
Mechanisms and Clinical Applications of Glucocorticoid Steroids in Muscular Dystrophy
Abstract
Glucocorticoid steroids are widely used as immunomodulatory agents in acute and chronic conditions. Glucocorticoid steroids such as prednisone and deflazacort are recommended for treating Duchenne Muscular Dystrophy where their use prolongs ambulation and life expectancy. Despite this benefit, glucocorticoid use in Duchenne Muscular Dystrophy is also associated with significant adverse consequences including adrenal suppression, growth impairment, poor bone health and metabolic syndrome. For other forms of muscular dystrophy like the limb girdle dystrophies, glucocorticoids are not typically used. Here we review the experimental evidence supporting multiple mechanisms of glucocorticoid action in dystrophic muscle including their role in dampening inflammation and myofiber injury. We also discuss alternative dosing strategies as well as novel steroid agents that are in development and testing, with the goal to reduce adverse consequences of prolonged glucocorticoid exposure while maximizing beneficial outcomes.
GLUCOCORTICOID STEROIDS ACT THROUGH THE GLUCOCORTICOID RECEPTOR TO REGULATE GENE EXPRESSION
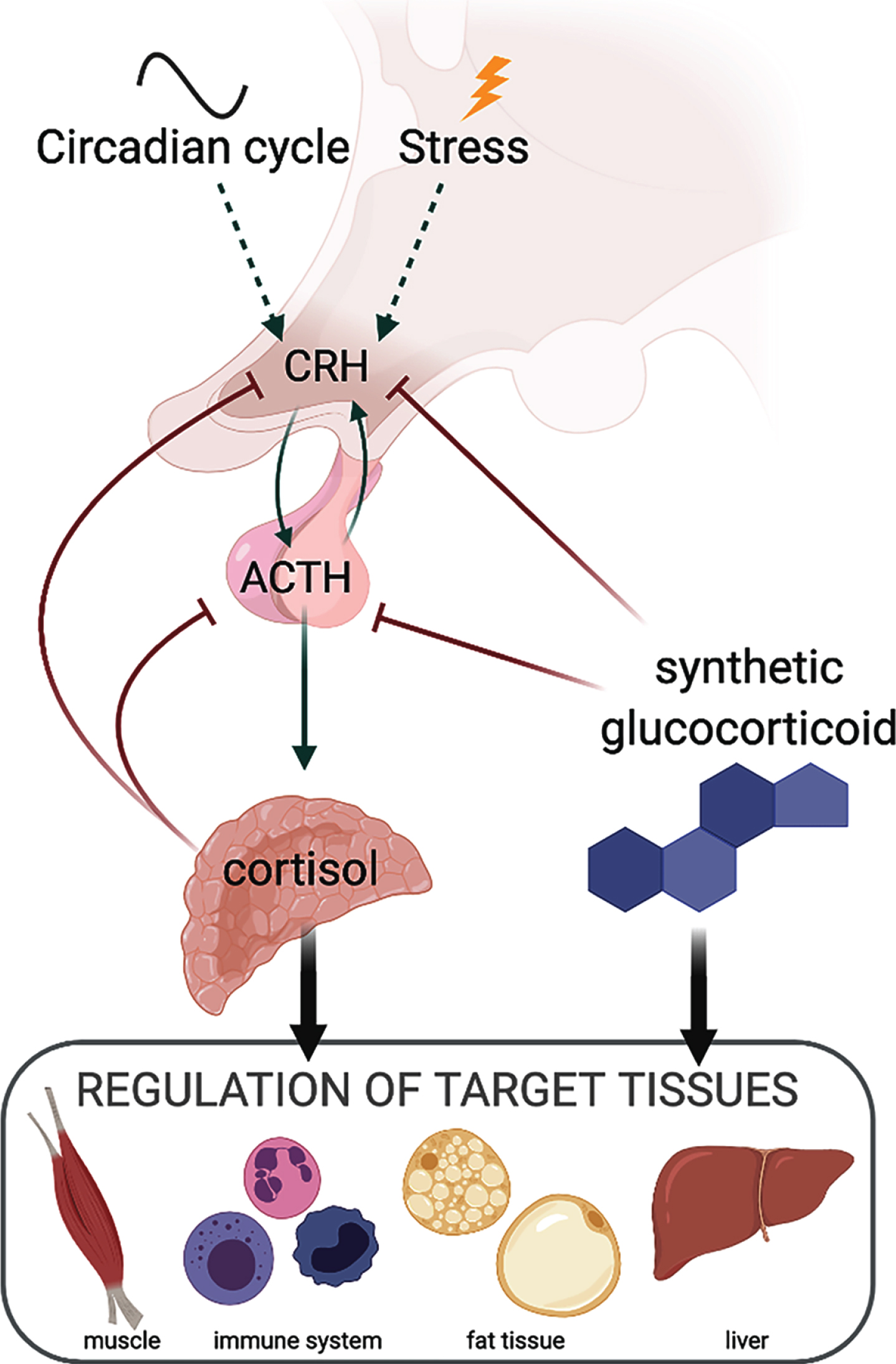
Glucocorticoid steroids are endogenous hormones that coordinate basal and stress responses by directing tissue-specific transcriptional programs. In humans, the primary endogenous glucocorticoid is cortisol, while in mice, corticosterone is the predominant form. The two compounds share the basic four ring steroid structure and are closely related differing by a single hydroxyl group. However, the two compounds differ in synthetic pathways where only corticosterone is a precursor to the mineralocorticoid, aldosterone. Cortisol and corticosterone are produced by the adrenal cortex in response to stress and circadian stimuli (Fig. 1). Activated by the corticotropin releasing hormone (CRH), the adrenocorticotropic hormone (ACTH) from the anterior pituitary stimulates the adrenal gland to secrete cortisol. In turn, cortisol activates the glucocorticoid receptor (GR) to antagonize production of CRH by the hypothalamus and ACTH by the pituitary gland in a negative feedback loop [1, 2]. Circulating endogenous glucocorticoid levels peak just prior to the beginning of the active phase each day. Synthetic glucocorticoids are classified as short- or long-acting depending upon the duration of ACTH suppression they elicit [3], although most have a serum half-life of approximately 1–3 hrs [4–6]. The most commonly prescribed synthetic glucocorticoids include dexamethasone, deflazacort, and prednisone, and these agents are widely used clinically to treat autoimmune and other conditions.
Fig. 1
Diagram summarizing relationships between endogenous and synthetic glucocorticoids and the hypothalamic-pituitary-adrenal axis.
In response to ligand binding, GR drives transcriptional changes to directly alter gene expression in target cells and tissues. Upon ligand binding, GR translocates into the nucleus where it binds glucocorticoid response elements (GREs) in DNA either by itself or in concert with co-factors to regulate gene expression. GRE binding by GR can lead to activation or repression of target genes, so-called “trans-activation” and “trans-repression” functions of the GR as reviewed in [7], depending on GR interactions with co-factors. As a member of the nuclear receptor superfamily, GR interacts with a diverse group of coactivators and co-repressors, orchestrating tissue-specific transcriptional responses [8–10]. In muscle, few specific GR co-factors have been identified, and the best studied is FOXO1, which mediates steroid-induced atrophy [11]. Traditionally, GR is thought to bind DNA as either homodimer or monomer in conjunction with co-factors, but a recent study suggested GR formed a tetramer, as two dimers, after binding DNA [12]. The significance of this conformation requires further study to determine whether it regulates precise transcriptional processes. Because GRs function within complexes, GR binding to a GRE, on its own, is not a strong predictor of GR-dependent gene regulation. The likelihood of an occupied GRE driving transcriptional regulation of a gene increases the closer that the GRE is to the gene’s transcriptional start site [13, 14]. GR binding sites can also work concordantly, with clusters of GREs mediating GR-dependent transcription [14]. GR binding can further control gene expression by modulating the epigenetic landscape around its target genes [15–17]. This epigenetic remodeling is likely a crucial component of GR-induced gene regulation, although more investigation is required to better decipher how loss of chromatin-modifying co-factors impacts expression of GR target genes.
In addition to GR, glucocorticoids can interact with structurally similar nuclear receptors including the mineralocorticoid receptor (MR) and the androgen receptor (AR) [18]. In the presence of glucocorticoid, these receptors can form heterodimers with stronger transactivation capacity than the individual receptors [19, 20]. Endogenous corticosteroids bind MR with 5- to 10-fold higher affinity than GR [21], so it is likely that basal circulating cortisol binds MR preferentially with GR occupancy during circadian peaks or stress [22, 23]. Synthetic glucocorticoids such as deflazacort [24], prednisone [25], and dexamethasone [26] have less affinity for MR [27]. AR is also structurally similar to GR, and the two proteins can form heterodimers [28]. AR and GR have substantial overlap in their agonist-dependent interactomes, indicating shared regulatory features [29]. AR and GR are known to interact in non-muscle tissues, and AR has critical roles in skeletal muscle development and function. However, the physiological effects of AR-GR heterodimerization are not fully understood, as data supports both competitive inhibition [28] and coordination [29].
Glucocorticoids as anti-inflammatory agents
Glucocorticoids both suppress proinflammatory signaling and activate anti-inflammatory responses [30, 31] (Fig. 2). Glucocorticoids inhibit the inflammatory cascades that cause acute tissue damage through the binding of GR to transcription factors NF-kB and AP-1, which inhibits their activity [32, 33]. In monocytes, dexamethasone is known to increase transcription and protein synthesis of the NF-kB inhibitor, IkBα [34]. Similarly, dexamethasone activates glucocorticoid-induced leucine zipper (GILZ), which inhibits AP-1 to lower downstream cytokine synthesis [35, 36]. Glucocorticoid-mediated trans-repression of NF-kB and AP-1 acts on multiple downstream gene targets, including genes encoding inflammatory cytokines and chemokines such as IL-6, IL-12, IL-1, TNFα, and COX-2 [37, 38]. Activated GR not only binds NF-kB to prevent its activation, but it is also known to displace the NF-kB coactivator CBP from the DNA-binding subunits of NF-kB, preventing its transcriptional activity, and adding an additional layer of immunosuppression [39, 40]. Similar to NF-kB, activated GR binds AP-1 to prevent its DNA binding and activity [32, 41]. A partial reduction in immune cells, especially T cell infiltration, into muscle has been observed in steroid-treated human DMD and mdx muscle [42–44].
Fig. 2
Glucocorticoids act through the glucocorticoid receptor (GR). GR activation promotes degradation of transcripts mediating proinflammatory signals through, among other mechanisms, RNA-binding proteins like tristetrapolin. GR activation also stimulates the expression of annexin A1 which serves to orchestrate termination of inflammation and avoid adverse prolonged activation. GR activation also acts directly to limit the action of key proinflammatory mediators.
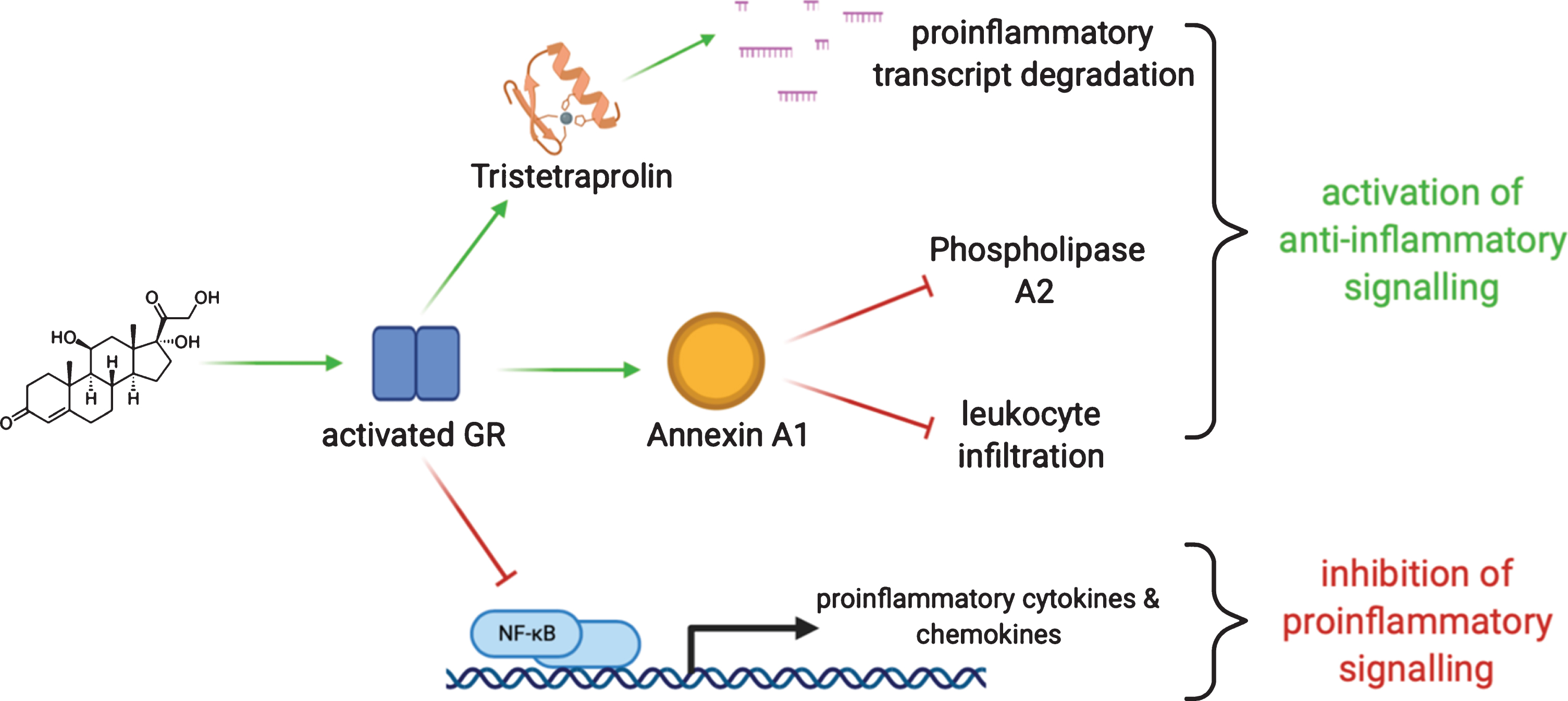
Lipocortin 1 (also known as Annexin A1) is a strong anti-inflammatory effector whose expression is driven by glucocorticoids [45]. High levels of annexin A1 suppress proinflammatory genes including IL-6, COX-2, and iNOS [46–49]. In the presence of glucocorticoids, annexin A1 is upregulated in myeloid cells where it dynamically hinders inflammatory responses. Annexin A1 is known to suppress phospholipase A2 to prevent synthesis of inflammatory eicosanoids [50–53]. Furthermore, enhanced production of annexin A1 in neutrophils inhibits leukocyte transmigration thereby limiting acute tissue injury [48, 49, 54, 55]. Glucocorticoids can also dictate annexin A1 localization within the cell [56, 57]. After exposure to dexamethasone, annexin A1 translocates to the plasma membrane, where it is then secreted to promote leukocyte detachment [52, 57]. Through these mechanisms, annexin A1 has been implicated in quelling acute inflammation to limit local tissue injury.
Metabolic modulation by glucocorticoids
Glucocorticoids also promote the “fight or flight” stress response through metabolic modulation [58]. Acutely, glucocorticoids trigger the liberation of glucose, amino acids, and fatty acids into circulation so these substrates are available for rapid energy production to fuel stress responses [59–61]. Glucocorticoids broadly stimulate metabolic cascades via enhanced cAMP signaling and PKA activation to amplify energy-producing pathways [62–64]. Substrate availability is orchestrated by extensive upregulation of gluconeogenesis and degradation of hepatic glycogen to generate glucose production. At the same time, lipolysis generates free fatty acids and glycerol, and proteolysis liberates amino acids to drive energy availability [65–68]. Chronic stimulation of these pathways, for example, from chronic use of synthetic glucocorticoids cause adverse metabolic effects with tissue-specific outcomes, including within the liver, pancreas, adipose tissue, and skeletal muscle [69–73].
Glucocorticoids exert their metabolic effects on multiple tissues and organs. In liver, the effects of glucocorticoids on gluconeogenesis and glycogen content can unfavorably shift hepatic metabolism when chronically stimulated. Persistent gluconeogenesis and glucose production directly cause hyperglycemia and lead to insulin resistance, a hallmark of the metabolic syndrome and diabetes mellitus. Glucocorticoid use can lead to clinical diabetes in non-diabetics [74, 75]. Furthermore, glucocorticoids exacerbate glucose control in those who have diabetes mellitus, often resulting in a greater need for insulin [74, 76, 77]. Prolonged hyperglycemia and insulin resistance promote lipogenesis in hepatic tissue, which can lead to non-alcoholic fatty liver disease and steatosis [75, 78].
In the pancreas, glucocorticoids stimulate glucagon production from α-cells [79]. Additionally, glucocorticoids suppress pancreatic β-cell activity, lowering rates of insulin secretion [80]. Similar to liver, the pancreas exhibits maladaptive responses when chronically exposed to glucocorticoids. Glucocorticoid-mediated inhibition of insulin production occurs concomitant with the inhibition of glucose uptake by other tissues, raising overall serum glucose [81]. The block of insulin production is partially attributed to the cytotoxic effects that glucocorticoids exert on β-cells [73, 82]. It has been shown that chronic dexamethasone exposure is associated with oxidative stress and pro-apoptotic effects in β-cells [83].
Adipose tissue is also a glucocorticoid target. With acute glucocorticoid exposure, enhanced lipolysis occurs, increasing glycerol and free fatty acids [62, 84]. In response to chronic glucocorticoid exposure, insulin-responsive lipolysis decreases and adipogenesis increases [62, 84–86]. Moreover, chronic glucocorticoid use alters adipokine levels, decreasing adiponectin [70] and increasing resistin and leptin secretion, which can influence food intake and insulin responses [87, 88]. Collectively, this hormonal dysregulation contributes to the onset of glucocorticoid-induced obesity and diabetes.
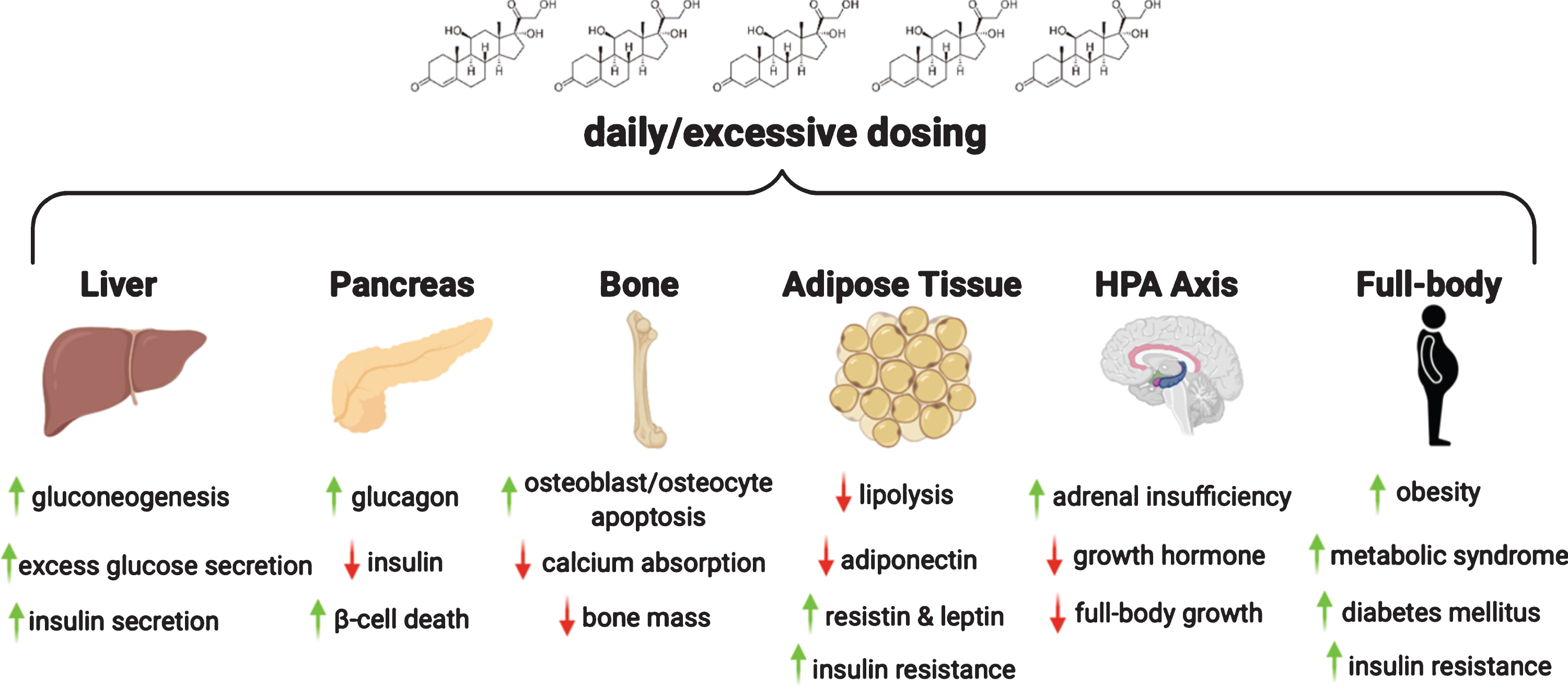
Synthetic glucocorticoids are powerful regulators of systemic metabolism. The effect of acute glucocorticoid exposure is adaptive since it supplies multiple substrates for enhanced energy production. However, chronic excess glucocorticoid levels produce adverse metabolic reprogramming, mediated through multiple peptide hormone pathways (Fig. 3). As a consequence, chronic, long-term glucocorticoids result in insulin resistance, fatty liver, obesity, and even diabetes mellitus. In the setting of muscular dystrophy, where muscle mass is already depleted, these consequences can be further exacerbated.
Fig. 3
Chronic intake of glucocorticoids results in glucocorticoid excess which, in turn, has adverse effects on liver, pancreas, bone, adipose tissue and hypothalamus-pituitary-adrenal axis. In DMD, chronic steroid use through puberty also leads to growth suppression and poor bone health. Long term steroid use is linked to metabolic syndrome and insulin resistance.
Effects of glucocorticoids on muscle
Glucocorticoids are pleiotropic regulators of striated muscle function. Genetic ablation of GR exclusively in heart and skeletal muscle significantly alters metabolism and striated muscle performance [89, 90]. Glucocorticoids are prohibited as performance enhancing drugs, although the supporting evidence and the mechanistic rationale are equivocal [91–93]. An important open question is how glucocorticoids regulate muscle contractility. Several studies have pointed to a direct activating role of glucocorticoids in store-operated calcium entry (SOCE). In cultured myotubes, dexamethasone increased SOCE by 15–25% [94]. In isolated rat ventricular cardiomyocytes, a short-term pre-treatment with dexamethasone increased contractile force, calcium transient amplitude, and SOCE magnitude through serum and glucocorticoid-regulated kinase 1 (SGK1) [95]. Genetic ablation of GR in murine hearts decreased t-tubule system density and increased the distance between ryanodine receptors and L-type calcium channels [96]. Conversely, dexamethasone decreased the physical distance and improved synchrony of intracellular calcium release through GR-mediated activation of the autophagic flux [96]. These steroid-driven changes in contractility are highly relevant in neuromuscular disease settings. Eight weeks of prednisolone dosing in 5-month-old mdx mice increased specific force of the extensor digitorum longus muscle by 26% [97]. Thus, glucocorticoids appear to improve muscle contractility, especially after acute short term exposure.
Nonetheless, prolonged and especially high-dose intake of glucocorticoids promotes muscle wasting and weakness. In a mechanical model of muscle contusion in rats, a high single-dose of 25 mg/kg methylprednisolone resulted in significant benefits in force recovery in the short term (24 hours), but promoted weakness and tissue disorganization at later time points (7–14 days) [98]. Daily administration (60–1,200μg/kg) of dexamethasone for 5 days induced a rapid dose- and GR-dependent induction of myostatin, promoting loss of muscle mass and myosin type II in rats [99]. Chronic glucocorticoid intake upregulates atrogenes like Fbxo32 and Trim63, which is reduced after muscle-specific GR ablation [100]. Glucocorticoid-induced muscle atrophy leads to sustained and unbalanced activation of the FOXO3 transcription factor. Dexamethasone increases FOXO3 phosphorylation and activity in muscle [67], and FOXO3 inhibition prevents glucocorticoid-induced atrophy in cultured myotubes [101]. Another proposed mechanism linking sustained GR activation to loss of muscle mass is the cross-inhibition between GR and the insulin-responsive anabolic factor mTORC1 [102]. In healthy human subjects, six days of daily prednisone at 0.8 mg/kg was sufficient to induce an acute state of muscle insulin resistance and depressed protein anabolism [103]. The long-term wasting effects are relevant for dystrophic muscle. Chronic continuous exposure to prednisone using sub-cutaneous pellets in food or water in mdx mice for 50 weeks resulted in early benefits for approximately ∼2 months and then showed exacerbation of dystrophic progression and weakness [104]. Thus, the atrophy-inducing effects from chronic daily glucocorticoids may counteract the benefits from anti-inflammatory and pro-performance effects.
GLUCOCORTICOIDS IN DYSTROPHIC MUSCLE
The chronic and ongoing injury state seen in DMD is one of the targets of glucocorticoids [105]. A comparative study of deflazacort versus prednisone in mdx mice studied the response to cardiotoxin-mediated injury and found that both drugs increased fiber diameter in tibialis anterior and diaphragm. In addition, deflazacort increased many features of regeneration [106]. The same group evaluated glucocorticoids in mdx limb muscles using NMR and found increased levels of taurine and creatine, metabolic biomarkers of muscle energy [107]. Glucocorticoids drive functional improvement in dystrophic muscle through activation of transcription factors like KLF15. KLF15 is a GR-activated factor shown to mediate nutrient utilization in glucocorticoid-treated mdx muscle [108]. Genetic manipulation of Klf15 showed that increased KLF15 is beneficial to dystrophic muscle [108]. KLF15, glucocorticoids, and branched chain amino acids may also be relevant to other models of neuromuscular diseases. For example, prednisone, Klf15 overexpression, and BCAA supplementation improved pathophysiology of a murine model of spinal muscular atrophy [109].
Glucocorticoids also play a direct role in the susceptibility of the sarcolemma to injury. Glucocorticoid exposure accelerated sarcolemmal resealing and repair cap formation at the site of sarcolemmal injury in normal myofibers and also in multiple models of muscular dystrophy including mdx mice and two models of limb girdle muscular dystrophy, Dysf-null and Sgcg-null mice [110, 111]. The effects of membrane stabilization in mdx mice and LGMD-2B patient (DYSF mutation) cells were particularly striking for vamorolone, a novel compound designed to minimize GR trans-activation while retaining anti-inflammatory action [112, 113]. Although the causal relationship between anti-inflammatory and anti-injury effects are still unclear, the benefits of glucocorticoids on membrane repair likely extend beyond dystrophinopathies.
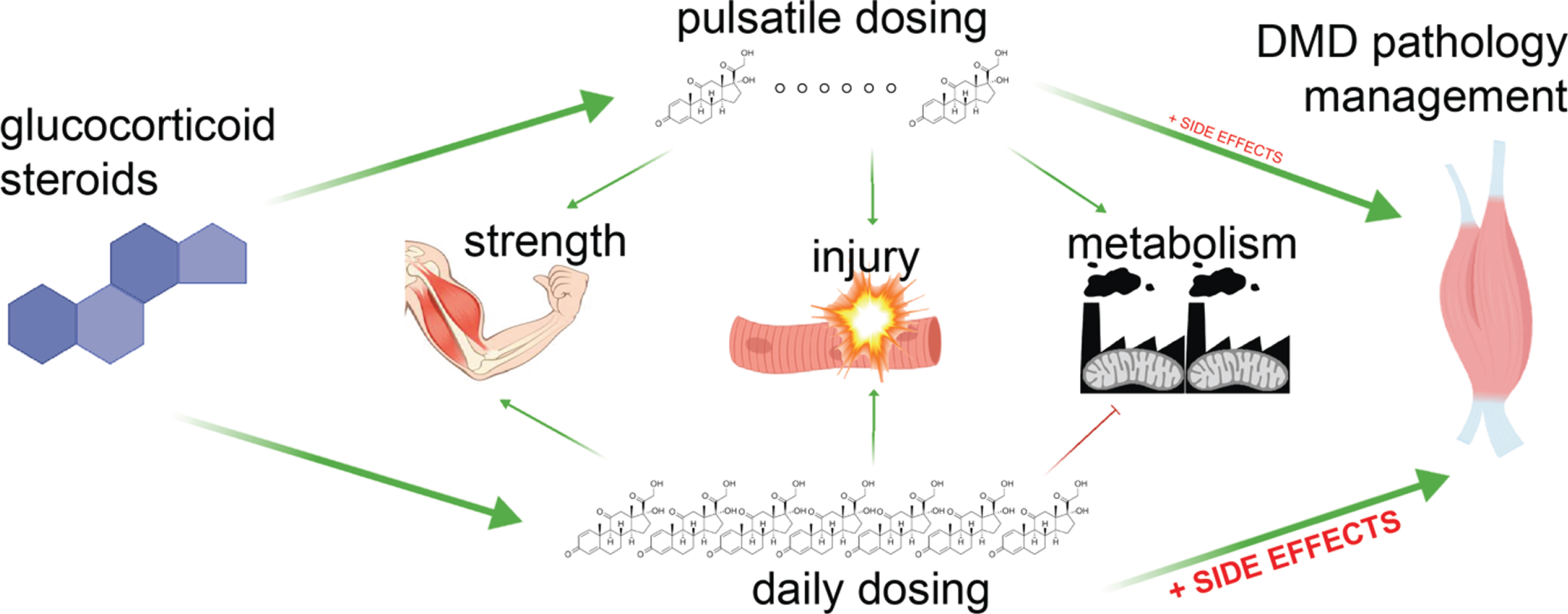
Because of the adverse consequences of chronic steroid use, multiple approaches are being pursued to reduce these side effects, such as novel compounds (e.g. vamorolone) and alternative dosing strategies. These alternative dosing strategies have primarily relied on having intervals (days) where steroids are not given. In both pilot and longitudinal studies of DMD patients receiving alternative dosing strategies, benefit was observed without the same magnitude of side effects [114, 115]. An intermittent regimen of oral prednisolone for two consecutive days per week was tested in mdx mice; treated mdx mice showed an increased strength over time and improved survival between 80 and 104 weeks of age [116]. Intermittent injection of prednisone or deflazacort at a minimal dose of once-weekly comparably benefitted sarcolemmal repair, fibrosis, and immune infiltrations as daily steroids in short term experiments [110, 111]. Once-weekly versus daily prednisone induced opposite epigenetic and metabolic programs in dystrophic mdx muscle. While the daily prednisone activated the GR-FOXO axis and drove muscle atrophy and insulin resistance, once-weekly prednisone activated the GR-KLF15-MEF2C axis and enhanced insulin sensitivity, nutrient uptake, and oxidative catabolism in dystrophic muscle, leading to long-term benefits (32–40 weeks) in both mdx and Dysf-null mice. Therefore, glucocorticoid dosing strategies can improve muscle physiology while minimizing adverse consequences (Fig. 4).
Fig. 4
Daily glucocorticoids improve DMD pathology but induce untoward metabolic side effects. Studies in dystrophic mice and DMD patients suggest that the metabolic benefits of intermittent glucocorticoids can reduce side effects, while maintaining benefits.
Clinical use of glucocorticoids in Duchenne muscular dystrophy
Glucocorticoid steroids are considered standard of care in DMD [117]. Early clinical trials of prednisone demonstrated a clear benefit compared to natural history. In a double-blind, randomized clinical trial of daily prednisone, DMD patients in both low and high dose groups improved muscle strength and function [118]. Follow-up studies confirmed long-term benefits on ambulation and pulmonary function [119, 120]. In a prospective multi-center cohort study of 440 DMD patients were followed for ten years (NCT-00468832), steroid treatment improved upper and lower extremity muscle strength across all ages and prolonged life expectancy. In 2016, glucocorticoids were recommended to treat DMD patients by the American Academy of Neurology (AAN) [121] and by a Cochrane review [122], but specific dosing recommendations were not given.
Although steroids improve outcome in DMD, it remains unclear if prednisone or deflazacort is superior. A multi-center double-blind randomized study compared prednisone (daily 0.75 mg/kg) or deflazacort (daily 0.9 mg) for 12 months (N = 18). Using a natural history cohort as steroid-naïve control, the study reported similar muscle benefits with both steroids, with deflazacort causing less weight gain than prednisone [123]. A larger study of 340 DMD boys indicated boys on deflazacort were able to maintain ambulation longer at the cost of increased adverse effects including short stature, Cushingoid appearance, and cataracts compared to prednisone [124]. The Finding the Optimum Regimen For Duchenne Muscular Dystrophy (FOR-DMD) study randomized 196 DMD boys to daily deflazacort 0.9 mg/kg, daily deflazacort 1.2 mg/kg, and daily prednisone 0.75 mg/kg, and reported that patients on all regimens showed similar muscle benefits, but deflazacort had less weight gain and fewer behavior-related adverse events as compared to prednisone (NCT-01603407). A post-hoc analysis from the placebo arm of the DMD ACT trial (ataluren) reported patients on deflazacort had improved 6 minute walk distances (6MWD) and 4 stair climb times (4SCT) compared to prednisone [125]. A similar post hoc analysis from the placebo groups of the phase 3 ataluren trial and tadalafil studies (N = 231) showed that deflazacort improved 6MWD and rise-from-supine time significantly more than prednisone, while no difference was found in 10-meter run and North Star Ambulatory Assessment (NSAA) scores between the steroid treated groups [126]. A retrospective study (N = 330) over 13 years reported that patients on deflazacort ambulated for a longer period of time (15.6 years) then on prednisone (13.5 years). It is unclear if the prolonged benefit is due to earlier average age at initiation (deflazacort, 6.5 y; prednisone 8.1 y) or steroid type. Additional reported benefits from deflazacort included increased lean body mass, lower weight gain, and decreased risk of scoliosis [127].
Steroids dosing is an active area of research interest with the hope to maximize the benefits/side effects ratio. In DMD steroid treatment is better than no steroid treatment, but the optimal dose and dosing regimen remains unclear [128]. Current dosing regimens include daily, 10 days on followed by 10 days off (10/10), and twice per week (two consecutive weekend days of high dose). Daily dosing causes many side effects including but not limited to weight gain, bone fractures, behavioral disturbances, and Cushingoid features [122]. The 10/10 regimen was initially reported to maintain muscle benefits with fewer side effects and no change in quality of life (N = 17), although the study lacked a daily treated control cohort [129]. A later study compared 10/10 to daily dosing (N = 25/group) for two years and reported that patients on daily steroids remained ambulatory for longer but had shorter stature, higher BMI, and higher rates of vertebral fractures than patients on 10/10 [130]. Two other studies looking at weekend high dose steroids in DMD showed similar trends. A year-long randomized study 4–10 y patients (N = 64; prednisone daily at 0.75 mg/kg versus high dose weekend at 10 mg/kg) showed no significant differences between treatment groups, concluding that weekend prednisone dosing was as effective as daily dosing [114]. A later study in infants/toddlers treated with weekend 10 mg/kg prednisone for 12 months (N = 23; 0.4–2.4 y) reported a slight improvement on the Bayley-III gross motor-scaled score and excessive weight gain for 56% of patients [115]. Recently, we reported data from a retrospective cross-sectional study (N = 24) comparing daily versus weekend steroid use over five years in groups with comparable body mass indices. Despite similar cumulative doses, DMD patients in the weekend steroid cohort showed lower levels of glycemia, insulinemia, and fat mass and higher lean mass than patients in the daily group [131]. Additionally, morning cortisol levels were higher in the weekend cohort than in the daily cohort, consistent with the concept of lower suppression of the hypothalamic-pituitary-adrenal axis with intermittent dosing [131]. Thus, intermittent steroid dosing appears to mediate steroid benefit with a lower side effect profile, even in the chronic setting.
Efforts to generate novel glucocorticoid derivatives are ongoing and seek to reduce side effects or improve targeted aspects of glucocorticoid function, such as activation of GR, as reviewed in [132, 133]. Examples of novel synthetic derivatives include CpdX, an anti-inflammatory GR agonist, as reviewed in [134, 135], and vamorolone (also known as VBP-15), a dissociative GR ligand and MR antagonist that improves membrane stability [112, 136]. Vamorolone was developed to help alleviate insulin resistance by selecting compounds with Δ9–11 and R1/R3 modifications to promote NF-KB activity and reduce a specific transcriptional cascade mediated by the glucocorticoid receptor [24, 112, 137, 138]. A phase IIA trial (N = 48; 4–7 y and steroid-naïve at start) tested daily vamorolone at multiple doses including 2.0 and 6.0 mg/kg/day and reported no evidence of insulin resistance or adrenal suppression after two weeks of treatment [136]. In the open label extension phase of the study, clinical improvement in time-to-stand (primary endpoint) was reported for the 2.0 and 6.0 mg/kg doses, which also associated with BMI Z-score averages comparable to the prednisone control group (0.493 vs. 0.543) [139]. Vamorolone is currently in a phase IIB clinical trial (NCT-03439670; 4–7 y, ambulatory and steroid-naïve at start), which will compare daily vamorolone 2.0 and 6.0 mg/kg to daily prednisone 0.75 mg/kg/day or placebo for 48 weeks.
Cardiomyopathy is a leading cause of death in DMD, and effects of glucocorticoids on the dystrophic heart require more study. Retrospective studies of steroid use in DMD found significant benefits to onset of cardiomyopathy and systolic function decline [140, 141]. In the study comparing weekend to daily glucocorticoid regimens, no differences were found between treatments on electrocardiography parameters, myocardial thickness or fractional shortening [131], suggesting intermittent dosing might match daily dosing for cardiac benefits. No data are yet available for vamorolone trials in DMD boys, although heart rate has been included as secondary outcome measure in the ongoing trial (NCT03439670). Despite some promising indications, dedicated clinical studies are still required to define the longterm cardiovascular effects of glucocorticoid steroids in DMD.
Currenlty, serum biomarkers and muscle imaging are being investigated to define longitudinal predictive biomarkers of responsiveness to steroids and side effect development. A study using aptamer technology for serum protein identification found seventeen DMD-associated potential biomarkers that responded to steroid intake, including lumican and osteomodulin [142]. A multi-center study found that glucocorticoid use partially counteracted the effects of disease progression on the circulating levels of malate dehydrogenase 2 and ankyrin repeat domain 2 proteins [143]. Objective measures of treatment response and overall muscle health are critical to the success of clinical trials to provide adjuncts to functional testing. In addition to chemical measures, muscle imaging using magnetic resonance is emerging as a promising biomarker [144]. In a year long MRI study, there was less intramuscular fat in steroid-treated versus steroid-naïve DMD (N = 30) [145]. Longitudinal MRI and magnetic resonance spectroscopy were used to quantify DMD disease progression and the effect of glucocorticoids in rapidly (vastus lateralis) versus slowly (soleus) degenerating muscles [146]. These data indicate several potential pharmacodynamic biomarkers that can be used to optimize steroid dosing.
Potential of glucocorticoids in other conditions and new treatments
Although there is relatively good data on the use of glucocorticoids in DMD, the use of these agents in other forms of muscular dystrophy, like the milder Becker Muscular Dystrophy (BMD) is less well studied. Examining glucocorticoids in BMD takes on new importance given the anticipated clinical outcomes from gene therapy with micro-dystrophins. There have been few clinical trials assessing glucocorticoid effects in BMD. One study tested daily prednisone for 6 months in a limited number of BMD patients (N = 6) and reported significant improvement in overall motor disability and myofiber necrosis [147], but further and larger studies are still required to consolidate these encouraging trends. Similarly, there are few trials examining the effect of steroids in the limb-girdle muscular dystrophies (LGMDs); the rare nature of these disorders challenges having sufficiently powered clinical trials. In a case report, two siblings with β-sarcoglycan limb-girdle muscular dystrophy (LGMD) showed clinical improvement in quantitative muscle testing with 22 months of 0.9 mg/kg/day deflazacort [148]. Prior case reports had shown similar benefits of prednisone in patients with δ-sarcoglycan LGMD [149, 150]. A double-blind placebo-controlled cross-over trial in dysferlin-mediated LGMD tested deflazacort (daily 1.0 mg/kg in month one, followed by 1 mg/kg every other day in months two through six) versus placebo (NCT-00527228; N = 25). Deflazacort associated with decreased muscle strength, per % CIDD score, and Neuromuscular Symptom Score, and the study concluded that off-label use of deflazacort is not warranted in LGMD-2B [151]. Currently, there are two clinical trials testing glucocorticoids in LGMD patients. In one study (NCT-03783923; enrolling N = 100) daily deflazacort 0.6 mg/kg will be tested versus placebo in FKRP related LGMD for 26 weeks with 4SCT as primary outcome. In another study (NCT-04054375; enrolling N = 30), once-weekly prednisone 0.75 mg/kg will be tested in patients with all forms of LGMD, with safety as primary endpoint and muscle function improvement as a secondary outcome. Clinical case studies and animal models point to some efficacy of glucocorticoids in a broader range of muscular dystrophies. The results of open label clinical trials will help shed light on potential and disease-specific strategies of glucocorticoid use beyond DMD.
Gene correction/replacement therapies are advancing. At least two oligonucleotides for skipping mutated exons of the DMD gene have been approved by the FDA (Exondys51, Vyondys53). Gene therapy to express mini-dystrophin in DMD muscle is being evaluated in ongoing clinical trials (NCT00428935, NCT02376816 and NCT03362502). Moreover, gene therapy to correct the endogenous mutant dystrophin through CRISPR/Cas9 has shown promising results in pre-clinical large animal models [152]. With the progress in virally-mediated gene therapies, it is likely that life expectancy, symptom development and duration of treatment will change considerably for DMD. It is expected that the gene therapies will significantly slow disease progression, therefore creating the clinical need for long term management. In this respect, it will be imperative to advance our precise understanding of mechanisms and biomarkers of glucocorticoid steroid action in dystrophic muscle physiology. This knowledge will be key to maximize steroid efficacy in supporting genetic/functional rescue, while minimizing the adverse effects associated with virtually life-long intake of these drugs.
CONCLUSIONS
Glucocorticoid steroids are powerful agents to regulate inflammation, metabolism and muscle physiology. Their clinical use in neuromuscular diseases will be further informed by integrative studies that evaluate the effect of these agents on immune cells, muscle tissue, and metabolic homeostasis. It will be critical to adapt glucocorticoid regimens to specific subtypes of neuromuscular disease, perhaps tailored using imaging or serum biomarkers. Therefore, a deeper understanding of direct versus indirect mechanisms of action will be critical to revise and expand to optimize the use of glucocorticoids in DMD and possibly in other neuromuscular conditions.
CONFLICT OF INTEREST STATEMENT
MQ and EMM are listed as co-inventors on a patent application related to intermittent glucocorticoid use filed by Northwestern University.
ACKNOWLEDGMENTS
Supported by NIH U54 AR052646, NIH RO1 NS047726, NIH F31 AR073655 (IMS), American Heart Association 20PRE35210837 (JAF), the Parent Project for Muscular Dystrophy. MQ is supported by NIH K01 DK121875 (NIDDK).
REFERENCES
[1] | Spencer RL , Chun LE , Hartsock MJ , Woodruff ER . Glucocorticoid hormones are both a major circadian signal and major stress signal: How this shared signal contributes to a dynamic relationship between the circadian and stress systems. Front Neuroendocrinol. (2018) ;49: :52–71. |
[2] | Erkut ZA , Pool C , Swaab DF . Glucocorticoids suppress corticotropin-releasing hormone and vasopressin expression in human hypothalamic neurons. J Clin Endocrinol Metab. (1998) ;83: (6):2066–73. |
[3] | Axelrod L . Glucocorticoid therapy. Medicine (Baltimore). (1976) ;55: (1):39–65. |
[4] | Disanto AR , Desante KA . Bioavailability and pharmacokinetics of prednisone in humans. J Pharm Sci. (1975) ;64: (1):109–12. |
[5] | Mollmann H , Hochhaus G , Rohatagi S , Barth J , Derendorf H . Pharmacokinetic/pharmacodynamic evaluation of deflazacort in comparison to methylprednisolone and prednisolone. Pharm Res. (1995) ;12: (7):1096–100. |
[6] | Queckenberg C , Wachall B , Erlinghagen V , Di Gion P , Tomalik-Scharte D , Tawab M , et al. Pharmacokinetics, pharmacodynamics, and comparative bioavailability of single, oral 2-mg doses of dexamethasone liquid and tablet formulations: A randomized, controlled, crossover study in healthy adult volunteers. Clin Ther. (2011) ;33: (11):1831–41. |
[7] | Newton R , Holden NS . Separating transrepression and transactivation: A distressing divorce for the glucocorticoid receptor? Mol Pharmacol. (2007) ;72: (4):799–809. |
[8] | Lonard DM , O’Malley BW . Nuclear receptor coregulators: Judges, juries, and executioners of cellular regulation. Mol Cell. (2007) ;27: (5):691–700. |
[9] | Lonard DM , O’Malley BW . Expanding functional diversity of the coactivators. Trends Biochem Sci. (2005) ;30: (3):126–32. |
[10] | Lim HW , Uhlenhaut NH , Rauch A , Weiner J , Hubner S , Hubner N , et al. Genomic redistribution of GR monomers and dimers mediates transcriptional response to exogenous glucocorticoid in vivo. Genome Res. (2015) ;25: (6):836–44. |
[11] | Waddell DS , Baehr LM , van den Brandt J , Johnsen SA , Reichardt HM , Furlow JD , et al. The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy-associated MuRF1 gene. Am J Physiol Endocrinol Metab. (2008) ;295: (4):E785–97. |
[12] | Presman DM , Ganguly S , Schiltz RL , Johnson TA , Karpova TS , Hager GL . DNA binding triggers tetramerization of the glucocorticoid receptor in live cells. Proc Natl Acad Sci U S A. (2016) ;113: (29):8236–41. |
[13] | Thormann V , Glaser LV , Rothkegel MC , Borschiwer M , Bothe M , Fuchs A , et al. Expanding the repertoire of glucocorticoid receptor target genes by engineering genomic response elements. Life Sci Alliance. (2019) ;2: (2). |
[14] | Thormann V , Rothkegel MC , Schopflin R , Glaser LV , Djuric P , Li N , et al. Genomic dissection of enhancers uncovers principles of combinatorial regulation and cell type-specific wiring of enhancer-promoter contacts. Nucleic Acids Res. (2018) ;46: (6):2868–82. |
[15] | Clark EA , Wu F , Chen Y , Kang P , Kaiser UB , Fang R , et al. GR and LSD1/KDM1A-Targeted Gene Activation Requires Selective H3K4me2 Demethylation at Enhancers. Cell Rep. (2019) ;27: (12):3522–32. e3 |
[16] | Guo B , Huang X , Cooper S , Broxmeyer HE . Glucocorticoid hormone-induced chromatin remodeling enhances human hematopoietic stem cell homing and engraftment. Nat Med. (2017) ;23: (4):424–8. |
[17] | Wu JN , Pinello L , Yissachar E , Wischhusen JW , Yuan GC , Roberts CWM . Functionally distinct patterns of nucleosome remodeling at enhancers in glucocorticoid-treated acute lymphoblastic leukemia. Epigenetics Chromatin. (2015) ;8: :53. |
[18] | Arriza JL , Weinberger C , Cerelli G , Glaser TM , Handelin BL , Housman DE , et al. Cloning of human mineralocorticoid receptor complementary DNA: Structural and functional kinship with the glucocorticoid receptor. Science. (1987) ;237: (4812):268–75. |
[19] | Liu W , Wang J , Sauter NK , Pearce D . Steroid receptor heterodimerization demonstrated in vitro and in vivo. Proc Natl Acad Sci U S A. (1995) ;92: (26):12480–4. |
[20] | Trapp T , Rupprecht R , Castren M , Reul JM , Holsboer F . Heterodimerization between mineralocorticoid and glucocorticoid receptor: A new principle of glucocorticoid action in the CNS. Neuron. (1994) ;13: (6):1457–62. |
[21] | Reul JM , Gesing A , Droste S , Stec IS , Weber A , Bachmann C , et al. The brain mineralocorticoid receptor: Greedy for ligand, mysterious in function. Eur J Pharmacol. (2000) ;405: (1-3):235–49. |
[22] | Reul JM , de Kloet ER . Two receptor systems for corticosterone in rat brain: Microdistribution and differential occupation. Endocrinology. (1985) ;117: (6):2505–11. |
[23] | Spencer RL , Young EA , Choo PH , McEwen BS . Adrenal steroid type I and type II receptor binding: Estimates of in vivo receptor number, occupancy, and activation with varying level of steroid. Brain Res. (1990) ;514: (1):37–48. |
[24] | Heier CR , Yu Q , Fiorillo AA , Tully CB , Tucker A , Mazala DA , et al. Vamorolone targets dual nuclear receptors to treat inflammation and dystrophic cardiomyopathy. Life Sci Alliance. (2019) ;2: (1). |
[25] | Grossmann C , Scholz T , Rochel M , Bumke-Vogt C , Oelkers W , Pfeiffer AF , et al. Transactivation via the human glucocorticoid and mineralocorticoid receptor by therapeutically used steroids in CV-1 cells: A comparison of their glucocorticoid and mineralocorticoid properties. Eur J Endocrinol. (2004) ;151: (3):397–406. |
[26] | Rupprecht R , Reul JM , van Steensel B , Spengler D , Soder M , Berning B , et al. Pharmacological and functional characterization of human mineralocorticoid and glucocorticoid receptor ligands. Eur J Pharmacol. (1993) ;247: (2):145–54. |
[27] | Brookes JC , Galigniana MD , Harker AH , Stoneham AM , Vinson GP . System among the corticosteroids: Specificity and molecular dynamics. J R Soc Interface. (2012) ;9: (66):43–53. |
[28] | Chen S , Wang J , Yu G , Liu W , Pearce D . Androgen and glucocorticoid receptor heterodimer formation. A possible mechanism for mutual inhibition of transcriptional activity. J Biol Chem. (1997) ;272: (22):14087–92. |
[29] | Lempiainen JK , Niskanen EA , Vuoti KM , Lampinen RE , Goos H , Varjosalo M , et al. Agonist-specific Protein Interactomes of Glucocorticoid and Androgen Receptor as Revealed by Proximity Mapping. Mol Cell Proteomics. (2017) ;16: (8):1462–74. |
[30] | Coutinho AE , Chapman KE . The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. (2011) ;335: (1):2–13. |
[31] | Barnes PJ . Anti-inflammatory actions of glucocorticoids: Molecular mechanisms. Clin Sci (Lond). (1998) ;94: (6):557–72. |
[32] | Jonat C , Rahmsdorf HJ , Park KK , Cato AC , Gebel S , Ponta H , et al. Antitumor promotion and antiinflammation: Down-modulation of AP-1 (Fos/Jun) activity by glucocorticoid hormone. Cell. (1990) ;62: (6):1189–204. |
[33] | Nelson G , Wilde GJ , Spiller DG , Kennedy SM , Ray DW , Sullivan E , et al. NF-kappaB signalling is inhibited by glucocorticoid receptor and STAT6 via distinct mechanisms. J Cell Sci. (2003) ;116: (Pt 12):2495–503. |
[34] | Scheinman RI , Cogswell PC , Lofquist AK , Baldwin AS Jr. . Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids. Science. (1995) ;270: (5234):283–6. |
[35] | Eddleston J , Herschbach J , Wagelie-Steffen AL , Christiansen SC , Zuraw BL . The anti-inflammatory effect of glucocorticoids is mediated by glucocorticoid-induced leucine zipper in epithelial cells. J Allergy Clin Immunol. (2007) ;119: (1):115–22. |
[36] | Mittelstadt PR , Ashwell JD . Inhibition of AP-1 by the glucocorticoid-inducible protein GILZ. J Biol Chem. (2001) ;276: (31):29603–10. |
[37] | Liu T , Zhang L , Joo D , Sun SC . NF-kappaB signaling in inflammation. Signal Transduct Target Ther. 2017;2. |
[38] | Srivastava M , Baig MS . NOS1 mediates AP1 nuclear translocation and inflammatory response. Biomed Pharmacother. (2018) ;102: :839–47. |
[39] | Shenkar R , Yum HK , Arcaroli J , Kupfner J , Abraham E . Interactions between CBP, NF-kappaB, and CREB in the lungs after hemorrhage and endotoxemia. Am J Physiol Lung Cell Mol Physiol. (2001) ;281: (2):L418–26. |
[40] | Lu NZ , Cidlowski JA . Glucocorticoid receptor isoforms generate transcription specificity. Trends Cell Biol. (2006) ;16: (6):301–7. |
[41] | Li MD , Yang X . A Retrospective on Nuclear Receptor Regulation of Inflammation: Lessons from GR and PPARs. PPAR Res. (2011) ;2011: :742785. |
[42] | Wehling-Henricks M , Lee JJ , Tidball JG . Prednisolone decreases cellular adhesion molecules required for inflammatory cell infiltration in dystrophin-deficient skeletal muscle. Neuromuscular disorders: NMD. (2004) ;14: (8-9):483–90. |
[43] | Kissel JT , Burrow KL , Rammohan KW , Mendell JR . Mononuclear cell analysis of muscle biopsies in prednisone-treated and untreated Duchenne muscular dystrophy. CIDD Study Group. Neurology. (1991) ;41: (5):667–72. |
[44] | Kissel JT , Lynn DJ , Rammohan KW , Klein JP , Griggs RC , Moxley RT 3rd , et al. Mononuclear cell analysis of muscle biopsies in prednisone- and azathioprine-treated Duchenne muscular dystrophy. Neurology. (1993) ;43: (3 Pt 1):532–6. |
[45] | Gavins FN , Hickey MJ . Annexin A1 and the regulation of innate and adaptive immunity. Front Immunol. (2012) ;3: :354. |
[46] | Han G , Lu K , Xu W , Zhang S , Huang J , Dai C , et al. Annexin A1-mediated inhibition of inflammatory cytokines may facilitate the resolution of inflammation in acute radiation-induced lung injury. Oncol Lett. (2019) ;18: (1):321–9. |
[47] | Parente L , Solito E . Annexin More than an anti-phospholipase protein. Inflamm Res. (2004) ;53: (4):125–32. |
[48] | Minghetti L , Nicolini A , Polazzi E , Greco A , Perretti M , Parente L , et al. Down-regulation of microglial cyclo-oxygenase-2 and inducible nitric oxide synthase expression by lipocortin 1. Br J Pharmacol. (1999) ;126: (6):1307–14. |
[49] | Wu CC , Croxtall JD , Perretti M , Bryant CE , Thiemermann C , Flower RJ , et al. Lipocortin 1 mediates the inhibition by dexamethasone of the induction by endotoxin of nitric oxide synthase in the rat. Proc Natl Acad Sci U S A. (1995) ;92: (8):3473–7. |
[50] | Solito E , Mulla A , Morris JF , Christian HC , Flower RJ , Buckingham JC . Dexamethasone Induces Rapid Serine-Phosphorylation and Membrane Translocation of Annexin 1 in a Human Folliculostellate Cell Line via a Novel Nongenomic Mechanism Involving the Glucocorticoid Receptor, Protein Kinase C, Phosphatidylinositol 3-Kinase, and Mitogen-Activated Protein Kinase. Endocrinology. (2003) ;144: (4):1164–74. |
[51] | Flower RJ , Blackwell GJ . Anti-inflammatory steroids induce biosynthesis of a phospholipase A2 inhibitor which prevents prostaglandin generation. Nature. (1979) ;278: (5703):456–9. |
[52] | Sheikh MH , Solito E . Annexin A1: Uncovering the Many Talents of an Old Protein. Int J Mol Sci. (2018) ;19: (4). |
[53] | Solito E , de Coupade C , Parente L , Flower RJ , Russo-Marie F . Human annexin 1 is highly expressed during the differentiation of the epithelial cell line A Involvement of nuclear factor interleukin 6 in phorbol ester induction of annexin 1. Cell Growth Differ. (1998) ;9: (4):327–36. |
[54] | Guido BC , Zanatelli M , Tavares-de-Lima W , Oliani SM , Damazo AS . Annexin-A1 peptide down-regulates the leukocyte recruitment and up-regulates interleukin-10 release into lung after intestinal ischemia-reperfusion in mice. J Inflamm (Lond). (2013) ;10: (1):10. |
[55] | Sugimoto MA , Vago JP , Teixeira MM , Sousa LP . Annexin A1 and the Resolution of Inflammation: Modulation of Neutrophil Recruitment, Apoptosis, and Clearance. J Immunol Res. (2016) ;2016: :8239258. |
[56] | Solito E , Christian HC , Festa M , Mulla A , Tierney T , Flower RJ , et al. Post-translational modification plays an essential role in the translocation of annexin A1 from the cytoplasm to the cell surface. FASEB J. (2006) ;20: (9):1498–500. |
[57] | Solito E , Mulla A , Morris JF , Christian HC , Flower RJ , Buckingham JC . Dexamethasone induces rapid serine-phosphorylation and membrane translocation of annexin 1 in a human folliculostellate cell line via a novel nongenomic mechanism involving the glucocorticoid receptor, protein kinase C, phosphatidylinositol 3-kinase, and mitogen-activated protein kinase. Endocrinology. (2003) ;144: (4):1164–74. |
[58] | Korte SM . Corticosteroids in relation to fear, anxiety and psychopathology. Neurosci Biobehav Rev. (2001) ;25: (2):117–42. |
[59] | Christiansen JJ , Djurhuus CB , Gravholt CH , Iversen P , Christiansen JS , Schmitz O , et al. Effects of cortisol on carbohydrate, lipid, and protein metabolism: Studies of acute cortisol withdrawal in adrenocortical failure. J Clin Endocrinol Metab. (2007) ;92: (9):3553–9. |
[60] | Macfarlane DP , Forbes S , Walker BR . Glucocorticoids and fatty acid metabolism in humans: Fuelling fat redistribution in the metabolic syndrome. J Endocrinol. (2008) ;197: (2):189–204. |
[61] | Olefsky JM , Kimmerling G . Effects of glucocorticoids on carbohydrate metabolism. Am J Med Sci. (1976) ;271: (2):202–10. |
[62] | Xu C , He J , Jiang H , Zu L , Zhai W , Pu S , et al. Direct effect of glucocorticoids on lipolysis in adipocytes. Mol Endocrinol. (2009) ;23: (8):1161–70. |
[63] | Nunez FJ , Johnstone TB , Corpuz ML , Kazarian AG , Mohajer NN , Tliba O , et al. Glucocorticoids rapidly activate cAMP production via Galphas to initiate non-genomic signaling that contributes to one-third of their canonical genomic effects. FASEB J. (2020) ;34: (2):2882–95. |
[64] | Dwivedi Y , Pandey GN . Adrenal glucocorticoids modulate [3H]cyclic AMP binding to protein kinase A (PKA), cyclic AMP-dependent PKA activity, and protein levels of selective regulatory and catalytic subunit isoforms of PKA in rat brain. J Pharmacol Exp Ther. (2000) ;294: (1):103–16. |
[65] | Kuo T , McQueen A , Chen TC , Wang JC . Regulation of Glucose Homeostasis by Glucocorticoids. Adv Exp Med Biol. (2015) ;872: :99–126. |
[66] | Smith OL , Wong CY , Gelfand RA . Influence of glucocorticoids on skeletal muscle proteolysis in normal and diabetic-adrenalectomized eviscerated rats. Metabolism. (1990) ;39: (6):641–6. |
[67] | Wang R , Jiao H , Zhao J , Wang X , Lin H . Glucocorticoids Enhance Muscle Proteolysis through a Myostatin-Dependent Pathway at the Early Stage. PLoS One. (2016) ;11: (5):e0156225. |
[68] | Sistare FD , Haynes RC , Jr. . Acute stimulation by glucocorticoids of gluconeogenesis from lactate/pyruvate in isolated hepatocytes from normal and adrenalectomized rats. J Biol Chem. (1985) ;260: (23):12754–60. |
[69] | Di Dalmazi G , Pagotto U , Pasquali R , Vicennati V . Glucocorticoids and type 2 diabetes: From physiology to pathology. J Nutr Metab. (2012) ;2012: :525093. |
[70] | Fallo F , Scarda A , Sonino N , Paoletta A , Boscaro M , Pagano C , et al. Effect of glucocorticoids on adiponectin: A study in healthy subjects and in Cushing’s syndrome. Eur J Endocrinol. (2004) ;150: (3):339–44. |
[71] | Ferris HA , Kahn CR . New mechanisms of glucocorticoid-induced insulin resistance: Make no bones about it. J Clin Invest. (2012) ;122: (11):3854–7. |
[72] | Geer EB , Islam J , Buettner C . Mechanisms of glucocorticoid-induced insulin resistance: Focus on adipose tissue function and lipid metabolism. Endocrinol Metab Clin North Am. (2014) ;43: (1):75–102. |
[73] | Ranta F , Avram D , Berchtold S , Dufer M , Drews G , Lang F ,et al.. Dexamethasone induces cell death in insulin-secreting cells, an effect reversed by exendin-4. Diabetes. (2006) ;55: (5):1380–90. |
[74] | Suh S , Park MK . Glucocorticoid-Induced Diabetes Mellitus: An Important but Overlooked Problem. Endocrinol Metab (Seoul). (2017) ;32: (2):180–9. |
[75] | Woods CP , Hazlehurst JM , Tomlinson JW . Glucocorticoids and non-alcoholic fatty liver disease. J Steroid Biochem Mol Biol. (2015) ;154: :94–103. |
[76] | Clore JN , Thurby-Hay L . Glucocorticoid-induced hyperglycemia. Endocr Pract. (2009) ;15: (5):469–74. |
[77] | Dirlewanger M , Schneiter PH , Paquot N , Jequier E , Rey V , Tappy L . Effects of glucocorticoids on hepatic sensitivity to insulin and glucagon in man. Clin Nutr. (2000) ;19: (1):29–34. |
[78] | Marino JS , Stechschulte LA , Stec DE , Nestor-Kalinoski A , Coleman S , Hinds TD Jr. . Glucocorticoid Receptor beta Induces Hepatic Steatosis by Augmenting Inflammation and Inhibition of the Peroxisome Proliferator-activated Receptor (PPAR) alpha. J Biol Chem. (2016) ;291: (50):25776–88. |
[79] | Wise JK , Hendler R , Felig P . Influence of glucocorticoids on glucagon secretion and plasma amino acid concentrations in man. J Clin Invest. (1973) ;52: (11):2774–82. |
[80] | Delaunay F , Khan A , Cintra A , Davani B , Ling ZC , Andersson A ,et al.. Pancreatic beta cells are important targets for the diabetogenic effects of glucocorticoids. J Clin Invest. (1997) ;100: (8):2094–8. |
[81] | Fichna M , Fichna P . Glucocorticoids and beta-cell function. Endokrynol Pol. (2017) ;68: (5):568–73. |
[82] | Sadr-Azodi O , Mattsson F , Bexlius TS , Lindblad M , Lagergren J , Ljung R . Association of oral glucocorticoid use with an increased risk of acute pancreatitis: A population-based nested case-control study. JAMA Intern Med. (2013) ;173: (6):444–9. |
[83] | Guo B , Zhang W , Xu S , Lou J , Wang S , Men X . GSK-3beta mediates dexamethasone-induced pancreatic beta cell apoptosis. Life Sci. (2016) ;144: :1–7. |
[84] | Lee RA , Harris CA , Wang JC . Glucocorticoid Receptor and Adipocyte Biology. Nucl Receptor Res. 2018;5. |
[85] | Lee MJ , Pramyothin P , Karastergiou K , Fried SK . Deconstructing the roles of glucocorticoids in adipose tissue biology and the development of central obesity. Biochim Biophys Acta. (2014) ;1842: (3):473–81. |
[86] | Cha JY , Kim HJ , Yu JH , Xu J , Kim D , Paul BD ,et al.. Dexras1 mediates glucocorticoid-associated adipogenesis and diet-induced obesity. Proc Natl Acad Sci U S A. (2013) ;110: (51):20575–80. |
[87] | Udden J , Bjorntorp P , Arner P , Barkeling B , Meurling L , Rossner S . Effects of glucocorticoids on leptin levels and eating behaviour in women. J Intern Med. (2003) ;253: (2):225–31. |
[88] | Leivo-Korpela S , Lehtimaki L , Vuolteenaho K , Nieminen R , Kankaanranta H , Saarelainen S ,et al.. Adipokine resistin predicts anti-inflammatory effect of glucocorticoids in asthma. J Inflamm (Lond). (2011) ;8: :12. |
[89] | Oakley RH , Ren R , Cruz-Topete D , Bird GS , Myers PH , Boyle MC ,et al.. Essential role of stress hormone signaling in cardiomyocytes for the prevention of heart disease. Proc Natl Acad Sci U S A. (2013) ;110: (42):17035–40. |
[90] | Shimizu N , Maruyama T , Yoshikawa N , Matsumiya R , Ma Y , Ito N ,et al.. A muscle-liver-fat signalling axis is essential for central control of adaptive adipose remodelling. Nat Commun. (2015) ;6: :6693. |
[91] | Duclos M . Evidence on ergogenic action of glucocorticoids as a doping agent risk. Phys Sportsmed. (2010) ;38: (3):121–7. |
[92] | Duclos M . Glucocorticoids: A doping agent? Endocrinol Metab Clin North Am. (2010) ;39: (1):107–26, ix-x. |
[93] | Heuberger J , Cohen AF . Review of WADA Prohibited Substances: Limited Evidence for Performance-Enhancing Effects. Sports Med. (2019) ;49: (4):525–39. |
[94] | Itagaki K , Menconi M , Antoniu B , Zhang Q , Gonnella P , Soybel D ,et al.. Dexamethasone stimulates store-operated calcium entry and protein degradation in cultured L6 myotubes through a phospholipase A(2)-dependent mechanism. Am J Physiol Cell Physiol. (2010) ;298: (5):C1127–39. |
[95] | Wester M , Heller A , Gruber M , Maier LS , Schach C , Wagner S . Glucocorticoid stimulation increases cardiac contractility by SGK1-dependent SOCE-activation in rat cardiac myocytes. PLoS One. (2019) ;14: (9):e0222341. |
[96] | Seidel T , Fiegle DJ , Baur TJ , Ritzer A , Nay S , Heim C ,et al.. Glucocorticoids preserve the t-tubular system in ventricular cardiomyocytes by upregulation of autophagic flux. Basic Res Cardiol. (2019) ;114: (6):47. |
[97] | Baltgalvis KA , Call JA , Nikas JB , Lowe DA . Effects of prednisolone on skeletal muscle contractility in mdx mice. Muscle Nerve. (2009) ;40: (3):443–54. |
[98] | Beiner JM , Jokl P , Cholewicki J , Panjabi MM . The effect of anabolic steroids and corticosteroids on healing of muscle contusion injury. Am J Sports Med. (1999) ;27: (1):2–9. |
[99] | Ma K , Mallidis C , Bhasin S , Mahabadi V , Artaza J , Gonzalez-Cadavid N ,et al.. Glucocorticoid-induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. Am J Physiol Endocrinol Metab. (2003) ;285: (2):E363–71. |
[100] | Watson ML , Baehr LM , Reichardt HM , Tuckermann JP , Bodine SC , Furlow JD . A cell-autonomous role for the glucocorticoid receptor in skeletal muscle atrophy induced by systemic glucocorticoid exposure. Am J Physiol Endocrinol Metab. (2012) ;302: (10):E1210–20. |
[101] | Sandri M , Sandri C , Gilbert A , Skurk C , Calabria E , Picard A ,et al.. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. (2004) ;117: (3):399–412. |
[102] | Shimizu N , Yoshikawa N , Ito N , Maruyama T , Suzuki Y , Takeda S ,et al.. Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab. (2011) ;13: (2):170–82. |
[103] | Short KR , Bigelow ML , Nair KS . Short-term prednisone use antagonizes insulin’s anabolic effect on muscle protein and glucose metabolism in young healthy people. Am J Physiol Endocrinol Metab. (2009) ;297: (6):E1260–8. |
[104] | Sali A , Guerron AD , Gordish-Dressman H , Spurney CF , Iantorno M , Hoffman EP ,et al.. Glucocorticoid-treated mice are an inappropriate positive control for long-term preclinical studies in the mdx mouse. PLoS One. (2012) ;7: (4):e34204. |
[105] | Tidball JG . Mechanisms of muscle injury, repair, and regeneration. Compr Physiol. (2011) ;1: (4):2029–62. |
[106] | Anderson JE , McIntosh LM , Poettcker R . Deflazacort but not prednisone improves both muscle repair and fiber growth in diaphragm and limb muscle in vivo in the mdx dystrophic mouse. Muscle Nerve. (1996) ;19: (12):1576–85. |
[107] | McIntosh L , Granberg KE , Briere KM , Anderson JE . Nuclear magnetic resonance spectroscopy study of muscle growth, mdx dystrophy and glucocorticoid treatments: Correlation with repair. NMR Biomed. (1998) ;11: (1):1–10. |
[108] | Morrison-Nozik A , Anand P , Zhu H , Duan Q , Sabeh M , Prosdocimo DA ,et al.. Glucocorticoids enhance muscle endurance and ameliorate Duchenne muscular dystrophy through a defined metabolic program. Proc Natl Acad Sci U S A. (2015) ;112: (49):E6780–9. |
[109] | Walter LM , Deguise MO , Meijboom KE , Betts CA , Ahlskog N , van Westering TLE ,et al.. Interventions Targeting Glucocorticoid-Kruppel-like Factor 15-Branched-Chain Amino Acid Signaling Improve Disease Phenotypes in Spinal Muscular Atrophy Mice. EBioMedicine. (2018) ;31: :226–42. |
[110] | Quattrocelli M , Barefield DY , Warner JL , Vo AH , Hadhazy M , Earley JU ,et al.. Intermittent glucocorticoid steroid dosing enhances muscle repair without eliciting muscle atrophy. J Clin Invest. (2017) ;127: (6):2418–32. |
[111] | Quattrocelli M , Salamone IM , Page PG , Warner JL , Demonbreun AR , McNally EM . Intermittent Glucocorticoid Dosing Improves Muscle Repair and Function in Mice with Limb-Girdle Muscular Dystrophy. Am J Pathol. (2017) ;187: (11):2520–35. |
[112] | Heier CR , Damsker JM , Yu Q , Dillingham BC , Huynh T , Van der Meulen JH ,et al.. VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO Mol Med. (2013) ;5: (10):1569–85. |
[113] | Sreetama SC , Chandra G , Van der Meulen JH , Ahmad MM , Suzuki P , Bhuvanendran S ,et al.. Membrane Stabilization by Modified Steroid Offers a Potential Therapy for Muscular Dystrophy Due to Dysferlin Deficit. Mol Ther. (2018) ;26: (9):2231–42. |
[114] | Escolar DM , Hache LP , Clemens PR , Cnaan A , McDonald CM , Viswanathan V ,et al.. Randomized, blinded trial of weekend vs daily prednisone in Duchenne muscular dystrophy. Neurology. (2011) ;77: (5):444–52. |
[115] | Connolly AM , Zaidman CM , Golumbek PT , Cradock MM , Flanigan KM , Kuntz NL ,et al.. Twice-weekly glucocorticosteroids in infants and young boys with Duchenne muscular dystrophy. Muscle Nerve. (2019) ;59: (6):650–7. |
[116] | Keeling RM , Golumbek PT , Streif EM , Connolly AM . Weekly oral prednisolone improves survival and strength in male mdx mice. Muscle Nerve. (2007) ;35: (1):43–8. |
[117] | Drachman DB , Toyka KV , Myer E . Prednisone in Duchenne muscular dystrophy. Lancet. (1974) ;2: (7894):1409–12. |
[118] | Mendell JR , Moxley RT , Griggs RC , Brooke MH , Fenichel GM , Miller JP ,et al.. Randomized, double-blind six-month trial of prednisone in Duchenne’s muscular dystrophy. N Engl J Med. (1989) ;320: (24):1592–7. |
[119] | Henricson EK , Abresch RT , Cnaan A , Hu F , Duong T , Arrieta A ,et al.. The cooperative international neuromuscular research group Duchenne natural history study: Glucocorticoid treatment preserves clinically meaningful functional milestones and reduces rate of disease progression as measured by manual muscle testing and other commonly used clinical trial outcome measures. Muscle Nerve. (2013) ;48: (1):55–67. |
[120] | Waldrop MA , Flanigan KM . Update in Duchenne and Becker muscular dystrophy. Curr Opin Neurol. (2019) ;32: (5):722–7. |
[121] | Gloss D , Moxley RT 3rd , Ashwal S , Oskoui M . Practice guideline update summary: Corticosteroid treatment of Duchenne muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. (2016) ;86: (5):465–72. |
[122] | Matthews E , Brassington R , Kuntzer T , Jichi F , Manzur AY . Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2016 (5):CD003725. |
[123] | Bonifati MD , Ruzza G , Bonometto P , Berardinelli A , Gorni K , Orcesi S ,et al.. A multicenter, double-blind, randomized trial of deflazacort versus prednisone in Duchenne muscular dystrophy. Muscle Nerve. (2000) ;23: (9):1344–7. |
[124] | Bello L , Gordish-Dressman H , Morgenroth LP , Henricson EK , Duong T , Hoffman EP ,et al.. Prednisone/prednisolone and deflazacort regimens in the CINRG Duchenne Natural History Study. Neurology. (2015) ;85: (12):1048–55. |
[125] | Shieh PB , McIntosh J , Jin F , Souza M , Elfring G , Narayanan S ,et al.. Deflazacort versus prednisone/prednisolone for maintaining motor function and delaying loss of ambulation: A post HOC analysis from the ACT DMD trial. Muscle Nerve. (2018) ;58: (5):639–45. |
[126] | McDonald CM , Sajeev G , Yao Z , McDonnell E , Elfring G , Souza M ,et al.. Deflazacort vs prednisone treatment for Duchenne muscular dystrophy: A meta-analysis of disease progression rates in recent multicenter clinical trials. Muscle Nerve. (2020) ;61: (1):26–35. |
[127] | Marden JR , Freimark J , Yao Z , Signorovitch J , Tian C , Wong BL . Real-world outcomes of long-term prednisone and deflazacort use in patients with Duchenne muscular dystrophy: Experience at a single, large care center. J Comp Eff Res. (2020) ;9: (3):177–89. |
[128] | Guglieri M , Bushby K , McDermott MP , Hart KA , Tawil R , Martens WB ,et al.. Developing standardized corticosteroid treatment for Duchenne muscular dystrophy. Contemp Clin Trials. (2017) ;58: :34–9. |
[129] | Beenakker EA , Fock JM , Van Tol MJ , Maurits NM , Koopman HM , Brouwer OF ,et al.. Intermittent prednisone therapy in Duchenne muscular dystrophy: A randomized controlled trial. Arch Neurol. (2005) ;62: (1):128–32. |
[130] | Crabtree NJ , Adams JE , Padidela R , Shaw NJ , Hogler W , Roper H ,et al.. Growth, bone health & ambulatory status of boys with DMD treated with daily vs. intermittent oral glucocorticoid regimen. Bone. (2018) ;116: :181–6. |
[131] | Quattrocelli M , Zelikovich AS , Jiang Z , Peek CB , Demonbreun AR , Kuntz NL ,et al.. Pulsed glucocorticoids enhance dystrophic muscle performance through epigenetic-metabolic reprogramming. JCI Insight. (2019) ;4: (24). |
[132] | Bromberg MB , Carter O . Corticosteroid use in the treatment of neuromuscular disorders: Empirical and evidence-based data. Muscle Nerve. (2004) ;30: (1):20–37. |
[133] | Desmet SJ , De Bosscher K . Glucocorticoid receptors: Finding the middle ground. J Clin Invest. (2017) ;127: (4):1136–45. |
[134] | Hua G , Zein N , Daubeuf F , Chambon P . Glucocorticoid receptor modulators CpdX and CpdX-D3 exhibit the same in vivo antiinflammatory activities as synthetic glucocorticoids. Proc Natl Acad Sci U S A. (2019) ;116: (28):14191–9. |
[135] | Hua G , Zein N , Paulen L , Chambon P . The glucocorticoid receptor agonistic modulators CpdX and CpdX-D3 do not generate the debilitating effects of synthetic glucocorticoids. Proc Natl Acad Sci U S A. (2019) ;116: (28):14200–9. |
[136] | Conklin LS , Damsker JM , Hoffman EP , Jusko WJ , Mavroudis PD , Schwartz BD ,et al.. Phase IIa trial in Duchenne muscular dystrophy shows vamorolone is a first-in-class dissociative steroidal anti-inflammatory drug. Pharmacol Res. (2018) ;136: :140–50. |
[137] | Damsker JM , Dillingham BC , Rose MC , Balsley MA , Heier CR , Watson AM ,et al.. VBP15, a glucocorticoid analogue, is effective at reducing allergic lung inflammation in mice. PLoS One. (2013) ;8: (5):e63871. |
[138] | Reeves EKM , Hoffman EP , Nagaraju K , Damsker JM , McCall JM . VBP Preclinical characterization of a novel anti-inflammatory delta 9,11 steroid. Bioorganic & Medicinal Chemistry. (2013) ;21: (8):2241–9. |
[139] | Hoffman EP , Schwartz BD , Mengle-Gaw LJ , Smith EC , Castro D , Mah JK ,et al.. Vamorolone trial in Duchenne muscular dystrophy shows dose-related improvement of muscle function. Neurology. (2019) ;93: (13):e1312–e23. |
[140] | Schram G , Fournier A , Leduc H , Dahdah N , Therien J , Vanasse M ,et al.. All-cause mortality and cardiovascular outcomes with prophylactic steroid therapy in Duchenne muscular dystrophy. J Am Coll Cardiol. (2013) ;61: (9):948–54. |
[141] | Barber BJ , Andrews JG , Lu Z , West NA , Meaney FJ , Price ET ,et al.. Oral corticosteroids and onset of cardiomyopathy in Duchenne muscular dystrophy. J Pediatr. (2013) ;163: (4):1080–4 e1. |
[142] | Hathout Y , Liang C , Ogundele M , Xu G , Tawalbeh SM , Dang UJ ,et al.. Disease-specific and glucocorticoid-responsive serum biomarkers for Duchenne Muscular Dystrophy. Sci Rep. (2019) ;9: (1):12167. |
[143] | Signorelli M , Ayoglu B , Johansson C , Lochmuller H , Straub V , Muntoni F ,et al.. Longitudinal serum biomarker screening identifies malate dehydrogenase 2 as candidate prognostic biomarker for Duchenne muscular dystrophy. J Cachexia Sarcopenia Muscle. (2020) ;11: (2):505–17. |
[144] | McDonald CM , Henricson EK , Abresch RT , Duong T , Joyce NC , Hu F ,et al.. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: A prospective cohort study. Lancet. (2018) ;391: (10119):451–61. |
[145] | Arpan I , Willcocks RJ , Forbes SC , Finkel RS , Lott DJ , Rooney WD ,et al.. Examination of effects of corticosteroids on skeletal muscles of boys with DMD using MRI and MRS. Neurology. (2014) ;83: (11):974–80. |
[146] | Rooney WD , Berlow YA , Triplett WT , Forbes SC , Willcocks RJ , Wang DJ ,et al.. Modeling disease trajectory in Duchenne muscular dystrophy. Neurology. (2020) ;94: (15):e1622–e33. |
[147] | Hussein MR , Hamed SA , Mostafa MG , Abu-Dief EE , Kamel NF , Kandil MR . The effects of glucocorticoid therapy on the inflammatory and dendritic cells in muscular dystrophies. Int J Exp Pathol. (2006) ;87: (6):451–61. |
[148] | Wong-Kisiel LC , Kuntz NL . Two siblings with limb-girdle muscular dystrophy type 2E responsive to deflazacort. Neuromuscular disorders: NMD. (2010) ;20: (2):122–4. |
[149] | Connolly AM , Pestronk A , Mehta S , Al-Lozi M . Primary alpha-sarcoglycan deficiency responsive to immunosuppression over three years. Muscle Nerve. (1998) ;21: (11):1549–53. |
[150] | Angelini C , Fanin M , Menegazzo E , Freda MP , Duggan DJ , Hoffman EP . Homozygous alpha-sarcoglycan mutation in two siblings: One asymptomatic and one steroid-responsive mild limb-girdle muscular dystrophy patient. Muscle Nerve. (1998) ;21: (6):769–75. |
[151] | Walter MC , Reilich P , Thiele S , Schessl J , Schreiber H , Reiners K ,et al.. Treatment of dysferlinopathy with deflazacort: A double-blind, placebo-controlled clinical trial. Orphanet J Rare Dis. (2013) ;8: :26. |
[152] | Amoasii L , Hildyard JCW , Li H , Sanchez-Ortiz E , Mireault A , Caballero D ,et al.. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. (2018) ;362: (6410):86–91. |


