
Modifier Gene Candidates in Charcot-Marie-Tooth Disease Type 1A: A Case-Only Genome-Wide Association Study
Abstract
Background:
Charcot-Marie-Tooth disease type 1A (CMT1A) is caused by a uniform 1.5–Mb duplication on chromosome 17p, which includes the PMP22 gene. Patients often present the classic neuropathy phenotype, but also with high clinical variability.
Objective:
We aimed to identify genetic variants that are potentially associated with specific clinical outcomes in CMT1A.
Methods:
We genotyped over 600,000 genomic markers using DNA samples from 971 CMT1A patients and performed a case-only genome-wide association study (GWAS) to identify potential genetic association in a subset of 644 individuals of European ancestry. A total of 14 clinical outcomes were analyzed in this study.
Results:
The analyses yielded suggestive association signals in four clinical outcomes: difficulty with eating utensils (lead SNP rs4713376, chr6 : 30773314, P = 9.91×10–7, odds ratio = 3.288), hearing loss (lead SNP rs7720606, chr5 : 126551732, P = 2.08×10–7, odds ratio = 3.439), decreased ability to feel (lead SNP rs17629990, chr4 : 171224046, P = 1.63×10–7, odds ratio = 0.336), and CMT neuropathy score (lead SNP rs12137595, chr1 : 4094068, P = 1.14×10–7, beta = 3.014).
Conclusions:
While the results require validation in future genetic and functional studies, the detected association signals may point to novel genetic modifiers in CMT1A.
INTRODUCTION
Charcot-Marie-Tooth disease (CMT) is a group of inherited neuropathies that affect peripheral motor and/or sensory nerves in human. As the most common inherited neurological disorder, CMT has an estimated prevalence of 1 in 2,500 worldwide [1]. CMT is both clinically and genetically heterogeneous. So far over 90 genes have been implicated in the disease, and all Mendelian inheritance patterns have been reported [2]. Based on patients’ nerve conduction velocity (NCV), CMT is divided into several subgroups: demyelinating type (CMT1) with slowed NCV, axonal type (CMT2) with normal NCV but reduced amplitude of conduction, and intermediate type (CMTi) with NCV in the medium range. While most CMT conditions are autosomal dominant, some CMT cases are inherited in X-linked or autosomal recessive forms, known as CMTX and CMT4 respectively. The most common subtype of CMT is CMT type 1A (CMT1A), an autosomal dominant demyelinating form of CMT caused by a uniform 1.5 Mb tandem duplication on chromosome 17p [3]. It accounts for 70% of demyelinating CMT cases and one third of all CMT cases [4]. Within this duplicated chromosomal region, the gene peripheral myelin protein 22 (PMP22) is the monogenic cause of CMT1A [5]. Patients with CMT1A show typical clinical features of CMT with both motor and sensory involvement, but the exact phenotypic presentation and disease severity often vary greatly. The cause of the clinical variability is largely unknown. Various factors, both environmental and genetic, may contribute to the phenotypic differences among patients. Thus far, only a few studies [6–8] have reported potential genetic modifiers in CMT1A, including for the variant I92V (rs4280262) in the gene LITAF and polymorphism rs2292832 in MIR149. Neither locus was correlated with subphenotypes in the present study [33].
To identify potential genetic modifiers in CMT1A on a genome-wide scale, we have recruited 971 CMT1A patients from 12 clinical centers in Europe, Asia, and the US. We previously identified signal induced proliferation associated 1 like 2 (SIPA1L2) as a genetic modifier for foot dorsiflexion strength in CMT1A using the same dataset [32]. In this present study, our aim is to scan for potential genetic modifiers for additional clinical features (subphenotypes) in CMT1A, applying a genome-wide association study (GWAS) approach. Analyses were performed in a subset of 644 patients of European ancestry from the initial cohort of 971 individuals. We analyzed total of 14 subphenotypes, including overall disease severity, motor symptoms, sensory symptoms, muscle strength, and other clinical features such as foot deformity, scoliosis, and hearing loss. The analyses identified several suggestive association signals, indicating that genes located in the vicinity of these markers may be promising candidates of genetic modifiers in CMT1A for future studies.
MATERIALS AND METHODS
Sample collection
A total of 971 CMT1A patients (705 probands and 266 affected family members) were recruited by the Inherited Neuropathies Consortium (INC) between 2011 and 2016. Institutional review board (IRB) approval was obtained from all participating clinical sites. Informed consents were obtained from all participants and/or their legal guardians. Patients with a phenotype consistent with CMT1A had to meet at least one of two criteria to be included in the study: (1) the participant has a documented PMP22 duplication evaluated by a clinical genetic test, and/or (2) the participant has a first-or second-degree relative with a documented PMP22 duplication evaluated by a clinical genetic test.
Clinical evaluations
Total of 14 clinical outcomes were collected from patients. Disease severity was measured by CMT neuropathy score (CMTNS) [9], a composite score that grades severity on a scale from 0 (mild) to 36 (severe) based on clinical symptoms, signs and neurophysiology tests. Muscle strength of foot dorsiflexion, foot plantar flexion, and first dorsal interosseous (FDI) was measured using Medical Research Council (MRC) standards: grade 0 = no contraction; grade 1 = slight contraction without movement; grade 2 = movement with gravity eliminated; grade 3 = movement against gravity, but not against resistance; grade 4 = movement against gravity and some resistance (4–, 4 and 4+ were used to indicate slight, moderate, and submaximal movement, respectively); grade 5 = normal contraction. Patients’ demographics data (including age at exam, sex, self-reported race and ethnicity) and additional subphenotypes (Table 1) were also collected.
Table 1
Clinical data and top associated SNPs in 14 subphenotypes
| Subphenotype | Total | Mild | Severe | OR (95% CI) | P | OR (95% CI) | P | Top SNP | P value |
| cases | cases | cases | (Age/year) | (Age/year) | (Sex/female) | (Sex/female) | |||
| Arthritic-like (aching) paina | 409 | 212 (51.8%) | 197 (48.2%) | 1.01 (1–1.02) | 0.062 | 1.76 (1.19–2.61) | 5.14×10–3 | rs964051 | 9.82×10–6 |
| Burning or tingling in feet or hands | 406 | 265 (65.3%) | 141 (34.7%) | 1.01 (0.99–1.02) | 0.323 | 1.49 (0.98–2.26) | 0.061 | rs7780160 | 6.14×10–6 |
| CMTNSb | 471 | 0.17 (0.13–0.2)f | 3.02×10–22 | – 0.04 (– 1.05–0.97)f | 0.940 | rs12137595 | 1.14×10–7 | ||
| Decreased ability to feelb | 420 | 112 (26.7%) | 308 (73.3%) | 1.03 (1.01–1.04) | 2.10×10–4 | 1.03 (0.66–1.6) | 0.890 | rs17629990 | 1.63×10–7 |
| Difficulties with balancec | 413 | 61 (14.8%) | 352 (85.2%) | 1.03 (1.01–1.05) | 4.97×10–4 | 1.84 (1.06–3.26) | 0.033 | rs11067360 | 1.22×10–6 |
| Difficulties with buttons, zippers, fasteners, bottlesc | 423 | 146 (34.5%) | 277 (65.5%) | 1.03 (1.02–1.05) | 1.17×10–6 | 2.03 (1.33–3.09) | 9.94×10–4 | rs11203943 | 2.32×10–6 |
| Difficulties with eating utensilsc | 420 | 288 (68.6%) | 132 (31.4%) | 1.04 (1.02–1.05) | 2.83×10–7 | 1.62 (1.05–2.52) | 0.030 | rs4713376 | 9.91×10–7 |
| Difficulty walkingc | 424 | 62 (14.6%) | 362 (85.4%) | 1.04 (1.02–1.06) | 4.71×10–6 | 1.98 (1.13–3.52) | 0.017 | rs8046213 | 9.30×10–7 |
| Foot dorsiflexion strengthc, d | 291 | 117 (40.2%) | 174 (59.8%) | 1.08 (1.06–1.11) | 1.39×10–14 | 2.86 (1.62–5.15) | 3.54×10–4 | rs17066814 | 1.31×10–5 |
| FDI strengthc, d | 200 | 80 (40.0%) | 120 (60.0%) | 1.11 (1.08–1.14) | 1.15×10–12 | 2.27 (1.09–4.91) | 0.031 | rs17698132 | 4.17×10–5 |
| Foot deformitya | 417 | 49 (11.8%) | 368 (88.2%) | 1 (0.98–1.01) | 0.611 | 0.46 (0.24–0.86) | 0.017 | rs16967483 | 8.13×10–7 |
| Hearing lossb | 364 | 303 (83.2%) | 61 (16.8%) | 1.06 (1.04–1.09) | 8.67×10–8 | 0.61 (0.34–1.1) | 0.104 | rs7720606 | 2.08×10–7 |
| Foot plantar flexion strengthb, e | 412 | 276 (67.0%) | 136 (33.0%) | 1.07 (1.05–1.09) | 3.50×10–14 | 1.31 (0.83–2.07) | 0.251 | rs2249498 | 5.13×10–7 |
| Scoliosis | 409 | 318 (77.8%) | 91 (22.2%) | 1 (0.99–1.01) | 0.949 | 1.33 (0.83–2.14) | 0.239 | rs10908366 | 2.86×10–6 |
aSex was included as a covariate in association analysis. bAge at exam was included as a covariate in association analysis. cAge at exam and sex were included as covariates in association analysis. dExtreme phenotype approach was used to separate mild and severe cases. Mild cases were defined as patients with strength of 5. Severe cases were defined as patients with strength of 0 to 3. eMild cases were defined as patients with strength of 5. Severe cases were defined as patients with strength of 0 to 4. fBeta coefficient (95% CI) of the linear regression analysis.
Genotyping and quality control (QC)
Genotyping and data QC have been described previously in [32, 33]. Patient DNA samples were genotyped using Illumina OmniExpress beadchip or Illumina OmniExpress Exome beadchip. Standard GWAS QC was performed using PLINK v1.07 [10] to remove monomorphic SNPs and SNPs with low call rate (<95%) or deviating from Hardy-Weinberg equilibrium (HWE P < 0.00001). Samples were checked for the classic 1.5 Mb duplication on chromosome 17p using Illumina cnvPartition v3.2.1. Population stratification was assessed by principal component analysis (PCA) using EigenStrat [11].
Phenotype analyses
Logistic regression or linear regression was performed to test the effect of patients’ age and sex on the subphenotypes. As a continuous trait, CMTNS was analyzed using linear regression: CMTNS =β0 +β1 age at exam +β2 sex +ɛ, where β1 and β2 show the effect of age and sex on the clinical outcome, while β0 and ɛ represent the intercept and the error term, respectively. All other clinical outcomes were analyzed as binary traits using logistic regression: logit (subphenotype) =β0 +β1 age at exam +β2 sex +ɛ. For analysis purpose, muscle strength measures (foot dorsiflexion, foot plantar flexion and FDI) were dichotomized into severe cases and mild cases. Foot dorsiflexion and FDI were dichotomized using an extreme-phenotype approach: patients with minimal strength of 5 were grouped into mild cases, and patients with minimal strength of 0 to 3 were grouped into severe cases. In foot plantarflexion, patients with strength of 5 were grouped into mild cases, and patients with strength of 0 to 4 were grouped into severe cases. All analyses were performed in R.
GWAS analyses
GWAS statistical analyses were performed with PLINK v1.07 [10] using an additive genetic model, where each SNP genotype was coded as the number of minor alleles. Logistic regression was used to analyze the association of binary traits (presence vs. absence of a subphenotype, or severe cases vs. mild cases). Linear regression was used to analyze the association of CMTNS. Patients’ sex and/or age at exam were included as covariates if they showed significant correlation with the traits in phenotype analyses. Association results were visualized using the R package qqman. Regional association plots were generated using LocusZoom [12].
RESULTS
The study cohort
The initial study cohort included 971 CMT1A patients recruited from Europe, Asia, and the US. CNV analysis identified eight patients (0.8%) with an extended duplication and 17 patients (1.8%) with a shorter-sized duplication at the PMP22 locus, and these samples were removed from the analysis to keep a genetically uniform sample set and guard against CMT1A chromosomal locus variability [32]. Population stratification analysis identified 644 individuals of European ancestry, and 213 individuals of Asian ancestry after data quality control (QC). Since the European cohort had a larger sample size, phenotype analyses and follow-up association tests were performed in the European subset to avoid bias from population-specific differences. In addition, only unrelated individuals were included in the analyses, so that the results would not be biased by family relatedness.
We analyzed 14 subphenotypes, including overall disease severity (CMTNS), motor symptoms (difficulties with buttons, zippers, fasteners and bottles, difficulties with eating utensils, difficulty walking, difficulties with balance), sensory symptoms (arthritic-like (aching) pain, burning or tingling in feet or hands, decreased ability to feel), muscle strength (foot dorsiflexion, FDI, foot plantar flexion), and additional clinical features such as foot deformity, scoliosis, and hearing loss (Table 1). As expected [4], most of the subphenotypes were positively correlated with patient age (Table 1), suggesting that disease severity increases with age. The exceptions were arthritic-like pain, burning or tingling in feet or hands, scoliosis and foot deformity, which did not show significant correlation with patient age. Sex was also associated with many of the subphenotypes (Table 1). Females were more likely to present the symptoms or develop severe forms of the following clinical signs: arthritic-like pain, difficulties with balance, difficulties with buttons, zippers, fasteners and bottles, difficulties with eating utensils, difficulty walking, foot dorsiflexion strength, FDI strength. Males had a higher likelihood to present with foot deformity.
Genome-wide association analyses
GWAS analyses were performed to identify SNPs associated with subphenotypes in the European cohort. Patient age at exam and/or sex were included as covariates in the association tests when they were shown to correlate with the clinical outcome from phenotype analyses. Due to the small sample size of the study cohort, we anticipated that GWAS had only limited power in identifying potential associations. Instead of adopting a stringent genome-wide significance threshold (P < 5×10–8), we looked for suggestive evidence of association based on a relaxed P value threshold (P < 10–6), multiple clustered supporting SNPs, and behavior of association signals on QQ plots (Figs. 1 and 2). We observed suggestive association signals in ‘difficulty with eating utensils’, CMTNS, hearing loss, and ‘decreased ability to feel’ (Fig. 1, Table 2). Table 1 contains the top associated SNPs in each subphenotype.
Fig. 1
Manhattan plots and QQ plots of genome-wide association analyses. Results of (A) ‘difficulties with eating utensils’, (B) CMTNS, (C) hearing loss, (D) ‘decreased ability to feel’ in the European population are shown. Association peaks (100–kb around the lead SNPs) are highlighted in green. The dashed lines show the suggestive significance thresholds at P = 1×10–6 and P = 1×10–4.
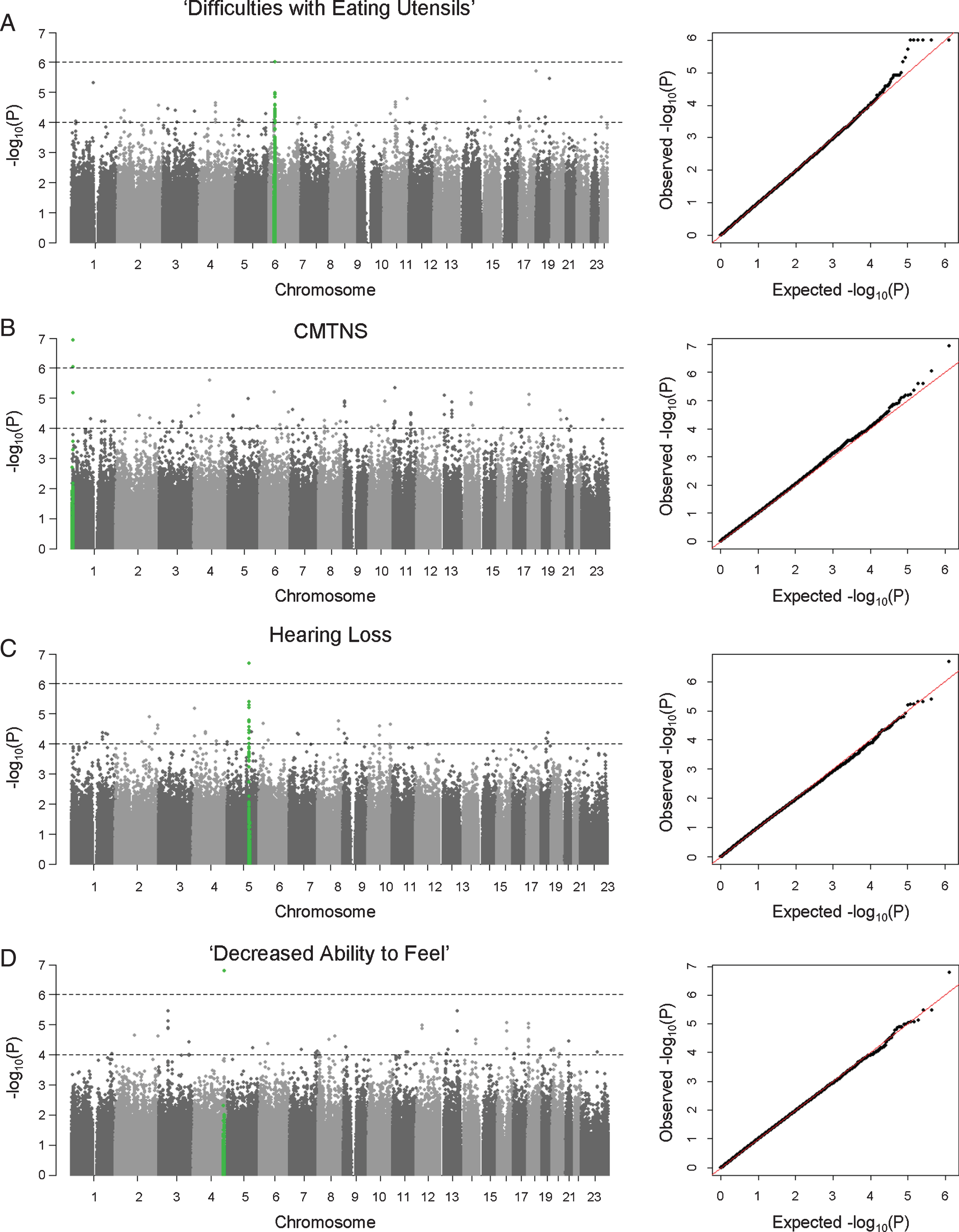
Fig. 2
Regional association plots.Results of (A) ‘difficulties with eating utensils’, (B) CMTNS, (C) hearing loss, (D) ‘decreased ability to feel’ in the European population are shown. The purple dots show the most significantly associated SNPs. The color scheme shows R2 values of the top SNPs with other SNPs (1000 Genomes Project, Nov 2014, EUR).
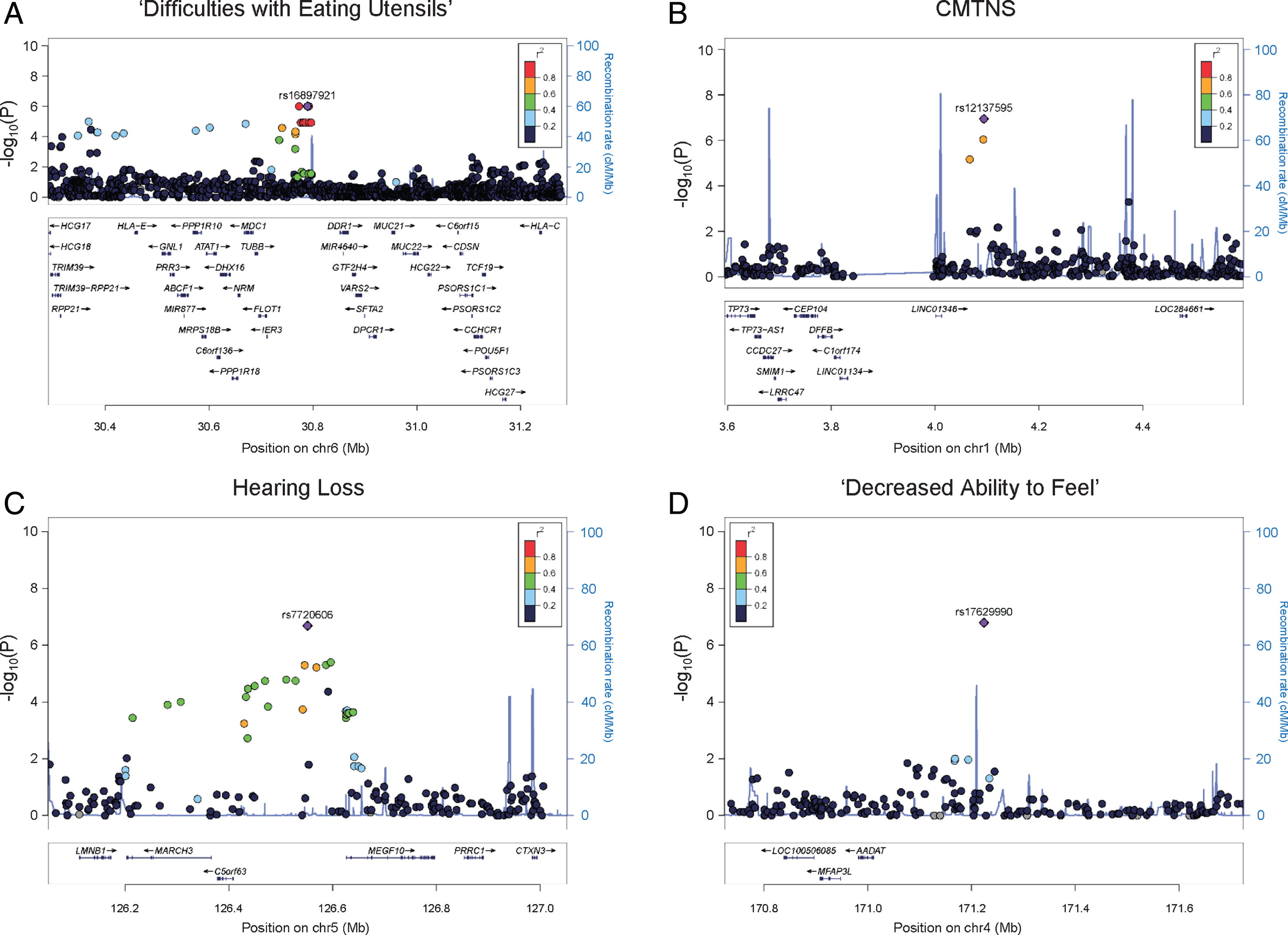
Table 2
Top SNPs with suggestive significance identified from GWAS
| Subphenotype | SNP | Chr | BP | A1 | A2 | European HapMap | P | OR | Neighboring |
| (effect allele) | (major allele) | Freq (%) | (95% CI) | genes | |||||
| Difficulties | rs4713376 | 6 | 30773314 | C | A | 17.4 | 9.91×10–7 | 3.288 (2.041–5.297) | HCG20, |
| with eating | rs16897921 | 6 | 30789526 | A | G | 17.9 | 9.91×10–7 | 3.288 (2.041–5.297) | LINC00243, |
| utensils | rs12195964 | 6 | 30790481 | G | T | 17.7 | 9.91×10–7 | 3.288 (2.041–5.297) | TUBB, FLOT1, |
| rs12203797 | 6 | 30790807 | G | A | 13.2 | 9.91×10–7 | 3.288 (2.041–5.297) | IER3 | |
| rs4713391 | 6 | 30792235 | C | T | 17.9 | 9.91×10–7 | 3.288 (2.041–5.297) | ||
| rs12192704 | 6 | 30792270 | A | G | 17.9 | 9.91×10–7 | 3.288 (2.041–5.297) | ||
| CMTNS | rs12137595 | 1 | 4094068 | T | C | 10.0 | 1.14×10–7 | 3.014 (1.917–4.111)a | LINC01346 |
| Hearing loss | rs7720606 | 5 | 126551732 | A | G | 48.2 | 2.08×10–7 | 3.439 (2.157–5.482) | MARCH3, C5orf63, MEGF10 |
| Decreased | rs17629990 | 4 | 171224046 | A | G | 18.3 | 1.63×10–7 | 0.336 (0.223–0.505) | LINC01612 |
| ability to feel |
aBeta coefficient (95% CI) of the linear regression analysis.
Fine motor functions
We identified an association peak on chromosome 6 with ‘difficulty with eating utensils’ (Fig. 1A). This clinical outcome is closely related to patients’ fine motor functions in the hands. In the study cohort, 132 individuals reported having difficulty with eating utensils, while 288 individuals reported no difficulty. The association signal contains six lead SNPs with the smallest P values of 9.91×10–7. The SNPs are in LD, and their allele combinations represent two haplotypes (rs4713376/rs16897921/rs12195964/rs12203797/rs4 713391/rs12192704 AGTATG and CAGGCA). Carriers of the CAGGCA haplotype are more likely to have difficulties with eating utensils (OR = 3.288, 95% CI = 2.041–5.297). The association was also supported by multiple additional SNPs in LD with the lead SNPs. The associated SNPs do not fall into any coding region of the genome. Several genes are located around the association signal within 200 kb, including HLA complex group 2 (HCG20), long intergenic non-protein coding RNA 243 (LINC00243), tubulin beta class I (TUBB), flotillin 1 (FLOT1), and immediate early response 3 (IER3) (Fig. 2A).
Disease severity
CMTNS is commonly used to assess disease severity in CMT. This composite score is graded on a scale from 0 (mild) to 36 (severe) based on symptoms (12 points), signs (16 points), and neurophysiology exams (8 points) [9]. In our cohort of 471 patients with CMTNS data, the score ranges from 2 to 34, with a mean (SD) of 14.83 (6.15). Consistent with previous reports [4], age is positively correlated with CMTNS with R2 of 0.18, suggesting that age-related physiological changes contribute to CMT disease severity. There is no significant difference in CMTNS between males and females. Applying association analysis, we identified a suggestive association signal on chromosome 1 (lead SNP rs12137595, chr1 : 4094068, P = 1.14×10–7, beta = 3.014, 95% CI = 1.917–4.111) (Fig. 1B). The minor allele T (rs12137595) is associated with increased CMTNS. Two markers (rs10915659 and rs4128222) in LD with the lead SNP showed P values of 9.17×10–7 and 6.83×10–7, respectively, further supporting the association signal. The association peak is located in an intergenic region close to long intergenic non-protein coding RNA 1346 (LINC01346), a non-coding gene with unknown functions (Fig. 2B).
Hearing loss
As part of the sensory nervous system, the cochlear nerves may also be affected in CMT. Hearing loss has been reported in many CMT subtypes including CMT1A [13, 14]. In our study cohort, 61 individuals (16.8%) reported having partial or complete hearing loss, while 303 individuals reported having no hearing loss. Applying GWAS analysis, we identified a suggestive association signal on chromosome 5 associated with hearing loss in patients (lead SNP rs7720606, chr5 : 126551732, P = 2.08×10–7, odds ratio = 3.439, 95% CI = 2.157–5.482) (Fig. 1C). Carriers of the minor allele A (rs7720606) were at an increased risk of having hearing loss. The association signal is further supported by multiple SNPs at the locus. The closet genes around the locus include membrane associated ring-CH-type finger 3 (MARCH3), chromosome 5 open reading frame 63 (C5orf63), and multiple epidermal growth factor like domains 10 (MEGF10) (Fig. 2C). None of these genes has previously been implicated in hearing loss.
Patient reported sensory functions
We also identified an intergenic SNP associated with ‘decreased ability to feel’ (rs17629990, chr4 : 171224046, P = 1.63×10–7, odds ratio = 0.336, 95% CI = 0.223–0.505) (Fig. 1D). The minor allele A of the marker confers lower risk of having sensory issues. However, there was no other SNP in LD to support this association signal. The closest gene to the associated SNP is a non-coding gene long intergenic non-protein coding RNA 1612 (LINC01612) (Fig. 2D).
DISCUSSION
Genetic modifiers play important roles in many genetic disorders including CMT1A. Patients with CMT1A often present high clinical variability, although they share the same genetic cause of a uniform PMP22 duplication. Thus far, the role of genetic modifiers in CMT1A is still not well studied. In this study, we aimed to perform a preliminary but comprehensive approach to identify potential association in specific subphenotypes in CMT1A. This case-only GWAS analyzed the association of over 600,000 SNPs with 14 clinical outcomes of CMT1A in a cohort of patients with European-ancestry. The SNPs identified in the European cohort do not show significant association in our Asian cohort. This may be caused by 1) the different LD structure in the two populations and 2) the smaller sample size of the Asian cohort, which also prevented us from further analyses.
Still, for most of the subphenotypes, the actual sample size for association tests was over 400 individuals. We identified association signals with suggestive significance in four subphenotypes: difficulties with eating utensils, CMTNS, hearing loss, and decreased ability to feel.
The first suggestive association was identified in difficulties with eating utensils. This clinical symptom is closely associated with patients’ fine motor functions in the hands. The associated haplotype is located in an intergenic region and is supported by multiple SNPs in LD with the lead SNPs. Neighboring genes include HCG20, LINC00243, TUBB, FLOT1, and IER3. HCG20 and LINC00243 are both noncoding RNA genes with unknown functions. TUBB encodes a beta tubulin protein, which is a major component of microtubules. Beta tubulins form heterodimers with alpha tubulins, and then assemble into microtubules. Microtubules are part of the cytoskeleton and are involved in many cellular processes such as intracellular transport in axons [15]. Mutations in TUBB have been reported to cause complex cortical dysplasia with other brain malformations 6 [16], and congenital symmetric circumferential skin creases [17]. FLOT1 encodes flotillin 1, a protein located in microdomains of the inner cell membrane [18]. It is involved in many cellular processes such as cytoskeletal regulation and endocytosis [19]. IER3, also known as IEX-1, encodes a member of the NF-kappa-B family. It is involved in protecting cells from apoptosis [20]. Among these genes, TUBB is a potential candidate because of its involvement in microtubule dynamics. Several CMT genes, such as heat shock protein family B (small) member 1 (HSPB1) [21], are known to affect microtubule functions. Microtubule-stabilizing agents, which are used in treatment of many cancers, are known to cause neuropathy in cancer patients as a major adverse effect [22, 23]. Interestingly, it has been reported that axonal loss is associated with hand dysfunction in CMT1A [24]. In addition, mutations in TUBB3, a paralogous gene to TUBB, have been reported to cause phenotypes of axonal sensorimotor polyneuropathy [25]. Based on the role of microtubule dynamics in peripheral neuropathies, TUBB is a potential modifier gene for hand dysfunction in CMT1A.
For hearing loss, we identified an associated SNP between genes C5orf63 and MEGF10. The association signal is supported by multiple SNPs around the lead SNP, thus the association peak further extends to neighboring genes including lamin B1 (LMNB1), MARCH3, C5orf63, and MEGF10. Interestingly, LMNB1 causes autosomal dominant leukodystrophy (ADLD) [26]. ADLD is a late-onset demyelinating disease affecting the central nervous system white matter. The major clinical feature of ADLD is autonomic dysfunction, but sensorineural hearing loss may be present in rare cases [27]. LMNB1 encodes a lamin protein, which is a component of the nuclear lamina [28]. MARCH3 encodes a member of the membrane-associated RING-CH (MARCH) family. The protein is an E3 ubiquitin-protein ligase and may play roles in the endosomal transport pathway [29]. C5orf63 encodes a glutaredoxin-like protein localized to mitochondria, but the function of the gene is still unknown [30]. MEGF10 encodes a member of the multiple epidermal growth factor-like domains protein family. Mutations in MEGF10 have been reported to cause myopathy, areflexia, respiratory distress, and dysphagia [31]. Together the results suggest this 800 kb genomic region may be a locus of interest to look for novel genetic modifiers.
Two more potential associations were identified in CMTNS and ‘decreased ability to feel’. Both association signals are located in intergenic regions close to non-coding RNA genes. The SNPs identified in the two subphenotypes have not been reported in any diseases. At this point it is highly speculative to develop specific hypotheses for non-coding RNA involvement in CMT; however future functional genomics studies in this areas might be able to take our findings into account. Due to lack of biological knowledge on these genomic regions, we are not certain of the biological relevance of these association signals.
While of great translational interest, the presented findings are likely not applicable for biomarkers at this point. We have not identified any likely pathogenic variants in CMT1A families in the reported candidate genes. Additional molecular studies; however, might be able to develop some of the suggested proteins or pathways into valid and highly correlated biomarkers.
In conclusion, our study identified several potential genetic associations with subphenotypes in CMT1A in a European population. More genomic studies are needed to replicate the associations in Europeans, and also in populations of different genetic ancestries. Experimental evidence is required to further elucidate the roles of the candidate genes.
CONFLICT OF INTEREST
The authors declare no competing interests.
DISCLOSURE
Nothing to disclose.
ACKNOWLEDGMENTS
This work was supported by NIH (R01NS43174, U54NS065712 to M.E.S., M.M.R., S.S.S., S.Z.), NIH (R01NS075764, U54NS065712 to S.Z. and M.S., R01NS075269 to J.S. and U54HD090256), NIH (R01NS094388 to B.O.C.), the Charcot-Marie-Tooth Association, and the Muscular Dystrophy Association. M.M.R. is grateful to the Medical Research Council (MRC), MRC Centre grant (G0601943), and the National Institutes of Neurological Diseases and Stroke and office of Rare Diseases (U54NS065712) for their support. This research was also supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
REFERENCES
[1] | Skre H . Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin Genet (1974) ;6: :98–118. |
[2] | Pareyson D , Saveri P , Pisciotta C . New developments in Charcot-Marie-Tooth neuropathy and related diseases. Curr Opin Neurol (2017) ;30: :471–480. |
[3] | Lupski JR , de Oca-Luna RM , Slaugenhaupt S , et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell (1991) ;66: :219–32. |
[4] | Fridman V , Bundy B , Reilly MM , et al. CMT subtypes and disease burden in patients enrolled in theInherited Neuropathies Consortium natural history study: a cross-sectional analysis. J Neurol Neurosurg Psychiatry (2015) ;86: :873–8. |
[5] | Patel PI , Roa BB , Welcher AA , et al. The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot-Marie-Tooth disease type 1A. Nat Genet (1992) ;1: :159–65. |
[6] | Meggouh F , de Visser M , Arts WF , De Coo RI , van Schaik IN , Baas F . Early onset neuropathy in a compound form of Charcot-Marie-Tooth disease. Ann Neurol (2005) ;57: 589–91. |
[7] | Sinkiewicz-Darol E , Lacerda AF , Kostera-Pruszczyk A , et al. The LITAF/SIMPLE I92V sequence variant results in an earlier age of onset of CMT1A/HNPP diseases. Neurogenetics (2015) ;16: :27–32. |
[8] | Nam SH , Kanwal S , Nam DE , et al. Association of miR-149 polymorphism with onset age and severity in Charcot-Marie-Tooth disease type 1A. Neuromuscul Disord (2018) ;28: :502–507. |
[9] | Murphy SM , Herrmann DN , McDermott MP , et al. Reliability of the CMT neuropathy score (second version) in Charcot-Marie-Tooth disease. J Peripher Nerv Syst (2011) ;16: :191–8. |
[10] | Purcell S , Neale B , Todd-Brown K , et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet (2007) ;81: :559–75. |
[11] | Price AL , Patterson NJ , Plenge RM , Weinblatt ME , Shadick NA , Reich D . Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet (2006) ;38: :904–9. |
[12] | Pruim RJ , Welch RP , Sanna S , et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics (2010) ;26:2336–7. |
[13] | Rance G , Ryan MM , Bayliss K , Gill K , O'Sullivan C , Whitechurch M . Auditory function in children with Charcot-Marie-Tooth disease. Brain (2012) ;135: :1412–22. |
[14] | Choi JE , Seok JM , Ahn J , et al. Hidden hearing loss in 0patients with Charcot-Marie-Tooth disease type 1A. Sci Rep (2018) ;8: :10335. |
[15] | Millecamps S , Julien JP . Axonal transport deficits and neurodegenerative diseases. Nature reviews. Neuroscience (2013) ;14: :161–76. |
[16] | Breuss M , Heng JI , Poirier K , et al. Mutations in the beta-tubulin gene TUBB5 cause microcephaly with structural brain abnormalities. Cell reports (2012) ;2: :1554–62. |
[17] | Isrie M , Breuss M , Tian G , et al. Mutations in Either TUBB or MAPRE2 Cause Circumferential Skin Creases Kunze Type. Am J Hum Genet (2015) ;97: :790–800. |
[18] | Bickel PE , Scherer PE , Schnitzer JE , Oh P , Lisanti MP , Lodish HF . Flotillin and epidermal surface antigen define a new family of caveolae-associated integral membrane proteins. J Biol Chem (1997) ;272: :13793–802. |
[19] | Otto GP , Nichols BJ . The roles of flotillin microdomains—endocytosis and beyond. J Cell Sci (2011) ;124: :3933–40. |
[20] | Schilling D , Pittelkow MR , Kumar R . IEX-1, an immediate early gene, increases the rate of apoptosis in keratinocytes. Oncogene (2001) ;20: :7992–7. |
[21] | Almeida-Souza L , Timmerman V , Janssens S . Microtubule dynamics in the peripheral nervous system: A matter of balance. Bioarchitecture (2011) ;1: :267–270. |
[22] | Lee JJ , Swain SM . Peripheral neuropathy induced by microtubule-stabilizing agents. Journal of clinical oncology : official journal of the American Society of Clinical Oncology (2006) ;24: :1633–42. |
[23] | Beutler AS , Kulkarni AA , Kanwar R , et al. Sequencing of Charcot-Marie-Tooth disease genes in a toxic polyneuropathy. Ann Neurol (2014) ;76: :727–37. |
[24] | Videler AJ , van Dijk JP , Beelen A , de Visser M , Nollet F , van Schaik IN . Motor axon loss is associated with hand dysfunction in Charcot-Marie-Tooth disease 1a. Neurology (2008) ;71: :1254–60. |
[25] | Tischfield MA , Baris HN , Wu C , et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell (2010) ;140: :74–87. |
[26] | Padiath QS , Saigoh K , Schiffmann R , et al. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet (2006) ;38: :1114–23. |
[27] | Schwankhaus JD , Katz DA , Eldridge R , Schlesinger S , McFarland H . Clinical and pathological features of an autosomal dominant, adult-onset leukodystrophy simulating chronic progressive multiple sclerosis. Arch Neurol (1994) ;51: :757–66. |
[28] | Goldman RD , Gruenbaum Y , Moir RD , Shumaker DK , Spann TP . Nuclear lamins: building blocks of nuclear architecture. Genes Dev (2002) ;16: :533–47. |
[29] | Fukuda H , Nakamura N , Hirose S . MARCH-III Is a novel component of endosomes with properties similar to those of MARCH-II. Journal of biochemistry (2006) ;139: :137–45. |
[30] | Schulten HJ , Hussein D , Al-Adwani F , et al. Microarray Expression Data Identify DCC as a Candidate Gene for Early Meningioma Progression. PLoS One (2016) ;11: :e0153681. |
[31] | Logan CV , Lucke B , Pottinger C , et al. Mutations in MEGF10, a regulator of satellite cell myogenesis, cause early onset myopathy, areflexia, respiratory distress and dysphagia (EMARDD). Nat Genet (2011) ;43: :1189–92. |
[32] | Tao F , Beecham GW , Rebelo AP , et al. Inherited Neuropathy Consortium. Variation in SIPA1L2 is Correlated with Phe-notype Modification in CMT Type 1A. Ann Neurol. (2019) Jan 31. |
[33] | Tao F ; Inherited Neuropathy Consortium, Ziichner S . Replication studies of MIR149 association in Charcot-Marie-Tooth disease type 1A in a European population. Neuromuscul Disord. (2018) ; pii: S0960-8966(18)31138-6. |

