
Infants Diagnosed with Spinal Muscular Atrophy and 4 SMN2 Copies through Newborn Screening – Opportunity or Burden?1
Abstract
Although the value of newborn screening (NBS) for early detection and treatment opportunity in SMA patients is generally accepted, there is still an ongoing discussion about the best strategy in children with 4 and more copies of the SMN2 gene. This gene is known to be the most important but not the only disease modifier.
In our SMA-NBS pilot project in Germany comprising 278,970 infants screened between January 2018 and November 2019 were 38 positive cases with a homozygous SMN1 deletion. 40% of them had 4 or more SMN2 copies. The incidence for homozygous SMN1 deletion was 1 : 7350, which is within the known range of SMA incidence in Germany.
Of the 15 SMA children with 4 SMN2 copies, one child developed physical signs of SMA by the age of 8 months. Reanalysis of the SMN2 copy number by a different test method revealed 3 copies. Two children had affected siblings with SMA Type III, who were diagnosed only after detection of the index patient in the NBS. One had a positive family history with an affected aunt (onset of disease at the age of 3 years). Three families were lost to medical follow up; two because of socioeconomic reasons and one to avoid the psychological stress associated with the appointments.
Decisions on how to handle patients with 4 SMN2 copies are discussed in the light of the experience gathered from our NBS pilot SMA program.
INTRODUCTION
Spinal muscular atrophy (SMA) is an autosomal recessive inherited neuromuscular disease with an incidence between 1 : 6,000 to 1 : 11,000 in newborns [1]. It is the most frequent genetic cause of death in infancy. The disease is caused by a lack of survival motor neuron (SMN) protein, which leads to an irreversible loss of motor neurons. SMA is classified by age of onset and the motor milestones achieved [2]. The clinical spectrum of severity is broad. In the most serious form of disease (type 0), children show severe weakness and respiratory insufficiency at birth. In SMA type 1 (Werdnig-Hoffmann disease) onset is during the first six months of life, and children are unable to sit and die of respiratory failure. SMA type 2 patients present clinically between 6 and 18 months. They achieve the ability to sit but cannot walk. SMA type 3 manifests after ambulation, but the ability of walking is frequently lost over time [1, 3].
The number of SMN2 copies was the first SMA modifier identified and is still the strongest known [4, 5]; higher SMN2 copy number correlates with a milder clinical phenotype [6–8]. However, the SMN2-copy number does not allow to precisely predict the course of the disease in a given individual [6, 9, 10]. An intermediate SMA type 2 phenotype is seen in 3–11% of patients with 4 SMN2 copies [6, 7].
Since a number of SMN-dependent and SMN-independent therapies are available [11], the timing of treatment is crucial for a good outcome [12, 13] and available data show that presymptomatic treatment is superior to treatment after onset of symptoms [14] experts agree that newborn screening should be established [15, 16–23]. Currently first pilot projects for a genetic NBS in SMA are underway [18, 24–28]. It has been shown that children with less than 4 SMN2 copies clearly have a benefit from an early presymptomatic treatment [14, 24].
There is an ongoing discussion about the management of children detected by NBS with 4 and more copies of SMN2. Genetic data of SMA patients’ relatives show that single cases exist [7] with a late onset of the disease although carrying a homozygous deletion of the SMN1-gene. Based on this finding, the American SMA NBS Multidisciplinary Working Group [16] recommended applying a strict follow-up strategy in these patients, well knowing that the overwhelming majority of patients with 4 and more SMN2 copies are expected to develop a loss of motoneurons which could be prevented by early treatment.
In this paper, we discuss the limitations and the medical and ethical issues associated with the recommended procedure, based on our experience with a cohort of patients with 4 SMN2 copies, who were detected in our NBS pilot project between January 2018 and November 2019. Specific problems are highlighted by 6 short case reports.
METHODS
The screening procedure has already been published in a previous manuscript, presenting the clinical and electrophysiological outcome after one year of newborn screening for SMA [24]. In short, the screening was performed in two federal states of Germany, Bavaria and North Rhine Westphalia, starting in January 2018. The investigation was performed from Guthrie-cards (dried blood spot test) taken for the routine NBS. An Informed consent especially for this investigation was given in advance and it could be rejected separately. Quantitative PCR was performed from DNA extracted from dried blood spots to screen for a homozygous deletion of exon 7 [29].
In case of a positive result, the neuromuscular center (Munich, Essen or Münster), which was closest to the hometown of the patient, contacted the parents and made an appointment within 1–2 days. MLPA-test from a new blood sample was used to confirm homozygous deletion of SMN1 gene and to determine the number of SMN2 copies. Since this is a semiquantitative method, the investigation was repeated by a second lab, in order to define the number of SMN2 copies as reliable as possible. MLPA was repeated again in 2020 with an improved MPLA-test. The patients were advised according to the treatment algorithm published by the American SMA NBS Multidisciplinary Working Group [16].
RESULTS
Of 278,970 newborns screened between January 15th 2018 and end of November 2019, 38 were identified to have a homozygous deletion of the SMN1 gene. In all but one, the number of SMN2 copies could be defined. The incidence for homozygous SMN1-deletion therefore was 1 : 7350. 15 of the 37 patients (40 %) had 4 SMN2 copies (none had > 4 copies). At the first examination all children were unaffected and had a normal muscle tone. All but one had present deep tendon reflexes. (in this patient, electromyography of the thigh excluded neurogenic changes) and a CHOP INTEND result > 35 points in the first two weeks of life. Similarly, the ulnar compound muscle action potential (CMAP) amplitudes were > 1 mV in all patients at that time. A strict follow-up strategy with regular clinical, electrophysiological and muscle ultrasound examinations was proposed to the families.
In 6 of the 15 “4 SMN2 copy”-patients, events with an impact on this proposed strategy occurred:
Case 1: Repeated neurological and neurophysiological examinations at intervals of 2 months were normal. At the age of 8 months, the parents noticed proximal weakness in the legs. The child was no longer able to hop when being held. Neurological exam revealed a proximal weakness of the legs with absent deep tendon reflexes. Electromyography showed no pathological spontaneous activity at rest but a neurogenic pattern with large amplitude long duration motor unit action potentials (MUAPs) was observed on voluntary activation. At this point, neurography of the ulnar and tibial nerve revealed normal compound muscle action potential. Muscle ultrasound was still normal.
Due to a delay in the confirmation of the insurance company to reimburse the treatment, it took another 5 weeks until medication with nusinersen could be started. By that time, the child was able to sit but unable to bear any weight on the legs. Sonographic examination showed a neurogenic pattern with atrophy and increased focal echogenicity (Fig. 1).
Fig. 1
Sonographic findings of quadriceps in case 1: (A) Age 2 months: normal echogenicity and clearly visible internal septa. (B) Age 14 months, 6 months after onset of disease: a pathologic increase in background echogenicity in comparison to subcutaneous fat tissue and less definable intramuscular septa. (C) Age 17 months, after 8 months of nusinersen therapy: partial recovery with increase in volume and normalisation of echogenicity in the rectus femoris.
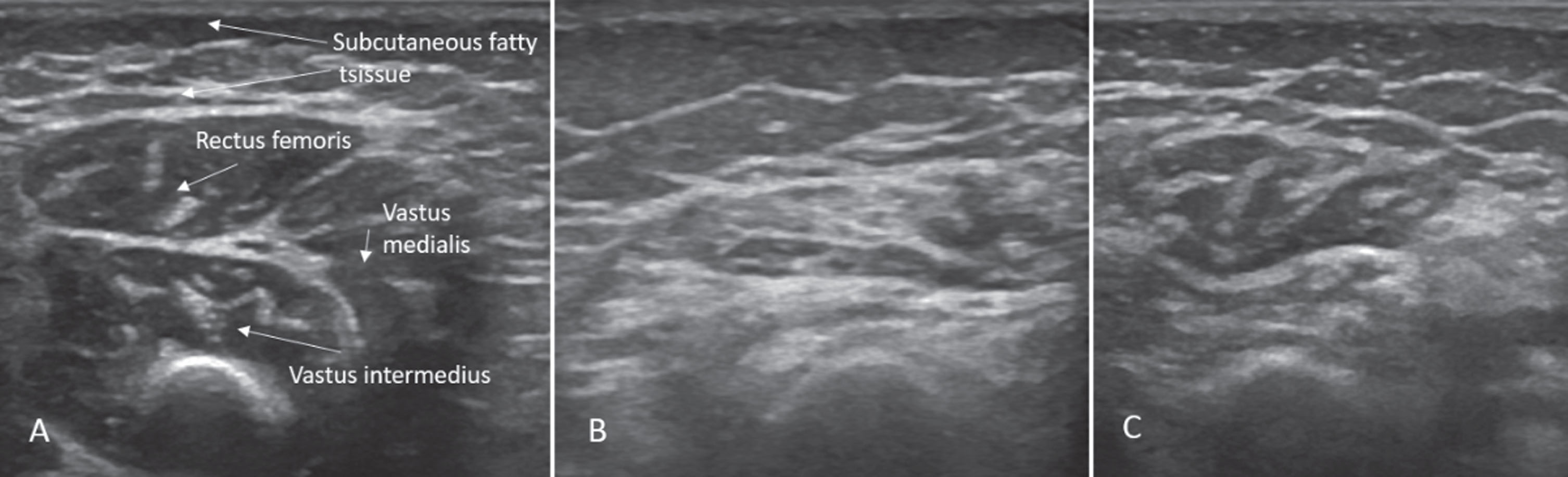
After the treatment was started the child started to develop new motor abilities again. Crawling and pushing up into a kneeing position was possible at 14 months, maintaining the kneeing position at 16 months, and walking with assistance with 17 months. This time, the sonographic picture showed improvement (Fig. 1) although still not completely normal.
Case 2: The patient was lost to follow up after 8 months. The family declined participation in the program despite extensive information about the rational of the suggested follow-up.
Case 3: The family agreed after repeated inquiries to perform confirmation diagnostics by the local pediatrician. They came to the neuromuscular center for information about the disease and the planned procedure. Nevertheless, they did not attend any recommended follow-up appointments despite repeated requests.
Case 4: The family announced their leave from the program at the age for the 13-month check-up, after the child had learned to walk independently. The reason for this was the parent’s extreme psychological stress that preceded the medical appointments.
Case 5: These parents reported that the child’s aunt suffered from a genetically proven SMA type 3 disease. Her number of SMN2 copies was unknown. She showed onset of symptoms at the age of 3 years. Based on the knowledge about the impact of SMA in daily life, the family was not willing to accept solely a strict-follow up strategy in the screened index patient. After approval of treatment by the insurance company, treatment was initiated at 6 months.
Cases 6: Initially, the family history seemed unremarkable. During the follow-up, the family reported that the now 5-year-old brother had a proximal weakness and a tremor of two years duration of unknown etiology. Genetic examination revealed a homozygous deletion in the SMN1 gene and therefore the diagnosis of SMA in this sibling. Treatment was initiated; the index patient is still untreated.
Case 7: Similar to case 6, the family history was initially regarded as negative. In the course of the follow-up, the family reported that the 6-year-old brother of the index patient had a tendency to tiptoe walk and to experience muscle fatigue. He was not able to run as fast as his peers. Genetic examination of the affected brother showed a homozygous deletion in the SMN1 gene proving the diagnosis of SMA. Clinical examination revealed an evolving proximal weakness with gait instability in the first minute of the 6-minute walk test. Electromyography showed a neurogenic pattern in both quadriceps’ muscles on voluntary activation (no spontaneous activity at rest). Although the muscle ultrasound was normal, treatment was initiated; the index patient is still untreated.
DISCUSSION
With the availability of new treatments for SMA and the technical feasibility of a population based newborn screening program, presymptomatic treatment of children with SMA has become a realistic option. As with many genetic disorders, a growing amount of data suggests presymptomatic treatment leading to far better outcomes than post-symptomatic therapy [30] does. However, in contrast to nearly all children with 2 or 3 copies of the SMN2 gene who develop the severe or intermediate phenotypes of SMA type 1 or type 2, the situation in patients with 4 and more copies is less clear. The high number of patients with 4 copies in the screened population emphasizes the necessity for a rational decision. The cases, which we present in this manuscript, highlight the specific challenges in this group.
The first patient (case 1), who developed an SMA type 2 age by 8 months encapsulates the ethical dilemma facing patients with 4 copies of SMN2. The risk of potential irreversible loss of motor neurons has to be balanced against the costs and risks of a possibly unnecessary treatment. Data from the early access program of nusinersen have shown that an early start of treatment is the most important factor influencing the outcome [31]. Based on the mechanism of SMN2-splicing modifiers like nusinersen, it is likely that patients with a high number of SMN2 copies profit most from therapy. The NURTURE study demonstrated that children with 3 copies of SMN2 have a better outcome than those with 2 SMN2 copies [30]. This gives hope that treatment in patients with 4 and more SMN2 copies will produce enough SMN-protein to live a normal life. On the other hand, an unnecessary therapeutic burden for the children, their families and the insurance system should be avoided.
Whereas most patients with 4 and more copies of SMN2 develop either type 2 or type 3 SMA, there are 32 reported individuals belonging to 22 families in the literature who show no signs despite a homozygous deletion in the SMN1 gene [32–38]. Most of these patients have 3-5 SMN2 copies and other known but separate SMA modifiers as well.
Currently, the initial attempt to solve this dilemma was to find consensus on the opinion of experts assessed via the Delphi questionnaire [16]. Unfortunately, the expert group did not find a definite answer to the question of which children should be treated. They propose that, when deciding on treatment initiation, the attending physician’s clinical judgment as well as the patient’s and/or the patient’s family’s wishes, should be factored in [16]. In cases where treatment for individuals with 4 SMN2 copies was deferred, the advice was to monitor closely and to start treatment as soon as weakness was detected.
The authors suggest that CMAPs might be used as a surrogate parameter to detect motor unit loss. It has been shown that CMAPs can reliably be measured and correlate well with the Hammersmith Functional Motor Scale Expanded for SMA [39]. It is known that CMAP amplitude clearly correlates with the severity of SMA in severe cases. The CMAPs have been used in clinical trials to characterize the amount of preserved motor neuron reserve at the time of treatment initiation in children with early onset SMA [40]. However, in patients with SMA type 3 the amplitudes are frequently in a normal range [41]. Considering this, our findings indicate that CMAPs are not a useful surrogate parameter when determining at what point treatment should be initiated in the presymptomatic patients. Whereas low CMAP is an indicator of a poor response to treatment in type 1 patients, in contrast this parameter is not sufficiently sensitive in late onset patients due to proximal muscle involvement in the milder forms of SMA. Other neurophysiological parameter like MUNE, which are used in ALS patients, are only informative in a very young pediatric group if done by very experienced examiners.
In contrast, electromyography was able to detect the neurogenic process in our patients, but our experience shows, that the families do not accept this invasive method during a long-term follow up with examinations every 3-6 months.
Non-compliant patients who fail to observe the follow-up program (cases 2-4) represent a particular challenge. The decision not to follow the recommended examinations may be based on an insufficient understanding of the problem in a hitherto healthy child, economic challenges and distance to the expert center. In two families, socioeconomic status and psychological stress in another mainly contributed to the lack of follow through. Similar problems are also observed in long-term established screening programs, which have to deal with clinical phenomena that are not immediately apparent to the caregivers. In phenylketonuria between 5 and 15 % of the patients leave the preventative treatment program during the first two years of life [42, 43]. They do so for a wide range of reasons including sociocultural, economic and religious beliefs, which have to be addressed, but which sometimes nonetheless cannot be solved or vanished [44].
Cases 5, 6 and 7 highlight aspects occurring to the “wait and see”-strategy with other family members suffering from SMA. Parents with known SMA cases in their family will naturally elect to treat their SMN-/- child. The expectation to treat the children as early as possible was one of the most important arguments for parents of children with SMA to vote for a NBS in SMA [45].
In clinical practice, the strict follow-up leads to a significant psychological stress for the family, which might detrimentally influence the psychosocial development of the child. One possible solution might be to not report patients with four SMN2 copies a priori. However, our case 1 is a clear argument against such a strategy. Due to the rigorous monitoring, the time between onset of symptoms and start of treatment was less than 2 months. In contrast, review of the literature shows the median latency between symptom onset and correct diagnosis to be 11 months in children with SMA type 2 and even longer in type 3 patients [46]. It is clearly possible, even likely, that the partial recovery in our patient was due to the early intervention. Experimental data suggest that a partial recovery of motor neurons or motor endplates might be possible if treatment is started early enough [47].
The question remains whether it is prudent to await the onset of symptoms before starting treatment in the subgroup with 4 SMN2 copies. The most important reason for this strategy is to avoid unnecessary, potentially harmful treatment for a relevant number of children who might stay asymptomatic if untreated. If this is the case, the incidence rates following NBS should be much higher than the incidence, which is known in the general population. The incidence in our cohort was 1 : 7350 which is nearly identical with the figure of 1 : 7353 which was found in symptomatic patients in Germany in 2014 [48]. This is a strong argument that the majority of patients with a homozygous SMN1 deletion detected in the NBS will become symptomatic regardless of their SMN2 copy number. With this in mind, early treatment in the first year of life should be considered.
Looking at the distribution of the SMN2 copy numbers, 40% of our patients had 4 SMN2 copies. Over the past two years this number has stabilized and seems to reflect the prevalence of this subgroup in a German population. This number is slightly higher than the figures, which have been published in cohorts describing symptomatic patients and their relatives. In a large cohort of mainly German patients 11.4% (43/375) showed 4 SMN2 copies of which 18.6% developed SMA type 2 and 81.4% SMA type 3 [7], In another cohort of Spanish patients, 8 % showed 4 and more SMN2 copies whereas 42% had 3 copies [6]. A meta-analysis of all patients in the literature revealed that 38 % of SMA patients had 4 and more copies of SMN2 [6].
One of the major reasons to explain this difference might be the fact that, especially when estimation of the SMN2 copy number was introduced into clinical practice, the interrater reliability between different labs was quite low. A recent paper showed that in 45% of the patients the results were discordant when the test was repeated by a single lab with the now available methods [49].
The methodological problem with the secure determination of the number of SMN2 copies has to be addressed. A recent study shows the discrepancy between standard qPCR and the more accurate ddPCR [50, 51]. In this pilot project, the problem was circumvented by carrying out confirmation diagnostics via MLPA from whole blood (multiple determination in case of uncertainties), and independent confirmation of the result by a second laboratory. Nevertheless in one case the initial copy number had to be revised after a different method was applied. As long as the number of SMN2 copies is part of the treatment algorithm, care has to be taken to standardize the estimation of the number of SMN2 copies between all laboratories, which are involved in national screening programs.
Cases 6 and 7 highlight the important issue of missed diagnoses in mildly affected patients. In both cases, siblings with clear neuromuscular signs and symptoms were not correctly diagnosed before positive findings in the NBS screening for first-degree relatives. The number of unreported cases in patients with less pronounced symptoms is unknown. Not infrequently, such children may be considered as unmotivated even lazy when compared with their peers. Keeping in mind that SMA is a progressive disorder [52] valuable time will be lost.
Moreover, with new therapies such as gene therapy coming along, a therapeutic intervention is only possible in early childhood [53]. AAV based SMA gene therapy (Onasemnogene abeparvovec) is already approved in US for children below 2 years [54] and it is expected to be approved by the EMA in 2020. Furthermore, the older the children are at the time of application the higher is the risk that they have developed antibodies against scAAV9 and therefore effectively precludes this special therapy [55].
It is also well known that SMA is particularly affecting the neuromuscular junction (NMJ). From animal and human studies, we know that SMN is particularly important in NMJ maturation and maintenance and SMN expression and requirement is high early in development (<P17 in mice and < 2 years in human) [56, 57]. This lets us assume that an early treatment limits the damage and helps the recovery, which is another argument for early treatment.
SUMMARY AND CONCLUSION
While the value of NBS for early detection and treatment of SMA patients is generally well accepted, there is still an ongoing discussion about the best strategy in SMA children with 4 copies of the SMN2 gene, the most important modifier of the disease. Up to now, a strict follow-up strategy still can be justified. Nevertheless, compliance problems, decisions based on personal experience of families and the fact that the prognosis of part of the children might be worsened by this strategy, will presumably modify this strategy in the future.
CONFLICT OF INTEREST/FINANCIAL DISCLOSURES
1. Serving on a scientific advisory board or data safety monitoring board
2. Gifts (other than travel or compensation for consulting or for educational efforts) worth more than USD $1000
3. Funding for travel or speaker honoraria to the individual from a commercial or non-profit entity not included in the study funding [Exclude CME activities and Grand Rounds].
4. Serving as a journal editor, an associate editor, or editorial advisory board member. This may include a journal published by your national medical/scientific organization. Please include regardless of whether you receive compensation.
5. Patents issued or pending.
6. Publishing Royalties (do not include honoraria for occasional writing)
7. Employment. If you are currently employed by a commercial entity, please disclose below. In addition, if your past employment at a commercial entity is directly related to this manuscript, please disclose below.
8. Consultancies
9. Speakers’ bureau
10. Other activities not covered in designations above (if in doubt, provide full disclosure)
11. Some published work has potential for financial gain for the study investigators or the sponsor. The following question seeks to provide transparency regarding any financial benefits to investigators or sponsors.
Heike Kölbel, Oliver Schwartz, Astrid Blaschek, Bernhard Olgemöller, Uta Nennstiel, Christine Müller, Katja Eggermann and Iris Hannibal have nothing to declare.
Siegfried Burggraf, Wulf Röschinger, Jürgen Durner and Marc Becker are employed by/owner of a commercial entity (Laboratory Becker and colleagues MVZ GbR, Führichstraße 70, 81871 München, Germany).
Dieter Gläser: employed by/owner of a commercial entity (Genetikum ®, Wegenerstr. 15, 89231 Neu-Ulm, Germany).
Ulrika Schara is serving on a scientific advisory board or data safety monitoring board for Biogen, Avexis and Novartis.
Brunhilde Wirth is serving on a scientific advisory board or data safety monitoring board for SMA Europe, and received travel and speaker honoraria from Biogen.
Katharina Vill received travel and speaker honoraria from Biogen.
Wolfgang Müller-Felber is serving on a scientific advisory board or data safety monitoring board for Biogen, Avexis, PTC, Sanofi-Aventis and Cytokinetics and received travel and speaker honoraria from Biogen, Avexis, PTC and Sanofi-Aventis.
ACKNOWLEDGMENTS
Until May 2019, the NBS-study was funded by the German Cystinosis Foundation (Cystinose Stiftung, DSZ-Regional Office Munich, Widenmayerstr. 10, 80538 Munich, Germany). The funders had no influence in study design, interpretation and publication of these data.
REFERENCES
[1] | Mercuri E , Finkel RS , Muntoni F , Wirth B , Montes J , Main M , Mazzone ES , Vitale M , Snyder B , Quijano-Roy S , et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscular disorders: NMD. (2018) ;28: :103–15. |
[2] | Wang CH , Finkel RS , Bertini ES , Schroth M , Simonds A , Wong B , Aloysius A , Morrison L , Main M , Crawford TO , et al. Consensus statement for standard of care in spinal muscular atrophy. Journal of Child Neurology. (2007) ;22: :1027–49. |
[3] | Finkel RS , Mercuri E , Meyer OH , Simonds AK , Schroth MK , Graham RJ , Kirschner J , Iannaccone ST , Crawford TO , Woods S , et al. Diagnosis andmanagement of spinalmuscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscular disorders : NMD. (2018) ;28: :197–207. |
[4] | Wirth B , Garbes L , Riessland M . How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic approaches. Current Opinion in Genetics & Development. (2013) ;23: :330–8. |
[5] | Prior TW , Krainer AR , Hua Y , Swoboda KJ , Snyder PC , Bridgeman SJ , Burghes AHM , Kissel JT . A positive modifier of spinal muscular atrophy in the SMN2 gene. American Journal of Human Genetics. (2009) ;85: :408–13. |
[6] | Calucho M , Bernal S , Alias L , March F , Vencesla A , Rodriguez-Alvarez FJ , Aller E , Fernandez RM , Borrego S , Millan JM , et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscular disorders: NMD. (2018) ;28: :208–15. |
[7] | Feldkotter M , Schwarzer V , Wirth R , Wienker TF , Wirth B , Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. American Journal of Human Genetics. (2002) ;70: :358–68. |
[8] | Wirth B , Brichta L , Schrank B , Lochmuller H , Blick S , Baasner A , Heller R . Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Human Genetics. (2006) ;119: :422–8. |
[9] | Cusco I , Barcelo MJ , Rojas-Garcia R , Illa I , Gamez J , Cervera C , Pou A , Izquierdo G , Baiget M , Tizzano EF . SMN2 copy number predicts acute or chronic spinal muscular atrophy but does not account for intrafamilial variability in siblings. Journal of Neurology. (2006) ;253: :21–25. |
[10] | Ogino S , Gao S , Leonard DG , Paessler M , Wilson RB . Inverse correlation between SMN1 and SMN2 copy numbers: evidence for gene conversion from SMN2 to SMN1. European Journal of Human Genetics: EJHG. (2003) ;11: :275–7. |
[11] | Shorrock HK , Gillingwater TH , Groen EJN . Overview of Current Drugs and Molecules in Development for Spinal Muscular Atrophy Therapy. Drugs. (2018) ;78: :293–305. |
[12] | Tizzano EF , Finkel RS . Spinal muscular atrophy: A changing phenotype beyond the clinical trials. Neuromuscular disorders: NMD. 2017. |
[13] | Dangouloff T , Servais L . Clinical evidence supporting early treatment of patients with spinal muscular atrophy: Current perspectives. Ther Clin Risk Manag. (2019) ;15: :1153–61. |
[14] | De Vivo DC , Bertini E , Swoboda KJ , Hwu WL , Crawford TO , Finkel RS , Kirschner J , Kuntz NL , Parsons JA , Ryan MM , et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul Disord. (2019) ;29: :842–56. |
[15] | Phan HC , Taylor JL , Hannon H , Howell R . Newborn screening for spinal muscular atrophy: Anticipating an imminent need. Semin Perinatol. (2015) ;39: :217–29. |
[16] | Glascock J , Sampson J , Haidet-Phillips A , Connolly A , Darras B , Day J , Finkel R , Howell RR , Klinger K , Kuntz N , et al. Treatment algorithm for infants diagnosed with spinal muscular atrophy through newborn screening. J Neuromuscul Dis. 2018. |
[17] | Boardman FK , Sadler C , Young PJ . Newborn genetic screening for spinal muscular atrophy in the UK: The views of the general population. Molecular Genetics & Genomic Medicine. (2018) ;6: :99–108. |
[18] | Kraszewski JN , Kay DM , Stevens CF , Koval C , Haser B , Ortiz V , Albertorio A , Cohen LL , Jain R , Andrew SP , et al. Pilot study of population-based newborn screening for spinal muscular atrophy in New York state. Genet Med. (2018) ;20: :608–13. |
[19] | Govoni A , Gagliardi D , Comi GP , Corti S . Time is motor neuron: Therapeutic window and its correlation with pathogenetic mechanisms in spinal muscular atrophy. Molecular Neurobiology. (2018) ;55: :6307–18. |
[20] | Tizzano EF and Zafeiriou D . Prenatal aspects in spinalmuscular atrophy: Fromearly detection to early presymptomatic intervention. European journal of paediatric neurology : EJPN : Official Journal of the European Paediatric Neurology Society. 2018. |
[21] | Farrar MA , Park SB , Vucic S , Carey KA , Turner BJ , Gillingwater TH , Swoboda KJ , Kiernan MC . Emerging therapies and challenges in spinal muscular atrophy. Annals of Neurology. (2017) ;81: :355–68. |
[22] | Vill K , Blaschek A , Schara U , Kolbel H , Hohenfellner K , Harms E , Olgemoller B , Walter MC , Muller-Felber W . [Spinal muscular atrophy: Time for newborn screening?]. Nervenarzt. (2017) ;88: :1358–66. |
[23] | Boardman FK , Young PJ , Griffiths FE . Newborn screening for spinal muscular atrophy: The views of affected families and adults. American Journal of Medical Genetics Part A. (2017) ;173: :1546–61. |
[24] | Vill K , Kolbel H , Schwartz O , Blaschek A , Olgemoller B , Harms E , Burggraf S , Roschinger W , Durner J , Glaser D , et al. One year of newborn screening for SMA –results of a German Pilot Project. J Neuromuscul Dis. (2019) ;6: :503–15. |
[25] | Boemer F , Caberg JH , Dideberg V , Dardenne D , Bours V , Hiligsmann M , Dangouloff T , Servais L . Newborn screening for SMA in Southern Belgium. Neuromuscular disorders: NMD. (2019) ;29: :343–9. |
[26] | Chien YH , Chiang SC , Weng WC , Lee NC , Lin CJ , Hsieh WS , Lee WT , Jong YJ , Ko TM , Hwu WL . Presymptomatic Diagnosis of Spinal Muscular Atrophy Through Newborn Screening. The Journal of Pediatrics. 2017 |
[27] | Czibere L , Burggraf S , Fleige T , Glück B , Keitel LM , Landt O , Durner J , Röschinger W , Hohenfellner K , Wirth B , et al. High-throughput genetic newborn screening for spinal muscular atrophy by rapid nucleic acid extraction from dried blood spots and 384-well qPCR. European journal of human genetics: EJHG. (2020) ;28: :23–30. |
[28] | Chien YH , Chiang SC , Weng WC , Lee NC , Lin CJ , Hsieh WS , Lee WT , Jong YJ , Ko TM , Hwu WL . Presymptomatic diagnosis of spinal muscular atrophy through newborn screening. J Pediatr. (2017) ;190: :124–29 e121. |
[29] | Czibere L , Burggraf S , Fleige T , Gluck B , Keitel LM , Landt O , Durner J , Roschinger W , Hohenfellner K , Wirth B , et al. High-throughput genetic newborn screening for spinal muscular atrophy by rapid nucleic acid extraction from dried blood spots and 384-well qPCR. European Journal of Human Genetics : EJHG. 2019. |
[30] | De Vivo DC , Bertini E , Swoboda KJ , Hwu WL , Crawford TO , Finkel RS , Kirschner J , Kuntz NL , Parsons JA , Ryan MM , et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscular disorders : NMD. 2019. |
[31] | Pechmann A , Langer T , Schorling D , Stein S , Vogt S , Schara U , Kolbel H , Schwartz O , Hahn A , Giese K , et al. Evaluation of children with SMA Type 1 under treatment with nusinersen within the expanded access program in Germany. J Neuromuscul Dis. (2018) ;5: :135–143. |
[32] | Cobben JM , van der Steege G , Grootscholten P , de Visser M , Scheffer H , Buys CH . Deletions of the survival motor neuron gene in unaffected siblings of patients with spinal muscular atrophy. American Journal of Human Genetics. (1995) ;57: :805–8. |
[33] | Cuscó I , Barceló MJ , Rojas-García R , Illa I , Gámez J , Cervera C , Pou A , Izquierdo G , Baiget M , Tizzano EF . SMN2 copy number predicts acute or chronic spinal muscular atrophy but does not account for intrafamilial variability in siblings. Journal of Neurology. (2006) ;253: :21–25. |
[34] | Hahnen E , Forkert R , Marke C , Rudnik-Schoneborn S , Schonling J , Zerres K , Wirth B . Molecular analysis of candidate genes on chromosome 5q13 in autosomal recessive spinal muscular atrophy: Evidence of homozygous deletions of the SMN gene in unaffected individuals. Human Molecular Genetics. (1995) ;4: :1927–33. |
[35] | Oprea GE , Krober S , McWhorter ML , Rossoll W , Muller S , Krawczak M , Bassell GJ , Beattie CE , Wirth B . Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. (2008) ;320: :524–7. |
[36] | Riessland M , Kaczmarek A , Schneider S , Swoboda KJ , Löhr H , Bradler C , Grysko V , Dimitriadi M , Hosseinibarkooie S , Torres-Benito L , et al. Neurocalcin delta suppression protects against spinal muscular atrophy in humans and across species by restoring impaired endocytosis. American Journal of Human Genetics. (2017) ;100: :297–315. |
[37] | Jedrzejowska M , Borkowska J , Zimowski J , Kostera-Pruszczyk A , Milewski M , Jurek M , Sielska D , Kostyk E , Nyka W , Zaremba J , et al. Unaffected patients with a homozygous absence of the SMN1 gene. European Journal of Human Genetics: EJHG. (2008) ;16: :930–4. |
[38] | Heesen L , Peitz M , Torres-Benito L , Hölker I , Hupperich K , Dobrindt K , Jungverdorben J , Ritzenhofen S , Weykopf B , Eckert D , et al. Plastin 3 is upregulated in iPSC-derived motoneurons from asymptomatic SMN1-deleted individuals. Cell Mol Life Sci. (2016) ;73: :2089–2104. |
[39] | Lewelt A , Krosschell KJ , Scott C , Sakonju A , Kissel JT , Crawford TO , Acsadi G , D’Anjou G , Elsheikh B , Reyna SP , et al. Compound muscle action potential and motor function in children with spinal muscular atrophy. Muscle & Nerve. (2010) ;42: :703–8. |
[40] | Finkel RS , Mercuri E , Darras BT , Connolly AM , Kuntz NL , Kirschner J , Chiriboga CA , Saito K , Servais L , Tizzano E , et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. (2017) ;377: :1723–32. |
[41] | Kang PB , Gooch CL , McDermott MP , Darras BT , Finkel RS , Yang ML , Sproule DM , Chung WK , Kaufmann P , de Vivo DC , et al. The motor neuron response to SMN1 deficiency in spinal muscular atrophy. Muscle & Nerve. (2014) ;49: :636–44. |
[42] | Jurecki ER , Cederbaum S , Kopesky J , Perry K , Rohr F , Sanchez-Valle A , Viau KS , Sheinin MY , Cohen-Pfeffer JL . Adherence to clinic recommendations among patients with phenylketonuria in the United States. Mol Genet Metab. (2017) ;120: :190–7. |
[43] | García MI , Araya G , Coo S , Waisbren SE , de la Parra A . Treatment adherence during childhood in individuals with phenylketonuria: Early signs of treatment discontinuation. Mol Genet Metab Rep. (2017) ;11: :54–58. |
[44] | MaCdonald A , van Rijn M , Feillet F , Lund AM , Bernstein L , Bosch AM , Gizewska M , van Spronsen FJ . Adherence issues in inherited metabolic disorders treated by low natural protein diets. Ann Nutr Metab. (2012) ;61: :289–95. |
[45] | Wood MF , Hughes SC , Hache LP , Naylor EW , Abdel-Hamid HZ , Barmada MM , Dobrowolski SF , Stickler DE , Clemens PR . Parental attitudes toward newborn screening for Duchenne/Becker muscular dystrophy and spinal muscular atrophy. Muscle & Nerve. (2014) ;49: :822–8. |
[46] | Lin CW , Kalb SJ , Yeh WS . Delay in diagnosis of spinal muscular atrophy: A systematic literature review. Pediatric Neurology. 2015. |
[47] | Govoni A , Gagliardi D , Comi GP , Corti S . Time is motor neuron: therapeutic window and its correlation with pathogenetic mechanisms in spinal muscular atrophy. Mol Neurobiol. (2018) ;55: :6307–18. |
[48] | Konig K , Pechmann A , Thiele S , Walter MC , Schorling D , Tassoni A , Lochmuller H , Muller-Reible C , Kirschner J . De-duplicating patient records from three independent data sources reveals the incidence of rare neuromuscular disorders in Germany. Orphanet Journal of Rare Diseases. (2019) ;14: :152. |
[49] | Schorling DC , Becker J , Pechmann A , Langer T , Wirth B , Kirschner J . Discrepancy in redetermination of SMN2 copy numbers in children with SMA. Neurology. (2019) ;93: :267–9. |
[50] | Stabley DL , Harris AW , Holbrook J , Chubbs NJ , Lozo KW , Crawford TO , Swoboda KJ , Funanage VL , Wang W , Mackenzie W , et al. SMN1 and SMN2 copy numbers in cell lines derived from patients with spinal muscular atrophy as measured by array digital PCR. Mol Genet Genomic Med. (2015) ;3: :248–57. |
[51] | Stabley DL , Holbrook J , Harris AW , Swoboda KJ , Crawford TO , Sol-Church K , Butchbach MER . Establishing a reference dataset for the authentication of spinal muscular atrophy cell lines using STR profiling and digital PCR. Neuromuscul Disord. (2017) ;27: :439–46. |
[52] | Wadman RI , Wijngaarde CA , Stam M , Bartels B , Otto LAM , Lemmink HH , Schoenmakers M , Cuppen I , van den Berg LH , van der Pol WL . Muscle strength and motor function throughout life in a cross-sectional cohort of 180 patients with spinal muscular atrophy types 1c-4. European Journal of Neurology. (2018) ;25: :512–8. |
[53] | Mendell JR , Al-Zaidy S , Shell R , Arnold WD , Rodino-Klapac LR , Prior TW , Lowes L , Alfano L , Berry K , Church K , et al. Single-dose gene-replacement therapy for spinal muscular atrophy. The New England Journal of Medicine. (2017) ;377: :1713–22. |
[54] | Hoy SM . Onasemnogene abeparvovec: First global approval. Drugs. (2019) ;79: :1255–62. |
[55] | Nidetz NF , McGee MC , Tse LV , Li C , Cong L , Li Y , Huang W . Adeno-associated viral vector-mediated immune responses: Understanding barriers to gene delivery. Pharmacol Ther. (2019) ;107453. |
[56] | Kariya S , Obis T , Garone C , Akay T , Sera F , Iwata S , Homma S , Monani UR . Requirement of enhanced survival motoneuron protein imposed during neuromuscular junction maturation. The Journal of Clinical Investigation. (2014) ;124: :785–800. |
[57] | Sumner CJ , Crawford TO . Two breakthrough gene-targeted treatments for spinal muscular atrophy: challenges remain. The Journal of Clinical Investigation. (2018) ;128: :3219–27. |


