
The Added Value of Cardiac Magnetic Resonance in Muscular Dystrophies
Abstract
Muscular dystrophies (MD) represent a heterogeneous group of rare genetic diseases that often lead to significant weakness due to progressive muscle degeneration. In many forms of MD, cardiac manifestations including heart failure, atrial and ventricular arrhythmias and conduction abnormalities can occur and may be a predominant feature of the disease. Cardiac magnetic resonance (CMR) can assess cardiac anatomy, global and regional ventricular function, volumes and mass as well as presence of myocardial inflammation, infiltration or fibrosis. The role for cardiac MRI has been well-established in a wide range of muscular dystrophies related cardiomyopathies. CMR is a more sensitive technique than echocardiography for early diagnosis of cardiac involvement. It has also great potential to improve the prediction of long-term outcome, particularly the development of heart failure and arrhythmic events; however it still has to be validated by longitudinal studies including large populations. This review will outline the utility of CMR in patients with muscular dystrophies for assessment of myocardial involvement, risk stratification, and in guiding therapeutic management.
INTRODUCTION
Muscular dystrophies (MD) represent a heterogeneous group of rare genetic diseases that often lead to significant weakness due to progressive muscle degeneration. In many forms of MD, cardiac manifestations including heart failure, atrial and ventricular arrhythmias and conduction abnormalities can occur and may be a predominant feature of the disease [1]. Patients with MD may have subclinical involvement for years prior to symptom onset and even otherwise asymptomatic genetic carriers of muscular dystrophies may develop cardiac manifestations [2]. Many patients with cardiac involvement are at higher risk for advanced heart failure and/or sudden cardiac death [3, 4]. It is important to identify early on markers of cardiac involvement to initiate cardio-protective therapies, to limit progression and to attenuate symptoms of heart failure [5, 6].
While echocardiography is widely used to assess cardiac structure and function given portability, low cost and availability, it has limitations in terms of satisfactory acoustic window, image quality and inter-observer variability [7, 8].
Cardiac magnetic resonance (CMR) is the non-invasive gold standard method for the assessment of ventricular volumes, mass and global systolic function due to its high reproducibility and low variability [9]. In addition, CMR gives information regarding tissue characterization, and has been used for over two decades in different populations with muscular dystrophies to assess for global and regional function, presence of fibrosis or fatty infiltration [10, 11]. CMR can also uncover early cardiac involvement when standard cardiac assessment appears unrevealing [12, 13]. This review will outline the utility of CMR in patients with muscular dystrophies for assessment of myocardial involvement, risk stratification and in guiding therapeutic management.
COMPLETE CARDIAC MAGNETIC RESONANCE EVALUATION
CMR provides anatomic and functional information, as well as tissue characterization in one scan. It has high spatial and temporal resolution and no ionizing radiation is used (Table 1). Basic sequences of black blood imaging with fast spin echo pulses can provide rapid anatomic information [14]. Cardiac function and ejection fraction measurement is assessed by bright blood cine imaging, mostly by steady-state free precession (SSFP) technique, which has higher contrast-to-noise and signal-to-noise ratio than gradient-echo (GRE) [14]. Post processing feature-tracking analysis from cine SSFP images can also provide myocardial deformation assessment with strain measurement [15].
Table 1
Cardiac MRI advantages and disadvantages
| Advantages | Disadvantages |
| Non-invasive | Acquisition can be long. Claustrophobia and inability to breath-hold are limiting factors. |
| No ionizing radiation | Contraindicated in patients with brain clips or ferromagnetic objects in the eyes. |
| No iodinated contrast. Gadolinium better tolerated and less nephrotoxic | Contraindicated if GFR * less than 30 mL/min/1.73 m2 given risk of NSF**. |
| Can assess anatomy, function, perfusion, viability, tissue characterization in one scan | Arrhythmias and poor breath-holding can reduce image quality and limit assessment |
| Good spatial and temporal resolution |
* Glomerular filtration rate.
** Nephrogenic systemic fibrosis.
Myocardial edema and inflammation can be assessed visually and semi-quantitatively by standard T2 weighted sequences and quantitatively with parametric T2 mapping.
With gadolinium contrast administration, the presence and extent of myocardial fibrosis can be assessed [16]. Gadolinium-based contrast agents distribute in the extracellular space and when there is cardiomyocyte damage and cell loss, gadolinium distribution will be of greater volume than in normal myocardium. Gadolinium is used usually with T1-weighted sequences and it decreases the tissue proton relaxation time, providing a hyperintense signal in the areas with fibrosis [17], that remains detectable for around 20–30 minutes after contrast has already washed out of normal myocardial segments.
The presence of late gadolinium enhancement (LGE) in most chronic conditions indicates myocardial fibrosis and the pattern of LGE can differentiate between an ischemic versus non-ischemic myocardial damage. Moreover, in non-ischemic cardiomyopathies, LGE pattern can help identify the type of cardiomyopathy [18].
Parametric T1 mapping techniques provide quantification of native and post-contrast T1 values as well as estimation of extracellular volume fraction (ECV). ECV measures the interstitial expansion that occurs due to diffuse myocardial processes - e.g. fibrosis or amyloid deposition, with gadolinium relative to the plasma. While changes in these values are not disease specific, they can serve as biomarkers in the context of specific scenarios, as they reflect alterations in myocardial tissue composition (Fig. 1). They can be abnormal in areas even before LGE is detected [19, 20].
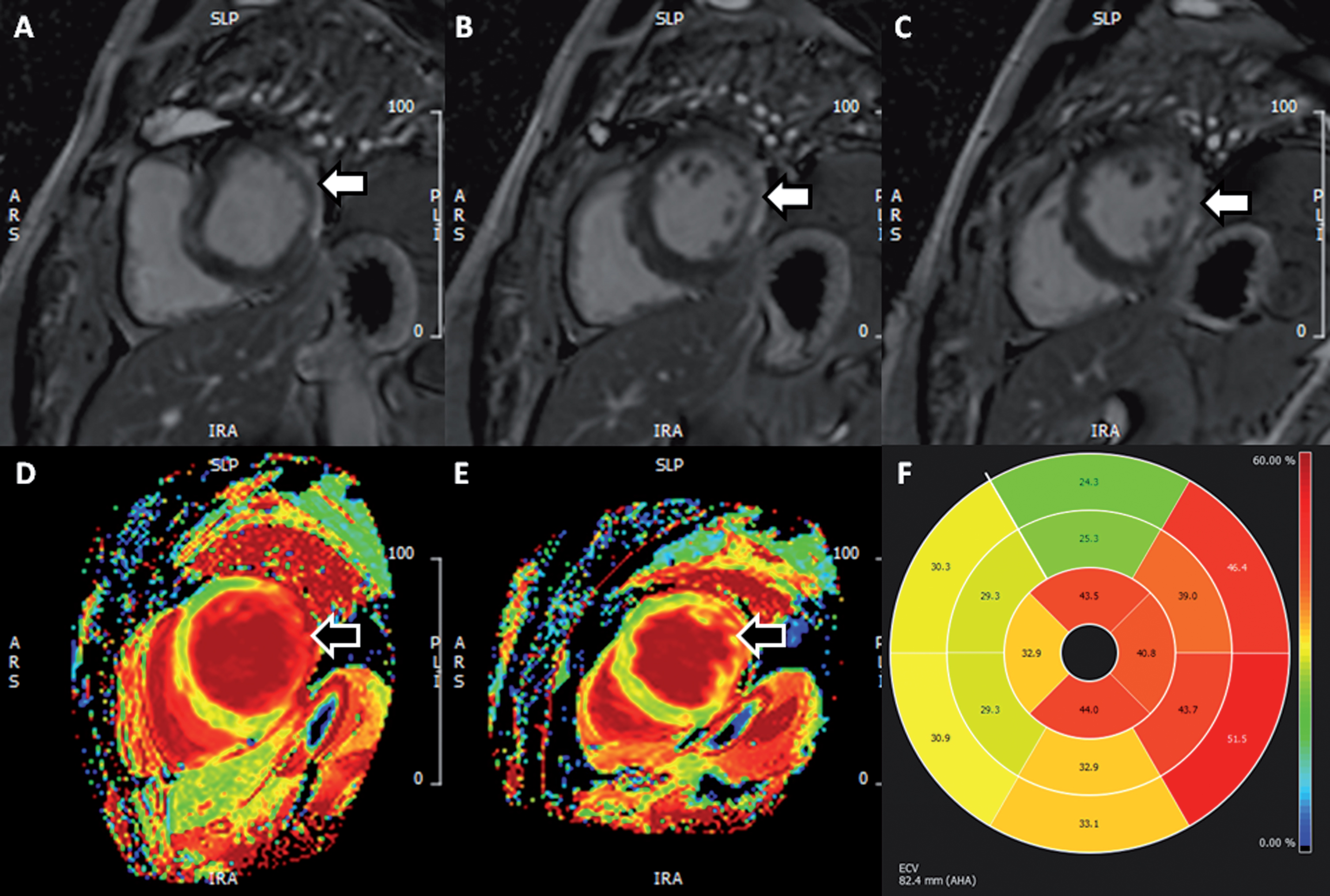
Fig. 1
Presence of scar in 53-year old man with myotonic dystrophy type 1. (A-C) Sequential basal and mid-ventricular slices in short-axis from LGE images demonstrating dense subepicardial scar in the left ventricular lateral wall (white arrows). (D-E) Matching basal and mid-ventricular ECV maps providing a qualitative impression of expanded extracellular space in this region (black arrows). (F) Polar plot of segmental ECV values – normal range 20–30%. Here we see significant elevations in ECV, not only in regions of dense scar (lateral wall) but also in the septal and inferior segments, suggesting the presence of diffuse fibrosis in these (LGE-free) segments. (LGE, late gadolinium enhancement; ECV, extracellular volume). Permission for submission granted.
A complete CMR scan with contrast takes usually around 40 min to be acquired. General contraindications for CMR are intracranial aneurysm clips and iron particles in the eye [21]. Claustrophobia is also a limitation for the scan as well the inability of breath holding, which can be an issue for some of these patients. CMR can present unique challenges given rapid and complex motion of the heart in addition to respiratory motion. However, this is usually compensated by electrocardiogram (ECG) gated images as well as breath-hold and motion correction techniques. Additionally, there exist sequences with real time free breathing or motion correction to facilitate acquisitions.
Tolerance of supine position should be systematically assessed beforehand, as it may be poorly tolerated in some patients with muscular dystrophy. In patients with restrictive respiratory insufficiency and particularly in those who are ventilated, MRI feasibility should be discussed with pneumologist and primary care physician and an alternative imaging modality might be considered. During the procedure, pulse oxygen saturation monitoring is recommended in patients with restrictive respiratory insufficiency and excessive oxygen administration should be avoided.
Regarding devices such as pacemaker and defibrillator, currently there are magnetic resonance imaging (MRI) conditional devices and patients can be safely scanned at 1.5T field strength, if followed specific recommendations from the device company [22, 23].
CMR IN MUSCULAR DYSTROPHY
In patients with cardiomyopathy related to muscular dystrophies, CMR can identify left and right ventricular dilatation, hypertrophy, presence of fatty infiltration, diffuse or focal fibrosis, and ventricular dysfunction [24–26].
Some patients with muscular dystrophies (i.e. myotonic dystrophy) can present mainly with arrhythmias and no structural abnormalities seen on echocardiogram. These patients can still have fibrosis identified on CMR, which can be a substrate for arrhythmias as well as provide prognostic information [10, 27, 28]. These patients can also have subclinical signs of ventricular dysfunction even with preserved left ventricular ejection fraction (LVEF) [26]. Strain analysis can help identify early regional dysfunction [29] (Fig. 2).
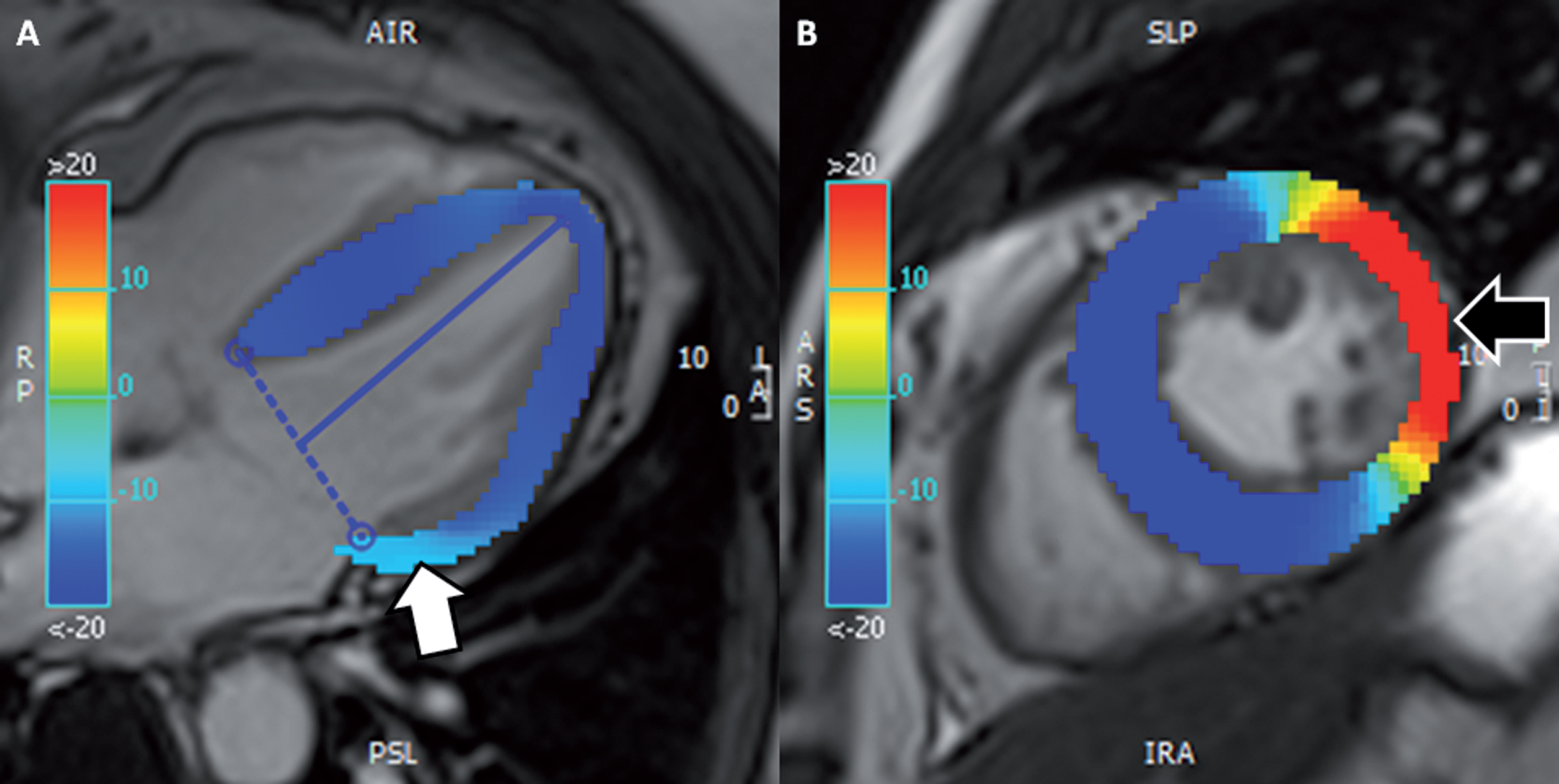
Fig. 2
(same patient as Fig. 1). CMR tissue tracking. (A) Longitudinal strain map in 4-chamber view cine demonstrating a reduction in regional longitudinal strain in the basal lateral wall, of – 10% (white arrow; normal values around – 20%). (B) Circumferential strain map of mid-ventricular short-axis cine demonstrating a more obvious reduction in deformation of around +10% (black arrow; normal values around – 20%). (CMR, cardiac magnetic resonance). Permission for submission granted.
DYSTROPHINOPATHIES: DUCHENNE AND BECKER, ISOLATED CARDIOMYOPATHY
DMD gene
More than 2000 mutations in the DMD gene have been identified in patients with either Duchenne or Becker muscular dystrophy. With 79 exons and 2.6 million base pairs of the genomic sequence, DMD-gene is the largest in the human genome, which is approximately 1.5% of the entire X chromosome [30]. Dystrophin is a sarcolemmal protein of skeletal and cardiac muscle cells, responsible for connecting the cytoskeleton with the extracellular matrix maintaining muscle integrity [31]. Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) are X-linked recessive disorders primarily affecting males with affected female carriers presenting with milder weakness or isolated cardiomyopathy [32]. Dystrophinopathies are caused by mutations in the dystrophin gene causing nearly absence in DMD and reduced or abnormal in BMD [33].
The mechanism thought to be responsible for cardiac involvement in both DMD and BMD is dysfunctional sarcolemma, stretch-activated channels and fragile membrane. That results in an increase influx of intracellular calcium, leading to impairment of myocyte contraction, mitochondrial deregulation, inflammation and eventually myocyte death [34]. The death of myocytes activates an inflammatory cascade that ultimately leads to fibrosis and scar. Increased fibrous deposition reduces normal heart contraction and function. Myocardial fibrosis can also lead to conduction defects and arrhythmias [34].
Cardiomyopathy is the primary cause of death in a high proportion of patients with BMD and DMD. The frequency of cardiac involvement can reach 60% to 75% in BMD [33, 35, 36] and almost every DMD patient will have some form of cardiac involvement after 18 years of age [1, 37]. Thus, prophylactic use of ACE-inhibitors (ACEi) is recommended by the age 10 years in DMD regardless to their cardiac function. Nevertheless, there are still unanswered questions regarding the optimal timing of initiation of cardio-protective therapies, e.g. ACEi, particularly in BMD patients. Cardiac MRI has the potential to help identify patients with early cardiac abnormalities in order to start on more aggressive therapeutic measures; however such approach has not been validated yet [36].
Randomized controlled trials have assessed ACEi in patients with DMD and showed slower progression of disease and lower mortality rate when treatment started early, while LVEF still normal [5, 24, 38–40]. A prospective study using this time CMR assessment of LGE and LVEF in patients with DMD or BMD under steroids treatment showed preserved LVEF and lack of LGE when treatment was associated with perindopril [41]. Another study compared lisinopril versus losartan (an angiotensin receptor blocker – ARB) in patients with DMD and recent diagnosis of heart involvement. It showed that there was no difference in terms of efficacy to improve or preserve LVEF [39].
A multicenter randomized placebo-controlled trial is ongoing to establish evidence of prophylactic use of ACEi and beta-blocker (BB) in DMD patients with normal LVEF [42].
A multicenter controlled trial added a mineralocorticoid receptor antagonist (MRA) in the treatment of DMD patients, treated with ACEi or ARB, with early cardiomyopathy (positive LGE) but preserved LVEF, compared with placebo. CMR demonstrated that patients treated with Eplerenone had slower rate of decline in LV circumferential strain and LVEF by CMR [43, 44]. Beta-blocker administration has also been studied in this population, usually added to ACEi or ARB and could help reduce arrhythmias and cardiac events [40, 42, 45].
The cardiac assessment with CMR has great implication on the diagnosis of subclinical and/or overt cardiomyopathy and can help identify patients that could benefit from therapy. Florian et al. (2014) studied the correlation between the cardiac phenotype of DMD and BMD patients based on CMR and genotype. Patients with deletions of exons 50/51 showed absence of late gadolinium enhancement (LGE) more frequently than patients with dystrophin duplications. The presence of LGE in this population is a sensitive early detection tool, as it usually precedes LV systolic dysfunction and the extent of fibrosis is related to the degree of dysfunction. These patients present a characteristic, myocarditis-like pattern of non-ischemic LGE starting in the subepicardium/mid-wall of the lateral/inferolateral wall and usually extending over years towards transmurality and to other myocardial segments (i.e. septum) (Table 2). Transmural LGE has also been observed in these patients and this correlates with lower LV ejection fraction (LVEF) and worse prognosis [36, 46]. In addition, in a retrospective study including DMD patients, more extensive LGE in the lateral wall and/or mid-wall LGE in septum were associated with a higher burden of ventricular arrhythmias and death [47]. The use of parametric mapping can also provide quantitative biomarkers related to fibrosis and inflammation, can depict subtle diffuse fibrosis in areas with normal appearing myocardial on LGE images and could potentially stratify disease severity [46, 48–50].
Table 2
Most common muscular dysthrophy genes associated with cardiomyopathy
| Gene | Phenotype | CMR LGE | Arrhythmias/ECG |
| DMD | Duchenne, Becker, Carrier (isolated cardiomyopathy) | Subepicardial to mid-myocardial, in the infero-/lateral wall | Sinus arrhythmias, atrial or ventricular arrhythmias, bundle branch block, AV conduction abnormalities with short or prolonged PR* |
| DMPK | Myotonic type 1 | Mid-myocardial of the septum and basal inferolateral wall | Atrial Flutter and atrial fibrillation, AV block, Bundle branch block |
| ZNF9 | Myotonic type 2 | Mid-myocardial of inferolateral wall | Atrial Flutter and atrial fibrillation, AV block, Bundle branch block** |
| LMNA | LGMD, EDMD, DCM, FPLD, L-CMD | Pronounced mid-myocardial, in thebasal- to mid-septal wall | Atrial or ventricular arrhythmias, AV block, Bundle branch block**, Sudden cardiac death |
| TTN | TMD, HMERF, LGMD, EDMD, DCM, HCM | Mid-myocardial | Atrial or ventricular arrhythmias |
* Please note arrhythmias are relatively rare in DMD and BMD and occur late in the disease course when compared to other muscular dystrophies.
** In Myotonic Distrophies and LMNA mutation, arrhythmias are dominant and pose importance.
Importantly, patients with cardiac involvement on CMR may have normal transthoracic echocardiograms. In a group of patients with DMD, 7 out of 10 of them presented LGE while only 2 had abnormal LVEF on echocardiogram [51]. Similarly, in another study, 73% of BMD patients displayed LGE and only 53% an impaired LVEF on echocardiography [52]. A more recent CMR study showed a correlation between LGE and progression of LV dysfunction in a large DMD cohort. LGE was present in 30% of patients with normal LVEF (>55%) reaching 84% in patients with LVEF <55% [53].
Isolated cardiomyopathy can occur in female carriers of the dystrophin gene mutations [54, 55]. A systematic review showed prevalence of dilated cardiomyopathy without muscle weakness in 7% to 16.7% of female carriers for DMD and up to 13% of BMD carriers. Most commonly these patients had deletions (54%), mostly located between exons 44 and 55. Some patients had duplications (16%), point mutations (17%), translocations (3%) or other mutations (7%). Eight large CMR studies assessed LGE and LVEF in female carriers. LV dysfunction was present in 14 to 40% of DMD carriers and 6% of BMD carriers. LGE was present in 35–65% of DMD carriers and 19–20% of BMD carriers. The pattern demonstrated is the same as male relatives and mostly in the inferolateral/lateral wall [32].
MYOTONIC DYSTROPHIES
DMPK gene
Myotonic Dystrophy type 1 (DM1) is caused by expansion of a CTG triplet repeat in the 3’ non-coding region of DMPK, the gene encoding the DM protein kinase in chromosome 19q13.3 [56]. Myotonic dystrophy type 1 (DM1) is one of the most common lethal monogenic disorders in populations of European descent and DM1 prevalence estimates in Europe ranged from 1 in 8,300 to 1 in 10,700 [57, 58]. The highest known prevalence has been reported in the French Canadian population (1 in 475) because of a founder effect [59–61].
Clinical severity of DM1 roughly correlates with the trinucleotide expansion of CTG-repeats with higher number of repeats associated with worsening severity of clinical manifestations [62]. DM1 is a multisystemic disease (myotonia, cataracts, diabetes, hypersomnolence, etc.). Cardiac involvement in DM1 primarily includes conduction abnormalities, such as first-degree and advanced atrioventricular blocks, right/left bundle branch blocks, and nonspecific intraventricular conduction delay [33]. Updated guidelines have recognized an earlier indication for permanent pacing in patients with DM1 when second or third degree AV block (class of recommendation (COR) I) or infrahissian conduction defects on electrophysiologic study with HV interval >70 ms, or first degree with PR interval >240 ms or QRS >120 ms or fascicular block (COR IIb) [6]. Atrial arrhythmias such as atrial fibrillation and atrial flutter and LV dysfunction are also common [63].
Patients with DM1 and cardiac involvement are at higher risk for sudden cardiac death and therefore determination of cardiac involvement is essential [64]. Cardiac MRI can demonstrate myocardial fibrosis even in the absence of conduction abnormalities on ECG [25]. A study with 80 DM1 patients showed that 16% of patients had abnormal CMR, out of those 39% with normal ECG. Of those 61% with abnormal ECG, 61% had abnormal CMR. Functional or structural abnormalities on cardiac MRI included LV systolic dysfunction (25%), LV dilatation (9%), LV hypertrophy (8%), RV (right ventricle) systolic dysfunction (5%) and RV dilatation (1%). Myocardial fibrosis was found in 13% of all patients, mostly as mid-myocardial LGE in the septum and basal inferolateral wall. Most patients with LGE had abnormal ECG [65]. The predictive value of CMR for arrhythmias and advanced LV dysfunction remains unknown due to lack of studies with long-term follow-up. An additional cohort analysis of 57 DM1 patients showed abnormal CMR in 44%, with non-ischemic LGE detected in 32%, with the same, above mentioned distribution. The presence of LGE was the only independent predictor for occurrence of atrial tachyarrhythmias and the authors suggest a more intensified monitoring for arrhythmias in these patients [28].
A recent research including DM1 patients highlights the high burden of subclinical cardiac involvement in asymptomatic patients without rest- or Holter ECG abnormalities. DM patients had significantly lower LVEF and strain values compared to controls, and LGE could be depicted in 29% of them [66]. The prognostic utility of these findings needs to be evaluated in future, follow-up studies.
CMR also can detect diffuse fibrosis with parametric mapping. Post-contrast myocardial T1 values were found to be lower than healthy controls, suggesting the presence of diffuse fibrosis even when focal fibrosis with LGE was not found - and was associated with PR and QRS interval progression during long-term follow-up [67].
ZNF9 gene
DM type 2 (DM2) is caused by a (CCTG)n expansion mutation in the first intron of zinc-finger protein 9 (ZNF9) in chromosome 3q21 [68, 69]. Patients with DM2 usually have more subtle presentation of symptoms and less cardiac involvement than DM1. However, DM2 can be also associated with atrioventricular and intraventricular conduction defects, arrhythmias, cardiomyopathy, and sudden cardiac death (SCD) [70].
CMR can demonstrate subclinical involvement of the myocardium in patients with DM2 and preserved LVEF [26]. Just as histopathological abnormalities in DM2 include fibrosis and fatty infiltration in the skeletal muscle, so too the autopsy data have shown the presence of fibrosis and fatty infiltration in the heart [71]. The pattern of LGE described is subepicardial in the basal inferolateral wall and increased native T1 values and extracellular volume were also demonstrated in the inferolateral segments [26].
As in DM1, a great number of patients can have cardiac abnormalities by CMR without ECG alterations [66]. Furthermore, specific CMR techniques can also detect fat infiltration beyond presence of fibrosis, which is an additional marker of cardiac disease [26].
LMNA gene
Genetic mutations in LMNA have been linked to dilated cardiomyopathy (DCM) with variable myopathy, limb girdle muscular dystrophy (LGMD), autosomal variant of Emery-Dreifuss muscular dystrophy (EDMD), LMNA-related congenital muscular dystrophy (L-CMD) and familial partial lipodystrophy (FPLD) [72]. Lamin A/C gene mutation (LMNA) is identified in up to 10% of familial DCM [73]. Male gender, LVEF ≤50% at first clinical contact and non-missense LMNA mutations have been identified as independent predictors of life threatening ventricular tachy-arrhythmias [3, 74]. These patients are at higher risk for SCD and this might be their initial clinical presentation. Most patients with LMNA mutations have a malignant long-term disease course and will experience atrial and/or ventricular arrhythmias, conduction blocks, embolic events or heart failure [75]. Moreover, a high percentage of patients will require the implantation of an antiarrhythmic device [75]. Cardiac manifestations can be isolated or associated with neuromuscular diseases [76].
CMR can detect focal and diffuse myocardial fibrosis in patients with pathogenic LMNA mutations prior to clinical symptoms or LV dysfunction. This, in turn, can predict functional and electrical abnormalities, as further shown. A mid-myocardial LGE pattern in the basal interventricular septum is a common and early CMR finding in patients with LMNA mutations, with similar distribution to fibrosis identified on autopsy from affected relatives [77, 78]. Positive LGE was correlated with diastolic dysfunction and these patients had significant left atrial enlargement when compared to controls [79]. In addition, Fontana et al. showed that LMNA carriers have increased ECV values compared to healthy controls, even in absence of LGE [73]. Septal myocardial fibrosis was associated with conduction abnormalities in asymptomatic or mildly symptomatic LMNA-mutation carriers [78]. Additionally, it has been correlated with reduced regional septal function by strain analysis, and to prolonged PR interval on ECG, which has been previously correlated with ventricular arrhythmias [80, 81].
TTN gene – cardiac phenotype
TTN has the longest coding sequence in the human gene (>100kb) with 364 exons and encodes the largest protein (titin) [82]. Patients with TTN mutations have various phenotypes (Table 2). Truncating titin variants (TTNtv) are the most prevalent genetic cause of dilated cardiomyopathy DCM [83]. TTN mutations can also cause core myopathies associated with primary cardiac disease. CMR identifies DCM with LV or biventricular dysfunction and diffuse LGE, as well as other forms of cardiac disease such as left ventricular non-compaction (LVNC) [84]. Patients with DCM associated with TTN mutations are more susceptible to ventricular arrhythmias and the presence of interstitial fibrosis [83].
MITOCHONDRIAL MYOPATHIES
Mitochondrial diseases are complex and heterogeneous disorders due to either nuclear DNA or mitochondrial DNA pathogenic gene mutations, leading to a decrease in oxidative phosphorylation and cellular energy (ATP) production. Increasing knowledge about molecular, biochemical, and genetic abnormalities related to mitochondrial dysfunction has expanded the cardiac imaging phenotypes of mitochondrial disorders. These disorders usually present with multi-organ involvement and have a slow progressive course. Cardiac involvement is mostly described as conduction disorders and arrhythmias but can also affect ventricular function and can present as dilated or hypertrophic cardiomyopathy [85]. Gadolinium-contrast enhanced CMR demonstrates presence of fibrosis by LGE as well as perfusion defects [86]. CMR-based phenotype patterns were also identified in different mitochondrial myopathy syndromes [87].
CONCLUSION
The role for cardiac MRI has been well-established in a wide range of muscular dystrophies related cardiomyopathies. CMR can provide a complete assessment of function, anatomy, perfusion and tissue characterization. CMR is a more sensitive technique than echocardiography for early diagnosis of cardiac involvement. It has also great potential to improve the prediction of long-term outcome, particularly the development of heart failure and arrhythmic events; however it still has to be validated by longitudinal studies including larger populations.
The pattern and extension of myocardial fibrosis as identified by LGE and with more sensitive techniques such as parametric mapping can assist with the diagnosis and provide prognostic information. During the same image acquisition, both global and regional function can be accurately assessed.
CMR is bound to play a future increasing role in clinical trials for patient selection with regard to their cardiac status and assessment of the efficacy of treatments on the heart.
Although CMR has class I indication for assessment in various cardiac diseases (i.e., ischemiccardiomyopathy, non-ischemic cardiomyopathy, valvular disease, congenital disease, pericardial disease, hypertrophic cardiomyopathy, heart failure, arrhythmogenic right ventricular cardiomyopathy, etc.) [88, 89], there are no current specific guidelines for the indications of cardiac MRI in the muscular dystrophy population. However, every patient with muscular dystrophy that is at risk for cardiac involvement should benefit from at least one diagnostic CMR scan to identify the extent of cardiac involvement and further follow up with an individualized frequency, according to baseline findings and further cardiac status (usually, in 1- to 3-year intervals). Although the gadolinium-based contrast is relatively safe from a nephrotoxicity perspective, being contraindicated only in patients with renal failure, specially when GFR is below 30 mL/min/1.73 m2 given the risk of nephrogenic systemic fibrosis [90], follow up CMR can also be done without contrast if there is no need to reassess scar. Diffuse fibrosis, as previously mention, can also be assessed contrast-free with native T1 mapping. CMR can be also combined with whole body MRI for diagnostic assessment of the muscular disease and natural history studies for clinical trials.
Particularly in dystrophinopathies, further randomized clinical trials using CMR as a tool for assessment of (early) cardiac involvement and response to therapies are needed. This could more clearly establish the optimal timing for initiation of early cardio-protective therapies with the purpose of delaying progression of cardiomyopathy, deterioration of ventricular function and, improving survival.
CONFLICTS OF INTEREST
None of the authors has any conflict of interest to declare.
REFERENCES
[1] | Beynon RP , Ray SG. Cardiac involvement in muscular dystrophies. QJM. (2008) ;101: (5):337–44. |
[2] | Adachi K , Hashiguchi S , Saito M , Kashiwagi S , Miyazaki T , Kawai H , et al. Detection and management of cardiomyopathy in female dystrophinopathy carriers. J Neurol Sci. (2018) ;386: :74–80. |
[3] | Wahbi K , Ben Yaou R , Gandjbakhch E , Anselme F , Gossios T , Lakdawala NK , et al. Development and validation of a new risk prediction score for life-threatening ventricular tachyarrhythmias in laminopathies. Circulation. (2019) . |
[4] | Van Ruiten HJ , Marini Bettolo C , Cheetham T , Eagle M , Lochmuller H , Straub V , et al. Why are some patients with Duchenne muscular dystrophy dying young: An analysis of causes of death in North East England. Eur J Paediatr Neurol. (2016) ;20: (6):904–9. |
[5] | Bourke JP , Bueser T , Quinlivan R. Interventions for preventing and treating cardiac complications in Duchenne and Becker muscular dystrophy and X-linked dilated cardiomyopathy. Cochrane Database Syst Rev. (2018) ;10: :CD009068. |
[6] | Kusumoto FM , Schoenfeld MH , Barrett C , Edgerton JR , Ellenbogen KA , Gold MR , et al. 2018 ACC/AHA/HRS guideline on the evaluation and management of patients with bradycardia and cardiac conduction delay. Circulation. (2018) :CIR0000000000000628. |
[7] | Malik SB , Chen N , Parker RA 3rd , Hsu JY. Transthoracic echocardiography: Pitfalls and limitations as delineated at cardiac CT and MR imaging-erratum. Radiographics. (2017) ;37: (3):1004. |
[8] | Thavendiranathan P , Popovic ZB , Flamm SD , Dahiya A , Grimm RA , Marwick TH. Improved interobserver variability and accuracy of echocardiographic visual left ventricular ejection fraction assessment through a self-directed learning program using cardiac magnetic resonance images. Journal of the American Society of Echocardiography: Official publication of the American Society of Echocardiography. (2013) ;26: (11):1267–73. |
[9] | Pattynama PM , De Roos A , Van der Wall EE , Van Voorthuisen AE. Evaluation of cardiac function with magnetic resonance imaging. American Heart Journal. (1994) ;128: (3):595–607. |
[10] | De Ambroggi L , Raisaro A , Marchiano V , Radice S , Meola G. Cardiac involvement in patients with myotonic dystrophy: Characteristic features of magnetic resonance imaging. European Heart Journal. (1995) ;16: (7):1007–10. |
[11] | Miyoshi K , Fujikawa K. Comparison of thallium-201 myocardial single-photon emission computed tomography and cine magnetic resonance imaging in Duchenne's muscular dystrophy. The American Journal of Cardiology. (1995) ;75: (17):1284–6. |
[12] | Blaszczyk E , Grieben U , von Knobelsdorff-Brenkenhoff F , Kellman P , Schmacht L , Funk S , et al. Subclinical myocardial injury in patients with Facioscapulohumeral muscular dystrophy 1 and preserved ejection fraction - assessment by cardiovascular magnetic resonance. J Cardiovasc Magn Reson. (2019) ;21: (1):25. |
[13] | Wexberg P , Avanzini M , Mascherbauer J , Pfaffenberger S , Freudenthaler B , Bittner R , et al. Myocardial late gadolinium enhancement is associated with clinical presentation in Duchenne muscular dystrophy carriers. J Cardiovasc Magn Reson. (2016) ;18: (1):61. |
[14] | Ginat DT , Fong MW , Tuttle DJ , Hobbs SK , Vyas RC. Cardiac imaging: Part 1, MR pulse sequences, imaging planes, and basic anatomy. AJR American Journal of Roentgenology. (2011) ;197: (4):808–15. |
[15] | Lamacie MM , Houbois CP , Greiser A , Jolly MP , Thavendiranathan P , Wintersperger BJ. Quantification of myocardial deformation by deformable registration-based analysis of cine MRI: Validation with tagged CMR. European Radiology. (2019) . |
[16] | Kim RJ , Wu E , Rafael A , Chen EL , Parker MA , Simonetti O , et al. The use of contrast-enhanced magnetic resonance imaging to identify reversible myocardial dysfunction. The New England Journal of Medicine. (2000) ;343: (20):1445–53. |
[17] | Muller W , Kramer G , Roder RG , Kuhnert A. Balance of T1-weighted images before and after application of a paramagnetic substance (Gd-DTPA). Neurosurg Rev. (1987) ;10: (2):117–22. |
[18] | Ordovas KG , Higgins CB. Delayed contrast enhancement on MR images of myocardium: Past, present, future. Radiology. (2011) ;261: (2):358–74. |
[19] | Messroghli DR , Moon JC , Ferreira VM , Grosse-Wortmann L , He T , Kellman P , et al. Clinical recommendations for cardiovascular magnetic resonance mapping of T1, T2, T2* and extracellular volume: A consensus statement by the Society for Cardiovascular Magnetic Resonance (SCMR) endorsed by the European Association for Cardiovascular Imaging (EACVI). Journal of Cardiovascular Magnetic Resonance: Official Journal of the Society for Cardiovascular Magnetic Resonance. (2017) ;19: (1):75. |
[20] | Puntmann VO , Peker E , Chandrashekhar Y , Nagel E. T1 mapping in characterizing myocardial disease: A comprehensive review. Circ Res. (2016) ;119: (2):277–99. |
[21] | Boxt LM. Cardiac MR imaging: A guide for the beginner. Radiographics. (1999) ;19: (4):1009–25; discussion 26-8. |
[22] | Nazarian S , Hansford R , Rahsepar AA , Weltin V , McVeigh D , Gucuk Ipek E , et al. Safety of magnetic resonance imaging in patients with cardiac devices. The New England Journal of Medicine. (2017) ;377: (26):2555–64. |
[23] | Bhuva AN , Kellman P , Graham A , Ramlall M , Boubertakh R , Feuchter P , et al. Clinical impact of cardiovascular magnetic resonance with optimized myocardial scar detection in patients with cardiac implantable devices. International Journal of Cardiology. (2019) ;279: :72–8. |
[24] | Duboc D , Meune C , Lerebours G , Devaux JY , Vaksmann G , Becane HM. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. Journal of the American College of Cardiology. (2005) ;45: (6):855–7. |
[25] | Petri H , Ahtarovski KA , Vejlstrup N , Vissing J , Witting N , Kober L , et al. Myocardial fibrosis in patients with myotonic dystrophy type 1: A cardiovascular magnetic resonance study. Journal of Cardiovascular Magnetic Resonance: Official Journal of the Society for Cardiovascular Magnetic Resonance. (2014) ;16: :59. |
[26] | Schmacht L , Traber J , Grieben U , Utz W , Dieringer MA , Kellman P , et al. Cardiac involvement in myotonic dystrophy type 2 patients with preserved ejection fraction: Detection by cardiovascular magnetic resonance. Circulation Cardiovascular Imaging. (2016) ;9: (7). |
[27] | Kono AK , Ishii K , Kumagai H , Taniguchi Y , Kajiya T , Sugimura K. Late gadolinium enhancement on cardiac magnetic resonance imaging: Is it associated with a higher incidence of nonsustained ventricular tachycardia in patients with idiopathic dilated cardiomyopathy? Jpn J Radiol. (2010) ;28: (5):355–61. |
[28] | Chmielewski L , Bietenbeck M , Patrascu A , Rosch S , Sechtem U , Yilmaz A , et al. Non-invasive evaluation of the relationship between electrical and structural cardiac abnormalities in patients with myotonic dystrophy type 1. Clin Res Cardiol. (2019) . |
[29] | Winther S , Williams LK , Keir M , Connelly KA , Bradley TJ , Rakowski H , et al. Cardiovascular magnetic resonance provides evidence of abnormal myocardial strain and primary cardiomyopathy in marfan syndrome. J Comput Assist Tomogr. (2019) . |
[30] | Kaspar RW , Allen HD , Montanaro F. Current understanding and management of dilated cardiomyopathy in Duchenne and Becker muscular dystrophy. J Am Acad Nurse Pract. (2009) ;21: (5):241–9. |
[31] | Blake DJ , Weir A , Newey SE , Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. (2002) ;82: (2):291–329. |
[32] | Ishizaki M , Kobayashi M , Adachi K , Matsumura T , Kimura E. Female dystrophinopathy: Review of current literature. Neuromuscul Disord. (2018) ;28: (7):572–81. |
[33] | Rajdev A , Groh WJ. Arrhythmias in the muscular dystrophies. Card Electrophysiol Clin. (2015) ;7: (2):303–8. |
[34] | van Westering TL , Betts CA , Wood MJ. Current understanding of molecular pathology and treatment of cardiomyopathy in duchenne muscular dystrophy. Molecules. (2015) ;20: (5):8823–55. |
[35] | Ho R , Nguyen ML , Mather P. Cardiomyopathy in becker muscular dystrophy: Overview. World Journal of Cardiology. (2016) ;8: (6):356–61. |
[36] | Florian A , Ludwig A , Engelen M , Waltenberger J , Rosch S , Sechtem U , et al. Left ventricular systolic function and the pattern of late-gadolinium-enhancement independently and additively predict adverse cardiac events in muscular dystrophy patients. Journal of Cardiovascular Magnetic Resonance: Official Journal of the Society for Cardiovascular Magnetic Resonance. (2014) ;16: :81. |
[37] | Cheeran D , Khan S , Khera R , Bhatt A , Garg S , Grodin JL , et al. Predictors of death in adults with duchenne muscular dystrophy-associated cardiomyopathy. J Am Heart Assoc. (2017) ;6: (10). |
[38] | Duboc D , Meune C , Pierre B , Wahbi K , Eymard B , Toutain A , et al. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years’ follow-up. American Heart Journal. (2007) ;154: (3):596–602. |
[39] | Allen HD , Flanigan KM , Thrush PT , Dvorchik I , Yin H , Canter C , et al. A randomized, double-blind trial of lisinopril and losartan for the treatment of cardiomyopathy in duchenne muscular dystrophy. PLoS Curr. (2013) ;5. |
[40] | Dittrich S , Graf E , Trollmann R , Neudorf U , Schara U , Heilmann A , et al. Effect and safety of treatment with ACE-inhibitor Enalapril and beta-blocker metoprolol on the onset of left ventricular dysfunction in Duchenne muscular dystrophy - a randomized, double-blind, placebo-controlled trial. Orphanet J Rare Dis. (2019) ;14: (1):105. |
[41] | Mavrogeni S , Giannakopoulou A , Papavasiliou A , Markousis-Mavrogenis G , Pons R , Karanasios E , et al. Cardiac profile of asymptomatic children with Becker and Duchenne muscular dystrophy under treatment with steroids and with/without perindopril. BMC Cardiovasc Disord. (2017) ;17: (1):197. |
[42] | Bourke JP , Watson G , Muntoni F , Spinty S , Roper H , Guglieri M , et al. Randomised placebo-controlled trial of combination ACE inhibitor and beta-blocker therapy to prevent cardiomyopathy in children with Duchenne muscular dystrophy? (DMD Heart Protection Study): A protocol study. BMJ Open. (2018) ;8: (12):e022572. |
[43] | Raman SV , Hor KN , Mazur W , Halnon NJ , Kissel JT , He X , et al. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. (2015) ;14: (2):153–61. |
[44] | Raman SV , Hor KN , Mazur W , He X , Kissel JT , Smart S , et al. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: Results of a two-year open-label extension trial. Orphanet J Rare Dis. (2017) ;12: (1):39. |
[45] | Matsumura T. Beta-blockers in children with duchenne cardiomyopathy. Rev Recent Clin Trials. (2014) ;9: (2):76–81. |
[46] | Marty B , Gilles R , Toussaint M , Behin A , Stojkovic T , Eymard B , et al. Comprehensive evaluation of structural and functional myocardial impairments in Becker muscular dystrophy using quantitative cardiac magnetic resonance imaging. European Heart Journal Cardiovascular Imaging. (2018) . |
[47] | Menon SC , Etheridge SP , Liesemer KN , Williams RV , Bardsley T , Heywood MC , et al. Predictive value of myocardial delayed enhancement in Duchenne muscular dystrophy. Pediatr Cardiol. (2014) ;35: (7):1279–85. |
[48] | Toussaint M GR , Greiser A , Marty B , Carlier P , Wahbi K. Myocardial characterisation in Becker muscular dystrophy using T1 and T2 mapping. Journal of Cardiovascular Magnetic Resonance: Official Journal of the Society for Cardiovascular Magnetic Resonance. (2015) ;17: ((Suppl 1)):P267. |
[49] | Florian A , Ludwig A , Rosch S , Yildiz H , Sechtem U , Yilmaz A. Myocardial fibrosis imaging based on T1-mapping and extracellular volume fraction (ECV) measurement in muscular dystrophy patients: Diagnostic value compared with conventional late gadolinium enhancement (LGE) imaging. European Heart Journal Cardiovascular Imaging. (2014) ;15: (9):1004–12. |
[50] | Wansapura JP , Hor KN , Mazur W , Fleck R , Hagenbuch S , Benson DW , et al. Left ventricular T2 distribution in Duchenne muscular dystrophy. Journal of Cardiovascular Magnetic Resonance: Official Journal of the Society for Cardiovascular Magnetic Resonance. (2010) ;12: :14. |
[51] | Silva MC , Meira ZM , Gurgel Giannetti J , da Silva MM , Campos AF , Barbosa Mde M , et al. Myocardial delayed enhancement by magnetic resonance imaging in patients with muscular dystrophy. Journal of the American College of Cardiology. (2007) ;49: (18):1874–9. |
[52] | Yilmaz A , Gdynia HJ , Baccouche H , Mahrholdt H , Meinhardt G , Basso C , et al. Cardiac involvement in patients with Becker muscular dystrophy: New diagnostic and pathophysiological insights by a CMR approach. Journal of Cardiovascular Magnetic Resonance: Official Journal of the Society for Cardiovascular Magnetic Resonance. (2008) ;10: :50. |
[53] | Hor KN , Taylor MD , Al-Khalidi HR , Cripe LH , Raman SV , Jefferies JL , et al. Prevalence and distribution of late gadolinium enhancement in a large population of patients with Duchenne muscular dystrophy: Effect of age and left ventricular systolic function. Journal of Cardiovascular Magnetic Resonance: Official Journal of the Society for Cardiovascular Magnetic Resonance. (2013) ;15: :107. |
[54] | Florian A , Ludwig A , Ong P , Klingel K , Kandolf R , Bornemann A , et al. Cause of cardiac disease in a female carrier of Duchenne muscular dystrophy: Myocarditis versus genetic cardiomyopathy without skeletal myopathy? Circulation. 2014;129: (18):e482–4. |
[55] | Florian A , Rosch S , Bietenbeck M , Engelen M , Stypmann J , Waltenberger J , et al. Cardiac involvement in female Duchenne and Becker muscular dystrophy carriers in comparison to their first-degree male relatives: A comparative cardiovascular magnetic resonance study. European Heart Journal Cardiovascular Imaging. (2016) ;17: (3):326–33. |
[56] | Mahadevan M , Tsilfidis C , Sabourin L , Shutler G , Amemiya C , Jansen G , et al. Myotonic dystrophy mutation: An unstable CTG repeat in the 3’ untranslated region of the gene. Science. (1992) ;255: (5049):1253–5. |
[57] | Siciliano G , Manca M , Gennarelli M , Angelini C , Rocchi A , Iudice A , et al. Epidemiology of myotonic dystrophy in Italy: Re-apprisal after genetic diagnosis. Clin Genet. (2001) ;59: (5):344–9. |
[58] | Magee A , Nevin NC. The epidemiology of myotonic dystrophy in Northern Ireland. Community Genet. (1999) ;2: (4):179–83. |
[59] | Pratte A , Prevost C , Puymirat J , Mathieu J. Anticipation in myotonic dystrophy type 1 parents with small CTG expansions. Am J Med Genet A. (2015) ;167A: (4):708–14. |
[60] | Wenninger S , Montagnese F , Schoser B. Core clinical phenotypes in myotonic dystrophies. Front Neurol. (2018) ;9: :303. |
[61] | Bouchard G , Roy R , Declos M , Mathieu J , Kouladjian K. Origin and diffusion of the myotonic dystrophy gene in the Saguenay region (Quebec). Can J Neurol Sci. (1989) ;16: (1):119–22. |
[62] | Chong-Nguyen C , Wahbi K , Algalarrondo V , Becane HM , Radvanyi-Hoffman H , Arnaud P , et al. Association between mutation size and cardiac involvement in myotonic dystrophy type 1: An analysis of the DM1-heart registry. Circ Cardiovasc Genet. (2017) ;10: (3). |
[63] | Wahbi K , Babuty D , Probst V , Wissocque L , Labombarda F , Porcher R , et al. Incidence and predictors of sudden death, major conduction defects and sustained ventricular tachyarrhythmias in 1388 patients with myotonic dystrophy type 1. European Heart Journal. (2017) ;38: (10):751–8. |
[64] | Wahbi K , Meune C , Porcher R , Becane HM , Lazarus A , Laforet P , et al. Electrophysiological study with prophylactic pacing and survival in adults with myotonic dystrophy and conduction system disease. Jama. (2012) ;307: (12):1292–301. |
[65] | Hermans MC , Faber CG , Bekkers SC , de Die-Smulders CE , Gerrits MM , Merkies IS , et al. Structural and functional cardiac changes in myotonic dystrophy type 1: A cardiovascular magnetic resonance study. Journal of Cardiovascular Magnetic Resonance: Official Journal of the Society for Cardiovascular Magnetic Resonance. (2012) ;14: :48. |
[66] | Luetkens JA , von Landenberg C , Isaak A , Faron A , Kuetting D , Gliem C , et al. Comprehensive cardiac magnetic resonance for assessment of cardiac involvement in myotonic muscular dystrophy type 1 and 2 without known cardiovascular disease. Circulation Cardiovascular Imaging. (2019) ;12: (6):e009100. |
[67] | Turkbey EB , Gai N , Lima JA , van der Geest RJ , Wagner KR , Tomaselli GF , et al. Assessment of cardiac involvement in myotonic muscular dystrophy by T1 mapping on magnetic resonance imaging. Heart Rhythm. (2012) ;9: (10):1691–7. |
[68] | Bachinski LL , Udd B , Meola G , Sansone V , Bassez G , Eymard B , et al. Confirmation of the type 2 myotonic dystrophy (CCTG)n expansion mutation in patients with proximal myotonic myopathy/proximal myotonic dystrophy of different European origins: A single shared haplotype indicates an ancestral founder effect. Am J Hum Genet. (2003) ;73: (4):835–48. |
[69] | Liquori CL , Ricker K , Moseley ML , Jacobsen JF , Kress W , Naylor SL , et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. (2001) ;293: (5531):864–7. |
[70] | Wahbi K , Meune C , Becane HM , Laforet P , Bassez G , Lazarus A , et al. Left ventricular dysfunction and cardiac arrhythmias are frequent in type 2 myotonic dystrophy: A case control study. Neuromuscul Disord. (2009) ;19: (7):468–72. |
[71] | Nguyen HH , Wolfe JT 3rd , Holmes DR Jr , Edwards WD. Pathology of the cardiac conduction system in myotonic dystrophy: A study of 12 cases. Journal of the American College of Cardiology. (1988) ;11: (3):662–71. |
[72] | Arbustini E , Pilotto A , Repetto A , Grasso M , Negri A , Diegoli M , et al. Autosomal dominant dilated cardiomyopathy with atrioventricular block: A lamin A/C defect-related disease. Journal of the American College of Cardiology. (2002) ;39: (6):981–90. |
[73] | Fontana M , Barison A , Botto N , Panchetti L , Ricci G , Milanesi M , et al. CMR-verified interstitial myocardial fibrosis as a marker of subclinical cardiac involvement in LMNA mutation carriers. JACC Cardiovascular imaging. (2013) ;6: (1):124–6. |
[74] | van Rijsingen IA , Arbustini E , Elliott PM , Mogensen J , Hermans-van Ast JF , van der Kooi AJ , et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. Journal of the American College of Cardiology. (2012) ;59: (5):493–500. |
[75] | Kumar S , Baldinger SH , Gandjbakhch E , Maury P , Sellal JM , Androulakis AF , et al. Long-term arrhythmic and nonarrhythmic outcomes of lamin A/C mutation carriers. Journal of the American College of Cardiology. (2016) ;68: (21):2299–307. |
[76] | Boriani G BE , Wahbi K , Duboc D. Cardiac involvement in laminopathies. Orphanet J Rare Dis. (2015) . |
[77] | Captur G , Arbustini E , Bonne G , Syrris P , Mills K , Wahbi K , et al. Lamin and the heart. Heart. (2018) ;104: (6):468–79. |
[78] | Holmstrom M , Kivisto S , Helio T , Jurkko R , Kaartinen M , Antila M , et al. Late gadolinium enhanced cardiovascular magnetic resonance of lamin A/C gene mutation related dilated cardiomyopathy. Journal of Cardiovascular Magnetic Resonance: Official Journal of the Society for Cardiovascular Magnetic Resonance. (2011) ;13: :30. |
[79] | Raman SV , Sparks EA , Baker PM , McCarthy B , Wooley CF. Mid-myocardial fibrosis by cardiac magnetic resonance in patients with lamin A/C cardiomyopathy: Possible substrate for diastolic dysfunction. Journal of Cardiovascular Magnetic Resonance: Official Journal of the Society for Cardiovascular Magnetic Resonance. (2007) ;9: (6):907–13. |
[80] | Hasselberg NE , Edvardsen T , Petri H , Berge KE , Leren TP , Bundgaard H , et al. Risk prediction of ventricular arrhythmias and myocardial function in Lamin A/C mutation positive subjects. Europace. (2014) ;16: (4):563–71. |
[81] | Haugaa KH , Hasselberg NE , Edvardsen T. Mechanical dispersion by strain echocardiography: A predictor of ventricular arrhythmias in subjects with lamin A/C mutations. JACC Cardiovascular Imaging. (2015) ;8: (1):104–6. |
[82] | Oates EC , Jones KJ , Donkervoort S , Charlton A , Brammah S , Smith JE 3rd , et al. Congenital Titinopathy: Comprehensive characterization and pathogenic insights. Ann Neurol. (2018) ;83: (6):1105–24. |
[83] | Verdonschot JAJ , Hazebroek MR , Derks KWJ , Barandiaran Aizpurua A , Merken JJ , Wang P , et al. Titin cardiomyopathy leads to altered mitochondrial energetics, increased fibrosis and long-term life-threatening arrhythmias. European Heart Journal. (2018) ;39: (10):864–73. |
[84] | Chauveau C , Bonnemann CG , Julien C , Kho AL , Marks H , Talim B , et al. Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum Mol Genet. (2014) ;23: (4):980–91. |
[85] | Kuno T , Imaeda S , Asakawa Y , Nakamura H , Takemura G , Asahara D , et al. Mitochondrial cardiomyopathy presenting as dilated phase of hypertrophic cardiomyopathy diagnosed with histological and genetic analyses. Case Rep Cardiol. (2017) ;2017: :9473917. |
[86] | Nakanishi M , Harada M , Tadamura E , Kotani H , Kawakami R , Kuwahara K , et al. Images in cardiovascular medicine. Mitochondrial cardiomyopathy evaluated with cardiac magnetic resonance. Circulation. (2007) ;116: (2):e25–6. |
[87] | Florian A , Ludwig A , Stubbe-Drager B , Boentert M , Young P , Waltenberger J , et al. Characteristic cardiac phenotypes are detected by cardiovascular magnetic resonance in patients with different clinical phenotypes and genotypes of mitochondrial myopathy. Journal of Cardiovascular Magnetic Resonance: Official Journal of the Society for Cardiovascular Magnetic Resonance. (2015) ;17: :40. |
[88] | von Knobelsdorff-Brenkenhoff F , Pilz G , Schulz-Menger J. Representation of cardiovascular magnetic resonance in the AHA / ACC guidelines. Journal of Cardiovascular Magnetic Resonance: Official Journal of the Society for Cardiovascular Magnetic Resonance. (2017) ;19: (1):70. |
[89] | von Knobelsdorff-Brenkenhoff F , Schulz-Menger J. Role of cardiovascular magnetic resonance in the guidelines of the European Society of Cardiology. Journal of Cardiovascular Magnetic Resonance: Official Journal of the Society for Cardiovascular Magnetic Resonance. (2016) ;18: :6. |
[90] | Ramalho M , Ramalho J , Burke LM , Semelka RC. Gadolinium retention and toxicity-an update. Adv Chronic Kidney Dis. (2017) ;24: (3):138–46. |


