
Evaluation of Genotypes and Epidemiology of Spinal Muscular Atrophy in Greece: A Nationwide Study Spanning 24 Years
Abstract
Background:
Promising genetic treatments targeting the molecular defect of severe early-onset genetic conditions are expected to dramatically improve patients’ quality of life and disease epidemiology. Spinal Muscular Atrophy (SMA), is one of these conditions and approved therapeutic approaches have recently become available to patients.
Objective:
Analysis of genetic and clinical data from SMA patients referred to the single public-sector provider of genetic services for the disease throughout Greece followed by a retrospective assessment in the context of epidemiology and genotype-phenotype associations.
Methods:
Molecular genetic analysis and retrospective evaluation of findings for 361 patients tested positive for SMA- and 862 apparently healthy subjects from the general population. Spearman rank test and generalized linear models were applied to evaluate secondary modifying factors with respect to their impact on clinical severity and age of onset.
Results:
Causative variations- including 5 novel variants- were detected indicating a minimal incidence of about 1/12,000, and a prevalence of at least 1.5/100,000. For prognosis a minimal model pertaining disease onset before 18 months was proposed to include copy numbers of NAIP (OR = 9.9;95% CI, 4.7 to 21) and SMN2 (OR = 6.2;95% CI, 2.5–15.2) genes as well as gender (OR = 2.2;95% CI, 1.04 to 4.6).
Conclusions:
This long-term survey shares valuable information on the current status and practices for SMA diagnosis on a population basis and provides an important reference point for the future assessment of strategic advances towards disease prevention and health care planning.
INTRODUCTION
Spinal Muscular Atrophy (SMA) is considered the third most common severe autosomal recessive disease in Greece, following Haemoglobinopathies and Cystic Fibrosis [1]. The estimated incidence of SMA among Caucasians ranges between 1:6,000 –1:11,000 live births, while carrier frequency is estimated to range between 1/38 and 1/50 [2–4]. The most distinct disease characteristic is the progressive wasting of lower motor neurons situated in the anterior horn of the spinal cord resulting mainly in muscle weakness and atrophy [5, 6]. Given the extreme clinical spectrum of the disease (from floppy infant to mild adulthood weakness) five main categories (SMA 0, I, II, III, IV) have been generally adapted so far [7].
The gene involved in all forms of SMA is the Survival of Motor Neuron 1 (SMN1) gene located within an inverted repeat region on chromosomal locus 5q13.2, containing the highly homologous SMN2 gene and other genes such as NAIP, GTF2H2 and H4F5 [4, 8]. The SMN protein appears to have a multifunctional role. It has the ability to bind with small nuclear ribonucleoproteins (snRNPs) and drive the biogenesis of small nuclear RNAs, particularly in discreet foci nuclear structures called “gems” [9].The SMN1 gene paralog, SMN2, contains a C > T transition in exon 7 (c.840C>T), which alters the splicing pattern of SMN2 mRNA, thus severely reducing the levels of full-length SMN protein produced [10].
In about 95% of SMA patients the disease is caused by a typical homozygous deletion of exon 7, or exons 7 and 8, of the SMN1 gene [8]. In a minority of patients, the disease is caused by compound heterozygosity of a typical SMN1 deletion and a small gene lesion within the SMN1 gene. The extreme clinical heterogeneity noticed is attributed to disease modifiers, several of which are located within and near to the SMN gene locus [11]. The most prominent involves the number of SMN2 copies, whereby multiple SMN2 genes contribute to the synthesis of more (supplementary) functional protein, ameliorating the clinical outcome [12–14]. However, as this is not universal, the implication of additional disease-modifying factors and variants within other syntenic genes, such as NAIP, GTF2H2, and H4F5 has been suggested [4, 11]. SMN2 gene variants, such as c.859G>C (p.Gly287Arg), or newly occurring gene conversion events have also been proposed to contribute to the overall clinical outcome, but data are currently limited [15, 16].
This study reports on the molecular genetics of a nationwide cohort of patients diagnosed with Spinal Muscular Atrophy over a period of 24 years. It provides insights into genotype -phenotype correlations in SMA patients, the disease epidemiology and the efficacy of the networks between tertiary health centers in Greece. This serves as a basis for evaluating the natural history of the disease and provides a key resource in an era when the implementation of genetic therapeutics and expansion of carrier screening for SMA in the general population are being introduced.
MATERIALS AND METHODS
Subjects
This study focuses on the molecular genetic analysis of the SMN1 gene locus of healthy and affected subjects along with a retrospective evaluation of genetic findings, phenotypes and disease frequency.
Genotyping was performed in the Laboratory of Medical Genetics (National and Kapodistrian University of Athens) spanning a period of 24 years (September 1995 to December 2018). The phenotype-genotype data reported are restricted to the 361 individual patients who tested positive for SMA. In order to establish the carrier rate of SMA in Greece, 862 individuals from the general population were also evaluated, anonymously. Informed consent for genetic analysis was provided by all individuals tested (or their legal guardians) to the laboratory or the referring clinicians. In the context of the retrospective evaluation of patients’ records, data were mined for statistical purposes and/or phenotype –genotype correlations. The data were de-identified and stored in an electronic file using numeric codes for each individual (approved by “Aghia Sophia” Children’s Hospital Scientific and Ethics Committee) to facilitate retrospective data analysis, for which according to Greek legislation and Recital 50 of the new EU General Data Protection Regulation (Directive 95/46/EC) no additional consent is required.
All patients, even those born in private maternity hospitals, were eventually referred to public tertiary care centers in various cities throughout Greece (Athens, Thessaloniki, Larissa, Ioannina, Herakleion, Patras) (Fig. 1). As indicated from the medical records, patients from remote areas (including the Ionian and Aegean islands) were referred from their local hospitals or private pediatricians to the 23 major and highly specialized hospital centers in Greece, rather than to a genetic center directly.
Fig.1
Geographical distribution of the main national medical centers for SMA referrals.
For every case (even for those before 1995), the referring clinicians sent a blood specimen and clinical information, the latter enabling classification into one of the 5 subtypes of SMA. Both the clinical information and the genetic results were stored in the laboratory medical records for each patient and updated if with any new clinical and or laboratory information, including DNA retesting to evaluate SMN2 copy number estimation. The disease classification was based on the age of onset, the motor milestones achieved and the maximal motor function [7]. The age of onset was the age at which one of the disease symptoms was reported by the clinician and in some cases by the patients/families themselves (type III and IV).
Methods
Genomic DNA was extracted from peripheral blood lymphocytes, amniotic cells or chorionic villi, following standard procedures (Qiagen Bio-robot M48, QIAamp DNA Blood mini Kit, QIAGEN, Cat No 51104).
The previously described method of Restriction Fragment Length Polymorphism analysis (RFLPs) was applied to investigate the presence of a homozygous exon 7 or/and exon 8 deletion in SMN1 [17].
The number of exon copies deriving from both the SMN1 and SMN2 genes, was evaluated using Multiplex Ligation-dependent Probe Amplification (MLPA) analysis, as per the manufacturer’s instructions (SALSA MLPA probe-mix P021-B1, MRC Holland). GeneMarker®V1.15 software was used for the assessment of MLPA findings, allowing both the calculation of SMN1/SMN2/NAIP copy numbers as well as an evaluation of the deletion size. For the investigation and characterization of either single exonic deletions or gene conversion events, a previously described modified protocol was performed [15].
In patients identified with heterozygous deletions, SMN1 analysis was extended to direct sequencing of exonic and flanking sequences, analyzed on ABI Prism 3500 Genetic Analyzer (Applied Biosystem, USA). The primers used were specifically designed to amplify the SMN1 coding region (Ref seq. NG_008691.1) and sequences are available upon request. The possible impact of c.859G>C variant as a modifying factor, either isolated, or in association to the SMN2 copies, was also evaluated [16].
Variant nomenclature followed the HUGO Gene Nomenclature Committee standard guidelines [18] and the reference sequence used was NM_000344.3 (ENST00000380707). “In silico” evaluation of nucleotide substitutions was performed using several prediction tools, including Mutation Taster, Polyphen, Provean, Human Splicing Finder and SIFT. The distinction of variants as SMN1 or SMN2 was not feasible; however, all variants were defined as bi-allelic (trans) versus mono-allelic (cis) through family segregation analysis.
For the retrospective analysis all patient records were reviewed to allow correlation of data including the deletion extent, the number of SMN2 and NAIP copies, the presence of additional variants and the clinical presentation of symptoms. For epidemiological evaluation, SMA incidence was based on the number of SMA patients born each year per recorded number of live births for the same period (1995-2018). The estimation of the prevalence of SMA in Greece per 100,000 persons of the general population (www.statistics.gr), is based on the number of patients who were confirmed as still alive during the biennium 2017-2018. This was facilitated since all patients contacted the laboratory for further information or/and an updated DNA report, a mandatory prerequisite from the health system for drug eligibility”.
Statistical analyses
All statistical analyses were conducted using the STATA version 15.0 software (Stata Corporation, College Station, TX, USA) in 213 patients for whom full data was available. Descriptive statistics were calculated for all measures as mean, unless stated otherwise. The Spearman rank test was performed to test the correlation of individual SMN2 and NAIP genes to SMA type. Generalized linear model and Collett selection was used to explain the joint effect of SMN2-NAIP-gender- deletion scale with disease age of onset. The age of onset was modeled as a categorical variable (>540 days and ≤540 days) as the most severe types (SMA0, SMAI and SMAII) manifest up to 18 months. For inclusion into the statistical model, SMN2 and NAIP-gene copy numbers were grouped as follows: ≥3 copies vs ≤2 copies, ≥2 copies vs 0-1 copies. With respect to the variables of gender and size of deletion in the SMN1 locus, the female sex and SMN1 small deletion size were used respectively as the reference category.
RESULTS
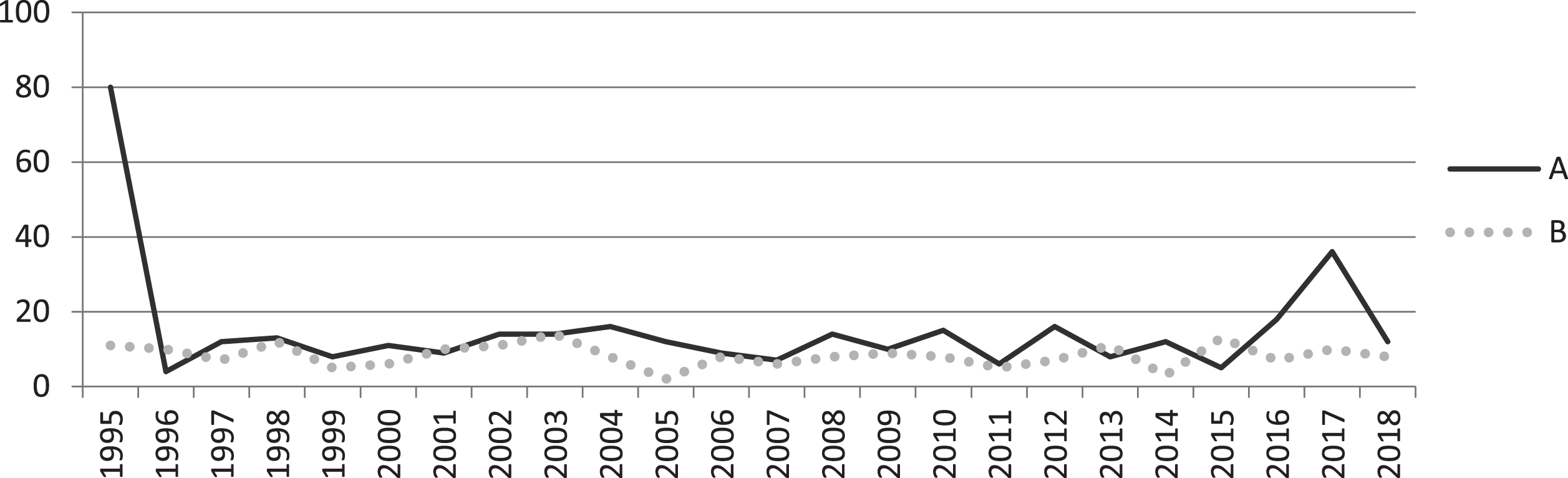
Over a period of 24 years (1995-2018), the molecular basis of SMA was confirmed in 361 individual patients (342 unrelated and 19 siblings) (Fig. 2). A mean number of about 12 new cases per year was estimated (Fig. 3). It should be noted that following the description of the newly discovered SMN1 gene in 1995, increased demand for genetic testing (from families or clinicians) resulted in an overrepresentation of patients and data from this year (1995, 80 patients) was excluded from the calculation of the annual number for new cases (Fig. 3).”
Fig.2
Distribution of SMA types included in the present survey.
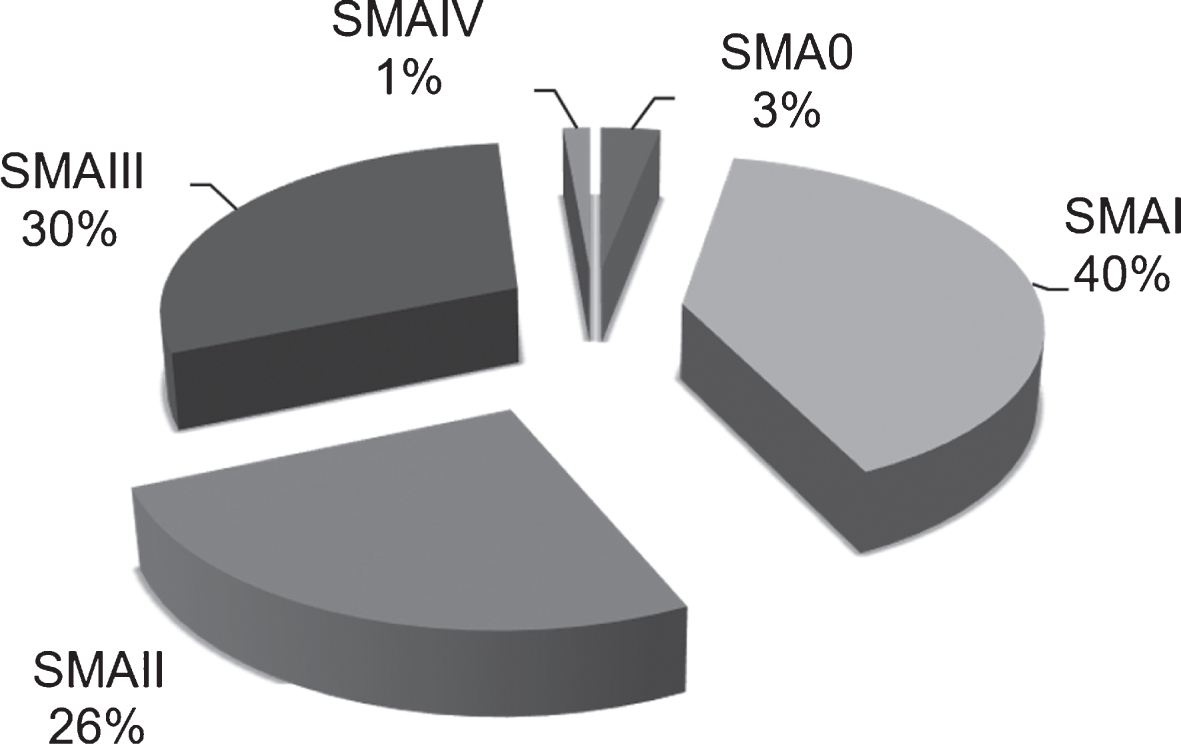
Fig.3
Annual numbers of: (A) new patients genetically diagnosed with SMA and (B) SMA births* for the years 1995 to 2018. Following the description of the newly discovered SMN1 gene in 1995 and the introduction of Spinraza in 2018, extreme numbers were recorded (A). *Based on the Date Of Birth of positive SMA cases of the cohort.
The majority of patients in this study, were classified as SMA type I (144/361 or 40%) followed by 108 with SMA III (29.6%), 93 with SMAII (25.8%), 11 with SMA0 (2.5%) and 5 with SMAIV (1.4%) (Fig. 2).
For assessing the incidence, the number of live SMA births was recorded based on the patients’ Dates of Births (DOBs) as reported in their case histories. A total of at least 200 positive SMA cases were born among 2,437,348 live births in Greece (www.statistics.gr) between 1995 to 2018 allowing an estimation of a minimum incidence at 1 per 12,000 live-births (∼8.2 per 100,000 livebirths).
From the medical records and natural history data, the age of onset ranged between 0 to 50 years. For SMA I patients in specific, the mean age of onset was 3 months (data from 121 patients) and the mean lifespan 8.7 months (data from 54 patients). During this 24-year period 221 prenatal tests were performed and 35 affected singleton fetuses were identified.
Following the passing of legislation approving the treatment of patients with Spinraza, a substantial number of surviving patients was recorded based on the number of requests for retesting to evaluate the SMN2 copy numbers. Combined with a review of the medical records available in the Laboratory of Medical Genetics, at least 161 SMA patients (aged between 2–72 years and of types II–IV) are currently living. Considering this number, along with the most recent Greek census of May 2011 (10 816 286 habitants/www.statistics.gr/en/2011-census-pop-hous), a minimum prevalence of SMA in Greece can be estimated at about 1.5/100,000.
In most of the cases (343/361 or 95%) a homozygous deletion of both SMN1 exons 7 and 8 was detected. A gene conversion event resulting in a homozygous deletion of exon 7 was ascertained in 10 patients (2.8%). One patient was identified with a homozygous deletion restricted to SMN1 exon 7 (∼0.3%) and the remaining seven patients (3 sporadic and 2 pairs of siblings) (2%) were compound heterozygotes for the common deletion of both SMN1 exons 7 and 8 in trans to a small intragenic alteration. The latter included frameshift variants, a missense substitution and a splice site variation, all confined to exons 5–7 in the SMN1 gene (Table 1).
Table 1
Novel intragenic lesions detected in this study
| Patient | Type | Age of onset | Clinical features | Genetic variant (NM_000344.3) | SMN2/NAIP COPIES |
| S-1 | SMAI | 40 d | Deceased at the age of 8 m | Exon 5: c.489_493delCCAA G p. Gln164Phefs*4 | 2/2 |
| S-2 | SMAII | 11 m | Respiratory support and gastrostomy at the age of 5,5 y | Exon 7: c.637_658del p.Lys213His fs*23 | 1/2 |
| Twins S-3a S-3b | SMAII | 15 m | Both twins were able to stand at the age of 15 m, never ambulant | Exon 5: c.551 dupA p.Pro185Alafs*71 | 3/2 |
| Siblings | |||||
| S-4a | SMAIII | 25 y | Ambulant at the age of 53 y*, independent but with difficulties | 2/2 | |
| S-4b | SMAIII | 18 y* | Ambulant at the age of 42 y, independent but with difficulties | Exon 5: c.475–2A>T | 2/2 |
| S-5 | SMAIII | 3,5 y | Progressive deterioration noted at the age of 8 y | Exon 7: c.767A>T, p.Asp256Val | 2/1 |
*Following a severe accident d: days, m: months, y: years.
The SMN2 copy number was determined in 215/361 patients of all clinical categories, including 6 patients with SMA0, 58 with SMAI, 65 with SMAII, 82 with SMAIII and 4 with SMAIV (Fig. 4).
Fig.4
Distribution of SMN2 copies among SMA types of the studied cohort and among individuals from the general population.
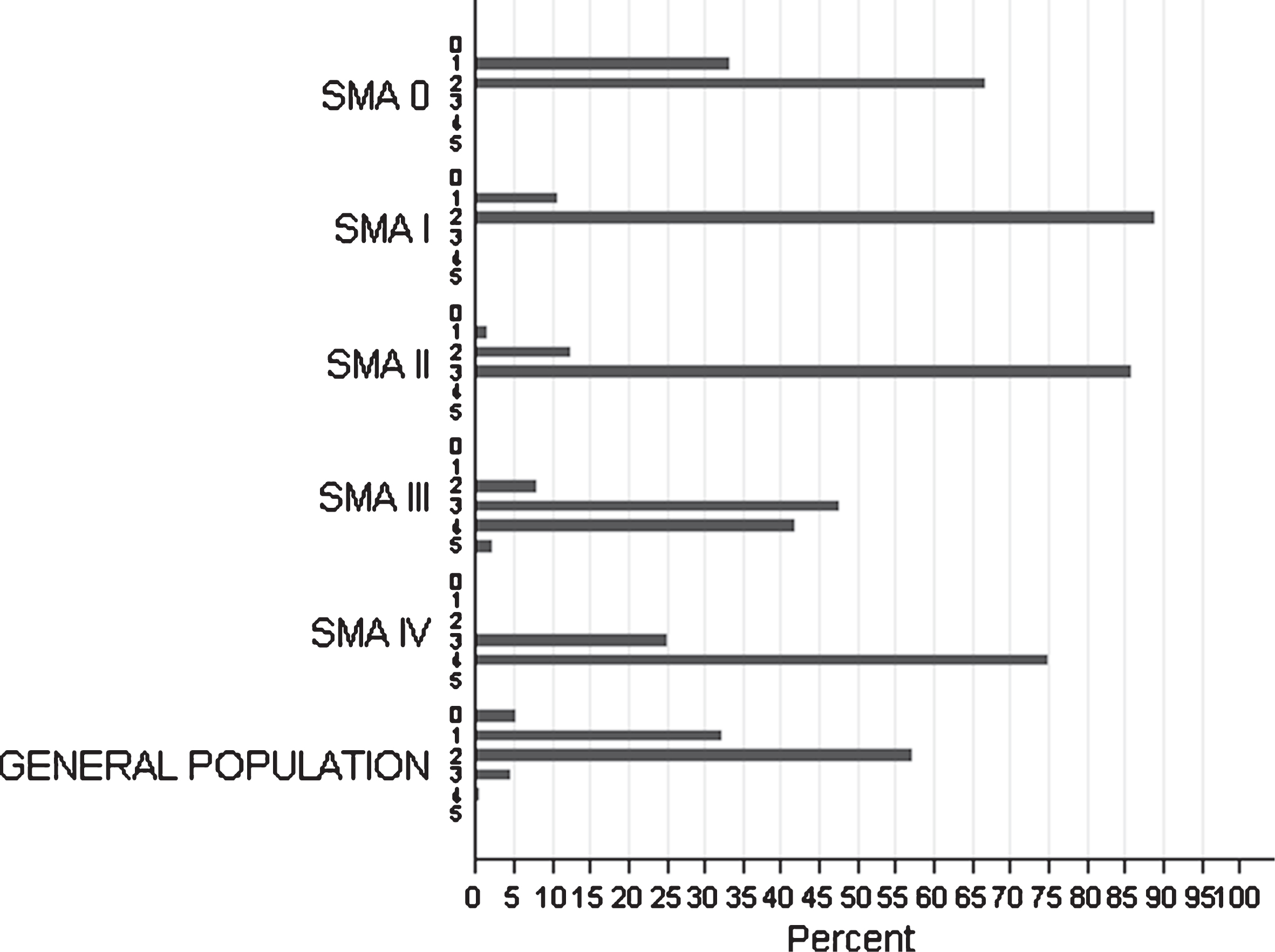
Nine out of these 215 patients also carried the SMN2: c.859G>C substitution, previously characterized as a protective variant. Of note, this variant was not detected in any SMA0 or SMAI patients.
The distribution of SMN1/SMN2 copies was evaluated in both the general and the patient population. In SMA patients with the severe SMA subtypes (SMA0 or SMAI) most carried 1 or 2 SMN2 copies, whereas patients with intermediate or milder SMA subtypes frequently had ≥3 SMN2 copies (Fig. 4). In patients homozygous for the SMN1 exon 7 and 8 deletion, the number of SMN2 copies was strongly correlated with SMA severity type (p < 0.001). More specifically, the Spearman rank test demonstrated a positive correlation concerning SMN2 (r = 0.79), as patients with the severe SMA0, SMAI types (100%) had ≤2 SMN2 copies, the intermediate SMAII (85.25%) had, in main, 3 SMN2 copies and the majority of patients with mild SMA forms of (SMAIII, SMAIV) were observed to carry ≥3 copies. However, 5 out of 209 patients (∼2.4%) showed discordance with the “SMN2 gene-copy rule”. These patients were classified as SMAII or SMAIII, despite a homozygous deletion on SMN1 gene, 2 SMN2 copies and absence of the ameliorating c.859G>C variant. It is interesting that, according to his medical record one 48-year-old SMAII male patient with 3 SMN2 copies, had a condition which dramatically worsened following a polio vaccination at the age of 7 months. Similarly, a strong correlation was also observed between the number of NAIP copies and SMA severity, with severely affected patients bearing fewer NAIP copies (0–1) in comparison to those with mild SMA types (r = 0.58, p < 0.001).
The best minimal model concerning early onset (<540 days) includes a combination of both NAIP and SMN2 copies and the individuals’ gender (Table 2). Specifically, the odds ratio for early onset (<540 days) pertaining SMN2 copy number was 6.2 (95% CI, 2.5 to 15.2) adjusted for NAIP copy number and gender. Patients with 0–1 NAIP copy present 9.9 higher odds for early onset (95% CI, 4.7 to 21). Finally, males showed 2.2 (95%CI, 1.04 to 4.6) higher odds. In search for the optimal model we showed an interaction between deletion size and NAIP copies with the greater effect in the presence of small deletions (odds ratio 21.6; 95% CI, 6.4 to 72.3) adjusted for SMN2 copy number and gender. In contrast a smaller effect was observed in cases with large deletions (odds ratio 2.4; 95% CI, 0.02 to 46.2) adjusted for NAIP copy number and gender.
Table 2
OR and 95%CI for onset before 540 days. Outcome associated with NAIP number, copies, SMN2 number copies and gender
| OR(95%CI) | |
| NAIP copies | |
| ≥2 copies* | |
| 0–1 copies | 9.9(4.7 to 21) |
| SMN2 copies | |
| ≥3 copies* | |
| 1–2 copies | 6.2(2.5 to 15.2) |
| Gender | |
| Females * | |
| Males | 2.2(1.04 to 4.6) |
Results of multivariable logistic regression analysis each variable adjusted for the other two. *Baseline category.
In this study, cases with an interesting intra-familial genetic or clinical heterogeneity were also observed. Specifically, in 7 families with more than one affected sibling, different SMA subtypes were recognized between siblings. In one family, a single exon 8 deletion detected in a mother with SMA IV was observed to be expanded to include both exons 7 and 8 following transmission to her male child who presented with SMAIII.
DISCUSSION
Although, there are a number of epidemiological studies in various populations worldwide which present data on SMA, only a few focus on findings ascertained from nationwide molecular analysis of the broader 5q chromosomal region [4, 19]. This long-term survey was carried out by the Laboratory of Medical Genetics the only provider of genetic services for SMA in the Greek public sector. The 5q13.2 genetic profile was assessed by MLPA analysis which in contrast to other methodologies- like HRMA or Real Time PCR- allows for the evaluation of all SMN1/SMN2/NAIP copy numbers and the characterization of the SMN1 deletion, either as restricted to exons 7 and 8 or extended to the entire gene. In terms of repeatability, patients with duplicate MLPA tests showed no discrepancies between the numbers of SMN2 copies disclosed on each sample. A sole exception was the patients with more than four or five copies where the exact number of copies could not be defined.
Consistent to previous studies, low SMN2 copies were strongly correlated with higher severity in patients with homozygous SMN1 deletions (r = 0.78) [7, 12, 14, 19]. The secondary influence of low NAIP copies on the severity is also discussed in several studies; however data on the relative impact on the age of onset is not so well established) [20, 21]. With our study a statistically significant effect of NAIP gene copies was demonstrated when 0–1 copies were considered low, adjusted for SMN2 copies and gender.
The prognostic value of subtle mutations is also of crucial importance to support the prediction of disease progression, especially in pre-symptomatic patients. The novel SMN1 variations identified in this study were absent in the gnomAD database, and provide further evidence on the importance of each specific protein region (Table 1). The SMN1: c.767A>T, (p. Asp256Val) variant, located within a highly conserved region of the SMN1 gene, was detected in an SMA type III patient (Table 1). The substituted Aspartic acid residue is located within the Tyrosine/Glycine rich region coded by exon 6, already known to be prone to small variations [22]. According to previous studies this conserved region affects the self-oligomerization and stabilization capacity of the SMN protein [23]. “In silico” prediction tools indicate a possible damaging effect for this variant. For segregation analysis only the maternal sample was available, since the patient’s father was deceased. The apparently healthy mother was shown to carry the deletion but not the p.Asp256Val variant, supporting the trans inheritance of the variants (compound heterozygote) in the patient who, at the age of 8yrs, is still ambulant and presents with a mild phenotype. The patient carries only 2 SMN2 copies indicating that his SMA type III phenotype is likely attributed to the mild impact of the p. Asp256Val missense variant. On the other hand, frameshift mutations identified in this study appear to be linked to more severe phenotypes even in the presence of 3 SMN2 copies (see patients S- 3a and b).
In view of timely intervention, a recent Newborn Screening Multidisciplinary Working Group suggested a genotype-based therapeutic algorithm [24]. It proposes the immediate treatment of pre-symptomatic patients upon identification of up to 3 copies of SMN2 gene even if others claim that various SMA types present with overlapping copy numbers (especially 2 and 3 copies) [19, 21] From our cohort intermediate to more severe phenotypes (onset before 18 months) were observed in patients with low copies of both SMN2 and NAIP genes and without a protective subtle variant. In addition, the number of NAIP copies was more significant in male patients bearing small deletions. Both observations indicate that an extended algorithm integrating all parameters (SMN2/NAIP copies, variants gender) might serve better as a prognostic indicator, if confirmed in larger cohort studies.
Further phenotypic variation is evident amongst the milder cases of SMA types (II, III, IV) indicating the presence of additional trans genetic modifiers, or other epigenetic or external factors influencing disease progression [11]. Intra-familial genomic rearrangements should also be taken into consideration especially when handling prenatal tests where novel findings resulting from the innate instability of the region may complicate the final diagnosis [15].
According to the literature, the incidence of SMA concerning pan-ethnic population studies is estimated at 1:11,000 births [2]. Recent high-throughput genetic newborn screenings in Germany (213.279 samples) and Taiwan (120.267 samples) revealed an incidence of 1:7,109 and 1:17,181 respectively. In both studies the rate indicated was much lower than that expected according to previous carrier-based procedures, ascertaining an incidence of 1:4,900 in Germany and 1:9,600 in Taiwan [25, 26]. In our study patients were recruited from throughout the country and it is presumed that almost no SMA patients born over the last 24 years have been missed from the corresponding estimations. Minimal SMA incidence in Greece was appraised at 1 in 12,000 or ∼8.3 new cases per year and the minimal prevalence at 1.5 per 100,000. According to the overall carrier frequency of 1/43 (2.3 %) and the high percentage (95%) of subjects carrying ≤2 SMN2 copies, one would expect an incidence of ∼1/7,400 live births and a higher percentage (>40%) of severe SMA 0 and I types. Given the discrepancies noted, it is likely that some severe cases were lost and may include spontaneous pregnancy losses or neonatal deaths. In addition, the study may have missed a few cases referred to genetics centers in the private sector, or asymptomatic patients (types III-IV). Further population-based studies such as newborn screening would be most appropriate for a more precise estimation of the incidence in Greece for all SMA subtypes.
The first therapeutic treatment for SMA with Spinraza (Nusinersen) was approved in Greece in December of 2017 (OGG/: N.4506/2017) and reimbursed for types 0, I, II and in exceptional type III cases. Valid and accurate diagnosis, as early as possible, preferably in newborns, infants and toddlers, potentially facilitates a timely intervention that is expected to positively impact morbidity, mortality, associated disabilities and prevalence [27, 28]. As indicated by the current cohort, newborn analysis should not be restricted to the number of SMN2 copies but extended to include additional genetic parameters such as NAIP copies, deletion size or certain ameliorating variants. Finally, this survey raises awareness towards the disease progression and management, and highlights the need for systematic population screening for SMA carriers following the successful example of the continuously improving prevention program for hemoglobinopathies in Greece [29].
DECLARATIONS OF CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
The authors would like to thank all SMA families and referring clinicians who collaborated in this study as well as the technician Mrs Sophia Vretta for her excellent technical assistance. The authors acknowledge the contribution of Dr Lina Florentin and Dr Chrysanthy Bili in collecting samples and establishing the genetic analysis of SMA in the Department of Medical Genetics (then A’ Department of Pediatrics) before 1995, and the Emeritus Professors Ekaterini Metaxotou and Emmanuel Kanavakis for their valuable support. The study was partially co-funded by the Muscular Dystrophy Association in Greece (MDA Hellas) and the Research Institute for the Study of Genetic and Malignant Disorders in Childhood.
REFERENCES
[1] | Manoli I , Fryssira H . Medical genetics and genomic medicine in Greece: Achievements and challenges. Molecular genetics & genomic medicine. (2015) ;3: (5):383–90. |
[2] | Sugarman EA , Nagan N , Zhu H , Akmaev VR , Zhou Z , Rohlfs EM , et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: Clinical laboratory analysis of >72,400 specimens. European journal of human genetics: EJHG. (2012) ;20: (1):27–32. |
[3] | Konig K , Pechmann A , Thiele S , Walter MC , Schorling D , Tassoni A , et al. De-duplicating patient records from three independent data sources reveals the incidence of rare neuromuscular disorders in Germany. Orphanet journal of rare diseases. (2019) ;14: (1):152. |
[4] | Verhaart IEC , Robertson A , Wilson IJ , Aartsma-Rus A , Cameron S , Jones CC , et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet journal of rare diseases. (2017) ;12: (1):124. |
[5] | Schorling DC , Pechmann A , Kirschner J . Advances in Treatment of Spinal Muscular Atrophy - New Phenotypes, New Challenges, New Implications for Care. Journal of neuromuscular diseases. (2020) ;7: (1):1–13. |
[6] | Kolb SJ , Kissel JT . Spinal Muscular Atrophy. Neurologic clinics. (2015) ;33: (4):831–46. |
[7] | Russman BS . Spinal muscular atrophy: Clinical classification and disease heterogeneity. J Child Neurol. (2007) ;22: (8):946–51. |
[8] | Lefebvre S , Burglen L , Reboullet S , Clermont O , Burlet P , Viollet L , et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. (1995) ;80: (1):155–65. |
[9] | Liu Q , Dreyfuss G . A novel nuclear structure containing the survival of motor neurons protein. EMBO J. (1996) ;15: (14):3555–65. |
[10] | Burglen L , Lefebvre S , Clermont O , Burlet P , Viollet L , Cruaud C , et al. Structure and organization of the human survival motor neurone (SMN) gene. Genomics. (1996) ;32: (3):479–82. |
[11] | Wirth B , Garbes L , Riessland M . How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic approaches. Current opinion in genetics & development. (2013) ;23: (3):330–8. |
[12] | Ruhno C , McGovern VL , Avenarius MR , Snyder PJ , Prior TW , Nery FC , et al. Complete sequencing of the SMN2 gene in SMA patients detects SMN gene deletion junctions and variants in SMN2 that modify the SMA phenotype. Human genetics. (2019) ;138: (3):241–56. |
[13] | Le TT , Pham LT , Butchbach ME , Zhang HL , Monani UR , Coovert DD , et al. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Human molecular genetics. (2005) ;14: (6):845–57. |
[14] | Butchbach ME . Copy Number Variations in the Survival Motor Neuron Genes: Implications for Spinal Muscular Atrophy and Other Neurodegenerative Diseases. Frontiers in molecular biosciences. (2016) ;3: :7. |
[15] | Kekou K , Sofocleous C , Konstantinidis G , Fryssira H , Mavrou A , Kitsiou S , et al. SMA prenatal diagnosis: A modified protocol to help differentiation between deletions and gene conversion. Mol Cell Probes. (2015) ;29: (1):71–3. |
[16] | Prior TW , Krainer AR , Hua Y , Swoboda KJ , Snyder PC , Bridgeman SJ , et al. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet. (2009) ;85: (3):408–13. |
[17] | Scheffer H , Cobben JM , Matthijs G , Wirth B . Best practice guidelines for molecular analysis in spinal muscular atrophy. European journal of human genetics: EJHG. (2001) ;9: (7):484–91. |
[18] | Eyre TA , Ducluzeau F , Sneddon TP , Povey S , Bruford EA , Lush MJ . The HUGO Gene Nomenclature Database, 2006 updates. Nucleic acids research. (2006) ;34: (Database issue):D319–21. |
[19] | Wadman RI , Stam M , Gijzen M , Lemmink HH , Snoeck IN , Wijngaarde CA , et al. Association of motor milestones, SMN2 copy and outcome in spinal muscular atrophy types 0-4. Journal of neurology, neurosurgery, and psychiatry. (2017) ;88: (4):365–7. |
[20] | Zhang Y , He J , Zhang Y , Li L , Tang X , Wang L , et al. The analysis of the association between the copy numbers of survival motor neuron gene 2 and neuronal apoptosis inhibitory protein genes and the clinical phenotypes in 40 patients with spinal muscular atrophy: Observational study. Medicine (Baltimore). (2020) ;Jan;99: (3):e18809. |
[21] | Taylor JE , Thomas NH , Lewis CM , Abbs SJ , Rodrigues NR , Davies KE , et al. Correlation of SMNt and SMNc gene copy number with age of onset and survival in spinal muscular atrophy. European journal of human genetics: EJHG. (1998) ;6: (5):467–74. |
[22] | Sun Y , Grimmler M , Schwarzer V , Schoenen F , Fischer U , Wirth B . Molecular and functional analysis of intragenic SMN1 mutations in patients with spinal muscular atrophy. Human mutation. (2005) ;25: (1):64–71. |
[23] | Praveen K , Wen Y , Gray KM , Noto JJ , Patlolla AR , Van Duyne GD , et al. SMA-causing missense mutations in survival motor neuron (Smn) display a wide range of phenotypes when modeled in Drosophila. PLoS genetics. (2014) ;10: (8):e1004489. |
[24] | Glascock J , Sampson J , Haidet-Phillips A , Connolly A , Darras B , Day J , et al. Treatment Algorithm for Infants Diagnosed with Spinal Muscular Atrophy through Newborn Screening. Journal of neuromuscular diseases. (2018) ;5: (2):145–58. |
[25] | Czibere L , Burggraf S , Fleige T , Gluck B , Keitel LM , Landt O , et al. High-throughput genetic newborn screening for spinal muscular atrophy by rapid nucleic acid extraction from dried blood spots and 384-well qPCR. European journal of human genetics: EJHG. (2019) . |
[26] | Chien YH , Chiang SC , Weng WC , Lee NC , Lin CJ , Hsieh WS , et al. Presymptomatic Diagnosis of Spinal Muscular Atrophy Through Newborn Screening. The Journal of pediatrics. (2017) ;190: :124-9 e1. |
[27] | Riessland M . Is there hope for spinal muscular atrophy synthetic pharmacotherapy? Expert opinion on pharmacotherapy. (2019) ;20: (9):1049–52. |
[28] | Saffari A , Kolker S , Hoffmann GF , Weiler M , Ziegler A . Novel challenges in spinal muscular atrophy - How to screen and whom to treat? Annals of clinical and translational neurology. (2019) ;6: (1):197–205. |
[29] | Theodoridou S , Prapas N , Balassopoulou A , Boutou E , Vyzantiadis TA , Adamidou D , et al. Efficacy of the National Thalassaemia and Sickle Cell Disease Prevention Programme in Northern Greece: 15-Year Experience, Practice and Policy Gaps for Natives and Migrants. Hemoglobin. (2018) ;42: (4):257–63. |


