
Results from a 3-year Non-interventional, Observational Disease Monitoring Program in Adults with GNE Myopathy
Abstract
BACKGROUND:
GNE myopathy is a rare, autosomal recessive, muscle disease caused by mutations in GNE and is characterized by rimmed vacuoles on muscle biopsy and progressive distal to proximal muscle weakness.
OBJECTIVE:
Investigate the clinical presentation and progression of GNE myopathy.
METHODS:
The GNE Myopathy Disease Monitoring Program was an international, prospective, observational study in subjects with GNE myopathy. Muscle strength was assessed with hand-held dynamometry (HHD), with upper extremity (UE) and lower extremity (LE) composite scores reflecting upper and lower extremity muscle groups, respectively. The GNE myopathy–Functional Activity Scale (GNEM-FAS) was used to further assess impairment in mobility, upper extremity function, and self-care.
RESULTS:
Eighty-seven of 101 enrolled subjects completed the trial until study closure by the sponsor; 60 completed 36 months. Mean (SD) HHD UE composite score decreased from 34.3 kg (32.0) at baseline to 29.4 kg (32.6) kg at month 36 (LS mean change [95%CI]: –3.8 kg [–5.9, –1.7]; P = 0.0005). Mean (SD) HHD LE composite score decreased from 32.0 kg (34.1) at baseline to 25.5 kg (31.2) at month 36 (LS mean change [95%CI]: –4.9 [–7.7, –2.2]; P = 0.0005). GNEM-FAS scores were more severe at baseline in subjects who walked <200 meters versus ≥200 meters in 6 minutes; in both groups, GNEM-FAS total, mobility, UE, and self-care scores decreased from baseline through month 36.
CONCLUSIONS:
These findings demonstrate progressive decline in muscle strength in GNE myopathy and provide insight into the appropriate tools to detect clinically meaningful changes in future GNE myopathy interventional trials.
INTRODUCTION
GNE myopathy (also known as hereditary inclusion body myopathy, Nonaka Distal Myopathy or Rimmed Vacuole Myopathy) is a rare, autosomal recessive, muscle disease caused by mutations in GNE, the gene coding for the bifunctional UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase [1–3]. This enzyme is the first step in the biosynthesis of sialic acid (SA) required for the glycosylation of proteins and lipids. GNE myopathy is characterized by rimmed vacuoles on a muscle biopsy and usually presents with often severe muscle weakness that slowly progresses distally to proximally, eventually compromising arm and leg muscle function, but sparing the quadriceps muscles. Wheelchair use may be required in 10 to 20 years after symptom onset. The prevalence of GNE myopathy is estimated at 1 to 9/1,000,000 [2]. While there are currently no approved treatments for GNE myopathy, increasing SA in a GNE myopathy mouse model with oral supplementation reduced muscle weakness and pathology [4]; proof-of-concept gene therapy research has also shown promise in mice [5].
Two previous studies have provided insight into the natural history of GNE myopathy [6, 7]. Mori-Yoshimura et al, 2014 followed 24 Japanese patients with GNE myopathy for one year noting declines in pulmonary function and muscle testing, including the 6-minute Walk Test and grip power. In an effort to understand disease progression over a longer period of time, Quintana et al, 2019 developed a Bayesian model of GNE myopathy based upon natural history data from 38 subjects followed for an average of 14 months that showed declines in muscle strength and physical functioning as a function of estimated disease progression.
As a part of a larger clinical program investigating the safety and efficacy of an extended—release formulation of aceneuramic acid as supplementation therapy for GNE myopathy, Ultragenyx Pharmaceutical Inc. developed a Disease Monitoring Program (GNEM-DMP; Study UX001-CL401). The GNEM-DMP comprises two non-interventional studies: an online survey, which was reported previously [8], and an in-clinic study. This report describes the results of the in-clinic component of the GNEM-DNP in 101 subjects with GNE myopathy followed for 3 years.
MATERIALS AND METHODS
Study design
The GNEM-DMP was an international, non-interventional, observational study to better understand the presentation, progression, geographic distribution, and prevalence of GNE myopathy. Additionally, the study aimed to provide connection for subjects to the broader GNE myopathy community and share the clinical outcomes of this study in an effort to further develop potential GNE therapies. The GNEM-DMP was comprised of an online, patient- or caregiver—reported component and an in-clinic, physician—reported component, and subjects were allowed to participate in either or both components of the program. This report summarizes the in-clinic component of the study; the online component was previously published [8]. After completing a consent form, subjects were assessed at baseline, 6 months (±1 month), 12 months, 24 months, and 36 months (±3 month). In-clinic visits lasted a full day and assessed medical history or interval history for subsequent visits, prior and concomitant medications, muscle strength, patient-reported outcomes, and safety.
The institutional review board at each participating center approved the protocol. The study was conducted in accordance with the Declaration of Helsinki and the Good Clinical Practice guidelines developed by the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. This study is registered on ClinicalTrials.gov (NCT01784679).
Subjects and assessments
Key inclusion criteria included age ≥18 years, willingness to release access to medical information for the study, and diagnosis of GNE myopathy (genetic confirmation was not required for the online component, but was required for the in-clinic component). Subjects with an unrelated, comorbid disease or condition that may have interfered with study participation or affected safety, in the opinion of the investigator, were excluded from participation.
The following were assessed by hand-held dynamometry (HHD) or manual muscle testing (MMT) using a microFET®2 or Jamar hydraulic hand dynamometer: gross grip, pinch, shoulder abductors, elbow flexors, elbow extensors, hip flexors, hip extensors, hip abductors, hip adductors, knee flexors, and knee extensors. Composite HHD scores were calculated for the sum of the upper (grip, shoulder abductors, elbow flexors, and elbow extensors) and lower (hip flexors, hip extensors, hip abductors, hip adductors, and knee flexors) muscle groups. Ambulatory subjects completed the 6-minute Walk Test (6MWT) and Stair Climb Test. All subjects completed the pulmonary function test. The GNE myopathy - Functional Activity Scale (GNEM-FAS), assessed in both ambulatory and non-ambulatory subjects, is a validated instrument with qualitative and quantitative measures designed to assess functional impairment in GNE myopathy including mobility, upper extremity (UE) function, and self-care; higher GNEM-FAS scores indicate a lower level of impairment [9, 10]. The incidence and severity of adverse events (AEs) were collected to assess safety. Additional assessments are listed in the study protocol available at ClinicalTrials.gov (identifier: NCT01784679) and clinicaldata.ema.europa.eu (procedure number: EMEA/H/C/004176/0000).
Statistical analysis
For continuous variables, the data were described using descriptive summaries such as median, mean, standard deviation, standard error of the mean, and range. For categorical variables, descriptive summaries include counts and proportions. For repeated measures, least squares (LS) mean change from baseline and 95%confidence intervals (CI) were calculated using the generalized estimating equation (GEE) model. Subjects receiving N-acetyl-D-mannosamine (ManNAc) and/or intravenous immune globulin (IVIG) at baseline were excluded from the analysis. As described above, some assessments were only feasible in ambulatory subjects, and therefore, only analyzed in ambulatory subjects. A ad-hoc analysis examining clinical endpoints in Bulgarian subjects, accounting for nearly half of all participants (47/101) in the study, was conducted to evaluate potential differences in a subgroup with a homogenous mutation. Bulgaria is home to a cluster of patients with GNE myopathy thought to express the same founder mutation [11, 12].
Individual de-identified participant data will be available for 12 months from the time of publication upon request to researchers providing a methodologically sound proposal for analysis for research that contributes to the field of GNE myopathy. Proposals should be directed to [email protected]. To gain access, data requestors will need to sign a data access and use agreement. Data will be shared via a secure portal.
Results
Baseline characteristics
Between April 5, 2013, and January 11, 2018, 101 subjects were enrolled in the study, 87 (86.1%) of whom completed the study up to the point of early closure by the sponsor on January 11, 2018. The remaining 14 subjects discontinued due to death (n = 3), consent withdrawn (n = 2), principal investigator decision (n = 2), non-compliance (n = 1), other (n = 1), and no reason given (n = 5). Ninety-eight subjects (97%) completed 6 months, 92 (91%) completed 12 months, 81 (80%) completed 24 months, 60 (59%) completed 36 months, and 9 (9%) completed 48 months. Six subjects who were receiving ManNAc and/or IVIG were excluded from this analysis. Because so few subjects completed 48 months due to early study closure, data is reported through month 36.
The majority of enrolled (N = 101) subjects were male, white, and Roma (Table 1). Forty-seven subjects (47%) reported a misdiagnosis, 56 (55%) reported their first GNE myopathy symptom as “tripping/foot-drop/toe-drop,” and 16 (16%) reported “walking funny/unsteady gait/difficulty walking.” At baseline, 40 subjects (40%) were using lower extremity (LE) orthotics, 27 (27%) were using an assistive walking device, and 39 (39%) were using a wheelchair/scooter. Approximately one-half of the subjects (n = 48; 48%) reported previous use of medications specifically for GNE myopathy (or HIBM), including SA, ManNAc, IVIG, sialyllactose, St. John’s Wort, and creatine. At enrollment, the most frequent medications for GNE myopathy were SA (n = 39; 39%) and creatine (n = 17; 17%), and the most frequent non—medications for GNE myopathy were physical therapy (n = 48; 48%) and massage (n = 16; 16%). Concomitant use of creatine was reported for 7 of 76 (9%) subjects at month 24 and 4 of 58 (7%) subjects at month 36. No concomitant use of SA was reported at months 24 and 36.
Table 1
Baseline Demographics and Characteristics
| Demographic or Characteristic | All Subjects (N = 101) |
| Male, n (%) | 55 (54.5) |
| Age, years, mean (SD) | 39.2 (12.2) |
| Height, (cm), mean (SD) | 168.9 (9.7) |
| Weight, (kg), mean (SD) | 73.8 (16.8) |
| Race, n (%) | |
| White (Hispanic/Latino) | 2 (2.0) |
| White (Non-Hispanic/Latino) | 87 (86.1) |
| Black of African American (Non-Hispanic/Latino) | 1 (1.0) |
| Asian | 10 (9.9) |
| Othera | 1 (1.0) |
| Ethnicity, n (%) | |
| Persian Jewish descent | 3 (3.0) |
| Indian (subcontinent) | 10 (9.9) |
| Italian | 4 (4.0) |
| French | 5 (5.0) |
| Roma | 47 (46.5) |
| Othera | 32 (31.7) |
| 6MWT Category, n (%) | |
| Able to Complete ≥200 Meters | 46 (45.5) |
| Completed <200 Meters or Unable to Complete Test | 55 (54.5) |
| Age at First Symptom, years, mean (SD) | 26.0 (8.5) |
| Age at GNE Myopathy Diagnosis, years, mean (SD) | 33.7 (10.8) |
| Medical History and Symptoms, n (%) | |
| Tripping/foot-drop/toe-drop | 91 (90.1) |
| Walking funny/unsteady gait/difficulty walking | 86 (85.1) |
| Frequent falls | 58 (57.4) |
| Weakness in legs/feet | 84 (83.2) |
| Weakness in arms/hands | 74 (73.3) |
| Fatigue | 48 (47.5) |
| Muscle twitching/spasms | 16 (15.8) |
| Muscle pain | 39 (38.6) |
| Difficulty climbing stairs | 83 (82.2) |
| Difficulty moving and changing positions | 56 (55.4) |
| Othera | 18 (17.8) |
| Creatine kinase, U/L, mean (SD)b | 414.1 (439.5) |
| Free Sialic Acid Level, μ#x03BC;#x03BC;g/mL, mean (SD)b | 0.17 (0.08) |
| HHD Upper Extremity Composite Score, kg, mean (SD)b | 34.3 (32.0) |
| HHD Lower Extremity Composite Score, kg, mean (SD) b | 32.0 (34.1) |
aIncludes British (n = 10), Scottish (n = 4), Non-Persian Jewish (n = 3), Dutch (n = 2), Algerian (n = 1), Canadian (n = 1), French and British (n = 1), French and UK (n = 1), Irish (n = 1), Irish and Norwegian (n = 1), Jewish Karaite (n = 1), Scottish (n = 4), Senegalese (n = 1), South American (n = 1), Spanish (n = 1), UK (n = 1), and Unknown (n = 1). bExcludes 6 subjects who were taking ManNAc and/or IVIG at baseline. 6MWT, 6-minute Walk Test; HHD, hand held dynamometry.
Hand held dynamometry upper extremity and lower extremity composite scores
Mean HHD UE composite scores decreased from baseline through month 36, with a decreased LS mean change from baseline to month 36 of –3.8 kg (95%CI: –5.9, –1.7; P = 0.0005; Fig. 1A, Supplemental Table 1). Mean HHD LE composite scores decreased from baseline through month 36, with a decreased LS mean change from baseline to month 36 of –4.9 kg (95%CI: –7.7, –2.2; P = 0.0005; Fig. 1B, Supplemental Table 1). Mean raw strength scores for individual muscle groups included in the HHD UE and LE composites likewise declined, with at least one statistically significant LS mean change from baseline all individual muscle groups (Table 2).
Fig. 1
Mean±SE change from baseline to month 36 in hand-held dynamometry upper extremity and lower extremity composite scores. Subject numbers are shown above the x-axis. **P < 0.01, **P < 0.001, and ***P < 0.0001 using the GEE model. GEE, generalized estimating equation; LS, least squares; SE, standard error.
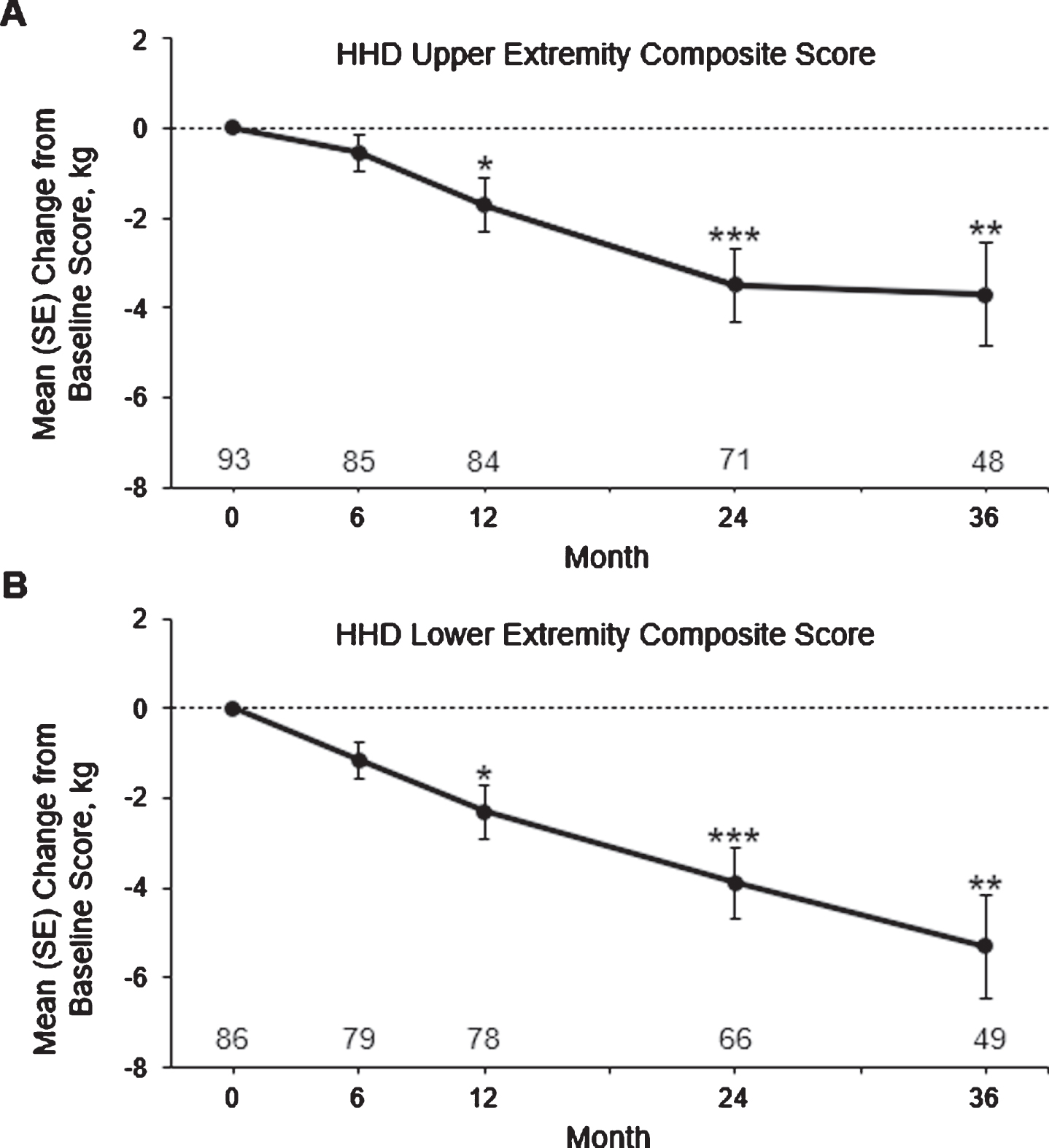
Table 2
Change in Individual Muscle Groups with Hand-Held Dynamometry
| Muscle Groups | All Subjects | ||||
| Baseline | Month 6 | Month 12 | Month 24 | Month 36 | |
| Gross Grip, kg | |||||
| Mean (SD) [n] | 12.6 (13.7) [95] | 12.8 (14.0) [92] | 12.6 (14.2) [90] | 11.9 (14.0) [76] | 10.8 (13.9) [67] |
| LSM (95%CI) change from baseline | — | 0.01 (–0.5, 0.5) | –0.4 (–0.9, 0.2) | –0.7 (–1.4, 0.1) | –1.3 (–2.1, –0.4) |
| P value | — | 0.9532 | 0.1627 | 0.0949 | 0.0051 |
| Shoulder Abductors, kg | |||||
| Mean (SD) [n] | 7.8 (7.3) [93] | 7.8 (7.3) [89] | 7.7 (7.3) [87] | 6.3 (6.5) [74] | 5.6 (5.8) [61] |
| LSM (95%CI) Change from Baseline | — | –0.2 (–0.6, 0.3) | –0.4 (–0.8, 0.1) | –1.2 (–1.6, –0.8) | –1.3 (–1.9, –0.8) |
| P value | — | 0.4661 | 0.0920 | <0.0001 | <0.0001 |
| Elbow Flexors, kg | |||||
| Mean (SD) [n] | 7.9 (7.7) [95] | 7.7 (8.1) [88] | 7.5 (8.0) [86] | 6.8 (7.3) [74] | 7.1 (7.8) [57] |
| LSM (95%CI) Change from Baseline | — | –0.2 (–0.6, 0.2) | –0.5 (–0.9, –0.1) | –0.8 (–1.3, –0.3) | –0.9 (–1.6, –0.2) |
| P value | — | 0.3847 | 0.0181 | 0.0015 | 0.0082 |
| Elbow Extensors, kg | |||||
| Mean (SD) [n] | 6.1 (5.6) [94] | 5.9 (5.6) [88] | 5.9 (5.7) [86] | 5.6 (5.4) [74] | 5.6 (5.5) [57] |
| LSM (95%CI) Change from Baseline | — | –0.2 (–0.5, 0.04) | –0.3 (–0.6, –0.1) | –0.5 (–0.9, –0.1) | –0.6 (–1.0, –0.2) |
| P value | — | 0.0928 | 0.0029 | 0.0077 | 0.0089 |
| Hip Abductors, kg | |||||
| Mean (SD) [n] | 10.2 (7.9) [95] | 9.5 (7.7) [89] | 9.9 (8.2) [86] | 9.3 (8.4) [75] | 9.4 (8.7) [58] |
| LSM (95%CI) Change from Baseline | — | –0.7 (–1.2, –0.2) | –0.5 (–1.0, 0.1) | –0.7 (–1.4, –0.009) | –0.8 (–1.5, 0.02) |
| P value | — | 0.0049 | 0.0880 | 0.0472 | 0.0559 |
| Hip Adductors, kg | |||||
| Mean (SD) [n] | 4.3 (6.2) [94] | 3.9 (5.7) [87] | 4.0 (5.8) [85] | 3.5 (6.0) [74] | 4.0 (7.0) [58] |
| LSM (95%CI) Change from Baseline | — | –0.1 (–0.4, 0.2) | –0.4 (–0.8, 0.1) | –0.6 (–1.1, –0.1) | –0.4 (–1.2, 0.4) |
| P value | — | 0.5106 | 0.1078 | 0.0163 | 0.2969 |
| Hip Flexors, kg | |||||
| Mean (SD) [n] | 4.9 (7.9) [93] | 5.0 (8.3) [88] | 4.5 (7.3) [86] | 3.5 (6.4) [73] | 3.3 (5.8) [52] |
| LSM (95%CI) Change from Baseline | — | 0.1 (–0.4, 0.5) | –0.5 (–0.9, –0.1) | –1.0 (–1.5, –0.5) | –1.3 (–1.9, –0.6) |
| P value | — | 0.7232 | 0.0148 | 0.0001 | 0.0001 |
| Hip Extensors, kg | |||||
| Mean (SD) [n] | 8.6 (9.3) [88] | 8.5 (9.7) [84] | 8.2 (9.8) [82] | 7.0 (9.4) [72] | 6.1 (8.0) [51] |
| LSM (95%CI) Change from Baseline | — | –0.04 (–0.8, 0.7) | –0.5 (–1.3, 0.4) | –1.3 (–2.4, –0.3) | –2.1 (–3.3, –1.0) |
| P value | — | 0.9140 | 0.2711 | 0.0154 | 0.0002 |
| Knee Flexors, kg | |||||
| Mean (SD) [n] | 3.8 (5.8) [93] | 4.0 (7.0) [88] | 3.7 (6.3) [85] | 3.0 (5.8) [75] | 2.8 (5.4) [59] |
| LSM (95%CI) Change from Baseline | — | –0.1 (–0.4, 0.3) | –0.1 (–0.5, 0.2) | –0.7 (–1.1, –0.3) | –1.0 (–1.4, –0.5) |
| P value | — | 0.7125 | 0.4873 | 0.0019 | <0.0001 |
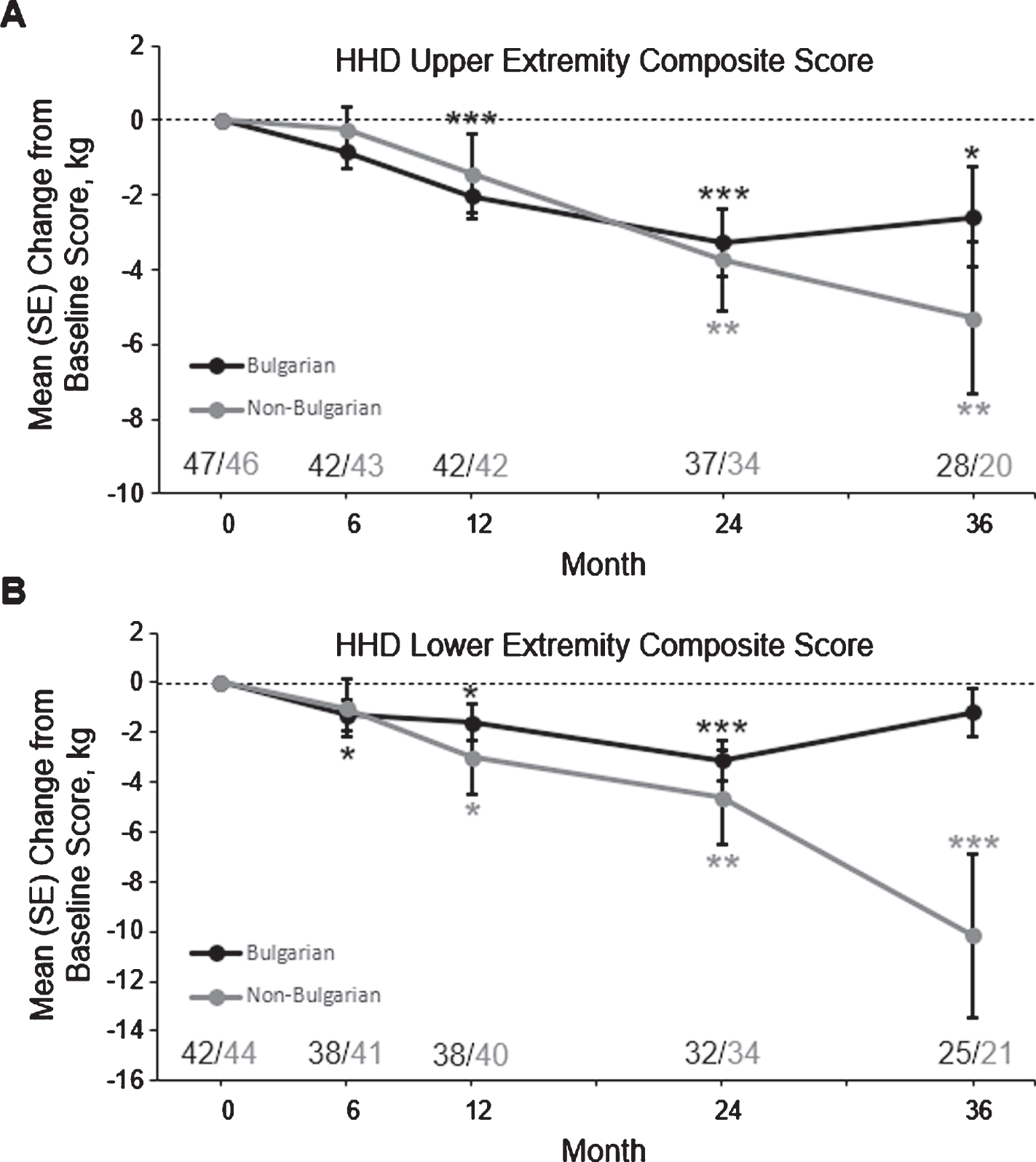
In an ad hoc subgroup analysis, mean HHD UE and LE scores were more severe at baseline and throughout the study in 47 Bulgarian subjects compared with 46 non-Bulgarian subjects who had baseline and post-baseline data (Supplemental Table 2). The mean (SD) age of first GNEM symptom was earlier in Bulgarian subjects compared with non-Bulgarian subjects (23.7 [6.4] vs 28.0 [9.6] years), as was mean (SD) age of GNEM diagnosis (31.0 [9.0] vs 36.1 [11.8] years). Mean HHD UE composite scores decreased from baseline through month 36 in Bulgarian and non-Bulgarian subjects, with a decreased in LS mean change from baseline to month 36 in Bulgarian subjects of –2.9 kg (95%CI: –5.2, –0.6; P = 0.0145) and –5.1 kg (95%CI, –8.8, –1.3; P = 0.0085) in non-Bulgarian subjects (Fig. 2A). Similarly, mean HHD LE composite scores decreased from baseline through month 36 in Bulgarian and non-Bulgarian subjects, with a decrease in LS mean change from baseline to month 36 in Bulgarian subjects of –1.5 kg (95%CI: –3.4, –0.3; P = 0.1028) and –8.8 kg (95%CI: –14.0, –3.6; P = 0.0009) in non-Bulgarian subjects (Fig. 2B).
Fig. 2
Mean±SE change from baseline to month 36 in hand-held dynamometry upper extremity and lower extremity composite scores in Bulgarian and non-Bulgarian subjects. Subject numbers are shown above the x-axis. *P < 0.05, **P < 0.01, and ***P < 0.001 using the GEE model. GEE, generalized estimating equation; LS, least squares; SE, standard error.
GNE myopathy –functional activity scale
As expected, GNEM-FAS scores (total, mobility, UE, and self-care) were more severe at baseline and throughout the study in subjects who walked <200 meters in baseline 6MWT (ie, “<200-meter subjects”) than in subjects who walked ≥200 meters (ie, “≥200-meter subjects”) (Supplemental Table 1). Mean GNEM-FAS total scores decreased from baseline through month 36, with a decrease in LS mean for from baseline to month 36 in ≥200-meter subjects of –6.3 units (95%CI: –9.4, –3.2; P < 0.0001) and –12.3 units (95%CI: –18.3, –6.3; P < 0.0001) in <200-meter subjects (Fig. 3A).
Fig. 3
Mean±SE change from baseline to month 36 in GNEM-FAS total score (A), mobility score (B), upper extremity score (C), and self-care score (D) in subjects who completed ≥200 and <200 meters at baseline. Subject numbers are shown above the x-axis. *P < 0.05, **P < 0.01, and ***P < 0.001 using the GEE model. GEE, generalized estimating equation; LS, least squares; SE, standard error.
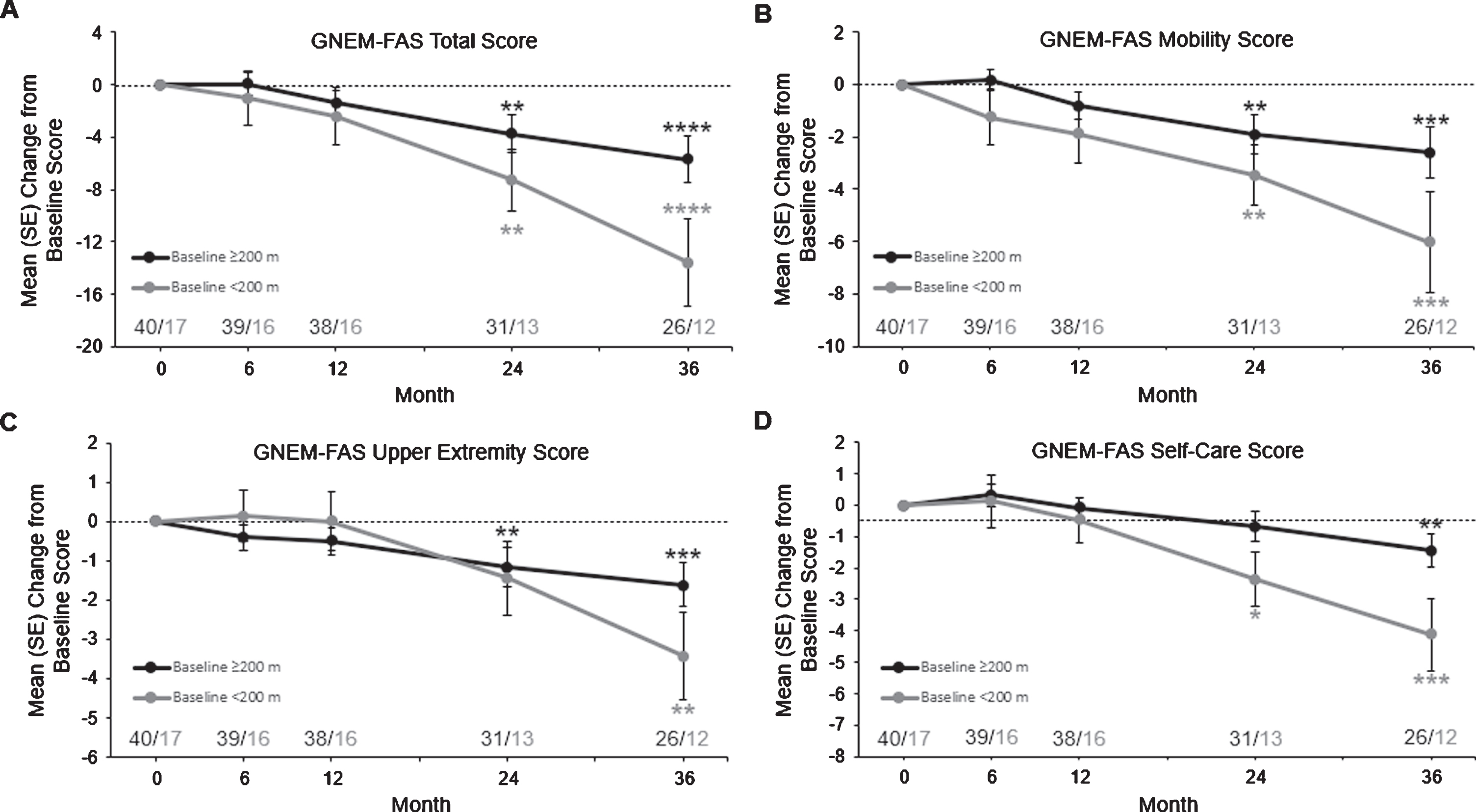
Among the GNEM-FAS subscores, mean GNEM FAS mobility scores decreased from baseline through month 36, with a decrease in LS mean change from baseline to month 36 for ≥200-meter subjects of –2.8 units (95%CI: –4.5, –1.2; P = 0.0007) and –5.7 units (95%CI: –8.9, –2.5; P = 0.0005) in <200-meter subjects (Fig. 3B). Mean GNEM-FAS UE scores decreased from baseline through month 36, with a decrease in LS mean change from baseline to month 36 in ≥200-meter subjects of –1.8 units (95%CI: –2.9, –0.7; P = 0.0009) and –3.1 units (95%CI: –5.1, –1.0; P = 0.0033) in in <200-meter subjects (Fig. 3C). Mean GNEM-FAS self-care scores decreased from baseline through month 36, with a decrease in LS mean change from baseline to month 36 in ≥200-meter subjects of –1.6 units (95%CI: –2.6, –0.6; P = 0.0015) and –3.7 units (95%CI: –5.6, –1.7; P = 0.0003) in <200-meter subjects (Fig. 3D).
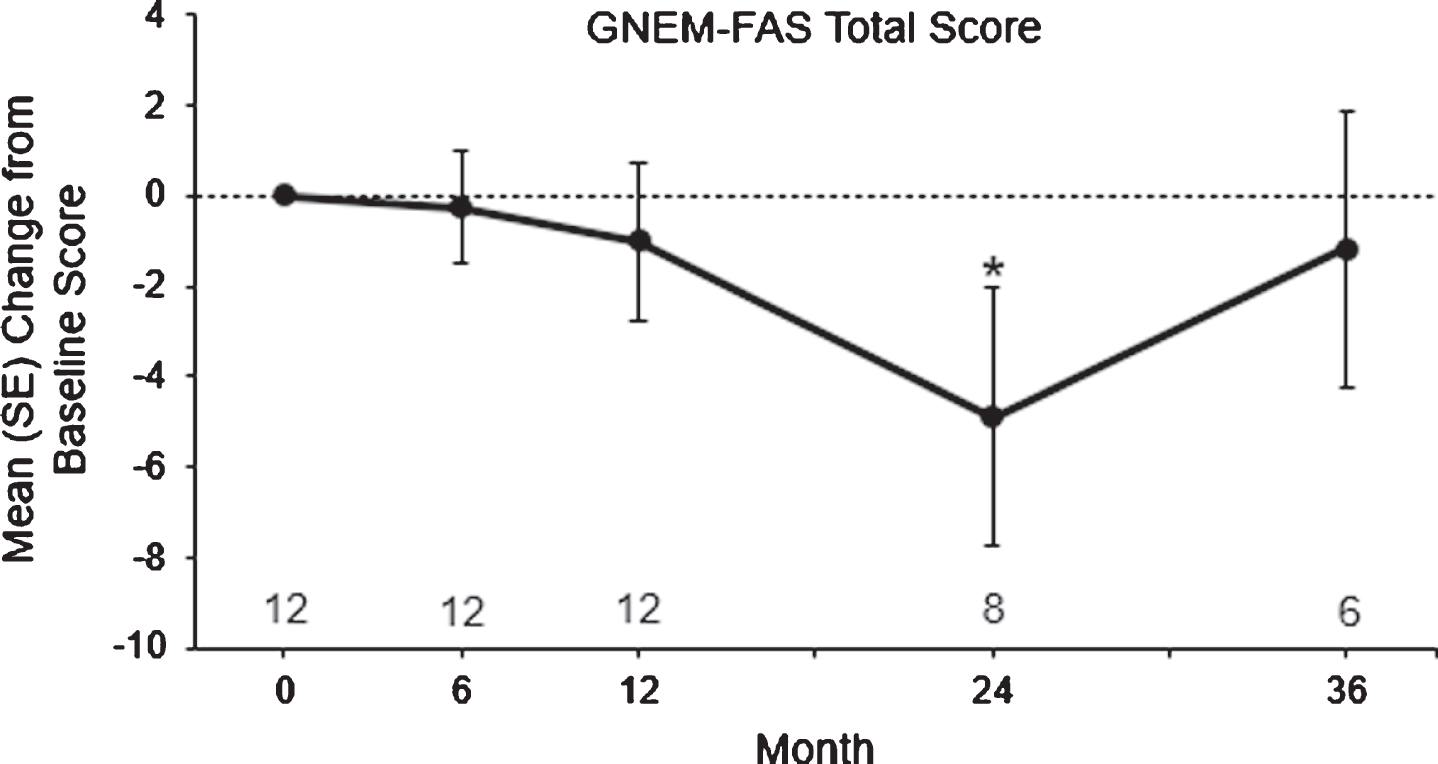
In the ad hoc subgroup analysis, mean GNEM-FAS total scores in ≥200-meter Bulgarian subjects decreased from baseline through month 36, with a decrease in LS mean change from baseline to month 36 of –2.0 units (95%CI: –5.9, 1.9; P = 0.3134; Fig. 4).
Fig. 4
Mean±SE change from baseline to month 36 in GNEM-FAS total score in Bulgarian subjects. Subject numbers are shown above the x-axis. *P < 0.05 using the GEE model. GEE, generalized estimating equation; LS, least squares; SE, standard error.
Six-minute walk test
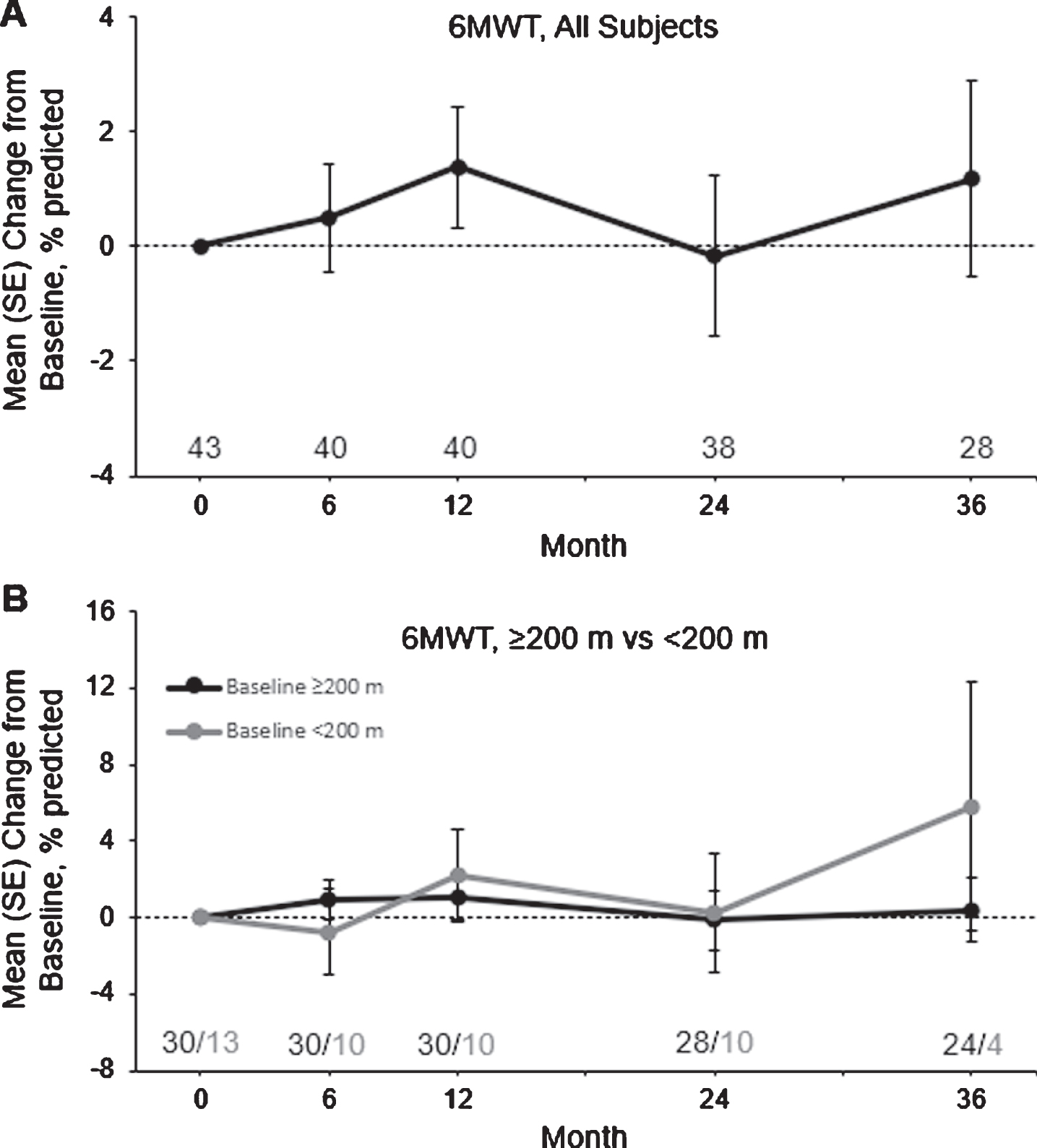
For all subjects, the mean distance walked in the 6MWT and the percentage of predicted distance walked in the 6MWT increased from baseline through month 36 (Supplemental Table 1). However, variability was high among ≥200-meter subjects and <200-meter subjects, and no statistically significant LS mean changes from baseline were observed at any time point (Fig. 5).
Fig. 5
Mean±SE change from baseline to month 36 in the 6MWT in all subjects (A) and in subjects who completed ≥200 and <200 meters at baseline (B). Subject numbers are shown above the x-axis. *P < 0.01 using the GEE model. 6MWT, six-minute walk test; GEE, generalized estimating equation; LS, least squares; SE, standard error.
Additional clinical outcomes
Pulmonary function assessments were added to the study protocol after the study was initiated. Therefore, only three subjects were evaluated at baseline for forced vital capacity, with a mean (SD) of 3.1 L (0.3). Over time, there was little change in this score, with a mean (SD) of 3.0 L (0.7) in six subjects at month 6, 3.5 L (1.1) in six subjects at month 12, 3.8 L (1.0) in 19 subjects at month 24, and 3.5 L (0.9) in 22 subjects at month 36.
Most subjects (∼90%) who remained in the study through month 36 continued to report new GNE myopathy symptoms, complications, or conditions following their baseline visit (Supplemental Table 3). Excluding the six subjects receiving ManNAc or IVIG at baseline, mean (SD) free SA was 0.2μ#x03BC;g/mL (0.1) at baseline and at months 12, 24, and 36. Mean (SD) serum creatine kinase was 414 U/L (439) at baseline, 380 U/L (393) at month 12, 292 U/L (300) at month 24, and 229 U/L (216) at month 36.
Safety
No serious study procedure-specific events occurred. Nine (9%) subjects experienced study procedure-specific AEs, all of which were mild (grade 1) in severity (Supplemental Table 4). Three deaths occurred during the course of the study, all of which were unrelated to participation in the study: poisoning, colon cancer, and cardiac insufficiency. There were no noteworthy changes throughout the study in vital signs or standard safety laboratory values.
DISCUSSION
To our knowledge, this in-clinic portion of the GNEM-DMP was the first large multicenter, multinational, prospective natural history study using validated measurements in GNE myopathy. Overall, subjects with GNE myopathy showed a slow and progressive decline in clinical outcomes. Subjects with GNE myopathy showed significant declines in upper and lower muscle strength as early as 12 months, with continued declines through months 24 and 36. After month 24, there was a greater decline in the GNEM-FAS total score in subjects who had greater disease severity at baseline, as determined by the ability to walk ≥200 meters in the 6MWT. Finally, because there was no change over the study period, free SA levels and creatine kinase levels were not meaningful as markers of disease progression.
Although the HHD UE and LE composite scores showed gradual declines throughout the study, the rate of decline for each muscle group varies. Use of the composite score to assess declines in muscle strength overtime may be limited by muscle groups that show a smaller rate of decline, such as the hip adductors. Similarly, it may be difficult to detect declines in muscle strength for subjects with varying disease severity based on genotype. Although both subgroups demonstrated declines in UE and LE composite scores overtime, Bulgarian subjects had reduced muscle strength at baseline and post-baseline assessments compared to non-Bulgarian subjects, similar to what has been observed in a recent analysis of the literature [13]. The change from baseline in HHD UE scores was similar between Bulgarian and non-Bulgarian subjects, whereas the decline in HHD LE scores appeared greater in non-Bulgarian subjects versus Bulgarian subjects.
Overall, subjects displayed significant declines by month 24 in the GNEM-FAS, a validated tool that aims to capture changes in signs and symptoms relevant to GNE myopathy, including mobility, upper extremity function, and self-care. A study validating this tool reported that the clinically meaningful difference in the GNEM-FAS total score is a change of approximately 3.6–8.0 units [10]. Subjects who were able and unable to complete ≥200 meters in the 6MWT at baseline showed a decline greater than 3.6 units by month 24. Such findings support GNEM-FAS as an appropriate tool to measure changes in GNE myopathy. More research may help determine the sensitivity of GNEM-FAS in subjects with greater disease severity.
This study is not without limitations. The study, originally planned to last up to 15 years, was discontinued before all subjects reached later time points; therefore, fewer subjects reached visits beyond 36 months and results beyond this timepoint were not reported due to the limited interpretability with such a small number of subjects. Still, data reported to month 36 provide valuable insight into the natural disease progression of GNE myopathy.
Our findings are consistent with the two studies that have evaluated disease progression in smaller groups of subjects with GNE myopathy, demonstrating significant declines in muscle strength and less significant declines in the 6MWT within roughly one year [6, 7]. Although there are only two studies examining the natural progression of GNE myopathy, we can compare the results of subjects in this natural history study who walked ≥200 meters at baseline to those of 43 subjects with GNE myopathy who received placebo in a phase 3 trial evaluating an extended release formulation of aceneuramic acid [13]. Subjects in the phase 3 study were required by an inclusion criterion to be capable of walking ≥200 meters. Comparisons between these studies should be made with caution, as 1) participants in the phase 3 trial were assessed with much more regularity and could therefore have improved in clinical outcomes with practice, and 2) the phase 3 trial was double-blind, and therefore, placebo-treated participants did not know if they were potentially benefiting from active drug. Furthermore, the relatively small number of subjects who completed the 6MWT at baseline and at later timepoints limited the to determine the usefulness of the 6MWT as a measure of disease progression.
In general, greater declines were observed after 11 months in the placebo-treated group of the phase 3 trial compared with changes observed over 12 months in this natural history study for the HHD UE composite score, GNEM-FAS total score, and 6MWT, though the reverse was true for the HHD LE composite score. In the previous phase 3 study, a LS mean change of –3.0 at roughly 11 months was observed in the HHD UE composite score in 42 placebo—treated subjects; this decline was greater than what was observed in this natural history study at month 12 (–1.8). Similarly, the decline in GNEM-FAS total score was greater in placebo-treated subjects from the previous phase 3 study at roughly 11 months (–4.05) compared with subjects in this natural history study at month 12 (–1.3). Declines in the 6MWT were also greater in placebo-treated subjects in the previous phase 3 trial at approximately 11 months (–6.8 m) compared with declines in this natural history study (–1.5 and 3.2 at months 24 and 36, respectively). In contrast, the decline in HHD LE composite score was greater at month 12 (–3.2) in this natural history study compared with the decline observed in subjects from the earlier phase 3 trial after approximately 11 months (–0.4).
Notably, a large subset of Bulgarian subjects, all carrying the same homozygous GNE mutation, had lower scores and further declines in all outcome measures compared with the non-Bulgarian subset of subjects in our natural history study. This is in keeping with previous estimates that 20%of the clinical variability in GNE myopathy may be explained by the primary mutation [13]. The GNE genotype should be considered when designing clinical trials; treatment groups in future trials may benefit from stratification by genotype or by populations associated with highly prevalent founder mutations.
Although declines in clinically relevant outcomes typically occurred within a year in this natural history study, in some situations, it may be informative for trials investigating new therapeutic options for GNE myopathy to evaluate clinical outcomes over a longer durations (eg, 2 years). It may be beneficial to evaluate individual muscle groups in addition to UE and LE composite scores, as well as clinical outcomes in subjects with a specific mutation to determine disease progression. The GNEM-FAS remains a valuable tool to document disease progression in GNE myopathy.
CONFLICT OF INTEREST
HL, AB, IT, MT, RH, OP, and TM served as investigators for this study, sponsored by Ultragenyx Pharmaceutical Inc. TK, AS, EK, and CLB are employees and shareholders of Ultragenyx Pharmaceutical Inc. JS was an employee and shareholder of Ultragenyx Pharmaceutical Inc when this work was performed.
ACKNOWLEDGMENTS
The authors would like to thank all study site personnel, as well as the subjects and their families, for making this study possible. Stan Krolczyk, DO (Ultragenyx Pharmaceutical Inc.), provided invaluable expertise. This manuscript was written by the authors with medical writing support from Catherine Woods, PhD, an employee of Ultragenyx Pharmaceutical Inc., and Ben Scott, PhD (Scott Medical Communications, LLC), whose work was funded by Ultragenyx Pharmaceutical Inc. This study was funded by Ultragenyx Pharmaceutical Inc.
REFERENCES
[1] | Pogoryelova O , Coraspe JAG , Nikolenko N , Lochmüller H , Roos A . GNE myopathy: From clinics and genetics to pathology and research strategies. Orphanet J Rare Dis. (2018) ;13: (1):70. |
[2] | Carrillo N , Malicdan MC , Huizing M . GNE myopathy: Etiology, diagnosis, and therapeutic challenges. Neurotherapeutics. (2018) ;15: (4):900–14. |
[3] | Nishino I , Carrillo-Carrasco N , Argov Z . GNE myopathy: Current update and future therapy. Journal of Neurology, Neurosurgery, and Psychiatry. (2015) ;86: (4):385–92. 10.1136/jnnp-2013-307051. |
[4] | Malicdan MC , Noguchi S , Tokutomi T , Goto Y , Nonaka I , Hayashi YK , et al. Peracetylated N-acetylmannosamine, a synthetic sugar molecule, efficiently rescues muscle phenotype and biochemical defects in mouse model of sialic acid-deficient myopathy. J Biol Chem. (2012) ;287: (4):2689–705. 10.1074/jbc.M111.297051. |
[5] | Mitrani-Rosenbaum S , Yakovlev L , Cohen MB , Telem M , Elbaz M , Yanay N , et al. Sustained expression and safety of human GNE in normal mice after gene transfer based on AAV8 systemic delivery. Neuromuscul Disorders. (2012) ;22: (11):1015–24. |
[6] | Mori-Yoshimura M , Hayashi YK , Yonemoto N , Nakamura H , Murata M , Takeda S , et al. Nationwide patient registry for GNE myopathy in Japan. Orphanet J Rare Dis. (2014) ;9: (150):1–9. |
[7] | Quintana M , Shrader J , Slota C , Joe G , McKew J , Fitzgerald M , et al. Bayesian model of disease progression in GNE myopathy. Stat Med. (2019) ;38: (8):1459–74. |
[8] | Pogoryelova O , Cammish P , Mansbach H , Argov Z , Nishino I , Skrinar A , et al. Phenotypic stratification and genotype-phenotype correlation in a heterogeneous, international cohort of GNE myopathy patients: First report from the GNE myopathy Disease Monitoring Program, registry portion. Neuromuscular disorders: NMD. (2017) ;28: (2):158–68. 10.1016/j.nmd.2017.11.001. |
[9] | Argov Z , Bronstein F , Esposito A , Feinsod-Meiri Y , Florence JM , Fowler E , et al. Characterization of Strength and Function in Ambulatory Adults With GNE Myopathy. Journal of clinical neuromuscular disease. (2017) ;19: (1):19–26. 10.1097/cnd.0000000000000181. |
[10] | Mayhew J , Bonner N , Arbuckle R , Turnbull A , Bowden A , Skrinar A . Development and preliminary evidence of the psychometric properties of the GNE myopathy functional activity scale. Journal of Comparative Effectiveness Research. (2017) ;7: (4):381–95. 10.2217/cer-2017-0062. |
[11] | Argov Z , Mitrani Rosenbaum S . GNE myopathy: Two clusters with history and several founder mutations. J Neuromuscul Dis. (2015) ;2: (s2):S73–S6. |
[12] | Chamova T , Guergueltcheva V , Gospodinova M , Krause S , Cirak S , Kaprelyan A , et al. GNE myopathy in Roma patients homozygous for the p.I618T founder mutation. Neuromuscular disorders: NMD. (2015) ;25: (9):713–8. 10.1016/j.nmd.2015.07.004. |
[13] | Lochmüller H , Behin A , Caraco Y , Lau H , Mirabella M , Tournev I , et al. A phase 3 randomized study evaluatingsialic acid extended-release for GNE myopathy. Neurol. 2019:10.1212/WNL. 0000000000006932. |


