
Therapies for Genetic Forms of Parkinson’s Disease: Systematic Literature Review
Abstract
Parkinson’s disease (PD) is a disabling neurological condition characterized by the loss of dopaminergic neurons. Currently, the treatment for PD is symptomatic and compensates for the endogenous loss of dopamine production. In cases where the pharmacological therapy is only partly beneficial or results in major wearing-off complications, surgical interventions such as deep brain stimulation may be an alternative treatment. The disease cause often remains unknown, but in some patients, a monogenic cause can be identified. Mutations in at least six genes, LRRK2, SNCA, and VPS35 (dominant forms) or Parkin/PRKN, PINK1, and DJ1/PARK7 (recessive forms) have been unequivocally linked to PD pathogenesis. We here systematically screened 8,576 publications on these monogenic PD forms. We identified 2,226 mutation carriers from 456 papers. Levodopa was the most widely applied treatment; only 34 patients were indicated to be untreated at the time of reporting. Notably, detailed treatment data was rarely mentioned including response quantification (good, moderate, minimal) in 951 and/or dose in 293 patients only. Based on available data, levodopa showed an overall good outcome, especially in LRRK2, VPS35, Parkin, and PINK1 mutation carriers (“good” response in 94.6–100%). Side effects of levodopa therapy were reported in ∼15–40%of levodopa-treated patients across genes with dyskinesias as the most frequent one. Non-levodopa medication was indicated to be administered to <200 patients with mainly good outcome. Only a few reports were available on outcomes of brain surgery. Here, most mutation carriers showed a good response. Importantly, none of the available treatments is harmful to one genetic form but effective in another one. In the light of different medication schemes, the progressive nature of PD, and side effects, an improvement of therapeutic options for PD is warranted including a treatabolome database to guide clinicians in treatment decisions. Further, novel disease-cause-modifying drugs are needed.
Parkinson’s disease (PD) is a complex and neurodegenerative disorder with diverse pathogenic traits [1]. Clinically, PD is characterized by bradykinesia, resting tremor, rigidity, and postural instability. The pathological hallmark is loss of dopaminergic neurons in the substantia nigra. Currently, the treatment for PD is symptomatic and usually consists of dopamine-based therapies with the aim to improve motor but also some non-motor signs and symptoms [2]. So far, disease-modifying pharmacologic treatments are not available. Occupational, physical and speech therapy as well as regular exercise complement pharmacologic treatments [3]. Most non-motor symptoms require additional non-dopaminergic approaches (such as selective serotonin reuptake inhibitors for psychiatric symptoms, cholinesterase inhibitors for cognition) [3]. In cases where the pharmacological therapy does not appropriately control PD tremor, is accompanied by intolerable side effects, or results in major wearing-off complications, a surgical intervention such as deep brain stimulation (DBS) may be an alternative treatment option [3, 4].
For most patients, the disease cause remains unknown (idiopathic PD) but in some patients, a monogenic cause can be identified. Mutations in at least six genes, LRRK2, SNCA, and VPS35 in dominantly inherited forms as well as Parkin/PRKN, PINK1, and DJ1/PARK7 in recessive forms, have been unequivocally linked to PD pathogenesis [5]. Of note, while PD is a frequent disorder with ∼6.1 million people affected worldwide [6], monogenic forms of PD comprise <5%of all patients and are individually rare [5, 7]. Within the past years, the number of identified mutation carriers has grown, triggered by an increasing availability of genetic testing due to technological advances including next generation sequencing approaches. Many review articles on genetic forms of PD have been published but the vast majority of these papers is focused on genetic data and molecular mechanisms as well as accompanying signs and symptoms. Beyond that, treatment options are only rarely systematically discussed and then focused on levodopa or deep brain stimulation (DBS).
In general, the current problem for non-specialist physicians is that - even when patients are genetically diagnosed - they often do not receive the best treatment for their specific mutation. There is a need for a systematic analysis of treatment options and outcomes including idiopathic but also genetically stratified, monogenic cases. Due to the rarity of genetic PD, single center studies are not suitable to compare treatment for hundreds or thousands of patients [7]. However, reviewing published data in a systematic fashion may collect enough data to guide treatment. A recent proof-of-principle has been provided for myasthenic syndromes [8]. Clinical diagnoses have to be matched with genetic-based decision-support systems for treatment guidance. Creating a treatabolome database is intended to link the genetic and clinical diagnosis with the best possible therapy and gain easier access to available data and the evidence they need to consider. This review aims to systematically collect clinical data of published articles on hereditary PD patients and to evolve a mutation-based treatment compass. It follows a recently published guide for systematic literature reviews [9]. Therefore, we here provide an overview of the currently available phenotypic and genotypic data on autosomal-dominant and autosomal-recessive PD-causing mutations, comparing published treatment-related data across the six genes, analyzing pharmacological and surgical therapy options with outcomes in each case.
METHODS
Literature search and eligibility criteria
The literature search and data extraction protocol have been adapted to serve the requirements for a systematic literature review in building a treatabolome [9] from MDSGene (available at http://www.mdsgene.org), which is a database that summarizes and quantifies phenotypic and genotypic data from the literature for hereditary movement disorders. While MDSGene focuses on genotype-phenotype correlations [10, 11], we here specifically looked for detailed treatment and outcome information in patients with genetic PD.
In brief, we performed a systematic literature search for publications on PD patients with autosomal-dominant SNCA, LRRK2, VPS35 mutations or autosomal-recessive Parkin, PINK1, DJ1 mutations using NCBI’s PubMed database ( https://www.ncbi.nlm.nih.gov/pubmed) and standardized search terms (Supporting Information Table S1 as previously reported [10, 11]). The literature search was limited to the time from the last MDSGene update (in 2019) until April 2020. Titles, abstracts, and, where applicable, full text of peer-reviewed, original articles in English were screened for inclusion in the systematic literature review. Older articles (published before 2019), that have been included in MDSGene, were screened for information on treatment. Treatment data were extracted as presented and interpreted in the original publication. This applies for response quantification as well as for the presence of levodopa-induced side effects. Quantification of response was divided into three groups 1) “good” including reports of “good” and/or “excellent” response, i.e. remarkable reduction of PD symptoms, 2) “moderate”, i.e. some but limited response on treatment, and 3) “minimal” including “minimal” or “intermittent”, i.e. very limited or short-lasting response according to the assertion in screened papers. Treatment-related side effects are difficult to disentangle from disease progression effects. For instance, we here categorized motor fluctuations as a treatment-related effect but they can also be considered as a sign of advanced disease progression. Further, dystonia can be part of the phenotypic spectrum of PD, especially in PARK-Parkin [10], and in the context of OFF dystonia in the morning that usually responds well to levodopa. Due to the lack of data, however, the distinction between treatment-related side effect and accompanying clinical PD feature could not be made for dystonia. Frequently reported side effects include dyskinesias; in contrast, neuropsychiatric symptoms (e.g. hallucinations, psychosis, impulse control disorders) have rarely been addressed in the publications. Decarboxylase inhibitors such as benserazide and carbidopa were not listed as additional drugs as they are usually combined with levodopa although this is often not specifically mentioned in the reports.
Inclusion and exclusion criteria for patients and genetic variants
A prerequisite for inclusion in this systematic treatabolome was reporting of individual genetic (exact variant), phenotypic, and treatment data. Mutation carriers were only included if they had definite PD designated as such in the publication or if they presented with at least one cardinal parkinsonian sign (i.e., resting tremor, rigidity, or postural instability) in addition to bradykinesia. This included patients with and without a family history of PD. This review covers three autosomal-dominant and three autosomal-recessive PD genes. For the autosomal-dominant gene LRRK2, patients with heterozygous and homozygous mutations were included since no phenotypic differences have been reported for carriers of different mutational load [10]. For the autosomal-recessive genes, only patients with biallelic mutations were included [11]. In addition, reviews on any of the six genes of interest were screened for additional data and for additional, potentially eligible articles. For an overview of the literature search as well as the filtering procedure, see Fig. 1. A list of all eligible articles can be found in Supporting Information Table S2.
Fig. 1
Flow chart of systematic literature review. The flow diagram shows the steps and respective numbers of papers.
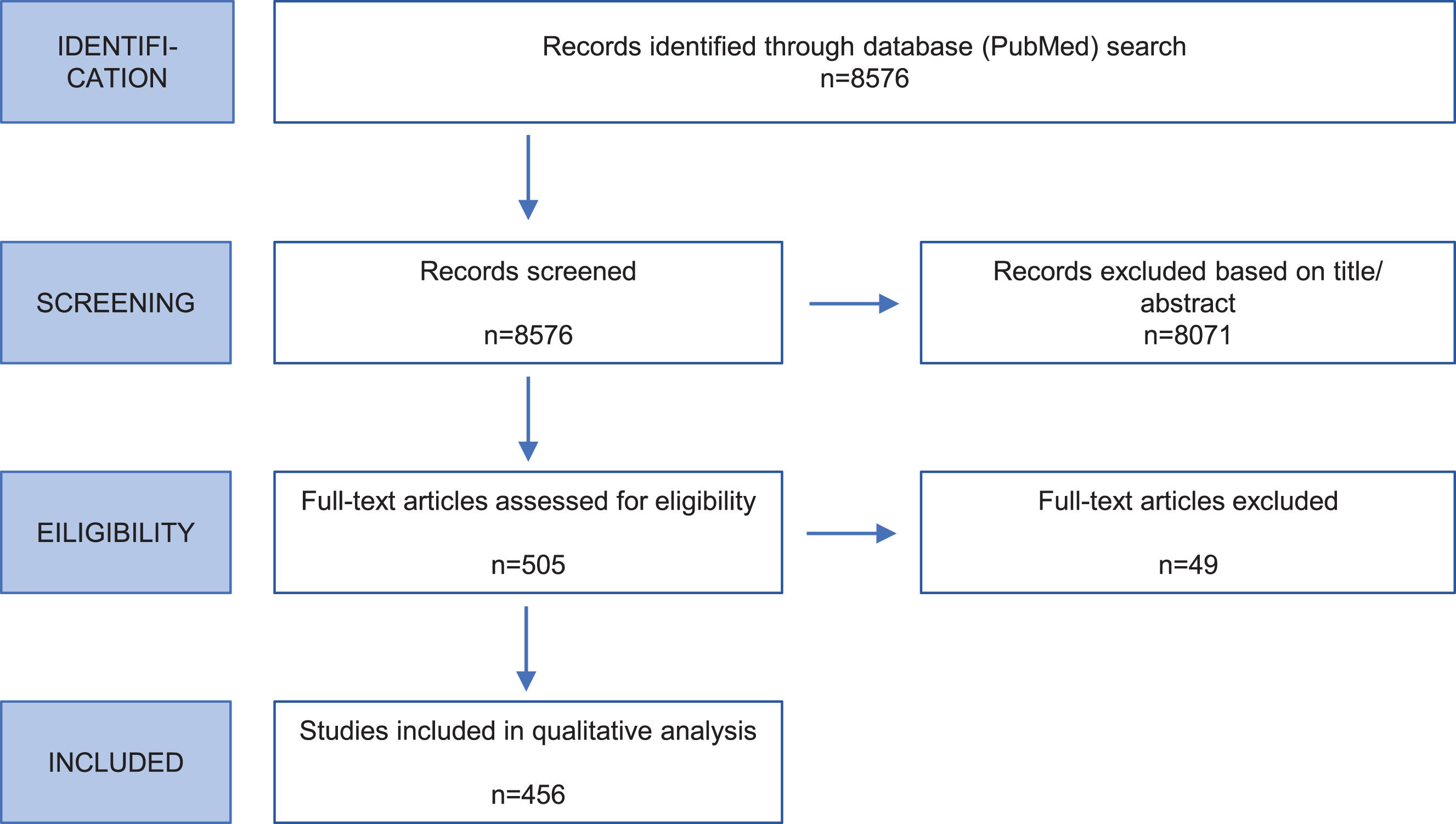
Carriers of variants considered to be non-pathogenic (benign) were excluded as well as carriers of mutations in more than one PD gene. Variants were excluded if they had a minor allele frequency (MAF) ≥ 1%based on ethnicity with the maximal MAF in the ExAC Browser (https://exac.broadinstitute.org) or gnomAD Browser (https://gnomad.broadinstitute.org/), and/or in at least 100 unaffected control individuals screened for the variant of interest in the respective publication.
Pathogenicity scoring
Degree of pathogenicity of a genetic variant was assessed as previously described (www.mdsgene.org/methods) [10]. Mutations were classified as definitely (score > 14), probably (score 10–14), or possibly pathogenic (score 5–9), or as benign (score < 5) based on segregation, MAF in databases, CADD score (Combined Annotation Dependent Depletion, https://cadd.gs.washington.edu/), and evidence from functional studies.
Data collection process
Characteristics of each publication were captured in standardized data extraction form, using Microsoft Excel. This format allows automated input into a treatabolome database [9]. We applied the above-described modified MDSGene data extraction protocol to all eligible articles. For each publication, data on demographic, genetic, clinical variables (as previously reported [10, 11]), and information on treatment were extracted (see Supporting Information Table S3 for the list of extracted treatment variables). Treatment variables comprised dose and response to levodopa, brain surgery, and alternative medications (i.e. dopamine agonists, MAO-B inhibitors). Genetic nomenclature was harmonized and curated from the given information in the publication, wherever possible, using the Ensemble (https://www.ensembl.org/) and MutationTaster database ( https://www.mutationtaster.org/) [10]. All mutations were mapped to GRCh37/hg19.
Statistical analysis
We have calculated mean values for relevant variables (i.e. age at onset, levodopa dose, duration levels) and provide minimum and maximum values (range). For group comparisons, we used one-way ANOVA tests (https://www.socscistatistics.com/tests/anova/default2.aspx). Confidence intervals were calculated using Z statistics (n > 30) or T statistics.
RESULTS
Articles and study types
The PubMed literature search resulted in 8,576 citations (Supporting Information Table S1). After evaluating in a two-step procedure abstracts and full text (Fig. 1), 456 publications contained information on mutation carriers that were eligible for inclusion in the database (49 for SNCA, 154 for LRRK2, 11 for VPS35, 172 for Parkin, 52 for PINK1, 18 for DJ1) (Supporting Information Table S2). Most frequently included study types were case series/reports, mutational screens, and family studies.
Included patients, pathogenicity of mutations, and data missingness
We included 2,226 patients with possible, probably, or definitely pathogenic scored SNCA (146 patients), LRRK2 (820 patients), VPS35 (74 patients), Parkin (1002 patients), PINK1 (151 patients), or DJ1 (33 patients) variants. These patients carried 320 different variants (10 in SNCA, 17 in LRRK2, 10 in VPS35, 192 in Parkin, 69 in PINK1, 22 in DJ1). Information on response to levodopa was available for a total of 1,373 patients (61.7%) while treatment with brain surgery (mostly DBS) has been reported for 67 individuals (3.0%) (Figs. 2–7).
Fig. 2
Results of the systematic literature review for SNCA mutation carriers. A) The chart shows information on carriers of SNCA mutations with levodopa treatment, respective response and side effects, subdivided by different mutations. B) Information on brain surgery and response.
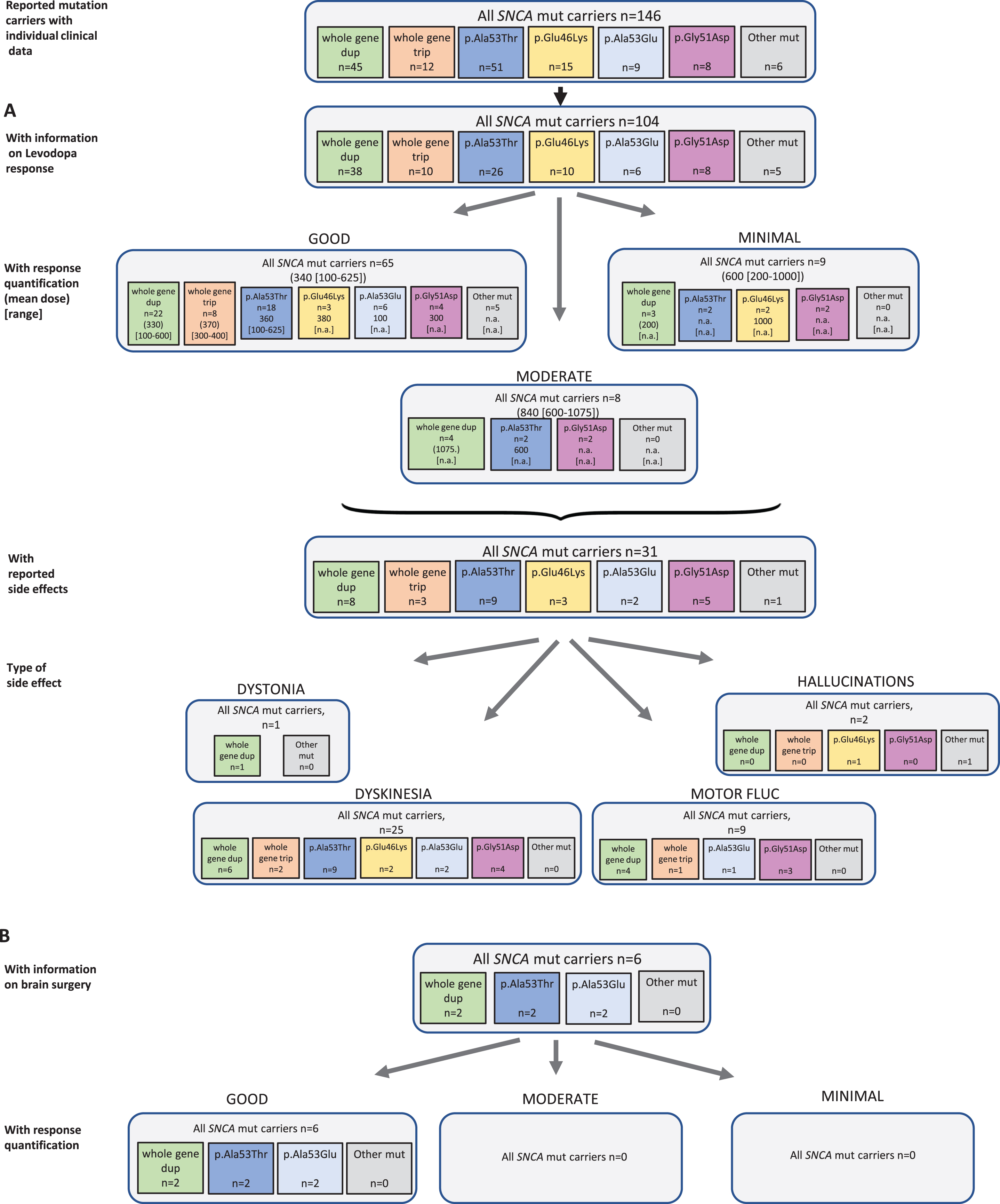
Fig. 3
Results of the systematic literature review for LRRK2 mutation carriers. A) The chart shows information on carriers of LRRK2 mutations with levodopa treatment, respective response and side effects, subdivided by the most frequent mutations. B) Information on brain surgery and response.
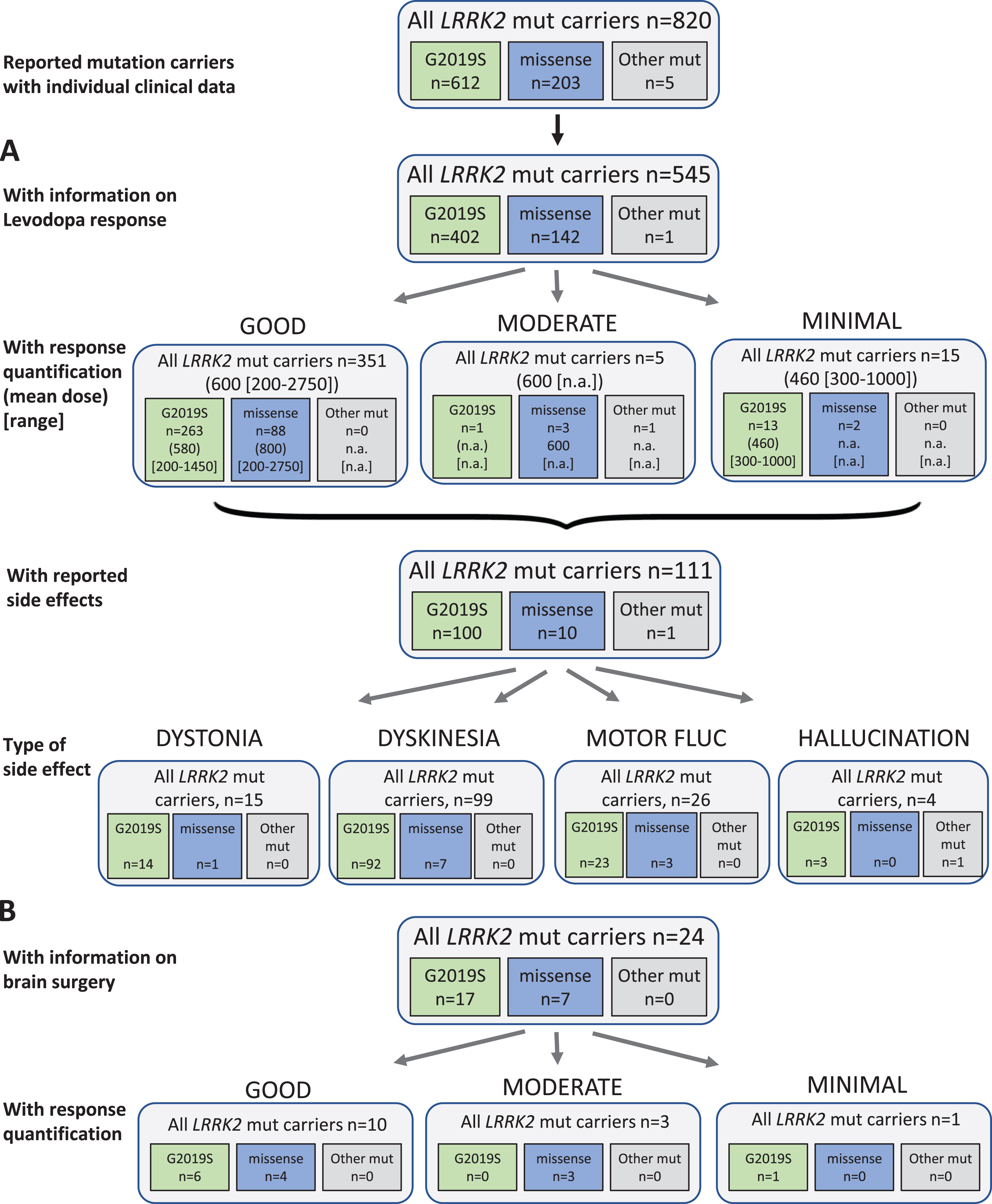
Fig. 4
Results of the systematic literature review for VPS35 mutation carriers. A) The chart shows information on carriers of VPS35 mutations with levodopa treatment, respective response and side effects, subdivided by the most frequent mutations. B) Information on brain surgery and response. *: Other side effects have not been reported.
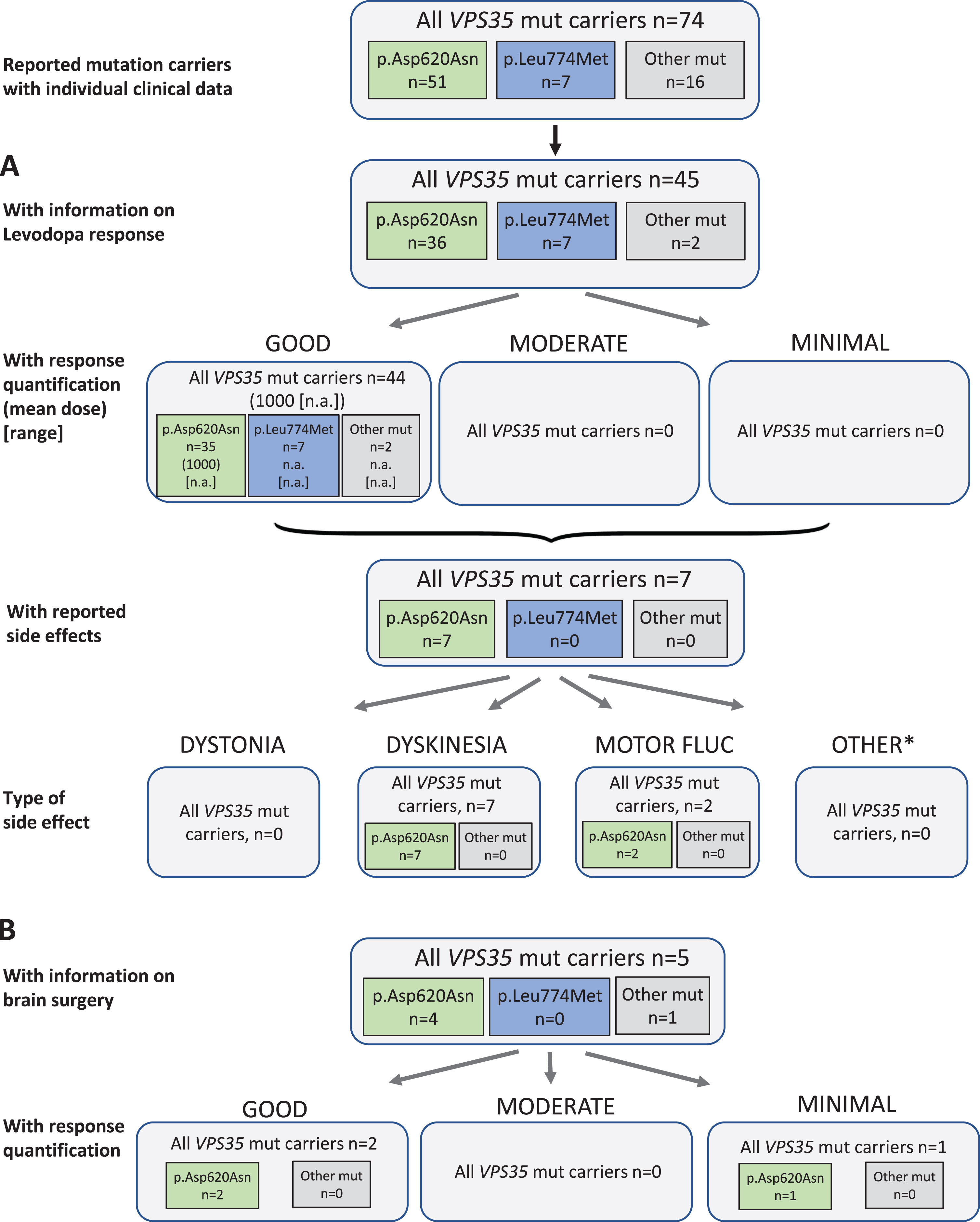
Fig. 5
Results of the systematic literature review for Parkin mutation carriers. A) The chart shows information on carriers of Parkin mutations with levodopa treatment, respective response and side effects, subdivided by type of mutation. B) Information on brain surgery and response.
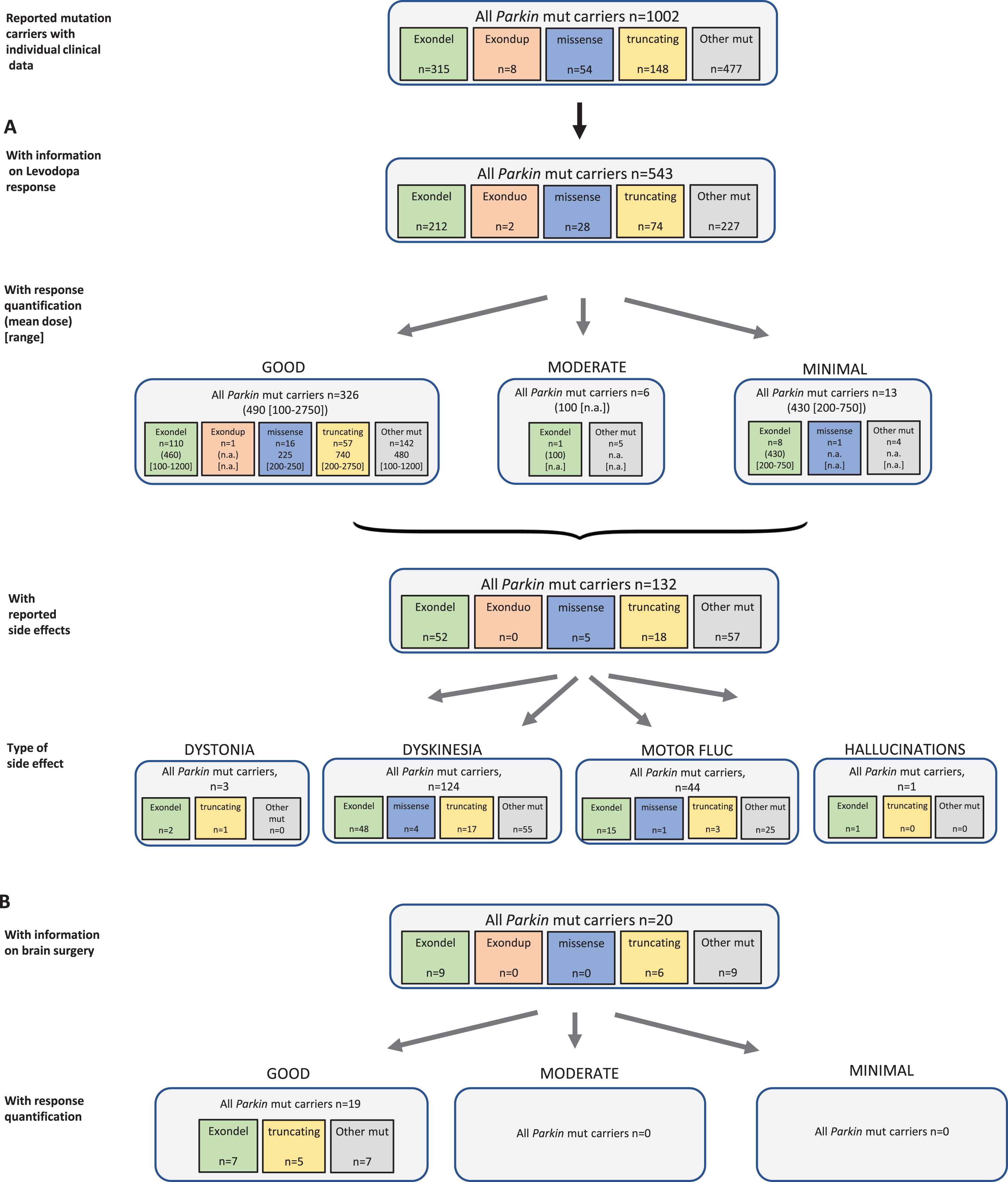
Fig. 6
Results of the systematic literature review for PINK1 mutation carriers. A) The chart shows information on carriers of PINK1 mutations with levodopa treatment, respective response and side effects, subdivided by the most frequent mutations. B) Information on brain surgery and response. *: Other side effects have not been reported.
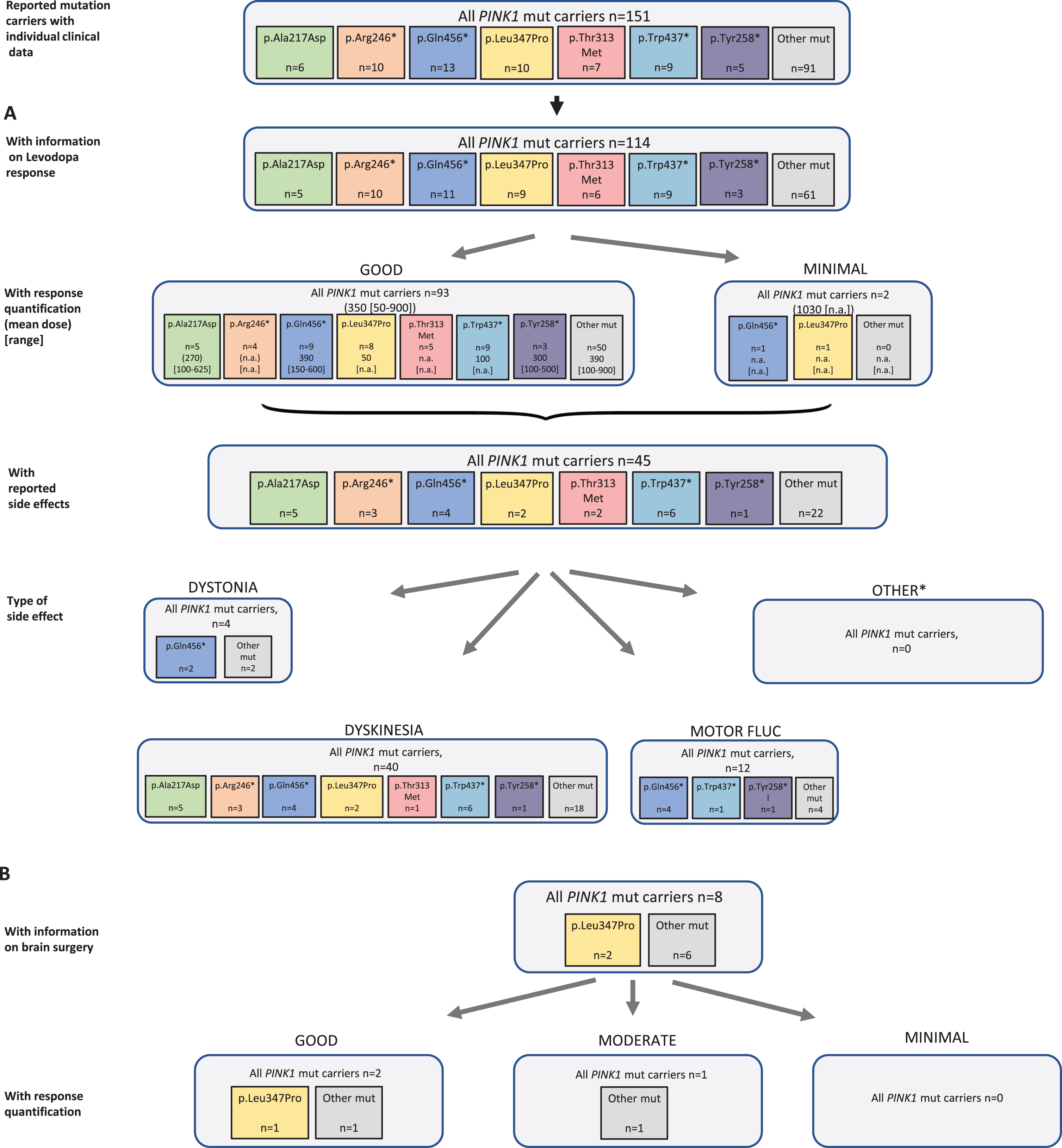
Fig. 7
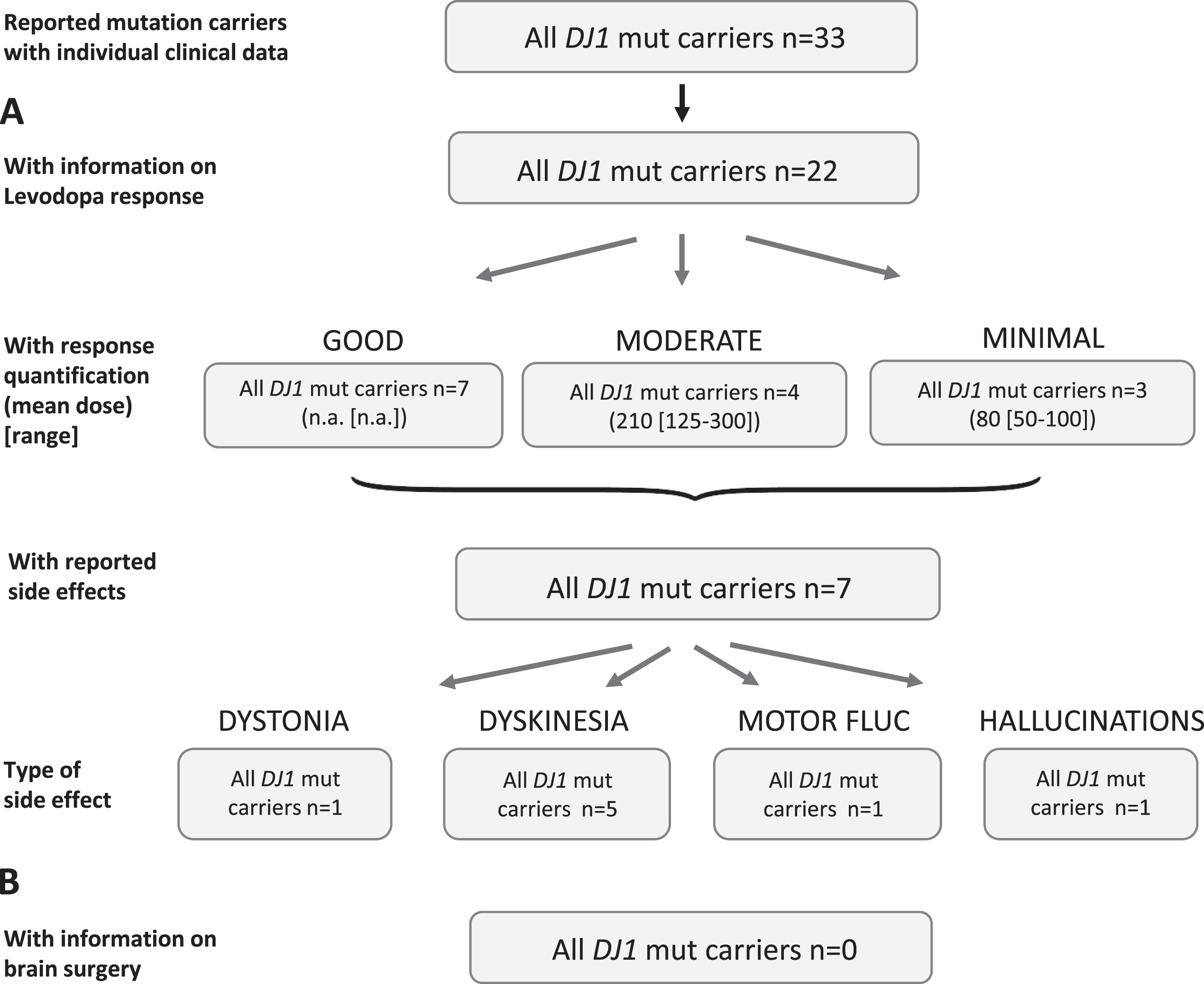
Results of the systematic literature review for DJ1 mutation carriers. A) The chart shows information on carriers of DJ1 mutations with levodopa treatment, respective response and side effects. Due to the low number of recurrent mutations, no subdivision for the most frequent mutations has been included. B) No patient with brain surgery has been reported.
A major challenge concerning the preparation of this systematic review was the missing detailed data on pharmacological and surgical treatment, duration of treatment, and side effects. In case of reported side effects, differentiation between the mentioned symptom as a side effect or as an original PD symptom was a challenge. This large amount of missing data, and even when reported then not clearly, made it difficult to make statistically valuable statements.
Current therapeutic options for Parkinson’s disease
In the following paragraph, we introduce the different treatments that are applied to improve the motor signs of PD. In the current literature, there are only guidelines for idiopathic PD available [2]. In expert’s opinion, levodopa, which is a prodrug that is metabolized in the brain to active dopamine, is still the most efficient medication although most guidelines recommend dopamine agonists as a first-line therapy in younger patients [2, 3]. Levodopa treatment is typically combined with a decarboxylase inhibitor to prevent premature degradation of levodopa.
In addition, there are other pharmacological treatment options besides levodopa, which can be divided into dopaminergic and non-dopaminergic [12]. Dopamine agonists, COMT inhibitors and MAO-B inhibitors are counted among dopaminergic drugs, NMDA antagonists and anticholinergics are non-dopaminergic. A variety of non-ergoline dopamine agonists (i.e. piribedil, rotigotine, pramipexole, ropinirole) is available on the market whereas ergoline agonists are nowadays only rarely prescribed due to their association with myocardial fibrosis. They act agonistic at postsynaptic striatal dopamine receptors and thus mimic the effects of levodopa but may show a different pattern of receptor subtype affinity. COMT inhibitors (i.e. entacapone, tolcapone, opicapone) act by inhibiting the enzyme catechol-O-methyltransferase, which is involved in dopamine degradation to methyldopa, the result is an increase in the available amount of dopamine. MAO-B inhibitors (i.e. selegiline, rasagiline, safinamide) provide another therapeutic addition. They also inhibit dopamine degradation and thus increase the amount of dopamine in the striatum. Another effect comprises the inhibition of the re-uptake of dopamine localized in the presynapse of neurons. Another option for the therapy of PD is the administration of the NMDA antagonist amantadine, which has many different mechanisms. It attenuates the overactivity of cholinergic striatal interneurons and, as a weak NMDA receptor antagonist, slows down the influence of glutamatergic projections from the cortex. Thus, it also acts indirectly agonistically on dopamine receptors in the brain by increasing dopamine release and by inhibiting dopamine reuptake into the presynaptic nerve cells. Anticholinergics (i.e. trihexyphenidyl, biperiden, benzatropine) are a group of drugs, which act by suppressing the effect of acetylcholine in the central nervous system.
Nowadays, DBS is the most widely used type of surgery; pallidotomy and thalamotomy are also rarely applied. These lesional procedures currently undergo a renaissance due to the recent introduction of transcranial magnetic resonance-guided focused ultrasound [13]. Indications for DBS are disturbing, treatment-related motor fluctuations and levodopa-induced dyskinesias or treatment-resistant PD tremor despite an optimized medical treatment. Intolerable side effects of medication, e.g. impulse control disorders, are another indication for DBS [4]. Established targets for DBS are the subthalamic nucleus (STN), globus pallidus internus (GPi) and thalamus (nucleus ventralis intermedius (VIM)).
PARK-SNCA
Among 146 reported SNCA mutation carriers, 104 received levodopa and seven patients were reportedly untreated. There was no information for the other patients (24.0%missing data). Among the 82 patients for whom quantitative information on response was available, 65 patients (79.3%) showed a good response at a mean dose of 340 (reported range in the patients with available information: 100–625) mg/d. Rather higher doses were used in eight patients with moderate response (mean dose 840 mg/d, range: 600–1075), and nine patients with minimal response (mean dose 600 [200–1000] mg/d (Fig. 2A). Looking at individual mutations, there was no obvious difference, but numbers are small and differences cannot be excluded. For instance, while all carriers with response quantification of a whole gene triplication and almost all of the duplication carriers (22/29, 75.9%) showed a good levodopa response, only 4/8 (50.0%) of the carriers of the p.Gly51Asp missense variant showed that outcome. In SNCA, 31 patients reported side effects (30.1%) including 25 patients with dyskinesia, one patient with dystonia, and two patients with other side effects (i.e. hallucinations). Nine patients were reported with motor fluctuations. The duration of levodopa treatment could not be specified based on the publications.
Regarding non-levodopa medication, 36 patients were reported who received dopamine agonists, COMT inhibitors, MAO-B inhibitors, anticholinergics, and/or NMDA antagonist, respectively. Most of the patients benefitted from the non-levodopa medication.
Six patients received brain surgery, such as DBS (n = 4) or thalamotomy/pallidotomy (n = 1 each). For all patients, a good response was reported (Fig. 2B).
PARK-LRRK2
Among 820 evaluated LRRK2 mutation carriers, 545 patients received levodopa therapy and 17 patients were reportedly untreated (258 without information, 31.5%). The mean duration of treatment in LRRK2 carriers was 17 years, however it should be noted that duration was only given for a very small number of patients (n = 50). Among the 371 patients for whom quantitative information on response was available, the vast majority (94.6%; 351 patients) showed a good outcome with a mean daily levodopa dose of 600 (200–2750) mg/d. Only five patients had a moderate outcome at a dose of 600 (range not available) mg/d; 15 patients had minimal outcome (mean dose: 460 [range: 300–1000] mg/d). The vast majority of patients carried the p.G2019S mutation and no mutation specific response rate could be evaluated (Fig. 3A). Side effects of levodopa therapy occurred in 111 patients (20.4%), comprising 15 patients with dystonia, 99 with dyskinesia, and 4 patients with other side effects (i.e. hallucinations). Motor fluctuations as a treatment related effect were reported in 26 patients.
Regarding non-levodopa medication, 76 patients were reported to receive non-levodopa medications including dopamine agonists, COMT inhibitors, MAO-B inhibitors, NMDA antagonist amantadine, and/or the trihexyphenidyl. The treatment was often beneficial. The use of amantadine had mixed outcome.
Twenty-four patients received brain surgery (18 DBS, 5 pallidotomy, 1 thalamotomy). Ten of them (10/14) with reported outcome showed a good response, three had a moderate, and one minimal benefit, respectively. Numbers were too low to evaluate a mutation-specific effect (Fig. 3B).
PARK-VPS35
Among the three dominantly inherited PD genes, least information was available for VPS35. Among 74 patients with a VPS35 mutation causing PD, 45 (60.8%) have been indicated to receive levodopa therapy, one patient was reported to be untreated (37.8%missing data). The outcome was reported as “good” for all patients with response quantification. The mean daily levodopa dose in the good responders was 1000 mg/d, noteworthy dose was given for only one patient. Seven patients (15.6%) were reported to have side effects on levodopa therapy. While none of them had dystonia, all seven patients developed dyskinesia, and two patients showed also motor fluctuations as a treatment related effect (Fig. 4A). Duration of Levodopa treatment was not further specified.
Concerning non-levodopa therapy, two patients received a dopamine agonist, anticholinergics, and/or tolcapone as a COMT inhibitor. When reported, the patients benefitted from this forms of treatment. There were no reports on other non-levodopa medications.
Five patients received brain surgery (all deep brain stimulation) as treatment; two of them had a good outcome, one was reported with minimal outcome. For the other two the outcome was not reported (Fig. 4B).
PARK-Parkin
Among the six PD genes addressed in this systematic review, most mutation carriers harbored biallelic Parkin mutations (n = 1002); 543 of them (54.2%) have been indicated to receive levodopa therapy, three were reported as untreated (45.3%missing data). Mean duration of levodopa administration was 13 years (SD 3 years) in the cases with respective information. For 345 of these mutation carriers, response quantification was available and 326 patients (94.5%) were reported with good outcome at an average dose of 490 (range: 100–2750) mg/d. Another six patients were published with moderate outcome at, notably, lower levodopa dose (mean 100 [range: n.a.] mg/d), and 13 patients showed minimal outcome at a mean dose of 430 (range: 200–750) mg/d. The mutation carriers with response quantification information carried 76 different Parkin variants. For a sub-analysis, we grouped variants according to mutation type (exon deletion, exon duplication, missense variants, truncating variants, and other). In all groups, >94%of patients showed a good response to the levodopa therapy (Fig. 5A). A total of 132 patients (24.3%) showed side effects with the levodopa therapy including 124 mutation carriers with dyskinesia, three with reportedly levodopa induced dystonia, one with hallucinations, and 44 with motor fluctuations as an indicated treatment related effect (Fig. 5A).
Different groups of non-levodopa therapy were administered in a total of 91 patients including dopamine agonists, COMT inhibitors, MAO-B inhibitors, anticholinergics and/or amantadine. Outcome was mostly beneficial, when provided.
Brain surgery was reported in 20 Parkin mutation carriers, and a good outcome was reported in all patients (19/19) with response quantification (Fig. 5B). Most of the patients had DBS and three patients thalamotomy or pallidotomy.
PARK-PINK1
In all eligible publications, 151 patients with biallelic PINK1 mutations were found including 114 carriers who were reported to receive levodopa therapy (75.5%). There were two untreated patients reported and for 35 (23.3%) of patients no information was available. The duration level of levodopa treatment can be specified with 13 years (SD 7 years). Among the 95 patients with response quantification, the vast majority (n = 93, 97.9%) showed a good outcome at a mean dose of 350 (range: 50–900) mg/d; the other two had minimal response at a higher mean dose of 1030 mg/d (Fig. 6A). The mutation carriers with information about response quantification carried 42 different PINK1 variants. For none of the mutations, >15 datasets were available. Therefore, response quantification could not be stratified for individual mutations. Side effects of the levodopa therapy were seen in 45 patients (39.5%) including 40 mutation carriers with dyskinesia and four with dystonia; motor fluctuations as a treatment related effect were mentioned in 12 mutation carriers.
Looking at non-levodopa therapy options, 31 patients were treated with dopamine agonists, COMT inhibitors, MAO-B inhibitors, anticholinergics, and/or amantadine. Outcome was usually beneficial when information on response was provided.
Among the eight reported patients who received brain surgery (DBS: n = 6; thalamotomy: n = 1, unspecified: n = 1), outcome was only reported in three patients (all treated with DBS) who were indicated to be responsive (two good, one moderate) (Fig. 6B).
PARK-DJ1
Least mutation carriers have been reported for DJ1 (n = 33) and 22 of them were reported to receive levodopa (66.7%), four patients were untreated (21.2%missing data). Response quantification was available for 14 patients, seven of whom (50%) showed a good response while four were moderate and three minimal responders (Fig. 7A). Information on dose was mostly not available and was rather low in the group of moderate (mean: 210 [range: 125–300] mg/d) and minimal responders (mean: 80 [range: 50–100]. Seven patients (31.8%) were reported with side effects including dyskinesia in five patients, dystonia in one, one patient showed hallucinations, and one showed motor fluctuations as a treatment related effect.
Non-levodopa medication was mentioned in the publications for 11 patients including dopamine agonists, NMDA antagonists, and/or anticholinergics, respectively. For about half of the patients a good outcome was noticed, otherwise it was mostly unspecified.
None patient was reported who was treated with brain surgery (Fig. 7B).
Comparison of clinical characteristics in autosomal-dominant mutation carriers
An overview of key figures for the genes SNCA, LRRK2, and VPS35 is provided in Table 1. Although the number of reported mutation carriers for all three genes is >1,000 in total, information on therapy on an individual level has been rather rarely provided. Levodopa is the most widely used treatment with a good response in ∼80%of SNCA and ∼95–100%in LRRK2 and VPS35 mutation carriers. However, the numbers should be interpreted with caution since response quantification was only available for about half of the patients (497/1,040). Even less information was available for side effects on levodopa therapy. Overall, side effects have been mentioned in 152 mutation carriers with an occurrence in ∼15–40%of VPS35, LRRK2, and SNCA mutation carriers in increasing order. However, the absolute numbers especially for VPS35 are small and 95%confidence interval (CI) is 3.4–23.2%. The most frequent side effect in all three forms was dyskinesia (∼80–100%of patients with reported side effects per gene), followed by motor fluctuations (∼20–30%) as a treatment related effect.
Table 1
Overview of treatment information per gene
| SNCA | LRRK2 | VPS35 | Parkin | PINK1 | DJ-1 | Total | |
| Reported mutation carriers | 146 | 820 | 74 | 1002 | 151 | 33 | 2226 |
| Reportedly untreated mutation carriers | 7 | 17 | 1 | 3 | 2 | 4 | 34 |
| (4.8%) | (2.1%) | (1.4%) | (0.3%) | (1.3%) | (12.1%) | (1.5%) | |
| Information on Levodopa dose | 32 | 117 | 1 | 98 | 40 | 5 | 293 |
| “Good” benefit from Levodopa (%of pat. with response quantification) | 65/82 | 351/371 | 44/44 | 326/345 | 93/95 | 7/14 | 886/951 |
| (79.3%) | (94.6%) | (100%) | (94.5%) | (97.9%) | (50.0%) | (93.2%) | |
| No Levodopa response | 0 | 4 | 0 | 6 | 1 | 0 | 11 |
| Published side effects on Levodopa (%of pat. with reported Levodopa response) | 31/104 | 111/545 | 7/45 | 132/543 | 45/114 | 7/22 | 336/1373 |
| (41.7%) | (20.4%) | (15.6%) | (24.3%) | (39.5%) | (31.8%) | (24.5%) | |
| “Good” benefit from brain surgery (%of pat. with response quantification) | 6/6 | 10/14 | 2/3 | 19/19 | 2/3 | n.a. | 39/45 |
| (100%) | (71.4%) | (66.7%) | (100%) | (66.7%) | (86.7%) |
With respect to non-levodopa medications, the spectrum of treatments was most diverse in SNCA mutation carriers. Independent of the gene, most mutation carriers benefitted from the non-levodopa medication.
Data on brain surgery were only available for a few mutation carriers. While all SNCA mutation carriers (6/6) with reported response quantification had a good outcome, about 71.4%of the LRRK2 mutations carriers (10/14) and 66.7%of the VPS35 mutation carriers (2/3) were reported to respond well.
Comparison of clinical characteristics of autosomal-recessive mutation carriers
An overview of key figures for the genes Parkin, PINK1, and DJ1 is provided in Table 1. Although the number of reported mutation carriers for all three genes well exceeds 1,000 individuals, information on therapy on an individual level has been rarely indicated. Levodopa is the most widely used treatment with a good response in ∼95%of Parkin and PINK1 but only in ∼50%(95%CI: 33.5–86.4%) of DJ1 mutation carriers. Of note, there is a significant difference in the response rate among the three genes (descriptive p < 0.0001 [ANOVA]). However, the numbers should be interpreted with caution since response quantification was only available for 38.8%of the patients (454/1186). Similarly, only little information was available for side effects on levodopa therapy. Overall, side effects have been indicated in 184 mutation carriers with an occurrence in ∼25–40%of carriers with recessive PD gene mutations (highest in PINK1 mutation carriers). As for the dominant PD genes, the most frequently reported side effect in all three forms were dyskinesias in ∼70–95%of patients with reported side effects per gene; most often in Parkin mutation carriers. In addition, motor fluctuations have been reported as treatment related effects in ∼15–30%of Parkin, PINK1, and DJ1 mutation carriers.
With respect to non-levodopa medications, the number of patients and diversity of treatments reflected the number of reported mutation carriers. Independent from the gene, most mutation carriers benefitted from the non-levodopa medication. Numbers on individual substances are too small for meaningful comparisons.
Data on brain surgery were only available for Parkin and PINK1 mutation carriers. When reported, all Parkin and 2 of 3 PINK1 patients had a good outcome.
DISCUSSION
This systematic review provides an overview of previously published information about pharmacological and surgical treatment options for autosomal-dominant (SNCA, LRRK2, VPS35) and autosomal-recessive (Parkin, PINK1, DJ1) forms of PD. It aims to be a compass for clinicians to treat their patients. Since the first PD gene has been reported in 1997 [14], SNCA, the review covers articles published in the past >20 years. We screened ∼9,000 publications and included 456 eligible studies with ∼1,000 mutation carriers for whom treatment information was given. To our knowledge, this is a first systematic review on published treatment and outcome in monogenic PD.
The most important and less surprising result is that treatment of PD is mainly independent from the genetic cause and the overall strategy is comparable to idiopathic PD. With this, the situation is different from other diseases such as myasthenic syndromes where one drug might be beneficial in a given genetic form but harmful in other genetic settings [8].
Another important finding is the overall lack of detailed reports on medication, respective response, and treatment-related complications, e.g. exact numbers of dyskinesias, hallucinations, psychosis, or impulse control disorders. The available figures for the latter were so small that it could not be analyzed. Lack of data represented a challenge in the evaluation of the various treatment options. Thus, results and conclusions based on the analysis of data should be interpreted with caution. It might well be that there is publication bias towards unusual cases neglecting reporting good responders which might be the case for response to DBS in LRRK2 mutation carriers. The genetic diversity (many different disease-causing mutations in most of the PD genes) did hamper mutation specific analyses and allowed only for gene-based comparisons to some extent.
Our review reveals that levodopa is beneficial for most patients but also that carriers of DJ1 and SNCA mutations may benefit less. A potential bias entails differences in disease stages independent of the genotype. It is thus difficult to draw conclusions and establish recommendations due to patient-specific factors. Treatment can be highly variable in respect to drug (levodopa or dopamine agonist) and dose. Similarly, also for DBS, many parameters can be modified including location of the electrode, active contacts, mode and steering of stimulation, amplitude, pulse width and frequency. On the other hand, patients vary in more than just their PD-causing mutations. There are other (genetic) effects that influence outcome of pharmacotherapy, and disease duration and co-morbidities also impact on treatment options. With most of the available articles being case reports/studies or mutational screens, it becomes clear that individual treatment for each patient is most important but difficult to predict. In general, the clinician adjusts the treatment according to the clinical phenotype, e.g. if a patient is more prone to dementia as in PARK-SNCA, the clinician will prescribe less dopamine agonists, lower levodopa doses, and DBS will rather not be performed as dementia is a contraindication for DBS.
Of note, reports on brain surgery in monogenic PD are rare independent of the gene, and the mutation, and approaches vary. However, based on quite small numbers, most of the reported patients experienced a good outcome. In the future, additionally reported mutation carriers treated with brain surgery will allow for more powerful and reliable evaluations.
Taking a more detailed look on the information given about side effects of levodopa therapy, the lack of information here is remarkable. For clinicians and patients, it would be useful to know, whether gene specific side effects may occur and whether this differs between different mutations. Notably, differentiation between side effect-caused symptoms and symptoms of PD progression are almost impossible to disentangle. This makes it a challenge creating for each PD patient with him/her personal mutation a specified therapy that combines best working pharmacological and surgical treatment options.
So far, there is still a lack of disease-modifying or –preventing treatment strategies for PD. The application of levodopa, a classic treatment, alleviates the signs and symptoms of PD, but it is also associated with the occurrence of dyskinesias, motor fluctuations and neuropsychiatric symptoms, which are limitations in this treatment for PD [15, 16]. Newly gained molecular insights might contribute to the development of novel therapeutic targets focusing not only on signs and symptoms but also on causes and thereby attenuating neurodegeneration and disease progression. The immune system for instance gains increasing interest in PD research [17]. The death of dopaminergic neurons is accompanied by astrocytic dysfunction, microglia hyper-activation and activation of various inflammatory networks in the substantia nigra of PD [17]. Since the role of neuroinflammation is increasingly recognized in PD [18, 19], potentially new therapeutics may target neuroinflammatory processes in PD [20]. Other approaches focus on compensating mutation-specific effects. For instance, the formation of alpha-synuclein oligomers and fibrils, enhanced by PD-causing SNCA mutations, results in alpha-synuclein aggregation and Lewy body pathology, a hallmark of PD. The first active and passive alpha-synuclein immunizations are currently tested in randomized clinical trials in patients with idiopathic PD [21]. Another example are LRRK2 inhibitors as most LRRK2 mutations increase phosphorylation of LRRK2 and other targets in the sense of a gain-of-function mechanism. Thus, LRRK2 kinase inhibitors can counteract the increased kinase activity to due PD-causing mutations [21]. Promising preclinical studies with LRRK2 inhibitors but also with an antisense oligonucleotide are the basis for first clinical trials in the early clinical phase.
Thus, to increase treatment outcome in PD two important approaches are taken: First, two systematically evaluate outcomes of symptomatic treatments and second, to further develop drugs targeting underlying disease mechanisms including gene-specific pathways in selected, monogenic patients and thus to attenuate neurodegeneration in this disabling disease.
CONCLUSION
Several conclusions can be drawn from this systematic review on treatment of monogenic PD with dominant mutations in SNCA, LRRK2, or VPS35 or with recessive mutations in Parkin, PINK1, and DJ1. First, there is no treatment that is harmful to one genetic form but beneficial in another one. Second, the report of detailed treatment data including dose and response quantification is rare. Third, based on the available data from about half of >2,000 mutation carriers, it can be concluded that most patients, independent of the genetic cause, respond well to levodopa. The percentage of good responders seems to be lowest for DJ1 and SNCA mutation carriers, respectively. Thus, levodopa is the most efficient and most frequently prescribed drug in monogenic PD as is for idiopathic PD. Fourth, accordingly, brain surgery is rather rarely applied and reported. Fifth, there is a wide spectrum of (additional) non-levodopa drugs that often show an additive beneficial effect. Sixth, reported outcomes on brain surgery comprise good outcome in all mutation carriers but the benefit might be limited in some LRRK2 patients [22]. Finally, information on treatment of specific mutations is rather anecdotal and cannot (yet) be statistically analyzed. However, to achieve the goal of best treatment in neurodegenerative disorders such as PD, it is necessary that clinicians have access to mutation-related treatment outcomes and can share their experience via a treatment database (treatabolome) containing all available information in a standardized format.
ACKNOWLEDGMENTS
This work was funded by the German Research Foundation (FOR2488) and the Damp foundation. L.O. is the recipient of a stipend for her medical thesis (“Excellenzstipendium”) from the University of Lübeck.
CONFLICT OF INTEREST
The authors do not have any conflict of interest.
N.B. served as a consultant for Centogene. NB is funded by the DFG (BR4328.2-1/2-2, GRK1957), the Collaborative Center for X-linked Dystonia-Parkinsonism and the Else-Kröner Fresenius-Stiftung (HA17_2017).
K.L. received funding from the German Research Foundation, the International Parkinson’s disease and Movement Disorder Society (MDS), and the Damp foundation. She is further an active member of the Global Parkinson’s Genetics Program (GP2) initiative.
REFERENCES
[1] | Obeso JA , Stamelou M , Goetz CG , Poewe W , Lang AE , Weintraub D , Burn D , Halliday GM , Bezard E , Przedborski S , Lehericy S , Brooks DJ , Rothwell JC , Hallett M , DeLong MR , Marras C , Tanner CM , Ross GW , Langston JW , Klein C , Bonifati V , Jankovic J , Lozano AM , Deuschl G , Bergman H , Tolosa E , Rodriguez-Violante M , Fahn S , Postuma RB , Berg D , Marek K , Standaert DG , Surmeier DJ , Olanow CW , Kordower JH , Calabresi P , Schapira AHV , Stoessl AJ . Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy. Mov Disord. (2017) ;32: :1264–310. |
[2] | Fox SH , Katzenschlager R , Lim SY , Barton B , de Bie RMA , Seppi K , Coelho M , Sampaio C , Movement Disorder Society Evidence-Based Medicine C. International Parkinson and movement disorder society evidence-based medicine review: Update on treatments for the motor symptoms of Parkinson’s disease. Mov Disord. (2018) ;33: :1248–66. |
[3] | Armstrong MJ , Okun MS . Diagnosis and Treatment of Parkinson Disease: A Review. JAMA. (2020) ;323: :548–60. |
[4] | Bronstein JM , Tagliati M , Alterman RL , Lozano AM , Volkmann J , Stefani A , Horak FB , Okun MS , Foote KD , Krack P , Pahwa R , Henderson JM , Hariz MI , Bakay RA , Rezai A , Marks WJ Jr , Moro JL , Vitek A , Weaver FM , Gross RE , DeLong MR . Deep brain stimulation for Parkinson disease: An expert consensus and review of key issues. Arch Neurol. (2011) ;68: :165. |
[5] | Domingo A , Klein C . Genetics of Parkinson disease. Handb Clin Neurol. (2018) ;147: :211–27. |
[6] | Collaborators GBDPsD. Global, regional, and national burden of Parkinson’s disease,1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. (2018) ;17: :939–53. |
[7] | Vollstedt EJ , Kasten M , Klein C , Group MGGPsDS.Using global team science to identify genetic parkinson’s disease worldwide. Ann Neurol. (2019) ;86: :153–7. |
[8] | Thompson R , Bonne G , Missier P , Lochmuller H . Targeted therapies for congenital myasthenic syndromes: Systematic review and steps towards a treatabolome. Emerg Top Life Sci. (2019) ;3: :19–37. |
[9] | Atalaia A , Thompson R , Corvo A , Carmody L , Piscia D , Matalonga L , Macaya A , Lochmuller A , Fontaine B , Zurek B , Hernandez-Ferrer C , Rheinard C , Gomez-Andres D , Desaphy JF , Schon K , Lohmann K , Jennings MJ , Synofzik M , Riess O , Yaou RB , Evangelista T , Ratnaike T , Bros-Facer V , Gumus G , Horvath R , Chinnery P , Laurie S , Graessner H , Robinson P , Lochmuller H , Beltran S , Bonne G . A guide to writing systematic reviews of rare disease treatments to generate FAIR-compliant datasets: Building a Treatabolome. Orphanet J Rare Dis. (2020) ;15: :206. |
[10] | Kasten M , Hartmann C , Hampf J , Schaake S , Westenberger A , Vollstedt EJ , Balck A , Domingo A , Vulinovic F , Dulovic M , Zorn I , Madoev H , Zehnle H , Lembeck CM , Schawe L , Reginold J , Huang J , Konig IR , Bertram L , Marras C , Lohmann K , Lill CM , Klein C . Genotype-Phenotype Relations for the Parkinson’s Disease Genes Parkin, PINK1, DJ MDSGene Systematic Review. Mov Disord. (2018) ;33: :730–41. |
[11] | Trinh J , Zeldenrust FMJ , Huang J , Kasten M , Schaake S , Petkovic S , Madoev H , Grünewald A , Almuammar S , König IR , Lill CM , Lohmann K , Klein C , Marras C . Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS MDSGene systematic review. Mov Disord. (2018) ;33: :1857–70. |
[12] | Oertel W , Schulz JB . Current and experimental treatments of Parkinson disease: A guide for neuroscientists. J Neurochem. (2016) ;139: (Suppl 1):325–37. |
[13] | Gallay MN , Moser D , Jeanmonod D . Safety and accuracy of incisionless transcranial MR-guided focused ultrasound functional neurosurgery: Single-center experience with 253 targets in 180 treatments. J Neurosurg. (2018) ;1–10. |
[14] | Polymeropoulos MH , Lavedan C , Leroy E , Ide SE , Dehejia A , Dutra A , Pike B , Root H , Rubenstein J , Boyer R , Stenroos ES , Chandrasekharappa S , Athanassiadou A , Papapetropoulos T , Johnson WG , Lazzarini AM , Duvoisin RC , Di Iorio G , Golbe LI , Nussbaum RL . Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. (1997) ;276: :2045–7. |
[15] | Tarakad A , Jankovic J . Diagnosis and Management of Parkinson’s Disease. Semin Neurol. (2017) ;37: :118–26. |
[16] | Zahoor I , Shafi A , Haq E . Pharmacological Treatment of Parkinson’s Disease. In: Stoker TB, Greenland JC (eds) Parkinson’s Disease: Pathogenesis and Clinical Aspects. Brisbane (AU) (2018). |
[17] | Yang L , Mao K , Yu H , Chen J . Neuroinflammatory Responses and Parkinson’ Disease: Pathogenic Mechanisms and Therapeutic Targets. J Neuroimmune Pharmacol. (2020). |
[18] | Joshi N , Singh S . Updates on immunity and inflammation in Parkinson disease pathology. J Neurosci Res. (2018) ;96: :379–90. |
[19] | Sliter DA , Martinez J , Hao L , Chen X , Sun N , Fischer TD , Burman JL , Li Y , Zhang Z , Narendra DP , Cai H , Borsche M , Klein C , Youle RJ . Parkin and PINK1 mitigate STING-induced inflammation. Nature. (2018) ;561: :258–62. |
[20] | Tan EK , Chao YX , West A , Chan LL , Poewe W , Jankovic J . Parkinson disease and the immune system - associations, mechanisms and therapeutics. Nat Rev Neurol. (2020) ;16: :303–18. |
[21] | Sardi SP , Cedarbaum JM , Brundin P . Targeted Therapies for Parkinson’s Disease: From Genetics to the Clinic. Mov Disord. (2018) ;33: :684–96. |
[22] | Kuusimaki T , Korpela J , Pekkonen E , Martikainen MH , Antonini A , Kaasinen V . Deep brain stimulation for monogenic Parkinson’s disease: A systematic review. J Neurol. (2020) ;267: :883–97. |


