
Advances in Treatment of Spinal Muscular Atrophy – New Phenotypes, New Challenges, New Implications for Care
Abstract
Spinal Muscular Atrophy (SMA) is caused by autosomal recessive mutations in SMN1 and results in the loss of motor neurons and progressive muscle weakness. The spectrum of disease severity ranges from early onset with respiratory failure during the first months of life to a mild, adult-onset type with slow rate of progression. Over the past decade, new treatment options such as splicing modulation of SMN2 and SMN1 gene replacement by gene therapy have been developed. First drugs have been approved for treatment of patients with SMA and if initiated early they can significantly modify the natural course of the disease. As a consequence, newborn screening for SMA is explored and implemented in an increasing number of countries. However, available evidence for these new treatments is often limited to a small spectrum of patients concerning age and disease stage. In this review we provide an overview of available and emerging therapies for spinal muscular atrophy and we discuss new phenotypes and associated challenges in clinical care. Collection of real-world data with standardized outcome measures will be essential to improve both the understanding of treatment effects in patients of all SMA subtypes and the basis for clinical decision-making in SMA.
INTRODUCTION
The SMA landscape has changed considerably since the first reports more than a century ago of patients with spinal muscular atrophy (SMA) by Werdnig and Hofmann in 1891 [1] and 1893 [2]. Decoding the disease’s genetic background, first in linkage analyses [3, 4] and later by identifying mutations in SMN1 as disease-causing [5], paved the way for targeted medical approaches. In this review we provide an overview of both the latest therapeutic options and emerging therapies for SMA. We also discuss new topics and challenges arising with the availability of drug treatments that alter the known trajectories of disease. These include changing phenotypes, new medical decisions, and newborn screening for SMA.
BACKGROUND
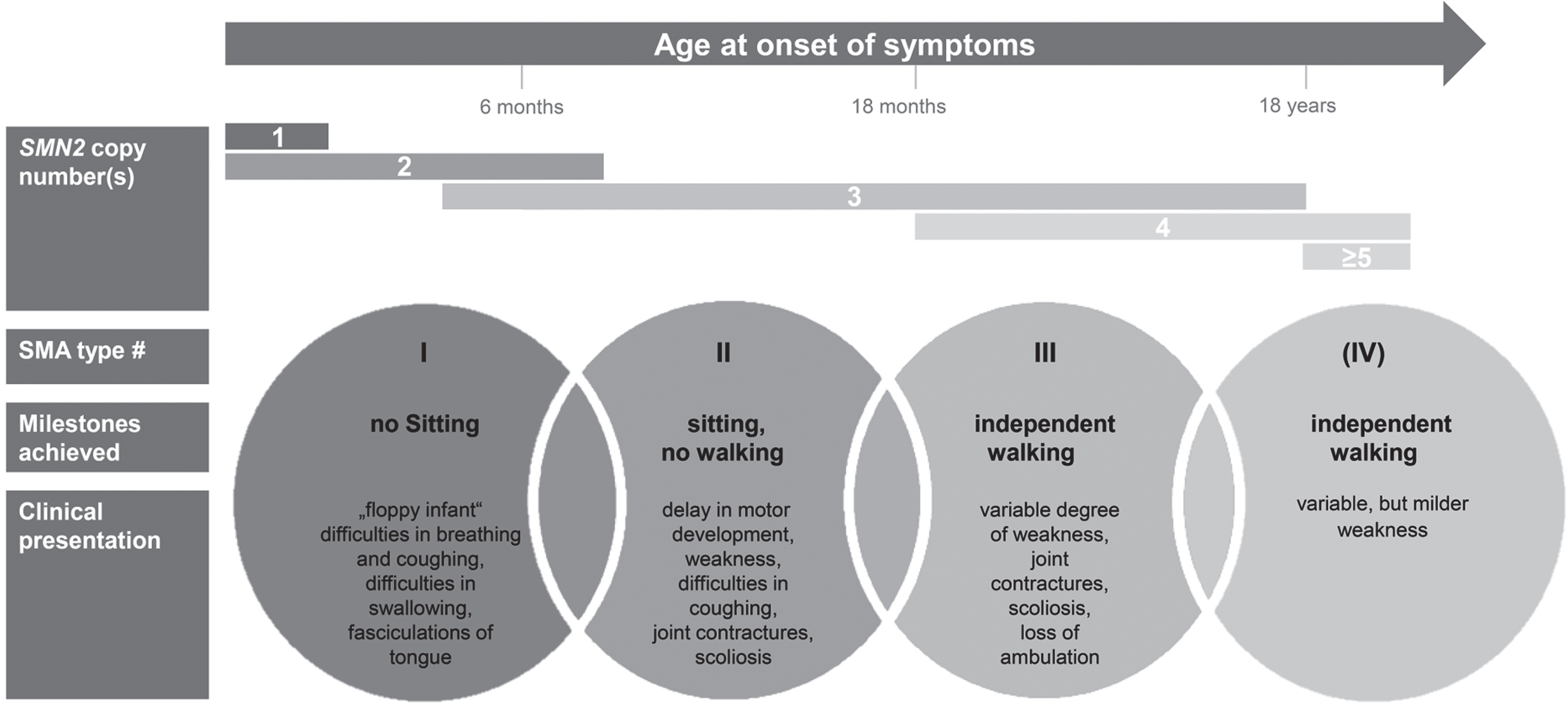
SMA is one of the most frequent monogenic neurodegenerative diseases with an incidence estimated to be around 1 : 6,000 to 1 : 10,000 in newborns [6–9]. SMA encompasses a wide clinical continuum of disease severity and has been classified into subtypes according to age at onset and the motor milestones achieved [10]. More than half of patients have the severe phenotype of SMA type 1 with onset of symptoms within the first 6 months of age. A ‘floppy infant’ presentation, reduced spontaneous movements and a paradoxical breathing pattern are characteristic; these infants fail to achieve the free-sitting milestone. Without drug treatment and ventilator support, SMA type 1 is the leading genetic cause of death in early infancy with a life expectancy of under 2 years [11, 12]. SMA type 2 is characterized by a milder course with onset of symptoms between the ages of 6 and 18 months. Per definition, these patients do manage free sitting, but not independent walking. The latter is achieved (at least temporarily) in patients with SMA type 3, whose symptoms’ onset is during infancy or adolescence. In addition, some classifications define SMA type 0 and SMA type 4 with prenatal onset or a very mild phenotype entailing an adult onset of symptoms, respectively. The disease’s hallmark is the degeneration of anterior horn cells in the spinal cord, leading to the characteristic symptom of progressive, proximal weakness involving varying degrees of muscle atrophy. Whereas all types of SMA are progressive, the rate of progression differs: SMA type 1 typically follows a rapidly progressing course, while type 3 progresses slowly [13].
Molecular genetics: About 95% of SMA cases are caused by homozygous deletions and less frequently point mutations in the SMN1 gene (survival of motor neuron 1) on the long arm of chromosome 5 (5q-SMA) [5, 14], whereas SMA mutations in other genes can also be causative (non-5q-SMA) [15]. Disease-causing mutations in SMN1 inhibit the production of functional SMN protein from this gene. SMA’s highly variable phenotypic spectrum is mainly attributable to variable copy numbers of the neighboring SMN2 gene [16, 17]. This gene is almost homologous to SMN1 except for few nucleotides [18, 19] and is of no relevance in healthy individuals. A single nucleotide transition of SMN2 causes predominant exon 7 skipping and mainly results in an unstable protein (SMNΔ7).
In patients with SMA on the other side, small amounts of full-length and fully functional SMN-protein can be produced by SMN2 [20], thus higher numbers of SMN2-copies are associated with milder phenotypes [21, 22]. Figure 1 summarizes SMA subtypes and displays typically associated SMN2 copy numbers.
Fig.1
Clinical classification of SMA subtypes according to onset, milestones achieved, and clinical presentation. Typically associated SMN2 copy numbers are displayed.
THERAPEUTIC APPROACHES – SYMPTOMATIC TREATMENT
While being a monogenetic neuromuscular disease, the resulting phenotypic spectrum is complex and SMA is generally perceived as a systemic disease [23]. Accordingly, caring for patients with SMA requires the interdisciplinary management of respiratory, nutritional and gastroenterological, orthopedic, and psychosocial issues. General treatment recommendations were published in 2007 in the first consensus statement on standards of care in SMA [24]. Nevertheless, the implementation of standards of care is highly variable and is influenced by cultural perspectives, socioeconomic factors, and the availability of regional resources [25]. Due to advances and improvements in care over the last decade, an updated version of recommendations on diagnosing SMA and patient care was published only recently [26, 27].
THERAPEUTIC APPROACHES – DRUG TREATMENT
Several different compounds have been investigated in randomized controlled trials in the last few decades, including approaches to increase muscle strength and function by (1) hyperacetylating agents such as valproic acid [28–30] or phenylbutyrate [31], (2) anabolic agents such as albuterol [32], thyreotropin-releasing hormone [33] or growth-hormone [34] and (3) neuroprotective agents such as gabapentin [35, 36], riluzol [37] and olesoxime [38]. Despite negative results regarding primary endpoints, those investigations validated outcome measures and yielded key information about trial designs and the feasibility of patient recruitment.
Actual therapeutic developments can be subdivided into therapies aiming to modify the splicing of SMN2, replacing the SMN1 gene, or upregulating muscle growth. Figure 2 summarizes the therapeutic approaches discussed in the following sections and illustrates the respective molecular mechanisms of action; Table 1 illustrates the current status of development of specific drugs.
Fig.2
Illustration of therapeutic approaches in SMA involving molecular mechanisms of action (modified illustration based on Farrar et al. 2017 [101] and Pechmann et al. 2017 [102]). FSTA = Fast Troponin Activator.
![Illustration of therapeutic approaches in SMA involving molecular mechanisms of action (modified illustration based on Farrar et al. 2017 [101] and Pechmann et al. 2017 [102]). FSTA = Fast Troponin Activator.](https://content.iospress.com:443/media/jnd/2020/7-1/jnd-7-1-jnd190424/jnd-7-jnd190424-g002.jpg)
Table 1
Synopsis of selected ongoing and recently finished clinical trials of medical treatments in spinal muscular atrophy (SMA). AAV-9 = Associated Adenovirus 9; 6MWT = six minute walking test; FSTA = Fast Skeletal Muscle Troponin Activator; IT = intrathecal; PO = oral intake; IV = intravenous application
| Drug | Sponsor | Mechanism of action | Route of application | Phase | FDA Approval | Comment | ||
| I | II | III | ||||||
| Splicing modification of SMN2: | ||||||||
| Nusinersen | Biogen-Ionis | Antisense-oligonucleotide | IT | x | x | x | x | Approval by FDA (Dec. 2016) and EMA (Jul. 2017) for all subtypes of SMA |
| RG7916 (Risdiplam) | Roche | Small molecule/splicing modifier | PO | x | x | (x) | SMA type 1: After 15 months of treatment inde-pendent sitting in 33% | |
| LMI070 (Branaplam) | Novartis | Small molecule/splicing modifier | PO | x | x | Recruitment temporarily halted (safety concerns), now completed | ||
| Replacement of SMN1-gene | ||||||||
| AVXS-101 (Zolgensma) | Avexis/Novartis | AAV-9-Vector | IV | x | x | x | x | FDA approval for SMA patients <2 years of age (May 2019) |
| AVXS-101 (Zolgensma) | Avexis/Novartis | AAV-9-Vector | IT | x | Study in children <6 years of age with 3 SMN2-copies | |||
| Upregulation of muscle growth | ||||||||
| CK-2127107 (Reldesemtiv) | Cytokinetics | FSTA | PO | x | x | Mild improvement in 6MWT after 4–8 weeks of treatment in SMA 2 and 3 | ||
| SRK-015 | Scholar Rock | Myostatin Inhibotor | IV | x | x | Positive results in animal models | ||
| Neuroprotection | ||||||||
| Olesoxime | Hoffmann-La Roche | Apoptosis-inhibitor | PO | x | x | Development stopped in 2018 | ||
Splicing modification of SMN2
The first drug approved for SMA treatment was nusinersen (former IONIS-SMNRX), an antisense-oligonucleotide (ASO) that enhances the inclusion of exon 7 in mRNA transcripts of SMN2. Nusinersen binds to an intronic splice-silencing-site in intron 7 of SMN2 and thereby suppresses the binding of other splice-factors [39]. This results in an increased proportion of SMN2-mRNA with included exon 7 and consecutively more functional full-length SMN2 protein [40, 41]. After promising results for nusinersen in phase I and phase II studies with children with SMA type 2 and 3 [42, 43], three phase III studies were initiated subsequently: In the ENDEAR study, 121 infants with SMA type 1 and younger than 7 months of age underwent either repeated intrathecal injections of nusinersen or a sham-intervention entailing no drug application. Those receiving nusinersen demonstrated a prolonged time to death or need for permanent ventilation compared to the sham-control group [44]. The criteria for being a “motor-milestone-responder” (achievement of motor milestones in HINE-2 scale; Hammersmith Infant Neurological Examination) were fulfilled by 51% in the verum group but by 0% in the sham-control group. Although the verum group’s motor development differed strongly from the disease’s natural history, only a minority of patients (6/73) achieved independent sitting during the nusinersen treatment period lasting about one year [44]. In the CHERISH study, the effects of nusinersen were studied in 126 older children (median age 4 years) with SMA type 2 and onset of symptoms after the age of 6 months. Again, the nusinersen group exhibited a gain in motor functions (mean + 4.0 points in HFMSE scale; Hammersmith Functional Motor Scale Expanded version), whereas the sham control group deteriorated slightly (–1.9 points in HFMSE scale) [45]. Both studies were terminated prematurely after these results became apparent in interim analysis and all participants were switched to the treatment group. The effects of pre-symptomatic nusinersen treatment were studied in the NURTURE study in 25 infants under 6 weeks of age with 2 (n = 15) or 3 (n = 10) SMN2 copies. All 25 patients acquired the ability to sit independently and 22/25 achieved independent walking [46, 47]. Nusinersen was approved by the Federal Drug Agency (FDA) in December 2016 and by the European Medicines Agency (EMA) in May 2017. The first patients with SMA type 1 had been treated beforehand within an Expanded Access Program (EAP) in some countries.
An approach to altering the splicing of SMN2 and thus increasing the amount of functional SMN-protein is also taken by small molecules such as RG7916 (risdiplam) and LMI070 (branaplam). These compounds are taken orally, cross the blood-brain barrier, and have been shown to increase the amount of full length SMN-protein [48]. The most advanced compound is the pyridazine derivative RG7916 now being investigated in several trials: In the open-label FIREFISH-study, 21 infants with SMA type 1 between 1 to 7 months of age received low-dose risdiplam (Part 1, n = 4) with the primary objective of safety-assessment, or a high dose (following Part 2, n = 17) with the primary objective of assessing efficacy (independent sitting after 12 months of treatment). Interim results were presented recently [49]: After a medium treatment duration of 14.8 months, the primary endpoint of independent sitting was attained by 33% of all infants (n = 7/21), and by 41% of those infants receiving the higher dose in Part 2 (n = 7/17). No treatment-related safety concerns were reported. Part 2 of the study is ongoing. Older patients with SMA type 2 and 3 received RG7916 in the SUNFISH-study. Again, that study was divided in Part 1 (dose-finding) and double-blind, placebo-controlled Part 2 (confirmatory). Of the 43 patients included in Part 1, 58% revealed an improvement in at least 3 points in the Motor Function Measure-32 (MFM32) scale [49]. Further ongoing studies include the JEWELFISH-study, including patients with all types of SMA (age 6 months to 60 years) treated previously with SMN-targeting therapies, gene therapy, or olesoxime, and the RAINBOWFISH-study for pre-symptomatic babies with SMA. LMI070 is now being investigated in a phase I/II study (that recently finished recruiting) with SMA type 1 patients after an almost two-year halt because of safety concerns in animal data.
Replacement of SMN1-gene
Gene therapy of SMA is the most advanced medical approach that directly targets the dysfunctional SMN1-gene in SMA. Studies employing an Adeno-Associated Viral serotype 9 (AAV9) vector to deliver an intact copy of wild-type SMN in murine models showed that these constructs cross the brain-blood barrier and lead to prolonged survival of treated SMA-mice [50–52]. The first clinical trial with zolgensma (AVXS-101) included 15 infants with SMA type 1 with 2 SMN2 copies (<8 months of age) [53]. All patients received a single intravenous dose of the compound in either low (n = 3) or high-dose (n = 12). Transient elevation of liver-enzymes occurred in two patients, all patients received steroid treatment. Marked improvement in CHOP INTEND scores (Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders) was observed in the high-dose cohort, with 11 patients attaining scores >40 points – a cutoff not usually attained in the natural history of SMA 1. During the follow-up period, 9 of the 12 children receiving high-dose zolgensma were able to sit without support for >30 seconds. One patient in the low-dose cohort needed permanent ventilation at age 29 months. Comparison to a natural history cohort confirmed the improvement of survival, motor function and milestones by AVXS-101 treatment [54]. Safety and efficacy are now being investigated in several ongoing studies: the phase-3 STR1VE study involves 20 patients with SMA type 1 under 6 months of age at the time of infusion with the primary endpoint of achieving independent sitting; similar studies for Europe and Asia are ongoing or planned. The SPR1NT study will investigate the pre-symptomatic treatment of SMA patients of all subtypes (<6 weeks of age). Zolgensma was approved by the FDA for intravenous application in patients with SMA under 2 years of age in May 2019. While infants undergo the systemic intravenous application of gene therapy, intrathecal application might be necessary for older patients to achieve sufficient transduction of motor neurons [55]. Initial trials addressing intrathecal gene therapy in mice and pigs have demonstrated improved gene expression that was achieved with a lower dose of viral vectors [56, 57]. The effects of intrathecal application of zolgensma in children with SMA type 2 (<6 years of age) are now being examined in the STRONG study.
Upregulation of muscle growth
Therapeutic approaches that do not directly target the genetic cause of SMA include the improvement of muscle mass and function. Two compounds are the most advanced: Myostatin-inhibitors and F ast Skeletal Muscle Troponin Activators (FSTA). Myostatin is a member of the TGFβ superfamily of growth factors that inhibits muscle over-growth and is primarily expressed in skeletal muscle. Myostatin-deficient animals are known to have considerably increased muscle mass and strength, and the use of the myostatin-inhibitor SRK-015 in SMA-mice resulted in improved muscle mass and function [58]. The safety of SRK-015 is being studied in a phase II trial whose first results are pending. FSTAs like CK-2127107 (reldesemtiv), on the other hand, slow the release of calcium leading to improved contractibility of muscle fibers and thus muscle function [59]. Its use in SMA was studied in 70 patients with SMA type 2–4 with official results also pending. The interim analyses reported a mild but statistically significant improvement in the six minute walk test (6MWT) after 4 and 8 weeks of treatment.
Therapeutic approaches in preclinical development
A growing number of compounds are in preclinical or early clinical development. Those comprise small molecules aiming to stabilize the SMN-protein and other types of ASOs targeting SMN2, but also SMN-independent approaches. The latter include myostatin-inhibition via Activin Receptor Type IIB-antagonists [60] and stand-alone approaches like inhibition of the p38MAPK pathway [61].
BIOMARKERS IN SMA
Apart from SMN2 copy numbers, a variety of other possible biomarkers are currently discussed and investigated [62]. Within the ENDEAR study population, symptomatic SMA type 1 patients showed higher levels of plasma phosphorylated neurofilament heavy chain (pNF-H) than healthy controls. Furthermore, higher pNF-H levels correlated positively with earlier onset of symptoms and inversely with motor function at start of nusinersen treatment. Under treatment with nusinersen, these levels decreased faster in the verum group than in sham control group. This decrease was more pronounced the earlier the therapy was started [63]. In CSF of an adult SMA cohort, levels of pNF-H in CSF were below detection limit, but levels of NSE and pTAU-protein showed a significant decrease under treatment [64]. Electrophysiological biomarkers include the examination of the compound muscle action potential (CMAP) and the motor unit number estimation (MUNE), which have already been used in clinical trials [42]. Availability of validated biomarkers would ideally allow predicting the clinical course of disease and the response to any drug treatment. This would improve clinical decision-making and significantly reduce the time and resources for clinical drug development.
EMERGING NEWBORN SCREENING
A consistent finding across clinical trials for both SMN2 splicing modification and gene therapy is the fact that the effect size depends on the age at treatment initiation: the earlier treatment is started, the greater the clinical benefit is. The most impressive results have been observed when treatment is initiated before the first clinical symptoms become apparent. As we know that denervation progresses rapidly during the first 6 months of life [65], the ‘rescue’ of these motoneurons before clinical deterioration appears to be essential. Nevertheless, the mean age of diagnosis in SMA type 1 is around 6 months of age [66]. Newborn screening (NBS) thus enables us to identify these patients at a pre-symptomatic stage. Four pilot projects of NBS programs in SMA have been conducted and published so far, all using quantitative polymerase chain reaction (qPCR) assays detecting homozygous deletions in either exon 7 [67–69] or intron 7 [70] of SMN1 via dried blood spot analysis. Only one of these assays was validated as also detecting heterozygous carrier deletions [67], and none of the assays was able to detect point mutations or quantify SMN2 copy numbers. In the NBS pilot studies in Taiwan, New York State and Germany, verification of NBS results by sequencing yielded a positive predictive value of 100% [67, 69, 70]. To lower the costs of analysis, different PCR-based assays have been developed that allow simultaneous screening for SMA (with or without SMN2 copy number quantification) and severe combined immunodeficiency (SCID) [71, 72]. SMA was added to the Recommended Uniform Screening Panel (RUSP) in July 2018; NBS for SMA is being implemented in a few US states and southern Belgium, and pilot screening projects are ongoing in other states and countries. Nevertheless, the issue regarding who should be treated is highly controversial [73]. The correlation between SMN2 copy numbers and disease severity was recently examined in a larger Spanish cohort of 625 patients with SMA of all subtypes [17]. Two SMN2 copies were associated with SMA type1 in almost 90% of patients. In patients presenting three and more copies, the individual age of onset and severity are more difficult to predict, but those factors still correlate with the copy number. An algorithm for treatment decisions for children diagnosed with SMA by NBS has been proposed by the SMA NBS Multidisciplinary Working Group, supported by CureSMA. There was consensus among the experts participating in this delphi-technique-based process, namely that treatment should be initiated immediately in truly asymptomatic infants with one SMN2 copy and in infants with two or three copies with or without symptoms, while those with four or more copies should be closely monitored and only treated after the onset of signs or symptoms [74]. However, this pragmatic proposition does not incorporate the presence of possible genetic modifiers in SMA other than the number of SMN2 copies that can mitigate or exacerbate the clinical course [75]. This is also reflected in the observation that disease severity can differ even in siblings possessing the same SMA genotype. The fact that parents of an apparently healthy baby are confronted by a severe diagnosis and difficult treatment decisions furthers adds to the complexity of NBS programs. To address these problems, greater awareness for SMA in the public and the availability of qualified genetic counseling are necessary to help parents make an informed decision [76, 77].
NEW PHENOTYPES, NEW CHALLENGES
Since the introduction of new drug treatments for SMA, we have observed disease trajectories that differ significantly from the known natural history of the disease. These new phenotypes now also cross the traditional subtypes of SMA. For example, patients with onset before six months of age (typical for SMA type 1) might achieve independent sitting (SMA type 2 by definition) if treatment is initiated early. It is now more appropriate to rely on a combination of age of onset, number of SMN2 copies, and age at start of drug treatment rather than the traditional subtypes to define a clinical phenotype of SMA.
These new disease trajectories also mean we must modify and adapt the clinical approach taken. For example, longer survival without ventilatory support following the initiation of drug treatment needs to be considered when counseling the parents of patients with early-onset types of SMA. On the one end of the spectrum, namely in very severe cases entailing a prenatal onset (SMA type 0), drug treatment is not likely to lead to any relevant improvement in motor function, nor will it prevent the need for permanent ventilation; it might therefore be inadvisable. On the other end, initiating treatment in a presymptomatic patient might result in almost normal motor development.
Additional organ involvement, including occurrence of cardiac defects [78], autonomic dysregulation [79] or abnormal fatty acid metabolism [80, 81] has been reported in SMA. SMN protein is known to be highly expressed prenatally in most organs, so that a significant role in organogenesis has been discussed [82]. Further research is needed to understand if systemic treatment of SMN deficiency is of clinical benefit compared to restricted treatment of the central nervous system.
Clinical trials and real world data
In the context of rare diseases, it is almost inevitable that drug approval will be based on weaker evidence than is the case with drugs for common diseases. This issue concerns the disease spectrums investigated in clinical trials, patient numbers, and observation periods. For example, nusinersen was approved for all types and disease stages of SMA despite the fact that the two randomized controlled trials covered only a small proportion of the total SMA population, namely infants and young children presenting a relatively early disease stage. Several centers published their experiences with nusinsersen treatment of SMA type 1 patients of different age groups in the early-access program (EAP) and delivered similar results [83–88]: Age at the beginning of treatment is the most important factor that determines motor response to treatment. Interestingly, no marked difference in motor response between patients with 2 or 3 copies of SMN2 was observed [84, 87]. In contrast to the clinical trials, patient cohorts were heterogeneous regarding ventilation-dependency, need for nutritional support and age; data on patients up to 19 years of age were analyzed in the Italian cohort [85]. More conflicting than the data on motor response are the findings regarding ventilation and nutritional support: In both the German and French cohorts, a significant proportion of patients exhibiting motor improvement had started permanent ventilation or underwent tracheostomy or the placement of a feeding tube. It is unclear whether this reflects a poor treatment effect on respiratory and bulbar function, or a more proactive approach in the participating centers to provide ventilator support or tube feeding.
A mild improvement in the 6MWT was seen in an adult cohort undergoing nusinersen treatment [64]. Overall, there is very little data available extending beyond the cohort of SMA type 1 patients, and it remains difficult for us to predict ‘which effect can be expected in which subtype and at which stage of the disease’ and ‘how long treatment-effects persist’. Furthermore, there are serious logistical and ethical factors that make additional placebo-controlled trials difficult if not impossible to carry out, now that effective treatment options are available.
Considering the limited evidence on the long-term efficacy and safety of novel drug treatments for SMA and their high costs, it is necessary to systematically collect real-world data to improve the basis for clinical decision-making and reimbursement for patients with SMA. Various international initiatives, including the International SMA Consortium Spinal Muscular Atrophy Patient Registry (iSMAC), the SMArtCARE project and the TREAT-NMD network have now established disease-specific registries or are aiming to collect data from different national registries. These projects share standardized data sets for the longitudinal assessment of SMA patients with or without drug treatment [89–91]. Ideally, these initiatives should involve patient representatives in the governance and follow the IRDiRC-recognized “FAIR Guiding Principles” in order to make the collected data Findable, Accessible, Interoperable and Reusable and thus maximise their utility for research [92].
Need for standardized outcome measures
The application of standardized outcome measures is crucial to harmonize real-world data from different registries and to enable the comparison of results. The ideal group of assessments covers and reflects all SMA subgroups, so that selecting the most suitable outcome measures relies on both the functional levels and age of SMA patients. Requirements should accommodate the developmental state of patients: whereas the CHOP INTEND might be best suited for documenting the functional status of severely affected SMA type 1 patients, some items are inappropriate for adolescent or adult patients. Another important issue is the problem of ceiling effects in certain assessments: children exhibiting improved motor function may attain the maximum score in the CHOP INTEND before the age of 2 years, but the HFMSE requires a certain degree of cooperation and has only been validated for application in children above 2 years of age. The motor domains of the Bayley Scales III might be an option to close this gap. As an example, the current SMArtCARE recommendations for standardized evaluation according to age and disease stage are shown in Table 2.
Table 2
Recommendations for the evaluation of patients with SMA by the SMArtCARE-project. RULM: revised-upper-limb-module; 6-MWT: six-minute-walking-test
| Baseline data (First visit only) |
| Current medical history and clinical examination |
| •including WHO motor milestones |
| Physiotherapeutic assessments: |
| CHOP INTEND |
| •All children <2 years of age |
| •All patients >2 years of age without ability to sit |
| Bayley-III Scale (motor part) |
| •Only for children <2 years of age with CHOP INTEND score >50 |
| HFMSE |
| •All patients >2 years of age with ability to sit |
| •If CHOP INTEND score >50: CHOP INTEND and HFMSE** |
| •If CHOP INTEND score >60: HFMSE instead of CHOP INTEND |
| RULM |
| •All patients >2 years of age with ability to sit (in a wheelchair) |
| 6-MWT |
| •All ambulant patients >3 years of age |
| ALS Functional Rating Scale (in adult patients) |
| Pulmonary function |
| Documentation of drug treatment and adverse events |
Challenges in clinical care
The conventional disease trajectories of the pre-treatment-era are now often modified by new drug treatments. This involves unprecedented challenges and issues regarding motor and non-motor symptoms. In many aspects, this requires that we reconsider earlier blueprints to enable individualized and the most appropriate decision-making.
Despite the improved survival and motor development of symptomatic patients with early onset SMA, these children also exhibit a higher rate of scoliosis during the first years of live. Greater awareness of this risk, and close monitoring of spinal deformities appear crucial to react early and enable the spine to be stabilized via medical orthoses. As many braces interfere with breathing in the more severely affected patients, choosing the ideal device can be difficult. Surgical interventions entailing ‘growing rod’ systems have been reported to be feasible in children with SMA type 1 as young as 4 to 6 years of age and might be an option for younger children with severe scoliosis [93, 94]. However, further experience in this field is needed to balance the risks and benefits of these interventions. Certain orthopedic devices – such as standing frames – have not been used in most SMA type 1 patients, but they appear promising for the prophylaxis of joint contractures and to allow age-appropriate positioning even in more severely affected patients.
Intrathecal application of drugs like nusinersen can be difficult in patients with severe scoliosis [95, 96]. Fluoroscopy may be necessary for lumbar access in these patients, but that involves high cumulative radiation exposure in potentially lifelong therapy in case of nusinersen [97]. Lumbar puncture is especially challenging in patients who have already undergone spinal fusion; some surgeons suggest creating artificial bone gaps during spinal surgery for later lumbar puncture, but we are still waiting for their long-term data [98]. Alternative routes of application, via intrathecal catheter systems [99] or even via cervical puncture [100] have been suggested, despite the fact that nusinersen has only been approved for application via lumbar puncture.
PERSPECTIVE
For the first time in the history of SMA, new treatments like splicing modification and gene therapy are allowing the clinical course to be substantially modified. Additional therapeutic approaches are currently being taken at advanced stages of clinical development and are likely to expand the spectrum of drug treatment options for SMA. This will add to the complexity of care for patients with SMA. To achieve maximum treatment effects, a timely diagnosis and treatment initiation are particularly important. Standard newborn screening seems to be an appropriate tool to attain this goal, although it remains unclear when treatment should be initiated in patients presenting high numbers of SMN2 copies.
When medications for rare diseases come up for approval, there is often only limited evidence available on its long-term effects and safety, and conducting randomized investigations to deliver such evidence is often impossible. Therefore, the only way to generate additional evidence is to collect and analyze real-world data via high-quality, well-monitored patient registries that attempt to avoid bias, so that they provide meaningful results.
Keeping in mind the recent success of drug treatment in SMA, it is important that we do not disregard individual interdisciplinary clinical management, which remains the backbone of SMA treatment, since many patients are left with a significant disease burden despite drug treatment.
DISCLOSURES
Dr. David C. Schorling, participated in workshops sponsored by Biogen and Roche.
Dr. Astrid Pechmann, received compensation for presentations and training activities from Biogen, received research funding from Biogen.
Prof. Dr. Janbernd Kirschner, received research funding and/or compensation for presentations and consulting services from Avexis, Biogen, Ionis Pharmaceuticals, Novartis, and Roche.
REFERENCES
[1] | Werdnig G . Two early infantile hereditary cases of progressive muscular atrophy simulating dystrophy, but on a neural basis. 1891. Arch Neurol. (1971) ;25: :276–8. |
[2] | Hoffmann J . Ueber chronische spinale Muskelatrophie im Kindesalter, auf familiärer Basis. Dtsch Z Für Nervenheilkd. (1893) ;3: :427–70. doi: 10.1007/BF01668496 |
[3] | Brzustowicz LM , Lehner T , Castilla LH , Penchaszadeh GK , Wilhelmsen KC , Daniels R , et al. Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2-13.3. Nature. (1990) ;344: :540–1. doi: 10.1038/344540a0 |
[4] | Melki J , Abdelhak S , Sheth P , Bachelot MF , Burlet P , Marcadet A , et al. Gene for chronic proximal spinal muscular atrophies maps to chromosome 5q. Nature. (1990) ;344: :767–8. doi: 10.1038/344767a0 |
[5] | Lefebvre S , Bürglen L , Reboullet S , Clermont O , Burlet P , Viollet L , et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. (1995) ;80: :155–65. |
[6] | Ogino S , Leonard DGB , Rennert H , Ewens WJ , Wilson RB . Genetic risk assessment in carrier testing for spinal muscular atrophy. Am J Med Genet. (2002) ;110: :301–7. doi: 10.1002/ajmg.10425 |
[7] | Lunn MR , Wang CH . Spinal muscular atrophy. Lancet Lond Engl. (2008) ;371: :2120–33. doi: 10.1016/S0140-6736(08)60921-6 |
[8] | Verhaart IEC , Robertson A , Wilson IJ , Aartsma-Rus A , Cameron S , Jones CC , et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy – a literature review. Orphanet J Rare Dis. (2017) ;12: :124. doi: 10.1186/s13023-017-0671-8 |
[9] | König K , Pechmann A , Thiele S , Walter MC , Schorling D , Tassoni A , et al. De-duplicating patient records from three independent data sources reveals the incidence of rare neuromuscular disorders in Germany. Orphanet J Rare Dis. (2019) ;14: :152. doi: 10.1186/s13023-019-1125-2 |
[10] | Munsat TL . International SMA Collaboration. Neuromuscul Disord. (1991) ;1: :81. doi: 10.1016/0960-8966(91)90052-T |
[11] | Zerres K , Rudnik-Schöneborn S . Natural history in proximal spinal muscular atrophy. Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch Neurol. (1995) ;52: :518–23. |
[12] | Farrar MA , Vucic S , Johnston HM , du Sart D , Kiernan MC . Pathophysiological insights derived by natural history and motor function of spinal muscular atrophy. J Pediatr. (2013) ;162: :155–9. doi: 10.1016/j.jpeds.2012.05.067 |
[13] | Zerres K , Rudnik-Schöneborn S , Forrest E , Lusakowska A , Borkowska J , Hausmanowa-Petrusewicz I . A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J Neurol Sci. (1997) ;146: :67–72. |
[14] | Hahnen E , Forkert R , Marke C , Rudnik-Schöneborn S , Schönling J , Zerres K , et al. Molecular analysis of candidate genes on chromosome 5q13 in autosomal recessive spinal muscular atrophy: Evidence of homozygous deletions of the SMN gene in unaffected individuals. Hum Mol Genet. (1995) ;4: :1927–33. |
[15] | Peeters K , Chamova T , Jordanova A . Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies. Brain J Neurol. (2014) ;137: :2879–96. doi: 10.1093/brain/awu169 |
[16] | Wirth B , Brichta L , Schrank B , Lochmüller H , Blick S , Baasner A , et al. Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum Genet. (2006) ;119: :422–8. doi: 10.1007/s00439-006-0156-7 |
[17] | Calucho M , Bernal S , Alías L , March F , Venceslá A , Rodríguez-Álvarez FJ , et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul Disord NMD. (2018) ;28: :208–15. doi: 10.1016/j.nmd.2018.01.003 |
[18] | Lorson CL , Hahnen E , Androphy EJ , Wirth B . A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. (1999) ;96: :6307–11. |
[19] | Feldkötter M , Schwarzer V , Wirth R , Wienker TF , Wirth B . Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. (2002) ;70: :358–68. doi: 10.1086/338627 |
[20] | Butchbach MER . Copy Number Variations in the Survival Motor Neuron Genes: Implications for Spinal Muscular Atrophy and Other Neurodegenerative Diseases. Front Mol Biosci. (2016) ;3: :7. doi: 10.3389/fmolb.2016.00007 |
[21] | Prior TW , Swoboda KJ , Scott HD , Hejmanowski AQ . Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am J Med Genet A. (2004) ;130A: :307–10. doi: 10.1002/ajmg.a.30251 |
[22] | Mailman MD , Heinz JW , Papp AC , Snyder PJ , Sedra MS , Wirth B , et al. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med Off J Am Coll Med Genet. (2002) ;4: :20–6. doi: 10.1097/00125817-200201000-00004 |
[23] | Lipnick SL , Agniel DM , Aggarwal R , Makhortova NR , Finlayson SG , Brocato A , et al. Systemic nature of spinal muscular atrophy revealed by studying insurance claims. PloS One. (2019) ;14: :e0213680. doi: 10.1371/journal.pone.0213680 |
[24] | Wang CH , Finkel RS , Bertini ES , Schroth M , Simonds A , Wong B , et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. (2007) ;22: :1027–49. doi: 10.1177/0883073807305788 |
[25] | Bladen CL , Thompson R , Jackson JM , Garland C , Wegel C , Ambrosini A , et al. Mapping the differences in care for 5,000 spinal muscular atrophy patients, a survey of 24 national registries in North America, Australasia and Europe. J Neurol. (2014) ;261: :152–63. doi: 10.1007/s00415-013-7154-1 |
[26] | Mercuri E , Finkel RS , Muntoni F , Wirth B , Montes J , Main M , et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord NMD. (2018) ;28: :103–15. doi: 10.1016/j.nmd.2017.11.005 |
[27] | Finkel RS , Mercuri E , Meyer OH , Simonds AK , Schroth MK , Graham RJ , et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute caremedications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord NMD. (2018) ;28: :197–207. doi: 10.1016/j.nmd.2017.11.004. |
[28] | Krosschell KJ , Kissel JT , Townsend EL , Simeone SD , Zhang RZ , Reyna SP , et al. Clinical trial of L-Carnitine and valproic acid in spinal muscular atrophy type I. Muscle Nerve. (2018) ;57: :193–9. doi: 10.1002/mus.25776 |
[29] | Kissel JT , Elsheikh B , King WM , Freimer M , Scott CB , Kolb SJ , et al. SMA valiant trial: A prospective, double-blind, placebo-controlled trial of valproic acid in ambulatory adults with spinal muscular atrophy. Muscle Nerve. (2014) ;49: :187–92. doi: 10.1002/mus.23904 |
[30] | Kissel JT , Scott CB , Reyna SP , Crawford TO , Simard LR , Krosschell KJ , et al. SMA CARNIVAL TRIAL PART II: a prospective, single-armed trial of L-carnitine and valproic acid in ambulatory children with spinal muscular atrophy. PloS One. (2011) ;6: :e21296. doi: 10.1371/journal.pone.0021296 |
[31] | Mercuri E , Bertini E , Messina S , Solari A , D’Amico A , Angelozzi C , et al. Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology. (2007) ;68: :51–5. doi: 10.1212/01.wnl.0000249142.82285.d6 |
[32] | Kinali M , Mercuri E , Main M , De Biasia F , Karatza A , Higgins R , et al. Pilot trial of albuterol in spinal muscular atrophy. Neurology. (2002) ;59: :609–10. |
[33] | Tzeng AC , Cheng J , Fryczynski H , Niranjan V , Stitik T , Sial A , et al. A study of thyrotropin-releasing hormone for the treatment of spinal muscular atrophy: a preliminary report. Am J Phys Med Rehabil. (2000) ;79: :435–40. |
[34] | Kirschner J , Schorling D , Hauschke D , Rensing-Zimmermann C , Wein U , Grieben U , et al. Somatropin treatment of spinal muscular atrophy: a placebo-controlled, double-blind crossover pilot study. Neuromuscul Disord NMD. (2014) ;24: :134–42. doi: 10.1016/j.nmd.2013.10.011 |
[35] | Merlini L , Solari A , Vita G , Bertini E , Minetti C , Mongini T , et al. Role of gabapentin in spinal muscular atrophy: results of a multicenter, randomized Italian study. J Child Neurol. (2003) ;18: :537–41. doi: 10.1177/08830738030180080501 |
[36] | Miller RG , Moore DH , Dronsky V , Bradley W , Barohn R , Bryan W , et al. A placebo-controlled trial of gabapentin in spinal muscular atrophy. J Neurol Sci. (2001) ;191: :127–31. |
[37] | Russman BS , Iannaccone ST , Samaha FJ . A phase 1 trial of riluzole in spinal muscular atrophy. Arch Neurol. (2003) ;60: :1601–3. doi: 10.1001/archneur.60.11.1601 |
[38] | Bertini E , Dessaud E , Mercuri E , Muntoni F , Kirschner J , Reid C , et al. Safety and efficacy of olesoxime in patients with type 2 or non-ambulatory type 3 spinal muscular atrophy: A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. (2017) ;16: :513–22. doi: 10.1016/S1474-4422(17)30085-6 |
[39] | Singh NK , Singh NN , Androphy EJ , Singh RN . Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. (2006) ;26: :1333–46. doi: 10.1128/MCB.26.4.1333-1346.2006 |
[40] | Rigo F , Hua Y , Krainer AR , Bennett CF . Antisense-based therapy for the treatment of spinal muscular atrophy. J Cell Biol. (2012) ;199: :21–5. doi: 10.1083/jcb.201207087 |
[41] | Passini MA , Bu J , Richards AM , Kinnecom C , Sardi SP , Stanek LM , et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. (2011) ;3: :72ra18. doi: 10.1126/scitranslmed.3001777 |
[42] | Chiriboga CA , Swoboda KJ , Darras BT , Iannaccone ST , Montes J , De Vivo DC , et al. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology. (2016) ;86: :890–7. doi: 10.1212/WNL.0000000000002445 |
[43] | Finkel RS , Chiriboga CA , Vajsar J , Day JW , Montes J , De Vivo DC , et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet Lond Engl. (2016) ;388: :3017–26. doi: 10.1016/S0140-6736(16)31408-8 |
[44] | Finkel RS , Mercuri E , Darras BT , Connolly AM , Kuntz NL , Kirschner J , et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med. (2017) ;377: :1723–32. doi: 10.1056/NEJMoa1702752 |
[45] | Mercuri E , Darras BT , Chiriboga CA , Day JW , Campbell C , Connolly AM , et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N Engl J Med. (2018) ;378: :625–35. doi: 10.1056/NEJMoa1710504 |
[46] | Bertini E , Hwu W-L , Reyna SP , Farwell W , Gheuens S , Sun P , et al. Efficacy and safety of nusinersen in infants with presymptomatic spinal muscular atrophy (SMA): Interim results from the NURTURE study. Eur J Paediatr Neurol. (2017) ;21: :e14. doi: 10.1016/j.ejpn.2017.04.1218 |
[47] | De Vivo DC , Bertini E , Swoboda KJ , Wuh-Liang H , Crawford TO , Finkel R , et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul Disord (2019) ; accepted. |
[48] | Poirier A , Weetall M , Heinig K , Bucheli F , Schoenlein K , Alsenz J , et al. Risdiplam distributes and increases SMN protein in both the central nervous system and peripheral organs. Pharmacol Res Perspect. (2018) ;6: :e00447. doi: 10.1002/prp2.447 |
[49] | Media release ROCHE (2019) . |
[50] | Dayton RD , Wang DB , Klein RL . The advent of AAV9 expands applications for brain and spinal cord gene delivery. Expert Opin Biol Ther. (2012) ;12: :757–66. doi: 10.1517/14712598.2012.681463 |
[51] | Valori CF , Ning K , Wyles M , Mead RJ , Grierson AJ , Shaw PJ , et al. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci Transl Med. (2010) ;2: :35ra42. doi: 10.1126/scitranslmed.3000830 |
[52] | Foust KD , Wang X , McGovern VL , Braun L , Bevan AK , Haidet AM , et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol. (2010) ;28: :271–4. doi: 10.1038/nbt.1610 |
[53] | Mendell JR , Al-Zaidy S , Shell R , Arnold WD , Rodino-Klapac LR , Prior TW , et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med. (2017) ;377: :1713–22. doi: 10.1056/NEJMoa1706198 |
[54] | Al-Zaidy SA , Kolb SJ , Lowes L , Alfano LN , Shell R , Church KR , et al. AVXS-101 (Onasemnogene Abeparvovec) for SMA1: Comparative Study with a Prospective Natural History Cohort. J Neuromuscul Dis. (2019) . doi: 10.3233/JND-190403 |
[55] | Hardcastle N , Boulis NM , Federici T . AAV gene delivery to the spinal cord: Serotypes, methods, candidate diseases, and clinical trials. Expert Opin Biol Ther. (2018) ;18: :293–307. doi: 10.1080/14712598.2018.1416089 |
[56] | Federici T , Taub JS , Baum GR , Gray SJ , Grieger JC , Matthews KA , et al. Robust spinal motor neuron transduction following intrathecal delivery of AAV9 in pigs. Gene Ther. (2012) ;19: :852–9. doi: 10.1038/gt.2011.130 |
[57] | Snyder BR , Gray SJ , Quach ET , Huang JW , Leung CH , Samulski RJ , et al. Comparison of adeno-associated viral vector serotypes for spinal cord and motor neuron gene delivery. Hum Gene Ther. (2011) ;22: :1129–35. doi: 10.1089/hum.2011.008 |
[58] | Long KK , O’Shea KM , Khairallah RJ , Howell K , Paushkin S , Chen KS , et al. Specific inhibition of myostatin activation is beneficial in mouse models of SMA therapy. Hum Mol Genet. (2019) ;28: :1076–89. doi: 10.1093/hmg/ddy382 |
[59] | Andrews JA , Miller TM , Vijayakumar V , Stoltz R , James JK , Meng L , et al. CK-2127107 amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve. (2018) ;57: :729–34. doi: 10.1002/mus.26017 |
[60] | Liu M , Hammers DW , Barton ER , Sweeney HL . Activin Receptor Type IIB Inhibition Improves Muscle Phenotype and Function in a Mouse Model of Spinal Muscular Atrophy. PloS One. (2016) ;11: :e0166803. doi: 10.1371/journal.pone.0166803 |
[61] | Simon CM , Dai Y , Van Alstyne M , Koutsioumpa C , Pagiazitis JG , Chalif JI , et al. Converging Mechanisms of p53 Activation Drive Motor Neuron Degeneration in Spinal Muscular Atrophy. Cell Rep. (2017) ;21: :3767–80. doi: 10.1016/j.celre2017.12.003 |
[62] | Kariyawasam DST , D’Silva A , Lin C , Ryan MM , Farrar MA . Biomarkers and the Development of a Personalized Medicine Approach in Spinal Muscular Atrophy. Front Neurol. (2019) ;10: :898. doi: 10.3389/fneur.2019.00898 |
[63] | Darras BT , Crawford TO , Finkel RS , Mercuri E , De Vivo DC , Oskoui M , et al. Neurofilament as a potential biomarker for spinal muscular atrophy. Ann Clin Transl Neurol. (2019) ;6: :932–44. doi: 10.1002/acn3.779 |
[64] | Walter MC , Wenninger S , Thiele S , Stauber J , Hiebeler M , Greckl E , et al. Safety and Treatment effects of Nusinersen in longstanding adult 5q-SMA type 3 – a prospective observational study. J Neuromuscul Dis (2019) ;6: :453–465. |
[65] | Swoboda KJ , Prior TW , Scott CB , McNaught TP , Wride MC , Reyna SP , et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol. (2005) ;57: :704–12. doi: 10.1002/ana.20473 |
[66] | Lin C-W , Kalb SJ , Yeh W-S . Delay in Diagnosis of Spinal Muscular Atrophy: A Systematic Literature Review. Pediatr Neurol. (2015) ;53: :293–300. doi: 10.1016/j.pediatrneurol.2015.06.002 |
[67] | Kraszewski JN , Kay DM , Stevens CF , Koval C , Haser B , Ortiz V , et al. Pilot study of population-based newborn screening for spinal muscular atrophy in New York state. Genet Med Off J Am Coll Med Genet. (2018) ;20: :608–13. doi: 10.1038/gim.2017.152 |
[68] | Boemer F , Caberg J-H , Dideberg V , Dardenne D , Bours V , Hiligsmann M , et al. Newborn screening for SMA in Southern Belgium. Neuromuscul Disord NMD. (2019) ;29: :343–9. doi: 10.1016/j.nmd.2019.02.003 |
[69] | Vill K , Kölbel H , Schwartz O , Olgemöller B , Harms E , Burggraf S , et al. One year of newborn screening for SMA – Results of a German pilot project. J Neuromuscul Dis (2019) ;6: :503–515. |
[70] | Chien Y-H , Chiang S-C , Weng W-C , Lee N-C , Lin C-J , Hsieh W-S , et al. Presymptomatic Diagnosis of Spinal Muscular Atrophy Through Newborn Screening. J Pediatr. (2017) ;190: :124–129.e1. doi: 10.1016/j.jpeds.2017.06.042 |
[71] | Taylor JL , Lee FK , Yazdanpanah GK , Staropoli JF , Liu M , Carulli JP , et al. Newborn blood spot screening test using multiplexed real-time PCR to simultaneously screen for spinal muscular atrophy and severe combined immunodeficiency. Clin Chem. (2015) ;61: :412–9. doi: 10.1373/clinchem.2014.231019 |
[72] | Gutierez-Mateo C , Baker M , Mochal S , Moore K , Filippov G , Dallaire S . A five-plex qPCR assay that measures copy numbers of SMN1, SMN2, TREC, KREC and RPP30. Am. Coll. Med. Genet. Genomics, vol. March 21-25, Phoenix, AZ: (2017) . |
[73] | Saffari A , Kölker S , Hoffmann GF , Weiler M , Ziegler A . Novel challenges in spinal muscular atrophy – How to screen and whom to treat? Ann Clin Transl Neurol. (2019) ;6: :197–205. doi: 10.1002/acn3.689 |
[74] | Glascock J , Sampson J , Haidet-Phillips A , Connolly A , Darras B , Day J , et al. Treatment Algorithm for Infants Diagnosed with Spinal Muscular Atrophy through Newborn Screening. J Neuromuscul Dis. (2018) ;5: :145–58. doi: 10.3233/JND-180304 |
[75] | Wirth B , Garbes L , Riessland M . How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic approaches. Curr Opin Genet Dev. (2013) ;23: :330–8. doi: 10.1016/j.gde.2013.03.003 |
[76] | Archibald AD , Smith MJ , Burgess T , Scarff KL , Elliott J , Hunt CE , et al. Reproductive genetic carrier screening for cystic fibrosis, fragile X syndrome, and spinal muscular atrophy in Australia: outcomes of 12,000 tests. Genet Med Off J Am Coll Med Genet. (2018) ;20: :513–23. doi: 10.1038/gim.2017.134 |
[77] | Wood SL , Brewer F , Ellison R , Biggio JR , Edwards RK . Prenatal Carrier Screening for Spinal Muscular Atrophy. Am J Perinatol. (2016) ;33: :1211–7. doi: 10.1055/s-0036-1593347 |
[78] | Rudnik-Schöneborn S , Heller R , Berg C , Betzler C , Grimm T , Eggermann T , et al. Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J Med Genet. (2008) ;45: :635–8. doi: 10.1136/jmg.2008.057950 |
[79] | Bach JR . Medical considerations of long-term survival of Werdnig-Hoffmann disease. Am J Phys Med Rehabil. (2007) ;86: :349–55. doi: 10.1097/PHM.0b013e31804b1d66 |
[80] | Tein I , Sloane AE , Donner EJ , Lehotay DC , Millington DS , Kelley RI . Fatty acid oxidation abnormalities in childhood-onset spinal muscular atrophy: primary or secondary defect(s)? Pediatr Neurol. (1995) ;12: :21–30. doi: 10.1016/0887-8994(94)00100-g |
[81] | Crawford TO , Sladky JT , Hurko O , Besner-Johnston A , Kelley RI . Abnormal fatty acid metabolism in childhood spinal muscular atrophy. Ann Neurol. (1999) ;45: :337–43. |
[82] | Novelli G , Calzà L , Amicucci P , Giardino L , Pozza M , Silani V , et al. Expression study of survival motor neuron gene in human fetal tissues. Biochem Mol Med. (1997) ;61: :102–6. |
[83] | Messina S , Pane M , Sansone V , Bruno C , Catteruccia M , Vita G , et al. Expanded access program with Nusinersen in SMA type I in Italy: Strengths and pitfalls of a successful experience. Neuromuscul Disord NMD. (2017) ;27: :1084–6. doi: 10.1016/j.nmd.2017.09.006 |
[84] | Pechmann A , Langer T , Schorling D , Stein S , Vogt S , Schara U , et al. Evaluation of Children with SMA Type 1 Under Treatment with Nusinersen within the Expanded Access Program in Germany. J Neuromuscul Dis. (2018) ;5: :135–43. doi: 10.3233/JND-180315 |
[85] | Pane M , Palermo C , Messina S , Sansone VA , Bruno C , Catteruccia M , et al. Nusinersen in type 1 SMA infants, children and young adults: Preliminary results on motor function. Neuromuscul Disord NMD. (2018) ;28: :582–5. doi: 10.1016/j.nmd.2018.05.010 |
[86] | Farrar MA , Teoh HL , Carey KA , Cairns A , Forbes R , Herbert K , et al. Nusinersen for SMA: expanded access programme. J Neurol Neurosurg Psychiatry. (2018) ;89: :937–42. doi: 10.1136/jnnp-2017-317412 |
[87] | Aragon-Gawinska K , Seferian AM , Daron A , Gargaun E , Vuillerot C , Cances C , et al. Nusinersen in patients older than 7 months with spinal muscular atrophy type 1: A cohort study. Neurology. (2018) ;91: :e1312–8. doi: 10.1212/WNL.0000000000006281 |
[88] | Walter MC , Dräger B , Günther R , Hermann A , Hagenacker T , Kleinschnitz C , et al. [Treatment evaluation in patients with 5q-associated spinal muscular atrophy: Real-world experience]. Nervenarzt. (2019) ;90: :343–51. doi: 10.1007/s00115-018-0653-7 |
[89] | International SMA Patient Registry – Full Text View – ClinicalTrials.gov n.d. https://clinicaltrials.gov/ct2/show/NCT00466349 (accessed June 18, 2019). |
[90] | TREAT-NMD : National SMA registries n.d. http://www.treat-nmd.eu/sma/patient-registries/sma/ (accessed June 18, 2019). |
[91] | Pechmann A , König K , Bernert G , Schachtrup K , Schara U , Schorling D , et al. SMArtCARE – A platform to collect real-life outcome data of patients with spinal muscular atrophy. Orphanet J Rare Dis. (2019) ;14: :18. doi: 10.1186/s13023-019-0998-4 |
[92] | Lochmüller H , Evans D , Farwell W , Finkel R , Goemans N , de Lemus M , et al. Position Statement: Sharing of Clinical Research Data in Spinal Muscular Atrophy to Accelerate Research and Improve Outcomes for Patients. J Neuromuscul Dis n.d. 5: :131–3. doi: 10.3233/JND-180325 |
[93] | Chandran S , McCarthy J , Noonan K , Mann D , Nemeth B , Guiliani T . Early treatment of scoliosis with growing rods in children with severe spinal muscular atrophy: a preliminary report. J Pediatr Orthop. (2011) ;31: :450–4. doi: 10.1097/BPO.0b013e31821722b1 |
[94] | Lenhart RL , Youlo S , Schroth MK , Noonan KJ , McCarthy J , Mann D , et al. Radiographic and Respiratory Effects of Growing Rods in Children With Spinal Muscular Atrophy. J Pediatr Orthope. (2017) ;37: :500–4. doi: 10.1097/BPO.0000000000000867 |
[95] | Stolte B , Totzeck A , Kizina K , Bolz S , Pietruck L , Mönninghoff C , et al. Feasibility and safety of intrathecal treatment with nusinersen in adult patients with spinal muscular atrophy. Ther Adv Neurol Disord. (2018) ;11: :1756286418803246. doi: 10.1177/1756286418803246 |
[96] | Wurster CD , Winter B , Wollinsky K , Ludolph AC , Uzelac Z , Witzel S , et al. Intrathecal administration of nusinersen in adolescent and adult SMA type 2 and 3 patients. J Neurol. (2019) ;266: :183–94. doi: 10.1007/s00415-018-9124-0 |
[97] | Oldenburg D , Guberina N , Stolte B , Kizina K , Stenzel E , Radbruch A , et al. Radiation exposure of image-guided intrathecal administration of nusinersen to adult patients with spinal muscular atrophy. Neuroradiology. (2019) ;61: :565–74. doi: 10.1007/s00234-019-02189-x |
[98] | Ko D , Blatt D , Karam C , Gupta K , Raslan AM . Lumbar laminotomy for the intrathecal administration of nusinersen for spinal muscular atrophy: technical note and outcomes. J Neurosurg Spine. (2019) :1–5. doi: 10.3171/2019.2.SPINE181366 |
[99] | Strauss KA , Carson VJ , Brigatti KW , Young M , Robinson DL , Hendrickson C , et al. Preliminary Safety and Tolerability of a Novel Subcutaneous Intrathecal Catheter System for Repeated Outpatient Dosing of Nusinersen to Children and Adults With Spinal Muscular Atrophy. J Pediatr Orthope. (2018) ;38: :610–7. doi: 10.1097/BPO.0000000000001247 |
[100] | Veerapandiyan A , Pal R , D’Ambrosio S , Young I , Eichinger K , Collins E , et al. Cervical puncture to deliver nusinersen in patients with spinal muscular atrophy. Neurology. (2018) ;91: :e620–4. doi: 10.1212/WNL.0000000000006006 |
[101] | Farrar MA , Park SB , Vucic S , Carey KA , Turner BJ , Gillingwater TH , et al. Emerging therapies and challenges in spinal muscular atrophy. Ann Neurol. (2017) ;81: :355–68. doi: 10.1002/ana.24864 |
[102] | Pechmann A , Kirschner J . Diagnosis and New Treatment Avenues in Spinal Muscular Atrophy. Neuropediatrics. (2017) ;48: :273–81. doi: 10.1055/s-0037-1603517 |

