
Elucidation of the Genetic Cause in Dutch Limb Girdle Muscular Dystrophy Families: A 27-Year’s Journey
Abstract
Background:
A Dutch cohort of 105 carefully selected limb girdle muscular dystrophy (LGMD) patients from 68 families has been subject to genetic testing over the last 20 years. After subsequent targeted gene analysis around two thirds (45/68) of the families had received a genetic diagnosis in 2013.
Objective:
To describe the results of further genetic testing in the remaining undiagnosed limb girdle muscular dystrophy families in this cohort.
Methods:
In the families of the cohort for whom no genetic diagnosis was established (n = 23) further testing using Sanger sequencing, next generation sequencing with gene panel analysis or whole-exome sequencing was performed. In one case DNA analysis for facioscapulohumeral dystrophy type 1 was carried out.
Results:
In eight families no additional genetic tests could be performed. In 12 of the remaining 15 families in which additional testing could be performed a genetic diagnosis was established: two LGMDR1 calpain3-related families with CAPN3 mutations, one LGMDR2 dysferlin-related family with DYSF mutations, three sarcoglycanopathy families (LGMDR3-5 α-, β- and γ-sarcoglycan-related) with SGCA/SGCB/SGCG mutations, one LGMDR8 TRIM 32-related family with TRIM32 mutations, two LGMDR19 GMPPB-related families with GMPPB mutations, one family with MICU1-related myopathy, one family with FLNC-related myopathy and one family with facioscapulohumeral dystrophy type 1. At this moment a genetic diagnosis has been made in 57 of the 60 families of which DNA was available (95%).
Conclusion:
A genetic diagnosis is obtained in 95% of the families of the original Dutch LGMD cohort of which DNA was available.
INTRODUCTION
A well-defined Dutch cohort of 105 patients with limb girdle muscular dystrophy (LGMD) from 68 families has been subject to subsequent genetic testing over the last 20 years [1–3]. Since the cohort was established in 1993 over 30 different genetic subtypes of LGMD have been described [4]. In 2013 around two thirds of the patients in this cohort had received a genetic diagnosis using immunohistochemical (IHC) staining and quantitative multiplex Western blotting as prescreening on muscle tissue followed by targeted gene sequencing [3]. (Fig. 1) Using the same techniques the detection rate in a cohort of LGMD patients in Italy and Denmark is comparable, respectively around 60% and 75% [5–8]. Using novel techniques such as next generation sequencing (NGS) of specific gene panels or whole exome sequencing (WES) with a virtual filter for known myopathy genes may increase this diagnostic yield as all known genes related to muscle disease can be screened simultaneously for pathogenic variants. Furthermore, there is evidence that NGS and WES is cost effective in pediatric muscle disease as compared to the traditional diagnostic protocol including open muscle biopsy [9]. Besides cost effectiveness a decrease in the number of invasive muscle biopsies can also lessen the burden of the diagnostic process in muscle disease.
Fig. 1
Flowchart of DNA and protein analysis of the Dutch limb girdle muscular dystrophy population.
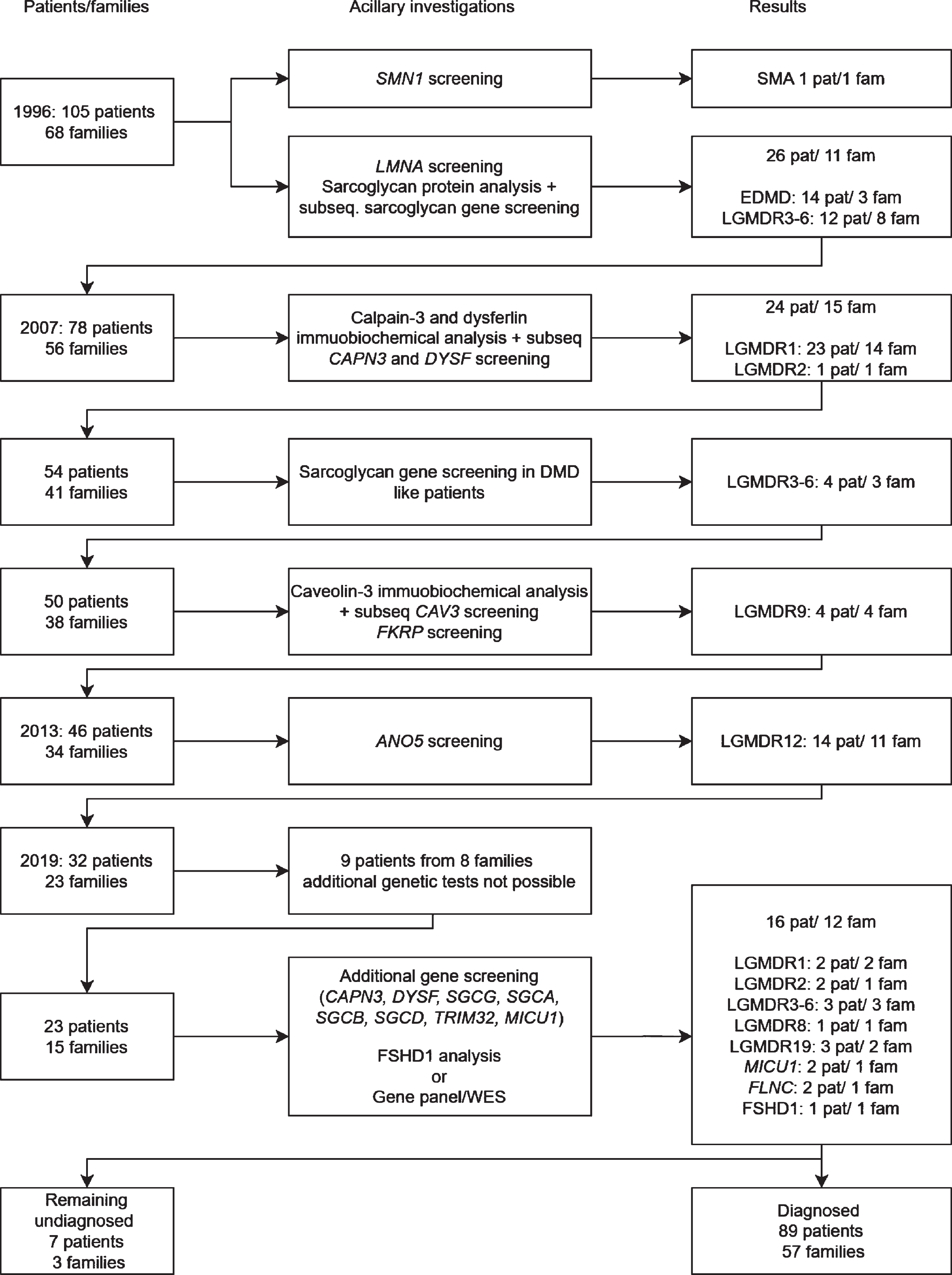
As the WES procedure allows analyzing still unknown genes the continued use of WES can also lead to the discovery of novel causative genes for muscle disease and may lead to an increased understanding of pathogenesis of muscle disease and future therapeutic targets.
However, there are some important drawbacks of NGS/WES based techniques as these techniques will not detect all types of monogenetic causes of diseases presenting with limb girdle weakness. Despite ongoing technical developments in NGS/WES based techniques diseases caused by repeat expansions (i.e. myotonic dystrophy type 2, spinal muscular atrophy, Kennedy disease), deletions or duplications (two thirds of the Duchenne/Becker muscular dystrophy patients), contraction of a repeat array (FSHD1) or variants in the mitochondrial DNA (some mitochondrial myopathies) might not be detected [10–15]. Besides not being able to detect some of the most prevalent causes of limb girdle weakness the diagnostic turnaround time of NGS techniques is, with a few months, quite lengthy and the more genes that are analyzed the more variants of unknown significance (VUS) can be found. VUS are difficult to interpret and can lead to diagnostic uncertainty for the clinician and patient [16].
The aim of this study is to describe how genetic testing has been performed in the Dutch LGMD cohort over the last 27 years and to propose a diagnostic algorithm for genetic testing in LGMD. We describe the results of additional Sanger sequencing and next generation sequencing (NGS) with a gene panel analysis of known myopathy genes or whole exome sequencing (WES) with a virtual filter for known myopathy genes in the remaining undiagnosed patients in this cohort. We describe the clinical and genetic spectrum of the newly diagnosed LGMD patients and provide an update on the total number of patients diagnosed.
METHODS
The patients were recruited from all neuromuscular centers in the Netherlands and from the national patients’ association and selected according to previously described inclusion and exclusion criteria [1]. These criteria included symmetric proximal muscle weakness and ancillary investigations (serum creatine kinase activity (CK), electromyography (EMG) and muscle biopsy) consistent with a primary myogenic disorder, with the exclusion of all other possible causes of a limb girdle syndrome. The study conforms with the World Medical Association Declaration of Helsinki. We received approval from an institutional ethical standards committee on human experimentation (METC AMC) for any experiments using human subjects and informed consent was obtained from all patients or their guardians participating in the study.
Patients who did not have a genetic diagnosis were subjected to various DNA techniques as described below. All undiagnosed patients were screened for variants in TRIM32. If patients were reassessed prior to the introduction of NGS or if a specific diagnosis (calpainopathy, dysferlinopathy, sarcoglycanopathy or FSHD1) was suspected we performed DNA analysis by means of targeted Sanger sequencing and MLPA or Southern blotting. If no diagnosis could be ascertained NGS was performed with either a gene panel of known myopathy genes if analyzed before 2018 or with WES with a virtual filter for known myopathy genes if analyzed after 2018.
Genomic sequencing, MLPA and Southern blotting
Genomic DNA was extracted from patients’ whole blood or muscle using standard procedures and screened for (likely) pathogenic variants in CAPN3 (NM_000070.2), DYSF (NM_003494.3), SGCG (NM_000231.2), SGCA (NM_000023.2), SGCB (NM_000232.4), SGCD (NM_000337.5), TRIM32 (NM_012210.3) and MICU1 (NM_006077.3) by direct sequencing or by NGS with a gene panel analysis of 55 known myopathy genes or by WES with a virtual filter for 148 known myopathy genes (Supplementary file 1). Multiplex ligand-dependent probe amplification (MLPA) analysis was performed according to the manufacturer’s instructions for screening of whole-exon deletions/duplications of CAPN3, DYSF, SGCG, SGCA, SGCB, SGCD using MRC Holland kits SALSA P176, P268-A1 and P116-A1 SGC, respectively. For one patient suspected of facioscapulohumeral dystrophy type 1 (FSHD1) after clinical reassessment Southern blotting was carried out as described previously [17]. Prediction of the pathogenicity of novel variants was performed by segregation analysis in the family and “in silico” analysis using the tools provided by Alamut Visual version 2.8.1 (Interactive Biosoftware, Rouen, France) [18]. All novel variants were submitted to the Leiden Open Variation Database (LOVD).
Clinical data and ancillary investigations
Detailed history and neurological examination were retrieved from patient files. We report age of onset of muscle weakness, age at last examination, and reconstructed age at loss of ambulation from follow up data if available. Age at start of non-invasive ventilation was reconstructed from patient files. Serum CK is expressed as international units/liter (IU/L). The upper limit of normal (ULN) for serum CK activity of our laboratory is 145 IU/L for women and 171 IU/L for men. Available muscle biopsy specimens had already been analyzed with IHC for expression of the sarcoglycans or by quantitative multiplex Western blotting for studying calpain-3 and dysferlin expression [19, 20]. Results of muscle imaging and cardiological investigations including electrocardiography, echocardiography and 24 hours Holter cardiography were retrieved [21].
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
RESULTS
A total of 105 patients from 68 families were originally included in this study. Seventy-three patients from 45 families had a previously established genetic diagnosis including Emery Dreifuss muscular dystrophy due to LMNA variants, calpainopathy, dysferlinopathy, sarcoglycanopathy, FKRP and ANO5 related LGMD and one erroneously in the study recruited SMA patient [2, 3]. Five patients from four families had passed away and no DNA was available for testing, three patients from three families could not give consent for further genetic testing (two passed away and one emigrated) and one patient declined further genetic testing. A genetic diagnosis was established in 16 of the remaining 23 patients from 12 out of 15 families (Fig. 1). This resulted in a genetic diagnosis in 95% of the families (57/60) and 93% of the patients (89/96) in whom genetic testing was still possible (Fig. 2). None of the patients had pathogenic variants in multiple muscle related genes. All patients originated from the Netherlands. Clinical data, serum CK activity and cardiac and respiratory data are summarized in Table 1.
Fig. 2
The frequencies of families (n = 60) with different neuromuscular disorders in the Dutch limb girdle muscular dystrophy cohort.
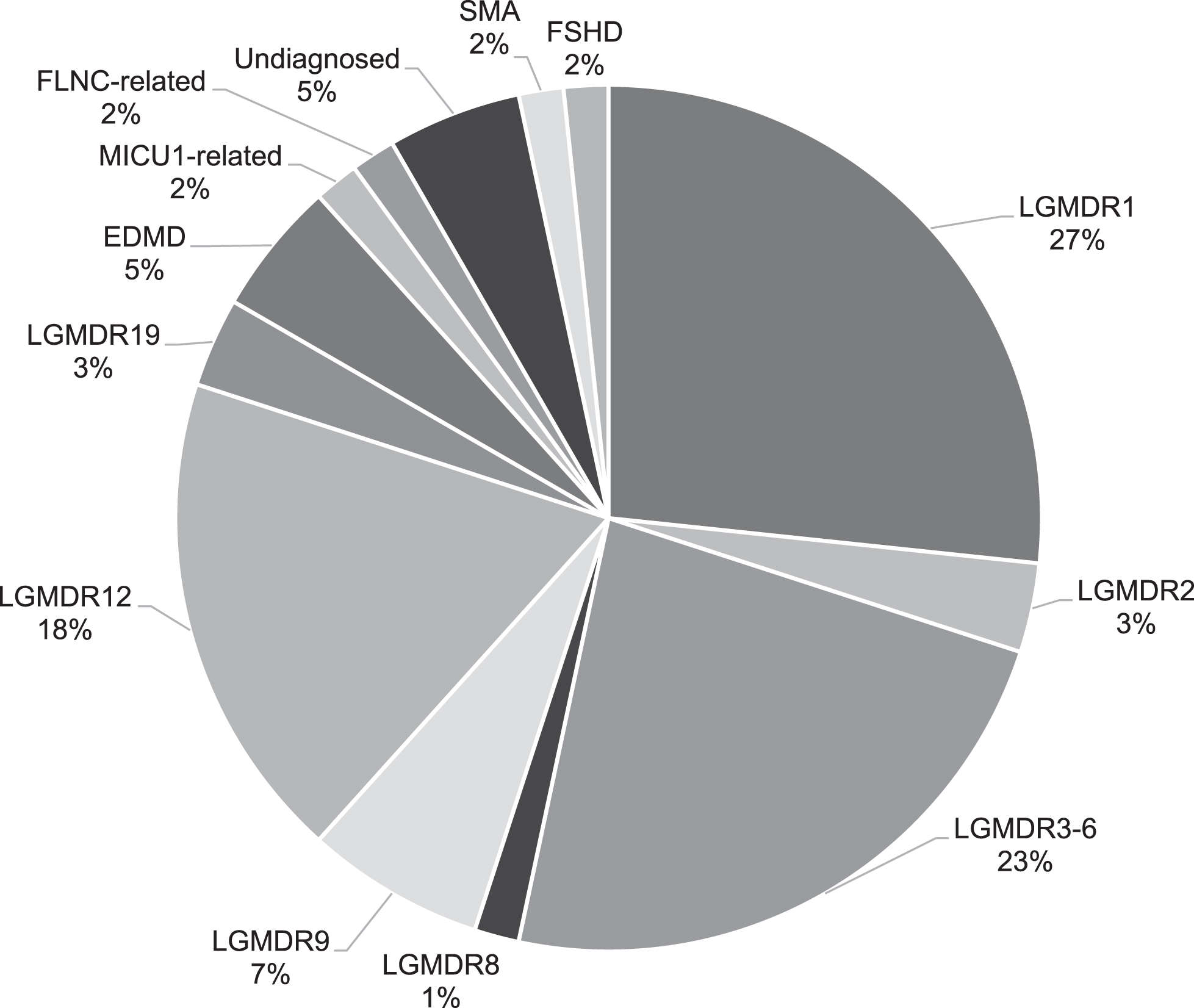
Table 1
Previously undiagnosed LGMD patients
| Gene symbol | Reference sequence | Mutation | Sex | Age of onset (years) | Serum CK activity (U/L) | Age at last examination (years) | Age at loss of ambulation (years) | Disease duration until loss of ambulation (years) | Cardiac involvement | Age at start of non-invasive ventilation (years) | ||
| 1 | CAPN3 | NM_000070.2 | c.1746-20C>G p.(Glu583Cysfs*9)¶ | Compound heterozygous | F | 12 | 473 | 63 | 61 | 49 | No | NR |
| c.598_612del p.(Phe200_Leu204del) | ||||||||||||
| 2 | CAPN3 | c.1328C>T p.(Ser443Phe) | Compound heterozygous | F | adolescence | 2066 | 58 | NR | NR | No | NR | |
| c.1746-20C>G, p.(Glu583Cysfs*9)¶ | ||||||||||||
| 3 | DYSF | NM_003494.3 | c.3118C> T p.(Arg1040Trp) | Compound heterozygous | M | 18 | 1649 | 34 | NR | NR | No | NR |
| c.4907dupC p.(Leu1637Serfs*13) | ||||||||||||
| 4 | DYSF | c.3118C> T p.(Arg1040Trp) | Compound heterozygous | M | 14 | 7020 | 29 | NR | NR | No | NR | |
| c.4907dupC p.(Leu1637Serfs*13) | ||||||||||||
| 5 | SGCG | NM_000231.2 | c.581T>C p.(Leu194Ser) | Homozygous | F | 6 | 1460 | 52 | 44 | 29 | No | 45 |
| 6 | SGCA | NM_000023.2 | c.101G>A p.(Arg34His) | Compound heterozygous | F | 5 | 248 | 27 | 11 | 6 | No | NI |
| c.402C>G p.(Tyr134*) | ||||||||||||
| 7 | SGCB | NM_000232.4 | c.341C>T p.(Ser114Phe) | Homozygous | F | 12 | 707 | 48 | 46 | 34 | Yes | NR |
| 8 | TRIM32 | NM_012210.3 | c.1184T>C p.(Ile395Thr) | Homozygous | F | 14 | 125 | 65 | 41 | 27 | Yes | 64 |
| 9 | GMPPB | NM_013334.3 | c.332T>C,p.(Val111Ala) | Compound heterozygous | M | 10 | 1200 | 70 | NR | NR | No | NR |
| c.516C>G p.(Asn172Lys) | ||||||||||||
| 10 | GMPPB | c.332T>C p.(Val111Ala) | Compound heterozygous | F | 15 | 642 | 57 | NR | 39 | No | NR | |
| c.516C>G p.(Asn172Lys) | ||||||||||||
| 11 | GMPPB | c.79G>C p.(Asp27His) | Compound heterozygous | F | 10 | 921 | 53 | NR | NR | No | NR | |
| c.1053dup p.(Val352Serfs*4) | ||||||||||||
| 12 | MICU1 | NM_006077.3 | c.741 + 1G>A p.(Val248Thrfs*9) | Homozygous | F | 6 | 11200 | 10 | NR | NR | No | NR |
| 13 | MICU1 | c.741 + 1G>A p.(Val248Thrfs*9) | Homozygous | F | childhood | 5390 | 7 | NR | NR | No | NR | |
| 14 | FLNC | NM_001458.4 | c.2791_2805del p.(Lys931_Val935del) | Heterozygous | F | 3 | normal | 66 | 34 | NI | +/- | 63 |
| 15 | FLNC | c.2791_2805del p.Lys931_Val935del) | Heterozygous | M | 25 | 1020 | 60 | 50 | 25 | No | No | |
| 16 | DUX4*(inappropriate reactivation) | F | 25 | 4.5xULN | 53 | NR | NR | No | No |
Novel variants are displayed in bold, family members are displayed in italics. F = Female, M = Male, CK = creatine kinase, IU/L = international units/liter, NR = Not relevant, NI = No information ULN = Upper limit of normal, ¶ = Variant described in (22).
CALPAINOPATHY (LGMDR1 CALPAIN3-RELATED)
Two patients from two families were diagnosed with calpainopathy.
One female patient (patient 1) had difficulty running as a child and muscle weakness was noted in adolescence. Loss of ambulation occurred after a disease duration of 49 years and cardiological examination at age 61 revealed mild left ventricular dysfunction. A strong reduction of calpain-3 expression in her initial muscle biopsy was noted. However, pre-screening by multiplex denaturing gradient gel electrophoresis (DGGE) and subsequent Sanger sequencing of CAPN3 performed in 2000 showed a heterozygous pathogenic variant (c.598_612del p.(Phe200_Leu204del)) and an, at that time as polymorphism classified, intronic variant c.1746-20C>G. The final diagnosis was established after direct Sanger sequencing of the patient’s DNA and meanwhile classification of c.1746-20C>G p.(Glu583Cysfs*9) as a pathogenic variant [22, 23]. Segregation analysis in the family showed that the variants were present on different alleles.
Another female patient (patient 2) had difficulty with running as a child and developed progressive muscle weakness of the lower extremities in late adolescence. During the last follow up at age 58 she was able to walk a few meters in her house without the use of an aid. She needed a wheelchair for longer distances. This family was initially erroneously classified as autosomal dominant (AD) LGMD since the father and several siblings were reported to have a waddling gait. This assumption was probably false as none of the family members had developed muscle weakness when the patient was reassessed 20 years after her initial visit. Muscle biopsy of the proband showed mild dystrophic changes with variation in fiber size, internal nuclei and fiber splitting. Multiplex Western blotting of the muscle tissue detected severe reduction of calpain-3 expression. Gene panel analysis showed compound heterozygosity for a likely pathogenic missense variant (class 4) c.1328C>T p.(Ser443Phe) in addition to a pathogenic variant c.1746-20C>G p.(Glu583Cysfs*9) in CAPN3.
DYSFERLINOPATHY (LGMDR2 DYSFERLIN-RELATED)
Two male patients from one family were diagnosed with dysferlinopathy. Both patients showed onset of muscle weakness in adolescence and were able to walk unaided at the time of the last examination at the age of 30. No muscle tissue was available for multiplex Western blotting analysis. In both patients Sanger sequencing revealed a pathogenic frameshift variant c.4907dup p.(Leu1637Serfs*13) in addition to a pathogenic missense variant c.3118C>T p.(Arg1040Trp) in DYSF. Segregation analysis in the family showed that the variants were present on different alleles.
SARCOGLYCANOPATHIES (LGMDR3-6 α-, β- AND γ-SARCOGLYCAN-RELATED)
Three patients from three families were diagnosed with a sarcoglycanopathy.
One female patient (patient 5) was diagnosed with LGMDR5 γ-sarcoglycan-related. Onset of muscle weakness was in childhood and she lost ambulation at age 44. Non-invasive ventilation was started at age 45. A muscle biopsy was performed at age 30 and IHC analysis was only performed for alpha-sarcoglycan expression which was normal. A gene panel analysis was performed at age 52 and showed that the patient carried a pathogenic homozygous missense variant c.581T>C p.(Leu194Ser) in SGCG. Her parents were first cousins and she had an affected sister who carried the same homozygous variant.
Another female patient (patient 6) was diagnosed with LGMDR3 α-sarcoglycan-related. Onset of muscle weakness was in childhood and loss of ambulation occurred at age 11. Last follow up was at age 27. She passed away at age 43 due to recurrent pneumonias as complications of non-invasive ventilation. A muscle biopsy showed dystrophic changes and IHC analysis was only performed for alpha-sarcoglycan expression which was normal. Sanger sequencing of SGCA showed compound heterozygosity for the pathogenic variants c.101G>A p.(Arg34His) and c.402C>G p.(Tyr134*).
The third sarcoglycanopathy patient (patient 7) who was diagnosed with LGMDR4 β-sarcoglycan-related had an age of onset of muscle weakness in adolescence and loss of ambulation at age 46. Cardiac analysis showed a cardiomyopathy with a diffuse hypokinetic left ventricle. No muscle biopsy specimen was available for IHC analysis. NGS with a gene panel analysis of known limb-girdle and cardiomyopathy genes showed that the patient carried a pathogenic homozygous missense variant c.341C>T p.(Ser114Phe) in SGCB. Her parents were second cousins and she had a brother with muscle weakness who passed away at the age of 19 due to a cardiomyopathy.
TRIM32 RELATED MUSCULAR DYSTROPHY (LGMDR8 TRIM 32-RELATED)
One female patient (patient 8) was diagnosed with LGMDR8 TRIM 32-related. In her teens she developed proximal muscle weakness and loss of ambulation occurred in her early forties. She underwent surgery for an aortic valve stenosis and started non-invasive ventilation because of respiratory insufficiency at age 64. Her parents were first cousins. Sanger sequencing revealed that the patient carried a pathogenic homozygous missense variant c.1184T>C p.(Ile395Thr) in TRIM32. The amino acid isoleucine is evolutionary highly conserved and located in a highly conserved area of the first NHL repeat domain. In the same NHL domain a missense mutation was found in two patients from Southern Italy [24]. The first mutation described in TRIM32 originated from the Manitoba Hutterite population and was found in the third NHL repeat domain of TRIM32 [25]. Mutations in the conserved NHL domains are supposed to prevent TRIM32 self-interaction.
GMPPB-RELATED MUSCULAR DYSTROPHY (LGMDR19 GMPPB-RELATED)
Three patients from two families were diagnosed with LGMDR19 GMPPB-related also known as limb-girdle muscular dystrophy-dystroglycanopathy type C14 (MDDGC14). In one family both patients had an age of onset of muscle weakness in adolescence. The male patient (patient 9) was able to walk a few meters indoors without the use of an aid at his last examination at age 70. The female patient (patient 10) used a wheelchair for outdoors at her last follow up at age 57, but was reported to be wheelchair bound at a later age. Both patients complained of significant fluctuations in muscle weakness during the day. There were no ocular symptoms. Repetitive nerve stimulation of the accessory nerve with recording from the trapezius muscle showed an abnormal decrement of 14% in the compound muscle action potential in the male patient. Muscle imaging (computed tomography) of this patient showed fatty degeneration of the erector spinae, hamstrings and gastrocnemius muscles which is consistent with the pattern described previously (Fig. 3) [26]. WES with a virtual filter for known myopathy genes showed two novel variants of uncertain significance (class 3) c.332T>C p.(Val111Ala) and c.516C>G p.(Asn172Lys) in the GMPPB gene (NM_013334.3). Both the amino acid valine and asparagine are evolutionary highly conserved and located in the nucleotidyl transferase domain. These variants were not found in 251143 and 281873 alleles respectively of the Genome Aggregation Database (GnomAD). An older affected sister also carried these variants. DNA analysis of four unaffected siblings showed they either carried one or none of these variants. A muscle biopsy for evaluation of alpha-dystroglycan or fibroblasts for protein studies were not available [27].
Fig. 3
Muscle computed tomography of patient 9. CT-scan at the level of the lumbar spine (A), the pelvis (B), the thigh (C) and lower leg (D) of a male patient with GMPPB-related muscular dystrophy (LGMDR19) showing prominent fatty degeneration of the erector spinae (ES), rectus abdominis (RA), transversus abdominis (TrA), adductor magnus (AM) and hamstring (BB = biceps femoris short head, ST = semitendinosus, SM = semimembranosus) muscles and to a lesser extent of the medial (GM) and lateral gastrocnemius (GL) muscles. The gastrocnemius and gracilis (G) muscles are also increased in size (pseudohypertrophy).
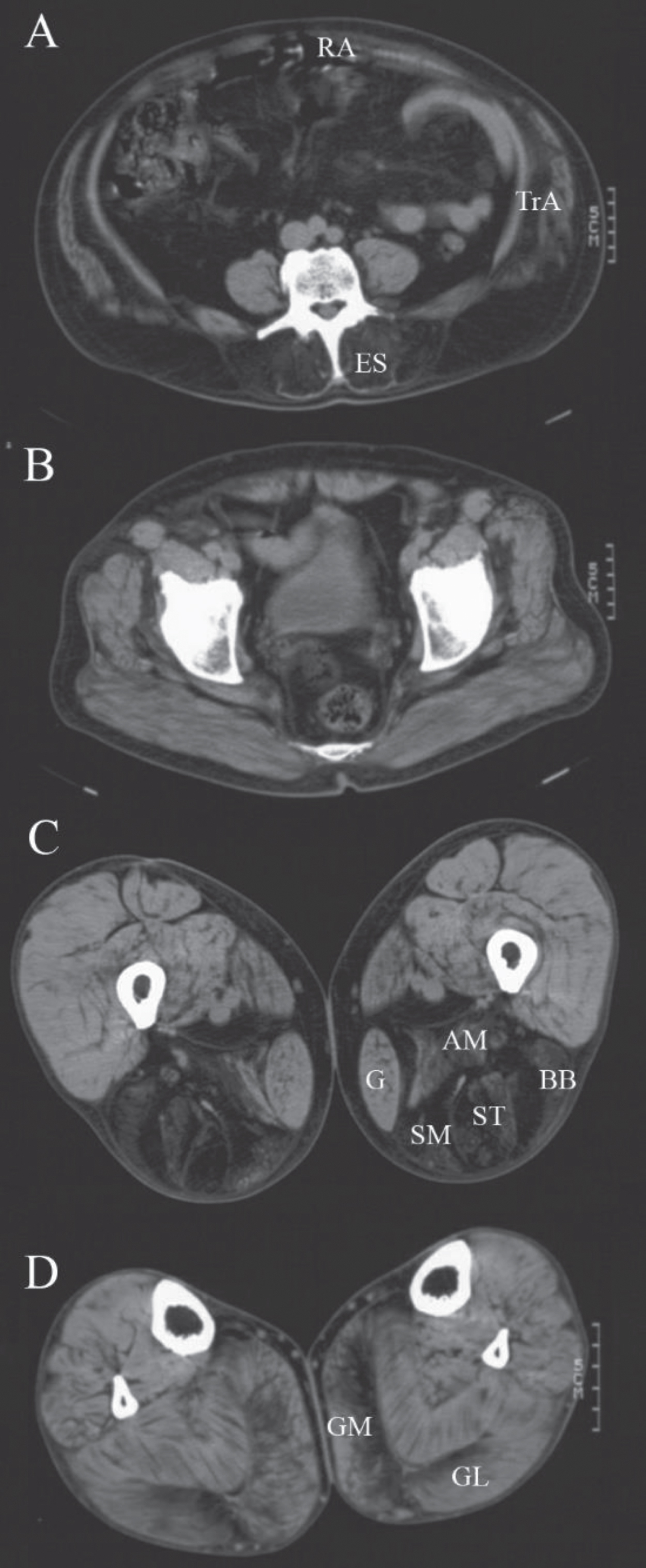
In the second family onset of muscle weakness in an isolated female patient (patient 11) was in early adolescence. Although she started using a wheelchair for travelling longer distances at age 44 she was still able to walk unaided for 25 meters at the time of the last examination at age 53. She did not complain of fluctuations in muscle strength. WES showed a pathogenic variant c.79G>C p.(Asp27His) in addition to a novel, likely pathogenic (class 4), frameshift variant c.1053dup p.(Val352Serfs*4) in GMPPB. The novel variant is present in the last exon and was found twice in 282678 alleles of the GnomAD. Unfortunately no DNA from siblings or parents was available.
MICU1-RELATED MYOPATHY
Two patients from one family were diagnosed with a MICU1-related myopathy. This family was analyzed after elevated serum CK activity (5390 IU/L) was found when the youngest sibling (patient 13) at the age of 5 presented at the emergency room because of an injury. A muscle biopsy showed a severe dystrophic pattern. There was decreased exercise tolerance in early childhood and proximal muscle weakness. At the age of 7 (patient 13) and 10 (patient 12), respectively, both patients were able to walk unaided for over 2 kilometers. Both patients subsequently developed learning disabilities and ataxia. Sanger sequencing revealed a pathogenic homozygous splice-site variant in MICU1 c.741 + 1G>A p.(Val248Thrfs*9) in both patients.
FILAMINOPATHY (FLNC-RELATED MYOPATHY)
Two patients from one family were diagnosed with FLNC-related myopathy. The female patient (patient 14) had difficulty running as a child and muscle weakness was noted at age 3. There was both proximal and distal muscle weakness on examination. She lost ambulation at age 34. She complained about heart palpitations and an electrocardiogram showed sinus tachycardia. Cardiac ultrasound showed no structural abnormalities. Non-invasive ventilation was started at age 63. The male patient (patient 15) presented with gait difficulty in adulthood and he lost ambulation at the age of 50. On initial examination there was more muscle weakness proximal than distal. No cardiac or respiratory involvement was found. A muscle biopsy from the deltoid muscle of patient 14 showed non-specific myopathic changes and electron microscopy showed no changes suggestive of a myofibrillary myopathy. No muscle biopsy was taken in patient 15. The patients were classified as AD LGMD because the mother and her sister also had limb girdle weakness. WES showed a novel heterozygous variant c.2791_2805del p.(Lys931_Val935del) in the FLNC gene (NM_001458.4) in both patients. This variant was classified as pathogenic (class 5) as it leads to an in-frame deletion of 5 highly conserved amino acids and was not found in > 246000 alleles of the GnomAD. Overlapping in frame deletions have been described previously in patient with FLNC-related myopathy [28–30]. DNA of the mother and sister was not available for testing. A son and daughter of patient 15 presented with limb girdle weakness and had the same variant.
FACIOSCAPULOHUMERAL DISEASE (FSHD1)
A female patient (patient 16) was diagnosed with facioscapulohumeral disease (FSHD) type 1. Onset of proximal muscle weakness was in adulthood and serum CK activity was only mildly elevated (4.5 times the upper limit of normal). No facial weakness was noted on initial investigation. Muscle biopsy showed predominantly myopathic changes. Skeletal muscle CT performed at age 26 and showed more or less symmetrical fatty degeneration of both the anterior and posterior compartment of the thigh and the gluteus maximus and abdominal muscles. No fatty degeneration was noted at the level of the shoulder or lower leg or of the paraspinal muscles. Screening of multiple LGMD related genes (e.g. CAPN3, FKRP, TCAP, TRIM32, MYOT, SGA, ANO5, DMD, SMCHD1) revealed no pathogenic variants. When the patient was reassessed at age 53 she had a facies myopathica and asymmetric weakness of the upper extremities with atrophy of the pectoralis major muscles. Although there was moderate to severe proximal muscle weakness of the lower limbs she was able to walk unaided for approximately 2 kilometers. This patient was initially classified as sporadic LGMD, however, when the mother of the patient was examined at the age of 73 she was also found to have a facies myopathica and moderate proximal muscle weakness on neurological examination. Southern blotting and hybridization with P13E-11 probe of the patient’s and her mother’s DNA showed an EcoRI fragment of ∼38 kb (about 10 copies of the D4Z4 repeat array) which, after digestion with BlnI, contracted further to ∼26 kb. This may indicate a short hybrid band (between 6–10 D4Z4 copies) in which D4Z4 repeats are partly BlnI sensitive and which is associated with a mild FSHD1 phenotype [12].
DISCUSSION
With the use of Sanger sequencing, a targeted gene panel or WES analysis, 12 previously undiagnosed LGMD families from our original cohort comprising 68 families could be diagnosed. This resulted in a genetic diagnosis in 95% of the families and 93% of the patients in whom genetic testing was still possible. Diagnosis included calpainopathy, dysferlinopathy, sarcoglycanopathy, LGMDR8 TRIM 32-related, LGMDR19 GMPPB-related, MICU1-related myopathy, filaminopathy and FSHD1.
A diagnosis was ascertained in 12 of the 15 families (80%). This diagnosis was dependent on NGS techniques in 3 of the 12 families. All families diagnosed with either dysferlinopathy or sarcoglycanopathy could potentially have been diagnosed earlier if complete immunohisto- and biochemical analysis had been performed. In one patient with LGMDR3 α-sarcoglycan-related due to a mutation in SGCA IHC analysis of alpha-sarcoglycan had been performed when staining became first available and it was considered normal. However, later IHC analysis of the other sarcoglycans could not be performed because muscle tissue was not available any more. In a previous study IHC analysis of alpha-sarcoglycan was found to be normal in 5 out of 9 LGMDR3 α-sarcoglycan-related patients. In all 5 patients at least one of the other sarcoglycans was considered abnormal on IHC analysis [31]. Pre-screening for sarcoglycanopathy by IHC analysis should therefore include all four sarcoglycans and if one of the sarcoglycan subunits is reduced in expression sequencing of all sarcoglycan genes should be performed [19]. In the other two sarcoglycanopathy patients analysis of the sarcoglycan genes was not performed as they had a mild phenotype remaining ambulant until their forties. The final diagnosis in the calpainopathy families was due to reclassification of a variant as pathogenic which was only possible when more information about the variant or additional family member information became available. One calpainopathy patient was initially classified as AD LGMD because of reported subjective symptoms of muscle weakness in multiple siblings and her father but was eventually diagnosed with LGMDR1 calpain3-related after reclassification of a second variant in CAPN3 as pathogenic.
Three patients from two families had a total of 3 novel variants in the GMPPB gene. Mutations in this gene lead to hypoglycosylation of the alpha-dystroglycan and can give rise to a spectrum of disorders ranging from congenital muscular dystrophy, congenital myasthenic syndrome to late onset LGMD [32, 33]. Two siblings complained of fluctuations in muscle weakness during the day and one had slight attenuation on repetitive nerve stimulation which are common findings in GMPPB-related muscular dystrophies. The muscle weakness was relatively mild with all patients remaining ambulatory until the last follow up. Although no cardiac or respiratory abnormalities were noted in our patients cardiomyopathy has been described in patients with a congenital or childhood onset of symptoms [27]. The prevalence of GMPPB-related muscular dystrophy (LGMDR19 GMPPB-related) in the Dutch LGMD cohort (3%) is similar to what is reported in a Danish cohort (1.5%) [26].
Genetic testing revealed diagnoses such as FSHD1 and myofibrillar myopathy that are not commonly associated with LGMD. Patients were initially classified as LGMD due to an atypical phenotype with the absence of classical clinical features such as facial weakness in the FSHD1 patient and the absence of myofibrillar changes on muscle biopsy in the FLNC-related myopathy patients. Atypical phenotypes of FSHD1 such as facial sparing scapulohumeral disease (SHD) and predominant lower limb muscle weakness have been previously reported [34]. Like in our patient SHD is associated with a higher age of onset of muscle weakness and fewer patients report loss of ambulation compared to the classical FSHD1 phenotype [35]. In contrast to our patient none of the SHD patients in a Chinese cohort who did show progressive limb weakness developed facial weakness on follow up [36]. The abnormalities on muscle imaging were not typical of FSHD1 [37, 38]. The patient with FSHD1 was initially classified as sporadic LGMD, instead of an AD muscular dystrophy, because the family history was negative due to late onset of symptoms in her mother. Limb girdle weakness in addition to a non-specific muscle biopsy in filaminopathy patients has been described especially in biopsies taken from a proximal muscle [39, 40]. As mutations in a single gene can lead to multiple phenotypes a LGMD phenotype of filaminopathy and FSHD1 should be considered in the diagnosis of muscle disorders with proximal muscle weakness. The patients with MICU1-related myopathy were included in the LGMD cohort because they did not yet manifest symptoms of brain involvement, i.e. learning disabilities and ataxia, at the time of inclusion.
As has been published previously it can be cumbersome to differentiate between an LGMD and a slowly progressive acquired myopathy (e.g. autoimmune necrotizing myopathy due to anti-SRP or anti-HMGCR antibodies) [41]. However, in none of our patients an erroneous diagnosis of myositis was considered prior to genetic testing. In the remaining 3 undiagnosed families WES with a virtual filter for 148 known myopathy genes showed no possible pathogenic variants.
The diagnostic rate of NGS techniques in LGMD described in the literature is still limited with 33% of cases solved by gene panel analysis, between 40% and 68.5% by WES and 37% by WES in LGMD patients in which previous Sanger sequencing did not yield a diagnosis [42–47]. The largest study of targeted WES in 1001 European and Middle Eastern patients with unexplained limb girdle weakness showed an overall diagnostic yield of 52% [48]. The diagnostic rate was dependent on whether analysis of the most prevalent LGMD subtypes had been performed prior to WES as was often the case in Western Europe (diagnostic rate of 35%) as compared to Eastern Europe and the Middle East (diagnostic rate 95%) [48]. The diagnostic yield is also highly dependent on patient selection and ranges between 18% in a group of patients with a suspected neuromuscular disorder based on medical history and/or neurological examination and/or abnormal ancillary investigations (i.e. muscle biopsy or laboratory findings) to 68.5% in a group of patients with medical history and neurological examination and muscle biopsy indicative of LGMD [47, 49]. As we present a description of various diagnostic methods a direct comparison of diagnostic yield is not feasible. As we have shown that mutations in multiple genes can result in clinically indistinguishable LGMD a (trio-based) WES with a virtual filter for known myopathy genes can be used as a first tier test. An exception on this can be made for example if there are clues, such as a positive family history, that points to a specific LGMD, or when there is a high level of suspicion of a disorder not covered and/or detected by WES. When using the (genotype) first approach of genotyping precise phenotyping can still be necessary somewhere in the diagnostic process to correctly interpret the results of molecular diagnostics. To ascertain more information on pathogenicity of a VUS or when variants in multiple muscle related genes are found guided muscle biopsy with immunohistochemical/biochemical analysis, additional RNA studies or blood-based protein assay, whole body MRI and neurological examination and genetic testing of family members in co-operation with the clinical geneticist can be useful. Preferably a trio-based WES is performed which compares the exome of the patient to that of the asymptomatic parents. This allows detection of de novo variants and in suspected AR muscle disease it can ascertain if two variants in one gene are present on different alleles. When no pathogenic variants are found additional genetic analyses such as sequencing of mitochondrial DNA and analysis of deletions/duplications or repeat expansions/contractions can be considered. Re-examination of WES data after 3 to 5 years may also improve the diagnostic yield as novel causative genes are identified. In selected cases additional techniques such as analysis of the whole exome or whole genome or transcriptome sequencing could be performed to ascertain a genetic diagnosis [50].
CONFLICT OF INTEREST
L. ten Dam reports no conflict of interest
M. de Visser is a member of the Data Safety Monitoring Board with AveXis.
H.B. Ginjaar reports no conflict of interest
H.A. van Duyvenvoorde reports no conflict of interest
S. van Koningsbruggen reports no conflict of interest
A.J. van der Kooi reports a grant from CSL Behring outside the submitted work.
ACKNOWLEDGMENTS
We would like to thank A.G.H. Langerak for obtaining additional DNA samples of patients. The authors wish to thank the Prinses Beatrix Spierfonds, The Hague for their financial support and C.S. Straathof, C.G. Faber, J.B.M. Kuks, E. Brusse, N.C. Voermans and J.E. Hoogendijk for their collaboration.
REFERENCES
[1] | van der Kooi AJ , Barth PG , Busch HF , de Haan R , Ginjaar HB , van Essen AJ , et al. The clinical spectrum of limb girdle muscular dystrophy. A survey in The Netherlands. Brain. (1996) ;119: (Pt 5):1471–80. |
[2] | van der Kooi AJ , Frankhuizen WS , Barth PG , Howeler CJ , Padberg GW , Spaans F , et al. Limb-girdle muscular dystrophy in the Netherlands: Gene defect identified in half the families. Neurology. (2007) ;68: (24):2125–8. |
[3] | van der Kooi AJ , ten Dam L , Frankhuizen WS , Straathof CSM , van Doorn PA , de Visser M , et al. ANO5 mutations in the Dutch limb girdle muscular dystrophy population. Neuromuscul Disord. (2013) ;23: :456–60. |
[4] | Straub /.V , Murphy A , Udd B . 229th ENMC international workshop: Limb girdle muscular dystrophies - Nomenclature and reformed classification Naarden, the Netherlands, 17-19 March 2017. Neuromuscul Disord. (2018) ;28: (8):702–10. |
[5] | Fanin M , Nascimbeni AC , Aurino S , Tasca E , Pegoraro E , Nigro V , et al. Frequency of LGMD gene mutations in Italian patients with distinct clinical phenotypes. Neurology. (2009) ;72: (16):1432–5. |
[6] | Magri F , Del Bo R , D’Angelo MG , Sciacco M , Gandossini S , Govoni A , et al. Frequency and characterisation of anoctamin 5 mutations in a cohort of Italian limb-girdle muscular dystrophy patients. Neuromuscul Disord. (2012) ;22: (11):934–43. |
[7] | Sveen ML , Schwartz M , Vissing J . High prevalence and phenotype-genotype correlations of limb-girdle muscular dystrophy type 2I in Denmark. Ann Neurol. (2009) ;59: (5):808–15. |
[8] | Witting N , Duno M , Petri H , Krag T , Bundgaard H , Kober L , et al. Anoctamin 5 muscular dystrophy in Denmark: Prevalence genotypes, phenotypes, cardiac findings and muscle protein expression. J Neurol. (2013) ;260: :2084–93. |
[9] | Schofield D , Alam K , Douglas L , Shrestha R , MacArthur DG , Davis M , et al. Cost-effectiveness ofmassively parallel sequencing for diagnosis of paediatricmuscle diseases. NPJ Genom Med. 2017;2. |
[10] | Bouman A , Brooks AS , van Slegtenhorst MA . [Whole Exome Sequencing in daily practice: The possibilities and impossibilities of this diagnostic test]. Ned Tijdschr Geneeskd. 2018;162. |
[11] | Chaturvedi LS , Mukherjee M , Srivastava S , Mittal RD , Mittal B . Point mutation and polymorphism in Duchenne/Becker muscular dystrophy (D/BMD) patients. Exp Mol Med. (2001) ;33: (4):251–6. |
[12] | Daxinger L , Tapscott SJ , van der Maarel SM . Genetic and epigenetic contributors to FSHD. Curr Opin Genet Dev. (2015) ;33: :56–61. |
[13] | Ahmed ST , Craven L , Russell OM , Turnbull DM , Vincent AE . Diagnosis and Treatment of Mitochondrial Myopathies. Neurotherapeutics. (2018) ;15: (4):943–53. |
[14] | Hehir-Kwa JY , Tops BBJ , Kemmeren P . The clinical implementation of copy number detection in the age of next-generation sequencing. Expert Rev Mol Diagn. (2018) ;18: (10):907–15. |
[15] | Halman A , Oshlack A . Accuracy of short tandem repeats genotyping tools in whole exome sequencing data. FRes. (2020) ;9: :200. |
[16] | Hoffman-Andrews L . The known unknown: The challenges of genetic variants of uncertain significance in clinical practice. J Law Biosci. (2017) ;4: (3):648–57. |
[17] | van Deutekom JC , Bakker E , Lemmers RJ , van der Wielen MJ , Bik E , Hofker MH , et al. Evidence for subtelomeric exchange of 3.3 kb tandemly repeated units between chromosomes 4q35 and 10q Implications for genetic counselling and etiology of FSHD1. Hum Mol Genet. (1996) ;5: (12):1997–2003. |
[18] | ten Dam L , Frankhuizen WS , Linssen W , Straathof CS , Niks EH , Faber CG , et al. Autosomal recessive limb-girdle and Miyoshi muscular dystrophies in the Netherlands: The clinical and molecular spectrum of 244 patients. Clin Genet. (2019) ;96: (2):126–33. |
[19] | Ginjaar HB , van der Kooi AJ , Ceelie H , Kneppers AL , van Meegen M , Barth PG , et al. Sarcoglycanopathies in Dutch patients with autosomal recessive limb girdle muscular dystrophy. J Neurol. (2000) ;247: (7):524–9. |
[20] | Anderson LV , Davison K . Multiplex Western blotting system for the analysis of muscular dystrophy proteins. Am J Pathol. (1999) ;154: (4):1017–22. |
[21] | van der Kooi AJ , de Voogt WG , Barth PG , Busch HF , Jennekens FG , Jongen PJ , et al. The heart in limb girdle muscular dystrophy. Heart. (1998) ;79: (1):73–7. |
[22] | Nascimbeni AC , Fanin M , Tasca E , Angelini C . Transcriptional and translational effects of intronic CAPN3 gene mutations. Hum Mutat. (2010) ;31: (9):E1658–69. |
[23] | Piluso G , Politano L , Aurino S , Fanin M , Ricci E , Ventriglia VM , et al. Extensive scanning of the calpain-3 gene broadens the spectrum of LGMD2A phenotypes. J Med Genet. (2005) ;42: (9):686–93. |
[24] | Saccone V , Palmieri M , Passamano L , Piluso G , Meroni G , Politano L , et al. Mutations that impair interaction properties of TRIM32 associated with limb-girdle muscular dystrophy 2H. Hum Mutat. (2008) ;29: (2):240–7. |
[25] | Frosk P , Weiler T , Nylen E , Sudha T , Greenberg CR , Morgan K , et al. Limb-girdle muscular dystrophy type 2H associated with mutation in TRIM32, a putative E3-ubiquitin-ligase gene. Am J Hum Genet. (2002) ;70: (3):663–72. |
[26] | Oestergaard ST , Stojkovic T , Dahlqvist JR , Bouchet-Seraphin C , Nectoux J , Leturcq F , et al. Muscle involvement in limb-girdle muscular dystrophy with GMPPB deficiency (LGMD2T). Neurol Genet. (2016) ;2: (6):e112. |
[27] | Carss KJ , Stevens E , Foley AR , Cirak S , Riemersma M , Torelli S , et al. Mutations in GDP-mannose pyrophosphorylase B cause congenital and limb-girdle muscular dystrophies associated with hypoglycosylation of alpha-dystroglycan. Am J Hum Genet. (2013) ;93: (1):29–41. |
[28] | Luan X , Hong D , Zhang W , Wang Z , Yuan Y . A novel heterozygous deletion-insertion mutation -del/GTTTGT ins) in exon 18 of the filamin C gene causes filaminopathy in a large Chinese family. Neuromuscul Disord. (2010) ;20: (6):390–6. |
[29] | Evila A , Arumilli M , Udd B , Hackman P . Targeted next-generation sequencing assay for detection of mutations in primary myopathies. Neuromuscul Disord. (2016) ;26: (1):7–15. |
[30] | Kley RA , Serdaroglu-Oflazer P , Leber Y , Odgerel Z , van der Ven PF , Olive M , et al. Pathophysiology of protein aggregation and extended phenotyping in filaminopathy. Brain. (2012) ;135: (Pt 9):2642–60. |
[31] | Klinge L , Dekomien G , Aboumousa A , Charlton R , Epplen JT , Barresi R , et al. Sarcoglycanopathies: Can muscle immunoanalysis predict the genotype? Neuromuscul Disord. (2008) ;18: (12):934–41. |
[32] | Astrea G , Romano A , Angelini C , Antozzi CG , Barresi R , Battini R , et al. Broad phenotypic spectrum and genotype-phenotype correlations in GMPPB-related dystroglycanopathies: An Italian cross-sectional study. Orphanet J Rare Dis. (2018) ;13: (1):170. |
[33] | Luo S , Cai S , Maxwell S , Yue D , Zhu W , Qiao K , et al. Novel mutations in the C-terminal region of GMPPB causing limb-girdle muscular dystrophy overlapping with congenital myasthenic syndrome. Neuromuscul Disord. (2017) ;27: (6):557–64. |
[34] | Krasnianski M , Eger K , Neudecker S , Jakubiczka S , Zierz S . Atypical phenotypes in patients with facioscapulohumeral muscular dystrophy 4q35 deletion. Arch Neurol. (2003) ;60: (10):1421–5. |
[35] | Ricci G , Cammish P , Siciliano G , Tupler R , Lochmuller H , Evangelista T . Phenotype may predict the clinical course of facioscapolohumeral muscular dystrophy. Muscle Nerve. (2019) ;59: (6):711–3. |
[36] | He JJ , Lin XD , Lin F , Xu GR , Xu LQ , Hu W , et al. Clinical and genetic features of patients with facial-sparing facioscapulohumeral muscular dystrophy. Eur J Neurol. (2018) ;25: (2):356–64. |
[37] | Dahlqvist JR , Vissing CR , Thomsen C , Vissing J . Severe paraspinal muscle involvement in facioscapulohumeral muscular dystrophy. Neurology. (2014) ;83: (13):1178–83. |
[38] | Leung DG , Carrino JA , Wagner KR , Jacobs MA . Whole-body magnetic resonance imaging evaluation of facioscapulohumeral muscular dystrophy. Muscle Nerve. (2015) ;52: (4):512–20. |
[39] | Kley RA , Hellenbroich Y , van der Ven PF , Furst DO , Huebner A , Bruchertseifer V , et al. Clinical and morphological phenotype of the filamin myopathy: A study of 31 German patients. Brain. (2007) ;130: (Pt 12):3250–64. |
[40] | van den Bogaart FJ , Claeys KG , Kley RA , Kusters B , Schrading S , Kamsteeg EJ , et al. Widening the spectrum of filamin-C myopathy: Predominantly proximal myopathy due to the A193T mutation in the actin-binding domain of FLNC. Neuromuscul Disord. (2017) ;27: (1):73–7. |
[41] | Benveniste O , Romero NB . Myositis or dystrophy? Traps and pitfalls. Presse Med. (2011) ;40: (4 Pt 2):e249–55. |
[42] | Ozyilmaz B , Kirbiyik O , Ozdemir TR , Kaya Ozer O , Kutbay YB , Erdogan KM , et al. Impact of next-generation sequencing panels in the evaluation of limb-girdle muscular dystrophies. Ann Hum Genet. (2019) ;83: (5):331–47. |
[43] | Kuhn M , Glaser D , Joshi PR , Zierz S , Wenninger S , Schoser B , et al. Utility of a next-generation sequencing-based gene panel investigation in German patients with genetically unclassified limb-girdle muscular dystrophy. J Neurol. (2016) ;263: (4):743–50. |
[44] | Ghaoui R , Cooper ST , Lek M , Jones K , Corbett A , Reddel SW , et al. Use of Whole-Exome Sequencing for Diagnosis of Limb-Girdle Muscular Dystrophy: Outcomes and Lessons Learned. JAMA Neurol. (2015) ;72: (12):1424–32. |
[45] | Harris E , Topf A , Barresi R , Hudson J , Powell H , Tellez J , et al. Exome sequences versus sequential gene testing in the UK highly specialised Service for Limb Girdle Muscular Dystrophy. Orphanet J Rare Dis. (2017) ;12: (1):151. |
[46] | Reddy HM , Cho KA , Lek M , Estrella E , Valkanas E , Jones MD , et al. The sensitivity of exome sequencing in identifying pathogenic mutations for LGMD in the United States. J Hum Genet. (2017) ;62: (2):243–52. |
[47] | Fichna JP , Macias A , Piechota M , Korostynski M , Potulska-Chromik A , Redowicz MJ , et al. Whole-exome sequencing identifies novel pathogenic mutations and putative phenotype-influencing variants in Polish limb-girdle muscular dystrophy patients. Hum Genomics. (2018) ;12: (1):34. |
[48] | Töpf A , Johnson K , Bates A , Phillips L , Chao KR , England EM , et al. Sequential targeted exome sequencing of 1001 patients affected by unexplained limb-girdle weakness. Genetics in Medicine. 2020; https://doi.org/10.1038/s41436-020-0840-3. |
[49] | Westra D , Schouten MI , Stunnenberg BC , Kusters B , Saris CGJ , Erasmus CE , et al. Panel-Based Exome Sequencing for Neuromuscular Disorders as a Diagnostic Service. J Neuromuscul Dis. (2019) ;6: (2):241–58. |
[50] | Gonorazky HD , Naumenko S , Ramani AK , Nelakuditi V , Mashouri P , Wang P , et al. Expanding the Boundaries of RNA Sequencing as a Diagnostic Tool for Rare Mendelian Disease. Am J Hum Genet. (2019) ;104: (3):466–83. |


