
Diagnosing X-linked Myotubular Myopathy – A German 20-year Follow Up Experience
Abstract
X-linked myotubular myopathy (XLMTM) is a life-threatening rare neuromuscular disease, which is caused by pathogenic variants in the MTM1 gene. It has a large phenotypic heterogeneity, ranging from patients, who are able to walk independently to immobile patients who are only able to bring hand to mouth and depend on a respirator 24 hours a day every day. This suggests that ventilator requirements may not illustrate the full clinical picture of patients with XLMTM. At present, there is no curative therapy available, despite first promising results from ongoing gene therapy studies.
In this study, we evaluated in detail the data from 13 German XLMTM patients, which was collected over a period of up to 20 years in our university hospital. We compared it to the international prospective longitudinal natural history study (NHS) data from 45 patients (containing 11 German patients). To highlight the broad phenotypic spectrum of the disease, we additionally focused on the clinical presentation of three cases at a glance.
Comparing our data with the above mentioned natural history study, it appears the patients of the present German cohort seem to be more often severely affected, with higher frequency of non-ambulatory patients and patients on ventilation (and for longer time) and a higher proportion of patients needing a percutaneous endoscopic gastrostomy. Another key finding is a potential gap in time between first clinical presentation and final diagnosis, showing a need for patients to be treated in a specialized center for neuromuscular diseases.
INTRODUCTION
X-linked myotubular myopathy (XLMTM, OMIM #310400) is a rare neuromuscular disorder caused by pathogenic variants in the MTM1 gene with an estimated incidence of 1:40–50.000 in newborn males [1, 6, 7]. The first patient was described by A. Spiro in 1966 based on the clinical study of a 12-year-old boy presenting a general muscular weakness, ophthalmoplegia and ptosis [2].
The MTM1 gene encodes the ubiquitous phosphatase myotubularin. Myotubularin is involved in the phosphatidylinositol 3-kinase pathway that regulates intracellular membrane trafficking and vesicular transport processes [3, 4]. The underlying pathology leads to a noticeable muscle weakness and hypotonia of skeletal muscles, respiratory symptoms, external opthalmoplegia and a unique clinical appearance with a long forehead [7].
Based on the duration of ventilatory support, patients with XLMTM are classified into mild (no ventilatory support), intermediate or severe phenotype (ventilatory support < 12 hours/day and ≥ 12 hours/day) [3, 6]. Motor development, muscle weakness and ambulatory status are proposed additional items for classification into these categories [1, 4, 12]. Based on available classifications from literature [1, 6], 15% to 29% of patients from existing international cohorts could be classified as mild, 6–16% as moderate and 55–79% as severe.
In the neonatal period most of the patients present with hypotonia and respiratory insufficiency, leading to death due to respiratory failure in the first months of life. Although patients with milder forms usually present with delayed motor milestones and in the best case reach the ability to walk, the majority of patients remains non-ambulant and need ventilatory and feeding support. Approximately half of the patients do not survive the first year of life and only a quarter reached 10 years of life in a European cohort [1, 9]. The most common cause of death among patients with XLMTM is respiratory failure and cardiorespiratory arrest [1, 5, 8, 10]. No curative treatment is available for XLMTM, yet. Standard of care comprises a multidisciplinary approach with focus on respiratory and bulbar symptoms, e.g. ventilation support, management of infections and tube feeding.
Recently, an international prospective longitudinal natural history study was published, summarizing 45 patients with XLMTM, assessing motor and respiratory function [6]. In this article we present the German cohort, which was part of the above-mentioned study, with new analyses, complemented by two additional German patients, and compare our findings with those obtained in the whole cohort. To highlight the broad phenotypic spectrum of the disease, we present three illustrative cases at a glance.
METHODS
Patients and data
The study group consisted of 13 German XLMTM patients followed up at the Centre for Neuromuscular Diseases being part of the Department of Neuropediatrics of the Children’s Hospital at the University Children’s Hospital Essen between the years 2000 and 2020.
Clinical records were retrospectively reviewed for demographic data, such as age and gender, as well as for clinical features and other disease-related elements: age of first symptom, type of first symptom, age at diagnosis, age at tracheostomy, ventilatory support, motor milestone development, ambulatory status, feeding support, heart, liver and skeletal manifestation and other comorbidities and interventions, as well as carrier status of relatives and performance of a muscle biopsy.
Concerning patients 1–11 (see Table 1), data was extracted from study files of their participation in the international prospective and longitudinal natural history study (NHS) “NatHis-MTM”[6] between September 2015 and May 2017 and complemented from medical routine files from May 2017 and March 2020. Regarding the two patients not included in the NHS (patients 12 and 13), data was extracted exclusively from their medical routine files.
Table 1
Overview of current age, age at diagnosis, genetic, clinical and genealogical information of all 13 patients included in this study. Part of the data (including the pathogenic variants of the MTM1 gene) of patients 1-11 has already been published in the NHS cohort, data of patients 12 and 13 has not been published before
| Patient | Age at diagnosis (months) | MTM1 gene variants (NM_000252.2) | Initial symptoms | Current age (years) | Current clinical status | Other clinical manifestations and interventions | Identified relatives |
| 1 | 5 | c.139_141del; p.(Lys47del) | respiratory insufficiency, hypotonia, no reflexes | 20 | non-ambulatory, invasive ventilation, 24h/day | paroxysmal supraventricular tachycardia, hearing impairment (hearing device), subluxation of both hips, scoliosis (spondylodesis Th4 L5) cryptorchism (orchidopexy), reflux (fundoplicatio), entropium/trichiasis (several corrections) | mother carrier |
| 2 | 13 | c.342 + G>A; p.(Ser79_Aspl15del) | respiratory insufficiency, hypotonia, contractures of hips and knees | 11 | non-ambulatory, invasive ventilation, 14h/day | scoliosis, osteoporosis, cryptorchism | mother carrier |
| 3 | 22 | c.1505T > A; p.(lle502Lys) | respiratory insufficiency, hypotonia, no reflexes | 5 | non-ambulatory, invasive ventilation, 24h/day | scoliosis | de novo |
| 4 | 3 | c.1505T > A; p.(lle502Lys) | respiratory insufficiency, hypotonia, no reflexes | 5 | non-ambulatory, invasive ventilation, 24 h/day | de novo | |
| 5 | 12 | Exon 8: c.595C > T; p.(Pro199Ser) | dyspnea, hypotonia, contractures of knees | 16 | non ambulatory, non-invasive ventilation, BIPAP, 10 h/day | scoliosis, cryptorchism (orchidopexy) | mother carrier |
| 6 | 8 | c.63 + 2T > C; p.(?) | dyspnea, hypotonia | 9 | non-ambulatory, non-nivasive ventilation, BIPAP, 8 h/day | hip luxation, laryngotracheomalacia | mother and grandmother carriers |
| 7 | 2 | c.141_144del; p.(Glu48Leufs*24) | respiratory insufficiency, hypotonia, no reflexes, arthrogryposis multiplex congenita | 6 | non-ambulatory, invasive ventilation, 12 h/day | scoliosis, ultrasound of the liver: inhomogeneous liver texture | de novo |
| 8 | 5 | c.32C > A; p.(Ser11*) | respiratory insufficiency, hypotonia, no reflexes, contractures of knees, ankles, wrists and toes | 2 | non-ambularoty, invasive ventilation, 24 h/day | scoliosis, cardio-pulmonary insufficiency leading to death | mother carrier |
| 9 | 4 | c.205C > T; p.(Arg69Cys) | respiratory insufficiency, hypotonia, no reflexes, contractures of hips and knees | 5 | non-ambulatory, invasive ventilation, 18 h/day | scoliosis | mother carrier |
| 10 | 16 | c.1210G > A; p.(Glu404Lys) | respiratory insufficiency, hypotonia, no reflexes | 16 | ambulatory breathing independently | both sisters confirmed carriers, grandfather confirmed XLMTM, mother not tested | |
| 11 | 3 | c.1406A > G; p.(His469Arg) | respiratory insufficiency, hypotonia, no reflexes | 3 | non-ambulatory, non-invasive ventilation, BIPAP, 12 h/day | cryptorchism (orchidopexy) | de novo |
| 12 | 4 | c.1261-10A > G; p.(Ser420_Arg421ins PhelleGln) | reduced fetal movements | 2 | non-ambulatory, invasive ventilation, 8-12 h/day | rachitis, hydrocele, thrombocytopenia, hemoptysis, hemolytic anemia, cholestasis, liver failure leading to death | mother carrier |
| 13 | 3 | c.1190A > G; p.(Tyr397Cys) | polyhydramnios, reduced fetal movements, hypotonia | 12 | non-ambulatory, invasive ventilation, 8-12 h/day | hydrocephalus internus (VP-shunt), craniosynostosis (cranioplasty), ulcerative colitis, kidney stones, cryptorchism, scoliosis, ultrasound of the heart: pericardial effusion, paradoxical septal movement | mother carrier |
Age at diagnosis was defined as day of genetic confirmation, adopting the date of the laboratory’s confirmation letter. We used the classification published in Annoussamy et al. 2019 for defining a mild phenotype (no ventilatory support), an intermediate phenotype (ventilatory support less than 12 hours a day) and a severe phenotype (ventilation support 12 or more hours a day) [6].
A comprehensive literature comparison of published cohorts [3, 4, 8, 9] was foregone, as the comparability of the data is limited due to different study designs, classification criteria, survey periods and changed standards of care.
Muscle biopsy
Histological analyses of a muscle biopsy (derived from patient 5 at the age of 12 months) as shown in Fig. 2, include hematoxylin and eosin (HE) and nicotinamide adenine dinucleotide (NADH) staining.
Statistical analysis
Descriptive data analyses were performed by calculation of absolute frequencies and percentages with Microsoft Excel for Office 365 MSO.
Standard protocol approvals, registrations, and patient consents
The international, prospective longitudinal study was approved by local ethics/institutional review boards and was registered on clinicaltrials.gov (NCT02057705). For all enrolled patients a signed informed consent exists (by patients or parents for patients younger than 18 years). Data concerning the two additional patients was extracted from their medical routine files, so no further consent was required according to German law. This approach was approved by our local ethics committee.
RESULTS
General results
Demographics
Thirteen male patients aged from 2.0 to 19.6 years (mean age 8.62 years) were included in the study. Out of these patients, two (15.4%) died at some point in the observation period: patient 8 died at 26 months of age due to acute respiratory insufficiency and patient 12 died at 24 months of age due to liver failure (Table 1).
Molecular genetic diagnostics
All 13 male patients presented with genetically confirmed pathogenic MTM1 variants. Mean age at diagnosis was 246 days (Fig. 1). Seven (53.8%) had a single base exchange causing a missense mutation, three (23.1%) showed an intronic mutation causing a splice site alteration. Moreover, two (15%) had a deletion of several bases resulting in a frameshift. One (7.7%) had a nonsense mutation. Precise pathogenic variants are listed in Table 1. The variants shown are described using the MTM1 gene NM_000252.2 transcript reference sequence.
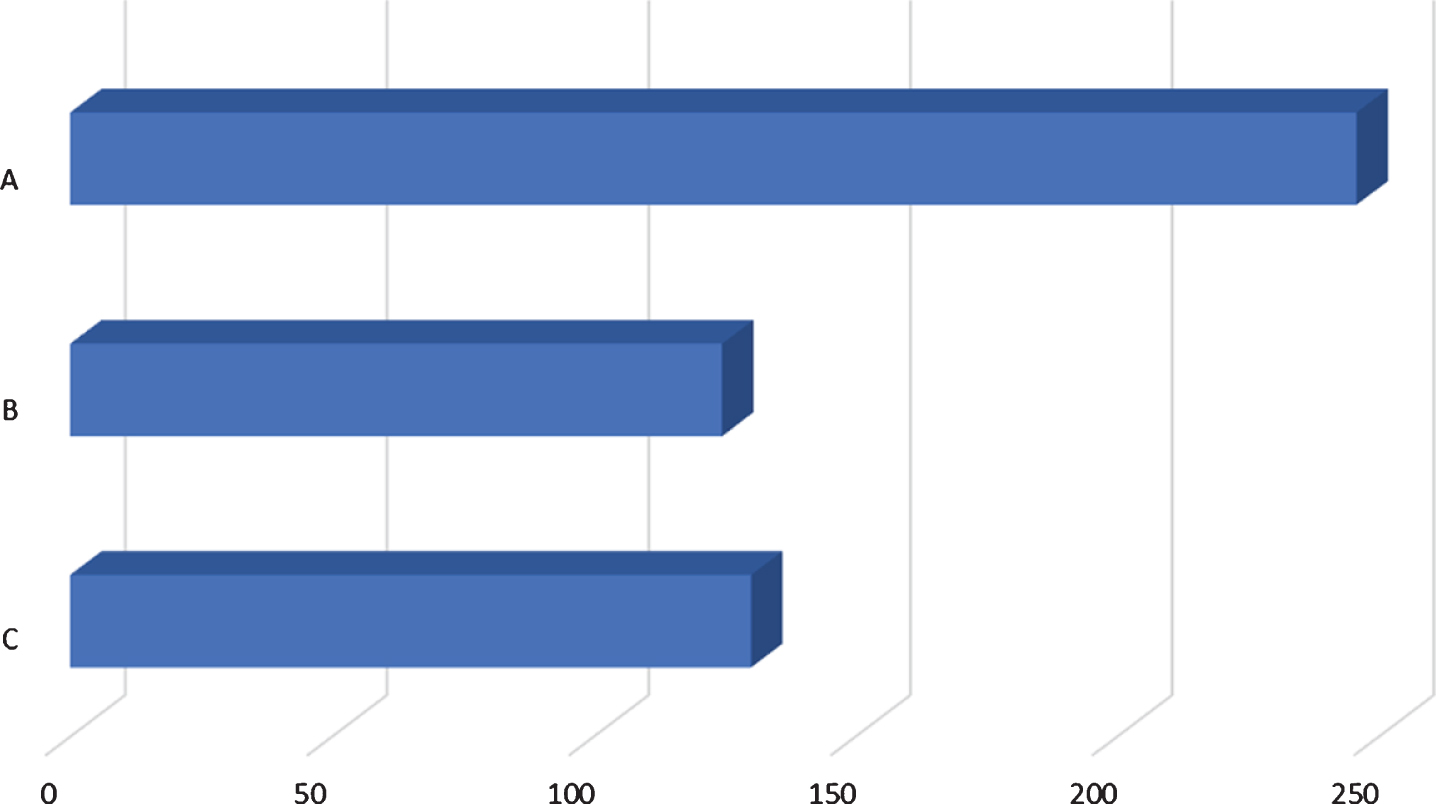
Fig. 1
Comparison of time of diagnosis and time of tracheostomy. A: average number of days from birth to genetic diagnosis (246; 13/13 patients), B: average number of days from birth to muscle biopsy (125; 7/13 patients), C: average number of days from birth to tracheostomy (130; 9/13 patients).
Muscle biopsy findings
Seven of the patients in our cohort (53.8%) underwent a muscle biopsy before genetic testing. Mean age at muscle biopsy was 125 days (Fig. 1). Histopathological findings accord with the presence of a congenital centronuclear myopathy showing (i) >5% centralized myonuclei, (ii) myofiber hypotrophy, (iii) pale peripheral halos seen with NADH staining, (iv) central aggregations of organelles including mitochondria, lysosomes, and sarcoplasmatic reticulum and (v) perinuclear vacuole-like areas [17]. Exemplary findings are shown in Fig. 2.
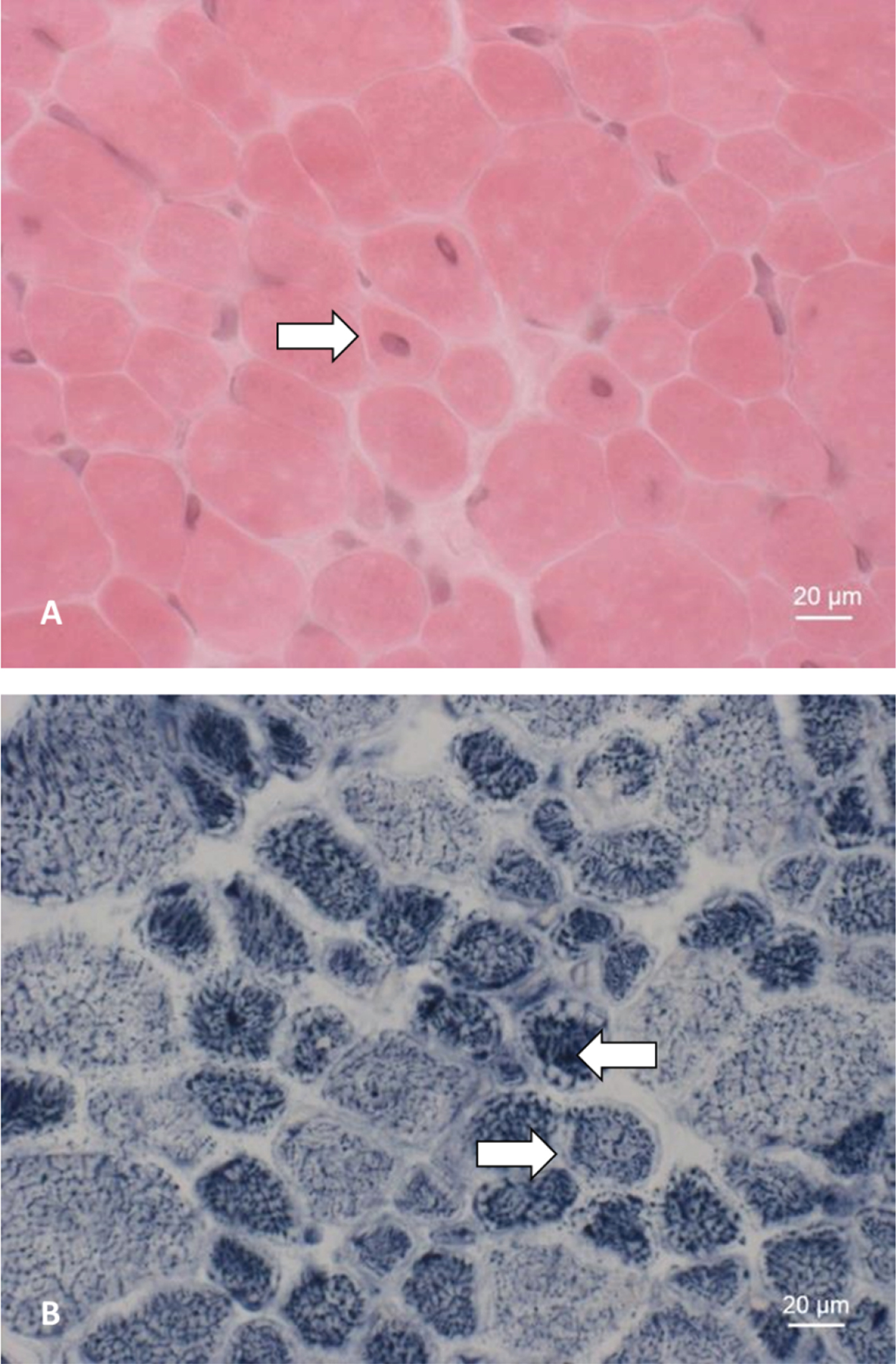
Fig. 2
Muscle biopsy of patient 5. A: HE staining showing an increase in centralized myonuclei (⇒). B: NADH staining showing pale peripheral halos (⇒) and increased central enzyme activity (⇐).
Clinical characteristics
Based on the clinical classification system mentioned in the introduction section, in our cohort consisting of 13 patients one patient (7.7%) had a mild, four (30.8%) a moderate and eight (61.5%) a severe phenotype. Six patients (46.2%) had prenatal onset (reduced fetal movements or congenital contractures) and the remaining seven (53.8%) presented at birth. Nine patients (69.2%) had respiratory insufficiency at birth and two (15.4%) dyspnea, but did not require ventilatory support. In twelve cases (92.3%) neonatal hypotonia was noted and nine (69.2%) had areflexia from birth onward. Furthermore, twelve patients (92.3%) achieved early motor milestones (independent head control, ability to roll and to sit) either with a considerable delay or not at all, remaining non-ambulant. Remarkably, one patient (7.7%) had only a mild motor delay (independent walking at 26 months of age) and is ambulatory until today (age of 15 years). Two patients (15.4%) reached the ability to transfer objects between hands as best motor function, one reached the ability to roll to one side, and one (7.7%) to roll over completely. Five patients (38.5%) reached the ability to sit independently when placed in sitting position and one to get into a sitting position by himself. One (7.7%) was able to crawl, two (15.4%) were able to walk as their best achieved motor function, one of them with assistance and one independently. Two (15.4%) patients lost their best achieved motor function in the course of the disease (one lost the ability to walk with assistance, one lost the ability to crawl). Ages at reaching best motor function are presented in Fig. 3.
Fig. 3
Motor milestones. Bars for each patient indicate current age, best achieved motor milestone pictured at age reached and (if applicable) age of loosing the ability. The grey bar indicates infant motor milestone range as time frame for achieving most milestones (adapted from Adolph et al.[14]).
![Motor milestones. Bars for each patient indicate current age, best achieved motor milestone pictured at age reached and (if applicable) age of loosing the ability. The grey bar indicates infant motor milestone range as time frame for achieving most milestones (adapted from Adolph et al.[14]).](https://content.iospress.com:443/media/jnd/2021/8-1/jnd-8-1-jnd200539/jnd-8-jnd200539-g003.jpg)
Twelve patients (92.3%) required ventilatory support and nine (69.2%) also underwent tracheostomy. Mean age at tracheostomy was 130 days (Fig. 1). A classification based only on ventilation hours is highly dependend on the cut-offs chosen. Therefore, we performed two analyses concerning ventilation support, based on a 12- and 8-hours-scheme (Fig. 4). Comparing the two schemes, we found 8 patients classified as severe in the 12-hours-scheme, but only 5 patients classified as severe referring to the 8-hours-scheme.
Fig. 4
Analysis of length of ventilatory support, based on A: 12-hours-scheme[6] and B: 8-hours-scheme[13].
![Analysis of length of ventilatory support, based on A: 12-hours-scheme[6] and B: 8-hours-scheme[13].](https://content.iospress.com:443/media/jnd/2021/8-1/jnd-8-1-jnd200539/jnd-8-jnd200539-g004.jpg)
Twelve patients (92.3%) needed feeding support, two (15.4%) only transient via nasogastric tube (NGT) during their first months of life and 10 (76.9%) continously via PEG. In eight (61.5%) patients adeno-associated virus serotype 8 (AAV8) was measured, showing a negative titer (<1 capsids/ml) in all of them.
Comparison of the German cohort to NHS data
The German cohort presented here consists of 13 male patients aged 2.0 to 19.6 years, compared to 45 male patients aged 3.5 months to 56.8 years published as the NHS cohort [6]. Interestingly, with only one mildly affected patient in our cohort, the number of cases from the “mild clinical spectrum of the disease” seems to be lower in Germany compared to patients summarized in the recently published NHS cohort (7.7% versus 28.9%). Along this line, only one German patient (7.7%) learned to walk independently, compared to ten patients (22.2%) of the NHS cohort. In contrast, four patients (30.8%) of the German cohort had a moderate phenotype, compared to seven patients (15.6%) published in the context of the NHS cohort. The percentage of patients receiving ventilatory support is 84.6% (n = 11) in the German cohort and therefore slightly higher than the 71.1% (n = 32) in the NHS cohort. In contrast, the percentage of patients presenting with a severe spectrum of the disease is almost comparable in our and the international cohort (55.6% versus 61.5%; 8/13 versus 25/45).
Phenotypical variance – Cases at a glance
To provide a more comprehensive overview of the clinical spectrum of our cohort, clinical findings of three patients from the mild, moderate and severe spectrum are presented more detailed:
The mild case – Patient 10
Patient 10 is a 154/12 year old boy and the third child of healthy, unrelated parents of Caucasian descent. His mother, currently 51 years of age, reports problems when climbing stairs since a few years ago. In addition, the maternal grandfather presented with weakness at birth, problems during growth spurts and chronic fatigue. At 66 years he developed progressive exercise intolerance, was no longer able to climb stairs and fell ill with a severe pneumonia. Muscle biopsy performed at this age showed results consistent with congenital structure myopathy. Currently, at the age of 83 years he is still able to walk, but dependent on a walker.
Our patient’s delivery was vaginal at term with normal birth dimensions (birth weight 3180 g [57. percentile, 0.18 z], length 52 cm [77. percentile, 0.75 z], head circumference 36 cm [86. percentile, 1.07 z]). At birth, he presented with respiratory insufficiency, hypotonia and areflexia (Apgar score 10’ 10, umbilical cord pH 7,26). Non-invasive ventilation was needed for the first 4 days of life, afterwards the patient started to breath spontaneously and unassisted ongoing until today. Regarding family history and neonatal clinical manifestation, XLMTM was suspected. At 16 months of age, genetic testing led to the identification of the same pathogenic variant in the MTM1 gene (c.1210G>A, p.[Glu404Lys]) in the patient and in his grandfather. A muscle biopsy of the index patient was not performed.
Although motor development was delayed, he achieved the ability to walk independently considerably delayed at the age of 26 months without losing this ability until today (Fig. 5E). However, our patient was never able to run in his life and since two years his motor function deteriorated as he is no longer able to climb stairs. Speech and cognitive development are normal. Feeding support via nasogastric-tube or PEG-tube was never needed. He shows mild ptosis and partial ophthalmoplegia until today (Fig. 5A-D). He did not develop scoliosis and has only mild contractures of both ankles. Echocardiography, ECG and abdominal ultrasound are performed on a regular basis and – so far – show no pathological findings. He has been receiving oral pyridostigmine to improve muscle function since 4 years, but did only show a moderate self-reported response and experienced side effects, e.g. circulation problems.
Fig. 5
Patient 10. A–D: at age of 13 years, mild ptosis and mild ophthalmoplegia, A: view to the left, B: view to the right, C: upward view, D: downward view, E: at age of 17 years, achieved the ability to walk independently at 26 months, without losing this ability until today (permission granted by parents).

The moderate case – Patient 7
Patient 7 is a 64/12 year old son of healthy, unrelated parents of Caucasian descent with unremarkable family history. Due to breech position, he was delivered via cesarean section at term in a peripheral maternity clinic. He showed low birth weight (2300 g, 1. percentile, – 2.32 z) normal length (52 cm, 65. percentile, 0,39 z) and head circumference (33,5 cm, 16. percentile, 1 z). At birth, he presented with respiratory insufficiency, hypotonia, generalized muscle weakness, areflexia and arthrogryposis multiplex congenita. Due to respiratory insufficiency, ventilatory support (CPAP, continuous positive airway pressure) via nose mask was installed directly after birth (Apgar score 1’ 3, 5’ 6, 10’ 8, umbilical cord pH 7,38) and intubation was necessary shortly after (Fig. 6A). Notably, several weaning attempts were unsuccessful due to recurrent respiratory exhaustion. Within the neonatal period the patient had to be resuscitated twice due to aspiration. Quadriceps muscle biopsy was performed on day 8 of life and histological examinations revealed a pathological pattern with some atrophic fibers, central nuclei and structural alterations, such as presence of halos, consistent with congenital structure myopathy. At the age of 2 months, genetic analysis was performed and revealed a causative pathogenic variant in the MTM1 gene (c.141_144del; p.[Glu48Leufs*24]). Segregation analysis revealed a de novo origin of base pair deletion thus excluding the mother from being an MTM1 carrier.
Fig. 6
Patient 7. A: At age of 1 month: presenting as floppy infant with legs in frog position. B: At age of 3 months: showing lack of head control and typical appearance with long forehead. C: At age of 6 months: first signs of motility and power in lower limbs and forearms. D: At age of 10 months: able to sit with support only when placed. E: At age of 22 months: able to sit independently and showing full head control. F: At age of 6 years (permission granted by parents).
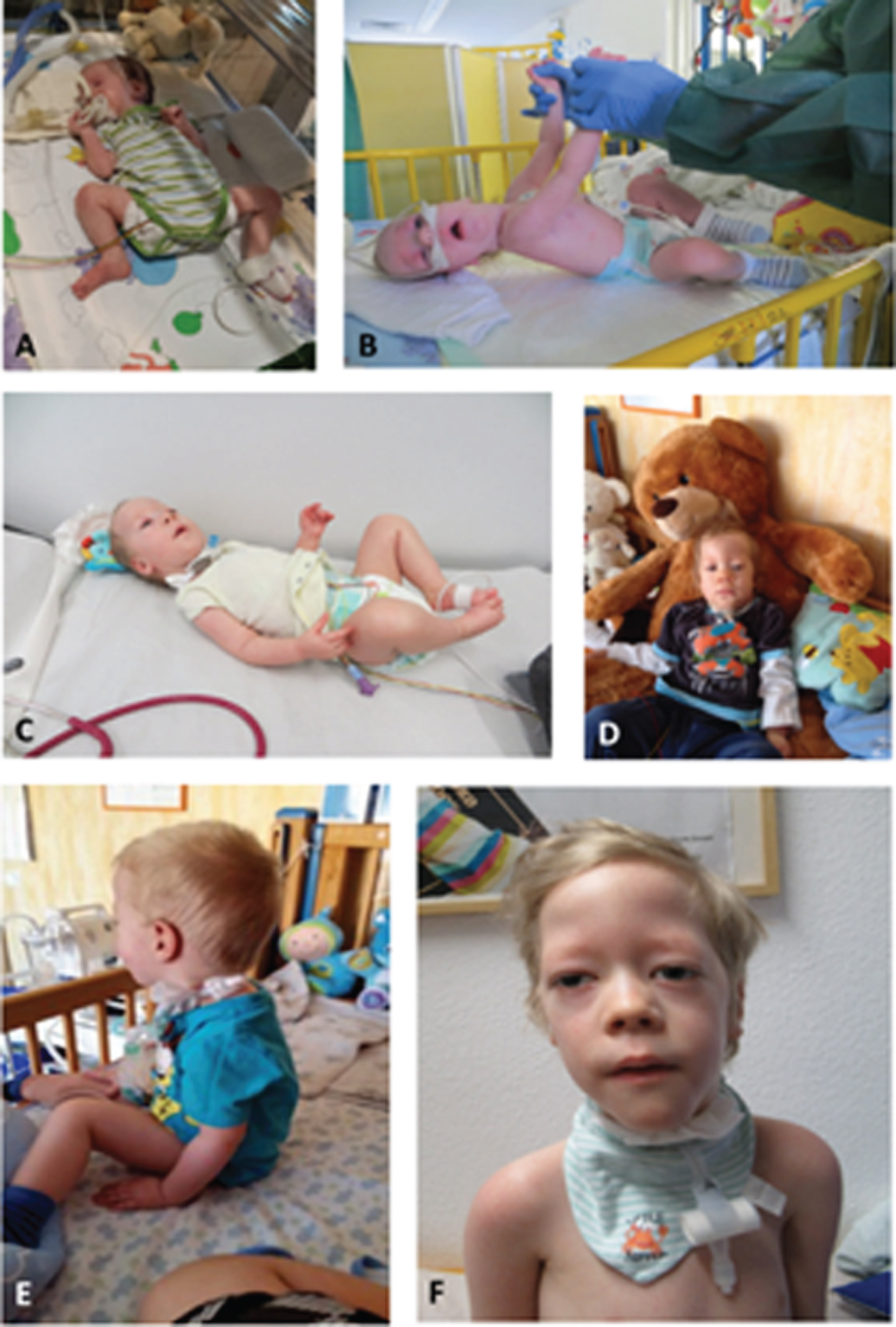
After the diagnosis was made, the patient was transferred to our clinic for further respiratory and neuromuscular care. Because of laryngotracheomalacia a supraglottoplasty was performed at 3 months of age to stabilize the airway. Due to persisting feeding difficulties a PEG-tube (Fig. 6B) was inserted and is still needed until today. Not having achieved the desired effect of a stable airway in combination with persisting respiratory insufficiency, a tracheostomy was carried out at 4 months of age. During the further clinical course, the boy showed delayed psychomotor development with generalized hypotonia and muscle weakness (Fig. 6C and 6D). Best motor milestone achieved until today (64/12 years) is the ability to sit independently when placed, possible since the age of 22 months (Fig. 6E). Early speech development was severely impaired due to the tracheostoma and did slightly improve through the use of a speech canula. The boy presents with ptosis and ophthalmoplegia. Currently, he requires 12 hours of invasive respiratory support via tracheostoma overnight; he is able to breath independently via a passive humidifier for the rest of the day (Fig. 6F). At the age of 47/12 years a mild scoliosis (Cobb angle 11°) with rib hump (left side) was noted for the first time. Echocardiography, ECG and abdominal ultrasound are performed on a regular basis and show no pathological findings so far.
The severe case – Patient 12
Patient 12 became 2 0/12 years old and was the only child of healthy, unrelated parents of Caucasian descent. Family history showed no indication of neuromuscular disorders. Notably, manifestation of first symptoms was already prenatal with reduced fetal movements. Birth was at term with normal dimensions (2760 g [9. percentile, – 1.32 z], length 50 cm [30. percentile, – 0.53 z], head circumference 35 cm [51. percentile, 0.03 z]) via cesarean section due to breech position. Because of respiratory insufficiency at birth (Apgar score 5’ 8, 10’ 9, umbilical cord pH 7,2) and ongoing ventilator dependency, tracheostomy was performed at the age of 5 months. A muscle biopsy was not performed. A genetic diagnosis was established at the age of 3 months by trio-based exome sequencing, identifying the same splice site mutation (c.1261-10A>G, p.[Ser420_Arg421insPheIleGln]) in the MTM1 gene in patient 12 and his mother. The mother is asymptomatic, except for mildly elevated transaminases. Due to impaired swallowing and sucking, a PEG-tube was inserted at 6 months and was needed for our patient’s whole life for 100% of his caloric intake. Psychomotor development was severely delayed and the best motor milestone achieved was to transfer objects between the hands (reached at 6 months of age). Head control, sitting, crawling or walking were never possible. Speech development was severely impaired due to the tracheostoma and he was never able to babble or speak. He showed mild ptosis and partial ophthalmoplegia.
At 5 months of age, mild thrombocytopenia (150/nl [normal value 200–360/nl]) was present for the first time. At the age of 12 months, he developed fractures of both femurs and multiple ribs without adequate trauma. In combination with hypocalcemia and hypophosphatemia the diagnosis of rachitis was made. Therapy was started with calcium gluconate and high dose vitamin D. During the same hospitalization, the diagnosis of autoimmune thrombocytopenia was made (platelet count reduced to 36/nl [normal value 200–360/nl]). At the age of 15 months he developed hemoptysis during a pneumonia with total atelectasis of the right lung (platelet count reduced to 19/nl [normal value 200–360/nl]). During this time hepatomegaly was first detected via abdominal ultrasound and a liver hemangioma was suspected. Because of the suspicion of an autoimmune thrombocytopenia, a first cortisone pulse for three days was initiated. By this intervention, hemoptysis finally stopped and the patient could be discharged.
At the age of 16 months, patient 12 had to be hospitalized due to severe cholestasis (Fig. 7, max. elevated bilirubin value 39,9 mg/dl [normal value 0,2–1,0 mg/dl]) and beginning liver failure (Quick 48% [normal value 53–100% ], INR 1,44 [no normal value], albumin 2,2 g/dl [normal value 3,6–5,0 g/dl], ammonia 90μg/dl [normal value 19–55μg/dl]). Under suspicion of an autoimmune hepatitis, another cortisone pulse therapy was performed, improving his general condition.
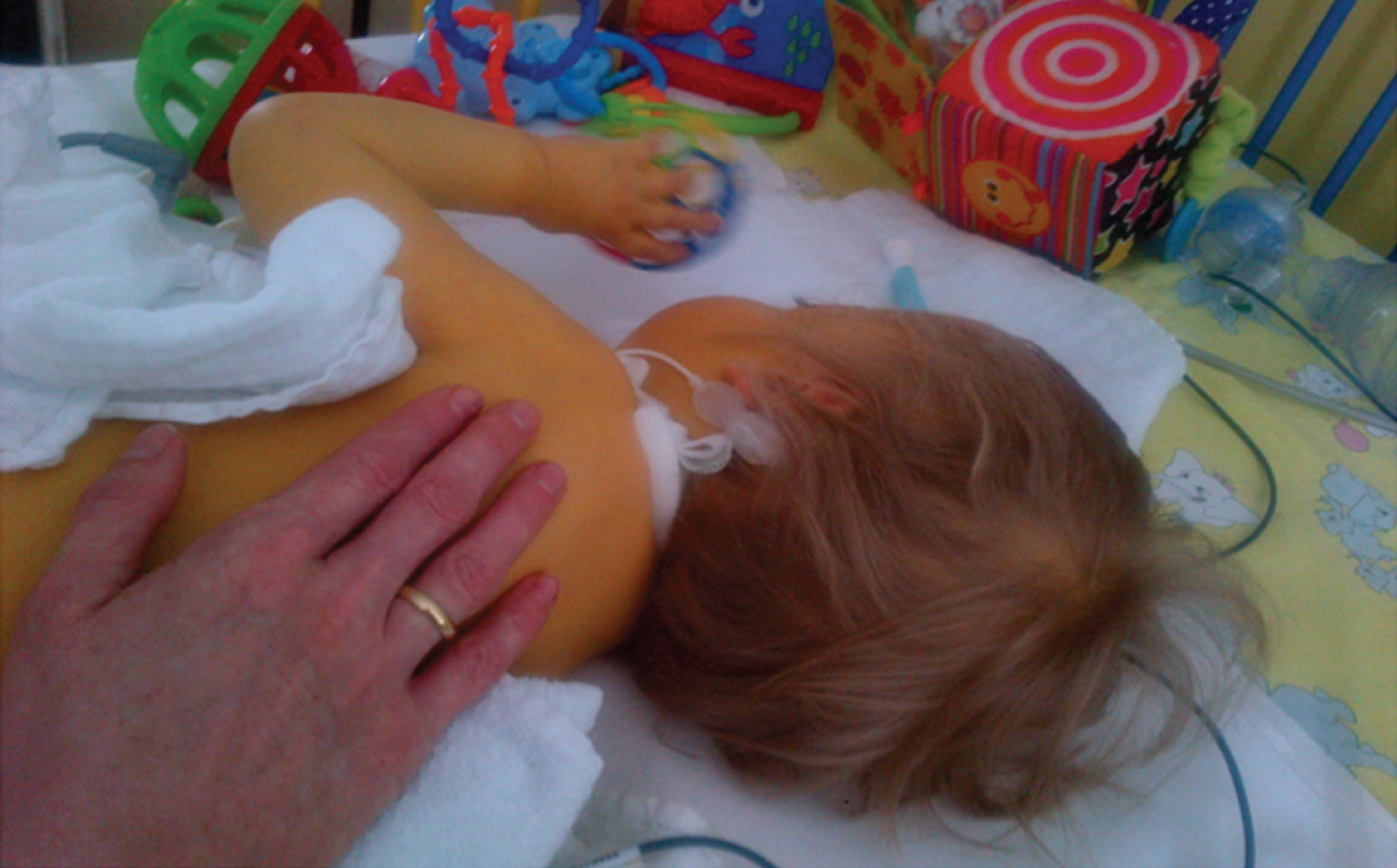
Fig. 7
Patient 12. At 16 months of age with significant icterus due to a total bilirubin value of 39.9 mg/dl (normal value 0.2–1.0 mg/dl), only able to transfer objects between hands (permission granted by parents).
During the following months, several hospital admissions were necessary due to hepatopathy with cholestasis (max. elevated bilirubin value at 34,8 mg/dl [normal value 0,2–1,0 mg/dl]) and deterioration of his general condition. At this time ultrasound of the liver only showed accentuated bile ducts (1 mm), a hemangioma was no longer suspected. He received four cortisone pulse therapies in total, each with low respondence. A liver biopsy was not performed due to thrombocytopenia and the risk of death from liver hemorrhage in patients with XLMTM reported in literature [4]. Finally, a palliative therapy concept was installed at 20 months of age.
At 2 years of age he was admitted to the hospital due to severe deterioration of his general condition. The mother reported him as being weak, lethargic and pale. His waist circumference had increased over the last 4 weeks. At the day of hospital admission, he had diarrhea and anuria. At examination he was cardiorespiratorily stable while on his home ventilator via tracheostoma, but somnolent and very weak. Blood tests showed a severe electrolyte imbalance, liver failure (ammonia 425μg/dl [normal value 19–55μg/dl], Quick 31% [normal value 53–100% ], INR 2,07 [no normal value], GOT 138 U/l [normal value < 50 U/l]), as well as kidney failure (creatinine 55μmol/l [normal value 35–62μmol/l], urea 18,9 mmol/l [normal value 1,8–59 mmol/l], uric acid 470,1μmol/l [normal value 101–339μmol/l]) and anemia (hemoglobin 6,6 g/dl [normal value 9,2–15,5]). During the same night of hospital admission, he developed bradycardia (40 bpm). Further escalation of therapy was not considered. Before his death at 2 years of age, the patient was ventilated 24 hours per day via tracheostoma.
4DISCUSSION
We present a German cohort of 13 XLMTM patients and compare our findings with natural history data summarizing 45 patients with XLMTM, focusing on motor and respiratory function, milestone achievement and other clinical manifestations.
One interesting finding in this analysis is the diagnostic workup. It reveals a time gap between manifestation, the severity of respiratory distress and related management, interventions done in the course of the disease before final diagnosis was confirmed. Despite presenting with an onset of clinical symptoms suspicious for a neuromuscular disorder, either intrauterine or at birth, the final diagnosis by genetic confirmation was delayed in all patients. The clinical presentation led to a muscle biopsy in 7 of 13 patients on day 125 on average and a tracheostomy in 9 of 13 patients on day 130 on average. Despite the interventions, genetic diagnosis was only verified on day 246 on average. These data may hint towards an incomplete workup of the differential diagnosis and therefore a delayed referral to a center of expertise.
Despite a similar early clinical onset with muscle hypotonia and muscle weakness, as well as a motor delay, other symptoms varied, showing a broad spectrum of the disease, which is not linked to the pathogenic variants found in each patient. To emphasize this finding, we presented three cases at a glance: while patient 10 is still able to walk independently at the age of 15 years, but was delayed in developing motor milestones and has lost abilities (additionally his affected grandfather is still alive at 83 years), patient 7 showed the typical course of the disease with severe muscle hypotonia and weakness, never being able to walk, and patient 13 already died at the age of 2 years due to liver failure. The cause for his liver failure remains unknown as a liver biopsy was not performed due to thrombocytopenia and risk of death from liver haemorrhage in patients with XLMTM reported in literature and the NHS [4, 6, 10]. A better understanding of liver dysfunction in XLMTM remains an unmet need. Whole exome sequencing performed in this patient did neither reveal pathogenic variants in genes known to be causative for thrombocytopenia, nor in such causative for genetic forms of liver disease. In addition, his family history did not show any evidence for increased bleeding propensity. Of note, the only symptom reported by his mother (a proven MTM1 carrier) is a mild elevation of liver enzymes without any apparent reason (no obesity & no indication of alcohol or drug abuse, no liver disease such as hepatitis, cirrhosis or liver tumour). Although this MTM1 carrier does not present with symptoms of muscle weakness, the elevated liver enzymes might indicate a mild subclinical manifestation of the pathogenic variant in the MTM1 gene, as gastrointestinal and liver manifestations were occasionally reported in XLMTM patients [4].
The varied genetic distribution of the pathogenic variants found does not reveal a remarkable genotype-phenotype correlation. However, more comprehensive functional and biochemical studies are needed to further elucidate the vulnerability of tissues beyond skeletal muscle to pathogenic variants in the MTM1 gene.
Comparing our cohort with the NHS cohort [6], it appears that German patients less often show a mild phenotype (7.7% versus 22.2%) and more often present with a moderate phenotype (30.8% versus 15.6%), with no clear difference concerning the amount of patients severly affected (55.6% versus 61.5%).
The higher percentage of patients showing a moderate phenotype may indicate a pronounced clinical vulnerability in our German cohort. This concept is further supported by the percentage of patients requiring ventilatory support, with 84.6% (n = 11) in the German cohort, compared to 71.1% (n = 32) in the NHS cohort. This information might be important in defining clinical outcome-measures for the testing of therapeutic intervention concepts. However, a description of a larger cohort of XLMTM patients of German origin would be needed to draw a final conclusion.
Both cohorts did not show an obvious link between the mutational spectrum and clinical presentations in terms of a reliable genotype-phenotype correlation [6]. Until now only small case series concentrate on MTM1 molecular genetics and associated clinical phenotypes [15, 16], which is why the need for a larger cohort to unravel potential statistically significant correlations remains.
The common classification for XLMTM is based on respiratory support only, not taking into consideration that these patients often have delayed, transient, or completely absent motor milestones, need feeding support or exhibit other comorbidities [3, 6]. A classification based only on ventilation hours is highly dependened on the cut-offs chosen. The current classification in XLMTM is based on a 12-hours-scheme whereas in other neuromuscular diseases other cut-offs are used. For example Finkel and colleagues defined ventilation up to 8 hours overnight as mild, whereas ventilation with more than 16 hours over night and daytime was defined as severe [13]. Comparing the two schemes, we found a slight shift in severity grading with more severe cases using the 12-hours-scheme, leading to some inconsistency in outcome of classification by ventilation status only. Hours on ventilation is one of the key markers for burden of disease, progression and prognosis in neuromuscular diseases. A classification only by ventilation hours does not reveal a comprehensive clinical picture of the patients. Most patients remain non-ambulatory (92.3%), receive feeding support (92.3%), display scoliosis (62%), contractures (32%) or other clinical symptoms (62%), e.g. cryptorchism, cholesteatoma, laryngotracheomalacia and autoimmune thrombocytopenia, which are also contributing to disease severity. A comprehensive and balanced classification – potentially including biomarker-based stratification – is a potential need for future assessments and access to potential therapeutic developments, as there is no curative treatment available for XLMTM, yet. Standard of care comprises a multidisciplinary approach with focus on motor, respiratory and bulbar symptoms, e.g. ventilation support, management of infections, tube feeding, physiotherapy, provision of orthoses, wheelchair and other devices. Conservative or surgical orthopedic interventions for contractures and/or scoliosis are also part of the interventional management of XLMTM patients. In single cases some slight improvement of motor function and respiratory distress was reported for pyridostigmine bromide treatment addressing the functionality of the neuromuscular junction [11]. Promising investigational treatments emerged comprising enzyme replacement therapy, dynamin-2 modulation, PIK3C2B inhibition and gene therapy (NCT03199469; ASPIRO). Therefore, it becomes increasingly important to have reliable natural history data to define outcome measures and severity grading. In our cohort, 92.3% of the patients were non-ambulatory, 84,6% were dependent on ventilatory and feeding support, showing that these parameters are suitable clinical outcome measures for future clinical trials. To enable access to clinical trials and newly discovered treatments, patients need to receive an immediate diagnosis and comprehensive care in specialised neuromuscular centers.
In the natural history study (NHS) mentioned above, neutralizing antibodies against adeno-associated virus type 8 were tested, to ensure clinical trial readiness for potential upcoming gene therapy [6]. Fortunately, AAV8 antibodies (possibly impeding gene therapy) were not present in our cohort, not preventing access to this therapeutic option for our cohort.
Our study has some limitations: as emphasized by the cases presented at a glance, the spectrum of the disease is broad and thereby our cohort heterogeneous and distinctly smaller in comparison to the recently published NHS cohort, thus limiting the statistic possibilities. We compared the German with the overall NHS cohort adding two new cases, but we did not extract the German cohort from the overall cohort. The overall NHS did not take into consideration different standards of care. In addition, follow-up for a longer time period and multicentric study design are always favourable. A potential selection bias could be related to the referral pattern, referring only more severe XLMTM patients to specialised neuromuscular centers. Additionally, it is to be expected that a number of patients remain without diagnosis up to a higher age, as the case of our patient’s grandfather demonstrates. This might especially apply to patients with atypical or mild clinical presentation. Our analysis demonstrated a disease burden for all XLMTM patients thoughout the heterogenenous clinical spectrum of the disease. When comparing the German cohort to the NHS, a slight shift to the severe spectrum of the disease becomes apparent and further assessing the potential need for a new or revised classification considering all aspects of XLMTM remains a future task.
CONFLICT OF INTEREST
Andrea Gangfuss, Andreas Roos, Melanie Annoussamy and Frederik Braun report no disclosures relevant to the manuscript. Dirk Schmitt is employee of Audentes, a company developing treatment for patients with XLMTM. Ulrike Schara-Schmidt and Laurent Servais received consultant fees from Dynacure, which is developing treatment for patients with XLMTM. Laurent Servais is coinventor of MoviPlate device and received consultancy fees from Audentes.
REFERENCES
[1] | Vandersmissen I , Biancalana V , Servais L , Dowling JJ , Vander Stichele G , Van Rooijen S , et al. An integrated modelling methodology for estimating the prevalence of centronuclear myopathy. Neuromuscul Disord. (2018) ;28: (9):766–77 10.1016/j.nmd.2018.06.012 |
[2] | Spiro AJ , Shy GM , Gonatas NK . Myotubular myopathy. Persistence of fetal muscle in an adolescent boy. Arch Neurol. (1966) ;14: (1):1–14. doi: 10.1001/archneur.1966.00470070005001 |
[3] | McEntagart M , Parsons G , Buj-Bello A , Biancalana V , Fenton I , Little M , et al. Genotype-phenotype correlations in X-linked myotubular myopathy. Neuromuscul Disord. (2002) ;12: (10):939–46. doi: 10.1016/s0960-8966(02)00153-0 |
[4] | Herman GE , Finegold M , Zhao W , de Gouyon B , Metzenberg A . Medical complications in long-term survivors with X-linked myotubular myopathy. J Pediatr. (1999) ;134: (2):206–14. doi: 10.1016/s0022-3476(99)70417-8 |
[5] | Amburgey K , Tsuchiya E , de Chastonay S , Glueck M , Alverez R , Nguyen CT , et al. A natural history study of X-linked myotubular myopathy. Neurology. (1355) ;89: (13):26–64. doi: 10.1212/WNL.0000000000004415 |
[6] | Annoussamy M , Lilien C , Gidaro T , Gargaun E , Chê V , Schara U , et al. X-linked myotubular myopathy: A prospective international natural history study. Neurology. (2019) ;16;92: (16):e1852–e1867. doi: 10.1212/WNL.0000000000007319 |
[7] | Jungbluth H , Wallgren-Pettersson C , Laporte J . Centronuclear (myotubular) myopathy. Orphanet J Rare Dis. (2008) ;3: 26 10.1186/1750-1172-3-26 |
[8] | Graham RJ , Muntoni F , Hughes I , Yum SW , Kuntz NL , Yang ML , et al. Mortality and respiratory support in X-linked myotubular myopathy: a RECENSUS retrospective analysis. Arch Dis Child. (2020) ;105: (4):332–8. doi: 10.1136/archdischild-2019-317910 |
[9] | Beggs AH , Byrne BJ , De Chastonay S , Haselkorn T , Hughes I , James ES , et al A multicenter, retrospective medical record review of X-linked myotubular myopathy: The recensus study. Muscle Nerve.550-60. (2018) ;57: (4)–10.1002/mus.26018 |
[10] | Funayama K , Shimizu H , Tanaka H , Kawachi I , Nishino I , Matsui K , et al. An autopsy case of peliosis hepatis with X-linked myotubular myopathy. Leg Med (Tokyo).77-82. (2019) ;38: :77–82. doi: 10.1016/j.legalmed.2019.04.005 |
[11] | Trippe H , Lutz S , Bouikidis A , Kaiser O , Della Marina A , Schara U . X-Linked Myotubular Myopathy Clinical Improvement by Use of Pyridostigmine in an Infant. Neuropediatrics. . (2015) ):46 10.1055/s-0035-1550749 |
[12] | Cocanougher BT , Flynn L , Yun P , Jain M , Waite M , Vasavada R , et al Adult MTM1-related myopathy carriers: Classification based on deep phenotyping. Neurology. . (1542) ;93: (16):e1535–e1542. doi: 10.1212/WNL.0000000000008316 |
[13] | Finkel RS , McDermott MP , Kaufmann P , Darras BT , Chung WK , Sproule DM , et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. (2014) ;83: (9):810–7. doi: 10.1212/WNL.0000000000000741 |
[14] | Adolph KE . Motor and Physical Development: Locomotion, New York University (NY), USA, Elsevier Inc., (2008) , pp. 359–373. |
[15] | Longo G , Russo S , Novelli G , Sangiuolo F , D’Apice MR . Mutation spectrum of the MTM1 gene in XLMTM patients: 10 years of experience in prenatal and postnatal diagnosis. Clin Genet. (2016) ;89: (1)93–8. doi: 10.1111/cge.12674 |
[16] | Laporte J , Hu LJ , Kretz C , Mandel JL , Kioschis P , Coy JF , et al A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet. (1996) ;13: (2)175–82. doi: 10.1038/ng0696-175 |
[17] | Lawlor MW , Beggs AH , Buj-Bello A , Childers MK , Dowling JJ , James ES , et al. Skeletal Muscle Pathology in X-Linked Myotubular Myopathy: Review With Cross-Species Comparisons. J Neuropathol Exp Neurol. (2016) ;75: (2)102–10. doi: 10.1093/jnen/nlv020 |

