
Current Therapeutic Approaches in FSHD
Abstract
Facioscapulohumeral muscular dystrophy (FSHD) is one of the most common muscular dystrophies. Over the last decade, a consensus was reached regarding the underlying cause of FSHD allowing—for the first time—a targeted approach to treatment. FSHD is the result of a toxic gain-of-function from de-repression of the DUX4 gene, a gene not normally expressed in skeletal muscle. With a clear therapeutic target, there is increasing interest in drug development for FSHD, an interest buoyed by the recent therapeutic successes in other neuromuscular diseases. Herein, we review the underlying disease mechanism, potential therapeutic approaches as well as the state of trial readiness in the planning and execution of future clinical trials in FSHD.
INTRODUCTION
Facioscapulohumeral dystrophy (FSHD) is the third most common muscular dystrophy after Duchenne muscular dystrophy and myotonic dystrophy, with a prevalence of ∼12–15 per 100,000 [1, 2]. Age of onset is variable with presentations at birth to late in life. On average, males tend to present earlier in their late teen years to mid-twenties whereas females present in their late twenties to early thirties. Classically, the disease presents with facial and proximal arm weakness with winged scapula followed by weakness of foot dorsiflexion and hip girdle muscles. Additionally, truncal muscles including the paraspinals and abdominal muscle are variably affected. Asymmetric involvement is frequent and often very prominent [3]. Bulbar, cardiac, and extraocular muscles are spared. Neuromuscular restrictive lung disease occurs in about 15% of individuals with a minority needing non-invasive ventilatory support [4]. Symptomatic hearing loss and retinal vascular disease (Coats disease) are infrequent occurring exclusively in infantile-onset disease [4]. Nevertheless, FSHD can result in significant morbidity with 20% of the patients becoming wheelchair dependent after the age of 50 [5]. As the wide spectrum of age at disease onset suggests, the rate of disease progression is variable but generally slow [6, 7].
MOLECULAR PATHOPHYSIOLOGY OF FSHD
Genetics of FSHD
Over the past decade, consensus was reached regarding the primary cause of FSHD, the inappropriate expression of the DUX4 gene on chromosome 4q35 in skeletal muscle (see Figure 1). There are multiple tandem copies of the DUX4 gene, each contained in a 3.3 kb repeat unit, known as a D4Z4 macrosatellite repeat [8]. Unlike microsatellite repeats that consist of a few base pairs, macrosatellite repeats are several kilobases in size. Tandem repeated DNA composes a significant portion (>50%) of the human genome and this type of copy number variation accounts for much of human phenotypic variation. The D4Z4 repeats are located at the telomeric end of chromosomes 4q and 10q with copy numbers from 11 to greater than 150 repeats [8]. FSHD results from a change in the non-permissive, highly methylated chromatin structure of the D4Z4 repeats to a more permissive euchromatic structure, allowing the expression of the DUX4 gene from the most distal D4Z4 repeat. There are two sequence variants distal to the last repeat termed A and B. Only the A variant on 4q35 contains has a polyadenylation signal, allowing the production of a stable DUX4 mRNA.
Fig. 1
DUX4 genetics. The production of DUX4 in human muscles requires the breakdown of the multiple genetic safeguards evolved to suppress its expression in somatic cells: 1) The presence of more than 10 tandem repeat units on 4q that allow for heterochromatin condensation [91]; 2) GC-rich sequence (73%) in the repeat that allow for methylation [92]; 3) a polyadenylation signal that cannot be used in somatic/muscle cells in ∼50% of the European population [93]; 4) histone modification H3K9me3 to cause a repressive chromatin state. The utilization of the 4qA polyadenylation signal seems to be specific in somatic cells and may be aided by muscle-specific enhancers [94] in the proximal end of 4q that may aid in the transcription of DUX4 and the stabilization of the mRNA. The polyadenylation signal is critical for pre-mRNA processing and allows for DUX4 pre-mRNA cleavage and extension of polyadenylation to the mRNA [95]. The pathomechanism of derepression of DUX4 as the cause of FSHD was discovered because of careful study of the genetic structure of the 4q locus and the many naturally occurring cross-over events with the 10q subtelomere and the conclusions are: 1) A single repeat containing DUX4 is required; as an individual with complete loss of 4q subtelomeric region did not have FSHD [96]; 2) The region proximal to DUX4 on 4q and absent on 10q, including the upstream region with FRG1, SLC25A4 (ANT1) and DUX4c genes, is not required because a translocation of the most distal end of 4q to 10q resulted in FSHD; 3) 10q contraction (as found in ∼10% of the normal population) does not result in FSHD [97–99]—most likely because while 10q has similarity to the permissive 4qA alleles with the presence of 6.2-kb β-satellite sequence, it lacks the polyadenylation signal—similar to the non-permissive 4qB alleles.
![DUX4 genetics. The production of DUX4 in human muscles requires the breakdown of the multiple genetic safeguards evolved to suppress its expression in somatic cells: 1) The presence of more than 10 tandem repeat units on 4q that allow for heterochromatin condensation [91]; 2) GC-rich sequence (73%) in the repeat that allow for methylation [92]; 3) a polyadenylation signal that cannot be used in somatic/muscle cells in ∼50% of the European population [93]; 4) histone modification H3K9me3 to cause a repressive chromatin state. The utilization of the 4qA polyadenylation signal seems to be specific in somatic cells and may be aided by muscle-specific enhancers [94] in the proximal end of 4q that may aid in the transcription of DUX4 and the stabilization of the mRNA. The polyadenylation signal is critical for pre-mRNA processing and allows for DUX4 pre-mRNA cleavage and extension of polyadenylation to the mRNA [95]. The pathomechanism of derepression of DUX4 as the cause of FSHD was discovered because of careful study of the genetic structure of the 4q locus and the many naturally occurring cross-over events with the 10q subtelomere and the conclusions are: 1) A single repeat containing DUX4 is required; as an individual with complete loss of 4q subtelomeric region did not have FSHD [96]; 2) The region proximal to DUX4 on 4q and absent on 10q, including the upstream region with FRG1, SLC25A4 (ANT1) and DUX4c genes, is not required because a translocation of the most distal end of 4q to 10q resulted in FSHD; 3) 10q contraction (as found in ∼10% of the normal population) does not result in FSHD [97–99]—most likely because while 10q has similarity to the permissive 4qA alleles with the presence of 6.2-kb β-satellite sequence, it lacks the polyadenylation signal—similar to the non-permissive 4qB alleles.](https://content.iospress.com:443/media/jnd/2021/8-3/jnd-8-3-jnd200554/jnd-8-jnd200554-g001.jpg)
In FSHD type 1 (FSHD1), which constitutes about 95% of patients with FSHD, contraction of the D4Z4 repeat number to between 1–10 repeats results in chromatin relaxation. When a contraction occurs on a 4q35 with an A variant, stable DUX4 mRNA and protein is produced leading to a toxic gain-of-function. In FSHD1, the shorter the number of residual repeats is broadly associated with younger disease onset, overall severity, and increase penetrance. Individuals with 1–3 repeats tend to have earlier onset and more severe muscle weakness and non-muscular manifestations such as symptomatic hearing loss, retinal vascular disease and more likely to develop restrictive lung disease [9]. Most individuals with FSHD1 have between 4–7 repeats and tend to have, as a group, more moderate disease. Contractions with 8–10 repeats have later onset, milder disease, and a higher frequency of non-penetrance (not developing symptoms). Despite the relationship between disease severity and repeat size, the wide intra-familial variability points to the presence of other factors influencing disease severity.
The remaining 5% of patients with FSHD have FSHD type 2 (FSHD2). FSHD2 is a digenic disease requiring the co-occurrence of two events: 1) at least one 4q35 D4Z4 with an A polymorphism and a contracted array, and 2) mutation in a gene that plays a role in the epigenetic repression of the D4Z4 repeats. Whereas the contraction of the D4Z4 array is the main reason for derepression of that D4Z4 array (in cis) for FSHD1, the mutations in FSHD2 result in derepression of D4Z4 repeats in trans, (of all D4Z4 arrays even the ones on chromosome 10 in addition to those on chromosome 4). About 95% of individuals with FSHD2 have concomitant mutation in the SMCHD1 gene. SMCHD1 protein is involved in DNA hypermethylation and plays a role in X-inactivation [10–13]. The SMCHD1 mutation results in chromatin hypomethylation of the repeats on chromosome 4 resulting in DUX4 expression from the 4q35 with a permissive A polymorphism. Another gene implicated in FSHD2 is DNA (cytosine-5-)-methyltransferase 3 beta (DNMT3B), a methyltransferase gene responsible for epigenetic repression [14]. Heterozygous mutations in DNMT3B also result in hypomethylation; but only those patients with relatively short 9–13 units of D4Z4 on a permissive allele develop FSHD. Most recently, a homozygous mutation in a third gene, Ligand-Dependent Nuclear Receptor Interacting Factor 1 (LRIF1 [also known as HBiX1]), was found in a patient with an FSHD phenotype and hypomethylation of the D4Z4 repeats consistent with FSHD2 [15]. The number of D4Z4 repeats in FSHD2 is on the lower range of what is considered normal, possibly less than 16 [16]. There is also an overlap between FSHD1 and FSHD2 where the penetrance of symptoms in patients with 8–10 repeats is increased due to hypomethylation at the D4Z4 array by mutations in trans such as SMCHD1 [16]. There is no differences between FSHD1 and FSHD2 clinical phenotypes [17].
Normal function of DUX4 in humans
DUX4 is highly conserved among primates such as humans, chimpanzees, and orangutans. DUX4 is a transcription factor important in zygotic genome activation at the two-/four-cell embryo stage as well as in regulation of spermatagonia in adult male testes [18–20]. It is not clear how DUX4 is activated during embryogenesis, however, DUX4 is produced from the most distal monomer of both 4q35 and 10q26 in the embryo [19]. Downstream DUX4 targets are expressed in human testis [21]. Other that testes, DUX4 is known to be expressed in the thymus but has not been shown to be in any other somatic cells [22].
Pathophysiologic consequences of DUX4 expression in human skeletal muscle
DUX4 expression in skeletal muscle tissue activates a similar transcriptional program as in the embryo—inducing many of the DUX4 target genes before zygotic genome activation at the four-cell stage of the human embryo [18]. However, DUX4 protein is toxic to muscle fibers or cultured myocytes [12, 23–26]. In FSHD patient-derived myoblasts, DUX4 is expressed in stochastic bursts in a small number of myonuclei as opposed to a steady low-level expression [19, 27]. With differentiation into myotubes, DUX4 expression is more pronounced resulting in cell death [28]. Once activated, DUX4 induces a number of genetic programs that lead to initiation of the inflammatory cascade, muscle atrophy, oxidative stress, and disrupted myogenesis [21, 28–32]. The expression of these genes is undetectable or nearly undetectable in control muscle samples but increased in FSHD1 and 2 muscle samples or DUX4-transfected cell lines [33].
THERAPEUTIC APPROACHES
A number of non-targeted therapeutic interventions were tried in FSHD. These include an open label trial of prednisone [34], several randomized control trials of albuterol [35–37], an intravenous myostatin inhibitor (MYO-029) [38], and a trial of oral antioxidants [39]. In none of those studies did the primary outcome measure show positive results. A more recent study evaluated the effects of an intramuscularly-administered myostatin inhibitor, ACE-083, in FSHD [40]; however, the phase 2 study was stopped as no functional benefit was demonstrated despite increasing muscle mass. One could speculate that muscle mass was only increased in good muscle and the lack of recovery of already-affected muscle prevents an improvement in functionality; this seems to be a lesson for not just FSHD but also myostatin inhibitors in other muscle diseases such as inclusion body myositis.
More targeted approaches are now possible. FSHD is an attractive target pharmaceutically because it is a relatively common muscle disease. Moreover, whereas most muscular dystrophies result from loss-of-function mutations in genes coding for critical skeletal muscle proteins, FSHD is the result of the deleterious gain-of-function due to the expression of a gene not expressed in somatic cells. Consequently, effective blocking of DUX4 expression could potentially be curative in FSHD. Possible therapeutic approaches include: 1) epigenetic silencing of the D4Z4 repeats; 2) blocking DUX4 mRNA production; 3) targeting one of several identified downstream pathologic pathways triggered by DUX4 expression. (For overview, see Figure 2.)
Fig. 2
Targeting DUX4. Possible targeted therapeutic approaches to FSHD include: 1) epigenetic silencing of the D4Z4 repeats; 2) blocking DUX4 mRNA production by inhibiting DUX4 promoter or DUX4 mRNA formation; 3) targeting one of several identified downstream pathologic pathways triggered by DUX4 expression.
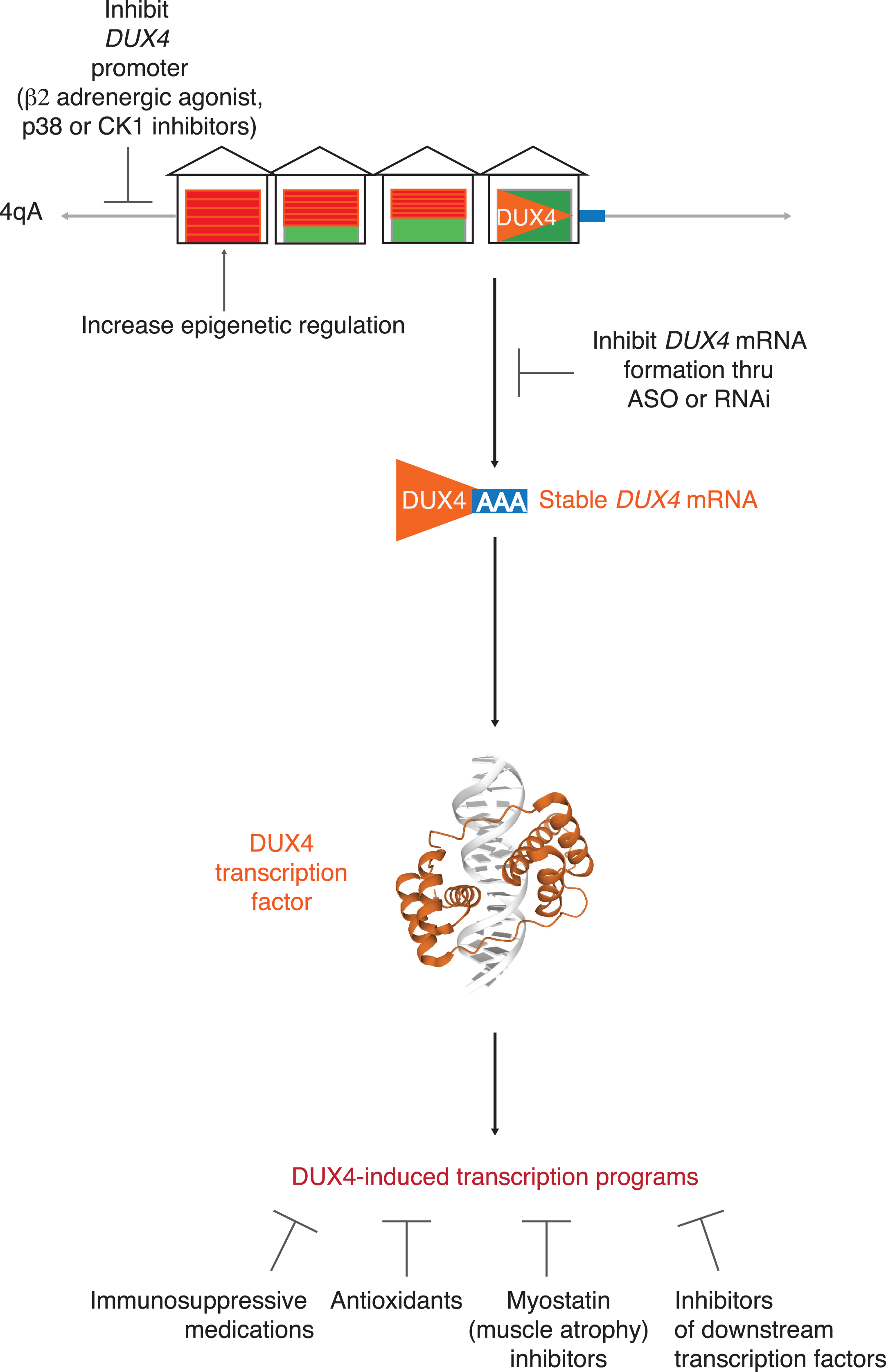
Targeting DUX4 upstream
Multiple approaches have shown decreased DUX4 expression by either enhancing epigenetic repression of D4Z4 repeats or inhibiting upstream signals. Enhancing epigenetic repression can be achieved by targeting methylation, SCHMD1 activity, or other signals that repress the chromatin.
Delivering non-coding RNAs of the D4Z4 repeats into muscle cells may help with regulation of D4Z4 repeats by facilitating DICER/AGO-dependent epigenetic silencing of the D4Z4 repeat arrays [41, 42]. SMCHD1 overexpression in FSHD1 and FSHD2 myotubes suppresses DUX4 expression [43] and small molecules are being developed to augment SMCHD1 activity. The caveat would the unintended effects of increased SMCHD1 activity as it regulates and inactivates other loci such as the X chromosome. Viral delivery of SMCHD1 under a muscle-specific promoter could be an alternative but would not be as elegant of a solution as a small molecule whose effect one can titrate.
For patients with FSHD2 secondary to SCHMD1 mutations, genome editing of intronic mutation was attempted in muscle cell culture and could be a therapeutic approach in patients with FSHD2 secondary to SCHMD1 mutations [44]. However, it is limited by the current low efficiency rate of genome editing in a whole organism. Other signals that repress the chromatin through histone modification such as inhibiting acetylation of histone or increasing methylation by the polycomb repressive complex 2, may suppress DUX4 expression [27]. Agents that increase the activity and/or expression of NuRD/MBD2 and/or MBDl/CAF-1 complex members that modify the chromatin/histone have also been patented for the treatment of FSHD.
Increasing methylation results in repression of the D4Z4 region, and molecular therapies that increase DNA methylation through DNMT3B may inhibit DUX4 expression. DNMTs utilize S-Adenosylmethionine as a co-factor, which is synthesized from methionine using co-factors such as folate, choline, betaine, and vitamins B2, B6 and B12. However, indiscriminate dietary supplementation has not been documented to effect epigenetic modification beyond the perinatal period [45] and therefore unlikely to increase DNMT3B activity in muscle specifically. This was borne out in a small study utilizing folic acid and methionine to try to increase methylation [46].
In a screen of immortalized myoblasts derived from patients with FSHD1 or FSHD2 transfected with a reporter of DUX4 activity, inhibitors of bromodomain and extra-terminal domain (BET) family of proteins were found to suppress DUX4 activity by blocking binding of BET family proteins to acetylated histones allowing class I histone deacetylases (HDACs) to suppress DUX4 expression [47].
Other potential drugs identified by screening immortalized FSHD patient-derived myoblasts are beta2 adrenergic receptor agonists, such as clenbuterol and albuterol, which decreased DUX4 mRNA synthesis [47]. Interestingly, β2 adrenergic agonists are known to be powerful anabolic agents that trigger skeletal muscle hypertrophy and have been tried for treatment of muscle wasting as well as FSHD (for review see Joassard et al. [48]). They activate adenylyl cyclase to increase cellular cyclic adenosine monophosphate (cAMP) levels and subsequently the protein kinase A (PKA) pathway. Clinical trials of β2 adrenergic agonists (albuterol/salbutamol) failed to improve strength or function [36, 37]. However, Kissel et al. reported that albuterol caused a modest, dose-dependent, but statistically significant increase lean body mass compared to placebo. Whether clenbuterol, a more potent β2 adrenergic agonist, can achieve a more marked increase in lean body mass resulting in improved strength or function remains to be seen.
A systematic search for small-molecule inhibitors of the PKA pathway led to the discovery of p38α/β inhibitors as inhibitors of DUX4 expression in immortalized myoblasts [49]. In xenograft transplant, RNA levels for DUX4 and its targets were decreased by approximately 80% with the p38α/β inhibitor losmapimod. P38 is a subgroup of the mitogen-activated protein kinases (MAPKs) that regulates multiple processes. Of the four family members, p38α is ubiquitously expressed, p38β is more restricted in expression and expressed at a lower level in muscle [50]. When muscle satellite stem cells are activated and divide, one cell with active p38α/β becomes the proliferating daughter cell (myoblast) and the other cell with inactive p38α/β replenishes the quiescent satellite stem cell pool. Further down the myogenesis process, p38α/β MAPKs are also important for activating transcriptional and epigenetic programs that result in differentiation of the myoblast into the myocyte [51]. Losmapimod appears to work in the myoblasts and the differentiated myocytes.
However, one must take into context the ubiquity of the p38 pathway with p38α being expressed in most cells. Tissue-specific knockout of p38α show that it is important for proliferation of lung epithelial cells and cardiomyocytes [52, 53]. Activation of p38 leads to activation of inflammatory cytokines. This led to therapeutic trials of losmapimod in chronic obstructive lung disease, and pain, none of which showed a beneficial effect of losmapimod [54–56]. Currently, Fulcrum Therapeutics is conducting a phase II trial to address whether p38α/β inhibitors decrease DUX4 expression and preserve muscle bulk on MRI [57]. However, the ubiquitous role of p38 in cell functions will make long-term surveillance important.
Another compound that inhibits DUX4 expression is casein kinase I (CK1) inhibitors which suppress DUX4 expression in FSHD myotubes and in the xenograft model [58]. The authors consider their drug-screening assay to be superior as it can simultaneously measure DUX4 repression and myoblast fusion index at various drug concentrations. Since DUX4 expression occurs when myoblasts fuse into myotubes, a drug inhibiting myoblast fusion will result in a false positive measure of DUX4 inhibition.
Another therapeutic approach to inhibit DUX4 upstream is to target the promoter of DUX4 on 4q. Himeda et al. fashioned a dominant negative inhibitor with a catalytically dead Cas9 loaded with a guide RNA to the promoter region of DUX4 linked to a protein that blocked transcription activation (the catalytically dead Cas9) [59]—suggesting a clever way to utilize our understanding of the 4q35 genetic architecture to block DUX4 expression.
Any approach to DUX4 repression should consider possible off-target effects on tissues that normally express DUX4, such as the thymus and testes. DUX4 repression in the thymus is not likely to cause untoward side effects but possible effects on spermatogenesis need to be considered.
Targeting DUX4 directly at the RNA level
Targeting DUX4 directly at the RNA level is appealing because it targets a transcript that should not be expressed and there is well-described chemistry, antisense oligonucleotide (ASOs) or inhibitory RNA (RNAi) therapies, to target the RNA.
Inhibitory RNA (RNAi) therapies using small interfering RNA (siRNA) were used to target the 3′ untranslated region transcribed from pLAM [60], the coding region [21], well as the region upstream of the DUX4 transcription start site [41]. The last is an endogenously produced siRNA that may be part of the cell’s regulatory mechanism of the D4Z4 region. One of the limitations of RNA interference approach is its high dose cytotoxicity derived from its off-target effects [61, 62].
Similarly, ASOs have been successful in targeting portions of the 3′ untranslated region of the DUX4 pre-mRNA to inhibit the polyadenylation in immortalized FSHD cells [63], myotubes derived from FSHD muscle cultures and xenografts [64]. A poly-adenosine tail is extended on the pre-mRNA and is beyond the polyadenylation signal and not encoded in the D4Z4 DNA. ASOs interfere with transcript termination and 3’ end processing to cause mRNA degradation and decrease DUX4 protein expression. ASOs have also been made to interfere with DUX4 mRNA splicing [65].
Locked nucleic acid (LNA) gapmer antisense oligonucleotides also have been engineered to bind to DUX4 mRNA and be knocked down through RNAse H-mediated degradation and shown to be successful in tissue culture and injection into mouse models [66].
However, these types of technologies are bedeviled by the electrostatic nature or bulk of the compounds which prevents efficient uptake through the lipid bilayer and the muscle cells when delivered systemically; as have been found in myotonic dystrophy and Duchenne muscular dystrophy [61, 62]. Some of this is being addressed by using adeno-associated virus (AAV) as a delivery vector. AAV vectors delivering artificial microRNAs targeting the DUX4 mRNA were able to direct the transcript toward an RNAi degradation pathway [67]. In addition, ASOs modified with a carrier that targets muscle cells and facilitates uptake are being tried. Finally, modifying the chemistry of the backbone could be beneficial as locked nucleic acid (LNA) gapmer antisense oligonucleotides.
Targeting downstream effects of DUX4
DUX4 is a powerful inducer of myriad genetic programs that lead to initiation of the inflammatory cascade, muscle atrophy, oxidative stress, disrupted myogenesis. As a transcription factor, DUX4 uncovers a vast, complex gene regulatory network. Inhibition of DUX4 can be achieved by utilizing DNA aptamers, short oligonucleotides, engineered bind to the DNA binding site of DUX4 and thus inhibiting DUX4 from binding to its transcriptional activator sites [68].
To exert its transcriptional activity, DUX4 recruits histone acetyltransferases (HATs) p300 and CBP (CREB binding protein) [69]. Selective inhibitors of p300 can inhibit the transcriptional activity of DUX4 in cell culture [70]. One such transcription factor is PITX1, another double homeobox transcription factors that activates pathways that lead to muscle atrophy (through atrogin-1 and MuRF-1) and inflammatory features [30]. PITX1 suppression with ASOs can ameliorate the pathological features of the muscle-specific PITX1 transgenic mouse [71].
DUX4 induction in FSHD myoblast model results in accumulation of the glycosaminoglycan hyaluronic acid and mediates a few of the downstream pathways that DUX4 is known to activate [72]. 4-methylumbelliferone, a well-characterized competitive inhibitor of HA biosynthesis prevents DUX4-induced accumulation of hyaluronic acid and subsequent downstream pathways. It is an already approved drug in Europe and Asia called “hymecromone” where it is used to treat biliary spasm.
Oxidative stress, with its resultant production of free radicals and reactive oxygen species, results in cellular damage can be important in the pathophysiology of FSHD and several known antioxidants have been identified to inhibit DUX4-induced toxicity in myoblasts [73]. A small subset of antioxidants were studied in adults with FSHD [39] and no follow-up studies have since been done to look at more specific antioxidants.
It is not clear, however, that inhibiting specific downstream pathways of DUX4 will completely abrogate all the damage caused by the myriad genetic programs that DUX4 uncovers in muscle cells.
CLINICAL TRIAL READINESS IN FSHD
Work on various aspects of trial readiness is ongoing for the last decade as consensus on the FSHD disease mechanism was reached [74, 75]. Critical components of trial readiness include facilitating patient access to clinical trials, establishing research centers familiar with FSHD assessments, having a good understanding of the natural history of the disease and developing a multitude of relevant outcome measures for early and late phase trials.
Patient access to research studies is facilitated by the presence of a number of FSHD patient registries in the US and in several European countries [76–79]. The oldest is the National Registry for Facioscapulohumeral Dystrophy in the US which prospectively collected yearly clinical data on patients with FSHD for almost two decades, data that proved valuable in understanding aspects of functional progression [80].
The first prospective natural history study of FSHD followed 80 patients for up to three years [6]. The study was limited by absence of genetic testing in all subjects and evaluations were restricted to manual muscle testing and quantitative myometry. Nevertheless, both outcome measures showed a slight 12-month decline in strength. A large, multi-national, natural history study, the ReSolve study, is currently in its third year and will exam a change in a variety of outcome measures over a span of 24 months [81]. These outcome measures include, in addition to strength testing, a composite functional outcome measure (FSHD-COM), reachable workspace as a quantitative measure of shoulder function an FSHD-specific patient reported health index (FSHD HI) (90, 91, 92, 93). Additionally, DEXA scan to assess changes in lean body mass and electrical impedance myography (EIM), a measure of muscle composition, to look for changes in individual muscles are being investigated as potential biomarkers [82]. Separate studies have investigated the utility of MRI and muscle ultrasound as a biomarker in FSHD [83–86].
Early phase 2 trials seeking to test safety and target engagement of DUX4 will require either a tissue or validated circulating biomarkers. To date, there are no validated FSHD circulating biomarkers [87, 88]. However, as DUX4 is expressed stochastically and at very low levels, it is difficult to measure quantitatively in muscle samples. Fortunately, as DUX4 is a transcription factor, a large number of genes are turned on and act as reliable surrogates of DUX4 activity. [21, 33, 47, 89]. Moreover, quantitating a subset of four DUX4-regulated genes (LEUTX, KHDC1L, PRAMEF2 and TRIM43) may increase sensitivity of detecting DUX4 activity. A recent study, using MRI to select muscles for biopsy showed that muscles with T2 STIR positive changes and fatty infiltration on T1 sequences showed the highest levels of DUX4-target expression [90]. These findings suggest MRI guidance is crucial in the selection of the optimal muscle to biopsy in early phase 2 trials. This concept is being validated in the current losmapimod trials, as is the use of downstream DUX4-targets as a marker of DUX4 activity.
CONCLUSION
For almost two decades, the underlying disease mechanism in FSHD, one of the most common muscular dystrophies, was an enigma. With consensus reached on disease mechanism, targeted treatments are now possible resulting in heightened interest from pharmaceutical companies. Simultaneously, active clinical research studies are reexamining FSHD natural history, vetting a number of novel disease-specific clinical outcome measures as well as imaging, circulating and tissue biomarkers.
FINANCIAL DISCLOSURES
Dr. Wang reports consultancy for Biogen. Dr. Tawil reports consultancy with Fulcrum Therapeutics and Acceleron Pharma.
REFERENCES
[1] | Deenen JC , Arnts H , van der Maarel SM , Padberg GW , Verschuuren JJ , Bakker E , et al. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology. (2014) ;83: (12):1056–9. 10.1212/WNL0000000000000797. |
[2] | Flanigan KM , Coffeen CM , Sexton L , Stauffer D , Brunner S , Leppert MF . Genetic characterization of a large, historically significant Utah kindred with facioscapulohumeral dystrophy. Neuromuscular Disorders. (2001) ;11: (6-7):525–9. 10.1016/s0960-8966(01)00201-2. |
[3] | Pastorello E , Cao M , Trevisan CP . Atypical onset in a series of 122 cases with FacioScapuloHumeral Muscular Dystrophy. Clin Neurol Neurosurg. (2011) ;114: (3):230–4. 10.1016/j.clineuro.2011.10.022. |
[4] | Tawil R , Kissel JT , Heatwole C , Pandya S , Gronseth G , Benatar M , et al. Evidence-based guideline summary: Evaluation, diagnosis, and management of facioscapulohumeral muscular dystrophy. Neurology. (2015) ;85: (4):357–528. 10.1212/wnl.0000000000001783. |
[5] | Tawil R , Van Der Maarel SM . Facioscapulohumeral muscular dystrophy. Muscle Nerve. (2006) ;34: (1):1–15. 10.1002/mus.20522. |
[6] | A prospective, quantitative study of the natural history of facioscapulohumeral muscular dystrophy (FSHD): Implica-tions for therapeutic trials. The FSH-DY Group. Neurology. (1997) ;48: (1):38–46. 10.1212/wnl.48.1.38. |
[7] | Statland JM , McDermott MP , Heatwole C , Martens WB , Pandya S , van der Kooi EL , et al. Reevaluating measures of disease progression in facioscapulohumeral muscular dystrophy. Neuromuscul Disord. (2013) ;23: (4):306–12. 10.1016/j.nmd.2013.01.008. |
[8] | Warburton PE , Hasson D , Guillem F , Lescale C , Jin X , Abrusan G . Analysis of the largest tandemly repeated DNA families in the human genome. BMC Genomics. (2008) ;9: :533. 10.1186/1471-2164-9-533. |
[9] | Goselink RJM , Schreuder THA , van Alfen N , de Groot IJM , Jansen M , Lemmers R , et al. Facioscapulohumeral Dystrophy in Childhood: A Nationwide Natural History Study. Ann Neurol. (2018) ;84: (5):627–37. 10.1002/ana.25326. |
[10] | de Greef JC , Lemmers RJLF , Camano P , Day JW , Sacconi S , Dunand M , et al. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology. (2010) ;75: (17):1548–54. |
[11] | Lemmers RJ , Goeman JJ , van der Vliet PJ , van Nieuwenhuizen MP , Balog J , Vos-Versteeg M , et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum Mol Genet. (2015) ;24: (3):659–69. 10.1093/hmg/ddu486. |
[12] | Lemmers RJ , Tawil R , Petek LM , Balog J , Block GJ , Santen GWE , et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nature Genetics. (2012) ;44: (12):1370–4. 10.1038/ng.2454. |
[13] | van Overveld PG , Lemmers RJ , Sandkuijl LA , Enthoven L , Winokur ST , Bakels F , et al. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat Genet. (2003) ;35: (4):315–7. 10.1038/ng1262. |
[14] | van den Boogaard ML , Lemmers R , Balog J , Wohlgemuth M , Auranen M , Mitsuhashi S , et al. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am J Hum Genet.. (2016) ;98: (5):1020–9. 10.1016/j.ajhg.2016.03.013. |
[15] | Hamanaka K , Sikrova D , Mitsuhashi S , Masuda H , Sekiguchi Y , Sugiyama A , et al. Homozygous nonsense variant in LRIF1 associated with facioscapulohumeral muscular dystrophy. Neurology. (2020) . 10.1212/WNL.0000000000009617. |
[16] | Sacconi S , Briand-Suleau A , Gros M , Bau–n C , Lemmers R , Rondeau S , et al. FSHD1 and FSHD2 form a disease continuum. Neurology. (2019) ;92: (19):e2273–e85. 10.1212/WNL.0000000000007456. |
[17] | de Greef JC , Lemmers RJLF , van Engelen BGM , Sacconi S , Venance SL , Frants RR , et al. Common Epigenetic Changes of D4Z4 in Contraction-Dependent and Contraction-Independent FSHD. Human Mutation. (2009) ;30: (10):1449–59. 10.1002/humu.21091. |
[18] | De Iaco A , Planet E , Coluccio A , Verp S , Duc J , Trono D . DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat Genet. (2017) ;49: (6):941–5. 10.1038/ng.3858. |
[19] | Snider L , Geng LN , Lemmers RJ , Kyba M , Ware CB , Nelson AM , et al. Facioscapulohumeral dystrophy: Incomplete suppression of a retrotransposed gene. PLoS Genet. (2010) ;6: (10):e1001181. 10.1371/journal.pgen.1001181. |
[20] | Wu SL , Tsai MS , Wong SH , Hsieh-Li HM , Tsai TS , Chang WT , et al. Characterization of genomic structures and expression profiles of three tandem repeats of a mouse double homeobox gene: Duxbl. Dev Dyn. (2010) ;239: (3):927–40. 10.1002/dvdy.22210. |
[21] | Geng LN , Yao Z , Snider L , Fong AP , Cech JN , Young JM , et al. DUX4 activates germline genes, retroelements, and immune mediators: Implications for facioscapulohumeral dystrophy. Dev Cell. (2011) ;22: (1):38–51. 10.1016/j.devcel.2011.11.013. |
[22] | Das S , Chadwick BP . Influence of Repressive Histone and DNA Methylation upon D4Z4 Transcription in Non-Myogenic Cells. PLoS One. (2016) ;11: (7):e0160022. 10.1371/journal.pone.0160022. |
[23] | Giesige CR , Wallace LM , Heller KN , Eidahl JO , Saad NY , Fowler AM , et al. AAV-mediated follistatin gene therapy improves functional outcomes in the TIC-DUX4 mouse model of FSHD. JCI Insight. (2018) ;3: (22). 10.1172/jci.insight.123538. |
[24] | Jones TI , Chen JC , Rahimov F , Homma S , Arashiro P , Beermann ML , et al. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: Evidence for disease modifiers and a quantitative model of pathogenesis. Hum Mol Genet. (2012) ;21: (20):4419–30. 10.1093/hmg/dds284. |
[25] | Wallace LM , Garwick SE , Mei W , Belayew A , Coppee F , Ladner KJ , et al. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann Neurol. (2011) ;69: (3):540–52. 10.1002/ana.22275. |
[26] | Bosnakovski D , Lamb S , Simsek T , Xu Z , Belayew A , Perlingeiro R , et al. DUX4c, an FSHD candidate gene, interferes with myogenic regulators and abolishes myoblast differentiation. Exp Neurol. (2008) ;214: (1):87–96. 10.1016/j.expneurol.2008.07.022. |
[27] | Haynes P , Bomsztyk K , Miller DG . Sporadic DUX4 expression in FSHD myocytes is associated with incomplete repression by the PRC2 complex and gain of H3K9 acetylation on the contracted D4Z4 allele. Epigenetics Chromatin. (2018) ;11: (1):47. 10.1186/s13072-018-0215-z. |
[28] | Rickard AM , Petek LM , Miller DG . Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum Mol Genet. (2015) ;24: (20):5901–14. 10.1093/hmg/ddv315. |
[29] | Knopp P , Krom YD , Banerji CR , Panamarova M , Moyle LA , den Hamer B , et al. DUX4 induces a transcriptome more characteristic of a less-differentiated cell state and inhibits myogenesis. J Cell Sci. (2016) ;129: (20):3816–31. 10.1242/jcs.180372. |
[30] | Dixit M , Ansseau E , Tassin A , Winokur S , Shi R , Qian H , et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc Natl Acad Sci U S A. (2007) ;104: (46):18157–62. 10.1073/pnas.0708659104. |
[31] | Bosnakovski D , Xu Z , Gang EJ , Galindo CL , Liu M , Simsek T , et al. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. Embo J. (2008) ;27: (20):2766–79. 10.1038/emboj.2008.201. |
[32] | Winokur ST , Barrett K , Martin JH , Forrester JR , Simon M , Tawil R , et al. Facioscapulohumeral muscular dystrophy (FSHD) myoblasts demonstrate increased susceptibility to oxidative stress. Neuromuscul Disord. (2003) ;13: (4):322–33. 10.1016/s0960-8966(02)00284-5. |
[33] | Yao Z , Snider L , Balog J , Lemmers RJ , Van Der Maarel SM , Tawil R , et al. DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Hum Mol Genet. (2014) ;23: (20):5342–52. 10.1093/hmg/ddu251. |
[34] | Tawil R , McDermott MP , Pandya S , King W , Kissel J , Mendell JR , et al. A pilot trial of prednisone in facioscapulohumeral muscular dystrophy. FSH-DY Group. Neurology. (1997) ;48: (1):46–9. |
[35] | Kissel JT , McDermott MP , Mendell JR , King WM , Pandya S , Griggs RC , et al. Randomized, double-blind, placebo-controlled trial of albuterol in facioscapulohumeral dystrophy. Neurology. (2001) ;57: (8):1434–40. |
[36] | van der Kooi EL , Kalkman JS , Lindeman E , Hendriks JC , van Engelen BG , Bleijenberg G , et al. Effects of training and albuterol on pain and fatigue in facioscapulohumeral muscular dystrophy. J Neurol. (2007) ;254: (7):931–40. 10.1007/s00415-006-0432-4. |
[37] | van der Kooi EL , Vogels OJ , van Asseldonk RJ , Lindeman E , Hendriks JC , Wohlgemuth M , et al. Strength training and albuterol in facioscapulohumeral muscular dystrophy. Neurology. (2004) ;63: (4):702–8. |
[38] | Wagner KR , Fleckenstein JL , Amato AA , Barohn RJ , Bushby K , Escolar DM , et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol. (2008) ;63: (5):561–71. 10.1002/ana.21338. |
[39] | Passerieux E , Hayot M , Jaussent A , Carnac G , Gouzi F , Pillard F , et al. Effects of vitamin C, vitamin E, zinc gluconate, and selenomethionine supplementation on muscle function and oxidative stress biomarkers in patients with facioscapulohumeral dystrophy: A double-blind randomized controlled clinical trial. Free Radic Biol Med. (2015) ;81: :158–69. 10.1016/j.freeradbiomed.2014.09.014. |
[40] | Pearsall RS , Davies MV , Cannell M , Li J , Widrick J , Mulivor AW , et al. Follistatin-based ligand trap ACE-083 induces localized hypertrophy of skeletal muscle with functional improvement in models of neuromuscular disease. Sci Rep. (2019) ;9: (1):11392. 10.1038/s41598-019-47818-w. |
[41] | Lim JW , Snider L , Yao Z , Tawil R , Van Der Maarel SM , Rigo F , et al. DICER/AGO-dependent epigenetic silencing of D4Z4 repeats enhanced by exogenous siRNA suggests mechanisms and therapies for FSHD. Hum Mol Genet. (2015) ;24: (17):4817–28. 10.1093/hmg/ddv206. |
[42] | Snider L , Asawachaicharn A , Tyler AE , Geng LN , Petek LM , Maves L , et al. RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: New candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum Mol Genet. (2009) ;18: (13):2414–30. 10.1093/hmg/ddp180. |
[43] | Balog J , Thijssen PE , Shadle S , Straasheijm KR , van der Vliet PJ , Krom YD , et al. Increased DUX4 expression during muscle differentiation correlates with decreased SMCHD1 protein levels at D4Z4. Epigenetics. (2015) ;10: (12):1133–42. 10.1080/15592294.2015.1113798. |
[44] | Goossens R , van den Boogaard ML , Lemmers R , Balog J , van der Vliet PJ , Willemsen IM , et al. Intronic SMCHD1 variants in FSHD: Testing the potential for CRISPR-Cas9 genome editing. J Med Genet. (2019) ;56: (12):828–37. 10.1136/jmedgenet-2019-106402. |
[45] | Zhang N . Epigenetic modulation of DNA methylation by nutrition and its mechanisms in animals. Anim Nutr. (2015) ;1: (3):144–51. 10.1016/j.aninu.2015.09.002. |
[46] | van der Kooi EL , de Greef JC , Wohlgemuth M , Frants RR , van Asseldonk RJ , Blom HJ , et al. No effect of folic acid and methionine supplementation on D4Z4 methylation in patients with facioscapulohumeral muscular dystrophy. Neuromuscul Disord. (2006) ;16: (11):766–9. 10.1016/j.nmd.2006.08.005. |
[47] | Campbell AE , Oliva J , Yates MP , Zhong JW , Shadle SC , Snider L , et al. BET bromodomain inhibitors and agonists of the beta-2 adrenergic receptor identified in screens for compounds that inhibit DUX4 expression in FSHD muscle cells. Skelet Muscle. (2017) ;7: (1):16. 10.1186/s13395-017-0134-x. |
[48] | Joassard OR , Durieux AC , Freyssenet DG . beta2-Adrenergic agonists and the treatment of skeletal muscle wasting disorders. Int J Biochem Cell Biol. (2013) ;45: (10):2309–21. 10.1016/j.biocel.2013.06.025. |
[49] | Oliva J , Galasinski S , Richey A , Campbell AE , Meyers MJ , Modi N , et al. Clinically Advanced p38 Inhibitors Suppress DUX4 Expression in Cellular and Animal Models of Facioscapulohumeral Muscular Dystrophy. J Pharmacol Exp Ther. (2019) ;370: (2):219–30. 10.1124/jpet.119.259663. |
[50] | Cuadrado A , Nebreda AR . Mechanisms and functions of p38 MAPK signalling. Biochem J. (2010) ;429: (3):403–17. 10.1042/BJ20100323. |
[51] | Segales J , Perdiguero E , Munoz-Canoves P . Regulation of Muscle Stem Cell Functions: A Focus on the p38 MAPK Signaling Pathway. Front Cell Dev Biol. (2016) ;4: :91. 10.3389/fcell.2016.00091. |
[52] | Engel FB , Schebesta M , Duong MT , Lu G , Ren S , Madwed JB , et al. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. (2005) ;19: (10):1175–87. 10.1101/gad.1306705. |
[53] | Ventura JJ , Tenbaum S , Perdiguero E , Huth M , Guerra C , Barbacid M , et al. p38alpha MAP kinase is essential in lung stem and progenitor cell proliferation and differentiation. Nat Genet.750-8. 10./ng. (2007) ;39: (6):750–8. 10.1038/ng2037. |
[54] | Newby LK , Marber MS , Melloni C , Sarov-Blat L , Aberle LH , Aylward PE , et al. Losmapimod, a novel p38 mitogen-activated protein kinase inhibitor, in non-ST-segment elevation myocardial infarction: A randomised phase 2 trial. Lancet. 384. (2014) ;384: (9949):1187–95. 10.1016/S0140-6736(14)60417-7. |
[55] | O’Donoghue ML , Glaser R , Cavender MA , Aylward PE , Bonaca MP , Budaj A , et al. Effect of Losmapimod on Cardiovascular Outcomes in Patients Hospitalized With Acute Myocardial Infarction: A Randomized Clinical Trial. JAMA. (2016) ;315: (15):1591–9. 10.1001/jama.2016.3609. |
[56] | Ostenfeld T , Krishen A , Lai RY , Bullman J , Baines AJ , Green J , et al. Analgesic efficacy and safety of the novel p38 MAP kinase inhibitor, losmapimod, in patients with neuropathic pain following peripheral nerve injury: A double-blind, placebo-controlled study. Eur J Pain. (2013) ;17: (6):844–57. 10.1002/j.1532-2149.2012.00256.x. |
[57] | Mellion M , Ronco L , Thompson D , Hage M , Brooks S , Van Brummelen E , et al. O.25Phase 1 clinical trial of losmapimod in FSHD: Safety, tolerability and target engagement.World Muscle Society. Neuromuscular Disorders; (2019) . p. S123. |
[58] | Available from: https://www.facio-therapies.com/wp-content/uploads/2019/09/Facio-WMS-October-2019.pdf.. |
[59] | Himeda CL , Jones TI , Jones PL . CRISPR/dCas9-mediated Transcriptional Inhibition Ameliorates the Epigenetic Dysregulation at D4Z4 and Represses DUX4-fl in FSH Muscular Dystrophy. Mol Ther. (2016) ;24: (3):527–35. 10.1038/mt.2015.200. |
[60] | Vanderplanck C , Ansseau E , Charron S , Stricwant N , Tassin A , Laoudj-Chenivesse D , et al. The FSHD atrophic myotube phenotype is caused by DUX4 expression. PLoS One. (2011) ;6: (10):e26820. 10.1371/journal.pone.0026820. |
[61] | McBride JL , Boudreau RL , Harper SQ , Staber PD , Monteys AM , Martins I , et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: Implications for the therapeutic development of RNAi. Proc Natl Acad Sci U S A. (2008) ;105: (15):5868–73. 10.1073/pnas.0801775105. |
[62] | Grimm D , Streetz KL , Jopling CL , Storm TA , Pandey K , Davis CR , et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. (2006) ;441: (7092):537–41. 10.1038/nature04791. |
[63] | Marsollier AC , Ciszewski L , Mariot V , Popplewell L , Voit T , Dickson G , et al. Antisense targeting of 3’ end elements involved in DUX4 mRNA processing is an efficient therapeutic strategy for facioscapulohumeral dystrophy: A new gene-silencing approach. Hum Mol Genet. (2016) ;25: (8):1468–78. 10.1093/hmg/ddw015. |
[64] | Chen JC , King OD , Zhang Y , Clayton NP , Spencer C , Wentworth BM , et al. Morpholino-mediated Knockdown of DUX4 Toward Facioscapulohumeral Muscular Dystrophy Therapeutics. Mol Ther. (2016) ;24: (8):1405–11. 10.1038/mt.2016.111. |
[65] | Ansseau E , Vanderplanck C , Wauters A , Harper SQ , Coppee F , Belayew A . Antisense Oligonucleotides Used to Target the DUX4 mRNA as Therapeutic Approaches in FaciosScapuloHumeral Muscular Dystrophy (FSHD). Genes (Basel).. (2017) ;8: (3). 10.3390/genes8030093. |
[66] | Lim KRQ , Maruyama R , Echigoya Y , Nguyen Q , Zhang A , Khawaja H , et al. Inhibition of DUX4 expression with antisense LNA gapmers as a therapy for facioscapulohumeral muscular dystrophy. Proc Natl Acad Sci U S A. (2020) ;117: (28):16509–15. 10.1073/pnas.1909649117. |
[67] | Wallace LM , Liu J , Domire JS , Garwick-Coppens SE , Guckes SM , Mendell JR , et al. RNA interference inhibits DUX4-induced muscle toxicity in vivo: Implications for a targeted FSHD therapy. Mol Ther. (2012) ;20: (7):1417–23. 10.1038/mt.2012.68. |
[68] | Klingler C , Ashley J , Shi K , Stiefvater A , Kyba M , Sinnreich M , et al. DNA aptamers against the DUX4 protein reveal novel therapeutic implications for FSHD. FASEB J. (2020) ;34: (3):4573–90. 10.1096/fj.201902696. |
[69] | Choi SH , Gearhart MD , Cui Z , Bosnakovski D , Kim M , Schennum N , et al. DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res. (2016) ;44: (11):5161–73. 10.1093/nar/gkw141. |
[70] | Bosnakovski D , da Silva MT , Sunny ST , Ener ET , Toso EA , Yuan C , et al. A novel P300 inhibitor reverses DUX4-mediated global histone H3 hyperacetylation, target gene expression, and cell death. Sci Adv. (2019) ;5: (9):eaaw7781. 10.1126/sciadv.aaw7781. |
[71] | Pandey SN , Lee YC , Yokota T , Chen YW . Morpholino treatment improves muscle function and pathology of Pitx1 transgenic mice. Mol Ther. (2014) ;22: (2):390–6. 10.1038/mt.2013.263. |
[72] | DeSimone AM , Leszyk J , Wagner K , Emerson CP Jr . Identification of the hyaluronic acid pathway as a therapeutic target for facioscapulohumeral muscular dystrophy. Sci Adv. (2019) ;5: (12):eaaw7099. 10.1126/sciadv.aaw7099. |
[73] | Bosnakovski D , Choi SH , Strasser JM , Toso EA , Walters MA , Kyba M . High-throughput screening identifies inhibitors of DUX4-induced myoblast toxicity. Skelet Muscle. (2014) ;4: (1):4. 10.1186/2044-5040-4-4. |
[74] | Tawil R , Padberg GW , Shaw DW , van der Maarel SM , Tapscott SJ , Participants FW . Clinical trial preparedness in facioscapulohumeral muscular dystrophy: Clinical, tissue, and imaging outcome measures 29-30 May 2015, Rochester, New York. Neuromuscul Disord. (2016) ;26: (2):181–6. 10.1016/j.nmd.2015.10.005. |
[75] | Tawil R , Shaw DW , van der Maarel SM , Tapscott SJ . Clinical trial preparedness in facioscapulohumeral dystrophy: Outcome measures and patient access: 8-9 April 2013, Leiden, The Netherlands. Neuromuscul Disord. (2014) ;24: (1):79–85. 10.1016/j.nmd.2013.07.009. |
[76] | Evangelista T , Wood L , Fernandez-Torron R , Williams M , Smith D , Lunt P , et al. Design, set-up and utility of the UK facioscapulohumeral muscular dystrophy patient registry. J Neurol. (2016) ;263: (7):1401–8. 10.1007/s00415-016-8132-1. |
[77] | Guien C , Blandin G , Lahaut P , Sanson B , Nehal K , Rabarimeriarijaona S , et al. The French National Registry of patients with Facioscapulohumeral muscular dystrophy. Orphanet J Rare Dis. (2018) ;13: (1):218. 10.1186/s13023-018-0960-x. |
[78] | Hilbert JE , Kissel JT , Luebbe EA , Martens WB , McDermott MP , Sanders DB , et al. If you build a rare disease registry, will they enroll and will they use it? Methods and data from the National Registry of Myotonic Dystrophy (DM) and Facioscapulohumeral Muscular Dystrophy (FSHD). Contemp Clin Trials. (2012) ;33: (2):302–11. 10.1016/j.cct.2011.11.016. |
[79] | Mul K , Kinoshita J , Dawkins H , van Engelen B , Tupler R , Consortium F . 225th ENMC international workshop:: A global FSHD registry framework, 18-20 2016, November Heemskerk, The Netherlands. Neuromuscul Disord. (2017) ;27: (8):782–90. 10.1016/j.nmd.2017.04.004. |
[80] | Statland JM , Tawil R . Risk of functional impairment in facioscapulohumeral muscular dystrophy. Muscle Nerve. (2014) ;49: (4):520–7. 10.1002/mus.23949. |
[81] | LoRusso S , Johnson NE , McDermott MP , Eichinger K , Butterfield RJ , Carraro E , et al. Clinical trial readiness to solve barriers to drug development in FSHD (ReSolve): Protocol of a large, international, multi-center prospective study. BMC Neurol. (2019) ;19: (1):224. 10.1186/s12883-019-1452-x. |
[82] | Mul K , Heatwole C , Eichinger K , Dilek N , Martens WB , Van Engelen BGM , et al. Electrical impedance myography in facioscapulohumeral muscular dystrophy: A 1-year follow-up study. Muscle Nerve. (2018) ;58: (2):213–8. 10.1002/mus.26127. |
[83] | Ferguson MR , Poliachik SL , Budech CB , Gove NE , Carter GT , Wang LH , et al. MRI change metrics of facioscapulohumeral muscular dystrophy: STIR and T1. Muscle Nerve. (2018) ;57: (6):905–12. 10.1002/mus.26038. |
[84] | Andersen G , Dahlqvist JR , Vissing CR , Heje K , Thomsen C , Vissing J . MRI as outcome measure in facioscapulohumeral muscular dystrophy: 1-year follow-up of 45 patients. J Neurol. (2017) ;264: (3):438–47. 10.1007/s00415-016-8361-3. |
[85] | Monforte M , Laschena F , Ottaviani P , Bagnato MR , Pichiecchio A , Tasca G , et al. Tracking muscle wasting and disease activity in facioscapulohumeral muscular dystrophy by qualitative longitudinal imaging. J Cachexia Sarcopenia Muscle. (2019) ;10: (6):1258–65. 10.1002/jcsm.12473. |
[86] | Fatehi F , Salort-Campana E , Le Troter A , Bendahan D , Attarian S . Muscle MRI of facioscapulohumeral dystrophy (FSHD): A growing demand and a promising approach. Rev Neurol (Paris). (2016) ;172: (10):566–71. 10.1016/j.neurol.2016.08.002. |
[87] | Petek LM , Rickard AM , Budech C , Poliachik SL , Shaw D , Ferguson MR , et al. A cross sectional study of two independent cohorts identifies serum biomarkers for facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul Disord. (2016) ;26: (7):405–13. 10.1016/j.nmd.2016.04.012. |
[88] | Statland J , Donlin-Smith CM , Tapscott SJ , van der Maarel S , Tawil R . Multiplex Screen of Serum Biomarkers in Facioscapulohumeral Muscular Dystrophy. J Neuromuscul Dis. (2014) ;1: (2):181–90. 10.3233/JND-140034. |
[89] | Yao G , Albon E , Adi Y , Milford D , Bayliss S , Ready A , et al. A systematic review and economic model of the clinical and cost-effectiveness of immunosuppressive therapy for renal transplantation in children. Health Technol Assess. (2006) ;10: (49):iii-iv, ix-xi, 1–157. |
[90] | Wang LH , Friedman SD , Shaw D , Snider L , Wong CJ , Budech CB , et al. MRI-informed muscle biopsies correlate MRI with pathology and DUX4 target gene expression in FSHD. Hum Mol Genet. (2019) ;28: (3):476–86. 10.1093/hmg/ddy364. |
[91] | Lunt PW . 44th ENMC International Workshop: Facioscapulohumeral Muscular Dystrophy: Molecular Studies 19-21 July Naarden, The Netherlands. Neuromuscul Disord. (1998) ;8: (2):126–30. 10.1016/s0960-8966(98)00012-1. |
[92] | Hewitt JE , Lyle R , Clark LN , Valleley EM , Wright TJ , Wijmenga C , et al. Analysis of the tandem repeat locus D4Z4 associated with facioscapulohumeral muscular dystrophy. Hum Mol Genet. (1994) ;3: (8):1287–95. 10.1093/hmg/3.8.1287. |
[93] | Lemmers RJ , de Kievit P , Sandkuijl L , Padberg GW , van Ommen GJ , Frants RR , et al. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat Genet. (2002) ;32: (2):235–6. 10.1038/ng999. |
[94] | Himeda CL , Debarnot C , Homma S , Beermann ML , Miller JB , Jones PL , et al. Myogenic enhancers regulate expression of the facioscapulohumeral muscular dystrophy-associated DUX4 gene. Mol Cell Biol. (2014) ;34: (11):1942–55. 10.1128/MCB.00149-14. |
[95] | Peart N , Wagner EJ . A distal auxiliary element facilitates cleavage and polyadenylation of Dux4 mRNA in the pathogenic haplotype of FSHD. Hum Genet. (2017) ;136: (9):1291–301. 10.1007/s00439-017-1813-8. |
[96] | Tupler R , Berardinelli A , Barbierato L , Frants R , Hewitt JE , Lanzi G , et al. Monosomy of distal 4q does not cause facioscapulohumeral muscular dystrophy. J Med Genet. (1996) ;33: (5):366–70. 10.1136/jmg.33.5.366. |
[97] | Lemmers RJL , de Kievit P , van Geel M , van der Wielen MJ , Bakker E , Padberg GW , et al. Complete allele information in the diagnosis of facioscapulohumeral muscular dystrophy by triple DNA analysis. Ann Neurol. (2001) ;50: (6):816–9. 10.1002/ana.10057. |
[98] | Wijmenga C , Sandkuijl LA , Moerer P , van der Boorn N , Bodrug SE , Ray PN , et al. Genetic linkage map of facioscapulohumeral muscular dystrophy and five polymorphic loci on chromosome 4q35-qter. Am J Hum Genet. (1992) ;51: (2):411–5. |
[99] | Zhang Y , Forner J , Fournet S , Jeanpierre M . Improved characterization of FSHD mutations. Ann Genet. (2001) ;44: (2):105–10. 10.1016/s0003-3995(01)01075-9. |


