological treatment strategies in McArdle highlighted two trials of low dose creatine [118, 119] and one of oral sucrose supplementation [120] prior to exercise to be efficacious in attenuating exercise-induced myalgias. However, for all three trials, the genetic variant data was not available and therefore these articles were not included in the treatabolome for this disease.
RCTs
Two RCTs were retrieved pertaining to McArdle disease [19] and long-chain FAOD [121]. A 2008 RCT by Martinuzzi and colleagues [19] sought to investigate the effect of an angiotensin-converting enzyme (ACE) inhibitor, Ramipril, on exercise capacity and quality of life in 10 patients with McArdle disease. The rationale for this double-blind, randomized, placebo-controlled trial was based on previous work from this group [122] demonstrating that a common insertion/deletion polymorphism in the ACE gene is associated with disease severity, where patients with McArdle disease carrying at least one allele with a deletion mutation have higher ACE activity and lower exercise capacity. Overall, there was no difference in exercise capacity measures performed after treatment with placebo and the ACE inhibitor. Sub-analysis suggests that the three patients with two copies of the deletion mutation in the ACE gene improved significantly compared to the seven harbouring an insertion/deletion mutation. These patients did not carry unique genetic variants in the PYGM gene, the cause of McArdle disease.
Gillingham and colleagues [121] utilized a double-blind RCT to examine the effect of traditional medium-chain triglyceride (MCT) supplementation in patients with long-chain FAOD. This study enrolled patients diagnosed with carnitine palmitoyl-transferase-2, very long-chain Acyl-Coenzyme A dehydrogenase (VLCAD), long-chain 3-hydroxy Acyl-Coenzyme A dehydrogenase or trifunctional protein deficiencies, with confirmatory genetic mutations in the CPT2, ACADVL, HADHA and HADHB genes, respectively. This study demonstrated both structural, physiological and functional cardiac improvements with triheptanoin, a seven-carbon fatty acid triglyceride, compared to the eight-carbon fatty acid triglyceride, trioctanoin.
Several RCTs related to these metabolic myo-pathies were not included in this review because there was either no confirmatory genetic diagnosis or it was not stated in the manuscript. Most notably, the hallmark RCT for ERT in patients with LOPD was not included [123]. Despite stating that all the included participants had a confirmatory genetic diagnosis of Pompe disease, the specific variant information was not provided. This was also the case in another RCT examining the cardiovascular effects of ERT and an RCT examining bezafibrate in carnitine palmitoyltransferase (CPT) II and VLCAD deficiencies [124, 125].
Case reports, case series and open-label trials
The remaining 85 articles were either observational studies or open-label and/or nonrandomized clinical trials. The number of each observational and clinical trials studied for each disease and pharmacological intervention is stated in Table 3. The most commonly studied GSD was Pompe disease with 45 research articles. 28 studies focused on IOPD and 27 studied LOPD. 36 research articles examined the effectiveness of ERT with the majority being alglucosidase alfa (marketed as Myozyme, FDA approved in 2010) [108] at a dose of 20–40 mg/kg every 1-2 weeks. 8 studies examined combination ERT with ITI in CRIM-negative IOPD (those devoid of endogenous GAA, increasing their likelihood of developing high sustained antibody titers), and finally one study evaluated the effectiveness of albuterol as an additive therapy to ERT. 182 different mutations in the GAA gene were identified in 630 patients. The majority of patients improved in clinical severity and increased life expectancy with ERT and ITI if CRIM-negative or a high sustained antibody titer developed. There were 40 patients across these studies who did not report a clear benefit. The majority of these patients (16/40) were CRIM-negative and did not receive ITI in accordance with current guidelines [110]. The other GSD studied was McArdle, which in addition to the RCT evaluating Ramipril, one case report treated a patient with vitamin B6 supplementation.
The second most commonly studied pathology was MCADD due to ETFDH mutations. 15 observational studies with 52 different ETFDH gene mutations from 103 patients showed clinical improvements with riboflavin supplementation at a dose of 50–100 mg, 3 times a day which can further be supplemented with coenzyme Q10, a secondary associated muscle deficiency seen in the later-onset forms of the disease. Riboflavin was also studied in a Short-chain acyl-CoA dehydrogenase deficiency case series as well as a case series and report of MCADD due to electron transfer flavoprotein A (ETFA gene) and riboflavin transporter 1 mutations (SLC52A1), respectively. L-Carnitine supplementation was an effective management strategy in treating primary systemic carnitine deficiency and carnitine cycle defects related to carnitine-acylcarnitine translocase and carnitine palmitoyltransferase 2 mutations, one case series found deleterious effects in the treatment of very-long-chain acyl-CoA dehydrogenase deficiency. Both patients developed a secondary carnitine deficiency and rhabdomyolysis that normalized once treatment was withdrawn. Treatment with gentamycin in patients with McArdle disease was the other research article that produced negative results [49]. Short-term gentamycin treatment does not normalize the disease signature nor cellular metabolism and energy metabolism.
DISCUSSION
This systematic review summarizes and evaluates the evidence for pharmacological therapeutic strategies for metabolic myopathies related to glycogen storage and lipid metabolism. It makes use of the treatabolome format adding to an on-going international collaborative effort to collate and appraise all the evidence of pharmacological treatment strategies for rare diseases at the level of the genetic variant [4, 5]. Our initial search identified 1,069 potentially relevant research articles, of which 87 were included in our final analysis. The three most studied metabolic myopathies were: Pompe disease (GAA gene, 45 studies); MADD due to ETFDH gene mutation (15 studies); and primary systemic carnitine deficiency (SLC22A5 gene, eight studies). The three most studied pharmacological management strategies were: ERT for Pompe disease; riboflavin supplementation for MADD; and carnitine supplementation related to FAOD. Overall, this treatabolome project identified 307 pathogenic variants in 14 different genes that resulted in a metabolic myopathy related to a perturbation in glycogen storage or lipid metabolism.
Alglucosidase alfa, developed by Sanofi Genzyme and commercially sold as Myozyme and Lumizyme [4], has been demonstrated to be an effective management strategy for treating both infantile-onset and symptomatic late-onset Pompe disease. This was the most heavily studied pharmacological intervention, following its FDA approval in 2006 for LOPD and expansion into all ages, including infantile-onset in 2010 [110]. In 2010, a larger 4,000-liter scale of recombinant human GAA, Lumizyme, was approved and most recently demonstrated improvements in cardiomyopathy, no increase requirement for assisted ventilation and preserved motor function in IOPD [127]. The standard ERT dose across the included studies was 20 mg/kg, administered intravenously every two weeks. If there was a minimal response, or the disease phenotype worsened, dosing was increased to 40 mg/kg, in accordance with the current guidelines [13]. Additionally, the earlier the treatment initiation in patients with IOPD the better the response in halting the disease progression [12, 114, 128]. Early intervention has resulted in increased life-expectancy and rescuing the loss of ambulation. Evidence from this treatabolome project also supports the use of ITI (rituximab and methotrexate with or without intravenous immune globulin) in CRIM-negative IOPD patients to attenuate the development of high sustained antibody titers, leading to poor response to ERT [114, 129]. When examining the genetic variants who had minimal responses to ERT, the majority of these patients (16/40) were CRIM-negative and did not receive ITI in accordance with current guidelines.
Across the 43 studies examining the effects of ERT on both IOPD and LOPD, 42 patients, encompassing 36 unique genetic variants, did not respond optimally to ERT. Upon reviewing the individual response in each of the included studies we concluded that none of the responses are variant-specific and therefore do not constitute a reason to not administer ERT for a particular genetic mutation. This is an important conclusion for this particular review, in which our aim is to link treatments with causative genetic variants. The majority of non-responders highlighted in this review (n = 16) were CRIM-negative but did not receive concurrent ITI therapy. For 8 other patients, the dosing regimen was less than the recommended guidelines [13] or there was a significant delay between diagnosis and treatment initiation. This included patients with both IOPD and LOPD. Finally, these seemingly poor responders may be confounded by the length of follow-up and outcome measures used. While ERT can completely rescue the associated cardiomyopathy and reduce risk of death by 99% in infants, gross motor milestones and reduction in risk of invasive ventilation is achieved by a smaller proportion [13]. The rescuing of the cardiomyopathy and normalization of other cardiac function measures were seen in all 13 of the patients who declined in terms of muscle and/or respiratory function. Moreover, with the increased life-expectancy in patients with IOPD, the risk of becoming ventilator-dependent or regressing in motor development and muscle strength is further exacerbated. This can be seen in the clinical trials and observational studies with longer follow-up [113, 128]. This is observed in three of these patients who improved in motor development but later regressed and/or became ventilator-dependent. Assessment of only one facet of the disease phenotype, such as muscle performance or cardiomyopathy, can obscure response to ERT. Therefore, we recommend that clinical trials employ multisystemic measures when evaluating ERT, especially in the context of IOPD, It is essential that we evaluate the reversal of the disease phenotype with measures such as left ventricular mass index, motor milestone achievement and decrease in ventilation requirement. Moreover, a distinction needs to be made that with increased survival, a new phenotype emerges that is characterized by progressive muscle weakness, which can lead to the need for oxygen supplementation, assisted feeding and increased predisposition to aspiration pneumonias and viral infections. While these outcomes are unfavorable, treatment success is nevertheless reflected by the increase in life expectancy. A second-generation recombinant GAA ERT, avalglucosidase alfa or neoGAA, was biochemically designed to increase uptake into skeletal muscle. There were minimal improvements in pulmonary and muscle endurance measures as well as no change from baseline in measured functional ability. Additionally, the plasma concentration of the drug exponentially dropped following the end of infusion [130]. A Phase III RCT comparing the safety and efficacy of neoGAA and alglucosidase alfa in untreated LOPD was initiated in 2016 and is currently ongoing (NCT02782741).
As with all treatabolome reviews, a significant limitation of our review is that our full analysis is limited to papers providing the precise genetic variant data for the patients receiving treatment, but unfortunately this data is frequently not provided as part of the original study. All evidence regarding the effectiveness of ERT in both IOPD and LOPD included in this review thus comes from open-label, non-RCT or observational studies which all correspond to Level 3 and lower on the Oxford CEBM 2011 Levels of Evidence [17]. Despite our search initially identifying both of the major RCTs examining the effects of ERT in LOPD [123, 124], neither could be included in the treatabolome dataset as the genetic variant data of the study participants was not provided. Although both RCTs stated that a confirmed genetic diagnosis of Pompe disease was an inclusion criterion for participants, the precise genetic data was not stated in any table nor any supplementary data file. Moreover, the one RCT examining the effects of different doses of ERT in IOPD was not identified by our search as it was not a requirement to have a confirmed genetic diagnosis for study eligibility [131].
This was also the case for many of the industry-sponsored clinical trials for ERT in Pompe disease [123, 132–134]. It is difficult to elucidate the reason for this, whether it be due to ethical concerns about sharing genetic information or whether it was simply felt that this information is not required in a loss-of-function mutation disease. This is a major challenge in the study of treatments for rare genetic diseases: in the absence of genetic data, variant-specific responses to therapeutic treatments cannot be examined. This lack of this information limits future reuse of the datasets as in our treatabolome example, and in cases where there is a possibility that specific variants respond differently, the broader field of research would benefit from the genetic data being made available as a standard practice. The development of secure sharing databases such as RD-Connect (2) for this genetic information is a crucial advance that allows secure sharing of full genomic data. However, the causative variant itself is not generally considered sensitive data and it is therefore possible to share this in a publication or supplementary data. Making it common practice to provide causative variants in publications, even where it is not the primary purpose of the publication, and also encouraging the upload of sensitive genetic data to a secure platform, regardless of the intervention studied, could eliminate this problem and allow for the collating of evidence on that particular rare genetic disease.
The only other GSD included in this treatabolome project was GSD V, also known as McArdle disease. Generally, there is little consensus over management strategies for this disease, with most of the recommendations supported by low-quality evidence. This finding is reflected in the three included articles studying McArdle’s disease, which all examine a different intervention: vitamin B6, ramipril, and gentamicin. Only one case reported treating a patient with McArdle disease with vitamin B6, showing reduction in creatine kinase levels and renal function recovery following acute rhabdomyolysis. This outcome corresponds to a weak recommendation that was supported by low-quality evidence. Ramipril was a recommendation from high-quality evidence (an RCT) but was contingent on the patient harboring two deletion copies of the ACE gene in addition to the PGYM mutation. Finally, gentamycin did not have any effect on muscle energy metabolism and myophosphorylase activity in patients with nonsense mutations, despite pre-clinical data suggesting that aminoglycosides facilitate read-through of premature stop codons [135].
As summarized in a review on myopathic GSDs [14], there is low-quality evidence to support that exercise can be a safe and effective treatment for these metabolic myopathies. The majority of the literature studied is in patients with McArdle disease. Small observational and cohort studies suggest that routine moderate aerobic exercise, while avoiding strenuous anaerobic activity, can increase cardiac output, muscle strength and function as well as increase the threshold for triggering myalgia and cramps. All of the evidence currently supporting exercise as a treatment option for McArdle disease is found within two open studies and one case report [136–139]. Higher quality evidence is required to fully evaluate the therapeutic potential of exercise. Furthermore, there is high-quality evidence from RCTs and placebo-controlled cross over studies that found dietary modifications responsible for attenuating symptoms and exercise intolerance [140–143]. A higher-carbohydrate diet and oral sucrose administration prior to exercise resulted in improvements in exercise tolerance. Examples include decreased heart rate with maximum exertion and maximal oxidative work capacity. However, creatine and triheptanoin did not have any effect on metabolic markers indicative of improved exercise intolerance [118, 119, 144].
L-carnitine supplementation was an effective therapeutic intervention in the context of metabolic myopathies related to defects in the carnitine-based transport of fatty acids. This included primary systemic carnitine deficiency and carnitine cycle defects related to SLC25A20, SLC22A5 and CPT2 mutations. In VLCAD deficiency, it still remains controversial whether L-carnitine supplementation is an appropriate therapy [145]. Preclinical murine research has suggested that supplementation does not raise free levels of carnitine and instead raise an intermediary metabolite that may cause toxic side effects [146]. This is supported by a case series [147] included in this review, where both siblings with ACADVL mutations experienced recurrent rhabdomyolysis upon carnitine supplementation. More recently, it was suggested that the peroxisome proliferator activated receptor alpha agonist bezafibrate could be an effective treatment strategy for long-chain-FAOD [148]. The one research article included in this review examining this treatment option showed varied responses in changes in biochemical profile and number of myopathic attacks [149]. As of now, treatment options for this cohort of FOADs is centered on dietary management including avoidance of fasting, dietary fat restriction, and MCT supplementation.
CONCLUSION
Our systematic review has collated evidence for pharmacologic-based interventions in genetically confirmed metabolic myopathies related to glycogen storage and lipid metabolism and created a dataset linking the treatments with the causative genetic variant. We summarize all the evidence, the majority being open-label trials and observational studies, on ERT in both infantile- and late-onset Pompe disease, riboflavin supplementation in MADD due to ETFDH mutations and carnitine supplementation in FAOD related to carnitine deficiency. While most evidence is classed as CEBM level 3 or 4, this is not unexpected, as cumulatively, the overall incidence of all GSD subtypes is 1:40,000 [106], while the most common lipid-metabolism related myopathy, MCAD, has an incidence of 1:9,000 [107]. Although large RCTs and other clinical trials do not exist for the majority of these rare genetic diseases, summaries such as this treatabolome review can still provide clinicians with guidance about possible therapeutic options and the level of evidence for each at the time of a patient’s genetic diagnosis.
Overall, this systematic review will aid in the ongoing populating of a readily accessible database, the treatabolome, that aims to enable clinicians to easily acquire evidence on therapeutic options for rare diseases based on genetic findings. By streamlining and summarizing the available data and feeding it into an online decision-support system, the lag between diagnosis and treatment initiation should decrease, leading to better outcomes, decrease in disease burden and increase in life expectancy. It is our goal that this treatabolome review on metabolic myopathies will provide the foundation for a system pointing clinicians to the evidence for the management of these rare genetic diseases, providing them with the data they need to make informed treatment choices for their newly diagnosed patients.
ACKNOWLEDGMENTS
AM received a Summer Studentship stipend from the Faculty of Medicine at the University of Ottawa. HL receives support from the Canadian Institutes of Health Research (Foundation Grant FDN-167281), the Canadian Institutes of Health Research and Muscular Dystrophy Canada (Network Catalyst Grant NG2-170044 for NMD4C), the Canada Foundation for Innovation (CFI-JELF 38412), and the Canada Research Chairs program (Canada Research Chair in Neuromuscular Genomics and Health, 950-232279). RT receives support from the Canadian Institutes of Health Research (postdoctoral fellowship award MFE-171275). The authors thank Dr Antonio Atalaia (Institut de Myologie and INSERM, Paris, France) and Dr Leslie Matalonga (Centro Nacional de Análisis Genómico, Barcelona, Spain) for detailed guidance on the treatabolome procedure and templates. This review is part of the Solve-RD treatabolome initiative. The Solve-RD project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 779257.
CONFLICTS OF INTEREST
The Authors declare that there are no competing interests associated with the manuscript.
REFERENCES
[1] | Allies in Rare Disease - Global Genes [Internet]. [cited 2020 Oct 14]. Available from: https://globalgenes.org/ |
[2] | RD-Connect Dashboard [Internet]. [cited 2020 Oct 14]. Available from: https://platform.rd-connect.eu/ |
[3] | Atalaia A , Thompson R , Corvo A , Carmody L , Piscia D , Matalonga L , et al. A guide to writing systematic reviews ofrare disease treatments to generate FAIR-compliant datasets: Building a Treatabolome. Orphanet J Rare Dis. (2020) ;15: (1):206. |
[4] | Thompson R , Bonne G , Missier P , Lochmüller H . Targeted therapies for congenital myasthenic syndromes:Systematic review and steps towards a treatabolome. Emerg Top Life Sci. (2019) ;3: (1):19–37. |
[5] | Jennings MJ , Lochmüller A , Atalaia A , Horvath R . Targeted Therapies for Hereditary Peripheral Neuropathies: Systematic Review and Steps Towards a ‘treatabolome.’ J Neuromuscul Dis. 2020 Jul 31;epub ahead of print. |
[6] | Tarnopolsky MA . Metabolic Myopathies. Vol. 22, CONTINUUM Lifelong Learning in Neurology. Lippincott Williams and Wilkins; 2016. pp. 1829-51. |
[7] | Chen Y-T . Glycogen storage diseases. In: The metabolic and molecular bases of inherited disease. (1995) . pp. 935–65. |
[8] | Castro-Gago M , Gómez-Lado C , Pérez-Gay L , Eirís-Puñal J , Pintos Martínez E , García-Consuegra I , et al. Primary adenosine monophosphate (AMP) deaminase deficiency in a hypotonic infant. J Child Neurol. (2011) ;26: (6):734–7. |
[9] | Tarnopolsky MA . What can metabolic myopathies teach us about exercise physiology?. Applied Physiology, Nutritionand Metabolism. Appl Physiol Nutr Metab. (2006) ;31: :21–30. |
[10] | Drugs@FDA: FDA-Approved Drugs. |
[11] | Prater SN , Banugaria SG , Dearmey SM , Botha EG , Stege EM , Case LE , et al. The emerging phenotype of long-termsurvivors with infantile Pompe disease. Genetics in Medicine. Genet Med. (2012) ;14: :800–10. |
[12] | Yang CF , Yang CC , Liao HC , Huang LY , Chiang CC , Ho HC , et al. Very Early Treatment for Infantile-Onset PompeDisease Contributes to Better Outcomes. J Pediatr. (2016) ;169: :174–180.e1. |
[13] | Kohler L , Puertollano R , Raben N . Pompe Disease: From Basic Science to Therapy. Neurotherapeutics. (2018) ;15: :928–42. |
[14] | Tarnopolsky MA . Myopathies Related to Glycogen Metabolism Disorders. Vol. 15, Neurotherapeutics. Springer New York LLC; 2018. pp. 915-27 |
[15] | Finsterer J . Update review about metabolic myopathies. Life. (2020) ;10: (4):43. |
[16] | Higgins , Julian PT and GreenS. Cochrane Handbook for Systematic Reviews of Interventions | Cochrane Training. Handbook. (2011) . pp. 649. |
[17] | Howick J , Chalmers I , Glasziou P , Greenhalgh T , Heneghan C , Liberati A , et al. The Oxford 2011 Levels of Evidence. Oxford Centre Evidence-Based Medicine. Vol. 1, Group. 2011. |
[18] | Wilkinson MD , Dumontier M , Aalbersberg IjJ , Appleton G , Axton M , Baak A , et al. Comment: The FAIR GuidingPrinciples for scientific data management and stewardship. Sci Data. (2016) ;3: :160018. |
[19] | Martinuzzi A , Liava A , Trevisi E , Frare M , Tonon C , Malucelli E , et al. Randomized, placebo-controlled,double-blind pilot trial of ramipril in McArdle’s disease. Muscle and Nerve. (2008) ;37: (3):350–7. |
[20] | Israeli S , Pessach Y , Sarig O , Goldberg I , Sprecher E . Beneficial effect of acitretin in Chanarin-Dorfmansyndrome. Clin Exp Dermatol. (2012) ;37: (1):31–3. |
[21] | Melegh B , Bene J , Mogyorósy G , Havasi V , Komlósi K , Pajor L , et al. Phenotypic manifestations of theOCTN2 V295X mutation: Sudden infant death and carnitine-responsive cardiomyopathy in Roma families. Am J Med Genet. (2004) ;131 A(2):121–6. |
[22] | Korpela MP , Paetau A , Löfberg MI , Timonen MH , Lamminen AE , Kiuru-Enari SMK . A novel mutation of the GAA genein a finnish late-onset Pompe disease patient: Clinical phenotype and follow-up with enzyme replacement therapy. Muscle and Nerve. (2009) ;40: (1):143–8. |
[23] | Yoon YA , Lee DH , Ki CS , Lee SY , Kim JW , Lee YW , et al. SLC22A5 mutations in a patient with systemic primarycarnitine deficiency: The first Korean case confirmed by biochemical and molecular investigation. Ann Clin Lab Sci. (2012) ;42: (4):422–3. |
[24] | But WM , Lee SH , Chan AOK , Lau GTC . Enzyme replacement therapy for infantile Pompe disease during the criticalperiod and identification of a novel mutation. Hong Kong Med J. (2009) ;15: (6):474–7. |
[25] | Ishii K , Komaki H , Ohkuma A , Nishino I , Nonaka I , Sasaki M . Central nervous system and muscle involvement in anadolescent patient with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Brain Dev. (2010) ;32: (8):669–72. |
[26] | Liang WC , Ohkuma A , Hayashi YK , López LC , Hirano M , Nonaka I , et al. ETFDH mutations, CoQ10 levels, andrespiratory chain activities in patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Neuromuscul Disord. (2009) ;19: (3):212–6. |
[27] | Shima A , Yasuno T , Yamada K , Yamaguchi M , Kohno R , Yamaguchi S , et al. First Japanese case of carnitinepalmitoyltransferase II deficiency with the homozygous point mutation S113L. Intern Med. (2016) ;55: (18):2659–61. |
[28] | Hamdan MA , El-Zoabi BA , Begam MA , Mirghani HM , Almalik MH . Antenatal diagnosis of pompe disease by fetalechocardiography: Impact on outcome after early initiation of enzyme replacement therapy. J Inherit Metab Dis. (2010) ;33: (SUPPL. 3):S333–9. |
[29] | Del Rizzo M , Fanin M , Cerutti A , Cazzorla C , Milanesi O , Nascimbeni AC , et al. Long-term follow-up results inenzyme replacement therapy for Pompe disease: A case report. J Inherit Metab Dis. (2010) ;33: (SUPPL. 3):S389–93. |
[30] | Cho A , Kim SJ , Lim BC , Hwang H , Park JD , Kim GB , et al. Infantile Pompe disease: Clinical and geneticcharacteristics with an experience of enzyme replacement therapy. J Child Neurol. (2012) ;27: (3):319–24. |
[31] | Yang CC , Chien YH , Lee NC , Chiang SC , Lin SP , Kuo YT , et al. Rapid progressive course of later-onset Pompedisease in Chinese patients. Mol Genet Metab. (2011) ;104: (3):284–8. |
[32] | Yilmaz BS , Kor D , Mungan NO , Erdem S , Ceylaner S . Primary systemic carnitine deficiency: A Turkish case with anovel homozygous SLC22A5 mutation and 14 years follow-up. J Pediatr Endocrinol Metab. (2015) ;28: (9-10):1179–81. |
[33] | Papadimas GK , Spengos K , Konstantinopoulou A , Vassilopoulou S , Vontzalidis A , Papadopoulos C , et al. Adult Pompedisease: Clinical manifestations and outcome of the first Greek patients receiving enzyme replacement therapy. Clin Neurol Neurosurg. (2011) ;113: (4):303–7. |
[34] | Shioya A , Takuma H , Yamaguchi S , Ishii A , Hiroki M , Fukuda T , et al. Amelioration of acylcarnitine profile usingbezafibrate and riboflavin in a case of adult-onset glutaric acidemia type 2 with novel mutations of the electrontransfer flavoprotein dehydrogenase (ETFDH) gene. J Neurol Sci. (2014) ;346: (1-2):350–2. |
[35] | Işıkay S , Yaman A , Ceylaner S . An infant with glutaric aciduria type IIc diagnosed with a novel mutation. Turk J Pediatr. (2017) ;59: (3):315–7. |
[36] | Wen B , Li D , Li W , Zhao Y , Yan C . Multiple acyl-CoA dehydrogenation deficiency as decreased acyl-carnitineprofile in serum. Neurol Sci. (2015) ;36: (6):853–9. |
[37] | Iacobazzi V , Pasquali M , Singh R , Matern D , Rinaldo P , Di San Filippo CA , et al. Response to Therapy inCarnitine/Acylcarnitine Translocase (CACT) Deficiency Due to a Novel Missense Mutation. Am J Med Genet. (2004) ;126A(2):150–5. |
[38] | Austin SL , Chiou A , Sun B , Case LE , Govendrageloo K , Hansen P , et al. Alglucosidase alfa enzyme replacementtherapy as a therapeutic approach for a patient presenting with a PRKAG2 mutation. Mol Genet Metab. (2017) ;120: (1-2):96–100. |
[39] | Vieira P , Myllynen P , Perhomaa M , Tuominen H , Keski-Filppula R , Rytky S , et al. Riboflavin-Responsive MultipleAcyl-CoA Dehydrogenase Deficiency Associated with Hepatoencephalomyopathy and White Matter Signal Abnormalitieson Brain MRI. Neuropediatrics. (2017) ;48: (3):194–8. |
[40] | Fu Liong H , Abdul Wahab SA , Yakob Y , Lock Hock N , Thong WK , Viswanathan S . Late-Onset Glycogen Storage DiseaseType II (Pompe’s Disease) with a Novel Mutation: A Malaysian Experience. Case Rep Neurol Med. (2014) ;2014: (PG-926510):1–6. |
[41] | Montagnese F , Barca E , Musumeci O , Mondello S , Migliorato A , Ciranni A , et al. Clinical and molecular aspects of30 patients with late-onset Pompe disease (LOPD): Unusual features and response to treatment. J Neurol. (2015) ;262: (4):968–78. |
[42] | Prater SN , Patel TT , Buckley AF , Mandel H , Vlodavski E , Banugaria SG , et al. Skeletal muscle pathology ofinfantile Pompe disease during long-term enzyme replacement therapy. Orphanet J Rare Dis. (2013) ;8: (1):90. |
[43] | Lahrouchi N , Lodder EM , Mansouri M , Tadros R , Zniber L , Adadi N , et al. Exome sequencing identifies primarycarnitine deficiency in a family with cardiomyopathy and sudden death. Eur J Hum Genet. (2017) ;25: (6):783–7. |
[44] | Moravej H , Amirhakimi A , Showraki A , Amoozgar H , Hadipour Z , Nikfar G . A new mutation causing severeinfantile-onset pompe disease responsive to enzyme replacement therapy. Iran J Med Sci. (2018) ;43: (2):218–22. |
[45] | Wen B , Dai T , Li W , Zhao Y , Liu S , Zhang C , et al. Riboflavin-responsive lipid-storage myopathy caused by ETFDHgene mutations. J Neurol Neurosurg Psychiatry. (2010) ;81: (2):231–6. |
[46] | Saral NY , Aksungar FB , Aktuglu-Zeybek C , Coskun J , Demirelce O , Serteser M . Glutaric acidemia type II patientwith thalassemia minor and novel electron transfer flavoprotein-A gene mutations: A case report and review ofliterature. World J Clin Cases. (2018) ;6: (14):786–90. |
[47] | Papadopoulou-Legbelou K , Gogou M , Dokousli V , Eboriadou M , Evangeliou A . Dilated Cardiomyopathy as the OnlyClinical Manifestation of Carnitine Transporter Deficiency. Indian J Pediatr. (2017) ;84: (3):231–3. |
[48] | Komlósi K , Magyari L , Talián GC , Nemes É , Káposzta R , Mogyorósy G , et al. Plasma carnitineester profile in homozygous and heterozygous OCTN2 deficiency. J Inherit Metab Dis. (2009) ;32: (SUPPL. 1):S15–9. |
[49] | Schroers A , Kley RA , Stachon A , Horvath R , Lochmüller H , Zange J , et al. Gentamicin treatment in McArdledisease: Failure to correct myophosphorylase deficiency. Neurology. (2006) ;66: (2):285–6. |
[50] | Izumi R , Suzuki N , Nagata M , Hasegawa T , Abe Y , Saito Y , et al. A case of late onset riboflavin-responsivemultiple acyl-CoA dehydrogenase deficiency manifesting as recurrent rhabdomyolysis and acute renal failure. Intern Med. (2011) ;50: (21):2663–8. |
[51] | Lamhonwah AM , Olpin SE , Pollitt RJ , Vianey-Saban C , Divry P , Guffon N , et al. Novel OCTN2 mutations: Nogenotype-phenotype correlations: Early carnitine therapy prevents cardiomyopathy. Am J Med Genet. (2002) ;111: (3):271–84. |
[52] | Kim YJ , Ko JM , Song J , Lee KA . Clinical features of multiple acyl-CoA dehydrogenase deficiency with ETFDHvariants in the first Korean cases. Ann Lab Med. (2018) ;38: (6):616–8. |
[53] | Gempel K , Topaloglu H , Talim B , Schneiderat P , Schoser BGH , Hans VH , et al. The myopathic form of coenzyme Q10deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain. (2007) ;130: (8):2037–44. |
[54] | Satoh A , Hirashio S , Arima T , Yamada Y , Irifuku T , Ishibashi H , et al. Novel Asp511Thr mutation in McArdledisease with acute kidney injury caused by rhabdomyolysis. CEN case reports. (2019) ;8: (3):194–9. |
[55] | Srinivasaraghavan R , Krishnamurthy S , Chandar R , Cassandrini D , Mahadevan S , Bruno C , et al. Acitretin-responsiveichthyosis in chanarin-dorfman syndrome with a novel mutation in the ABHD5/CGI-58 gene. Pediatr Dermatol. (2014) ;31: (5):612–4. |
[56] | Olsen RKJ , Olpin SE , Andresen BS , Miedzybrodzka ZH , Pourfarzam M , Merinero B , et al. ETFDH mutations as a majorcause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain. (2007) ;130: (8):2045–54. |
[57] | Chen W , Zhang Y , Ni Y , Cai S , Zheng X , Mastaglia FL , et al. Late-onset riboflavin-responsive multiple acyl-CoAdehydrogenase deficiency (MADD): Case reports and epidemiology of ETFDH gene mutations. BMC Neurol. (2019) ;19: (1):330. |
[58] | Mosegaard S , Bruun GH , Flyvbjerg KF , Bliksrud YT , Gregersen N , Dembic M , et al. An intronic variation in SLC52A1causes exon skipping and transient riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Mol Genet Metab. (2017) ;122: (4):182–8. |
[59] | Kim YM , Cheon CK , Park KH , Park SW , Kim GH , Yoo HW , et al. Novel and recurrent ACADS mutations and clinicalmanifestations observed in korean patients with short-chain Acyl-coenzyme a dehydrogenase deficiency. Ann ClinLab Sci. (2016) ;46: (4):360–6. |
[60] | Rairikar M V , CaseLE, BaileyLA, KaziZB, DesaiAK, BerrierKL, et al. Insight into the phenotype of infantswith Pompe disease identified by newborn screening with the common c.-32-13T >G “late-onset” GAA variant. . MolGenet Metab. (2017) ;122: (3):99–107. |
[61] | Al-Hassnan ZN , Khalifa OA , Bubshait DK , Tulbah S , Alkorashy M , Alzaidan H , et al. The phenotype, genotype, andoutcome of infantile-onset Pompe disease in 18 Saudi patients. Mol Genet Metab Reports. (2018) ;15: (PG-50-54):50–4. |
[62] | Goh LL , Lee Y , Tan ES , Lim JSC , Lim CW , Dalan R . Patient with multiple acyl-CoA dehydrogenase deficiency diseaseand ETFDH mutations benefits from riboflavin therapy: A case report. BMC Med Genomics. (2018) ;11: (1):37. |
[63] | Hong D , Yu Y , Wang Y , Xu Y , Zhang J . Acute-onset multiple acyl-CoA dehydrogenase deficiency mimickingGuillain-Barré syndrome: Two cases report. BMC Neurol. (2018) ;18: (1):219. |
[64] | van der Meijden JC , Kruijshaar ME , Harlaar L , Rizopoulos D , van der Beek NAME , van der Ploeg AT . Long-termfollow-up of 17 patients with childhood Pompe disease treated with enzyme replacement therapy. J Inherit Metab Dis. (2018) ;41: (6):1205–14. |
[65] | Tegtmeyer LC , Rust S , van Scherpenzeel M , Ng BG , Losfeld M-E , Timal S , et al. Multiple Phenotypes inPhosphoglucomutase 1 Deficiency. N Engl J Med. (2014) ;370: (6):533–42. |
[66] | Peng SSF , Hwu WL , Lee NC , Tsai FJ , Tsai WH , Chien YH . Slow, progressive myopathy in neonatally treated patientswith infantile-onset Pompe disease: A muscle magnetic resonance imaging study Dr. Segolene Ayme Dr. Segolene Ayme. Orphanet J Rare Dis. (2016) ;11: (1):63. |
[67] | Liu XY , Wang ZQ , Wang DN , Lin MT , Wang N . A Historical Cohort Study on the Efficacy of Glucocorticoids andRiboflavin Among Patients with Late-onset Multiple Acyl-CoA Dehydrogenase Deficiency. Chin Med J (Engl). (2016) ;129: (2):142–6. |
[68] | Banugaria SG , Prater SN , Patel TT , DeArmey SM , Milleson C , Sheets KB , et al. Algorithm for the Early Diagnosisand Treatment of Patients with Cross Reactive Immunologic Material-Negative Classic Infantile Pompe Disease: AStep towards Improving the Efficacy of ERT. PLoS One. (2013) ;8: (6):e67052. |
[69] | Suyama T , Shimura M , Fushimi T , Kuranobu N , Ichimoto K , Matsunaga A , et al. Efficacy of bezafibrate in twopatients with mitochondrial trifunctional protein deficiency. Mol Genet Metab Reports. (2020) ;24: (PG-100610):100610. |
[70] | De Biase I , Champaigne NL , Schroer R , Pollard LM , Longo N , Wood T . Primary carnitine deficiency presentsatypically with long QT syndrome: A case report. JIMD Rep. (2012) ;2: (PG-87-90):87–90. |
[71] | Chien YH , Hwu WL , Lee NC , Tsai FJ , Koeberl DD , Tsai WH , et al. Albuterol as an adjunctive treatment to enzymereplacement therapy in infantile-onset Pompe disease. Mol Genet Metab Reports. (2017) ;11: (PG-31-35):31–5. |
[72] | Parini R , De Lorenzo P , Dardis A , Burlina A , Cassio A , Cavarzere P , et al. Long term clinical history of anItalian cohort of infantile onset Pompe disease treated with enzyme replacement therapy. Orphanet J Rare Dis. (2018) ;13: (1):32. |
[73] | Winkel LPF , Van Den Hout JMP , Kamphoven JHJ , Disseldorp JAM , Remmerswaal M , Arts WFM , et al. Enzyme ReplacementTherapy in Late-Onset Pompe’s Disease: A Three-Year Follow-up. Ann Neurol. (2004) ;55: (4):495–502. |
[74] | Van den Hout JMP , Kamphoven JHJ , Winkel LPF , Arts WFM , De Klerk JBC , Loonen MCB , et al. Long-term intravenoustreatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics. (2004) ;113: (5):e448–57. |
[75] | Kishnani PS , Nicolino M , Voit T , Rogers RC , Tsai ACH , Waterson J , et al. Chinese hamster ovary cell-derivedrecombinant human acid α-glucosidase in infantile-onset Pompe disease. J Pediatr. (2006) ;149: (1):89–97. |
[76] | Chien YH , Lee NC , Peng SF , Hwu WL . Brain development in infantile-onset pompe disease treated by enzymereplacement therapy. Pediatr Res. (2006) ;60: (3):349–52. |
[77] | Case LE , Koeberl DD , Young SP , Bali D , DeArmey SM , Mackey J , et al. Improvement with ongoing Enzyme ReplacementTherapy in advanced late-onset Pompe disease: A case study. Mol Genet Metab. (2008) ;95: (4):233–5. |
[78] | Chien YH , Lee NC , Thurberg BL , Chiang SC , Zhang XK , Keutzer J , et al. Pompe disease in infants: Improving theprognosis by newborn screening and early treatment. Pediatrics. (2009) ;124: (6):e1116–25. |
[79] | Bonnefont JP , Bastin J , Laforêt P , Aubey F , Mogenet A , Romano S , et al. Long-term follow-up of bezafibratetreatment in patients with the myopathic form of carnitine palmitoyltransferase 2 deficiency. Clin Pharmacol Ther. (2010) ;88: (1):101–8. |
[80] | van Capelle CI , van der Beek NAME , Hagemans MLC , Arts WFM , Hop WCJ , Lee P , et al. Effect of enzyme therapy injuvenile patients with Pompe disease: A three-year open-label study. Neuromuscul Disord. (2010) ;20: (12):775–82. |
[81] | Orlikowski D , Pellegrini N , Prigent H , Laforêt P , Carlier R , Carlier P , et al. Recombinant human acidalpha-glucosidase (rhGAA) in adult patients with severe respiratory failure due to Pompe disease. Neuromuscul Disord. (2011) ;21: (7):477–82. |
[82] | Messinger YH , Mendelsohn NJ , Rhead W , Dimmock D , Hershkovitz E , Champion M , et al. Successful immune toleranceinduction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genet Med. (2012) ;14: (1):135–42. |
[83] | Banugaria SG , Patel TT , Mackey J , Das S , Amalfitano A , Rosenberg AS , et al. Persistence of high sustainedantibodies to enzyme replacement therapy despite extensive immunomodulatory therapy in an infant with Pompedisease: Need for agents to target antibody-secreting plasma cells. Mol Genet Metab. (2012) ;105: (4):677–80. |
[84] | Regnery C , Kornblum C , Hanisch F , Vielhaber S , Strigl-Pill N , Grunert B , et al. 36 months observational clinicalstudy of 38 adult Pompe disease patients under alglucosidase alfa enzyme replacement therapy. J Inherit Metab Dis. (2012) ;35: (5):837–45. |
[85] | De Vries JM , Van Der Beek NA , Hop WC , Karstens FP , Wokke JH , De Visser M , et al. Effect of enzyme therapy andprognostic factors in 69 adults with Pompe disease: An open-label single-center study. Orphanet J Rare Dis. (2012) ;7: (1):73. |
[86] | Van Rijt WJ , Heiner-Fokkema MR , Du Marchie Sarvaas GJ , Waterham HR , Blokpoel RGT , Van Spronsen FJ , et al. Favorable outcome after physiologic dose of sodium-D,L-3-hydroxybutyrate in severe MADD. Pediatrics. (2014) ;134: (4):e1224–8. |
[87] | Chien YH , Lee NC , Chen CA , Tsai FJ , Tsai WH , Shieh JY , et al. Long-term prognosis of patients withinfantile-onset pompe disease diagnosed by newborn screening and treated since birth. J Pediatr. (2015) ;166: (4):985–991.e2. |
[88] | Yang CF , Yang CC , Liao HC , Huang LY , Chiang CC , Ho HC , et al. Very Early Treatment for Infantile-Onset PompeDisease Contributes to Better Outcomes. J Pediatr. (2016) ;169: (PG-174-80.e1):174–180.e1. |
[89] | van Gelder CM , Poelman E , Plug I , Hoogeveen-Westerveld M , van der Beek NAME , Reuser AJJ , et al. Effects of ahigher dose of alglucosidase alfa on ventilator-free survival and motor outcome in classic infantile Pompedisease: An open-label single-center study. J Inherit Metab Dis. (2016) ;39: (3):383–90. |
[90] | Kazi ZB , Prater SN , Kobori JA , Viskochil D , Bailey C , Gera R , et al. Durable and sustained immune tolerance toERT in Pompe disease with entrenched immune responses. JCI Insight. (2016) ;1: (11). |
[91] | Byrne BJ , Geberhiwot T , Barshop BA , Barohn R , Hughes D , Bratkovic D , et al. A study on the safety and efficacy ofreveglucosidase alfa in patients with late-onset Pompe disease. Orphanet J Rare Dis. (2017) ;12: (1):144. |
[92] | Papadopoulos C , Orlikowski D , Prigent H , Lacour A , Tard C , Furby A , et al. Effect of enzyme replacement therapywith alglucosidase alfa (Myozyme®) in 12 patients with advanced late-onset Pompe disease. Mol Genet Metab. (2017) ;122: (1-2):80–5. |
[93] | Gillingham MB , Heitner SB , Martin J , Rose S , Goldstein A , El-Gharbawy AH , et al. Triheptanoin versus trioctanoinfor long-chain fatty acid oxidation disorders: A double blinded, randomized controlled trial. J Inherit Metab Dis. (2017) ;40: (6):831–43. |
[94] | Kuperus E , Kruijshaar ME , Wens SCA , De Vries JM , Favejee MM , Van Der Meijden JC , et al. Long-term benefit ofenzyme replacement therapy in Pompe disease: A 5-year prospective study. Neurology. (2017) ;89: (23):2365–73. |
[95] | van Capelle CI , Poelman E , Frohn-Mulder IM , Koopman LP , van den Hout JMP , Régal L , et al. Cardiac outcome inclassic infantile Pompe disease after 13-years of treatment with recombinant human acid alpha-glucosidase. Int JCardiol. (2018) ;269: (PG-104-110):104–10. |
[96] | Kazi ZB , Desai AK , Troxler RB , Kronn D , Packman S , Sabbadini M , et al. An immune tolerance approach usingtransient low-dose methotrexate in the ERT-naïve setting of patients treated with a therapeutic protein:Experience in infantile-onset Pompe disease. Genet Med. (2019) ;21: (4):887–95. |
[97] | Elder ME , Nayak S , Collins SW , Lawson LA , Kelley JS , Herzog RW , et al. B-Cell depletion and immunomodulationbefore initiation of enzyme replacement therapy blocks the immune response to acid alpha-glucosidase ininfantile-onset pompe disease. J Pediatr. (2013) ;163: (3):847–54.e1. |
[98] | Furusawa Y , Mori-Yoshimura M , Yamamoto T , Sakamoto C , Wakita M , Kobayashi Y , et al. Effects of enzyme replacementtherapy on five patients with advanced late-onset glycogen storage disease type II: A 2-year follow-up study. JInherit Metab Dis. (2012) ;35: (2):301–10. |
[99] | Matsuoka T , Miwa Y , Tajika M , Sawada M , Fujimaki K , Soga T , et al. Divergent clinical outcomes ofalpha-glucosidase enzyme replacement therapy in two siblings with infantile-onset Pompe disease treated in thesymptomatic or pre-symptomatic state. Mol Genet Metab Reports. (2016) ;9: (PG-98-105):98–105. |
[100] | Angelini C , Semplicini C , Ravaglia S , Bembi B , Servidei S , Pegoraro E , et al. Observational clinical study injuvenile-adult glycogenosis type 2 patients undergoing enzyme replacement therapy for up to 4 years. J Neurol. (2012) ;259: (5):952–8. |
[101] | van Gelder C , Poelman E , Plug I , Hoogeveen-Westerveld M , Kuperus E , van der Beek N , et al. A higher dose ofalglucosidase alpha in classic infantile Pompe disease positively affects ventilator-free survival and motoroutcome: An open-label single-center study. Neuromuscul Disord. (2015) ;25: (PG-S189-S190):S189–90. |
[102] | Banugaria SG , Prater SN , McGann JK , Feldman JD , Tannenbaum JA , Bailey C , et al. Bortezomib in the rapid reductionof high sustained antibody titers in disorders treated with therapeutic protein: Lessons learned from Pompedisease. Genet Med. (2013) ;15: (2):123–31. |
[103] | Yamada K , Shiraishi H , Oki E , Ishige M , Fukao T , Hamada Y , et al. Open-label clinical trial of bezafibratetreatment in patients with fatty acid oxidation disorders in Japan. Mol Genet Metab Reports. (2018) ;15: (PG-55-63):55–63. |
[104] | Watanabe K , Yamada K , Sameshima K , Yamaguchi S . Two siblings with very long-chain acyl-CoA dehydrogenase (VLCAD)deficiency suffered from rhabdomyolysis after L-carnitine supplementation. Mol Genet Metab Reports. (2018) ;15: (PG-121-123):121–3. |
[105] | Shiraishi H , Yamada K , Oki E , Ishige M , Fukao T , Hamada Y , et al. Open-label clinical trial of bezafibratetreatment in patients with fatty acid oxidation disorders in Japan; 2nd report QOL survey. Mol Genet Metab Reports. (2019) ;20: (PG-100496):100496. |
[106] | Therrell BL , Lloyd-Puryear MA , Camp KM , Mann MY . Inborn errors of metabolism identified via newborn screening: Ten-year incidence data and costs of nutritional interventions for research agenda planning. Vol. 113, Molecular Genetics and Metabolism. Academic Press Inc.; 201. pp. 14-26. |
[107] | Pennisi E , Garibaldi M , Antonini G . Lipid Myopathies. J Clin Med. (2018) Nov 23 [cited 2020 Nov 24];7: (12):472. |
[108] | Schoser B , Stewart A , Kanters S , Hamed A , Jansen J , Chan K , et al Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: A systematic review and meta-analysis. Vol. 264, Journal of Neurology. Dr. Dietrich Steinkopff Verlag GmbH and Co. KG; 2017. pp. 621-30. |
[109] | Kemper AR , Hwu WL , Lloyd-Puryear M , Kishnani PS . Newborn screening for pompe disease: Synthesis of the evidenceand development of screening recommendations. Pediatrics. (2007) ;120: (5):e1327–34. |
[110] | Kronn DF , Day-Salvatore D , Hwu WL , Jones SA , Nakamura K , Okuyama T , et al. Management of confirmednewborn-screened patients with pompe disease across the disease spectrum. Pediatrics. (2017) ;140: (Supplement1):1098–4275. |
[111] | Rairikar M V , CaseLE, BaileyLA, KaziZB, DesaiAK, BerrierKL, et al. Insight into the phenotype of infantswith Pompe disease identified by newborn screening with the common c.-32-13T >G “late-onset” GAA variant. MolGenet Metab. (2017) ;122: (3):99–107. |
[112] | Chien YH , Lee NC , Thurberg BL , Chiang SC , Zhang XK , Keutzer J , et al. Pompe disease in infants: Improving theprognosis by newborn screening and early treatment. Pediatrics. (2009) ;124: (6). |
[113] | Chien YH , Lee NC , Chen CA , Tsai FJ , Tsai WH , Shieh JY , et al. Long-term prognosis of patients withinfantile-onset pompe disease diagnosed by newborn screening and treated since birth. J Pediatr. (2015) ;166: (4):985–991.e2. |
[114] | Banugaria SG , Prater SN , Patel TT , DeArmey SM , Milleson C , Sheets KB , et al. Algorithm for the Early Diagnosisand Treatment of Patients with Cross Reactive Immunologic Material-Negative Classic Infantile Pompe Disease: AStep towards Improving the Efficacy of ERT. PLoS One. (2013) ;8: (6). |
[115] | Kazi ZB , Desai AK , Troxler RB , Kronn D , Packman S , Sabbadini M , et al. An immune tolerance approach usingtransient low-dose methotrexate in the ERT-naïve setting of patients treated with a therapeutic protein:Experience in infantile-onset Pompe disease. Genet Med. (2019) ;21: (4):887–95. |
[116] | van der Ploeg AT , Kruijshaar ME , Toscano A , Laforêt P , Angelini C , Lachmann RH , et al. European consensus forstarting and stopping enzyme replacement therapy in adult patients with Pompe disease: A 10-year experience. EurJ Neurol. (2017) ;24: (6):768-e3. |
[117] | Quinlivan RM , Beynon RJ . Pharmacological and nutritional treatment trials in McArdle disease. Acta Myol. (2007) ;26: (1):58–60. |
[118] | Vorgerd M , Grehl T , Jäger M , Müller K , Freitag G , Patzold T , et al. Creatine therapy in myophosphorylasedeficiency (McArdle disease): A placebo-controlled crossover trial. Arch Neurol. (2000) ;57: (7):956–63. |
[119] | Vorgerd M , Zange J , Kley R , Grehl T , Hüsing A , Jäger M , et al. Effect of high-dose creatine therapy onsymptoms of exercise intolerance in McArdle disease: Double-blind, placebo-controlled crossover study. ArchNeurol. (2002) ;59: (1):97–101. |
[120] | Haller RG , Vissing J . Spontaneous “second wind” and glucose-induced second “second wind” in McArdle diseaseoxidative mechanisms. Arch Neurol. (2002) ;59: (9):1395–402. |
[121] | Gillingham MB , Heitner SB , Martin J , Rose S , Goldstein A , El-Gharbawy AH , et al. Triheptanoin versus trioctanoinfor long-chain fatty acid oxidation disorders: A double blinded, randomized controlled trial. J Inherit Metab Dis. (2017) ;40: (6):831–43. |
[122] | Martinuzzi A , Sartori E , Fanin M , Nascimbeni A , Valente L , Angelini C , et al. Phenotype modulators inmyophosphorylase deficiency. Ann Neurol. (2003) ;53: (4):497–502. |
[123] | van der Ploeg AT , Clemens PR , Corzo D , Escolar DM , Florence J , Groeneveld GJ , et al. A Randomized Study ofAlglucosidase Alfa in Late-Onset Pompe’s Disease. N Engl J Med. (2010) ;362: (15):1396–406. |
[124] | Forsha D , Li JS , Smith PB , Van Der Ploeg AT , Kishnani P , Pasquali SK . Cardiovascular abnormalities in late-onsetPompe disease and response to enzyme replacement therapy. Genet Med. (2011) ;13: (7):625–31. |
[125] | Ørngreen MC , Madsen KL , Preisler N , Andersen G , Vissing J , Laforêt P . Bezafibrate in skeletal muscle fattyacid oxidation disorders: A randomized clinical trial. Neurology. (2014) ;82: (7):607–13. |
[126] | Drug Approval Package: Myozyme (Alglucosidase Alfa) NDA #125141. [cited 2020 Nov 24]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/125141s000_MyozymeTOC.cfm |
[127] | Hahn SH , Kronn D , Leslie ND , Pena LDM , Tanpaiboon P , Gambello MJ , et al. Efficacy, safety profile, andimmunogenicity of alglucosidase alfa produced at the 4,000-liter scale in US children and adolescents with Pompedisease: ADVANCE, a phase IV, open-label, prospective study. Genet Med [Internet]. (2018) ;20: (10):1284–94. |
[128] | Kishnani PS , Corzo D , Leslie ND , Gruskin D , Van Der Ploeg A , Clancy JP , et al. Early treatment with alglucosidasealfa prolongs long-term survival of infants with pompe disease. Pediatr Res. (2009) ;66: (3):329–35. |
[129] | Chen M , Zhang L , Quan S . Enzyme replacement therapy for infantile-onset Pompe disease. Vol. 2017, Cochrane Database of Systematic Reviews. John Wiley and Sons Ltd; 2017. |
[130] | Pena LDM , Barohn RJ , Byrne BJ , Desnuelle C , Goker-Alpan O , Ladha S , et al. Safety, tolerability,pharmacokinetics, pharmacodynamics, and exploratory efficacy of the novel enzyme replacement therapyavalglucosidase alfa (neoGAA) in treatment-naïve and alglucosidase alfa-treated patients with late-onsetPompe disease: A phase 1, o. Neuromuscul Disord. (2019) ;29: (3):167–86. |
[131] | Kishnani PS , Corzo D , Nicolino M , Byrne B , Mandel H , Hwu WL , et al. Recombinant human acidα-glucosidase: Major clinical benefits in infantile-onset Pompe disease. Neurology. (2007) ;68: (2):99–109. |
[132] | Van der Ploeg AT , Barohn R , Carlson L , Charrow J , Clemens PR , Hopkin RJ , et al. Open-label extension studyfollowing the Late-Onset Treatment Study (LOTS) of alglucosidase alfa. Mol Genet Metab. (2012) ;107: (3):456–61. |
[133] | Case LE , Bjartmar C , Morgan C , Casey R , Charrow J , Clancy JP , et al. Safety and efficacy of alternativealglucosidase alfa regimens in Pompe disease. Neuromuscul Disord. (2015) ;25: (4):321–32. |
[134] | Rairikar M , Kazi ZB , Desai A , Walters C , Rosenberg A , Kishnani PS . High dose IVIG successfully reduces high rhGAAIgG antibody titers in a CRIM-negative infantile Pompe disease patient. Mol Genet Metab. (2017) ;122: (1-2):76–9. |
[135] | Birch KE , Quinlivan RM , Morris GE . Cell models for McArdle disease and aminoglycoside-induced read-through of apremature termination codon. Neuromuscul Disord. (2013) ;23: (1):43–51. |
[136] | Quinlivan R , Vissing J , Hilton-Jones D , Buckley J . Physical training for McArdle disease. Cochrane Database Syst Rev. (2011) ;(12). |
[137] | Haller RG , Wyrick P , Taivassalo T , Vissing J . Aerobic conditioning: An effective therapy in McArdle’s disease. Ann Neurol. (2006) ;59: (6):922–8. |
[138] | Maté-Muñoz JL , Moran M , Pérez M , Chamorro-Viña C , Gómez-Gallego F , Santiago C , et al. Favorable responses to acute and chronic exercise in McArdle patients. Clin J Sport Med. (2007) ;17: (4):297–303. |
[139] | Pérez M , Foster C , González-Freire M , Arenas J , Lucia A . One-Year Follow-Up in a Child With McArdleDisease: Exercise is Medicine. Pediatr Neurol. (2008) ;38: (2):133–6. |
[140] | O’Reilly DSJ . The effect of oral sucrose on exercise tolerance in patients with McArdle’s disease. Ann ClinBiochem. (2004) ;41: (Pt 3):255. |
[141] | Andersen ST , Vissing J . Carbohydrate- and protein-rich diets in McArdle disease: Effects on exercise capacity. J Neurol Neurosurg Psychiatry. (2008) ;79: (12):1359–63. |
[142] | Andersen ST , Haller RG , Vissing J . Effect of oral sucrose shortly before exercise on work capacity in McArdle disease. Arch Neurol. (2008) ;65: (6):786–9. |
[143] | Quinlivan R , Martinuzzi A , Schoser B . Pharmacological and nutritional treatment for McArdle disease (Glycogen Storage Disease type V). Vol. 2014, Cochrane Database of Systematic Reviews. John Wiley and Sons Ltd; 2014. |
[144] | Madsen KL , Laforêt P , Buch AE , Stemmerik MG , Ottolenghi C , Hatem SN , et al. No effect of triheptanoin onexercise performance in McArdle disease. Ann Clin Transl Neurol. (2019) ;6: (10):1949–60. |
[145] | Arnold GL , Van Hove J , Freedenberg D , Strauss A , Longo N , Burton B , et al. A Delphi clinical practice protocolfor the management of very long chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab. (2009) ;96: (3):85–90. |
[146] | Primassin S , Ter Veld F , Mayatepek E , Spiekerkoetter U . Carnitine supplementation induces acylcarnitineproduction in tissues of very long-chain Acyl-CoA dehydrogenase-deficient mice, without replenishing low freecarnitine. Pediatr Res. (2008) ;63: (6):632–7. |
[147] | Watanabe K , Yamada K , Sameshima K , Yamaguchi S . Two siblings with very long-chain acyl-CoA dehydrogenase (VLCAD)deficiency suffered from rhabdomyolysis after L-carnitine supplementation. Mol Genet Metab Reports. (2018) ;15: :121–3. |
[148] | Spiekerkoetter U , Lindner M , Santer R , Grotzke M , Baumgartner MR , Boehles H , et al. Treatment recommendations inlong-chain fatty acid oxidation defects: Consensus from a workshop. J Inherit Metab Dis. (2009) ;32: (4):498–505. |
[149] | Yamada K , Shiraishi H , Oki E , Ishige M , Fukao T , Hamada Y , et al. Open-label clinical trial of bezafibratetreatment in patients with fatty acid oxidation disorders in Japan. Mol Genet Metab Reports. (2018) ;15: :55–63. |


