
Laminopathies’ Treatments Systematic Review: A Contribution Towards a ‘Treatabolome’
Abstract
Background:
Variants in the LMNA gene, encoding lamins A/C, are responsible for a growing number of diseases, all of which complying with the definition of rare diseases. LMNA-related disorders have a varied phenotypic expression with more than 15 syndromes described, belonging to five phenotypic groups: Muscular Dystrophies, Neuropathies, Cardiomyopathies, Lipodystrophies and Progeroid Syndromes. Overlapping phenotypes are also reported. Linking gene and variants with phenotypic expression, disease mechanisms, and corresponding treatments is particularly challenging in laminopathies. Treatment recommendations are limited, and very few are variant-based.
Objective:
The Treatabolome initiative aims to provide a shareable dataset of existing variant-specific treatment for rare diseases within the Solve-RD EU project. As part of this project, we gathered evidence of specific treatments for laminopathies via a systematic literature review adopting the FAIR (Findable, Accessible, Interoperable, and Reusable) guidelines for scientific data production.
Methods:
Treatments for LMNA-related conditions were systematically collected from MEDLINE and Embase bibliographic databases and clinical trial registries (Cochrane Central Registry of Controlled Trials, clinicaltrial.gov and EudraCT). Two investigators extracted and analyzed the literature data independently. The included papers were assessed using the Oxford Centre for Evidence-Based Medicine 2011 Levels of Evidence.
Results:
From the 4783 selected articles by a systematic approach, we identified 78 papers for our final analysis that corresponded to the profile of data defined in the inclusion and exclusion criteria. These papers include 2 guidelines/consensus papers, 4 meta-analyses, 14 single-arm trials, 15 case series, 13 cohort studies, 21 case reports, 8 expert reviews and 1 expert opinion. The treatments were summarized electronically according to significant phenome-genome associations. The specificity of treatments according to the different laminopathic phenotypical presentations is variable.
Conclusions:
We have extracted Treatabolome-worthy treatment recommendations for patients with different forms of laminopathies based on significant phenome-genome parings. This dataset will be available on the Treatabolome website and, through interoperability, on genetic diagnosis and treatment support tools like the RD-Connect’s Genome Phenome Analysis Platform.
INTRODUCTION
Variants in the LMNA gene, encoding A-type lamins, are responsible for a growing number of rare monogenic diseases. A unique characteristic of the LMNA pathogenic variants is that they lead to a myriad of phenotypic expressions although they arise from the same gene. A complete explanation for this phenotypic variability still lacks at present despite the ever-growing amount of data from research [1–6]. A-type Lamins (lamins A and C) are intermediate filaments that build a meshwork at the inner face of the nuclear membrane after polymerization. They are also present in the nucleoplasm. They interact with the DNA, histones and chromatin in the nucleus, protect it from mechanical stress [7] and help in the maintenance of the nuclear shape while providing an anchorage to the endoplasmic reticulum through their interaction with other proteins like SUN1/SUN2 and the outer layers of the nuclear membrane [8].
The disorders that arise from changes to the LMNA gene have a varied phenotypic expression with more than 15 syndromes already described belonging to five phenotypic groups of pathologies, i.e. Muscular Dystrophies, Neuropathies, Cardiomyopathies, Lipo-dystrophies and Progeroid Syndromes [9]. Phenotypic overlaps are also reported between one or several laminopathic entities. The ubiquitous LMNA expression and the major role of A-type lamins in the functional organization of chromatin and the subsequent regulation of developmental genes probably play important roles in the pathophysiology of the different tissue-specific laminopathies [10]. In addition, the variable phenotypic expression arising from pathological LMNA variants could also result from epigenetic factors, modifier genes, altered expression levels and defective protein processing. Consequently, connecting gene and variants with phenotypic expression, disease mechanisms, and corresponding treatments is challenging in laminopathies. However, this approach could provide useful data to improve the guidelines and recommendations for the clinical management of these diseases, which remain under-recognized.
A recent paper on congenital myasthenia syndro-mes served as proof of concept of an innovative idea that consists of assembling a knowledge database of gene and variant-specific treatments for a significant entity group while preparing its future integration into electronic decision-support systems. This concept was baptized “Treatabolome” by the authors [11]. Subsequently, a standard methodology has been defined for other disease groups writing systematic literature reviews (SLR) of treatments in their expertise area [12]. The Treatabolome concept is developed within the Solve-RD EU project and addresses the need to identify and improve the visibility of the existing specific treatments for rare diseases. Several teams have collected gene and variant-specific treatments for different rare diseases in Findable, Accessible, Interoperable, and Reusable (FAIR)-compliant datasets [13]. This information will be freely available through the Treatabolome website to complement existing diagnostic tools and support clinical management.
The current paper is an attempt to collect know-ledge of specific treatments for laminopathies. However, since pathogenic LMNA variants may trigger varied phenotypical presentations, laminopathies do not display univocal genome-phenome relationships, thus hindering the collection of variant-specific treatments. To adapt to these circumstances, we have decided to flag significant phenome-genome associations that trigger laminopathies’ treatment recommendations.
We first collected 4783 papers through a systematic approach, then selected 78 studies reporting treatments for the diverse forms of laminopathies. From these data, we generated FAIR-compatible datasets to feed the Laminopathies’ Treatabolome knowledge base. The corresponding complete dataset is provided as a Supplementary File S1.
List of Abbreviations
| ARVC | Arrhythmogenic Right Ventricular Cardiomyopathy |
| ChEBI https://www.ebi.ac.uk/chebi/ | Chemical Entities of Biological Interest |
| CENTRAL (Cochrane Central Registry of Controlled Trials) https://www.cochranelibrary.com/central/about-central | The Cochrane Central Register of Controlled Trials (CENTRAL) is a highly concentrated source of reports of randomized and quasi-randomized controlled trials |
| CHADS-VASC score | The CHADS2 score and its updated version, the CHA2DS2-VASc score, are clinical prediction rules for estimating the risk of stroke in patients with non-rheumatic atrial fibrillation (AF), a common and serious heart arrhythmia associated with thromboembolic stroke |
| Clinicaltrials.gov | ClinicalTrials.gov is a database of privately and publicly funded clinical studies conducted around the world |
| CMD1A | Familial Dilated Cardiomyopathy, type 1A (i.e. related to LMNA) |
| CRT-D | Cardiac Rehabilitation Therapy - Defibrillator |
| DCM | Dilated cardiomyopathy |
| Embase www.embase.com | Embase is the most comprehensive source for biomedical literature (36 + million records) from peer reviewed journals and conference abstracts |
| EDMD2 | Emery-Dreifuss Muscular Dystrophy type 2 |
| EudraCT https://eudract.ema.europa.eu/ | EudraCT (European Union Drug Regulating Authorities Clinical Trials Database) is the European database for all interventional clinical trials on medicinal products authorized in the European Union (EEA) and outside the EU/EEA if they are part of a Pediatric Investigation Plan (PIP) from 1 May 2004 onwards |
| EU | European Union |
| FAIR | Findable, Accessible, Interoperable, and Reusable. “The principles emphasize machine-actionability (i.e., the capacity of computational systems to find, access, interoperate, and reuse data with none or minimal human intervention) because humans increasingly rely on computational support to deal with data as a result of the increase in volume, complexity, and creation speed of data” (see http://go-fair.org) |
| FPLD2 | Familial Partial Lipodystrophy type 2, Dunnigan Syndrome |
| HGPS | Hutchinson-Guilford Progeria Syndrome |
| ICD | Implantable Cardioversion Defibrillator |
| LMNA-CMD | LMNA-related congenital muscular dystrophy |
| MADA | Mandibulo Acral Dysplasia with Type A Lipodystrophy |
| MEDLINE https://www.nlm.nih.gov/bsd/medline.html | MEDLINE is the U.S. National Library of Medicine® (NLM) premier bibliographic database that contains more than 26 million references to journal articles in life sciences with a concentration on biomedicine. A distinctive feature of MEDLINE is that the records are indexed with NLM Medical Subject Headings (MeSH®) |
| OEBML https://www.cebm.ox.ac.uk/resources/levels-of-evidence | Oxford Evidence-Based Medicine Level |
| PCOS | Polycystic Ovary Syndrome |
| PRISMA http://www.prisma-statement.org | PRISMA is an evidence-based minimum set of items for reporting in systematic reviews and meta-analyses. |
| PROSPERO https://www.crd.york.ac.uk/prospero/ | Website from the University of York, UK, that accepts registrations for systematic reviews, rapid reviews and umbrella reviews. |
| PubMed https://pubmed.ncbi.nlm.nih.gov/ | PubMed® comprises more than 30 million citations for biomedical literature from MEDLINE, life science journals, and online books. |
| RD-Connect | The RD-Connect Project was a multidisciplinary project running from 2012 to 2018 that united partners from the EU and beyond to create an integrated global infrastructure for Rare Disease research. |
| SLR | Systematic literature review |
| Solve-RD https://www.solve-rd.eu | European research project aiming to solve the NGS-unsolved rare disease cases |
| Treatabolome | Publicly-available database of gene and variant-specific treatments, to be designed within the Solve-RD project |
METHODS
Published treatments for LMNA-related conditions were collected and appraised following a research question shared by all Treatabolome systematic literature reviews [12]: “What treatments have been described for this condition/gene/variant; on which specific genetic variants have they been tested; and what is the strength of the associated supporting evidence?”. This review follows the recommendations from the Cochrane Collaboration systematic reviews handbook [14] and the Centre for Reviews and Dissemination, namely by adopting the Systematic Review Protocol template of the PROSPERO tool [15]. The reporting of our findings follows the PRISMA reporting guidelines [16].
Search methods
We have searched the Cochrane Central Registry of Controlled Trials, clinicaltrial.gov and EudraCT (https://eudract.ema.europa.eu/eudract-web/login/login.faces) for clinical trials on LMNA-related diseases treatments. Simultaneously, we accessed MED-LINE and Embase through PubMed to extract any publications on the same subject. We did not impose a starting date for data collection that has included all references up to 31/12/2019. The searches were made in English, French, Spanish, Italian and Portuguese. We ran recurrent searches with the same search strategy that consisted of de-duplicating independent searches by each one the following expressions (all fields): “LMNA”, “Lamin A/C”, “A type Lamin”, “Lamin A”, “Lamin C” and “Laminopathy OR Lam-inopathies”.
The search results were then reviewed by title and abstract, followed by a selective full-text data extraction. Inclusion and exclusion criteria are listed in Table 1. An electronic data capture form was built for this purpose by one of the authors (AA) using Filemaker Pro version 12 Software. This form was inspired by a publicly-available template from the Cochrane Collaboration Project [17] and followed the Methodological Expectations of Cochrane Intervention Reviews - the MECIR Standards [18]. We also complied with the Treatabolome Systematic Reviews’ Methodology paper [12].
Table 1
Inclusion and exclusion criteria
| Inclusion Criteria | Exclusion Criteria |
| Papers with any report of clinical use of a treatment for a LMNA gene-related disease, from single case reports to meta-analysis | Papers reporting preclinical treatments for LMNA gene-related diseases |
RESULTS
The PRISMA flowchart (see Fig. 1) details the publication numbers at each stage of our selection.
Fig. 1
Laminopathies’ Treatabolome PRISMA Flow Diagram.
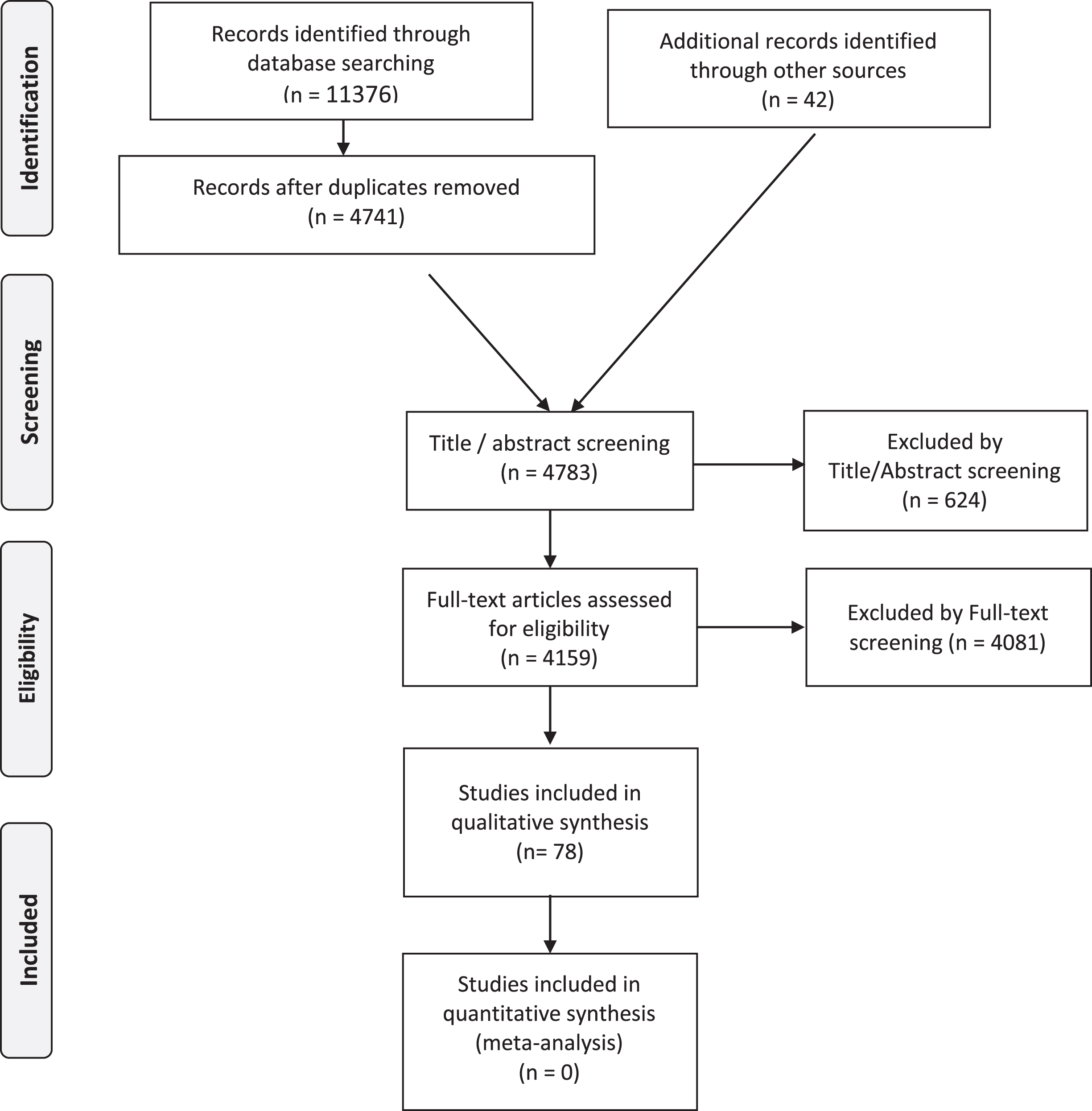
After applying the described search strategy in PubMed, the initial starting number of papers was 11376 and the number went down to 4741 after de-duplication of entries with the following number of references for each search term:
– “LMNA”: 1536 references
– “Lamin A/C”: 2585 (932 duplicates eliminated: 1653)
– “A type Lamin”: 3213 (2145 duplicates eliminated: 1068)
– “Lamin A”: 2891 (2587 duplicates eliminated: 304)
– “Lamin C”: 450 (437 duplicates eliminated: 13)
– “Laminopathy OR Laminopathies”: 701 (534 duplicates eliminated: 167).
We then added 42 papers from additional sources (mainly expert bibliography references, besides ClinicalTrials.gov, EudraCT and Cochrane Library), reaching 4783 references eligible for Title/Abstract screening. At this stage, we excluded 624 references, mainly because they were unrelated to the LMNA gene. We full-text reviewed 4159 papers. We excluded 4081 papers, mainly for not addressing treatment findings or presenting only preclinical therapies in animal models and/or cell-culture experiments (see inclusion and exclusion criteria in Table 1). At the end of the process, 78 articles ended up in the qualitative analysis. These papers include 2 guidelines/consensus papers, 4 meta-analyses, 14 single-arm trials, 15 case series, 13 cohort studies, 21 case reports, 8 expert reviews and 1 expert opinion.
Two investigators extracted and analyzed the literature data independently. The treatments were summarized electronically according to significant phenome-genome associations. A complete list of the reported treatments is provided in Table 2.
Table 2
Summary of reported laminopathy treatments
| Treatment or intervention database | Treatment or intervention name | Treatment or intervention ID | Main Phenotype | Pubmed # |
| ChEBI | Corticosteroid | CHEBI: 50858 | LMNA-CMD | 26034236 |
| MeSH | Anesthesia (Total Intravenous Anesthesia TIVA) | D000758 | EDMD2 | 22973525 |
| MeSH | Implantable Cardiac Defibrillator (ICD) | D017147 | CMD1A | 23811080, 17605093, 29173404, 26835025, 23946316, 22019351, 30287275, 12854972, 18926329, 30482687, 15551023, 22281253, 31155932, 28696268, 20627339, 23483212, 26385533, 30586772, 30518714, 15598919, 27993908, 27884249, 17605093, 29173404, 26835025, 23946316, 22019351, 30287275, 12854972, 18926329, 30482687, 15551023, 22281253, 31155932, 28696268, 20627339, 23483212, 26385533, 30586772, 30518714, 15598919, 27993908 |
| MeSH | Transplant (heart) | D019737 | CMD1A | 31060954, 30287275, 18926329, 30482687 |
| MeSH | Catheter Ablation | D017115 | CMD1A | 31060954, 29759522, 27506821 |
| MeSH | Cardiac Pacing, Artificial | D002304 | CMD1A | 26620845 |
| MeSH | CRT-D Cardiac Resynchronization Therapy | D058406 | CMD1A | 30891417 |
| ChEBI | Anticoagulation | CHEBI: 50249 | CMD1A | 30191544, 30518714, 23073275 |
| MeSH | rt-PA (alteplase) | D010959 | CMD1A | 30191544, 30518714, 23073275 |
| MeSH | Percutaneous atrial appendage occlusion | D020517; Q000601£ | CMD1A | 29570041 |
| ChEBI | Insulin | CHEBI: 145810 | FPLD2 | 21168376 |
| ChEBI | Pioglitazone | CHEBI: 8228 | FPLD2 | 18728124 |
| ChEBI | Pioglitazone | CHEBI: 8228 | FPLD2 | 18728124 |
| Metformin | CHEBI: 6801 | |||
| Flutamide | CHEBI: 5132 | |||
| ChEBI | Pioglitazone | CHEBI: 8228 | FPLD2 | 17936664 |
| Metformin | CHEBI: 6801 | |||
| ChEBI | Pioglitazone | CHEBI: 8228 | FPLD2 | 19249234 |
| Metformin | CHEBI: 6801 | |||
| Insulin | CHEBI: 145810 | |||
| ChEBI | Fenofibrate | CHEBI: 5001 | FPLD2 | 19249234 |
| ChEBI | Nicotinamide | CHEBI: 17154 | FPLD2 | 12766116 |
| ChEBI | Rosiglitazone | CHEBI: 50122 | FPLD2 | 16241930, 22274718, 14510863 |
| ChEBI | Liraglutide | CHEBI: 71193 | FPLD2 | 29044799 |
| MeSH | Roux en Y Gastric Bypass | D015390 | FPLD2 | 27778252 |
| MeSH | Noninvasive Ventilation | D063087 | FPLD2 | 17893350, 19418082 |
| ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | FPLD2 with hypoleptinemia | 19727665, 31135595, 31194872, 30296183, 26584826, 25734254, 27710244, 30370487, 31620670, 24926953, 27692500, 30990519, 27207511, 31300002, 30539782, 23439261, 15791619, 22068254, 29644599, 27642538, 30805888, 29267953 |
| MeSH | Surgery, Plastic | D013518 | FPLD2 | 21561824, 21306965 |
| ChEBI | Troglitazone (No market authorization for safety reasons) | CHEBI: 9753 | FPLD2 | 10929166 |
| ChEBI | Lonafarnib | CHEBI: 47097 | HGPS | 23012407, 29710166 |
| ChEBI | Lonafarnib | CHEBI: 47097 | HGPS | 27400896 |
| Pravastatin | CHEBI: 63618 | |||
| Zoledronic acid | CHEBI: 46557 | |||
| ChEBI | Alendronic acid / biphosphonates in general | CHEBI: 2567 | HGPS | 27400896 |
| ChEBI | Growth Hormone (GH) | CHEBI: 37845 | HGPS | 31199775, 17642424, 9258264 |
CMD1A: Familial Dilated Cardiomyopathy, type 1A; EDMD2: Autosomal Dominant Emery-Dreifuss Muscular Dystrophy 2; FPLD2: Familial Partial Lipodystrophy, Dunningan Type; HGPS: Hutchinson-Gilford Progeria Syndrome; LMNA-CMD: LMNA-related congenital muscular dystrophy. £D020517 code for atrial appendage, Q000601 qualifier for surgery, no qualifier was found for percutaneous procedures.
The specificity of treatments according to the different LMNA-related diseases is variable. Some therapeutic approaches are specific for a unique phenotypical presentation. Others apply for laminopathic phenotypes that share a common clinical feature, as it happens regarding the risk of cardiac arrhythmia and sudden death, present both in cardiomyopathies and in several other phenotypic groups of laminopathies. The Tables 3 to 4, specific of laminopathic phenotypes, are ordered according to the alphabetic order of the treatment or intervention names.
Table 3
LMNA-related muscular syndromes treatment
| Pubmed | Ref. | Clinical diagnosis ORDO | ORPHA code | Type of study | OCEBM | Number LMNA patients | HGVS cDNA | HGVS protein | Treatment database | Treatment or intervention name | Treatment or intervention ID | Clinical effect | Comments |
| 22973525 | Schuster et al., 2012 [90] | EDMD2 | 98853 | Case report | 5 | 1 | NA | NA | MeSH | Anesthesia (Total Intravenous Anesthesia TIVA) | D000758 | large | safe in this patient, presumed LMNA from phenotype |
| 30518714 | Wang et al., 2019 [91] | EDMD2 | 98853 | Expert review | 5 | NA | NA | NA | ChEBI | Anticoagulation | CHEBI: 50249 | large | prevention of stroke |
| 26034236 | Moraitis et al., 2015 [31] | LMNA-CMD | 157973 | Case report | 5 | 1 | c.91_93 | p.Glu31del | ChEBI | Corticosteroid | CHEBI: 50858 | small | motor improvement |
| delGAG | |||||||||||||
| 17605093 | Antoniades et al., 2007 [34] | EDMD2 | 98853 | Case series | 4 | 15 | c.908_909 | p.Ser303 | MeSH | ICD | D017147 | large | sudden cardiac death prevention |
| delCT | CysfsX27 | ||||||||||||
| 30518714 | Wang et al., 2019 [91] | EDMD2 | 98853 | Expert review | 5 | NA | NA | NA | MeSH | ICD | D017147 | large | prevention of stroke |
EDMD2: Autosomal Dominant Emery-Dreifuss Muscular Dystrophy 2; LMNA-CMD: LMNA-related Congenital Muscular Dystrophy.
Table 4
LMNA-related sudden cardiac death preventive treatment
| Pubmed | Ref. | Clinical diagnosis ORDO | ORPHA code | Type of study | OCEBM evidence | Number of LMNA patients | HGVS cDNA | HGVS protein | Treatment or intervention database | Treatment or intervention name | Treatment or intervention ID | Clinical effect | Comments |
| 23073275 | van Rijsingen et al., 2013 [50] | CMD1A | 300751 | Case-control study | 3 | 76 | NA | NA | ChEBI | Anticoagulation | CHEBI: 50249 | large | NA |
| 30191544 | Homma et al., 2018 [92] | CMD1A | 300751 | Case report | 5 | 1 | NA | NA | ChEBI | Anticoagulation | CHEBI: 50249 | large | NA |
| 27506821 | Kumar et al., 2016 [49] | CMD1A | 300751 | Cohort study | 4 | 25 | NA | NA | MeSH | Catheter Ablation | D017115 | moderate | NA |
| 29759522 | Roberts et al., 2017 [48] | CMD1A | 300751 | Case report | 5 | 1 | c.979C > G | p.Leu327Val | MeSH | Catheter Ablation | D017115 | moderate | NA |
| 31060954 | Hasebe et al., 2019 [46] | CMD1A | 300751 | Cohort study | 4 | 6 | IVS3–10A > G | p.?p.Asp272 | MeSH | Catheter Ablation | D017115 | moderate | transient effects |
| 815_818 delins | AlafsX203 | ||||||||||||
| CCAGAC | |||||||||||||
| 26620845 | Kato et al., 2016 [36] | CMD1A | 300751 | Case series | 4 | 2 | c.2T > A | p.Met1? | MeSH | Cardiac Pacing, Artificial | D002304 | small | does not prevent sudden cadiac death |
| ARVD | 293910 | c.1542G > A | p.Trp514* | ||||||||||
| 30891417 | Rudbeck-Resdal et al., 2019 [93] | CMD1A | 300751 | Case report | 5 | 1 | c.1411C.T | p.Arg471Cys | MeSH | CRT-D Cardiac Resynchronization Therapy | D058406 | moderate | NA |
| 12854972 | MacLeoad et al., 2003 [94] | CMD1A | 300751 | Case report | 5 | 1 | c.908_909delCT | p.Ser303 | MeSH | ICD | D017147 | large | NA |
| CysfsX27 | |||||||||||||
| 15598919 | Desai et al., 2004 [95] | CMD1A | 300751 | Meta-analysis | 1 | 1854 | NA | NA | MeSH | ICD | D017147 | large | NA |
| 15551023 | van Berlo et al., 2005 [32] | CMD1A | 300751 | Meta-analysis | 1 | 299 | NA | NA | MeSH | ICD | D017147 | large | NA |
| 18926329 | Pasotti et al., 2008 [38] | CMD1A | 300751 | Cohort study | 4 | 94 | NA | NA | MeSH | ICD | D017147 | large | NA |
| 20627339 | Meune et al., 2011 [96] | CMD1A | 300751 | Cohort study | 4 | 19 | NA | NA | MeSH | ICD | D017147 | large | NA |
| 26835025 | Charron et al., 2012 [40] | CMD1A | 300751 | Expert review | 5 | NA | NA | NA | MeSH | ICD | D017147 | large | NA |
| EDMD2 | 264 | ||||||||||||
| EDMD2 | 98853 | ||||||||||||
| 22019351 | Keller et al., 2012 [35] | CD1A | 300751 | Case report | 5 | 1 | c.367_369delAAG | p.Lys123del | MeSH | ICD | D017147 | large | NA |
| 22281253 | van Rijsingen et al., 2012 [41] | CMD1A | 300751 | Case series | 3 | 149 | NA | NA | MeSH | ICD | D017147 | large | NA |
| 23811080 | Anselme et al., 2013 [42] | CMD1A | 300751 | Case series | 4 | 47 | c.16C > T | p.Gln6* | MeSH | ICD | D017147 | large | Innapropriate shocks |
| c.748G > C | p.Arg249Pro | ||||||||||||
| c.1129C > T | p.Arg377Cy | ||||||||||||
| c.1130G > A | p.Arg377His | ||||||||||||
| c.1145G > A | p.Arg482Gl | ||||||||||||
| c.1589T > C | p.Leu530Pro | ||||||||||||
| 23946316 | Disertori et al., 2013 [43] | CMD1A | 300751 | Expert review | 5 | NA | NA | NA | MeSH | ICD | D017147 | large | NA |
| 23483212 | Ng &Kaye, 2013 [97] | CMD1A | 300751 | Case report | 5 | 1 | NA | NA | MeSH | ICD | D017147 | large | NA |
| 27884249 | Kumar et al., 2016 [45] | CMD1A | 300751 | Cohort study | 4 | 87 | NA | NA | MeSH | ICD | D017147 | large | NA |
| 26385533 | Olde Nordkampe et al., 2016 [88] | CMD1A | 300751 | Meta-analysis | 1 | 462 | NA | NA | MeSH | ICD | D017147 | large | ICD implantation carries a significant risk of inappropriate shocks and in hospital &post discharge complications in relatively young patients with inherited arrhythmia syndromes |
| 29173404 | Atteya et al., 2017 [98] | CMD1A | 300751 | Expert review | 5 | NA | NA | NA | MeSH | ICD | D017147 | large | NA |
| 27993908 | Golwhala et al., 2017 [99] | CMD1A | 300751 | Meta-analysis | 1 | 1854 | NA | NA | MeSH | ICD | D017147 | large | NA |
| 28696268 | Halliday et al., 2017 [44] | CMD1A | 300751 | Expert review | 5 | NA | NA | NA | MeSH | ICD | D017147 | large | NA |
| 30586772 | Kusumoto et al., 2019 [100] | CMD1A | 300751 | Cohort study | 1 | NA | NA | NA | MeSH | ICD | D017147 | large | NA |
| 30287275 | Kwapich et al., 2019 [101] | CMD1A | 300751 | Case-control study | 4 | 58 | c.139G > T | p.Asp47Tyr | MeSH | ICD | D017147 | large | NA |
| c.310C > G | p.Leu104Val | ||||||||||||
| c.398G > T | p.Arg133Leu | ||||||||||||
| c.448A > G | p.Thr150Ala | ||||||||||||
| c.467G > A | p.Arg156His | ||||||||||||
| c.481G > A | p.Glu161Lys | ||||||||||||
| c.694G > C | p.Gly232Arg | ||||||||||||
| c.751C > T | p.Gln251* | ||||||||||||
| c.860del | p.Ala287Valfs*193 | ||||||||||||
| c.949G > A | p.Glu317Lys | ||||||||||||
| c.1157G > C | p.Arg386Thr | ||||||||||||
| c.1173dup | p.Ser392Glnfs*34 | ||||||||||||
| c.1238del; | p.Gly413Alafs*67 | ||||||||||||
| c.1315C > T | p.Arg439Cys | ||||||||||||
| c.1357C > T | p.Arg453Trp | ||||||||||||
| c.1444C > T | p.Arg482Trp | ||||||||||||
| c.1445G > A | p.Arg482Gln | ||||||||||||
| c.1445G > T | p.Arg482Leu | ||||||||||||
| c.1930C > T | p.Arg644Cys | ||||||||||||
| 30482687 | Peters et al., 2019 [47] | CMD1A | 300751 | Expert review | 5 | NA | NA | NA | MeSH | ICD | D017147 | large | NA |
| 31155932 | Wahbi et al., 2019 [39] | CMD1A | 300751 | Cohort study | 3 | 444 | NA | NA | MeSH | ICD | D017147 | large | innapropriate implantation of ICD |
| 29570041 | De Roeck et al., 2019 [102] | CMD1A | 300751 | Case report | 5 | 1 | c.235C > G | p.Leu85Val | MeSH | percutaneous atrial appendage occlusion | D020517 SU | large | NA |
| 23360689 | Chen et al., 2013[51] | CMD1A | 300751 | Case report | 5 | 1 | c.513 + 1 G > A | p.Lys152Lys | MeSH | rt-PA (alteplase) | D010959 | large | NA |
| 18926329 | Pasotti et al., 2008 [38] | CMD1A | 300751 | Observational study | 3 | 94 | NA | NA | MeSH | Transplant (heart) | D019737 | large | NA |
| 31060954 | Hasebe et al., 2019 [46] | CMD1A | 300751 | Cohort study | 4 | 6 | IVS3–10A > G | p.? | MeSH | Transplant (heart) | D019737 | large | NA |
| 815_818 delins | p.Asp272 | ||||||||||||
| CCAGAC | AlafsX203 | ||||||||||||
| 30287275 | Kwapich et al., 2019 [101] | CMD1A | 300751 | Case-control study | 3 | 58 | c.139G > T | p.Asp47Tyr | MeSH | Transplant (heart) | D019737 | large | NA |
| c.310C > G | p.Leu104Val | ||||||||||||
| c.398G > T | p.Arg133Leu | ||||||||||||
| c.448A > G | p.Thr150Ala | ||||||||||||
| c.467G > A | p.Arg156His | ||||||||||||
| c.481G > A | p.Glu161Lys | ||||||||||||
| c.694G > C | p.Gly232Arg | ||||||||||||
| c.751C > T | p.Gln251* | ||||||||||||
| c.860del | p.Ala287Valfs*193 | ||||||||||||
| c.949G > A | p.Glu317Lys | ||||||||||||
| c.1157G > C | p.Arg386Thr | ||||||||||||
| c.1173dup | p.Ser392Glnfs*34 | ||||||||||||
| c.1238del; | p.Gly413Alafs*67 | ||||||||||||
| c.1315C > T | p.Arg439Cys | ||||||||||||
| c.1357C > T | p.Arg453Trp | ||||||||||||
| c.1444C > T | p.Arg482Trp | ||||||||||||
| c.1445G > A | p.Arg482Gln | ||||||||||||
| c.1445G > T | p.Arg482Leu | ||||||||||||
| c.1930C > T | p.Arg644Cys | ||||||||||||
| 30482687 | Peters et al., 2019 [47] | CMD1A | 300751 | Expert review | 5 | NA | NA | NA | MeSH | Transplant (heart) | D019737 | large | NA |
ARVD: Familial isolated arrhythmogenic ventricular dysplasia, right dominant form; CMD1A: Familial dilated cardiomyopathy with conduction defect due to LMNA mutation; FPLD2: Familial Partial Lipodystrophy, Dunnigan Type.
Treatabolome data for LMNA-related muscular phenotypes
The LMNA-related muscular phenotypes comprise a range of muscular dystrophies, i.e., the congenital muscular dystrophy (LMNA-CMD) [19], the Emery-Dreifuss muscular dystrophy (EDMD2) [20] and the Limb-Girdle Muscular Dystrophy type 1B [21]. These LMNA-muscular dystrophies differ in the age at onset of the muscular symptoms, the degree of joint contractures, when present, and, the severity, progression rate and topology of muscle wasting and weakness. But they all share a common feature, i.e. a life-threatening cardiac disease characterized by conduction and/or rhythm defects associated with dilated cardiomyopathy resulting in a high frequency of cardiac sudden death [1]. Of note, the cardiac involvement of LMNA-related muscular phenotypes is highly similar to the isolated LMNA-related cardiomyopathy presentation (CMD1A) [22, 23].
Currently, there are no specific treatments for LMNA related muscle weakness/wasting. Those treatments are common to all muscular dystrophies and neuropathies and, for that reason, are not included in the LMNA Treatabolome dataset. However, a frequent question asked by physicians following these patients pertains the management of joint contractures. Early joint contractures observed in the LMNA-related Emery-Dreifuss disease, which are not necessarily linked to muscle deficit, may benefit from direct surgical procedures when severe or responsible for high disability [24]. The most common joint contractures localizations are Achilles tendons, elbows and post-cervical muscles. There is published evidence on the surgical management of severe extension deformity of the cervical spine associated or not with scoliosis [24, 25] and also of severe upper extremity contractures. These were treated successfully with contracture release and musculotendinous lengthening [26] that improved range of motion without a significant sacrifice of strength. This literature is however limited. Some peri- or postoperative complications have been reported [27] and these patients should be managed by specific anesthetic and per operatory protocols [25, 28–30], preceded by careful full-spine analysis and preoperative evaluation.
In the case of LMNA-related congenital muscular dystrophy, there is scarce evidence that ste-roid therapy may bring some motor improvement [31]. Nevertheless, it has been included in our Treatabolome dataset but with a weak evidence-level (see Table 3). We have additional entries whose treatment is related to prevention of sudden cardiac death and that were included in Table 3, as the main phenotype is muscular. The prevention of sudden cardiac death is quite similar whether skeletal muscle is present or not (see Table 4).
Treatabolome data for LMNA-related sudden cardiac death prevention
A major LMNA-associated clinical problem is represented by the phenotypes that induce the risk of sudden cardiac death due to malignant arrhythmia (Table 4). Phenotypically, these arise either as isolated dilated cardiomyopathy or dilated cardiomyopathy associated with skeletal muscular dystrophy [32]. In principle, all laminopathies involving heart muscle bear a risk of cardiac arrhythmia and sudden death as demonstrated in a 2005 meta-analysis [32] and on published case series as well [33–37]. It is also established that mutations leading to haploinsufficiency (nonsense, indel, truncating insertions/deletions and splice site) carry the highest risk of sudden cardiac death [38]. An updated list of these mutations is supplied as Supplementary File S2. Defining the precise risk level has fueled different risk models published in the literature [39–45]. The different papers converge on an agreement that pacing does not prevent sudden cardiac death occurrence and the need for early cardiac defibrillator implantation (with or without resynchronization therapy) to improve patient’s vital prognosis. The treatment does not delay progression to heart failure though, and when arrhythmia occurs under the latter condition, only cardiac transplantation extends survival [46]. Early referral for heart transplant is therefore advised in laminopathies [47].
There is evidence of some efficacy of radiofrequency catheter ablation for ventricular tachyarrhythmias [48, 49], which should delay referral to heart transplantation.
Atrial fibrillation and other atrial arrhythmias are common manifestations of laminopathies. They have been associated with high risk of stroke and other cardioembolic complications, therefore requiring the systematic use of curative anticoagulation, regardless to CHADS-VASC score [45, 50, 51].
Treatabolome data for LMNA-related lipodystrophies
The LMNA-related lipodystrophies central entity is the Familial Partial Lipodystrophy Type 2, also known as Dunnigan type lipodystrophy. It is characterized by loss of subcutaneous adipose tissue from the trunk, buttocks and limbs and accumulation of fat around face, neck, pelvis and axillae coexisting with muscle hypertrophy later accompanied by metabolic perturbations such as hypertriglyceridemia, low HDL cholesterol, hepatic steatosis, insulin-resistant diabetes, and early atherosclerosis. The phenotype is more marked in females, who also frequently develop ovarian hyperandrogenia leading to hirsutism, menstrual disturbances and decreased fertility [52]. A prevalence of the Dunnigan syndrome below 1/100 000 was reported, but is probably underestimated, since partial lipodystrophy is largely underdiagnosed [53, 54]. LMNA-related lipodystrophies are the most common forms of genetic lipodystrophies in Europe. In the great majority of cases they are inherited in an autosomal dominant fashion. The characteristic hotspot results from heterozygous mutations at the 482nd codon of the gene (p.Arg482Trp/Gln or Leu). However, other LMNA pathogenic variants can be found rarely as well, that may lead to typical partial lipodystrophic syndromes or mixed laminopathic phenotypes [55, 56].
These patients have severe cardiovascular risk through atherosclerosis. Female patients may suffer from Polycystic Ovarian Syndrome (PCOS), associated with reduced fertility, hirsutism and menstrual disturbances. Due to the multiple comorbidities associated with LMNA-related lipodystrophic syndromes, patients require multidisciplinary management. The first-line management of diabetes and dyslipidemia mainly follows the general population’s guidelines, with dietary and lifestyle rules being fundamental. No cure is available for lipodystrophy itself (Table 5).
Table 5
LMNA-related lipodystrophic syndromes treatment
| Pubmed | Ref. | Clinical diagnosis ORDO | ORPHA code | Type of study | OCEBM evidence | Number of LMNA patients | HGVS cDNA | HGVS protein | Treatment or intervention database | Treatment or intervention name | Treatment or intervention ID | Clinical effect | Biomarker Effect$ | Comments |
| 23073275 | van Rijsingen et al., 2013 [50] | CMD1A | 300751 | Case-control study | 3 | 76 | NA | NA | ChEBI | Anticoagulant | CHEBI: 50249 | large | NA | NA |
| 27506821 | Kumar et al., 2016 [49] | CMD1A | 300751 | Cohort study | 4 | 25 | NA | NA | MeSH | Catheter Ablation | D017115 | moderate | NA | NA |
| 12766116 | Herbst et al., 2003 [103] | FPLD2 | 2348 | Case series | 4 | 13 | NA | NA | ChEBI | Fenofibrate | CHEBI: 5001 | NA | moderate | NA |
| 17642424 | Sadeghi-Nejad et al., 2007 [84] | HGPS | 740 | Case report | 5 | 1 | c.1822G > A | p.G608S | ChEBI | Growth Hormone | CHEBI: 37845 | small | NA | NA |
| 9258264 | Abdenur et al., 1997 [85] | HGPS | 740 | Case series | 4 | 3 | NA | NA | ChEBI | Growth Hormone; Nutritional Intervention | CHEBI: 37845 | small | NA | does not stop athero-sclerosis |
| 15598919 | Desai et al., 2004 [95] | CMD1A | 300751 | Meta-analysis | 1 | ### | NA | NA | MeSH | ICD | D017147 | large | NA | NA |
| 23483212 | Ng &Kaye, 2013 [97] | CMD1A | 300751 | Case report | 5 | 1 | NA | NA | MeSH | ICD | D017147 | large | NA | NA |
| 27884249 | Kumar et al., 2016 [45] | CMD1A | 300751 | Cohort study | 4 | 87 | NA | NA | MeSH | ICD | D017147 | large | NA | NA |
| 27993908 | Golwala et al., 2017 [99] | CMD1A | 300751 | Meta-analysis | 1 | ### | NA | NA | MeSH | ICD | D017147 | large | NA | NA |
| 31155932 | Wahbi et al., 2019 [39] | CMD1A | 300751 | Cohort study | 3 | 444 | NA | NA | MeSH | ICD | D017147 | large | NA | innapropriate implantation of ICD |
| 21168376 | Cardona-Hernandez et al., 2011 [104] | FPLD2 | 2348 | Case series | 5 | 1 | c.29C > T | p.Thr10Ileu | ChEBI | Insulin | CHEBI: 145810 | large | large | NA |
| 15791619 | Javor et al., 2005 [105] | FPLD2 | 2348 | Case series | 4 | 2 | NA | NA | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | moderate | NA |
| 19727665 | Chong et al., 2010 [70] | FPLD2 | 2348 | Observational study | 3 | 48 | NA | NA | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | small | NA |
| 22068254 | Chan et al., 2011 [106] | FPLD2 | 2348 | Case series | 4 | 19 | NA | NA | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | moderate | NA |
| 23439261 | Safar Zadeh et al., 2013 [107] | FPLD2 | 2348 | Cohort study | 3 | 27 | NA | NA | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | moderate | NA |
| 24926953 | Joseph et al., 2014 [108] | FPLD2 | 2348 | Cohort study | 5 | 82 | NA | NA | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | small | NA |
| 25734254 | Diker-Cohen et al., 2015 [71] | FPLD2 | 2348 | Cohort study | 4 | 31 | c.1444C > T | p.Arg482Trp | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | small | NA |
| c.1445G > A | p.Arg482Gln | |||||||||||||
| c.1445G > T | p.Arg482Leu | |||||||||||||
| 27642538 | Ajluni et al., 2016 [109] | FPLD2 | 2348 | Cohort study | 4 | 23 | NA | NA | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | small | NA |
| 27710244 | Brown et al., 2016 [63] | FPLD2 | 2348 | Expert review | 5 | NA | NA | NA | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | small | NA |
| 27207511 | Schlogl et al., 2016 [110] | FPLD2 | 2348 | Cohort study | 5 | 9 | NA | NA | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | small | NA |
| 26584826 | Vatier et al., 2016 [72] | FPLD2 | 2348 | Case-control study | 4 | 9 | NA | NA | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | small | NA |
| 27692500 | Vatier et al., 2017 [111] | FPLD2 | 2348 | Cohort study | 10 | 16 | NA | NA | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | moderate | NA |
| 30370487 | Akinci et al., 2018 [69] | FPLD2 | 2348 | Expert opinion | 5 | NA | NA | NA | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | small | NA |
| 29644599 | Brown et al., 2018 [112] | FPLD2 | 2348 | Cohort study | 4 | 66 | NA | NA | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | moderate | NA |
| 29267953 | Hussain et al., 2018 [113] | NA | NA | Cohort study | 4 | 7 | c.29C > T | p.T10I | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | large | NA |
| 31620670 | Kinzer et al., 2019 [114] | FPLD2 | 2348 | Cohort Study | 4 | 5 | NA | NA | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | small | NA |
| 30990519 | Lee et al., 2019 [115] | FPLD2 | 2348 | Cohort study | 4 | 42 | NA | NA | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | moderate | NA |
| 31135595 | Melvin et al., 2019 [116] | FPLD2 | 2348 | Expert review | 5 | NA | NA | NA | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | small | NA |
| 30805888 | Oral et al., 2019 [117] | FPLD2 | 2348 | Cohort study | 4 | 41 | NA | NA | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | large | NA |
| 30539782 | Puschel et al., 2019 [118] | FPLD2 | 2348 | Cohort study | 4 | 10 | NA | NA | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | small | NA |
| 31194872 | Sekizkardes, et al., 2019 [75] | FPLD2 | 2348 | Cohort study | 4 | 22 | c.1444C > T | pArg482Trp | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | small | NA |
| c.1445G > A | p.Arg482Gln | |||||||||||||
| c.IVS8 + 5G > C | p.Ileu497Valfs*20 | |||||||||||||
| c.1543A > G | p.Lys515Glu | |||||||||||||
| c.1662G > C | p.Arg541Pro | |||||||||||||
| c.1751G > A | p.Arg584His | |||||||||||||
| 30296183 | Vatier et al., 2019a [76] | FPLD2 | 2348 | Case series | 4 | 1 | c.1444C > T | p.Arg482Trp | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | small | oestrogens contra-indicated |
| 31300002 | Vatier et al., 2019b [119] | FPLD2 | 2348 | Cohort study | 4 | 10 | NA | NA | ChEBI | Leptin (Metreleptin Myalept) | CHEBI: 81571 | small | small | NA |
| 29044799 | Banning et al., 2017 [62] | FPLD2 | 2348 | Case report | 5 | 1 | c.1445G > A | p.Arg482Gln | ChEBI | Liraglutide | CHEBI: 71193 | small | large | NA |
| 12766116 | Herbst et al., 2003 [103] | FPLD2 | 2348 | Case series | 4 | 13 | NA | NA | ChEBI | Nicotinamide | CHEBI: 17154 | NA | small | NA |
| 17893350 | Hegele et al., 2007 [65] | FPLD2 | 2348 | Case series | 4 | 2 | c.1445G > A | p.Arg482Gln | MeSH | Noninvasive Ventilation | D063087 | moderate | moderate | NA |
| 19418082 | Patel et al., 2009 [66] | FPLD2 | 2348 | Case report | 5 | 1 | c.1445G > A | p.Arg482Gln | MeSH | Noninvasive Ventilation | D063087 | moderate | large | NA |
| 18728124 | Gambineri et al., 2008 [64] | FPLD2 | 2348 | Case report | 4 | 2 | c.1445G > A | p.Arg482Gln | ChEBI | Pioglitazone 30 mg/d | CHEBI: 8228 | moderate | NA | NA |
| 17936664 | Moreau et al., 2007 [59] | FPLD2 | 2348 | Case report | 5 | 1 | NA | NA | ChEBI | Pioglitazone | CHEBI: 8228 | small | large | NA |
| Metformin | CHEBI: 6801 | |||||||||||||
| 18728124 | Gambineri et al., 2008 [64] | FPLD2 | 2348 | Case report | 4 | 2 | c.1445G > A | p.Arg482Gln | ChEBI | Pioglitazone 30 mg/d; Metformin 1700 mg/d; Flutamide 250 mg/d | CHEBI: 5132 | moderate | NA | NA |
| 19249234 | Collet-Gaudillat et al., 2009 [60] | FPLD2 | 2348 | Case report | 5 | 1 | NA | NA | ChEBI | Pioglitazone | CHEBI: 8228 | small | large | NA |
| Metformin | CHEBI: 6801 | |||||||||||||
| Insulin | CHEBI: 45810 | |||||||||||||
| 14510863 | Owen et al., 2003 [58] | FPLD2 | 2348 | Case report | 5 | 1 | c.1444C > T | p.Arg482Trp | ChEBI | Rosiglitazone | CHEBI: 50122 | small | NA | NA |
| 16241930 | Ludtke et al., 2005 [120] | FPLD2 | 2348 | Case report | 5 | 1 | c.1444C > T | p.Arg482Trp | ChEBI | Rosiglitazone | CHEBI: 50122 | small | moderate | NA |
| 22274718 | Luedtke et al., 2012 [61] | FPLD2 | 2348 | Cohort study | 3 | 5 | c.1444C > T | p.Arg482Trp | ChEBI | Rosiglitazone | CHEBI: 50122 | small | moderate | NA |
| c.1445G > A | p.Arg482Gln | |||||||||||||
| 27778252 | Grundfest-Broniatowski et al., 2017 [121] | FPLD2 | 2348 | Case report | 5 | 1 | c.1444C > T | p.Arg482Trp | MeSH | Roux en Y Gastric Bypass | D015390 | moderate | NA | NA |
| 23360689 | Chen et al., 2013 [51] | CMD1A | 300751 | Case report | 5 | 1 | c.513 + 1G > A | p.Lys171Lys | MeSH | rt-PA (alteplase) | D010959 | large | NA | NA |
| + splice defect ? | ||||||||||||||
| 21561824 | Calderoni et al., 2011 [67] | FPLD2 | 2348 | Case report | 5 | 1 | NA | NA | MeSH | Surgery, Plastic | D013518 | moderate | NA | NA |
| 21306965 | Hughes et al. 2011 [68] | FPLD2 | 2348 | Case report | 5 | 1 | NA | NA | MeSH | Surgery, Plastic | D013518 | moderate | NA | NA |
| 10929166 | Arioglu et al., 2000 [57] | FPLD2 | 2348 | Cohort study | 3 | 7 | NA | NA | ChEBI | Troglitazone | CHEBI: 9753 | small | small | NA |
FPLD2: Familial Partial Lipodystrophy, Dunnigan Type; CMD1A: Familial dilated cardiomyopathy with conduction defect due to LMNA mutation. $Biomarkers effects are indicated only in this table V, as some were reported only for these class of phenotypes/symptoms/treatments. # # #Not possible to find how many patients with LMNA-related disease were reported in these meta-analyses.
Rare studies report the effects of nonspecific antidiabetic medications such as metformin, thiazolidinediones and glucagon-like peptide-1 (GLP-1) receptor agonists, and insulin in some patients with LMNA-related lipodystrophies. Usually, these are case reports of different combinations with low evidence level. One open-label prospective trial with the thiazolidinedione troglitazone, which is withdrawn from the market since 2000, found that the drug lowered HbA1C levels in FPLD patients [57]. Additional anecdotal evidence exists from case reports [58–61] regarding thiazolidinediones (pioglitazone, rosiglitazone) in different associations with insulin and/or metformin, that improve metabolic markers (leptin levels, HbA1C levels, insulin sensitivity), but could exacerbate faciocervical fat accumulation [58]. To note, all thiazolidinediones were withdrawn from the market in France, so checking locally their availability is advisable. GLP-1 receptor agonists have shown promises as a glucose-lowering therapy in a case report [62].
Lipid-lowering drugs are also used in accordance to guidelines for the general population in LMNA-related lipodystrophies [63].
A case report has shown that thiazolidinediones could improve PCOS in women with FPLD2 [64]. Obstructive Sleep Apnea Syndrome is a known complication of LMNA-related lipodystrophies that should benefit from Non-Invasive Ventilation as treatment [65, 66]. We suspect that more systematic sleep studies in these populations will potentially disclose sleep disturbed breathing as a frequent feature. Dunnigan lipodystrophy syndrome is also a stigmatizing disease and plastic surgery can be useful for some patients (liposuction of lipohypertrophic areas and/or reconstructive procedures for lipoatrophic areas). A few case reports have described such surgical treatments [67, 68]. Bariatric surgery [69] has been occasionally used in cases of Dunnigan syndrome associated with obesity.
LMNA-related lipodystrophic syndromes, especially when lipoatrophic features are prominent, are associated with decreased leptin levels which contribute to the metabolic alterations and their associated comorbidities. The hormone replacement therapy’s efficiency using the orphan drug Metre-leptin, a recombinant leptin agonist, has not been studied in placebo-controlled studies. Still, several reports suggest that Metreleptin can be useful to improve glucose and lipid homeostasis and decrease hepatic steatosis in lipodystrophic syndromes, at least partly independently from its anorexigenic effects. Leptin-replacement therapy with Metreleptin has been assessed in two single-arm open-label trials [70–75]. They addressed heterogeneous populations with different genome-phenome associations, and Metreleptin seems to have some benefit in low-leptin populations in reducing triglycerides. Raised triglycerides are associated with cardiovascular risk and incidence of acute pancreatitis in these patients. No risk reduction figures of such outcomes are provided, though. A practice guideline reaches similar treatment recommendation for Metreleptin [63] as well as some case series and reports [72, 75, 76]. Although Metreleptin is more efficient in generalized than partial lipodystrophy, it could be useful in Dunnigan lipodystrophy, especially when metabolic alterations are severe and leptin levels very low at baseline [63, 71].
The increased cardiovascular risk in lipodystrophy should also lead to early screening and treatment of atherosclerotic events and rhythm and conduction disturbances. This has been mentioned in case reports, but specific recommendations are needed. [56].
Treatabolome data for LMNA-related progeroid syndromes
Although some progeroid syndromes still do not have a specific Orpha code, that is not the case of the archetypal LMNA-related progeroid presentation Hutchinson-Guilford Progeria Syndrome (HGPS) [77, 78]. It is an accelerated ageing developmental disorder that affects children at a young age, markedly reducing their life expectancy. Despite being born in apparent health, affected children fail to thrive before the first year of life and go on to develop the characteristic features that spare the cognitive development and that result in early cardiovascular death from a heart attack or stroke [79, 80]. Treatments described for the condition are summarized in Table 6.
Table 6
LMNA-related progeroid syndromes treatment
| Pubmed | Ref. | Clinical diagnosis ORDO | ORPHA code | Type of study | OCEBM | Number LMNA patients | HGVS cDNA | HGVS protein | Treatment or intervention name | Treatment or intervention ID | Clinical effect |
| 17935239 | Kosho et al., 2007 [87] | MADA | 90153 | Case report | 5 | 1 | c.1585G > A | p.Ala529Thr | Alendronic acid biphosphonates in general | CHEBI: 2567 | small |
| 31199775 | Toni et al., 2019 [122] | HGPS | 740 | Case report | 5 | 1 | c.433G > A | p.Glu145Lys | Growth Hormone | CHEBI: 37845 | small |
| 17642424 | Sadeghi-Nejad et al., 2007 [84] | HGPS | 740 | Case report | 5 | 1 | c.1822G > A | p.Gly608Ser | Growth Hormone | CHEBI: 37845 | small |
| 9258264 | Abdenur et al., 1997 [85] | HGPS | 740 | Case series | 4 | 3 | NA | NA | Growth Hormone; Nutritional Intervention (= no code) | CHEBI: 37845 | small |
| 23012407 | Gordon et al., 2012 [81] | HGPS | 740 | Cohort study | 3 | 26 | c.1824C > T | p.Gly608Gly | Lonafarnib | CHEBI: 47097 | small |
| 29710166 | Gordon et al., 2018 [82] | HGPS | 740 | Cohort study | 3 | 63 | c.1824C > T | p.Gly608Gly | Lonafarnib | CHEBI: 47097 | small |
| 27400896 | Gordon et al., 2016 [83] | HGPS | 740 | Cohort study | 3 | 37 | c.1824C > T | p.Gly608Gly | Lonafarnib | CHEBI: 47097; | small |
| Pravastatin | CHEBI: 63618 | ||||||||||
| Zoledronic acid | CHEBI: 46557 |
MADA: Mandibulo Acral Dysplasia Type A with Lipodystrophy; HGPS: Hutchinson-Gilford Progeria Syndrome.
Two clinical trials involving a farnesyl transferase inhibitor, named lonafarnib, have risen great expectations. The initial 2012 trial (ClinicalTrials.gov, NCT02579044) enrolled 26 subjects and was a non-randomized controlled trial [81]. At the conclusion, treated patients had improved weight, vascular stiffness, bone structure and audiological state. The treatment seemed to have a beneficial effect on survival but the findings were limited by the observational design [82]. A second trial (Clinical Trials.gov, NCT00879034) involving 37 patients followed, employing a combination of lonafarnib, pravastatin and zoledronic acid in which comparisons with lonafarnib monotherapy treatment revealed additional bone mineral density benefit [83]. There was no added cardiovascular benefit, leaving small hope that such an approach can improve survival. There is an ongoing Phase I/II trial combining lonafarnib and everolimus that estimates enrolling 80 patients and being completed by December 2021 (ClinicalTrials.gov, NCT02579044).
Growth hormone (GH) has been mentioned as a treatment that may favor growth in HGPS patients. An initial 3 cases report of GH and nutritional therapy as well as a more recent case report suggest that it brings mild transient benefits [84, 85]. A mixed case with empty sella has found no improvement in long term outcome [86]. The evidence, therefore, remains weak for recommending this therapy in HGPS.
Finally, a case report of LMNA-related case of Mandibulo Acral Dysplasia (MADA) recommends bisphosphonates to prevent the clastic activity with a rationale based on mechanism, so with a low level of evidence supporting the suggestion [87]. This entity is sometimes found in association with lipodystrophy.
DISCUSSION
The current systematic literature review of the Treatabolome pilot study research question (“What treatments have been described for this condition/gene/variant; on which specific genetic variants have they been tested; and what is the strength of the associated supporting evidence?”) did not provide any accessible list of LMNA variant-specific treatments. As an example, LMNA variants reported as “malignant” because of their association with a high risk of sudden cardiac death require the same cardiological management as “unlabelled” variants, as they share the same potential risk. However, we could identify a list of inactivating mutations conferring a major risk of sudden cardiac death (Supplementary File S2). We recommend having in mind that although many papers based the assertion of variant pathogenicity on existing functional studies, a sizeable number have not indicated what scientific validation has been done for some of the previously undescribed variants. Keep in mind that variants reported in our Supplementary File S2 and this paper may have less than complete evidence regarding their pathogenicity.
In LMNA-triggered conditions, the specific treatment indications thus rather relate to significant genome-phenotype pairings. The evidence regarding these pairings are summarised in Tables 3 (LMNA-related muscular phenotypes treatment), 4 (Sudden Cardiac Death Prevention), 5 (LMNA-related lipodystrophy treatment) and 6 (LMNA-related progeroid syndrome treatment). Regarding the corresponding gene and variant information, we have included reported variants from case reports and series and some genetic hotspots for several diseases, bearing in mind there is no variant-specific relationship with the listed treatments.
Our view is that the data assembled in our tables are of relevance for the Treatabolome database. A growing number of non-specialized clinicians gain access to genetic results and the Treatabolome database provides fundamental information for the management of patients. The integration of a treatment-related early warning system in the context of the genetic diagnosis tools has the potential to reduce management delays and to improve standards of care for patients with rare diseases.
Regarding the risk of sudden cardiac death, al-though superficially solved by a blanket indication of implantation of a defibrillator, the timing and risk assessment for that therapy have yet to achieve a clear consensus. The use of a “risk factors” approach, derived from the study by van Rijsingen et al. [41], has been implemented in the European and North American guidelines from cardiology scientific societies on sudden death prevention. A recent publication [39] has proposed an algorithm that is available as an online calculator (https://lmna-risk-vta.fr/) and could reduce the risk for patients to die suddenly, along with the number of patients having unnecessary device placements. It is also noticeable that although life-saving, implantable cardioversion defibrillators (ICD) have unpleasant side-effects when patients receive inappropriate shocks. A meta-analysis estimates that about a fifth of patients have these complications [88] and analysis continues on the mechanisms that originate this unwanted side effect of treatment. This requires that a risk stratification strategy is clearly laid down, namely for asymptomatic candidates. Overall, the ratio between the benefit and risk of prophylactic ICD placements appear to be extremely favorable in the very arrhythmogenic condition and we recommend using the online tool developed as described by Wahbi et al. [39] to assess the risk of sudden cardiac death prior to ICD implantation. The risk factors were identified in cardiology tertiary centers’ LMNA patients, some of whom with neuromuscular involvement. The score was derived from a French nationwide cohort including all phenotypes, so one can reasonably conclude that the resulting sudden death risk stratification applies to any LMNA variant carriers. Therefore, we believe that it improves patient selection for implantation of ICD. Still, we recognize a limitation in Wahbi et al. paper’s approach [39] because it has not been specifically addressed whether the clinical presentation (myopathy, neuropathy, lipodystrophy...) influences the risk for cardiac events beyond the genetic and cardiac risk factors, although the authors intend to study this in the future.
Another clear point is that pacing is inadequate for these patients and should be replaced by ICD [33]. Cardiac Rehabilitation Therapy coupled with defibrillator (CRT-D) may support patients awaiting transplant and is a valid treatment option [39, 41, 42] but its wide use is limited by this population’s modest cardiac function response to CRT. There is no specific arrhythmogenic phenotype linked to the LMNA gene or its variants but general cardiological guidance for anticoagulation applies also in these cases.
In LMNA-related lipodystrophies, diet and exercise have to be strongly encouraged for prevention and treatment of metabolic complications. Nonspecific antidiabetic and lipid lowering treatments are largely used, and the numerous comorbidities (liver disease, cardiovascular risk, polycystic ovary syndrome, muscular symptoms, morphological and psychological consequences of the disease) require a multidisciplinary care. There was some success in reducing triglyceride levels and improving insulin resistance and glucose parameters by administering the orphan drug Metreleptin in patients with low leptin levels and severe metabolic alterations.
The literature about Progeroid Syndromes includes two non-randomized non-blinded controlled studies on the use of a farnesyl transferase inhibitor, lonafarnib, either in monotherapy or in association with pravastatin and zoledronic acid. The results show that weight, vascular stiffness, bone structure and audiological state (and bone density in the association trial) improve, but little or no effect on survival was observed. Reports on the use of Growth Hormone (GH) and nutritional measures unfortunately show only a transient advantage.
CONCLUSION
We have performed a systematic literature review to extract ‘uploadable’ data for Treatabolome data-set and to trigger the discussion on information management of laminopathies treatments. The corresponding dataset will integrate the Treatabolome platform and will be shared with interoperable data platforms like the Genome-Phenome Analysis Platform (https://platform.rd-connect.eu/), allowing its incorporation in this and other clinical support tools. As examples of platforms that may consider looking into how to make interoperability with Treatabolome happen in the future, we have considered OPALE [89] the National French Registry for Laminopathies, UMD-LMNA (available at www.umd.be/LMNA/), LOVD (available at http://www.dmd.nl/lmna_home.html) and CMDIR (available at https://www.cmdir.org). We are confident that others will arise in time, as the treatment component of all rare diseases is a concern of researchers, clinicians and patients alike.
ACKNOWLEDGMENTS
This publication is part of the Solve-RD project (http://solve-rd.eu/). The Solve-RD project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 779257.
The authors of this publication are members of the European Reference Network for Neuromuscular Diseases - Project ID N° 870177.
CONFLICTS OF INTEREST
None to declare.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-200596.
REFERENCES
[1] | Worman HJ , Bonne G . “Laminopathies”: a wide spectrum of human diseases. Exp Cell Res. (2007) ;313: (10):2121–33. |
[2] | Bonne G , Quijano-Roy S . Chapter 142 - Emery–Dreifuss muscular dystrophy, laminopathies, and other nuclear envelopathies. In: Dulac O, Lassonde M, Sarnat HB, editors. Handbook of Clinical Neurology. 113: Elsevier; 2013. pp. 1367-76. |
[3] | Worman HJ . Nuclear lamins and laminopathies. J Pathol. (2012) ;226: (2):316–25. |
[4] | Lattanzi G , Maggi L , Araujo-Vilar D . Laminopathies. Nucleus. (2018) ;9: (1):543–4. |
[5] | Brull A , Morales Rodriguez B , Bonne G , Muchir A , Bertrand AT . The Pathogenesis and Therapies of Striated Muscle Laminopathies. Front Physiol. (2018) ;9: :1533. |
[6] | Osmanagic-Myers S , Foisner R . The structural and gene expression hypotheses in laminopathic diseases-not so different after all. Mol Biol Cell. (2019) ;30: (15):1786–90. |
[7] | Ho R , Hegele RA . Complex effects of laminopathy mutations on nuclear structure and function. Clin Genet. (2019) ;95: (2):199–209. |
[8] | Dittmer TA , Misteli T . The lamin protein family. Genome Biol. (2011) ;12: (5):222. |
[9] | Camozzi D , Capanni C , Cenni V , Mattioli E , Columbaro M , Squarzoni S , et al. Diverse lamin-dependent mechanisms interact to control chromatin dynamics. Focus on laminopathies. Nucleus. (2014) ;5: (5):427–40. |
[10] | Briand N , Collas P . Laminopathy-causing lamin A mutations reconfigure lamina-associated domains and local spatial chromatin conformation. Nucleus. (2018) ;9: (1):216–26. |
[11] | Thompson R , Bonne G , Missier P , Lochmuller H . Targeted therapies for congenital myasthenic syndromes: Systematic review and steps towards a treatabolome. Emerg Top Life Sci. (2019) ;3: (1):19–37. |
[12] | Atalaia A , Thompson R , Corvo A , Carmody L , Piscia D , Matalonga L , et al. A guide to writing systematic reviews of rare disease treatments to generate FAIR-compliant datasets: Building a Treatabolome. Orphanet J Rare Dis. (2020) ;15: (1):206. |
[13] | Wilkinson MD , Dumontier M , Aalbersberg IJ , Appleton G , Axton M , Baak A , et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci Data. (2016) ;3: :160018. |
[14] | Cochrane Collaboration. Cochrane Handbook for Systematic Reviews of Interventions version 5.1 2011 [10/05/2019]. Available from: https://training.cochrane.org/handbook. |
[15] | CRD. PROSPERO - international register of systematic reviews 2019 [Available from: https://www.crd.york.ac.uk/PROSPERO/. |
[16] | Moher D , Shamseer L , Clarke M , Ghersi D , Liberati A , Petticrew M , et al. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) statement. Syst Rev. (2015) ;4: :1. |
[17] | Collaboration. C. Data extraction forms: Cochrane; 2020 [Available from: https://dplp.cochrane.org/sites/dplp.cochrane.org/files/public/uploads/CDPLPG%20data%20collection%20form%20for%20intervention%20reviews%20for%20RCTs%20and%20non-RCTs.doc. |
[18] | Cochrane-Methods. Methodological Expectations of Cochrane Intervention Reviews (MECIR) 2019 [cited 2020 07/05/2020]. Available from: https://methods.cochrane.org/sites/default/files/public/uploads/pleacs_2019.pdf. |
[19] | Quijano-Roy S , Mbieleu B , Bonnemann CG , Jeannet PY , Colomer J , Clarke NF , et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann Neurol. (2008) ;64: (2):177–86. |
[20] | Bonne G , Di Barletta MR , Varnous S , Bécane HM , Hammouda EH , Merlini L , et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. (1999) ;21: (3):285–8. |
[21] | Muchir A , Bonne G , van der Kooi AJ , van Meegen M , Baas F , Bolhuis PA , et al. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum Mol Genet. (2000) ;9: (9):1453–9. |
[22] | Fatkin D , MacRae C , Sasaki T , Wolff MR , Porcu M , Frenneaux M , et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. (1999) ;341: (23):1715–24. |
[23] | Bonne G , Mercuri E , Muchir A , Urtizberea A , Becane HM , Recan D , et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann Neurol. (2000) ;48: (2):170–80. |
[24] | Diebo BG , Shah NV , Messina JC , Naziri Q , Post NH , Riew KD , et al. Restoration of Global Sagittal Alignment After Surgical Correction of Cervical Hyperlordosis in a Patient with Emery-Dreifuss Muscular Dystrophy: A Case Report. JBJS Case Connect. (2020) ;10: (1):e0003. |
[25] | Choudhry DK , Mackenzie WG . Anesthetic issues with a hyperextended cervical spine in a child with Emery-Dreifuss syndrome. Anesthesia and analgesia. (2006) ;103: (6):1611–3. |
[26] | Fishman FG , Goldstein EM , Peljovich AE . Surgical treatment of upper extremity contractures in Emery-Dreifuss muscular dystrophy. J Pediatr Orthop B. (2017) ;26: (1):32–5. |
[27] | Poulter GT , Garton HJ , Blakemore LC , Hensinger RN , Graziano GP , Farley FA . Mortality and morbidity associated with correction of severe cervical hyperextension. Spine (Phila Pa 1976). (2009) ;34: (4):378–83. |
[28] | Aldwinckle RJ , Carr AS . The anesthetic management of a patient with Emery-Dreifuss muscular dystrophy for orthopedic surgery. Canadian journal of anaesthesia=Journal canadien d’anesthesie. (2002) ;49: (5):467–70. |
[29] | Shende D , Agarwal R . Anaesthetic management of a patient with Emery-Dreifuss muscular dystrophy. Anaesthesia and Intensive care. (2002) ;30: (3):372–5. |
[30] | Funnell A , Morgan J , McFadzean W . Anaesthesia and orphan disease: Management of cardiac and perioperative risks in a patient with Emery-Dreifuss muscular dystrophy. Eur J Anaesthesiol. (2012) ;29: (12):596–8. |
[31] | Moraitis E , Foley AR , Pilkington CA , Manzur AY , Quinlivan R , Jacques TS , et al. Infantile-onset LMNA-associated Muscular Dystrophy Mimicking Juvenile Idiopathic Inflammatory Myopathy. J Rheumatol. (2015) ;42: (6):1064–6. |
[32] | van Berlo JH , de Voogt WG , van der Kooi AJ , van Tintelen JP , Bonne G , Yaou RB , et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: Do lamin A/C mutations portend a high risk of sudden death? J Mol Med (Berl). (2005) ;83: (1):79–83. |
[33] | Meune C , Van Berlo JH , Anselme F , Bonne G , Pinto YM , Duboc D . Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med. (2006) ;354: (2):209–10. |
[34] | Antoniades L , Eftychiou C , Kyriakides T , Christodoulou K , Katritsis DG . Malignant mutation in the lamin A/C gene causing progressive conduction system disease and early sudden death in a family with mild form of limb-girdle muscular dystrophy. J Interv Card Electrophysiol. (2007) ;19: (1):1–7. |
[35] | Keller H , Finsterer J , Steger C , Wexberg P , Gatterer E , Khazen C , et al. Novel c.367_369del LMNA mutation manifesting as severe arrhythmias, dilated cardiomyopathy, and myopathy. Heart Lung. (2012) ;41: (4):382–6. |
[36] | Kato K , Takahashi N , Fujii Y , Umehara A , Nishiuchi S , Makiyama T , et al. LMNA cardiomyopathy detected in Japanese arrhythmogenic right ventricular cardiomyopathy cohort. J Cardiol. (2016) ;68: (4):346–51. |
[37] | Kwapich M , Lacroix D , Espiard S , Ninni S , Brigadeau F , Kouakam C , et al. Cardiometabolic assessment of lamin A/C gene mutation carriers: A phenotype-genotype correlation. Diabetes Metab. 2018 |
[38] | Pasotti M , Klersy C , Pilotto A , Marziliano N , Rapezzi C , Serio A , et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol. (2008) ;52: (15):1250–60. |
[39] | Wahbi K , Ben Yaou R , Gandjbakhch E , Anselme F , Gossios T , Lakdawala NK , et al. Development and Validation of a New Risk Prediction Score for Life-Threatening Ventricular Tachyarrhythmias in Laminopathies. Circulation. 2019. |
[40] | Charron P , Arbustini E , Bonne G . What Should the Cardiologist know about Lamin Disease? Arrhythm Electrophysiol Rev. (2012) ;1: (1):22–8. |
[41] | van Rijsingen IA , Arbustini E , Elliott PM , Mogensen J , Hermans-van Ast JF , van der Kooi AJ , et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J Am Coll Cardiol. (2012) ;59: (5):493–500. |
[42] | Anselme F , Moubarak G , Savoure A , Godin B , Borz B , Drouin-Garraud V , et al. Implantable cardioverter-defibrillators in lamin A/C mutation carriers with cardiac conduction disorders. Heart Rhythm. (2013) ;10: (10):1492–8. |
[43] | Disertori M , Quintarelli S , Mazzola S , Favalli V , Narula N , Arbustini E . The need to modify patient selection to improve the benefits of implantable cardioverter-defibrillator for primary prevention of sudden death in non-ischaemic dilated cardiomyopathy. Europace. (2013) ;15: (12):1693–701. |
[44] | Halliday BP , Cleland JGF , Goldberger JJ , Prasad SK . Personalizing Risk Stratification for Sudden Death in Dilated Cardiomyopathy: The Past, Present, and Future. Circulation. (2017) ;136: (2):215–31. |
[45] | Kumar S , Baldinger SH , Gandjbakhch E , Maury P , Sellal JM , Androulakis AF , et al. Long-Term Arrhythmic and Nonarrhythmic Outcomes of Lamin A/C Mutation Carriers. J Am Coll Cardiol. (2016) ;68: (21):2299–307. |
[46] | Hasebe Y , Fukuda K , Nakano M , Kumagai K , Karibe A , Fujishima F , et al. Characteristics of ventricular tachycardia and long-term treatment outcome in patients with dilated cardiomyopathy complicated by lamin A/C gene mutations. J Cardiol. 2019. |
[47] | Peters S , Kumar S , Elliott P , Kalman JM , Fatkin D . Arrhythmic Genotypes in Familial Dilated Cardiomyopathy: Implications for Genetic Testing and Clinical Management. Heart Lung Circ. (2019) ;28: (1):31–8. |
[48] | Roberts JD , Gollob MH , Young C , Connors SP , Gray C , Wilton SB , et al. Bundle Branch Re-Entrant Ventricular Tachycardia: Novel Genetic Mechanisms in a Life-Threatening Arrhythmia. JACC Clin Electrophysiol. (2017) ;3: (3):276–88. |
[49] | Kumar S , Androulakis AF , Sellal JM , Maury P , Gandjbakhch E , Waintraub X , et al. Multicenter Experience With Catheter Ablation for Ventricular Tachycardia in Lamin A/C Cardiomyopathy. Circ Arrhythm Electrophysiol. (2016) ;9: (8). |
[50] | van Rijsingen IA , Bakker A , Azim D , Hermans-van Ast JF , van der Kooi AJ , van Tintelen JP , et al. Lamin A/C mutation is independently associated with an increased risk of arterial and venous thromboembolic complications. Int J Cardiol. (2013) ;168: (1):472–7. |
[51] | Chen CH , Tang SC , Su YN , Yang CC , Jeng JS . Cardioembolic stroke related to limb-girdle muscular dystrophy 1B. BMC Res Notes. (2013) ;6: :32. |
[52] | Guenantin AC , Briand N , Bidault G , Afonso P , Bereziat V , Vatier C , et al. Nuclear envelope-related lipodystrophies. Semin Cell Dev Biol. (2014) ;29: :148–57. |
[53] | Dutour A , Roll P , Gaborit B , Courrier S , Alessi MC , Tregouet DA , et al. High prevalence of laminopathies among patients with metabolic syndrome. Hum Mol Genet. (2011) ;20: (19):3779–86. |
[54] | Gonzaga-Jauregui C , Ge W , Staples J , Van Hout C , Yadav A , Colonie R , et al. Clinical and Molecular Prevalence of Lipodystrophy in an Unascertained Large Clinical Care Cohort. Diabetes. (2020) ;69: (2):249–58. |
[55] | Guillin-Amarelle C , Fernandez-Pombo A , Sanchez-Iglesias S , Araujo-Vilar D . Lipodystrophic laminopathies: Diagnostic clues. Nucleus. (2018) ;9: (1):249–60. |
[56] | Mosbah H , Vatier C , Boccara F , Jeru I , Lascols O , Vantyghem MC , et al. Looking at New Unexpected Disease Targets in LMNA-Linked Lipodystrophies in the Light of Complex Cardiovascular Phenotypes: Implications for Clinical Practice. Cells. (2020) ;9: (3). |
[57] | Arioglu E , Duncan-Morin J , Sebring N , Rother KI , Gottlieb N , Lieberman J , et al. Efficacy and safety of troglitazone in the treatment of lipodystrophy syndromes. Ann Intern Med. (2000) ;133: (4):263–74. |
[58] | Owen KR , Donohoe M , Ellard S , Hattersley AT . Response to treatment with rosiglitazone in familial partial lipodystrophy due to a mutation in the LMNA gene. Diabet Med. (2003) ;20: (10):823–7. |
[59] | Moreau F , Boullu-Sanchis S , Vigouroux C , Lucescu C , Lascols O , Sapin R , et al. Efficacy of pioglitazone in familial partial lipodystrophy of the Dunnigan type: A case report. Diabetes Metab. (2007) ;33: (5):385–9. |
[60] | Collet-Gaudillat C , Billon-Bancel A , Beressi JP . Long-term improvement of metabolic control with pioglitazone in a woman with diabetes mellitus related to Dunnigan syndrome: A case report. Diabetes Metab. (2009) ;35: (2):151–4. |
[61] | Luedtke A , Boschmann M , Colpe C , Engeli S , Adams F , Birkenfeld AL , et al. Thiazolidinedione response in familial lipodystrophy patients with LMNA mutations: A case series. Horm Metab Res. (2012) ;44: (4):306–11. |
[62] | Banning F , Rottenkolber M , Freibothe I , Seissler J , Lechner A . Insulin secretory defect in familial partial lipodystrophy Type 2 and successful long-term treatment with a glucagon-like peptide 1 receptor agonist. Diabet Med. (2017) ;34: (12):1792–4. |
[63] | Brown RJ , Araujo-Vilar D , Cheung PT , Dunger D , Garg A , Jack M , et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J Clin Endocrinol Metab. (2016) ;101: (12):4500–11. |
[64] | Gambineri A , Semple RK , Forlani G , Genghini S , Grassi I , Hyden CS , et al. Monogenic polycystic ovary syndrome due to a mutation in the lamin A/C gene is sensitive to thiazolidinediones but not to metformin. Eur J Endocrinol. (2008) ;159: (3):347–53. |
[65] | Hegele RA , Al-Attar SA , Rutt BK . Obstructive sleep apnea in 2 women with familial partial lipodystrophy due to a heterozygous LMNA R482Q mutation. Cmaj. (2007) ;177: (7):743–5. |
[66] | Patel K , Roseman D , Burbank H , Attarian H . Obstructive sleep apnea in familial partial lipodystrophy type 2 with atypical skin findings and vascular disease. Sleep Breath. (2009) ;13: (4):425–7. |
[67] | Calderoni DR , Ramos TM , de Castro JR , Kharmandayan P . Surgical management of phenotypic alterations related to the Dunnigan variety of familial partial lipodystrophy. Journal of Plastic, Reconstructive & Aesthetic Surgery: JPRAS. (2011) ;64: (9):1248–50. |
[68] | Hughes JM , Stephen C , Johnson AB , Wilson S . Breast augmentation in Familial Partial Lipodystrophy: A case report. Journal of Plastic, Reconstructive & Aesthetic Surgery: JPRAS. (2011) ;64: (5):e121–4. |
[69] | Akinci B , Meral R , Oral EA . Update on Therapeutic Options in Lipodystrophy. Curr Diab Rep. (2018) ;18: (12):139. |
[70] | Chong AY , Lupsa BC , Cochran EK , Gorden P . Efficacy of leptin therapy in the different forms of human lipodystrophy. Diabetologia. (2010) ;53: (1):27–35. |
[71] | Diker-Cohen T , Cochran E , Gorden P , Brown RJ . Partial and generalized lipodystrophy: Comparison of baseline characteristics and response to metreleptin. J Clin Endocrinol Metab. (2015) ;100: (5):1802–10. |
[72] | Vatier C , Fetita S , Boudou P , Tchankou C , Deville L , Riveline J , et al. One-year metreleptin improves insulin secretion in patients with diabetes linked to genetic lipodystrophic syndromes. Diabetes Obes Metab. (2016) ;18: (7):693–7. |
[73] | Simha V , Subramanyam L , Szczepaniak L , Quittner C , Adams-Huet B , Snell P , et al. Comparison of efficacy and safety of leptin replacement therapy in moderately and severely hypoleptinemic patients with familial partial lipodystrophy of the Dunnigan variety. J Clin Endocrinol Metab. (2012) ;97: (3):785–92. |
[74] | Park JY , Javor ED , Cochran EK , DePaoli AM , Gorden P . Long-term efficacy of leptin replacement in patients with Dunnigan-type familial partial lipodystrophy. Metabolism. (2007) ;56: (4):508–16. |
[75] | Sekizkardes H , Cochran E , Malandrino N , Garg A , Brown RJ . Efficacy of Metreleptin Treatment in Familial Partial Lipodystrophy Due to PPARG vs LMNA Patho-genic Variants. J Clin Endocrinol Metab. (2019) ;104: (8):3068–76. |
[76] | Vatier C , Vantyghem MC , Storey C , Jeru I , Christin-Maitre S , Feve B , et al. Monogenic forms of lipodystrophic syndromes: Diagnosis, detection, and practical management considerations from clinical cases. Curr Med Res Opin. (2019) ;35: (3):543–52. |
[77] | De Sandre-Giovannoli A , Bernard R , Cau P , Navarro C , Amiel J , Boccaccio I , et al. Lamin A Truncation in Hutchinson-Gilford Progeria. Science. (2003) ;300: :2055. |
[78] | Eriksson M , Brown WT , Gordon LB , Glynn MW , Singer J , Scott L , et al. Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature. (2003) ;423: (6937):293–8. |
[79] | Hennekam RC . Hutchinson-Gilford progeria syndrome: Review of the phenotype. Am J Med Genet A. (2006) ;140: (23):2603–24. |
[80] | Merideth MA , Gordon LB , Clauss S , Sachdev V , Smith AC , Perry MB , et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. (2008) ;358: (6):592–604. |
[81] | Gordon LB , Kleinman ME , Miller DT , Neuberg DS , Giobbie-Hurder A , Gerhard-Herman M , et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. (2012) ;109: (41):16666–71. |
[82] | Gordon LB , Shappell H , Massaro J , D’Agostino RB Sr , Brazier J , Campbell SE et al. Association of Lonafarnib Treatment vs No Treatment With Mortality Rate in Patients With Hutchinson-Gilford Progeria Syndrome. Jama. (2018) ;319: (16):1687–95. |
[83] | Gordon LB , Kleinman ME , Massaro J , D’Agostino RB Sr , Shappell H , Gerhard-Herman M et al. Clinical Trial of the Protein Farnesylation Inhibitors Lonafarnib, Pravastatin, and Zoledronic Acid in Children With Hutchinson-Gilford Progeria Syndrome. Circulation.. (2016) ;134: (2):114–25. |
[84] | Sadeghi-Nejad A , Demmer L . Growth hormone therapy in progeria. J Pediatr Endocrinol Metab. (2007) ;20: (5):633–7. |
[85] | Abdenur JE , Brown WT , Friedman S , Smith M , Lifshitz F . Response to nutritional and growth hormone treatment in progeria. Metabolism. (1997) ;46: (8):851–6. |
[86] | Toni L , Dušátková P , Novotná D , Zemková D , Průhová Š , Lebl J . Short stature in a boy with atypical progeria syndrome due to LMNA c. 433G>A [p.(Glu145Lys)]: Apparent growth hormone deficiency but poor response to growth hormone therapy. J Pediatr Endocrinol Metab. (2019) ;32: (7):775–9. |
[87] | Kosho T , Takahashi J , Momose T , Nakamura A , Sakurai A , Wada T , et al. Mandibuloacral dysplasia and a novel LMNA mutation in a woman with severe progressive skeletal changes. Am J Med Genet A. (2007) ;143a: (21):2598–603. |
[88] | Olde Nordkamp LR , Postema PG , Knops RE , van Dijk N , Limpens J , Wilde AA , et al. Implantable cardioverter-defibrillator harm in young patients with inherited arrhythmia syndromes: A systematic review and meta-analysis of inappropriate shocks and complications. Heart Rhythm. (2016) ;13: (2):443–54. |
[89] | Yaou RB , Vigouroux C , Quijano-Roy S , Campanna-Salort E , Cintas P , Cuisset J , et al. OPALE: A patient registry for laminopathies and emerinopathies in France. Neuromuscular Disorders. (2016) ;26: :S138-S. |
[90] | Schuster F , Wessig C , Schimmer C , Johannsen S , Lazarus M , Aleksic I , et al. In vitro contracture test results and anaesthetic management of a patient with emery-dreifuss muscular dystrophy for cardiac transplantation. Case Rep Anesthesiol. (2012) ;2012: :349046. |
[91] | Wang Z , Dong Y , Yang J , He Y , Lin X , Wu F , et al. A new laminopathy caused by an Arg133/Leu mutation in lamin A/C and the effects thereof on adipocyte differentiation and the transcriptome. Adipocyte. (2019) ;8: (1):280–91. |
[92] | Homma K , Nagata E , Hanano H , Uesugi T , Ohnuki Y , Matsuda S , et al. A Young Patient with Emery-Dreifuss Muscular Dystrophy Treated with Endovascular Therapy for Cardioembolic Stroke: A Case Report. Tokai J Exp Clin Med. (2018) ;43: (3):103–5. |
[93] | Rudbeck-Resdal J , Nielsen JC , Bundgaard H , Jensen HK . Appropriate use of genetics in a young patient with atrioventricular block and family history of sudden cardiac death. HeartRhythm Case Rep. (2019) ;5: (3):169–72. |
[94] | MacLeod HM , Culley MR , Huber JM , McNally EM . Lamin A/C truncation in dilated cardiomyopathy with conduction disease. BMC Med Genet. (2003) ;4: :4. |
[95] | Desai AS , Fang JC , Maisel WH , Baughman KL . Implantable defibrillators for the prevention of mortality in patients with nonischemic cardiomyopathy: A meta-analysis of randomized controlled trials. JAMA. (2004) ;292: (23):2874–9. |
[96] | Meune C , Wahbi K , Gobeaux C , Duboc D , Pecker F , Bonne G . N-terminal Pro brain natriuretic peptide is a reliable biomarker of reduced myocardial contractility in patients with lamin A/C gene mutations. Int J Cardiol. (2011) ;151: (2):160–3. |
[97] | Ng KK , Kaye G . A case of Lamin C gene-mutation with preserved systolic function and ventricular dysrrhythmia. Australas Med J. (2013) ;6: (2):75–8. |
[98] | Atteya G , Lampert R . Sudden Cardiac Death in Genetic Cardiomyopathies. Card Electrophysiol Clin. (2017) ;9: (4):581–603. |
[99] | Golwala H , Bajaj NS , Arora G , Arora P . Implantable Cardioverter-Defibrillator for Nonischemic Cardiomyopathy: An Updated Meta-Analysis. Circulation. (2017) ;135: (2):201–3. |
[100] | Kusumoto FM , Schoenfeld MH , Barrett C , Edgerton JR , Ellenbogen KA , Gold MR , et al. 2018 ACC/AHA/HRS Guideline on the Evaluation and Management of Patients With Bradycardia and Cardiac Conduction Delay: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Circulation. (2019) ;140: (8):e382–e482. |
[101] | Kwapich M , Lacroix D , Espiard S , Ninni S , Brigadeau F , Kouakam C , et al. Cardiometabolic assessment of lamin A/C gene mutation carriers: A phenotype-genotype correlation. Diabetes Metab. 2018. |
[102] | De Roeck F , Scott B , Verheye S , Vermeersch P . Percutaneous left atrial appendage occlusion in a lamin A/C gene mutation related cardiomyopathy patient with persistent left atrial appendage thrombus: A case report. Acta Cardiol. (2019) ;74: (2):182–3. |
[103] | Herbst KL , Tannock LR , Deeb SS , Purnell JQ , Brunzell JD , Chait A . Kobberling type of familial partial lipodystrophy: An underrecognized syndrome. Diabetes Care. (2003) ;26: (6):1819–24. |
[104] | Cardona-Hernandez R , Suarez-Ortega L , Torres M . [Difficult to manage diabetes mellitus associated with generalized congenital lipodystrophy. Report of two cases]. An Pediatr (Barc). (2011) ;74: (2):126–30. |
[105] | Javor ED , Ghany MG , Cochran EK , Oral EA , DePaoli AM , Premkumar A , et al. Leptin reverses nonalcoholic steatohepatitis in patients with severe lipodystrophy. Hepatology. (2005) ;41: (4):753–60. |
[106] | Chan JL , Lutz K , Cochran E , Huang W , Peters Y , Weyer C , et al. Clinical effects of long-term metreleptin treatment in patients with lipodystrophy. Endocr Pract. (2011) ;17: (6):922–32. |
[107] | Safar Zadeh E , Lungu AO , Cochran EK , Brown RJ , Ghany MG , Heller T , et al. The liver diseases of lipodystrophy: The long-term effect of leptin treatment. J Hepatol. (2013) ;59: (1):131–7. |
[108] | Joseph J , Shamburek RD , Cochran EK , Gorden P , Brown RJ . Lipid regulation in lipodystrophy versus the obesity-associated metabolic syndrome: The dissociation of HDL-C and triglycerides. J Clin Endocrinol Metab. (2014) ;99: (9):E1676–80. |
[109] | Ajluni N , Dar M , Xu J , Neidert AH , Oral EA . Efficacy and Safety of Metreleptin in Patients with Partial Lipodystrophy: Lessons from an Expanded Access Program. J Diabetes Metab. (2016) ;7: (3). |
[110] | Schlogl H , Muller K , Horstmann A , Miehle K , Puschel J , Villringer A , et al. Leptin Substitution in Patients With Lipodystrophy: Neural Correlates for Long-term Success in the Normalization of Eating Behavior. Diabetes. (2016) ;65: (8):2179–86. |
[111] | Vatier C , Arnaud L , Prieur X , Guyomarch B , Le May C , Bigot E , et al. One-year metreleptin therapy decreases PCSK9 serum levels in diabetic patients with monogenic lipodystrophy syndromes. Diabetes Metab. (2017) ;43: (3):275–9. |
[112] | Brown RJ , Oral EA , Cochran E , Araujo-Vilar D , Savage DB , Long A , et al. Long-term effectiveness and safety of metreleptin in the treatment of patients with generalized lipodystrophy. Endocrine. (2018) ;60: (3):479–89. |
[113] | Hussain I , Patni N , Ueda M , Sorkina E , Valerio CM , Cochran E , et al. A Novel Generalized Lipodystrophy-Associated Progeroid Syndrome Due to Recurrent Heterozygous LMNA T10I Mutation. J Clin Endocrinol Metab. (2018) ;103: (3):1005–14. |
[114] | Kinzer AB , Shamburek RD , Lightbourne M , Muniyappa R , Brown RJ . Advanced Lipoprotein Analysis Shows Atherogenic Lipid Profile That Improves After Metre-leptin in Patients with Lipodystrophy. J Endocr Soc. (2019) ;3: (8):1503–17. |
[115] | Lee HL , Waldman MA , Auh S , Balow JE , Cochran EK , Gorden P , et al. Effects of metreleptin on proteinuria in patients with lipodystrophy. J Clin Endocrinol Metab. 2019. |
[116] | Melvin A , Stears A , Savage DB . Recent developments in lipodystrophy. Curr Opin Lipidol. 2019. |
[117] | Oral EA , Gorden P , Cochran E , Araujo-Vilar D , Savage DB , Long A , et al. Long-term effectiveness and safety of metreleptin in the treatment of patients with partial lipodystrophy. Endocrine. (2019) ;64: (3):500–11. |
[118] | Puschel J , Miehle K , Muller K , Villringer A , Stumvoll M , Fasshauer M , et al. Beneficial effects of leptin substitution on impaired eating behavior in lipodystrophy are sustained beyond 150weeks of treatment. Cytokine. (2019) ;113: :400–4. |
[119] | Vatier C , Kalbasi D , Vantyghem MC , Lascols O , Jeru I , Daguenel A , et al. Adherence with metreleptin therapy and health self-perception in patients with lipodystrophic syndromes. Orphanet J Rare Dis. (2019) ;14: (1):177. |
[120] | Lüdtke A , Heck K , Genschel J , Mehnert H , Spuler S , Worman HJ , et al. Long-term treatment experience in a subject with Dunnigan-type familial partial lipodystrophy: Efficacy of rosiglitazone. Diabet Med. (2005) ;22: (11):1611–3. |
[121] | Grundfest-Broniatowski S , Yan J , Kroh M , Kilim H , Stephenson A . Successful Treatment of an Unusual Case of FPLD The Role of Roux-en-Y Gastric Bypass-Case Report and Literature Review. J Gastrointest Surg. (2017) ;21: (4):739–43. |
[122] | Toni L , Dusatkova P , Novotna D , Zemkova D , Pruhova S , Lebl J . Short stature in a boy with atypical progeria syndrome due to LMNA c.433G>A [p.(Glu145Lys)]: Apparent growth hormone deficiency but poor response to growth hormone therapy. J Pediatr Endocrinol Metab. 2019. |


