
A Canadian Adult Spinal Muscular Atrophy Outcome Measures Toolkit: Results of a National Consensus using a Modified Delphi Method
Abstract
Background:
Spinal Muscular Atrophy (SMA) is a rare disease that affects 1 in 11 000 live births. Recent developments in SMA treatments have included new disease-modifying therapies that require high quality data to inform decisions around initiation and continuation of therapy. In Canada, there are no nationally agreed upon outcome measures (OM) used in adult SMA. Standardization of OM is essential to obtain high quality data that is comparable among neuromuscular clinics.
Objective:
To develop a recommended toolkit and timing of OM for assessment of adults with SMA.
Methods:
A modified delphi method consisting of 2 virtual voting rounds followed by a virtual conference was utilized with a panel of expert clinicians treating adult SMA across Canada.
Results:
A consensus-derived toolkit of 8 OM was developed across three domains of function, with an additional 3 optional measures. Optimal assessment frequency is 12 months for most patients regardless of therapeutic access, while patients in their first year of receiving disease-modifying therapy should be assessed more frequently.
Conclusions:
The implementation of the consensus-derived OM toolkit will improve monitoring and assessment of adult SMA patients, and enrich the quality of real-world evidence. Regular updates to the toolkit must be considered as new evidence becomes available.
INTRODUCTION
Spinal Muscular Atrophy (SMA) is a recessive neuromuscular genetic disorder which affects 1 in 11 000 live births and has a prevalence of approximately 1-2 per 100,000 persons [1, 2]. There are at least 33 causative genes identified for conditions with SMA manifestations, with the most common causative gene being the SMN1 gene located on the long arm of chromosome 5, which produces a variety of SMA known as 5q SMA [3]. 5q SMA encompasses 92%of all SMA, of which 95%have a homozygous deletion of SMN1 resulting in a significant decrease in the amount of functional SMN protein [2, 4]. 5q SMA disease severity is modulated by the number of copies of SMN2, a low-functioning homologue of SMN1 which produces a lesser amount of functional SMN protein [2, 4]. The number of copies of SMN2 naturally varies within the general population and is inversely related to disease severity of 5q SMA [2–4].
Recently, there have been significant advancements in SMA treatments which utilize therapeutic mechanisms such as SMN1 gene replacement via viral vectors [5], synthetic olignoucleotides and small molecules which encourage production of SMN1 protein via altered SMN2 splicing [6, 7], neuroprotection, and enhancement of nerve and/or muscle function [4, 8]. These therapeutic advances have widened the spectrum of clinical presentation and will lead to a higher disease prevalence due to longer life expectancies [6, 9]. Despite progress in treatments, there is a paucity of data on the natural history, prevalence, and progression patterns of SMA, particularly in adults [10].
Given the changing therapeutic environment of SMA and the crucial need for high quality real-world data, routine use of a standardized toolkit of nationally agreed upon outcome measures (OM) will advance our ability to elucidate important patterns of disease trajectory and response to therapy, enabling a deeper understanding of both the disease and the effectiveness of emerging treatments [11]. Furthermore, the creation of a standard consensus-derived toolkit will support the development of best practices. Due to the heterogenous nature of disease progression and multi-system involvement, this toolkit shall include a diverse set of OM which capture, but are not limited to, patient reported outcome measures (PROM), and domains of gross motor function, fine motor function, quality of life, and respiratory function. McGraw et al. and Sansone et al. both advocate for the inclusion of OM, which assess a wide range of activities and capture meaningful clinical changes [12, 13]. To date, few efforts have been made to derive consensus on which OM should be used to assess adult SMA. The purpose of this study was to derive a national consensus on a set of recommended OM and frequency of assessment for adult SMA.
MATERIALS AND METHODS
A modified delphi method was chosen as the process to achieve a national consensus on OM. The delphi method is an anonymized serial survey methodology to elicit expert opinion in an iterative voting process to distill judgements into a consensus [14, 15]. The delphi method has been widely used in healthcare for making decisions with conflicting evidence, poor availability of evidence, or when judgemental information is necessary [15–18]. The study participants (the study authors with exception to JS, VH, and JL) were clinical leads or directors from clinics treating adults with SMA in Canada, and members of the Canadian Neuromuscular Disease Network. Participants were encouraged to discuss the OM project with their teams throughout the process. The delphi process began in June 2019 and was completed in April 2020. This study modified the delphi method by including a final virtual conference following the survey-based voting rounds, during which all participants could discuss and interpret the results of the voting rounds (Fig. 1) and achieve a final consensus.
Fig. 1
Flowchart of modified delphi methodology.
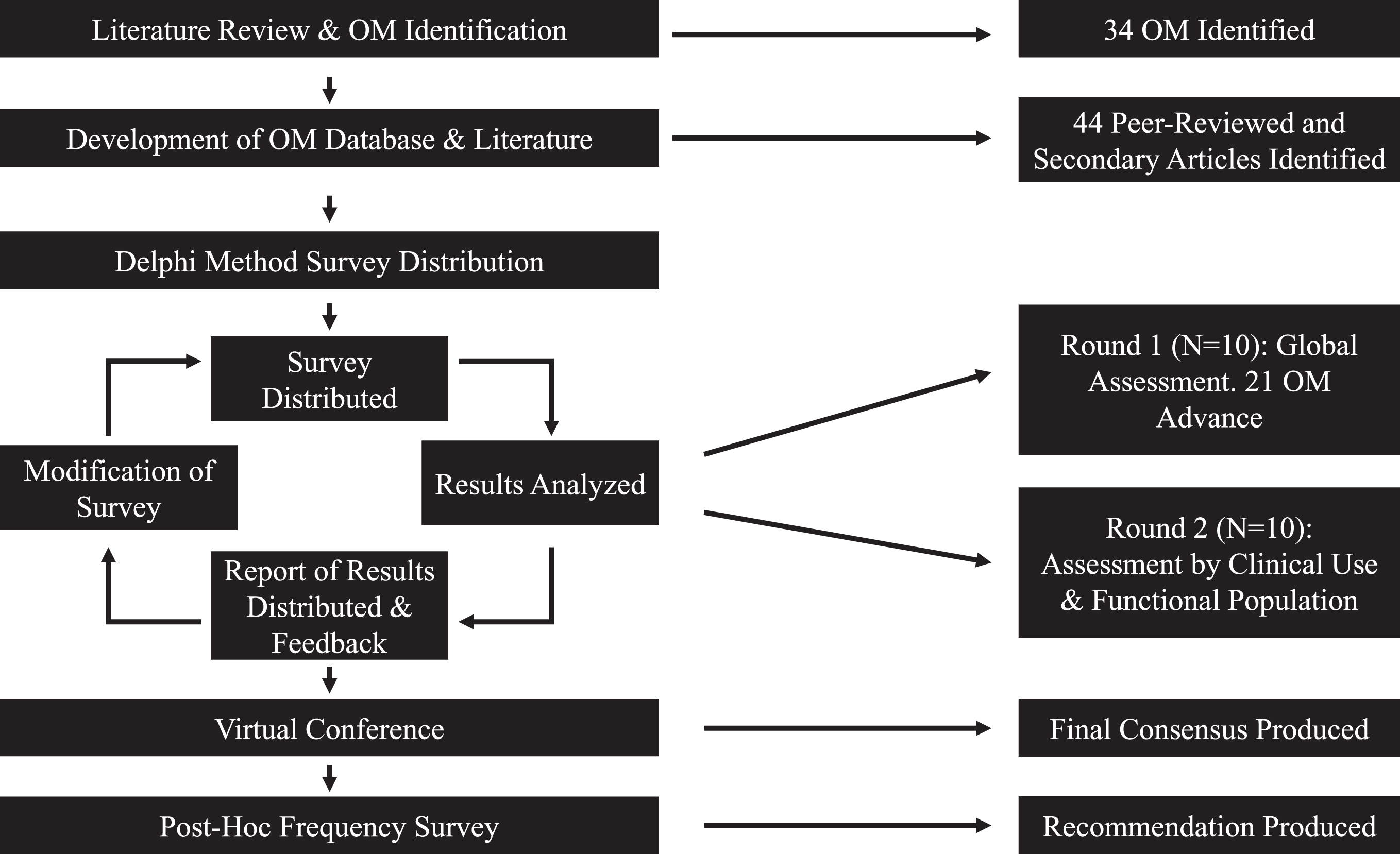
In order to effectively vote on appropriateness of outcome measures, an initial table was developed to provide information and background on validity and reliability of currently available OM. Firstly, a literature review of OM in SMA was conducted, and a questionnaire sent to the participants requesting input on OM for consideration. Following the literature review and participant feedback, a comprehensive table of all potential OM was generated. The OM table included available evidence of validity, reliability, clinical application and domain (e.g. adult/paediatric, gross motor/fine motor), and any additional information (such as time to completion, resources required, and references) for each of the OM (Supplementary Table 1). The voting surveys were developed and delivered through both google forms (first voting round) [19] and SurveyMonkey (second voting round) [20], and were distributed through the Canadian Neuromuscular Disease Registry (CNDR) Network [11].
The first voting round asked the participant to assess the quality and the clinical utility of each of the OM. Participants were provided with and instructed to refer to the comprehensive table of OM, including the embedded links to available publications and OM instruction manuals. This round served to integrate all of the clinical expertise of the participants into a global rating. The survey used a 5-point likert scale from ‘very weak’ (1) to ‘very strong’ (5). The first voting round was available for 8 weeks, after which the results were analyzed. The OM were grouped based on cut point values of the mean and lower bounds of the standard deviation. A cut point of a mean score ≥3 (neither weak nor strong) was determined for inclusion in the next round of voting. A report was produced (Supplementary Report 1) outlining the results of the voting, which was then distributed to the participants prior to the next voting round.
The second round of voting rated the remaining OM across stages of disease, and their frequency of use. Participants were instructed to consider the quality and utility of the measures, and not necessarily the OM currently used in their practice. The OM were assessed for each of 3 functional populations of patients (non-sitters, sitters, and walkers). The frequency of use ratings identified which OM were used in practice, and thus could more easily be implemented if included in the toolkit. The response options were also expanded to allow respondents who were unfamiliar with a particular outcome measure to opt out of that question. This modification allowed for an informed ranking of OM by limiting the responses to participants familiar with the OM in question. The second round of voting was available for 10 weeks, after which results were analyzed and a second report was compiled and distributed to the participants (Supplementary Report 2). Once all participants had reviewed the report, a virtual conference was held with the participants to discuss the results of both rounds and to generate a final consensus-derived toolkit of recommended OM. This last step allowed for participants to discuss the merits and/or barriers of the selected measures, and included agreement to re-visit recommendations in 1-2 years, as new evidence becomes available.
As a secondary initiative that evolved from the virtual conference, the participants requested a post-hoc survey to determine the optimal frequency of clinical assessment of OM, in part due to a range of provincial regulatory criteria tied to reimbursement and drug approvals. The post-hoc survey aimed to describe clinically appropriate assessment timing and frequency of each of the recommended OMs while considering both treated (i.e –on a disease-modifying medication) and untreated (i.e –natural history) populations. Additionally, the post-hoc survey aimed to gather baseline information on interprovincial variance of assessment frequency practice.
RESULTS
A total of 34 OM were identified through literature review and questionnaire of Canadian SMA adult clinicians. From these 34 OM, 44 peer-reviewed articles and supporting literature were identified which provided supplementary information for the OM included in the voting process (Supplementary Table 1).
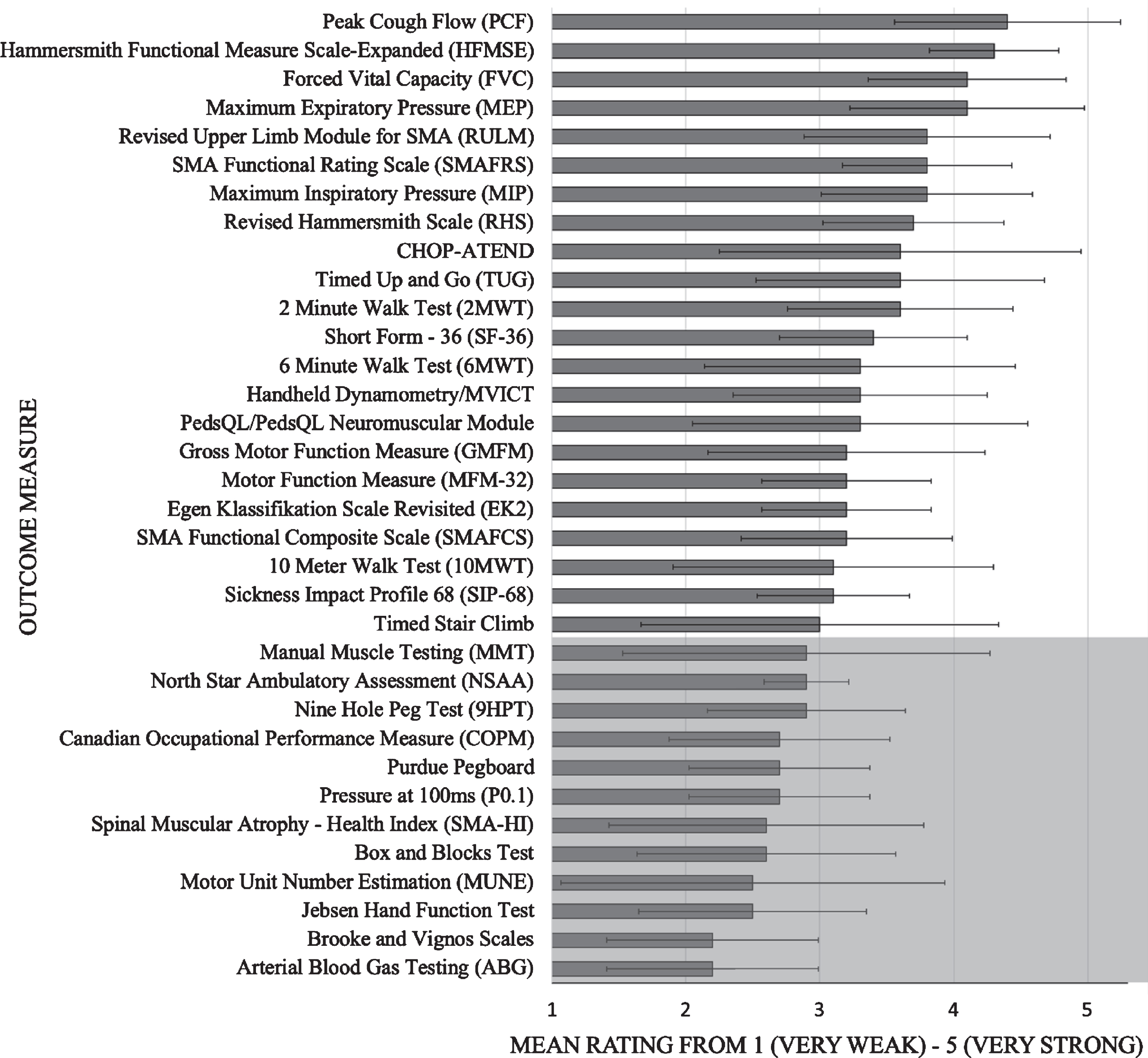
Health care providers from ten clinics treating adults with SMA (neurologists, physiatrists, physiotherapists) across Canada participated in each of the first two rounds of online voting. After the first round, the cut point (mean score ≥3) narrowed the number of OM from 34 to 21 (Table 1; Fig. 2). The results from the second round of voting for each of the functional populations are outlined in Table 1. The most frequently used OM were Forced Vital Capacity (FVC), Peak Cough Flow (PCF), Maximum inspiratory and expiratory pressures (MIP and MEP), Hammersmith Functional Motor Scale –Expanded (HFMSE), Revised Upper Limb Module (RULM), 6 Minute Walk Test (6MWT), and Timed Up and Go Test (TUG). Other assessed measures in the second round were used only sporadically.
Table 1
Round 1 and 2 results of OM which were included in 2nd voting round. The mean is scored on a scale of 1-5 with 5 (very strong), 3 (neither weak nor strong), and 1 (very weak)
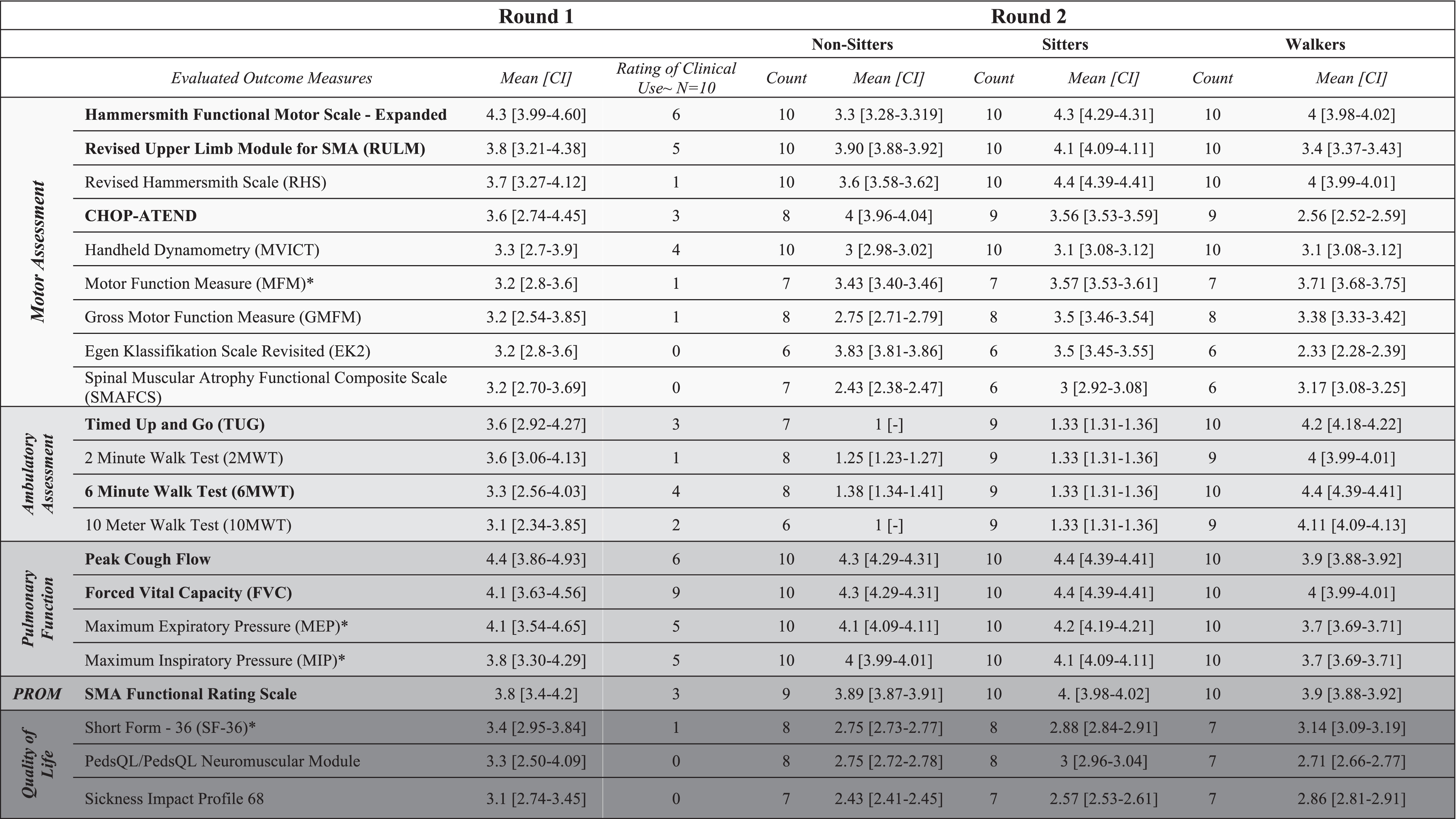 |
*Included for future considerations; ∼Combined count of Always and Often responses; Bolded OM are those included in the final consensus; Shading indicates type of OM. CHOP-ATEND: Childrens Hospital of Philadelphia – Adult Test of Neuromuscular Disorders.
Fig. 2
Mean ratings of outcome measures from round 1. Grey shading indicates outcome measures below cut point of 3 “Neither weak nor strong”. Error bars indicate standard deviation.
During the final virtual conference, consensus was achieved to recommend the routine use of 8 OM across each of the functional populations (Table 2). The final 8 OM have administration time estimates between 3 minutes (TUG), and 30–40 minutes (HFMSE) with a total estimated time required to administer the OM being between 70 and 85 minutes for each of the three populations. Although the SF-36, Motor Function Measure (MFM), and MIP/MEP were not included in the final consensus group, they were recommended as optional as the participants determined them to be highly important and with suggestion to re-consider within a 2 year period as evidence and clinical needs evolve.
Table 2
Outcome measures selected in the final consensus from the modified delphi method
| Non-sitters (85 Minutes)* | Sitters (85 Minutes)* | Walkers (70 Minutes)* | |
| Pulmonary function | FVC | FVC | FVC |
| PCF | PCF | PCF | |
| Motor | RULM | RULM | HFMSE |
| CHOP-ATEND | HFMSE | ||
| Functional | SMAFRS | SMAFRS | SMAFRS |
| TUG | |||
| 6MWT | |||
| Optional OM | SF-36 | ||
| MFM | |||
| MIP | |||
| MEP |
*Total estimated time required to complete all OM within the population, excluding optional outcome measures. FVC: Forced Vital Capacity, PCF: Peak Cough Flow, RULM: Revised Upper Limb Module for SMA, CHOP-ATEND: Children’s Hospital of Philadelphia –Adult Test of Neuromuscular Disorders, HFMSE: Hammersmith Functional Motor Scale –Expanded, SMAFRS: Spinal Muscular Atrophy Functional Rating Scale, TUG: Timed Up and Go, 6MWT: Six Minute Walk Test, SF-36: Short Form –36, MFM: Motor Function Measure, MIP: Maximum Inspiratory Pressure, MEP: Maximum Expiratory Pressure.
The post-hoc frequency survey was completed by 9 clinics across Canada (Ontario, Alberta, New Brunswick, Quebec, Saskatchewan and British Columbia). The survey identified that adult SMA patients should be evaluated on a yearly basis, while patients receiving disease-modifying therapy should be assessed every 6 to 12 months. All OM included in the final consensus are best assessed on a yearly basis, with exceptions to the FVC and PCF which both had a mixed response of a 6 to 12 month frequency (Table 3). Additionally, the post-hoc frequency survey identified two major barriers for clinicians in conducting outcome assessments: 1. Availability of and access to allied health professionals and 2. lack of available clinical time.
Table 3
Optimal frequencies of assessment for patient population and OM included in the consensus-derived toolkit
| Patient population | Frequency of assessment |
| All patients regardless of treatment | 12 Months |
| Patients receiving disease-modifying treatment: 1 year post-therapy initiation | 6–12 Months |
| Patient receiving disease-modifying treatment: 2+ years post-therapy initiation | 6–12 Months |
| Outcome measures | |
| Outcome measure | Frequency of assessment |
| HFMSE | 12 Months |
| CHOP-ATEND | 12 Months |
| RULM | 12 Months |
| TUG | 12 Months |
| 6MWT | 12 Months |
| SMAFRS | 12 Months |
| FVC | 6–12 Months |
| PCF | 6–12 Months |
HFMSE: Hammersmith Functional Motor Scale –Expanded, CHOP-ATEND: Children’s Hospital of Philadelphia –Adult Test of Neuromuscular Disorders, RULM: Revised Upper Limb Module for SMA, TUG: Timed Up and Go, 6MWT: Six Minute Walk Test, SMAFRS: Spinal Muscular Atrophy Functional Rating Scale, FVC: Forced Vital Capacity, PCF: Peak Cough Flow.
DISCUSSION
In rare diseases such as SMA, the use of several different OM nationally and internationally creates challenges in monitoring disease progression, ability to pool data, or assessing therapeutic response and resource utilization. This study provides a consensus-derived toolkit developed via a modified delphi method with national experts who treat adults with SMA. The national OM toolkit can lead to improved monitoring and larger, robust datasets which are of great importance in the adult SMA population [1, 2]. Operationalizing a national toolkit with a patient registry such as the Canadian Neuromuscular Disease Registry (CNDR) [11] provides a valuable data resource for clinicians and researchers to facilitate further insight into SMA [13].
The final OM are specific to each of the three functional populations and were chosen with careful consideration to their practicality, feasibility, supporting evidence, domain of function, and clinical importance. Among the 8 OM included in the final consensus, there are 5 functional measures (3 gross and/or fine motor, 2 ambulation), 2 respiratory measures, and 1 PROM. Each of the included OM take less than 30 minutes to conduct, with a total estimated administration time of less than two hours.
The SF-36 was included in the optional group of OM and recommended for future consideration, as there is a limited pool of OM which assess quality of life in adult SMA population. Additional quality of life measures for SMA should be developed to better capture these needs [12, 21]. The MFM was felt by the participants to be comparable to the HFMSE but was excluded from the final consensus due to a lack of familiarity or experience with the measure, but participants recognize the evolving field. The MFM is currently widely used in Europe [22], and has been the primary motor measure selected for some clinical trial programs [23]. Over time, additional evidence for the MFM will be generated and continued monitoring of the MFM is important, with future reconsideration in the toolkit. The MIP and MEP were included as optional OM as they were considered by the participants to have high intra-individual variability due to reliance on patient effort [24]. The intra-individual variability and the availability of more reliable alternatives such as the FVC and PCF, led the participants to select FVC and PCF as the recommended respiratory measures.
The results of the present study are fundamentally in accordance with recent research, which have largely included the same OM as those in the present consensus. Sansone et al. recently recommended OM in adult SMA based upon expert opinion of international neuromuscular experts [13] and their results align with the findings of the present study, with differences only in the selection of the SMAFRS and the TUG. Similarly, Hagenacker et al., Walter et al. and Maggi et al. studied the effects of nusinersen (an SMN2-targeted anti-sense oligonucleotide [6]) in an adult SMA population and all utilized the HFMSE, RULM, and 6MWT [25–27] in evaluation of therapy effectiveness and safety. In 2016, Mercuri et al. selected the HFMSE as the primary outcome measure when reporting upon the natural progression of SMA in children and adults [28]. In a natural history study by Chabanon et al. the MFM, Pulmonary Function Tests (including FVC, PCF, MIP and MEP), and the 6MWT were included among additional OM [22]. A 2019 report by the Institut National d’Excellence en Santé et en Services Sociaux (INESSS) in Québec, recommended the CHOP-ATEND, HFMSE, RULM, 6MWT and the SMAFRS as adult SMA OM [29]. The SF-36 was chosen as an optional measure over the PedsQL 4.0 in the present study, although neither OM is particularly well suited to the adult SMA population.
The final consensus-derived toolkit of recommended OM is not fully comprehensive, and there remain recognized gaps in the assessment of adult SMA. Consistent with the literature, the participants in our study indicated poor availability of OM that assess important domains of quality of life, bulbar function (speech and swallowing), fine motor skills, and endurance (particularly in non-sitter and sitter populations) [4, 10, 12, 25, 30]. Capturing additional domains is important to further increase sensitivity to detect changes and to better assess other aspects of life [12]. For example, Sansone et al. described the importance of finger and hand strength, while Kizina et al. and Binz et al. examined the presence of fatigue [13, 31, 32]. Bulbar function was previously identified by Rouault et al. as a priority for stabilization for persons with SMA II and III [30]. Additional domains such as bone health, metabolic effects and participation reflect larger aspects of a persons health. In addition to standardized OM, PROM are integral in capturing individual needs and goals and can provide unique person-focused data [13, 33]. Development of additional standardized OM and PROM is needed to capture the breadth of changes and systemic nature of the disease [12].
Within Canada, at the time of publication, provincial drug reimbursement requirements ranged from assessments every 4 to 12 months, or after every injected drug dose, with varying OM province to province [34]. The post-hoc frequency survey identified an evaluation frequency of one year for adults with SMA, with those receiving disease-modifying therapy being assessed every 6 to 12 months. This recommendation is in keeping with the consensus findings of Hodgkinson et al. which suggested visits should occur at 6 months, and annually thereafter [34]. Advocating for a unified assessement recommendation that is based on clinical expertise and outcome measure sensitivity is a needed step towards providing evidence-based care.
There are a number of barriers to routine use of OM in the clinic setting; some OM require specific training or specialized equipment, certain measures are time consuming and resource heavy to administer (patients may need to be transferred with a lift), others may be burdensome or fatiguing for the patient to complete. Such factors can lead to variable time commitment for OM completion, as well as poor compliance with OM administration. An efficient system for performing OM depends on having the necessary support for required resources: equipment, time, space and health professionals. Further, poor sensitivity to change coupled with floor and ceiling effects may limit the utility of any particular outcome measure. Developing OM which are easily administered, sensitive and reliable can greatly facilitate their uptake [13, 35]. Alternatively, introducing additional means to provide access to allied health professionals and increased clinical time is also important. The finding of limited access to appropriate health professionals and clinic time underscores the importance of interdisciplinary and interspecialty care in the treatment of adult SMA, and need for valid and reliable PROM and OM which do not require intensive use of clinical resources. Another consideration is the timing of outcome collection. Due to the slow progression of adult SMA, too frequent evaluations may be burdensome and yield little change; data collection schedules informed by long-term real-world studies could aid in optimizing data quality.
Due to the evolving nature of the field, it is possible that some standardized OM may have been missed. While this study did not formally correct for the potential bias of multiple respondants within a province, all CNDR adult SMA sites were engaged throughout the process, representing a majority of clinical adult SMA expertise in Canada. It is possible that factors such as trial participation experience, resource availability, and regulatory criteria could have influenced individual OM ratings despite instructions to rate the OM based on clinical experience, review of the literature, and the evidence available, rather than strictly current practice or regulations. Lastly, this study did not separate OM selections into patients with 5q SMA, and those with non-5q SMA; despite this limitation, 5q SMA represents the vast majority of all SMA patients (96%) [2], and clinicians with non-5q SMA patients may require additional or different OM.
The implementation of this toolkit as standard practice in SMA clinics will help capture standard real-world evidence and allow a deeper understanding of both the natural history of adult SMA and the effectiveness of emerging treatments. While this Canadian toolkit is largely in agreement with existing studies (mostly from Europe), an international consensus study could greatly benefit alignment of patient assessments globally, facilitating data pooling through partners such as the TREAT-NMD Neuromuscular Network. Furthermore, an international consensus would support advocacy efforts for improved availability of clinical services.
This work represents a important first step in achieving an accessible, standardized assessment for adults living with SMA. As a living document, this toolkit will be revisited and refined, with incorporation of SMA-specific measures as they evolve, and much needed measures to capture what is meaningful for persons with lived experience.
ACKNOWLEDGMENTS
The authors would like to thank the Dalhousie University Faculty of Medicine, and the New Brunswick Health Research Foundation (NBHRF) for funding support. The authors would also like to thank the CNDR for their assistance in facilitating this work.
CONFLICTS OF INTEREST
Victoria Hodgkinson reports personal fees from Biogen and Hoffman-La Roche. Bernard Brais contributes to teaching sessions organized by Biogen, and participates in medical consultant meetings with Biogen and Hoffman-La Roche. Kristine Chapman serves on the Expensive Drugs for Rare Diseases Working Group. Angela Genge reports grants from Biogen; and consulting fees from AL-S Pharma, Alexion, Quralis, Cytokinetics, Mitsubishi-Tanabe, and Hoffman-La Roche. Aaron Izenberg reports personal fees Biogen, Mitsubishi-Tanabe, Hoffman-La Roche, Takeda, Akcea, Alnylam, Genzyme. Wendy Johnston reports personal fees from Biogen and Mitsubishi-Tanabe. Erin O’Farrell reports personal fees from Biogen, and grants from Sanofi Genzyme, Acceleron, and Grifols. Xavier Rodrigue reports personal fees from Hoffman-La Roche and Biogen. Christen Shoesmith serves on advisory boards for Mitsubishi-Tanabe and Orion Pharmaceuticals and is also a primary investigator for a clinical trial sponsored by Cytokinetics; a family member is employed by Hoffman-La Roche. Monique Taillon reports serving on Advisory Boards for Biogen, Hoffman-La Roche and Novartis. Lawrence Korngut reports grants and personal fees from Biogen and Hoffman-La Roche; and personal fees from Novartis. Colleen O’Connell reports grants from the New Brunswick Health Research Foundation (NBHRF) and personal fees from Hoffman-La Roche.
Jeremy Slayter, Josh Lounsberry, Hanns Lochmüller, Gerald Pfeffer, Stephanie Plamondon, Kerri Schellenberg, Christine Stables and Jodi Warman-Chardon have nothing to disclose.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-200617.
REFERENCES
[1] | Sugarman EA , Nagan N , Zhu H , Akmaev VR , Zhou Z , Rohlfs EM , et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: Clinical laboratory analysis of > 72,400 specimens. Eur J Hum Genet. (2012) ;20: :27–32. doi: 10.1038/ejhg.2011.134. |
[2] | Verhaart IEC , Robertson A , Wilson IJ , Aartsma-Rus A , Cameron S , Jones CC , et al. Prevalence, incidence and carrier frequency of 5q–linked spinal muscular atrophy –a literature review. Orphanet J Rare Dis (2017) ;12: :124. doi: 10.1186/s13023-017-0671-8 |
[3] | Farrar MA , Kiernan MC . The Genetics of Spinal Muscular Atrophy: Progress and Challenges. Neurotherapeutics; New York. (2015) ;12: :290–302. doi: 10.1007/s13311-014-0314-x |
[4] | Farrar MA , Park SB , Vucic S , Carey KA , Turner BJ , Gillingwater TH , et al. Emerging therapies and challenges in spinal muscular atrophy. Annals of Neurology. (2017) ;81: :355–68. doi: 10.1002/ana.24864 |
[5] | Mendell JR , Al-Zaidy S , Shell R , Arnold WD , Rodino-Klapac LR , Prior TW , et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. New England Journal of Medicine. (1713) ;377: :13–22. doi: 10.1056/NEJMoa1706198 |
[6] | Mercuri E , Darras BT , Chiriboga CA , Day JW , Campbell C , Connolly AM , et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. The New England Journal of Medicine; Boston. (2018) ;378: :625–35. doi: 10.1056/NEJMoa1710504 |
[7] | Poirier A , Weetall M , Heinig K , Bucheli F , Schoenlein K , Alsenz J , et al. Risdiplam distributes and increases SMN protein in both the central nervous system and peripheral organs. Pharmacology Research & Perspectives. (2018) ;6: (6):e00447. doi: 10.1002/prp2.447 |
[8] | Messina S , Sframeli M . New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges. J Clin Med. (2020) ;9: . doi: 10.3390/jcm9072222 |
[9] | Strauss KA , Farrar MA , Swoboda KJ , Saito K , Chiriboga CA , Finkel RS , et al. Onasemnogene Abeparvovec-xioi Gene-Replacement Therapy in Presymptomatic Spinal Muscular Atrophy: SPR1NT Study Update (2384). Neurology. (2020) ;94: :2384. |
[10] | Mercuri E , Sansone V . Nusinersen in adults with spinal muscular atrophy: New challenges. The Lancet Neurology. (2020) ;19: :283–4. doi: 10.1016/S1474-4422(20)30068-5 |
[11] | Hodgkinson VL , Oskoui M , Lounsberry J , et al. A National Spinal Muscular Atrophy Registry for Real-World Evidence. Can J Neurol Sci. (2020) ;47: (6):810–815. doi: 10.1017/cjn.2020.111 |
[12] | McGraw S , Qian Y , Henne J , Jarecki J , Hobby K , Yeh W-S . A qualitative study of perceptions of meaningful change in spinal muscular atrophy. BMC Neurol. (2017) ;17: :68. doi: 10.1186/s12883-017-0853-y |
[13] | Sansone VA , Walter MC , Attarian S , Delstanche S , Mercuri E , Lochmüller H , et al. Measuring Outcomes in Adults with Spinal Muscular Atrophy - Challenges and Future Directions –Meeting Report. Journal of Neuromuscular Diseases. 2020:1-12. doi: 10.3233/JND-200534 |
[14] | Skulmoski GJ , Hartman FT , Krahn J . The Delphi Method for Graduate Research. Journal of Information Technology Education: Research. (2007) ;6: :1–21. |
[15] | Steurer J . The Delphi method: An efficient procedure to generate knowledge. Skeletal Radiol. (2011) ;40: :959–61. doi: 10.1007/s00256-011-1145-z |
[16] | Tsiamis E , Millar J , Baxi S , Borg M , De Ieso P , Elsaleh H , et al. Development of quality indicators to monitor radiotherapy care for men with prostate cancer: A modified Delphi method. Radiotherapy and Oncology. (2018) ;128: :308–14. doi: 10.1016/j.radonc.2018.04.017 |
[17] | Evered L , Silbert B , Knopman DS , Scott DA , DeKosky ST , Rasmussen LS , et al. Recommendations for the Nomenclature of Cognitive Change Associated with Anaesthesia and Surgery—2018. Journal of Alzheimer’s Disease. (2018) ;66: :1–10. doi: 10.3233/JAD-189004 |
[18] | Eubank BH , Mohtadi NG , Lafave MR , Wiley JP , Bois AJ , Boorman RS , et al. Using the modified Delphi method to establish clinical consensus for the diagnosis and treatment of patients with rotator cuff pathology. BMC Med Res Methodol. (2016) ;16: :56. doi: 10.1186/s12874-016-0165-8 |
[19] | Google LLC. Google Forms: Free Online Surveys for Personal Use. Mountain View, California, United States. |
[20] | SurveyMonkey Inc. SurveyMonkey: The World’s Most Popular Free Online Survey Tool. https://www.surveymonkey.com/; San Mateo, California, USA |
[21] | Mercuri E , Mazzone E . Choosing the right clinical outcome measure: From the patient to the statistician and back. Neuromuscular Disorders. (2011) ;21: :16–9. doi: 10.1016/j.nmd.2010.09.003 |
[22] | Chabanon A , Seferian AM , Daron A , Péréon Y , Cances C , Vuillerot C , et al. Prospective and longitudinal natural history study of patients with Type 2 and 3 spinal muscular atrophy: Baseline data NatHis-SMA study. PLoS ONE. (2018) ;13: :e0201004. doi: 10.1371/journal.pone.0201004 |
[23] | Hoffmann-La Roche. A Two Part Seamless, Multi-Center Randomized, Placebo-Controlled, Double-Blind Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of RO7034067 in Type 2 and 3 Spinal Muscular Atrophy Patients. clinicaltrials.gov; 2020. |
[24] | Caruso P , de Albuquerque ALP , Santana PV , Cardenas LZ , Ferreira JG , Prina E , et al. Diagnostic methods to assess inspiratory and expiratory muscle strength. J Bras Pneumol. (2015) ;41: :110–23. doi: 10.1590/S1806-37132015000004474 |
[25] | Hagenacker T , Wurster CD , Günther R , Schreiber-Katz O , Osmanovic A , Petri S , et al. Nusinersen in adults with 5q spinal muscular atrophy: A non-interventional, multicentre, observational cohort study. The Lancet Neurology. (2020) ;19: :317–25. doi: 10.1016/S1474-4422(20)30037-5 |
[26] | Walter MC , Wenninger S , Thiele S , Stauber J , Hiebeler M , Greckl E , et al. Safety and Treatment Effects of Nusinersen in Longstanding Adult 5q-SMA Type 3 - A Prospective Observational Study. J Neuromuscul Dis. (2019) ;6: (4):453–65. doi: 10.3233/JND-190416 |
[27] | Maggi L , Bello L , Bonanno S , Govoni A , Caponnetto C , Passamano L , et al. Nusinersen safety and effects on motor function in adult spinal muscular atrophy type 2 and 3. J Neurol Neurosurg Psychiatry. (1166) ;91: (11):74. doi: 10.1136/jnnp-2020-323822 |
[28] | Mercuri E , Finkel R , Montes J , Mazzone ES , Sormani MP , Main M , et al. Patterns of disease progression in type 2 and 3 SMA: Implications for clinical trials. Neuromuscul Disord. (2016) ;26: :126–31. doi: 10.1016/j.nmd.2015.10.006 |
[29] | Recommendations concerning the information required to monitor nusinersen use in real-world settings. Quebec: L’Institut national d’excellence en sante et en service sociaux (INESSS); 2020. |
[30] | Rouault F , Christie-Brown V , Broekgaarden R , Gusset N , Henderson D , Marczuk P , et al. Disease impact on general well-being and therapeutic expectations of European Type II and Type III spinal muscular atrophy patients. Neuromuscul Disord. (2017) ;27: :428–38. doi: 10.1016/j.nmd.2017.01.018 |
[31] | Kizina K , Stolte B , Totzeck A , Bolz S , Schlag M , Ose C , et al. Fatigue in adults with spinal muscular atrophy under treatment with nusinersen. Scientific Reports. (2020) ;10: :11069. doi: 10.1038/s41598-020-68051-w |
[32] | Binz C , Schreiber-Katz O , Kumpe M , Ranxha G , Siegler H , Wieselmann G , et al. An observational cohort study on impact, dimensions and outcome of perceived fatigue in adult 5q-spinal muscular atrophy patients receiving nusinersen treatment. J Neurol. 2020 Oct 7. doi: 10.1007/s00415-020-10227-5 |
[33] | Ciafaloni E , Sansone V . Outcome measures in adults living with SMA: Standardization versus personalization 2020. |
[34] | Hodgkinson VL , Chapman K , Izenberg A , Lochmüller H , O’Connell C , O’Ferrall EK , et al. Response to provincial governments’ decisions regarding monitoring for adults with Spinal Muscular Atrophy. Canadian Journal of Neurological Sciences undefined/ed:1-10. doi: 10.1017/cjn.2020.161 |
[35] | Montes J , Gordon AM , Pandya S , De Vivo DC , Kaufmann P . Clinical Outcome Measures in Spinal Muscular Atrophy. Journal of Child Neurology. (2009) ;24: :968–78. doi: 10.1177/0883073809332702 |


