
Pseudoexons of the DMD Gene
Abstract
The DMD gene is the largest in the human genome, with a total intron content exceeding 2.2Mb. In the decades since DMD was discovered there have been numerous reported cases of pseudoexons (PEs) arising in the mature DMD transcripts of some individuals, either as the result of mutations or as low-frequency errors of the spliceosome. In this review, I collate from the literature 58 examples of DMD PEs and examine the diversity and commonalities of their features. In particular, I note the high frequency of PEs that arise from deep intronic SNVs and discuss a possible link between PEs induced by distal mutations and the regulation of recursive splicing.
CHARACTERISTICS OF THE DMD GENE
The DMD gene is the largest gene in the human genome. Situated on the p-arm of the X chromosome, DMD spans over 2.22Mb, more than 99% of which is intronic sequence, with the coding sequence of its largest isoform totalling 11,058 bases across 79 exons. Eight unique alternative promoters [1], alternatively spliced exons, and an alternative polyadenylation site [2] produce at least 17 DMD transcript variants [3], one or more of which are expressed and translated in all types of muscle as well as various other cell types throughout the body, including myoblasts, lymphocytes and retinal cells.
The Dp427 m transcript of DMD encodes the muscle isoform of dystrophin, the DMD protein. In XY individuals, who carry just a single DMD copy, mutations that fully disrupt the function of the DMD gene (resulting in functionless or absent dystrophin protein) give rise to Duchenne muscular dystrophy, while mutations that only partially diminish the gene’s function and/or quantity of product give rise to Becker Muscular Dystrophy.
THE MAJOR SPLICEOSOME
The vast majority of RNA splicing in humans is achieved via the major spliceosome, a ribonucleoprotein complex responsible for excising introns from pre-mRNA molecules [4]. In order for the spliceosome to process a transcript correctly it first must accurately recognize the transcript’s exon-intron boundaries. This recognition is achieved through a network of mechanisms, including sequence-specific interaction with conserved acceptor and donor splice site motifs in the RNA, silencer and enhancer binding motifs both proximal and distal to the splice junctions, and RNA secondary structure [5, 6]. Mutations to a gene that alter the interactions of these factors with its transcripts can lead to errors in the processing of those transcripts, such as the expansion, truncation or loss of canonical exons, or the initiation of pseudoexons (PEs) within its introns [7]. These incorrectly spliced transcripts may be degraded prior to translation or may be translated to less functional or even harmful protein isoforms, with deleterious consequences for the health of the patient.
Thirty-six of the 78 introns in DMD are more than ten times the human median intron length of 1334bp [4], and of these 36, three are more than 100 times the median size. A transcript of this size and complexity presents a unique challenge to the major spliceosome, and as a result is arguably more vulnerable to splicing errors such as pseudoexons.
PSEUDOEXONS: ONE DEFINITION OF MANY
In the literature, the terms “pseudoexon” and “cryptic exon” are often applied interchangeably and inconsistently in reference to a wide range of splicing errors. For the sake of clarity, I hereby define a pseudoexon as: Any continuous tract of a transcribed gene that: (1) does not overlap, adjoin or duplicate any sense-strand sequence of that gene’s canonical exons; (2) bears an acceptor splice site motif at its 5′ end and a donor splice site motif at its 3′ end; and, (3) via both these motifs, is spliced into a measurable proportion of the mature transcripts of that gene in at least one proband.
Though this definition of “pseudoexon” may not agree with every prior usage of the term, it includes the majority of prior use while excluding splicing events that are better described by other terms, such as cryptic splice sites and whole exon duplications.
While some PEs are observable as rare splicing events in normal individuals, the majority are created by mutations that give the PE site an exon-like profile, resulting in the spliceosome falsely recognizing it and splicing it into an increased proportion of transcripts. When PE-splicing levels are high compared to normal splicing, these inclusions are likely to bear negative consequences for the phenotype of the affected organism, as the majority of PEs will disrupt the transcript’s open reading frame and/or encode premature stop codons. Consequently, the resulting transcript, if it is not degraded by nonsense-mediated decay, will be translated to a non-functional or truncated protein. Even in cases where a pseudoexon preserves the reading frame and does not encode a premature stop codon, it is likely that the amino acids it encodes will disrupt the secondary structure of the protein and thereby abrogate its function.
Numerous DMD PEs have been reported over the last few decades, perhaps more than have been described for any other single gene. When considered as a body of research, these reports comprise a unique opportunity for generating new insights into the splicing of DMD and other large genes.
In this review, I catalog the characteristics of 58 reported DMD PEs. Where possible, I describe the origins of these rare splicing events and draw inferences from their common features.
PSEUDOEXONS OF THE DMD GENE
Following a thorough search of the literature, I compiled a catalog of all 58 known PEs of the DMD gene (Table 1). In order to consistently record highly similar PEs, I adopted the criteria of unique local sequence: I assigned a separate catalog entry to each PE that was unique in at least one nucleotide of its sequence or splice motifs (eg. PE09 vs. PE10, PE15 vs. PE16); and listed as single entries all PEs with locally identical sequence (eg. PEs 11 and 12).
Table 1
Details of 58 known pseudoexons (PEs) arising from mutations within the human DMD gene. Bold, upper-case sequence denotes the PE itself, while regular lower-case sequence indicates flanking intron. Inverted sequence is italicized. Duplications and insertions are underlined. Single base transitions are shown in the form of “X > Y,” where X is the reference nucleotide and Y is the mutant. Shapiro-Senapathy splice scores are also shown, before and after mutation where appropriate (see supplementary materials for splice score calculator). “RNA Source” lists the cell or tissue types where the PE was detected. Genomic co-ordinates pertain to human genome assembly accession GCA_000001405.27, chromosome X, except where otherwise stated. Transcript co-ordinates pertain to DMD reference sequence NG_012232.1(DMD_v001)
| PE # | Intron | Sequence | Genomic Co-ordinates (chrX.) | Size (bp) | Acceptor Score | Donor Score | Mutation(s) | Orientation of Mutation to PE and Splice Sites | ORF | RNA Source | Reference |
| 01 | 1 | tctcttccttggttttgc(g>a)gCTTCTCGAGTTCATAGGAGACTTTCAGTTTCCAGTGACCTGGAAACTCACCATTCCTCATCACCATCCTTTACTGTAGTAACTTCCTTTTACCTGACCACCCTGCATAGTCACAGAAGATGCACTCCTGACAAGTGATCCTCAAAACAGgtagtaatctctttgagaag | 33174185-33174333 | 149 | 71.37 ->87.94 | 72.72 | c.31+36947G>A | Proximal | Frameshift | Skeletal muscle | [9] |
| 02 | 1 | ctcaattaaaatatatttagTCAAAGCCAGATGCAGTGGCTCACACCTGTGATCCCAGAACTTTGGGAGGCGGAGGCGGATGGATCATTTGAAGTCAGGA>GTTCGACACCAGCTTGATCAACATGgtgagacccctgtctctact | 33088244-33088348 | 105 | 62.92 | 81.59 | c.exon3_6del | Distal (3′) | Premature stop codon | Heart, skeletal muscle | [10] |
| 03 | 1 | tttttacatcatggcataagATCAACACTAAATTTTAGGAGTGCATAgtaagtattaccatagtgta | 33085901-33085927 | 27 | 58.89 | 78.81 | c.exon3_6del | Distal (3′) | Premature stop codon | Heart, skeletal muscle | [10] |
| 04 | 1 | tctgttttcttttgtaacagGACACCATTGGAGAAATTGGTCATTTTACCAAGGCTTTGACTGGAATGGCATGCTTCCTTTAAAGAATCAAAGTTGACTTATAGAGCCATTTAAAGCCCGTTGGGGAATCTGGCCTCATACCTTGTCCACACAGAGTCCCTGTACAAGGTTCCTGACCTGTGgtaagtaaagaatgtcactt | 33078186-33078347 | 162 | 96.38 | 88.39 | Multiple mutations, also observedin normalcells (see main text) | Distal (3′) | Premature stop codon | Lymphocytes | [10 - 14] |
| 05 | 2 | ttccttgtttctctacat(t>a)gGTTGAATCTGTTCCTGCAGCAACTAGTAACCCCAACAATCTGACTGgtgagaaattcaatctccgt | 33014502-33014547 | 46 | 68.17 ->84.74 | 82.07 | c.93+5590T>A | Proximal | Frameshift | Lymphocytes | [15] |
| 06 | 2 | ttccttgtttctctacat(t>a)gGTTGAATCTGTTCCTGCAGCAACTAGTAACCCCAACAATCTGACTGGTGAGAAATTCAATCTCCGTAACATTTTGCATTCTTCTACTTCTTCCCACTTCCATTCCAAAGGGCCAGGTAGAGTCCAGAATACTgtaagtgtcctggttttcag | 33014416-33014547 | 132 | 68.17 ->84.74 | 77.7 | c.93+5590T>A | Proximal | ORF preserved | Lymphocytes, skeletal muscle | [15] |
| 07 | 2 | tgttagtgttttttgttcagGTCTGTGCAGGCCAAGGTTATGGCCTAGCTGAGAAGAGGGCTCAGAGGAGCCTAGCTAAAGTTTGGTCAAGGGGAGCGTCTTGGTGGAGCCTCACTCCTGTTCAAGTAAAGAAGACACATTATTCATCCTTCTGAACAGTgtaagtccatttgtcattcg | 32960208-32960347 | 140 | 100 | 77.84 | Multiple deletions and duplications [17]; also observed in normal cells [16] | Distal (3′) | Frameshift | Skeletal muscle | [16, 17] |
| 08 | 2 | agtcttggtctgtcgcctagGCTAGAGTGCAGTGGTGCTGTCTCAACTCAATGCAACCTCCGCCTCCCGGGTTCAAGCAACTCTCCTGCCTCAGCCTCCCCAGTAGCTGGGATTACAGgtacctgccactgtgcccgg | 32878883-32878980 | 98 | 72.84 | 76.65 | c.exon2dup; also observed in normal cells | Distal (3′) | Frameshift | Multiple tissue types | [13] |
| 09 | 2 | tgatttggaacttcctgtagACAGACCCTTACAGGCATGGAAGAAGAAAGAATGAATAAACCAAGGATGACTTCCCAGTAGGTGGAGGATGGGAATAATTAGGAAGAAGCTGTGTCTTGTCACCTATATTGTCCATACACAACTGCACCGTAGGGTAATTGAGAAATTATAAGAATGgtaataaatacgttttacat | 32863759-32863915 | 157 | 70.85 | 73.68 | c.exon8_11dup | Distal (3′) | Frameshift | Lymphocytes | [18] |
| 10 | 2 | ttggaacttcctgtagacagACCCTTACAGGCATGGAAGAAGAAAGAATGAATAAACCAAGGATGACTTCCCAGTAGGTGGAGGATGGGAATAATTAGGAAGAAGCTGTGTCTTGTCACCTATATTGTCCATACACAACTGCACCGTAGGGTAATTGAGAAATTATAAGAATGgtaataaatacgttttacat | 32863759-32863911 | 154 | 79.35 | 73.68 | c.exon8_11dup | Distal (3′) | Frameshift | Lymphocytes | [18] |
| 11 | 3 | aatcttactctgtcgcccagGCTGGAGTCAGCGACATGATCTTGGCTCACTGCAACCTCTGCCTCCTGGGTTCAAGCAATTTTCCTGCCTCGGCCTCCCAAGTAGCTGAGACTACAGGCATGTTCCATCATGCCTGGCTAAGTTTTGTATTTTTAGTAGACATGGGGTTTCACCATGTTGGCCAGGCTGGTCTTGAGCTCCTGATCTCAATGAGCCACCTGCCTCAGCCTCCCCAAATGTCTGGCTGTTGCTTTTTTTTTTTTTTTTTTTTTTTTAAATCTAAAGTCTTATTTTTTTCCTCTTTTTGGTGGAAAGTGGGAGAAATATCAGAATGTAAACCAACATCATTCTGACATTCTTGGAGGAAAATCTAAGAGgtaaaaatggagattctggt | 32846622-32846978 | 357 | 81.06 | 79.88 | c.exon8_11dup; also observed in normal cells | Distal (3′) | Premature stop codon | Lymphocytes | [18] |
| 12 | 3 | ttcctctttttggtggaaagTGGGAGAAATATCAGAATGTAAACCAACATCATTCTGACATTCTTGGAGGAAAATCTAAGAGgtaaaaatggagattctggt | 32846622-32846683 | 62 | 72.62 | 79.88 | c.280Adel [12], c.exon 8_11 dup [18] | Distal (3′) | Frameshift | Lymphocytes | [12, 18] |
| 13 | 4 | ttcatatgcttgcttttcagATGCTTGTGTTTAACTTTACTCCACCTTAAACATTTGAGGAGTGTGAAGGACAGGAGACACAGAGATTTGCCTTGATTAAGGCAAATAAAACCCTGCCAGATTTTCATTTCCAAACACAGTCCTAGACAGAG(a>g)taagagagctggcagtttg | 32823851-32823982 | 132 | 90.56 | 74.59 ->91.82 | c.265-463A>G | Proximal | Premature stop codon | Skeletal muscle | [19] |
| 14 | 7 | atattctaattgaatttcag(A>C)GTGAATTTCACACCTCTCCTTTTGAAAGATTCATTTCTATGAATTTGGGACAGCTTCCTAGTATGATATTCCATCT(a>g)taagtatatccatatcatc | 32738792-32738868 | 77 | 79.91 ->78.04 | 56.78 ->74.00 | c.650-39575A>C; c.650-39498A>G | Proximal | Frameshift | Skeletal muscle | [20] |
| 15 | 9 | ttcattttcttcattta(a>c)agAGATTGATCATATTGGGATAAAGACGTGTTTTGGAATCCAAACAACCTGGTTTTAAGTCCCAGAACCACCAGTTACCTTTGTGACCTTTGgcaagtcatctaatttttct | 32650985-32651074 | 90 | 83.55 ->93.28 | 70.04 | c.961-5925A>C | Proximal | Premature stop codon | Skeletal muscle | [21] |
| 16 | 9 | ttcattttcttcatttaaagAGATTGATCATATTGGGATAAAGACGTGTTTTGGAATCCAAACAACCTGGTTTTAAGTCCCAGAACCACCAGTTACCTTTGTGACCTTTGg(c>t)aagtcatctaatttttct | 32650985-32651074 | 90 | 83.55 | 70.28 ->87.27 | c.961-5831C>T | Proximal | Premature stop codon | Skeletal muscle | [9] |
| 17 | 11 | cttttctctctctacctaagACAGGGTTTGGATAGATCCAGTCGGAAGCCATTATTTCCTGCCTAAGCC[del:11631bp]AAACCTTTTCTACAAGAAATGGTAAAGGGCGTTCTTCAATCTTAAAGAAAAGGATGTTAATGAGCAATGAGTCATCATCTGAAGTAACAAAACTCACTGGTGATAGTAAGgtttgttactattctgttac | 32641751-32641799; 32630010-32630119 | 159 | 73.92 | 79.54 | c.1331+2382_1331+14010del | Proximal | Premature stop codon | Multiple tissue types | [22 - 25] |
| 18 | 11 | ctctttttttttcccccaagTGTCTATTTGACTCTGGAAATAAGATGGCATATGTGAGAGTGGATAGAGAAAAGGAGGTGCTGAACAAATGAGTGGGTTATTTTCTCCCAAGTGCAGTGAAGTTTGCGTATGTGAATATATGAATGAATAGATGAACAACTAAATTAAGTTATTCAGgtaattccaatcttggaatt | 32638732-32638888 | 157 | 79.76 | 87.83 | c.1336_1337del | Distal (3′) | Frameshift | Lymphocytes | [26] |
| 19 | 11 | agttttgttctttcacccagGCTGGAGTGAAGTGGTGTGATCTCAGCTCACTGCAACCTCTGTCCCCCCGGGTTCAAGTGATTCTCCTGCCTCAGCCTC(c>g)tgagtagccgggattaaaa | 32626363-32626441 | 79 | 90.08 | 56.56 ->73.78 | c.1332-11909C>G | Proximal | Frameshift | Skeletal muscle | [27] |
| 20 | 18 | tttttttccacctgccttagTGGAAGAGGCTATCCTTCACCTGGTTGAGGCTCATCTCTGGGTGTGTGTTCTCAGCAGCATCACTGACTATGTATTAAGCCACCTGGTTCCATTCAGCTGTATATCCAGATTGTCAAAAATCTACATCCCAGgtctttatcattagctttta | 32514091-32514222 | 132 | 68.21 | 67.88 | c.2622+1G>A | Distal (3′) | Premature stop codon | Lymphocytes | [10] |
| 21 | 21 | tttatttttaatacatctagGCAAAGGAATGTTTTGTTCCAGTAGACACATAATCTGTTGCATTGCTCCTTCACTCCAGAAACTAGgtaaactgtttgtaaatgtt | 32480880-32480945 | 66 | 80.03 | 78.22 | None | N/A | ORF preserved | Skeletal muscle | [14] |
| 22 | 25 | tatctgtgcttttcctaca(a>g)GTATCACTCTGGCCATGTTCTGACTTTGTAGCCAAATGAGTTAGGTTGTAAAAGGAAGGAACAATGGCGCTCAAGGAGAAGAAGAAGACGATGCGgtaaaaacaaggaagccata | 32461308-32461402 | 95 | 79.15 ->95.72 | 70.75 | c.3432+2036A>G | Proximal | Frameshift | Skeletal muscle | [28] |
| 23 | 25 | atctgtgcttttcctacaagTATCACTCTGGCCATGTTCTGACTTTGTAGCCAAATGAGTTAGGTTGTAAAAGGAAGGAACAATGGCGCTCAAGGAGAAGAAGAAGACGATGCGGTAAAAACAAGGAAGCCATATGTGAATATTGTTACCAATTCAGCATTCCAGAGAGAATAATGGAAATGAAGTGTAAATCTATGCATTACAGAAATATCTACAGACAAA(a>g)taagtgtgtgatacactct | 32461200-32461401 | 202 | 76.71 | 70.14 ->87.36 | c.3432+2240A>G | Proximal | Frameshift | Skeletal muscle | [29] |
| 24 | 25 | gccatgttctgactttgtagCCAAATGAGTTAGGTTGTAAAAGGAAGGAACAATGGCGCTCAAGGAGAAGAAGAAGACGATGCGGTAAAAACAAGGAAGCCATATGTGAATATTGTTACCAATTCAGCATTCCAGAGAGAATAATGGAAATGAAGTGTAAATCTATGCATTACAGAAATATCTACAGACAAA(a>g)taagtgtgtgatacactct | 32461200-32461371 | 172 | 82.72 | 70.14 ->87.36 | c.3432+2240A>G | Proximal | Frameshift | Skeletal muscle | [29] |
| 25 | 26 | cttctctctctgcttatcagAAAACTGCAATTCCCAGATA(G>C)GTCAAATGATTTAGCCATAGTCACAGACTTTATTTGTGGTAGAGCCCACAGGATTGAAGgtattttattctatttcatc | 32452550-32452629 | 80 | 86.2 | 77.21 | c.3603+2053G>C | Proximal | Frameshift | Skeletal muscle | [30] |
| 26 | 27 | catattcttatgtgttacagATGTTGCAGTTGTTTTCTTCTATTTTGAGTCTGTTTTTCAATTACTTGATGGTGTCCTTTGATGAATAGAAGGTCCTAATTTTAATGTAGTCGAATGTATTCATCTCTTTTTTCTTACGgt(c>a)agttttttacgagatgt | 32442160-32442278 | 119 | 87.46 | 80.58 ->90.39 | c.3787-843C>A | Proximal | Frameshift | Lymphocytes or skeletal muscle (unspecified) | [31] |
| 27 | 29 | cattttttttctatcaacagAGCTGAATGAGTGCCAGGAAGCTGCGAAATCTGTCTTACAAAAAGgtgattgtggaagagtctag | 32412019-32412063 | 45 | 90.17 | 83.09 | c.3613delG | Distal (5′) | Premature stop codon | Lymphocytes | [10] |
| 28 | 34/42 | caaatgttcccgttttatagATGTTCCCGTTTTATAGATGAACAAATACAAATACAATA[del:78031bp]CAGCCAAGAAGATATGTTGGTGCACGTTTCTGGTACCTGACCTAATCAGGCTAGCGGAGTAGCCCTAAACATTCCACCCAAGACTCCAGGCTTGGAGCCATCAGAAGATGGTAGTAAAATTCTACCAGgtaaataaaagtaaagaaat | 32372085-32372098; 32293942-32294069 | 167 | 83.07 | 88.06 | c.4846-6885_6118-6369delinsATACAATA;c.4846-6900_4846-6899ins17 | Proximal | Frameshift | Unspecified | [32] |
| 29 | 37 | gaatttgctctccaccacagATGTGACAGACCCAGCCAATACAAGTTTGTGACCAAGACAAGTTTGTGAGTTTTCATTTCACATTCTGCATGAATTTgtaagtatacttctggaaat | 32360933-32361009 | 77 | 77.72 | 74.58 | c.5325+1740_5325+1757del | Distal (5′) | Frameshift | MyoD transformed fibroblasts | [33] |
| 30 | 37 | tgaattgtactatcttgca(t>g)GTATGTTCAGCTCTGGTGATGTGAAATGTTTCTCCTTATTTGCATCCTCAGgtacttttccagttgtattt | 32348692-32348742 | 51 | 71.85 ->88.42 | 77.01 | c.5326-215T>G | Proximal | Premature stop codon | Skeletal muscle | [34] |
| 31 | 43 | agtgattttgtcaccaccagGCCTGGCTTACAAGAGCTCCTGAAGGAATCACTAAACATGGAAAGGAAAAACCGGTACCAGCCACTGAGAGAAACATACCAAATTGTAAAGACCATCGACCCTATGAAGAAACTGCCTCAACTAACAGg(c>t)aaaataactaaccaacat | 32256577-32256704 | 128 | 86.94 | 65.72 ->82.94 | c.6290+30954C>T | Proximal | Frameshift | Skeletal muscle | [35] |
| 32 | 43 | ttttatttattttattctagATTTCCTGCTCTTCTAAGACACAGTGATTTAGAATTTCTGTTCAAGCAAGAGAACTAAAGACTTCTTTAAAGAAGAGAAGgtaagtgcaagagattaaaa | chr4.182041664-182041743 | 80 | 90.26 | 99.52 | Complex insertion/deletion (see cited work) | Encompassing | Frameshift | Skeletal muscle | [36] |
| 33 | 43 | tgtctcccatcaccccccagATGGGACCATCTAGCTGCAGGAAAGGAAGCTCAAGGCTCCCACTGAATCTACATTATGgtgagttgtataattatttc | 32235147-32235204 | 58 | 76.18 | 86.71 | c.6291-21015_6438+98743dupinsA; c.6291-21008_6291-21007insCTCCCCTGAACATGG | Distal (5′ and 3′) | Frameshift | Unspecified | [32] |
| 34 | 44 | ttttttttaattgcaatcagATTCCAGATGGACCCTAAGAGCTCAGTTACGGAAGTGGAAGGGGAAAAACAGCAGCAGCAGCAGCAACAACAACAACGACAACAACAACAACGACAACAACAAACCGAAGAGGAAAAGAGAGCTGAGTGGAAAAAGTCTTTGAAAATTTAGTCCTAAAACTTATATTTACATTCTCATGCTCTCCTGAAGTCACAGAAAGAGGTTCGGAACTTCTGCTTTAAGAGGACATTTGAATCGACTGGAGAGGAACAAAGCATTGAAGAGAGAGGAAGTGAACTCCTCAGATAAAGCTCTAATATGAGAAACATAGTTCCAGAGAGAAGTTAATGTTACTTCATTTTTCTGTCTCATTTCCTTCAAGCTCAAAGCATGAGTAAGTGATAATGgtaagtaaatatgtattctc | 32074802-32075188 | 387 | 75.04 | 90.97 | Complex inversion (see cited work) | Encompassing | Premature stop codon | Skeletal muscle | [37] |
| 35 | 44 | tttttttttttttttttgagAGGGAGTTTCGCTCTTTCGCCCAGGCTGGAGTGAGGTGGCGCAATCTCAGCTGACTGTAACCTCTGCCCCCACCAGGTTCGAgt[del:121167bp]aagtatgaaaaaaataaa | 32024402-32024483 | 82 | 85.52 | 62.43 ->78.86 | c.6439-55921_6912+26400del | Proximal | Frameshift | MyoD transformed fibroblasts | [38] |
| 36 | 45 | tttcttctggagtattctagGAGAAGACATACCAGTCGAGGGGTTCTGGGGAGCCAGGCCTTCAAGCAATGGATTGCTGACAACATAATGAAGAGGATTTTACTTAGAATAATGTCAGTTGATAAAAGTTTGAATGGGAGACGGAAGCAAGGCAGTGg(g>t)aagtggaattcctaaatt | 31965031-31965167 | 137 | 73.52 | 71.16 ->88.39 | c.6614+3310G>T | Proximal | Frameshift | Skeletal muscle | [27] |
| 37 | 47 | aattatggtaatccccaca(t>g)GTCAAGGGTGGAGCCAGGTGCAGATAATTGAATCATGGAAGAGGATCCCCCATACTGTTCTCCTGATAATCAgtaagtttcacaagatctga | 31879338-31879409 | 72 | 61.61 ->78.18 | 74.53 | c.6913-4037T>G | Proximal | Premature stop codon | Skeletal muscle | [27] |
| 38 | 48 | aaagctgctctttccgctagGGTCCTGGGATGAGAAGACATTTAGAGCAGATCCATATTCTAGAATGAAAAGACCTTAACTTGATTCATAGTGTGGAGTAAAGCTGCAGCTGATCTGCAGACACATGAgtgagtaataaatgtttgtt | 31850637-31850744 | 108 | 81.97 | 70.41 | Complex inversion (see cited work) | Encompassing | Premature stop codon | Cultured myogenic cells | [39] |
| 39 | 48 | gacagtgggctctattttagGCAATGTGAGCACAGAAGACTGCTCCTCTCTTCAGGAAATGGAATGAAGTGAAGAAATGAGTCATGTCAACATACTGAAGACTATTTCAGGCAGAGGGAAAAGCAAGTTCCATTGCCAAAAGGCGgtgaggagttttgcttgtat | 31845152-31845276 | 125 | 75.76 | 78.66 | Complex inversion (see cited work) | Encompassing | Frameshift | Cultured myogenic cells | [39] |
| 40 | 49 | atgtatttgaccttttacagGAAGTCCTCAAAGAAAATAAAGAAGTTGGTTAGTCAGAGATTTTAAAAAAGAAACCAGAAATATACCATCTTTGAAAATAAAAGAAATCTTAAAGAAGTTGACGAAGTGAAGGAAAGAAGAAAGAGGAAAGAAACTGAGCAGGGATGAACCCAGCTTCCAATCAGAACTCAGCTTGGGAGgtgagcgtaggagctggaca | 31833539-31833718 | 180 | 93.4 | 87.02 | Complex inversion (see cited work) | Encompassing | Premature stop codon | Cultured myogenic cells | [39] |
| 41 | 49 | atgatgctgtcattttgcagTTCATATCTGAGGAAATGATGCCTGGAAGATTATAATTGCCCTCTTGACCATTTGTCTACAGCAATTATTTTGTGTTAAAGGTTGGGATGGAACGCTTTGGCCAAATACAGAAAGTTCCTCACACCTTTTTCTGTTACATTGAAAAATAATTCAAGAGCAgtaagtaaaacatattacta | 31832500-31832659 | 160 | 87.27 | 75.66 | Complex inversion (see cited work) | Encompassing | Frameshift | Cultured myogenic cells | [39] |
| 42 | 49 | tattgttatcttctttctagAAGTCCTCTCCCAACTTATTCTGGTGTTATTGGCATGGAAATTGTGCTCAATAAATGAGTCAGTCAAAATAGACTTGTTCATTTCAGGGGACAATTTTTGTCTGTTATCTCTCTATTGATTTCTGTTGCTACAAGTGAAGTTACACCAGgtaaatgagattgtacgcat | 31831990-31832138 | 149 | 88.15 | 88.06 | Complex inversion (see cited work) | Encompassing | Frameshift | Cultured myogenic cells | [39] |
| 43 | 49 | ctttctgaattcctccttagAATCATGCAGCCAGCTGAACTCTGTAAGAAGCTTTTTCCAATGCCCTCTTACTGAGATGCCCACACTCTGAAGTGTTTTACGCTCTTGCTGCAATAAGgtaaatattcttaatttttt | 31831041-31831138 | 98 | 73.62 | 87.58 | Complex inversion (see cited work) | Encompassing | Frameshift | Cultured myogenic cells | [39] |
| 44 | 51 | aaatggtcttttggttacagGGTGAAAGAGACCCAACAATACACCTTTCCCACTTCCGGAGGCCTTTGGTTAAACCATGTCTGCCACAAGGACACAGGAGCCTGgtatgactggttgttttttg | 31752441-31752524 | 84 | 96.74 | 76.2 | None | N/A | ORF preserved | Skeletal muscle | [14] |
| 45 | 51 | tgcttttatgatttttaaagAATTCGGAGGAGGAGCCAAGATGGCCGAATAGGAACAGCTCCGGTCTACAGCTCCCAGCGTGAGCGACGCAGAAGACGGGTGATTTCTGCATTTCCATCTGAGgtaccgggttcatctcaata... | 31765009-31765013 | 103 | 79.68 | 68.7 | c.7542+8951_7542+8952ins6091 | Proximal | Frameshift | Skeletal muscle | [40] |
| 46 | 53 | tttatcctgaggaattatagACTACTAAGCAGACAGATATTTGAGGAAATTTCAGAGGAAAGAGAAAGAAGAGAAGAATGAGCTGGGCTGGAGgtaagagaatggggggagaa | 31678558-31678630 | 73 | 64.29 | 91.82 | Complex inversion (see cited work) | Encompassing | Frameshift | Skeletal muscle | [41] |
| 47 | 55 | ttttacttttctatttaca(a>g)AAATGGAACACCACCAGAAAAACAAGAATTTGAAAGACGAGATGAGAAAAgtaagttgtaattggaaaca | 31609571-31609620 | 50 | 79.61 ->96.18 | 87.36 | c.8217+18052A>G | Proximal | Frameshift | Skeletal muscle | [33] |
| 48 | 55 | tttttgtttttcctttttagAGTTCTTGCTAATGATGGGCCCAAAGTTATATTAAGAACTGCAAAGTAAATTTCAACCAATTACTTTATTCAGg(g>t)gagtcattaaattgaggt | 31595572-31595644 | 73 | 90.26 | 78.52 ->95.74 | c.8217+32103G>T | Proximal | Frameshift | Skeletal muscle | [35] |
| 49 | 56 | catccaattttttctaccagAAATATTAAGAATTGTTGACTACAACAGTATGGAAAAGCAATAGATTCCAGTGTGTATTTCATGCCAAAAGTCTCAGCATTCTGCATGTGGAAATAAACATATGGCTAAACACTGCCTTTTCTCAAAATTGCCATCAAACTATCCTCTGTTTTGTGGCTCTCAAAAgtaagtagccagatttttat | 31497079-31497244 | 166 | 94.32 | 87.36 | c.8391-1419_8391-828del; c.8391-102_8391-76del | Distal (5′ and 3′) | Frameshift | Skeletal muscle | [37] |
| 50 | 60 | ttgattgattattgttgcagGTTGAGTCCTCCAAGAAGCAGATGCCAGGGCAGATTTATTGCAGAATAACACCTGTGAAGAAATAGGGGTGGAAGCAGAATTGAACAAGg(g>t)aagccatcagatcacaat | 31364155-31364243 | 89 | 88.65 | 76.64 ->93.87 | c.9085-15519G>T | Proximal | Frameshift | Skeletal muscle | [9] |
| 51 | 62 | ttttgttctttttgtggcagGAGCTGATAGCCAGCAACCACACTTCAAGAAATGGAAGACAGCTGTGAATGCTTCATTCAGGCCCA(A>G)gtaaatataggaagaggtgt | 31261663-31261729 | 67 | 96.61 | 75.91 ->88.06 | c.9225-647A>G | Proximal | Frameshift | Skeletal muscle | [9, 42] |
| 52 | 62 | tgtcggtgtcctttctgtagTGTTCCACATTGGATGGGTGAAGAAGTCCTGATAGTCGATTATTGATCACATAACAAGgtca(a>g)tttatcataactgaa | 31261306-31261363 | 58 | 81.83 | 77.76 ->89.70 | c.9225-285A>G | Proximal | Frameshift | Skeletal muscle [28]; Lymphocytes or skeletal muscle (unspecified) [31] | [28, 31] |
| 53 | 63 | tgtttccactacatctgcagTTACATCCTCCACTGAAGTCTTGAACCCCTGACAGTCATCCATGAGGGTTGGAATCAACGTCTTCCAAACTCCTGgtaatgttgatattttgacg | 31247694-31247768 | 75 | 76.44 | 74.39 | Unknown | Unknown | Premature stop codon | Lymphocytes | [10] |
| 54 | 65 | tggcactgttttctttgcagATGACATGTGAATGCATTCTGAATGTATAACTTCCTTCTACCTGACTGAAAAGTATTTGGTGACAATTTTAACTCCTTGAAGACCTGAGTTGCTGTATAAAGTGGATTTGTTAAATTTTGATCTACCTTTTCTTAAGAGGGAGAAAG(a>g)taagaaaattttccagtga | 31208284-31208430 | 147 | 96.18 | 77.17 ->94.40 | c.9563+1215A>G | Proximal | Premature stop codon | Skeletal muscle | [33, 43] |
| 55 | 65 | attaattttctttcaataagGGGCAATCTGATGAAGATCTGAGCATTTAAGAGGGCTGAGCAGTTAGTTGCTGgtaa(t>g)ttttttggcttccat | 31207099-31207151 | 53 | 83.88 | 79.28 ->91.45 | c.9564-427T>G | Proximal | Frameshift | Skeletal muscle | [19, 45] |
| 56 | 67 | ttgctttttctctgtcgtagTCAGTGCCCCAGAAACACCCTCTTCTTCCCAAATGCCTGAGCACTCCACTGgtagacattcatcttgttct | 31203344-31203394 | 51 | 81.16 | 63.87 | Unknown | Unknown | ORF preserved | Lymphocytes | [10] |
| 57 | 67 | gtgttttgttttgttttcagAAGGGGTCTAACTTCGTCACCCAGGCTGGAGTGCAGTGGCACGATCACAGCTCATTGCAGCCTCGACCTCTGGGCTCAAGTGATCCTCCCACCTCAGCCTCCTGAGTAGCTGGGACTACAGg(c>t)atggaccgccacgcctgg | 31201249-31201369 | 121 | 96.18 | 67.75 ->84.98 | c.9807+2714C>T | Proximal | Frameshift | Lymphocytes or skeletal muscle (unspecified) | [31] |
| 58 | 77 | gtacttatggtcaattgcagATACAGAACTCCAAAGAAATTCAAATCACAGTACAATCATTGATTACTGTATGATCCTGGCCCCTTGAACTGGCAGCTTGCTTACCTGCTCTGGAAAGTTGCTGGCTGCCCTGTTGGCACCCTGGGCATTTCTTCCACATCTAAACAAGAGgttagtagagaagaagctac | 31132483-31132633 | 151 | 76.72 | 87.1 | Unknown | Unknown | Frameshift | Lymphocytes | [10] |
PSEUDOEXONS ARISING WITHOUT MUTATIONS
Six DMD pseudoexons have been reported as low-frequency splicing events in normal cells lacking any known mutation: PEs 04, 07, 08, 11, 21 and 44. Interestingly, four of these PEs are of lengths that do not shift the reading frame of the transcript – 162bp for PE04, 357 bp for PE11, 66bp for PE21 and 84bp for PE44 – and of these four, only PEs 04 and 11 contain stop codons.
In addition to their splicing profile in normal cells, PEs 04, 07, 08 and 11 are also spliced at much higher frequencies in the cells of some patients with other DMD mutations. For PEs 08 and 11, this behavior was observed for the cells of only a single patient each (see below, subsection ‘Pseudoexons arising from duplications’). However, the behavior of PEs 04 and 07 – referred to in some prior literature as exons 1a [10–14] and 2a [16, 17] respectively – is somewhat more complex. Though the inclusion of PE04 in muscle cell DMD transcripts is rare [14], this PE is included in approximately 50% of DMD transcripts in lymphocytes [11], and is included at higher frequencies in both cell types as a result of a frame-shifting single nucleotide deletion in exon 5 [12] and, in a different proband, an exon 2 tandem duplication [13]. Given the frequency of its inclusion in mature DMD transcripts, especially in normal lymphocytes, it may be that PE04 would be better classified as a canonical, alternatively spliced exon rather than a pseudoexon. However, at the time of writing no functional role for PE04 has been conclusively determined [10]. Similarly, PE07 has also been observed as a predominant inclusion in the muscle DMD transcripts of multiple patients with deletions and duplications in the 5′ exons of the gene – though, as with PE04, it is yet to be determined whether these inclusions indicate a functional role for PE07 [17].
PSEUDOEXONS OF UNKNOWN ORIGIN
Underlying genomic mutations were not identified for three of the pseudoexons catalogued (PEs 53, 56 and 58). However, as these pseudoexons were exclusively detected in the RNA of specific DMD patient cells, they are believed to be pathogenic and therefore were not classed as arising sans mutation.
PSEUDOEXONS ARISING FROM DELETIONS
Of the 54 known mutation-obligate DMD PEs, ten arose from genomic deletions: PEs 02, 03, 12, 17, 18, 27, 28, 29, 35, and 49. For some of these cases, PE initiation can easily be explained as a direct result of the deletion event bringing into conjunction tracts of sequence that, when transcribed, present a strong exon signal to the spliceosome. Pseudoexons appearing to fit this description are PEs 17, 28 and 35, though it should be noted that PE28 also has two small insertions (17bp and 8bp) near its junction site. Two additional but less obvious examples can be seen with PEs 29 and 49 – in these cases, the sequence of the pseudoexons and their splice sites are unaltered from normal individuals, but their inclusions in mature transcripts are initiated by deletions of immediately flanking intronic regions, which presumably contain essential splicing silencers.
For the remaining five deletion-initiated PEs, the link between mutation and pseudoexon is less clear. PEs 12 (i3), 18 (i11) and 27 (i29) all arose from frame-shifting deletions of one, two and one bases in exons 5, 12 and 27 respectively, and PE02 (i1) and PE03 (i1) (both from the same patient) were purportedly initiated by a deletion of exons 3 to 6. Though a more detailed explanation of these PEs may not be possible at present, they appear to support the general theory that splicing of a given DMD intron is often interdependent on the correct processing of distant elements of the same transcript [46].
PSEUDOEXONS ARISING FROM DUPLICATIONS
Five DMD pseudoexons arose from genomic duplications: PEs 08, 09, 10, 11, and 33. PE08 (i2), which has also been observed as a low-frequency inclusion in normal skeletal muscle RNA, was converted to a pseudoexon by a tandem duplication of exon 2. PE09 (i2), PE10 (i2) and PE11(i3) were reported in the same proband as a result of an exon 8–11 duplication, and PE33 (i43) arose from an exon 44 duplication. These cases offer further support to the theory of correct DMD splicing occurring through coordination of distant elements. At this point, however, it is not clear whether these PEs are induced specifically by alterations to the canonical exon order, or whether they arise from disruptions to intronic sequences that would normally act as distal pseudoexon silencers.
PSEUDOEXONS ARISING FROM INVERSIONS
Eight DMD pseudoexons arose from inversion mutations: PEs 34, 38, 39, 40, 41, 42, 43, and 46. In all these cases, each PE was completely internal to the inverted region. PE34 arose from an inversion internal to intron 44 – i.e. no canonical exons were directly affected. PEs 38 to 43 (i48 and i49) were reported from a single patient with a complex inversion of exons 49 and 50, while PE46 (i53) arose in a patient with a deletion of exons 48–52 and an inversion of exon 53. It is perhaps unsurprising that such dramatic rearrangements of large tracts of transcribed sequence would result in splicing disturbances of some kind, but these cases nevertheless serve to illustrate that, in addition to recognition of canonical exons, the silencing of pseudoexons is an equally essential component of spliceosome function, and one that is likely to be achieved through orientation-dependent sequence motifs in the intron.
PSEUDOEXONS ARISING FROM INSERTIONS
Two DMD pseudoexons arose from insertion mutations, PEs 32 and 45. PE32 was created by an insertion into intron 43 of two large tracts (88.0kb and 2.6kb) of intragenic sequence from chromosome 4, the PE itself originating within the larger of these two tracts, while PE45 was created by a 6096bp LINE-1 retrotransposon with a potential donor site at its 5′ end inserting immediately 3′ of a latent acceptor site in DMD intron 51.
PSEUDOEXONS ARISING FROM SINGLE BASE-PAIR SUBSTITUTIONS
Single base-pair substitutions were the most commonly observed cause of DMD PEs, accounting for 26 of the 58 catalogued, through 24 unique mutations. In most of these cases, the etiology of the PE appears to stem from the creation or enhancement of a latent mid-intron splice site – of the 24 unique mutations, 7 created new acceptor splice sites (PEs 01, 05/06, 15, 22, 30, 37 and 47) and 15 created new donor splice sites (PEs 13, 14, 16, 19, 23/24, 26, 31, 36, 48, 50, 51, 52, 54, 55 and 57). All 22 of these acceptor-motif and donor-motif mutations greatly enhanced the Shapiro-Senapathy splice score of the mutated site, and in every case the new nucleotide was the most common consensus base for that position in the splice site (Fig. 1). While a possible exception to this rule was noted at the acceptor site of PE14 (c.650-39575A>C), this mutation was found to be a common SNP (rs113593006, dbSNP build 151 – see ref. 44) that only marginally decreased the Shapiro-Senapathy score of the acceptor site (from 79.91 to 78.04). I therefore judged that this SNP was likely to be incidental to the pathology of this pseudoexon and did not constitute a true counterexample to the prevailing pattern of splice site enhancement.
Fig. 1
Locations of pseudoexon-initiating single-nucleotide variations in the DMD gene, relative to acceptor and donor splice site consensus sequences. Numbers above each nucleotide indicate the exemplar pseudoexons. Lower-case letters indicate intron sequence, upper-case letters indicate exon sequence. Dash-line boxes highlight the essential “ag” and “gt” of the acceptor and donor site motifs respectively.
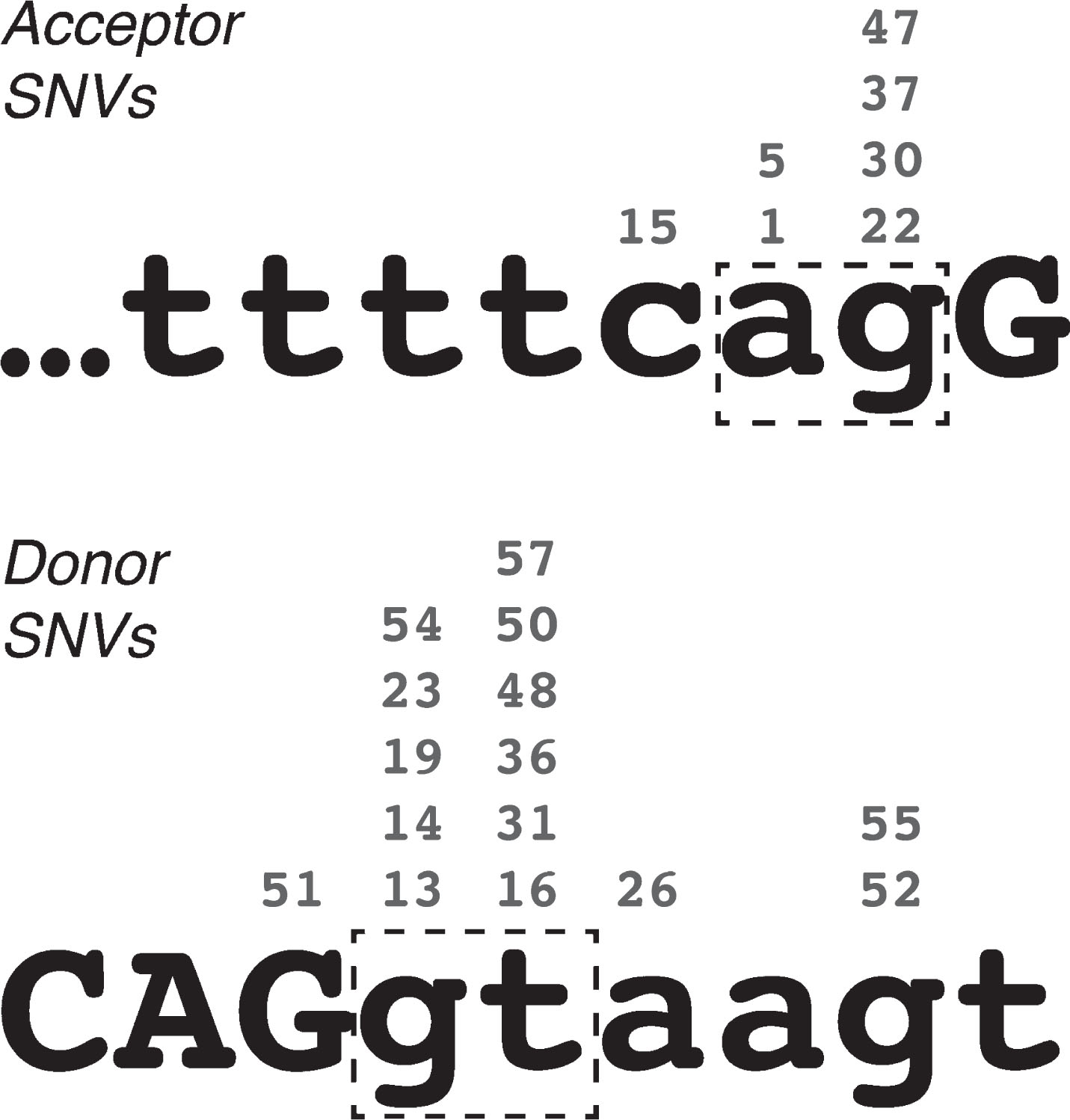
Only two PEs arose from SNVs outside of the PE consensus splice sites, PE20 and PE25. PE20 (i18) arose from a G-to-A substitution at the first base of intron 20, suggesting that the correct splicing of these two introns may be interdependent. In this way, PE20 is qualitatively similar to PEs 12 and 18, which also arose from small mutations distal to the pseudoexon, although the mutation that initiated PE20 did not directly alter the DMD coding sequence. PE25 (i26) is a unique case that arose from a G-to-C substitution internal to the PE that altered the predicted binding of splicing enhancer SRp55 [30].
PSEUDOEXONS AND RECURSIVE SPLICING
Multi-step or recursive splicing was first described in Drosophila in 2005 [47] and has more recently been discovered to be prevalent in the genes of the human transcriptome [48, 49], including DMD. While conventionally spliced introns are removed with a single splicing event, recursively spliced introns are excised from their maturing transcripts in two or more segments, via intronic acceptor splice sites called ‘ratchet points’. Sibley et al. [48] have also reported that recursive splicing in vertebrates is facilitated by recognition of evolutionarily conserved donor-like splice sites downstream of acceptor-like ratchet points.
Georgomanolis et al. [49] have postulated that some of the low-frequency pseudoexons observed in the transcripts of normal cells may be a natural byproduct of the spliceosome incorrectly recognizing exon-like intronic ratchet points. I suggest that this hypothesis can reasonably be extended to include mutation-induced PEs – i.e. mutations that enhance the exon-like characteristics of intronic ratchet points may thereby convert them into pathogenic PEs. Evidence supporting this hypothesis has already been described by Bouge et al. [14], who noted the alignment of six pseudoexon splice sites with six of the DMD intron ratchet points predicted by Gazzoli et al. [51]. Seeking to expand upon these observations, I cross-referenced the splice sites of all eligible PEs in Table 1 with all of the intronic ratchet points predicted by Gazzoli et al. This analysis excluded the splice sites of the DMD inversion PEs (34, 38, 39, 40, 41, 42, 43 and 46), the chromosome 4 insertion PE (32), and the de novo donor site for PE45, as these sites could not be sensibly compared to any part of the DMD reference sequence. Splice sites shared by multiple PEs (acceptor sites for PEs 5 and 6, 15 and 16, and donor sites for PEs 9 and 10, 11 and 12, and 15 and 16) were included but were counted only once each to avoid bias. Using these criteria, including the matches noted by Bouge et al. I confirmed 12 Gazzoli matches out of 47 unique acceptor sites and 14 Gazzoli matches out of 44 unique donor sites (Table 2).
Table 2
DMD pseudoexon splice sites coinciding with recursive splicing ratchet points predicted by Gazzoli et al. (2016). Co-ordinates listed are for genomic reference sequence NC_000023.10, as used by the cited authors. Dotted-line boxes enclose pairs of split reads that match to the same pseudoexon splice site. Asterisks (*) indicate the six coinciding splice sites previously noted by Bouge et al. [14]
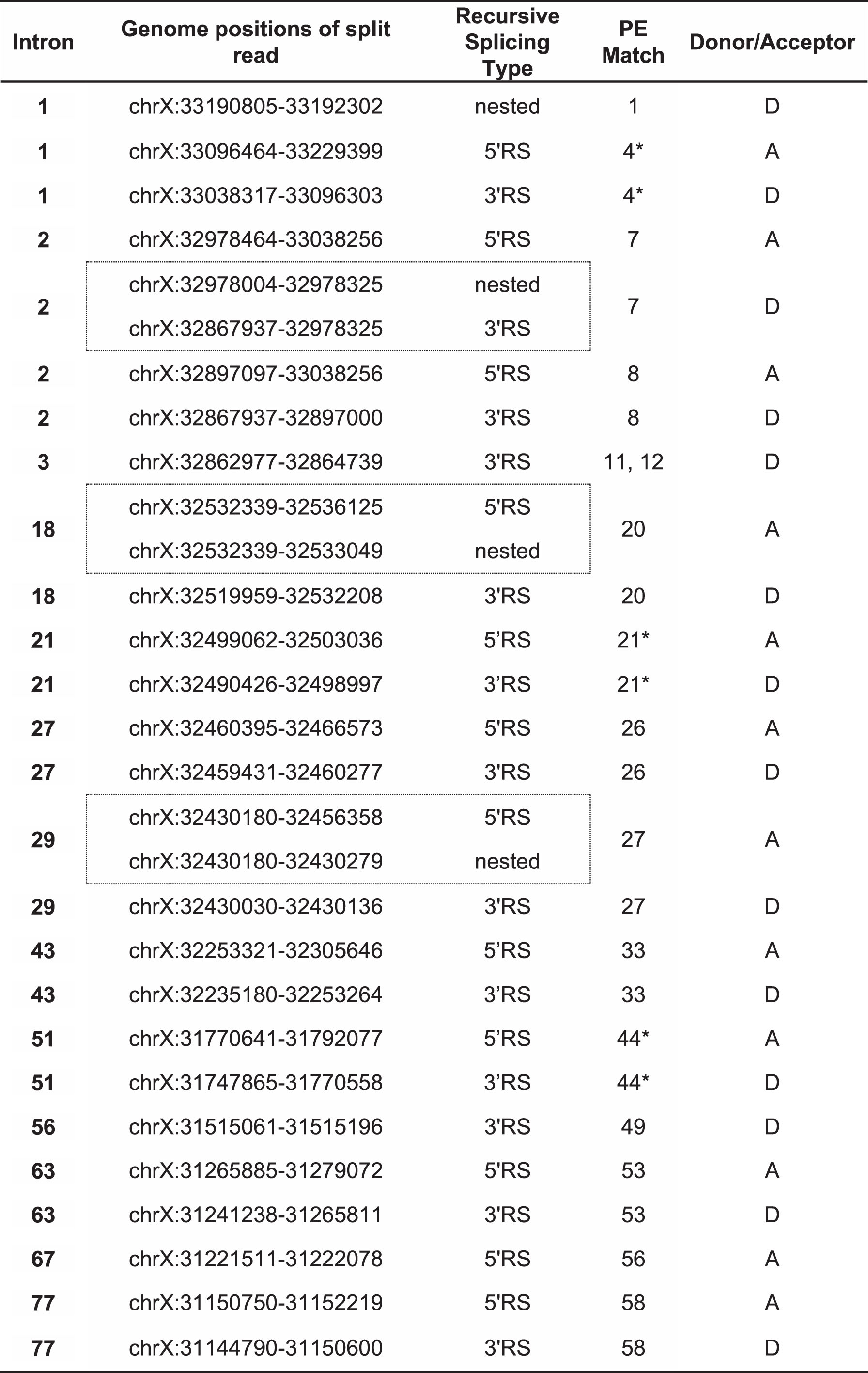 |
Several interesting features were apparent in this set of Gazzoli-matched splice sites. Firstly, most of the matched PEs matched at both their acceptor and donor splice sites. Only PEs 1, 11/12 and 49 matched at their donor sites alone, and only PE56 matched at its acceptor site alone. Secondly, a clear bias was evident in the mutation categories of the matched PEs, as the majority of the Gazzoli-matched sites were from PEs induced either without mutations or by mutations distal to the PE and its splice motifs, PEs 1 and 26 being the only exceptions to this rule. Lastly, of the 15 PEs where inducing distal mutations were identified, 11 arose exclusively from mutations that were 3′ to the PE (PEs 2, 3, 4, 7, 8, 9, 10, 11, 12, 18 and 20). Only PEs 27 and 29 were induced exclusively by 5′ mutations, while PEs 33 and 49 were each induced by flanking mutations.
PSEUDOEXONS AND RECURSIVE SPLICING REGULATION
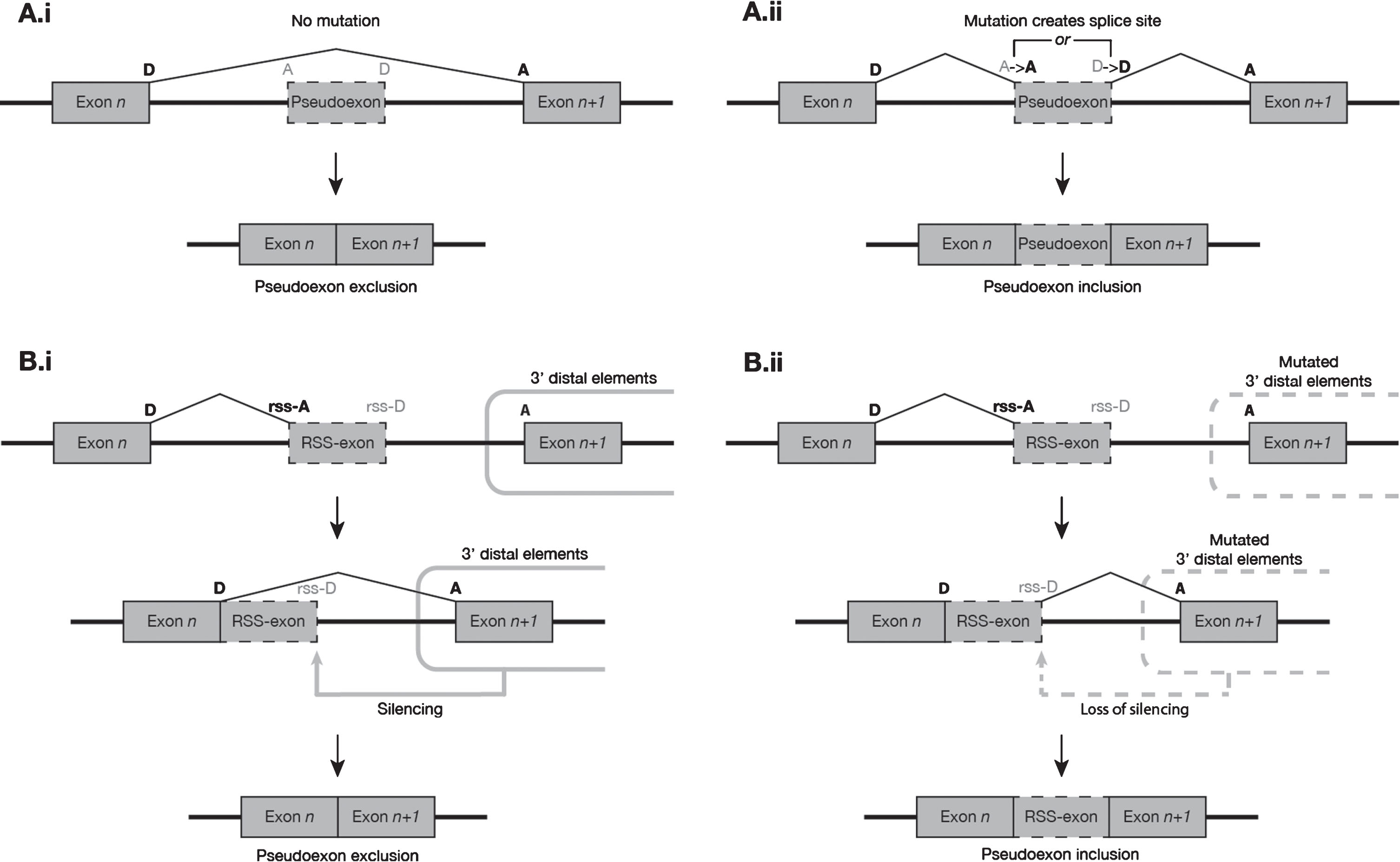
Canonical splicing of a donor-acceptor pair is often dependent on distal regulatory elements, including but not limited to other canonical splice sites [51, 52, 53]. Mutations that alter or destroy these distal elements can impede exon definition and decrease the frequency of inclusion of the affected exons in the mature transcript [42, 43]. It may be that spliceosome recognition of recursive splice sites (which necessarily exhibit a strong exon-like profile in their local motifs) is regulated by a similar system of mostly 3′ distal elements, but a system that acts to silence rather than promote the inclusion of its targets. Mutations that impaired these distal silencing elements might thereby permit an increase in the erroneous processing of recursive splice sites, converting them to PEs via a distinctly different pathway than PEs created by proximal mutation (Fig. 2). If valid, this model would explain the high coincidence of Gazzoli predicted recursive splice sites with the splice sites of PEs induced by distal mutations. However, while the mutations collated in this report may offer broad clues as to the locations of some such suppressive distal elements in the DMD gene, any further analysis of their common features awaits the assembly and analysis of a much larger dataset of PEs and recursive splice sites – one that will have to encompass multiple other genes besides DMD.
Fig. 2
Suggested model of the two most common modes of pseudoexon initiation observed in the DMD gene. (A) Proximal mutations at non-RS sites. (i) In the absence of mutation, a putative pseudoexon presents a weak exon-like profile to the spliceosome and is predominantly excluded from mature transcripts. (ii) The presence of a mutation, usually a splice-site-creating SNV, increases the exon-like profile of the putative pseudoexon, resulting in its inclusion in a much higher proportion of transcripts. (B) Mutations 3′ distal to RS sites. (i) In the absence of mutation, the exon n donor site and RS-acceptor site are used to excise the 5′ segment of an intron. Silencing elements distal (usually 3′) to the RS-exon prevent spliceosome recognition of its donor-like motif, and the RS-exon is subsequently removed from the transcript along with the rest of the 3′ intron segment. (ii) When mutations to the distal silencing elements impair their function, the intron segment 5′ to the RS-exon is spliced as normal, but the RS-exon donor-like site escapes silencing and is more readily recognised by the spliceosome, leading to a much higher frequency of inclusion of the RS-exon in the mature transcript population.
CONCLUSIONS
The 58 DMD pseudoexons collated from published reports exhibit great diversity in their sizes, locations, and pathologies. Surprisingly, PEs arising either from no mutation, or from mutations distal to the pseudoexon and its splice sites, exhibited a high coincidence with predicted recursive splice sites in the DMD introns, suggesting that some such pseudoexons may arise from disruptions to recursive splicing regulation. This finding may represent an important new insight into the etiology of pseudoexons in DMD specifically and human disease genes generally.
CONFLICT OF INTEREST
The author has no conflict of interest to declare.
ACKNOWLEDGMENTS
I would like to thank the Enid and Arthur Home Scholarship and Murdoch University for the provision of funding and resources; my reviewers, for their thoroughness and insightful suggestions; and my colleagues at the Centre for Molecular Medicine and Innovative Therapeutics, especially professors Sue Fletcher and Steve Wilton, for their advice and constructive criticism.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-190431.
REFERENCES
[1] | Nudel U . Alternative Promoters: Duchenne Muscular Dystrophy (DMD) Gene. In eLS, (Ed.). (2005) . |
[2] | Feener CA , Koenig M , Kunkel LM . Alternative splicing of human dystrophin mRNA generates isoforms at the carboxy terminus. Nature. (1989) ;338: (6215):509–11. |
[3] | Doorenweerd N , Mahfouz A , van Putten M , Kaliyaperumal R , t’Hoen PAC , Hendriksen JGM , et al. Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenn muscular dystrophy. Sci Rep. (2017) ;7: (12575). |
[4] | Wahl MC , Will CL , Lührmann R . The Spliceosome: Design Principles of a Dynamic RNP Machine. Cell. (2009) ;136: :701–18. |
[5] | Berget SM . Exon recognition in vertebrate splicing. J Biol Chem. (1995) ;270: (6):2411–4. |
[6] | De Conti LM , Baralle M , Buratti E . Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip Rev RNA. (2013) ;4: (1):49–60. |
[7] | Anna A , Gos M . Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genet. (2018) ;59: (3):253–68. |
[8] | Hong X , Scofield DG , Lynch M . Intron size, abundance, and distribution within untranslated regions of genes. Mol Biol Evol. (2006) ;23: (12):2392–404. |
[9] | Beroud C , Carrie A , Beldjord C , Deburgrave N , Llense S , Carelle N , et al. Dystrophinopathy caused by mid-intronic substitutions activating cryptic exons in the DMD gene. Neuromuscular disorders: NMD. (2004) ;14: (1):10–8. |
[10] | Zhang Z , Habara Y , Nishiyama A , Oyazato Y , Yagi M , Takeshima Y , et al. Identification of seven novel cryptic exons embedded in the dystrophin gene and characterization of 14 cryptic dystrophin exons. Journal of human genetics. (2007) ;52: (7):607–17. |
[11] | Roberts RG , Bentley DR , Bobrow M . Infidelity in the structure of ectopic transcripts: a novel exon in lymphocyte dystrophin transcripts. Human mutation. (1993) ;2: (4):293–9. |
[12] | Suminaga R , Takeshima Y , Adachi K , Yagi M , Nakamura H , Matsuo M . A novel cryptic exon in intron 3 of the dystrophin gene was incorporated into dystrophin mRNA with a single nucleotide deletion in exon 5. Journal of human genetics. (2002) ;47: (4):196–201. |
[13] | Tran VK , Zhang Z , Yagi M , Nishiyama A , Habara Y , Takeshima Y , et al. A novel cryptic exon identified in the 3′ region of intron 2 of the human dystrophin gene. Journal of human genetics. (2005) ;50: (8):425–33. |
[14] | Bouge AL , Murauer E , Beyne E , Miro J , Varilh J , Taulan M , et al. Targeted RNA-Seq profiling of splicing pattern in the DMD gene: exons are mostly constitutively spliced in human skeletal muscle. Sci Rep. (2017) ;7: :39094. |
[15] | Yagi M , Takeshima Y , Wada H , Nakamura H , Matsuo M . Two alternative exons can result from activation of the cryptic splice acceptor site deep within intron 2 of the dystrophin gene in a patient with as yet asymptomatic dystrophinopathy. Human genetics. (2003) ;112: (2):164–70. |
[16] | Dwi Pramono ZA , Takeshima Y , Surono A , Ishida T , Matsuo M . A novel cryptic exon in intron 2 of the human dystrophin gene evolved from an intron by acquiring consensus sequences for splicing at different stages of anthropoid evolution. Biochemical and biophysical research communications. (2000) ;267: (1):321–8. |
[17] | Gualandi F , Rimessi P , Trabanelli C , Spitali P , Neri M , Patarnello T , et al. Intronic breakpoint definition and transcription analysis in DMD/BMD patients with deletion/duplication at the 5′ mutation hot spot of the DMD gene. Gene. (2006) ;370: :26–33. |
[18] | Ishibashi K , Takeshima Y , Yagi M , Nishiyama A , Matsuo M . Novel cryptic exons identified in introns 2 and 3 of the human dystrophin gene with duplication of exons 8-11. The Kobe journal of medical sciences. (2006) ;52: (3-4):61–75. |
[19] | Sedláčková J , Vondráček P , Hermanová M , Zámečník J , Hrubá Z , Haberlová J , et al. Point mutations in Czech DMD/BMD patients and their phenotypic outcome. Neuromuscular Disorders. (2009) ;19: (11):749–53. |
[20] | Zaum AK , Stuve B , Gehrig A , Kolbel H , Schara U , Kress W , et al. Deep intronic variants introduce DMD pseudoexon in patient with muscular dystrophy. Neuromuscular disorders: NMD. (2017) ;27: (7):631–4. |
[21] | Tuffery-Giraud S , Saquet C , Thorel D , Disset A , Rivier F , Malcolm S , et al. Mutation spectrum leading to an attenuated phenotype in dystrophinopathies. European journal of human genetics: EJHG. (2005) ;13: (12):1254–60. |
[22] | Ferlini A , Muntoni F . The 5′ region of intron 11 of the dystrophin gene contains target sequences for mobile elements and three overlapping ORFs. Biochemical and biophysical research communications. (1998) ;242: (2):401–6. |
[23] | Ferlini A , Galie N , Merlini L , Sewry C , Branzi A , Muntoni F . A novel Alu-like element rearranged in the dystrophin gene causes a splicing mutation in a family with X-linked dilated cardiomyopathy. American journal of human genetics. (1998) ;63: (2):436–46. |
[24] | Nasim MT , Chernova TK , Chowdhury HM , Yue BG , Eperon IC . HnRNP G and Tra2beta: opposite effects on splicing matched by antagonism in RNA binding. Human molecular genetics. (2003) ;12: (11):1337–48. |
[25] | Rimessi P , Fabris M , Bovolenta M , Bassi E , Falzarano S , Gualandi F , et al. Antisense modulation of both exonic and intronic splicing motifs induces skipping of a DMD pseudo-exon responsible for x-linked dilated cardiomyopathy. Human gene therapy. (2010) ;21: (9):1137–46. |
[26] | Malueka RG , Takaoka Y , Yagi M , Awano H , Lee T , Dwianingsih EK , et al. Categorization of 77 dystrophin exons into 5 groups by a decision tree using indexes of splicing regulatory factors as decision markers. BMC genetics. (2012) ;13: :23. |
[27] | Gurvich OL , Tuohy TM , Howard MT , Finkel RS , Medne L , Anderson CB , et al. DMD pseudoexon mutations: splicing efficiency, phenotype, and potential therapy. Annals of neurology. (2008) ;63: (1):81–9. |
[28] | Tuffery-Giraud S , Saquet C , Chambert S , Claustres M . Pseudoexon activation in the DMD gene as a novel mechanism for Becker muscular dystrophy. Human mutation. (2003) ;21: (6):608–14. |
[29] | Ikezawa M , Minami N , Takahashi M , Goto Y , Miike T , Nonaka I . Dystrophin gene analysis on 130 patients with Duchenne muscular dystrophy with a special reference to muscle mRNA analysis. Brain & development. (1998) ;20: (3):165–8. |
[30] | Trabelsi M , Beugnet C , Deburgrave N , Commere V , Orhant L , Leturcq F , et al. When a mid-intronic variation of DMD gene creates an ESE site. Neuromuscular Disord. (2014) ;24: (12):1111–7. |
[31] | Takeshima Y , Yagi M , Okizuka Y , Awano H , Zhang Z , Yamauchi Y , et al. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. Journal of human genetics. (2010) ;55: (6):379–88. |
[32] | Ishmukhametova A , Van Kien PK , Mechin D , Thorel D , Vincent MC , Rivier F , et al. Comprehensive oligonucleotide array-comparative genomic hybridization analysis: new insights into the molecular pathology of the DMD gene. European Journal of Human Genetics. (2012) ;20: (10):1096–100. |
[33] | Bovolenta M , Neri M , Fini S , Fabris M , Trabanelli C , Venturoli A , et al. A novel custom high density-comparative genomic hybridization array detects common rearrangements as well as deep intronic mutations in dystrophinopathies. BMC genomics. 2008;9. |
[34] | Gonorazky H , Liang M , Cummings B , Lek M , Micallef J , Hawkins C , et al. RNAseq analysis for the diagnosis of muscular dystrophy. Ann Clin Transl Neurol. (2016) ;3: (1):55–60. |
[35] | Cummings BB , Marshall JL , Tukiainen T , Lek M , Donkervoort S , Foley AR , et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med. (2017) ;9: (386):eaal5209. |
[36] | Baskin B , Gibson WT , Ray PN . Duchenne muscular dystrophy caused by a complex rearrangement between intron 43 of the DMD gene and chromosome 4. Neuromuscular Disorders. (2011) ;21: :178–82. |
[37] | Khelifi MM , Ishmukhametova A , Khau Van Kien P , Thorel D , Mechin D , Perelman S , et al. Pure intronic rearrangements leading to aberrant pseudoexon inclusion in dystrophinopathy: a new class of mutations? Human mutation. (2011) ;32: (4):467–75. |
[38] | Greer K , Mizzi K , Rice E , Kuster L , Barrero RA , Bellgard MI , et al. Pseudoexon activation increases phenotype severity in a Becker muscular dystrophy patient. Molecular genetics & genomic medicine. (2015) ;3: (4):320–6. |
[39] | Madden HR , Fletcher S , Davis MR , Wilton SD . Characterization of a complex Duchenne muscular dystrophy-causing dystrophin gene inversion and restoration of the reading frame by induced exon skipping. Human mutation. (2009) ;30: (1):22–8. |
[40] | Goncalves A , Oliveira J , Coelho T , Taipa R , Melo-Pires M , Sousa M , et al. Exonization of an Intronic LINE-1 Element Causing Becker Muscular Dystrophy as a Novel Mutational Mechanism in Dystrophin Gene. Genes. (2017) ;8: (10). |
[41] | Cagliani R , Sironi M , Ciafaloni E , Bardoni A , Fortunato F , Prelle A , et al. An intragenic deletion/inversion event in the DMD gene determines a novel exon creation and results in a BMD phenotype. Human genetics. (2004) ;115: (1):13–8. |
[42] | Juan-Mateu J , Gonzalez-Quereda L , Rodriguez MJ , Verdura E , Lazaro K , Jou C , et al. Interplay between DMD point mutations and splicing signals in Dystrophinopathy phenotypes. PloS one. (2013) ;8: (3):e59916. |
[43] | Deburgrave N , Daoud F , Llense S , Barbot JC , Recan D , Peccate C , et al. Protein- and mRNA-based phenotype-genotype correlations in DMD/BMD with point mutations and molecular basis for BMD with nonsense and frameshift mutations in the DMD gene. Human mutation. (2007) ;28: (2):183–95. |
[44] | Sherry ST , Ward MH , Kholodov M , Baker J , Phan L , Smigielski EM , et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. (2001) ;29: :308–11. |
[45] | Magri F , Del Bo R , D’Angelo MG , Govoni A , Ghezzi S , Gandossini S , et al. Clinical and molecular characterization of a cohort of patients with novel nucleotide alterations of the Dystrophin gene detected by direct sequencing. BMC medical genetics. (2011) ;12: :37. |
[46] | Suzuki H , Aoki Y , Kameyama T , Saito T , Masuda S , Tanihata J , et al. Endogenous Multiple Exon Skipping and Back-Splicing at the DMD Mutation Hotspot. International Journal of Molecular Sciences. (2016) ;17: (10). |
[47] | Burnette JM , Miyamoto-Sato E , Schaub MA , Conklin J , Lopez AJ . Subdivision of large introns in Drosophila by recursive splicing at nonexonic elements. Genetics. (2005) ;170: (2):661–74. |
[48] | Sibley CR , Emmett W , Blazquez L , Faro A , Haberman N , Briese M , et al. Recursive splicing in long vertebrate genes. Nature. (2015) ;521: (7552):371–5. |
[49] | Georgomanolis T , Sofiadis K , Papantonis A . Cutting a Long Intron Short: Recursive Splicing and Its Implications. Front Physiol. (2016) ;7: :598. |
[50] | Suzuki H , Kameyama T , Ohe K , Tsukahara T , Mayeda A . Nested introns in an intron: evidence of multi-step splicing in a large intron of the human dystrophin pre-mRNA. FEBS Lett. (2013) ;587: (6):555–61. |
[51] | Gazzoli I , Pulyakhina I , Verwey NE , Ariyurek Y , Laros JF , t Hoen PA , et al. Non-sequential and multi-step splicing of the dystrophin transcript. RNA Biol. (2016) ;13: (3):290–305. |
[52] | Ke S , Chasin LA . Intronic motif pairs cooperate across exons to promote pre-mRNA splicing. Genome Biol. (2010) ;11: :R84. |
[53] | Kim SW , Taggart AJ , Heintzelman C , Cygan KJ , Hull GH , Wang J , et al. Widespread intra-dependencies in the removal of introns from human transcripts. Nucleic Acids Res. (2017) ;45: (16):9503–13. |
[54] | Shapiro MB , Senapathy P . RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res. (1987) ;15: (17):7155–74. |
[55] | Sheth N , Roca X , Hastings ML , Roeder T , Krainer AR , Sachidanandam R . Comprehensive splice-site analysis using comparative genomics. Nucleic Acids Res. (2006) ;34: (14):3955–67. |


