
A Novel Mutation in MARS in a Patient with Charcot-Marie-Tooth Disease, Axonal, Type 2U with Congenital Onset
Abstract
Charcot-Marie-Tooth disease is a phenotypically and genetically heterogeneous group of disorders affecting both motor and sensory neurons. Exome sequencing has driven discovery of genes responsible for Charcot-Marie-Tooth disease with more than 70 genes now associated with this neuromuscular disease. The MARS gene was recently reported as the cause of Charcot-Marie-Tooth 2U, a slowly progressive axonal sensorimotor polyneuropathy with adult-onset reported in six patients. We report here a patient with a progressive, early childhood-onset, motor-predominant form of Charcot-Marie-Tooth disease. Exome sequencing identified a novel MARS variant (c.1189G>A; p.Ala397Thr) that was not present in her unaffected mother; her unaffected father was unavailable. Further studies using structural modeling and a yeast humanization assay support pathogenicity of the variant. Our study expands the phenotype of Charcot-Marie-Tooth 2U, while highlighting the utility of functional assays to evaluate variant pathogenicity.
INTRODUCTION
Charcot-Marie-Tooth (CMT) disease is a genetically and phenotypically heterogeneous group of inherited peripheral neuropathies that affects motor and sensory nerves. CMT can present from birth to late adulthood and varies in rate of disease progression [1]. Charcot-Marie-Tooth disease type 2 (CMT2) is an axonal sensorimotor polyneuropathy with an autosomal dominant inheritance pattern.
More than 70 genes have been linked to CMT, including seven genes belonging to the family of aminoacyl-tRNA synthetases (ARS) [2–6]. The enzymes encoded by the ARS genes link specific tRNAs to their corresponding amino acids. CMT phenotypes caused by mutation of ARS genes include: KARS and YARS which result in a sensorimotor polyneuropathy and; AARS, HARS, GARS, and WARS that give rise to a motor-predominant CMT [7–9].
Mutations in methionyl-tRNA synthetase (MARS) have been identified as the cause of CMT type 2U (CMT2U) [10]. The initial family had two affected members who presented with adult-onset distal muscle wasting and lower extremity weakness [10]. Subsequent studies have reported four additional patients from three families with CMT2U and MARS variants [2, 11, 12]. However, given the small number of patients reported to date and incomplete genetic evidence for the role of MARS in neuropathy, additional clinical, functional, and genetic characterization is required to gain a full appreciation of MARS related neuropathies. Herein, we report a patient with early-onset progressive motor neuropathy secondary to a novel mutation in MARS, c.1189G>A, p.Ala397Thr.
METHODS
Exome capture and high-throughput sequencing was performed at McGill University and Genome Québec Innovation Centre (Montréal, Canada) from total genomic DNA extracted from blood of the patient and her mother. Exonic DNA was selected using the SureSelect 50Mb (V5) All ExonKit (Agilent, Santa Clara, California). Sequencing (Illumina HiSeq 2000 Systems, San Diego, California) generated approximately 6 Gbp of 100 bp, paired-end reads per sample. Read alignment, variant calling [single nucleotide variants (SNV) and insertions/deletions], and annotation were performed as outlined for previous FORGE and Care4Rare Canada projects (additional information on variant filtering available in supplementary materials) [13]. The mean sequencing coverage was 122x with 97.5% of bases in the consensus coding sequences (CCDS) covered by at least 20 reads. PCR amplification followed by bi-directional Sanger sequencing was used to validate the identified mutation and its absence in the patient’s mother. The patient’s father was not available for testing; there were no SNPs in proximity of the variant to facilitate a haplotype analysis.
Yeast complementation assays to study the functional consequences of MARS variants were performed as previously described [10]. The p.Ala397Thr missense variant was modeled in the human MARS open-reading frame (primers available upon request) (additional information on clone selection methods available in supplementary materials). Four independent transformations were performed, using one of four independently generated expression clones (1–4 in Fig. 2). One colony from each transformation was selected for analysis and grown to saturation for two days at 37°C in liquid media lacking uracil and leucine. A 10μl aliquot of each culture was spotted undiluted or diluted 1:10, 1:100, or 1:1,000 in H2O, plated on medium containing 0.1% 5-FOA (Teknova), and incubated at 30°C for 72 hours. Survival was determined by visual inspection of growth.
The structure of the human methionyl-tRNA synthetase (MARS) complexed with methionine and bound to two zinc ions has been reported by X-ray crystallography at 2.278 Å and deposited in the Protein Data Bank (PDB IDs: 5GOY and 5GL7) [14]. The coordinate files were downloaded and 5GOY was used as template to model the structure of the MARS variants relevant to CMT (Ala397Thr, Arg618Cys and Pro800Thr) using Modeller 9.19 (homology modeling) [15]. Modeller DOPE score was used to rank models and the best predicted model was selected for further refinement. Coot (version 0.8.6.1) [16] was used to manually refine the models, fix steric clashes, improper angles and other modeling issues. Prosa-web [17] and Ramachandran plot analysis module of Coot was used to validate the quality of the models [19]. Intramolecular contacts were calculated using Arpeggio [18]. Duet was used to calculate protein structure stability changes upon mutation [19]. Images were prepared using PyMOL (https://pymol.org/2/).
CASE REPORT
An 11 year-old girl was first noted to have gross motor difficulties in her first year of life. She began to roll at 7 months sat independently at 11 months and walked at 2 years of age. Her gross motor development plateaued at 2.5 years of age. She was never able to run and fell frequently. Her birth and family history were unremarkable.
At 4 years of age she would walk but could not rise up on her heels or toes. Cranial nerve examination was normal. Distal muscle wasting was noted and her Gower maneuver was positive. Deep tendon reflexes were absent. Sensory testing was unreliable due to her age. Serum creatine kinase was 139 U/L (normal<175 U/L). Nerve conduction studies (Supplemental Table 1) were consistent with a diffuse motor neuropathy or neuronopathy. Her right median and bilateral sural nerve sensory responses were intact; however, all motor responses showed reduced or absent amplitudes. Needle EMG revealed abundant fibrillation potentials and positive sharp waves in her right iliopsoas and tibialis anterior muscles with large, rapidly-firing motor units observed.
Genetic testing for SMA, distal-SMA type 4, and a next-generation-sequencing panel of CMT genes (MFN2, RAB7A, LMNA, MPZ, HSPB1, NEFL, GADP1, TRPV4, HSPB8 and GJB1) was unremarkable. Muscle biopsy at 6 years of age revealed neurogenic atrophy. MRI brain and spine were unremarkable.
By 9 years of age she had further decline in her strength. If placed in a standing position she could take several hesitant steps with her knees locked. She was unable to rise from a seated to standing position. Cranial nerves remained intact. Muscle strength revealed (MRC scale): biceps 3, triceps 3, abductor pollicis brevis 0, iliopsoas 4+, tibialis anterior 2, extensor hallicus longus 0. Sensory testing noted a mild decrease in vibration and cold sensation in her lower extremities. By 11 years of age she was non-ambulatory. She could no longer feed or dress independently. She remained cognitively normal and had no weakness of her bulbar, facial or extraocular muscles.
Due to the lack of a molecular diagnosis the patient was enrolled in the Care4Rare Canada research study for exome sequencing. Ethics approval was received from the Children’s Hospital of Eastern Ontario Research Ethics Board (REB Protocol#11/04E) and consent was obtained from the patient’s mother.
RESULTS
Duo exome sequencing was performed for the patient and her mother, as the patient’s father was unavailable. We first assessed for variants in known recessive disease genes: no variants were identified which could explain the patient’s condition. Next, we assessed rare heterozygous variants in known disease genes and identified MARS (NM_004990.3) c.1189G>A, p.Ala397Thr. This variant has never been observed in affected individuals or control databases (EVS, 1000Genomes, ExAC, gnomAD). The p.Ala397 residue is highly conserved (down to yeast) and in silico analyses (SIFT (0), CADD (36), and PolyPhen-2 (0.99)) all predict this change to be deleterious. Sanger sequencing confirmed the presence of the variant in the patient and its absence in her mother. Given the previous association of mutations in MARS with CMT, the deleterious-appearing nature of the p.Ala397Thr, the unavailability of the patient’s father for testing, and that no other changes in neuropathy associated genes were identified we set out to further evaluate the functional consequences using structural modeling and a yeast complementation assay.
To investigate the effect of the p.Ala397Thr variant identified in this study, as well as the previously identified CMT-associated variants p.Arg618Cys and p.Pro800Thr, we modeled their tridimensional structure based on the crystallographic structure of the human MARS protein (PDB ID 5GOY) (Fig. 1A) [20]. The residues p.Ala397 and p.Arg618 are invariant and the p.Pro800 is conserved in most organisms, including plants (Fig. 1B). Ala397 is located within the catalytic domain, adjacent to one of the zinc fingers (Fig. 1A), likely important for the linker and/or zinc finger motif structure and, therefore, to MARS catalytic activity. Substitution to threonine can alter the network of contacts established by residue 397, particularly with regard to water interactions and novel hydrogen bonds (Fig. 1C-D). Arg618 is completely buried in the core of the helical domain, adjacent to the predicted tRNA-binding sites, and its replacement by a cysteine residue abolishes most of the important contact network of the all-helical domain, especially hydrogen bonds and dipole interactions (Fig. 1E-F). Pro800 is an exposed residue located on the helical domain, facing the predicted position of the anticodon (Fig. 1A). A structurally adjacent residue to p.Pro800, p.Ser662 has been shown to be directly implicated in tRNA-Met binding [21]. A hydrogen bond established between p.Pro800 and p.Asp655 (Fig. 1G) is lost in the p.Pro800T model (Fig. 1H) which may impact the proper orientation of the adjacent key anticodon recognition residues.
Fig.1
Structural studies of CMT-associated missense variants. (A) Structure of the human MARS protein (PDB ID 5goy) complexed with methionine (green) and docked with a tRNA molecule. Residues Ala 397, Arg 618 and Pro 800, found mutated in CMT affected individuals, are shown in yellow spheres. (B) Sequence alignment showing conservation of residues 397, 618 and 800. Internal contacts calculated using the software Arpeggio for each residue are shown: (C) A397, (D) T397, (E) R618, (F) C618, (G) P800, (H) T800. Contacts in C-H are shown in dotted lines: black lines indicate hydrophobic interactions (short distance, <5 Å), red show hydrogen bonds, yellow polar interactions and grey carbon-π-aromatic interactions. The catalytic domain of MARS, as well as of other class I aminoacyl-tRNA synthetases, is composed by a Rossmann fold domain and a connective polypeptide (CP) domain formed by β ribbon with two zinc fingers (A) [20]. The tRNA binding is established in an all-helical domain in the C-terminal portion of the protein (A). An extrapolation of the predicted tRNA binding position is shown (A), based on superimposition with tRNA-bound Ile synthetase (1FFY).
![Structural studies of CMT-associated missense variants. (A) Structure of the human MARS protein (PDB ID 5goy) complexed with methionine (green) and docked with a tRNA molecule. Residues Ala 397, Arg 618 and Pro 800, found mutated in CMT affected individuals, are shown in yellow spheres. (B) Sequence alignment showing conservation of residues 397, 618 and 800. Internal contacts calculated using the software Arpeggio for each residue are shown: (C) A397, (D) T397, (E) R618, (F) C618, (G) P800, (H) T800. Contacts in C-H are shown in dotted lines: black lines indicate hydrophobic interactions (short distance, <5 Å), red show hydrogen bonds, yellow polar interactions and grey carbon-π-aromatic interactions. The catalytic domain of MARS, as well as of other class I aminoacyl-tRNA synthetases, is composed by a Rossmann fold domain and a connective polypeptide (CP) domain formed by β ribbon with two zinc fingers (A) [20]. The tRNA binding is established in an all-helical domain in the C-terminal portion of the protein (A). An extrapolation of the predicted tRNA binding position is shown (A), based on superimposition with tRNA-bound Ile synthetase (1FFY).](https://content.iospress.com:443/media/jnd/2019/6-3/jnd-6-3-jnd190404/jnd-6-jnd190404-g001.jpg)
Fig.2
The p.Ala397Thr MARS allele displays a loss-of-function effect in yeast complementation studies. Yeast lacking endogenous Mes1 (the yeast ortholog of MARS) were transformed with vectors containing wild-type or p.Ala397Thr MARS, or a vector with no MARS insert (‘Empty’). Four independent p.Ala397Thr MARS expression constructs were transformed, grown in selective media, and plated undiluted or diluted (1 : 10, 1 : 100, or 1 : 1,000) on media containing 5-FOA. This system allows for assessment of the ability of experimental alleles to support yeast cell growth in vivo (in the absence of the yeast MARS ortholog, Mes1). The haploid yeast strain deleted for the endogenous Mes1 locus was transformed with a pRS315 experimental vector containing the LEU2 gene and either: (1) no MARS insert (‘Empty’); (2) wild-type MARS; or (3) p.Ala397Thr MARS. Wild-type MARS supported robust cellular growth, while the vector with no MARS insert did not. These data indicate that the experimental vector harbors a functional wild-type copy of MARS and that Mes1 is an essential gene, respectively. In contrast, p.Ala397Thr MARS supported very little yeast cell growth, consistent with this being a loss-of-function allele.
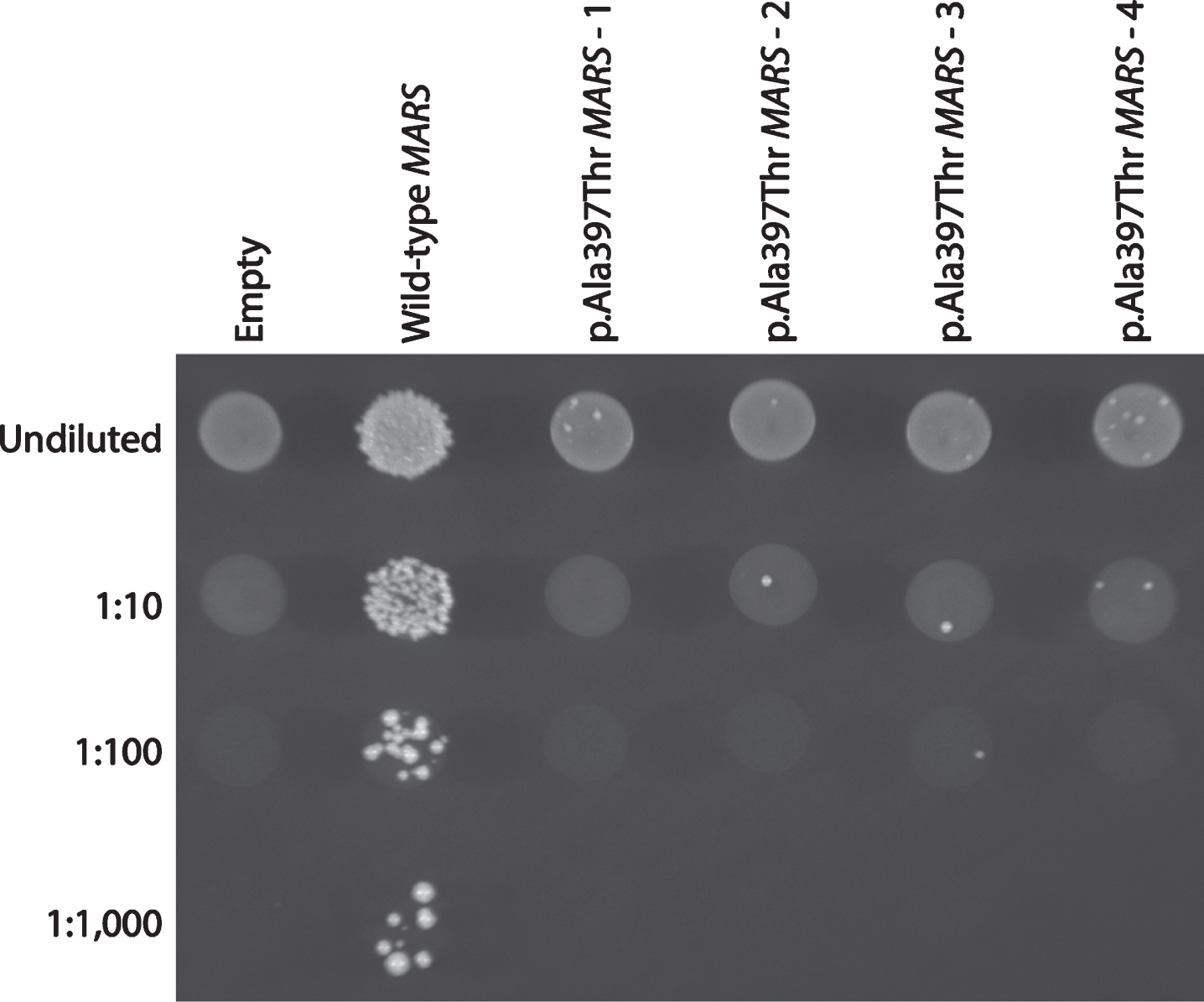
Previous studies revealed that the MARS yeast ortholog Mes1, is an essential gene for cell survival [10]. Complementation assays have been previously employed to study the functional consequences of ARS variants of unknown significance, including variants identified in methionyl-tRNA synthetase [22]. To determine the functional consequences of p.Ala397Thr we utilized this previously established system to model the variant in the human MARS open-reading frame. The p.Ala397Thr MARS variant resulted in severely depleted cellular growth (Fig. 2), indicating that p.Ala397Thr negatively affects enzymatic function. This is consistent with other CMT-associated ARS mutations, which decrease function in vitro and in vivo [5]. However, the effects of impaired ARS function on neuronal health are currently unclear. It should be noted that the other ARS enzymes implicated in CMT function as homodimers, suggesting a dominant-negative effect. MARS, however, functions as a monomer, raising the possibility of multiples mechanisms for ARS-mediated neuropathy.
DISCUSSION
We report an 11 year-old girl with an early-onset and rapidly progressive motor-predominant neuropathy due to a novel mutation in MARS (c.1189G>A, p.Ala397Thr). The clinical presentation reported here is distinct from the previously documented phenotypic spectrum of patients with MARS mutations and suggests that the spectrum of neuropathy and rate of progression for this condition is broader than previously thought.
CMT2U was described in 2013 as a late-onset neuropathy with incomplete penetrance [10]. Using exome sequencing, a heterozygous p.Arg618Cys MARS variant was identified in a proband (onset at 45 years of age) (Table 1) and his affected maternal uncle (onset at 67 years of age), as well as his unaffected mother (NCS not reported). The authors postulated that the late and variable age of onset in this family may have contributed to the apparent incomplete penetrance in this family. Yeast complementation assays for this variant showed a detrimental effect on growth suggesting p.Arg618Cys is a loss-of-function allele. Two additional unrelated patients with late-onset CMT associated with a p. Pro800Thr MARS variant have been reported (Table 1) [2, 12]. Age-of-onset varied from 50 to 66 years and the patients’ severity also ranged from mild weakness in the absence of muscle atrophy to distal muscle wasting and sensory loss (Table 1). The same p.Pro800Thr MARS variant has been reported in a third family, with an earlier age of onset at 5 and 16 years of age, no additional clinical data was provided [11]. Although the p.Pro800Thr MARS variant has been identified in multiple unrelated families, the pathogenicity of this variant remains unclear.
Table 1
Clinical features of CMT2U patients with mutations in MARS
| This report | Hirano et al. 2016 | Hyun et al. 2014 | Nam et al. 2016 | Nam et al. 2016 | Gonzalez et al. 2013 | Gonzalez et al. 2013 | |
| Family | 1 | 2 | 3 | 4 | 5 | ||
| Variant (NM_004990.3) | c.1189G>A p.Ala397Thr | c.2398C>A p.Pro800Thr | c.2398C>A p.Pro800Thr | c.2398C>A p.Pro800Thr | c.2398C>A p.Pro800Thr | c.1852C>T p.Arg618Cys | c.1852C>T p.Arg618Cys |
| Age at diagnosis (yrs) | 11 | 69 | 68 | N/A | N/A | 50 | 81 |
| Age at onset (years) | <1 | 66 | 50 | 5 | 16 | 45 | 67 |
| Sex | F | F | M | N/A | N/A | M | M |
| Motor Symptoms | Distal>proximal muscle wasting. Loss of ambulation | Very mild toe weakness without muscle atrophy | Distal muscle wasting | N/A | N/A | Distal muscle wasting | Distal muscle wasting |
| Sensory Symptoms | Mild sensory symptoms | Dysesthesia in soles | Sensory Loss | N/A | N/A | Stabbing pain in lower limbs | Distal sensory loss |
| Sural Nerve Biopsy | N/A | Axonal neuropathy | N/A | N/A | N/A | Axonal neuropathy | N/A |
The early-onset and rapidly progressive motor-predominant neuropathy for the patient reported here is distinct from previous sensorimotor presentations reported for patients with CMT2U. A similar spectrum of clinical presentation has been reported in patients with GARS mutations associated with CMT2D [9]. While the majority of patients with CMT2D have onset of sensorimotor symptoms and signs in adolescence associated with slow progression, patients have also been reported with infantile onset of a distal-SMA type phenotype. The reason for phenotypic variability in patients with MARS and GARS mutations remains unclear. Structurally, the residues p.Arg616 and p.Pro800, mutated in the late-onset form of CMT2U, locate in the all-helical domain important for anticodon recognition and proper MARS function. The p.Ala397 residue, identified in our patient, locates adjacently to the Zn2+ binding site, crucial for the catalytic activity of methionyl-tRNA synthetase. How different classes of mutations cause different clinical presentations of CMT remains to be elucidated and is an area of future investigation.
This patient illustrates the variable phenotype of CMT2U and highlights the value of robust functional assays in supporting pathogenicity of variants.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to report.
ACKNOWLEDGMENTS
This work was supported by the Care4Rare Canada Consortium (Enhanced Care for Rare Genetic Diseases in Canada) funded by Genome Canada, the Canadian Institutes of Health Research (CIHR), the Ontario Genomics Institute, Ontario Research Fund, Genome Quebec, and Children’s Hospital of Eastern Ontario Foundation. A.A. is supported by a grant from the National Institute of General Medical Sciences (GM118647). R.M. is supported by the Michigan Pre-doctoral Training in Genetics Program (GM007544) and the Ruth L. Kirschstein Predoctoral Individual National Research Service Award (F31 NS108510-01). I.A.P is supported by a CIHR fellowship award. K.M.B. is supported by a CIHR Foundation grant (FDN-154279).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JND-190404.
REFERENCES
[1] | Murphy SM , Laura M , Fawcett K , Pandraud A , Davidson GL , Rossor AM , et al. Charcot-Marie-Tooth disease: Frequency of genetic subtypes and guidelines for Genetic Testing. J Neurol Neurosurg Psychiatry. (2012) ;83: (7):706–10. doi: 10.1136/jnnp-2012-302451 |
[2] | Hyun YS , Park HJ , Heo SH , Yoon BR , Nam SH , Kim SB , et al. Rare variants in methionyl- and tyrosyl-tRNA synthetase genes in late-onset autosomal dominant Charcot-Marie-Tooth neuropathy. Clin Genet. (2014) ;86: (6):592–4. doi:10.1111/cge.12327 |
[3] | Jordanova A , Irobi J , Thomas FP , Van Dijck P , Meerschaert K , Dewil M , et al. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat Genet. (2006) ;38: (2):197–202. |
[4] | McLaughlin HM , Sakaguchi R , Giblin W , NISC Comparative Sequencing Program, Wilson TE , Biesecker L , et al. A recurrent loss-of-function alanyl-tRNA synthetase (AARS) mutation in patients with Charcot-Marie-Tooth disease type 2N (CMT2N). Hum Mutat. (2012) ;33: (1):244–53. doi:10.1002/humu.21635 |
[5] | Meyer-Schuman R , Antonellis A . Emerging mechanisms of aminoacyl-tRNA synthetase mutations in recessive and dominant human disease. Hum Mol Genet. (2017) ;26: (R2):R114–27. doi: 10.1093/hmg/ddx231 |
[6] | Wallen RC , Antonellis A . To charge or not to charge: Mechanistic insights into neuropathy-associated tRNA synthetase mutations. Curr Opin Genet Dev. (2013) ;23: (3):302–9. doi: 10.1016/j.gde.2013.02.002 |
[7] | Rossor AM , Tonaselli PJ , Reilly MM . Recent advances in genetic neuropathies. Curr Opin Neurol. (2016) ;29: (5):537–48. doi: 10.1097/WCO.0000000000000373 |
[8] | Tsai PC , Soong BW , Mademan I , Huang YH , Liu CR , Hsiao CT , et al. A recurrent WARS mutation is a novel cause of autosomal dominant distal hereditary motor neuropathy. Brain. (2017) ;140: (5):1252–66. doi: 10.1093/brain/awx058 |
[9] | James PA , Cader MZ , Muntoni F , Childs AM , Crow YJ , Talbot K . Severe childhood SMA and axonal CMT due to anticodon binding domain mutations in the GARS gene. Neurology. (2006) ;67: (9):1710–2. doi: 10.1212/01.wnl.0000242619.52335.bc |
[10] | Gonzalez M , McLaughlin H , Houlden H , Guo M , Liu YT , Hadjivassilious M , et al. Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J Neurol Neurosurg Psychiatry. (2013) ;84: (11):1247–9. doi: 10.1136/jnnp-2013-305049 |
[11] | Nam SH , Hong YB , Hyun YS , Nam da E , Kwak G , Hwang SH , et al. Identification of genetic causes of inherited peripheral neuropathies by targeted gene panel sequencing. Mol Cells. (2016) ;39: (5):382–8. doi: 10.14348/molcells.2016.2288 |
[12] | Hirano M , Oka N , Hashiguchi A , Ueno S , Sakamoto H , Takashima H , et al. Histopathological features of a patient with Charcot-Marie-Tooth disease type 2U/ AD-CMTax-MARS. J Peripher Nerv Syst. (2016) ;21: (4):370–4. doi: 10.1111/jns.12193 |
[13] | Beaulieu CL , Majewski J , Schwartzentruber J , Samuels ME , Fernandez BA , Bernier FP , et al. FORGE Canada Consortium: Outcomes of a 2-year national rare-disease gene-discovery project. Am J Hum Genet. (2014) ;94: (6):809–17. doi: 10.1016/j.ajhg.2014.05.003 |
[14] | Lee HJ , Cho HY , Kang BS . The crystal structure of human cytosolic methionyl-tRNA synthetase in complex with methionine. Protein Data Bank (2016) . https://www.rcsb.org/structure/5GOY [accessed 1 May 2018]. |
[15] | Webb B , Sali A . Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. (2016) ;86: :1–5. doi: 10.1002/cpps.20 |
[16] | Emsley P , Lohkamp B , Scott WG , Cowtan K . Features and development of Coot. Acta Crystallogr D Biol Crystallogr. (2010) ;66: (Pt 4):486–501. doi: 10.1107/S0907444910007493 |
[17] | Wiederstein M , Sippl MJ . ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. (2007) ;35: :W407–10. doi: 10.1093/nar/gkm290 |
[18] | Jubb HC , Higueruelo AP , Ochoa-Montaño B , Pitt WR , Ascher DB , Blundell TL . Arpeggio: A web server for calculating and visualising interatomic interactions in protein structures. J Mol Biol. (2017) ;429: (3):365–71. doi: 10.1016/j.jmb.2016.12.004 |
[19] | Pires DE , Ascher DB , Blundell TL . DUET: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. (2014) ;42: :W314–9. doi: 10.1093/nar/gku411 |
[20] | Sugiura I , Nureki O , Ugaji-Yoshikawa Y , Kuwabara S , Shimada A , Tateno M , et al. The 2.0 Å crystal structure of Thermus thermophilus methionyl-tRNA synthetase reveals two RNA-binding modules. Structure. (2000) ;8: (2):197–208. |
[21] | Kwon NH , Kang T , Lee JY , Kim HH , Kim HR , Hong J , et al. Dual role of methionyl-tRNA synthetase in the regulation of translation and tumor suppressor activity of aminoacyl-tRNA synthetase-interacting multifunctional protein-3. Proc Natl Acad Sci. (2011) ;108: (49):19635–40. doi: 10.1073/pnas.1103922108 |
[22] | Oprescu SN , Griffin LB , Beg AA , Antonellis A . Predicting the pathogenicity of aminoacyl-tRNA synthetase mutations. Methods. (2017) ;113: :139–51. doi: 10.1016/j.ymeth.2016.11.013 |
[23] | Ng PC , Henikoff S . SIFT: Predicting amino acid changes that affect protein function. Nucleic Acid Res. (2003) ;31: (13):3812–4. |
[24] | Kircher M , Witten DM , Jain P , O’Roak BJ , Cooper GM , Shendure J . A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. (2014) ;46: (3):310–5. doi: 10.1038/ng.2892 |
[25] | Chien CI , Chen YW , Wu YH , Chang CY , Wang TL , Wang CC . Functional substitution of a eukaryotic glycyl-tRNA synthetase with an evolutionarily unrelated bacterial cognate enzyme. PLoS One. (2014) ;9: (4):e94659. doi: 10.1371/journal.pone.0094659 |


