
Defects in Axonal Transport in Inherited Neuropathies
Abstract
Axonal transport is a highly complex process essential for sustaining proper neuronal functioning. Disturbances can result in an altered neuronal homeostasis, aggregation of cargoes, and ultimately a dying-back degeneration of neurons. The impact of dysfunction in axonal transport is shown by genetic defects in key proteins causing a broad spectrum of neurodegenerative diseases, including inherited peripheral neuropathies. In this review, we provide an overview of the cytoskeletal components, molecular motors and adaptor proteins involved in axonal transport mechanisms and their implication in neuronal functioning. In addition, we discuss the involvement of axonal transport dysfunction in neurodegenerative diseases with a particular focus on inherited peripheral neuropathies. Lastly, we address some recent scientific advances most notably in therapeutic strategies employed in the area of axonal transport, patient-derived iPSC models, in vivo animal models, antisense-oligonucleotide treatments, and novel chemical compounds.
COMMON ABBREVIATIONS
AF | actin filament |
ALS | amyotrophic lateral sclerosis |
CMT | Charcot-Marie-Tooth |
dHMN | distal hereditary motor neuropathy |
ER | endoplasmic reticulum |
HDAC6 | histone deacetylase 6 |
HSAN | hereditary sensory and autonomic neuropathy |
HSP | hereditary spastic paraplegia |
IPN | inherited peripheral neuropathies |
iPSC | induced pluripotent stem cell |
MIM | mitochondrial inner membrane |
MOM | mitochondrial outer membrane |
MT | microtubule |
MTA | microtubule-targeting agents |
NEFL | neurofilament light |
NEFM | neurofilament medium |
NEFH | neurofilament heavy |
NF | neurofilament |
NMJ | neuromuscular junction |
PS | Perry syndrome |
PTM | post-translational modifications |
RBP | RNA-binding protein |
RNP | ribonucleoprotein particles |
SARM1 | Sterile Alpha and TIR Motif Containing 1 |
sHSP | small heat shock protein |
SMA | spinal muscular atrophy |
SMALED | spinal muscular atrophy with lower limb predominance |
Axonal transport is a highly complex process allowing movement of molecules and organelles within neurons over tremendous distances towards presynaptic nerve termini (anterograde transport), and transferring material from the periphery back to the neuronal soma (retrograde transport). This intracellular trafficking is crucial for sustaining proper neuronal functioning as well as clearing misfolded proteins or damaged organelles to avoid accumulation of harmful aggregates. Neuronal homeostasis depends on an efficient axonal transport allowing neuronal growth (axonal outgrowth), repair and regeneration upon injury, endocytosis and exocytosis of large and small molecules. The complex morphology and length (which can reach over one meter) of neurons make them particularly vulnerable to changes in axonal trafficking. Transport of a molecule or organelle (cargo) within the axon implies its recognition and binding to motor proteins, followed by an ATP-dependent movement of the motors along the cytoskeleton, direction of the cargo to the correct destination and release of the cargo upon reaching its destination. Axonal transport is regulated at all of these stages in a number of different ways. Impaired axonal transport can result in an altered neuronal homeostasis, aggregation of cargoes, and ultimately a dying-back degeneration of neurons.
Here we review the cytoskeletal components, molecular motors and adaptor proteins involved in axonal transport mechanisms and their implication in neuronal functioning. Furthermore, we discuss how genetic mutations, associated with inherited peripheral neuropathies (IPN), impact on axonal transport. Finally, we focus on some of the underlying mechanisms of axonal transport disturbances causing neurodegeneration and highlight future therapeutic prospects.
CYTOSKELETAL COMPONENTS OF AXONAL TRANSPORT
Neurons become post-mitotic cells in early development and need to remain functional for a lifetime, requiring a solid structural cytoskeleton. The neuronal cytoskeleton consists of microtubules (MTs), intermediate filaments and actin filaments (AFs), each having their own intracellular distribution (Fig. 1).
Fig.1
Schematic overview of the peripheral nerve and the mechanisms directly involved in typical and pathogenic axonal transport in anterograde and retrograde direction. (A) Transport of mitochondria occurs along microtubules by dynein (purple) and kinesin (blue) motors. Mitochondria undergo a coordinated balance between mitochondrial fission and fusion. Mitochondria-ER tethering and mitochondrial Ca2+ flux are maintained to sustain proper axonal transport. (B) Formation of autophagosomes and endosomes with subsequent fusion with lysosomes. Anterograde transport of lysosomes and retrograde transport of endosomes and autophagosomes occurs along microtubules by kinesin and dynein motors, respectively. (C) mRNA are bound by RNA-binding proteins (RBPs) and heterogeneous nuclear ribonuclear proteins (hnRNPs), and subsequently formed into ribonucleotide particles (RNPs) capable of being transported by kinesin and dynein motors along the microtubules. Increased aggregation of RBPs causes formation of stress granules. (D) The neuronal cytoskeleton provides the tracks on which the molecular motors move to facilitate axonal transport. The dynein and kinesin motors move along the microtubules, whereas the myosin motors move along actin filaments. Both actin filaments and microtubules undergo dynamic polymerization and depolymerization mediated by adaptor proteins. (E) Synaptic vesicles are transported in anterograde direction by binding to kinesin motors, which move along microtubules. Fusion of synaptic vesicles at the nerve terminal is mediated by v-SNARE (purple) and t-SNARE (green) complexes. Here the t-SNARE complex is depicted as Syntaxin (light green) and SNAP25 (dark green).
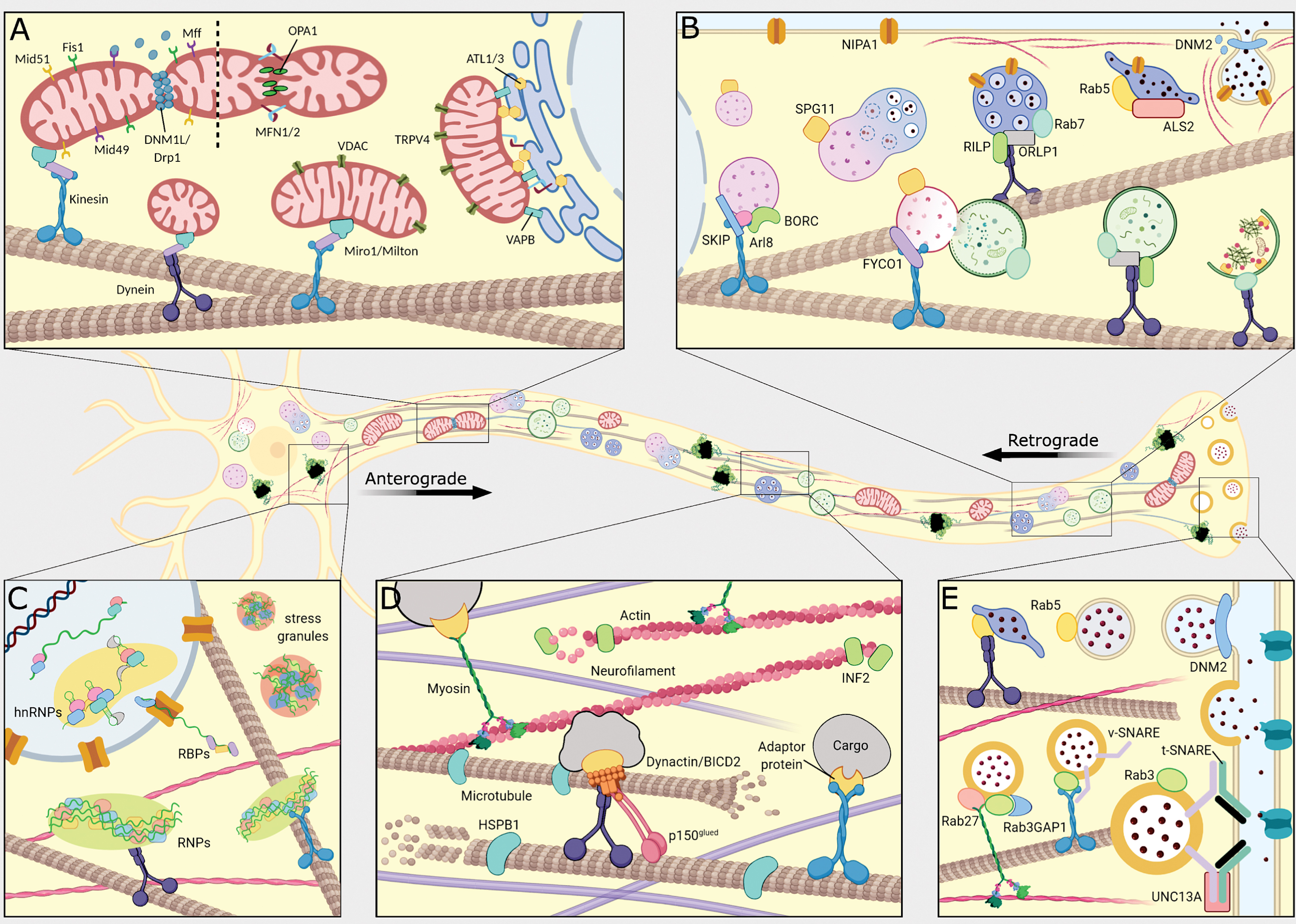
The MTs originate from the centrosome and are the major component of the neuronal cytoskeleton. MTs consist of α- and β-tubulin polymers that form polarized tubular structures, which are subjected to a dynamic process of polymerization and depolymerization [1]. Parallel MTs form unipolar arrays with the ‘minus-ends’ orientated towards the soma and the ‘plus-ends’ towards the axon, directing motor proteins in antero- or retrograde direction respectively. The tubulin polymers are involved in spatial organization and cell shape maintenance, as well as in many features of cytoplasmic structure, including organization of specific signaling pathways [1]. Microtubule-associated proteins, motor proteins, post-translational tubulin modifications and plus-end tracking proteins regulate MT dynamics.
The major intermediate filaments of neurons are neurofilaments (NFs), which control axon diameter and thereby axonal conductance [2]. Mature NFs contain neurofilament light (NEFL), medium (NEFM) and heavy (NEFH) chains. However, NFs in the peripheral nervous system can also contain peripherin. NF monomers share a common structural organization: a central α-helical rod domain, flanked by a N-terminal head, and a C-terminal tail domain [2]. In addition to their function in maintenance of the axonal architecture, NFs are fundamental to maintain Schwann cell-axon interactions, NF complex assembly and axonal transport [3].
The AFs consist of polymers of globular actin units and have a growing end (plus-end) where actin monomers are attached, while monomers are dissociated at the minus end. AF dynamics sustain a balance between polymerization and depolymerization and provide a structured cytoskeletal network for cell support, shape and migration. Within the cell, the function of AFs is regulated by a range of actin-binding proteins such as profilins and formins.
Disturbance or alteration of cytoskeleton stability and dynamics occurs in various neurodegenerative diseases, including IPN, and can be caused by mutations in the cytoskeletal proteins themselves, in their adaptor proteins or in other regulators. Mutations in INF2, encoding formin, cause autosomal dominant focal and segmental glomerulosclerosis (FSGS) as well as dominant intermediate Charcot-Marie-Tooth (CMT) type E [4, 5].
Mutations in tubulin genes are commonly referred to as tubulinopathies, such as mutations in β-tubulin known to cause polymicrogyria, congenital fibrosis of extraocular muscle (CFEOM) or malformation of cortical development. Mutations in TUBB3 are not only associated with CFEOM, e.g. the D417N mutation can also cause a polyneuropathy by reducing kinesin localization to microtubule plus-ends and affecting the axonal transport. TUBB3 mutations can lead to microtubule overstabilization and increased depolymerization [6].
Mutations in NEFL were identified in several subtypes of axonal CMT (CMT2) neuropathies, with dominant mutations causing their aggregation and recessive mutations resulting in a loss-of-function [7–9]. NEFL mutations can target different domains in the protein affecting the NF assembly, e.g. the Q333P mutation leads to destabilization of coiled–coil interactions resulting in reduced self-interaction and dimerization of NFs [10]. A mutation located in the NEFL head domain, P8R, affects phosphorylation and destabilization of the NF complex resulting in NF aggregation [11]. Furthermore, phosphorylation of the NEFL head domain not only regulates NF assembly and disassembly, but also its transport in the axon [11, 12]. Dominant mutations in NEFH can also cause CMT2 [7, 13]. All reported NEFH mutations are frameshifts leading to the translation of additional 3’UTR regions encoding a cryptic amyloidogenic element. This leads to toxic protein aggregation similar to mutational mechanisms in NEFL and FUS, which when recognized by the autophagic pathway in motor neurons, trigger caspase 3 activation resulting in apoptosis [7, 13].
Several small heat shock proteins (sHSP), HSPB1 and HSPB8 are associated with cytoskeletal abnormalities and neuromuscular dysfunction [14–16]. Dominant mutations in HSPB1 are associated with CMT type 2F and distal hereditary motor neuropathy (dHMN) [16]. The S135F mutation results into a higher affinity of HSPB1 for α-tubulin and overstabilization of MTs, and several HSPB1 mutants reduce α-tubulin acetylation, affecting axonal transport [16]. HSPB1 mutations can also affect the assembly and transport of NFs [17]. Moreover, spinal motor neurons, differentiated from patient-derived induced pluripotent stem cells (iPSC), carrying HSPB1 mutations showed a significant reduction of the mitochondrial mobility along the axons [18].
MOLECULAR MOTORS AND ADAPTORS IN AXONAL TRANSPORT
Neurons depend on efficient transport of cargoes, which is enforced by several different classes of molecular motor proteins (Fig. 1) [19]. Microtubule based molecular motors are grouped into the kinesins (for anterograde transport) and cytoplasmic dyneins (for retrograde transport) [19]. The kinesin family consists of 45 genes classified into 15 subfamilies of which the kinesin-1, kinesin-2 and kinesin-3 families contribute to axonal transport [20]. Kinesin motor domains homo- and heterodimerize, and bind additional kinesin light chains forming the kinesin motor complex [20]. The dynein family is much smaller, and the main component -dynein heavy chain- is encoded by only one gene (DYNC1H1). Dynein functions as a complex consisting of two dynein heavy chains that dimerize at their tail domains. Additional dynein intermediate chains and dynein light chains will bind to these tails by forming a cargo-binding domain [20].
Many axonal cargoes have multiple motor types that can bind simultaneously, even cargoes that move steadily in either antero- or retrograde direction [21, 22]. This shows that molecular motors do not function independently from each other when pulling an organelle in opposite directions. Rather they are co-dependent, as shown by impairment of minus-end motors resulting in severe suppression of plus-end motility [22]. This simultaneous application of opposite forces does not necessarily result in stalled cargoes. Instead, it likely provides additional support for transport coordination to overcome mechanical obstacles on MT tracks [21].
The third type of molecular motors are myosins, specifically involved in the transport over actin filaments [19]. Mammals have 40 myosin genes [19]. The most essential differences are found in the C-terminal globular tail domain, which recognizes various cargoes through direct interactions or via adaptor proteins [19]. For instance, Myosin Va is able to interact with kinesin heavy chains, with MTs through its tail domain and with NEFL through its head motor domain. Doing so, Myosin Va plays a crucial role in coupling microtubule- and actin-based transport mechanisms and regulates distribution of cargoes across the cytoskeletal network [23, 24].
Post-translational modification (PTM) of cytoskeletal components includes polyglutamylation, polyglycylation, detyrosination, acetylation, phosphorylation and palmitoylation [1]. For MTs, these preferentially occur on tubulin subunits already incorporated into microtubules. Molecular motors can recognize different PTMs as signature for their recruitment; e.g. polyglutamylation recruits the kinesin-3 family member KIF1A motor, whereas tyrosination recruits kinesin-1 family member KIF5 motor proteins [25, 26]. Additionally, protein kinases regulate axonal transport through direct phosphorylation of motors, adapters and cargoes, or protein kinases can phosphorylate several factors involved in the regulation of microtubule stability [27].
Dynactin is the best known adaptor protein of the cytoplasmic dynein complex. Dynactin-dynein interaction expands the range of cargoes that dynein can move, and increases the dynein motor processivity. Independently of cytoplasmic dynein, dynactin can anchor microtubules at the centrosome. The bicaudal D homologue (BICD) proteins represent another group of activating adaptors. In mammals, there are two BICD proteins, BICD1 and BICD2, as well as two related proteins, BICDR1 and BICDR2. The BICD functioning is diverse; BICD2 mostly functions in a complex of one dynein and one dynactin, whereas BICDR1 can recruit two dynein dimers to a single dynactin, further enhancing the force and velocity of the motor complex [28–30]. BICD2 also interacts with the dynein heavy chain and dynactin, enhancing the affinity of the dynein–dynactin interaction. Another class of microtubule adaptors are the Hook proteins, which are involved in motor-microtubule interactions as well as in cargo binding [31]. However, mutations in Hook proteins have so far not been associated with IPN or other neurodegenerative diseases.
Where the cytoskeleton functions as the tracks, the molecular motors are equally important for proper axonal transport. Disturbances in these motor complexes and their adaptors are a recognized causal mechanism for IPN and related diseases. Dominant mutations in dynein heavy chain (DYNC1H1) are linked to several distinct neurodegenerative phenotypes, e.g. CMT type 2O, spinal muscular atrophy with lower limb predominance (SMALED), hereditary spastic paraplegia (HSP) and intellectual disability with neuronal migration defects (MRD13) [reviewed in [32]].
Several dynein adaptors and modulators are associated with neurological disease; dynactin (DCTN1), Huntingtin (HTT), LIS1, BICD1 and BICD2. Mutations in DCTN1 can cause Perry Syndrome (PS) and dHMN, and susceptibility to develop amyotrophic lateral sclerosis (ALS) [33–35]. DCTN1 mutations found in PS patients affect amino acid residues within or immediately adjacent to the p150glued CAP-Gly GKNDG motif of dynactin. A G59S variant in DCTN1 causing HMN occurs centrally in the p150glued CAP-Gly domain. However, both PS and HMN associated variants induce a modest decrease in MT binding. Cells transfected with DCTN1 mutants have a dramatic redistribution of dynactin and more p150Glued aggregates [34, 35]. It remains unclear why mutations localized in different part of the DCTN1 sequence manifest such different disease phenotypes [34, 35].
BICD2 mutations cause a spectrum of phenotypes including SMALED, HSP, and distal myopathy [36–40]. BICD2 joins dynein and dynactin in a motor protein complex capable of processive movement. Modeling of SMALED-BICD2 mutations in cells showed an increased retrograde transport, suggesting that an imbalance of anterograde and retrograde dynein motor complex motility could be the underlying pathomechanism for these mutations [41, 42]. In addition, BICD2 has been implicated in Golgi functioning as the BICD2 coiled-coil (CC) structure allows interaction with the small GTPase RAB6A located at the Golgi apparatus and fragmentation of the Golgi apparatus has been shown in BICD2-patient fibroblasts [43].
Mutations in the other major group of molecular motors (kinesins) can also give rise to a spectrum of neurodegenerative diseases. Variants in KIF1A have been associated with HSP, hereditary sensory and autonomic neuropathy (HSAN) and complex phenotypes (combining HSP, HSAN and ataxia) [44, 45]. KIF5A variants can cause CMT2, HSP and ALS [46–49]. Genotype/phenotype correlations revealed that the site of KIF5A mutations determine the clinical phenotype. Mutations in the kinesin motor or neck domain cause CMT2 and HSP. Mutations affecting splicing (exon 27) at the C-terminus cause atypical ALS with an earlier onset and a slower disease progression [46–49]. KIF5A mutations in HSP alter processivity and directionality of kinesin by changing order–disorder transition of the neck linker, or affecting ATPase activity/microtubule gliding. Both mechanisms can alter MT-dependent transport and net anterograde transport of cargoes [50–52]. KIF1Bβ mutations were first implicated in CMT2A in 2001, but as only few patients were identified, the association has been controversial [37, 53, 54]. Functional evidence for KIF1Bβ mutations in neuropathy phenotypes suggests that different mechanisms may be at play. The initially reported Q98L mutation resides in the conserved ATP-binding site and significantly reduced ATPase activity and perinuclear accumulation of the mutant KIF1Bβ protein. Only recently a novel CMT2-associated Y1087C mutation in KIF1Bβ was identified. It specifically impairs KIF1Bβ binding capacity and transport of Insulin-like growth factor 1 receptor (IGF1R) down the axons affecting IGF-I/IGF1R signaling, which is essential for neuronal survival and axonal development [54].
CARGOES IN AXONAL TRANSPORT
Synaptic vesicles, NFs, and cytosolic proteins are cargoes transported in anterograde fashion. Signaling endosomes, autophagosomes and proteins involved in injury signaling are transported towards the cell body. Lastly, mitochondria, a variety of endosomes, lysosomes and mRNA are transported in a bi-directional manner [31]. Depending on the cargo and its destination, specific regulatory proteins and motor complexes are recruited (Fig. 1). While cargoes make use of different transport pathways with their specific components, there is clear interdependence between these pathways and many of them converge [55]. In addition to the cargoes discussed below, axons also provide possibilities to transport viruses [reviewed in [56]].
mRNA and cytosolic proteins
Axons constitute the connections between neurons and their targets allowing them to communicate and respond to environmental stimuli. A large and diverse pool of cytosolic proteins is transported slowly from the soma towards the axon [57]. With a couple of examples studied so far, current models suggest that these cytosolic proteins form spontaneously aggregated complexes that undergo ‘dynamic recruitment’ to allow short bursts of anterograde transport by hitching a ride on passing vesicles [58, 59]. However, the fast-acting mechanisms of the axon, required to promptly respond to external stimuli, could not depend solely on slow transport of proteins from the soma. As such processes in the distal axons rely on localized protein synthesis to spatially and temporally regulate protein content by localized mRNA translation. In addition, local translation allows for differential PTM variants of translated proteins according local requirements.
The mRNAs subjected to local translation bind to RNA-binding proteins (RBPs) before undergoing active transport by molecular motors. While RBPs are reported to bind to both anterograde and retrograde motor complexes, it is unclear whether these RBPs have bound mRNAs, which would allow for differentiation between the two supposed functions: (I) relocation of mRNAs within the axon or (II) delivery of RBPs to the cell body for reuse [60]. Binding of mRNAs to RBPs regulates PTMs and stress responses ensuring mRNA stability, translation efficiency, and localization to cytoplasmic granules [61]. mRNA association with RBPs also guarantees the formation of transport-competent ribonucleoprotein particles (RNPs), which are protein complexes commonly referred to as ‘RNA transport granules’. Specificity of RBP binding to mRNAs is dictated by sequences within the 5′ and 3′ UTR of mRNAs, which are recognized by RBPs. Although more rarely, RBP regulatory sequences can also be present in protein coding regions. RBPs can cooperate or compete for a regulatory outcome at more than one of these sites per mRNA and specific RBP binding sequences are often present in many different mRNAs. Interestingly, mRNAs encoding proteins with complementary functions were shown to bind similar sets of RBPs leading to the concept of ‘RNA regulons’, which could be involved in subcellular compartment specific transport and translation.
Mutation in genes encoding RBPs are linked with several neuromuscular diseases such as ALS, SMA, multisystem proteinopathy (MSP) and frontotemporal lobar degeneration (FTLD). Well-known examples are TDP-43 mutations causative for ALS and FTLD. TDP-43 mutations affect its subcellular localization causing accumulation of RNP granules in the cell soma and proximal axons, they show disturbance in RNP complex transport and alter the axonal content of both mRNAs and miRNAs [62]. A major subtype of RBPs are the heterogeneous nuclear ribonuclear proteins (hnRNPs), wherein mutations are responsible for a number of cases of familial ALS and FTD [63]. Purice et al., (2018) reviewed the subset of disease-causing RBPs that are hnRNPs, namely TDP-43, FUS, hnRNPA1, hnRNPA2B1, matrin-3, and TIA1 [63]. While hnRNPs have an intrinsic tendency to aggregate, a common causal mechanism for these genes is the presence of mutations in the low complexity prion-like domain, which exacerbate the propensity to form self-seeding fibrils, resulting in accumulation of persistent stress granules [64, 65].
Mitochondrial transport
Neurons require functional mitochondria that need to travel long distances to provide support, such as adequate ATP-production, at specific sites of the neuron, like synaptic termini. Mutations affecting the balance between either division (known as fission), or collision and fusion of mitochondria, can cause alterations in the mitochondrial morphology. Subsequently, a more fragmented or dense mitochondrial network impacts axonal transport. The most prominent axonal CMT subtype (CMT2A) consists of mutations in MFN2 affecting mitochondrial transport [66]. Together with the MFN1 and OPA1, MFN2 plays a major role in the mitochondrial fusion process. Mitochondrial fusion is a unique process involving two membranes, i.e. the mitochondrial outer membrane (MOM) and mitochondrial inner membrane (MIM), that requires rearrangement in a coordinated manner in order to maintain the organelle’s integrity [67]. Mitochondrial fission is mediated by the recruitment of the DNM1L/Drp1 on the MOM [68, 69]. This happens through the interaction with the mitochondrial fission factor (Mff), mitochondrial dynamics protein Mid49 and Mid51, and in a minor interaction with mitochondrial fission 1 (Fis1) [70–72]. Mutations in GDAP1, causative for several types of CMT, alter the regulation of mitochondrial fission activity dependent on the fission factors Drp1 and Fis1. Interestingly, the effect of GDAP1 mutations is dependent on the mode of inheritance. Recessive mutations cause a reduction in the fission activity, whereas the dominant inherited mutations hamper mitochondrial fusion events [73]. Mitochondrial function and localization can be indirectly affected by mutations in the Miro/Milton complex, which mediate MT interaction, and by mutations in NEFL, which in turn alter the mitochondrial distribution [74]. Interestingly, CMT associated mutations in small heat shock proteins HSPB1 and HSPB8 indirectly affect mitochondrial function transport by altering cytoskeletal properties. Furthermore, mitochondrial functioning in neurons can also be affected by mutations causing loss of contacts between the mitochondria and the endoplasmic reticulum (ER) or ER network stability. As a consequence, the ER is hampered to initiate mitochondria to promote fission, e.g. mutations in VAPB induce the formation of abnormal ER-derived inclusions [75]. Mutations in REEP1, associated with HSP and dHMN, were shown to disrupt ER network and promote ER fragmentation [76]. Whereas, decrease of VAPB MOM protein, causes a perturbation of the uptake of Ca2+ by mitochondria, which is required to maintain an intracellular homeostasis as well as mitochondrial transport. Ca2+ is an important factor for various other functions such as cell signaling as well as regulating mitochondrial function and structure [77]. Therefore, mutations in Ca2+-channels such VDAC or TRPV4 lead to mitochondrial dysfunction in IPN [77].
Membrane-bound dynamin like GTPases known as atlastins (ATLs) mediate the formation of ER-mitochondria contact sites essential for calcium communication [78]. Mutations in the human isoforms ATL1 and ATL3 are associated with HSP and HSAN. Transmission electron microscopy studies reported higher ER-mitochondria contact sites upon expression of mutant ATL3, resulting in increased Ca2+ uptake into the mitochondria [78]. Moreover, aberrant calcium signaling affects mitochondrial trafficking through the Rho GTPases Miro1 and Miro2. Atlastin mutations demonstrate a reduced mobility of mitochondria in the cytoplasm together with altered mitochondrial localization, where the mitochondria are retained within the soma rather than distributed to the neuronal processes [78]. Furthermore, mutations in SPTLC1 and SPTLC2 produce atypical sphingolipids observed in HSAN type I. Alecu et al., (2017) demonstrated that these toxic deoxysphingolipids could localize into mitochondria disrupting the mitochondrial integrity [79].
Mutations in tRNA synthetases (e.g. GARS, HARS or KARS) also affect mitochondrial function. Mitochondrial dysfunction could contribute to neuromuscular junctions (NMJ) degeneration. E.g. a CMT2 type D mouse model expressing mutant GARS mutant displayed affected NMJs as well as muscle atrophy prior to synaptic degeneration [80].
Vesicular transport
Vesicular transport in axons occurs in different forms depending on the origin of intracellular components. Late endosomes and autophagosomes are subjected to retrograde transport to reach the lysosome, which mainly have a perinuclear localization. In contrast, synaptic vesicle precursors are produced in the soma and transported in anterograde direction towards the axon. These, mature synaptic vesicles are essential for proper neuronal growth, function and survival as they contain neurotransmitters, contribute to synapse formation and location, as well as help to sustain a balance between exo- and endocytosis [81, 82]. The transport of all vesicle types occurs through conserved mechanisms. Defects in the formation of endosomes, autophagosomes, lysosomes and synaptic vesicles, as well as impairments in their antero- and retrograde trafficking, may cause axonal degeneration [83, 84].
Rab GTPases regulate vesicular trafficking at different levels from vesicle formation, vesicle movement along actin and tubulin networks, to membrane fusion. Members of this protein family are involved in autophagy, lysosome and synaptic vesicle transport. Relevant for IPN and related neurodegenerative diseases is the small GTPase Rab5, which mediates transport and fusion of early endosomes, inducing neurite outgrowth and dendritic branching [85]. ALS2 encodes a Guanine Nucleotide Exchange Factor for Rab5 and is involved in endosomal dynamics. Mutations in ALS2, associated with the onset of HSP, ALS and primary lateral sclerosis, produce truncated forms of alsin causing alterations in Rab5 and Rab7 signaling and Rab5-to-Rab7 conversion [86]. Mutations in NIPA1 or SPG11 similarly affect endosomal trafficking. A list of other HSP genes, divided by functionality has been reviewed in [87]. In addition to Rab5, Rab7 plays a crucial role in trafficking of late endosomes to lysosomes and is responsible for bidirectional transport of autophagosomes as well as for autophagosome-lysosome fusion in all cells. Rab7 binds to effector proteins like FYCO1, responsible for plus-end, or to ORP1L and RILP, responsible for minus-end-directed transport [88].
Other classes of GTPases are involved in membrane trafficking and in vesicle formation in the endo-lysosome pathway. Dynamin2 (DNM2) polymers work by wrapping the neck of budding membranes, promoting membrane fission. Mutations in DNM2 are associated with autosomal dominant centronuclear myopathy (ADCNM) and intermediate CMT [89]. DNM2 contains actin binding sites suggesting that it may regulate actin dynamics during membrane tabulation [90].
An ensemble of kinesin-1, SKIP, Arl8, and subunits of the BLOC-one-related complex (BORC) direct anterograde transport of lysosomes into the axon. These eight BORC subunits consist of BLOS1, BLOS2, Snapin, KXD1, LOH12CR1 (myrlysin), C17orf59 (lyspersin), C10orf32 (diaskedin) and LOC729991 (MEF2BNB). Interference of the BORC function decreases lysosome transport in the axon, and its function is required for maintenance of axonal growth cone dynamics and autophagosome clearance [91].
The different functions of Rab GTPases correlated with the pathological conditions was reviewed by [86]. Nian et al., (2019) demonstrated that primary neurons from Rab–/– mice showed a reduction in lysosomal trafficking together with the accumulation of autophagosomes, suggesting an altered autophagic vesicle transport [92]. Rab3 and Rab27 are important in synaptic vesicle exocytosis, while Rab5 is a key regulator of synaptic vesicles retrieval/endocytosis. Warburg Micro Syndrome (WARBM) and CMT2 type B neuropathy are associated with mutations in RAB3GAP1 and RAB7 respectively. Zebrafish Rohon-Beard spinal sensory neurons expressing Rab7 mutations reported defects in neurite outgrowth and branching, and a marked decrease in the speed of K157N Rab7 containing vesicles, underscoring the importance of Rab7 in endosome transport [93]. Furthermore, mutations in CHMP2B cause a reduced Rab7-endosome recruitment and are linked to a FTD phenotype with partial overlap with ALS.
Another aspect is the fusion of synaptic vesicles at the nerve terminals. The neuronal v-SNAREs, vesicle Soluble N-ethylmaleimide-sensitive factor Attachment protein Receptors, are essential for these fusion processes. Mutations in VAMP2 encoding one of the v-SNAREs, cause aberrant synaptic vesicle morphology and vesicle endocytosis often characterized by axial hypotonia [94]. Other mutations such as those in UNC13A can affect synaptic regulators and are known to be involved in neurodevelopmental disorders often in combination with involuntary movements [95].
RESEARCH AND THERAPEUTICS PROSPECTS
Axonal transport deficits are one of the most common and recurrent pathomechanisms in IPN and other neurodegenerative diseases (Table 1 and Fig. 2). For this reason, axonal transport studies are crucial, not only to understand its physiological role, but also to determine the effect of mutations that impair the network of axonal transport process and its regulation. Of note is that similar disturbances of axonal transport, or one of its key components, are also at play in other far more prevalent acquired disorders of the PNS [96]. Axonal transport in human peripheral nerves is difficult to investigate, as the tissue of interest is inaccessible. Therefore, animal models have been used especially for real-time monitoring of cargoes trafficking and axonal transport defects in order to develop novel therapeutic strategies. The wings of the fruit fly Drosophila melanogaster offer an in vivo model to study axonal cargo dynamics [97] and specific targeting of fluorescently-labelled organelles in the nematode Caenorhabditis elegans allow studying of axonal growth and synaptogenesis [98]. Furthermore, in transgenic MFN2 zebrafish (Danio rerio) it is possible to genetically label mitochondria in motor neurons [99]. Other platforms for in vivo axonal transport studies compared to ex vivo tools have been reviewed by [100].
Fig.2
Overview of the subcellular components and genes (illustrated by gene symbols) involved in axonal transport mechanisms and in which defects are associated with IPN and related neurodegenerative diseases.
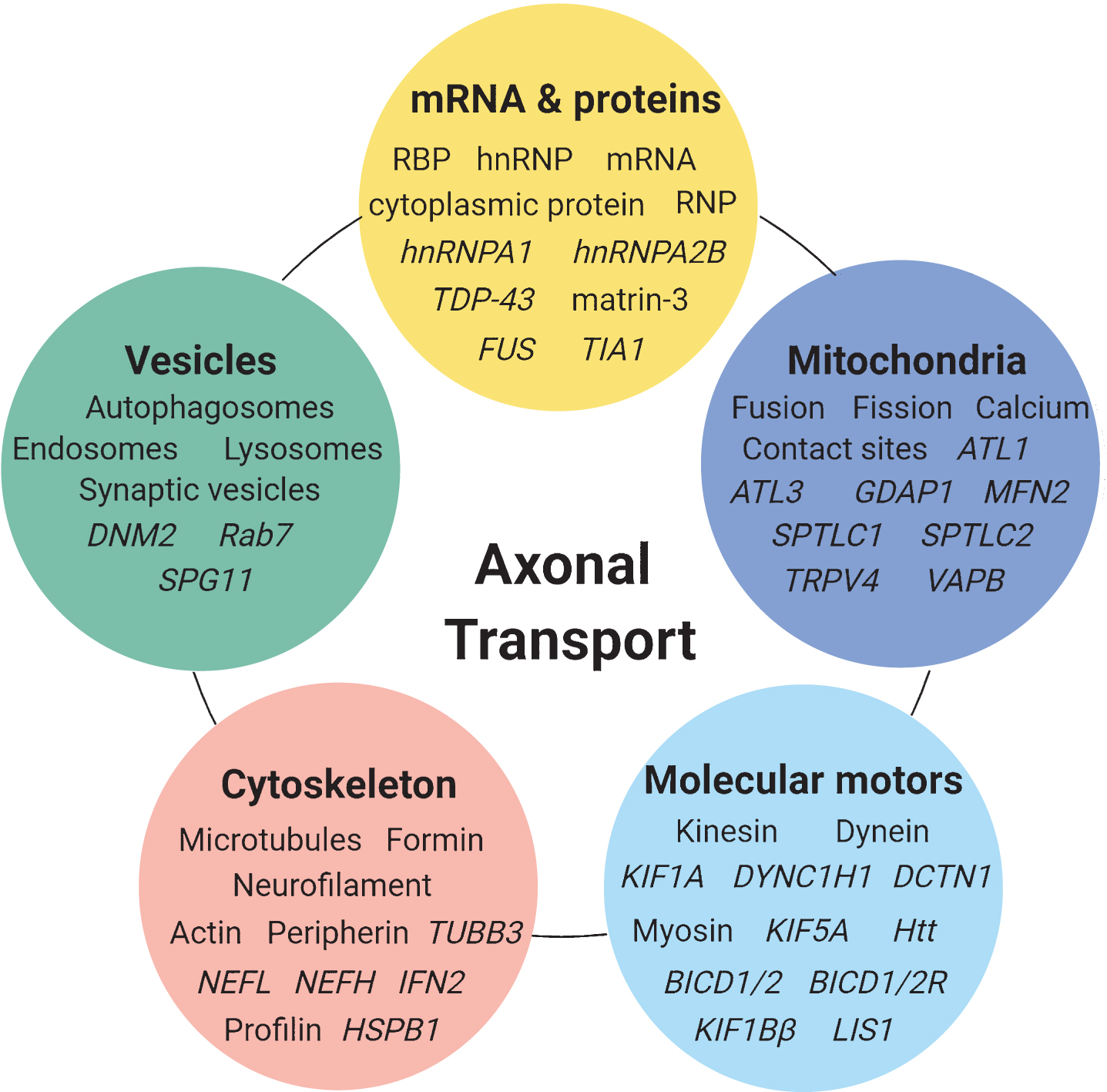
Table 1
Overview of genes causative for IPN-related disorders involved in axonal transport
| Gene symbol | Protein name | Function | Mutational effects | Mode of inheritance | Disease associations | Refs | |
| Cytoskeleton | TUBB3 | Tubulin Beta 3 Class III | Structural component of neurofilaments | Impairment of tubulin heterodimer formation. | AD | CMT2 | [1, 6] |
| Congenital fibrosis of extraocular muscles 3 | |||||||
| NEFL | Neurofilament Light | Structural component of neurofilaments | Reduced self-interaction and neurofilament dimerization. | AD/AR | CMT1F | [2, 3, 7–12] | |
| Neurofilament aggregation. | CMT2E | ||||||
| CMT2B5 | |||||||
| Nemaline rod myopathy | |||||||
| NEFH | Neurofilament Heavy | Structural component of neurofilaments | Translation of cryptic amyloidogenic element causing protein aggregation. | AD | CMT2CC | [2, 3, 7, 13] | |
| HSPB1 | Heat Shock Protein Family B (Small) Member 1 | Molecular chaperone for reducing protein aggregation and misfolding | Microtubule overstabilitization. | AD | CMT2F | [16–18, 114] | |
| Impaired mitochondrial transport. | dHMN2B | ||||||
| HSPB8 | Heat Shock Protein Family B (Small) Member 8 | Molecular chaperone for reducing protein aggregation and misfolding | Cytoskeletal destabilization. | AD | CMT2L | [14, 15] | |
| Reduced mitochondrial membrane potential. | dHMN2 | ||||||
| Impairment of aggregate degradation via autophagy. | |||||||
| INF2 | Inverted formin-2 | Stimulating actin filament growth and mitochondrial fission | Perturbation of actin filament dynamics and cytoskeletal networks. | AD | Focal and segmental glomerulosclerosis | [4, 5] | |
| CMT intermediate type E | |||||||
| Motor complexes and adaptors | |||||||
| DYNC1H1 | Dynein Cytoplasmic 1 Heavy Chain 1 | Dynein subunit for retrograde axonal transport | Inhibition of microtubule gliding. | AD | CMT2O | [31, 32] | |
| Compromised dynein processive movement activation. | SMALED | ||||||
| MRD13 | |||||||
| DCTN1 | Dynactin subunit 1 | Dynein adaptor protein for retrograde axonal transport | Altered localization of dynactin. | AD | HMN7B | [31, 33–35] | |
| Reduced dynactin-microtubule binding. | ALS susceptibility | ||||||
| BICD2 | Protein bicaudal D homolog 2 | Dynein adaptor protein for retrograde axonal transport | Imbalance in anterograde and retrograde dynein motor complex motility. | AD | SMALED2A | [28–31, 36–43] | |
| SMALED2B | |||||||
| Spastic paraparesis | |||||||
| Distal myopathy | |||||||
| KIF1A | Kinesin-like protein KIF1A | Motor protein for anterograde axonal transport | Impaired microtubule binding. | AD/AR | HSN2C | [19, 20, 25, 44, 45] | |
| Impaired movement along microtubules. | CMT2 with acrodystrophy | ||||||
| SPG30 | |||||||
| KIF1Bβ | Kinesin family member 1Bbeta isoform III | Motor protein for anterograde axonal transport | Reduced ATPase activity and perinuclear localization. | AD | CMT2A1 | [19, 20, 37, 53, 54] | |
| Impairment of binding capacity and transport of IGF1R. | |||||||
| KIF5A | Kinesin heavy chain isoform 5A | Motor protein for anterograde axonal transport | Altered processivity and directionality of kinesin dependent transport. | AD | CMT2 with pyramidal signs | [19, 20, 46–50, 52] | |
| SPG10 | |||||||
| ALS25 | |||||||
| mRNA and proteins | |||||||
| HNRNPA1 | Heterogeneous nuclear ribonucleoprotein A1 | Packages mRNA into RNP particles | Increased fibrillization and self-aggregation. | AD | Myopathy (IBM) with Paget disease of Bone without Dementia (IBMPFD3) | [61, 63–65] | |
| Increased formation of cytoplasmic stress granules. | ALS20 | ||||||
| HMN (unpublished) | |||||||
| Mitochondria | |||||||
| ATL1 | Atlastin-1 | Formation of ER-mitochondria contact sites | Impairment of GTPase activity and dimer formation. | AD | HSN1D | [78] | |
| Reduced ER-mitochondia fusion effect. | SPG3A | ||||||
| ATL3 | Atlastin-3 | Formation of ER-mitochondria contact sites | Higher ER-mitochondria contact sites. | AD | HSN1F | [78] | |
| Increased Ca2+ uptake into the mitochondria. | |||||||
| GDAP1 | Ganglioside-induced differentiation-associated protein 1 | Mediates mitochondrial fission | Impairment of mitochondrial fusion (AD). | AD/AR | CMT2K | [67, 73, 74] | |
| Reduction in mitochondrial fission activity (AR). | CMT4A | ||||||
| MFN2 | Mitofusin-2 | Mediates mitochondrial clustering and fusion | Aberrant mitochondrial morphology and altered mitochondria-ER tethering. | AD/AR | CMT2A2A | [67, 99, 115] | |
| CMT2A2B | |||||||
| REEP1 | Receptor Expression Enhancing Protein 1 | Stabilization of ER tubules | Destabilization of ER tubules. | AD/AR | HSP | [76] | |
| ER fragmentation. | dHMN | ||||||
| Accumulation of synaptic vesicle proteins. | Congenital axonal neuropathy with diaphragm palsy | ||||||
| SPTLC1 | Serine palmitoyltransferase 1 | Production of sphingolipids | Production of toxic deoxysphingolipids that disrupt mitochondrial integrity. | AD | HSAN1A | [79] | |
| SPTLC2 | Serine palmitoyltransferase 2 | Production of sphingolipids | Production of toxic deoxysphingolipids that disrupt mitochondrial integrity. | AD | HSAN1C | [79] | |
| TRPV4 | Transient receptor potential cation channel subfamily V member 4 | Non-selective calcium permeant cation channel | Abnormal TRPV4-regulated Ca2+ influx. | AD | HMN8 | [77, 128–130] | |
| Higher basal intracellular Ca2+ levels. | SMA | ||||||
| SMALED | |||||||
| CMT2C | |||||||
| VAPB | Vesicle-associated membrane protein-associated protein B/C | Formation of membrane contact sites between ER and other organelles | Formation of intracellular aggregates not associated with membranes. | AD | Proximal SMA | [75] | |
| Vesicular transport | |||||||
| DNM2 | Dynamin-2 | Production of microtubule bundles | Increased GTPase activity and oligomerization. | AD | CMT intermediate type B | [89, 90] | |
| Impaired autophagic degradation. | CMT2M | ||||||
| Impaired DNM2 lipid binding and GTPase activity. | Centronuclear myopathy | ||||||
| Lethal congenital contracture syndrome 5 | |||||||
| GAN | Gigoxonin | Involved in crosstalk of the cytoskeletal architecture and E3 ligase | Inhibition of autophagosome synthesis and altered fusion to the lysosome. | AR | GAN | [131, 132] | |
| RAB7 | Ras-related protein Rab-7 | Regulation of endo-lysosomal trafficking | Increased nucleotide exchange rates. | CMT2B | [93] | ||
| Reduced hydrolysis of GTP. | |||||||
| SPG11 | Spatacsin | Endosomal trafficking | Alters Rab5 and Rab7 signaling. | AR | CMT2X | [87, 133] | |
| Alters Rab5-to-Rab7 conversion. | HSP | ||||||
| ALS5 |
AD - autosomal dominant; ALS - amyotrophic lateral sclerosis; AR - autosomal recessive; CMT - Charcot-Marie-Tooth; (d)HMN - (distal) hereditary motor neuropathy; GAN - giant axonal neuropathy; HSAN - hereditary sensory and autonomic neuropathy; HSN - hereditary sensory neuropathy; HSP - hereditary spastic paraplegia; MRD13 - mental retardation, autosomal dominant 13; SMA - spinal muscular atrophy; SMALED - spinal muscular atrophy with lower limb predominance; SPG - spastic paraplegia.
Anatomical, metabolic and physiological differences between small animal models and human complicate the translation of many therapies into clinical trials. However, iPSC technology offers the possibility to reprogram patient-derived cell lines into pluripotent cells before differentiation into a specific cell type relevant for the disease [101]. This provides new possibilities for IPN, allowing the generation of patient-derived motor and sensory neurons, potentially Schwann cells as well, without using peripheral nerve biopsies or post-mortem tissues. Nevertheless, further improvements are required to more closely model the in vivo situation to have a more representative environment and to take the necessary steps in optimization of co-cultures to study motor neuron-Schwann cell interactions.
Working with 2D-monolayers, microfluidic chambers provide the possibility to investigate retrograde and anterograde transport of organelles with live-cell imaging. Moreover, microfluidic chambers are a flexible tool to mimic the generation of neuromuscular junctions (NMJ), when motor neurons are co-cultured with iPSC-derived muscle cells. Nowadays, bioengineering and iPSC-research work together to develop functional 3D-models to properly reconstruct the immunological, biochemical and anatomical feature of specific organs. A 3D-printed heart-like structure was recently reported using personalized hydrogel, combined with patient-derived cells to print thick, vascularized and perfusable cardiac patches [102]. This suggest that we are not far from the generation of 3D models which could include iPSC-derived peripheral nerves, Schwann cells, blood vessels and myofibers, supported by 3D-scaffolds obtained with new biomaterials and methods of fabrication [103], being entirely supplied by a regulated oxygen transfer with flow pumps. The iPSC-derived models are therefore moving closer to a preclinical application, by providing a pre-screening platform for candidate drugs before testing them in vivo. However, animal models will still remain indispensable to investigate translational research in a whole organism [104, 105].
To date, treatment of IPN is limited to supportive measures that have a partial benefit in relieving the symptoms, such as neuropathic pain or gait impairment. However, no effective interventions exist targeting the causative pathway. Thanks to the understanding of genetic causes, together with the development of more appropriate cellular and animal models, axonal transport emerged as candidate drugable pathway to restore the neuronal function. This will apply to IPN, however many of these insights will also be relevant for the more common acquired diseases of the peripheral nervous system.
The first attempts in targeting axonal degeneration have come from the field of drug repurposing of small molecules. To reduce protein aggregation and improve axonal transport, chemical compounds target MTs through direct binding, PTMs, or chaperone upregulation, aiming to reach the right balance between highly stable and hyperdynamic MTs. In HSP patient-derived neurons, low doses of microtubule-targeting agents (MTA), such as taxol and vinblastine, increased the acetylated α-tubulin levels and restored peroxisome trafficking speeds and distance travelled, effectively improving the mutant phenotype [106]. Other MTA like Epothilone D improved microtubule density, axonal density and cognition in an AD mouse model [107]. However, these compounds, already approved as anticancer drugs, have been discontinued in clinical trials for neurodegenerative diseases due to neurotoxicity [108]. This suggests that new strategies are required to effectively intervene in the treatment of IPN.
Recent studies reported that histone deacetylase 6 (HDAC6) inhibitors such as ACY-1215 (Ricolinostat), currently in clinical trials for cancer, or ACY-1083 showing a higher selectivity towards HDAC6, are able to restore nerve function. They also affect microtubule dynamics by increasing the acetylation of α-tubulin, which re-established the transport function of MTs and offer neuroprotection [109]. Miro1 has been identified and shown to act directly as a novel target of HDAC6, in which Miro1 is deacetylated, at lysine 105, resulting in reduction of mitochondrial transport and outgrowth of the axonal cone. Therefore, HDAC6 inhibitors significantly correct anterograde transport of mitochondria, supporting the therapeutic use of this class of molecules for IPN [110].
Axonal transport involves specific kinase cascade activation, which is altered in neurodegenerative diseases [111]. In ALS, overactivation of p38 MAPKα causes excessive phosphorylation of molecular motors preventing their movement along MTs and p38 MAPKα phosphorylation of NF subunits altering their transport and inducing bundling, effectively inhibiting retrograde transport. The use of p38 MAPKα inhibitors reverses these transport deficits in SOD1-G93A motor neurons, representing a novel therapeutic strategy in ALS. This represents a promising perspective in the treatment of AD, as well as in IPN [112, 113]. Failure in the protein quality control system can lead to deficits in axonal trafficking and aggregate clearance, which can drastically affect neuronal function. Therefore, upregulation of chaperones could be beneficial. Dual treatment using celastrol and arimoclomol increases the expression of a set of sHSPs (HSPA6, HSPA1A, DNAJB1, HO-1, HSPB1) in differentiated SH-SY5Y neuronal cells [114]. Despite the ability of celastrol in reducing the percentage of neuronal inclusions in the transgenic SOD1 mouse model of ALS, or in neurons expressing aggregation prone NEFL mutants, celastrol shows a motor neuron specific effect, with no effect in sensory neurons, limiting its use in IPN with sensory involvement [10]. As drugs can act on different levels of interactions (e.g. level of target, pathway, processes), co-administration of pharmaceutical active molecules should be considered to target the complexity and diversity of affected molecular pathways in IPN [115].
Despite the benefit of small molecules to rescue axonal degeneration, the risk for side effects has limited the use of these treatment strategies. However, a new class of molecules active on microtubule, molecular motors and autophagy [116], as well as antisense oligonucleotides (ASO) or gene therapy directly targeting the affected genes, have emerged and offer promising tools to treat IPN with axonal transport defects. ASO therapies have recently been developed and approved in the treatment of patients with spinal muscular atrophy (SMA) [117]. Similarly, ASO therapies could be applied to reduce PMP22 transcription levels in the CMT1A duplication [118]. Further advancements have been made in the delivery of vector-based gene therapies using adeno-associated viruses (AAV) in the treatment of SMA, consisting of delivery of a functional copy of the human SMN1 gene into motor neuron cells [119, 120]. Likewise, intrathecal injection of lentiviral vectors for Schwann cell-targeted expression has been used to restore the nodal architecture in demyelinating neuropathies. In Sh3tc2–/– mice, a genetic model of CMT4C, or in the mutant GJB1 mouse model for CMTX1, gene delivery respectively of hSH3TC2 or WT Cx32, resulted in amelioration of motor performances together with a reduced myelin pathology [120, 121]. These few examples show that understanding dysfunctional genes operating in the axon, together with optimization of gene delivery methods, including vectors and administration routes, opens promising prospects to treat IPN and related axonopathies.
Deficits in axonal transport are inevitably linked with progressive axonal degeneration, a common feature of IPN and other neurodegenerative diseases. SARM1 (Sterile Alpha and TIR Motif Containing 1) is a mediator of axonal degeneration and initiates cellular self-destruction. Deletion of SARM1 or expression of dominant negative SARM1 mutations, both impairing its activation, showed a reduction in axonal degeneration after axonal transection, the most rapid and aggressive trigger of axonal degeneration [122]. Furthermore, Turkiew et al., (2017) reported that Sarm1–/– mice are resistant to distal axonal degeneration in a model of chemotherapy induced peripheral neuropathy and in high fat diet induced metabolic neuropathy [123]. AAV-mediated delivery of dominant negative SARM1 in mice induces long-lasting axon protection following nerve transection. This approach may provide a new strategy to slow axon loss in chronic neurodegenerative diseases [124].
For neuropathies with toxic aggregations, therapeutic strategies are currently in development making use of gene replacement or silencing in a cell-specific manner. Recently, for CMT2A caused by dominant heterozygous mutations in MFN2, a combined therapy was tested on iPCS-derived spinal motor neurons which include the simultaneous use of RNA interference to silence the mutant allele and insertion of a mutagenized MFN2 gene, resistant to shRNA activity, encoding for the native protein [125]. Next to therapeutic strategies, the identification of biomarkers for disease is relevant to diagnose and treat pre-symptomatic patients or to follow-up ongoing treatments. Currently, measuring NEFL levels in plasma is correlated with disease severity in multiple forms of CMT neuropathies [126], and also PFN2 and GAMT were identified as molecular determinants for CMT2 neuropathy, with a possible role of PFN2 in disease progression [127].
CONCLUSION
Axonal transport is a highly dynamic process involving the movement of different types of cargoes (mRNA, proteins, mitochondria, lysosomes and synaptic vesicles) that are essential to sustain healthy neuronal functions. In this review, we have highlighted how deficits in cargo transportation and related factors affect axonal transport. The identification of numerous genetic causes for IPN-related disorders provides important insights into the underlying mechanisms of axonal degeneration. This knowledge allows the design of targeted therapeutic approaches, some of which have taken up a lead role and moved into clinical trials. Despite this, not all components and mechanisms of axonal transport have been unraveled, novel research strategies have emerged and will move towards patient-derived model systems (e.g. iPSC derived neurons in 2D and 3D-cultures) and in vivo animal models. These models will create new platforms to study and test therapeutic strategies for axonal degeneration.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
The research in our laboratories is supported by the University of Antwerp, Flanders Fund for Scientific Research (FWO-Vlaanderen), Medical Foundation Queen Elisabeth (GSKE), Association Belge contre les Maladies Neuromusculaires (ABMM), Association Française contre les Myopathies (AFM), Muscular Dystrophy Association (MDA) and the European Union’s Horizon 2020 project Solve-RD (Solving the unsolved Rare Diseases) under grant agreement N°779257. These granting organizations provided in part support to PhD students D.B., A.S. and J.V.L. JB is supported by a Senior Clinical Researcher mandate of the Research Fund - Flanders (FWO) under grant agreement number 1805016N.
REFERENCES
[1] | Chakraborti S , Natarajan K , Curiel J , Janke C , Liu J . The emerging role of the tubulin code: From the tubulin molecule to neuronal function and disease, Cytoskeleton (Hoboken). (2016) ;73: (10):521–50. doi: 10.1002/cm.21290 |
[2] | Grant P , Pant HC . Neurofilament protein synthesis and phosphorylation, J Neurocytol. (2000) ;29: (11-12):843–72. doi: |
[3] | Lancaster E , Li J , Hanania T , Liem R , Scheideler MA , Scherer SS . Myelinated axons fail to develop properly in a genetically authentic mouse model of Charcot-Marie-Tooth disease type 2E, Exp Neurol. (2018) ;308: :13–25. doi: 10.1016/j.expneurol.2018.06.010 |
[4] | Brown EJ , Schlondorff JS , Becker DJ , Tsukaguchi H , Tonna SJ , Uscinski AL , et al. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis, Nat Genet. (2010) ;42: (1):72–6. doi: 10.1038/ng.505 |
[5] | Boyer O , Nevo F , Plaisier E , Funalot B , Gribouval O , Benoit G , et al. INF2 mutations in Charcot-Marie-Tooth disease with glomerulopathy, N Engl J Med. (2011) ;365: (25):2377–88. doi: 10.1056/NEJMoa1109122 |
[6] | Hong YB , Lee JH , Park HJ , Choi YR , Hyun YS , Park JH , et al. A family with axonal sensorimotor polyneuropathy with TUBB3 mutation, Mol Med Rep. (2015) ;11: (4):2729–34. doi: 10.3892/mmr.2014.3047 |
[7] | Rebelo AP , Abrams AJ , Cottenie E , Horga A , Gonzalez M , Bis DM , et al. Cryptic amyloidogenic elements in the 3’ UTRs of neurofilament genes trigger axonal neuropathy, Am J Hum Genet. (2016) ;98: (4):597–614. doi: 10.1016/j.ajhg.2016.02.022 |
[8] | Sainio MT , Ylikallio E , Maenpaa L , Lahtela J , Mattila P , Auranen M , et al. Absence of NEFL in patient-specific neurons in early-onset Charcot-Marie-Tooth neuropathy, Neurol Genet. (2018) ;4: (3):e244. doi: 10.1212/NXG.0000000000000244 |
[9] | Jordanova A , De Jonghe P , Boerkoel CF , Takashima H , De Vriendt E , Ceuterick C , et al. Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot-Marie-Tooth disease, Brain. (2003) ;126: (Pt 3):590–7. doi: 10.1093/brain/awg059 |
[10] | Gentil BJ , Mushynski WE , Durham HD . Heterogeneity in the properties of NEFL mutants causing Charcot-Marie-Tooth disease results in differential effects on neurofilament assembly and susceptibility to intervention by the chaperone-inducer, celastrol, Int J Biochem Cell Biol. (2013) ;45: (7):1499–508. doi: 10.1016/j.biocel.2013.04.009 |
[11] | Brownlees J , Ackerley S , Grierson AJ , Jacobsen NJ , Shea K , Anderton BH , et al. Charcot-Marie-Tooth disease neurofilament mutations disrupt neurofilament assembly and axonal transport, Hum Mol Genet. (2002) ;11: (23):2837–44. doi: 10.1093/hmg/11.23.2837 |
[12] | Yates DM , Manser C , De Vos KJ , Shaw CE , McLoughlin DM , Miller CC . Neurofilament subunit (NFL) head domain phosphorylation regulates axonal transport of neurofilaments, Eur J Cell Biol. (2009) ;88: (4):193–202. doi: 10.1016/j.ejcb.2008.11.004 |
[13] | Jacquier A , Delorme C , Belotti E , Juntas-Morales R , Sole G , Dubourg O , et al. Cryptic amyloidogenic elements in mutant NEFH causing Charcot-Marie-Tooth 2 trigger aggresome formation and neuronal death, Acta Neuropathol Commun. (2017) ;5: (1):55. doi: 10.1186/s40478-017-0457-1 |
[14] | Bouhy D , Juneja M , Katona I , Holmgren A , Asselbergh B , De Winter V , et al. A knock-in/knock-out mouse model of HSPB8-associated distal hereditary motor neuropathy and myopathy reveals toxic gain-of-function of mutant Hspb8, Acta Neuropathol. (2018) . doi: |
[15] | Irobi J , Holmgren A , De Winter V , Asselbergh B , Gettemans J , Adriaensen D , et al. Mutant HSPB8 causes protein aggregates and a reduced mitochondrial membrane potential in dermal fibroblasts from distal hereditary motor neuropathy patients, Neuromuscul Disord. (2012) . doi: |
[16] | d’Ydewalle C , Krishnan J , Chiheb DM , Van Damme P , Irobi J , Kozikowski AP , et al. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease, Nat Med. (2011) ;17: (8):968–74. doi: 10.1038/nm.2396 |
[17] | Ackerley S , James PA , Kalli A , French S , Davies KE , Talbot K . A mutation in the small heat-shock protein HSPB1 leading to distal hereditary motor neuronopathy disrupts neurofilament assembly and the axonal transport of specific cellular cargoes, Hum Mol Genet. (2006) ;15: (2):347–54. doi: 10.1093/hmg/ddi452 |
[18] | Kalmar B , Innes A , Wanisch K , Kolaszynska AK , Pandraud A , Kelly G , et al. Mitochondrial deficits and abnormal mitochondrial retrograde axonal transport play a role in the pathogenesis of mutant Hsp27-induced Charcot Marie Tooth Disease, Hum Mol Genet. (2017) ;26: (17):3313–26. doi: 10.1093/hmg/ddx216 |
[19] | Xiao Q , Hu X , Wei Z , Tam KY . Cytoskeleton molecular motors: Structures and their functions in neuron, Int J Biol Sci. (2016) ;12: (9):1083–92. doi: 10.7150/ijbs.15633 |
[20] | De Vos KJ , Hafezparast M . Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiol Dis. (2017) ;105: :283–99. doi: 10.1016/j.nbd.2017.02.004 |
[21] | Hancock WO . Bidirectional cargo transport: Moving beyond tug of war, Nat Rev Mol Cell Biol. (2014) ;15: (9):615–28. doi: 10.1038/nrm3853 |
[22] | Gross SP , Welte MA , Block SM , Wieschaus EF . Coordination of opposite-polarity microtubule motors, J Cell Biol. (2002) ;156: (4):715–24. doi: 10.1083/jcb.200109047 |
[23] | Cao TT , Chang W , Masters SE , Mooseker MS . Myosin-Va binds to and mechanochemically couples microtubules to actin filaments, Mol Biol Cell. (2004) ;15: (1):151–61. doi: 10.1091/mbc.e03-07-0504 |
[24] | Rao MV , Mohan PS , Kumar A , Yuan A , Montagna L , Campbell J , et al. The myosin Va head domain binds to the neurofilament-L rod and modulates endoplasmic reticulum (ER) content and distribution within axons, PLoS One. (2011) ;6: (2):e17087. doi: 10.1371/journal.pone.0017087 |
[25] | Ikegami K , Heier RL , Taruishi M , Takagi H , Mukai M , Shimma S , et al. Loss of alpha-tubulin polyglutamylation in ROSA22 mice is associated with abnormal targeting of KIF1A and modulated synaptic function, Proc Natl Acad Sci U S A. (2007) ;104: (9):3213–8. doi: 10.1073/pnas.0611547104 |
[26] | Konishi Y , Setou M . Tubulin tyrosination navigates the kinesin-1 motor domain to axons, Nat Neurosci. (2009) ;12: (5):559–67. doi: 10.1038/nn.2314 |
[27] | Gibbs KL , Greensmith L , Schiavo G . Regulation of axonal transport by protein kinases, Trends Biochem Sci. (2015) ;40: (10):597–610. doi: 10.1016/j.tibs.2015.08.003 |
[28] | Urnavicius L , Lau CK , Elshenawy MM , Morales-Rios E , Motz C , Yildiz A , et al. Cryo-EM shows how dynactin recruits two dyneins for faster movement, Nature. (2018) ;554: (7691):202–6. doi: 10.1038/nature25462 |
[29] | Grotjahn DA , Chowdhury S , Xu Y , McKenney RJ , Schroer TA , Lander GC . Cryo-electron tomography reveals that dynactin recruits a team of dyneins for processive motility, Nat Struct Mol Biol. (2018) ;25: (3):203–7. doi: 10.1038/s41594-018-0027-7 |
[30] | Schlager MA , Serra-Marques A , Grigoriev I , Gumy LF , Esteves da Silva M , Wulf PS , et al. Bicaudal d family adaptor proteins control the velocity of Dynein-based movements, Cell Rep. (2014) ;8: (5):1248–56. doi: 10.1016/j.celre2014.07.052 |
[31] | Olenick MA , Holzbaur ELF . Dynein activators and adaptors at a glance, J Cell Sci. (2019) ;132: (6). doi: 10.1242/jcs.227132 |
[32] | Schiavo G , Greensmith L , Hafezparast M , Fisher EM . Cytoplasmic dynein heavy chain: The servant of many masters. Trends Neurosci. (2013) ;36: (11):641–51. 10.1016/j.tins.2013.08.001 |
[33] | Farrer MJ , Hulihan MM , Kachergus JM , Dachsel JC , Stoessl AJ , Grantier LL , et al. DCTN1 mutations in Perry syndrome, Nat Genet. (2009) ;41: (2):163–5. doi: 10.1038/ng.293 |
[34] | Levy JR , Sumner CJ , Caviston JP , Tokito MK , Ranganathan S , Ligon LA , et al. A motor neuron disease-associated mutation in p150Glued perturbs dynactin function and induces protein aggregation, J Cell Biol. (2006) ;172: (5):733–45. doi: 10.1083/jcb.200511068 |
[35] | Puls I , Jonnakuty C , LaMonte BH , Holzbaur EL , Tokito M , Mann E , et al. Mutant dynactin in motor neuron disease, Nat Genet. (2003) ;33: (4):455–6. doi: 10.1038/ng1123 |
[36] | Neveling K , Martinez-Carrera LA , Holker I , Heister A , Verrips A , Hosseini-Barkooie SM , et al. Mutations in BICD2, which encodes a golgin and important motor adaptor, cause congenital autosomal-dominant spinal muscular atrophy, Am J Hum Genet. (2013) ;92: (6):946–54. doi: 10.1016/j.ajhg.2013.04.011 |
[37] | Drew AP , Zhu D , Kidambi A , Ly C , Tey S , Brewer MH , et al. Improved inherited peripheral neuropathy genetic diagnosis by whole-exome sequencing, Mol Genet Genomic Med. (2015) ;3: (2):143–54. doi: 10.1002/mgg3.126 |
[38] | Koboldt DC , Kastury RD , Waldrop MA , Kelly BJ , Mosher TM , McLaughlin H , et al. In-frame de novo mutation in BICD2 in two patients with muscular atrophy and arthrogryposis, Cold Spring Harb Mol Case Stud. (2018) ;4: (5). doi: 10.1101/mcs.a003160 |
[39] | Kropatsch R , Schmidt HM , Buttkereit P , Epplen JT , Hoffjan S . BICD2 mutational analysis in hereditary spastic paraplegia and hereditary motor and sensory neuropathy, Muscle Nerve. (2019) ;59: (4):484–6. doi: 10.1002/mus.26394 |
[40] | Storbeck M , Horsberg Eriksen B , Unger A , Holker I , Aukrust I , Martinez-Carrera LA , et al. Phenotypic extremes of BICD2-opathies: From lethal, congenital muscular atrophy with arthrogryposis to asymptomatic with subclinical features, Eur J Hum Genet. (2017) ;25: (9):1040–8. doi: 10.1038/ejhg.2017.98 |
[41] | Huynh W , Vale RD . Disease-associated mutations in human BICD2 hyperactivate motility of dynein-dynactin, J Cell Biol. (2017) ;216: (10):3051–60. doi: 10.1083/jcb.201703201 |
[42] | Martinez Carrera LA , Gabriel E , Donohoe CD , Holker I , Mariappan A , Storbeck M , et al. Novel insights into SMALED2: BICD2 mutations increase microtubule stability and cause defects in axonal and NMJ development, Hum Mol Genet. (2018) ;27: (10):1772–84. doi: 10.1093/hmg/ddy086 |
[43] | Martinez-Carrera LA , Wirth B . Dominant spinal muscular atrophy is caused by mutations in BICD2, an important golgin protein, Front Neurosci. (2015) ;9: :401. doi: 10.3389/fnins.2015.00401 |
[44] | Esmaeeli Nieh S , Madou MR , Sirajuddin M , Fregeau B , McKnight D , Lexa K , et al. De novo mutations in KIF1A cause progressive encephalopathy and brain atrophy, Ann Clin Transl Neurol. (2015) ;2: (6):623–35. doi: 10.1002/acn3.198 |
[45] | Riviere JB , Ramalingam S , Lavastre V , Shekarabi M , Holbert S , Lafontaine J , et al. KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2, Am J Hum Genet. (2011) ;89: (2):219–30. doi: 10.1016/j.ajhg.2011.06.013 |
[46] | Brenner D , Yilmaz R , Muller K , Grehl T , Petri S , Meyer T , et al. Hot-spot KIF5A mutations cause familial ALS, Brain. (2018) ;141: (3):688–97. doi: 10.1093/brain/awx370 |
[47] | Citrigno L , Magariello A , Pugliese P , Di Palma G , Conforti FL , Petrone A , et al. Kinesins in neurological inherited diseases: A novel motor-domain mutation in KIF5A gene in a patient from Southern Italy affected by hereditary spastic paraplegia, Acta Neurol Belg. (2018) ;118: (4):643–6. doi: 10.1007/s13760-018-1039-0 |
[48] | Filosto M , Piccinelli SC , Palmieri I , Necchini N , Valente M , Zanella I , et al. A novel mutation in the stalk domain of KIF5A causes a slowly progressive atypical motor syndrome, J Clin Med. (2018) ;8: (1). doi: 10.3390/jcm8010017 |
[49] | Nam DE , Yoo DH , Choi SS , Choi BO , Chung KW . Wide phenotypic spectrum in axonal Charcot-Marie-Tooth neuropathy type 2 patients with KIF5A mutations, Genes Genomics. (2018) ;40: (1):77–84. doi: 10.1007/s13258-017-0612-x |
[50] | Dutta M , Diehl MR , Onuchic JN , Jana B . Structural consequences of hereditary spastic paraplegia disease-related mutations in kinesin, Proc Natl Acad Sci U S A. (2018) ;115: (46):E10822–E9. doi: 10.1073/pnas.1810622115 |
[51] | Jennings S , Chenevert M , Liu L , Mottamal M , Wojcik EJ , Huckaba TM . Characterization of kinesin switch I mutations that cause hereditary spastic paraplegia, PLoS One. (2017) ;12: (7):e0180353. doi: 10.1371/journal.pone.0180353 |
[52] | Ebbing B , Mann K , Starosta A , Jaud J , Schols L , Schule R , et al. Effect of spastic paraplegia mutations in KIF5A kinesin on transport activity, Hum Mol Genet. (2008) ;17: (9):1245–52. doi: 10.1093/hmg/ddn014 |
[53] | Zhao C , Takita J , Tanaka Y , Setou M , Nakagawa T , Takeda S , et al. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta, Cell. (2001) ;105: (5):587–97. doi: |
[54] | Xu F , Takahashi H , Tanaka Y , Ichinose S , Niwa S , Wicklund MP , et al. KIF1Bbeta mutations detected in hereditary neuropathy impair IGF1R transport and axon growth, J Cell Biol. (2018) ;217: (10):3480–96. doi: 10.1083/jcb.201801085 |
[55] | Jean S , Kiger AA . Coordination between RAB GTPase and phosphoinositide regulation and functions, Nat Rev Mol Cell Biol. (2012) ;13: (7):463–70. doi: 10.1038/nrm3379 |
[56] | Taylor MP , Enquist LW . Axonal spread of neuroinvasive viral infections, Trends Microbiol. (2015) ;23: (5):283–8. doi: 10.1016/j.tim.2015.01.002 |
[57] | Roy S . Seeing the unseen: The hidden world of slow axonal transport, Neuroscientist. (2014) ;20: (1):71–81. doi: 10.1177/1073858413498306 |
[58] | Scott DA , Das U , Tang Y , Roy S . Mechanistic logic underlying the axonal transport of cytosolic proteins, Neuron. (2011) ;70: (3):441–54. doi: 10.1016/j.neuron.2011.03.022 |
[59] | Tang Y , Scott D , Das U , Gitler D , Ganguly A , Roy S . Fast vesicle transport is required for the slow axonal transport of synapsin, J Neurosci. (2013) ;33: (39):15362–75. doi: 10.1523/JNEUROSCI.1148-13.2013 |
[60] | van Niekerk EA , Willis DE , Chang JH , Reumann K , Heise T , Twiss JL . Sumoylation in axons triggers retrograde transport of the RNA-binding protein La, Proc Natl Acad Sci U S A. (2007) ;104: (31):12913–8. doi: 10.1073/pnas.0611562104 |
[61] | Harvey R , Dezi V , Pizzinga M , Willis AE . Post-transcriptional control of gene expression following stress: The role of RNA-binding proteins, Biochem Soc Trans. (2017) ;45: (4):1007–14. doi: 10.1042/BST20160364 |
[62] | Alami NH , Smith RB , Carrasco MA , Williams LA , Winborn CS , Han SSW , et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations, Neuron. (2014) ;81: (3):536–43. doi: 10.1016/j.neuron.2013.12.018 |
[63] | Purice MD , Taylor JP . Linking hnRNP function to ALS and FTD pathology, Front Neurosci. (2018) ;12: :326. doi: 10.3389/fnins.2018.00326 |
[64] | Geuens T , Bouhy D , Timmerman V . The hnRNP family: insights into their role in health and disease, Hum Genet. (2016) ;135: (8):851–67. doi: 10.1007/s00439-016-1683-5 |
[65] | Li YR , King OD , Shorter J , Gitler AD . Stress granules as crucibles of ALS pathogenesis, J Cell Biol. (2013) ;201: (3):361–72. doi: 10.1083/jcb.201302044 |
[66] | Verhoeven K , Claeys KG , Zuchner S , Schroder JM , Weis J , Ceuterick C , et al. MFN2 mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth type 2, Brain. (2006) ;129: (Pt 8):2093–102. doi: 10.1093/brain/awl126 |
[67] | Hoppins S , Lackner L , Nunnari J . The machines that divide and fuse mitochondria, Annu Rev Biochem. (2007) ;76: :751–80. doi: 10.1146/annurev.biochem.76.071905.090048 |
[68] | Smirnova E , Griparic L , Shurland DL , van der Bliek AM . Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells, Mol Biol Cell. (2001) ;12: (8):2245–56. doi: 10.1091/mbc.12.8.2245 |
[69] | Wangler MF , Assia Batzir N , Robak LA , Koenig MK , Bacino CA , Scaglia F , et al. The expanding neurological phenotype of DNM1L-related disorders, Brain. (2018) ;141: (4):e28. doi: 10.1093/brain/awy024 |
[70] | Gandre-Babbe S , van der Bliek AM . The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells, Mol Biol Cell. (2008) ;19: (6):2402–12. doi: 10.1091/mbc.E07-12-1287 |
[71] | Hu C , Huang Y , Li L . Drp1-Dependent mitochondrial fission plays critical roles in physiological and pathological progresses in mammals, Int J Mol Sci. (2017) ;18: (1). doi: 10.3390/ijms18010144 |
[72] | Palmer CS , Osellame LD , Laine D , Koutsopoulos OS , Frazier AE , Ryan MT . MiD49 and MiD51, new components of the mitochondrial fission machinery, EMBO Rep. (2011) ;12: (6):565–73. doi: 10.1038/embor.2011.54 |
[73] | Niemann A , Wagner KM , Ruegg M , Suter U . GDAP1 mutations differ in their effects on mitochondrial dynamics and apoptosis depending on the mode of inheritance, Neurobiol Dis. (2009) ;36: (3):509–20. doi: 10.1016/j.nbd.2009.09.011 |
[74] | Ni HM , Williams JA , Ding WX . Mitochondrial dynamics and mitochondrial quality control, Redox Biol. (2015) ;4: :6–13. doi: 10.1016/j.redox.2014.11.006 |
[75] | De Vos KJ , Morotz GM , Stoica R , Tudor EL , Lau KF , Ackerley S , et al. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis, Hum Mol Genet. (2012) ;21: (6):1299–311. doi: 10.1093/hmg/ddr559 |
[76] | Yalcin B , Zhao L , Stofanko M , O’Sullivan NC , Kang ZH , Roost A , et al. Modeling of axonal endoplasmic reticulum network by spastic paraplegia proteins, Elife. (2017) ;6. doi: 10.7554/eLife.23882 |
[77] | Kumar A , Kumar Majhi R , Kanta Acharya T , Smalla K , Gundelfinger ED , Goswami C . [PREPRINT] TRPV4 interacts with mitochondrial proteins and acts as a mitochondrial structure-function regulator, BioRxiv. (2018) . doi: 10.1101/330993 |
[78] | Krols M , Asselbergh B , De Rycke R , De Winter V , Seyer A , Muller FJ , et al. Sensory neuropathy-causing mutations in ATL3 affect ER-mitochondria contact sites and impair axonal mitochondrial distribution, Hum Mol Genet. (2019) ;28: (4):615–27. doi: 10.1093/hmg/ddy352 |
[79] | Alecu I , Tedeschi A , Behler N , Wunderling K , Lamberz C , Lauterbach MA , et al. Localization of 1-deoxysphingolipids to mitochondria induces mitochondrial dysfunction, J Lipid Res. (2017) ;58: (1):42–59. doi: 10.1194/jlr.M068676 |
[80] | Spaulding EL , Sleigh JN , Morelli KH , Pinter MJ , Burgess RW , Seburn KL . Synaptic deficits at neuromuscular junctions in two mouse models of charcot-marie-tooth type 2d, J Neurosci. (2016) ;36: (11):3254–67. doi: 10.1523/JNEUROSCI.1762-15.2016 |
[81] | Kratsios P , Pinan-Lucarre B , Kerk SY , Weinreb A , Bessereau JL , Hobert O . Transcriptional coordination of synaptogenesis and neurotransmitter signaling, Curr Biol. (2015) ;25: (10):1282–95. doi: 10.1016/j.cub.2015.03.028 |
[82] | Lou X . Sensing exocytosis and triggering endocytosis at synapses: Synaptic vesicle exocytosis-endocytosis coupling, Front Cell Neurosci. (2018) ;12: :66. doi: 10.3389/fncel.2018.00066 |
[83] | Frake RA , Ricketts T , Menzies FM , Rubinsztein DC . Autophagy and neurodegeneration, J Clin Invest. (2015) ;125: (1):65–74. doi: 10.1172/JCI73944 |
[84] | Tammineni P , Ye X , Feng T , Aikal D , Cai Q . Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons, Elife. (2017) ;6. doi: 10.7554/eLife.21776 |
[85] | Deinhardt K , Salinas S , Verastegui C , Watson R , Worth D , Hanrahan S , et al. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway, Neuron. (2006) ;52: (2):293–305. doi: 10.1016/j.neuron.2006.08.018 |
[86] | Agola JO , Jim PA , Ward HH , Basuray S , Wandinger-Ness A . Rab GTPases as regulators of endocytosis, targets of disease and therapeutic opportunities, Clin Genet. (2011) ;80: (4):305–18. doi: 10.1111/j.1399-0004.2011.01724.x |
[87] | Blackstone C . Cellular pathways of hereditary spastic paraplegia, Annu Rev Neurosci. (2012) ;35: :25–47. doi: 10.1146/annurev-neuro-062111-150400 |
[88] | Wijdeven RH , Janssen H , Nahidiazar L , Janssen L , Jalink K , Berlin I , et al. Cholesterol and ORP1L-mediated ER contact sites control autophagosome transport and fusion with the endocytic pathway, Nat Commun. (2016) ;7: :11808. doi: 10.1038/ncomms11808 |
[89] | Zuchner S , Noureddine M , Kennerson M , Verhoeven K , Claeys K , De Jonghe P , et al. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease, Nat Genet. (2005) ;37: (3):289–94. doi: 10.1038/ng1514 |
[90] | Gonzalez-Jamett AM , Baez-Matus X , Olivares MJ , Hinostroza F , Guerra-Fernandez MJ , Vasquez-Navarrete J , et al. Dynamin-2 mutations linked to Centronuclear Myopathy impair actin-dependent trafficking in muscle cells, Sci Rep. (2017) ;7: (1):4580. doi: 10.1038/s41598-017-04418-w |
[91] | Snouwaert JN , Church RJ , Jania L , Nguyen M , Wheeler ML , Saintsing A , et al. A Mutation in the Borcs7 subunit of the lysosome regulatory BORC complex results in motor deficits and dystrophic axonopathy in mice, Cell Rep. (2018) ;24: (5):1254–65. doi: 10.1016/j.celre2018.06.118 |
[92] | Nian FS , Li LL , Cheng CY , Wu PC , Lin YT , Tang CY , et al. Rab18 collaborates with Rab7 to modulate lysosomal and autophagy activities in the nervous system: An overlapping mechanism for warburg micro syndrome and charcot-marie-tooth neuropathy type 2B, Mol Neurobiol. (2019) . doi: 10.1007/s12035-019-1471-z |
[93] | Ponomareva OY , Eliceiri KW , Halloran MC . Charcot-Marie-Tooth 2b associated Rab7 mutations cause axon growth and guidance defects during vertebrate sensory neuron development, Neural Dev. (2016) ;11: :2. doi: 10.1186/s13064-016-0058-x |
[94] | Salpietro V , Malintan NT , Llano-Rivas I , Spaeth CG , Efthymiou S , Striano P , et al. Mutations in the neuronal vesicular SNARE VAMP2 affect synaptic membrane fusion and impair human neurodevelopment, Am J Hum Genet. (2019) ;104: (4):721–30. doi: 10.1016/j.ajhg.2019.02.016 |
[95] | Lipstein N , Verhoeven-Duif NM , Michelassi FE , Calloway N , van Hasselt PM , Pienkowska K , et al. Synaptic UNC13A protein variant causes increased neurotransmission and dyskinetic movement disorder, J Clin Invest. (2017) ;127: (3):1005–18. doi: 10.1172/JCI90259 |
[96] | Prior R , Van Helleputte L , Benoy V , Van Den Bosch L . Defective axonal transport: A common pathological mechanism in inherited and acquired peripheral neuropathies, Neurobiol Dis. (2017) ;105: :300–20. doi: 10.1016/j.nbd.2017.02.009 |
[97] | Vagnoni A , Bullock SL . A simple method for imaging axonal transport in aging neurons using the adult Drosophila wing, Nat Protoc. (2016) ;11: (9):1711–23. doi: 10.1038/nprot.2016.112 |
[98] | Lipton DM , Maeder CI , Shen K . Rapid assembly of presynaptic materials behind the growth cone in dopaminergic neurons is mediated by precise regulation of axonal transport, Cell Rep. (2018) ;24: (10):2709–22. doi: 10.1016/j.celre2018.07.096 |
[99] | Bergamin G , Cieri D , Vazza G , Argenton F , Mostacciuolo ML . Zebrafish Tg(hb9:MTS-Kaede): A new in vivo tool for studying the axonal movement of mitochondria, Biochim Biophys Acta. (2016) ;1860: (6):1247–55. doi: 10.1016/j.bbagen.2016.03.007 |
[100] | Sleigh JN , Vagnoni A , Twelvetrees AE , Schiavo G . Methodological advances in imaging intravital axonal transport, F1000Res. (2017) ;6: :200. doi: 10.12688/f1000research.10433.1 |
[101] | Han SS , Williams LA , Eggan KC . Constructing and deconstructing stem cell models of neurological disease, Neuron. (2011) ;70: (4):626–44. doi: 10.1016/j.neuron.2011.05.003 |
[102] | Noor N , Shapira A , Edri R , Gal I , Wertheim L , Dvir T . 3D Printing of personalized thick and perfusable cardiac patches and hearts, Adv Sci (Weinh). (2019) ;6: (11):1900344. doi: 10.1002/advs.201900344 |
[103] | Jammalamadaka U , Tappa K . Recent advances in biomaterials for 3D printing and tissue engineering, J Funct Biomater. (2018) ;9: (1). doi: 10.3390/jfb9010022 |
[104] | Little D , Luft C , Mosaku O , Ketteler R , Devine MJ , Gissen P . High-content autophagy analysis in iPSC-derived neurons using immunofluorescence, Methods Mol Biol. (1994) :165–74. doi: 10.1007/978-1-4939-9477-9_15 |
[105] | Marrone L , Poser I , Casci I , Japtok J , Reinhardt P , Janosch A , et al. Isogenic FUS-eGFP iPSC reporter lines enable quantification of FUS stress granule pathology that is rescued by drugs inducing autophagy, Stem Cell Reports. (2018) ;10: (2):375–89. doi: 10.1016/j.stemcr.2017.12.018 |
[106] | Fan Y , Wali G , Sutharsan R , Bellette B , Crane DI , Sue CM , et al. Low dose tubulin-binding drugs rescue peroxisome trafficking deficit in patient-derived stem cells in Hereditary Spastic Paraplegia, Biol Open. (2014) ;3: (6):494–502. doi: 10.1242/bio.20147641 |
[107] | Ballatore C , Brunden KR , Huryn DM , Trojanowski JQ , Lee VM , Smith AB 3rd . Microtubule stabilizing agents as potential treatment for Alzheimer’s disease and related neurodegenerative tauopathies, J Med Chem. (2012) ;55: (21):8979–96. doi: 10.1021/jm301079z |
[108] | Hung SY , Fu WM . Drug candidates in clinical trials for Alzheimer’s disease, J Biomed Sci. (2017) ;24: (1):47. doi: 10.1186/s12929-017-0355-7 |
[109] | Krukowski K , Ma J , Golonzhka O , Laumet GO , Gutti T , van Duzer JH , et al. HDAC6 inhibition effectively reverses chemotherapy-induced peripheral neuropathy, Pain. (2017) ;158: (6):1126–37. doi: 10.1097/j.pain.0000000000000893 |
[110] | Kalinski AL , Kar AN , Craver J , Tosolini AP , Sleigh JN , Lee SJ , et al. Deacetylation of Miro1 by HDAC6 blocks mitochondrial transport and mediates axon growth inhibition, J Cell Biol. (2019) ;218: (6):1871–90. doi: 10.1083/jcb.201702187 |
[111] | Spurrier J , Shukla AK , McLinden K , Johnson K , Giniger E . Altered expression of the Cdk5 activator-like protein, Cdk5alpha, causes neurodegeneration, in part by accelerating the rate of aging, Dis Model Mech. (2018) ;11: (3). doi: 10.1242/dmm.031161 |
[112] | Roy SM , Grum-Tokars VL , Schavocky JP , Saeed F , Staniszewski A , Teich AF , et al. Targeting human central nervous system protein kinases: An isoform selective p38alphaMAPK inhibitor that attenuates disease progression in Alzheimer’s disease mouse models, ACS Chem Neurosci. (2015) ;6: (4):666–80. doi: 10.1021/acschemneuro.5b00002 |
[113] | Gibbs KL , Kalmar B , Rhymes ER , Fellows AD , Ahmed M , Whiting P , et al. Inhibiting p38 MAPK alpha rescues axonal retrograde transport defects in a mouse model of ALS, Cell Death Dis. (2018) ;9: (6):596. doi: 10.1038/s41419-018-0624-8 |
[114] | Deane CA , Brown IR . Induction of heat shock proteins in differentiated human neuronal cells following co-application of celastrol and arimoclomol, Cell Stress Chaperones. (2016) ;21: (5):837–48. doi: 10.1007/s12192-016-0708-2 |
[115] | Pemovska T , Bigenzahn JW , Superti-Furga G . Recent advances in combinatorial drug screening and synergy scoring, Curr Opin Pharmacol. (2018) ;42: :102–10. doi: 10.1016/j.coph.2018.07.008 |
[116] | White JA , Banerjee R , Gunawardena S . Axonal transport and neurodegeneration: How marine drugs can be used for the development of therapeutics, Mar Drugs. (2016) ;14: (5). doi: 10.3390/md14050102 |
[117] | Wood MJA , Talbot K , Bowerman M . Spinal muscular atrophy: antisense oligonucleotide therapy opens the door to an integrated therapeutic landscape, Hum Mol Genet. (2017) ;26: (R2):R151–9. doi: 10.1093/hmg/ddx215 |
[118] | Zhao HT , Damle S , Ikeda-Lee K , Kuntz S , Li J , Mohan A , et al. PMP22 antisense oligonucleotides reverse Charcot-Marie-Tooth disease type 1A features in rodent models, J Clin Invest. (2018) ;128: (1):359–68. doi: 10.1172/JCI96499 |
[119] | Food Drug Administration Center for Drugs Evaluation Research. BLA APPROVAL BL 125694/0 ZOLGENSMA (onasemnogene abeparvovec-xioi) AveXis, Inc. FDA Maryland; 2019. doi: |
[120] | Schiza N , Georgiou E , Kagiava A , Medard JJ , Richter J , Tryfonos C , et al. Gene replacement therapy in a model of Charcot-Marie-Tooth 4C neuropathy, Brain. (2019) ;142: (5):1227–41. doi: 10.1093/brain/awz064 |
[121] | Kagiava A , Karaiskos C , Richter J , Tryfonos C , Lapathitis G , Sargiannidou I , et al. Intrathecal gene therapy in mouse models expressing CMT1X mutations, Hum Mol Genet. (2018) ;27: (8):1460–73. doi: 10.1093/hmg/ddy056 |
[122] | Gerdts J , Summers DW , Milbrandt J , DiAntonio A . Axon self-destruction: New links among SARM1, MAPKs, and NAD+Metabolism, Neuron. (2016) ;89: (3):449–60. doi: 10.1016/j.neuron.2015.12.023 |
[123] | Turkiew E , Falconer D , Reed N , Hoke A . Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy, J Peripher Nerv Syst. (2017) ;22: (3):162–71. doi: 10.1111/jns.12219 |
[124] | Geisler S , Huang SX , Strickland A , Doan RA , Summers DW , Mao X , et al. Gene therapy targeting SARM1 blocks pathological axon degeneration in mice, J Exp Med. (2019) ;216: (2):294–303. doi: 10.1084/jem.20181040 |
[125] | Rizzo F , Bono S , Salani S , Bordoni A , Melzi V , Ruepp M , et al. RNAi/gene therapy combined approach as therapeutic strategy for Charcot-Marie-Tooth 2A (S58.005), Neurology. (2019) ;92. doi: |
[126] | Sandelius A , Zetterberg H , Blennow K , Adiutori R , Malaspina A , Laura M , et al. Plasma neurofilament light chain concentration in the inherited peripheral neuropathies, Neurology. (2018) ;90: (6):e518–e24. doi: 10.1212/WNL.0000000000004932 |
[127] | Juneja M , Burns J , Saporta MA , Timmerman V . Challenges in modelling the Charcot-Marie-Tooth neuropathies for therapy development JNNP. (2018) . doi: |
[128] | Auer-Grumbach M , Olschewski A , Papic L , Kremer H , McEntagart ME , Uhrig S , et al. Alterations in the ankyrin domain of TRPV4 cause congenital distal SMA, scapuloperoneal SMA and HMSN2C, Nat Genet. (2010) ;42: (2):160–4. doi: 10.1038/ng.508 |
[129] | Deng HX , Klein CJ , Yan J , Shi Y , Wu Y , Fecto F , et al. Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4, Nat Genet. (2010) ;42: (2):165–9. doi: 10.1038/ng.509 |
[130] | Landoure G , Zdebik AA , Martinez TL , Burnett BG , Stanescu HC , Inada H , et al. Mutations in TRPV4 cause Charcot-Marie-Tooth disease type 2C, Nat Genet. (2010) ;42: (2):170–4. doi: 10.1038/ng.512 |
[131] | Scrivo A , Codogno P , Bomont P . Gigaxonin E3 ligase governs ATG16L1 turnover to control autophagosome production, Nat Commun. (2019) ;10: (1):780. doi: 10.1038/s41467-019-08331-w |
[132] | Bomont P , Cavalier L , Blondeau F , Ben Hamida C , Belal S , Tazir M , et al. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy, Nat Genet. (2000) ;26: (3):370–4. doi: 10.1038/81701 |
[133] | Branchu J , Boutry M , Sourd L , Depp M , Leone C , Corriger A , et al. Loss of spatacsin function alters lysosomal lipid clearance leading to upper and lower motor neuron degeneration, Neurobiol Dis. (2017) ;102: :21–37. doi: 10.1016/j.nbd.2017.02.007 |


