
Targeted Therapies for Hereditary Peripheral Neuropathies: Systematic Review and Steps Towards a ‘treatabolome’
Abstract
Background:
Hereditary peripheral neuropathies are inherited disorders affecting the peripheral nervous system, including Charcot-Marie-Tooth disease, familial amyloid polyneuropathy and hereditary sensory and motor neuropathies. While the molecular basis of hereditary peripheral neuropathies has been extensively researched, interventional trials of pharmacological therapies are lacking.
Objective:
We collated evidence for the effectiveness of pharmacological and gene-based treatments for hereditary peripheral neuropathies.
Methods:
We searched several databases for randomised controlled trials (RCT), observational studies and case reports of therapies in hereditary peripheral neuropathies. Two investigators extracted and analysed the data independently, assessing study quality using the Oxford Centre for Evidence Based Medicine 2011 Levels of Evidence in conjunction with the Jadad scale.
Results:
Of the 2046 studies initially identified, 119 trials met our inclusion criteria, of which only 34 were carried over into our final analysis. Ascorbic acid was shown to have no therapeutic benefit in CMT1A, while a combination of baclofen, naltrexone and sorbitol (PXT3003) demonstrated some efficacy, but phase III data are incomplete. In TTR-related amyloid polyneuropathy tafamidis, patisiran, inotersen and revusiran showed significant benefit in high quality RCTs. Smaller studies showed the efficacy of L-serine for SPTLC1-related hereditary sensory neuropathy, riboflavin for Brown-Vialetto-Van Laere syndrome (SLC52A2/3) and phytanic acid-poor diet in Refsum disease (PHYH).
Conclusions:
The ‘treatable’ variants highlighted in this project will be flagged in the treatabolome database to alert clinicians at the time of the diagnosis and enable timely treatment of patients with hereditary peripheral neuropathies.
INTRODUCTION
Rare diseases (RD) are individually rare but collectively common, affecting 6– 8% of the global population [1]. Their rarity poses unique problems to clinicians and their patients, resulting in significant delays in both diagnosis and treatment. One problem is that clinicians dealing with the relevant patients are largely unaware or unable to keep up with the increasing evidence base associated with these conditions. Considering that approximately 72% of rare disease cases are caused by Mendelian genetic variants [2], an ability to accurately and quickly inform clinicians of existing gene and variant-specific therapeutics would represent a quantum leap in clinical care. The ‘treatabolome’ project aims to systematically collate and evaluate evidence for the effectiveness of rare disease treatments to provide a database of ‘treatable’ variants. Currently, as part of the multi-national ‘Solve-RD’ project, several expert-led teams across the rare disease community are working towards building that database. This knowledgebase will be integrated into an interoperable rare disease genomics platform to facilitate clinicians’ access to the latest clinical data and enable timely treatment of rare disease patients. The first element of this project has been completed and published for congenital myasthenic syndromes [3], and a standardised protocol to be applied across the different arms of the treatabolome project has been submitted. We now aim to undertake an element of this project to define the treatabolome of hereditary peripheral neuropathies.
Hereditary peripheral neuropathies are a group of inherited disorders affecting the peripheral nervous system. These are divided into four major subgroups: hereditary sensory and motor neuropathies, distal hereditary motor neuropathies, hereditary sensory and autonomic neuropathies and more complex hereditary neuropathies. The most common form is the hereditary sensory and motor neuropathy, also known as Charcot-Marie-Tooth disease (CMT), with a prevalence of around 1/2500 [4, 5]. CMT can be further subdivided into demyelinating and axonal forms, depending on their pathophysiology. In demyelinating forms, myelin sheath degeneration precedes axon degeneration, whereas axon degeneration is the primary pathology in axonal forms.
As of January 2020, 93 genes have been identified to cause CMT [6]. These genes cover a wide range of molecular functions and intracellular processes, which can be broadly categorised as affecting intracellular transport in the axon, organelle dynamics and protein synthesis (axonal CMT), and cellular processes causing disruption of myelin production or organisation (demyelinating CMT). Inheritance patterns include all modes of Mendelian inheritance, maternal inheritance (mitochondrial DNA variants) and more complex patterns.
All CMT-related disorders are characterised by a length-dependent degeneration of the peripheral neurons and differ by the groups of neurons affected, with CMT affecting both sensory and motor neurons. Motor symptoms include weakness, atrophy or deformity, and sensory symptoms include numbness, paraesthesia or pain [7]. Some may involve the autonomic nervous system, resulting in features like impaired sweating, postural hypotension or insensitivity to pain [7]. Patients may also present with other neurological symptoms (e.g. spastic paraparesis, ataxia, cranial nerve abnormalities) and have non-neurological features (e.g. cataract, scoliosis, cardiac symptoms). It is important to note that the phenotype may be overlapping across the disease subgroups [5].
The disease typically follows a slowly progressive course. The prognosis of individuals depends on their genotype, but it is modified by other so far unknown genetic, epigenetic and environmental factors [8]. Curative pharmacological treatment is currently only available for some subtypes of hereditary peripheral neuropathies, but supportive management is useful in all cases. Supportive treatments involve physical therapy, occupational therapy, genetic counselling, orthotics (splints, braces, walking aids) and surgery [9].
METHODS
The systematic review was designed using Cochrane Collaboration methodology [10], as guided by the treatabolome project principles for generating FAIR-compliant datasets (soon to be published). Studies of randomised and non-randomised controlled trials, observational studies, case series and case reports of pharmacological treatment for patients with genetically confirmed hereditary peripheral neuropathies were considered for inclusion in the full systematic review. Reviews including systematic and expert reviews were screened for relevant primary data not included in our original search. All appropriate outcome measures clearly indicating a positive, neutral or negative (toxic) response to treatment were considered, including change in measures of muscle strength, functional ability and clinical examination results. Studies were assessed for quality using the Oxford Centre for Evidence-Based Medicine (OCEBM) 2011 Levels of Evidence [11] in conjunction with the Jadad scale [12]. All steps were carried out by two investigators (A.L. and M.J.) independently and reviewed by the expert team lead (R.H.), with regular meetings to establish consensus.
Inclusion criteria
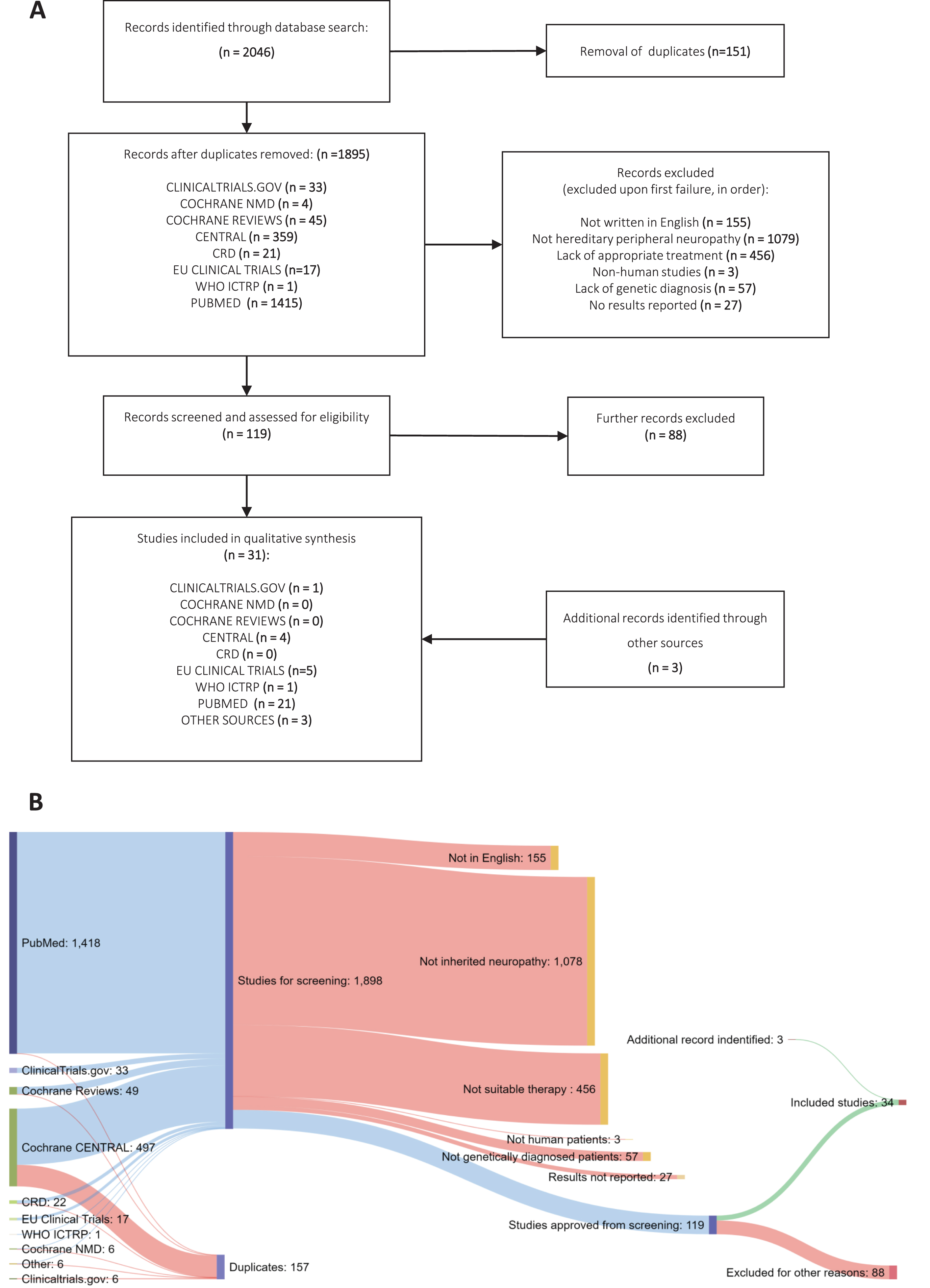
Fig.1
Systematic Review methodology. (A) PRISMA flowchart of studies identified under the search strategy, showing filtering steps to produce study database for codification of data (B) Sankey diagram showing numbers of studies from each source and progressive exclusion of studies according to inclusion criteria.
The condition studied was genetically confirmed hereditary peripheral neuropathy. We considered only studies involving patients with genetically confirmed mutations in known neuropathy genes. Hereditary peripheral neuropathies diagnosed by techniques other than formal genetic testing were excluded due to the phenotypic overlap between the different types of neuropathies, which could confound clinical or electrophysiological diagnoses. Participants with any level of severity, age of onset and level of penetrance of phenotypic features were included. Pharmacological therapies or genotype-related dietary changes specifically targeting the neuropathy were included in this study. Non-pharmacological interventions such as physiotherapy, surgery and genetic counselling were excluded.
Literature and trial database search
Several electronic databases were searched in order to cover all available evidence regarding pharmacological treatment for hereditary peripheral neuropathies. Firstly, to discover studies of the highest OCEBM evidence levels, we searched for randomised controlled trials and systematic reviews of genetic neuropathies using the Cochrane Central Register of Controlled Trials (CENTRAL), The Cochrane Neuromuscular Disease Group Specialized Register, ClinicalTrials.gov, European Clinical Trials Database (EudraCT), WHO International Clinical Trials Registry (ICTRP) and the Centre for Reviews and Dissemination (CRD) database. Secondly, we carried out a wider search using the PubMed database, intending to capture a greater range of relevant literature. Full details of search terms and filters used for each database can be seen in the Supplementary Table 1. Database specific filters were applied to ensure only interventional studies with results available were included, and that major “miss-hit” diseases (e.g. Guillain-Barré syndrome) were excluded. Additionally, a filter for publication date after 01/01/1991 was applied where necessary, as causative genes for inherited peripheral neuropathies were discovered after this date [13, 14].
Data selection
Studies identified from each database were compiled and screened for duplicates. Our inclusion criteria progressively determined studies (1) written in English, with (2) human patients, who were (3) genetically diagnosed with (4) hereditary peripheral neuropathy, involving a (5) pharmacological intervention and which (6) declare their results. Additionally, relevant articles were extracted from the reference sections of systematic or expert reviews and screened for possible inclusion as above.
Data extraction and analysis
Characteristics of each study that fulfilled our inclusion criteria were captured in a standardised data extraction table (Supplementary Table 2). Information was collected on participants (number of participants, phenotype, age, affected gene and variant), interventions (type, dose, delivery, frequency and duration of treatment) and both molecular (biomarker) and clinical (functional) outcome measures.
The quality of each study was assessed using both the OCEBM 2011 Levels of Evidence and the Jadad scale, to accommodate for smaller yet higher quality studies commonly seen in rare disease research. Data extraction was carried out by A.L. and M.J., prior to second assessment by R.H. As the OCEBM and Jadad scores incorporate risk of bias in their scoring, we did not carry out a formal risk of bias assessment. We must assume a high risk of bias for all non-randomised studies, and the OCEBM and Jadad scores are correspondingly low.
RESULTS
Our search identified 2043 potentially relevant entries across all databases (Fig. 1). Following duplicate analysis with manual confirmation and elimination of duplicates, 1892 individual studies remained. The titles and abstracts of these studies were then screened for inclusion using the stated inclusion criteria in progressive steps (see Data Selection). 119 studies met all the criteria on initial screening, whereas 1776 studies failed to satisfy at least one aspect of the criteria (Fig. 1). Next, studies which passed this stage were read in full by two assessors to confirm their suitability according to the criteria, excluding a further 88 studies. The characteristics and findings of these studies were recorded in full in Supplementary Table 2. At this stage, systematic reviews and expert reviews included in our search were examined for any additional primary data that was missed. Then, the expert team lead (R.H.) reviewed the process and the 34 studies remained for detailed analysis. Additionally, 1 study which was not included in the original dataset was flagged for inclusion [15], giving a total of 36 included studies. The final studies are presented as grouped by the causative genes involved (Tables 1– 3). For studies involving more than two different gene mutations, all described mutations are listed in Supplementary Table 3.
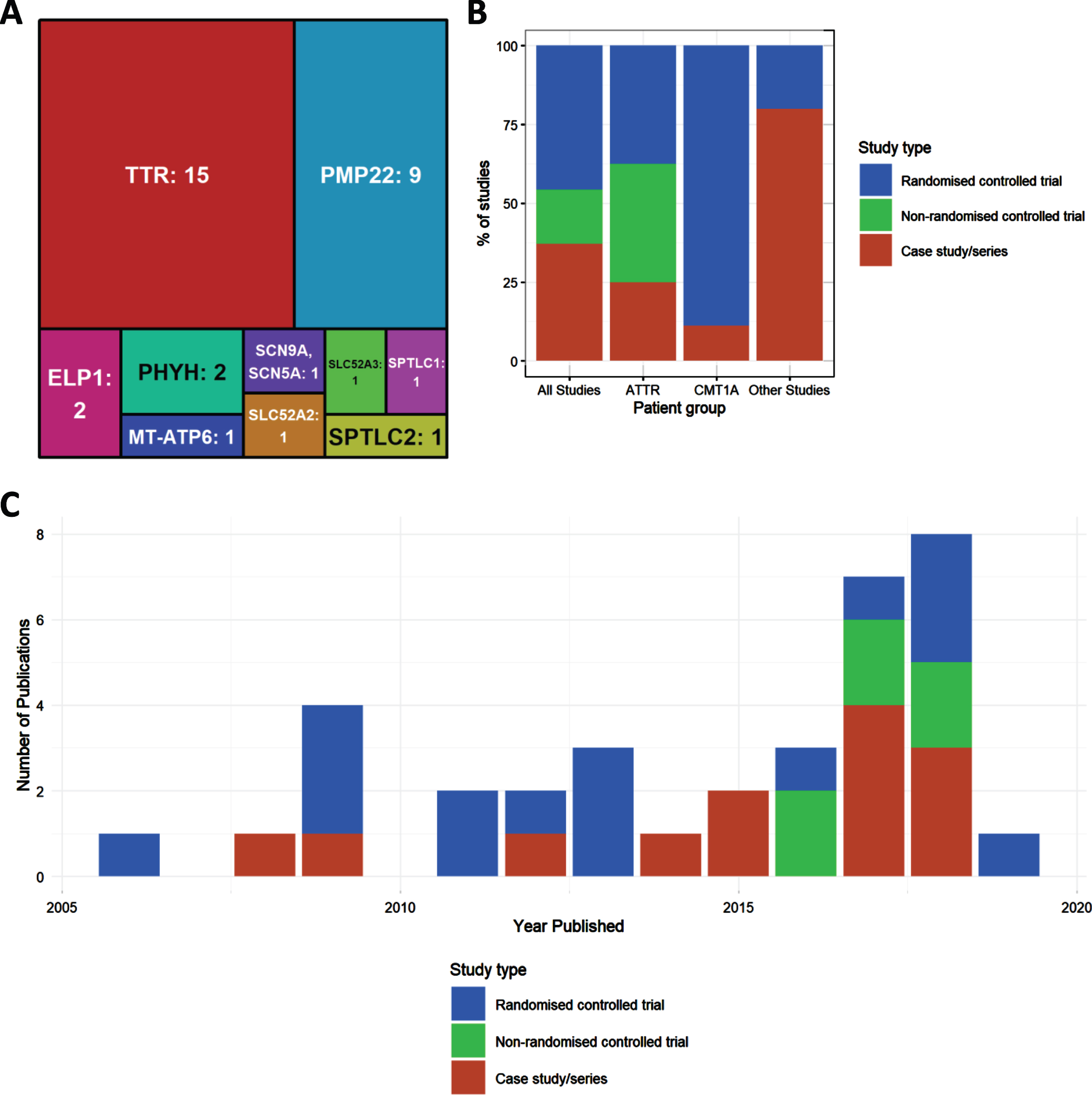
Of all included studies (n = 34) the largest two genetic groups were amyloid neuropathy caused by mutations in the TTR gene (n = 15) and CMT1A, due to the duplication of PMP22 (n = 8) and point mutation (n = 1) respectively. Numbers of studies were much fewer for other genotypes. Four studies looked at HSAN due to mutations in ELP1 (n = 2), SPTLC1 (n = 1) and SPTLC2 (n = 1). Two studies examined Brown-Vialetto-Van Laere Syndrome caused by mutation in SLC52A2 (n = 1) or SLC52A3 (n = 1). The genes MT-ATP6 (n = 1) and SCN9A (n = 1) each had 1 study. The remaining studies looked at neuropathies due to metabolic causes associated with mutations in PHYH (Refsum disease, n = 2).
Randomised controlled clinical trials
We identified 18 randomised controlled trials (RCTs). The most investigated treatment was oral ascorbic acid for CMT1A, with 6 independent studies. The studies varied in dose between 1.5 g and 4 g per day, for either 1 or 2 years. Most studies used the Charcot-Marie-Tooth Neuropathy Score (CMTNS) [16] as the primary outcome measure, with the possible addition of muscle strength, sensory or nerve conduction studies. However, none resulted in a statistically significant clinical improvement. Likewise, measurements of target effect (e.g. PMP22 mRNA levels) showed no changes on treatment with ascorbic acid. On quality assessment, all ascorbic acid RCTs were scored at 5 according to the Jadad criteria, indicating that these were well designed and properly reported studies. Two studies investigated the use of PTX3003, a combination therapy of baclofen, naltrexone and sorbitol, to treat CMT1A. These were large phase II and III studies, both scoring 5 by Jadad criteria. The phase II study showed no change in Overall Neuropathy Limitation Scale (ONLS) [16] in the group treated with low doses, but a modest improvement in the highest dose group. This result prompted further investigation of the treatment in a phase III study. However, inconsistencies were found in the stability of the high dose formulations in the phase III study, which led to early termination of this arm of the study. Despite this, there was a small, significant improvement in neuropathy severity (ONLS) prior to termination. We conclude that although there is some evidence that PTX3003 is effective in CMT1A, doubts remain about the validity of the incomplete data from this study.
We identified 6 RCTs with four different compounds (tafamidis, diflunisal, patisiran, inotersen) which showed positive results in ATTR-familial amyloid polyneuropathy [17]. Two of these investigated the treatment of the common c.148 G>A, p.Val50Met (previously called p.Val30Met) variant with tafamidis, a TTR stabiliser drug preventing TTR protein complex dissociation and thereby inhibiting amyloid formation [18, 19]. Both studies used 20 mg oral tafamidis daily for 18 months. The larger of these studies (n = 128) [18] did not show a significant improvement in the primary endpoints of Neuropathy Impairment Score in the Lower Limbs (NIS-LL) or Total Quality of life (TQOL), although some improvement was shown in neurological impairment (evidenced by reduced muscle weakness overall, specifically at the distal muscle sites). The smaller study (n = 63), however, did show a significant improvement in NIS-LL [19]. One other study in 130 patients treated over 2 years with diflunisal, a non-steroid anti-inflammatory drug which has been shown to stabilise TTR, reduced the rate of progression of neurological impairment and preserved quality of life compared to placebo [20].
The third study (APOLLO) examined the effect of patisiran, a double-stranded synthetic ribonucleic acid molecule (RNAi) that targets mutant and wild type TTR, after 18 months treatment in patients carrying the c.148 G>A (p.Val50Met) (∼50% of patients) or other TTR mutations [21, 22]. There was an improvement detected by the modified Neuropathy Impairment Score (mNIS+7).
Inotersen, a single-stranded deoxynucleotide analogue (DNA) complementary to TTR pre-mRNA has also been implemented to silence TTR in a 15-month multi-centre RCT (NEURO-TTR) in 172 patients with TTR-related peripheral neuropathy, of which approximately 50% had the p.Val50Met mutation [23]. This trial met both of its primary endpoints, as there was significantly less decline in the neuropathy and quality of life measures in the inotersen group compared with the placebo group.
Table 1
Summary of results in Charcot-Marie-Tooth type 1A (CMT1A)
| Gene, | Title | First Author, | Phenotype | Study | Number of | Drug | Duration | Target effect | Clinical effect | JADAD | OCEBM | UK | ||
| Mutation | Journal | target HPO | type | patients | name | (+/– / =), | (+/– / =), | score | score | License | ||||
| (Year) | term(s) | description | description | (1– 5) | (1– 5) | (Y/N) | ||||||||
| PMP22, dup | Ascorbic acid for CMT type 1A in children: a randomised, double-blind, placebo-controlled, safety and efficacy trial | Burns, Lancet Neurol (2009) [30] | Abnormal nerve conduction velocity | RCT | 81 (42 intervention, 39 placebo) | Ascorbic acid | 1 year | + | ↑plasma ascorbic acid concentration | = | No significant change in median NCV, strength, function, or QoL | 5 | 2 | *N |
| PMP22, dup | Effect of ascorbic acid in patients with CMT type 1A: a multicentre, randomised, double-blind, placebo-controlled trial | Micallef, Lancet Neurol (2009) [36] | Muscle weakness, Distal sensory impairment | RCT | 179 (intervention 61 dose 3 g, 56 dose 1 g, 62 placebo) | Ascorbic acid | 1 year | Not described | = | No significant change in CMTNS | 5 | 2 | *N | |
| PMP22, dup | Oral high dose ascorbic acid treatment for one year in young CMT1A patients: a randomised, double-blind, placebo-controlled phase II trial | Verhamme, BMC Med (2009) [37] | Muscle weakness, Distal sensory impairment | RCT | 13 (6 intervention, 7 placebo) | Ascorbic acid | 1 year | Not described | = | No significant change in median NCVs, strength, sensation, CMTNS or disability | 5 | 2 | *N | |
| PMP22, dup | Randomised controlled trial with ascorbic acid in CMT type 1A: results of the CMT-TRIAAL/CMT-TRAUK Ascorbic Acid in Charcot-Marie-Tooth Disease Type 1A (CMT-TRIAAL and CMT-TRAUK): A Double-Blind Randomised Trial | Pareyson, JPNS (2011) Pareyson, Lancet neurol (2011) [36] | Muscle weakness, Distal sensory impairment | RCT | 277 (138 intervention, 133 placebo) | Ascorbic acid | 24 months | Not described | = | No significant change in CMTNS | 5 | 2 | *N | |
| PMP22, dup | High-dosage ascorbic acid treatment in CMT type 1A: results of a randomized, double-masked, controlled trial | Lewis, JAMA Neurol (2013) [37] | Muscle weakness, Distal sensory impairment | RCT | 85 (69 intervention, 16 placebo) | Ascorbic acid | 2 years | = | No change in PMP22 mRNA levels | = | No significant change in CMTNS | 5 | 2 | *N |
| PMP22, dup | Phase II, Randomized, Placebo-controlled Trial in Patients With CMT type 1A | ClinicalTrials.gov Identifier: NCT01401257 (2017) [38] | Muscle weakness, Distal sensory impairment | RCT | 80 | PXT3003 | 12 months | Not described | + | Decreased CMTNS &ONLS in high dose group | 5 | 2 | N | |
| PMP22, dup | A multicenter, double-blind, placebo controlled, pivotal phase III study (PLEOCMT) of a fixed combination of baclofen, naltrexone and sorbitol (PXT3003), for the treatment of CMT1A | Attarian, Muscle &nerve (2017) NCT02579759 (2020) [39] | Muscle weakness, Distal sensory impairment | RCT | 323 | PXT3003 | 15 months | Not described | +/= | Significant ↓ONLS for dose 2, non-significant ↓ONLS for dose 1 | 5 | 2 | N | |
| PMP22, dup | Effects of exercise and creatine on myosin heavy chain isoform composition in patients with Charcot-Marie-Tooth disease | Smith, Muscle Nerve (2006) [40] | Muscle weakness | RCT | 18 | Creatine monohydrate + resistance training | 12 weeks | + | Significant decrease in MHC type I | + | ↓Chair-rise-time, ↑muscle strength | 5 | 2 | N |
Summary of results in Charcot-Marie-Tooth type 1A (CMT1A). Key: + positive change, - negative change, = no change, NCV nerve conduction velocity, RCT randomised controlled trial, CMTNS Charcot-Marie-Tooth Neuropathy Score, ONLS Overall Neuropathy Limitations Scale, QoL Quality of Life, *Licensed for other indication.
Two studies looked at treatment options in hereditary sensory and autonomic neuropathies (HSAN). Treatment with L-serine in 18 patients with SPTLC1 variants resulted in significant decrease in CMTNS scores compared to placebo. Additionally, there was a corresponding decrease in deoxysphinganine levels, confirming target effect.
Non-randomised trials
We identified 5 non-randomised trials, all of which were open-label extensions of RCTs in patients with TTR-related neuropathy treated with patisiran, tafamidis and revusiran. These resulted in useful data on the long-term impact of these treatments. The APOLLO phase II trial showed a significant improvement in neuropathy in p.Val50Met patients following treatment with patisiran for 24 months [24], and formed the basis for the APOLLO phase III RCT, which gives better evidence of the efficacy of patisiran. Another trial (Pfizer) investigated the use of tafamidis in the non-p.Val50Met subpopulation of patients and demonstrated TTR stabilisation, but it did not investigate changes in the neuropathy phenotype [25]. Revusiran treatment (Alnylam Pharmceuticals) was administered in patients who had previously received liver transplants, but the study was terminated due to increased mortality rate in the treated patients [26], similar to another study in TTR-related cardiomyopathy [27]. The increased mortality was unlikely to be related to the treatment, but rather the effect of studying an older and more severely affected patient population.
Case series
Table 2
Summary of results in Transthyretin Familial amyloid polyneuropathy (TTR-FAP)
| Gene, Mutation | Title | First Author, | Phenotype | Study | Number | Drug | Duration | Target effect | Clinical effect | JADAD | OCEBM | UK | ||
| (RefSeq) (*see | Journal | target HPO | type | of | name | (+/– / =) & | (+/– / =) & | score | score | License | ||||
| Supplement 3 | (Year) | term(s) | Patients | description | description | (1– 5) | (1– 5) | (Y/N) | ||||||
| for other | ||||||||||||||
| mutations) | ||||||||||||||
| TTR, c.148 G>A, p.Val50Met (NM_000371.3) +*21 other mutations | Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial | Berk, JAMA (2013) [20] | Muscle weakness, Distal sensory impairment | RCT | 130 (71 V30M, 59 other mutations) | Diflunisal | 2 years | Not described | + | ↑NIS+7, ↓Progression of neuropathy, ↑QoL (↓SF-36 physical, ↑SF-36 mental) | 1 | 2 | *N | |
| TTR, c.148 G>A, p.Val50Met (NM_000371.3) | Tafamidis for Transthyretin Familial Amyloid Polyneuropathy: A Randomized, Controlled Trial | Coelho, Neurology (2012) [18] | Muscle weakness, sensory impairment | RCT | 128 | Tafamidis | 18 months | + | TTR stabilisation | =/+ | No change in NIS-LL or total QoL, ↓progression neuropathy | 5 | 2 | Y |
| TTR, c.148 G>A, p.Val50Met (NM_000371.3) +*8 other mutations | Long-term safety and efficacy of tafamidis for the treatment of hereditary transthyretin amyloid polyneuropathy: results up to 6 years | Barroso, Amyloid (2017) [41] | Muscle weakness, Distal sensory impairment | Open label extension study | 93 (75 V30M + 18 non-V30M) | Tafamidis | 6 years | Not described | =/+ | Significant delay neuropathy progression | 1 | 3 | Y | |
| TTR, c.224T>C, p.Leu75Pro (NM_000371.3) | Effects of liver transplantation and tafamidis in hereditary transthyretin amyloidosis caused by transthyretin Leu55Pro mutation: a case report | Kon, Amyloid (2015) [42] | Muscle weakness, Distal sensory impairment, Cardiomyopathy | Case study | 1 | Tafamidis | 1 year | Not described | =/+ | ↓Progression of polyneuropathy &cardiac amyloidosis | 0 | 4 | Y | |
| TTR, c.148 G>A, p.Val30Met (NM_000371.3) +*8 other mutations | Monitoring effectiveness and safety of Tafamidis in transthyretin amyloidosis in Italy: a longitudinal multicenter study in a non-endemic area | Cortese, J Neurol (2016) [43] | Muscle weakness, Distal sensory impairment, Cardiomyopathy | Cohort study | 61 | Tafamidis | 3 years | Not described | =/+ | No effect in V30M, ↓neuropathy progression in non-V30M &high disability | 1 | 3 | Y | |
| TTR, c.173A>C, p.Asp58Ala (NM_000371.3) | Tafamidis for the Treatment of Hereditary Transthyretin Amyloid Cardiomyopathy: A Case Report | Fujita, Cardiology (2017) [44] | Muscle weakness, Distal sensory impairment, Cardiomyopathy | Case study | 1 | Tafamidis meglumine | 2 years | Not described | =/– | No change in nerve conduction (axonopathy), progression of amyloid cardiomyopathy | 0 | 4 | Y | |
| TTR, c.290 C>A, p.Ser97Tyr (NM_000371.3) + *13 other mutations | The Effects of Fx-1006A on Transthyretin Stabilization and Clinical Outcome Measures in Patients with Non-V30M Transthyretin Amyloidosis | EudraCT number: 2007-006791-12 (2016) [25] | Muscle weakness, Distal sensory impairment, Cardiomyopathy | Open label study | 21 | Tafamidis | 43– 76 years | + | TTR stabilisation | N/A | 1 | 3 | *N | |
| TTR, c.401A>G, p.Tyr134Cys (NM_000371.3) | Transthyretin-related hereditary amyloidosis in an Argentinian family with TTR Tyr114Cys mutation | M. A. Aguirre, Amyloid (2017) [45] | Muscle weakness, Distal sensory impairment, Cardiomyopathy | Case study | 1 | Tafamidis | 65 years | Not described | N/A | Discontinued due to adverse effects | 0 | 4 | Y | |
| TTR, c.148 G>A, p.Val30Met (NM_000371.3) | Safety and Efficacy of Orally Administered Fx-1006A in Patients with Familial Amyloid Polyneuropathy (FAP): a Phase II/III, Randomised, Double-Blind, Placebo-Controlled Study | EudraCT number: 2006-002792-41 (2009) [19] | Muscle weakness, Distal sensory impairment, Cardiomyopathy | RCT | 63 | Tafamidis | 25– 74 years | Not described | + | ↓change from baseline NSLL LSMean score | 3 | 2 | Y | |
| TTR (V30M and others) | The Effect Of Tafamidis For The Transthyretin Amyloid Polyneuropathy Patients With V30M Or Non-V30M Transthyretin | Clinicaltrials.gov Identifier: NCT01435655 (2015) | Muscle weakness, Distal sensory impairment, Cardiomyopathy | RCT | 10 | Tafamidis | 20– 75 years | Not described | = | No observable change in clinical outcomes | 0 | 4 | Y | |
| TTR, c.148 G>A, p.Val30Met (NM_000371.3) + *38 other mutations | APOLLO: A Phase 3 Multicenter, Multinational, Randomized, Double-blind, Placebo-controlled Study to Evaluate the Efficacy and Safety of Patisiran (ALN-TTR02) in Transthyretin (TTR)-Mediated Polyneuropathy (Familial Amyloidotic Polyneuropathy-FAP) + Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis | EudraCT number: 2013-002987-17 (2017) [22] Adams, Neurology (2018) [21] | Muscle weakness, Distal sensory impairment, Cardiomyopathy | RCT | 77 | Patisiran | 24– 83 years | + | ↓serum TTR | + | ↓change from baseline in mNIS+7 neuropathy score, ↓change from baseline in Norfolk QOL-DN score | 5 | 2 | Y |
| TTR (Not described) | A Phase 2, Multicenter, Open-Label, Extension Study to Evaluate the Long-Term Safety, Clinical Activity, and Pharmacokinetics of ALN-TTR02 in Patients With Familial Amyloidotic Polyneuropathy Who Have Previously Received ALN-TTR02 | EudraCT number: 2013-001644-65 (2017) [24] | Muscle weakness, Distal sensory impairment, Cardiomyopathy | RCT | 27 | Patisiran | 18– 84 years | + | 92% decrease in serum TTR | + | Significant improvement in neuropathy | 1 | 3 | Y |
| TTR (Not described) | An Open-Label Study To Evaluate The Efficacy And Safety Of Revusiran In Patients With Transthyretin-Mediated Familial Amyloidotic Polyneuropathy With Disease Progression Post Orthotopic Liver Transplant | EudraCT number: 2015-002603-29 (2018) [26] | Muscle weakness, Distal sensory impairment, Cardiomyopathy | RCT | 12 | Revusiran | 18– 84 years | Not described | N/A | Study terminated | 1 | 3 | N | |
| TTR, c.148 G>A, p.Val30Met (NM_000371.3) + *26 other mutations | Inotersen Treatment for Patients With Hereditary Transthyretin Amyloidosis | M. D. Benson, N Engl J Med (2018) [23] | Muscle weakness, Distal sensory impairment, Cardiomyopathy | RCT | 172 | Inotersen | 18– 83 years | + | ↓serum TTR | + | ↓change from baseline in mNIS+7 neuropathy score, ↓change from baseline Norfolk QOL-DN score | 5 | 2 | Y |
Summary of results in Transthyretin Familial amyloid polyneuropathy (TTR-FAP). Key: + positive change, - negative change, = no change, NCV nerve conduction velocity, RCT randomised controlled trial, V30M p.Val30Met mutation, NIS-LL Neuropathy Impairment Score in the Lower Limbs, QoL Quality of Life, mNIS+7 modified Neuropathy Impairment Score (+7 neurophysiologic tests), SF-36 Short Form 36 Health Survey Questionnaire. (*licensed for other indications).
Fourteen case studies or case series were identified, all of which included only a few or single patients. These represented incidental findings on patients with unusual presentations or reported on treatments already established to be effective in larger studies (Tables 1–3). However, there were several interesting examples of seemingly effective genotype-specific targeted treatments of the molecular defect, which should be considered in patients with causative mutations in these genes. One case study highlighted a positive effect of riboflavin in a single patient with Brown-Vialetto-Van-Laere syndrome (BVVL) due to mutations in the riboflavin transporter (SLC52A2/3). Another study investigating riboflavin treatment for BVVL, not identified in literature but known to R.H., was also included [28]. This publication was not detected in our original search as it did not include search terms ‘genetic’ or ‘inherited’ in the title or abstract. High dose riboflavin was shown to have a positive effect on neurological symptoms in both SLC52A2 and SLC52A3 genotypes, and early treatment of these patients may prevent neurological deterioration [28].
Other targeted treatments with positive results include low phytanic acid diet in Refsum disease, an inborn error of metabolism in which high phytanic acid results in neurological symptoms [29, 30], and a case study with L-serine in a patient with HSAN due to SPTLC2 mutation decreased deoxysphinganine levels [31].
Several case studies were not included in Tables 1-3 due to case-specific reasons given below, as decided by the expert team lead (R.H.). Four case studies investigating immune therapies were excluded, as they were thought to be treating secondary immune-mediated neuropathies rather than the primary hereditary neuropathy and were therefore not gene specific. Furthermore, these treatments may even be harmful in hereditary neuropathies. Another case study of potential benefit was reported in a patient with neuropathy caused by docosahexaenoic acid (DHA) deficiency, which was treated with cod liver oil (high levels of DHA), however the causative mutation was not identified [32]. Four other studies were excluded on the basis of treating non-gene specific symptoms of neuropathies, such as foot deformity in CMT1A and CMT4A, erectile dysfunction in TTR and fatigue in CMT1A. One uncontrolled trial investigating algasidase alfa in Fabry disease and another study on treatment with betain, folinic acid, B12 and B6 vitamins to restore the folate pathway in MTHFR mutations were excluded because these are complex diseases and cannot be listed as a specific neuropathy treatment.
Table 3
Summary of results in other CMT subtypes and metabolic neuropathies
| Gene, Mutation | Descriptive | First Author, | Phenotype | Study type | Drug | Target effect | Clinical effect | JADAD | OCEBM | ||
| (Ref Seq) (*see | name | Journal | target HPO | (number of | name | (+/– / =) & | (+/– / =) & | score | score | ||
| Supplement 3 | (Year) | term(s) | patients) | description | description | (1– 5) | (1– 5) | ||||
| for other | |||||||||||
| mutations) | |||||||||||
| ELP1, c.2204 + 6T>C, splice site mutation (NM_003640.5) | HSAN3 | Norcliffe-Kaufmann, Neurology (2013) [46] | Nausea &vomiting | RCT (12) | Carbidopa | Not described | + | Decreased nausea &retching | 5 | 2 | |
| ELP1, c.2204 + 6T>C, splice site mutation (NM_003640.5) | HSAN3 | Bar-Aluma, Lung (2018) [47] | Functional respiratory abnormality | RCT (14) | Albuterol sulfate, Ipratropium bromide | Not described | + | Increase in FEV1 &FVC, decrease in airway resistance, decrease in airway obstruction | 5 | 4 | |
| MT-ATP6, m.9185T>C, p.Leu220Pro (NC_012920.1) | CMT2 | Panosyan, Muscle Nerve (2017) [48] | Episodic flaccid weakness | Case series (2) | Acetazolamide | Not described | + | Decreased frequency &severity of episodic weakness | 0 | 4 | |
| PHYH, c.830 C>A, p.Ala277Glu (NM_001037537.1) | Refsum disease | Kohlschutter, J Child Neurol (2012) [30] | Muscle weakness, Distal sensory impairment, Visual impairment | Case study (1) | Phytanic acid-poor diet + extracorporeal lipid apharesis | + | Decrease in blood phytanic acid levels | + | Decrease in neurological &ophthalmological disease progression | 0 | 4 |
| PHYH, c.135-2A>G, splice site mutation (NM_006214.4) | Refsum disease | Finsterer, J Neurol Sci (2008) [29] | Not described (Peripheral neuropathy?) | Case study (1) | Phytanic acid-poor diet (Chelsea diet) | + | Slight decrease in blood phytanic acid levels | + | Decrease in symptoms (subjective) | 0 | 4 |
| SCN9A, c.2428 G>A, p.Val810Met (NM_002977.3) | Painful peripheral neuropathy | Adi, Mol Pain (2018) [49] | Distal sensory impairment, Pain insensitivity | Case study (1) | Carbamazepine | + | Use-dependent channel inhibition | + | Decrease in burning pain | 0 | 4 |
| SLC52A3, c.639 G>C, p.Tyr213Ter + c.374 C>A, p.Thr125Asn (NM_001370085.1) | BVVL | Chaya, Semin Pediatr Neurol (2018) [31] | Muscle weakness | Case study (1) | Riboflavin | + | Increased EGRAC &riboflavin levels | +/= | Slight increase in motor function | 0 | 4 |
| SLC52A2, c.916 G>A, p.Gly306Arg (NM_001253815.2) +*7 other mutations | BVVL | Foley, Brain (2014) [28] | Patient I1: Muscle weakness, Functional respiratory abnormality; Patient E1: Same as I1 + Visual impairment, Hearing impairment | Case series (2) | Riboflavin | + | Decreased (normalised) acylcarnitine levels | + | Patient I1: Increased motor &respiratory function in patient I1; Patient E1: Same as I1 + decreased visual &hearing impairment in patient | 1 | 4 |
| SPTLC1, N/A | HSAN1 | Fridman, Neurology (2019) [50] | Muscle weakness, Distal sensory impairment | RCT (18) | L-serine | + | Significant decrease in deoxysphinganine levels | + | Significant decrease in CMTNS compared with placebo | 5 | 2 |
| SPTLC2, c.547 C>T, p.Arg183Trp (NM_004863.3) | HSAN1C | Auranen, Cold Spring Harb Mol Case Stud (2017) [51] | Muscle weakness, Distal sensory impairment | Case study (1) | L-serine | + | Decreased deoxysphinganine levels | = | Slight increase in CMTNS score | 0 | 4 |
| PMP22, c.215 C>T, p.Ser72Leu (NM_000304.4) | CMT3 | Burns, Pediatr Neurol (2009) [52] | Muscle weakness, Functional respiratory abnormality | Case study (1) | Curcumin | Not described | = | No change in neurophysiology, muscle strength, respiratory function, disability or QoL | 0 | 4 | |
Summary of results in other CMT subtypes and metabolic neuropathies. Key: HSAN3 Hereditary sensory and autonomic neuropathy type 3, CMT2 Charcot-Marie-Tooth type 2, CMT2 Charcot-Marie-Tooth type 3, MTHFR Methylenetetrahydrofolate reductase deficiency, HNPP Hereditary neuropathy with liability to pressure palsies, BVVL Brown-Violetto-Van-Laare syndrome, HSAN1 Hereditary Sensory and Autonomic Neuropathy type 1, HSAN1 C Hereditary Sensory and Autonomic Neuropathy type 1 C, DHA Docosahexaenoic acid, + positive change, - negative change, = no change, NCV nerve conduction velocity, CMAP compound muscle action potential, RCT randomised controlled trial, V50M p.Val50Met mutation, NIS-LL Neuropathy Impairment Score in the Lower Limbs, QoL Quality of Life, FEV1 forced expiratory volume in 1 second, FVC forced vital capacity, eGFR estimated glomerular filtration rate, EGRAC erythrocyte glutathione reductase activity coefficient, mNIS+7 modified Neuropathy Impairment Score (+7 neurophysiologic tests), *Licensed for other indications.
Distribution and relationships of Jadad and OCEBM scores
Both Jadad and OCEBM scores aim to give an estimate of the scientific credit of a study, illustrated by a very high degree of correlation between the two scores (Fig. 3B), with 34/47 studies belonging to 2 clusters of studies. Studies with low OCEBM (≤2) and high Jadad (5) are well conducted RCTs, while studies with high OCEBM (4) and low Jadad (0) represent case studies or case series lacking most of the desired features of a good clinical trial.
DISCUSSION
Fig.2
Characteristics of included studies. (A) Treemap showing number of studies by mutated gene. (B) Percentages of study type for studies overall, and then broken down by disease type (amyloid TTR neuropathy, ATTR; Charcot-Marie-Tooth type 1A, CMT1A; all other studies). (C) Distribution of publications by year, colours indicating study types.
We performed a systematic review to evaluate the evidence for pharmacological and gene-based treatments in inherited peripheral neuropathies. Our original search identified 1892 independent publications, of which 34 were included in our final analysis. The evidence for the efficacy of the studied treatments was weighted based on the quality of the clinical trials or case studies. We identified 15 treatments, which should be initiated in patients with some genetic forms of inherited neuropathies.
However, our study has some limitations. Firstly, we may have missed some papers reporting positive effect of a treatment as part of a larger study on novel disease genes or describing large cohorts of diverse patients. However, search terms including these would have produced many times more results, thereby precluding careful consideration of studies. The future inclusion of machine-readable variant-level genotype information in clinical trial registration and reporting would improve data collation in the rare disease field. Indeed, we had to add one paper based on our expertise in inherited neuropathies on riboflavin-responsive neuropathy (BVVL) [28, 31]. Secondly, although there are supporting data on the rationale and efficacy of some of these treatments, the degree of evidence is not satisfactory in many studies. The reasons include insufficient study design, inappropriate outcome measures, low number of participants and short study duration. Our review highlights these limitations and demonstrates that we must improve our standards in planning clinical trials. Careful consideration of study size and duration, number of participants, appropriate outcome measures and international collaboration (multi-centre studies) are essential to improve the development of treatments in inherited neuropathies. Thirdly, many studies did not give gene and variant data of study participants in sufficient detail, which meant they were of limited use to our study. Lastly, this database needs regular updating in the future, as the number of clinical trials and relevant case studies are steadily increasing in inherited peripheral neuropathies. This is a time intensive process and requires a dedicated expert team.
Most evidence has been gathered in CMT1A. Ascorbic acid has been extensively researched and proven to be ineffective in this disease subtype. More recently, a combination of baclofen, naltrexone and sorbitol (PXT3003) has shown significant improvement in CMT scores (CMTNS and ONLS) and may be a strong candidate for pharmacotherapy in the future. However, these results must first be confirmed in the phase III trial currently ongoing [1]. Therefore, based on the evidence presented in this review, no definitive recommendations can be made for first-line treatment at present.
In TTR-related amyloid neuropathy, four treatments (tafamidis, patisiran, inotersen and diflunisal) showed significant benefit in high quality RCTs and should be initiated in patients with pathogenic variants in TTR. These drugs have been approved in many different countries for treatment of TTR-related neuropathy. There are still uncertainties about what the long-term clinical benefits are, when to initiate treatment and how to incorporate these treatments into the current algorithms. The choice of treatment and time of commencement needs careful consideration in specialised centres [17], and is influenced by national regulatory and licensing guidelines [33]. Early diagnosis of this condition is essential for timely access to treatment. The availability of gene silencing treatments raises issues regarding genetic screening and management of asymptomatic individuals.
In other forms of CMT and metabolic neuropathies, treatment recommendations are based on a slightly weaker evidence base of case studies and smaller trials. A positive effect of L-serine supplementation in patients with HSAN due to mutations in SPTLC1 (and potentially SPTLC2) is emerging, and we highlighted the efficacy of targeted dietary supplementation and restriction in a few other rare genetic neuropathies (BVVL, Refsum and methylenetetrahydrofolate reductase deficiency). Therefore, if pathogenic mutations are identified in these genes, targeted treatments should be considered in clinical practice.
Fig.3
Meta-analysis of studies. (A) Comparison of OCEBM levels with Jadad scoring, the size of each point indicating the number of studies at that point, colour indicating the quality defined as the product of these two scores (lower OCEBM with higher Jadad being the highest quality studies). (B) Distribution of study outcomes, stratified by patient number and labelled by study type.
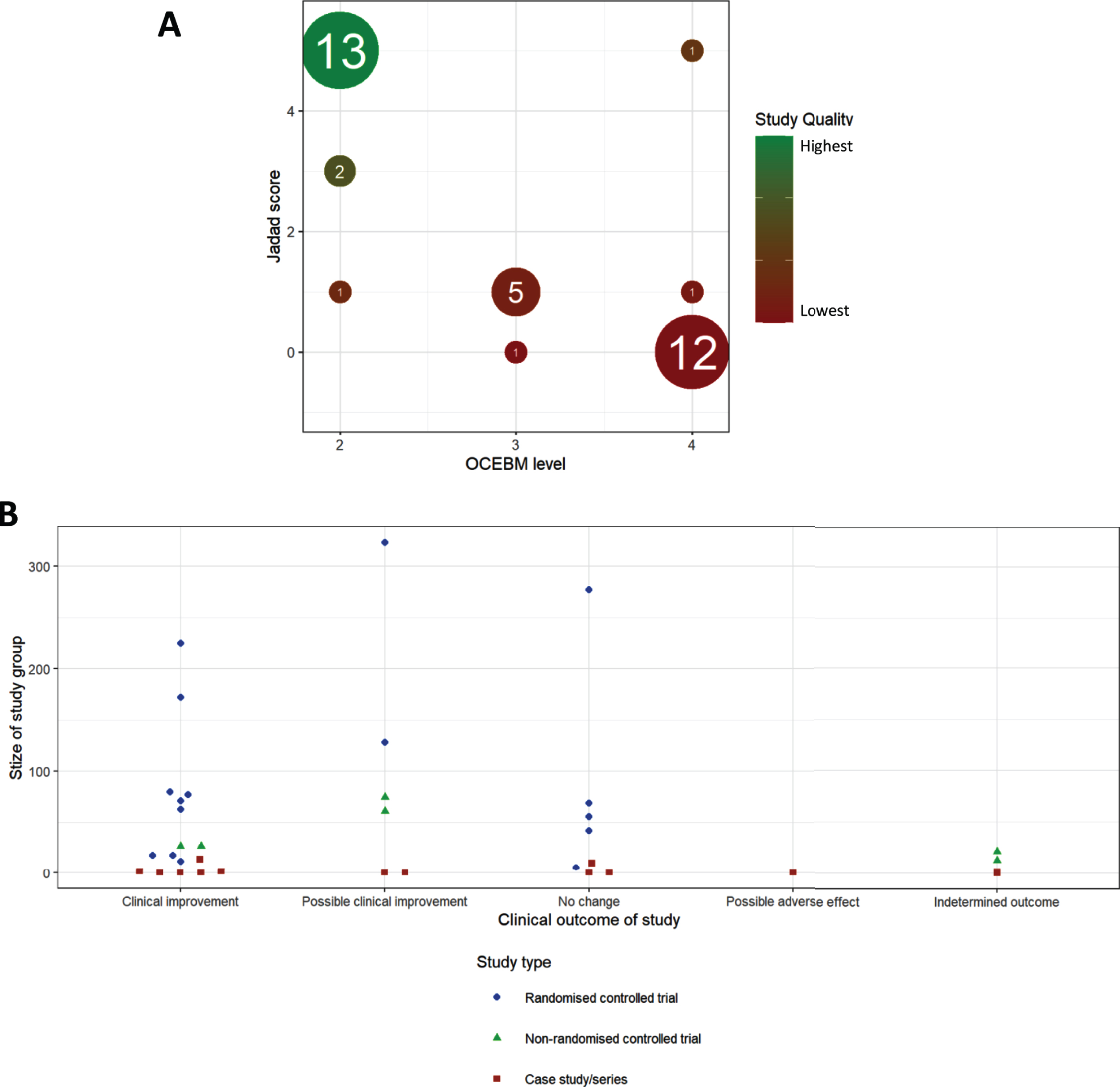
We detected an increase in the number of publications in recent years (Fig. 2B), although this increase was still driven mainly by case studies and case series. Well-designed RCTs were performed mostly in CMT1A and in amyloid neuropathy caused by TTR mutations (Fig. 2B). When plotting study size against clinical outcome (Fig. 3B), we observed that RCTs with the largest patient enrolments (n > 100) were more likely to demonstrate efficacy in TTR amyloid neuropathy or conclude lack of benefit in CMT1A. This reflects the greater statistical power of larger studies as compared to smaller studies. Case studies and small case series are more likely to report a clinical improvement, but this may be based on unsatisfactory evidence. For example, there is often reporting bias as unusual patient presentations are more likely to be reported. However, some case studies targeting the molecular defect in rare metabolic conditions should be applied to treat specific genetic neuropathies even without performing RCTs.
Although the number of effective treatments in inherited peripheral neuropathies is still very limited, the availability of large international patient cohorts, better outcome measures and more accurate natural history data will facilitate the development of further effective therapies. The recent identification of new genetic forms of CMT has launched research into developing novel therapies, which may in the future be added to the treatabolome database. Since the completion of our search, the discovery of mutations in SORD has unveiled how existing aldose reductase inhibitors have great potential to treat this common type of CMT [34]. Based on in vitro studies in patient-derived fibroblasts and in Drosophila, aldose reductase inhibitors normalized intracellular sorbitol levels and also ameliorated motor and eye phenotypes. These findings will form the basis for future clinical trials in patients with this form of CMT. Further progress is being made due to the technological advancements in molecular genetics, allowing scientists to generate diverse animal models that closely resemble the CMT phenotype. Additionally, it is now possible to culture patient-derived neurons in a dish using cellular reprogramming and differentiation techniques [35].
There is an ever-increasing need for data sharing in rare genetic diseases. This need is beginning to be met by the establishment and implementation of several large databases, which integrate next generation sequencing data with clinical phenotyping data (e.g. RD-CONNECT, DECIPHER, GENESIS, etc). These databases contain information on several thousands of patients and their family members, as well as being easily accessible to clinicians analysing their patients’ samples. On average, each human genome has approximately 30,000 variants compared to the standard assembly, which may be benign or pathogenic. For this reason, appropriate filtering for rare, causative variants and linking the available phenotypic information (HPO terms in RD-CONNECT) results in a higher diagnostic rate. Furthermore, to enable rapid translation from diagnosis to treatment, variants in the genes highlighted as part of the treatabolome project will be flagged in RD-CONNECT. This data will be immediately reported back to the clinician, together with the genetic diagnosis as determined by exome sequencing. Our work will allow this streamlined solution to be made available to clinicians dealing with inherited peripheral neuropathy patients. This approach facilitates clinicians’ access to the latest clinical data relevant to the genotype of their patients and enables timely treatment, ultimately leading to improved prognosis and a better quality of life of rare disease patients. The treatabolome project therefore offers a powerful tool, with significant advantages for both clinicians and patients with inherited peripheral neuropathies.
Funding
The authors of this study are supported by the Newton Fund UK/Turkey (MR/N027302/1 to RH), the Medical Research Council (UK) (MR/N025431/1 to RH), the Wellcome Investigator fund (109915/Z/15/Z to RH), the Lily Foundation UK (to RH), the European Research Council (309548 to RH) and the Evelyn Trust (19/14 to RH); and the Wellcome Trust Pathfinder Scheme (201064/Z/16/Z to RH). The study was further supported by the Horizon 2020 research and innovation program via grant 779257 “Solve-RD”, the RD-Connect Genome-Phenome Analysis platform developed under FP7/2007-2013 funded project (grant agreement nº 305444).). Authors of this publication are members of the European Reference Network for Rare Neuromuscular Diseases EURO-NMD (AA, RH).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JND-200546.
REFERENCES
[1] | Dawkins HJS , et al. Progress in Rare Diseases Research 2010-2016: An IRDiRC Perspective. Clin Transl Sci. (2018) ;11: (1):11–20. |
[2] | Nguengang Wakap S , Lambert DM . Estimating cumulative point prevalence of rare diseases: Analysis of the Orphanet database ((2020) ;28: (2):165–73. |
[3] | Thompson R , et al. Targeted therapies for congenital myasthenic syndromes: Systematic review and steps towards a treatabolome. Emerging Topics in Life Sciences. (2019) ;3: (1):19–37. |
[4] | Skre H . Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin Genet. (1974) ;6: (2):98–118. |
[5] | Bansagi B , et al. Genetic heterogeneity of motor neuropathies. Neurology. (2017) ;88: (13):1226–34. |
[6] | Benarroch L , et al. The 2020 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul Disord. (2019) ;29: (12):980–1018. |
[7] | Reilly MM , Murphy SM , Laura M . Charcot-Marie-Tooth disease. J Peripher Nerv Syst. (2011) ;16: (1):1–14. |
[8] | Bis-Brewer DM , Fazal S , Zuchner S . Genetic modifiers and non-Mendelian aspects of CMT. Brain Res. (1726) )146459. |
[9] | McCorquodale D , Pucillo EM , Johnson NE . Management of Charcot-Marie-Tooth disease: Improving long-term care with a multidisciplinary approach. J Multidiscip Healthc. (2016) ;9: :7–19. |
[10] | Higgins J J.P.T.T.J.C.J.C.M.L.T.P.M.J.W.V.A . Cochrane Handbook for Systematic Reviews of Interventions. Cochrane, version 6.0 (updated July 2019) availible from. www.training.cochrane.og/handbook. |
[11] | Group, O.L.o.E.W. The Oxford Levels of Evidence 2 2011 [cited 2019; Available from: https://www.cebm.net/index.aspx?o=5653. |
[12] | Jadad AR , et al. Assessing the quality of reports of randomized clinical trials: Is blinding necessary? Control Clin Trials ((1996) ;17: (1):1–12. |
[13] | Lupski JR , et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. (1991) ;66: (2):219–32. |
[14] | Raeymaekers P , et al. Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a). The HMSN Collaborative Research Group. Neuromuscul Disord. (1991) ;1: (2):93–7. |
[15] | FoleyAR, et al. Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain. (2014) ;137: (Pt 1):44–56. |
[16] | Graham RC , Hughes RAC . A modified peripheral neuropathy scale: The Overall Neuropathy Limitations Scale. Journal of Neurology, Neurosurgery, and Psychiatry. (2006) ;77: (8):973–6. |
[17] | S.A., Phase III Trial Assessing the Efficacy and Safety of PXT3003 in CMT1A Patients (PLEO-CMT) (PLEO-CMT). ClinicalTrials.gov, 2020. |
[18] | Berk JL , et al. Repurposing diflunisal for familial amyloid polyneuropathy: A randomized clinical trial. Jama. (2013) ;310: (24):2658–67. |
[19] | Aguirre MA , et al. Transthyretin-related hereditary amyloidosis in an Argentinian family with TTR Tyr114Cys mutation. Amyloid. (2017) ;24: (sup1):102. |
[20] | Smith CA , et al. Effects of exercise and creatine on myosin heavy chain isoform composition in patients with Charcot-Marie-Tooth disease. Muscle Nerve. (2006) ;34: (5):586–94. |
[21] | Alnylam Pharmaceuticals I . A Phase 2, Multicenter, Open-Label, Extension Study to Evaluate the Long-Term Safety, Clinical Activity, and Pharmacokinetics of ALN-TTR02 in Patients With Familial Amyloidotic Polyneuropathy Who Have Previously Received ALN-TTR02. EU Clinical Trials, (2017) . |
[22] | Limited FP . Safety and Efficacy of Orally Administered Fx-1006A in Patients with Familial Amyloid Polyneuropathy (FAP): A Phase II/III, Randomised, Double-Blind, Placebo-Controlled Study. EU Clinical Trials. (2009) . |
[23] | Norcliffe-Kaufmann L , et al. Hyperdopaminergic crises in familial dysautonomia: A randomized trial of carbidopa. Neurology. (2013) ;80: (17):1611–7. |
[24] | Alnylam Pharmaceuticals I . An Open-Label Study To Evaluate The Efficacy And Safety Of Revusiran In Patients With Transthyretin-Mediated Familial Amyloidotic Polyneuropathy With Disease Progression Post Orthotopic Liver Transplant. EU Clinical Trials 2018. |
[25] | Barroso FA , et al. Long-term safety and efficacy of tafamidis for the treatment of hereditary transthyretin amyloid polyneuropathy: Results up to 6 years. Amyloid. (2017) ;24: (3):194–204. |
[26] | Benson MD , et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med. (2018) ;379: (1):22–31. |
[27] | Burns J , et al. Effect of oral curcumin on Déjérine-Sottas disease. Pediatr Neurol. (2009) ;41: (4):305–8. |
[28] | Chaya S , et al. The First Case of Riboflavin Transporter Deficiency in sub-Saharan Africa. Semin Pediatr Neurol. (2018) ;26: :10–14. |
[29] | Kohlschütter A , et al. A child with night blindness: Preventing serious symptoms of Refsum disease. J Child Neurol. (2012) ;27: (5):654–6. |
[30] | Burns J , et al. Ascorbic acid for Charcot-Marie-Tooth disease type 1A in children: A randomised, double-blind, placebo-controlled, safety and efficacy trial. Lancet Neurol. (2009) ;8: (6):537–44. |
[31] | Adi T , et al. A novel gain-of-function Na(v)1. 7 mutation in a carbamazepine-responsive patient with adult-onset painful peripheral neuropathy. Mol Pain. (2018) ;14: :1744806918815007–. |
[32] | Alnylam Pharmaceuticals I . APOLLO: A Phase 3 Multicenter, Multinational, Randomized, Double-blind, Placebo-controlled Study to Evaluate the Efficacy and Safety of Patisiran (ALN-TTR02) in Transthyretin (TTR)-Mediated Polyneuropathy (Familial Amyloidotic Polyneuropathy-FAP). EU Clinical Trials (2018) . |
[33] | Kapoor M , et al. Clinical Presentation, Diagnosis and Treatment of TTR Amyloidosis. J Neuromuscul Dis. (2019) ;6: (2):189–99. |
[34] | Cortese A , et al. Biallelic mutations in SORD cause a common and potentially treatable hereditary neuropathy with implications for diabetes. Nat Genet. (2020) ;52: (5):473–81. |
[35] | Juneja M , et al. Challenges in modelling the Charcot-Marie-Tooth neuropathies for therapy development. J Neurol Neurosurg Psychiatry. (2019) ;90: (1):58–67. |
[36] | Micallef J , et al. Effect of ascorbic acid in patients with Charcot-Marie-Tooth disease type 1A: A multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. (2009) ;8: (12):1103–10. |
[37] | Verhamme C , et al. Oral high dose ascorbic acid treatment for one year in young CMT1A patients: A randomised, double-blind, placebo-controlled phase II trial. BMC Med. (2009) ;7: :70. |
[38] | Lewis RA , et al. High-dosage ascorbic acid treatment in Charcot-Marie-Tooth disease type 1A: Results of a randomized, double-masked, controlled trial. JAMA Neurol. (2013) ;70: (8):981–7. |
[39] | Attarian S , et al. Phase II, Randomized, Placebo-controlled Trial in Patients With Charcot-marie-tooth Disease Type 1A. ClinicalTrials.gov, (2014) . |
[40] | Pareyson D , et al. Ascorbic acid in Charcot-Marie-Tooth disease type 1A (CMT-TRIAAL and CMT-TRAUK): A double-blind randomised trial. Lancet Neurol. (2011) ;10: (4):320–8. |
[41] | Coelho T , et al. Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology. (2012) ;79: (8):785–92. |
[42] | Kon T , et al. Effects of liver transplantation and tafamidis in hereditary transthyretin amyloidosis caused by transthyretin Leu55Pro mutation: A case report. Amyloid. (2015) ;22: (3):203–4. |
[43] | Cortese A , et al. Monitoring effectiveness and safety of Tafamidis in transthyretin amyloidosis in Italy: A longitudinal multicenter study in a non-endemic area. J Neurol. (2016) ;263: (5):916–24. |
[44] | Fujita T , et al. Tafamidis for the Treatment of Hereditary Transthyretin Amyloid Cardiomyopathy: A Case Report. Cardiology. (2017) ;137: (2):74–77. |
[45] | Limited FP . The Effects of Fx-1006A on Transthyretin Stabilization and Clinical Outcome Measures in Patients with Non-V30M Transthyretin Amyloidosis. EU Clinical Trials. (2016) . |
[46] | Bar-Aluma BE , et al. A Controlled Trial of Inhaled Bronchodilators in Familial Dysautonomia. Lung. (2018) ;196: (1):93–101. |
[47] | Panosyan FB , Tawil R , Herrmann DN . Episodic weakness and Charcot-marie-tooth disease due to a mitochondrial MT-ATP6 mutation. Muscle Nerve. (2017) ;55: (6):922–7. |
[48] | Perna A , et al. Severe 5,10-methylenetetrahydrofolate reductase deficiency: A rare. Treatable Cause of Complicated Hereditary Spastic Paraplegia. (2018) ;25: (3):602–5. |
[49] | Finsterer J , Regelsberger G , Voigtländer T . Refsum disease due to the splice-site mutation c.135-2A>G before exon 3 of the PHYH gene, diagnosed eight years after detection of retinitis pigmentosa. J Neurol Sci. (2008) ;266: (1-2):182–6. |
[50] | Foley AR , et al. Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain. (2014) ;137: (Pt 1):44–56. |
[51] | Fridman V , et al. Randomized trial of l-serine in patients with hereditary sensory and autonomic neuropathy type 1. Neurology. (2019) ;92: (4):e359–e370. |
[52] | Auranen M , et al. Clinical and metabolic consequences of L-serine supplementation in hereditary sensory and autonomic neuropathy type 1C. Cold Spring Harb Mol Case Stud. (2017) ;3: (6). |


