
The Canadian Neuromuscular Disease Registry 2010–2019: A Decade of Facilitating Clinical Research Througha Nationwide, Pan-NeuromuscularDisease Registry
Abstract
We report the recruitment activities and outcomes of a multi-disease neuromuscular patient registry in Canada. The Canadian Neuromuscular Disease Registry (CNDR) registers individuals across Canada with a confirmed diagnosis of a neuromuscular disease. Diagnosis and contact information are collected across all diseases and detailed prospective data is collected for 5 specific diseases: Amyotrophic Lateral Sclerosis (ALS), Duchenne Muscular Dystrophy (DMD), Myotonic Dystrophy (DM), Limb Girdle Muscular Dystrophy (LGMD), and Spinal Muscular Atrophy (SMA). Since 2010, the CNDR has registered 4306 patients (1154 pediatric and 3148 adult) with 91 different neuromuscular diagnoses and has facilitated 125 projects (73 academic, 3 not-for-profit, 3 government, and 46 commercial) using registry data. In conclusion, the CNDR is an effective and productive pan-neuromuscular registry that has successfully facilitated a substantial number of studies over the past 10 years.
INTRODUCTION
Neuromuscular diseases are a heterogenous group of conditions that are individually rare, but together they comprise a relatively common group of conditions with an overall combined prevalence of 160 per 100,000 [1]. As with all rare diseases, research in neuromuscular disease is limited by patient numbers in any given clinic necessitating collaboration between centres, regions, and countries. Canada is a diverse country with broad geographic spread. At 9.984 million square kilometres, Canada is the second largest country in the world by total area after Russia, and has a population of just over 37 million people, equating to a population density of 4 people per square kilometre (or 223rd in the world) [2, 3]. Canada has two national languages, English and French. Healthcare in Canada is universal, publicly funded, and regulated by provincial and territorial systems. The national landscape, both geographic and political creates barriers to studying rare disease.
Rare disease registry platforms enable research to generate real-world evidence (RWE) to evaluate outcome measures, natural history of disease, and standards of care, as well as facilitating clinical trial feasibility evaluations [4, 5].
The Canadian Neuromuscular Disease Registry (CNDR) recruits individuals from across Canada with a diagnosed neuromuscular disease [6]. Initially, the primary goal of the registry was to increase efficient patient access to cutting edge research and clinical trials. In addition, secondary objectives were to increase the understanding of the natural history and epidemiology of neuromuscular disease across Canada and facilitate a more collaborative research community. Over the past decade the registry has evolved with additional goals of improving health outcomes, pharmacoeconomic analyses, and most recently collection of post-approval real world evidence (RWE) for novel therapies.
METHODS
The CNDR began enrollment in September 2010. Patients are consented to participate in the registry in CNDR- affiliated clinics across Canada or, in limited cases, self-registered through the National Office. In order to register, patients are required to have a confirmed diagnosis (clinical and/or genetic) according to the World Federation of Neurology classification [7] and must provide informed consent. For indexed diseases (ALS, DMD, DM, LGMD, and SMA), a full disease-specific clinical dataset and patient demographics are collected and updated annually by trained site coordinators and/or physicians. For all other neuromuscular diseases (non-indexed diseases), only a confirmed diagnosis and patient demographics (name, contact information, date of birth, and gender) are collected. Ethnicity is currently collected in some disease-specific datasets, but not routinely across all patient populations. The distinction between indexed and non-indexed diseases results from the availability of funding to support the development and collection of a full dataset. Study data are collected and managed using the REDCap (Research Electronic Data Capture) system hosted by the University of Calgary [8]. The data collection system is built using a ‘quality by design’ approach, and includes data validation measures such as appropriate ranges, auto-calculations, and missing value flags to ensure quality control. Additional quality control measures include concise case report forms, centralized training of data entry personnel across clinical sites, and systematic remote auditing by the national office to flag missing values and data outliers. From inception the CNDR has been designed to enable approved data sharing; however, the registry is currently undergoing a process to ensure FAIR (findable, accessible, interoperable, reusable) principles are being adhered to. As of December 2019, the CNDR Network consists of 36 clinical sites in 15 academic centres across Canada (Supplemental Table 1). The CNDR network includes coordination and operations of the registry through a national office based at the University of Calgary. Each CNDR-affiliated site is responsible for consenting patients and data entry, while the national site coordinates local site onboarding, data quality control, funding, and data utilization. Historically, participating sites received a general pot of funding to support data entry. More recently, funding is provided to sites on a per patient per visit basis to help support consenting and data entry. Patients are recruited in accordance with local ethics approval processes, following ethics approval at each participating site. Patients are consented to the registry at their local clinic by physicians, members of their care team, or research staff. Nationally, patients may be consented by the national office staff. All neuromuscular diagnoses are recruited in affiliated adult and pediatric clinics, while ALS patients are recruited from multi-disciplinary ALS clinics (Supplemental Table 1). The CNDR is supported by an advisory committee comprised of adult and pediatric clinician experts and is under the responsibility of the national principal investigator.
Requests for research projects using CNDR data are reviewed by the advisory committee and are required to demonstrate local ethics approval as part of the review process. Typical projects include clinical trial feasibility and planning; trial notifications and targeted recruitment letters to eligible registrants; survey and questionnaire-based studies; data analyses; and other informational mail-outs. Data is always provided in a de-identified manner and typically as aggregate data. Inquiries from academic-based individuals are completed on a cost recovery basis (e.g., postage) while fees charged on industry-based inquiries are used to offset operating costs of the registry.
The CNDR is a member of the TREAT-NMD global registry network and contributes de-identified data to international studies upon request from vetted and approved projects (https://treat-nmd.org).
RESULTS
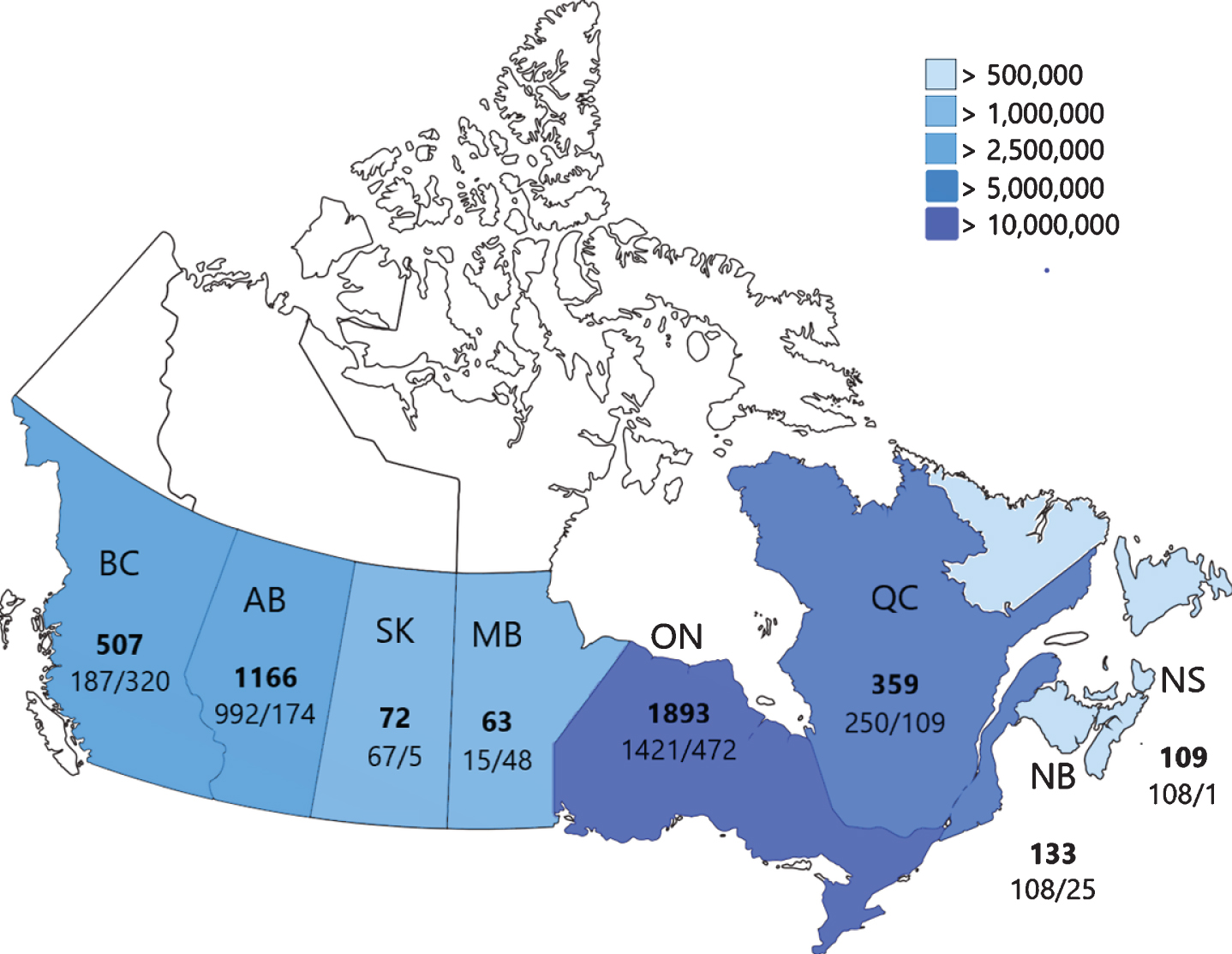
As of December 1, 2019, the CNDR has recruited 4310 patients from all 13 provinces and 3 territories across Canada. Of the enrolled individuals, 3066 (71%) have indexed diseases (ALS, DMD, DM, LGMD, and SMA), while the other 1244 (29%) are non-indexed representing 78 additional neuromuscular diseases (Supplemental Table 2). The most common non-indexed diseases in the registry are Charcot-Marie-Tooth (CMT) (170, 4%), Myasthenia Gravis (MG), (169, 4%), Facioscapulohumeral dystrophy (FSHD) (141, 3%), Inclusion Body Myositis (IBM) (74, 2%), and Oculopharyngeal Muscular Dystrophy (OPMD) (59, 1%). Detailed registry composition and patient demographics are reported in Table 1. Provincial recruitment statistics of adult and pediatric patients are outlined in Fig. 1, alongside overall provincial population [9].
Table 1
Demographics of registry participants
| N (% of total) | N by type (% of total) | % Male | Median age* (range) | % Deceased | % Pediatric | ||||||
| ALS | 1555 (36.1) | N/A | 59.6 | 67.5 (27.5–95.3) | 55.7 | 0 | |||||
| DMD | 574 (13.3) | DMD | BMD | IMD | FC | 96.3 | 17.4 (2.4–72.0) | 3.0 | 52.6 | ||
| 446 (80.7) | 93 (16.8) | 3 (0.5) | 11 (2.0) | ||||||||
| DM | 451 (10.5) | I | II | Congenital | 48.6 | 44.7 (0.4–82.4) | 5.1 | 18.3 | |||
| 291 (70.5) | 60 (14.5) | 62 (15.0) | |||||||||
| LGMD | 257 (5.9) | 1 (A–Q) | 2 (A–Z) | Undefined | Congenital Myopathies | Congenital MD’s | Pompe | 54.0 | 33.5 (0.3–81.9) | 0.0 | 27.0 |
| 1 (0.4) | 34 (13.2) | 115 (44.7) | 91 (35.4) | 7 (2.7) | 9 (3.5) | ||||||
| SMA | 229 (5.3) | I | II | III | IV | Distal | 51.3 | 16.1 (0.3–75.8) | 3.8 | 54.7 | |
| 46 (22.7) | 91 (44.8) | 59 (29.1) | 1 (0.5) | 3 (1.5) | |||||||
| Non-indexed | 1244 (28.9) | See Supplemental Table 2 | 54.6 | 51.5 (1.3–97.3) | 2.0 | 21.1 | |||||
| Total | 4310 | N/A | 61.1 | 45.8 (0.3–97.3) | 17.8 | 24.3 |
*Active patients only. ALS = Amyotrophic Lateral Sclerosis; DMD = Duchenne Muscular Dystrophy; DM = Myotonic Dystrophy; LGMD = Limb Girdle Muscular Dystrophy; SMA = Spinal Muscular Atrophy; BMD = Becker Muscular Dystrophy; IMD = Intermediate Muscular Dystrophy; FC = Female Carrier; MD = Muscular Dystrophy.
Fig. 1
CNDR recruitment by province. Total number of registered individuals is shown in bold, followed by adult cases (left) and pediatric cases (right). Relative provincial and territorial populations are shown through color-coding for reference (Statistics Canada, 2018). BC = British Columbia; AB = Alberta; SK = Saskatchewan; MB = Manitoba; ON = Ontario; QC = Quebec; NB = New Brunswick; NS = Nova Scotia.
The CNDR has collected genetic test results ranging from 16.9% (sporadic ALS) to 86.4% (DM) of the indexed patient population (Table 2). Additional patients have a confirmed genetic diagnosis; however, not all testing results are currently available to the registry. Between 8.3% (LGMD) to 28.4% (DMD) of CNDR registrants are participating or have participated in clinical trials (Table 2).
Table 2
Genetic testing and clinical trial participation
| N | % genetic results | % clinical trial participation | |
| ALS | |||
| Negative family Hx | 1096 | 16.9 | 24.5 |
| Positive family Hx | 124 | 57.3 | |
| DMD | 520 | 84.8 | 28.4 |
| DM | 398 | 86.4 | Not collected |
| LGMD | 56 | 64.3 | 8.3 |
| SMA | 178 | 74.7 | 19.6 |
Hx = history. ALS = Amyotrophic Lateral Sclerosis; DMD =Duchenne Muscular Dystrophy; DM = Myotonic Dystrophy; LGMD = Limb Girdle Muscular Dystrophy; SMA = Spinal Muscular Atrophy.
Using calculations of expected cases based on published prevalence estimates [1, 5, 10, 11], the CNDR currently has an estimated ascertainment of affected individuals with indexed neuromuscular diseases ranging from 9.0% for DM to 83.5% of DMD cases (Table 3).
Table 3
Recruitment ascertainment
| Active # in CNDR | Prevalence | Projected # in Canada | % Ascertainment | |
| ALS | 624 | 10/100,000 | 3630 | 17.2 |
| DMD | 497 | 13/100,000 (5–24 y.o. males)[11] | 595 | 83.5 |
| DM | 410 | 12.5/100,000 | 4537 | 9.0 |
| LGMD | 240 | 1–9/100,000 | 363–3267 | 7.3–66.1 |
| SMA | 207 | 1–2/100,000[10] | 544 | 38.1 |
Based on Canadian population of 36.3 million, and 4.315 million boys 5–24 years old [9]. ALS = Amyotrophic Lateral Sclerosis; DMD = Duchenne Muscular Dystrophy; DM = Myotonic Dystrophy; LGMD = Limb Girdle Muscular Dystrophy; SMA = Spinal Muscular Atrophy.
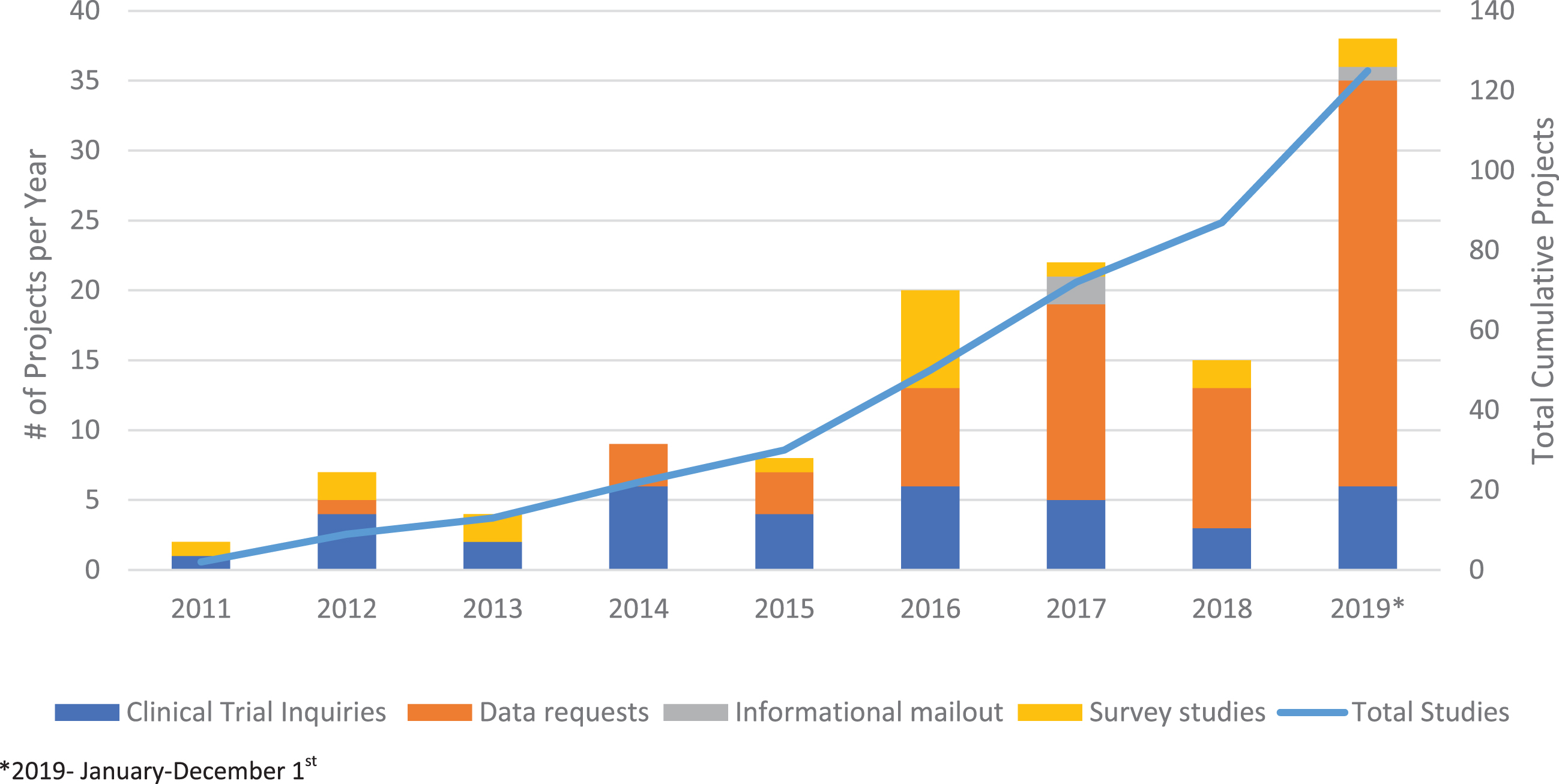
Since 2011, the CNDR has facilitated 125 research projects: 37 clinical trial inquiries and recruitment notifications, 67 data releases, 18 questionnaire-based studies, and 3 informational mail-outs (Fig. 2). Questionnaire-based studies included: multiple quality-of-life studies for DMD patients and SMA patients (including 1 longitudinal study) [12, 13], access to computer technology for DM patients [14], disease burden in myotonic dystrophy [15], barriers and motivators for research participation for ALS patients; a global study of risk factors for ALS [16], a shared decision making respiratory needs assessment for DMD and SMA patients, respiratory practices for neuromuscular health care practitioners, and outcome measure assessments for adult SMA neuromuscular health care practitioners. Other studies included provincial comparisons of time to diagnosis and use of riluzole in ALS [17], feeding tube use in ALS [18], exon-skipping in DMD [19], and global studies of DMD and SMA [10, 20–23]. The CNDR is currently undertaking a natural history study in SMA and is investigating natural history/post-marketing surveillance studies in other diseases. Twenty-four clinical trial inquiries were coordinated in Canada and another 11 through data sharing with the global TREAT-NMD network of registries.
Fig. 2
CNDR Projects. Total projects completed by the CNDR is shown by the line graph (right axis). Type of projects completed per year are shown by bar graph (left axis).
DISCUSSION
The CNDR is a pan-neuromuscular disease registry with clinic-based data entry. This multi-disease methodology is an efficient and comprehensive model that has also been implemented by other NM registries such as Australia and New Zealand [24], Belgium [25], and the Netherlands [26], but differs from the disease-specific registries across Europe and the United States (e.g., United Kingdom SMA, DM [27], and DMD registries, or Duchenne Connect in the United States) and worldwide registries for ultra-rare NMD such as Pompe [28] and GNE myopathy. Clinic-based recruitment and data entry, along with close working relationships with patient organizations, has allowed the CNDR to recruit individuals from many regions across Canada. However, there are some neuromuscular clinics not participating in the CNDR. For instance, recruitment in Quebec is lower than expected given its population (Fig. 1). This is in part due to the phased expansion of the CNDR, whereby institutions in Alberta and Ontario joined the registry prior to those in Quebec. The addition of affiliated clinics in these regions and expansion of French language materials used by the CNDR is expected to improve recruitment. Adult neuromuscular patients are slightly underrepresented (Fig. 1) in the CNDR. This may be in part due to increased participation of affiliated pediatric hospitals versus adult clinics. Expansion of the registry to additional adult clinics will improve ascertainment of this patient population. Additionally, as adult neuromuscular disorders are often less severe than pediatric cases the number of individuals routinely followed by neurologists and physiatrists in our network is fewer, resulting in lower recruitment. Expansion of recruitment through direct-to-patient avenues should help to mitigate this.
A rapid series of developments in the Canadian therapeutic landscape have recently led to improved case ascertainment in the registry. The recent approval of a new therapy for SMA, for example, has resulted in enrollment numbers almost doubling over a 6-month period. Patients likely see value in registering to receive information from the network of clinicians, while clinics are more likely to encourage registration to track outcome measures on treatment. Similarly, case ascertainment is quite high in DMD likely due to the large number of ongoing clinical trials. In contrast, case ascertainment is much lower for individuals with DM, likely in part due to the fewer evolving clinical options for treatment, along with psychosocial factors. Additionally, disease-specific registries exist both globally (e.g. The Duchenne Registry) and within Canada (Quebec DM registry), that may be competing for and/or duplicating registrations. As the current therapeutic landscape for neuromuscular disease is evolving, so too are the aims and use of the registry. Currently, the CNDR is expanding activities to capture more rigorous longitudinal RWE clinical outcomes for novel therapies to evaluate effectiveness in the Canadian population.
Clinical trials of novel therapies (e.g., those for familial ALS) will drive increased accessibility to genetic testing in certain diseases, and the CNDR has been instrumental in encouraging clinics to assist registered patients in obtaining proper genetic testing for both the index and non-index diseases. As there has been an increase in the number of disorders with available diagnostic genetic testing and available genetic-based therapies, awareness of and access to formal genetic counseling is an essential part of clinician patient discussions. The limited availability of genetic testing for some patient populations in the registry impacts both patient care and also the utility of registry data. Access to the results of genetic testing can also be limiting as these results are often not recorded at some of our affiliated-rehabilitation clinics. The CNDR is developing a direct-to-patient portal, to allow patient reporting of genetic results, when applicable, to foster improved patient care and data utility.
In this new era of therapeutic discovery for rare NM diseases, national disease registries and the associated network of affiliated clinics are essential tools to facilitate research and improve care. Ultimately, the availability of comprehensive real-world data in Canada has been an asset to all stakeholders in the NM community, and will likely become more so, as we strive to provide the best evidence-based management for individuals affected by NM disorders in the era of novel and emerging therapies.
FUNDING
The CNDR was supported by the ALS Society of Canada, Biogen, CureSMA Canada, Jesse’s Journey, the Marigold Foundation, Sanofi Genzyme, and the Starratt Family Foundation. The sponsors were not involved in study design, data collection, or any analysis or interpretation of data, or writing of this article.
DECLARATIONS
VH, JL, SD and LK wrote the manuscript. All authors reviewed and provided critical feedback on the manuscript. All authors reviewed and approved the final article.
DISCLOSURES
Dr. Hodgkinson reports personal fees from Biogen, Roche, Sarepta, outside the submitted work. Mr. Lounsberry reports personal fees from Biogen Canada Limited, outside the submitted work; Dr. M’Dahoma has nothing to disclose. Ms. Russell has nothing to disclose. Dr. Jewett has nothing to disclose. Dr. Benstead has nothing to disclose. Dr. Campbell reports grants, personal fees and other from Biogen, grants, personal fees and other from PTC Therapeutics, other from Acceleron, AMO, Catabasis, Pfizer, Sarepta, Wave, BMS. Scholar Rock, Roche, outside the submitted work; Dr. McCormick has nothing to disclose. Dr. Johnston declares personal fees from Mitsubishi Tanabe, and clinical trials from Alexion, Cytokinetics, Biogen, Orion, Mallinkrodt, Apellis, Alexion, AB Science; Dr. Nguyen reports other from Pfizer, other from Catabasis outside the submitted work. Dr. O’Ferrall reports personal fees from PTC Therapeutics nmDMD Advisory Board; grants from Sanofi Genzyme, grants from Acceleron, grants from Sanofi Genzyme, grants from Grifols, outside the submitted work. Dr. Oskoui reports grants from Fonds de Recherche Sante du Québec, grants from Kids Brain Health Network, grants from Canadian Institutes of Health Research, grants from SickKids Foundation, grants from Cerebral Palsy Alliance Research Foundation, other from Ionis, other from Biogen, other from Roche, other from Cytokinetics, personal fees and non-financial support from American Academy of Neurology, grants from Fondation du Grand Defi Pierre Lavoie, outside the submitted work; Dr. Brais has nothing to disclose. Dr. Briemberg has nothing to disclose. Dr. Bourque has nothing to disclose. Dr. Botez has nothing to disclose. Dr. Cashman reports ownership of ProMIS Neurosciences, outside the submitted work. Dr. Chapman has nothing to disclose. Dr. Chrestian received financial support to prepare review courses about SMA for neurologists, Physical therapists and Paediatricians. Dr. Crone reports personal fees from Biogen, outside the submitted work. Dr. Dobrowolski has nothing to disclose. Dr. Dojeiji has nothing to disclose. Dr. Dowling has nothing to disclose. Dr. Dupré has nothing to disclose. Dr. Genge reports grants from Biogen, Sanofi-Genzyme, CSL Behring, MTPA, AL-S Pharma, AB Sciences, Novartis, Wave Life Sciences, Argenx, Alexion, Avexis, Ackea, Cytokinetics, Hoffman-La Roche, outside the submitted work. Dr. Gonorazky reports personal fees from Biogen. Dr. Grant has nothing to disclose. Dr. Hasal has nothing to disclose. Dr. Izenberg has nothing to disclose. Dr. Kalra has nothing to disclose. Dr. Katzberg reports grants from CSL Behring, Takaeda and Muscular Dystrophy Canada and personal fees from Takada, Grifols, CSL Behring, Terumo, Biogen, Pfizer, Akcea, Alexion, Octapharma outside the submitted work. Dr. Krieger has nothing to disclose. Dr. Leung has nothing to disclose. Dr. Linassi has nothing to disclose. Dr. Lochmüller reports grants and personal fees from AMO Pharma, Biogen, Desitin, GW Pharma, Pfizer, PTC Therapeutics, Roche, Santhera, Sarepta, Satellos, Ultragenyx, outside the submitted work;. Dr. MacKenzie reports grants from Biogen, outside the submitted work. Dr. Mah has nothing to disclose. Dr. Marrero has nothing to disclose. Dr. Massie has nothing to disclose. Dr. Matte reports personal fees from Mitsubishi Tanabe, Biogen outside the submitted work, and grants from Biogen, Cytokinetics, Orion, Mallinckrodt outside the submitted work. Dr. McAdam reports other from Italfarmaco, outside the submitted work. Dr. McMillan has nothing to disclose. Dr. Melanson has nothing to disclose. Dr. Mezei reports personal fees from Genzyme, Alnylam, Pfizer, Akcea, CSL Behring, outside the submitted work. Dr. O’Connell reports personal fees from MT Pharma, grants and personal fees from Canopy Growth, personal fees from IPSEN, grants from Cytokinetics, grants from Mallincrodtk, grants from Orion, personal fees from Shoppers Drug Mart, outside the submitted work. Dr. Pfeffer has nothing to disclose. Dr. Phan has nothing to disclose. Dr. Plamondon reports other from Biogen, outside the submitted work; Dr. Poulin has nothing to disclose. Dr. Rodrigue has nothing to disclose. Dr. Schellenberg received grants for educational activities: Genzyme, Allergan, Mitsubishi-Tanabe; speakers honoraria: Genzyme, EMD Serono, Akcea, and advisory board: Mitsubishi-Tanabe, Alexion, Roche, Biogen, Akcea. Dr. Selby reports grants from Biogen/IONIS, personal fees from Biogen, outside the submitted work. Dr. Sheriko reports personal fees from Biogen, outside the submitted work. Dr. Shoesmith reports other from Biogen, outside the submitted work. Dr. Smith reports personal fees from Shire Pharmaceuticals, personal fees from Purdue Pharmaceuticals, personal fees from Janssen Pharmaceuticals, outside the submitted work. Dr. Taillon reports grants from Allergan, personal fees from Biogen outside the submitted work. Dr. Taylor has nothing to disclose. Dr. Venance has nothing to disclose. Dr. Warman has nothing to disclose. Dr. Worley reports personal fees from Cytokinetics, outside the submitted work. Dr. Zinman reports personal fees from Mitsubishi Tanabe and Biogen outside the submitted work. Dr. Korngut reports grants from Biogen, Sanofi Genzyme, ALS Canada, Jesse’s Journey, during the study, and personal fees from Alexion, Alnylam, Novartis, Mitsubishi Tanabe, Sarepta, Biogen, CSL Behring, outside the submitted work.
ACKNOWLEDGMENTS
We would like to thank the patients who participate and make the registry possible and our global collaborators at TREAT-NMD.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-200538.
REFERENCES
[1] | Deenen JC , Horlings CG , Verschuuren JJ , Verbeek AL , van Engelen BG . The epidemiology of neuromuscular disorders: A comprehensive overview of the literature. J Neuromuscul Dis. (2015) ;2: (1):73–85. |
[2] | Population density (people per sq. km of land area) [Internet]. Data. [cited 2020 May 12]. Available from: https://data.worldbank.org/indicator/EN.POP.DNST |
[3] | World Population Prospects - Population Division [Internet]. United Nations. [cited 2020 May 12]. Available from: https://population.un.org/wpp/Download/Standard/Population/. |
[4] | Korngut LM , Johnston M , Pringsheim T , Jette N . The future of neurological patient registries. Clinical Practice. 2014. |
[5] | Gaskin J , Gomes J , Darshan S , Krewski D . Burden of neurological conditions in Canada. Neurotoxicology. (2017) ;61: :2–10. doi: 10.1016/j.neuro.2016.05.001. |
[6] | Korngut L , Campbell C , Johnston M , Benstead T , Genge A , Mackenzie A , et al. The CNDR: Collaborating to translate new therapies for Canadians. Can J Neurol Sci. (2013) ;40: 5:698–704. |
[7] | Rowland LP , McLeod JG . Classification of neuromuscular disorders. J Neurol Sci. (1994) ;124: Suppl:109–30. |
[8] | Harris PA , Taylor R , Thielke R , Payne J , Gonzalez N , Conde JG . Research electronic data capture (REDCap)–a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. (2009) ;42: (2):377–81. doi: 10.1016/j.jbi.2008.08.010. |
[9] | Statistics Canada. [Internet] Table 17-10-0005-01 Population estimates on July 1st, by age and sex, 2018. [cited 2020 May 12] Available from: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501 |
[10] | Verhaart IEC , Robertson A , Leary R , McMacken G , Konig K , Kirschner J , et al. A multi-source approach to determine SMA incidence and research ready population. J Neurol. (2017) ;264: (7):1465–73. doi: 10.1007/s00415-017-8549-1. |
[11] | Mah JK , Korngut L , Dykeman J , Day L , Pringsheim T , Jette N . A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord. (2014) ;24: (6):482–91. doi: 10.1016/j.nmd.2014.03.008. |
[12] | Wei Y , Speechley KN , Zou G , Campbell C . Factors associated with health-related quality of life in children with duchenne muscular dystrophy. J Child Neurol. (2016) ;31: (7):879–86. doi: 10.1177/0883073815627879. |
[13] | Wei Y , Speechley KN , Zou G , Campbell C . The relationship between quality of life and health-related quality of life in young males with Duchenne muscular dystrophy. Dev Med Child Neurol. (2017) ;59: (11):1152–7. doi: 10.1111/dmcn.13574. |
[14] | Climans SA , Piechowicz C , Koopman WJ , Venance SL . Survey of Canadian myotonic dystrophy patients’ access to computer technology. Can J Neurol Sci. (2017) ;44: (5):567–71. doi: 10.1017/cjn.2017.47. |
[15] | Johnson NE , Ekstrom AB , Campbell C , Hung M , Adams HR , Chen W , et al. Parent-reported multi-national study of the impact of congenital and childhood onset myotonic dystrophy. Dev Med Child Neurol. (2016) ;58: (7):698–705. doi: 10.1111/dmcn.12948. |
[16] | Kullmann J , Pamphlett R . Does the index-to-ring finger length ratio (2D:4D) differ in amyotrophic lateral sclerosis (ALS)? Results from an international online case–control study. 2017. doi: 10.1136/bmjopen-2017-016924. |
[17] | Hodgkinson VL , Lounsberry J , Mirian A , Genge A , Benstead T , Briemberg H , et al. Provincial differences in the diagnosis and care of amyotrophic lateral sclerosis. Can J Neurol Sci. (2018) ;45: (6):652–9. doi: 10.1017/cjn.2018.311. |
[18] | Jackson-Tarlton CS , Benstead TJ , Doucette S . Correlating factors in the recommendation of feeding tubes in the nutritional management of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2016) ;17: (7-8):515–21. doi: 10.1080/21678421.2016.1213851. |
[19] | Echigoya Y , Lim KRQ , Melo D , Bao B , Trieu N , Mizobe Y , et al. Exons 45-55 skipping using mutation-tailored cocktails of antisense morpholinos in the DMD gene. Molecular Therapy. (2019) ;27: (11):2005–17. doi: 10.1016/j.ymthe.2019.07.012. |
[20] | Verhaart IEC , Robertson A , Wilson IJ , Aartsma-Rus A , Cameron S , Jones CC , et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet J Rare Dis. (2017) ;12: (1):124. doi: 10.1186/s13023-017-0671-8. |
[21] | Bladen CL , Salgado D , Monges S , Foncuberta ME , Kekou K , Kosma K , et al. The TREAT-NMD DMD global database: Analysis of more than 7,000 duchenne muscular dystrophy mutations. Hum Mutat. (2015) ;36: (4):395–402. doi: 10.1002/humu.22758. |
[22] | Koeks Z , Bladen CL , Salgado D , van Zwet E , Pogoryelova O , McMacken G , et al. Clinical outcomes in duchenne muscular dystrophy: A study of 5345 patients from the TREAT-NMD DMD global database. J Neuromuscul Dis. (2017) ;4: (4):293–306. doi: 10.3233/jnd-170280. |
[23] | Bladen CL , Thompson R , Jackson JM , Garland C , Wegel C , Ambrosini A , et al. Mapping the differences in care for 5,000 spinal muscular atrophy patients, a survey of 24 national registries in North America, Australasia and Europe. J Neurol. (2014) ;261: (1):152–63. doi: 10.1007/s00415-013-7154-1. |
[24] | Rodrigues MJ , O’Grady GL , Hammond-Tooke G , Kidd A , Love DO , Baker RK , et al. The New Zealand neuromuscular disease patient registry; five years and a thousand patients. J Neuromuscul Dis. (2017) ;4: (3):183–8. doi: 10.3233/jnd-170240. |
[25] | Roy AJ , Van den Bergh P , Van Damme P , Doggen K , Van Casteren V . Early stages of building a rare disease registry, methods and 2010 data from the Belgian Neuromuscular Disease Registry (BNMDR). Acta Neurol Belg. (2015) ;115: (2):97–104. doi: 10.1007/s13760-014-0320-0. |
[26] | van Engelen BG , van Veenendaal H , van Doorn PA , Faber CG , van der Hoeven JH , Janssen NG , et al. The Dutch neuromuscular database CRAMP (Computer Registry of All Myopathies and Polyneuropathies): Development and preliminary data. Neuromuscul Disord. (2007) ;17: (1):33–7. doi: 10.1016/j.nmd.2006.09.017. |
[27] | Wood L , Cordts I , Atalaia A , Marini-Bettolo C , Maddison P , Phillips M , et al. The UK myotonic dystrophy patient registry: Facilitating and accelerating clinical research. J Neurol. (2017) ;264: (5):979–88. doi: 10.1007/s00415-017-8483-2. |
[28] | Byrne BJ , Kishnani PS , Case LE , Merlini L , Muller-Felber W , Prasad S , et al. Pompe disease: Design, methodology, and early findings from the Pompe Registry. Mol Genet Metab. (2011) ;103: (1):1–11. doi: 10.1016/j.ymgme.2011.02.004. |

