
Burden of Spinal Muscular Atrophy (SMA) on Patients and Caregivers in Canada
Abstract
Background:
Spinal muscular atrophy (SMA) is a rare neurodegenerative disease characterized by progressive muscular weakness, which occurs in one in 6,000 to 10,000 live births. The burden of SMA on Canadian patients and caregivers is not known.
Objective:
To characterize the burden of SMA in Canada as reported by patients and caregivers, including disease and treatment impacts, indirect costs, and caregiver burden.
Methods:
Surveys were distributed by Cure SMA Canada and Muscular Dystrophy Canada to individuals with SMA and their caregivers. The online surveys were anonymous and completed between January 28 and February 21, 2020.
Results:
965 patient and 962 caregiver responses met the eligibility criteria. Patients reported SMA subtypes as: type I (25.0%), type II (41.3%), type III (29.3%). Using the EQ-5D, patients were shown to have impaired quality of life with an average health utility index of 0.49 (SD: 0.26). The median expenditure was $4,500 CAD (IQR: $1,587 – $11,000) for assistive devices; $6,800 CAD (IQR: $3,900–$13,000) on health professional services; and $1,200 CAD (IQR: $600 –$3,100) on SMA-related travel and accommodation in the past 12 months. Caregivers reported needing respite care (45.7%), physiotherapy for an injury from a lift/transfer (45.7%), or other health impacts (63.3%). Caregivers reported changes to personal plans, sleep disturbances, and work adjustments, with a mean Caregiver Strain Index score of 7.5 [SD: 3.3].
Conclusion:
SMA in Canada is associated with a significant burden for patients and their caregivers.
INTRODUCTION
SMA is the most common fatal autosomal recessive disorder in childhood and the second most commonly acquired autosomal recessive disorder in children after cystic fibrosis [1, 2]. Worldwide, the estimated incidence of SMA ranges from one in 6,000 to one in 10,000 live births [2–5]. In Canada, the actual number of SMA cases still remains unknown, but it is estimated there are 700 to 2,140 cases of SMA based on various epidemiological data sources [6–8], with approximately 35 new cases per year, and 21 (60%) of those new cases being SMA type I [3, 9]. The Government of Canada, in collaboration with Novartis Canada, has also recently developed a National Newborn Screening in SMA program [10], and the Canadian Neuromuscular Disease Registry is also working to expand its SMA dataset [11] to better evaluate the epidemiology of SMA. Although the number of cases of SMA in Canada is quite small, the healthcare-related burden is substantial.
In Canada, approximately 70% of healthcare expenditures are covered by the public health system (e.g., government funding), with the remainder of expenses (30%) being covered by private health plans (e.g., employment insurance) or as an out-of-pocket expense for patients [12, 13]. Each provincial and territorial government administers and delivers services covered by the public health system (e.g., hospital and physician services) without any form of direct payment from the patient [14]. However, supplementary benefits such as prescription drug coverage, dental, or vision care are not covered by the health system, and are instead covered by private health plans or paid as an out-of-pocket expense [14, 15]. The federal government does provides benefits (e.g., tax credits) to individuals living with a disability or those caring for an individual with a disability [14, 16] such as SMA. However, there are specifications regulating the amount of expenses or the type of equipment/supports that can be claimed [16], and individuals or their caregivers would still initially be paying out-of-pocket for these services, which would be a significant financial burden. As noted by the Canadian Institute for Health Information, the average out-of-pocket health expenditure in 2017 for Canadians was $973 per person [17].
SMA is associated with substantial direct costs (e.g., the financial value associated with disease-specific utilization of medical and non-medical resources, such as hospital or physician visits, medication, rehabilitation, home modifications), informal care costs (e.g., time spent caregiving by relatives or unpaid caregivers), and indirect costs (e.g., loss of productivity to society due to absence from work, reduced productivity, or premature death) [18]. It is anticipated that these direct, informal, and indirect costs [18] result in much higher healthcare expenditures for individuals with SMA, and caregivers of individuals with SMA, compared to the expenditures for average Canadians. However, there are currently no published estimates of the direct and indirect costs associated with SMA in Canada.
In terms of the current treatment landscape, there have been recent breakthroughs for treating SMA, including nusinersen (Spinraza® Biogen), onasemnogene abeparvovec (Zolgensma® Novartis Gene Therapies), and risdiplam (Evrysdi® Roche). Spinraza was approved by Health Canada in June 2017 [19], however the Canadian Agency for Drugs and Technologies in Health (CADTH) initially recommended several restrictions for nusinersen access, while the Quebec-based Institut national d’excellence en santé et en services sociaux (INESSS) did not recommend access restrictions [20]. CADTH revised its decision for nusinersen in March 2019 by recommending reimbursement for a broader range of SMA diagnoses, but only for individuals 12 years of age or younger and those who have never achieved the ability to walk independently [21]. However, access to nusinersen for individuals with SMA in Canada varies between each province and territory. The treatment landscape in Canada will continue to evolve with onasemnogene abeparvovec very recently receiving approval from Health Canada in December 2020 [22], while risdiplam is currently under review with Health Canada [23].
The economic and societal costs of SMA, for both patients and caregivers, have been well documented in several studies outside of Canada, including Australia [24], the Netherlands [25], Germany [18], Spain [26], and the Czech Republic [27], which demonstrate the impacts of SMA on quality of life for patients and caregivers, caregiver burden, and indirect costs. Patient- and caregiver-reported outcomes are increasingly recognized as vital to our understanding of the effectiveness of SMA-related healthcare services and treatments. While the impact of SMA on patients and caregivers has been described in a handful of international studies, no such study has been conducted in Canada, where the healthcare and social security systems are different from other countries. The objective of this study was to characterize the burden of SMA in Canada among persons’ with SMA and their caregivers. As new disease-modifying therapies are emerging from clinical trials, it is imperative for Canadian insurance providers, thi-rd-party payers, governments, regulatory bodies, and society as a whole to have a better understanding of the burden of illness of SMA.
MATERIALS AND METHODS
Study population and design
Individuals with SMA and their caregivers living in Canada were surveyed. A caregiver was defined as someone with a personal relationship, such as a parent, spouse, partner, or adult relative who provides unpaid care for an individual with SMA. Two patient support groups (Cure SMA Canada and Muscular Dystrophy Canada) invited individuals with SMA and caregivers of individuals with SMA from their membership lists, via e-mail, to take part in this study. As the survey was anonymous, it was possible that individuals from the mailing list may have circulated the survey link to others who were not on the list (e.g., through Facebook). Surveys were completed from January 28, 2020 to February 21, 2020. At the start of the survey, participants were asked to complete a series of questions to self-assess their eligibility and to direct them to the correct questionnaire (e.g., patient questionnaire, self-completed or proxy-completed, versus caregiver questionnaire). Participants were asked the following screening questions: 1) if they were an individual with SMA or an unpaid caregiver of an individual with SMA; 2) if they had lived in Canada for the previous 12 months and; 3) if they were over the age of majority (≥18 year or ≥19 years old, depending on province of residence) at the time of survey completion. Respondents who had not resided in Canada for the previous 12 months were excluded, as were respondents who indicated they were not an individual with SMA or an unpaid caregiver of someone with SMA. Respondents that indicated they were an individual with SMA were taken to a different survey than individuals who indicated that they were an unpaid caregiver of a person with SMA. Respondents who noted that they were under the age of majority or that they needed help completing the survey were redirected to the proxy-completed survey. Individuals with SMA (or their proxy) were asked in the survey if the individual had been confirmed by genetic testing to have SMA, but proof of their genetic diagnosis of 5q-linked SMA was not required for participation. The caregiver questionnaire did not ask about confirmatory genetic testing.
Upon completion of the survey, additional exclusion criteria included: 1) respondents that were identified as potential “bots” or “ballot box stuffers” or; 2) patient survey respondents that indicated that they did not have genetic confirmation of 5q-linked SMA. Bots were defined as a software program that can complete routine tasks such as completing questionnaires. Bot identification was established based on methodology embedded in the Qualtrics© survey software (Qualtrics, Provo, UT). Using Google’s reCAPTCHA v3, Qualtrics rated every response on the probability that the respondent was a bot based on respondent interactions with the site using values ranging from 0 to 1 [28]. Potential bots are defined as responses that scored less than 0.5, which is the default threshold recommended by Google [28]. Ballot box stuffers were defined as a respondent who submitted more than one completed questionnaire. Duplicate, or ballot box stuffing, responses were also identified based on embedded Qualtrics software methodology. Qualtrics software places a cookie in the browser during the first survey session opened by a respondent. If the same respondent returns to the same browser and device without clearing any cookies, this subsequent response will be flagged as a duplicate, indicating potential ballot box stuffing [28].
The survey was based on a questionnaire developed for a study in the UK [29] and modified to the Canadian context, including adding two validated instruments: the EQ-5D-5L and Caregiver Strain Index (CSI). The study protocol was reviewed by clinical advisors for clinical relevance. The patient survey collected information on demographics, clinical characteristics, impact of SMA on education and employment, SMA-related healthcare resource utilization, SMA-related treatments, adaptations to living environment, average travel and accommodation costs for SMA-related appointments and treatments, costs of services and assistive devices, and quality of life. The patient survey was either self-completed by adult individuals with SMA or proxy-completed by caregivers of children or adults with SMA who needed assistance.
The caregiver survey collected information on demographics of the caregiver and individual with SMA in their care, as well as caregiver-related burden (e.g., number of hours caring for the person with SMA, travel to SMA-related appointments, impacts on caregiver sleep and employment, and quality of life). The caregiver survey was self-completed by adult caregivers of an individual with SMA.
In both the patient and caregiver surveys, quality of life was assessed using the EQ-5D-5L and associated visual analogue scale (VAS). The EQ-5D-5L includes five dimensions (mobility, self-care, usual activities, pain/discomfort, and anxiety/depression) and each dimension has five levels of responses (e.g., no problems, slight problems, moderate problems, severe problems, and extreme problems). The EQ-5D-5L VAS asks the respondent to rate their perceived health from 0 (the worst imaginable health) to 100 (the best imaginable health) [30]. The health utility index score can range from 0 (the value of a health state equivalent to dead) to 1 (the value of full health), with higher scores indicating higher quality of life; the health utility index was calculated using a published time trade-off-derived value set for Canada [31]. Caregiver strain was assessed using the CSI, a 13-item tool that includes questions about areas of life caregivers may find difficult (e.g., sleep) and requires a “Yes” or “No” response [32]. The CSI is scored from 0–13; the higher the number of “Yes” answers, the higher the strain on the caregiver, with a score equal to or greater than 7 indicating high stress and burden [32]. The CSI (Robinson BC, Validation of a Caregiver Strain Index, Journal of Gerontology, 1983;38(3):344–8) was used with permission by Oxford University Press.
The surveys were designed using Qualtrics© software (Qualtrics, Provo, UT). The surveys were available in both English and French (Canada). Upon completion of the survey, participants were invited to enter their email address to receive an e-gift card for Amazon.ca of $50 CAD value. The submission of an email address for gift card receipt was not linked to the patient or caregiver surveys.
This study was approved by the Health Research Ethics Board of Alberta Community Health Committee (HREBA-CHC) for the province of Alberta, and the Advarra Institutional Review Board for the rest of Canada (excluding Newfoundland and Labrador).
Statistical analysis
For the patient surveys, data from both patient-completed and proxy-completed surveys were combined. The patient survey results reported below excluded records identified as potential bots, ballot box stuffing, or responses where 5q-deletion was not confirmed by genetic testing as self-reported by the respondent. The caregiver survey results reported below excluded records identified as potential bots or ballot box stuffing.
Analyses were conducted using SAS 9.4 (SAS Institute, Cary, NC). Descriptive statistics were calculated for the responses received for each question. For continuous variables, descriptive statistics of central tendency (medians and means) and dispersion (standard deviation [SD], interquartile range [IQR]) were calculated. Categorical variables were described with frequencies and percentages. Non-responses were excluded per question from the analyses. In some cases, non-responses were driven by survey branch logic. In other cases, non-responses reflected respondents having omitted a response to a particular question that was presented. Results were stratified by self-reported SMA type (I, II, III, or other), based upon the diagnosis that the patient or their caregiver had communicated to them by their clinician.
Given the unexpected responses to the questions related to medication use in the patient survey, we conducted a post-hoc sensitivity analysis further res-tricting the respondents to a subgroup that indicated nusinersen use only or no use of any of the three novel therapeutics (nusinersen, risdiplam, or onasemnogene abeparvovec), since, at the time of study, only nusinersen had marketing authorization and was more widely available in Canada. We reported demographic characteristics, SMA-related cost estimates, and quality of life variables in this subgroup for comparison (see Supplementary Materials).
RESULTS
Overall, 3,077 surveys were received in Qualtrics. Empty records containing no data (N = 339) and surveys missing a respondent categorization such as patient, proxy, or caregiver (N = 21) were excluded. Of the 1,467 patient surveys completed by individuals with SMA or their adult proxy, we excluded 502 surveys that were identified as potential bots or ballot box stuffers (N = 257) or because respondents stated that they did not have genetically-confirmed 5q-linked SMA (N = 245). Surveys completed by individuals with SMA or their adult proxy will hereafter be referred to as “patient surveys”. Therefore, a total of 965 patient surveys met the study inclusion criteria. Of the 1,250 completed surveys received from unpaid caregivers, we excluded those that met the criteria for potential bots or ballot box stuffers (N = 288), resulting in 962 caregiver surveys that met inclusion criteria for the study (Fig. 1)
Fig. 1
Patient and caregiver survey respondent inclusion flow chart. Legend: Refer to Methods section for a more detailed description of bots and ballot box stuffers and the technique used to identify and exclude these surveys.

Patient survey
Demographics
One quarter of patient survey respondents (241/965) reported having SMA type I, while 41.3% (399/965) reported having SMA type II, and 29.3% (283/965) reported having SMA type III. The median age of all patients with SMA completing the survey was 8.50 years (IQR: 2.75–23.50), increasing with SMA type. Overall, 59.8% were male and 40.2% were female. The median age of initial symptom onset was 2.17 years (IQR: 0.83–8.25) while the median age at diagnosis was 2.67 years (IQR 1.00–10.50). Demographics, including age of symptom onset and age of diagnosis, by SMA type are reported in Table 1.
Table 1
Patient demographics: Overall and by self-reported SMA type
| Characteristics | Patient or adult proxy Respondents | Type I | Type II | Type III | Other |
| (N = 965) | (n = 241) | (n = 399) | (n = 283) | (n = 42) | |
| Age in years at time of survey response, n = 934 | |||||
| Mean (SD) | 13.70 (12.45) | 7.19 (11.67) | 12.38 (11.56) | 19.20 (10.55) | 28.74 (9.09) |
| Median (Q1 –Q3) | 8.50 (2.75–23.50) | 1.58 (1.25–7.00) | 7.50 (3.75–20.75) | 18.67 (10.75–26.42) | 27.67 (23.08–34.33) |
| Age in years at time of survey response, n (%) | |||||
| 0 – <6 months | <5 | <5 | 0 | 0 | 0 |
| 6 months – <18 months | 104 (11.1%) | 100 (42.0%) | <5 | 0 | 0 |
| 18 months – <18 years | 512 (54.8%) | 104 (43.7%) | 273 (70.5%) | 132 (48.7%) | <5 |
| ≥18 years | 317 (33.9%) | 33 (13.9%) | 110 (28.4%) | 139 (51.3%) | 35 (92.1%) |
| Non-respondents | 31 | <5 | 12 | 12 | <5 |
| Sex, n (%) | |||||
| Female | 387 (40.2%) | 115 (47.9%) | 154 (38.6%) | 105 (37.2%) | 13 (31.0%) |
| Male | 576 (59.8%) | 125 (52.1%) | 245 (61.4%) | 177 (62.8%) | 29 (69.0%) |
| Non-respondents | <5 | <5 | 0 | <5 | 0 |
| Age at first onset of symptoms, in years, n = 883 | |||||
| Mean (SD) | 5.95 (8.20) | 2.75 (7.03) | 3.93 (6.86) | 9.25 (6.40) | 23.58 (10.21) |
| Median (Q1 –Q3) | 2.17 (0.83–8.25) | 0.50 (0.25–1.17) | 1.25 (0.92–3.58) | 8.25 (4.33–12.50) | 22.83 (18.75–30.75) |
| Age at diagnosis, in years, n = 883 | |||||
| Mean (SD) | 6.57 (8.48) | 2.99 (7.13) | 4.49 (7.27) | 10.22 (6.54) | 24.16 (10.03) |
| Median (Q1 –Q3) | 2.67 (1.00–10.50) | 0.50 (0.42–1.42) | 1.42 (1.08–4.33) | 9.92 (4.75–13.92) | 23.21 (19.50–31.50) |
Note: All numbers are years or years old. Cells with fewer than 5 responses are masked using < 5 for privacy standards. SD = standard deviation; SMA = spinal muscular atrophy; Q1 = first quartile; Q3 = third quartile.
Among the 223 patient survey respondents to whom the questions pertaining to the impact of SMA on school were relevant, patients reported receiving a median of 32 hours (IQR: 24–36) of educational support per week; the most commonly reported spe-cialized support received by these patients were specialized seating (72.1%), wheelchair access (69.9%), and specialized desks (64.2%). Patients who responded to questions regarding the number of school days missed (n = 226) reported missing a median of 8 days (IQR: 4–15) of school for SMA-related reasons in the past 12 months.
Individuals with SMA may experience barriers to employment. Of employed patient survey respondents, 95 reported taking a median of 9 sick days per year (IQR: 7–18) due to SMA. Similarly, 100 patients noted their SMA was associated with a change in work responsibilities, where 49.0% (49/100) had reduced their working hours and 34.0% (34/100) had to take extra unpaid leave off work. Only 15.0% (15/100) reported no changes in this regard. Fatigue has been found to be particularly prevalent for individuals with SMA [33], although the cause of work schedule adjustment was not captured in our survey. Of the 90% of patient survey respondents that did not answer work-related questions, it is not known if they were too young (<18 years old), attending post-secondary school and/or not working due to disease progression.
Healthcare resource use
Almost all individuals with SMA (96.8%, 906/936) reported visiting an outpatient clinic at least once in the past year, with type II and III being associated with higher utilization of outpatient services compared to type I (data not shown). For patients with SMA type II and III, 81.2% reported more than one outpatient visit in the past year. This was in contrast to SMA type I, where 65.1% (149/229) reported more than one outpatient visit in the past 12 months. Overall, 76.8% of patients with SMA (717/934) had ≥1 hospital visit in the past year, and most patients visited a hospital once (35.7%, 333/934) or twice (26.0%, 243/934) within the past year. Patients with SMA type I reported visiting a hospital more frequently than those with type II and type III: 62.6% (144/230) of patients with type I visited a hospital two or more times in the past year, compared to 37.8% (147/389) with type II and 26.2% (72/275) with type III. While nearly half of the patient survey respondents overall had not visited an emergency department in the past year (41.7%, 388/931), most patients with SMA type I (90.4%, 208/230) had visited an emergency department in the past year, compared to 51.2% (199/389) with type II and 36.9% (101/273) with type III (data not shown).
When asked whether they had been prescribed an SMA-related treatment in the past 12 months, a higher-than-expected number and percentage of patients reported having been prescribed several medications including: nusinersen 40.8% (379/930); risdiplam 33.5% (312/930); onasemnogene abeparvovec 32.2% (299/930) and/or olesoxime 18.3% (170/930). As a larger number of patients than anticipated reported having been prescribed one or more of these medications, we did not further analyze these results (data not shown); see Discussion for further details.
Costs
All patient or caregiver expenditures are listed in Canadian dollars (CAD). As shown in Table 2, patients (n = 347) reported a median spend of $12,800 (IQR: $6,800–$20,000) on home modifications ever; patients with SMA type II and type III reported a higher median spend compared to those with type I. Patient survey respondents (n = 915) reported a median spend of $4,500 (IQR: $1,587–$11,000) on assistive devices in the past year, and a median personal, out-of-pocket spend on allied health professional services related to SMA in the past year of $6,800 (IQR: $3,900–$13,000, n = 919). Overall, 917 patients reported a median spend of $1,200 (IQR: $600–$3,100) on travel and accommodation relating to SMA-appointments in the past year, and the costs were higher for patients with SMA type I compared to type II and type III.
Table 2
Self-reported SMA costs: Overall and by self-reported SMA type
| Characteristics | Patient or adult proxy respondents | Type I | Type II | Type III | Other |
| (N = 965) | (n = 241) | (n = 399) | (n = 283) | (n = 42) | |
| Estimated total spent on assistive devices for SMA (any kind) in the last year, CAD$, n = 915 | |||||
| Mean (SD) | 13,397 | 8,555 | 14,011 | 16,360 | 14,422 |
| (25,031) | (15,760) | (28,413) | (26,453) | (19,771) | |
| Median (Q1 –Q3) | 4,500 | 3,000 | 4,785 | 3,225 | 6,000 |
| (1,587–11,000) | (1,542–6,250) | (2,000–13,400) | (1,300–18,500) | (2,000–18,000) | |
| Estimated total cost of home modifications, CAD$, n = 347 | |||||
| Mean (SD) | 19,570 | 9,617 | 26,195 | 18,927 | 16,658 |
| (26,830) | (17,145) | (35,397) | (19,129) | (14,803) | |
| Median (Q1–Q3) | 12,800 | 6,100 | 15,000 | 15,000 | 15,000 |
| (6,800–20,000) | (2,900–7,700) | (9,000–25,000) | (10,000–18,500) | (4,500–20,000) | |
| Estimated cost spent personally on travel and accommodation relating to SMA | |||||
| appointments in the last year (12 months), CAD$, n = 917 | |||||
| Mean (SD) | 4,164 | 3,731 | 4,533 | 3,813 | 5,495 |
| (9,991) | (7,035) | (11,187) | (10,369) | (9,612) | |
| Median (Q1–Q3) | 1,200 | 2,105 | 1,000 | 1,000 | 1,500 |
| (600–3,100) | (1,000–3,300) | (500–2,620) | (500–3,900) | (700–6,500) | |
| Estimated personal, out-of-pocket expenses for SMA-related, allied health | |||||
| professional services in the past 12 months, CAD$, n = 919 | |||||
| Mean (SD) | 14,665 | 12,972 | 14,067 | 14,103 | 33,240 |
| (34,844) | (53,865) | (23,249) | (19,965) | (58,466) | |
| Median (Q1 –Q3) | 6,800 | 6,961 | 7,500 | 5,800 | 8,000 |
| (3,900–13,000) | (5,000–9,000) | (4,000–16,000) | (3,500–10,000) | (2,000–32,000) | |
Note: All numbers reflect Canadian dollars ($ CAD). SD = standard deviation; SMA = spinal muscular atrophy; Q1 = first quartile; Q3 = third quartile.
Quality of life (EQ-5D-5L)
Patient quality of life was assessed using the EQ-5D-5L, shown in Fig. 2 (additional data is available in Table S-1). Quality of life was lower among patients with SMA type I, as indicated by worse scores on the mobility, washing and dressing, usual activities, pain or discomfort, and anxiety or depression domains of the EQ-5D-5L. Among patients with SMA type I, more than half reported they were unable to walk about; unable to wash or dress themselves; unable to do usual activities; and most commonly reported having moderate pain or discomfort; just under half of type I respondents reported being moderately anxious or depressed. Given the average age of the type I population, these findings may be age-related rather than a reflection of disability. The mean EQ-VAS score for all patients with SMA was 62.9 (SD: 19.3); VAS scores were lowest among those with type I and highest among those with type III.
Fig. 2
EQ-5D-5L Visual Analogue Scale: Overall and by self-reported SMA type. Legend: EQ-5D-5L visual analogue scale assesses (y-axis) the perceived health of the individual with SMA (or their adult proxy) from 0 (worst imaginable health) to 100 (the best imaginable health). Scores are reported for each self-reported SMA type with error bars representing one standard deviation from the mean. SMA = spinal muscular atrophy.
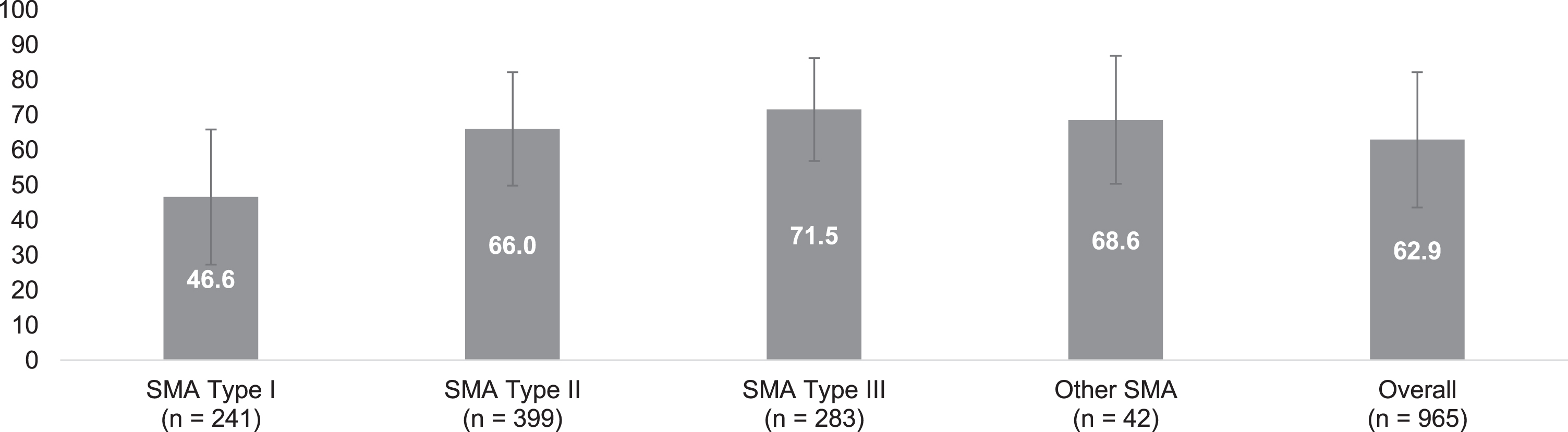
Across all patient survey respondents, patients with SMA reported significantly impaired quality of life with an average health utility index of 0.49 (SD: 0.26). Patients with SMA type I were more impaired (0.32, SD: 0.25) compared to those with type II (0.46, SD: 0.23) and type III (0.65, SD: 0.21).
Caregiver survey
Demographics
Overall, the median age of caregivers was 34.96 years (IQR: 31.17–39.17), 57.6% were female, and the most frequently reported relationship to the person with SMA was mother or father. Caregivers reported that the mean age of the person with SMA they were caring for was 11.71 (SD: 16.54) years old, with 29.7% of caregivers reporting that they cared for a person with SMA type I, and 44.2% and 25.2% of caregivers reporting caring for a person with type II or type III, respectively (Table 3).
Table 3
Caregiver demographics: Overall and by self-reported SMA type of person cared for
| Characteristics | Caregiver respondents | By SMA type of patient cared for | |||
| Type I | Type II | Type III | Unknown | ||
| (N = 962) | (n = 285) | (n = 423) | (n = 241) | (n = 9) | |
| Caregiver age, in years, at time of survey, n = 878 | |||||
| Mean (SD) | 35.25 (6.59) | 33.93 (5.72) | 35.03 (7.01) | 37.19 (6.62) | 37.02 (3.45) |
| Median (Q1–Q3) | 34.96 | 33.17 | 34.25 | 37.58 | 38.58 |
| (31.17–39.17) | (30.00–37.42) | (31.42–38.42) | (34.21–40.67) | (33.50–39.33) | |
| Age of patient receiving care, in years, at time of survey, n = 945 | |||||
| Mean (SD) | 11.71 (16.54) | 10.11 (19.67) | 10.90 (15.73) | 14.32 (12.80) | 33.58 (14.41) |
| Median (Q1–Q3) | 6.25 | 1.75 | 5.67 | 10.13 | 41.71 |
| (2.00–11.42) | (1.25–7.42) | (2.33–9.42) | (6.38–15.17) | (22.71–43.21) | |
| Sex, n (%) | |||||
| Female | 551 (57.6%) | 166 (58.5%) | 241 (57.2%) | 139 (58.2%) | <5 |
| Male | 406 (42.4%) | 118 (41.5%) | 180 (42.8%) | 100 (41.8%) | 5 (55.6%) |
| Non-respondents | 5 | <5 | <5 | <5 | 0 |
| Relationship to patient with SMA, n (%) | |||||
| Mother | 505 (52.5%) | 153 (53.7%) | 222 (52.5%) | 127 (52.7%) | <5 |
| Father | 365 (37.9%) | 112 (39.3%) | 164 (38.8%) | 86 (35.7%) | 0 |
| Spouse or partner | 44 (4.6%) | 10 (3.5%) | 11 (2.6%) | 18 (7.5%) | 5 (55.6%) |
| Other | 24 (2.5%) | 8 (2.8%) | 9 (2.1%) | 7 (2.9%) | 0 |
| Friend | 17 (1.8%) | <5 | 11 (2.6%) | <5 | <5 |
| Neighbor | 7 (0.7%) | 0 | 6 (1.4%) | <5 | 0 |
Note: Cells with fewer than 5 responses are masked using < 5 for privacy standards. SD = standard deviation; SMA = spinal muscular atrophy; Q1 = first quartile; Q3 = third quartile.
Caregivers reported that a median of two other people (IQR: 1–3) provided unpaid care for the person with SMA, and caregivers reported spending a median of 35 hours (IQR: 27–55) per week caring for the person with SMA. Additionally, 46.5% (442/951) reported that the person with SMA received paid support from a professional caregiver. When asked about employment, 37.0% (353/953) of unpaid caregivers reported being employed part-time and 27.8% (265/953) reported being employed full-time. While 15.4% (146/948) of caregivers reported no changes in work hours since caring for the person with SMA, 19.3% (183/948) reported an increased amount of care for the person with SMA outside of work hours, 39.9% (378/948) reported reduced working hours, and 18.6% (176/948) reported having to take extra unpaid leave off work or give up their job completely (data not shown).
Caregiver-related burden
When asked a series of questions regarding the impact of caregiving on their own health (Table 4), 31.1% (262/842) of caregiver survey respondents reported taking medications for anxiety and/or depression, 45.7% (385/842) reported receiving respite care for exhaustion, 36.5% (307/842) reported that they had received counseling, 45.7% (385/842) reported that they had received physiotherapy for an injury associated with lifting/transferring the patient and 63.3% (533/842) reported other impacts on their health. A higher percentage of caregivers for persons with SMA type I received respite care for exhaustion compared to those caring for persons with type II or type III. In contrast, a higher percentage of caregivers for those with type II and type III received counselling compared to caregivers of persons with SMA type I.
Table 4
Impact on caregiver health from caring for a person with SMA: Overall and by self-reported SMA type of person cared for
| Characteristics | Caregiver respondents | By SMA type of patient cared for | |||
| Type I | Type II | Type III | Unknown | ||
| (N = 962) | (n = 285) | (n = 423) | (n = 241) | (n = 9) | |
| Taken medications for anxiety and/or depression, n (%) | |||||
| Yes | 262 (31.1%) | 80 (31.5%) | 119 (30.6%) | 57 (29.8%) | 6 (75.0%) |
| No | 580 (68.9%) | 174 (68.5%) | 270 (69.4%) | 134 (70.2%) | <5 |
| Received respite care for exhaustion, n (%) | |||||
| Yes | 385 (45.7%) | 135 (53.1%) | 172 (44.2%) | 77 (40.3%) | <5 |
| No | 457 (54.3%) | 119 (46.9%) | 217 (55.8%) | 114 (59.7%) | 7 (87.5%) |
| Received counselling, n (%) | |||||
| Yes | 307 (36.5%) | 73 (28.7%) | 165 (42.4%) | 69 (36.1%) | 0 |
| No | 535 (63.5%) | 181 (71.3%) | 224 (57.6%) | 122 (63.9%) | 8 (100.0%) |
| Existing medical condition has worsened, n (%) | |||||
| Yes | 30 (3.6%) | 7 (2.8%) | 17 (4.4%) | 6 (3.1%) | 0 |
| No | 812 (96.4%) | 247 (97.2%) | 372 (95.6%) | 185 (96.9%) | 8 (100.0%) |
| Other, n (%) | |||||
| Yes | 533 (63.3%) | 160 (63.0%) | 238 (61.2%) | 133 (69.6%) | <5 |
| No | 309 (36.7%) | 94 (37.0%) | 151 (38.8%) | 58 (30.4%) | 6 (75.0%) |
| Received physiotherapy for an injury associated with lifting / transferring, n (%) | |||||
| Yes | 385 (45.7%) | 135 (53.1%) | 172 (44.2%) | 77 (40.3%) | <5 |
| No | 457 (54.3%) | 119 (46.9%) | 217 (55.8%) | 114 (59.7%) | 7 (87.5%) |
| Non-respondents | 120 | 31 | 34 | 50 | <5 |
Note: Cells with fewer than 5 responses are masked using < 5 for privacy standards. SMA = spinal muscular atrophy.
Caregiver strain index
As shown in Table 5, for each domain of the CSI, nearly half or more of the caregiver survey res-pondents indicated strain. The domains with the highest percentage of caregivers indicating strain were: changes in personal plans (77.2%), sleep being disturbed (70.9%), and there being work adjustments (70.5%). The overall mean CSI score was 7.5 (SD: 3.3), with a score of 7 or higher indicating high stress and burden. The mean CSI score varied by the type of SMA the person cared for had: type I was 6.8 (SD: 2.7), type II was 8.1 (SD: 3.0), type III was 7.3 (SD: 3.9).
Table 5
Caregiver Strain Index: overall and by self-reported SMA type of person cared for
| Characteristics, n (%) who responded ‘yes’ * | Caregiver respondents | By SMA type of patient cared for | |||
| Type I | Type II | Type III | Unknown | ||
| (N = 962) | (n = 285) | (n = 423) | (n = 241) | (n = 9) | |
| Sleep is disturbed (n = 951) | 674 (70.9%) | 187 (66.1%) | 338 (80.3%) | 143 (59.8%) | 6 (75.0%) |
| It is inconvenient (n = 949) | 518 (54.6%) | 148 (52.3%) | 241 (57.5%) | 121 (50.6%) | 8 (100.0%) |
| It is a physical strain (n = 950) | 611 (64.3%) | 164 (58.0%) | 308 (73.2%) | 132 (55.5%) | 7 (87.5%) |
| It is confining (n = 949) | 497 (52.4%) | 131 (46.5%) | 241 (57.4%) | 119 (49.8%) | 6 (75.0%) |
| There have been family adjustments (n = 945) | 508 (53.8%) | 130 (46.1%) | 242 (57.9%) | 129 (54.4%) | 7 (87.5%) |
| There have been changes in personal plans (n = 950) | 733 (77.2%) | 203 (71.7%) | 318 (75.7%) | 205 (85.8%) | 7 (87.5%) |
| There have been other demands on my time (n = 949) | 551 (58.1%) | 150 (53.0%) | 262 (62.4%) | 131 (55.0%) | 8 (100.0%) |
| There have been emotional adjustments (n = 950) | 471 (49.6%) | 123 (43.5%) | 243 (57.7%) | 98 (41.2%) | 7 (87.5%) |
| Some behaviour is upsetting (n = 949) | 433 (45.6%) | 124 (44.0%) | 196 (46.7%) | 106 (44.4%) | 7 (87.5%) |
| It is upsetting to find the person has changed | 471 (49.9%) | 123 (43.6%) | 225 (53.7%) | 116 (49.4%) | 7 (100.0%) |
| so much from his/her former self (n = 943) | |||||
| There have been work adjustments (n = 947) | 668 (70.5%) | 161 (57.3%) | 306 (73.0%) | 193 (80.8%) | 8 (100.0%) |
| It is a financial strain (n = 948) | 576 (60.8%) | 154 (54.4%) | 274 (65.2%) | 141 (59.5%) | 7 (87.5%) |
| Feeling completely overwhelmed (n = 950) | 451 (47.5%) | 129 (45.7%) | 214 (50.8%) | 103 (43.1%) | 5 (62.5%) |
* Each item is in response to a prompt at the beginning of the section “Here is a list of things that other people have found to be difficult in helping out after somebody comes home from the hospital. Would you please tell me whether any of these apply to you?” Note: Cells with fewer than 5 responses are masked using < 5 for privacy standards. SMA = spinal muscular atrophy.
Quality of life (EQ-5D-5L)
When asked to rate their quality of life using the EQ-5D-5L, most caregivers reported that they did not have any trouble with their own mobility (74.2%, 706/952), self-care (75.3%, 717/952), or their usual activities (69.3%, 660/952), and most caregivers reported having no pain or discomfort (62.3%, 593/952) (data not shown). However, most caregivers (65.2%, 621/962) experienced at least some degree of anxiety or depression, which was higher in caregivers of persons with SMA type I (74.2%) and type II (69.3%) compared to patients with type III (46.2%).
The mean EQ-VAS score among caregivers was 81.8 (SD: 13.3), and was relatively consistent despite their patient’s SMA type, ranging from 79.6 (SD: 12.5) among caregivers caring for someone with type I to 83.0 (SD: 13.3) among those caring for someone with type III. The mean health utility index among all caregiver survey respondents was 0.84 (SD: 0.17; data not shown).
DISCUSSION
Canadian patients with SMA and their caregivers reported significant health and economic burdens associated with this disease. Our study confirms substantial use of healthcare resources, with notable differences in the pattern of use by SMA type. Most (96.8%) Canadian patients with SMA reported visiting an outpatient clinic at least once annually, while 76.8% reported at least one annual visit to a hospital. Outpatient visits were stratified by SMA type, with approximately 80% of patients with type II or type III reporting at least one annual outpatient visit com-pared to 65.1% of patients with type I. It was unclear from the question if patient survey respondents vie-wed visits to hospital outpatient clinics (such as multidisciplinary neuromuscular clinics) as outpatient visits, since patients with SMA type I did report more frequent hospital visits compared to patients with other SMA types. Since patients with SMA type I are typically younger, and multidisciplinary clinics are often located at pediatric hospitals or rehabilitation centres, it is possible that the lower number of outpatient visits and relatively higher number of hospital visits may reflect the physical location of these clinics. Adults with SMA, in contrast, may be followed at private outpatient clinics or outpatient rehabilitation centres. To this point, the trend differed for the number of hospital visits, where 62.6% of patients with SMA type I reported visiting the hospital two or more times in the past year compared to 37.8% of those with type II and 26.2% of those with type III. Although 41.7% of patient survey respondents had not visited an emergency department in the last year, of those who had, 90.4% were patients with type I (n = 208), reflecting the trend towards younger age and potential for higher medical fragility in the more severe type of SMA.
This study showed a significant financial burden on patients, including home modifications (median spend of $12,800 [IQR: $6,800–20,000] ever), allied health professional services (median spend of $6,800 [IQR: $3,900–13,000] in the past year), assistive devices (median spend of $4,500 [IQR: $1,587–$11,000] in the past year), as well as travel and accommodation for SMA-related treatments (median spend of $1,200 [IQR: $600–$3,100] in the past year), some of which were paid out-of-pocket despite also receiving funding from other sources. The lower spending on home modifications in those with type I SMA relative to those with type II or type III SMA may be because type I patients are usually infants with a shortened lifespan, and therefore, home modifications are less frequently undertaken. When SMA is untreated, the mean survival is 8 to 10–1/2 months of age, with most patients having a life expectancy of less than 2 years [34, 35].
Quality of life among patients with SMA was assessed using the EQ-5D-5L in this study. At least some degree of problem was observed across each of the domains, and the results were aligned with anticipated motor abilities based on SMA type and age.
Our findings build on existing literature that has demonstrated substantial direct and indirect costs among patients with SMA, along with the significant impact on quality of life for both individuals with SMA and caregivers of individuals with SMA.
An Australian study used semi-structured interviews to understand the financial, opportunity and psychosocial costs among caregivers to patients with SMA type II and III [24]. The authors reported that along with substantial direct healthcare costs, caregivers noted indirect costs related to impeded opportunities for employment and career development as well as increased mental health burdens [24].
A German study evaluated the economic burden and disease-specific health-related quality of life (HRQoL) of SMA [18]. This cross-sectional study of 189 patients with SMA type I, II, and III reported an overall mean direct cost of illness of € 54,721/year per patient and an overall mean indirect cost of illness of € 15,845/year per patient [18]. This amount is greater than our cohort that reported a mean total cost for assistive devices and home modifications totaling $13,397 CAD and $19,570 CAD, respectively. In the German study, the authors noted that self-rated (or proxy-rated) HRQoL scores increased from SMA type I to III [18]. This trend was similar to the improvement in EQ-5D-5L observed in Canadian patients (or their adult proxy) in our study, which also showed an increase from SMA type I to III.
A Spanish study reported high costs of illness (direct and indirect costs) and decreased quality of life for patients with SMA and their caregivers [26]. That study reported average annual costs per patient of € 33,721 (€ 10,882 in direct healthcare costs and € 22,839 in direct non-healthcare costs) [26]. Among patients with SMA, the mean score on the EQ-5D VAS was 54.09 (SD: 26.30) compared to 69.10 (SD: 21.96) among caregivers [26]. The mean EQ-5D VAS score in the Spanish study [26] was slightly lower than what was observed in this current study (patient: 62.9 [SD: 19.3]; caregiver: 81.8 [SD: 13.3]). See Table 6 for a tabular summary of the current study findings and the two aforementioned studies that report indirect costs associated with SMA, converted to CAD for comparison.
Table 6
Indirect cost values reported among identified SMA publications
| Study reference | Country | Year data collected | Definition of SMA (and types) | Type of indirect costs included and reported | Indirect cost results* |
| Current study | Canada | 2020 | Patients or caregivers self-reporting genetically confirmed SMA via online survey | –Home modification spending ever | –Home modification median expenditure ever, overall: $12,800 |
| –Assistive device spending in the past year | ∘ SMA type I: $6,100 | ||||
| ∘ SMA type II and SMA type III: $15,000 | |||||
| –Personal, out-of-pocket spend on | –Assistive device median expenditure | ||||
| allied health professional services | in the last year, overall: $4,500 | ||||
| related to SMA in the past year | –Personal, out-of-pocket spend on allied | ||||
| –Travel and accommodation spending | professional health services in past | ||||
| relating to SMA appointments | 12 months, overall: $6,800 | ||||
| in the past year | –Travel and accommodation median | ||||
| spend in the last year, overall: $1,200 | |||||
| ∘ SMA type I: $2,105 | |||||
| ∘ SMA type II: $1,000 | |||||
| ∘ SMA type III: $1,000 | |||||
| Klug 2016 | Germany | 2013, Cross | Patients with a | –Overall mean annual indirect | Overall mean annual indirect |
| sectional | genetically confirmed | COI added up to € 15,845 per patient | COI: $24,566 per patient | ||
| study | SMA (types I to III) | –The overall mean total COI | –Overall mean total COI | ||
| were identified via | was estimated at € 70,566/y per patient. | estimate: $109,397/y per patient | |||
| the German SMA | –The most relevant disease | –Most relevant disease burden for | |||
| patient registry | burden was found in SMA I | SMA type 1 ($167,131/y per patient), | |||
| (€ 107,807/y per patient) with a | SMA type II ($139,939/y per patient), | ||||
| decrease towards SMA II (€ 90,267/y) | SMA type III ($81,291/y per patient) | ||||
| and SMA III (€ 52,440/y) and a | |||||
| significant difference for SMA | |||||
| I and II compared to SMA III (p < 0.05) | |||||
| Lopez-Bastida | Spain | 2015, Cross | Children or adolescents | –Average annual cost associated with | –Average annual cost associated |
| 2016 | sectional and | diagnosed with SMA | SMA reached € 33,721 (SD: 38,700) | with SMA: $52,275 | |
| retrospective | and their main caregiver; | ∘ The family caregiving costs represented | ∘ Family caregiving costs: $32,753 | ||
| study (costs: | patients with SMA were | the largest component reaching | |||
| (€, 2014)) | classified into three types | € 21,127 (62.7% of the total | |||
| based on age at disease | cost of the illness in Spain). | ||||
| onset and the clinical | |||||
| severity of the disease, | |||||
| as defined by the | |||||
| International SMA | |||||
| Consortium |
* Conversion to CAD (where applicable) was calculated using the exchange rate available on November 23rd 2020. COI = cost of illness, ICD-9-CM = International Classification of Diseases, Ninth revision, Clinical Modification; SD = standard deviation, SMA = spinal muscular atrophy.
A Dutch study of adults with SMA reported that patients had a lower physical quality of life, but nor-mal mental quality of life, when compared with a reference population from the Netherlands [36]. Patients with milder, later onset disease who reported feelings of anxiety and depression were at risk of experiencing reduced mental quality of life [36]. In a Dutch survey of mothers of home-living patients with SMA [25], three-quarters of mothers perceived a high caregiver burden, while a separate Dutch study [37] reported caregivers of cancer patients had a higher rate of disrupted activity schedules which placed them at a higher risk of developing depressive symptoms. Among our caregiver survey respondents, 77.2% reported the need to make changes in their personal plans and activities within the last 12 months, which was the most common response in our assessment of caregiver strain followed by disrupted sleep (70.9%).
A study of HRQoL from the Czech Republic of children and adolescents with SMA, as well as their parents, reported that both patients and parents had a lower quality of life compared to patients with SMA living in the United States [27]. The authors hypothesized that the lower quality of life may have been attributed to economic factors and a lack of social support [27]. Although there is some variability in results between existing studies and our current study, it is generally reported that patients with SMA and their caregivers experience significant health and economic burdens.
In line with what has been reported in the medical literature, caregiver-related burden was demonstrated in several sections of our survey, including the amount of unpaid care required by the patient, the impact of caring for a person with SMA on the caregiver’s own health, caregiver strain, as well as quality of life.
In each domain, almost half or more of caregivers responding to the CSI indicated strain. The domains with the highest percentage of caregivers indicating strain were reported for changes in personal plans, sleep being disturbed, and there being work adjustments. In our study, the mean CSI score was 7.5 (SD: 3.3) which was very similar to a CSI score of 8.1 in a Dutch study of caregivers [25]. The CSI in our study did vary by the type of SMA of the person cared for: type I was 6.8 (SD: 2.7), type II was 8.1 (SD: 3.0), type III was 7.3 (SD: 3.9). The CSI evaluates several different aspects of caregiver burden, including physical burden, time investment, emotional health, and financial pressure [25]. As such, the higher CSI scores for caregivers of persons with SMA type II or type III could be due to the relatively longer lifespan of these individuals. Many persons with SMA type II and type III survive into adulthood [3, 4, 38, 39], and would therefore require comprehensive care over a longer period of time compared to individuals with SMA type I who have a life expectancy of less than two years [3, 4, 38, 39].
The economic burden of SMA is significant, but it has largely been evaluated in the United States and Europe. Droege et al. (2020) evaluated the healthcare resource utilization and costs for patients with SMA who were treated with nusinersen in the United States [40]. Patients with SMA type I treated with nusinersen reported lower total mean healthcare costs compared to those without nusinersen treatment ($92,618 USD vs. $137,627 USD per-patient-per-year, excluding nusinersen-related costs) [40]. A different trend was observed among patients with other types of SMA, where those receiving nusinersen treatment reporting higher total mean healthcare costs compared to those not treated with nusinersen ($76,371 USD vs. $49,175 USD, excluding nusinersen-related costs) [40]. A retrospective analysis of pediatric and adult patients with SMA in the United States reported higher mean healthcare costs among patients under two years of age ($159,227 USD vs. $64,490 USD per-patient-per-year) [40, 41]. In Germany, the mean direct costs for patients with SMA were found to be 14 times higher compared to average healthcare spending [18, 42]. Our study provides context for understanding the significant economic burden on patients with SMA and their caregivers in Canada.
As a larger number of patients than anticipated reported having been prescribed one or more of the therapies available for SMA, and it was implausible that such a high proportion of patients would have received these medications through a clinical trial or sought care internationally, we hypothesized that some patient survey respondents may have misinterpreted this question pertaining to medication use, since only nusinersen was approved by Health Canada at the time the questionnaire was administered. To confirm this hypothesis, we conducted a post-hoc analysis on demographic characteristics, costs, and quality of life for a subset of patients (n = 442) that had indicated: 1) they had been prescribed nusinersen but not risdiplam or onasemnogene abeparvovec or; 2) that they had not been prescribed any of the medications. The post-hoc analysis of this subset showed comparable results to the full cohort of patient survey respondents. The subset did report lower median expenditure for assistive devices (e.g., $2,200 for the subset compared to $4,500 for the larger cohort) but a higher median expenditure for home modifications ($13,750 for the subset compared to $12,800 for the full cohort). The authors feel that the potential misinterpretation of the treatment question does not appear to influence answers in any other domain in the questionnaire. The post-hoc data analysis is provided (see Supplementary Materials).
This study has several strengths, including the fact it addresses a significant knowledge gap regarding the impact of SMA on Canadian patients and caregivers, since both pediatric and adult patients, as well as their caregivers, were surveyed, and several aspects of disease burden were measured.
In spite of the strengths of our study, there were several limitations to note. Since the survey relied on self-reported data, it was not possible to verify that all patient survey respondents were indeed genetically-confirmed to have 5q-linked SMA as they had indicated in the survey response. In the caregiver survey, the questionnaire asked the type of SMA, but did not ask whether the person they cared for had 5q-linked SMA. It was also not possible to confirm the patient SMA type as reported by patients, proxies, or caregivers. Similarly, it was not possible to verify if all unpaid caregivers did indeed care for individuals with SMA. Since identifying information was not collected from the participants (e.g., name, address, date of birth), it was not feasible to link patient and caregiver responses, nor was it possible to determine whether the same participant had completed the survey more than once, or if two caregivers for the same patient (e.g., mother and father) had both completed the survey. The $50 CAD gift card incentive to complete the survey may have created a paradigm where some survey respondents may have been bots or ballot box stuffers, completing the survey more than once to obtain this reward, or completing the survey even if they did not themselves have SMA or care for someone with SMA. The approach to identify and exclude potential fraudulent survey respondents was intended to address this, but it may not have captured all instances of bots or ballot box stuffing, particularly if the repeat surveys were completed through different internet provider (IP) addresses (e.g., work and home).
Second, like all surveys, this study is subject to potential biases involving recollection and self-reporting, such as recall bias and response bias. Recall bias should be noted as a limitation, and some questions may have been misconstrued or misunderstood. One such example was noted where most patient survey respondents who self-reported to have SMA type I indicated that they were non-ambulatory (65.5%), while a small proportion reported only a slight problem (3.8%) or no problem (4.2%) with mobility (Supplemental Table 1). We interpreted this as patients either perceiving their SMA type to be different from the historical typing system (since patients with SMA type I would not be predicted to sit or walk) or potentially a misinterpretation of the survey question. “Mobility” can also be interpreted as mobilizing using an assistive device such as an electronic wheelchair which might lead to a different response. One other explanation is the early effects of disease-modifying therapy altering the natural history of SMA, since nusinersen had been available for three years prior to the completion of this survey and may also have been associated with some SMA type I patients achieving motor outcomes that would not have otherwise been predicted. The EQ-5D was used to assess quality of life among all individuals with SMA. While the EQ-5D-5L proxy version is validated for adult populations, a large portion of the patient sample were under 18 and the domains captured may be less valid for infants and young children. Response bias is an important consideration, particularly regarding demand characteristics. That is, survey respondents may alter their responses or behavior because they know they are part of a study. In this study, it is noted within the informed consent form that the study is sponsored by Hoffmann-La Roche Limited, which could influence responses, particularly in regard to treatment-related questions. Due to the survey distribution method and the anonymous nature of the survey respondents, it was challenging to implement additional verification steps to avoid any potential response biases. Other, non-anonymous or non-incentivized approaches to the survey study design could be considered in the future.
Third, the number of patient survey respondents indicating having “been prescribed” various disease-modifying therapies for SMA (e.g., nusinersen, risdiplam or onasemnogene abeparvovec) was higher than expected. The number of patients indicating that they had been prescribed risdiplam or onasemnogene abeparvovec was not plausible, as neither of these therapies were approved for use in Canada at the time of questionnaire completion. As such, it is possible that patient survey respondents may have misinterpreted “been prescribed” as having “been recommended” each medication, or possibly as being related to awareness of whether they would want to have the medication prescribed. Another possibility may have been that patient survey respondents answered in a positive manner in the hope that they might have been selected for a clinical trial or increased the likelihood of clinically accessing these treatments. To assess if this effect was isolated to this question, we performed a post-hoc sensitivity analysis of the data, including only patients who stated that they had received no treatment, or nusinersen only. With this post-hoc sensitivity analysis, there was no meaningful change in the study results, strengthening the argument that it was this specific question that was misinterpreted and not an attempt to influence study responses on the part of a subset of patient survey respondents. It was also reassuring that while the treatment responses were implausible, the other responses were consistent with other published studies relating to SMA quality of life and burden.
Finally, it should also be noted that for all patients under the age of majority (either 18 or 19 years of age, depending on the province of residence) the surveys were proxy-completed by an adult caregiver. Among adults with SMA, 84.9% of patients completed the surveys themselves. Though there may be some concern regarding parent or caregiver-proxy responses, given that most questions pertained to objective measures (e.g., healthcare resource use, expenses, travel time) we believe there would be minimal bias from proxy-completed surveys [43]. While quality of life was assessed using the validated, proxy-version of the EQ-5D-5L, we acknowledge that proxy responses may vary from what the patient may have responded for these more subjective measures, as has been demonstrated in previous studies [44].
CONCLUSION
SMA affects the quality of life of individuals dia-gnosed with the disease and their caregivers as measured by the EQ-5D-5L. Other impacts include reduced attendance at work or school, increased sup-ports required for work or school, substantial SMA-related healthcare resource utilization and travel for appointments, along with costs associated with living adaptations and assistive devices.
Unpaid caregivers for individuals with SMA report significant caregiving-related burden, as demonstrated by the number of caregivers reporting changes in personal plans, disturbed sleep, and adjustments to work using the CSI. Other aspects of caregivers’ health reported to have been impacted by caregiving for a person with SMA included receiving respite care for exhaustion and undergoing physiotherapy for an injury associated with lifting/transferring the patient.
Until recently, the management of SMA was foc-used upon optimizing respiratory, orthopedic, rehabi-litation, and nutritional care [40, 45, 46]. In December 2016, the Food and Drug Administration (FDA) in the United States approved nusinersen, the first disease-modifying therapy for SMA [40, 42], which was subsequently approved by Health Canada in 2017 [19]. In 2019, the FDA approved the first gene replacement therapy, onasemnogene abeparvovec, for children with SMA under two years old, which was approved by Health Canada in late 2020, after completion of this survey [47, 48]. Most recently, in August 2020, the FDA approved risdiplam for children with SMA over two months old and this therapy is currently under review by Health Canada [23, 49]. These emerging treatment modalities are changing the management, outcomes, and costs associated with SMA. The full impact of these treatments upon disease burden and quality of life for Canadian patients with SMA will not become apparent for several years to come. As such, it would be beneficial to conduct a similar study in the future to evaluate the changing landscape of SMA management, healthcare costs and utilization for Canadian patients with SMA as well as their caregivers.
To minimize the burden on patients with SMA and their caregivers, more research is needed to imp-rove SMA-associated healthcare services and therapies, specifically in the Canadian context. This is particularly important given the recent approval of nusinersen and onasemnogene abeparvovec in Can-ada and the reality that one additional disease-mod-ifying therapy is currently under review for approval at the present time.
ACKNOWLEDGMENTS
This is a Hoffmann-La Roche Limited sponsored study. We thank Myka Estes, Khushboo Gurjar and Tayler Scory (Medlior) for their work in creating the Qualtrics surveys.
Conflict OF INTEREST
Gerber B, Cowling T, Khuu W and Mayer M are employed by Medlior Health Outcomes Research Ltd. which received funding for the study from Hoffmann-La Roche Limited. Cabalteja C, Wu J, Maturi B, and Klein-Panneton K are employed by Hoffmann-La Roche Limited who funded this study. Cabalteja C, Wu J, and Maturi B also hold Hoffmann-La Roche Limited stock. McMillan H reports fin-ancial support for research from Hoffmann-La Roche Limited and was a member of an advisory board for AveXis. Lochmuller H reports: consultancy and financial support for research projects and clinical trials from Amplo Biotechnology, AMO Pharma, Biogen, Desitin, Fulcrum Therapeutics, GW Pharma, Milo Biotechnology, Pfizer, PTC Therapeutics, Hof-fmann-La Roche Limited, Sanofi-Genzyme, Santhera, Sarepta, Satellos and Ultragenyx. Lochmuller H is the Editor-in-chief for the Journal of Neuromuscular Diseases (IOS Press). All study authors participated in the design of this study, interpretation of the results, and development of the manuscript.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-200610.
REFERENCES
[1] | Oskoui M , Ng P , Liben S , Zielinski D . Physician driven variation in the care of children with spinal muscular atrophy type 1. Pediatr Pulmonol. (2017) ;52: (5):662–8. |
[2] | Prior TW . Spinal muscular atrophy: newborn and carrier screening. Obstet Gynecol Clin North Am. (2010) ;37: (1):23–36. |
[3] | Verhaart IEC , Robertson A , Wilson IJ , Aartsma-Rus A , Cameron S , Jones CC , et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet J Rare Dis. (2017) ;12: (1):124. |
[4] | Lunn MR , Wang CH . Spinal muscular atrophy. Lancet. (2008) ;371: (9630):2120–33. |
[5] | Ogino S , Leonard DG , Rennert H , Ewens WJ , Wilson RB . Genetic risk assessment in carrier testing for spinal muscular atrophy. Am J Med Genet. (2002) ;110: (4):301–7. |
[6] | Isaac Foundation for Rare Diseases. Spinal Muscular Atrophy (SMA) - A Resource for Canadian Families Battling SMA. Available from: https://www.theisaacfoundation.com/sma/. |
[7] | Chen G , Sharif B , Gerber B , Farris M , Cowling T , Cabalteja C , Wu J , Maturi B , Klein-Panneton K , Mah J . A population-based study examining the epidemiologic burden, healthcare resource utilization and costs of spinal muscular atrophy in Alberta, Canada. 25th International Annual Congress of the World Muscle Society; 2020. October 1, 2020; Virtual Congress. |
[8] | Statistics Canada. Table 17-10-0009-01 Population estimates, quarterly. |
[9] | Mah JK , Haig T , Hodgkinson V , Innes M , Korngut L , Parboosingh J . 5q spinal muscular atrophy. January 22, 2021. Available from: https://www.cpsp.cps.ca/uploads/studies/5q-spinal-muscular-atrophy-protocol.pdf. |
[10] | Muscular Dystrophy Canada. Statement from Stacey Lintern, Chief Executive Officer, Muscular Dystrophy Canada on a Collaboration with Novartis for National Newborn Screening in Spinal Muscular Atrophy (SMA); 2020.Available from: https://muscle.ca/2020/12/04/mdc-statement/. |
[11] | The Neuromuscular Disease Network for Canada (NMD4C). A national spinal muscular atrophy registry for real world evidence; 2020. Available from: https://neuromuscularnetwork.ca/news/a-national-spinal-muscular-atrophy-registry-for-real-world-evidence/. |
[12] | Canadian Institute for Health Information. Health Care Cost Drivers: The Facts. Ottawa, Ontario, Canada: Canadian Institute for Health Information; 2011. |
[13] | Institute of Health Economics. IHE In Your Pocket 2014: A Handbook of Health Economic Statistics: Institute of Health Economics; 2014. Available from: https://www.ihe.ca/advanced-search/ihe-in-your-pocket-2014-a-handbook-of-health-economic-statistics. |
[14] | Health Canada. Canada’s Health Care System. Available from: https://www.canada.ca/en/health-canada/services/health-care-system/reports-publications/health-care-system/canada.html. |
[15] | MarchildonGP. Canada: Health system review. Health Systems in Transition. (2013) ;15: , 1–179. |
[16] | Canada Revenue Agency. Disability-Related Information. February 4, 2021. Available from: https://www.canada.ca/en/revenue-agency/services/forms-publications/publications/rc4064/disability-related-information-2016.html. |
[17] | Canadian Institute for Health Information. National Health Expenditure Trends 1975 to 2019. Ottawa, ON: Canadian Institute for Health Information; 2019. |
[18] | Klug C , Schreiber-Katz O , Thiele S , Schorling E , Zowe J , Reilich P , et al. Disease burden of spinal muscular atrophy in Germany. Orphanet J Rare Dis. (2016) ;11: (1):58. |
[19] | Health Canada. Regulatory Decision Summary - Spinraza 2017. Available from: https://hpr-rps.hres.ca/reg-content/regulatory-decision-summary-detail.php?lang=en&linkID=RDS00268. |
[20] | Muscular Dystrophy Canada. Spinraza Decision Signals the Need for a Canadian Rare Disease (or Orphan Drug) Framework; 2019. Available from: https://muscle.ca/2019/03/08/cadth-review-and-spinraza/. |
[21] | Canadian Agency for Drugs and Technologies in Health. CADTH Common Drug Review: Canadian Drug Expert Committee Recommendation - Nusinersen (Spinraza - Biogen Canada Inc.); 2019. January 22, 2021. Available from: https://www.cadth.ca/sites/default/files/cdr/complete/SR0576-Spinraza-Resubmission-Mar-1-19.pdf. |
[22] | Health Canada. Regulatory Decision Summary - Zolgensma. 2020. |
[23] | Hoffmann-La Roche. Roche presents new 2-year data for Evrysdi (risdiplam) in infants with Type 1 spinal muscular atrophy (SMA); 2020. Available from: https://www.roche.com/media/releases/med-cor-2020-09-28.htm. |
[24] | Farrar MA , Carey KA , Paguinto SG , Chambers G , Kasparian NA . Financial, opportunity and psychosocial costs of spinal muscular atrophy: An exploratory qualitative analysis of Australian carer perspectives. BMJ Open. (2018) ;8: (5):e020907. |
[25] | Cremers CH , Fischer MJ , Kruitwagen-van Reenen ET , Wadman RI , Vervoordeldonk JJ , Verhoef M , et al. Participation and mental well-being of mothers of home-living patients with spinal muscular atrophy. Neuromuscul Disord. (2019) ;29: (4):321–9. |
[26] | Lopez-Bastida J , Pena-Longobardo LM , Aranda-Reneo I , Tizzano E , Sefton M , Oliva-Moreno J . Social/economic costs and health-related quality of life in patients with spinal muscular atrophy (SMA) in Spain. Orphanet J Rare Dis. (2017) ;12: (1):141. |
[27] | Kocova H , Dvorackova O , Vondracek P , Haberlova J . Health-related quality of life in children and adolescents with spinal muscular atrophy in the Czech Republic. Pediatr Neurol. (2014) ;50: (6):591–4. |
[28] | Qualtrics. Fraud Detection 2020. Available from: https://www.qualtrics.com/support/survey-platform/survey-module/survey-checker/fraud-detection/. |
[29] | Rowell J , Vincent SA , Saberian S , Scoto M , Muntoni F . PRO22 A Real World Study Investigating the Resource Use and Burden Assocaited with Spinal Muscular Atrophy (SMA) from the Perspective of Patients and Carers in the UK. Value in Health. (2020) ;23: , S693. |
[30] | EuroQol Research Foundation. EQ-5D-5L | About 2017. Available from: https://euroqol.org/eq-5d-instruments/eq-5d-5l-about/. |
[31] | Xie F , Pullenayegum E , Gaebel K , Bansback N , Bryan S , Ohinmaa A , et al. A Time Trade-off-derived Value Set of the EQ-5D-5L for Canada. Med Care. (2016) ;54: (1):98–105. |
[32] | Robinson BC . Validation of a Caregiver Strain Index. J Gerontol. (1983) ;38: (3):344–8. |
[33] | Dunaway Young S , Montes J , Kramer SS , Podwika B , Rao AK , De Vivo DC . Perceived Fatigue in Spinal Muscular Atrophy: A Pilot Study. J Neuromuscul Dis. (2019) ;6: (1):109–17. |
[34] | Finkel RS , McDermott MP , Kaufmann P , Darras BT , Chung WK , Sproule DM , et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. (2014) ;83: (9):810–7. |
[35] | Kolb SJ , Coffey CS , Yankey JW , Krosschell K , Arnold WD , Rutkove SB , et al. Natural history of infantile-onset spinal muscular atrophy. Ann Neurol. (2017) ;82: (6):883–91. |
[36] | Kruitwagen-Van Reenen ET , Wadman RI , Visser-Meily JM , van den Berg LH , Schroder C , van der Pol WL . Correlates of health related quality of life in adult patients with spinal muscular atrophy. Muscle Nerve. (2016) ;54: (5):850–5. |
[37] | Nijboer C , Triemstra M , Tempelaar R , Sanderman R , van den Bos GA . Determinants of caregiving experiences and mental health of partners of cancer patients. Cancer. (1999) ;86: (4):577–88. |
[38] | Verhaart IEC , Robertson A , Leary R , McMacken G , König K , Kirschner J , et al. A multi-source approach to determine SMA incidence and research ready population. J Neurol. (2017) ;264: (7):1465–73. |
[39] | Mercuri E , Bertini E , Iannaccone ST . Childhood spinal muscular atrophy: Controversies and challenges. Lancet Neurol. (2012) ;11: (5):443–52. |
[40] | Droege M , Sproule D , Arjunji R , Gauthier-Loiselle M , Cloutier M , Dabbous O . Economic burden of spinal muscular atrophy in the United States: A contemporary assessment. J Med Econ. (2020) ;23: (1):70–9. |
[41] | Goble J , Dai D , Boulos F , Weng A , Johnson K . The Economic Burden of Spinal Muscular Atrophy Patients in a Commercially-Insured Population in the United States (P1.6-054). Neurology. (2019) ;92. |
[42] | Tan H , Gu T , Chen E , Punekar R , Shieh PB . Healthcare Utilization, Costs of Care, and Mortality Among Patients With Spinal Muscular Atrophy. J Health Econ Outcomes Res. (2019) ;6: (3):185–95. |
[43] | Shields M . Proxy reporting in the National Population Health Survey. Health Rep. (2000) ;12: (1):21–39. |
[44] | Liang Y , Che T , Zhang H , Shang L , Zhang Y , Xu Y , et al. Assessing the proxy response bias of EQ-5D-3L in general population: A study based on a large-scale representative household health survey using propensity score matching. Health Qual Life Outcomes. (2020) ;18: (1):75. |
[45] | Mercuri E , Finkel RS , Muntoni F , Wirth B , Montes J , Main M , et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. (2018) ;28: (2):103–15. |
[46] | Finkel RS , Mercuri E , Meyer OH , Simonds AK , Schroth MK , Graham RJ , et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. (2018) ;28: (3):197–207. |
[47] | FDA NEWS RELEASE: FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality [press release]. May 24, 2019. |
[48] | Wexler M . Zolgensma Approved to Treat Young SMA Patients in Canada 2020. Available from: https://smanewstoday.com/news-posts/2020/12/18/zolgensma-approved-pediatric-sma-patients-health-canada/. |
[49] | FDA NEWS RELEASE: FDA Approves Oral Treatment for Spinal Muscular Atrophy [press release]. August 7, 2020. |


