
15th International Congress on Neuromuscular Diseases, July 6 - 10, 2018 Vienna, Austria
CONTENTS
Plenary Sessions ................................................................................................................................S1
Scientifi c Sessions.............................................................................................................................S9
Workshop ...........................................................................................................................................S43
Teaching Courses ...............................................................................................................................S73
Overarching Courses...........................................................................................................................S89
Virtual Sessions .................................................................................................................................S93
ENMC ................................................................................................................................................S97
Poster Sessions....................................................................................................................................S101
Author Index .......................................................................................................................................S385
Plenary Sessions
PL 1.1 / #155PATHOMECHANISMS
Topic: 01. Muscle
Volker Straub
John Walton Muscular Dystrophy Research Centre, Newcastle University, Institute of Genetic Medicine, Newcastle Upon Tyne, GB
Abstract: Traditionally a skeletal muscle is viewed as a distinct anatomical structure that is attached to bone by its tendons, is innervated by a nerve and generates movement through contraction. This simplistic view does highlight the most important physiological function of muscle, force generation through activity of its main molecular structure, the sarcomere. But rather than being defined by its anatomical structure, skeletal muscle is now viewed as a complex organ system consisting of about 700 muscles that make up roughly 40% of our body weight. The skeletal muscular system doesn’t just permit movement of the body and maintains it posture, but supports vital metabolic functions, contributes to temperature control and circulation, interacts with focal and systemic hormonal pathways, and is a crucial contributor to respiratory function and the operation of our senses. Through the application of –omics technologies we have also learned that skeletal muscle tissue itself is far less homogenous than originally anticipated. Distinct skeletal muscles show different transcriptomes and proteomes, despite the fact that their mechanical functions may be very similar. The complexity of normal muscle function is reflected in the broad spectrum of clinical symptoms and muscle pathology when something goes wrong. Over the past 30 years, since the discovery of dystrophin and the characterisation of the DMD gene, >500 different genes leading to about 900 distinct genetic neuromuscular disease have been described. Trying to identify and understand the pathomechanisms that specifically underlie genetic muscle diseases has helped to better characterize the relevant molecular pathways and subcellular structures that are involved in maintaining muscle fibre integrity and function. Not surprisingly we distinguish metabolic muscle disease from structural muscle disease, including diseases that affect the integrity of the sarcolemma, the sarcomere or the nuclear lamina. But although classification systems based on pathomechanistic concepts have been helpful to both basic and clinical scientists, the application of a large array of new molecular imaging and –omics technologies have illustrated the complexity of disease mechanisms and continue to blur the boundaries between pathway-based modelling approaches and our anatomical view of muscle fibre architecture. The responsiveness of the muscular system to damage caused by pathogenetic mechanism is in the end limited and will result in muscle wasting and weakness, the main symptoms of genetic muscle diseases
PL 1.2 / #100INFLAMMATORY MECHANISMS
Topic: 01. Muscle
Andrew Mammen
NIAMS/NIH, Bethesda, US
Abstract not received.
PL 1.3 / #94GENETICS / EPIGENETICS
Topic: 01. Muscle
Hanns Lochmüller
Institute Of Genetic Medicine, Newcastle University, International Centre for Life, Newcastle Upon Tyne, US
Abstract not received.
PL 2.1 / #140INHERITED NEUROPATHIES: FROM GENES TO CLINICAL PHENOTYPE
Topic: 02. Neuropathy
Mary Reilly
Queen Square, Institue Of Neurology, London, GB
Abstract: The inherited neuropathies are a heterogeneous group of disorders encompassing those disorders in which the neuropathy is the sole manifestation and those neuropathies where the neuropathy is part of a more complex neurological or multisystem disorder. The first group includes Charcot Marie Tooth disease (CMT) and the related disorders, the hereditary sensory neuropathies (HSN) and the distal hereditary motor neuropathies (HMN). The second group includes a wide range of disorders examples of which include the leuokodystrophies and the mitochondrial diseases. We now know that there is overlap between the causative genes for CMT, HSN and HMN and often use the term CMT to include all of these diseases. Next generation sequencing especially whole exome and whole genome analysis has also revealed that genes that were associated with other neurological diseases e.g. hereditary spastic paraparesis and the ataxias can sometimes cause CMT or start with a CMT like syndrome before evolving to a more classical disease. Traditionally we have approached the diagnosis of an inherited neuropathy with careful phenotyping to guide us in selecting genetic tests. Although this is still important, increasingly we are using phenotyping to validate variants identified by next generation sequencing. As well as clinical examination and neurophysiology, we use MRI increasingly both of the CNS in complex neuropathies and neuromuscular protocols of the limbs looking for patterns of denervation associated with certain neuropathies (e.g. sparing of adductors and semitendinosus in the thighs of patients with BICD2 and Dynein mutations). The phenotypes associated with individual genes are expanding rapidly but for the common genes certain patterns are common and therefore helpful in validating variants (e.g. early ankle plantar flexion weakness in HSPB1 and HSPB8 mutations). Understanding the detailed phenotypes is critical in therapy development. It is an exciting time in inherited neuropathy therapy development with the advent of genetic therapies including antisense, silencing RNA, viral delivered gene therapies and various gene editing therapies. All of these are either in preclinical or clinical trials for various neuropathies. For gene therapies knowing both what tissues need to be targeted for efficacy and which organs needed to be avoided to prevent side effects is important. We also need better ways to monitor disease progression so responsive outcome measures can be developed for clinical trials. While we may have started with detailed phenotyping of families many decades ago to identify causative genes, the mechanistic insights from these genes and the subsequent therapies being developed have led us back to the patients to delineate and understand the phenotypes better in order to be able to perform clinical trials and deliver efficacious therapies to our patients safely.
PL 2.2 / #167IMMUNE MEDIATED NEUROPATHIES: AN EXPANDING FIELD OF TREATABLE NEUROPATHIES
Topic: 02. Neuropathy
Pieter Van Doorn
Center For Lysosomal And Metabolic Diseases, Erasmus MC, University Medical Center Rotterdam, Rotterdam, NL
Abstract: Immune mediated neuropathies comprise an extending field of disorders with specific subgroups. Patients with these disorders need to be diagnosed because they are treatable. It ranges from the Guillain-Barré syndrome (GBS), a heterogeneous acute polyneuropathy to the more chronic - often demyelinating -polyradiculoneuropathies, of which chronic inflammatory demyelinating polyneuropathy (CIDP) is the most prominent one. CIDP can be divided into the most often occurring symmetric sensory-motor variant, and the pure motor or sensory subgroups. Focal, or multifocal varieties of CIDP are known as Lewis-Sumner syndrome or multifocal acquired demyelinating sensory-motor neuropathy (MADSAM). There additionally is a variety named distal acquired demyelinating sensory neuropathy (DADS). Careful electrophysiological investigation (EMG) and the use of peripheral nerve ultrasound or MRI of the plexus can be helpful to diagnose these patients. EAN/Peripheral Nerve Society guidelines for the diagnosis and treatment of GBS and CIDP are available or under construction. Intravenous immunoglobulin (IVIg) and plasma exchange (PE) are effective in GBS. There is now an indication that complement inhibition using eculizumab potentially potentially is effective in GBS, but additional studies are required. The results of the second-dose IVIg RCT (SID-GBS) conducted in GBS patients with a poor prognosis - based upon the mEGOS prognostic model - are eagerly awaited. An interesting new study that especially may be helpful in low-income countries studied the feasibility of small volume plasma exchange as an alternative and simple method for treatment of GBS.Patients within the spectrum of CIDP can be treated with IVIg, PE or steroids. If one of these proven effective treatments fails, one of these other treatments is still likely to be successful. A recent RCT showed that patients with CIDP can also be treated with subcutaneous IgG (ScIG). There is debate what treatment for CIDP is best. IVIg acts fast but is expensive, and steroids are cheap but can induce major side effects. Pulse steroid treatment however may have a higher chance to induce remission of disease, which is now investigated in new studies. Under- and over treatment are important, therefore it needs to be investigated at least once every 6-12 months if a patient still requires treatment because the disease can get into remission either therapeutically induced or spontaneously. Multifocal motor neuropathy (MMN) is a disorder that may mimic motor neuron disease, but is an IVIg treatable condition. Surprisingly these patients do not improve after steroids or PE. Other important immune-mediated neuropathies are related to the presence of a paraprotein or for example due to vasculitis.A comprehensive overview will be given and the latest developments will be discussed.
PL 2.3 / #55ADVANCES IN THE TREATMENT OF PERIPHERAL NEUROPATHY
Topic: 02. Neuropathy
David Cornblath
Johns Hopkins Medicine, Baltimore, US
Abstract not received.
PL 3.1 / #97EMERGING THERAPIES FOR MOTOR NEURONE DISEASES
Topic: 03. Motor Neurone
Albert Ludolph
Universitätsklinikum Ulm, Ulm, DE
Abstract not received.
PL 3.2 / #164CLINICAL CONCEPT OF ALS
Topic: 03. Motor Neurone
Leonard Van Den Berg
Department Of Neurology, University Medical Center Utrecht, Utrecht, NL
Abstract not received.
PL 3.3 / #182ETHICAL ISSUES
Topic: 03. Motor Neurone
David J. Oliver
Tizard Centre, University of Kent, Canterbury, GB
Background: The care of a person with Motor Neurone Disease is often complex, as they face multiple issues – physical, psychosocial and spiritual. Within these issues are often complex ethical issues, involving difficult discussions with patient and family and within the multidisciplinary team. Careful discussion and consideration of the issues is needed, to enable patients and families to make the most appropriate decision. Methods: A review of the literature, combined with clinical experience, has been undertaken. Results: The ethical issues may occur throughout the disease progression: - At diagnosis The telling of the diagnosis may raise issues – particularly if the family is resistant to telling a patient, who has capacity and is asking for information, or the patient has lost capacity Many patients may now have genetic testing and there are issues as to how they react to knowing that they have a gene mutation.This may influence their future and family plans and they face a dilemma as to what to tell other family member who may be at risk of having the same mutation - When swallowing problems start and become more difficult consideration maybe given to the insertion of a gastrostomy. This discussion may occur when respiratory function is deteriorating. A gastrostomy may improve quality of life, and maybe survival but patients may find the change in feeding very difficult to cope with - Ventilatory support – non-invasive ventilation and, on occasions, invasive ventilation with a tracheostomy - may be considered if respiratory failure develops. Although these interventions may improve symptoms the disease progresses and the patient faces increasing disability, and even the risk of becoming locked in – with no communication. These issues need to be discussed before starting the intervention - At the end of life patients may request treatment withdrawal, such as withdrawal of ventilatory support. There may be complex discussion between family members and team members, who may understand these ethical dilemmas in a different way - Some patients, and families, may ask about assisted dying. In some countries this may be an option – of euthanasia or physician assisted suicide – and there are ethical issues balancing the wishes of the patient and family. In many countries there is no option, but the issue raises discussion for all concerned. Throughout the care of a person with MND there is the need to assess mental capacity to make decisions. As there is increasing evidence that up to 15% of patients may develop fronto-temporal dementia and a further 35% may show evidence of frontal lobe dysfunction, which may affect decision making, these issues have profound implications. Conclusion: Ethical issues need to be considered carefully for all patients with MND throughout the disease progression and involves careful discussion between patient and family and all members of the wider multidisciplinary team.
PL 4.1 / #47PHYSIOLOGY AND STRUCTURE
Topic: 04. Neuromuscular Junction
Steve Burden
Skirball Institute, Nyu Medical School, New York, US
Abstract not received.
PL 4.2 / #121CONGENITAL MYASTHENIC SYNDROMES - NEW GENES AND BETTER TREATMENTS / ANTIBODIES
Topic: 04. Neuromuscular Junction
Kinji Ohno
Nagoya University Graduate School of Medicine, Nagoya, JP
Abstract not received.
PL 4.3 / #171AUTOIMMUNITY
Topic: 04. Neuromuscular Junction
Angela Vincent
Dep. Of Clinical Neuroscience, University of Oxford, Nuffield, Oxford, GB
Abstract: In the 1970s, myasthenia gravis was shown to be caused by antibodies to the acetylcholine receptors (AChRs) on the postsynaptic surface of the neuromuscular junction (NMJ). The mechanisms of AChR antibodies include internalisation of AChRs caused by divalent antibodies, complement-mediated lysis of the postsynaptic membrane, and less common direct inhibition of AChR function. Together, and variably, these cause loss of AChR numbers or function. The weakness is caused by failure of the endplate potential to reach the critical threshold for activation of the compound muscle action potential. This is compounded by the loss of postsynaptic folds where the voltage gated sodium channels that initiate the action potential, are situated. Interestingly, the presynaptic nerve terminal and the postsynaptic muscle both ”compensate” for the defect in transmission by upregulating acetylcholine release and AChR synthesis respectively. There are different subtypes of MG, with thymectomy of established benefit in the young-onset patients, but an increasing number of late-onset patients (>50 years at onset) who often have co-morbidities and present a challenge. Some of the patients who do not have AChR antibodies by conventional assays, have antibodies that only bind detectably to ”clustered AChRs” expressed on cell lines. This emphasises the importance of clustering of AChRs at the NMJ and for synaptic function in general. Some of these patients do not have EMG evidence of MG and are less severe but respond well to immunotherapies. In 2001, antibodies to MuSK were found in some of the myasthenia patients who were negative for AChR antibodies (seronegative MG); these patients often have marked bulbar and facial involvement which can be difficult to treat with the immunotherapies as used for AChR-MG. The MuSK antibodies are predominantly, but not exclusively, IgG4 and the anti-CD20 drug, rituximab, appears to work well in many MuSK-MG patients. More recently antibodies to another NMJ protein, LRP4, have been found in a small number of additional patients. MuSK and LRP4 are both postsynaptic membrane proteins that are responsible for the agrin-induced clustering of the AChRs at the neuromuscular junction. When LRP4 binds agrin, MuSK is activated, DOK7 binds and this begins a sequence of intracellular events that maintain the high density of AChRs at the NMJ. Anything that interferes with LRP4 or MuSK membrane molecules would be expected to lead to dispersal of AChRs, but the detailed mechanisms are still not fully explored. A better understanding could lead to novel pharmacological treatments as will be described. Koneczny I, Cossins J, Vincent A. The role of muscle-specific tyrosine kinase (MuSK) and mystery of MuSK myasthenia gravis. J Anat. 2014 Jan;224(1):29-35. Huda S, Cao M et al. Increased phosphorylation of MuSK provides a potential therapy for MuSK-Ab myasthenia gravis. 2018 submitted for publication.
Scientific Sessions
SS 1.1 / #38TREATMENT STRATEGIES IN GNE MYOPATHY
Topic: 01. Muscle
Zohar Argov
Hebrew University of Jerusalem EKMD, Jerusalem, IL
Background: Introduction: GNE myopathy (previously known as HIBM or DMRV) is associated with mutations in the gne gene, a bifunctional key enzyme in synthesis of sialic acid (neuraminic acid).As a result a progressive distal myopathy with relative quadriceps sparing is observed. The typical histopathology is composed of ‘rimmed vacuoles’, which are autophagic vacuoles. Background: The degree of reduction in enzyme activity ranges between 30-60%. Hyposialylation of muscle is observed in transgenic animal model, but is not consistently found in affected human muscle. It is possible that GNE myopathy is caused by additional mechanisms apart from metabolic deficiency. Methods: The degree of reduction in enzyme activity ranges between 30-60%. Hyposialylation of muscle is observed in transgenic animal model, but is not consistently found in affected human muscle. It is possible that GNE myopathy is caused by additional mechanisms apart from metabolic deficiency. Results: 1. In the mouse model supplementation with sialic acid or its metabolic derivatives led to improvement. 2. In a phase 3 trial in patients receiving 6 gr/day of sialic acid for 1 year, no change in muscle power was found. A phase 2 trial with the metabolic intermediate ManNac is ongoing. 3. An AAV-GNE construct was shown to deliver normal human GNE to muscle in normal and diseased animals, with persistence for prolonged period. A human trial is in preparation. Conclusion: Discussion: supplementation of sialic acid or its derivatives may not suffice to correct the muscle disease. Viral mediated gene therapy may be more suitable at this stage when basic mechanism is still controversial. In the meantime trials with medication affecting autophagy or other downstream processes should be encouraged.
SS 1.2 / #283UPDATE OF RNA-BASED NMD THERAPIES
Topic: 01. Muscle
Kanneboyina Nagaraju
Pharmaceutical Sciences, SUNY-Binghamton University, Binghamton, NY, US
Background: Recent advances in genetics and genomics significantly enhanced our ability to manipulate gene expression at RNA level in vitro in cell culture conditions and in vivo in mouse models and in patients with neuromuscular diseases (NMDs). Methods: The US Food and Drug Administration (FDA) recently approved Exondys 51 (eteplirsen), a drug that specifically targets Exon 51 to induce its exclusion in dystrophin mRNA resulting in a truncated functional dystrophin protein. Efforts are currently underway to target different exons in Duchenne Muscular Dystrophy using different chemistries to enhance efficiency of dystrophin protein expression in skeletal and cardiac muscle. Results: This presentation will briefly cover various RNA targeting approaches (Exon exclusion, inclusion, exchange, altering mRNA decay and stop codon read through), stage of their current development and potential scientific, regulatory and ethical hurdles involved in developing these drugs for Neuromuscular diseases. Conclusion: The potential for RNA based approaches to treat NMDs is excellent.
SS 1.3 / #59PROGRESS IN THE TREATMENT OF IDIOPATHIC INFLAMMATORY MYOPATHIES?
Topic: 01. Muscle
Marianne De Visser
Department Of Neurology, Academic Medical Centre, Amsterdam, NL
Abstract: Idiopathic inflammatory myopathies (IIMs) can be distinguished into subacute onset and treatable disorders (dermatomyositis (DM), necrotizing immune mediated myopathy (IMNM), antisynthetase syndrome (ASS), non-specific myositis (NSM)) and chronic inclusion body myositis (IBM). The mainstay of treatment in the treatable IIMs is long-term oral glucosteroids, and second line immunosuppressants are often added not only to obtain better efficacy but also for its steroid-sparing effect. If patients do not respond quickly to oral steroids one might consider intravenous administration of methylprednisolone or add third line medication such as rituximab (RTX) of intravenous immunoglobuline (IVIg). A trial with RTX on DM/PM did not reach the primary and secondary outcome measures. However, most of the patients clearly improved. IVIg was investigated in two RCT’s and various open-label prospective trials on refractory DM/PM patients and was found to be clinically efficacious. Still, long-term outcome in most IIM patients is not favourable. IIM patients have a polycyclic or chronic continuous course in almost 70% of the cases with similar percentages of perceived disabilities and lower quality of life scores after long-term follow up. This requires other vigorous treatment modalities, which not only aim at suppressing inflammation but also at preventing muscle damage. Increased knowledge about the pathomechanisms of IIMs will likely advance new and targeted treatments. Recent data suggests that exercise could be an effective anti-inflammatory treatment albeit the underlying mechanisms are not fully understood. IBM is as yet considered to be treatment-resistant. Not only is IBM refractory to the conventional immunosuppressive and immunomodulating modalities, but recently a phase 2b/3 study with bimagrumab, a monoclonal antibody that acts via the myostatin-mediated induction of muscle growth failed its primary endpoint. The results of a proof-of-concept study with rapamycine were promising. Conducting clinical trials in IIMs is challenging for many reasons. First, classification seems to be a moving target with the ever-increasing number of myositis-specific autoantibodies giving rise to further subclassification. Second, consensus on assessment tools that are highly responsive to interventions is required. The International Myositis Assessment and Clinical Studies Group ‘response criteria’ are recently published and could serve as such. Third, patient reported outcome measures are indispensible to evaluate the efficacy of a therapy and currently, they are under development.
SS 2.1 / #224TISSUE ENGINEERING STRATEGIES FOR REPAIR OF PERIPHERAL NERVE
Topic: 11. Nerve Regeneration
Anthony Windebank
Mayo Clinic, Rochester, US
Abstract not received.
SS 2.2 / #1019USE OF PHYSICAL METHODS IN NERVE REGENERATION
Topic: 11. Nerve Regeneration
Thomas Hausner
Trauma Surgery, Lorenz Böhler Trauma Hospital, Vienna, AT
Abstract: Regeneration and recovery of peripheral nerves after injury still today is a long-lasting procedure. Human nerves elongate with an average speed of 1 mm per day. Transection of a nerve at the level of the forearm e.g. with 25cm distance to the finger-tips would stand for about 250 days of recovery, given that rapid surgery has been performed corresponding to the actual technical standard. Increasing the speed of elongation would be very helpful in the post-surgical treatment. Electric stimulation since long time is used in nerve regeneration, with quite good results. However, new physical methods to increase axonal recovery are in development. Extracorporeal shock wave treatment (ESWT) is an established method in wound regeneration, bone regeneration and integration of skin grafts as well as treatment of some painful conditions as plantar fasciitis or humero-radial epicondylitis. In the last few years ESWT has been examined extensively in the field of nerve regeneration. Different in vivo and in vitro studies have been performed using the Dermagold 100 device (MTS, Germany). It could be clearly shown that axonal elongation is increased by a single use of ESWT immediately after surgery in different rat experimental nerve injury models. ESWT in vitro enhances stemness and preserves multipotency of adipose derived stem cells. ESWT in vitro also increases Schwann cell survival and activation state. ESWT may augment and potentiate the axonal elongation and functional recovery in a regenerating peripheral nerve segment. It also can be seen as a promising supporting tool for tissue engineering in peripheral nerve regeneration.
SS 2.3 / #378NEW DEVELOPMENTS IN CONDUITS
Topic: 11. Nerve Regeneration
David Hercher
Ludwig Boltzmann Institut, Vienna, AT
Abstract not received.
SS 3.1 / #70THE ROLE OF HU PROTEINS IN THE DEVELOPMENT OF PARANEOPLASTIC NEUROPATHIES
Topic: 08. Cancer
Bruno Giometto
Universita die Padova, Padova, IT
Abstract not received.
SS 3.2 / #79PHYSIOPATHOLOGY OF PARANEOPLASTIC GANGLIONOPATHIES
Topic: 08. Cancer
Romana Hoeftberger
Institute Of Neurology, Medical University of Vienna, Vienna, AT
Background: Paraneoplastic ganglionopathies are acquired neuronopathies that are characterized by a primary degeneration either of sensory neurons in dorsal root ganglia (paraneoplastic sensory neuronopathy, PSN) or sympathetic and parasympathetic neurons in autonomic ganglia (paraneoplastic autonomic neuronopathy, PAN). Methods: Summary of relevant findings in paraneoplastic ganglionopathies with emphasis on physiopathology and clinical presentation. Results: The PSN is mainly associated with small cell lung cancer and antibodies directed against intracellular antigens, so-called onconeuronal antibodies. These antibodies can be found in up to 80% of cases, most frequently anti-Hu or anti-CV2/CRMP5, less common amphiphysin antibodies. The pathogenetic concept is considered to be an immune response that is initially triggered by an aberrant expression of the onconeuronal antigen by tumor cells and subsequently misdirected against the nervous system. Neuropathological investigation reveales moderate inflammation with diffuse endoneurial T-cell, B-cell- and plasma cell infiltration in the spinal ganglia. CD8-positive T-cells are tightly attached to the surface of neurons indicating a T-cell mediated attack resulting in irreversible neuronal death. Nerve biopsy usually shows an axonal neuropathy with loss of large myelinated fibers and interstitial macrophages containing myelin debris. The clinical presentation is characterized by a subacute onset and rapidly progressive disease course with asymmetrical or multifocal numbness in the upper or lower limbs, severe impairment of joint position and vibratory sensation, pain, paresthesias, and sensory ataxia. Cranial nerve involvement may present as loss of taste, numbness of the face or sensorineural hypoacusia. PSN associated with anti-Hu antibodies often presents with additional CNS involvement such as encephalomyelitis but may remain an isolated syndrome in about 25% of cases. PSN associated with anti-CV2/CRMP5 antibodies more often presents as a mixed axonal and demyelinating neuropathy, predominates in the lower limbs, may have motor involvement, and pain is less frequent. PSN associated with amphiphysin antibodies may present with additional symptoms of stiff person syndrome or encephalopathy. Patients with PAN may either present as chronic intestinal pseudo-obstruction due to affection of the enteric plexus, or less common as subacute pandysautonomia with dysfunction of the sympathetic and parasympathetic nervous system. Patients may harbor anti-Hu antibodies, up to 20% may have ganglionic acetylcholine receptor antibodies. The underlying tumor in PSN and PAN often remains occult for a long time and search for a malignancy should focus in particular on the detection of a SCLC. Conclusion: Early diagnosis of paraneoplastic ganglionopathy by the detection of onconeuronal antibodies is important to confirm the diagnosis and initiate tumor screening. Early treatment of the cancer gives the best chance of stabilizing the disorder.
SS 3.3 / #133PARANEOPLASTIC MUSCLE DISORDERS
Topic: 08. Cancer
Andrew Mammen
NIAMS/NIH, Bethesda, US
Abstract not received.
SS 3.4 / #170LAMBERT-EATON-SYNDROME (LEMS)
Topic: 08. Cancer
Jan Verschuuren
Leiden University Medical Center, Leiden, NL
Abstract not received.
SS 4.1 / #159UPDATE ON CLINICAL ASPECTS OF GLYCOGEN STORAGE DISORDERS
Topic: 01. Muscle
Antonio Toscano
University of Messina, Messina, IT
Abstract not received.
SS 4.2 / #188NOVEL ENTITIES IN CONGENITAL MYOPATHIES OF ADULT ONSET
Topic: 01. Muscle
Baziel Van Engelen
Radboud University Medical Centre, Nijmegen, NL
Abstract not received.
SS 4.3 / #161NEW PHENOTYPES IN TITIN DEFECTS
Topic: 01. Muscle
Bjarne Udd
Tampere Neuromuscular Center, Tampere, FI
Abstract not received.
SS 5.1 / #1037STANDARDS OF CARE: WHAT WE HAVE LEARNED FROM LONG TERM-MANAGEMENT IN CHILDREN WITH NMD’S
Topic: 13. Pediatric
Guenther Bernert
Department Of Paediatrics, Kaiser Franz Joseph Hospital, Vienna, AT
Abstract not received.
SS 5.2 / #832FUTURE OF AAV-BASED GENE THERAPY APPROACHES FOR NMDS
Topic: 13. Pediatric
Thomas Voit
UCL Great Ormond Street Institute of Child Health, London, GB
Abstract: After more than a decade of animal experimentation, AAV-based clinical trials are now under way addressing Spinal Muscular Atrophy (SMA), Duchenne Muscular Dystrophy (DMD) and myotubular myopathy. They are addressing patients from infancy to young adults as well as different organ systems (CNS, skeletal and cardiac muscle). While too early to judge results across disease applications, there are important commonalities particularly regarding route of administration and maximum dosage in vector genomes/kg. At the same time, important questions remain, which need to be addressed in the future, both in animal experiments and in the human. These include the issues of hepatic and even potentially systemic toxicity of AAV capsids; pre-existing and treatment-induced immunity to AAV; the need for re-administration depending on the target tissue and the degree of genetic correction achieved. In some diseases such as SMA the route of administration remains an issue and has important consequences on the total dose, but potentially also on the biodistribution. In order to fine-tune these treatment approaches in the human biomarkers are becoming increasingly important to monitor the treatment effect over time. Finally, both in SMA and in DMD there is a potential to combine AAV-based gene therapy with chemical antisense-treatment, even if these technologies at the moment are largely developed in competition. These questions will be discussed in light of the most recent clinical and experimental data followed by a discussion of possible future steps.
SS 5.4 / #856TRANSITION FROM PEDIATRIC INTO ADULT CARE: A LONG WAY TO GO?
Topic: 13. Pediatric
Thomas Serjensen
Karolinska Institute, Solna, SE
Abstract not received.
SS 6.1 / #151GENERAL PRINCIPLES AND CLINICAL ASPECTS OF CIPN
Topic: 08. Cancer
Nathan Staff
Neurology, Mayo Clinic, Rochester, MN, US
Background: Chemotherapy-induced peripheral neuropathy (CIPN) is a common dose-limiting side effect that follows administration of several chemotherapeutic classes. While most CIPN is due to direct neurotoxicity of the chemotherapeutic, the recent introduction of check-point inhibitor cancer therapies has also resulted in a variety of immune-mediated neuromuscular side effects. CIPN occurs in up to 50% of all patients receiving neurotoxic chemotherapy and has been associated with both acute and long-term morbidities. Methods: Literature review was performed. Results: The clinical manifestations and pathomechanisms of CIPN from the different neurotoxic chemotherapy classes will be reviewed. The epidemiology and disease burden of CIPN will be explored by highlighting a recent study from Olmsted County, MN. Treatment and preventative strategies for CIPN, which have been largely unsuccessful, will also be described. Finally, a discussion surrounding the discovery and validation of CIPN susceptibility factors will be provided. Conclusion: CIPN continues to be a significant problem despite the development of more targeted cancer therapies. Key outstanding questions in CIPN are: 1) What are the pathomechanisms of CIPN? 2) Why is there variable susceptibility to CIPN? Answers to these key questions are expected to lead to rational predictive, preventative, and treatment strategies for this entity.
SS 6.2 / #196BASIC MECHANISMS: AXONAL DEGENERATION AND CHEMOTHERAPY-INDUCED PERIPHERAL NEUROPATHY
Topic: 08. Cancer
Ahmet Hoke
Johns Hopkins University School of Medicine, Baltimore, US
Abstract not received.
SS 6.3 / #51ANIMAL MODELS OF CHEMOTHERAPY-INDUCED PERIPHERAL NEUROPATHY
Topic: 08. Cancer
Guido Cavaletti
Head, Experimental Neurology Unit And Phd Program In Neuroscience School Of Medicine, University of Milano-Bicocca, Bicocca, IT
Abstract not received.
SS 7.1 / #157NEW GENES AND NEW MECHANISMS IN INHERITED NEUROPATHIES
Topic: 02. Neuropathy
Vincent Timmerman
Biomedical Sciences, University Of Antwerp, Antwerpen, BE
Background: New genes and new mechanisms in inherited neuropathies Vincent Timmerman Peripheral Neuropathy Research Group, University of Antwerp, Belgium Inherited peripheral neuropathies (IPN) are genetic conditions affecting 1 in 2,500 individuals. Charcot-Marie-Tooth (CMT) neuropathy is the best known form characterized by a vast phenotypic and genetic heterogeneity. The identification of mutations in more than 100 known genes through gene screening panels, whole exome or genome sequencing to find variants in novel disease causing genes, in combination with the increasing availability of cell and animal models, has deepened our understanding in the disease mechanism. The heterogeneity is complicated by the fact that CMT associated genes have diverse functions; they regulate myelination through Schwann cells, take care of axonal transport and neuronal metabolism, or have essential cellular functions by regulating autophagy and protein quality control. However, despite this knowledge there is still no effective therapy for most patients with CMT and related disorders. Methods: More recently novel models, such as the use of patient derived induced pluripotent stem cells, and neurons differentiated from these cells, in combination with omics approaches has opened novel opportunities for identifying common disease mechanisms and pathways. Results: These new methods will identify mode-of-actions and potential targets for therapy that are applicable for multiple CMT phenotypes. Conclusion: This information will complement IPN mutation databases, improve clinical and molecular diagnosis, and hopefully allow treatment strategies for some of these rare to ultra-rare IPN phenotypes.
SS 7.2 / #148THERAPEUTIC OPTIONS IN CMT
Topic: 02. Neuropathy
Michael Sereda
Neurogenetics, Max-Planck-Institute of Experimental Medicine, Gottingen, DE
Abstract not received.
SS 7.3 / #126FAMILIAL AMYLOID NEUROPATHIES: A TREATABLE GENETIC NEUROPATHY
Topic: 02. Neuropathy
Davide Pareyson
Dept Of Clinical Neurosciences, IRCCS Foundation, C.Besta Neurological Institute, Milan, IT
Abstract: Inherited transthyretin amyloidosis (ATTR) is an autosomal dominant disorder due to mutations of the transthyretin (TTR) gene. TTR is synthetized mainly by the liver and released in plasma as a tetrameric transport protein. Mutations in TTR, of which Val30Met is the most common worldwide, cause transthyretin tetramer dissociation, monomer misfolding, and aggregation into insoluble fibrillar proteins in different tissues. Orthotopic liver transplantation (OLT), by removing the main site of mutated TTR production, proved able to halt or slow neurological progression and was until few years ago the standard-of-care treatment in patients aged <50 years with Val30Met mutation. OLT is associated however with not negligible mortality rate and is not curative, since cardiac disease tends to progress and leptomeningeal amyloid deposition may become an issue. Tafamidis meglumine is a small molecule which kinetically stabilizes the TTR tetramer and prevents its dissociation into amyloidogenic monomers. It proved able to slow down disease progression particularly in the early disease phases and is currently approved in several countries for treatment of symptomatic patients with polyneuropathy in disease stage I (independent ambulation). Responders were also described among patients with late-onset disease, non-Val30Met mutations, and in later disease stages. The old nonsteroidal anti-inflammatory drug diflunisal has also been reported to be effective as a TTR tetramers stabilizer and produced significant slowing in disease progression in treated patients. The antibiotic doxycycline and the taurine conjugate form of ursodeoxycholic acid (TUDCA) have a synergistic effect on fibril disruption and their use is currently under investigation with encouraging results. Monoclonal antibodies directed against either the serum amyloid P component (SAP) or the amyloidogenic forms of transthyretin constitute another strategy to clear the amyloid deposits. Antisense Oligonucleotides (Inoserten) or interfering RNA lipid nanoparticles (Patisiran) binding to wild type and mutated TTR mRNA proved able to reduce TTR production by more than 75% and both proved very effective in two recently completed phase III trials. Both trials reached the primary endpoints and multiple measures were significantly better in treated as compared to untreated patients: stabilization or improvement occurred for a good proportion of treated patients. Inoserten was administered subcutaneously once a week for 15 months; thrombocytopenia and renal problems were adverse events which required monitoring of platelet and renal function. Patisiran, administered intravenously every 3 weeks for 18 months, was well tolerated with infusion-related reactions and peripheral edema as the most relevant reported side effects. Both treatments were effective independently from disease stage, presence of cardiopathy, type of mutation. The development of such novel therapies is changing the natural history of ATTR-neuropathy from a relentlessly progressive disorder inexorably leading to death into an effectively treatable disorder.
SS 8.1 / #1028CRANIAL NERVES: WITHIN AND OUT OF THE SKULL, INCL. ANGIOSOMA
Topic: 07. Cranial Nerves
Wolfgang Grisold1, Anna Grisold2, Stefan Meng3
1Ludwig Boltzmann Institute for Experimental und Clinical Traumatology, Vienna, AT;2Dep. Neurology, Allgemeines Krankenhaus Vienna, Vienna, AT;3Institute for Radiology, KFJ hospital, Vienna, AT
Abstract: The course of cranial nerves (CN) is characterized by their intraparenchymal, intracranial part, the site of exit of the skull and their peripheral course. Thus dysfunction can be due to brainstem lesions, meningeal and CSF involvement, local changes at their exit of the skull, and a variety of conditions in their extracranial course. All parts of the cranial nerves are dependent on blood supply, which is provided by small arteries.The distribution of the blood supply follows the distribution of angiosomas, which follows a concept applicable for the whole body. According to this concept of blood supply, individual nerves receive the blood supply in different segments from adjacent angiosomas. As a practical consequence, intravascular interventions as embolizations of tumors of the base of the skull, can damage cranial nerves. As indirect signs of damage of motor nerves, also the MR visualization of muscles at the base of the skull is useful, which helps to detect asymmetries and neurogenic lesions. The tongue muscles as an example for strict unlilateral blood supply is a good example. Increasingly ultrasound can be used to demonstrate the extracranial parts of the nerves, where also thickening, and local tumors can be demonstrated.
SS 8.2 / #44INFECTIOUS AND INFLAMMATORY CRANIAL NERVE LESIONS
Topic: 07. Cranial Nerves
Nazha Birouk
Clinical Neurophysiology Rabat, Speciality Hospital, Rabat, MA
Abstract: Cranial nerves can be involved in various infectious and inflammatory diseases either as a mononeuropathy or multicranial neuropathy. The diagnosis can be easily made in conditions with other expressions of the infection or inflammations within the nervous system or in extra-neurologic organs with in some conditions typical presentations. The diagnosis might be more challenging when the cranial neuropathy is the revealing symptom. The most frequent infection worldwide remains an active infectious ganglioneuritis caused by varicella zoster virus, producing shingles. The commonly associated cranial nerve neuropathies are: herpes zoster ophthalmicus (10% to 20% of all zoster cases), the Ramsay Hunt syndrome (triad of ipsilateral facial paralysis, ear pain, and vesicles in the auditory canal and auricle) and optic neuritis. Facial nerve palsy can be due to Lyme borreliosis with a similar clinical presentation to Bell’s palsy. This infection is to be searched in endemic areas, in the presence of skin rash or if the facial palsy is bilateral and/or recurrent. Cranial neuropathies in HIV occur at various stages of infection and are sometimes multifocal. Facial nerve palsy, for example, can develop at the time of seroconversion or later in the disease course secondary to opportunistic infections. In immunocompetent patients, many infectious conditions can be accompanied by cranial neuropathies such as meningitis due to tuberculosis, syphilis, listeriosis, cryptococcus and various other infectious agents. Cranial nerves might be impaired by contiguous infection as otitis or sinusitis. Cranial neuropathy can be associated to systemic inflammatory disorders such as sarcoidosis (mostly nerves VII, VIII and II), Sjogren syndrome (mostly nerves V and II) or ANCA vasculatis (nerves III, IV, VI, VII and X) and others. IgG4-related disease has been newly identified with a wide spectrum of diseases that can involve nearly any organ system, including the central and peripheral nervous systems. The principal neurological manifestations result from orbital disease, pachymeningitis, and pituitary gland and stalk involvement. The common aspect is an inflammatory pseudotumor with possible impairment of oculomotor (as Tolosa Hunt syndrome), trigeminal and optic nerves. The diagnosis is based on the serum IgG4 elevation, the histological aspects with significant infiltration with IgG4 positive plasma cells, and the elimination of other inflammatory disorders like vasculitis. The disease responds well to treatment by corticosteroids. Specific inflammatory diseases of central nervous system such as multiple sclerosis and NMO imply cranial neuropathies, mostly optic neuritis but also other nerves such as oculomotor, facial and trigeminal nerves. We should keep in mind that an authentic Guillain Barre syndrome can rarely present with isolated bilateral facial nerve or multicranial nerves palsy.
SS 8.3 / #230PAINFUL CRANIAL NERVE LESIONS
Topic: 07. Cranial Nerves
Frank Thömke
Klinikum Worms gGmbH Fachbereich Neurologie, Worms, DE
Background: This presentation deals with cranial nerve palsies with persistent pain lasting days to weeks. Methods: Description of clinical signs, diagnosis and therapy. Results: Optic neuritis may be accompanied by persistent pain on the affected side, which occurs or increases with movements of the affected eye. Pain usually decreases with high does steroid treatment (usually 1000 mg methylprednisolone for 3 days). MRI should be done since optic neuritis is a frequent initial symptom of multiple sclerosis. Moreoever, one should look for aquaporin-4 antibodies (NMO-IgG) to detect the beginning of a neuromyelitis optica. Painful ocular motor nerve lesions are often summarized as ”painful ophthalmoplegia, a heterogenous group of diseases characterized by paresis of one or more ocular motor nerves associated with frontal or orbital pain. Vascular, inflammatory or tumorous diseases are main causes. Up to one third of ocular motor nerve palsies are attributed to microvascular nerve infarctions, a diagnosis by exclusion. Most of the patients complain ipsilateral pain, especially with 3rd nerve palsies. This must be differentiated from 3rd nerve palsies due to an aneurysm, which is easily done by MRI- or CT-angiography. Diagnosis of a Tolosa Hunt syndrome depends, among others, on the demonstration of granulomatous inflammation of the cavernous sinus, the superior orbital fissure or the orbit demonstrated by MRI or biopsy. Pain promptly resolves with steroid treatment (usually 1 mg prednisolone or methylprednisolone per kg body weight). Abnormal eye movements recover within several weeks, which seem to be equally probable with or without steroids. Diagnosis of recurrent painful ophthalmoplegic neuropathy (“ophthalmoplegic migraine”) may be considered after two attacks and exclusion of an orbital, parasellar or posterior fossa tumor. In some patients, MRI may disclose gadolinium-enhancement of a thickened 3rd nerve. Pain promptly decreases with steroid treatment (usually 1 mg prednisolone or methylprednisolone per kg body weight). Abnormal eye movements recover within several weeks, which seem to be equally probable with or without steroids. Persistent pain with trigeminal nerve lesions may occur with acute or previous Herpes zoster, previous trauma, multiple sclerosis, or space occupying lesions. Herpes zoster may be diagnosed by laboratory findings, otherases by MRI or (CT). Herpes Zoster is treated with aciclovir or valaciclovir, and persistent pain due to multiple sclerosis with high dose steroids (usually 1 000 mg methylprednisolone for 3 to 5 days). Pain is a prominent symptom in facial palsy due to Herpes Zoster (Ramsay Hunt syndrome) and treated with aciclovir or valaciclovir (and steroids) added by analgetics and/or anticonvulsant substances such as carbamazepine or pregabalin. Bell’s palsy may also be associated with retroauricular pain, which resolves with steroid treatment of Bell’s palsy. Painful lesions of one or multiple of these nerves may occur with space occupying lesions or meningeosis neoplastica. They are often associated with diffuse headache and focal signs like hemiparesis or hemiataxia. Treatment depends on the etiology and involves neurosurgical procedures, steroid and radiation. Conclusion: MRI (or CT) should be done in most patients with painful cranial nerve lesions, and MRI- or CT-angiography in some of them. Steroids are an effective treatment in most cases.
SS 8.4 / #190PAROXYSMAL DYSFUNCTIONS
Topic: 07. Cranial Nerves
Friedrich Zimprich
Department Of Neurology, Medical University of Vienna, Vienna, AT
Abstract: Neuromuscular specialists are often required to evaluate patients displaying symptoms of transient (upper) cranial nerve dysfunctions, with ocular myasthenia gravis being a main differential diagnosis. In fact there is a wide range of different diseases which might manifest as oculomotor dysfunction with or without ptosis either as a consequence of cranial nerve palsies (III, IV or VI) or because they mimic these conditions. While many of these disorders may eventually result in a typical, i.e. clearly recognizable clinical picture the presentation in the initial stages or in mild cases can take the shape of fluctuating or paroxysmal, transient symptoms, which obviously stands in the way of finding a fast and definite diagnosis. The possible differential diagnoses range from ocular myasthenia to other myopathies (e.g. mitochondrial disorders), autoimmune diseases (e.g. thyroid-associated ophthalmopathy), inflammatory neuropathies, diseases affecting the intracranial pressure (intracranial hypotension or pseudotumor cerebri), vascular ischaemic diseases, diabetes associated cranial nerve palsies, inflammatory lesions of the central nervous system, nerve sheath tumors or other space occupying lesions along the nerves to ocular neuromyotonia. This differential diagnoses will be discussed and illustrative cases will be presented.
SS 9.1 / #1013PROTEINOPATHIES (THE AUTONOMIC VIEW)
Topic: 05. Autonomic
G Wenning
Department Of Neurology, Medical University Innsbruck, Innsbruck, AT
Abstract not received.
SS 9.2 / #1014PROTEINOPATHIES (THE NEUROMUSCULAR VIEW)
Topic: 05. Autonomic
Walter Struhal
Department For Neurology, University Clinic Tulln, Karl Landsteiner University of Health Sciences, Tulln, AT
Abstract not received.
SS 9.3 / #1015HEREDITARY NEUROPATHIES (THE AUTONOMIC VIEW)
Topic: 05. Autonomic
Max J. Hilz
Neurology, Universitätsklinikum Erlangen, Erlangen, DE
Abstract not received.
SS 9.4 / #1016HEREDITARY NEUROPATHIES (THE NEUROMUSCULAR VIEW)
Topic: 05. Autonomic
Michaela Auer-Grumbach
Department Of Orthopaedics, Medical University of Vienna, Vienna, AT
Abstract not received.
SS 9.5 / #1017METABOLIC ANS DISEASE (DIABETES AND LIVER) (THE AUTONOMIC VIEW)
Topic: 05. Autonomic
R Freeman
Beth Israel Deaconess Medical Center, Boston, US
Abstract not received.
SS 9.6 / #1039METABOLIC ANS DISEASE (DIABETES AND LIVER) (THE NEUROMUSCULAR VIEW)
Topic: 05. Autonomic
Paola Sandroni
Mayo Clinic, Rochester, US
Abstract not received.
SS 10.1 / #462017 ADA POSITION STATEMENT ON DIABETIC NEUROPATHY: DOES IT CHANGE YOUR PRACTICE?
Topic: 02. Neuropathy
Vera Bril
Department Of Medicine (neurology), University Health Network, University of Toronto, Toronto, CA
Abstract not received.
SS 10.2 / #144SCIENTIFIC DISCOVERY LEADS TO CLINICAL TRIALS IN DIABETIC NEUROPATHY
Topic: 02. Neuropathy
James Russell
University of Maryland School of Medicine, Baltimore, US
Abstract: A complex constellation of pathways are directly or indirectly affected in diabetic neuropathy. Dissecting out these pathways can lead to development of new approaches to reverse neuropathy and treat neuropathic pain. Despite the focus on glycemic control in diabetes, the evidence from randomized clinical trials that improved glycemic control reduces the prevalence of neuropathy in the majority of diabetics is unclear. The evidence for type I diabetes is strong but weak or absent for type 2 diabetes mellitus. While many potential pathways have been recognized as potential targets for therapy, this lecture will focus on mechanisms that currently offer or may offer immediate translation into clinical practice. While many clinical trials in diabetic neuropathy have failed, increasingly trials are being initiated by investigators that understand the complexities of mechanism and trial design. They appreciate the importance of sensitive and rapidly responsive efficacy measures in mild neuropathy observed early in the course of diabetes, when response to therapy is most likely to occur. There is considerable interest in the role of lifestyle interventions in reversing diabetic neuropathy. However, what is not always appreciated is that the dietary and exercise manipulations have clear and measureable effects on specific biological pathways similar to the effect of a medication. These interventions affect critical signaling pathways that regulate mitochondrial function, generation of reactive oxygen species, lipid metabolism, and growth factor production that may lead to nerve regeneration. Lipidomic characterization may lead to not only understanding how lipid pathways are implicated in diabetic neuropathy (pathway characterization) but also how manipulation of abnormal lipid metabolism may lead to therapy. Already, there is evidence that modification of lipid metabolism can result in improvement in diabetic small fiber neuropathy in humans. A novel approach to therapy has resulted from the observation that muscarinic receptor antagonists are implicated in protection from axonal degeneration both in vitro and in vivo. Neurite outgrowth from sensory neurons is in part mediated by muscarinic receptor-dependent regulation of mitochondrial function through the AMPK signaling pathway. Consistent with these observations in vitro, pharmacological blockade of the muscarinic receptor, M1R, using specific or selective antagonists, prevented or reversed peripheral neuropathy and reduced diabetes-induced mitochondrial dysfunction in vitro and in vivo. Work in vivo is currently being translated into clinical trials. Finally, the implications of specific drugs that target the Peroxisome proliferator-activated receptor-gamma co-activator 1alpha (PGC-1alpha) signaling pathway, which critically regulates mitochondrial and lipid metabolism, and the potential for translation to clinical trials will be discussed. Common polymorphisms of PGC-1alpha are associated with conversion from impaired glucose tolerance to diabetes and loss of PGC-1alpha causes neuropathy that is worsened by diabetes. Thus, understanding the critical mechanisms that lead to diabetic neuropathy may result in improved therapy and management of neuropathic symptoms.
SS 10.3 / #68PAINFUL DIABETIC NEUROPATHY: A MANAGEMENT CENTERED APPROACH
Topic: 02. Neuropathy
Eva Feldman
University of Michigan, Ann Arbor, US
Abstract: Pain is a persistent and morbid complication of diabetic polyneuropathy (DPN). There are no compelling data supporting the concept that glycemic control alone improves DPN pain, and most physicians and patients turn toward pharmacologic treatment. The American Diabetes Association, the American Academy of Neurology and the Canadian Pain Society are among the organizations that have issued pain management guidelines for DPN. DPN is controlled by different classes of agents: monoamine reuptake inhibitors and anticonvulsants are the main stay of therapy. Two agents, duloxetine (a selective norepinephrine and serotonin reuptake inhibitor) and pregabalin (a calcium channel a2-δ subunit ligand anticonvulsant) are approved by the FDA, Health Canada and the European Medicines Agency for painful DPN treatment. Other effective monoamine reuptake inhibitors, not officially approved, include the tricyclic antidepressant amitriptyline, the secondary amines, nortriptyline and desipramine and the dual reuptake inhibitor venlafaxine. Among the effective anticonvulsants that lack formal agency approval, gabapentin (another ligand of the calcium channel a2-δ subunit) is most commonly used, followed by topiramate and less commonly carbamazepine or phenytoin. Following guidelines, either duloxetine or pregabalin are used in the initial treatment of painful DPN; if neither single agent provides pain relief, the drugs are taken together, depending on side effect profiles. A common final dosage of duloxetine is either 60 or 120 mg/day, depending on tolerability; pregabalin is 150 mg, either twice or thrice daily, again dependent on tolerability. When drug cost is an issue, an alternative to the above approach is to begin with gabapentin, reaching doses up to 1,800 mg to 3,600 mg/day in 3 to 4 divided doses. The tertiary amine amitriptyline can be added to gabapentin, or also begun as sole therapy, at nightly doses that range from 50 up to 100 mg. The secondary amines, nortriptyline and desipramine, are preferred over amitriptyline, if there is any history of cardiovascular disease or risk of urinary retention due to prostatism. While providing less pain relief that amitriptyline, the secondary amines have a more robust safety profile; finally, the dual reuptake inhibitor venlafaxine, also provides pain relief, although studies suggest pain reduction is less that with duloxetine. Dosages of 150 to 225 mg/day can be used alone or in combination with an anticonvulsant drug. Importantly, duloxetine or venlafaxine therapy is best combined with anticonvulsant therapy; combination with the tricyclic antidepressants should be used only with caution secondary to adverse patient outcomes. Tapentadol is a centrally acting opioid with a dual mechanism of action: inhibition of noradrenaline reuptake and activation of the μ-opioid receptor. While tapentadol is FDA approved, the International Association for the Study of Pain’s Special Interest Group on Neuropathic Pain (NeuPSIG) completed a systematic meta-analysis and determined that the efficacy of tapentadol in the treatment of painful DPN was uncertain. The use of tapentadol is therefore not recommended as a first or second line treatment, and opioids as a class of drugs should be avoided in the treatment of painful DPN.
SS 11.1 / #851JAMES LIND ALLIANCE PRIORITY SETTING PARTNERSHIP IN NEURO-ONCOLOGY (RESEARCH PRIORITIES)
Topic: 12. Patient Issues
Kathy R. Oliver
International Brain Tumour Alliance (IBTA), Tadworth, GB
Background: It is crucial that what patients value is reflected in research relating to treatment, care and support. This is particularly relevant in rare cancers, like brain and spinal cord tumours, where many clinical questions about these diseases remain unanswered. The James Lind Alliance (JLA - http://www.lindalliance.org/) was established in 2004 and is coordinated by the UK National Institute for Health Research (NIHR). It brings patients, caregivers and clinicians together in a ‘Priority Setting Partnership’ (PSP) to determine the top ten unanswered questions relating to a particular disease. Methods: The JLA PSP in neuro oncology (N-O) identified clinical research questions that were most important to people living with a brain or spinal cord tumour. The project’s scope was: clinical uncertainties of interventions for primary brain or spinal cord tumours, in people of any age, from diagnosis to terminal stages. The methods for determining the top ten uncertainties were: 1. identification of stakeholder representatives (28) from a cross-section of the brain tumour community; 2. obtaining funding from four brain tumour charities, Cochrane Neuro-Oncology, and Edinburgh and Lothian Health Foundation; 3. developing and producing a protocol and website (www.neuro-oncology.org.uk); 4. publicising the JLA PSP N-O survey to UK brain tumour communities and those abroad using multi-disciplinary professional and charity databases; 5. analysing first round survey results and eliminating duplicate questions and those already answered by previous research; 6. categorising and standardising questions into PICO (participants, interventions, comparisons, outcomes) format; 7. carrying out systematic Cochrane-style literature searches to ensure genuine uncertainties; 8. prioritising questions using standard JLA PSP methodology and unbiased facilitators; 9. conducting a second public survey on a short-list of prioritised questions; 10. holding a workshop to determine the final top ten questions using a modified Delphi and Nominal Group technique. Results: Over 600 individual questions were generated from the initial survey, a patient forum and the Database of Treatment Uncertainties (DUETS). A second public survey was distributed (44 PICO questions created from uncertainties resulting from the initial survey). Over 200 people responded and voted for their top ten research priorities for brain and CNS tumours. The final top ten uncertainties were agreed. Much of the work of the JLA PSP N-O evolved to form a core activity of the UK National Cancer Research Institute (NCRI) Brain and CNS Clinical Studies Subgroup on Supportive and Palliative Care, which is approaching UK governmental, neuroscience and charity funders to support research into the top ten uncertainties identified. Conclusion: The JLA PSP N-O collaboration was an enlightening and successful project. It will now be crucial to pro-actively promote the top ten research uncertainties for brain and CNS tumours in order to focus attention on what really matters to people affected by these diseases. It is vital to obtain reliable, evidenced-based information about these uncertainties through robust research focussed on the JLA PSP N-O top ten priorities.
SS 11.2 / #212HOW TO DO INVESTIGATOR-INITIATED TRIALS: PCORI
Topic: 12. Patient Issues
Richard J. Barohn
Neurology, University of Kansas Medical Center, Kansas City, KS, US
Abstract: In recent years there has been a great effort to get patients, families, community and patient advisory groups more involved in the clinical research process. When developing an idea for a clinical research project we get patients involved to identify their needs and what areas they believe we should focus on. We then place patients on the project steering committees, communication committees and Data Safety Monitoring Boards (DSMB). Outcome measures are increasingly becoming patient driven and the field of patient reported outcomes measures (PROM) is extremely important. We use various patient reported outcomes as either primary or secondary outcomes. In the USA such research is funded by the Patient Center for Outcomes Research Institute (PCORI). One such funding mechanism through PCORI is for comparative effectiveness research (CER). The goal is to compare the effectiveness of two or more interventions or approaches to health care by answering questions important to patients and other stakeholders. We have been successful in obtaining one of these awards to conduct a comparative effectiveness study of drugs for management of pain in cryptogenic sensory polyneuropathy (CSPN) called Patient Assisted Intervention for Neuropathy: Comparison of Treatment in Real Life Situations (PAIN CONTRoLS). We are comparing four medications (nortriptyline, duloxetine, pregabalin, mexiletine) at 45 sites in the USA and Canada. We enrolled 402 patients. The study assessed both effectiveness in reducing pain and quits due to various factors and utilized a novel Bayesian adaptive design. The results showed nortriptyline and duloxetine were more likely to have pain relief and fewer quits than pregabalin and mexiletine. We utilized patients and their caregivers in developing the concept and preparing the grant application. PCORI created an award that provides infrastructure in which the PCORI grants can operate through. This infrastructure called (PCORnet, the National Patient-Centered Clinical Research Network) goal is to foster a wide range of experimental and observational patient-centered studies. This network is broken down to Clinical Data Research Networks (CDRNs). University of Kansas Medical Center has a CDRN called the Greater Plains Collaborative (GPC). One of the first accomplishments by the GPC was to deploy the development of the Healthcare Enterprise Repository for Ontological Narration (HERON) system at KUMC. This is an i2B2 software program that allows use of electronic medical records to survey for potential research subjects. One of the first projects was to develop a survey and utilizing the sites EMR, identify patients and utilize this list to send out the survey. We took an existing functional status form (the Amyotrophic Lateral Sclerosis Functional Rating Scale- Revised (ALSFRS-R)) and asked patients what they wanted to change or what additional questions they wanted to see poised to them regarding their functional status. We then modified the form and then submitted a protocol to be able to send out this survey.
SS 11.3 / #213THE PATIENT VOICE IN MYASTHENIA GRAVIS TRIALS- PCORI, CER, AND MORE ALPHABET SOUP
Topic: 12. Patient Issues
Pushpa Narayanaswami
Neurology, Beth Israel Deaconess Medical Center/Harvard Medical School, Boston, MA, US
Background: The Patient Centered Outcomes Research Institute (PCORI) is a nonprofit, nongovernmental organization in Washington, DC, that funds patient-centered comparative effectiveness research (CER). CER is the generation and synthesis of evidence that compares the benefits and harms of two or more interventions or approaches to manage a condition or improve healthcare. The US Food and Drug Administration defines patient reported outcomes (PROs) as ”any report of the status of a patient’s health condition that comes directly from the patient, without interpretation by a clinician or anyone else”. PROs assess outcomes that are important to patients; they cannot be developed without actively engaging patients with the condition of interest. PROMISE-MG is a PCORI funded prospective, observational, comparative effectiveness trial of immunosuppressant treatments for myasthenia gravis (MG). We describe our patient engagement process to select a patient centered primary outcome measure. Methods: Patients were actively engaged to identify a PRO by 1. Questionnaire survey of 58 people with self-identified MG at the annual Myasthenia Gravis Foundation of America meeting. 2. An online, web-based focus group of 13 people with MG, using the results of the survey to develop the focus group script. 3. Selection of the outcome measure. 4. Evaluation of the measure in 30 patients with MG for completeness and accuracy in describing their experience with MG. Results: 1: Patient survey: Staying out of hospital was very important to 96% of respondents, followed by living independently (91.5%). Ability to ambulate normally, drive, groom themselves and normal speech, chewing and swallowing were very important to >80% of respondents. Fewer/less frequent side effects, fewer tablets/ less frequent dosing, less frequent physician visits/ laboratory monitoring were very important to over a third of respondents. Symptoms with the most significant impact were limb weakness (64%), bulbar and ocular symptoms (36-38%). The most burdensome adverse effects of treatment were weight gain (43%), mood swings (36%), insomnia (34%) and diarrhea (30%). 2. Focus group: the word ”fatigue” was most frequently associated with MG, followed by ”weakness” and ”double vision”. Participants endorsed frustration and isolation. Mobility limitations, visual symptoms and respiratory symptoms were most troublesome. Avoiding hospitalization was a high priority. The most concerning adverse effects were weight gain and long term risk of cancer. A good outcome was defined as ”fewer medication side effects and 80% improvement in symptoms”. 3: Based on the survey and focus group results, the MG-Quality of Life 15r scale (MG-QOL15r) was thought to capture outcomes of greatest importance to patients. 4. 29/30 patients to whom the MG-QOL15r was administered responded that it completely and accurately described their experience with MG. Conclusion: MG-QOL is a 60 item scale which was developed with input from patients. The MGQOL-15 was subsequently developed with rigorous psychometric methods. Rasch analysis resulted in the MG-QOL15r, a three option modification, recently validated in a large cohort. We confirmed that the MG-QOL15r accurately and completely reflects the patient voice and selected it as the primary PRO for PROMISE-MG. Acknowledgements: Donald Sanders, MD, Co-Primary Investigator, PROMISE-MG; Myasthenia Gravis Foundation of America
SS 11.4 / #308HOW CAN OUTCOMES OF CLINICAL STUDIES BE TRANSLATED FOR PATIENTS
Topic: 12. Patient Issues
Angela Genge
Montreal Neurological Institute and Hospital, Montreal, CA
Abstract not received.
SS 12.1 / #127PATHOPHYSIOLOGY OF THE NODE OF RANVIER
Topic: 02. Neuropathy
Antonino Uncini
Neuroscience, Imaging And Clinical Sciences, University “G. d’Annunzio”, Chieti-Pecsra, Chieti, IT
Background: The myelinated axons are organised in distinct domains characterised by specific molecular arrangements: nodes of Ranvier, paranodes, juxtaparanodes and internodes. The nodal region is a crucial evolutionary structure of the nervous system ensuring, by saltatory conduction, a fast transmission of impulses with the least expenditure of energy. It took almost eight decades, since the original description by Ranvier in 1871, to demonstrate that the nodes are the places where ionic currents generated action potentials for saltatory conduction, and only in the last two decades, the nodal region has been recognised as a possible site of specific autoimmune attack in peripheral neuropathies. For these neuropathies, the classification in demyelinating and axonal may be inadequate or even misleading, and the new category of nodo-paranodopathy has been recently proposed. Methods: In this lecture the process of saltatory conduction in myelinated and demyelinated axons will be firstly reviewed. Then the findings from serial electrophysiological studies, immunopathologic and ultrastructural evidence in humans, experimental models of autoimmune attack at the nodal region by antibodies against gangliosides, neurofascin 186, axoglial proteins (neurofascin 155, contactin 1, and contactin associated protein 1) will be examined. Results: The electrophysiological correlates of autoimmune attack at the excitable nodal axolemma and adhesion molecules at nodal and paranodal region ranging by a continuum from transitory nerve conduction failure to axonal degeneration, and the ”demyelinating” features, explainable just by the paranodal involvement, will be discussed. Conclusion: At last the utility of the autoimmune nodo-paranodopathy category will be re-emphasized as the term pointing to the site of nerve injury, reminds specific pathophysiological mechanisms, reconciles apparently contrasting electrophysiological and pathological findings, and avoids misdiagnosis and taxonomic confusion.
SS 12.2 / #172NEW PATHOGENETIC MECHANISMS UNDERLYING IMMUNE NEUROPATHIES
Topic: 02. Neuropathy
Hugh J. Willison
Institute Of Infection, Immunity And Inflammation, University of Glasgow, Glasgow, GB
Background: Guillain-Barré syndrome (GBS) is in part mediated by anti-GM1 ganglioside antibodies induced by preceding infections. Anti-GM1 antibodies target plasma membrane GM1 that is extensively distributed in both glial and axonal membranes, particularly at the node of Ranvier. Antibodies deposited at this site in models of GBS are associated with complement deposition, conduction block, structural disruption of ion channels and macrophage infiltration. The wide distribution of the GM1 ganglioside target leads to unwanted complexity in ascribing pathological outcomes to injury of cell-specific membranes, in particular unravelling the consequence of paranodal Schwann cell membrane injury on axonal function, and vice versa. Methods: To overcome this impasse, we have generated transgenic mice through glycosyltransferase manipulation that express GM1 exclusively in either neurons or glia, thus allowing us to very specifically target and injure axonal or glial membranes with a single anti-GM1 ganglioside antibody. Through this route we can create mouse models of both the axonal and demyelinating forms of GBS, induced by a single anti-GM1 antibody, thus creating otherwise highly comparable conditions. Results: Here, we show anti-GM1 antibody binding is restricted to the nodal axolemma in GalNAcT-/--Tg(neuronal) mice and conversely to paranodal loops in GalNAcT-/--Tg(glial) mice. When anti-GM1 antibody and a source of complement is added to a nerve-muscle ex vivo injury paradigm, there is a loss of axonal integrity (i.e. loss of neurofilament immunolabeling) when the neuronal membrane is targeted in GalNAcT-/--Tg(neuronal). Conversely, axonal integrity is maintained when the paranodal membranes are decorated by antibody and complemnt products ex vivo in GalNAcT-/--Tg(glial) mice. In an passive immunisation model in vivo, GalNAcT-/--Tg(neuronal) mice acutely develop weakness, respiratory dysfunction, associated complement deposition, and degenerative pathology in distal axons. In contrast, GalNAcT-/--Tg(glial) mice have significantly fewer abnormalities under the same acute conditions. Conclusion: These data indicate the high vulnerability of axonal membranes to acute injury and underline the importance of developing specific axonal projection strategies. In summary, targeting the nodal axolemmal or glial membranes allows us to study associated nodal pathology, and determine the downstream consequences on function and axon fate, currently a major area in GBS clinical research.
SS 12.3 / #137ANTIBODIES TO THE NODE OF RANVIER IN CIDP: A NEW CLUE TO THERAPY
Topic: 02. Neuropathy
Luis Querol
Neuromuscular Diseases Unit, Hospital de la Santa Creu i Sant Pau, Barcelona, ES
Abstract not received.
SS 13.1 / #216OVERVIEW – DRUG TREATMENT FOR SMA
Topic: 03. Motor Neurone
Janbernd Kirschner
Dept. Of Neuropediatrics And Muscle Disorders, Medical Center - University of Freiburg, Freiburg, DE
Background: Spinal muscular atrophy (SMA) is an autosomal-recessive, neuromuscular disorder that is characterized by degeneration of the anterior horn cells of the spinal cord, resulting in muscle atrophy and proximal muscle weakness. SMA is caused by a homozygous deletion in the survival motor neuron 1 (SMN1) gene on chromosome 5q13. The SMN gene region also comprises a centromeric copy containing the SMN2 gene. The severity of the disease correlates with age of onset and SMN2 copy number and varies from a severe muscle weakness with tetraplegia in infants to a mild proximal muscle weakness in ambulant children and adults. Affected patients need a multidisciplinary treatment approach to address the different manifestation of the disease. In addition to symptomatic treatment including nutrition and ventilatory suuport new pharmaceutical approaches have been developed to address different aspects of the pathophysiology. Methods: Targets of the most advanced approaches include survival of motor neurons, increase of SMN protein production from the SMN2 gene by splicing modifiers, and introduction of the missing SMN1 gene with viral vectors. Results: Currently, nusinersen is the only drug that has been approved for the treatment of SMA. It is an antisense oligonucleotide that is administered by intrathecal injections and modifies the splicing of the SMN2 gene. Phase III trials have shown shown clear improvement of motor function in infants with SMA type 1 and in young children with SMA type 2. Long term safety and treatment effect in adolescent and adult still need further evaluation. Other splicing modifiers have been indentified through screening methods, they can be administered orally and are currently evaluated in clinical trials. A gene therapy approach using a AAV9 virus with the SMN1 gene has shown promising results in a phase I study and a larger European phase III trial for SMA tpye 1 will start this year. Olesoxime is a drug that has been developed to improve survival of motor neurons, but results of an international placebo-controlled trial were not sufficient for approval. Additional explorations of the pharmacokinetics and its effect on functional outcomes are ongoing. Conclusion: Clinical trials show promising results for the treatment of SMA with splicing modifiers and gene therapy. Early initiation of treatment seems particularly effective and warrants the implementation of newborn screening programs. As SMA is a rare disease with a broad spectrum of severity clinical trials cannot cover all age groups and all stages of the disease. Patient registries are an important tool to collect additional information to document the long-term effect of any drug treatment in SMA.
SS 13.2 / #98TREATMENTS TO IMPROVE SURVIVAL OF MOTOR NEURONS
Topic: 03. Motor Neurone
Albert Ludolph
Universitätsklinikum Ulm, Ulm, DE
Abstract not received.
SS 13.3 / #73SPLICING MODIFICATION FOR SMA
Topic: 03. Motor Neurone
Nathalie Goemans
Treat-NMD Neuromuscular Network, Leuven, BE
Abstract not received.
SS 13.4 / #48GENE THERAPY FOR SMA
Topic: 03. Motor Neurone
Arthur H.m. Burghes1, Jerry Mendell2, Samiah Al-Zaidy2, Richard Shell3, W. D. Arnold4, Louise Rodino-Klapac2, Thomas W. Prior5, Linda Lowes2, Lindsay Alfano3, Kathleen Church3, John T. Kissel4, Sukumar Nagendran6, James L’Italien6, Douglas M. Sproule6, Courtney Wells6, Kevin Foust6, Kathrin Meyer2, Shibi Likhite2, Vicki Mcgovern1, Stephen Kolb4, Brian Kaspar6
1Biological Chemistry And Pharmacology, The Ohio State University, Columbus, OH, US;2Center For Gene Therapy, Nationwide Children’s Hospital, Columbus, OH, US;3Department Of Pediatrics, Nationwide Children’s Hospital, Columbus, OH, US;4Department Of Neurology, The Ohio State University, Columbus, OH, US;5Department Of Pathology, The Ohio State University, Columbus, OH, US;6AveXis, Inc., Bannockburn, IL, US
Abstract: Spinal Muscular Atrophy (SMA) is an autosomal recessive disorder caused by mutation or loss of the survival motor neuron 1 gene (SMN1) gene and retention of SMN2. The SMN2 gene produces insufficient SMN but increased copy number of SMN2 modulates phenotype and does result in immune tolerance to an immune response against SMN when it is introduced from exogenous sources. Spinal muscular atrophy has been modeled in both mice and pigs. Untreated SMA mice live for 14 days and show loss of motor neurons including electrophysiological defects of the neuromuscular junction and compound muscle action abnormalities (CMAP) and MUNE. Knockdown of SMN in pig results in similar motor neuron abnormalities. scAAV9 has the ability to cross the blood brain barrier and efficiently transduce motor neurons with either delivery through the blood stream or via CSF delivery. Early scAAV9-SMN treatment of SMA mice and pigs results in correction of survival and motor neuron defects including CMAP and MUNE. Latter introduction especially in the pig results in correction of CMAP but MUNE is reduced indicating that motor neurons once lost cannot be recovered but those remaining can compensate by sprouting. A phase 1 gene therapy trial of scAAV9-SMN (AVXS-101) was initiated in Type 1 SMA, the most severe form of SMA , with untreated children never gaining the ability to sit or maintain head control. All patients lacked SMN1 and had 2 copies of SMN2 and none contained the C859G variant which increases incorporation of SMN2 exon 7. Patients received an intravenous dose of AVXS-101 at the low dose (Cohort 1, n=3) or proposed therapeutic dose (Cohort 2, n=12). AVXS-101 had a favorable safety profile and improved survival when compared to natural history studies. All 15 patients were alive and event-free at 20 months of age and did not require permanent mechanical ventilation, compared with 8% in published natural history. Patients in Cohort 2 demonstrated improvements in motor function: 11/12 had achieved CHOP-INTEND scores >40 points and a mean increase of 24.6 points from a mean baseline of 28.2 points; 11/12 were able to sit unassisted for at least 5 seconds, 10 for at least 10 seconds, and 9 for at least 30 seconds; 11/12 achieved head control and 9 could roll over. Two patients were able to crawl, pull to stand, stand independently, and walk independently. None of the aforementioned motor function gains and milestone achievement is seen in untreated patients with SMA type 1 and 2 copies of SMN2. Asymptomatic elevated serum aminotransferase levels occurred in 4 patients and was attenuated by prednisolone treatment. (see Mendell et al this meeting for further details.) As in preclinical studies in animal models scAAV9-SMN (AVXS-101) has a dramatic impact on motor function and appeared to increase survival. This was particularly evident with early treatment in cohort 2 with achievement of motor milestones rarely or never seen in the type 1 SMA population.
SS 14.1 / #90MR NEUROGRAPHY: NEW APPROACHES AND TECHNIQUES FOR DIAGNOSING MONONEUROPATHIES
Topic: 06. Mononeuropathy
Jennifer Kollmer
Heidelberg University Hospital, Heidelberg, DE
Abstract not received.
SS 14.2 / #189NEW ULTRASENSITIVE ULTRASOUND METHODS
Topic: 06. Mononeuropathy
Stefan Meng
KFJ Hospital, Vienna, AT
Abstract not received.
SS 14.3 / #63OCT
Topic: 06. Mononeuropathy
Wolfgang Drexler
Medical University of Vienna, Vienna, AT
Abstract not received.
SS 15.1 / #150CHANGES IN THE NODAL COMPLEX IN PAINFUL NEUROPATHY
Topic: 12. Patient Issues
Claudia Sommer
Neurologische Klinik Und Poliklinik, Universitätsklinikum Würzburg, Würzburg, DE
Background: Autoantibodies to components of the paranodal protein complex have recently been described and are considered to characterize a subset of immune neuropathies that differs from other subtypes in clinical phenotype pathogenesis and response to therapy. Methods: We systematically screened sera of our patients with suspected immune neuropathies for the presence of autoantibodies to paranodal proteins. We analyzed skin and nerve biopsies for changes in the nodal/paranodal complex. Results: Pain was one of the predominant findings in two patients with anti-caspr antibodies, possibly reflected by binding of patients’ IgG to transient receptor potential vanilloid 1 immunoreactive dorsal root ganglia neurons. Severe disruption of the paranodal and nodal architecture was detectable in teased fibres of the sural nerve biopsy and in dermal myelinated fibres, supporting the notion of the paranodes being the site of pathology. Nodal/paranodal architecture was disrupted in patients with anti-contactin-1 antibodies, but pain was not a prominent feature in these patients. Conclusion: In contrast to other neuropathies with antibodies to the paranodal complex (anti-contactin-1, anti-neurofascin-155) two patients with auto-antibodies against caspr suffered from severe neuropathic pain. The mechanisms underlying this pain need to be uncovered.
SS 15.2 / #86THE USE OF NERVE EXCITABILITY TESTING IN UNDERSTANDING ION CHANNEL DYSFUNCTION IN NEUROPATHY
Topic: 12. Patient Issues
Matthew C. Kiernan
Brain & Mind Centre, University of Sydney, Sydney, AU
Abstract: Measurement of nerve excitability by threshold tracking provides complementary information to conventional nerve conduction studies and may be used to infer the activity of a variety of ion channels, energy-dependent pumps and ion exchange processes activated during the process of impulse conduction. While routine nerve conduction studies can document the presence of a neuropathy, they do not provide further insight into pathophysiology. This may be even more relevant across the range of neuropathic syndromes, where conventional nerve conduction studies are often normal. Following the validation of excitability testing protocols, clinical excitability techniques are being adopted to complement diagnostic nerve conduction studies. A number of recent clinical studies will be highlighted which have established mechanisms for neuropathy associated with systemic disease, likely to impact on the future management of patients, both acutely and chronically; provided insight into the mechanisms of painful neuropathies due to neurotoxicity associated with chemotherapy; determined mechanisms of hyperexcitability, further contributing to symptoms of neuropathy. While clinical nerve excitability studies are still in their relative infancy, and it is too early to know whether they have diagnostic value, there is growing evidence of their utility to provide novel insights into the pathophysiological mechanisms involved in a variety of neuropathic disorders.
SS 15.3 / #43HUMAN PAIN CHANNELOPATHIES
Topic: 12. Patient Issues
David Bennett
Nuffield Department Of Clinical Neuroscience, University of Oxford / Nuffield, Oxford, US
Abstract: There has been significant progress over the last decade in understanding the ion channels required by sensory neurons to transmit noxious stimuli. Over this same period we have recognized that mutations in such ion channels can result in primary neuropathic pain disorders. An excellent example is the voltage gated ion channel NaV 1.7 encoded by the gene SCN9a. Loss of function mutations in this ion channel result in congenital inability to experience pain and gain of function mutations can cause a number of distinct neuropathic pain disorders including erythromelalgia, paroxysmal extreme pain disorder and small fibre neuropathy. There is a correlation between the impact of mutations on the biophysical properties of the ion channel and the severity of the clinical phenotype. The fact that mutations in such channels can cause monogenic pain disorders makes them attractive analgesic drug targets. Secondary neuropathic pain disorders such as traumatic neuropathy or painful diabetic neuropathy are also associated with altered expression and function of multiple ion channels which contribute to hyperexcitability. Finally we are now beginning to recognize that auto-antibodies to ion channels and proteins with which they complex can cause acquired pain channelopathies. We are trying to improve the stratification of neuropathic pain patients by linking the clinical phenotype with genotype with the ultimate aim of optimizing analgesic drug selection on an individualized basis.
SS 16.1 / #138NIV AND ALS
Topic: 03. Motor Neurone
Marianne De Visser
Department Of Neurology, Academic Medical Centre, Amsterdam, NL
Abstract not received.
SS 16.2 / #109NUTRITION AND ALS
Topic: 03. Motor Neurone
Esther Hobson
University Of Sheffield, Sheffield Institute for Translational Neurosciences, HQ, GB
Background: Malnutrition and weight loss are well recognised poor prognostic factors in amyotrophic lateral sclerosis. Body mass index (BMI) is an independent predictor of survival in ALS, with mild obesity having a protective effect and lower BMI associated with a worse prognosis. In patients with weight loss of 5% or more at diagnosis there is a two-fold increase in risk of death, compared to those who have lost less than 5% of their usual pre-morbid weight. Poor nutrition in ALS is not simply as a result of bulbar dysfunction. Fatigue and poor upper limb dysfunction make feeding difficult whilst respiratory failure cause loss of appetite. As a consequence, eating is no longer a pleasurable and social activity, instead it becomes a burden on the patient and caregiver. The energy deficit caused by inadequate intake is compounded by observed hypermetabolism with resting energy expenditure observed to be, on average, 20% higher than healthy individuals. Patients with hypermetabolism have a greater level of lower motor neuron involvement, faster rate of functional decline and shorter survival compared with normometabolic patients with ALS. Methods: It is suggested that metabolic defects and the energy deficit are not simply a consequence of denervation and advanced disease but are an early feature of ALS pathology. They could therefore be potentially modifiable using dietary interventions. Patients themselves have not waited for evidence with more than half using some dietary supplementation. The consequences of these untested diets remains unclear but thereis some limited evidence that correction of the energy deficit with a high calorie diet extends life in an ALS mouse model.Moreover, a small pilot study of ALS patients indicated a potential survival advantage and tolerability of a high calorie diet. Future research will investigate the potential of calorie manipulation, particularly in the early stages of the disease. Results: Most patients later in the disease course opt for placement of a gastrostomy tube. Despite gastrostomy use being common practice, in the absence of comparative trials it remains difficult to determine the impacts for patient survival and quality of life. However, prospective and observational studies can provide some information. There are a variety of methods available for insertion, each likely to be broadly similar in its insertion risks. Longer term consequences and risk of tube damage or displacement are related to the robustness of the tube. Gastrostomy insertion with traditional feeding techniques alone does not typically reverse weight loss particularly in those who have already lost over 10% of their premorbid bodyweight, in whom, along with those in respiratory failure, the 30 day-mortality is significantly greater. Conclusion: This leads to the conclusion that gastrostomy insertion should be offered early in the disease. However there is often resistance to accepting this procedure due to various psychosocial factors, some of which may be overcome by better information (such as the www.mytube.mymnd.org website). What remains unclear is what is the best nutritional regime post-gastrostomy insertion, and how to communicate the individual risk and benefits, particularly those with a poorer prognosis or those without bulbar disease.
SS 16.3 / #52NUTRITION NIV AND COGNITION IN ALS
Topic: 03. Motor Neurone
Vincenzo Silani
University of Milan Medical School, Milan, IT
Abstract not received.
SS 17.1 / #187NEUROFIBROMATOSIS
Topic: 06. Mononeuropathy
Katharina Wimmer
Medical University of Innsbruck, Innsbruck, AT
Abstract not received.
SS 17.2 / #175PERIPHERAL NERVE TUMORS
Topic: 06. Mononeuropathy
Gelareh Zadeh
University Health Network, Toronto, CA
Abstract not received.
SS 17.3 / #106INTRANEURAL PERINEURIOMA: MAKING THE DIAGNOSIS AND CLUES TO THE PATHOGENESIS
Topic: 06. Mononeuropathy
Michelle Mauermann
Neurology, Mayo Clinic, Rochester, MN, US
Background: Intraneural perineurioma is a benign peripheral nerve neoplasm composed of whorls of perineurial cells surrounding nerve fibers and restricted to the boundaries of a peripheral nerve. It was previously unclear if these were caused by a genetic abnormality or were related to prior trauma. Methods: To review the clinical, electrophysiological, radiological and pathological features of intraneural perineurioma and its associated natural history. We will discuss how to differentiate these tumors from other benign nerve tumors and inflammatory nerve lesions. The recent advances in the underlying genetic causes will be discussed. Results: Intraneural perineurioma presents insidiously in children and young adults with a motor-predominant mononeuropathy or plexopathy. The sciatic nerve is most commonly affected. The electrophysiological findings demonstrate a chronic axonal process. The radiologic hallmark is fusiform nerve enlargement with T2 hyperintensity and contrast enhancement. Targeted biopsy of tumor demonstrating the typical pathological features is the gold standard for diagnosis. Pathologic features include nerve hypertrophy due to pseudo-onion bulb formations consisting of whorls of perineurial cells surrounding nerve axons that react for epithelial membrane antigen. A recent study led to the discovery of frequent recurrent missense somatic mutations in TRAF7. Rare patients have large somatic macrodeletions and duplications. Conclusion: Intraneural perineurioma is a benign hypertrophic peripheral nerve tumor that presents in young people with a motor-predominant mononeuropathy or plexopathy. It is slowly progressive and causes mild disability. Underlying genetic mutations in TRAF7 suggest a shared pathogenesis with intracranial meningiomas.
SS 17.4 / #1029CANCER AND PERIPHERAL NERVES
Topic: 06. Mononeuropathy
Wolfgang Grisold1, Anna Grisold2, Stefan Meng3, Elisabeth Lindeck-Pozza4
1Ludwig Boltzmann Institute for Experimental und Clinical Traumatology, Vienna, AT;2Dep. Neurology, Allgemeines Krankenhaus Vienna, Vienna, AT;3Institute for Radiology, KFJ hospital, Vienna, AT;4Dep. Neurology, KFJ hospital, Vienna, AT
Abstract: Peripheral nerve tumors are rare and are conventionally classified according to the WHO classification 2016, which adds several new entities as hybrid tumors, perineuroma and malignant peripheral nerve sheath tumors (MPNST). In addition to this classification also mesenchymal, vascular and tumor like conditions have to be considered. The neoplastic involvement of peripheral nerves occurs in cancer, lymphoma, leukemia and even more rarely in plasmocytoma. In all cancer patients, MPNSTs can be a side effect of radiation therapy (RT). In solid tumor nerve metastasis, or circular compression of individual nerves is rare, however in the peripheral parts of the cranial nerves infiltration and spread can be observed. Infiltration and compression can also be seen in nerve roots and the nerve plexus. Lymphoma and leukemia can present with isolated appearance or diffuse infiltration, at any time of the disease, also as a recurrence. Also solid manifestations in leukemia termed chloroma are rarely reported. Tumorous infiltration of peripheral nerve by plasmocytoma is are, and also local amyloidomas have been described. For the different tumor entities also different morphological changes and mechanisms are described. Therapy depends on the tumor type and in addition to surgical and RT also chemotherapy or biological therapies depending on the tumor type are used. As in MPNST the prognosis is usually poor.
SS 18.1 / #85MND: THERAPEUTIC LANDSCAPES
Topic: 03. Motor Neurone
Matthew C. Kiernan
Brain & Mind Centre, University of Sydney, Sydney, AU
Abstract: Recent evidence has emerged concerning unique pathophysiological processes linked to the development of motor neurone disease (MND), including glutamate-mediated excitotoxicity, which has resulted in development of novel diagnostic investigations and uncovered potential therapeutic targets. Advances in genetics, including the recently discovered C9orf72 gene, have radically changed the pathological mind-set, such that ALS forms part of a continuum with other primary neurodegenerative disorders, including frontotemporal dementia. At the simplest level, the management of MND remains focused on symptom control. Evidence-based management guidelines advise a multi-disciplinary model of care, led by a neurologist and clinical nurse consultant working together with physical therapists, occupational therapists, speech pathologists, respiratory physicians, gastroenterologists, psychologists and social workers to guide patient management, and such an approach has profoundly impacted patient quality of life and survival. In terms of emerging therapies, there have been several recent phase II/III trials that have recently shown promising results. Data concerning masitinib reported significant improvement from baseline to week 48 in the Revised ALS Functional Rating Scale scores (primary endpoint) and FVC (secondary endpoint). This new oral tyrosine kinase inhibitor regulates abnormal neuroglia activation and proliferation, processes linked to motor neurone death. A clinical trial of ultrahigh-dose methylcobalamin (E0302) significantly prolonged survival in a dose-dependent manner in MND patients who were treated within 1year of disease onset. Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), a potent free radical scavenger, is known to prevent neuronal damage inhibiting endothelial injury and is currently marketed for use after ischaemic stroke. Benefit from edavarone was established for a subgroup of MND patients, specifically younger patients with an FVC at least 80%, and with recent onset of symptoms. Other potential treatment targets awaiting confirmation with human clinical trials include CNS copper treatment and anti–human endogenous retrovirus K (HERV K) treatment. Stem cell therapy remains a theoretical possibility: previous attempts to grow anterior horn cells have been abandoned. Now the overall goal is prevention of further neuronal loss by supporting residual motor neurones. The optimal route of administration and specific type of stem cells needed, remain unanswered questions that are currently being considered through several phase II studies.
SS 18.2 / #114TREATMENT STRATEGIES FOR HEREDITARY ALS
Topic: 03. Motor Neurone
Timothy Miller
Washington University, St. Louis, US
Background: Recent discoveries have defined the genetic cause in more than 60% of hereditary ALS. Along with this increased genetic discovery has emerged increased tools to specifically target genes and pathways that cause ALS. Methods: These therapeutic strategies include viral delivered siRNA, antisense oligonucleotides, antibodies, and small molecules. Results: Novel treatment strategies for hereditary ALS will be discussed with a particular focus on antisense oligonucleotides. Conclusion: Treatment strategies for hereditary ALS have tremendous potential to positively impact ALS.
SS 18.3 / #131DIAGNOSIS AND TREATMENT OF MOTOR NEUROPATHIES
Topic: 03. Motor Neurone
Alan Pestronk
Department Of Neurology, Washington University School of Medicine, St. Louis, MO, US
Abstract not received.
SS 19.1 / #139DISTAL HEREDITARY MOTOR NEUROPATHIES (DHMN)
Topic: 03. Motor Neurone
Mary Reilly
Queen Square, Institue Of Neurology, London, GB
Abstract: The distal hereditary motor neuropathies (HMN) are a clinically and genetically heterogeneous group of slowly progressive neuropathies characterised by a distal predominant almost exclusive motor syndrome. There is overlap with the axonal forms of Charcot Marie Tooth disease (CMT2) such that causative genes are often described as causing both HMN and CMT2 (e.g. HSPB1 and HSPB8). The differentiation of these motor predominant syndromes into HMN and CMT2 is artificial as these conditions are motor predominant but especially with time there can be minimal sensory involvement. It may be more appropriate to use the term CMT to include HMN, CMT and the sensory predominant form, HSN, reflecting the spectrum of clinical disease from pure motor or motor predominant (HMN) through motor and sensory (CMT) to pure sensory or sensory predominant (HSN). As multiple causative genes are identified for HMN, it is clear that there is also overlap with other inherited neurological conditions with some genes causing both HMN and hereditary spastic paraparesis (HSP) (e.g. REEP1) and others such as the mitochondrial gene MT-ATP 6 causing a spectrum of neurological conditions including a motor predominant CMT2 like phenotype. Although the traditional definition of distal HMN is a length dependent motor neuropathy starting in the lower limbs many clinical variations are now recognised. Many forms of distal HMN do not present with the typical weak ankle dorsiflexion with relatively preserved ankle plantarflexion reflecting the anterior lower limb muscle involvement seen with CMT and instead present with equal involvement of anterior and posterior calf muscle resulting in equal ankle dorsiflexion and plantarflexion weakness (e.g. this is seen with HSPB1 and HSPB8 mutations). We also now recognise with these same two genes (HSPB1 and HSPB8) that a myopathy can be present as well as a neuropathy. Other genes cause a non length dependent lower limb predominant motor neuropathy (e.g. with Dynein and BICD2). A number of genes (BSCL2, GARS, REEP1) cause a motor syndrome which is much more pronounced in the upper limbs and can involve the upper motor neurone fibers in some cases (BSCL2 can cause Silver syndrome which is characterised by amyotrophy of the hands with spasticity of the lower limbs). The involvement of upper and lower motor neurone fibres is important as it underlies that the pathogenesis of both upper motor neurone motor syndromes (e.g. HSP) and lower motor neurone syndromes (e.g. HMN) is likely to involve common pathways like axonal transport or protein misfolding disorders with the possibility of developing pathway therapies for all axonopathies rather than therapies for each individual genetic condition. This talk will focus on the evolving genotypes and phenotypes and the progress towards therapy development.
SS 19.2 / #32DISTAL MYOPATHIES
Topic: 03. Motor Neurone
Anthony Amato
Dep. Of Neurology, Brigham and Women’s Hospital, Boston, US
Abstract: The distal myopathies are characterized clinically by progressive atrophy and weakness of distal arm or leg muscles and histologically by nonspecific myopathic features on muscle biopsy. The distal myopathies can be subdivided, based on the clinical features, age of onset, CK levels, muscle histology, and mode of inheritance. Myopathic causes of distal weakness are distal muscular dystrophies, of which we would include the myofibrillar myopathies and various, hereditary inclusion body myopathies, congenital myopathies, glycogen and lipid storage myopathies, and acquired forms such as seen in granulomatous myopathy.
SS 19.3 / #84ACQUIRED DISTAL MOTOR SYNDROMES
Topic: 03. Motor Neurone
Matthew C. Kiernan
Brain & Mind Centre, University of Sydney, Sydney, AU
Abstract: Distal or lower motor neuron syndromes are characterised by muscle atrophy, weakness and hyporeflexia without sensory involvement. These disorders may arise from disease processes affecting the anterior horn cell, the motor axon, or its surrounding myelin. Neuromuscular junction pathology and muscle disorders may mimic these disorders and form part of the differential diagnosis. Distal motor syndromes can be broadly classified as hereditary, sporadic or immune-mediated. Immune-mediated neuropathies, such as multifocal motor neuropathy and chronic inflammatory demyelinating polyneuropathy are important to distinguish from sporadic and hereditary forms, as effective treatments are available. Lower motor neuronal presentations of motor neuron disease (MND) are most often sporadic, but several genetic mutations have been described. Hereditary forms include distal hereditary motor neuropathies and spinal muscular atrophy (SMA). With the latter, onset typically occurs during childhood or teenage years, although adult onset is not uncommon. The increasing availability of next-generation sequencing, including the ability for multiple genes to be sequenced in parallel, has resulted in an increase in the discovery of novel genetic mutations. The clinical evaluation of a patient presenting with a distal motor syndrome includes a directed assessment of disease onset and progression. This is particularly important to ascertain, as a rapid rate of decline may suggest a diagnosis of MND and remains an important factor in distinguishing MND from other relatively indolent conditions, such as SMA and immune neuropathies. The pattern of weakness may also suggest a specific diagnosis, with considerations including (1) symmetry versus asymmetry, (2) proximal versus distal involvement, (3) upper versus lower limb predominance and (4) presence versus absence of bulbar involvement. Nerve conduction studies and electromyography are essential to confirm that the disorder is neurogenic and should focus on assessing the pattern of involvement, including symmetry and length dependence; the presence of focal motor conduction block or demyelinating features; and the presence or absence of subclinical sensory abnormalities. Neuromuscular imaging, genetic testing, assessment of antibody markers and advanced neurophysiological techniques are all useful adjuncts to determine a specific diagnosis and the best therapeutic approach.
SS 20.1 / #49EVOLUTION OF MONOCLONAL AB TREATMENT APPROACHES IN MG
Topic: 04. Neuromuscular Junction
Ted M. Burns
Department Of Neurology, University of Virginia, Charlottesville, US
Abstract not received.
SS 20.2 / #136OTHER NOVEL TREATMENT APPROACHES: EXERCISE
Topic: 04. Neuromuscular Junction
Anna R. Punga
Neuroscience, Uppsala University, Uppsala, SE
Background: The benefits of physical exercise for healthy individuals are well established, in particular to reduce the risks of life-style disorders. Furthermore, physical exercise provides beneficial effects in several chronic diseases such as rheumatoid arthritis, chronic obstructive pulmonary disease and multiple sclerosis, and is therefore recommended as part of the treatment regimen. It is reasonable to assume that MG patients achieve at least similar benefits from physical exercise, as do healthy adults. Yet, exercise-related research in the field of MG is sparse and does not provide any guidelines on how MG patients should perform physical exercise to obtain desirable benefits without the risk of causing disease deterioration or more pronounced muscle fatigue. Two recent pilot studies reported that MG patients with mild disease activity could adhere safely to general exercise recommendations including resistance and aerobic training regimen without subjective or objective disease deterioration. Overall, though, there is an obvious lack of literature in the field of physical exercise and MG that hampers the possibility to individualize training programs that could be of particular importance to avoid development of secondary disease conditions. Methods: Two studies were performed: 1) To objectively evaluate functional skeletal muscle parameters in MG patients who conducted a 12-week supervised physical therapy program, focusing on aerobic and high-resistance strength training. 2) A follow-up study on patients for 6 months with an individually tailored program from a personal trainer. The primary hypothesis was that a physical exercise program based on general exercise recommendations improves muscle status in MG patients. The secondary hypothesis was that moderate aerobic and high-resistance strength training improves the cardiovascular risk profile and self-assessed-well-being of MG patients. Both studies were prospective and unblinded, where each patient served as his/her own control. Outcome measures included clinical MG status, isometric muscle force, neurophysiological evaluations, neuromuscular ultrasound, physical performance-based measures, quality of life, fatigue severity and blood sample analysis. Wilcoxon signed-rank test was used to compare non-parametric data before and after training in each patient. A P-value < 0.05 was considered significant. Results: After the training program, functional muscle parameters (neurophysiological parameters, isometric muscle force and ultrasound muscle thickness) improved in the proximal leg muscles. Further, physical performance-based measures improved as well as the clinical MG composite score. The longer follow-up study also indicated positive adherence to individualized programs and additional beneficial effects. Conclusion: These findings indicate that MG patients can indeed improve their functional muscle status as a result of aerobic and high-resistance strength training, especially in proximal leg muscles. This is important knowledge in order to establish both collective as well as individualized guidelines of physical exercise for MG patients.
SS 20.3 / #174OVERVIEW OF TREATMENT GUIDELINES: GOING FORWARD
Topic: 04. Neuromuscular Junction
Gil I. Wolfe
Neurology, University at Buffalo Jacobs School of Medicine and Biomedical Sciences, Buffalo, NY, US
Background: The rarity of myasthenia gravis (MG) makes the planning and completion of randomized controlled trials (RCTs) logistically difficult with many trials failing to reach their recruitment goals. Due to restricted entry criteria that are often an essential element of well-designed RCTs, results from these studies may also have limited generalizability to the overall patient population. Furthermore, the majority of trials do not address the comparative effectiveness of different treatment modalities in a disease as heterogeneous as MG. As a result, most current treatments for MG are based on uncontrolled studies, with a high risk of bias. Recommendations based on a formalized consensus opinion of experts can help to guide treatment in such situations. Methods: To review treatment recommendation statements that have been produced recently by neuromuscular neurology experts in several European and Asian countries and to describe the process and results generated by an international panel that focused on several treatment domains related to MG. Plans for future revisions to the statements will also be presented. Results: Since 2012, treatment recommendations for MG have been established and published by several countries, beginning with Germany and including England, Japan and China. Of these national statements, only the Chinese employed a formal consensus process, using the RAND/UCLA appropriateness method (RAM). In October 2013, the Myasthenia Gravis Foundation of America (MGFA) appointed a Task Force to develop treatment guidance for MG, and a panel of 15 international experts was convened. The panel also included a facilitator with extensive experience in consensus methodology and an observer representing patient interests from the MGFA. RAM methodology was used to develop consensus guidance statements. To frame the treatment guidance the following definitions were established by the panel: goals of treatment, minimal manifestations, remission, ocular MG, impending crisis, manifest crisis, and refractory MG. An in-person meeting of the panel then determined 7 treatment topics to be addressed. Initial guidance statements were developed based on summaries of the literature for each topic. Three rounds of anonymous e-mail votes with modifications of the guidance statements, based on panel input were used to reach consensus. Guidance statements were developed for the following MG treatment domains: symptomatic and immunosuppressive treatments, intravenous immunoglobulin and plasma exchange, management of impending and manifest myasthenic crisis, thymectomy, juvenile MG, MG associated with antibodies to muscle specific tyrosine kinase and MG in pregnancy. Conclusion: Both national and international treatment recommendations have helped define best practices in the management of MG. Although there are common threads across the various recommendations, differences do exist that reflect national practice and the availability or approval of treatment modalities. The formal international consensus can serve as a guide for clinicians who wish to optimize function and quality of life for their MG patients. It may be of particular benefit to those clinicians who practice in countries that do not have the resources to generate a national treatment statement. The international guidance is a living document that will require revision every few years to reflect developments in the field.
SS 21.1 / #67NEUROLOGICAL COMPLICATIONS OF ZIKA VIRUS
Topic: 09. Systemic Diseases
John D. England
LSUHSC School of Medicine, New Orleans, US
Abstract: Zika virus (ZIV) is a mosquito-borne single-stranded RNA virus in the Flaviviridae family. On 1 February 2016 the World Health Organization (WHO) declared ZIV infection a Public Health Emergency of International Concern (PHEIC) due to reports from Brazil of a cluster of microcephaly cases and other neurologic disorders termed Congenital Zika Syndrome as well as clusters of Guillain-Barre Syndrome (GBS) from French Polynesia and South America. Since that time an expanding spectrum of neurological complications of Zika virus infection have been described. ZIV is now recognized as a new neuropathological agent associated with a wide variety of neurological complications. These neurological sequelae of ZIV infection represent both direct effects of virus invasion and autoimmune responses to the virus. The neurological complications of ZIV include the following: 1) Congenital Zika Syndrome associated with infection of neural progenitor cells in the brain; 2) Guillain-Barre Syndrome (GBS), which is most likely caused by a secondary autoimmune response directed against components of peripheral nerve; 3) Sensory polyneuropathy, which may be associated with direct infection of peripheral neurons; 4) Encephalitis/meningoencephalitis and myelitis, which may be associated with direct viral infection/inflammatory process in the central nervous system; 5) Acute Disseminated Encephalomyelitis; 6) Optic neuropathy; 7) Arterial Ischemic Stroke (AIS) in children due to probable vasculitis and possible direct ZIV infection of brain endothelial cells. Ongoing investigations are confirming these associations and exploring the pathogenesis of the complications of ZIV infection. Since there is no specific treatment for ZIV infection, public health measures are directed at prevention of primary infection.
SS 21.2 / #146LEPROUS NEUROPATHIES
Topic: 09. Systemic Diseases
Gerard Said
Institut Vernes, PARIS, FR
Abstract: Leprous neuropathy, which is due to infection of nerve cells by Mycobacterium leprae remains a health problem worldwide. In addition to being an infectious and potentially contagious disease, it induces irreversible damage to peripheral nerves, exposing patients to lifelong complications of sensory and motor denervation. The number of new cases reported globally in 2016 exceeds 200 000 with 135 485 in India, 25 218 in Brazil and 16 826 in Indonesia. Nasal secretions and skin from untreated multibacillary leprosy (ML) cases can contaminate contacts. The incubation period may be as long as 20 years. Factors such as nutrition, hygiene and house crowding play an important role.M. leprae is an obligatory intracellular parasite with tropism for macrophages and Schwann cells. The bacillus attaches to Schwann cells via PGL-1. Once inside the Schwann cell, the mycobacterium will multiply very slowly, triggering a subacute to chronic inflammatory reaction, with replacement of functional parenchymal tissue by connective tissue and fibrosis. In the multibacillary type bacilli are easily found in the skin, and in nerve cells including Schwann cells, endothelial cells and macrophages. Conversely, in the paucibacillary (or tuberculoid) type a strong cell mediated immune reaction leads to granulomas formation and destruction of cells harbouring bacilli and nerve fibres nearby. In many cases treatment of patients with multibacillary leprosy is complicated by early or late reversal reaction, and further nerve damage. Nerve lesions lead to a symmetrical, pseudo-polyneuritic pattern, in most cases of multibacillary leprosy (ML), usually associated with typical skin lesions. In 10% of patients however skin lesions are missing, then only nerve biopsy permits diagnosis. The multifocal pattern, is more common in paucibacillar leprosy. Clinical manifestations of leprous neuropathy include 1. Specific cutaneous lesions, which reveals the disease in half or more of the patients, depending on the type of leprosy. 2. Sensory loss is the most constant neurological finding, due to the involvement of mixed dermal nerves and nerve trunks. Proprioception is often preserved, so patients can still use their largely anaesthetic limbs effectively, which leads to painless trauma and trophic changes. The topographical distribution of sensory disturbances is extremely variable. 3. Motor involvement is a late event in the course of the disease. Amyotrophy and motor weakness usually progress pari passu. They predominate in the ulnar and median nerve territories, with characteristic claw hands, and in the territory of the peroneal nerve. 4. Tendon reflexes are often preserved 5. Nerve hypertrophy: Nerve trunks are palpably enlarged in one third of the patients with leprosy, sometimes before the occurrence of sensory loss in the corresponding territory. Doppler ultrasound can be useful in this setting. 6 There is no dysautonomia or sphincter disturbances 7. Facial palsy with lagophthalmos of one or both eyes, with sparing of the other muscles supplied by the facial nerve is a classical feature. 8. Trophic disturbances are the most spectacular manifestations of leprosy. Severe sensory loss is always found in those areas where ulcers occur. 9. Symptomatic neuropathy, which is usually preceded by alteration of conduction velocity due to demyelination of nerve fibers, is invariably associated with major axonal damage and loss of more than 90% of nerve fibres in corresponding territory. Treatment is currently based on multidrug therapy with dapsone, rifampicin and clofazimine. The use of corticosteroids can reduce or prevent nerve damage in reversal reactions. Residual sensory loss which is extremely common in affected areas, can lead to trophic changes related to repeated traumas in painless areas.
SS 21.3 / #301NEUROPATHIES IN INFECTIOUS DISEASES
Topic: 09. Systemic Diseases
Nazha Birouk
Clinical Neurophysiology Rabat, Speciality Hospital, Rabat, MA
Abstract: Infectious neuropathies are heterogeneous group of neuropathies regarding their multiple causes and numerous clinical presentations. They till represent an important world health issue. The clinical presentations are variable including symmetric distal neuropathy, mononeuropathy multiplex, radiculopathy or cranial neuropathy. The disorder process can be acute, sub acute or chronic axonal or demyelinating and is diagnosed by eletrodiagnostic or in some situations by nerve biopsy. The infectious agent can involve the peripheral nerve directly or indirectly by autoimmune and/or inflammatory process. The HIV related neuropathy became the major complication of HIV infection. The most frequent form is the painful distal and symmetric lent dependent polyneuropathy; it can be either HIV induced or antiretroviral toxic neuropathy. Inflammatory neuropathies such as acute or chronic polyradiculoneuropathies can be encountered at early stage of HIV infection. In the later stages, HIV infected patients can present with acute radiculopathies due to CMV infection. Peripheral neuropathy in patients with chronic infection with hepatitis C virus is mainly due to associated cryoglobulinemia and the associated vasculitis that leads to acute or sub acute mononeuritis multiplex. The polyneuropathy in this context can be also induced by direct neurotoxic effect of the treatment. In Lyme disease the involvement of peripheral nerves is part of meningoradiculitis with frequent involvement of cranial nerves. The most frequent infectious neuropathy in the world remains an active infectious ganglioneuritis caused by varicella zoster virus, producing shingles. The main issue with this condition is the post-herpetic neuralgia that can be severe in some patients. Infectious diseases are an important part of differential diagnosis of peripheral neuropathies because most of them are potentially treatable. The systematic diagnosis for the most frequent ones such as HIV and hepatitis C is highly recommended in a patient with polyneuropathy. Other infections are discussed regarding particular clinical presentations and in some geographic areas.
SS 21.4 / #866INFECTION RELATED NEUROMUSCULAR DISORDERS (NEUROPATHIES AND MYOPATHIES)
Topic: 09. Systemic Diseases
Chandrashekhar Meshram
Neurology, Brain and Mind Institute, Nagpur, IN
Background: Various bacterial, viral and parasitic infections directly or indirectly affect peripheral nerves and muscles. Neuromuscular disorders can be caused by three mechanisms : direct infection, infection related but toxin mediated and post infectious syndrome. Involvement of nerves and muscles secondary to systemic infections is relatively rare phenomenon. Some of these infections are sporadic, while some occur in outbreaks. Some infections are predominantly observed in tropical countries. During outbreaks, cases are observed in clusters in affected regions and one gets the opportunity to study the disorders. Methods: Personal experience and unpublished personal communication was supplemented by search of published literature on neurological manifestations of systemic infections with involvement of peripheral nerves and/or muscles. Special attention was given to evaluate neuromuscular involvement in Chikungunya, Dengue, Leptospirosis, Scrub Typhus and Rabies. Results: During the Chikungunya epidemic in India in 2006 , Peripheral Neuropathy was observed in 36 % patients with neurological involvement ,entrapment neuropathy was seen in 10 % while myositis was noted in 15 % patients.In many patients there was combination of central and peripheral manifestations. Polyradiculoneuropathy ,lumbosacral plexopathy ,GBS have been reported with Dengue virus infection. Autonomic symptoms are common and may occur in 30 % patients. These features are mainly immune mediated. Dengue associated hypokalaemic paralysis and myositis is also reported. A more benign and common, transient muscle weakness with moderately raised CPK is usually self limiting. But sometimes the myositis can be fulminant and require respiratory support. In Neuroleptospirosis, peripheral nervous system involvement is seen in 10 % patients in form of myositis, rhabdomyolysis, GBS like presentation , brachial neuritis and mononeuritis multiplex. However CPK is raised in 30- 50 % patients. Neurological involvement occurs in 25 % patients of Scrub Typhus. Neuromuscular involvement is in form of cranial nerve dysfunction, peripheral neuropathy, and myositis.In one study 39 % patients of Scrub typhus had muscle involvement in form of myalgia,weakness,raised CPK and myopathic EMG. Twenty percent patients of Rabies present with ascending paralysis. Pain and paraethesia at the site of bite are common symptoms. Both upper and lower limbs are involved. Sometimes weakness is limited to the bitten limb. Conclusion: Neuromuscular manifestations associated with systemic infections, though uncommon should be considered in diffential diagnosis. Involvement is variable in severity and sometimes they occur in combination. Early detection and appropriate treatment helps in reducing the morbidity and mortality.
SS 22.1 / #82THE HISTORY OF NEUROMUSCULAR DISEASES AND THEIR TREATMENT IN HABSBURG COUNTRIES
Topic: 10. History
Sonja Horn
University of Vienna, Vienna, AT
Abstract: Research on neuromuscular diseases and their therapy in history is usually covered under the notion of ”electrotherapy” – at least in the Habsburg inherited countries. Electrotherapy was used to treat soldiers of the Habsburg army already in the late 18thC., however there are also distinctive reports about the frequent use of electrotherapy at the Vienna clinic during the mid 18th C. Perhaps as a typical way of practicing medicine at Vienna, the treatment of patients at hospitals in was intensely linked to the training of physicians and surgeons, as well as to post mortem dissections and theoretical research. Therefore ”electrotherapy” has to be seen as strongly linked to theoretical and experimental research on the function of nerves and muscles as well as on the anatomy of the brain and the ”location” of intellectual functions. This concept of the production of medical and scientific knowledge was practiced not only at Vienna, but also at other universities in the Habsburg countries like Prague or Pavia, that are connected with researchers like Jan Evangelista Purkinje (1787 – 1869), Alessandro Volta (1745 – 1827) or Camillo Golgi (1843 – 1926). In my paper I will focus on concepts and topics of research on neuromuscular diseases, which is strongly linked to the practical use of electrotherapy.
SS 22.2 / #31HISTORY ON NM DISEASE
Topic: 10. History
Richard J. Barohn
Neurology, University of Kansas Medical Center, Kansas City, KS, US
Abstract: MG was suspected as a disease probably since Thomas Willis first wrote about ”the palsy” in the late 1600s. In the late 1800s, MG was defined as a clinical entity by German neurologists; Wilhelm Erb, Samuel Goldflam, Herman Oppenheim, and Friedrich Jolly described the decremental muscle response on electrophysiologic testing. In the first half of the twentieth century. The two discoveries that led to a better understanding of MG involved the thymus gland’s relation to MG and the drug treatment for MG using acetylcholinesterase inhibitors. Sauerbach first successfully removed an enlarged thymus gland in 1911. In the 1930s and 1940s, Blalock advocated and began performing thymectomy for MG patients with and without thymoma. Mary Walker in England was the first to observe the drugs physostigmine and neostigmine improved MG patients. In the second half of the twentieth century a number of discoveries occurred: (1) an understanding of the neuroanatomy and neurochemistry of the NMJ; (2) development of the animal model of experimental allergic MG; (3) the discovery of antibodies to the acetylcholine receptor (AChR) in humans with MG and passive transfer of MG to mice from human immunoglobulin of MG patients; (4) passive transfer of MG to mice from human immunoglobulin of MG patients; (5) the discovery that corticosteroids and other immunosuppressive agents, including plasmapheresis, can improve MG symptoms and signs; and finally, (6) the development of mechanical ventilation to keep patients alive when they go into respiratory failure from myasthenia crisis; (7) improvement in electrophysiologic techniques for diagnosing MG, and the development of single fiber EMG. In the 1950s, the disorder known as LEMS was described, initially in patients with cancer and later in noncancer patients. In the 1970s, the first congenital myasthenic syndrome was described. In the current era, new autoantibodies are being discovered that are responsible for NMJ disorders (voltage-gated P-/Q-type calcium channels in LEMS; to LRP4, rapsyn, agrin, and cortactin in MG). We now have a great deal of experience in clinical research in MG and have refined the endpoint measurements and trial designs. With the completion and publication of the landmark thymectomy trial in MG, we can now say with conviction that patients with MG who undergo thymectomy improve and their prednisone dose can be lowered more than patients who do not have a thymectomy. Prior to 2000, there were only seven randomized controlled published MG trials. As of 2018, 22 randomized clinical trials have been published for MG. Recently, this led to the US Food and Drug Administration approval of the complement inhibitor eculizumab (Soliris) for adult patients with generalized MG who are AChR antibody positive. Prior studies demonstrated increased complement in the serum of MG patients and the presence of complements at the NMJ in MG. MG indeed used to be a grave disease with 30%mortality. Now, with the advent of all the advances and others described above, everyone we see with MG should improve, and it should be a rare event indeed for a patient to die of MG.
SS 22.3 / #177ANDALUSIAN NEUROLOGY OVER 700 YEARS
Topic: 10. History
Raad Shakir
World Federation of Neurology, London, GB
Abstract: I will confine myself to the evolution of Medicine and Neurology in Andalusia 711-1492 AD. These centuries saw the massive proliferation of not only Sciences and Philosophy but were the vehicle of transfer of Greco-Roman literature back to Europe. There are numerous scholars and physicians who influenced our thinking and practice. Four will be discussed Averroes (1126-1198), Maimonides (1135-1209), Avenzoar (1094-1161) and Abulcasis (936-1013). The first three were contemporaneous and influenced each other. Each has contributed in a rather unique way to medicine with some emphasis on Neurology and the diseases of the nervous system. Averroes was a defender of the Aristotelian philosophy and is considered to be the father of secular thought in Europe. Maimonides in addition to being a first class physician and the private physician of Saladin was a most respected Torah scholar and theologian. Avenzoar was probably the first physician to practice surgery on animals before trying them on Humans. Abulcasis was a brilliant surgeon and his neurosurgical tools in treating hydrocephalus as an example, were ingenious to say the least. The Golden age of Andalusia was at its zenith during the 10th to the 12th century.
SS 23.1 / #42THYMECTOMY FOR MYASTHENIA GRAVIS
Topic: 04. Neuromuscular Junction
Richard J. Barohn
Neurology, University of Kansas Medical Center, Kansas City, KS, US
Abstract: Thymectomy has a central role in the treatment of MG. In thymomatous MG, the tumor should be removed. Along with thymoma, the entirety of the thymus tissue should be removed. Improvement in myasthenic symptoms may or may not follow. A systematic review of the existing thymectomy literature emphasized this knowledge gap and recommended the MG field perform a randomized, controlled trial. (Gronseth and Barohn, 2000) However, owing to the difficulty of performing controlled trials involving thoracic surgery in a rare disease, high-quality evidence about thymectomy had been lacking. A recently completed landmark international, randomized, rater-blinded clinical trial controlling for medical treatment was designed to address this uncertainty. One hundred twenty-six recently diagnosed patients, ages 18 through 65 with AChR antibody–positive generalized MG were randomized to receive either extended transsternal thymectomy plus prednisone versus medical management with prednisone. Over a 3-year follow-up period, the time-weighted average QMG score was lower in the patients who underwent thymectomy (6.15 vs 8.99; P<.001) Similarly, the thymectomy group had a lower time-weighted alternate-day prednisone dose requirement (initially reported at 44 mg vs 60 mg; P<.001), which was later corrected to 32 mg versus 54 mg (95% confidence interval, 12-32 mg; P<.001). Also in the thymectomy group, there were fewer patients requiring additional immunosuppression, fewer adverse events, and fewer admissions for myasthenic crises. These data provide support for thymectomy as a first-line treatment modality that can improve MG status and decrease the required dose and duration of immunotherapy in generalized MG. (Surprisingly, the effects of the thymectomy could be observed as early as 3 to 4 months and were maintained for the entire 3-year study.) Patients with persistent bulbar, respiratory, or limb weakness should be treated with PLEX before surgery. Thymectomy in MuSK, LRP4, and agrin antibody–positive patients is not supported by current evidence. Patients with MG with MuSK antibodies were not included in the recent thymectomy study. Nevertheless, MuSK and ”double-negative” antibody patients have undergone thymectomy and anecdotally have done well. Similarly, there is limited evidence to support thymectomy in patients with ocular MG, although if the patient is AChR antibody positive, it may be considered in refractory cases. The recently completed thymectomy trial mandated a sternal-splitting procedure. Several new less invasive procedures are now being used for thymus removal. Video-assisted thoracoscopic surgery and robotic approaches to thymectomy such as robotic video-assisted thoracoscopic surgery offer shorter hospital durations of stay and limited morbidity have emerged as alternatives to the classic transsternal approach. There are no trials comparing these surgical techniques, however, and available reports suggest comparable results.
SS 23.2 / #173RANDOMIZED CONTROLLED TRIAL OF THYMECTOMY: A DEEPER DIVE INTO DATA
Topic: 04. Neuromuscular Junction
Gil I. Wolfe1, Henry J. Kaminski2, Inmaculada B. Aban3, Greg Minisman4, Hui-Chien Kuo3, Joshua R. Sonett5, Alexander Marx6, Cleo-Aron Weis6, Gary R. Cutter7
1Neurology, University at Buffalo, Buffalo, NY, US;2Neurology, George Washington University, Washington, DC, US;3Biostatistics, University of Alabama at Birmingham, Birmingham, AL, US;4Univesity of Alabama at Birmingham, Birmingham, AL, US;5General Thoracic Surgery, Columbia University Medical Center, New York, NY, US;6Pathology, University Medical Centre Mannheim/University of Heidelberg, Mannheim, DE;7Biostatistics, UNiversity of Alabama at Birmingham, Birmingham, AL, US
Background: MGTX, the Thymectomy Trial in Non-thymomatous Generalized Myasthenia Gravis Patients Receiving Corticosteroids, was the first-ever randomized, rater-blinded study of thymectomy in myasthenia gravis (MG). The international trial addressed questions regarding the efficacy of thymectomy in non-thymomatous MG that had persisted for 75 years since use of the procedure was first reported by Blalock and colleagues in 1941. Methods: MGTX was designed to answer three questions: does the combination of alternate-day prednisone and extended transsternal thymectomy after 3 years compared to an identical dosing protocol of prednisone alone, (1) lead to better outcomes in MG as measured by a time-weighted average of the Quantitative MG Score (QMG), (2) reduce the time-weighted average alternate-day prednisone requirements, and (3) reduce side-effect burdens on patients primarily arising from medications used to treat the disease? Patients age 18 to 65 years with generalized non-thymomatous acetylcholine receptor antibody-positive MG of less than 5 years duration and MG Foundation of America Clinical Classification 2 to 4 were enrolled. Dual primary outcomes were the time-weighted average of the Quantitative MG (QMG) score and prednisone dose over 3 years. Results: A total of 126 MG patients underwent randomization at 36 sites. Subjects who underwent thymectomy showed significant improvements in MG status (time-weighted average QMG 6.15 vs. 8.99, p<0.001) and reduced alternate-day prednisone requirements (32 mg vs. 54 mg, p<0.001) at 3 years with benefits observed as early as 9 to 12 months. The ability to withdraw completely from prednisone and remain off it at 3 years also favored the surgical arm (58% vs. 20%, p<0.01). The ability to withdraw from prednisone did not correlate with duration of disease, or age at MG onset or trial enrollment. Histological analysis of thymic follicular hyperplasia, seen in 67% of surgical patients, failed to correlate with the time-weighted QMG or prednisone exposure. However, intra-thymic fat content directly correlated with time-weighted prednisone exposure (p=0.004). On secondary outcomes, fewer patients who had thymectomy required immunosuppression with azathioprine (17% vs. 48%, p<0.001) or were hospitalized for exacerbations (9% vs. 37%, p<0.001). The number of patients with treatment-associated complications did not differ significantly between groups (p=0.73), but the thymectomy group had fewer treatment-associated symptoms (p<0.001) and lower distress levels related to symptoms (p=0.003). MG-Activities of Daily Living scale and the proportion of patients reaching Minimal Manifestation Status also significantly favored the surgical arm. Conclusion: MGTX provided a definitive answer sought by the MG community since the middle of the last century. Extended transsternal thymectomy performed within the first 5 years of disease improves clinical outcomes, reduces immunosuppressive medication requirements, and decreases side-effect burdens in generalized acetylcholine receptor antibody- positive MG patients without thymoma. This group represents the largest subpopulation of MG patients. Although the MGTX trial did not test less-invasive approaches for removing the thymus gland which have become increasingly popular over the last two decades, one can surmise that less invasive approaches that completely replicate the maximal tissue resection performed in an extended transsternal thymectomy would confer similar benefits for patients.
SS 23.3 / #104PATHOLOGY OF THE THYMUS GLAND IN MG
Topic: 04. Neuromuscular Junction
Alexander Marx
Pathology, University Medical Centre Mannheim/University of Heidelberg, Mannheim, DE
Abstract not received.
SS 24.1 / #214SYNAPTIC STABILITY IN CMS
Topic: 04. Neuromuscular Junction
David Beeson
Nuffield Department Of Clinical Neurosciences, University of Oxford, Oxford, GB
Abstract not received.
SS 24.2 / #218RECENT PROGRESS IN UNDERSTANDING PRESYNAPTIC CONGENITAL MYASTHENIC SYNDROMES (CMS)
Topic: 04. Neuromuscular Junction
Hanns Lochmüller1, Emily O’Connor2, Grace Mcmacken2, Yoshiteru Azuma2, Sunitha Belaraju2, Teresinha Evangelista2, Roger Whittaker3, Ana Töpf4, Andreas Roos4, Rita Horvath4
1Department Of Neuropediatrics And Muscle Disorders, Freiburg University - Medical Center, Faculty of Medicine, Freiburg, DE;2Institute Of Genetic Medicine, Newcastle University, Newcastle, GB;3Institute Of Neuroscience, Newcastle University, Newcastle, GB;4John Walton Muscular Dystrophy Research Center, Newcastle University, Newcastle, GB
Background: Neuromuscular junction disorders, also called Myasthenic syndromes (MS), are a rare heterogeneous group of acquired (Myasthenia Gravis, MG) and inherited (Congenital Myasthenic Syndromes, CMS) neuromuscular disorders associated with distinctive clinical, electrophysiological, laboratory and ultrastructural abnormalities. The genetic defects in CMS either impair neuromuscular transmission directly or result in secondary impairments, which eventually compromise the safety margin of neuromuscular transmission and may affect proteins that are specific to the synapse or more widely expressed proteins. Methods: While the majority of patients with CMS harbours mutations in genes encoding postsynaptic proteins, defects in presynaptic proteins are increasingly discovered, often associated with additional symptoms of the PNS or CNS. Results: Mutations in SYT2, the gene encoding synaptotagmin 2 are dominantly inherited and show signs reminiscent of hereditary motor neuropathies and fatigability. Mutations in several presynaptic genes are causing severe childhood disorders with episodic apnea or intellectual disability. Loss of MYO9A, a recently identified CMS-associated protein, affects the neuronal cytoskeleton and leads to impaired transport of proteins, including agrin, which may provide a new and unexpected treatment option. Conclusion: We will cover the significant progress made in understanding the molecular pathogenesis of CMS, which is important for both patients and clinicians in terms of reaching a definite diagnosis and selecting the most appropriate treatment.
SS 24.3 / #225CLINICAL ASPECTS OF CMS
Topic: 04. Neuromuscular Junction
Hakan Cetin
Department Of Neurology, Medical University of Vienna, Vienna, AT
Background: Congenital myasthenic syndromes (CMS) are a heterogeneous group of inherited disorders of the neuromuscular junction (NMJ). They are caused by mutations in genes that code for proteins affecting NMJ structure and function. The genetic defect can be located at the presynaptic nerve terminal, the synaptic cleft and the postsynaptic membrane. More recently, next-generation sequencing techniques have also identified causative genes encoding ubiquitously expressed molecules, the functional defect of which, however, may be largely restricted to the NMJ. Methods: Postsynaptic NMJ defects including i) acetylcholine receptor (AChR) deficiency syndromes, ii) slow-channel CMS, iii) fast channel CMS, iv) low conductance CMS and v) CMS due to perturbation of the AChR clustering pathway are reviewed and correlated with clinical findings. Results: Mutations in the AChR subunit genes cause a primary AChR deficiency at the endplate. AChR deficiency syndromes show a recessive inheritance pattern. Disease severity is variable but weakness is usually evident at birth or within the first year of life. Feeding difficulties, ptosis, impaired eye movements and delayed motor milestones are frequent. The disease course is usually not progressive and the patients sometimes improve by early adulthood. Loss of endplate AChR may also be due to the recessive inheritance of RAPSN mutations. The more common ”early onset” form of the disease is associated with hypotonia, bulbar dysfunction and joint contractures of hands and ankles. Affected neonates often require assisted ventilation. By contrast, ”late onset” forms begin from early adulthood and are not associated with bulbar, speech and respiratory problems. They may be mistaken as seronegative myasthenia gravis, but weakness of ankle dorsiflexion may be a helpful clinical tool to differentiate CMS from autoimmune myasthenia gravis. Other AChR subunit mutations predominantly affect ion channel kinetics without a severe reduction of AChR numbers. Slow-channel CMS forms are characterised by longer AChR burst durations. This is associated with prolonged endplate potentials resulting in a depolarisation block of voltage-gated sodium channels. Slow-channel CMS shows a dominant inheritance pattern but there can be variable penetrance, age of onset and disease severity. However, weakness of cervical and scapular muscles and finger extensors is often an early feature. Repetitive compound muscle action potentials to single nerve stimuli in electrodiagnostic studies are characteristic. Fast-channel CMS, by contrast, is characterised by shorter AChR burst-duration and endplate potentials that fail to elicit action potentials in the muscle cell. They have recessive inheritance. Feeding difficulties, ptosis, impaired eye movements and delayed motor milestones are often present at birth with the patients generally more severely affected than in AChR deficiency syndromes. Low-conductance CMS is also inherited recessively. It is very rare and the phenotype is similar to fast-channel syndrome. Mutations in the AChR clustering pathway including agrin, LRP4, MuSK and DOK7 are characterised by a limb-girdle distribution of weakness. Patients may achieve normal walking milestones but then start to experience falls associated with proximal muscle weakness. Conclusion: A deep understanding of disease mechanisms underlying CMS is crucial in order to correlate diagnostic and clinical findings with corresponding molecular defects and thus direct genetic screening and effective treatment.
SS 24.4 / #312THERAPEUTIC STRATEGIES FOR CONGENITAL MYASTHENIC SYNDROMES
Topic: 04. Neuromuscular Junction
Jacqueline Palace
Neurology, Oxford univeristy Hospitals Trust, DU, GB
Abstract: The congenital myasthenic syndromes (CMS) are a heterogeneous group of genetic disorders which cause neuromuscular junction dysfunction. More than 30 different genes have been described as causing CMS and the treatment regimes vary, dependent upon the CMS subtype. For example pyridostigmine and 3,4-Diaminopyridine improve muscle strength in many genetic forms of CMS but make others worse. The mainstay of treatment remains anticholinesterases, in particular pyridostigmine, which improves strength in AChR deficiency syndromes due to epsilon subunit mutations, the fast channel syndromes and CMS due to mutations in RAPSN, CHAT, and glycosylation pathways genes. 3,4-diaminopyrodine (3-4DAP) can also be beneficial in the same conditions (although because acetylcholine is deficient in CHAT CMS increasing the presynaptic release with 3,4-DAP may not be a good strategy). Oral salbutamol or ephedrine are beneficial in DOK7 and COLQ CMS and other forms of CMS where the AChR clustering pathway is impaired. It can also improve strength in forms of CMS where pyridostigmine is being taken at higher doses such as in patients with AChR epsilon mutations causing deficiency of the receptor, and in patients with fast channel CMS. Fluoxetine (usually at doses of 40-100mg daily) or quinidine are the main treatment options for slow channel syndrome CMS. Prolonged QT interval may limit the dose and needs monitoring. It is important to note that pyridostigmine (and 3,4-DAP) usually worsen slow channel syndromes, COLQ and DOK7 CMS and thus where CMS is suspected as a diagnosis, genetic screening of these genes should be undertaken prior to initiating anticholinesterase medication.
Workshop
WS 1.1 / #181CHEMOTHERAPY INDUCED PNP
Topic: 06. Mononeuropathy
Anthony Windebank
Mayo Clinic, Rochester, US
Abstract not received.
WS 1.2 / #185IATROGENIC MONONEUROPATHIES AFTER SURGERY AND TRAUMA
Topic: 06. Mononeuropathy
Tatjana Paternostro-Sluga
Social Medical Center East - Donauspital, Institute of Physical Medicine and Rehabilitation, Vienna , AT
Background: Iatrogenic mononeuropathies after surgery and trauma occur accidentally or may be part of the necessary treatment. It is of utmost importance to diagnose the lesions immediately and to start the appropriate treatment early. Even so iatrogenic mononeuropathies have always occurred and will occur in the future, the delayed timing of diagnosis and treatment is highly prevalent. Mononeuropathies may interfere significantly with activities of daily living and reduce the quality of life. Patients may recover completely from the primary disease, but the sequelae of the iatrogenic nerve lesion leads to long-lasting impairment and loss of social participation. Methods: The aim of the workshop is to increase the knowledge on iatrogenic nerve lesions and therefore improve diagnostic and therapeutic procedures for the patients. Results: Iatrogenic nerve lesions can result from different surgical procedures. In the upper extremity median nerve, accessory nerve, ulnar nerve, radial nerve, cervical and brachial plexus, axillary nerve and phrenic nerve may be typically affected. Lesions of the median nerve occur most frequently during carpal tunnel surgey, accessory nerve lesions result from lymph node biopsy in the posterior neck triangle and even this is common knowledge it still occurs. The ulnar nerve is frequently associated with supracondylar humeral fractures in children. This has to be taken into account when children with supracondylar humeral fractures are examined. Ulnar nerv palsy in small children may be overlooked at the beginning, especially when atrophy of the intrinsic muscles is not yet visible and finger abduction is done by radial innervated extrinsic finger extensors. In the lower extremity peroneal nerve, femoral nerve, ischiadic nerve, tibial nerve , ilioinguinal and genitofemoral nerves, lateral cutaneous femoral nerve, lumbosacral plexus, saphenous nerve and infra-patellar branch and sural nerve may be typically affected. Orthopedic and gynecological procedures may affect the ischiadic nerve, orthopedic procedures may also affect the femoral nerve and/or the gluteal nerves in total hip replacement. Knee surgery may affect the tibial nerve or the infra-patellar branch of the saphenous nerve. Tourniquet pressure at the thigh during knee surgery might be associated with femoral nerve palsy and a partial lesion might be overlooked. Weakness and insecurity of walking is wrongly attributed to the knee surgery. Conclusion: Prognosis of iatrogenic mononeuropathies depends on the degree of the lesion. The timing for nerve reconstruction is very important to optimize the outcome in high grade lesions. Diagnostic procedure include history, clinical examination, clinical electrophysiology, nerve ultrasound and magnetic resonance imaging. Conservative treatment has to include pain managment, maintenace of range of motion of joints and tendons, motor and sensory reeducation, minimizing trophic disturbances and regaining function. Surgical treatment includes primary nerve repair, nerve reconstruction, neurolysis. Secondary reconstructive procedures, eg muscle transposition may be applied if nerve regneration even after nerve surgery is not sufficient for regaining function. Knowledge, expertise and dedication in diagnosing and treatment of peripheral nerve lesions as well as interdisciplinary and multiprofessional teamwork are the landmarks of best practice in the treatment of iatrogenc nerve lesions.
WS 2.1 / #156DUX4, ITS ROLE IN DEVELOPMENT AND MUSCLE DISEASE
Topic: 01. Muscle
Stephen J. Tapscott
Fred Hutchinson Cancer Research Center, Seattle, US
Abstract not received.
WS 2.2 / #152TRIALS READINESS IN FSHD: CLINICAL, IMAGING AND BIOMARKERS
Topic: 01. Muscle
Jeffrey M. Statland
Neurology, University of Kansas Medical Center, Kansas City, KS, US
Background: Facioscapulohumeral muscular dystrophy (FSHD) is one of the most common muscular dystrophies, and although it is slowly progressive, it can cause significant morbidity over the course of a lifetime, interfering with the ability to maintain a job or walk independently. There is at present no effective treatment for FSHD. Substantial evidence now implicates aberrant reactivation of DUX4 as the cause of FSHD providing a clear target for therapeutic intervention. The success in identifying the molecular and genetic mechanisms of FSHD provides a strong basis for drug development and therapeutic clinical trials. The key abnormality in FSHD, the aberrant expression of DUX4, is potentially amenable to RNA therapies making possible similar strategies to drugs already approved for spinal muscular atrophy and Duchenne muscular dystrophy, or may be amenable to other disease-directed strategies targeting epigenetic changes in FSHD. In addition, drugs not specifically targeting DUX4 but designed to ameliorate downstream pathology, such as muscle inflammation or muscle atrophy and fibrosis, are either underway now or proposed to start in the next few years. Methods: A prospective study of FSHD was performed in the 1990s that determined the multisite reliability and sensitivity to disease progression of manual muscle testing, quantitative myometry, and timed functional outcomes. However limitations to using strength or functional outcomes for FSHD were identified: the first is that the clinical relevance of relatively small changes in strength is unknown; and the second is that demonstrating arrest of disease progression would require large numbers of participants or long follow up times, creating a significant patient and financial burden. To best inform clinical trial design, it is now necessary to identify and validate measures of disease activity and progression. A conceptual framework for thinking about outcome development in FSHD is built on expert opinion about molecular pathology, disease progression, natural history studies, recent large MRI studies, and patient surveys and registries. It is likely we do not need a single outcome measure for FSHD but rather a clinical trial toolbox: in which clinical outcomes and biomarkers will play an important role. Results: Biomarkers can provide proof of concept a drug is reaching its target, can provide strategies to inform inclusion criteria or stratification, and can provide early signals of disease activity which will predict a later functional change. Here we will review the current state of biomarker development in FSHD, looking at the current experience with: 1) molecular biomarkers in muscle tissue; 2) serum biomarkers; 3) the use of MRI as a measure of disease progression, or for stratification; and 4) other techniques like electrical impedance myography or muscle ultrasound for tracking early changes in muscle quality. Conclusion: Development of robust FSHD-relevant biomarkers can hasten therapeutic development, and form a cornerstone of clinical trial preparedness.
WS 2.3 / #125FSHD’S CHANGING LANDSCAPE AND FUTURE PERSPECTIVES
Topic: 01. Muscle
George W. Padberg
Dep. Of Neurology, Radboud University Medical Center, Nijmegen, NL
Background: Facioscapulohumeral Dystrophy is an autosomal dominant disorder with a major myopathic phenotype and a variable, often incomplete minor phenotype. The myopathy has two components: an intrinsic muscle weakness, possibly congenital, on top of which develops a dystrophic proces with a specific descending sequence of muscle involvement which resembles a replay of embryological somite development. The age at onset and the rate of progression are extremely variable within and between families with FSHD and are used as measures of severity of the disease in individual cases. In earlier studies we estimated that 75% of the age at onset was related to environmental factors other than the primary gene defect. FSHD is caused by an epigenetic dysregulation of the D4Z4 macrosatellite repeat on 4q35 which leads to an aberrant expression of the Double-Homeobox gene Dux4 embedded in the terminal repeat unit. Dux4 can only be stably expressed if the repeat is followed by a PAS-sequence present in 4qA telomeric alleles to be found in 50% of the normal population. Dux4 is a double-homeodomain transcription factor normaly expressed in germ-cells and in the zygote-cleavage stage and repressed in later stages of development. Unknown at present is the rather basic question whether it has a function in myogenic stem-cells. The epigenetic repression of Dux4 has been related to the level of methylation proximal in the repeat. Hypomethylation is caused by a reduction of the number of repeats below 10 (FSHD1) or by mutations in SMCHD1 or DNMT3B (FSHD2), all leading to an identical muscle phenotype.The latter two are able to modulate FSHD1 as well. How the hypomethylation is established is presently under study but hypomethylation in itself appears not the only factor leading to Dux4 mRNA transcription. The upstream factors and modifyers of Dux4 expression and its downstream effects and pathways have to be evaluated for their risks and benefits in emerging therapies. Methods: Review of recent genetic and myo-biological literature in relationship to clinical features against the perspective of emerging therapies. Results: Knowledge of the epigenetic factors preventing Dux4 expression are invaluable for counselling (non-penetrance) and might lead to therapies. The downstream effects have to take into account the inflammatory response in muscles (possibly the onset of the dystrophic process), the distal to proximal fatty infiltration pattern as seen on MRI, the atrophic processes of the muscles, the early involved versus the relatively spared muscles and their genetic make-up and metabolic properties, the characteristic asymmetry of muscle involvement, possible gender differences and their metabolic background, and retinal vascular, cochlear, skeletal and possibly CNS involvement. Review of these aspects will lead to a translational research agenda. Conclusion: All the clinical aspects of the disease have to be explained before we fully understand the possible consequences of therapeutic interventions for this, probably most prevalent genetic risk factor for muscular dystrophy and at the same time indispensible gene for the primate lineage.
WS 3.1 / #123THE ROLE OF PALLIATIVE CARE €– EARLY INVOLVEMENT OR END OF LIFE CARE?
Topic: 12. Patient Issues
David J. Oliver
Tizard Centre, University of Kent, Canterbury, GB
Background: Palliative care is ”an approach that improves the quality of life of patients and their families facing problems associated with life-threatening illness, through the prevention and relief of suffering, early identification and impeccable assessment and treatment of pain and other problems, physical, psychosocial and spiritual”(WHO 2002). Thus within this definition the timing of palliative care involvement is open and should be related to the needs of the patient, and family, as assessed by an holistic assessment by a multidisciplinary team. Methods: A review of the literature was undertaken, together with clinical experience. Results: Although palliative care is often associated with care at the end of life the role is much greater. For a patient with progressive neuromuscular disease, such as ALS, it can be argued that the care should be palliative in it approach from the time of diagnosis. For instance the way the diagnosis is told to the patient and family can have significant effect on the later stages of care – a patient who understands that death may be distressing, from choking or breathlessness, may continue to fear these issues, despite any further discussion and explanation that these risks are small. The care of any patient with neuromuscular disease will need to include the multidisciplinary team assessment of all aspects of care: Physical – the symptoms and mobility issues Psychological – the fears and concerns of the person Social – most patients are part of wider families and have concerns for them or are the cause of concern to them Spiritual – the deeper issues that all people consider, particularly as they face progressive illness, of the meaning of illness and life Thus all professionals involved in the care of people with neuromuscular disease need to provide palliative care – aiming to maximize the quality of life of patients and families and listen and respond to the issues they face as individuals. This is usually from a multidisciplinary team, and may improve not only quality, but length, of life. Some patients and families will need increased care, from a specialist palliative care team because of very complex symptoms or psychosocial issues., perhaps at particular times – such as with the distress of diagnosis, consideration of gastrostomy, commencement of ventilatory support and at the end of life in ALS. At other times the care may be provided by the multidisciplinary team providing palliative care. Conclusion: Close collaboration and communication is essential among all the teams involved always responding to the individual patient’s needs. As the patient deteriorates and comes toward the end of life there may be increasing specialist palliative care, and hospice, involvement to support the patient, family, carers and professionals as death approaches. The care in the earlier stages will influence this time at the end of life and a careful team involvement, using specialist help as required, will allow patients and their families to approach death with confidence in their care and enable a less distressing terminal phase, and later bereavement for their family. Reference World Health Organization. Palliative care 2002 www.who.int/cancer/palliative/definition/en/
WS 3.2 / #110THE ROLE OF THE MULTIDISCIPLINARY TEAM IN NMD – THE EVIDENCE FROM ALS
Topic: 12. Patient Issues
Christopher Mcdermott
University Of Sheffield, Sheffield, US
Abstract not received.
WS 3.3 / #169THE RECOGNITION OF THE END OF LIFE PHASE IN NMD – THE SUPPORT OF PATIENTS, FAMILIES AND PROFESSIONALS
Topic: 12. Patient Issues
Stefan Lorenzl
Insitute of Nursing Science And Practice, Salzburg, DE
Abstract not received.
WS 3.4 / #854INTEGRATING PALLIATIVE CARE INTO SERVICE FOR ALS PATIENTS: REAL-LIFE EXPERIENCE
Topic: 12. Patient Issues
Lev Brylev1, Vadim Parshikov2, Yaroslava Krasnaya2, Ekaterina Dikhter2, Egor Larin3, Yana Batmanova2, Vasiliy Shtabnitskiy4, Sergey Avdeykin5, Anastasia Ataulina5, Daria Puzanok2, Anna Kasianova2, Vera Fominyh1, Margarita Fominyh2, Maria Ivanova6, Elena Lysogorskaya6, Anna Vorobyeva6, Vera S. Demeshonok7, Tatiana Leontyeva2, Timur Ivanov8, Maria Ilchenko2, Alisa Apreleva9, Inessa Zakroyshikova6, Kirill Gorbachev1, Artur Volov2, Timur Mukhametov2, Sergey Korneychuk2, Natalia Semina8, Maria Zakharova6
1Buyanov City Hospital, Moscow, RU;2ALS center Moscow, Moscow, RU;3Center of palliative medicine, Moscow, RU;4Chaika, Moscow, RU;5Buyanov city hospital, Moscow, RU;6Research center of neurology, Moscow, RU;7North-Western State Medical University named after I.I. Mechnikov, Saint Petersburg, RU;8Aprel clinic, Moscow, RU;9Anglia Ruskin University, Cambridge, GB
Background: The first specialized multidisciplinary outpatient center for ALS patients in Moscow started in 2012 and it is now a part of ENCALS network. The clinic follows about 200 families with ALS every year and is funded by charity foundation ”Live now”. Before 2012 patients with ALS had no access to palliative care, because hospices would not accept them and the national legislation which first mentioned the provision of palliative care for non-cancer patientswas only passed in 2011. Methods: Experience has shown that the main needs of patients and their caregivers are information, symptom management, and social and psychological support. To meet these needs palliative care has been gradually introduced into everyday practice. As we had no specialized education in palliative care for neurological patients in Russia, foreign experts were invited to teach our team and a palliative care physician, who studied palliative care abroad, was invited to the multidisciplinary discussions. Multidisciplinary outpatient consultations were started and the palliative care approach, with the emphasis on patient autonomy, dignity and quality of life, changed our practice completely. Results: Analyzing our data from 672 patients we found out that median disease duration was 1188 days (range 791-1787), the time from first symptom to diagnosis was 365 days (range 191 -587) and the time from diagnosis to referral to the team was 320 days (range 132 - 630) days. Patients on admission to our service had ALS for about 2 years and at this time the median vital capacity of 51% (range 38-80) and it was apparent that they had never talked about advance decision making and only 30% of them had ever talked to their families or physicians about prognosis and death. The training we received enhanced our communication skills and it was possible to talk with patients about prognosis and decisions about life sustaining interventions. This was particularly important if patient and families lack information and symptoms are inadequately controlled to prevent unwanted hospitalizations and prolonged ventilator support, that could affect quality of life. Moreover according to Russian legislation interventions, such as ventilation, cannot be withdrawn. One of the most prevalent end-of-life symptoms is dyspnea, -40% of our patients have persistent dyspnoea with median Borg score 5 out of 10. Dyspnoea is also one of the main reason for admission to Intensive care, especially where there has been limited discussion of the use of life sustaining interventions. We were the first team in Russia to use morphine for dyspnea in ALS patients. Our experience shows that morphine may be used safely, with evidence that there is no increase in pCO2 in arterial blood, and effectively – patients evaluated the response as good for 50%, partial for 40% and only 10% reported no help. Conclusion: There is continued close working between neurologists and palliative care specialists with the aim of integrating palliative care principles in standards of care for ALS patients. There are still many challenges on this way. The reported study was funded by ”Live now” charity foundation and RFBR according to the research project № 18-315-00228мол–а
WS 4.1 / #33UPDATE ON HISTOPATHOLOGICAL FEATURES OF INFLAMMATORY MYOPATHIES
Topic: 01. Muscle
Anthony Amato
Dep. Of Neurology, Brigham and Women’s Hospital, Boston, US
Abstract: The major types of inflammatory myopathies (IM), including dermatomyositis (DM), polymyositis (PM), immune-mediated necrotizing myopathy (IMNM), antisynthetase syndrome (ASS), and inclusion body myositis (IBM) The characteristic histopathological abnormality on muscle biopsy in DM is perifasicular atrophy, however, this is present in perhaps only 50% of patients. Immunohistochemical staining for myxovirus resistance protein A (MxA) is diagnostically more sensitive and highly specific. The inflammatory cell infiltrate is predominantly perivascular and in the perimysium and is composed primarily of macrophages, B cells, and plasmacytoid dendritic cells (PDCs). PM is a heterogeneous disorder and muscle pathology is variable. Most often, CD8+ T cells and macrophages are evident in perimysial and endomysial locations. These mononuclear inflammatory cells surround fibers with sarcolemmal MHC-I expression, however true invasion of non-necrotic muscle fibers is much more common in IBM than in PM. The major subtypes of IMNM are anti-SRP and anti-HMGCR necrotizing myopathies, though some are paraneoplastic, associated with other connective tissue diseases, or are idiopathic. The hallmark on muscle biopsy are the presence of multifocal necrotic and regenerating muscle fibers with a paucity of inflammatory cells. However, some patients with HMGCR myopathy have endomysial, macrophage-predominant infiltrates similar to what is seen in PM. Overexpression of MHC-I and membrane attack complex (MAC) deposition in capillaries may be evident on sarcolemma of non-necrotic fibers. In ASS, muscle biopsies demonstrate a predilection for perimysial damage including perimysial fragmentation and staining with alkaline phosphatase, plasmacytoid dendritic cells and macrophages in the perimysium and around blood vessels, and MAC deposition on capillaries. Similar to DM there is perifascicular muscle fiber damage, but with AAS there is more perifascicular muscle fiber necrosis compared to DM, while in perifascicular atrophy is more prominent. MHC-1 and MAC deposits on muscle fibers may be seen on sarcolemma of perifascicular muscle fibers. EM may show actin filaments within myonuclei.
WS 4.2 / #101UPDATE ON MYOSITIS SPECIFIC AUTOANTIBODIES / DISCUSSION ON UTILITY OF MSAS IN DIAGNOSIS AND PROGNOSIS
Topic: 01. Muscle
Andrew Mammen
NIAMS/NIH, Bethesda, US
Abstract not received.
WS 4.3 / #115UPDATE ON UTILITY OF SKELETAL MUSCLE MRI AND ULTRASOUND
Topic: 01. Muscle
Jasper M. Morrow
Queen Square Centre For Neuromuscular Diseases, University College London, London, GB
Background: Recent advances in inherited muscle diseases have led to two major challenges. The development of next generation sequencing has made interpretation of genetic results very difficult; whilst the development of potentially effective gene therapies has emphasised the need for robust outcome measures to determine their efficacy. Qualitative and quantitative skeletal muscle imaging, in particular MRI is emerging as useful tools to address these challenges. Methods: The speaker will update recent literature on the topic, in addition to discussing his own research and clinical experience through illustrative case studies. Results: Pattern of muscle involvement on qualitative MRI or ultrasound has an important role in clinical phenotyping to help guide and interpret genetic testing in inherited muscle diseases; most notably in the congenital myopathies. Whilst most research has focussed on the lower limbs, whole body MRI is becoming more common. In acquired myopathies, MRI is useful in diagnosis, targeted biopsy and in aiding treatment decisions. The use of imaging in diagnosis will be illustrated through a number of clinical vignettes. Quantitative muscle MRI is able to measure both acute and chronic changes in skeletal muscle providing potential biomarkers in clinical trials. Fat-fraction quantification using methods such as three-point Dixon is the most robust marker of disease progression and has now been applied across a wide range of diseases in both natural history and interventional studies showing superior responsiveness to other outcome measures. Acute changes in muscle water distribution are sensitive to a number of quantitative techniques with T2 quantification being the most commonly used. Recent studies have shown that these changes are reversible with effective therapy, such as corticosteorids in Duchenne. Conclusion: Skeletal muscle MRI and ultrasound are rapidly becoming essential tools in neuromusuclar diseases. Potential avenues for furture research will be discussed.
WS 5.1 / #120DIAGNOSIS AND TREATMENT OF CIDP: LESSON FROM THE DATABASES
Topic: 02. Neuropathy
Eduardo Nobile-Orazio, Giuseppe Liberatore, Pietro Doneddu
Neuromuscular And Neuroimmunology Service, Humanitas Research Hospital, Milan University, Rozzano, Milan, IT
Background: Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is a chronic disabling disease that often improves with immune therapy. A few variants of CIDP have been described but their frequency and possible evolution into typical CIDP are unsettled, possibly because of differences in the diagnostic criteria and in the time of follow-up. To date, the most reliable diagnostic criteria for CIDP are the 2010 EFNS/PNS revised criteria, with a reported sensitivity of 80% and specificity of 90%. Methods: We implemented a web-based database to collect data from patients with CIDP followed by Italian Centers with expertise on CIDP to determine the frequency and characteristic of CIDP and it variants, the diagnostic criteria used for their diagnosis, the possible evolution into typical CIDP, the association with specific anti-nerve antibodies, and their response to therapy. All the patients were evaluated at the time of inclusion and will be followed for two years to monitor their outcome and response to therapy. Results: By February 2018 we enrolled 493 patients in the database, 41 of whom were excluded for the presence of an alternative diagnosis (23 patients) or insufficient data (18 patients). The diagnosis of typical CIDP was made in 368 (81%) of the 452 included patients with 305 (83%) fulfilling the EFNS/PNS criteria. Eighty-four patients (19%) patients had a diagnosis of atypical CIDP, 64 (76%) of whom fulfilled the EFNS/PNS criteria. Thirty-six of these patients (8% of included patients) had distal acquired demyelinating symmetric neuropathy (DADS), 17 (4%) purely motor CIDP, 17 (4%) Lewis-Sumner syndrome (LSS) including two with focal CIDP, 14 (3%) purely sensory including two with chronic immune sensory polyradiculopathy (CISP). Based on the symptoms ad disease onset and for at least one year, 39% of the patients had an initial diagnosis of atypical CIDP that had evolved into typical CIDP in more than 50% of the patients (53.5%) at the time of inclusion after a mean disease duration of 6.5 years. Improvement after therapy was observed in 87% of the 289 treated patients with typical CIDP including response to IVIg in 74% of the patients, steroids in 52%, plasma exchange in 53% and immune suppressant in 37% with Rituximab being the most frequently effective (70%). Patients with DADS and LSS had a less frequent response to therapy than those with typical CIDP (p=0.0006; p=0.0185), mainly reflecting a less frequent response to IVIg (p=0.0440; p=0.0136). Patients with pure motor and sensory CIDP did not differ from typical CIDP. In 63 patients the diagnosis of CIDP was not supported by nerve conduction studies according to the EFNS/PNS diagnostic criteria. In 74% of them there were however at least two supportive criteria for the diagnosis of CIDP including response to immune therapies (94%) and increased CSF proteins (82%). Similarly to typical CIDP, 50% of the patients with clinical CIDP had a relapsing course increasing the reliability of the diagnosis for CIDP. Conclusion: This study on a large population of patients is providing useful information that may help to revise the current diagnostic criteria for CIDP.
WS 5.2 / #93PARAPROTEINEMIC NEUROPATHIES: WHEN AND HOW TO TREAT
Topic: 02. Neuropathy
Jean-Marc Léger
National Referral Center For Neuromuscular Diseases, University Hospital Pitié-Salpêtrière, Paris, FR
Abstract: Since the first description 25 years ago of a high prevalence of monoclonal gammopathy (MG) in a context of peripheral neuropathy (PN), an increased number of such associations has been reported. However, neuropathies associated with MG have heterogeneous clinical, neurophysiological, neuropathological, and haematological features. The more pertinent relationship seems that found in distal acquired demyelinating sensory (DADS) neuropathy associated with IgM MG of unknown significance (MGUS), and the presence of serum auto-antibodies reacting with myelin-associated-glycoprotein (MAG). However in the absence of adequate data concerning relationship in most of cases, the following good practice points may be agreed: (1) Patients with demyelinating neuropathy (DNP) associated with MG should be investigated for a malignant plasma cell dyscrasia. The MG is more likely to be causing the neuropathy if it is IgM, autoantibodies (mainly directed to MAG) are present in serum or on nerve biopsy, or the clinical presentation is chronic distal sensory neuropathy. DNP associated with IgM MGUS sometimes responds to immune therapies. Their potential benefit should be balanced against their possible side-effects. Rituximab, an anti-CD 20 monoclonal antibody, seems a promising therapy. DNP associated with IgG or IgA MGUS is indistinguishable from chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), on clinical, electrophysiologic, and therapeutic issues. For POEMS (polyneuropathy, organomegaly, endocrinopathy, M-protein, skin changes) syndrome, suited therapy may be followed by dramatical improvement of the neuropathy. (2) Patients with chronic axonal polyneuropathy associated with IgG MG should be investigated for Al amyloidosis. However, the majority of chronic axonal polyneuropathies associated with IgG or IgM MGUS are indistinguishable from chronic idiopathic axonal polyneuropathies and do not need any treatment.
WS 5.3 / #107POEMS SYNDROME: FROM DIAGNOSIS TO THERAPY
Topic: 02. Neuropathy
Michelle Mauermann
Mayo Clinic, Rochester, MN, US
Background: Monoclonal gammopathies affect 3-4% of the population older than 50 years and affect more than 5% of the population older than 70 years. They reflect a diverse group of disorders that share the secretion a monoclonal Immunoglobulin produced by the bone marrow. POEMS syndrome is a rare paraneoplastic syndrome associated with a monoclonal plasma cell disorder. It frequently presents with a subacute sensorimotor peripheral neuropathy that can progress to a severe polyradiculoneuropathy. The diagnostic criteria for POEMS syndrome require the presence of a monoclonal plasma cell proliferative disorder and a polyneuropathy. Other major criteria include elevated vascular endothelial growth factor (VEGF), Castleman disease and sclerotic bone lesions. There are several minor criteria including organomegaly, extravascular volume overload, endocrinopathy, skin changes, papilledema and thrombocytosis/polycythemia. Methods: In this course we will review the challenges in making the diagnosis of POEMS syndrome. We will also review the clinical, electrophysiological and pathologic features of the peripheral neuropathy associated with POEMS syndrome. We will discuss the rationale behind the treatments available for POEMS syndrome and the future directions in therapy. Results: POEMS syndrome typically presents with a demyelinating sensorimotor peripheral neuropathy that progresses to a polyradiculoneuropathy. It is often mistaken for chronic inflammatory demyelinating polyneuropathy (CIDP). Distinguishing features include older age of onset, infrequent cranial nerve involvement, more severe distal lower extremity involvement and frequent pain. The electrophysiological features show uniform demyelination with less frequent conduction block and temporal dispersion. Recent studies have showed the utility of blink reflexes in demyelinating neuropathies and its utility in assessing treatment responses in POEMS syndrome. The monoclonal plasma cell disorder associated with POEMS syndrome is lambda light chain restricted in greater than 95 percent of patients. Radiologic evaluation for the presence of sclerotic bone lesions is recommended, with CT providing increased sensitivity over skeletal survey. Currently there is no approved treatment for POEMS syndrome. Radiation is used for patients with limited bone lesions without evidence of systemic disease. For patients with greater numbers of bone lesions or bone marrow involvement, systemic therapy is recommended. Autologous stem cell transplantation has been shown to be beneficial in the improvement of overall survival and progression free survival. Studies of also shown that the peripheral neuropathy improves after autologous stem cell transplantation. In patients ineligible for radiation therapy or autologous stem cell therapy other systemic therapies are available and have been shown to be efficacious. Conclusion: POEMS syndrome typically presents with a subacute painful demyelinating polyradiculoneuropathy. It is differentiated from other monoclonal protein associated disorders by the presence of a lambda monoclonal protein, demyelinating features on nerve conduction studies as well as systemic symptoms. It is important to consider this diagnosis in cases of treatment refractory CIDP. Early diagnosis is necessary to prevent worsening disability. There are several therapies shown to be beneficial in the treatment of POEMS syndrome but there is lack of comparison between these treatments.
WS 6.1 / #195ULTRASOUND IN CRANIAL NERVES
Topic: 07. Cranial Nerves
Stefan Meng
KFJ Hospital, Vienna, AT
Abstract not received.
WS 6.2 / #141CLINICAL AND MICROSURGICAL SIGNIFICANCE OF CRANIAL NERVE VISUALIZATION
Topic: 07. Cranial Nerves
Andres Rodriguez
Uppsala University, Uppsala, SE
Abstract not received.
WS 6.3 / #89HIGH RESOLUTION MRI OF CRANIAL NERVES
Topic: 07. Cranial Nerves
Jennifer Kollmer
Heidelberg University Hospital, Heidelberg, DE
Abstract not received.
WS 7.1 / #71CACHEXIA AND LATE EFFECTS OF CANCER (FROM AN ONCOLOGICAL PERSPECTIVE)
Topic: 08. Cancer
Ioannis Gioulbasanis
Chemotherapy, Larissa General Clinic ”E Patsides”, Larissa, GR
Background: Cancer cachexia is a syndrome characterized by progressive loss of weight and muscle wasting that eventually leads to functional decline and adversely affects survival. When fully developed, even patients themselves can easily recognize it, as it is associated with major changes in body image. However, a key prerequisite for a successful therapy is the timely initiation of any intervention - before the syndrome becomes irreversible. An important limitation, however, is that there are no specific symptoms, signs or laboratory tests that could trigger well-timed clinical attention and diagnosis. Methods: According to our current perception, there is a pre-cachectic stage characterized by anorexia and ”minor metabolic changes”. Anorexia, however, is a non-specific symptom that may be present in about half of cancer patients at diagnosis and could be attributed to a variety of causative factors that may or may not be related with the pathogenesis of the syndrome. When the syndrome is progressing, reduced food intake (the unavoidable consequence of persistence anorexia) will lead to decrease energy intake. Moreover, systemic inflammation, measured in practice by increased levels of C-reactive protein, is usually present and is associated with increased metabolic rate and energy consumption. These conditions result in negative caloric and protein balance and they can be routinely identified in the context of a detailed nutritional assessment. They also lead to depletion of fat and muscle stores quantified as weight loss >5% or >2% when either body mass index is <20kg/m2 or sarcopenia is present. Patients at this stage of cachexia may frequently experience fatigue, a symptom causally related to cachexia and, together with anorexia, they composed one of the most prevalent symptom clusters. Results: In the last (refractory) phase, the most characteristic feature is the deterioration of the performance status and patients commonly suffer from asthenia (a subjective sensation of tiredness without any effort) and/or depression. Moreover, because the tumor is no longer responsive to anticancer treatment, many of them experience uncontrolled symptoms like pain and dyspnea and their quality of life deteriorates further. Conclusion: While these aforementioned phases of the syndrome usually cannot be clearly distinguished from one another, most patients with advanced cancer with ultimately develop cachexia.
WS 7.2 / #130SARCOPENIA
Topic: 08. Cancer
Alan Pestronk
Washington University School of Medicine, Neuromuscular Clinic, St. Louis, US
Abstract not received.
WS 7.3 / #30OUTLINE ON MYOPATHIES / NEUROMUSCULAR COMPLICATIONS ASSOCIATED WITH CANCER, INCL. STEROID MYOPATHY
Topic: 08. Cancer
Anthony Amato
Dep. Of Neurology, Brigham and Women’s Hospital, Boston, US
Abstract: Weakness in patients with cancer is often multifactorial. Most common etiology is sarcopenia related to cachexia and disuse. Corticosteroids used to treat edema (brain tumors) can cause type 2 muscle fiber atrophy leading to weakness as well. Dermatomyositis, polymyositis, and autoimmune necrotizing myopathy as well as Lambert-Eaton myasthenic syndrome can be paraneoplastic and these disorders be the presenting manifestation. Check-point inhibitors have recently been shown to be associated with autoimmune myopathies and neuropathies. Only rarely have chemotherapies been associated with a direct toxic effect on muscle.
WS 7.4 / #103CANCER REHABILITATION
Topic: 08. Cancer
Christine Marosi
Medical University of Vienna, Vienna, AT
Abstract not received.
WS 8.1 / #118SPECTRUM OF PARAPROTEINEMIC NEUROPATHIES
Topic: 02. Neuropathy
Eduardo Nobile-Orazio
Neuromuscular And Neuroimmunology Service, Humanitas Research Hospital, Milan University, Rozzano, Milan, IT
Background: A consistent proportion patients with neuropathy have a monoclonal gammopathy (or paraprotein) in their serum suggesting a possible pathogenetic link between the two abnormalities. In a series from an highly specialized Center for the diagnosis of neuropathy in the United States, 4% of patients with neuropathy had a monoclonal gammopathy being the third leading cause of neuropathy after diabetes (31%) and genetic disease (7%). A similarly high frequency was observed in an unselected series of patients with neuropathy from Sweden where monoclonal gammopathy was only less frequent than diabetes (19%), genetic diseases (12%), alcohol (10%), chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) (8%) other toxic neuropathies (6%) and connective tissue disorders (5%). On the other hand monoclonal gammopathy is frequent in the adult population being found in 3% of the population above 50 years of age and 7.5% above 85 years indicating that a coincidental association may also frequently occur. It is therefore important for the neurologist to clarify the pathogenetic significance of this association since the neuropathy may occur in patients with an otherwise asymptomatic monoclonal gammopathy not requiring for the hematologist any treatment. Methods: Neuropathy has been associated with several diseases characterized by the presence of a monoclonal gammopathy being reported in 3-14% of patients with multiple myeloma, 30-50% of those with Waldenström’s macroglobulinemia (WM), 2-8% with Lymphoma, and 10-20% with light chain amyloidosis (AL). In these conditions the treatment is mainly addressed by the hematologist while neurologist are mainly involved in the initial diagnosis, since neuropathy may be the presenting symptom, or in assessing the possible occurrence of neuropathy induced by the chemotherapy used for the hematological disorder. More relevant for the neurologist is the occurrence of neuropathy in patients with monoclonal gammopathy of undetermined significance (MGUS), where the prevalence of symptomatic neuropathy range form 8% to 36%. In all these studies the prevalence of neuropathy is higher in patients with IgM than with IgG or IgA MGUS, indicating that IgM monoclonal gammopathy are more likely to have a pathogenetic role in the neuropathy. This is also supported by the frequent occurrence of IgM antibodies against nerve antigens in patients with IgM monoclonal gammopathy that may be found in over 50% of the patients with neuropathy. Results: An additional condition presenting to the neurologist is the POEMS syndrome, a rare multisystem disease characterized by the presence polyneuropathy (P), organomegaly (O), endocrinopathy (E), monoclonal gammopathy (M) and skin changes (S). The pathogenesis of POEMS syndrome is poorly understood but overproduction of vascular endothelial growth factor (VEGF) is considered to play a major role. A severe neuropathy with electrophysiological signs of both axonal and myelin involvement is the predominant manifestation of this syndrome and requires aggressive therapy to prevent a disabling clinical course. Conclusion: We will address in this lecture the therapy used by the neurologist in the treatment of these neuropathies and only briefly mention the therapies used by the hematologist to treat the malignant forms of monoclonal gammopathy.
WS 8.2 / #153ANTI-MAG NEUROPATHY: CURRENT AND NOVEL TREATMENTS
Topic: 02. Neuropathy
Andreas Steck
Department Neurology, Basel, CH
Abstract: Anti-MAG (myelin-associated glycoprotein) neuropathy is a chronic disabling demyelinating peripheral neuropathy with an autoimmune etiology. Anti-MAG antibodies are detected in half of IgM paraproteinaemic neuropathy cases. Monoclonal IgM autoantibodies recognize the HNK-1 trisaccharide epitope that is present on MAG as well as on other glycoconjugates of the peripheral nervous system. Electrodiagnostic tests reveal a distal demyelinating polyneuropathy with marked prolonged distal motor latencies. Nerve biopsy shows demyelinating characteristics with pathognomonic features: IgM deposits in the myelin sheaths and widening of myelin lamellae. Anti-MAG antibodies are detected by ELISA. The goal of current therapies is the reduction of pathogenic autoantibodies, the decrease of expanded autoantibody-producing B cell clones, and/or the interference with antibody-effector mechanisms. We will review current and novel treatments. Cytostatics (e.g. cyclophosphamide), plasma exchange (for acute worsening), intravenous immunoglobulin, and the CD20+ B cell-depleting agent Rituximab have been used for the therapy of anti-MAG neuropathy. New anti- B cells drugs such as Ofatumumab, Ocrelizumab, Obinutuzumab and Ibrutinib have been proposed. However, these approaches lack selectivity, and thus so far no satisfactory treatment is available. Because clinical improvement of patients’ neuropathic symptoms correlates with reduced serum levels of anti-MAG antibodies and disease worsening is associated with increasing anti-MAG levels during treatment follow-up, a more efficient and safe therapy might be achieved with antigen-specific agents that selectively target anti-MAG IgM antibodies or antibody-producing B cells. We will present data showing that a synthetic glycomimetic of the natural HNK-1 glycoepitope efficiently remove pathogenic anti-MAG antibodies in an immunological mouse model for anti-MAG neuropathy thus opening a new avenue for treatment. References: Herrendorff R. et al. Selective in vivo removal of pathogenic anti-MAG autoantibodies, an antigen-specific treatment option for anti-MAG neuropathy. Proc Natl Acad Sci U S A. 2017;114(18):E3689-E3698 Pruppers M.H.J. et al. Improving future assessment and research in IgM anti-MAG peripheral neuropathy: A consensus collaborative effort. Neuromuscular Disorders 27 (2017) 1065–1072
WS 8.3 / #111CREATING PROPER OUTCOME MEASURES
Topic: 02. Neuropathy
Ingemar Merkies
Department Of Neurology, Maastricht University Medical Center, Maastricht, NL
Abstract not received.
WS 9.1 / #56NERVE FIBRE TRANSFER VERSUS NERVE RECONSTRUCTION
Topic: 06. Mononeuropathy
Robert Schmidhammer
Ludwig Boltzmann Institute for Experimental and Clinical Traumatology, Vienna, AT
Abstract not received.
WS 9.2 / #81NERVE REGENERATION
Topic: 06. Mononeuropathy
Ahmet Hoke
Johns Hopkins University School of Medicine, Baltimore, US
Abstract not received.
WS 9.3 / #179BRAIN PLASTICITY IN NERVE REGENERATION
Topic: 06. Mononeuropathy
Thomas Hausner
Trauma Surgery, Lorenz Böhler Trauma Hospital, Vienna, AT
Background: Injuries to the peripheral nervous system induces reorganization of the projections to the Central Nervous System (CNS). This reorganization occurs in the cortex and in different other levels of the CNS, such as the thalamus, brainstem relay nuclei, and spinal cord. In general, this reorganization is referred to as cerebral plasticity or as brain plasticity.Injuries to the peripheral nervous system induces reorganization of the projections to the Central Nervous System (CNS). This reorganization occurs in the cortex and in different other levels of the CNS, such as the thalamus, brainstem relay nuclei, and spinal cord. In general, this reorganization is referred to as cerebral plasticity or as brain plasticity. Methods: Functional recovery after nerve injury and repair relates to the plastic changes in the CNS. The reorganization of the cortex and subcortical structures occur very rapid and therefore influence the outcome of a peripheral nerve repair. To provide cortical reorganization different methods based on the phenomenon of transmodality - converging of a unimodal sensoric input to multimodal association areas of the cortex - have been established. Results: Rosen and Lundborg in 2007 published the ”Sensor Glove System” which mediates impulses to the somatosensory cortex through the hearing sense initiated early after surgery. Schmidhammer and his group also in 2007 extended the sensoric input by using both the hearing and the optical sense starting the treatment the day after surgery. The same group could demonstrate recovery of primary activation of the affected hemisphere in a longitudinal functional magnetic resonance imaging (fMRI) observation of cortical somatosensory reorganization. Conclusion: The use of brain plasticity thus is one part of the complex treatment strategies in the repair and rehabilitation after peripheral nerve injuries. The author will give a review of the literature and report clinical experience and results in this field.
WS 10.1 / #183HOW TRANSTHYRETIN BECOMES TOXIC
Topic: 02. Neuropathy
Laura Obici
Fondazione IRCCS Policlinico San Matteo, Pavia, IT
Abstract not received.
WS 10.2 / #198CHALLENGES IN DIAGNOSING AND MONITORING ATTR AMYLOIDOSIS
Topic: 02. Neuropathy
Davide Pareyson
Dept Of Clinical Neurosciences, IRCCS Foundation, C.Besta Neurological Institute, Milan, IT
Abstract: Inherited transthyretin amyloidosis (ATTR) has become a treatable disorder with effective therapies available. Early diagnosis is therefore fundamental for preventing progression in this disease, lethal if left untreated. Diagnosis is easier in familial cases in endemic regions, where Val30Met is the most frequent mutation, degree of awareness is high, and presentation is typical, with a predominantly small-fibre length-dependent sensory neuropathy with dysautonomia, only later involving motor fibres. On the other hand, diagnosis is often delayed in non-endemic region where the neuropathy occurs late in life, the spectrum of mutations is much wider, family history if often negative or misleading, signs of dysautonomia are subtle, presentation is atypical with a sensory-motor polyneuropathy involving all fibre types, and progression is faster. High degree of clinical suspicion, careful investigation of neuropathy features, of early autonomic manifestations, of carpal tunnel syndrome, and of cardiac, ocular or renal involvement allow prompt identification of the disease and early treatment. Careful monitoring of presymptomatic mutation carriers permits early detection of the first symptomatic phases of the disease. Systematic and homogenous follow up of treated patients, with common core protocols, will allow the collection of data useful to optimize treatment and select the best therapeutic options according to disease type and stage. Improvement of clinical and paraclinical outcome measures is a challenging task. The currently used types of Neuropathy Impairment Score (NIS) scores should be unified and simplified. Linear scales should be designed. Glomerular filtration rate (GFR) seems to be a reliable biomarker for disease staging and possibly for monitoring disease evolution, to be added to Brain Natriuretic Peptide (BNP) as a marker of cardiac disease. mBMI is another important parameter easy to calculate. Standardized nerve conduction studies, nerve echography, nerve MRI, skin biopsy, echocardiography, cardiac scintigraphy, cardiac MRI, autonomic function tests are all important tools for diagnosis and monitoring disease course during treatment.
WS 10.3 / #194THE NOVEL SCENARIO OF ATTR AMYLOIDOSIS TREATMENT
Topic: 02. Neuropathy
Teresa Coelho
Hospital de Santo Antonio, Porto, PT
Abstract not received.
WS 11.1 / #918VITAMIN DEFICIENCIES AND NEUROPATHY
Topic: 09. Systemic Diseases
Carrie K. Grouse
Neurology, University of California San Francisco, San Francisco, US
Background: Vitamin deficiencies may result in neurologic deficits, including neuropathy. Methods: Review of published literature on the topic of vitamin deficiencies and neuropathy. Results: Vitamin B12 (cobalamin), B1 (thiamine), B6 (pyridoxine), and vitamin E (alpha-tocopherol) are all essential vitamins that can become deficient, causing a peripheral neuropathy. Conclusion: It is important for neurologists to recognize the role of vitamin deficiencies in causing treatable sensory and motor neuropathies.
WS 11.2 / #217MINERAL DEFICIENCIES AND NEUROPATHY
Topic: 09. Systemic Diseases
Steven L. Lewis
Neurology, Lehigh Valley Health Network, ALLENTOWN, PA, US
Background: Mineral deficiencies may result in various neurologic manifestations including neuropathy. Methods: Review of published literature on the topic of mineral deficiencies and neuropathy. Results: Copper deficiency may cause a primarily sensory axonal polyneuropathy in addition to a myelopathy; optic neuropathy can also occur. Hypophosphatemia, occurring due to intravenous hyperalimentation without inorganic phosphate, has been associated with an acute sensorimotor polyneuropathy resembling Guillain–Barre syndrome. Molybdenum deficiency has been reported to be associated with a motor neuron disorder in sheep. Conclusion: Although less common than vitamin deficiencies as a cause of neuropathy, it is important for neurologists to recognize the role of some mineral deficiencies in causing treatable peripheral and optic neuropathies.
WS 11.3 / #223VITAMIN AND MINERAL TOXICITIES AND NEUROPATHY
Topic: 09. Systemic Diseases
Nathan Staff
Neurology, Mayo Clinic, Rochester, MN, US
Background: Peripheral nerves are uniquely susceptible to a variety of toxic insults, which is likely attributable to the metabolic demands on these neurons that span up to 1 meter in length. Peripheral neuropathy due to vitamin and mineral toxicities has been reported due a number of environmental, medicinal, and industrial exposures. While advances in public and occupational health have rendered many of these causes as very rare, these important and preventable etiologies may be overlooked in clinical practice if not considered in the differential diagnosis of peripheral neuropathy. Methods: Literature review was performed. Results: While most vitamins lead to neurological disease due to their deficiency, vitamin B6 leads to neurotoxicity from overuse and presents as a sensory-predominant axonal neuropathy or neuronopathy. Neurotoxicity from heavy metals arises from exposure to lead, mercury, inorganic arsenic, or thallium. Heavy metal neurotoxicity often has multisystem involvement, including abnormalities in the central nervous system (lead, mercury), integument (lead, arsenic, thallium), or gastrointestinal system (lead, arsenic, thallium). The peripheral neuropathy in heavy metal intoxication is variable and may range from motor-predominant (lead), sensory-predominant (mercury, arsenic, thallium), or polyradiculoneuropathy (arsenic, thallium). Treatment for heavy metal intoxication includes removal of exposure and chelation. Conclusion: Peripheral neuropathy due to vitamin and mineral toxicities continues to be a rare, but important cause of peripheral neuropathy. Oftentimes, these toxic neuropathies occur in the setting of multisystem disease, which may be a clue to the etiology. Furthermore, understanding the pathomechanisms of these rare neuropathies can provide important insights more broadly into peripheral nerve biology.
WS 12.1 / #165MULTIDISCIPLINARY CLINICS- WHAT IS THE EVIDENCE AND HOW CAN WE DO BETTER?
Topic: 03. Motor Neurone
Leonard Van Den Berg
Department Of Neurology, University Medical Center Utrecht, Utrecht, NL
Abstract not received.
WS 12.2 / #77MEASUREMENT OF DISEASE PROGRESSION, CAN WE DO BETTER?
Topic: 03. Motor Neurone
Orla Hardiman
Trinity College Dublin, Dublin, IE
Abstract not received.
WS 12.3 / #113REHABILITATION INTERVENTIONS THAT CHANGE OUTCOME IN ALS/MND
Topic: 03. Motor Neurone
Presenter TBD
WS 13.1 / #1030CRANIAL NERVES
Topic: 08. Cancer
Wolfgang Grisold1, Anna Grisold2, Stefan Meng3
1Ludwig Boltzmann Institute for Experimental und Clinical Traumatology, Vienna, AT;2Dep. Neurology, Allgemeines Krankenhaus Vienna, Vienna, AT;3Institute for Radiology, KFJ hospital, Vienna, AT
Abstract: In cancer patients the cranial nerves (CN) can be affected by several mechanisms as infections, therapy related, and neoplastic. They can present as singular, or multiple lesions. Infections: Herpes Zoster is the most prominent infection in immune compromised patients. Usually the trigeminal nerve is involved resulting in sensory loss and pain. In caudal CN lesions local mucosatoxic effects need to be excluded. Therapy related effects can be classified into radiotherapy (RT) induced damage, damage by surgical interventions and embolization and a spectrum of chemotherapy induced effects as dysgeusia, which is prominent for hedgehog pathway inhibitors. Motor dysfunction is rare, but has been observed in vinka alkaloids. Neoplastic involvement within the skull is often caused by neoplastic CSF involvement, but also focal tumor seeding. Metastases at the base of the skull, and also in the major cavities present with pain in combination with CN lesions. Also skin tumor may spread retrogradely into the CNS. A very important model for CN lesions is the neoplastic spread via CN, either antero- or retrogradely. This mechanism is most frequently observed in ENT tumors. In addition to clinical neurological examination imaging techniques are the most useful tool to determine the site and cause of the CN lesions. For the extracranial parts of the CN, increasingly High frequency ultrasound is useful.
WS 13.2 / #64PERIPHERAL NERVE LYMPHOMA
Topic: 08. Cancer
P. James B. Dyck
Mayo Clinic, Rochester, US
Background: Lymphoma can involve almost any organ in the body including the nervous system. Neurolymphomatosis (NL) is a rare presentation of lymphoma involving the peripheral nerves. NL can be subdivided into those patients in whom the lymphoma began in the peripheral nervous system (primary NL) and those in whom it began in another organ system but spread to peripheral nerves (secondary NL). Methods: We have studied NL in terms of clinical, electrophysiological, radiographical and pathological features in a cohort of Mayo Clinic patients with NL proven by nerve biopsy. Results: We identified 48 NL patients (28 primary and 20 secondary). The most common presentations were asymmetric, painful radiculoplexus neuropathy (37.5%), polyradiculoneuropathy (23%) and multiple or single mononeuropathy (18%). Primary and secondary NL present similarly with primary NL having longer symptoms duration (10 vs. 6 months, p<0.001), single disease episodes (96% vs. 35%, p<0.001), more disease remission (58% vs. 20%, p=0.056) and lower rates of mortality (25% vs 67%, p=0.054). 11 of the 42 patients had electrophysiology showing demyelination. Nerve biopsies diagnostic of lymphoma were distal cutaneous (sural 9/11) or proximal targeted fascicular (35/35) nerves. MRI was better identifying lymphoma than CT or PET (84% vs. 46% vs. 58%). The lymphoma types were B-cell (46/48) and rarely T-cell (2/48) with diffuse large B-cell most common. Teased fiber showed increased rates of demyelination (19/24), increased rates of axonal degeneration (18/24) and increased numbers of empty nerve strands (9/24). The location of nerve lymphoma within the nerve was most commonly in the endoneurium (43/48), followed by epineurial (29/33). Clinical and pathological findings of primary and secondary NL were similar except secondary NL had a shorter duration of neuropathic symptoms (8.5 vs 4.5 months, p=0.01), more frequent other organ involvement (p=0.045), more axonal degeneration (31% vs. 7%, p=0.005), less favourable response to treatment and a higher mortality rate. Conclusion: 1) peripheral nerve lymphoma (NL) is a multifocal painful neuropathy that causes endoneurial inflammatory demyelination; 2) primary NL presents as a single episode whereas secondary NL often presents after remission of the lymphoma. These findings suggest that the nerve may act as a ”safe-haven” for NL as most chemotherapeutic agents don’t cross the blood nerve barrier and 3) primary NL is less severe with longer duration, less axonal degeneration, more favourable response to treatment and higher survival rates.
WS 13.3 / #270CANCER-ASSOCIATED MYOSITIS
Topic: 08. Cancer
Yves Allenbach
Department Of Internal Medicine And Clinical Immunology, APHP Pitié-Salpêtrière Hospital, Paris, FR
Abstract not received.
WS 14.1 / #143MOLECULAR MECHANISMS AFFECTING NEUROMUSCULAR JUNCTION DEVELOPMENT AND MAINTENANCE
Topic: 04. Neuromuscular Junction
Markus Rüegg
University Of Basel, Basel, CH
Abstract not received.
WS 14.2 / #193SALBUTAMOL AND DOK-7 AS POTENTIAL TREATMENT OPTIONS FOR NEUROMUSCULAR DISORDERS
Topic: 04. Neuromuscular Junction
David Beeson
Nuffield Department Of Clinical Neurosciences, University of Oxford, Oxford, GB
Abstract not received.
WS 14.3 / #142SYMPATHETIC INNERVATION OF THE NMJ; A POSSIBLE EXPLANATION FOR THE THERAPEUTIC EFFECT OF SYMPATHICOMIMETICS IN NMDS?
Topic: 04. Neuromuscular Junction
Rüdiger Rudolf
Institute Of Molecular And Cell Biology, Mannheim University of Applied Sciences, Mannheim, DE
Background: Recently, sympathicomimetic drugs, such as albuterol or salbutamol, have been increasingly used to treat a wide range of congenital myasthenic syndromes. However, the rationale underlying the effectiveness of these drugs has remained elusive. Methods: The distribution of sympathetic neurons in skeletal muscle was investigated with immunofluorescence and confocal microscopy. Functional interaction between sympathetic neurons and skeletal muscle was investigated by combined electrical stimulation of sympathetic ganglia and in vivo two-photon microscopy. Effects of local chemical sympathectomy or congenital myasthenic syndrome on muscle atrophy and NMJ function and morphology as well as their rescue by systemic treatment with sympathicomimetics, were probed using in vivo confocal microscopy and measurement of compound muscle action potentials. Results: We found that sympathetic neurons form a dense network in skeletal muscle already in the newborn mouse and make close contact with neuromuscular junctions. Functionally, they seem to connect different targets including blood vessels, motor neurons, and muscle fibers. Electric stimulation of sympathetic neurons triggered immediate activation of muscle postsynaptic ß2-adrenoreceptor (ADRB2), cAMP production, and nuclear import of PGC1alpha. Systemic treatment with the sympathicomimetic clenbuterol rescued electrophysiological and morphological defects of neuromuscular junctions as well as muscle atrophy upon local chemical sympathectomy and in myasthenic mice. Conclusion: In summary, this work shows intense sympathetic innervation of skeletal muscle that plays functions beyond the control of blood flow. Indeed, it appears to be of fundamental importance for muscle trophism and regulates the functionality and maintenance of neuromuscular junctions. Sympathetic innervation of skeletal muscle could be a link to the understanding of the therapeutic effects of sympathicomimetics in congenital myasthenic syndromes.
WS 15.1 / #178PLECTIN: OVERVIEW AND LESSONS FROM MOUSE MODELS
Topic: 01. Muscle
Gerhard Wiche
Biochemistry And Cell Biology, University of Vienna - MFPL, Vienna, AT
Background: Plectin is the prototype of a family of proteins known as cytolinkers, or cytoskeletal linker proteins. Mainly associating with intermediate filaments (IFs) and acting as a cytoskeletal crosslinker, plectin strengthens cells mechanically. Plectin’s large size of >500 kDa, multi-modular surface structure, and oligomerization potential enable it to undergo a broad spectrum protein-protein interactions that dictate its spatial localization, regulate cellular dynamics, and create cytoskeleton based signaling scaffolds. Plectin binds not only to all types of IFs, the actomyosin network system, and microtubules, but also to transmembrane receptors, proteins of the subplasma membrane protein skeleton, components of the nuclear and mitochondrial envelopes, and signaling proteins such as kinases with key roles in migration, proliferation, and energy metabolism of cells. Due to alternative splicing, plectin is expressed as multiple isoforms bearing distinct N-terminal head domains that dictate the differential subcellular targeting of the isoforms. Through specific interactions with proteins at their target sites and their high-affinity binding to IFs of all types, distinct plectin isoforms provide site-specific and robust IF-docking sites. Plectin isoforms are thus central to IF network organization, compartmentalization, and connectivity. In fact, the functional repertoire of IF networks in different cell types is largely dependent on the type and expression levels of individual plectin isoforms. IF anchorage has profound effects on the cytoplasmic microenvironment, including the redirection of actomyosin-driven forces and the repression of microtubule networks. Moreover, plectin isoform-mediated linkage of peripheral junctional complexes, interior organelles, and specialized cellular machineries (e.g. the contractile apparatus of muscle cells) to the IF network creates a tension-sensitive communication system which is key to mechanotransduction. Methods: To decipher plectin’s diverse functions and role in disease, we have generated a large collection of genetically altered mouse lines, comprising full-knockouts (null-KOs), tissue/cell type-restricted conditional-KOs, isoform-specific KOs, double-KOs, and knock-ins. This unique assemblage of mouse models serves as a powerful tool for studying consequences of plectin gene defects on the organismal, cellular, and molecular levels. Results: As constituent and interaction partner of vital cellular elements, dysfunctional plectin leads to multiple disorders and diseases, some of which are multi-systemic, i.e. affecting more than one tissue and cell type. The most common disease caused by plectin deficiency is epidermolysis bullosa simplex (EBS)-MD, an autosomal-recessive skin blistering disorder with late-onset muscular dystrophy. Other subtypes of EBS, EBS-PA and EBS-Mys, manifest with pyloric atresia and congenital myasthenia, respectively, and a dominant subtype, EBS-Ogna, results in a skin-only phenotype. Mutations in isoform-specific sequences of the plectin gene can cause either muscular dystrophy without skin phenotype, or skin blistering without muscular phenotype, in accordance with the isoforms’ specific functions. Conclusion: The analysis of mouse mutants revealed plectin to play an essential role in maintaining the integrity of the neuromuscular system, including the contractile apparatus of myofibers, the neuromuscular synapse, myonuclei and mitochondrial networks, as well as motor neuron and Schwann cell dependent functions. Furthermore, the role and pathomechanisms involving individual isoforms could be dissected and advances were made towards the development of treatment and therapeutic strategies of muscle-related plectinopathies using mutant mice-derived immortalized cell cultures.
WS 15.2 / #184HUMAN PLECTIN AND CLINICAL ASPECTS
Topic: 01. Muscle
Rolf Schröder
Neuropathology, University Hospital Erlangen, Erlangen, DE
Background: Plectin, a multi-functional linker protein of exceptionally large size (> 500kDa), is abundantly expressed in a wide range of mammalian cells and tissues, most prominently in muscle, brain and stratified squamous epithelia. Mutations in the human plectin gene (PLEC) on chromosome 8q24 cause a variety of rare human disorders (referred to as ”plectinopathies”), namely autosomal recessive epidermolysis bullosa simplex with muscular dystrophy (EBS-MD), EBS-MD with myasthenic features (EBS-MD-MyS), limb girdle muscular dystrophy (LGMD) type 2Q (LGMD2Q), EBS with pyloric atresia (EBS-PA), and the autosomal dominant variant EBS-Ogna. Methods: Clinical, genetic, myopathological and biochemical analyses of patients with plectinopathies. Results: In addition to an overview on the broad spectrum of clinical presentations of plectinopathies, this lecture will focus on the essential features of plectin-related human skeletal muscle pathology. Furthermore, recent advances in our current understanding of the molecular pathogenesis of human plectinopathies will be discussed in relation to future targeted treatment concepts. Conclusion: Plectin has an essential role for the structural and functional integrity of human skeletal muscle tissue. To date, mutations in the plectin gene account for three distinct forms of human myopathies. Given the recent advances of next generation sequencing approaches, more plectin-related myopathies and cardiomyopathies are likely to be discovered.
WS 15.3 / #191PLECTIN AND MITOCHONDRIA (MICE AND MEN)
Topic: 01. Muscle
Lilli Winter
Center For Anatomy And Cell Biology, Medical University of Vienna, Vienna, AT
Background: Plectin, a 500-kDA multifunctional cytolinker protein, interlinks intermediate filaments (IFs) with each other and anchors them to sites of strategic importance for the organization and function of cells. Plectin’s versatility is in part due to complex splicing events in the N-terminal region of its gene (PLEC), giving rise to a multitude of alternatively spliced isoforms containing different first exons. In skeletal muscle, plectin is essential for myofiber integrity and function: by specifically targeting and anchoring desmin IFs to Z-disks (plectin 1d, P1d), costameres (P1f), mitochondria (P1b), and the nuclear/ER membrane system (P1), the muscle-specific set of plectin isoforms determines the cytoarchitecture of the IF network and thereby ensures the precise alignment of sarcomers. Accordingly, mutations in the human plectin gene cause epidermolysis bullosa simplex with muscular dystrophy (EBS-MD), an autosomal recessive skin blistering disorder associated with progressive muscle weakness. In addition, PLEC mutations lead to EBS-MD with myasthenic syndrome, EBS with pyloric atresia, limb-girdle muscular dystrophy type 2Q, or EBS-Ogna. With skeletal muscles harboring desmin-positive protein aggregates, degenerative changes of the myofibrillar apparatus, and mitochondrial abnormalities, most plectinopathies can be annotated among the expanding group of myofibrillar myopathies (MFM). All MFM show a progressive clinical course, lead to severe physical disability and premature death. The knowledge of the precise molecular mechanisms that translate MFM-causing gene mutations into the myopathic phenotype is still limited, but critical for the understanding of patients’ needs and the development of treatments. Methods: To assess the mitochondrial pathology of plectinopathy patients, skeletal muscle tissues derived from three EBS-MD patients were analyzed for mitochondrial abnormalities, including altered subcellular localization and expression of respiratory chain enzymes. In addition, to better understand the mechanisms behind mitochondrial defects in plectinopathies and to dissect the role of individual plectin isoforms, we comparatively analyzed morphological and functional characteristics of mitochondrial networks in skeletal muscle tissues and teased muscle fibers derived from wild-type, muscle-specific conditional plectin knockout (MCK-Cre/cKO), and plectin-isoform-specific knockout mice, lacking just one isoform (either P1b or P1d) while expressing all others. Results: In muscle sections from EBS-MD patients and plectin-deficient mice, mitochondrial content appeared reduced. Mitochondria were abnormally distributed within muscle fibers, aggregated, and no longer associated with the Z-disks. Mitochondrial clusters were observed in sarcoplasmic and subsarcolemmal regions. Additionally, decreased citrate synthase activity, decreased mitochondrial respiratory functions, and altered ADP kinetics were detected in plectin-deficient mouse muscles. By comparing mitochondrial networks in muscle sections and teased muscle fibers derived from isoform P1b or P1d-specific knock-out mice, we found plectin-isoform-dependent morphological alterations of mitochondria. In P1d-KO muscle, where Z-disks appeared misaligned, mitochondria lost their regular cross-striated staining pattern, but remained in close contact to the Z-disks. In contrast, mitochondria in P1b-KO muscle were more elongated and loosely arranged around the Z-disks; however, no Z-disk misalignment occurred in this case. Conclusion: Plectin (isoform) mutations dramatically affect the subcellular distribution and biochemical properties of mitochondria in men and mice. The depletion of distinct plectin isoforms affected mitochondrial network organization in different ways.
WS 15.4 / #192PLECTIN, CARDIOMYOPATHIES, AND THERAPEUTIC APPROACHES (MICE)
Topic: 01. Muscle
Oliver Mueller
University Of Kiel, Kiel, DE
Abstract not received.
WS 16.1 / #292THE CLINICAL PHENOTYPES OF CALPAINOPATHY
Topic: 01. Muscle
Andoni Urtizberea
Aphp - Filnemus, Hendaye, FR
Abstract: Primary calpain deficiency, or LGMD2A, is characterized by a selective, slowly progressive involvement of proximal muscles of pelvic and scapular girdles. In most LGMD2A patients, the typical overall picture corresponds to the clinical phenotype originally described by W. Erb in 1884 and revisited by M. Fardeau in the 1980s in a genetic isolate of patients living in the Reunion Island. The onset of weakness occurs in the early teens, in most cases in the lower limbs rather than in the upper limbs. Difficulties in running or in climbing stairs, tiptoe walking and scapular winging are classical symptoms/signs at presentation. Marked contractures in elbows or ankles, and muscle pseudo-hypertrophy may exist but are rarely observed in the first years. Muscle weakness and wasting are noted predominantly in the the scapular fixators, the biceps brachialis, the glutei muscles as well as in the posterior compartment of legs and thighs suggesting a high selectivity, a clue that is usually confirmed on muscle imaging. A gradual drop of the forced vital capacity (FVC) may be noted i a few patients especially after they turn wheelchair bound (on average 20-30 years after onset). Primary cardiac compromise has never been reported to date allowing these patients to live long. The pace of functional deterioration remains variable but overall seems slow as established by a natural history of disease studied on a four-year duration. A number of clinical variants of LGMD2A have been described in the literature. Pseudometabolic forms include exercice intolerance and myalgias. Unusual cases have been reported at both ends of the age spectrum: benign cases can present with very mild, late-onset non-progressive muscle weakness and moderately high CK levels whereas early onset, even before age 5 years, and rapid loss of ambulation are also observed. Eosinophilic myositis presentation is rare, often misleading and most probably not specific to LGMD2A. Although an autosomal recessive mode of inheritance remains the rule, a very few cases of dominantly transmitted calpain-related LGMD have recently been reported. Similarly cases where marked intra-familial variability are not that exceptional. Likewise, when it comes to compare the phenotypes between families harbouring the very same genotype suggesting the existence of modifying and/or epigenetic factors. With a wider, easier access to NGS studies, it is likely that the spectrum of these clinical exceptions to the rule will expand over time.
WS 16.2 / #54MUSCLE PATHOLOGY OF LIMB-GIRDLE MUSCULAR DYSTROPHIES (LGMD)
Topic: 01. Muscle
Kristl G. Claeys
Neurology, University Hospitals Leuven and KU Leuven, Leuven, BE
Abstract: Limb-girdle muscular dystrophies (LGMD) are a heterogeneous group of rare, genetic and progressive muscle disorders, characterized by weakness and atrophy of predominantly pelvic and shoulder girdle muscles. The inheritance pattern in LGMD is mostly autosomal recessive (LGMD2), and less frequently autosomal dominant (LGMD1). So far, 26 subforms of LGMD2 (LGMD2A-2Z) and 8 subtypes of LGMD1 (LGMD1A-1H), caused by respectively two or one mutation(s) in mostly distinct genes, have been identified. In most LGMD patients, creatine kinase (CK) level in serum is elevated. To date, pathomechanisms in LGMD are not well understood and curative treatments do not exist. Muscle biopsy in LGMD usually shows dystrophic changes consisting of necrotic and regenerating muscle fibers. Further histopathological abnormalities are increased variability in fiber diameter, elevated number of internalized nuclei and increased amount of endomysial connective tissue. Besides the general findings of necrosis and regeneration, additional pathological features can be identified in the muscle biopsy of some LGMD subforms, such as lobulated fibers in LGMD2A, eosinophils in LGMD2A, amyloid deposits in myofibers and the perivascular area in LGMD2B and LGMD2L, rimmed vacuoles in LGMD2G, LGMD1A, LGMD1D, LGMD1F and LGMD1G, and protein aggregates in LGMD1D. Perivascular and/or endomysial inflammation can be observed in some biopsies of LGMD2A, LGMD2B, LGMD2C, LGMD2I or LGMD1A patients. In some cases, the inflammatory infiltrates can even be associated with up-regulation of the major histocompatibility class 1 antigen (MHC-I), which might lead to erroneous treatment of these genetic muscle diseases with immunosuppressive agents. Also the presence of mitochondrial abnormalities in some LGMD subforms, such as LGMD1H, can lead to a wrong diagnosis of mitochondrial disease. Immunohistochemistry and immunoblotting with antibodies directed against distinct affected proteins are very helpful in the diagnosis of some LGMD subforms. Furthermore, ultrastructural findings can also add important information to the diagnosis in a few LGMD subtypes, such as the presence of sarcolemmal defects with replacement of the membrane by layers of small vesicles in LGMD2B and LGMD2L, or a reduction in caveolae in LGMD1C. With the novel massive parallel sequencing technologies rapidly entering clinical practice, there is more and more a shift in the order of diagnostic examinations to be expected. In the traditional diagnostic work-up of patients with muscle disease, a muscle biopsy is performed followed by Sanger sequencing of single candidate genes. Because of the high diagnostic rates of neuromuscular gene panels and whole-exome sequencing (WES) and their favorable cost-effectiveness, these novel massive parallel sequencing technologies are increasingly being used even before a muscle biopsy is performed. However, muscle histopathology will remain very important to study pathogenicity of the large number of variants that frequently evolve from these large genetic analyses.
WS 16.3 / #117THE ADVENT OF NGS IN LGMD DIAGNOSIS
Topic: 01. Muscle
Vincenzo Nigro1, Marco Savarese2, Annalaura Torella1
1Precision Medicine, Università degli Studi della Campania ”Luigi Vanvitelli”, Naples, IT;2Medical Genetics, Medicum, Folkhalsan Institute of Genetics, Uni Helsinki, Helsinki, FI
Background: Next generation sequencing (NGS) has revolutionized our approach to genetic disorders, from a single gene testing, to genomic studies. This is especially true for heterogeneous conditions with overlapping phenotypes, like limb girdle muscular dystrophies. Methods: Three different NGS strategies for DNA sequencing have been developed: a targeted re-sequencing of specific regions/genes of interest captured by specific probes or amplified by PCR; a whole exome sequencing (WES) targeting the whole set of coding regions (about 1.5% of the human genome); and a whole genome sequencing (WGS) which has no preselection of DNA fragments. Different targeted approaches have been described so far. In particular, we originally developed a strategy based on Haloplex selection MotorPlex) and capture of 89->270 genes characterized by a megabase size of the targeted regions. Using that approach about 50% of >500 undiagnosed LGMD were solved. However, the further reduction of sequencing costs provides us the opportunity to move towards WES: a unique test able to investigate, at the same time, both disease-genes and novel candidate ones. Results: Patients with undiagnosed LGMD undergo a standardized annotation using Phenotips and selected cases are recruited for trio/quartet whole exome analysis. We adopted a complete whole exome enrichment with a target of ~69Mb and a coverage of ~200x. Results are shared mainly using Phenome Central to recognize similar patients with the same genetic disease. We also optimized the bioinformatic pipeline thank to a collaboration with the Telethon Undiagnosed Program. In addition, we developed a next approach for the still negative cases. We designed a ”ultra exome” probe collection to be used in connection with the 10x Genomics. This produces a partitioning of high molecular weight DNA fragments (HMW-gDNA) into micelles, along with an adapter molecule and a barcode sequence. With this strategy, WES are fully covered and phased and even small structural variations may be detected. Conclusion: Unbiased NGS methods (like WES or WGS) may represent the first step of a comprehensive diagnostic workflow for LGMD, including traditional investigations, such as muscle biopsies and electromyography, and more specialized procedures like ultrasound muscle imaging and magnetic resonance imaging.
WS 16.4 / #35PERSPECTIVES IN CLINICAL TRIALS OF RECESSIVE AND DOMINANT LGMD
Topic: 01. Muscle
Corrado Angelini
IRCCS S.Camillo Hospital, Venice, IT
Background: The term Limb Girdle Muscular Dystrophy(LGMD) defines a progressive weakness with onset in the proximal limb girdle muscles, with age at onset of symptoms varying from early childhood(not congenital)to late adulthood. The progression of muscle weakness is usually symmetrical and variable among individuals and genetic type. These disorders present a wide spectrum of muscle involvement and wasting, spanning from very severe forms with childhood onset and rapid progression, to relatively benign forms with late onset. Methods: A consortium meeting, under the auspices of the European Neuromuscular Center, introduced a classification of LGMD based on molecular/genetic criteria. The autosomal dominant loci were designated as LGMD1, and the autosomal recessive loci were designated as LGMD2. LGMD nomenclature adopted a progressive alphabetical letter indicating the order of gene mapping identification. A new classification was proposed in an ENMC meeting held in 2017 naming autosomal dominant LGMD D and recessive forms R and numbering them. This new classification has included dystrophies with proximal or disto-proximal features, high CK presentation with histopathological evidence at biopsy of fiber degeneration/regeneration, fiber splitting, MRI imaging consistent with degenerative changes, fibro-fatty infiltration. In differential diagnosis appear the inflammatory myopathies, myofibrillar myopathies and metabolic myopathies, which can be excluded on the basis of clinical features muscle histopathology and laboratory exams. Results: LGMDs constitute a considerable fraction of all dystrophic patients and their prevalence ranges from about 8 to 70 cases per million inhabitants(1:123,000-1:14,500) depending on the geographical area and ethnic origin. The frequency of each form of LGMD is variable in different populations Calpainopathy is the most prevalent form in the majority of countries, its frequency ranges from about 10% of LGMD cases in the United States , to 80% in Basque country. Calpainopathy and Dysferlinopathy account for about 50% of total cases and primary sarcoglycanopathies are about 10-15% of cases.LGMD2I is the most common form in Northern Europe. To perform clinical trials similar pathomechanisms and homogeneous phenotypes are required. The clinical phenotypes due to mutation in the LGMD genes include instead severe childhood-onset forms, distal and proximal myopathies, pseudo-metabolic myopathies, eosinophilic myositis, and hyperCKemia. It is crucial to identify suitable selection criteria to be used in trials when treating patients after identifying defective for genes responsible for LGMD. Nowadays therapy is still an unresolved problem in LGMD mostly limited to rehabilitation, clinical follow-up of cardiological and respiratory complications.Several drugs have been tested such as corticosteroids and myostatin inhibitors with variable success. There is active pursue of new drugs and of genetic and cell therapy and a number of clinical trials are undergoing development. Conclusion: LGMDs are relatively rare and clinical trials should be done in homogeneous groups of patients after careful study of outcomes. About 8% of patients with a diagnosis of LGMD may actually have FSHD, and misdiagnosis can occur. Muscle imaging (CT-scan and MRI)may be helpful to characterize the severity at the pattern and distribution of muscle wasting and in follow up of LGMDs. Outcome measures for each clinical subtype should be carefully studied and tailored for each clinical trial.
WS 17.1 / #76PRIMARY AND SECONDARY VASCULITIC NEUROPATHY
Topic: 09. Systemic Diseases
Gerard Said
Institut Vernes, PARIS, FR
Abstract: Ischemic nerve lesions occur in primary or secondary vasculitis. The consequences of vascular inflammation and occlusion depend on the size and number of blood vessels affected. Clinical neuropathy occurs in more than 75 per cent of the patients with systemic vasculitis of the PAN group. Typically the clinical picture is that of an acute or subacute mononeuritis multiplex, with successive or simultaneous involvement of multiple nerve trunk territories over days, weeks or months. Distal symmetrical sensory or sensorimotor neuropathy also occurs. The peroneal nerve is the most commonly affected nerve. The onset of the neuropathy is typically abrupt and the deficit severe, but in many cases only partial deficit in a nerve territory is observed. Recovery from motor deficit due to ischemic neuropathy takes months, because of the axonal pattern of nerve lesions. Residual pains may be difficult to differentiate from relapses of the neuropathy. In polyarteritis nodosa (PAN) the ischemic neuropathy induced by NA can be observed as an isolated manifestation, or in the context of a multisystemic disorder. In classic PAN cutaneous vasculitis is the most common non neurological manifestation. Arthritis, renal involvement and asthma are present in 10% of patients on average. Biological markers of inflammation including CRP and ESR, increased platelet and white blood cell count with eosinophilia are often markedly elevated, but remain normal in nearly 30% of the patients seen in neurology. Churg and Strauss syndrome : In the CSS, disseminated necrotizing vasculitis occurs among asthmatic patients, with fever, eosinophilia, and a fulminant multisystem disease with pathology of NA, eosinophilic infiltration and extravascular granulomas. The vascular and nerve lesions observed in nerve biopsies from patients with this syndrome and response to treatment are actually similar to those observed in PAN, with sometimes presence of elevated ANCA. The occurrence of NA in the context of rheumatoid arthritis is associated with a poor outcome Granulomatous vasculitis (GV) is characterised by vasculitis of upper and lower respiratory tract with or without glomerulonephritis. Peripheral neuropathy has been observed in 25% of the patients with GV but peripheral neuropathy seldom is the first manifestation of the disease. Of major interest to neurologists is the occurrence of peripheral neuropathy as the only clinical manifestation of vasculitis. General signs or symptoms, usually minor, including fever and loss of weight are present in half of them. One fourth of the patients presented with a distal symmetrical neuropathy. The diagnosis of NA had seldom been considered before the results of the nerve and muscle biopsies. One third of patients with the so-called non systemic vasculitis subsequently developed systemic manifestations within an average 6 years, but the overall outcome remains better than in classic PAN. Vasculitic neuropathy is a frequent factor of disability in the elderly. The diagnosis of NA needs histological confirmation: the specific lesion can be found in the muscle, in the nerve, or both in the nerve and the muscle specimens, which must be studied on serial sections because NA is segmental. Nerve ischemia due to vasculitis neuropathy induces acute axonal degeneration. Secondary vasculitis: Symptomatic viral infection, by HIV infection, CMV; B and C virus hepatitis, and HTLV-1, can be associated with neuropathy and necrotizing vasculitis. Angiitis has been recognized at autopsy in the CNS and occasionally in patients with sarcoid neuropathy. A small proportion of diabetic patients over the age of 50, may present proximal neuropathy of the lower limbs characterised by a variable degree of pain and sensory loss associated with uni- or bilateral proximal muscle weakness and atrophy. Treatment: Prednisone is usually started at 1mg/Kg/day. Simultaneous treatment with cyclophosphamide, 2 mg/Kg/day, or Imuran may help reducing the doses of corticosteroids. Full dose of steroids is prescribed for 6-8 weeks and then tapered. Markers of disease activity must be checked periodically to monitor the treatment. Half of the patients relapse during tapering of prednisone or after treatment has been stopped. In some patients treatment with low dose prednisone must be pursued indefinitely. Spontaneous remissions of several years duration occur.
WS 17.2 / #102STATIN NEUROPATHIES
Topic: 09. Systemic Diseases
Hadi Manji
Mrc Centre For Neuromuscular Diseases, National Hospital for Neurology, London, GB
Background: In the United States, 37 milllion people are on statins; in the UK 13% of the population are on statins. Despite this huge number of patients taking this class of drug, reports of a peripheral neuropathy due to statins are relatively sparse when compared to the well-known myopathic side effects. It was the publication by Gaist et al, in Neurology in 2002, that the first red flag was raised regarding the risk of neuropathy with statins. This community based case control study found that stain users were 4 – 14 times more likely to develop a polyneuropathy.1 The study was subsequently critisised on the grounds of patient selection and the criteria used for a diagnosis of neuropathy. In 2017, Gaist and colleagues published their results from a similar but larger cohort study and concluded ”statin use was not associated with an increased risk of idiopathic neuropathy.2” Nevertheless, anecdotal reports of statin related neuropathic syndromes continue to be published in the literature which are difficult to ignore. For example, Rajabally et al reported a well studied case of ”a disorder resembling Guillain-Barre syndrome on initiation of statin therapy.”3 On the contrary, the Fremantle Diabetes Study into lipid lowering therapy and peripheral sensory neuropathy in type 2 diabetes suggested that statins may in fact protect patients against the development of diabetic neuropathy.4 Similarly, in HIV infected patients on anti-retroviral drugs, again statins were associated with a protective effect against the development of a neuropathy.5 More recent evidence suggests that statins may have a role to play in the treatment of neuropathic pain. Zhang et al generated experimental models in mice and rats that mimicked trauma associated neuropathic pain. Their results indicated that simvastatin and rosuvastatin could prevent or reverse mechanical and thermal hypersensitivity in a dose dependant manner.6 The underlying mechanism of action is hypothesized to be related to the reduction of the expression of proinflammatory cytokines and reducing the activation of spinal microglia and astrocytes. Conclusion: The risk of statin induced neuropathy is low although it is not always possible to exclude individual idiosyncratic reactions. Statins may have a role in the prevention of neuropathy in some situations such as diabetes and HIV infection. Statins may help in the treatment of neuropathic pain as a results of their anti-inflammatory properties. References: 1) Gaist D et al. Neurology 2002;58:1333-1337 Statins and risk of polyneuropathy. 2) De Koning Svendsen et al. British Journal of Clinical Pharmacology 2017(83) 2087-2095 Statins and polyneuropathy revisited: case-control study in Denmark, 1999-2013. 3) Rajabally Y et al. Muscle and Nerve 2004:30:663-666. Disorder resembling Guillain Barre Syndrome on initiation of statin therapy. 4) Davies TME et al Diabetologia 2008:51:562-566 Lipid lowering therapy and peripheral neuropathy in type 2 diabetes: the Fremantle study 5) Chen H et al. Peripheral neuropathy in ART –experienced patients: Prevalence and risk factors. J Neurovirol 2013:19(6):557-564. 6) Xiang Qun Shi et al. Pain 2011 (152): 1033-1043. Statins alleviate experimental nerve injury-induced neuropathic pain.
WS 17.3 / #154MGUS ASSOCIATED PERIPHERAL NEUROPATHY: DIAGNOSIS AND TREATMENT
Topic: 09. Systemic Diseases
Andreas Steck
Department Neurology, Basel, CH
Abstract: A monoclonal gammopathy of undetermined significance (MGUS) is a frequent diagnosis in an elderly patient with a peripheral neuropathy (PN). It is important for the clinician to be able to determine whether an association exists between the paraprotein and the PN. The clinical phenotypes of the neuropathy, as well as the type of monoclonal protein are important clues for the diagnosis and optimal management. The prevalence of PN in patients with MGUS is about 5% in IgG, 15% in IgA and up to 30–50% in IgM MGUS. We first discuss the broad concepts of diagnostic evaluation, before presenting specifics relating to each diagnosis. The evaluation for a monoclonal protein in a patient with a peripheral neuropathy should include a serum protein electrophoresis and serum immunofixation electrophoresis and quantitative immunoglobulins, and serum free light chains should be obtained. The nature of the symptoms, clinical course, and involvement of motor, sensory or autonomic systems helps in the differential diagnosis. Electrophysiology delineates specific features of the different paraprotein- associated PN. Anti-MAG and anti-ganglioside antibodies (GQ1b, GM1, GD1a, GD1b) are useful tests. We will discuss and present clinical vignettes of the major clinical syndromes such as the NP associated with anti-myelin-associated glycoprotein IgM antibodies and the chronic sensory ataxic neuropathy with monoclonal IgM anti-disialosyl antibodies. NP associated with IgG and IgA MGUS often present with a CIDP phenotype. Treatment strategy for these disorders will be presented. Finally important differential diagnoses will be mentioned. This clinically oriented presentation should help neurologists improve their knowledge and stay abreast of recent advances in the field of MGUS PN.
WS 18.1 / #69AUTOANTIBODIES IN MYASTHENIA GRAVIS
Topic: 04. Neuromuscular Junction
Nils Erik Gilhus
Department Of Clinical Medicine, University of Bergen, Bergen, NO
Abstract: Myasthenia gravis (MG) is an autoimmune disease characterized by autoantibodies. The autoantibodies are directed against acetylcholine receptors (AChR) in the postsynaptic muscle membrane, or alternatively against molecules nearby that interact with AChR. The antibodies bind in vivo leading to dysfunction and reduction in AChR and defective postsynaptic signaling in muscle. MG patients should always be classified into subgroups. This classification depends on autoantibody detection, as well as age, clinical manifestations and thymus pathology.
| MG subgroup | Antibody | Age at onset | Thymus |
| Easrly onset | AChR | < 50 years | Hyperplasia common |
| Late onset | AChR | > 50 years | Atrophy common |
| Thymoma | AChR | Any | Lymphoepithelioma |
| MuSK | MuSK | Any | Normal |
| LRP4 | LRP4 | Any | Normal |
| Seronegative | None detected | Any | Variable |
| Ocular | Variable | Any | Variable |
Antibodies against muscle-specific kinase appear in 1-10% of patients, and against LRP4 in 1-3%. MG in childhood is nearly always of the early onset or ocular type. Detection of AChR, MuSK or LRP4 antibodies has a near 100% specificity for MG. The sensitivity is around 80%, highest for thymoma and lowest for ocular MG. More sensitive techniques have been developed for antibody detection, reducing the seronegative MG subgroup with one half. However, it is not known if the most sensitive assays have the same high MG specificity, and these assays are not commercially available. There is no correlation between antibody concentration and MG severity. This reflects that pathogenicity varies between antibody isotypes and idiotypes. There is a tendency for some correlation between changes in antibody concentration and clinical development in the same patient over time, so that repeated antibody measurements can be of value. Some MG patients have additional antibodies against intracellular muscle antigens, titin and ryanodine receptor in particular. Presence of such antibodies indicates thymoma or late onset MG. Patients with the antibodies tend to have more severe disease and a greater need for long-term immunosuppressive treatment. Antibodies against agrin, cortactin, and collagen Q in muscle have been detected in some MG patients without other antibodies. The MG specificity of these antibodies is not known in detail. The disease-inducing antibodies against AChR, MuSK and LRP4 all pass placenta and into the fetus during pregnancy. In approximately 20% of the newborn of MG mothers they lead to a transient neonatal myasthenia, usually with only mild symptoms for some days or a few weeks. Arthrogryposis and spontaneous abortions induced by mother’s AChR antibodies have been described in rare cases. MG is the prototype of an antibody-mediated autoimmune disease. It usually responds well to immunosuppressive therapy, so that the great majority of MG patients have rather mild disease or are in remission.
WS 18.2 / #135CIRCULATING MICRO-RNA AS BIOMARKERS IN MYASTHENIA GRAVIS
Topic: 04. Neuromuscular Junction
Anna R. Punga
Neuroscience, Uppsala University, Uppsala, SE
Background: MicroRNAs (miRNAs) are small noncoding RNA molecules, which bind specifically to different mRNA targets and thereby regulate several important intracellular biological processes. Circulating (extracellular) miRNAs can be measured in most biofluids, and therefore they have emerged as easily accessible markers in various body fluids, including blood serum and plasma, with disease-specific profiles for example in autoimmune conditions. In myasthenia gravis (MG), there is a great need for reliable objective biomarkers to monitor the disease course and therapeutic response. Recent studies in MG patients have focused on early onset MG with generalized disease phenotype. In acetylcholine receptor antibodypositive (AChR+) MG, levels of the miRNAs miR-150-5p and miR-21-5p were elevated. Further, miR-150-5p levels were lower in patients receiving immunosuppression and those having undergone thymectomy. In MuSK+ MG, another profile of circulating miRNAs is present, including upregulation of the let-7 family of miRNAs. Methods: The aim was to determine disease specific pattern of miRNAs in other subgroups of MG, including late onset MG (LOMG) and ocular MG. Different discovery cohorts consisted of patients with generalized LOMG and ocular MG as well as age- and gender-matched healthy controls (HCs). We also included longitudinal validation sets of LOMG patients to correlate disease over time with miRNA pattern. For the ocular MG cohort, we had longitudinal data and aimed at determining whether certain miRNA levels would predict generalization of MG weakness. Total RNA was isolated from 200 μl blood serum and cDNA synthesis was performed by reverse transcription, followed by real-time PCR amplification. MicroRNA expression was analyzed using the Serum/Plasma Focus microRNA PCR Panel (Exiqon) in the discovery cohorts and in the validation cohorts we added selected primers detected from the discovery cohort to the qPCR plates. Internal technical controls included interplate calibration with UniSp3, quality of RNA isolation with UniSp2 and UniSp4, cDNA synthesis control with UniSp6 and hemolysis control ΔCq (miR-23a - miR-451a). miRNA data were converted to the logarithmic scale and paired two-tailed T-test or the Wilcoxon matched paired test was used to compare the values of miRNA over time. Spearman Rank correlation was performed to find correlation between continuous factors (age, disease duration, disease severity) and different miRNAs. Statistical significance was defined as p < 0.05. Results: A pattern of miRNAs was found to be strongly elevated in the LOMG cohort and in the ocular MG cohort another pattern of elevated miRNAs was found. A few miRNAs correlated with the clinical MG composite score (MGC) and were reduced in parallel with initiation of immunosuppression and clinical improvement at 1-year follow-up. Further, levels of some miRNAs differentiated generalized from ocular MG. Conclusion: In conclusion, we found a specific pattern of miRNA expression in the subgroup of MG patients with LOMG and in the cohort of ocular MG. The correlation between some of these miRNAs with the MGC score and the clinical course strengthen the potential role of these miRNAs as biomarkers in follow-up of MG.
WS 18.3 / #105THYMUS BIOMARKERS AND CLINICAL RELEVANCE
Topic: 04. Neuromuscular Junction
Alexander Marx
Pathology, University Medical Centre Mannheim/University of Heidelberg, Mannheim, DE
Abstract not received.
WS 19.1 / #147MUSCLE BIOPSIES
Topic: 01. Muscle
Rolf Schröder1, Monika Hofer2
1Neuropathology, University Hospital Erlangen, Erlangen, DE;2Oxford University Hospitals, Oxford, GB
Background: Although next generation sequences approaches have certainly revolutionised the diagnostics of human muscle and nerve diseases, there is still an essential role for muscle biopsies in the diagnostic work-up of patients with neuromuscular symptoms. However, appropriate indications, careful choice of muscle to biopsy and appropriate handling and work-up of the muscle tissue are of paramount importance to ensure a high diagnostic yield. Methods: Light and electron microscopic analysis of skeletal muscle tissue in conjunction with immunohistochemistry of essential disease-related muscle proteins. Results: Our talk will address the following issues: 1. Indications and contraindications 2. Choosing the right muscle for biopsy 3. Essentials for handling and further work-up of muscle tissue 4. Standard histochemical staining panel and basic interpretation of results 5. Standard immunohistochemical staining panel and basic interpretation of results 6. Indications for electron microscopy analysis and basic interpretation Conclusion: Diagnostic muscle biopsies are still an invaluable tool in the diagnostic work-up of neuromuscular patients. This presentation discusses the aspects mentioned above and is primarily aimed at neurologists looking after muscle patients. Selected cases will be used to demonstrate the principles.
WS 19.2 / #65NERVE BIOPSIES: THERE IS STILL A PLACE FOR NERVE BIOPSY IN MODERN CLINICAL PRACTICE
Topic: 01. Muscle
P. James B. Dyck
Mayo Clinic, Rochester, US
Background: Nerve biopsy is a useful tool for the diagnosis and treatment of peripheral neuropathies but the need for its continued use in the future has been questioned. With improvement in laboratory, radiographic and electrophysiological evaluations, nerve biopsy use has been on the decline. With better understanding of the pathophysiology, some neuropathies no longer need to be biopsied (inherited) whereas others can be recognized by the disease pattern (CIDP). Methods: Herein, we review our approach to doing nerve biopsies including selection of appropriate patients and nerve biopsy sites. Results: When deciding to do a nerve biopsy, there are some features that are helpful. A thorough neurological evaluation should be completed before nerve biopsy is performed. Patients with rapidly worsening, progressive neuropathies who have major clinical deficits are good biopsy candidates. Traditionally, whole distal cutaneous nerve biopsies are performed. These are taken from sensory nerves (e.g. sural, superficial peroneal, saphenous, superficial radial, etc.) and so are only useful in sensorimotor or pure sensory neuropathies. The nerve needs to be clinically affected in order for a nerve biopsy to show findings. Nerve biopsies laboratories should do a wide variety of preparations including teased fibers, paraffin and epoxy sections, immunohistochemistry and electron microscopy. Reports on the diagnostic yield of nerve biopsy vary and the findings are hard to validate because the quality of the evaluations and of the pathological specimens vary. In our experience, nerve biopsies from carefully selected patients frequently show important findings that change clinical management. For motor predominant, focal or proximal neuropathies the traditional distal cutaneous biopsy may be inadequate. With improved resolution of MRI, focal lesions along the nerve can now be identified and biopsied. We have extensive experience with targeted fascicular nerve biopsies and find that about 84% of these targeted biopsies reveal meaningful pathology from which 76% are potentially treatable. Such cases usually are focal or multifocal, proximal or motor predominant. The causes include inflammatory demyelinating, vasculitis, sarcoidosis, amyloidosis, leprosy, lymphoma, metastatic tumors, vascular malformations and others. Conclusion: There is strong evidence that nerve biopsy should still be performed in selected patients in modern neurological practice. For most typical length-dependent neuropathies whole distal cutaneous biopsy is the procedure of choice. In contrast, for focal motor predominant or proximal neuropathies targeted fascicular nerve biopsy is a superior approach. Patients should be carefully screened and have a thorough evaluation and only patients with severe neuropathies that have a high likelihood of being treatable should be biopsied to offset potential side effects. Biopsies should be performed at centers with expertise in peripheral nerve diseases with specialized neurologists, radiologists, peripheral nerve surgeons and pathologists and laboratories that offer a variety of nerve preparations to ensure maximal benefit with minimal morbidity.
WS 19.3 / #91SKIN BIOPSIES
Topic: 01. Muscle
Giuseppe Lauria
”Carlo Besta” Neurological Institute, Milan, IT
Abstract not received.
WS 20.1 / #99DIABETIC CRANIAL NEUROPATHIES
Topic: 09. Systemic Diseases
Tudor Lupescu
Department Of Neurology, ”Agrippa Ionescu” Hospital,, Bucharest, RO
Background: Diabetes mellitus is a frequent condition representing nowadays practically a pandemic with a prevalence of almost 1 in every 10 people. There is also a high prevalence of neuropathy among diabetic patients, of about 50%. The cranial nerves affected in diabetes are the oculomotor (3rd, 4th, and 6th) and the facial. Although cranial neuropathies represent 1-2% of the total diabetic neuropathies, due to the very high number of diabetic patients, most neurologists will encounter such cases. Methods: The presentation will deal with the two most frequent types of cranial neuropathies, namely those that involve the oculomotor and the facial nerves. Results: All the oculomotor nerves can be involved, but neuropathy of the 3rd cranial nerve is the most frequent. It is followed by involvement of the abducens and the trochlear nerve. The common oculomotor diabetic neuropathy is characterized by sudden onset of palpebral ptosis, diplopia, divergent strabismus, usually preceded by ocular pain. Sparing of the pupillary function is quite typical for diabetic palsy of the third cranial nerve. This feature is explained by the ischemic mechanism of this neuropathy, with involvement of the centrofascicular portion of the nerve, leaving intact the fibres concerned with pupillary function that are situated more peripherally. Abducens nerve palsy can be recognized by the convergent strabismus. Trochlear nerve palsies are rare. The differential diagnosis of these cranial neuropathies is very important, since other conditions in which the oculomotor nerves are affected can be very significant and dangerous. We must exclude an aneurysm of the posterior communicating artery, midbrain ischemia, or tumour. Therefore a thorough clinical examination and neuroimaging are very important in the management of these cases. The prognosis for recovery is good. The diabetic facial palsy is more frequent in older diabetics than in non-diabetic patients. The clinical presentation is similar to the common Bell’s palsy. There is an acute onset of unilateral weakness of facial muscles, often preceded by retroauricular pain. A study showed that in the majority of diabetic patients, there was no taste abnormality. Therefore it was assumed that the lesion in diabetic facial palsies is most often distal to the chorda tympani. Of course it is important to see that other signs of central nervous involvement are absent, otherwise we might misdiagnose important clinical conditions (stroke, multiple sclerosis). The clinical examination and neuroimaging are useful here. Sometimes, electrodiagnosis (nerve conduction studies, blink reflex) can add important functional data. As in idiopathic facial palsy, the prognosis for recovery is good. There are situations in which cranial nerve palsies accompany more serious conditions, associated or not with diabetes. There are cases with oculomotor nerve involvement in mucormycosis, and also cases with facial and cervical radiculoplexus neuropathy. Conclusion: Although rare among associated neuropathies, the high prevalence of diabetes mellitus provides a significant number of cases with cranial nerve neuropathies. Thus, knowledge regarding these conditions is important, since they present with an acute onset, pain, and may mimic other important and sometimes life-threatening diseases.
WS 20.2 / #75MUSCULOSKELETAL COMPLICATIONS OF DIABETES ARE COMMON AND UNDERDIAGNOSED
Topic: 09. Systemic Diseases
Anna Grisold
Medical University of Vienna, Vienna, AT
Background: Diabetes mellitus (DM) is a disease with an enormous socio-economic impact and a still increasing prevalence worldwide. The underlying pathophysiology of type I DM is an autoimmune destruction of pancreatic cells, whereas in type II DM insulin resistance, hyperglycemia and inadequate insulin secretion are the main mechanisms. Persistent hyperglycemia subsequently leads to microvascular complications such as nephropathy, neuropathy and retinopathy and macrovascular complications like stroke, coronary and peripheral artery diseases. Further, musculoskeletal disorders represent a broad spectrum of still underestimated comorbidities in diabetes. The exact pathomechanisms of musculoskeletal complications still remain elusive. Muscle inflammation, fibrosis, infarction, hemorrhage, necrosis, neuropathy and vasculopathy have been discussed as concomitant structural alterations. From an etiological point of view, disease duration as well as disease control, age and comorbidities play a crucial role in the development of musculoskeletal complications. Musculoskeletal complications in DM: Regarding complications of the skeleton, upper extremities are more often affected than lower extremities. Common manifestations include ”frozen shoulder” syndrome due to adhesive capsulitis, but also limited joint mobility, Dupuytren’s contractures, flexor tenosynovitis, carpal tunnel syndrome and diffuse idiopathic skeletal hyperostosis. In the lower extremities, especially in the feet, skeletal complications are usually more disabling, for example Charcot’s joint of the ankle and foot. Apart from the aforementioned skeletal involvement, also muscle tissue might be affected by DM. Sarcopenia and muscle atrophy as generalized presentations, but also painful muscle infarcts are characteristic. Additionally, infectious and inflammatory myositis may be observed. Given their generally impaired metabolic state, diabetic patients are particularly prone to infectious complications in articular structures. Also a decreased bone density has been described in the literature. Therapy: Glycemic control due to pharmacotherapy and diet are the most important therapeutic options. Besides, the anti-inflammatory and immune-mediating effect of corticosteroids may be used in the treatment of musculoskeletal disorders in DM patients. However, the indication has to be chosen carefully in consideration of potentially resulting metabolic perturbations. A regular and sensible physiotherapy program is an important cornerstone of DM management to prevent further musculoskeletal complications and strengthen the patient. Conclusion: Musculoskeletal complications are frequent, but still underestimated in patients with DM. They often present with reduced mobility and pain, which might be difficult to distinguish from peripheral nerve lesions. This lecture points out the most important and frequent musculoskeletal complications in patients with DM.
WS 20.3 / #128ATYPICAL NEUROPATHIES IN DIABETES: DIAGNOSIS AND MANAGEMENT
Topic: 09. Systemic Diseases
Amanda Peltier
Vanderbilt University, Nashville, US
Abstract not received.
WS 20.4 / #50TREATMENT INDUCED AND INFLAMMATORY NEUROPATHIES IN DIABETES
Topic: 09. Systemic Diseases
Brian Christopher Callaghan
University of Michigan School of Public Health, Ann Arbor, US
Abstract: Patients with diabetes may experience multiple types of periperhal neuropathies. The most common is distal symmetric polyneuropathy; however, other less common neuropathies such as treatment induced neuropathy (TIND), chronic inflammatory demyleinating polyneuropathy (CIDP), and diabetic lumbosacral radiculoplexus neuropathy (LSRPN) may also occur. In patients with diabetes, TIND is often unrecongnized. The neuropathy predominantly affects small nerve fibers leading to a length dependent, painful neuropathy as well as an autonomic neuropathy. This neuropathy usually occurs when the HA1C falls 3 or more points in 3 months. Patients with type 1 and type 2 diabetes can both have this complication. Recurrent events can lead to large fiber injury and worse patient outcomes. The more autonomic involvement that occurs, the less likely the disease is to be recognized. Only 5% of mild TIND and 41% of severe neuropathy is recognized by treating physicians. TIND should be considered in patients with diabetes that have abrupt changes in pain and/or autonomic symptoms. Typical diabetic polyneuropathy presents slowly; therefore acute/subacute presentations are a red flag for an atypical neuropathy such as TIND. Inflammatory neuropathies, such as CIDP may also be more common in patients with diabetes. Multiple groups have shown that the prevalence of CIDP is higher in patients with diabetes than in those without diabetes. Furthermore, another study demonstrated that the treatment responsiveness is not different between patients with CIDP with and without diabetes. However, patients with CIDP and diabetes require more EFNS criteria to have the same treatment responsiveness as those without diabetes. Fulfilling more EFNS criteria increases the likelihood of treatment response in those with diabetes. A separate study found that patient with CIDP and diabetes are less likely to be treatment than those without diabetes. Despite these studies, other investigators have found that the prevalence of CIDP is not higher in those with diabetes than those without. This group has also reported patients with painless motor neuropathy in diabetes that they believe is a LSRPN and not CIDP. LSRPN also occurs more commonly in patients with diabetes compared to those without. This peripheral neuropathy usually presents with severe pain, followed by weakness, followed by some degree of recovery. Patients typically have an asymmetric presentation with proximal weakness in one leg more than the other. The only clinical trial did not show benefit of weekly IV steroids on the primary outcomes, but did lead to reduced pain. A cervical variant of this condition also occurs, but is less common. Methods: Review of the literature Results: summarized in the background. Conclusion: Diabetes is associated with many manifestations of peripheral nerve injury. In addition, to distal symmetric polyneuropathy, TIND, CIDP, and LSRPN can also occur.
Teaching Courses
TC 1.1 / #58CLINICAL PHENOTYPES AND HOW TO MANAGE PATIENTS WITH HIGH HYPERCKEMIA, STATIN MYOPATHY
Topic: 01. Muscle
Marianne De Visser
Department Of Neurology, Academic Medical Centre, Amsterdam, NL
Abstract not received.
TC 1.2 / #145HOW TO DIAGNOSE A PATIENT WITH HYPERCKEMIA
Topic: 01. Muscle
Sabrina Sacconi
Centre de référence des Maladies Neuromusculaires, Hôpital Archet, France. CNRS UMR7277, INSERM U1091, IBV - Institute of Biology Valrose, UNS Université Nice Sophia- Antipolis, Faculté de Médecine, Nice, FR
Abstract: Rhabdomyolysis ranges to an asymptomatic illness to a life-threatening condition associated with electrolyte imbalances, acute renal failure and disseminated intravascular coagulation. It results from skeletal muscle injury followed by the release of muscle contents into the plasma. The most sensitive laboratory finding of muscle injury is an elevated plasma creatine kinase level. Creatine kinases (CK) is a dimeric enzyme catalyzing the reversible phosphorylation of creatine by adenosine triphosphate. The CK enzyme is composed of subunits derived from either muscle (M) or brain (B). Three isoenzymes have been identified, and they are present in several tissues: striated muscle (MM), heart tissue (MB), and brain (BB). As a consequence of cardiac or brain injury MB and BB isoforms may be elevated. Increase in CK can be also seen in several physiological conditions: CK levels transiently rise after exercise or heavy manual labor; they vary significantly by sex and race, possibly in relation to differences in muscle mass or total body mass, and/or inherited differences in the permeability of the sarcolemma to CK. There is also a small reduction in CK levels as people age. In pathological conditions, weakness, myalgia and tea-colored urine can be associated to increase in CK. Muscular trauma is one of the most common non-neuromuscular causes of rhabdomyolysis. Less common causes include electrolyte abnormalities, vessels occlusion, shock state, infectious causes, drugs, toxins, endocrinopathies, auto-immune diseases. A large spectrum of neuromuscular conditions can be diagnosed in presence of acute recurrent or chronic rhabdomyolysis including inflammatory and necrotizing myopathies, metabolic diseases as glycogenosis, B oxidation disorders, mitochondrial myopathy, lipidosis, pseudo metabolic presentation of muscular dystrophy or congenital myopathies. The diagnostic work-up may be complicated by the fact that, among the patients displaying increased CK, asymptomatic or pauci symptomatic carriers as well as early stages myopathies may be diagnosed. High CK can also be found in patients suffering from peripheral nervous system or motor neuron disease associated to extensive muscle wasting. The objective of this teaching course is to describe the aetiological spectrum of acute recurrent and chronic rhabdomyolysis and to provide an appraisal of the current strategies available to facilitate early diagnosis of this condition. Early diagnosis will allow early therapy when available, and prevention and/or the prompt management of acute and chronic complications. On this purpose, muscle biopsy is still the key exam in order to solve the large majority of myopathic-related causes of rhabdomyolisis, but it appears the more and more evident that new technologies are changing the way to address this issue and may, in the next future, reduce the need of this invasive approach. We will discuss the role of these new and powerful technologies, especially when unraveling genetic associated diseases, combined with more traditional approaches including an accurate clinical examination, standard biological exams, electrophysiology and muscle MRI, that are, despite all that, still necessary to guide the interpretation of the results. Since the field of new technologies is rapidly evolving, future researches are required to establish algorithms for cost-effective medical care.
TC 1.3 / #36HYPERCKEMIA IN MUSCULAR DISTROPHIES AND METABOLIC MYOPATHIES
Topic: 01. Muscle
Corrado Angelini
IRCCS S.Camillo Hospital, Venice, IT
Background: Hyperckemia is a common finding in asymptomatic and paucisymptomatic muscle patients, that can precede the onset of several myopathies. The first description was by Rowland in MDR Symposium held in Venice in 1980 and this generated an interest by clinicians and patients with either high CK or paucisymptomatic clinical presentation such as cramps or exercise intolerance. Methods: A literature review was performed using the following keywords: metabolic myopathies, Pompe disease, Mc Ardle disease, disorder of lipid metabolism, high CK, biopsy, MRI. Since then this entity has been covered by international guidelines in order to handle such patients and perform exams and in such guidelines need to include nowdays muscle CT/MRI imaging. Results: The most common muscular dystrophies that appear first as isolated hyperckemia are calpainopathy, disferlinopathy or alpha-sarcoglycanopathies. High CK in females evokes as first suspicion the carrier status for dystrophinopathy. An increase of CK in pauci/asymptomatic male can be due to juvenile onset Pompe disease, that is the first hereditary myopathy for which a registered treatment is available, it is therefore important to diagnose such cases. A similar screening was done in the LOPED study by DBS assay, that identified numerous late onset Pompe phenotypes presenting with hyperckemia or mild LGMD phenotype that subsequently were found to have pathogenetic mutations in GAA gene and underwent ERT treatment. Mc Ardle patients suffer from cramps and exercise intolerance. The timing of onset of symptoms in relation to exercise provides a clue to the nature of the underlying problem and guides further investigation. Glycolysis is the main source of energy at rest and during sustained moderate exercise. In early exercise, particularly during high-intensity exercise such as weight lifting or sprinting, and before adaptive changes, such as increased respiration and blood flow, muscle energy demands are met primarily by anaerobic glycolysis. In muscle glycogenolytic disorders myalgia develops early during exercise, in most cases the onset of symptoms starts in childhood. In these conditions, fixed weakness may develop later in life. Recurrent myoglobinuria is common in all these disorders, a history of a ‘second wind’ phenomenon is suggestive of McArdle disease. This disorder is caused by a lack of glycogen breakdown due to mutations in the muscle glycogen phosphorylase(myophosphorylase). The nonsense p.R50X stop codon mutation accounts for 43% of cases of myophosphorylase deficiency in caucasian populations and ACE I/D polymorphism influences severity of disease expression. Fatty acids are the major substrate at rest and in prolonged low-intensity exercise, such as during a marathon. In lipid storage disorders elevated CK can be observed in carnitine deficiency, in ETF dehydrogenase deficiency. Myalgia or myoglobinuria in fatty acid oxidation defects occur later in exercise than in glycogenolytic disorders. Conclusion: Serum CK varies greatly depending on prior physical activity level and is typically elevated in patients with complete enzymatic blocks (myophosphorylase and phosphofructokinase deficiency)or in muscular dystrophies. In fatty acid disorders recurrent myoglobinuria following prolonged aerobic exercise is common in a number of lipid disorders, especially if preceded by fasting. Between attacks CK is usually normal at difference with muscular dystrophy.
TC 1.4 / #87DIAGNOSTIC APPROACH AND GUIDELINES FOR OLIGOSYMPTOMATIC HYPERCKEMIA
Topic: 01. Muscle
Theodore Kyriakides
Clinic A, Cyprus Institute of Neurology and Genetics, Nicosia, CY
Background: Increased serum creatine kinase (CK) above the upper limit of normal (ULN) or hyperCKemia is a frequent finding among the normal population and prevalence rates of persistent hyperCKemia of 1.3% have been reported. The overwhelming majority of these individuals are oligo/asymptomatic and hyperCKemia is a chance finding. The dilemma often arises as to whether there is an underlying myopathy which could potentially evolve into clinically significant weakness in the future or whether such an individual could be at risk by certain drugs such as statins. Current evidence suggests that on a population basis the likelihood of discovering a significant cause for oligo/asymptomatic and hyperCKemia is extremely low and routine investigation of such individuals is not cost-effective. Potential tools for investigation include the muscle biopsy, specific DNA testing and muscle panel DNA testing made possible by Next Generation Sequencing. Methods: In the absence of RCTs to provide a diagnostic strategy, case-based observational studies could be interrogated in an attempt to provide posItive and negative predictive values of certain parameters so selected patients could be investigated.Such parameters could be ; age of the patient, symptoms, level of hyperCKemia and the EMG. Results: Data will be presented on the positive and negative predictive value on reaching a diagnosis in patients with hyperCKemia following biopsy according to patient’s age, level of hyperCKemia, symptoms and EMG result. Conclusion: Randomised control studies are not available to guide a diagnostic strategy in oligosymptomatic patients with hyperCKemia. An approach based on available evidence from observational studies has to be used to optimise investigations of such patients.
TC 2.1 / #132SYNAPTIC SIGNALLING PATHWAYS IN NEUROMUSCULAR JUNCTION DEVELOPMENT AND PATHOPHYSIOLOGY
Topic: 04. Neuromuscular Junction
William D. Phillips
Physiology, The University Of Sydney, Sydney, NSW, AU
Abstract: Myasthenia gravis provides the clearest example of impaired neuromuscular transmission, but compromised nerve-muscle communication can contribute to weakness in diverse diseases and conditions. Mouse models of Duchenne muscular dystrophy reveal relatively mild impairments of neuromuscular transmission. In contrast, loss of nerve terminals from motor endplates seems to begin early in the progression of motor neuron disease. A slower, progressive loss of neuromuscular connections occurs in old age. In elderly mice, at least, this loss of neuromuscular connections could be slowed by exercise or dietary restriction. Mouse studies also show that failure of two-way synaptic communication can contribute to failure of neuromuscular transmission, and/or loss of nerve-muscle connections. Most of what we know about the interactions between nerve and muscle has come from studying embryonic development of the neuromuscular junction (NMJ). Muscle specific kinase (MuSK), a transmembrane tyrosine kinase, is a central player in formation of the NMJ. A small patch of MuSK in the central region of the embryonic muscle fibre marks the site for the future NMJ. The intrinsic kinase activity of MuSK recruits acetylcholine receptors (AChR) and the cytoplasmic adaptor protein, rapsyn, so as to form specialized postsynaptic membrane. A cytoplasmic protein called DOK7 binds directly to MuSK dimers. Higher levels of DOK7 expression elevate the intrinsic kinase activity of MuSK. As motor axons invade embryonic muscles they secrete a proteoglycan called neural agrin. Neural agrin binds a complex of LRP4 and MuSK, which further increases MuSK activity. The agrin-LRP4-MuSK-rapsyn pathway stabilizes the AChR-rich postsynaptic membrane beneath the nerve terminal. Acetylcholine acts via postsynaptic AChR and calcium-activated proteases to drive an opposing system that tends to remove AChRs from the AChR patch. During normal development, agrin-driven AChR stabilization and acetylcholine-driven AChR disassembly balance each other. This helps to align postsynaptic AChRs beneath the growing presynaptic nerve terminal. Meanwhile postsynaptic LRP4 and synaptic-cleft laminins signal back to the overlying axon membrane to induce the assembly of presynaptic acetylcholine release machinery. The same molecular mechanisms that control development of the embryonic NMJ remain vital for sustaining effective neuromuscular connections through adult life. Loss-of-function mutations that affect neural agrin, LRP4, MuSK, DOK7, or rapsyn can each give rise to congenital myasthenias. A subset of myasthenia gravis patients display IgG4-type MuSK autoantibodies that inhibit the activation of LRP4-MuSK by neural agrin. When injected into adult mice these antibodies cause disassembly of the postsynaptic AChRs, leading to myasthenic weakness. The MuSK antibodies also disrupt the adaptive increase in presynaptic acetylcholine release. In this mouse model, where MuSK function is impaired, the acetylcholine-driven AChR disassembly pathway can be perilous. Pyridostigmine (a first line treatment for anti-AChR myasthenia gravis) exacerbated the loss of postsynaptic AChRs. Studies in mouse models neuromuscular disease suggest that enhancing MuSK activity can help prevent failure of neuromuscular control. A better understanding of how the MuSK system operates in mature muscle will be needed before the therapeutic potential of MuSK can be properly understood.
TC 2.2 / #524NEUROMUSCULAR TRANSMISSION AND ITS ASSESSMENT
Topic: 04. Neuromuscular Junction
Clarke Slater
Institute Of Neuroscience, Newcastle University, Newcastle Upon Tyne, GB
Abstract: Neuromuscular transmission provides the essential link between neural commands and the coordinated muscle contractions that turn them into useful actions. In a clinical setting, the efficacy of transmission can be assessed by recording the summed electrical activity of many muscle fibers, using either large electrodes on the muscle surface or needle electrodes inserted into the muscle. In both cases one records compound muscle action potentials (CMAPs) which reflect the activity of many muscle fibers. In normal subjects, during repetitive nerve stimulation at frequencies of up to 100 Hz, the amplitude of the CMAP remains constant, but in some patients there is a significant (>10%) ‘decrement’ of the response, indicating that some of the neuromuscular junctions (NMJ) are unable to follow faithfully the pattern of stimulation. To investigate the basis of such failure in detail, it is necessary to make intracellular recordings from individual NMJs in isolated nerve-muscle preparations. When the electrodes are placed within 1 mm or so of an NMJ, one records both evoked and spontaneous depolarizations from the resting potential of about -75 mV. The evoked endplate potentials (EPPs) are typically about 40 mV in amplitude, but to record them special precautions must be taken to block action potentials and contractions. Spontaneous miniature EPPs (mEPPs) occur at a frequency of about 1 Hz. These result from the action of multimolecular ‘quanta’ of acetylcholine (ACh), the contents of individual presynaptic vesicles (SVs), on the muscle fiber surface. A nerve impulse causes entry of Ca2+ into the nerve terminal which normally triggers the release of 20-200 quanta whose effects sum to make up the EPP. Because the effects of mEPPs do not add linearly, the number of quanta released, or ‘quantal content’ (QC), is best estimated from recordings of the synaptic currents (EPCs and mEPCs). This is done using 2 intracellular electrodes, one to record voltage and the other to pass current, in a ‘voltage-clamp’ arrangement. Usually substantially more quanta are released in response to a single nerve impulse that are required to activate the muscle fiber, giving the process of transmission a high ‘safety factor’. Muscles are normally activated by bursts of 5-10 nerve impulses at up to 100 Hz or longer trains at lower frequency. In response to repetitive stimulation, the QC at individual NMJs declines, but the high safety factor of transmission means that the EPP normally remains large enough to trigger an action potential. In patients with impaired neuromuscular transmission, the EPP may fall below threshold, resulting in a failure of muscle fiber activation. Different factors may cause a reduction of EPP amplitude. These include; reduced AChR density (e.g. MG, AChR deficiency), reduced QC resulting from impaired quantal release (e.g. LEMS, various forms of CMS), and reduced QC resulting from reduced NMJ area (e.g. DOK-7 myasthenia).
TC 2.3 / #158PRESYNAPTIC RECEPTORS AND NEUROMUSCULAR TRANSMISSION
Topic: 04. Neuromuscular Junction
Ana M. Sebastião
Faculty Of Medicine, Institute Of Pharmacology And Neurosciences And Institute Of Molecular Medicine, University of Lisbon, Lisboa, PT
Background: The neuromuscular junction has unique properties to evaluate presynaptic mechanisms and receptors: 1. It has a relatively simple and well known structure. 2. There is only one nerve terminal per each skeletal muscle fibre, each synaptic response thus corresponding to the activity of a single nerve terminal. 3. There is only one type of neurotransmitter being released, acetylcholine, though several neuromodulators are also released to control synaptic function, as it is the case of ATP and adenosine. Acetylcholine itself can also act as a neuromodulator, controlling its own release and function. 4. It is possible to record from the synaptic region, in contrast with what occurs in most central nervous system synapses. 5. Changes in the amount of neurotransmitter released per nerve impulse (quantal content) can be easily quantified. Methods: Using the neuromuscular junction as a model, it could be established that: Results: 1. ATP, which is released together with acetylcholine as well as from peri-synaptic Schwann cells (PSCs), is extracellularly converted into adenosine through a cascade of enzymes, denominated ecto-nucleotidases 2. Adenosine, by acting through G-protein coupled receptors, can either inhibit or facilitate acetylcholine release, through A1 (inhibitory) or A2A (excitatory) receptors that can co-exist in the same nerve terminal. 3. At low frequencies of stimulation the influence of adenosine is inhibitory, but at higher stimulation frequencies it can become excitatory. 4. Adenosine is removed from the synaptic cleft through equilibrative adenosine transporters. 5. ATP itself acts on PSCs through Gq-coupled ATP receptors, leading to calcium signals that induce the release of more ATP from these cells. 6. Calcium signals in PSCs can also be evoked by acetylcholine through Gq-coupled muscarinic receptors. 7. Muscarinic and nicotinic acetylcholine receptors are also present in the nerve terminal to control ACh release. During disease states this fine-tune control of neurotransmission may become dysfunctional, as it is the case of Amyotrophic Lateral Sclerosis. Using disease models, it was found that at very early stages, there is an unbalanced modulation of calcium homeostasis in the nerve terminal and PSCs, which reflects in enhanced but asynchronous ACh release. Concomitantly, there are alterations in muscarinic receptor function in PSCs as well as A1 and A2A receptor function in motor nerve terminals, which are already evident in the pre-symptomatic stage. Interestingly, some of these excitatory alterations fade away at symptomatic states of the disease, where a global inhibition of neuromuscular transmission becomes evident. Conclusion: Summing up, the simplicity of the neuromuscular junction allowed to unravel further levels of synaptic complexity, which extend to non-neuronal components and allow fine-tune modulation of synaptic activity. This fine-control fails in several pathologies, as we found in Amyotrophic Lateral Sclerosis model. Initially there is an exacerbated and unbalanced excitatory activity, which collapses at a later stage of disease progression, leading to a progressive failure of neuromuscular transmission. Work supported by LISBOA-01-0145-FEDER-007391, co-funded by FEDER through POR Lisboa 2020 (Programa Operacional Regional de Lisboa) from PORTUGAL 2020 and Fundação para a Ciência e Tecnologia (FCT) and by a Twinning action (SynaNet) from the EU H2020 programme (project number: 692340).
TC 2.4 / #95DIAGNOSIS AND THERAPY FOR NEUROMUSCULAR TRANSMISSION DISORDERS - MYASTHENIA GRAVIS AND CONGENITAL MYASTHENIC SYNDROMES
Topic: 04. Neuromuscular Junction
Hanns Lochmüller
Department Of Neuropediatrics And Muscle Disorders, Freiburg University - Medical Center, Faculty of Medicine, Freiburg, DE
Background: Neuromuscular junction disorders, also called Myasthenic syndromes (MS), are a rare heterogeneous group of acquired (Myasthenia Gravis, MG) and inherited (Congenital Myasthenic Syndromes, CMS) neuromuscular disorders associated with distinctive clinical, electrophysiological, laboratory and ultrastructural abnormalities. Methods: MG has been associated with auto-antibodies against the acetylcholine receptor (AChR) or MuSK, and more recently with other antibodies in rare cases. Therapy is based on esterase inhibitors, immune therapies and thymectomy. Recent advances in understanding and clinical research have led to treatment guidelines, which were developed by a subcommittee of the American Academy of Neurology which are under consideration for adoption by the European Reference Network for Rare Neuromuscular Disorders (EURO-NMD). Results: The genetic defects in CMS either impair neuromuscular transmission directly or result in secondary impairments, which eventually compromise the safety margin of neuromuscular transmission. More recently, we have identified two genes (DOK7, GFPT1) that cause fatigable weakness of muscles in a limb-girdle distribution, but rarely affecting facial or eye muscles. Next-generation sequencing and deep phenotyping, in combination with international data sharing, reveals new genetic causes of CMS, but also unusual, overlapping clinical phenotypes which blur the boundaries with primary myopathies and motor neuropathies (SYT2, GMPPB). Conclusion: We will cover the significant progress made in understanding the molecular pathogenesis of CMS and MG, which is important for both patients and clinicians in terms of reaching a definite diagnosis and selecting the most appropriate treatment. Moreover, new avenues of clinical and translational research will be addressed.
TC 3.1 / #40CMT NEUROPATHIES IN THE ELDERLY
Topic: 02. Neuropathy
Michaela Auer-Grumbach
Department Of Orthopaedics, Medical University of Vienna, Vienna, AT
Abstract: Axonal neuropathies are frequently observed in the elderly and may lead to progressive disability. The underlying causes often remain elusive. However, recent studies have identified pathogenic variants in genes known to be involved in Charcot-Marie-Tooth neuropathy type 2 (CMT2) disease in patients with and without family history. We present clinical and genetic data of 126 index patients with unexplained progressive axonal neuropathies and disease onset after age 35 years. Whole exome sequencing has been carried out in all probands. In more than 20% of the patients we observed one or two variants in MME, encoding the metalloprotease neprilysin. We detected heterozygous, homozygous and compound heterozygous loss-of-function and missense mutations in our probands and therefore confirm that MME mutations may segregate in an autosomal dominant and autosomal recessive fashion. Whenever possible, neprilysin levels in plasma were determined and correlated with particular variants and associated phenotypes. In further patients we identified pathogenetic or likely pathogenic variants in other genes known to cause late onset neuropathies (MPZ, LRSAM1, TTR, GARS, HARS, AARS, HMBS, HSPB8, MFN2, DHTKD1, WARS). Still more than 50% of the probands did not receive a genetic diagnosis and remain unexplained but further evaluation of WES data is ongoing.
TC 3.2 / #180ESSENTIAL CLUES TO EARLY RECOGNITION OF AMYLOID NEUROPATHIES
Topic: 02. Neuropathy
Davide Pareyson
Dept Of Clinical Neurosciences, IRCCS Foundation, C.Besta Neurological Institute, Milan, IT
Abstract: Inherited transthyretin amyloidosis (ATTR) is an autosomal dominant disorder due to mutations of the transthyretin (TTR) gene. TTR is synthetized mainly by the liver and released in plasma as a tetrameric transport protein. Mutations in TTR, of which Val30Met is the most common worldwide, cause transthyretin tetramer dissociation, monomer misfolding, and aggregation into insoluble fibrillar proteins in different tissues. Peripheral nerves and heart are the most frequently affected organs, but also eye, leptomeninges and kidneys can be involved. As there are effective treatments which are already available or will soon be marketed, and such treatments are more effective in the first disease phases, early diagnosis is fundamental for preventing disease progression in this otherwise lethal disorder. Early diagnosis is easier in familial cases in endemic regions, where Val30Met is by far the predominant mutation, degree of awareness is high, and presentation is typical, i.e., a predominantly small-fibre length-dependent sensory neuropathy with dysautonomia, only later involving motor fibres. Diagnosis is often delayed in non-endemic region where onset occurs much later in life, family history if often negative or misleading, dysautonomia is more subtle, presentation is atypical with a sensory-motor polyneuropathy involving all fibre types (or with other atypical presentations), and progression is definitely faster. ATTR-neuropathy must be considered in the differential diagnosis of any rapidly progressive neuropathy, particularly if later in life. Red flags are: association with carpal tunnel syndrome, early dysautonomia (particularly impotence in males), gastrointestinal involvement, neuropathic pain, loss of weight, fasciculations. When dealing with a patient with a possible TTR-related neuropathy, the clinician should look for evidence of clinical or subclinical involvement of other target organs: ocular involvement with vitreal opacities, glaucoma, pupillary anomalies; cardiac involvement with arrhythmia, conduction blocks, interventricular septum hypertrophy, overt cardiomyopathy; proteinuria, renal failure, recurrent urinary tract infections. Ecocardiography, Technetium-99m-diphosphonate (99mTc-DPD) scintigraphy showing abnormal cardiac tracer accumulation, cardiac MRI, tilt test, sudomotor function testing are all ancillary investigation which may give clues to amyloidosis. Increased brain natriuretic peptide (BNP) levels may reveal subclinical cardiopathy, glomerular filtration rate is an important disease biomarker. Biopsies can be helpful by demonstrating tissue amyloid accumulation in nerve, abdominal fat, salivary glands, skin, and gastro-intestinal tract, but may also fail in detecting amyloid. A careful nerve conduction study with EMG is important but results may be aspecific; nerve conduction slowing is not infrequent and may be misleading. CSF protein content may be increased, explaining the high rate of misdiagnosis with CIDP. Other incorrect diagnoses include lumbar stenosis and Charcot-Marie-Tooth disease. However, if the clinical picture is compatible with ATTR-neuropathy the clinician should ask for genetic testing, which provides a definite diagnosis.
TC 3.3 / #78SENSORY AND AUTONOMIC HEREDITARY NEUROPATHIES
Topic: 02. Neuropathy
Max J. Hilz
Neurology, University of Erlangen-Nuremberg, Erlangen, DE
Abstract: Hereditary sensory and autonomic neuropathies (HSAN), Fabry disease, transthyretin-related familial amyloid polyneuropathy (TTR-FAP) and acute intermittent porphyria are examples of hereditary autonomic dysfunction. So far, eight HSANs have been classified. HSAN I presents in adulthood with prominent ulcers/mutilations of the lower extremities, stress-fractures, osteomyelitis, osteolysis, distal anhidrosis, low-normal nerve conduction velocities, impaired cold and heat pain-perception. Already during infancy, HSAN II-patients have acral anhidrosis, tonic pupils, eating/swallowing difficulties, constipation, apneic episodes, and develop deformed distal phalanges, paronychia, finger and plantar ulcers, unrecognized fractures and mutilating acropathy. They have severely impaired sensory-perception including vibratory and thermal perception, abnormal sensory but normal motor nerve conduction. Autosomal-recessive HSAN III (Familial Dysautonomia) afflicts Ashkenazi Jewish children, is characterized by diminished deep tendon reflexes, absent overflow tears, fungiform papillae of the tongue and axon flare response following intradermal histamine injection, pronounced central and peripheral autonomic dysregulation, including severe orthostatic hypotension (OH) and autonomic crises with excessive arterial hypertension, profuse sweating, skin blotching, puffy hands and behavioral abnormalities. Children with HSAN IV (Congenital Insensitivity to Pain with Anhidrosis) have episodic high fever with anhidrosis, pain-insensitivity, unnoticed injuries, multiple fractures, neuropathic joints and limb mutilation. Children with HSAN V have a selective loss of pain-perception. Autosomal-recessive HSAN VI is due to a DST (Dystonin) gene mutation, presents with neonatal hypotonia, poor feeding, respiratory problems, joint contractions, dysmorphic features, early death by age 2 and was reported in one Ashkenazi Jewish family. HSAN VII was reported in three patients with mutations in the SCN11A (Sodium Voltage-Gated Channel Alpha Subunit 11) gene which is also involved in Episodic Pain Syndrome. Patients have pain-insensitivity, multiple injuries, fractures, muscle-weakness, gastrointestinal-dysfunction and hyperhidrosis. Autosomal-recessive HSAN VIII is due to mutations in the PRDM12 gene (encoding a protein regulating pain-perception) and presents with pain- and temperature-insensitivity, self-mutilation, reduced sweating and tear-formation, absent corneal reflex, etc. Neurological manifestations of Fabry disease include severe lancinating pain, burning paraesthesias in the extremities, often triggered by temperature changes, anhidrosis, gastrointestinal-problems, vasomotor and cardiovascular autonomic dysfunction, baroreflex alteration, etc. Early diagnosis and enzyme replacement therapy can be beneficial. Autosomal-dominant TTR-FAP causes life-threatening autonomic dysfunction, particularly in early-onset patients, and length-dependent peripheral polyneuropathy with prominent small nerve fiber dysfunction causing impaired thermal and pain-perception, neuropathic-pain, allodynia, painless injuries, Charcot joints, carpal tunnel syndrome, and cardiac conduction failure. Patients have peripheral blood flow and blood pressure dysregulation with OH, altered heart rate variability, and cardiac arrhythmia resulting in syncope and sudden death. Gastrointestinal dysfunction includes constipation and/or diarrhea, nausea, vomiting. There may be bladder and erectile dysfunction. Autosomal-dominant acute intermittent porphyria due to haem-biosynthetic enzyme hydroxymethylbilane synthase deficiency, causes life-threatening acute attacks with autonomic symptoms, particularly painful-abdominal crises, peripheral-neuropathy progressing to respiratory paralysis, and cerebral involvement including psychosis, seizures or posterior reversible encephalopathy syndrome. Splanchnic autonomic dysfunction causes abdominal pain, with/without constipation or diarrhoea. There may be nausea and vomiting, arterial hypertension or OH, excessive sweating, bladder-sphincter dysfunction and other features of autonomic dysfunction. Autopsies show demyelination/axonal degeneration of vagus nerve and loss of sympathetic ganglion cells.
TC 3.4 / #66NEUROPATHIC MANIFESTATIONS IN PORPHYRIA
Topic: 02. Neuropathy
P. James B. Dyck
Mayo Clinic, Rochester, US
Background: Porphyrias are rare disorder of heme metabolism that presents in different types and each of the different types are characterized by a defect in an enzyme required for heme synthesis. These disorders can produce disturbances of multiple organ systems including the skin, liver, and central and peripheral nervous systems. Methods: We present information on the natural history, laboratory findings, evaluation and treatment of the neurological syndromes occurring in porphyria patients. Results: The types of porphyria that typically cause neurological disease are acute intermittent porphyria, hereditary coproporphyria and variegate porphyria, which are all autosomal dominant inherited conditions. The porphyrias generally present with attacks of illness. These attacks are characterized by neuropsychiatric symptoms with mood disorder or psychosis, motor predominant peripheral neuropathy (which often involve proximal and distal nerve segments and so resemble CIDP with severe weakness). Autonomic involvement (tachycardia, constipation, hypertension) and gastrointestinal disturbance (abdominal pain) are common, and (in the case of variegate porphyria and some hereditary coproporphyria) photosensitivity and cutaneous manifestations occur. Classically, there is a reddish discoloration of urine during an attack (after prolonged observation oxidation of porphyria precursors occurs). Attacks in porphyria can be induced by hormonal factors, nutritional factors, alcohol and use of drugs which induce the heme production pathway leading to an overabundance of heme precursors. An accurate diagnosis can be made through routine testing for overproduction of porphyria during an acute attack but this is more difficult in the quiescent phase. The most important factor in making the diagnosis is a high index of suspicion and an awareness of the diversity of clinical manifestations. DNA testing is available for the diagnosis. The best management of the disorder is through the prevention of acute attacks (avoidance of cytochrome P450-inducing agents and avoidance of periods of fasting) but intravenous hematin and glucose are helpful and should be considered during an acute attack. Introduction of new agents for the treatment of the porphyrias is in development. Conclusion: The neurological manifestation of porphyria generally present with attacks of neuropsychiatric symptoms, motor predominant peripheral neuropathy, abdominal pain and autonomic symptoms. They occur in patients with acute intermittent porphyria, hereditary coproporphyria and variegate porphyria. The diagnosis is often made by measuring excess porphyrins during an attack.
TC 4.1 / #1033EPIDEMIOLOGY AND DIFFERENTIAL DIAGNOSIS
Topic: 13. Pediatric
Guenther Bernert
Department Of Paediatrics, Kaiser Franz Joseph Hospital, Vienna, AT
Abstract not received.
TC 4.2 / #1034CLINICAL ASSESSMENT AND MUSCLE IMAGING: TECHNIQUES AND PITFALLS
Topic: 13. Pediatric
Presenter TBD
TC 4.3 / #1035FLOPPY INFANT SYNDROME AND THE ROLE OF “NEXT GEN”
Topic: 13. Pediatric
James J. Dowling
Hospital for Sick Children, Toronto, ON, CA
Abstract not received.
TC 4.4 / #858WHAT YOU MAY MISS WITHOUT EMG
Topic: 13. Pediatric
Matthew Pitt
Department Of Clinical Neurophysiology, Great Ormond Street Hospital for Children NHS Foundation Trust, London, GB
Abstract not received.
TC 5.1 / #45ENTRAPMENT SYNDROMES IN THE UPPER EXTREMITY – ELECTROPHYSIOLOGY
Topic: 06. Mononeuropathy
Christian Bischoff
Neurologische Gemeinschaftspraxis, München, DE
Abstract not received.
TC 5.2 / #163ENTRAPMENT SYNDROMES IN THE UPPER EXTREMITY – SONOGRAPHY
Topic: 06. Mononeuropathy
Nens Van Alfen
Radboudumc, Nijmegen, NL
Background: Nerve ultrasound is a proven technique for assessment of peripheral neuropathies, with excellent resolution in superficial tissue layers. In experienced hands it is more sensitive than MRI in detecting peripheral nerve pathologies (93% versus 67%) and has comparable specificity (86%). Ultrasound is a valuable adjunct to electrophysiology for assessing entrapment neuropathies. It has similar sensitivity for detecting entrapments as EMG, but both techniques complement eachother by adding different types of information about the lesion, so one can describe both form and function. Methods: For nerve size measurement, the workhorse is the transverse cross-sectional area (CSA). Because peripheral nerves can have different shapes, for example, round, oval, oblong, or triangular, depending on their anatomic location, transverse CSA is more reliable and reproducible than nerve diameter measurement, and is the most robust measure to confirm nerve pathology. The CSA should be measured within the hyperechoic epineurial rim that encircles the nerve fascicles. As it does not take much time to scan along the length of a nerve, it is strongly advised to scan the whole nerve during an exam, instead of just scanning a few predefined points along its course. Results: It is important to use the right reference value for the right patient. Ideally, a disease specific reference is available for the clinical question that needs to be answered, but if not, a population-based reference such as the p95 for CSA can also be used. Reference values for different nerves and populations are available in the literature. Conclusion: This talk will focus on the use of nerve ultrasound in entrapment syndromes of the upper extremities, including carpal tunnel syndrome, interosseus anterior syndrome, ulnar entrapment at the wrist and elbow, Martin Gruber anastomosis, radial nerve compression in the upper arm and supinator region, the incisura scapulae syndrome and neurogenic thoracic outlet syndrome. Case examples will show scanning technique, and diagnostic values will be presented where available.
TC 5.3 / #134ENTRAPMENT SYNDROMES IN THE LOWER EXTREMITY – ELECTROPHYSIOLOGY
Topic: 06. Mononeuropathy
Simon Podnar
University Medical Centre Ljubljana, Ljubljana, SI
Abstract not received.
TC 5.4 / #74ENTRAPMENT SYNDROMES IN THE LOWER EXTREMITY €
Topic: 06. Mononeuropathy
Alexander Grimm
Neurology, Tuebingen University Hospital, Tuebingen, DE
Abstract: Entrapment syndromes in the lower limbs are rare compared to those of the upper limbs. However, to accurately diagnose them is even more difficult. Gold standard of diagnosis are nerve conduction studies and electromyography, e.g. in peroneal or tibial palsy. Nevertheless, its sensitivity in tarsal tunnel syndrome or meralgia paresthetica is restricted. Second, as already known from ulnar nerve palsy or carpal tunnel syndrome, anatomical variants and secondary reasons of entrapment cannot be detected by electrophysiology. In these cases, nerve ultrasound has proven its validity. With high resolution probes ranging from 10 MHz (for femoral and ischiatic nerve) to 20 MHz (for sural or superficial peroneal nerve) we can accurately visualize the nerve and its fascicles. Thereby one can not only perform cross-sectional area measurements, but also elucidate fascicular anatomy and echointensity. Further, surrounding tissue can be demonstrated, e.g. vessels, tendons, muscles, scare tissue, ganglion cysts and others. E.g. in peroneal palsy, one might often find intra- or extraneural ganglia as reasons of pathology. Consequences, which might arise out of the imaging, are outstanding. In my talk, I want to go on further details concerning common entrapments as tarsal tunnel syndrome and fibular head palsy of the peroneal nerve, but also demonstrate some interesting and astonishing cases of rare nerve pathology, in which ultrasound could visualize the underlying pathology and prepare the way to surgical or conservative procedure. Methods: Nerve ultrasound, electrophysiology. Results: Examples of secondary reasons of entrapments. Conclusion: Nerve ultrasound might be a useful technique to further analyze mononeuropathies
TC 6.1 / #41A PATTERN RECOGNITION APPROACH TO PATIENTS WITH A SUSPECTED MYOPATHY
Topic: 01. Muscle
Richard J. Barohn
Neurology, University of Kansas Medical Center, Kansas City, KS, US
Abstract: Myopathies are disorders affecting the channel, structure, or metabolism of skeletal muscle. Myopathies can be distinguished by characteristic clinical and laboratory features. The pattern recognition approach involves asking key questions and then based on the answer putting the patient in one of the ten patterns. The key questions are: 1.) Does the patient have “negative” or “positive” symptoms and signs? 2.) What is the temporal time line? What is the distribution of weakness or stiffness? 4.) Are there triggering events for episodic weakness, pain, stiffness? 5.) Is there a family history of a myopathic disorder? 6.) Are there associated systematic symptoms/signs? The ten patterns are: 1.) Proximal “limb-girdle” Weakness 2.) Distal Weakness 3.) Proximal arm/distal leg weakness (Scapuloperoneal) 4.) Distal arm/proximal leg weakness 5.) Eyeball Weakness-Ptosis/ophthalmoplegia 6.) Prominent neck and trunk extensor weakness 7) Bulbar / diaphragm weakness 8.) Episodic pain, weakness, dark urine 9.) Episodic weakness delayed or unrelated to exercise 10.) Stiffness/decreased ability to relax. Once a pattern has been identified, the next step is determining what laboratory tests are needed.
TC 6.2 / #61LABORATORY APPROACH TO MUSCLE DISORDERS
Topic: 01. Muscle
Mazen Dimachkie
The University of Kansas Medical Center, Kansas City, US
Abstract not received.
TC 6.3 / #116GENETIC APPROACH TO MUSCLE DISORDERS
Topic: 01. Muscle
Tahseen Mozaffar
Uc Irvine-mda Als And Neuromuscular Center, University of California, Irvine, Orange, US
Abstract not received.
TC 6.4 / #1031APPROACH TO TRANSLATIONAL RESEARCH AND CLINICAL TRIALS IN MUSCLE DISEASE
Topic: 01. Muscle
Michael Hanna
UCL Institute Of Neurology, London, GB
Abstract not received.
TC 7.1 / #34HOW DO WE DISCUSS THE GENETIC IMPLICATIONS WITH ALS
Topic: 03. Motor Neurone
Ryuji Kaji
Tokushima University Graduate School of Medicine, JP
Abstract not received.
TC 7.2 / #96COGNITIVE CHANGE – RECOGNITION AND MANAGEMENT
Topic: 03. Motor Neurone
Albert Ludolph
Universitätsklinikum Ulm, Ulm, DE
Abstract not received.
TC 7.3 / #53BREAKING THE NEWS OF THE DIAGNOSIS
Topic: 03. Motor Neurone
Dorothée Lulé
University of Ulm, Ulm, DE
Abstract not received.
TC 7.4 / #108THE ROLE OF MULTIDISCIPLINARY CARE IN ALS
Topic: 03. Motor Neurone
Christopher Mcdermott
University Of Sheffield, Sheffield, US
Abstract not received.
TC 8.1 / #72MRI AND US IN THE DIAGNOSIS OF NEUROPATHY
Topic: 02. Neuropathy
Stephan Goedee
Brain Centre Rudolph Magnus, Umc Utrecht, Utrecht, NL
Background: Diagnosis of chronic inflammatory neuropathies rely heavily on electrodiagnosis. In order to demonstrate sufficient proof of demyelination, elaborate studies are needed since the electrodiagnostic findings of demyelination are focal and patchy distributed. Neuro-imaging may complement this, as even extensive EDX studies may fail to identify these potentially treatable neuropathies. MRI imaging has been aknowledged as an important adjunctive diagnostic tool in the current sets of diagnostic criteria. More recently, ultrasound has gained importance as a powerful and practical bedside tool, that allows reliable identification of chronic inflammatory neuropathies. Methods: The role of MRI and ultrasound imaging in the diagnosis of neuropathy wil be discussed, including their part in complementing electrodiagnosis in the appropriate cinical context. Results: Standardization of MRI protocols, including diagnostically relevant sequences (T2/DIXON) and parameters (enlargment, T2 hyperintense signal nerve(root)), and recent developments such as DTI imaging and neurography will be discussed in detail. Ultrasound evaluation and diagnosis of chronic inflammatory neuropathies will also demonstrated in detail, including proposed scoring systems and a practical sonographic protocol en prediction rule.
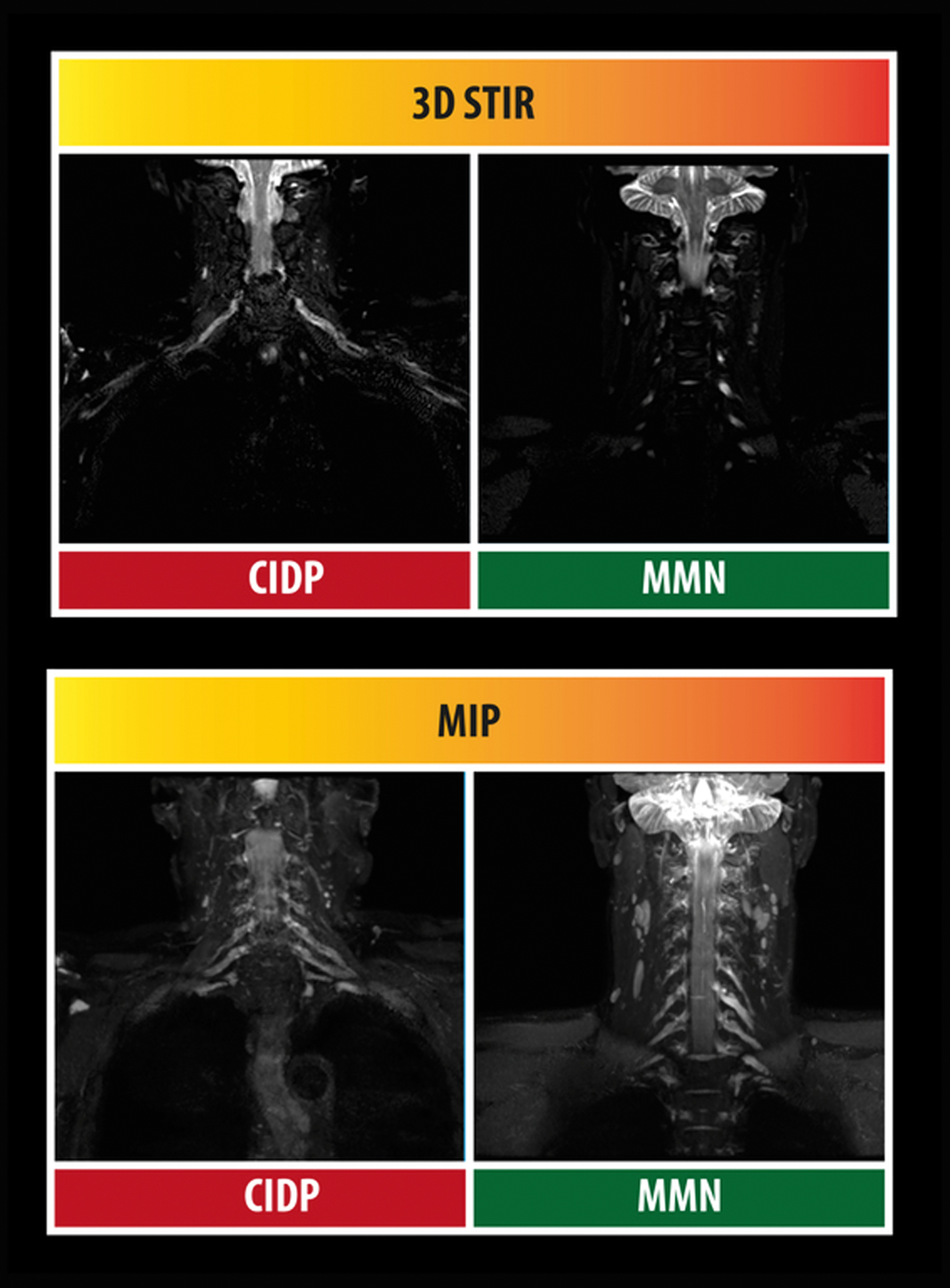
Conclusion: MRI and ultrasound imaging are important emerging diagnostic tools in neuromuscular practice, that can aid subtantially in the distinction of potentially treatable neuropathies.
TC 8.2 / #92SKIN BIOPSY: NOT JUST FOR DIAGNOSIS
Topic: 02. Neuropathy
Grazia Devigili
Fondazione Irccs Istituto Neurologico Carlo Besta, Milan, IT
Abstract not received.
TC 8.3 / #112OUTCOME MEASURES IN NEUROPATHY
Topic: 02. Neuropathy
Ingemar Merkies
Department Of Neurology, Maastricht University Medical Center, Maastricht, NL
Abstract not received.
TC 8.4 / #166ROLE OF ELECTROPHYSIOLOGY IN THE CHARACTERIZATION AND DIAGNOSIS OF NEUROPATHIES: NEW INSIGHTS
Topic: 02. Neuropathy
Peter Van Den Bergh
Neuromuscular Reference Centre, University Hospital Saint-Luc, Brussels, BE
Abstract: Electrophysiology plays a crucial role in the characterization and diagnosis of peripheral neuropathies. It provides insight in the type and mechanism of peripheral neuropathy by giving information on the spatial pattern (generalized, multifocal, focal), the fibre type involved (motor, sensory), pathology (axonal, demyelinating), and the severity and time course (acute, ongoing, chronic). Electrophysiological studies are key in the early detection and characterization of inflammatory demyelinating neuropathies and in differentiating these from primary axonal neuropathies. Inflammatory demyelinating neuropathies constitute a significant proportion of the acquired peripheral neuropathies. They include Guillain-Barré syndrome (GBS), chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), multifocal motor neuropathy (MMN), multifocal demyelinating neuropathy with persistent conduction block (Lewis-Sumner syndrome), and paraproteinemic neuropathies. A proper diagnosis as early as possible is very important because timely immune treatment can largely reduce morbidity and disability. The diagnosis is based on a constellation of clinical and laboratory features, including electrophysiological studies, spinal fluid examination, and in selected cases serological studies and peripheral nerve biopsy. In CIDP, electrophysiological criteria for demyelination are designed to exclude abnormalities that can be explained by axonal degeneration (Eur J Neurol 2010). Therefore, lesser degrees of demyelination cannot be defined with certainty. Optimised electrophysiological criteria are capable, however, to support the diagnosis with different levels of probability (possible, probable, definite) in the very large majority of cases. In GBS, much effort has gone into developing criteria which can distinguish axonal and demyelinating subtypes. The discovery of reversible conduction failure (RCF) has led to the concept of nodopathy/paranodopathy, where conduction slowing and conduction block are due to the immune attack mainly at the nodal axolemma level. There is no actual demyelinisation as defined pathologically and if the immune attack continues, conduction failure may not reverse and axonal degeneration will ensue. Recent electrophysiological studies support this pathophysiological mechanism and show that the dichotomous distinction between axonal and demyelinating in GBS is not tenable (Muscle nerve, 2018).
TC 9.1 / #1021REQUIREMENTS AND PITFALLS FOR SMALL MOLECULES
Topic: 01. Muscle
Mike Kelly
Rush University Medical Center, Chicago, US
Abstract not received.
TC 9.2 / #1022REQUIREMENTS AND PITFALLS FOR GENE THERAPY
Topic: 01. Muscle
Isabelle Richard
INTEGRARE, Généthon, Inserm, Univ Evry, Université Paris-Saclay, Evry, FR
Abstract not received.
TC 9.3 / #1023REQUIREMENTS AND PITFALLS FOR STEM CELLS
Topic: 01. Muscle
Miranda Grounds
University of Western Australia, Perth, AU
Abstract not received.
TC 9.4 / #1059PRECLINICAL TRIAL DESIGN
Topic: 01. Muscle
Annamaria De Luca
Dep Pharmacy & Drug Sciences, University of Bari, Bari, IT
Abstract not received.
TC 9.5 / #1024STATISTICAL POWER EVALUATION
Topic: 01. Muscle
Heather Gordish-Dressman
George Washington University School of Medicine and Health Sciences, Washington, DC, US
Abstract not received.
TC 9.6 / #732DATA INTERPRETATION, TRANSLATIONAL MEANING
Topic: 01. Muscle
Annamaria De Luca
Dep Pharmacy & Drug Sciences, University of Bari, Bari, IT
Abstract not received.
TC 9.7 / #1036REGULATORY REQUIREMENTS
Topic: 01. Muscle
Didier Caizergues
Genethon, Paris, FR
TC 9.8 / #1020CLINICAL TRIAL READINESS
Topic: 01. Muscle
Kathryn R. Wagner
The Johns Hopkins School of Medicine, Baltimore, MD, US
Abstract not received.
TC 9.9 / #1025CARE ASPECTS IN CLINICAL TRIAL PLANNING
Topic: 01. Muscle
Anna Mayhew
Newcastle University, Newcastle Upon Tyne, GB
Abstract not received.
TC 9.10 / #1026PATIENT PERSPECTIVE
Topic: 01. Muscle
Elizabeth Vroom
Duchenne Parent Project Netherlands, Veenendaal, NL
Abstract not received.
TC 9.11 / #1027BUSINESS MODEL
Topic: 01. Muscle
Cristina Csimma
Csimma Llc, Lincoln, US
Abstract not received.
Overarching Courses
OA 1.1 / #129STEM CELLS AS TOOLS FOR RESEARCH AND THERAPY IN NEUROMUSCULAR DISORDERS
Topic: Overarching Topic
Rita Perlingeiro
University of Minnesota, Minneapolis, US
Abstract not received.
OA 1.2 / #176STEM CELLS IN MUSCULAR DYSTROPHIES
Topic: Overarching Topic
Mayana Zatz
University Of São Paulo, São Paulo, BR
Abstract not received.
OA 1.3 / #83STEM CELLS IN ALS
Topic: Overarching Topic
Dimitrios Karussis
Hadassah Hmo, Jerusalem, IL
Abstract not received.
OA 2.1 / #60NEUROMUSCULAR DISORDERS WITH DYSPHAGIA: AN OUTLINE
Topic: Overarching Topic
Marianne De Visser
Department Of Neurology, Academic Medical Centre, Amsterdam, NL
Abstract not received.
OA 2.2 / #39HOW TO ASSESS AND MANAGE DYSPHAGIA
Topic: Overarching Topic
Zohar Argov
Hebrew University of Jerusalem EKMD, Jerusalem, IL
Background: The topic of dysphagia management and swallowing assessment receives low attention in the neuromuscular field. The main risks of dysphagia are reduced food intake and recurrent aspirations. Treatment trials in at least two muscle disorders (OPMD and sIBM) looked at dysphagia as an outcome, but there are no agreed upon clinical and investigational guidelines. Aims To review tests that can serve as treatment assessments for either clinical follow up or therapy trials and to present the main dysphagia management modalities. Observations 1. There are several patient reported outcome measures that were validated for dysphagia (e.g. EAT10 SWAL-QoL). 2. Drinking tests include the ‘classical’ 80 cc cold water test (useful and easy to use bedside test that has served as an outcome in OPMD trials). Its main shortcomings are test-test variability and lack of strong correlation with aspiration risk. Using different fluid consistencies may resolve these issues. 3. Videofluoscopy (VFS) of barium swallow using an 8 point penetration- aspiration scale has been practiced in neuromuscular and other neurological conditions. It was validated in some disorders, its main deficiency is the semi quantitative nature of the test and the inconsistency between swallow attempts. VFS can monitor and quantify the several stages of the swallowing process. It requires high technical skills and dedicated equipment. Radionuclide swallow test has been developed and is less technically complicated yet limited data are available. 4. Dysphagia management includes: change of diet and eating habits, mechanical dilatation (which usually needs to be repeated), Botox injection (minimal observations), cricopharyngeal myotomy (needs a very experienced surgeon using the right technique for prolonged effects) and feeding tubes. Conclusion: Neuromuscular experts should become more familiar with dysphagia assessment and management. New therapeutic trials, now being planned for several conditions, mandate the development of clinical and investigational guidelines.
OA 2.3 / #122ETHICAL ISSUES IN DYSPHAGIA TREATMENTS
Topic: 03. Motor Neurone
David J. Oliver
Tizard Centre, University of Kent, Canterbury, GB
Background: Dysphagia is a common symptom for people with neuromuscular disease – up to 90% in ALS. It is a distressing symptom for patients who face frustration at mealtimes, reduced nutrition, anxiety and fear of starvation, increased risks of aspiration and drooling. Moreover it is often a family issues as family carers experience the same emotional reaction and fears. Methods: A review of the literature has been undertaken. Results: There are several aspects of managing the symptoms – speech and language assessment allowing nutrition and hydration to be continued by careful feeding of foods with the most appropriate consistency; careful feeding by hand at risk, acknowledging the risk of aspiration but allowing the patient to retain the taste and activity of eating; consideration of enteral feeding, by naso-gastric tube or gastrostomy; intravenous or subcutaneous hydration to allow hydration. However there are many ethical issues raised in the assessment and discussion and management of these patients. In particular: - Whether to consider and use at risk feeding by hand – this may be appreciated by the patient and family but there is a risk of aspiration and possible deterioration and death - Whether to consider and discuss enteral feeding. Nasogastric tubes have a small risk of causing harm, and there is an appreciable morbidity and mortality of the placement of a gastrostomy. This may need to be considered earlier in the patient’s progression, as deterioration in the respiratory function may be a factor with greater importance for the safety of the procedure. - Whether to with-hold treatment – either the procedure such as gastrostomy, or the use of feeds and hydration – or withdraw treatment as the patient deteriorates and comes towards the end of life. - In some countries, where assisted dying is permitted, dysphagia may be one of the symptoms that can lead to patients requesting a hastened death, from physician assisted suicide or euthanasia. Conclusion: These issues are complex and require careful assessment and communication within teams, and closely involving patient and family.
OA 3.1 / #37IMPACT AND EVOLUTION OF COGNITIVE IMPAIRMENT IN MUSCULAR DYSTROPHIES
Topic: 01. Muscle
Corrado Angelini
Irccs S.Camillo Hospital, Venice, IT
Background: Cognitive involvement is an important feature in several types of muscular dystrophies such as myotonic dystrophy (DM1) or limb girdle dystrophies (LGMD) and needs to be addressed by physicians. DM1 is a multisystemic disease characterized by muscle, heart and CNS dysfunction. There are at least five sub-types of DM1 classified as congenital (onset at birth),childhood (1-10 years),juvenile (10-20 years), adult (20-40 years),late onset(>40 years). Methods: A literature review was performed using the following keywords: DM1, LGMD, cognitive impairment, MRI and white matter abnormalities. Results: Congenital DM1 presents at birth with polyhydramnios, reduced fetal movements; after birth the child has severe weakness, muscle hypotonia, respiratory involvement. There is then a gradual improvement although these children present cognitive deficit and learning abnormalities. Apathy seems to be associated with impaired cognitive status, fatigue, motor disability. Fatigue is a prominent sign and a major complaint in DM1. Besides cognitive and behavioural features reduced awareness of disease is defined as “anosognosia”, i.e.lack of awareness of disease burden can be studied by correlating patient’s perception of disease status to caregiver observation. A significant number of brain imaging studies were done in DM1. In young/adult patients a significant structural alteration is present in white matter with relevant abnormalities in frontal, temporo-parietal polar and occipital regions. Atrophy of cerebral grey matter was also observed suggesting a global network and neuronal impairment. In congenital DM1 hydrocephalus might be present on MRI. The juvenile form of DM1 with age of onset after 10 years has under-recognized school learning problems beside the classical signs of grip myotonia distal muscle weakness and atrophy,. In adult onset DM1 nocturnal episodes of natural apnoea and day-time sleepness are common. Loss of serotonin-containing neurons might be in relation to hypersomnia. In late onset forms there are behavioural abnormalities that range from inappropriate behaviour to depression. In DM1 the evolution of cognitive and white matter abnormalities are a matter of research seem to progress with time and studies report after 5-9 years a progressive dysfunction for verbal memory, attention tasks and visual-spatial construction. White matter abnormality seems to progress although with variable extent on tested patients and families. Few longitudinal studies have attempted to correlate MRI imaging with neuropsychological changes, further research on longitudinal DM1 cohorts are needed. In LGMD due to a dystroglycan involvement (LGMD 2I)important cognitive deficits and visual-spatial construction difficulties are present. In an observational study we found several changes in nine LGMD 2I patients who underwent brain MRI scanning: four showed non-specific white matter abnormalities; two showed moderate ventriculomegaly; three showed mild enlargement of subarachnoid spaces; cerebellar atrophy was present in one patient. Conclusion: These observations point-out to the need for of developing clinical protocols for LGMD and DM1 patients that address the CNS dysfunction and quality of life. Physician should remain vigilant about personal hygiene, child neglect, financial needs in muscular dystrophy patients. The use of drugs such as modafinil might help to keep DM1 patients in a better vigilance status. Behaviour cognitive abnormalities might be approached both with drugs or individual cognitive strategies.
OA 3.2 / #57BRAIN IMAGING TECHNIQUES FOR THE NEUROMUSCULAR PATIENTS
Topic: Overarching Topic
Nicola De Stefano
University Of Siena, Siena, IT
Abstract not received.
OA 3.3 / #62THE GAP BETWEEN COGNITIVE ASSESSMENT AND BRAIN IMAGING: LESSONS FROM DMD
Topic: 01. Muscle
Nathalie Doorenweerd
C.j. Gorter Center For High Field Mri, Leiden University Medical Center, Leiden, NL
Background: Cognitive assessments have demonstrated cognitive impairment, learning and behaviour difficulties and/or increased comorbidity of neuropsychiatric disorders in several different neuromuscular dystrophies. Neuroimaging has been used to elucidate the development of the healthy brain throughout a person’s lifespan and its relation to cognitive function. However, when applied to Duchenne muscular dystrophy patients, who have a range of cognitive abilities and also a range of quantified structural cerebral alterations, the relationship between the two is anything but straightforward. Methods: On a group level, the power of imaging to detect patterns in altered structure or metabolism that point towards the involvement of specific mechanistic pathways is remarkable. When the function of a protein or gene within the brain is largely unknown, this is an important relationship. For an individual assessment, however, it is important to note the variability in absolute values of quantitative MRI measures, and the partial overlap with values associated with healthy controls. Such variability is not restricted to neuromuscular dystrophies, but rather a common theme in neuroscience. Results: For example, cerebral atrophy has been reported in cognitively-normal Alzheimer’s patients, and a patient with severe autism can have a normal-appearing anatomical MRI. Conclusion: To discuss the gap between cognitive assessment and brain imaging, we will provide examples from our own research into the brain involvement in Duchenne muscular dystrophy. We will also review the recent literature and show examples from Myotonic Dystrophy and specific Congenital muscular dystrophies that highlight the wide spectrum of both cognitive ability and MRI abnormalities.
Virtual Sessions
ID:840NEUROPATHIES IN AFRICA
Topic: 02. Neuropathy
Riadh Gouider
Neurology, Razi Hospital, Tunis, TN
Background: Systematic studies about peripheral neuropathies (PN) in Africa are lacking. PN are common and their incidence was estimated at 2.1 to 31 per thousand in this continent. Methods: We reviewed epidemiology and etiologies of PN in Africa. We searched Medline, Scopus and science direct using the following search terms “Neuropathies”, “Africa”, “epidemiology” and “etiology”. Results: Diabetic neuropathy, one of the leading causes of PN worldwide, is becoming an increasing burden in African countries because of the poor quality of diabetes control and care. Conversely, etiologies of neuromuscular disorders vary considerably according to the African region considered. In Sub-Saharan Africa, infectious diseases are very prevalent causes. Neuropathies related to HIV, are frequently observed in South of Africa. They may be due to the direct effects of HIV, exposure to antiretroviral medications, advanced immune suppression and/or comorbid tuberculosis infection and exposure to antituberculosis medications. On another hand, leprosy is one of the main causes of PN in endemic countries such as in east and tropical Africa. Neuropathy is often clinically silent in its evolution making early diagnosis exceptionally challenging. Elsewhere, both deficiency in micronutrients related to malnutrition and toxin ingestion are mostly represented in western Africa. The occurrence of tropical ataxic neuropathy (TAN), due to the ingestion of poorly processed cassava, continues at a lesser prevalence than reported thirty years ago.Finally, hereditary neuropathies, especially Autosomal recessive forms of Charcot-Marie-Tooth disease (AR CMT) are frequent in North Africa because of high consanguinity in this region. They are characterized by an early onset, and quiet severe phenotype.Genetic analysis of large African families allowed discovering many new genes mutations such as LMNA/C gene mutations with variable phenotypes (CMT or Muscular dystrophy) and MTMR13 gene mutations with some clinical and histological characteristics as glaucoma with focally folded myelin sheaths aspect. Conclusion: Given this particular distribution of PN etiologies among regions in Africa, it is difficult to draw a diagnostic strategy without considering the patient origin. Lack of neurologists in some countries and of tools of diagnosis such an EMG in several countries makes the situation even more challenging.Although the handicap generated by such diseases, many patients still not accessing the level of health and social care services that could improve their health related quality of life.
ID:857VIRTUAL SESSION: HIV-ASSOCIATED NEUROMUSCULAR MANIFESTATIONS: EXPERIENCE FROM CAPE TOWN
Topic: 02. Neuropathy
Jeannine M. Heckmann, Kathleen J. Bateman, Melody Asukile, Sarvani Chetty, Herman Ekea, Saara Neshuku
Medicine (neurology), University of Cape Town, Cape Town, ZA
Background: HIV infection can affect multiple levels of the nervous system at the same time and at all stages of infection. This also applies to the neuromuscular system. South Africa remains a high prevalence region with >5 million people on government sponsored antiretroviral treatment (ART) programs. Methods: The aim of this virtual session is to present interesting cases which we have encountered in our resource-constrained, high HIV prevalence setting. In addition, we invite discussion of more commonly encountered management issues. Results: The commonest manifestation, distal sensory polyneuropathy shows increasing incidence with advancing age, despite effective antiretroviral therapy, and has been shown to associate with increasing functional disability when compared to those with one or less neuropathic signs. Acute inflammatory demyelinating polyradiculoneuropathy (AIDP) occur in patients with CD4 counts between 45 and 652 cells/mm3 and have a similar response to therapy compared to those with HIV-unaffected AIDP. Mimics of AIDP in our setting have included tuberculous lumbosacral radiculopathy, which may manifest as an immune reconstitution syndrome, diffuse infiltrative lymphocytic syndrome (DILS), and, rarely, syphilitic meningitis with radiculopathy. Since the earlier roll-out of ART, lower motor neuron syndromes are less frequently encountered including the segmental inflammatory motor radiculopathy, lymphomatous or cytomegalovirus radiculopathies. HIV-associated chronic inflammatory demyelinating polyradiculoneuropathy responds well overall to treatment with corticosteroids and antiviral agents, and may be monophasic, but second-line treatment may be required in the minority. Subacute HIV-associated “motor neuron disease” remains very rare. Myasthenia gravis is seen in patients infected with HIV, presenting either before or after HIV infection, but these cases are treated similarly to those without HIV. Several cases of HIV-associated inflammatory myopathy have been encountered and mostly respond to immune-therapy and ART. Although uncommon, we will also discuss a sporadic steroid-responsive, but steroid-dependent nemaline myositis. Conclusion: We describe a range of neuromuscular manifestations associated with HIV-infection. As has been observed in first world countries, the earlier initiation of ART has resulted in an overall decline in neuromuscular complications associated with more advanced HIV-infection.
ENMC
ID:321WHY OPT FOR AN ENMC WORKSHOP?
Topic: European Neuromuscular Centre (ENMC)
George W. Padberg1, Baziel Van Engelen2
1Dep. Of Neurology, Radboud University Medical Center, Nijmegen, NL
Abstract not received.
ID:322THE IMPACT OF 25 YEARS ENMC WORKSHOPS
Topic: European Neuromuscular Centre (ENMC)
Raffaella Willmann
Enmc Executive Committee, Swiss Foundation for Research on Muscle Diseases, Zürich, CH
Background: The European NeuroMuscular Centre (ENMC) was constituted in 1992 to encourage and facilitate collaborative research in Europe on neuromuscular disorders (NMD) to contribute to the eradication of these diseases. During its 25 years of activity, the ENMC established a network of more than 2500 clinicians and scientists from over 65 different countries and hosted/organized 236 workshops. Discussion topics, agreements and deliverables of the workshops have been regularly summarised in scientific reports, published on Neuromuscular Disorders and actively cited in the scientific community. During its first 10 years of activity (with about 100 workshop sponsored), the ENMC contributed to the achievement of several goals, such as the creation of several disease-specific consortia, publication of books on diagnostic criteria, definition of clinical and molecular features of NMD. These activities reflected the most urgent needs of the NMD community, but then it was also felt that future policies had to include other initiatives to improve neuromuscular cure and care (Rüdel et al., Neuromusc. Dis., 2000, 10:75-82; Emery et al., Clinical Medicine, 2001, 1:200-202). Particularly, the founders encouraged the ENMC to engage in additional activities in order to foster the development of protocols for therapeutic trials and management studies, to promote training and to establish new cooperative networks for clinical trials. Therefore, while continuing its support to the NMD community through the financing of focused workshops, the ENMC also invested in the promotion of new initiatives, such as the “Clinical Trial Network”, aimed at providing the evidence and guidance for developing treatments and for management of NMD. This activity contributed to set the ground for the TREAT-NMD network, funded from 2007 to 2011 within the EU FP6 Health program, to which ENMC was part, now continuing as Treat-NMD Alliance (www.treat-nmd.eu). In the most recent years, the ENMC refreshed the structure of its sponsored workshops, paying particular attention to the value of involving people affected by neuromuscular conditions in every meeting. Giving voice to patients often resulted in important contributions during the discussion and is now an essential component of each workshop’s program. In addition, the ENMC started to actively involve young researchers and clinicians in the workshops, thus contributing to the development of the next generation of professionals. Methods: To evaluate the long-term vision of improving quality of life of people affected by NMD, the ENMC conducted a survey and a detailed analysis of workshop topics and citation index in the last 25 years. Moreover, it retrospectively monitored the implementation in the NMD community of agreements, decisions and other deliverables derived from a recent five-years-cohort of ENMC workshops. Results: The evaluation identified and quantified pitfalls and challenges in fulfilling deliverables and goals, highlighting possible reasons for success or for delays or failure in translating the workshop outcomes into actions to improve patients’ quality of life. Conclusion: These data will be presented in occasion of the ENMC 25th anniversary symposium at the ICNMD meeting and will be used in future to support effective and constructive workshops for implementable research progresses.
ID:323CHARCOT-MARIE-TOOTH DISEASE AT ENMC WORKSHOPS
Topic: European Neuromuscular Centre (ENMC)
Mary Reilly
Institue Of Neurology, London, GB
Abstract: Charcot Marie Tooth disease (CMT) is the commonest inherited neuromuscular disorder affecting 1 in 2,500 people. It characteristically starts in the first two decades of life with distal wasting, weakness, foot deformity and sensory loss and progresses at different rates depending on the subtype. There are over 100 causative genes but in northern Europe, UK and the US about 50% of patients have the common CMT1A genotype due to a 1.4 megabase duplication of chromosome 17 containing the peripheral myelin protein 22 gene (PMP22). This is also the commonest genotype worldwide. The chromosome 17 duplication was the first causative gene to be identified for CMT in 1991 and the 25 years since then has seen an explosion in gene identification followed by natural history studies, outcome measure development, guideline development and clinical trials. Since it was founded in 1992, the ENMC has played a crucial role in all of the above developments in CMT and has enabled collaborative research and progress which would have been much more difficult without the CMT ENMC workshops. The ENMC has not only brought European researchers and patients together but it has also facilitated the inclusion of people from further afield including the US and Australia when beneficial. The ENMC was ahead of its time by truly giving patients a voice in research and developing therapies for their disease. The initial ENMC meetings facilitated gene discovery by enabling carefully phenotyped family details to be gathered, discussed and combined for further genetic studies. Many new types of CMT were identified in this way. In the early 2000s pre clinical studies suggested that ascorbic acid might be a beneficial therapy for CMT1A. A very important ENMC meeting on CMT1A in 2005 allowed us to look at the evidence and critically to agree a protocol for trials. This meant all three of the 2 year international trials (French, UK/Italian and US) used the same primary outcome measure (CMTNS) which allowed all the results to eventually be considered together in a Cochrane review. Although all the trials were negative, I suspect this may have been the first time all trials of a new therapy used an identical primary outcome measure. Following this a further ENMC meeting in 2009 to look critically at our outcome measures resulted in both agreeing modifications to the adult measure (CMTNS) and laid the grounds for developing the paediatric outcome measure (CMTPeds), which has since been published and validated. Most recently in response to the lack of evidence for the varying types of foot surgery for CMT, an ENMC meeting in 2016 resulted in the publication of preliminary guidelines and the identification of areas needing further research. CMT clinicians, researchers and most of all patients owe a lot to the ENMC.
ID:324ENMC TRANSLATIONAL RESEARCH WORKSHOP
Topic: European Neuromuscular Centre (ENMC)
Kanneboyina Nagaraju
School Of Pharmacy And Pharmaceutical Sciences, SUNY-Binghamton University, Binghamton, NY, US
Background: There are very few specific therapies for neuromuscular diseases (NMD) despite significant basic sciences advances for the last 2-3 decades in this field. There are several reasons why basic science research failed to translate into new therapies for these disorders. One of the reasons is the lack of rigor and reproducibility of preclinical research in animal models of these diseases. One way to address this issue is to bring various stakeholders such as basic scientists, pre-clinical scientists, journal editors, clinicians, clinical trialists, industry, funding agencies, parent groups, and regulatory agencies to formulate guidelines that enhance reproducibility of preclinical animal research in NMDs. Methods: The ENMC workshop assembled 27 stakeholder representatives from seven different countries (USA, Italy, Switzerland, Netherlands, Belgium, United Kingdom, Australia) in Heemskerk in the Netherlands, on the weekend of the 10th– 11th of February 2017 to help finalize a plan to guarantee quality in translational research for neuromuscular diseases. Results: After extensive one and half day discussions the participants agreed that there are significant gaps in a) understanding translational research both among basic scientists and clinicians, b) evaluating grant applications by funding agencies and c) publication of peer reviewed translational research in journals. Workshop participants agreed to 1) organize a teaching workshop for clinicians and basic researchers interested in the planning and conduct of NMD clinical trials, 2) promote rigorous scientific evaluation of grant applications and to enforce the requirement for strong if possible independent validation of pre-clinical data before human clinical trials and 3) encourage journal editors to implement the ARRIVE guidelines and publish negative data. Conclusion: This workshop for the first time highlighted that promoting robust and reproducible translational research would enhance success of future treatments for NMDs. By facilitating these discussions and by offering the adequate format, ENMC greatly contributed and continues to contribute to the progress in research for neuromuscular diseases.
ID:326THE POSITION OF NEUROMUSCULAR PATIENTS IN SHARED DECISION MAKING
Topic: European Neuromuscular Centre (ENMC)
Anna Ambrosini
Research And Development, Fondazione Telethon, Milan, IT
Background: In occasion of its 25th anniversary, the European Neuromuscular Centre (ENMC) organised a special workshop involving 45 participants from 15 countries, representing patients and patient organizations, health professionals, clinical researchers, regulatory agencies, social scientists, ethicists and industry. Aim of the meeting was the assessment of patient participation in research and care options for neuromuscular diseases, using Shared Decision Making (SDM) as a tool to drive discussions. Methods: SDM has been defined as “an approach where clinicians and patients share the best available evidence when faced with the task of making decisions, and where patients are supported to consider options, to achieve informed preferences”. In practice, SDM applies to the communication between a health professional and a patient about the options of prevention, screening, diagnostic tests and treatment, including the option of not intervening; the goals of the method being better-informed patients, more confidence in and satisfaction about the treatment, and more compliance. Results: During the meeting, the SDM concept was discussed at first in relation to its natural field of application, the bilateral interaction between patients and their clinician. Different moments of the delivery of care were addressed, from genetic screening and diagnosis, to the management of transition from child to adult, and implementation of standards of care that have major impact on daily life. Then, the participants borrowed the SDM concepts to investigate the position of patient/patient organizations with respect to their level of participation in decision processes dealing with clinical research, in particular, in relation to patient registries and biobanks, research on quality of life, clinical trials design, and regulatory and consenting processes. Expectations are that, thanks to a better knowledge and empowerment, the patient community is able to take an increasingly decisive role in clinical trials, dissemination, and relationship with pharmaceutical industry and public health institutions. For each field addressed during the workshop, working groups including both professionals and patient representatives analysed: i) opportunities and limitations of patient participation as well as wishes of the patient community; ii) positive and negative examples and availability of best practices; iii) feasibility of implementation of a greater patient involvement; iv) next steps to be taken. In the end, some common features among all topics became evident. In order to be active partners, patients need to be properly informed, hence formation is key. However, education and training concern all stakeholders, with coaching and proficient communication about wishes and challenges of implementing patients’ participation in the various areas addressed. The roles and preferences of all stakeholders should be made more explicit. Moreover, structural changes are necessary in order to include patient representatives/organizations in advisory boards, ethics committees and in every stage of clinical trials. Conclusion: Overall, the active inclusion of patients in delivery of care and research processes requires changing attitudes, and therefore, ambassadors are needed who inspire and empower others about the benefits of patients’ participation. It was emphasized that exchange at the European level is also needed. An ENMC white paper will identify more specific actions and goals for the six topics discussed.
Poster Sessions
PS1Group1-001 / #342A FAMILY-BASED STUDY INTO PENETRANCE IN FACIOSCAPULOHUMERAL MUSCULAR DYSTROPHY TYPE 1
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Mariëlle Wohlgemuth1, Richard J. Lemmers2, Marianne A. Jonker3, Elly L. Van Der Kooi4, Corinne G. Horlings5, Baziel G. Van Engelen5, Silvere M. Van Der Maarel2, George W. Padberg6, Nicol C. Voermans5
1Neurology, Radboud University Medical Center, Nijmegen, NL;2Human Genetics, leiden University medical Center, Leiden, NL;3Health Sciences, Radboud University Medical Center, Nijmegen, NL;4medical center leeuwarden, leeuwarden, NL;5Neurology, Radboud University Medical Center, Nijmegen, NL;6Dep. Of Neurology, Radboud University Medical Center, Nijmegen, NL
Background: Objective: An observational cross-sectional study in a national FSHD expertise center to estimate the penetrance of facioscapulohumeral muscular dystrophy (FSHD)1 and to evaluate phenotype-genotype correlations. Methods: Ten FSHD1 probands carrying 4-9 D4Z4 units alleles and 140 relatives were examined. All 150 subjects were genetically characterized, including D4Z4 methylation levels in the mutation carriers. Mutation carriers were classified as 1) symptomatic: with symptoms of muscle weakness on history and muscle FSHD signs on examination; 2) asymptomatic: without symptoms of muscle weakness but with muscle FSHD signs on examination; and 3) non-penetrant: without symptoms of muscle weakness on history and without muscle FSHD signs on examination. We assessed the relationship between age-corrected clinical severity score and repeat size, sex and D4Z4 methylation levels. Results: The maximum likelihood estimates of symptomatic and those of symptomatic plus asymptomatic FSHD showed that penetrance depends on repeat size and increases until late adulthood. We observed many asymptomatic carriers with subtle facial weakness with or without mild shoulder girdle weakness (25% (17/69)). Non-penetrance was observed less frequently than in recent population studies (17% (12/69)), and most asymptomatic patients reported some shoulder pain. Methylation tended to be lower in moderately to severely affected mutation carriers with longer repeats. Conclusion: This family-based study detected a lower overall non-penetrance than previously observed, probably due to many asymptomatic mutation carriers identified by careful examination of facial and shoulder muscles. The recognition of asymptomatic mutation carriers is essential for selection of participants for future trials, and the likelihood estimates are helpful in counseling.
PS1Group1-002 / #482EVALUATING THE USEFULNESS OF NEW LINE IMMUNOASSAYS FOR MYOSITIS ANTIBODIES IN CLINICAL PRACTICE: A RETROSPECTIVE STUDY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Federica Montagnese1, Haris Babacic2, Peter Eichhorn3, Benedikt Schoser4
1Neurology, Friedrich Baur Institut LMU, Munich, DE;2Neurology, Friedrich Baur Institut LMU, Munich, DE;3Institute Of Laboratory Medicine, Ludwig Maximilian University, Munich, DE;4Friedrich-baur Institute, Departement Of Neurology, Ludwigs-Maximilians University of Munich, Munich, DE
Background: The diagnosis of idiopathic inflammatory myopathies (IIM) relies on clinical and histological (inflammatory infiltrates on muscle biopsy) criteria. However, an increasing number of myositis-associated (MAA) and myositis-specific antibodies (MSA) are being detected in sera of IIM patients and are considered useful diagnostic biomarkers. New line immunoassays are nowadays widely used for MSA/MAA detection though some concerns on their reliability arise due to limited validation in comparison with the gold standard immunoprecipitation. Furthermore, the disease specificity of these antibodies has been mainly studied in myositis patients and data on screening in control patients are often limited. The aim of our study was to assess the accuracy of MSA and MAA in diagnosing IIM in our large cohort of patients. Methods: We have retrospectively analysed the sera of patients that between 2014 and 2017 have been tested for myositis antibodies. The “Euroline: myositis 16 Ag” Kit by EUROIMMUN has been used to assess the presence of the following MSA/MAA: Mi-2alpha,Mi-2beta,TIF1gamma,MDA5,NXP2,SAE1,Ku,PM-Scl100,PM-Scl75,Jo-1,SRP,PL-7,PL-12,EJ,OJ,Ro-52. Additional clinical data as symptoms at onset, CK, muscle biopsy and conclusive diagnosis were also analysed. We have calculated the sensitivity (Se), specificity (Sp), positive/negative predictive values (PPV, NPV) and positive/negative likelihood ratios (LR+, LR-) for MSA and MAA as well as group as for the single antibodies. Results: 1232 patients (1727 serum samples) were identified. Muscle biopsy was performed in 583 patients (47%). Conclusive diagnoses were: myopathy (n=356), IIM (n=148), no evidence of neuromuscular diseases (n=587) and other neuromuscular diseases (e.g. ALS, neuropathies; n=141). The overall Sp was for MSA 95% and for MAA 89%, whereas the Se was 21% and 22% for MSA and MAA respectively. Further analyses were performed considering only individuals who underwent muscle biopsy as gold standard for IIM diagnosis. This group comprised 134 myositis and 446 controls (55% myopathy, 11% other neuromuscular disease and 35% no neuromuscular diseases). In this subgroup of patients the Se and Sp were equal to those observed in the whole cohort. The PPV was higher for MSA (54%) compared to MAA (37%) whereas the NPV was the same for both MSA/MAA (80%). A positive test increases the post-test probability that the patient has myositis of about 30% (LR+=4) for MSA and 15% (LR+=2) for MAA, whereas a negative test does not significantly decrease the probability that the patient has a myositis (LR-=0,8 for MSA and 0,9 for MAA). In 154 patients the test was repeated at least twice, here we found a high agreement between repeated measurements as in only 18% of patients the test retrieved discrepant results. Conclusion: This study may help clinicians to understand the significance and proper use of MSA/MAA in clinical practice. Commercial immunoassays for myositis antibodies show low sensitivity and high specificity, supporting the claim that they should be used for confirmatory rather than screening purposes. Repeating the test does not seem necessary, considering the high agreement in repeated measurements. Combining antibody findings with clinical features will help building up specific diagnostic algorithms for IIM
PS1Group1-003 / #853EXPRESSION OF DP116 IS A PREDISPOSING FACTOR FOR CARDIAC DYSFUNCTION IN DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Tetsushi Yamamoto1, Awano Hiroyuki1, Takamitsu Imanishi1, Yuji Nakamachi1, Masafumi Matsuo2, Kazumoto Iijima3, Jun Saegusa1
1Department Of Clinical Laboratory, Kobe University Hospital, --, Kusunoki-cho, Chuo-ku, Kobe, JP;2Department Of Physical Therapy, Faculty Of Rehabilitation, Kobe Gakuin University, kobe, JP;3Department Of Pediatrics, Kobe University Graduate School of Medicine, kobe, JP
Background: BACKGROUND: Duchenne muscular dystrophy (DMD), the most common inherited muscular disease in childhood, is caused by dystrophin deficiency because of mutations in the DMD gene. Cardiomyopathy is the most important non-muscular symptom threatening the life of DMD patients. The correlation between cardiac involvement and dystrophin mutation has been not fully understood since it was analyzed only in the view of mutation position in the DMD gene. In this study, we focused dystrophin isoforms produced by tissue-specific promoters and analyzed relationship between cardiac involvement in DMD patients and disrupted dystrophin isoform. Methods: METHODS: The medical records of DMD patients registered at the Department of Pediatrics of Kobe University Hospital from August 2007 to January 2017 were retrospectively reviewed. All echocardiograms were obtained by 1 examiner (T. Yamamoto), with considerable experience in imaging of patients with DMD, using a commercially available echocardiographic system (Aplio XG; Toshiba Medical Systems, Tochigi, Japan). Total 1109 echocardiographic data were obtained from 181 DMD patients with confirmed mutations in the DMD gene. For Kaplan-Meier analysis, to estimate a cardiac dysfunction-free survival rate, 19 patients were excluded due to existing cardiac dysfunction at the first examination. As a result, the remaining 162 patients were analyzed. Results: RESULTS: A Kaplan-Meier curve revealed that at the age of 14 years, cardiac dysfunction (left ventricular ejection fraction <53%) was observed in nearly half of the DMD patients. To examine the impact of dystrophin isoforms on a cardiac dysfunction, patients were divided by patterns of dystrophin isoform deficiency into 5 groups based on the mutations affecting dystrophin isoforms. The Dp427 group consisted of all 162 patients who had mutations at any position in the DMD gene. The Dp260, Dp140, Dp116, and Dp71 groups consisted of 120, 102, 19, and 11 patients, respectively. The cardiac dysfunction-free survival rates of the Dp260 group was similar to that of the other. Furthermore, the survival rates of the Dp140 and Dp71 groups did not differ significantly from in the others. However, the survival rate was significantly higher in the Dp116 group than in the others (log-rank test, P=0.022). Moreover, patients in the Dp116 group were at lower risk of cardiac dysfunction than the other patients (hazard ratio, 2.89; 95% confidence interval, 1.038–8.045; P=0.042). At age 25 years, the survival rate was 0.6 in the Dp116 group, and 0.15 in the other patients. Left ventricular dilation-free survival rates were estimated in the 5 DMD patient groups and compared with those in other patients. None of these comparisons showed significant differences, including a comparison of patients in the Dp116 group with all other patients (log-rank test, P=0.23). Conclusion: CONCLUSIONS: Cardiac dysfunction was less frequent in mutation of Dp116 coding region than other patients with DMD. These results indicate that Dp116 is a predisposing factor for cardiac dysfunction in DMD.
PS1Group1-004 / #348RIMEPORIDE: RESULTS FROM A PHASE IB STUDY IN PATIENTS WITH DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Teresa Gidaro1, Laurent Servais1, Stefano Previtali2, Alberto Zambon2, Jackie Pitchforth3, Kate Maresh3, Jordi Diaz-Manera4, Pierre G. Carlier5, Pierre Yves Baudin6, Benjamin Marty5, Stéphanie Carnesecchi7, Christian Laveille8, Julian Gray9, Florence Porte Thome9, Delphine Labolle9, Melanie Annousamy1, Virginie Chê1, Maria Grazia Natali Sora2, Simonetta Gerevini10, Nuria Vidal4, Katie Groves11, Francesco Muntoni3
1I-motion, Hopital Trousseau, PARIS, FR;2Neurology, San Raffaele Scientific Institute, Milano, IT;3Dubowitz Neuromuscular Unit, UCL Great Ormond Street Institute of Child Health, London, GB;4Neuromuscular Disorders Unit, Hospital de la Santa Creu i Sant Pau, Barcelona, ES;5Aim & Cea Nmr Laboratory, Neuromuscular Investigation Center, Institute of Myology, Paris, FR;6CRIS, Froyennes, BE;7Department Of Pathology And Immunology, Faculty of Medicine, Geneva, CH;8Calvagone, Lyon, FR;9R&d, EspeRare, Plan les Ouates, CH;10Department Of Neuroradiology, San Raffaele Scientific Institute, Milano, IT;11Great Ormond Street Hospital For Children, Somers Clinical Research Facility, London, GB
Background: Duchenne muscular dystrophy (DMD) is an orphan disease affecting approximately 1 in 3500 male births worldwide. Children typically lose their ability to walk between 7-13 y and this is followed by progressive respiratory muscle weakness and complications. Cardiac disease is an increasingly important source of morbidity and mortality in these patients. Rimeporide is a first-in-class NHE-1 inhibitor originally developed for congestive heart failure (CHF) that is being repositioned for patients with DMD. Rimeporide demonstrated a beneficial effect in mdx mice and hamster with dilated cardiomyopathy. Its safety, tolerability, and pharmacokinetics were examined in 7 clinical pharmacology studies involving 145 healthy subjects and 21 patients with CHF treated. Rimeporide was well tolerated in healthy male subjects and in patients with CHF as single and multiple doses up to 600 mg. Methods: Rimeporide was tested in a multicentre, European, open-label, Phase 1b study in 6-11 years DMD boys to assess its safety, tolerability and pharmacokinetics and to explore effects on biomarkers (serum and MRI) after a 4-week treatment. 4 dose levels were assessed sequentially through 4 ascending dose cohorts (from 75 to 900 mg daily dose). Dosing in the next cohort being started only after safety was deemed satisfactory at the preceding dose by an independent Safety Monitoring Committee. The trial sites were in Paris, Milan, London and Barcelona. 20 patients were enrolled and completed the study. Results: Over the course of the study, there has been no significant safety signal. Safety and tolerability were good in all dose cohorts. Adverse events observed were mild, short-lived, and considered unrelated to study medication. There was one serious adverse event, (loose stools and vomiting requiring overnight hospitalisation in an 8y old patient in cohort 3 receiving 600 mg) on the last day of the dosing period. The SAE was considered to be due to food poisoning and unrelated to Rimeporide. No significant new risks associated with treating young patients with DMD (6 to 11 years old) were identified. The pharmacokinetic profile was in line with the plasma concentrations observed in adults. Rapid absorption, dose related increase in concentration and no accumulation were observed. Serum biomarkers of inflammation and cardiac/skeletal muscle are currently measured. In addition, skeletal muscle MRIs taken at baseline and end of study are currently being analysed centrally. Conclusion: Rimeporide was shown to provide beneficial effects in animal models of DMD & cardiomyopathy and was shown to be well tolerated in healthy adults and now in DMD boys. It has the potential to be a muscle-sparing agent that may alleviate long-term accumulated skeletal and cardiac muscle damage and inflammation. In contrast to many therapeutics under development, rimeporide is potentially applicable to all DMD patients, regardless of the mutation. As it was already shown to be well tolerated in adults and now in DMD boys, it is anticipated that it could be safely combined with other therapies (e.g. corticosteroids, exon skipping, ACE inhibitors..) to achieve synergistic effect. The next milestone is a phase II study to evaluate the efficacy of rimeporide in a larger group of patients.
PS1Group1-005 / #603A LONG TERM PREVENTIVE EFFICACY STUDY WITH RIMEPORIDE, A SODIUM-PROTON EXCHANGER INHIBITOR, IN GRMD DOGS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Inès Barthélémy1, Jinbo Su2, Yves Fromes3, Stéphanie Carnesecchi4, Florence Porte Thome5, Pierre G. Carlier3, Bijan Ghaleh2, Stephane Blot1
1U955 - Imrb, Inserm, Ecole Nationale Vétérinaire d’Alfort, UPEC, Maisons-Alfort, FR;2Inserm Umr 955, Institut Mondor de Recherche Biomédicale, Créteil, FR;3Aim & Cea Nmr Laboratory, Neuromuscular Investigation Center, Institute of Myology, Paris, FR;4Department Of Pathology And Immunology, Faculty of Medicine, Geneva, CH;5R&d, EspeRare, Plan les Ouates, CH
Background: While the concept around the NHE-1 exchanger inhibition and its cardioprotective effect in models of heart failure is known for over a decade, it is a new concept in Duchenne Muscular Dystrophy (DMD). Cardiac disease is the leading cause of death in DMD and myocardial fibrosis is a key pathology predicting worsening of heart function and cardiac events. Rimeporide, working through inhibition of NHE-1, is cardioprotective in cardiomyopathic hamsters and mdx mice with reduction in cardiac pathology including fibrosis. Rimeporide also showed positive effects on skeletal muscles and diaphragm in mdx mice. Golden Retriever Muscular Dystrophic (GRMD) dogs reproduce the full spectrum of DMD and allow the study of skeletal, respiratory and cardiac outcomes with technologies translatable to patients. This study was designed to collect further knowledge on the co-evolution of cardiac, respiratory and locomotion defects. Methods: This is the largest study ever conducted in GRMD dogs. 24 GRMD dogs received Rimeporide at 20 mg/kg/day or placebo. At 2 months, dogs were stratified using a previously validated method between severe and moderate phenotype. Treatment was blinded, started from the age of 2 months until the age of 10 months. The gait quality was assessed using 3D-accelerometry recording during spontaneous locomotion. Validated clinical scoring analogous to motor scores for DMD patients were performed monthly. Respiratory function was also evaluated monthly using respiratory inductance plethysmography and non-invasive tools including fluoroscopic assessments. Echocardiography, including Tissue Doppler Imaging and speckle tracking techniques (at 2, 4, 6, 9 and 12 months). MRIs (at 2, 6 and 12 months with Gadolinium enhancement for cardiac) were also used for cardiac and skeletal muscles assessments. Serum biomarkers and histopathology analysis are conducted in search of non-invasive biomarkers to be used in patients. Results: A pharmacokinetic study was conducted to select the dose prior to the study initiation. Based on cardiomyopathic hamster and mdx mice studies, therapeutic exposure is above 500 ng/mL. GRMD dogs received 10 mg/kg of Rimeporide as a capsule in the morning and in the evening. Despite a high variability in absorption and elimination, it was possible to confirm that 20 mg/kg/day given as 2 doses of 10 mg/kg/day was maintaining plasma concentration above the therapeutic exposure. 24 GRMD dogs were enrolled and subsequently stratified between moderate and severe forms. At 2-month of age, 6 GRMD dogs were found with decreased stride frequency and a reduced spontaneous speed together with an elevation number of circulating CD4+/CD49dhi T cells and were subsequently assigned as severe forms. 18 additional GRMD dogs were classified as moderate forms. Over the 10-month treatment period, the tolerability of rimeporide was good. As observed in toxicology studies, there was no safety flags. Analyses of skeletal, cardiac, respiratory functions are ongoing and will be discussed in the poster. Conclusion: Results from this translational study in GRMD dogs will contribute to design phase II/III pivotal study by guiding outcome criteria, dose and biomarker selections as well as increasing the understanding of the efficacy profile of Rimeporide in skeletal, respiratory and cardiac muscle and in general supporting DMD disease understanding.
PS1Group1-006 / #458PREDICTORS OF EARLY LEFT VENTRICULAR SYSTOLIC DYSFUNCTION IN DMD PATIENTS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Raphael H.D. Cirino1, Rosana H. Scola2, Renata D. Ducci3, Ana C.C. Wermelinger1, Claudia S.K. Kay2, Paulo J. Lorenzoni2, Lineu C. Werneck2, Eliane R. Carmes4, Claudio L.P. Da Cunha1
1Cardiology, Hospital de Clínicas - Universidade Federal do Paraná, Curitiba, BR;2Neurology, Hospital de Clínicas - Universidade Federal do Paraná, Curitiba, BR;3Neurology, Hospital de Clínicas - Universidade Federal do Paraná, Curitiba, BR;4Internal Medicine, Hospital de Clínicas - Universidade Federal do Paraná, Curitiba, BR
Background: The life expectancy of Duchenne muscular dystrophy (DMD) patients has improved in the last years, but the management of cardiac complications remains a challenge in DMD therapy. Most DMD patients develop cardiomyopathy, but the symptoms can be masked by severe musculoskeletal weakness. Myocardial strain is an emerging technique that measures myocardial deformation and is able to diagnose left ventricular systolic dysfunction (LVSD) even in the absence of reduced left ventricular ejection fraction (LVEF). Early detection of left ventricular systolic dysfunction (LVSD) is important for therapeutic strategies for DMD patients. We analyzed the myocardial strain by echocardiography as a main tool for the early detection of LVSD and determined the predictors of early LVSD. Methods: Cross-sectional study of forty DMD patients with normal LVEF followed at the Neuromuscular Disorders Service of the Hospital de Clínicas of the Federal University of Parana in Curitiba, Brazil between January 2014 and June 2016. LVEF was measured by 3-dimensional transthoracic echocardiography. Global longitudinal strain (GLS) was used to analyse subtle disturbances in the longitudinal contraction of the myocardium. Patients were determined to have early LVSD [GLS>-18] or normal left ventricular systolic function [GLS≤-18]. Independent samples Student’s t-test was used to compare continuous variables in the studied groups. For qualitative variables, the comparison was made using Fisher’s exact test. We adjusted bivariate logistic regression models for variables with p < 0.05 in the univariate analysis, when possible, including age as covariate. After adjustment, the Wald test was used to assess the significance of the variables included in the model, estimating odds ratios with 95% confidence intervals. P values < 0.05 were considered statistically significant. The study was approved by the Ethics Committee. Results: The study population consisted of 40 DMD male patients aged 2 to 19 years (median: 10.5 years; mean: 10.7 ± 3.9 years). Only 18 patients were still walking. Thirty-one patients used corticosteroids. Twenty (50.0%) patients had early LVSD. The remaining 20 patients had normal LVSF as determined by GLS measurements. Patients who had early LVSD were older and had a longer disease duration and a higher frequency of gait loss, corticosteroid therapy and of mutations in exons 45, 46, 47, 48, 49, 50 and 52. The logistic regression model was adjusted for the analysis of exons 45 (OR 9.6, 95% CI 0.90-102.8, p=0.053) and 50 (OR 36.1, 95% CI 2.15-604.0, p=0.010). It was not possible to adjust the bivariate model for the other variables. Conclusion: Echocardiographic myocardial strain analysis can detect LVSD before the reduction of the LVEF in a large number of DMD patients. The importance of the early detection of LVSD lies in the fact that early initiation of specific treatments could prevent the progression of cardiomyopathy and its complications. The results of this study also suggest that older age, longer disease duration, gait loss, use of corticosteroids and mutations within the “hot spot” region of the DMD gene are predictors of early LVSD.
PS1Group1-007 / #736GENE THERAPY BY CRISPR-CAS9 MEDIATED EXON SKIPPING IN A PRE-CLINICAL MODEL OF DMD, THE GRMD DOG
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Isabel Punzón1, Inès Barthélémy1, Frédéric Auradé2, Nicolas Blanchard-Gutton1, France Piétri-Rouxel3, Frédéric Relaix2, Stéphane Blot1
1Neurobiology, U955 - IMRB, INSERM, Ecole Nationale Vétérinaire d’Alfort, UPEC, Maisons Alfort, FR;2Henri Mondor Hospital School Of Medicine, INSERM-UPEC IMRB U955-E10, Créteil cedex, FR;3Sorbonne Université-UMRS974-Inserm-Institut de Myology, Paris, FR
Background: Duchenne muscular dystrophy (DMD) is a rare degenerative disease without any cure at present. It is an X linked disease due to mutations into the dystrophin gene, that causes the absence of the protein and the progressive degeneration of muscles including heart and diaphragm. Golden Retriever Muscular Dystrophy dog (GRMD) mimics perfectly the DMD and is the accepted pre-clinical large animal model for this disease. The GRMD presents a spontaneous splice site single mutation in the dystrophin gene that leads to the skipping of exon 7, disruption of the reading frame, absence of protein and consequently progressive muscle degeneration, weakness, heart and respiratory failure. A promising approach to cure DMD is gene therapy and specifically exon skipping which efficacy has already been proved restoring partially functional variants of dystrophin. Drugs start to be commercialized to skip exon 51, but a personalised medecine would be preferable. Our group showed that exon skipping strategy using antisense sequences vectorized into adenoassociated vectors (AAV) could rescue the dystrophin absence and significantly improves muscle function in GRMD dogs. However a longterm follow-up showed a decrease in dystrophin expression parallel to a loss of viral genome. To avoid the need of repeated injections of molecules for the exon skipping, with the associated inconvenience for patients and the possible immunoreactions side effects we propose the use of CRISPR tool. This emerging tool for genome editing, appears powerful to perform exon skipping therapy in a permanent way (genome edition of satellite cells) and it has been already successfully proved in mice and human cells. Methods: We started a translational study of genome edition based exon-skipping therapy in GRMD dog, using AAV. 2 pairs of RNA guides were cloned into an AAV-9 vector with tropism for skeletal muscles and heart. A CMV promoter was used for driving the Cas9 and mCherry expression and human and mouse U6 promoters for both gRNAs. A 7 month-old GRMD male dog was intramuscularly injected with an AAV vector containing two RNA guides flanking exons 6 and 9. Results: First, we showed efficacy of CRISPR-exon skipping (6 to 9) in vitro on primary GRMD myoblasts. In vivo, after intramuscular injections, the skipped transcript was detected and associated with dystrophin expression, in biopsies performed 2 months after AAV injection. This is the first proof of principle that genome editing-mediated multi-exon skipping can be performed in a large animal model of DMD, leading to an in-frame transcript and dystrophin expression. Conclusion: Further steps will include systemic delivery of AAVs, to evaluate the levels of dystrophin in targeted muscles and to correlate this to clinical and functional improvements. The genome edition of satellite cells, which would allow a sustained expression of dystrophin, will also be assessed. We also expect to answer crucial preclinical questions of feasibility, bio distribution, efficiency and safety before translation to humans. The GRMD model offers the unique opportunity to study long-term effects (several years) of such a strategy in a dystrophic context and will allow a relevant comparison of the CRISPR based strategy with pre-existing ones.
PS1Group1-008 / #734IN VIVO CELL TRACKING OF CANINE MYOBLASTS BY SODIUM/IODIDE SYMPORTER GENE EXPRESSION
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Isabel Punzón1, David Mauduit1, Inès Barthélémy1, Nicolas Blanchard-Gutton2, Jean-Laurent Thibaud3, Pauline De Fornel3, Bryan Holvoet4, Jean-Thomas Vilquin5, Maurilio Sampaolesi6, Stéphane Blot2
1Enva, Neurobiology, U955 - IMRB, INSERM, Maisons Alfort, FR;2Enva, Neurobiology, U955-IMRB, INSERM, Maisons Alfort, FR;358 Rue Auguste Perret,, MICEN Vet,, Créteil, FR;4Ku Leuven, Belgium, Translational Cardiomyology Lab, Stem Cell and Embryo Biology, Dept Development & Regeneration,, Leuven, BE;5Myology Research Center, Pitié Salpêtrière Hospital,, Sorbonne Université UPMC Univ Paris 06, Inserm UMRS974, CNRS FRE3617, Myology Research Center, Paris, FR;6Translational Cardiomyology Lab, Stem Cell and Embryo Biology, Dept Development & Regeneration, Leuven, BE
Background: Cell therapy emerged as a promising approach for the treatment of degenerative muscular diseases but due to disappointing results of clinical trials, efforts need to be done for the identification of the potential therapeutic cell (survival, homing to skeletal muscle, bio distribution, muscle regeneration…). For cell therapy improvement through a better understanding of therapeutic stem cells, we propose a non-invasive cell monitoring by medical imaging that would improve the understanding of the behavior of the cells following transplantation. Methods: We demonstrated the feasibility to track canine NIS+ myoblasts by SPECT/CT with specific uptake of 99mTc-pertechnetate (99mTcO4-) in the skeletal muscle of a large animal. The sodium/iodine symporter (NIS), a natural symporter of iodide in thyroid, salivary glands and stomach was used as a reporter gene. We used a lentiviral vector expressing canine NIS to transduce primary canine myoblasts. Results: NIS did not interfere with myotubes formation, and only cells expressing NIS (myoblasts and myotubes) were able to uptake radioactivity both in vitro and in vivo. We performed whole body scintigraphy imaging after intramuscular injections of different amounts of NIS+ and NIS– expressing myoblasts. SPECT/CT acquisitions at different timepoints until 30 days post cells’ injection were done and biopsies were taken at day 32 for histological analysis. Three millions of NIS expressing myoblasts were detected by scintigraphic imaging only at 48h, 10 millions were detected until one week, and 20 millions were still detected after one month. Histological analysis of injected muscles showed cNIS expression 1 month after cell injection and myoblasts expressing NIS fused with muscle fibers. Conclusion: We demonstrate that this is a non-invasive tool to track cells in vivo, that only functional cells trap radioactivity and that a long-term follow-up is possible being repeatable as many times as necessary. We propose a follow-up study using non-invasive cell tracking by scintigraphy imaging, in combination with functional studies to determine the migratory behavior and therapeutic properties of cells: muscle colonization, dystrophin expression, influences of cells’ environment on their migration and differentiation.
PS1Group1-009 / #928CLINICAL OUTCOME STUDY OF DYSFERLINOPATHY: RELATIONSHIP BETWEEN MUSCLE MRI AND PHYSIOTHERAPY OUTCOME MEASURES
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Jordi D. Manera1, Roberto Fernández-Torrón2, Meredith James2, Anna Mayhew2, Michele Eagle2, Robert Muni Lofra2, Fiona Smith3, Helen Sutherland2, Anne Marie Sawyer4, Carolina Tesi Rocha5, John W. Day5, Anthony Peduto6, Kirsti J. Jones7, Eva M. Coppenrath8, Maggie C. Walter9, Pierre G. Carlier10, Tanya Stojkovic11, Andrew Blamire3, Kate Bushby2, Volker Straub12
1Neuromuscular Unit, Neurology Department, Hospital de la Santa Creu i Sant Pau, Barcelona, ES;2The John Walton Muscular Dystrophy Research Centre, Newcastle Hospitals Trust, Newcastle University, Newcastle Upon Tyne, GB;3Magnetic Resonance Centre, Institute For Cellular Medicine, Newcastle University, Newcastle Upon Tyne, GB;4Lucas Centre For Imaging, Stanford University School of Medicine, Stanford, CA, US;5Department Of Neurology And Neurological Sciences, Stanford University School of Medicine, Stanford, CA, US;6Department Of Radiology, Westmead Hosptial, Faculty Of Health Sciences, University of Sydney, Sydney, AU;7Institute For Neuroscience And Muscle Research, Children’s Hospital At Westmead, University of Sydney, Sydney, AU;8Department Of Clinical Radiology, Ludwig-Maximilians-University Munich, Munich, DE;9Friedrich-baur Institute, Departement Of Neurology, Ludwigs-Maximilians University of Munich, Munich, DE;10Aim & Cea Nmr Laboratory, Neuromuscular Investigation Center, Institute of Myology, Paris, FR;11Ap-hp, G.h. Pitié-salpêtrière 47-83, Boulevard De L’hôpital, Institut de Myologie, Paris, FR;12John Walton Muscular Dystrophy Research Centre, Newcastle University, Institute of Genetic Medicine, Newcastle Upon Tyne, GB
Background: Dysferlinopathies are caused by mutations in the DYSF gene. Patients may present with isolated hyperCKemia, limb girdle muscle weakness or predominant weakness of the lower legs. The Jain Foundation is funding The Clinical Outcome Study for Dysferlinopathy, a multi-centre natural history study in a cohort of 203 dysferlinopathy patients. Previous smaller studies have reported an involvement of scapular muscles on muscle MRI but no longitudinal studies have been reported for upper limb involvement in muscle MRI in relation to physiotherapy functional outcome measures. Methods: Here we describe baseline and year 3 upper limb muscle MRI data in a large cohort of dysferlinopathy patients. To examine the longitudinal correlation between muscle MRI semiquantitative scoring and functional outcome measures. 203 patients (11 to 86 years old) were enrolled in the JAIN COS study across 15 centers (Europe, USA, Australia, Japan). Patients underwent physiotherapy, medical and MRI assessments. 74 patients underwent upper limb muscle MRI at the time of enrollment and 61 patients had upper limb muscle MRI at year 3 at the point this analysis was completed. Scans were acquired from different systems and manufacturers at 1.5T and 3T. Muscles were scored on axial T1-weighted sequences with the semiquantitative Mercuri visual scale modified by Fisher. Physiotherapy assessments included muscle strength (manual muscle testing; hand held dynamometry) and functional ability evaluations (Performance of Upper limb, Brooke test). Change between baseline and year 3 muscle MRI was assessed using Wilcoxon’s Signed Rank Tests and statistical significance was set at p=0.05. Results: The subscapularis (80.8%), latissimus dorsi (82.6%), infraspinatus (73.8%) and supraspinatus (72.8%) were the most affected scapular muscles at baseline. The subscapularis muscle was involved in some patients without proximal upper limb symptoms or dysfunction. The biceps brachii (57.1%) and the anterior muscles of the forearm (53.8%) were the most affected muscles from the arm and forearm at baseline. Baseline data identified the tongue (34.2%) and the cervical paraspinal muscles (24.6%) as the most commonly involved cranial and cervical muscles. Cranial and cervical muscles did not show longitudinal change apart for the tongue and cervical paraspinal muscles. Conclusion: Muscle semi-quantitative scoring correlated with functional outcome measures in upper limb in dysferlinopathy patients.
PS1Group1-010 / #10MUSCLE IMAGING AND CLINICAL OUTCOME MEASURES IN OCULOPHARYNGEAL MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
H. M.j.m. Kroon1, Corinne G. Horlings2, J. G. Kalf3, Baziel G. Van Engelen2
1Rehabilitation, Radboud University Medical Centre, Nijmegen, NL;2Neurology, Radboud University Medical Centre, Nijmegen, NL;3Rehabilitation, Radboud University Medical Centre, Nijmegen, NL
Background: Oculopharyngeal muscular dystrophy (OPMD) is a rare late onset progressive neuromuscular disorder. Currently, there is a profound lack of knowledge on muscle abnormalities in OPMD. Therefore we collected a large data set to present for the first time in OPMD (1) quantitative muscle ultrasound (QMUS) of the head-neck region and legs and its relation to clinical outcome measures and (2) a comparison between QMUS and muscle MRI. Methods: Forty eight OPMD patients (24 Women, mean age 60.5, SD 8.7) including asymptomatic patients, participated. MRI (DIXON, quantitative fat fractions) was used to measure muscle fatty infiltration of the rectus femoris, gastrocnemius, soleus and biceps femoris muscles. QMUS (echo-intensities) was used to measure fatty infiltration of the same leg muscles and of the tongue muscle and masseter muscle. Also, all participants were clinically tested by the Motor Function Measure (MFM) and muscle strength measures (MRC scale and dynamometry) of knee extension, knee flexion, and foot plantar flexion and by measuring the maximum isometric pressure of the tongue (MIP) and maximum bite force (Bite Force Gauge). Correlations were calculated between the echo intensities (EI) and the clinical measurements and between the EI and the mean MRI fat fractions, with accepting r <.30 being a weak, r.30-.70 being a moderate and r >.70 being a strong correlation. Results: Mean EI of all muscles correlated moderately with the MFM (r = -.584; p =.00). EI of rectus and biceps femoris correlated moderately with knee extension and knee flexion strength (r = -.660; p =.00 and r = -.526; p =.00), and moderately with dynamometry of knee extension and flexion (r = -.661; p =.00 and r= -.363; p =.02). EI of the soleus and gastrocnemius did not correlate with foot plantar flexion (r = -.241; p =.11). EI of tongue correlated moderately with the MIP (r = -.633; p =.00), but EI of the masseter did not correlate with the maximum bite force (r = -.271; p =.06). Mean EI and the mean MRI fat fractions are moderately correlated biceps femoris (r =.354; p =.02), rectus femoris (r =.502; p =.00) and gastrocnemius (r =.544; p =.00), except the soleus (r =.193; p =.23). Conclusion: In OPMD patients echo intensity correlates moderately with the measures of maximum motor functioning of the legs and the tongue, but not when using dynamometry or bite force, suggesting that the latter measures are less useful in OPMD. QMUS and MRI of the legs show moderate correlations, suggesting that QMUS of the leg muscles could be used instead of more invasive and expensive MRI.
PS1Group1-011 / #444DIGENIC SQSTM1-TIA1 MYOPATHY: CLINICAL AND PATHOLOGICAL FEATURES
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Zhiyv Niu1, Carly S. Pontifex2, Sarah Berini1, Leslie E. Hamilton2, Elie Naddaf1, Eric Wieben1, Ross A. Aleff1, Kristina Martens2, Angela Gruber3, Andrew G. Engel1, Gerald Pfeffer2, Margherita Milone4
1Mayo Clinic, Rochester, MN, US;2Clinical Neurosciences, University of Calgary, Calgary, AB, CA;3PreventionGenetics, Marshfield, WI, US;4Neurology, Mayo Clinic, Rochester, MN, US
Background: T-cell intracellular antigen-I (TIA1, chr.2) is a largely expressed RNA-binding protein required for the cytoplasmic stress granules formation, which is a crucial step in preventing misfolded protein accumulation. To date, a single TIA1 mutation (p.Glu384Lys) is known to cause a late-onset myopathy with rimmed vacuoles. TIA1 mutations were recently identified in amyotrophic lateral sclerosis (ALS) / frontotemporal dementia (FTD). Sequestosome-1 (SQSTM1, chr.5) is a scaffolding protein participating in apoptosis, cell survival, autophagy, and regulation of ubiquitinated protein turnover. Mutations in SQSTM1 can cause a multisystem proteinopathy manifesting as Paget disease of bone (PDB), ALS, FTD and myopathy with rimmed vacuoles but have incomplete penetrance. Methods: Review of clinical, serological, electrophysiological, muscle morphological, cardiac and radiological findings of three unrelated probands with late-onset distal myopathy and their affected and unaffected family members, including the affected fraternal twin of a proband. Next Generation Sequencing panel, whole exome sequencing, and targeted Sanger sequencing analyses of family members were performed to investigate the molecular genetic etiology of the myopathy. Results: Genetic analyses detected two variants in genes located on different chromosomes responsible for the myopathy in the three unrelated probands: a heterozygous pathogenic mutation in SQSTM1 (c.1175C>T, p.Pro392Leu) and a heterozygous variant in TIA1 (c.1070A>G, p.Asn357Ser). The affected fraternal twin of a proband carries both variants; the unaffected family members have one or no variants. Two probands have a distal myopathy with rimmed vacuoles that presented with weakness of the index extensor; the other proband has a distal myopathy with myofibrillar pathology accompanied by hypercapnic respiratory insufficiency. Some of the affected individuals in a family have cognitive impairment. Conclusion: 1) The affected individuals of three unrelated families have a myopathy associated with both variants, one in SQSTM1 and one in TIA1 (genes located on different chromosomes), suggesting that the myopathy is digenic. 2) The combined SQSTM1 and TIA1 variants can cause myofibrillar myopathy, and therefore, analysis of both genes should be considered in the molecular investigation of myopathy with rimmed vacuoles as well as in the genetic characterization of myofibrillar myopathy. 3) Respiratory insufficiency can occur in SQSTM1-TIA1 myopathy and, therefore, patient’s respiratory status should be closely monitored. 4) The cognitive impairment observed in a family is in keeping with the known deleterious effects of TIA1 and SQSTM1 mutations on central nervous system, previously reported in the form of FTD.
PS1Group1-012 / #26PATHOLOGICAL AND GENETIC STUDIES IN PATIENTS WITH MENDELIAN DISORDERS OF MTDNA MAINTENANCE
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Diana Lehmann1, Amy E. Vincent2, Hannah Rosa2, Mariana Rocha2, Stephan Zierz3, Robert W Taylor2, Douglass M. Turnbull2
1Department Of Neurology, University of Ulm, Ulm, DE;2Institute Of Neuroscience, Newcastle University Wellcome Trust Centre for Mitochondrial Research, Newcastle upon Tyne, GB;3Department Of Neurology, University of Halle/S., Halle/S., DE
Background: The underlying genetic defect in patients with mitochondrial PEO is either a primary mutation of the mitochondrial genome (single, large-scale mtDNA deletion or mtDNA point mutation) or recessively and dominantly-inherited mutations in nuclear genes involved in mtDNA maintenance leading to clonally-expanded multiple mtDNA deletions in muscle. The nuclear disease genes are largely implicated in the replication and stability of mtDNA, and as such a pathogenic mutation leads to secondary instability of the mitochondrial genome. Causal mtDNA deletions can be found in a heteroplasmic (mixture of mutated and wild type mtDNA) state. However, each tissue/cell has its own genetic defect and biochemical threshold of mutated mtDNA load which needs to be exceeded before focal respiratory chain deficiency becomes evident. Methods: To investigate this, muscle biopsies of 16 patients with genetically- and clinically-characterized mitochondrial disease of nuclear origin (9 POLG, 4 TWNK, 2 RRM2B and 1 SLC25A4 (ANT1)) and 4 healthy controls were analysed using quadruple OXPHOS immunohistochemistry, quantifying the biochemical phenotype in individual muscle fibres of patient muscle biopsies. This technique is based on quadruple immunofluorescence to detect structural components of complexes I (NDUFB8) and IV (COXI), as well as porin (a marker of mitochondrial mass) and laminin (a cell membrane marker to define the boundaries of muscle fibres). Further studies on 6/17 patients (2 POLG, 2 RRM2B, 1 TWNK, 1 SLC25A4 (ANT1)) included the correlation of the biochemical deficiency with the mtDNA abnormality in individual cells, following laser microcapture and determination of the size and level of clonally-expanded mtDNA deletion within fibres by real-time PCR, long-range PCR and sequencing of breakpoints. Results: The data from quadruple immunocytochemical studies show that the muscle biochemical phenotype is different in patients with multiple mtDNA deletions compared to other mtDNA mutations, but showed no difference between genotypes. Real-time PCR showed, that the level of deletion is increasing with the biochemical defect. In all patients, the levels of deletion of ND4/D Loop and ND4/ND1 are significantly increasing with the MRC profile. However, in all groups of fibres there seem to be very low levels of ND1 deletions. Conclusion: It has already been shown, that patients harbouring multiple deletions have a distinct MRC- (muscle respiratory chain) profile. Three different groups of fibres were found in these patients: cells without deficiency, cells with isolated complex I deficiency and cells with combined complex I and complex IV deficiency. The correlation of biochemical deficiency with level of deletion is arguing against any point mutations as cause of the deficiency.
PS1Group1-013 / #451EPILEPSY CHARACTERIZATION IN LAMA2-RELATED CONGENITAL MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Daniel Natera De Benito1, Debora Itzep2, Jordi Muchart2, Alia Ramirez2, Delia Yubero2, Laura Carrera2, Javier Aparicio2, Cristina Jou2, Cecilia Jimenez Mallebrera2, Jaume Colomer2, Carlos Ortez2, Andres Nascimento2, Victoria San Antonio Arce2
1Neuromuscular Disorders Unit, Hospital Sant Joan de Déu, Barcelona, ES;2Hospital Sant Joan de Déu, Barcelona, ES
Background: LAMA2-related congenital muscular dystrophy (CMD), is an autosomal recessive neuromuscular disorder resulting in deficiency of the laminin a2 chain, a component of the skeletal muscle extracellular matrix laminin-2. Laminin-2 is expressed in basement membranes of striated muscle, but also in the neuromuscular synapses, schwann cells and in brain where it is thought to have a role in neuronal migration. Although it is a muscular disease, it may be associated with peripheral neuropathy and a diversity of cerebral manifestations. The risk of epileting seizures is described in the literature up to 20% of the cases, even without cerebral structural anomalies on MRI. Although both focal and generalized seizures have been reported, the epilepsy phenotypic spectrum has not been defined. In this study we describe the epilepsy phenotype in a cohort of patients with LAMA2-related CMD who have experienced seizures in order to clarify the spectrum of phenotypes that may occur. Electroclinico-radiologic presentation, response to treatment, and long-term outcome are described. Methods: All patients with genetically and/or pathologically confirmed diagnosis of LAMA2-related CMD followed-up in our hospital were included. Medical records of patients with demographic information, clinical data, EEG records, and MRI studies were compiled. Seizures and epilepsy were described and classified according to the ILAE classifications. Results: Epilepsy was observed in eight of twenty patients with LAMA2-related CMD. A partial merosin deficiency was detected in five patients and a complete merosin deficiency in three. The mean age at the time of the study was 19.5 years (range 10-32 years). Mean age at seizure onset was 8 years (range 2-20 years). The most common presenting seizure type was focal onset seizure with or without impaired awareness in 5 of 8 subjects. Visual aura was described in four patients. Vomiting was also frequently reported (4/8).Three subjects had motor seizures with an unknown onset. Two subjects also had bilateral tonic-clonic seizures. One case had atypical absences and drop attacks. Long-term seizure control was assessed based on the Brodie classification: six patients (75%) had ongoing refractory seizures (D), and two (25%) had fluctuating course (C: periods of at least 12 months seizure-free, interspersed with relapses). Interictal EEGs showed focal or multifocal epileptiform abnormalities in seven of eight cases. Focal occipital abnormalities were the most commonly observed. Background activity was normal in six patients, and generalized background slowing was reported in two cases An increased signal intensity of subcortical and periventricular white matter on T2-weighted MRI was reported in all the patients. Four patients (50%) had suspected focal cortical dysplasia. Conclusion: The epilepsy in LAMA2-related CMD typically presents with childhood-onset focal seizures that are not usually prolonged and have prominent visual and autonomic features. EEG show focal interictal abnormalities that predominate in the posterior quadrants. MRI brain demonstrates a constellation of structural abnormalities, including focal cortical dysplasia, in addition to the well-known increased signal intensity of white matter on T2-weighted MRI.
PS1Group1-014 / #786A PHASE 1B/2 STUDY OF THE ANTI-MYOSTATIN ADNECTIN RG6206 (BMS-986089) IN AMBULATORY BOYS WITH DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Kathryn R. Wagner1, Brenda L. Wong2, Barry Byrne3, Lee Sweeney3, Leslie Jacobsen4, Giridhar Tirucherai4, Michael Rabbia5, Jeppe Buchbjerg6, Dukart Juergen6, Michelle Krishnan6, Clifford Bechtold4
The Johns Hopkins School of Medicine, Baltimore, MD, US;2Neurology, Cincinnati Children’s Hospital Medical Center, Cincinatti, OH, US;3University of Florida, Gainesville, FL, US;4Bristol-Myers Squibb, Princeton, NJ, US;5Roche Translational and Clinical Research Center, New York, NY, US;6F. Hoffmann-La Roche Ltd, Basel, CH
Background: Pharmacologic inhibition of myostatin, a negative regulator of muscle growth, has been shown to increase skeletal muscle mass in several species, including humans. RG6206 is a potent, novel anti-myostatin adnectin previously shown to inhibit myostatin activity in healthy adults. This Phase 1b/2 study (NCT02515669) assessed the safety, tolerability, pharmacokinetics and pharmacodynamics of the novel anti-myostatin adnectin RG6206 (BMS-986089) in ambulatory boys with Duchenne muscular dystrophy (DMD). Methods: Forty-three ambulatory boys with DMD aged 5–10 years were randomized to receive weekly subcutaneous doses of RG6206 (4–50 mg) or placebo. The primary endpoint was safety over 24 weeks. Secondary endpoints included pharmacokinetics of RG6206, frequency of anti-drug antibodies and pharmacodynamic effect on serum myostatin levels. Exploratory outcome measures included 4-stair climb velocity (4SC), 6-minute walk test (6MWT) and North Star Ambulatory Assessment (NSAA) score. Muscle volume and lean body mass were assessed by dual-energy X-ray absorptiometry (DXA) or magnetic resonance imaging (MRI). Results: No clinically significant changes in laboratory values, vital signs or echocardiogram parameters were observed over 24 weeks. The most common adverse events considered to be related to study drug were cutaneous injection site reactions (RG6206 n=10, 31.3%; placebo n=1, 9.1%), which were mild (except one event of moderate injection site discomfort). One patient tested positive for anti-drug antibodies; however, this patient had a positive titer at baseline. RG6206 serum concentrations increased with dose and were accompanied by dose-dependent reduction in free myostatin, with a ≥ 95% reduction from baseline with the highest dose. The study was not powered to demonstrate effects on exploratory functional endpoints and, as such, no efficacy on 4SC, 6-MWT and NSAA score was observed. However, DXA imaging data showed an increase in lean body mass in boys receiving RG6206 compared with placebo. In addition, MRI imaging of the right thigh showed positive effects on muscle composition due to RG6206, with an increase in contractile, and decrease in non-contractile, tissue. Conclusion: RG6206 was well tolerated by ambulatory boys with DMD, with no clinically significant safety concerns. Robust anti-myostatin target engagement was demonstrated, and DXA and MRI imaging outcomes suggest a positive effect of RG6206 on muscle over the 24-week study. A 48-week open-label phase of this study is ongoing and a Phase 2/3 study is underway to further evaluate RG6206 in ambulatory boys with DMD (NCT03039686). Acknowledgements Study sponsored by F. Hoffmann-La Roche.
PS1Group1-015 / #350IN VITRO FUNCTIONAL CHARACTERIZATION OF FKRP PATIENT MISSENSE MUTATIONS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Sara F. Dias Henriques, Evelyne Gicquel, Justine Marsolier, Isabelle Richard
INTEGRARE, Généthon, Inserm, Univ Evry, Université Paris-Saclay, Evry, FR
Background: Muscular dystrophies are monogenic diseases affecting the skeletal muscle, characterized by a progressive loss of the muscle mass, can lead to premature death by heart or respiratory complications and are currently without treatment. One group of muscle dystrophies termed “dystroglycanopathies” encompasses different genetic pathologies that lead to a change in the glycosylation status of the protein alpha-dystroglycan (alpha-DG). This protein is a crucial component of the dystrophin-associated glycoprotein complex (DGC) at the skeletal muscle whereby, through a complex and specific glycosylation, the plasma membrane is anchored to the extracellular-matrix. Loss of this glycosylation weakens this link and leads to the death of muscle fibers. Many proteins cooperate in the assembly of the final glycosylation moiety such as the Fukutin-Related Protein (FKRP). Mutations in FKRP lead to different pathologies, from the severe Congenital Muscular Dystrophy type 1C (MDC1C) to the mild Limb-Girdle Muscular Dystrophy type 2I (LGMD2I) and one of the most prevalent of the LGMD group. Most patient-mutations are missense mutations, some of which leading to a misfolded protein that is retained in the endoplasmic reticulum (ER) and prematurely degraded before reaching the functionality compartment, the Golgi (Esapa et al., 2002 and 2005; Torelli et al., 2005; Keramaris-Vrantsis et al., 2007). We have shown that some LGMD-associated misfolded proteins still retain partial function when rescued to the correct compartment (Soheili et al., 2011). It is therefore crucial to assess the functionality of FKRP mutant proteins in line with potential corrective treatments. Methods: Through transgene overexpression in a FKRP knockout (KO) cell line, we have characterized different patterns of cellular localization of the mutant proteins, some of which never described before. Results: Combination of these localization results with functional assays lead to the identification of some mutants that still bear function when properly addressed to the Golgi. Interestingly we have also identified mutants that, although correctly localized in the Golgi, where nonetheless severely functionally impaired, revealing that accurate cellular localization is not directly linked with function. Structural analysis is currently being pursued in understanding the function impairment of these mutants. Conclusion: Our results clearly show that cellular localization studies are not enough to fully understand the consequences of the mutations and a complementary functional assessment of the mutated proteins is required when curative treatments are envisioned.
PS1Group1-016 / #528IDENTIFICATION OF MUTATIONS IN A COHORT OF UNCLASSIFIED INHERITED MUSCLE DISORDERS BY TARGETED NEXT GENERATION SEQUENCING
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Kiran Polavarapu1, Aditi Joshi2, Veeramani Preethish-Kumar1, Aradhna Mathur2, Sushmita Nayak2, Sakshi Ambawat2, Saraswati Nashi3, Seena Vengalil3, Rashmi Santhosh4, Gayathri Narayanappa4, Mohammed Faruq2, Atchayaram Nalini5
1Clinical Neurosciences, Neurology, National Institute of Mental Health and Neurosciences (NIMHANS), Bangalore, IN;2Institute of Genomics and Integrative Biology, New Delhi, IN;3Neurology, National Institute of Mental Health and Neurosciences (NIMHANS), Bangalore, IN;4Neuropathology, National Institute of Mental Health and Neurosciences (NIMHANS), Bangalore, IN;5Neurology, National Institute of Mental Health and Neurosciences, IN
Background: Inherited myopathies and muscular dystrophies are a genetically heterogenous group of neuromuscular disorders which often present with overlapping clinical and pathological phenotypes. Although muscle histochemistry, immunostaining, western-blot and electron microscopy techniques offer important clues, these are often found insufficient to provide a conclusive diagnosis. With rapid evolution of gene discovery, accurate molecular diagnosis is essential for these disorders to provide appropriate genetic counselling, prognostication and identification of disease burden essential for future therapeutic research. Targeted Next generation sequencing (NGS) with gene panels are emerging as quick low-cost methods to diagnose mendelian disorders.
Methods: Randomly selected patients with clinical features suggestive of Limb girdle muscular dystrophies (LGMD), distal myopathies (DM) and congenital myopathies (CM) evaluated over last 10 years in our Neuromuscular clinic were taken up for genetic analysis by Targeted NGS with a gene panel constituting 53 known muscular dystrophy and myopathy genes. DNA extracted from blood was utilized for library preparation as per Agilent SureselectQXT protocol followed by sequencing on HIseq Illumina platform.
Results: We performed Target NGS in 45 patients with mean age of 21±11.7 (3-49 years) and male/female ratio of 3:2. Broad clinical phenotypes included LGMD: 31 (68.9%), CM: 9 (20%), DM: 5 (11.1%). Majority of cases were sporadic: 33 (73.3%), while family history was present in 12 (26.7%): Autosomal dominant inheritance in 7, recessive history in 3 and suspected X-linked inheritance in 2 families. Serum Creatine kinase was elevated in 82.2% (>5 times: 22, <5 times: 15). Muscle biopsy reports available in 42 patients showed muscular dystrophy in 25 (60.9%). Myopathic features were reported in 17 patients showing varying degree of cytoplasmic inclusions in 9 cases and rimmed vacuoles in 4. Putative disease-causing variants in 17 genes were identified in 31 patients (68.8%) and diagnosis was possible in 21 (46.66%) where clinical suspicion was supportive, and variants were either reported pathogenic or novel rare variants (MAF=0) recognized as damaging/probably damaging by in-silico predictions (Table 1). In another 10 cases, variants were identified with uncertain significance/not exactly correlating with phenotype and need to be further characterized. Most common genes affected were CAPN3 (Calpain-LGMD2A) in 5 patients followed by DYSF (Dysferlin-LGMD2B), EMD (Emerin-EDMD) in 4 cases each, RYR1 (Ryanodine receptor1-CM) in 3 and GMPPB (LGMD 2T), LMNA (LGMD 1B/EDMD) in 2 each.
Conclusion: Targeted gene panel NGS offers a quick cost-effective strategy with good diagnostic yield in inherited muscle disorders, when complimented with proper clinical phenotyping.
| S.No | Gender | Age | Clinical phenotype | Gene | Variant | Zygosity | Type |
| 1 | F | 19 | LGMD 2A (Agarwal community) | CAPN3 | exon19:c.2051-1G>T, exon22:c.G2338C:p.D780H | Compund heterozygous | Reported |
| 2 | F | 15 | LGMD | CAPN3 | exon17:c.1962delC:p.R655Gfs*6 | Homozygous | Novel |
| 3 | M | 17 | LGMD (consanguinity with dominant family history) | CAPN3 | exon17:c.1962delC:p.R655Gfs*6 | Homozygous | Novel |
| 4 | M | 18 | LGMD | CAPN3 | exon10:c.G1343A:p.R448H, exon20:c.A2120G:p.D707G | Compund heterozygous | Reported |
| 5 | F | 14 | LGMD 2A (Agarwal community) | CAPN3 | exon19:c.2051-1G>T, exon22:c.G2338C:p.D780H | Compund heterozygous | Reported |
| 6 | F | 26 | LGMD, early contractures, CK: Mild elevated (2X) | COL6A1 | exon29:c.1822+2T>C (splice site) | Homozygous | Novel |
| 7 | F | 12 | Cong. Myopathy with Dilated Cardiomyopathy | DES | exon1:c.C448T:p.R150X | Homozygous | Novel |
| 8 | M | 20 | ?BMD (X-linked history- multiple males affected in 3 generations) | DMD | exon72:c.C10255T:p.Q3419X | Hemizygous | Reported |
| 9 | M | 29 | HyperCkemia with mild limb girdle weakness | DYSF | exon18:c.1638+1G>A (splice site) | Homozygous | Novel |
| 10 | F | 24 | LGMD | DYSF | exon9:c.G895A:p.G299R | Homozygous | Reported |
| 11 | M | 20 | EDMD | EMD | exon2:c.C123A:p.Y41X | Hemizygous | Novel |
| 12 | M | 21 | EDMD (X-linked history) | EMD | exon6:c.651–655del:p.Q219Sfs*28 | Hemizygous | Novel |
| 13 | M | 9 | EDMD | EMD | exon4:c.266-2A>G (splice site) | Hemizygous | Reported |
| 14 | M | 13 | EDMD | FHL1 | exon7:c.C862G:p.R288G | Hemizygous | Novel |
| 15 | M | 8 | LGMD | FKRP | Exon4: c.C638T: p.P213L | Homozygous | Reported |
| 16 | F | 35 | LGMD with fatigability | GMPPB | exon9:c.G1000A:p.D334N | Homozygous | Reported |
| 17 | M | 49 | LGMD | GMPPB | exon9:c.G1000A:p.D334N | Homozygous | Reported |
| 18 | M | 38 | Proximo-distal weakness, Muscle MRI: severe involvement of medial gastrocnemius with sparing of soleus and rectus femoris | LMNA | exon2:c.369delG:p.E124Rfs*3, exon2:c.G370C:p.E124Q | Compund heterozygous | Novel |
| 19 | M | 20 | LGMD with axial contractures, Cardiomyopathy (AD family history), Muscle MRI: severe involvement of medial gastrocnemius with sparing of soleus and rectus femoris. | LMNA | exon7:c.G1318A:p.V440M | Heterozygous | Reported |
| 20 | F | 10 | Cong. Myopathy | RYR1 | exon27:c.C3725T:p.P1242L, exon39:c.G6502A:p.V2168M (reported) | Compund heterozygous | Novel |
| 21 | F | 19 | Cong. Myopathy, Muscle MRI: Selective semitendinosus involvement | TTN | exon308:c.98702–98703del:p.S32901fs*0 | Homozygous | Novel |
PS1Group1-017 / #849L-CITRULLINE AND METFORMIN DELAY MUSCLE DEGENERATION IN DUCHENNE MUSCULAR DYSTROPHY: RESULTS FROM A ANDOMISED CLINICAL TRIAL
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Patricia Hafner1, Ulrike Bonati1, Andrea Klein2, Daniela Rubino1, Vanya Gocheva1, Simone Schmidt1, Vincent Laugel3, Andrea Capone4, Monika Gloor5, Oliver Bieri5, Thomas Zumbrunn6, Nuri Gueven7, Dirk Fischer8
1Division Of Paediatric Neurology, University Children’s Hospital Basel, Basel, CH;2Division Of Paediatric Neurology, University of Berne Hospital, Berne, CH;3Strasbourg University Hospital, Strasbourg, FR;4Division Of Paediatric Neurology, Children’s Hospital Aarau, Aarau, CH;5Department Of Radiology, University of Basel Hospital, Basel, CH;6Department Of Clinical Research, University of Basel Hospital, Basel, CH;7Pharmacy, School Of Medicine, University of Tasmania, Hobart, AU;8Division Of Neurology, Kantonsspital Bruderhlz, Bruderholz, CH
Background: Sufficient therapeutic efficacy in Duchenne muscular dystrophy (DMD) has not been achieved yet and current therapeutic management still remains supportive. Recent evidence suggests that the loss of dystrophin in DMD not only disturbs cytoskeletal integrity but also cell metabolism resulting in reduced intracellular nitric monoxide (NO) levels. This phase IIa clinical trial assessed the efficacy and safety of the NO enhancing components L-citrulline and metformin to protect against muscle degeneration in ambulant patients with DMD. Methods: 49 ambulant DMD patients aged between 6.5 and 10 years were screened and 47 randomly allocated in a 1:1 ratio to receive daily treatment with oral L-citrulline (2.5g three times a day) and metformin (250mg three times a day) or placebo for 26 weeks. Co-medication with glucocorticoids was allowed if started 6 months prior to randomisation. Primary endpoint was the change of motor function measure (MFM) D1 subscore from baseline to week 26 in the modified intention to treat (mITT) patient set. Secondary endpoints included the total MFM score and its subscores; 6 min walking distance; timed function tests, quantitative muscle testing, quantitative muscle MRI assessments and biomarker laboratory analysis. Two subgroups were defined by a 6 min walking distance (6MWD) of ≥350m (stable) and < 350m (unstable) at baseline. Results: 45 of the 47 randomised patients (mean age 8.20 [1.07] years; 45 [100%] boys) completed the study. At week 26, L-citrulline and metformin treatment improved the MFM D1 decrease compared to placebo non-significantly (mean difference 5.54% [95% CI -1.00 to 12.09]; P =.095). Significant differences in favor of the L-citrulline and metformin treatment were observed in the stable subgroup with a baseline 6 MWD of ≥ 350m (mean MFM D1 difference of 6.81% [95% CI 1.00 to 12.26]; P =.024) and on both quantitative MRI sequences to assess muscle degeneration (mean fat fraction difference -1.79 [95% CI -3.26 to – 0.32]; P =.018; mean T2 relaxation time difference -1.33 [95% CI -2.40 to – 0.26]; P = 0.016). The treatment with L-citrulline and metformin was well tolerated, with stomach-ache, a known side effect of metformin, as the most frequent adverse event. Conclusion: The non-significant difference of the D1 in the mITT in favor of L-citrullin and metformin is clinically meaningful, but the study is underpowered.
PS1Group1-018 / #814MUTATION ANALYSIS IN MLPA NEGATIVE DUCHENNE MUSCULAR DYSTROPHY: NGS AS A DIAGNOSTIC TOOL PRIOR TO MUSCLE BIOPSY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Kiran Polavarapu1, M K. Saroja2, Veeramani Preethish-Kumar1, Deepah Sekar3, Saraswati Nashi4, Seena Vengalil4, Priya T. Thomas5, Sudha N. Rao2, Atchayaram Nalini6
1Clinical Neurosciences, Neurology, National Institute of Mental Health and Neurosciences (NIMHANS), Bangalore, IN;2R&d Division, Genotypic Technology, Bangalore, IN;3NIMHANS, Bengaluru, Bengaluru, IN;4Neurology, National Institute of Mental Health and Neurosciences (NIMHANS), Bangalore, IN;5Department Of Psychiatric Social Work, National Institute of Mental Health and Neuro Sciences, Bangalore, IN;6Department Of Neurology, National Institute of Mental Health and Neuro Sciences, Bangalore, IN
Background: Duchenne muscular dystrophy (DMD) is the most severe and common form of childhood muscular dystrophies resulting from mutations in Dystrophin gene (Xp21), affecting 1 in 3500 boys worldwide. While 75% patients have deletions/duplications of one or more exons, smaller mutations - point mutations, insertion/deletions (INDELs) etc., occur in 20-25% who require direct sequencing of entire gene or invasive muscle biopsy for diagnostic confirmation. Next generation sequencing (NGS) offers a cheaper and higher throughput alternative to traditional Sanger sequencing and can be developed as a single diagnostic platform to identify both copy number variations (CNVs) and small mutations in DMD patients. Methods: Clinically suspected and/or biopsy confirmed DMD children in whom MLPA (Multiplex ligation-dependent probe amplification) did not identify deletions/duplications of exons were recruited from our Neuromuscular clinic. Custom probe design for DMD gene was created using Agilent’s SureDesignTM tool with a total capture size of 2.077Mbp to cover entire gene (exons, introns and promoter regions) at least twice. After obtaining informed consent/assent, DNA extracted from blood was used to prepare libraries and sequenced on NextSeqTM(Illumina). Results: Mutational analysis was performed in 64 MLPA negative DMD children with mean age of 7.87±2.3 (range:3-13 years). Majority were sporadic cases and family history (X-linked recessive) is present in only six cases (9.37%). Biopsy was performed in 51 boys (79.7%) and showed loss of Dystrophin on immunohistochemistry. NGS identified hemizygous mutations of DMD gene in 58/64 children (90.6%). Nonsense mutations were most common at 54.7% (35/64) followed by frameshift mutations due to small INDELs 21.9% (14/64). Small INDELs of up to 10 bases were identified accurately. Variants affecting splicing occurred in 8/63 (12.5%) out of which six involved invariant GT and AG dinucleotides at the 5’ end (splice donor) and 3’ end (splice acceptor) of introns. Missense mutation was identified in only one patient. Except for one splice mutation where variation occurred nine bases upstream of exon 12, all other variants were classified as pathogenic (55) or likely pathogenic (2) as per ACMG guidelines. In total there were 57 unique variants among which 60% (34) were novel and only one mutation (p.Arg539*) recurred in two unrelated patients. Unlike larger CNVs, which occur predominantly in exons 45–55 and 2–10 for deletions and duplications respectively, small mutations lacked any hot spot regions and more uniformly spread across coding region with exons 30 and 44 having most number of mutations (4 each). In thirteen patients who did not undergo biopsy and clinical suspicion was high, we were able to accurately identify pathogenic mutations. Mutations were not identified in only six cases (9.5%), where possibility of deep intronic variants/complex rearrangements should be considered. Conclusion: In this study we describe spectrum of smaller mutations in a large cohort of DMD children from India. Accurate diagnosis of these mutations is important to identify potential cases who can benefit from mutation specific therapies like nonsense read-through and NGS offers a valuable diagnostic screening tool before contemplating invasive muscle biopsy in clinically suspected DMD patients. Acknowledgement: This study is funded by DST-SERB (Govt. of India) project: EMR/2014/000943
PS1Group1-019 / #756EZUTROMID SIGNIFICANTLY REDUCES MUSCLE DAMAGE IN DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Francesco Muntoni1, Gary Layton2, Indranil Bhattacharya3, Krista Vandenborne4, Crystal Faelan5, Anne Heatherington3, David Roblin6, Ruth Osahon6, Jon Tinsley7, Kay Davies8
1Dubowitz Neuromuscular Unit, UCL Great Ormond Street Institute of Child Health, London, GB;2Summit Therapeutics Inc., Abingdon, AL, GB;3Summit Therapeutics Inc., Cambridge, MA, US;4University of Florida, Gainsville, FL, US;5Flagship Biosciences, Westminster, CO, US;6Summit Therapeutics Inc., Abingdon, GB;7Summit Therapeutics Plc, Abingdon, GB;8university of oxford, oxford, GB
Background: To share interim 24-week safety and efficacy results from PhaseOut DMD evaluating ezutromid, a potential first-in-class utrophin modulator for the treatment of all patients with Duchenne muscular dystrophy (DMD). Utrophin is a naturally occurring protein that may act as a functional replacement for dystrophin. Increasing the production of utrophin in mature muscle fibres protects and improves outcome in preclinical models, and could modify the course of DMD. Methods: PhaseOut DMD is a Phase 2 open-label study of ezutromid administered to 40 ambulatory patients with DMD. Primary endpoints (48 week) are magnetic resonance spectroscopy (MRS) assessments. Key secondary endpoints are evidence of target engagement and quantification of muscle damage on muscle biopsies; collected at baseline (n=40) and week 24 (n=25) or 48 (n=15). Safety, pharmacokinetics, and functional measures are monitored throughout. Safety monitoring included collection of treatment emergent adverse events (TEAE), electrocardiogram (ECG), echocardiogram (ECHO), and laboratory parameters. An interim 24-week analysis was planned to assess efficacy and safety compared to baseline, with a focus on changes in the degree of muscle pathology as monitored by muscle biopsy. Results: A statistically significant reduction in %fibres expressing developmental myosin (MHCd) was observed (from 11.37% to 8.76%, least mean square difference (LSMD) -2.62% CI, -4.33, -0.90); a relative reduction of 23%. Mean utrophin intensity increased (relative +7%) (0.370 to 0.396, LSMD 0.026 95% CI, -0.005, 0.058). MRS indicated a statistically significant decrease in water relaxation time (T2) in the soleus muscle: the average reduction was -0.86 milliseconds (95% confidence intervals -1.44, -0.28), which was from 31.85 milliseconds at baseline to 30.99 milliseconds at 24 weeks, (n=38). Further, an increase in mean fat fraction (vastus lateralis: from 14.7% to 18.5%, n=37) was noted. No patient lost ambulation and there were no meaningful changes in functional performance (eg 6MWD: 404m at baseline, 395m at 24 weeks, n=39). All patients achieved ezutromid plasma levels sufficient to modulate utrophin. Up to Week 24, none of the subjects had any clinically significant TEAE, and no subject withdrew due to safety issues. Only one subject experienced an SAE, which resolved within a day with no dosage day missed, whilst the subject remained in the study. In addition, there was no relevant prolongation of the corrected QT interval on the ECG, or deterioration of left ventricular ejection fraction or fractional shortening. There were no clinically significant laboratory abnormalities related to ezutromid use. Ezutromid continues to be safe and well tolerated. Conclusion: Decreased MHCd, a marker of regeneration, indicates reduction in muscle damage. Utrophin levels were increased which, with decreasing MHCd, provides evidence of target engagement. MRS T2 results in this age group are consistent with a reduction in muscle inflammation and/or damage. Ezutromid has continued to be safe and well-tolerated after prolonged dosing across a wide exposure range. These interim results support utrophin modulation as a potential treatment for DMD patients irrespective of their mutation.
PS1Group1-020 / #698NUMBER NEEDED TO TREAT IN SPINAL MUSCULAR ATROPHY TYPE 1 WITH AVXS-101 RELATIVE TO NUSINERSEN
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Omar Dabbous1, Martin Cloutier2, Annie Guerin2, Irina Pivneva2, Eric Q. Wu2, Douglas M. Sproule3
1AveXis, Inc., Bannockburn, AL, US;2Analysis Group, Inc., Boston, MA, US;3AveXis, Inc., Bannockburn, IL, US
Background: To assess the number needed to treat (NNT) to prevent death and use of permanent assisted ventilation and to improve motor function with AVXS-101 compared to nusinersen in patients with spinal muscular atrophy type 1 (SMA1). Methods: Patients diagnosed with SMA1 were treated in clinical trials with AVXS-101 (NCT02122952; study cohort 2; N=12) or nusinersen (NCT02193074; N=80). Trial duration was up to 24 months for AVXS-101 (median = 24.1 months) and up to 13 months for nusinersen (median = 9.2 months). NNT with AVXS-101 compared to nusinersen was assessed for survival and event-free survival (absence of death and use of permanent assisted ventilation) at last visit, and for motor function (increase of ≥4 points in Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders [CHOP-INTEND] score from baseline) at last visit and at a median of 9 months. For the nusinersen trial, CHOP-INTEND score was measured for patients with ≥6 months of follow-up (N=73). The NNT to prevent one death, one event (death or use of permanent assisted ventilation), or for one patient to improve motor function relative to nusinersen was calculated as the reciprocal of the difference between AVXS-101 and nusinersen in event rates or motor function achievement rates. Results: Patient mean age at first dose was 3.4 (0.9-7.9) and 5.3 (1.7-7.9) months in the AVXS-101 and nusinersen trials. NNT analyses showed that treating 6.2 patients with AVXS-101 instead of nusinersen would prevent 1 more death over the course of the study interval; treating 2.6 patients with AVXS-101 versus nusinersen would prevent 1 more event; and treating 3.5 patients with AVXS-101 versus nusinersen would allow 1 more patient to improve motor function (at last visit and at a median of 9 months). Conclusion: Efficacy in preventing death and use of permanent assisted ventilation and in improving motor function is much higher with AVXS-101 versus nusinersen.
PS1Group1-021 / #821A NOVEL NEXT-GENERATION THERAPY FOR POMPE DISEASE WITH IMPROVED EFFICACY IN MICE
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Su Xu, Yi Lun, Rebecca Soska, Anju Nair, Michelle Frascella, Anadina Garcia, Abdul S. Ponery, Jessie Feng, Cecilia Della Valle, Russell Gotschall, Hung Do, Kenneth J. Valenzano, Richie Khanna
Amicus Therapeutics, Cranbury, US
Background: Pompe disease is a lysosomal storage disorder that results from deficiency in acid a-glucosidase (GAA) activity, and is characterized by progressive accumulation of lysosomal glycogen in cardiac and skeletal muscles. Currently, the only approved treatment is enzyme replacement therapy using biweekly infusions of a recombinant human GAA (rhGAA), alglucosidase alfa. Although alglucosidase alfa provides some clinical benefits, the infused enzyme shows insufficient uptake into key disease-relevant skeletal muscles, mainly due to sub-optimal levels of mannose-6-phosphate (M6P), a carbohydrate that binds cation-independent M6P receptors at the cell surface, resulting in enzyme internalization and lysosomal targeting. Methods: We have developed a proprietary, novel rhGAA, ATB200, that has optimized M6P content due to its combination of a specialized cell line and an optimized manufacturing/purification process. Moreover, we are developing ATB200 to be co-administered with a small molecule pharmacological chaperone, AT2221 (miglustat, N-butyl-deoxynojorimycin), as a next-generation therapy for Pompe disease. Results: ATB200 use led to substantially greater lysosomal targeting compared to alglucosidase alfa following intravenous injection to Gaa knock-out (KO) mice. Our data indicate that AT2221 stabilizes ATB200 in blood ex vivo and improves its pharmacokinetic properties in vivo. In Gaa KO mice, co-administration of ATB200 with AT2221 (ATB200/AT2221) showed significantly greater glycogen reduction in key disease-relevant muscles compared to alglucosidase alfa. Repeat administrations of ATB200/AT2221 also substantially reduced lysosomal proliferation, reversed autophagic buildup, and increased muscle fiber size. The effects seen at the biochemical and cellular levels ultimately translated into improved muscle function in Gaa KO mice compared to the standard of care. Conclusion: Collectively, these data demonstrate the superiority of ATB200/AT2221 over the current approved therapy in these preclinical studies, warranting further investigation as a treatment of Pompe disease.
PS1Group1-022 / #360THE MULTIPLE FACES OF ANTI-HMGCR ANTIBODY-RELATED MYOPATHIES
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Pegah Masrori1, Willem De Ridder2, Jonathan Baets1
1Neuromuscular Reference Centre, Departement Of Neurology, Antwerp University Hospital, Edegem, BE;2Neuromuscular Reference Centre, Departement Of Neurology, Antwerp University Hospital, Edegem, BE
Background: Anti-HMGCR antibody-associated myopathies define a distinct and clinically diverse subset of idiopathic inflammatory myopathies (IIM). The most typical clinical presentation consists of subacute progressive proximal muscle weakness with moderately to severely elevated serum creatine kinase (CK) levels and myopathic electromyography findings. The muscle biopsy usually shows features of a necrotising myopathy, evident by myofibre necrosis and regeneration, with minimal or absent lymphocytic infiltrates and major histocompatibility complex I (MHC-I) expression on non-necrotic muscle fibres. As for most other IIMs, high-dose corticosteroids is considered the first-line treatment but most cases are refractory to conventional steroid monotherapy. Other immunosuppressive therapy such as methotrexate, azathioprine, rituximab and cyclophosphamide are increasingly being used. Intravenous immunoglobulin (IVIg) and plasmapheresis may be required. Associations have been described with statin exposure and some genetic risk factors. Exact pathomechanisms and potential triggers of the disorder particularly still need to be identified in patients not exposed to statins. Methods: Myositis specific antibodies (immunodot assay covering 16 auto-antibodies, including anti-HMGCR) were systematically assessed in a cohort of 40 IIM patients. Patients with anti-HMGCR antibodies were selected for specific follow-up. CK levels were repeatedly measured as well as auxiliary laboratory findings and an electromyography (EMG) was performed. Muscle biopsies were obtained for all patients from quadriceps muscle and analysed following standard histologic and immunohistochemical techniques for light microscopy. Whole-body muscle magnetic resonance imaging (MRI) was performed on a 1.5T MRI platform. Results: We present five patients with an anti-HMGCR antibody-related myopathy. Firstly, we found that the presenting symptoms are highly variable ranging from subacute severe proximal weakness to more insidious complaints linked to subtle proximal weakness. Statin use is only associated in one patient but viral infection preceding the myopathy is noted in three patients. CK levels are consistently highly elevated (above > 5000 U/L). EMG confirmed myopathic findings, muscle MRI imaging illustrated oedema in different muscle groups and muscle biopsy showed often relatively subtle abnormalities mainly consisting of fibre necrosis with myophagic reaction in the absence of florid inflammatory changes. The immune-mediated aetiology of the disease could however be confirmed immunohistochemically in all patients. Initial treatment was based on conventional steroid monotherapy with highly variable response among the patients reported ranging from significant clinical and biochemical improvement to complete refractoriness in others. As needed, therapy was escalated with second and third-line agents and the responses will be discussed in detail. Conclusion: Our findings highlight the clinical, histopathological and prognostic heterogeneity of anti-HMGCR antibody-related myopathies and point-out potential diagnostic and therapeutic pitfalls of this treatable myopathy. Based on our findings, we explore the role of viral infections in the pathophysiology of this disorder.
PS1Group1-023 / #1010A PLURIPOTENT STEM CELL-DERIVED MODEL OF MEROSIN DEFICIENT CONGENITAL MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Kenyon Lyon1, Amanda Rickard2, Monica Hayhurst Bennett2, Uli Schmidt2
1College Of Engineering, California Polytechnic State University, San Luis Obispo, US;2Genea Biocells, San Diego, CA, US
Background: Congenital muscular dystrophies (CMD) are a group of early onset, often very severe forms of muscle disorders. Congenital muscular dystrophy type 1A (MDC1A) accounts for about 40% of cases of CMD and has an estimated prevalence of 7/1,000,000. MDC1A is an autosomal recessive disease caused by mutations in the LAMA2 gene encoding the a2 chain of laminin-211. Laminin-211 is a major component of the skeletal muscle basement membrane that stabilizes muscle and facilitates extracellular signaling. MDC1A is a life-threatening disease characterized by hypotonia, muscle weakness, scoliosis, joint contractures and respiratory difficulties. Currently, there is no treatment available for MDC1A. Human pluripotent stem cells (hPSCs) represent a renewable source of skeletal muscle cells for disease modeling and therapeutics screening and serve as an attractive alternative to primary cultures which are more variable and only undergo limited population doublings. In these studies, MDC1A affected hPSCs were differentiated to skeletal muscle and examined for disease-relevant phenotypes. Methods: Genea Biocells’ published protocol was used to differentiate both healthy and MDC1A affected hPSCs into mature skeletal muscle myotubes and examined for MDC1A-specific defects. Results: MDC1A-affected hPSCs differentiated into multinucleated myotubes and expressed myogenic markers. We have identified a variety of clinically relevant MDC1A phenotypes that are amenable to high-throughput screening and therapeutics testing, including impaired metabolic function and mitochondrial defects. We have also detected increased activation of apoptosis-related markers in MDC1A-affected cells. Moreover, we detected defects in myogenesis through high-content imaging for myogenic markers throughout the differentiation process. Conclusion: We have developed and characterized the world’s first pluripotent stem cell-derived model of MDC1A, a powerful tool for studying MDC1A pathophysiology and for use in the development of effective treatments for CMDs.
PS1Group1-024 / #1006AN IN VITRO MODEL OF MYOTONIC DYSTROPHY TYPE 1 USING HUMAN EMBRYONIC STEM CELL-DERIVED SKELETAL MUSCLE
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Ernesto Solana-Guizar1, Samuel Labarge2, Monica Hayhurst Bennett2, Uli Schmidt2
1California State University San Marcos, San Marcos, CA, US;2Genea Biocells, San Diego, CA, US
Background: Myotonic Dystrophy Type 1 (DM1) is the most common form of adult-stage onset muscular dystrophy with an estimated prevalence of 1/8,000 worldwide. Patients affected by DM1 display a range of symptoms (e.g. muscle wasting, intellectual disability, and infertility). DM1 is an autosomal-dominant, multisystemic genetic disorder characterized by CTG triplet repeat expansion in the 3’ untranslated region of the dystrophia myotonica protein kinase (DMPK) gene, with larger repeat size directly correlating to higher disease severity. The CTG repeat expansion leads to sequestration of splicing regulators (such as Muscleblind Like Splicing Regulator 1 [MBNL1]) in nuclear foci containing DMPK mRNA, and subsequent splicing defects (e.g. exon inclusions and exclusions). In this study, we differentiated unaffected control and DM1-affected human pluripotent stem cell lines to skeletal muscle using Genea Biocells’ published protocol and investigated their phenotypes with the aim to establish a clinically relevant stem cell-based disease model of DM1. Methods: Unaffected control and DM1-affected (Genea067 [1626/1773 CTG], Genea157 [1060 CTG], Genea158 [180 CTG]) human embryonic stem cell lines were differentiated into skeletal muscle (i.e. myoblasts and myotubes) according the manufacturer’s protocol and analyzed for the following: (1) Lineage commitment and differentiation kinetics of myoblasts to terminally mature myotubes was examined and quantified. (2) MBNL1 nuclear foci were visualized and quantitated by high content imaging. (3) At the molecular level, splice variants were analyzed with semi-quantitative PCR and Nanostring gene expression analysis technology. (4) We investigated cellular metabolism and energetics using Seahorse Technologies’ XF Cell Mito Stress test to understand ATP production, as well as basal and maximal respiration. Results: (1) We found that affected cells display aberrant myotube formation, as previously reported in animal models and the congenital (infant-onset) form of DM1. (2) We observed a direct correlation between disease severity and MBLN1 foci intensity. (3) We observed impaired splicing in ALPK3, ATP2A1, and CACNA1S between unaffected and Genea067 day 3 myotubes. (4) We found an inverse relationship between disease severity, i.e. number of CTG repeats, and mitochondrial respiration. Conclusion: Together, this work demonstrates the usefulness of DM1-affected human pluripotent stem cell-derived skeletal muscle to accurately model myotonic dystrophy type-1 in vitro, recapitulating known phenotypes that correlate with disease severity as well as tissue-specific mechanisms. This model represents a new and attractive tool to develop and test DM-1 muscle-targeted therapeutics.
PS1Group1-025 / #827DIMETHYLFUMARATE (DMF) DOWNMODULATES INFLAMMATION IN SKELETAL MUSCLE
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Karsten Schmidt1, Simranjeet Kaur1, Alice Faust1, Peter Balcarek2, Jens Schmidt1
1Department Of Neurology, University Medical Center Göttingen, Göttingen, DE;2Department Of Trauma Surgery, Orthopaedics And Plastic Surgery, University Medical Center Göttingen, Göttingen, DE
Background: The pathogenesis of IBM is caused by degenerative and inflammatory mechanisms. Various immunosuppressive therapies have failed to show a significant beneficial effect in clinical studies. DMF is an anti-inflammatory drug used in multiple sclerosis and psoriasis. It is known to diminish the cellular stress response to pro-inflammatory stimuli and some of these effects are mediated by activation and nuclear translocation of NRF-2. This study aims to evaluate the effect of dimethylfumarate (DMF) in in vitro and in vivo model systems for inclusion body myositis (IBM). Methods: The effect of DMF was studied in vitro in a well-established pro-inflammatory cell culture model system originated from a myoblast cell line and cultured primary muscle cells on RNA and protein level using quantitative PCR, immunocytochemistry and western blotting. For in vivo studies, a previously characterized double-transgenic mouse strain was used that harbours human mutated amyloid precursor protein (APP) and presenilin (PS)-1 under muscle specific control via the creatin kinase (CK) promotor. The mice were fed by oral gavage with 15, 30 or 45μg DMF or vehicle twice per day for 14 days, followed by a pro-inflammatory stimulus via injection of LPS compared to saline into the calf. Animals were sacrificed after 48 hours and the calf muscles were analyzed by qPCR, immunohistochemistry and ex vivo flow cytometry. Results: In muscle cell cultures, DMF led to an upregulation and nuclear translocation of NRF-2. DMF diminished the muscle inflammation in the mouse model of IBM (APP/PS1 transgenic mice) upon intramuscular injection of LPS compared to controls. mRNA expression analysis by qPCR demonstrated a modest effect on pro-inflammatory cytokines in DMF treated mice compared to controls. Conclusion: The data suggest that DMF is active in skeletal muscle by translocation of NRF-2 and upregulation of associated genes. Moreover, a significant reduction of LPS-induced inflammation was noted in mice treated with DMF compared to controls in double-transgenic mice (APP/PS1).
PS1Group1-026 / #878TAMOXIFEN IN DMD: RATIONALE AND PROTOCOL FOR A MULTICENTRE, RANDOMISED, DOUBLE-BLIND, PLACEBO-CONTROLLED, PHASE 3 TRIAL
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Simone Schmidt1, Anna-Lena Orsini1, Patricia Hafner1, Daniela Rubino-Nacht1, Olivier M. Dorchies2, Andres Nascimento3, U Schara4, Stefan Spinty5, Haluk Topaloglu6, Dirk Fischer1
1University Children’s Hospital Basel, Basel, CH;2School Of Pharmaceutical Sciences, University of Geneva, Geneva, CH;3Hospital Sant Joan de Déu, Barcelona, ES;4Universitaetsklinikum, Essen, DE;5Alder Hey Children’s Hospital, Liverpool, GB;6Hacettepe Children’s Hospital, Ankara, TR
Background: Currently, only symptomatic treatment with glucocorticoids is available in Duchenne muscular dystrophy (DMD) which have limited efficacy but many adverse effects. Most current research on therapeutics of DMD focuses on correcting the gene defect. However, as there are more than 250 mutations in the human dystrophin gene, this approach will treat only a small percentage of patients and will be expensive. Using the mouse DMD model it could be shown that tamoxifen (TAM), given orally for periods of 2 or 15 months at doses as low as 0.3mg/kg/day, results in almost full recovery of force and structure of muscles. TAM is one of the most efficacious drugs ever investigated in an animal model of DMD. Our aim is to investigate whether TAM treatment, compared to placebo, reduces the disease progression in DMD patients. Methods: We are setting up an international (France, Germany, Greece, Spain, Switzerland, United Kingdom, The Netherlands, Turkey) randomised double blind placebo controlled 48-week clinical trial with a core population (group A) of 79 ambulant 6.5 to 12 years old DMD patients that are on a stable standard treatment with glucocorticoids. Furthermore, we will include 16-20 non-ambulant patients age 10 to 16 years who do not receive glucocorticoids (parallel group B) to obtain efficacy and safety data in a broader DMD population. All patients will receive 20 mg of TAM or placebo once daily over 48 weeks. The primary outcome for ambulant DMD patients (group A) is the motor function measure (MFM) subscore D1 (standing and transfer). In the second patient population (non-ambulant patients, group B) the MFM D2 subscore is the primary endpoint, allowing extrapolation of D2 data from the group A (ambulant patient) population. Secondary outcomes include the total MFM and subscores D2 and D3, the North Star Ambulatory Assessment (NSAA), timed function tests (TFT) including 6 minute walking distance (6MWD), proximal upper limb function (PUL), and quantitative muscle testing (Grip force). In addition, to investigate whether longterm 48-week TAM treatment can slow muscle degeneration, quantitative thigh muscle magnetic resonance imaging will be performed. Results: The study aims to describe an efficacy and safety profile for tamoxifen in the treatment of DMD patients. Conclusion: According to the recommendations for trials in Duchenne and Becker muscular dystrophy released by the Committee for Medicinal Products for Human Use (CHMP) of the European Medicinal Agency (EMA/CHMP/236981/2011, Corr. 1), muscle function and muscle force should be evaluated to assess the effect of any studied intervention in muscular dystrophies. The purpose of this study is to evaluate if tamoxifen shows positive effects on muscle function and muscle force in comparison to placebo in patients with DMD. Recruitment is expected to start in May 2018.
PS1Group1-027 / #494MUSCLE HISTOPATHOLOGY IN INFANTILE DNM1L-RELATED MITOCHONDRIAL EPILEPTIC ENCEPHALOPATHY IS KEY FOR CLINICAL DIAGNOSIS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Enrico Bertini1, Daniela Verrigni2, Domenica Battaglia3, Lucia Fusco4, Lorenzo Figà Talamanca5, Rosalba Carrozzo2, Daria Diodato2, Adele D’Amico2, Laura Papetti4, Daniele Ghezzi6, Costanza Lamperti6
1Unit Of Neuromuscular Disorders, Laboratory Of Molecular Medicine, Bambino Gesu’ Children’s Hospital,, Rome, IT;2Unit Of Neuromuscular Disorders, Laboratory Of Molecular Medicine, Bambino Gesu’ Children’s Hospital,, Rome, IT;3Infantile Neuropsychiatric Unit, Fondazione Policlinico Universitario Agostino Gemelli, Rome, IT;4Division Of Neurology, Bambino Gesu’ Children’s Hospital,, Rome, IT;5Division Of Neuroradiology, Bambino Gesu’ Children’s Hospital,, Rome, IT;6Division Of Molecular Neurogenetics, Istituto Neurologico C. Besta, Milano, IT
Background: DNM1L (dynamin 1 like) encodes a member of the dynamin superfamily of GTPases and mediates membrane remodeling during a variety of cellular processes; in fact DNM1L has an important role in the maintenance of mitochondrial and peroxisomal morphology. Dysfunction of DNM1L is implicated in several neurological disorders and associated with either de novo-dominant mutations or compound heterozygous DNM1L mutations (MIM #603850),as well. Many patients develop refractory seizures, consistent with an epileptic encephalopathy, and thereafter show neurologic decline. The age at onset, features, and severity are variable, and some patients may not have clinical evidence of mitochondrial or peroxisomal dysfunction. Following the first description there has been increasing reports in the last 2 years on sporadic patients all affected by early onset encephalopathy with acquired microcephaly and drug-resistant seizures, progressive brain atrophy or abnormal brain development, optic atrophy, and persistent lactic acidemia related to de novo dominant-negative mutations. Methods: We collected 5 sporadic patients affected by early onset epileptic encephalopathy. Patients were studied by neuroimaging, skin and muscle biopsy and genomic DNA was analyzed by high-throughput sequencing using TruSight One panel (Illumina (San Diego, CA) in 3 patients while the remaing 2 patients were were analyzed using a custom gene panel for the screening of 230 genes associated with mitochondrial diseases.The muscle biopsies and cultures fibroblasts were processed by routine stainings, and immunofluorescence to detect the mitochondrial network arrangement. Results: Molecular genetic analysis disclosed heterozygous de novo DNML1 mutations in all 5 patients, and 3 of these mutations were novel. Unreported mutations were validated in a yeast model. Summarizing the clinical reports of the 5 patients, in all first symptoms appeared between the second and fifth year of life after normal developmental milestones occurring in the first year of life. The first manifestation consisted in prolonged treatment-resistant.generalized clonic seizures or hemiclonic seizures that shortly progressed into frequently recurrent epileptic mal status. The brain MRI showed at the beginning T2 hyperintensities in the posterior white matter regions and transient T2 hyperintensities in the cortical areas in 2 patients when performed serially. With time, the MRI showed moderate to severe global cerebral atrophy in all patients Histopathology of the muscle biopsy was revised in 4 patients and showed peculiar features: scattered fibers with a partial reduction of cytochrome c oxidase (COX) and succinate dehydrogenase (SDH) stain with aspects of polymorphic core like areas. These areas corresponded to reduced immunoreactive areas displayed by immunohistochemistry with antibodies recognizing TOMM20, defining abnormally distributed mitochondria. Biochemical spectrophotometric assay of the mitochondrial OXPHOS enzymes showed normal activities. In cultures fibroblasts we observed an increased filamentous network in mutant fibroblasts in 3 patients, while in one patient the mitochondrial network appeared as normal controls and in the last patient we observed a “chain-like” structure, not observed in controls but already reported in patients with biallelic mutations in DNM1L. Conclusion: We describe 5 sporadic patients affected by severe mitochondrial treatment-resistant seizures correlated with de novo heterozygous mutations in DNML1. The most consistent finding in all patients was a histopathological peculiar pattern
PS1Group1-028 / #731EFFECT OF A LONG-TERM TREATMENT WITH METFORMIN IN DYSTROPHIC MDX MICE: A RECONSIDERATION OF ITS THERAPEUTIC INTEREST IN DMD
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Paola Mantuano1, Roberta F. Capogrosso1, Francesca Sanarica1, Maria Grazia Morgese2, Michela De Bellis1, Anna Cozzoli1, Adriano Fonzino1, Elena Conte1, Giulia Maria Camerino1, Luigia Trabace2, Annamaria De Luca1
1Pharmacy - Drug Sciences, University of Bari “Aldo Moro”, Bari, IT;2Experimental And Clinical Medicine, University of Foggia, Foggia, IT
Background: The pharmacological stimulation of AMP-activated protein kinase (AMPK) via metabolic enhancers has recently emerged as a potential therapeutic strategy for Duchenne muscular dystrophy (DMD). Here we focused on metformin, a widely-prescribed anti-hyperglycemic drug which indirectly activates AMPK by inhibiting mitochondrial respiratory chain complex I. Recently, the potential synergistic action of metformin and NO-precursors has been proposed in clinical trials on DMD patients; a combination of metformin and L-arginine exerted positive effects on mitochondrial protein expression, in parallel slowing disease progression (NCT02516085). Despite these encouraging results, preclinical data supporting the rationale of metformin use in DMD are still poor. In addition, controversial actions of metformin on skeletal muscle have been reported. These observations made us questioning about the actual therapeutic potential of metformin as metabolic enhancer on dystrophic muscle in mdx mice and humans. In the present study, we assessed the effects of a long-term treatment with metformin in the exercised mdx mouse, characterized by severe mechanical-metabolic uncoupling and functional maladaptation, in order to gain more insight into the mechanisms of action of the drug and its potential efficacy in a metabolic state of dystrophic muscle closer to that observed in patients. Methods: Metformin was administered in drinking water at a dose of 200mg/kg for 20 weeks. A validated multidisciplinary in vivo and ex vivo approach was used to assess the impact of drug treatment on disease-related primary readouts. Results: We disclosed the ability of metformin to markedly ameliorate histopathology of mdx gastrocnemius (GC) muscle, significantly reducing total area of damage and, specifically, non-muscle area. This was accompanied by a significant reduction of pro-fibrotic TGF-ß1 in GC muscle (recovery score, r.s., 106%) and by a marked decrease of plasma MMP-9 level (r.s. 43%), both measured by ELISA tests. In parallel, metformin partially counteracted in vivo mdx weakness, detected by grip strength test, and significantly increased ex vivo specific twitch and tetanus of diaphragm (r.s. 28%), but not of EDL muscle, although this latter was partially protected by the treatment with respect to mechanical threshold, electrophysiological index of excitation-contraction coupling. However, these improvements were not related to a protective metformin action on dystrophic muscle metabolism, as shown by the modest effects exerted on the increase of pAMPK/AMPK ratio measured by western blot, on the expression of genes involved in mechanical-metabolic response measured by Real-time PCR, and by the lack of fast-to-slow fiber-type shift revealed by SDH staining on tibialis anterior muscle. Similar results were obtained in the milder phenotype of sedentary mdx mice. This limited efficacy could be related to the inability of metformin to increase the low muscle levels of L-arginine, L-citrulline and taurine in non-exercised/exercised mdx mice, as shown by HPLC analysis, suggesting the need of a synergic action on metabolic pathways for an efficient genetic or epigenetic effect of metformin. Conclusion: Our findings encourage the need to improve our knowledge on alternative, metabolism-independent mechanisms of action of metformin, to differently repurpose this drug for the treatment of DMD, further supporting its clinical use in combination with metabolism enhancers such as NO precursors.
PS1Group1-029 / #366IDENTIFICATION OF LATE-ONSET POMPE DISEASE AMONG HIGH-RISK POPULATION WITH NATIONWIDE SCREENING STUDY IN JAPAN
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Katsuhisa Ogata1, Motomichi Kosuga2, Eri Takeshita3, Tsuyoshi Matsumura4, Keiko Ishigaki5, Shiro Ozasa6, Hajime Arahata7, Kazuma Sugie8, Toshiaki Takahashi9, Satoshi Kuru10, Michio Kobayashi11, Hiroto Takada12, Ayako Hattori13, Masanori P. Takahashi14, Nobuyuki Tanaka15, Takashi Kimura16, Michinori Funato17, Torayuki Okuyama18, Hirofumi Komaki19
1Institute Of Clinical Research / Department Of Neurology, National Hospital Organization Higashisaitama Hospital, Hasuda, Saitama, JP;2Center For Lysosomal Storage Diseases / Department Of Clinical Laboratory Medicine / Division Of Medical Genetics, National Center for Child Health and Development, Setagaya, Tokyo, JP;3Department Of Child Neurology, National Center of Neurology and Psychiatry, Kodaira, Tokyo, JP;4Neurology, National Hospital Organization Toneyama National Hospital, Toyonaka, JP;5Department Of Pediatrics, Tokyo Women’s Medical University, Shinjuku, Tokyo, JP;6Department Of Pediatrics, Kumamoto University, Kumamoto, JP;7Department Of Neurology, National Hospital Organization Omuta Hospital, Omuta, Fukuoka, JP;8Department Of Neurology, Nara Medical University, Kashihara, Nara, JP;9Department Of Neurology, National Hospital Organization Sendai-Nishitaga National Hospital, Sendai, Miyagi, JP;10Department Of Neurology, National Hospital Organization Suzuka National Hospital, Suzuka, Mie, JP;11Department Of Neurology, National Hospital Organization Akita National Hospital, Yurihonjo, Akita, JP;12Department Of Neurology, National Hospital Organization Aomori Hospital, Aomori, JP;13Department Of Neonatology And Pediatrics, Nagoya City University, Nagoya, Aichi, JP;14Department Of Neurology, Osaka University, Suita, Osaka, JP;15Department Of Neurology, National Hospital Organization Shimoshizu National Hospital, Yotsukaido, Chiba, JP;16Department Of Neurology, National Hospital Organization Asahikawa Medical Center, Asahikawa, Hokkaido, JP;17Department Of Pediatrics, National Hospital Organization Nagara Medical Center, Gifu, JP;18Center For Lysosomal Storage Diseases / Department Of Clinical Laboratory Medicine, National Center for Child Health and Development, Setagaya, Tokyo, JP;19Department Of Clinical Research Promotion / Department Of Child Neurology, National Center Hospital, National Center of Neurology and Psychiatry, Kodaira, Tokyo, JP
Background: Late-onset Pompe disease (LOPD) is a rare, but treatable metabolic myopathy caused by deficiency of acid alpha-glucosidase (GAA). LOPD is characterized by progressive muscle weakness and/or respiratory failure, but some LOPD patients could be overlooked due to lack of typical signs. To evaluate the utility of dried blood spots (DBS) in the diagnostic work-up and assess the prevalence of LOPD within a Japanese high-risk population, we prospectively screened for LOPD in a Japanese cohort of undetermined myopathy patients aged one year or older with muscle weakness and/or elevated serum creatine kinase levels. Methods: Sixteen neuromuscular center hospitals, members of Muscular Dystrophy Clinical Trial Network (MDCTN), joined the Pompe disease high risk screening study in Japan (PHiRS-J). We tested GAA activity with DBS as first screening method. Patients with reduced GAA activity in DBS were re-analyzed for enzyme activity in lymphocytes, and investigated for GAA gene mutations. Results: Among 164 patients enrolled, two were incompatible with inclusion criteria. Eleven patients showed reduced GAA activity with DBS. Of these patients, three showed normal GAA enzyme activity in lymphocytes; five were revealed as pseudodeficiency; three with confirmed reduction of GAA activity in lymphocytes were identified to have homozygous or compound heterozygous mutations of the GAA gene. Conclusion: In a Japanese high-risk cohort, we found a prevalence of LOPD around 2%. This study suggests that screening of GAA activity with DBS is useful to diagnose patients with LOPD in a high-risk population.
PS1Group1-030 / #496CLINICAL COURSE OF ADULT POMPE DISEASE PATIENTS WHO DID NOT START OR DISCONTINUED ENZYME REPLACEMENT THERAPY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Harmke A. Van Kooten1, Laurike Harlaar1, Nadine A.m.e. Van Der Beek1, Pieter A. Van Doorn1, Ans T. Van Der Ploeg2, Esther Brusse1
1Department Of Neurology, Center For Lysosomal And Metabolic Diseases, Erasmus MC, University Medical Center Rotterdam, Rotterdam, NL;2Department Of Paedicatrics, Center For Lysosomal And Metabolic Diseases, Erasmus MC, University Medical Center Rotterdam, Rotterdam, NL
Background: Since the introduction of enzyme replacement therapy (ERT) for Pompe disease in 2006, the disease course of patients who did not start ERT, or who discontinued ERT has rarely been described. However, insight in the clinical course of these patients adds to the implementation and further development of guidelines on starting and stopping of ERT in adult Pompe patients. Methods: From our cohort of adult Pompe patients (n=127), we describe the clinical course of two groups; 1) those who have never been treated with ERT and 2) patients in whom ERT was discontinued for various reasons.
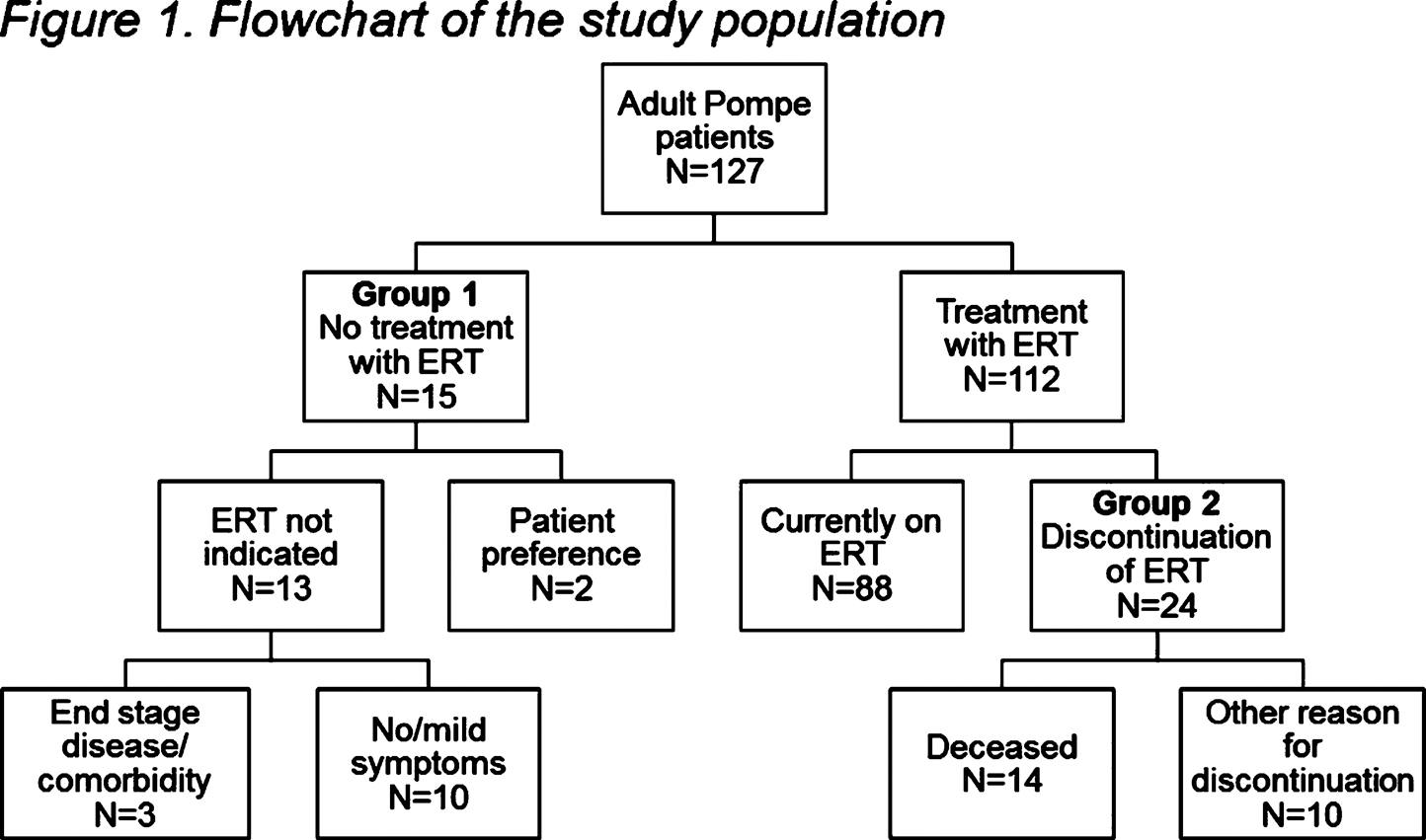
Figure 1 shows an overview of the study population. In group 1 (no ERT; n=15) 10 patients had no or only very mild symptoms; these patients remained clinically stable during follow-up (median 5.8 years, range 0.6 – 11.4 years). Two patients with moderate symptoms of Pompe disease preferred not to start ERT and three patients were too severely affected to start ERT or had life threatening comorbidity. In group 2 (discontinuation of ERT; n=24) ERT was ceased in 14 patients after death. In 10 patients, ERT was discontinued for other reasons: in five patients because of infusion related reactions, two patients chose to stop ERT because they experienced a high burden of the treatment regime, in one severely affected patient we stopped because of continued clinical deterioration under ERT, one patient stopped ERT to undergo treatment for lymphoma and in one patient we ended ERT because of non-compliance. Median treatment duration of the 10 patients who discontinued ERT was 2.1 years (range 0.3 – 14.6 years). ERT had been effective, improving or stabilizing motor and pulmonary function, in eight of them. For three patients sufficient follow up data after treatment cessation was available, with follow up duration of 1.3 – 8.0 years. After discontinuation of ERT we did not observe accelerated disease progression beyond the estimated natural progression rate in these three patients. Conclusion: Pompe patients with no or mild symptoms may show a stable disease course for many years, indicating that starting ERT could likely be postponed in this group of patients, under careful follow up. Importantly, in patients who discontinued ERT for various reasons, we did not observe accelerated disease progression beyond estimated disease progression rate. Both findings facilitate the process of shared decision making when considering start or discontinuation of ERT in adults with Pompe disease.
PS1Group1-031 / #314PROLONGED EXERCISE TEST IN PATIENTS WITH HISTORY OF THYROTOXICOSIS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Cheng-Yin Tan1, Hui-Ting Tan2, R. Jeyakantha Ratnasingam2, Khean-Jin Goh2
1University of Malaya, Kuala Lumpur, MY; 2University of Malaya, Kuala Lumpur, MY
Background: Thyrotoxic periodic paralysis (TPP) is characterised by recurrent episodes of reversible, severe proximal muscle weakness associated with hypokalaemia and hyperthyroidism. TPP is common in Asians and is a secondary hypokalaemic periodic paralysis, with clinical and electrophysiological characteristics similar to primary hypokalaemic periodic paralysis. Prolonged exercise test (PET) is an easy, non-invasive test evaluating abnormal muscle membrane excitability in periodic paralyses. Results of PET studies in TPP patients on their thyroid status have been variable with one study showing normalization of response while another did not. We aim to evaluate patients with history of thyrotoxicosis with PET and correlate it with the thyroid status. Methods: We prospectively recruited 35 patients with history of thyrotoxicosis (regardless of whether they had TPP) from Endocrinology Clinic. Of the 35 patients, 18 were hyperthyroid, 17 euthyroid or hypothyroid. The thyroid status was determined biochemically from the latest thyroid function test. All patients underwent PET as described by McManis. Compound muscle action potential (CMAP) amplitudes post-exercise were compared against pre-exercise amplitudes and recorded as percentage of mean baseline CMAP amplitude. Results: Compared to 17 euthyroid or hypothyroid patients, significant time-dependent declines in CMAP amplitudes at 20 minutes (88.3±11.9% vs 96.7±5.2%; p=0.012), 25 minutes (88.8±12.9% vs 96.4±8.6%; p=0.047) and 30 minutes (88.1±13.0% vs 97.1±10.3%; p=0.030) after exercise were observed in hyperthyroid patients. The calculated mean greatest decrement from immediate post exercise CMAP amplitudes were 22.5±13.0% in hyperthyroid patients and 15.5±13.0% in euthyroid/hypothyroid patients. Conclusion: CMAP decrement on PET was significantly greater in hyperthyroid patients compared to euthyroid/hypothyroid patients even without a history of TPP. Muscle membrane excitability is highly influenced by thyroid hormone level. TPP results from increased levels of thyroid hormone activity in genetically susceptible patients.
PS1Group1-032 / #882CLINICAL AND HISTOPATHOLOGICAL FINDINGS IN MYOTONIC MUSCULAR DYSTROPHY TYPE 2: RETROSPECTIVE REVIEW OF 50 DNA-CONFIRMED CASES
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Bhaskar Roy1, Qian Wu2, Charles Whitaker3, Kevin Felice3
1Neurology, Yale University, New Haven, CT, US;2 Pathology, University of Connecticut, Farmington, CT, US;3 Neurology, Hospital for Special Care, New Britain, CT, US
Background: Myotonic dystrophy type 2 (DM2) is an autosomal dominant multisystem disorder causing a progressive myopathy and myotonia due to (CCTG)n repeat expansion in intron 1 of the CNBP gene in chromosome 3q21. Focused study on a US-based population is limited. Methods: We reviewed the clinical presentation, muscle strength grading by MRC, Jamar hand grip dynamometry, electromyography (EMG), creatine kinase (CK) values, forced vital capacity (FVC), muscle histopathology, CCTG repeat expansion (bp) size for DM2 patients from 1992 to 2015 evaluated at the Muscular Dystrophy Care Center at Hospital for Special Care, CT, USA. Muscle histopathology was graded based on myofiber size variability, internal nucleation, pyknotic nuclear clumps, necrosis, myofiber hypertrophy and atrophy. Results: Fifty patients with DM2 were included in this study. Forty-six patients had positive genetic study and 4 were affected family members. Age of symptom onset ranged from 15 to 72 years (48.58 ± 13.12). Duration between symptom-onset and initial evaluation ranged from 1 to 42 years (7.3 ± 9.37). Majority of the patients (64%) had proximal lower extremity symptoms as the initial presentation. Ten patients did not have any weakness and 17 patients had severe weakness based on a composite MRC scoring system. Hip flexors were the most frequently and severely affected muscles, followed by knee extensors and elbow extensors. About 30% patients had finger muscles involvement or ankle dorsiflexion weakness. Though most patients had a milder and stable disease course, we noted significant progression of weakness in a few patients. For composite MRC score, the rate of mean decline per year was 0.91 ± 0.79. For hip flexors, the rate of mean decline in MRC score per year was 0.21 ± 0.19. FVC (% predicted) in 32 (64%) patients ranged from 36 to 100% (mean, 81.1 ± 18.3%). CK values in 36 patients ranged from 20 to 2000 units/L (mean, 581.7 ± 449.3). Nine patients had clinical myotonia and 35 patients had myotonia on EMG. Among the muscles examined, biceps and tibialis anterior had myotonic discharges most frequently, 91.6% and 85.7% times respectively. No association was noted between the presence of myotonia in EMG and muscle weakness. We noted significant correlation of severity of histopathology with weakness. There was no correlation between CCTG repeat numbers with age of onset, weakness, disease progression, CK value, hand grip strength or histopathological severity. Among systemic involvement, 24 patients had cataracts (48%) and 7 patients had hearing loss. Five patients had symptomatic cardiac arrhythmia and 3 of them required pacemaker. Eleven patients had diabetes and 8 patients had hypothyroidism. Conclusion: This large cohort of DM2, evaluated over a 24-year-period in a single center, showed variation in clinical presentation and disease severity.
PS1Group1-033 / #327ASCERTAINMENT OF THE ADULT PATIENT COHORT WITH MITOCHONDRIAL DISEASE IN GLASGOW
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Maria Elena Farrugia1, Cheryl Longman2, Lesley Snadden2, Grainne S. Gorman3, Andrew M. Schaefer3, Robert W Taylor3, Yi Shiau Ng3, Douglass M. Turnbull3, Robert Mcfarland3, Richard K. Petty4
1Neurology, Institute of Neurological Sciences, Glasgow, GB;2Genetics, West of Scotland Regional Genetics Service, Glasgow, GB;3Institute Of Neuroscience, Newcastle University Wellcome Trust Centre for Mitochondrial Research, Newcastle upon Tyne, GB;4Neurology, Institute of Neurological Sciences, Glasgow, GB
Background: Mitochondrial disorders are caused by gene mutations in nuclear or mitochondrial (mt) DNA, encoding structural mitochondrial proteins or proteins involved in mitochondrial function. Patients with mitochondrial disease have diverse phenotypes. Patients may present with ptosis and ocular motility disorders (which may be misdiagnosed as ocular myasthenia). Some suffer from myopathy, diabetes, migraine, deafness and seizures (common red flags for general neurologists). Others may present with ataxia, neuropathy, cognitive impairment, psychosis, or intestinal pseudo-obstruction. These symptoms may not be immediately recognised as part of the disease spectrum and may lead to mis-management. The muscle clinic in Glasgow captures a population of circa 2 million. We have a longstanding collaboration with the NHS Highly Specialised Service for Mitochondrial Disease and Wellcome Centre for Mitochondrial Research in Newcastle, where we refer muscle pathology and patients to help us with the molecular diagnoses and patient management. We wanted to determine the population of molecularly confirmed mitochondrial patients attending the adult muscle clinic in Glasgow and characterise their phenotypes. Methods: We retrospectively identified 57 adult patients (20 families) with molecularly confirmed mitochondrial mutations from a neuromuscular database held in Glasgow. Results: The cohort included 25 patients with m. 3243 A>G mutation (14 families), 9 with the m. 8344 A>G mutation (2 families), 11 with sporadic large-scale mtDNA deletions, 3 with POLG dominant mutations and 2 with YARS2 recessive mutations (reported in previous publication; Sommerville et al, JAMA Neurol 2017). Another 7 had various mutations (including mutations in mt-transfer RNA, defects in complex I, complex III, mt-ATP 6, a patient with m. 7497 G>A mutation with a contractural limb-girdle phenotype, and a patient with multiple mitochondrial deletions for which no nuclear defect has been identified). The common symptoms within the m. 3243 A>G cohort included diabetes, deafness and migraine. Four patients had sustained stroke-like episodes (including 2 from the same pedigree with recurrent events and including one which led to her demise). Within the m. 8344 A>G cohort, symptoms included hearing loss, myopathy and exercise intolerance. Two patients (mother and son) had abnormal EEGs with lateralising epileptiform features but no clear clinical seizures or myoclonus. Within the single large-scale mtDNA deletions all had ptosis with progressive external ophthalmoplegia (PEO), 2 had pacemakers, 2 had cognitive difficulties and one required feeding through a gastrostromy tube. All 3 patients with POLG mutations had ptosis with PEO and myopathy. One patient had an episode of intestinal pseudo-obstruction which settled with conservative management. We finally discuss the case of a lady with m. 3243 A>G mutation, who succumbed to complications from a stroke-like episode and the difficulties pertaining to her management. Such patients can present with super-refractory epileptic seizures, requiring aggressive management. We discuss the difficult decisions pertaining to prophylactic anti-seizure medication for her family members, who harbour the mutation at high heteroplasmy levels in urine and muscle. Conclusion: Patients with mitochondrial disorders suffer from a complex multi-system disease. Awareness of this is pertinent when assessing mitochondrial patients, discussing diagnosis and prognosis with patients and their families. Their management requires a multidisciplinary approach.
PS1Group1-034 / #373RESTING-STATE FMRI SHOWS ALTERED DEFAULT-MODE NETWORK FUNCTIONAL CONNECTIVITY IN DUCHENNE MUSCULAR DYSTROPHY PATIENTS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Nathalie Doorenweerd1, Mischa De Rover2, Chiara Marini-Bettolo3, Kieren G. Hollingsworth4, Erik H. Niks5, Jos G.m. Hendriksen6, Hermien E. Kan1, Volker Straub7
1C.j. Gorter Center For High Field Mri, Leiden University Medical Center, Leiden, NL;2Clinical Psychology Unit, Leiden University, Leiden, NL;3John Walton Muscular Dystrophy Research Center, Newcastle University, Newcastle Upon Tyne, GB;4Newcastle Magnetic Resonance Centre, Newcastle University, Newcastle Upon Tyne, GB;5Neurology, Leiden University Medical Center, Leiden, NL;6Neurological Learning Disabilities, Kempenhaeghe Epilepsy Center, Heeze, NL;7John Walton Muscular Dystrophy Research Centre, Newcastle University, Institute of Genetic Medicine, Newcastle Upon Tyne, GB
Background: Duchenne muscular dystrophy (DMD) is an X-linked neuromuscular disorder caused by mutations in the dystrophin encoding DMD gene. Dystrophin is expressed in muscle but also in brain and difficulties with attention/inhibition, working memory and learning are well described. Reduced functional connectivity in the motor cortex has been reported in DMD. However, the default mode network (DMN) remains unexplored despite being involved in switching between different stages of attention as well as information processing and self-reflection. Therefore, the aim of this study was to determine if there are differences in DMN connectivity between DMD and controls. Methods: Thirty-three DMD and twenty-four male age matched controls were recruited and scanned at two clinical sites (LUMC and NU). T1-weighted anatomical scans and resting state functional scans were obtained on a 3 Tesla Philips Achieva. FSL v.5.0.8 was used for all analyses which were performed in accordance with recent FSL recommendations, including ICA-AROMA motion correction.The DMN was first identified in the controls. Statistics were performed with FSL RANDOMISE to test for differences between DMD and controls in the DMN and the visual network, with age as covariate and threshold-free cluster enhancement multiple comparison correction. Results: There was significant hyper-connectivity in the precuneus and hippocampus and additional connections with the right thalamus in relation to the DMN in DMD compared to controls. This may be related to the often impaired learning abilities in DMD. Additional connectivity is also found in the sensori-motor cortex. As DMD is hallmarked by progressive muscle wasting, altered connectivity in this region is not surprising but the relationship with the default mode network is unclear. Reduced grey matter volume and perfusion known to be present throughout the brain in DMD could lead to lower signal to noise ratio which could have a confounding effect on the comparison. Therefore, the visual network was assessed as negative control and indeed, no significant differences were found between DMD and controls. Conclusion: Altered DMN connectivity was found in DMD with both hyper-connectivity within the DMN and reconfiguration of the network to include additional brain regions. As the DMN in general is active during rest and inactive during tasks, it will be important to assess in DMD if this extended DMN, which includes greater parts of the precuneus, hippocampus and thalamus, is also inactivated when a task is performed. If so, this may help explain the incidence of attention/inhibition, working memory and learning difficulties in DMD.
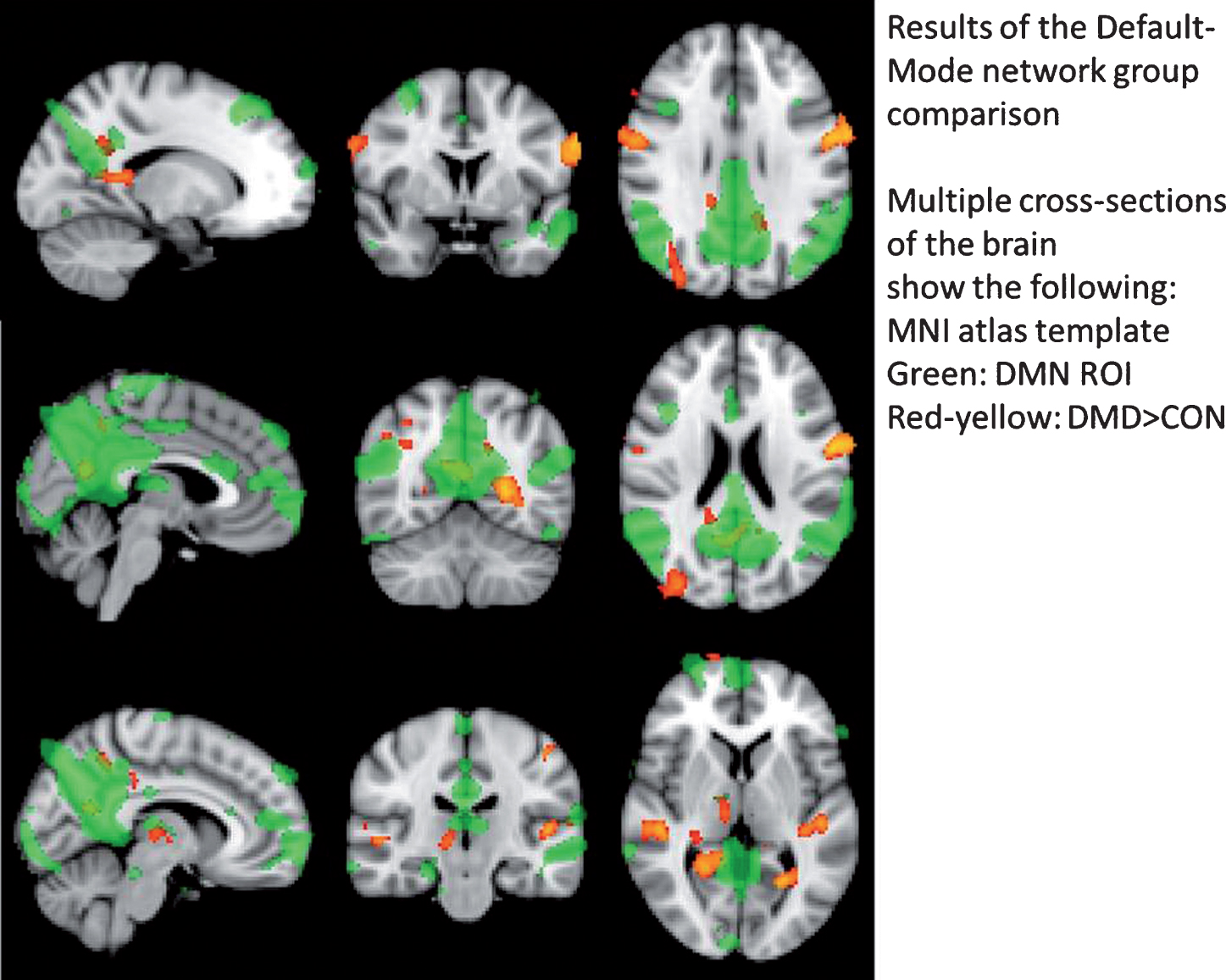
PS1Group1-035 / #930SPECIFIC MUTATIONS IN MYBPC1 CAUSE MYOPATHY AND “MYOGENIC TREMOR”
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Sandra Jackson1, Jochen Schaefer1, Annika Saak1, Janis Stavusis2, Baiba Lace3, Janelle Geist4, Inna Inashkina2, Dita Pelnena2, Sander Pajusalu5, Nathan Wright6, Aikaterini Kontrogianni-Konstantopoulos4, Carsten G. Bönnemann7
1Neurology, Uniklinikum C.G.Carus, Dresden, DE;2Latvian Biomedical Research Centre, Riga, LV;3Centre Hospitalier Universitaire de Quebec, Quebec, CA;4University of Maryland School of Medicine, Baltimore, US;5Tartu University Hospital, Tartu, EE;6James Madison University, Harrisonburg, US;7NIH Porter Neuroscience Research Center, Bethesda, MD, US
Background: Myosin-binding protein C1, a sarcomeric protein, is thought to stabilize the thick filaments interacting with myosin, actin and creatine kinase. To date, only few MYPBC1-mutations have been reported in human disease and lead to autosomal-dominant distal arthrogryposis and autosomal-recessive lethal contracture syndrome. Methods: We describe two multi-generation families with non-progressive, proximal and axial weakness, mild spinal rigidity and myalgia. In both families, postural fast-frequency tremor of hands and tongue segregated with the disease. Pathogenic mutations identified by panel sequencing were introduced into truncated GST-tagged recombinant mutant and wild-type proteins. Binding of the mutant protein to myosin was assessed with overlay and immunoblotting assays, and 3D-modelling of the MYBPC1-myosin interaction was performed. Results: Two mutations located in adjacent codons in the M-motif of MYPBC1 and segregating with the disease in both families, demonstrated significantly increased binding of the mutant to myosin compared to the wild-type protein. 3D-modelling and MD-simulation implicated increased electrostatic interactions of MYBPC1 with myosin. Neurophysiological testing excluded a peripheral generator of the tremor. Conclusion: The mutations, which segregated with the disease in both families were predicted by MD-modelling to increase the binding affinity of MYBPC1 to myosin. This was confirmed with in vitro binding-assays. Presumably, cross-bridge cycling of actin and myosin will be affected thus reducing force generation. The tremor associated with the disease was shown to be neither of peripheral nor of central origin, because MYBPC1 is exclusively expressed in muscle sarcomeres. Thus we postulate a novel class of tremor which we designated “myogenic tremor”.
PS1Group1-036 / #957DIAGNOSTIC APPROACH TO CHRONIC PROGRESSIVE EXTERNAL OPHTHALMOPLEGIA – FROM CLINICAL EVALUATION TO GENETIC CONFIRMATION
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Biruta Kierdaszuk1, Magdalena Kaliszewska2, Katarzyna Tonska2, Ewa Bartnik2, Anna M. Kaminska1, Anna Kostera-Pruszczyk1
1Department Of Neurology, Medical University of Warsaw, Warsaw, PL;2Institute Of Genetics And Biotechnology, Faculty Of Biology, University of Warsaw, Warsaw, PL
Background: Mitochondrial encephalomyopathies comprise a group of heterogeneous disorders which result from impaired oxidative phosphorylation. The respiratory chain complexes present in mitochondria are dependent both on mitochondrial (mtDNA) and nuclear genome (nDNA). Different age of onset, various clinical presentations and individual course of the disease make the diagnosis of mitochondrial disorders challenging. From a variety of symptoms progressive external ophthalmoplegia (PEO) seems to be the most common. The aim of this study was the clinical and genetic characteristics of Polish patients with progressive external ophthalmoplegia. Methods: Clinical, electrophysiological, neuroradiological and morphological data of 72 patients aged 11 to 76 years were analyzed. The muscle specimens were assessed on light and electron microscopy using routine procedures. Genetic studies of mtDNA were performed in all patients. Among nDNA genes POLG was studied in 26 patients, TWNK (C10orf2) in 3 patients and RNASEH1 in 2 patients. Results: 24 patients with ptosis and PEO were included to chronic progressive external ophthalmoplegia (CPEO) group and 19 with ptosis, PEO and limb or trunk muscles’ weakness to CPEO+ group. There were 22 patients with PEO and the central nervous system impairment classified as mitochondrial encephalomyopathy (ME), 6 patients with Kearns-Sayre syndrome (KSS) and one patient with sensory ataxic neuropathy, dysarthria, ophthalmoparesis (SANDO) syndrome. 7 patients had relatives with similar symptoms. Among various diagnostic procedures electromyography showed myopathic changes in the majority of cases. Ragged-red fibers were present in most of muscle biopsies. Magnetic resonance imaging showed atrophied ocular muscles in patients with pure myopathic phenotype and revealed cortical and subcortical atrophy in those with the central nervous system involvement. In single cases magnetic resonance spectroscopy showed elevated lactates in the brain. Genetic studies of mtDNA proved already known single or multiple mtDNA deletions in all patients and in most cases they were detected in the muscle tissue. Genetic analysis of nDNA genes confirmed mutations in POLG gene in 6 patients. There were 3 CPEO patients with p.[Arg309Leu];[Gln968Glu], p.[Ala518Thr];[=] and p.[Trp748Ser];[Ser998Pro] mutations, and 2 CPEO+ patients with p.[Thr251Ile;Pro587Leu];[Thr251Ile;Pro587Leu] and p.[Thr251Ile;Pro587Leu];[Lys1191Asn] mutations. In patient with SANDO syndrome the mutation p.[Arg290Cys];[Arg309Cys] in POLG gene was confirmed. Additionally the analysis of the TWNK (C10orf2) gene proved the mutation p.[Arg374Gln];[=] in 2 CPEO patients and one CPEO+ patient. Assessment of RNASEH1 gene revealed homozygous variant p.[Gly165Arg];[Gly165Arg] in two siblings with ME. Conclusion: Detailed patients’ history and careful assessment of family history are essential in the diagnostic work-up. Morphological evaluation of the muscle biopsy as well as magnetic resonance imaging are also important. Genetic studies of both mtDNA and nDNA are necessary for final diagnosis of chronic progressive external ophthalmoplegia and its genetic counseling.
PS1Group1-037 / #473FEMALE FERTILITY IN MYOTONIC DYSTROPHY TYPE 1 AND 2
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Olesja Parmova1, Iva Srotova1, Monika Hulova1, Eva Vlckova1, Livie Mensova2, Martina Podborska3, Petra Stradalova1, Eva Kralickova1, Igor Crha4, Radim Mazanec2, Stanislav Vohanka1, Josef Bednarik1
1Neurology, University Hospital Brno, Brno, CZ;2Neurology, Motol University Hospital, Prague, CZ;3Clinical Biochemistry, University Hospital Brno, Brno, CZ;4Obstetrics And Gynecology, University Hospital Brno, Brno, CZ
Background: Myotonic dystrophy (DM) represents the most common type of muscular dystrophy in adult age. Clinical signs and symptoms in type 1 (DM1) and type 2 (DM2) overlap. DM is a multisystem disorder that, among others, affects endocrine system and thus may have a negative impact on fertility. Female fertility impairment has already been shown in patients with DM1, while only few data are available on DM2 patients. The aim of our study was to compare the fertility characteristics in women with DM1, DM2 and healthy volunteers. Methods: Patients: 22 reproductive-age females (mean age 34.8 ± 8.0) with DM2 and 18 reproductive-age females with DM1 (mean age 33.9 ± 6.8) were included in this case control study. With respect to significant differences between these two groups in several relevant parameters (e.g. number of pregnancies), particular control groups were created for each of them: DM2 control group consisted of 22 age-matched healthy controls (mean age 34.1 ± 7.5) and DM1 control group of 18 age-matched healthy controls (mean age 33.2 ± 7.0). Methods: All participants completed a set of questionnaires used to evaluate fertility. Results: The time to get pregnant was significantly longer in DM1 females when compared with appropriate controls (p < 0.01) and the miscarriages were more frequent in this DM group (p = 0.02). Females with DM1 also had more often irregular menstrual cycle (p = 0.02) which was more frequently controlled by oral contraceptives in this group. Furthermore, patients with DM1 had higher incidence of uterine myomas and more frequently underwent abdominal gynecologic surgery (p < 0.01). In comparison with healthy controls, an unsignificant trend to more frequent investigation of fertility problems and their treatment was observed in women with DM1 (p = 0.08). Females with DM2 have more frequent miscarriages (p = 0.04) as the only statically significant difference when compared with healthy controls. Conclusion: Our study confirmed clear fertility impairment in women with DM1 and showed less pronounced (but still significant) fertility changes also in female patients with DM2.
PS1Group1-038 / #340EFFECT OF AHK, A NOVEL MODULATOR OF RYANODINE RECEPTORS, IN DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Haizpea Lasa Fernandez1, Garazi Aldanondo2, Jaione Lasa Elgarresta2, Aitziber Irastorza3, Jose Ignacio Miranda3, Jesus Maria Aizpurua3, Adolfo López De Munain4, Ainara Vallejo Illarramendi2
1Department Of Neuroscience, University of the Basque Country, Leioa, ES;2Neuroscience Area, Biodonostia Research Institute, Donostia, ES;3Faculty Of Chemistry, University of the Basque Country, Donostia, ES;4Neurology, Hospital Donostia, Donostia, ES
Background: In the mdx mouse model of Duchenne muscular dystrophy (DMD) the sarcoplasmic reticulum ryanodine receptor (RyR) is abnormally nitrosylated and this leads to calstabin depletion from the protein complex and subsequent calcium leak through the channel. RyR stabilizers, such as S107, enhance RyR-calstabin binding preventing calcium leak and the consequent damage. Methods: To study the in vitro effect of AHK we have evaluated RyR-calstabin colocalization using in situ proximity ligation assay (PLA) in dystrophin-deficient human myotubes treated overnight with AHK150 nM. We also have determined intracellular calcium levels in these myotubes using Fura-2AM, a ratiometric calcium indicator. For the in vivo studies, we have administered AHK to male mdx mice in the drinking water for 5 and 12 weeks. We have determined forelimb strength using a grip strength meter. Diaphragm muscle sections were used for histopathological analysis using immunofluorescence. The percentage of central nucleation was determined as a marker of muscle degeneration/regeneration. To study cardiac and cognitive deficits in vivo, we have used 4 month-old mdx mice. We have analyzed cardiomyocyte damage after beta-isoproterenol challenge, using Evans blue dye (EBD) uptake, as an indicator of cardiomyocyte plasma membrane integrity. Finally, we have measured mdx cognitive deficits by tracking the movement after an acute stress produced by a brief restrain. Results: The present work shows that treatment of 1 month-old mdx mice during 5 and 12 weeks with AHK, a novel RyR modulator, improves the dystrophic phenotype of mdx mice. After the treatment during 5 weeks the histopathological and biochemical evidence of skeletal muscle damage is reduced and the strength is improved. Since cardiac and cognitive impairments manifest in older mice, the treatment is prolonged for 12 weeks. Once we characterized the behavioral phenotype of mdx mice, we observe improvements in treated mice regarding to heart function and behavioral deficits. Conclusion: Overall, our results demonstrate that AHK is effective in improving not only skeletal muscle dystrophic phenotype but also the impairments observed in cardiac muscle and CNS in the mdx mouse model of DMD. Thus, they support AHK as a potential therapeutic drug to prevent the progression of muscular, cardiac and neurological defects in DMD patients.
PS1Group1-039 / #380ASPIRO PHASE 1/2 GENE THERAPY TRIAL IN X-LINKED MYOTUBULAR MYOPATHY (XLMTM): PRELIMINARY SAFETY AND EFFICACY FINDINGS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Nancy Kuntz1, Perry B. Shieh2, Barbara Smith3, Carsten G. Bönnemann4, James J. Dowling5, Michael W. Lawlor6, Wolfgang Müller-Felber7, Mo Noursalehi8, Sal Rico8, Laurent Servais9, Suyash Prasad8
1Ann & Robert H Lurie Children’s Hospital of Chicago, Chicago, IL, US;2University of California, Los Angeles, Los Angeles, CA, US;3University of Florida, Gainesville, FL, US;4NIH Porter Neuroscience Research Center, Bethesda, MD, US;5Hospital for Sick Children, Toronto, ON, CA;6Medical College of Wisconsin, Milwaukee, WI, US;7Klinikum der Universität München, Munich, DE;8Audentes Therapeutics, San Francisco, CA, US;9Hopital Armand Trousseau, Paris, FR
Background: X-linked myotubular myopathy (XLMTM) is a rare disease caused by mutations in the MTM1 gene and characterized by profound muscle weakness, respiratory failure and early death. A retrospective chart review of 112 boys with XLMTM (RECENSUS study [Beggs, 2017]) showed overall mortality of 44%. Additional natural history data are being collected in an ongoing, prospective, non-interventional, run-in study of male patients with XLMTM (the INCEPTUS study), to serve as a longitudinal baseline and within-patient control for the ASPIRO clinical study. Methods: Phase 1/2, open-label, randomized, ascending dose study to evaluate the safety and preliminary efficacy of an investigational gene therapy product (AT132) in patients with XLMTM. AT132 (rAAV8-Des-hMTM1) is designed to deliver functional copies of the MTM1 gene to skeletal muscle cells. Approximately 12 XLMTM patients less than 5 years of age are planned to be randomized into three ascending dose cohorts (n=4 per cohort) to receive a single AT132 administration (n=3) or to act as a delayed treatment control (n=1). Treatment-randomized Cohort 1 patients received an intravenous infusion of 1x1014 vector genomes (vg) per kg of AT132.
Results: A total of 6 adverse events (AEs) were reported, of which 3 were deemed probably or possibly related to drug and 2 were deemed serious AEs (SAEs). Both SAEs occurred in Patient 3, who is responding to intravenous steroids and supportive care. Efficacy assessments have shown notable improvements in both the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) scale and maximal inspiratory pressure (MIP) (Table 1). Whereas age-appropriate motor milestones had not been achieved at baseline, by week 12, Patient 1 (aged 1 year) had acquired several new developmental skills, including the ability to control head movements, roll over by himself and sit unassisted for >5 seconds. Patient 2 was observed for 8 weeks, and Patients 3 and 4 (Control) were observed for 4 weeks. All treated patients have demonstrated improvements in airway clearance and secretion management as evidenced by changes in the Parental Global Impression of Secretion Improvement and Severity Scales. Clinical Global Impression of Improvement reports demonstrate increased limb and trunk strength, improved velocity and accuracy of movement and increased loudness during vocalization and crying, improving their ability to communicate. Conclusion: Preliminary data from cohort 1 of the ASPIRO study are encouraging and based on these observations, the current plan is to enroll three additional patients at this dose level.
Table 1:
ASPIRO Cohort 1 Baseline Characteristics and Preliminary Efficacy Results
| Patient # | 1 | 2 | 3 | 4 (Delayed Treatment Control) | |
| Age at Baseline (Years) | 0.8 | 4.1 | 2.6 | 4.0 | |
| Ventilator Support | 12 h/day BiPAP | 17 h/day invasive ventilation | 24 h/day invasive ventilation | 12 h/day BiPAP | |
| CHOP-INTEND Score (a maximum score of 64 is typically attained in healthy infants between 3–6 months of age) | Median Score in INCEPTUS | 29 | 45 | 28 | 49 |
| Baseline Score in ASPIRO | 29 | 45 | 34 | 49 | |
| Most Recent Score in ASPIRO | 56 (week 12) | 56 (week 8) | 36 (week 4) | 46 (week 4) | |
| Change from Baseline (%) | 27 (93%) | 11 (24%) | 2 (6%) | 3 (-6%) | |
| Maximum Inspiratory Pressure (cm H2O; normal minimal pressures in healthy children <5 years are ≥ 80 cm H2O) | Median Pressure in INCEPTUS | 29 | 34 | 24 | 65 |
| Baseline Pressure in ASPIRO | 33 | 44 | 26 | 58 | |
| Most Recent Pressure in ASPIRO | 80 (week 12) | 77 (week 8) | 44 (week 4) | NA | |
| Change from Baseline (%) | 47 (142%) | 33 (76%) | 18 (70%) | NA | |
At time of data cut (21Dec17), individual patient follow-up ranged from 4 to 12 weeks.
PS1Group1-040 / #442MR IMAGING OF RESPIRATORY MUSCLE DYSFUNCTION IN POMPE DISEASE
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Laurike Harlaar1, Pierluigi Ciet2, Alice Pittaro2, Piotr A. Wielopolski2, Harmke A. Van Kooten1, Nadine A.m.e. Van Der Beek1, Esther Brusse1, Ans T. Van Der Ploeg3, Marleen De Bruijne4, Harm A.w.m. Tiddens5, Pieter A. Van Doorn1
1Department Of Neurology, Center For Lysosomal And Metabolic Diseases, Erasmus MC, Rotterdam, NL;2Department Of Radiology And Nuclear Medicine, Erasmus MC, Rotterdam, NL;3Department Of Paediatrics, Division Of Metabolic Diseases And Genetics, Center For Lysosomal And Metabolic Diseases, Erasmus MC-Sophia, Rotterdam, NL;4Biomedical Imaging Group Rotterdam, Departments Of Radiology And Medical Informatics, Erasmus MC, Rotterdam, NL;5Department Of Paediatrics, Respiratory Medicine And Allergology, Department Of Radiology And Nuclear Medicine, Erasmus MC, Rotterdam, NL
Background: Adult patients with Pompe disease present with a spectrum of symptoms, mainly characterized by progressive limb-girdle weakness and decreased pulmonary function. Limb-girdle weakness mostly improves or stabilizes after start of enzyme replacement therapy (ERT). Pulmonary function often benefits less well from ERT and may continue to deteriorate. Standard pulmonary function tests only give limited insight in the pathophysiological mechanisms of the pulmonary insufficiency. MRI enables to visualize the diaphragmatic and chest wall movements simultaneously. In a previous study, MRI showed very limited movement of the diaphragm in ten Pompe patients with severe respiratory muscle weakness. Our aim is to study diaphragmatic function in a large group of Pompe patients and to investigate the best outcome measures to evaluate the function of the diaphragm. Methods: We perform spirometer-controlled MRI scans in patients with Pompe disease and sex- and age-matched healthy controls. We acquired dynamic sagittal 2D images in left and right mid hemi-diaphragm during forced inspiration. After manual segmentation of these images, we measured the cranial-caudal and anterior-posterior distance, lung area, diaphragm height, diaphragmatic shape factor and diaphragm slope (figure). We calculated the ratios between maximal inspiration and maximal expiration. We tested intra- and interobserver variability of the measurements, and differences between Pompe patients and controls. Results: In our ongoing study we have now included 34 patients with Pompe disease (age range 15-70 years) with varying disease severity, and 18 controls. All thorax-related measurements showed a high reliability (intraclass correlation coefficient > 0.9). Diaphragm-related measurements showed a larger intra- and interobserver variability. On the right side, mean cranial-caudal ratio was 1.4 in Pompe patients and 1.7 in controls (p<0.01); mean lung area ratio was 2.1 in Pompe patients and 2.5 in controls (p<0.01). On the left side, mean cranial-caudal ratio was 1.4 in Pompe patients and 1.6 in controls (p<0.01); mean lung area ratio was 1.9 in Pompe patients and 2.1 in controls (p=0.03). In anterior-posterior ratio we found no significant difference between Pompe patients and controls. Conclusion: MRI has good possibilities to evaluate simultaneously diaphragmatic and chest wall movements, which both contribute to pulmonary function. Cranial-caudal ratio and lung area ratio are decreased in the group of patients with Pompe disease compared to controls. We are currently exploring more specific diaphragmatic measures to see whether these can serve as a biomarker for diaphragmatic function in the evaluation of disease progression and response to therapy.
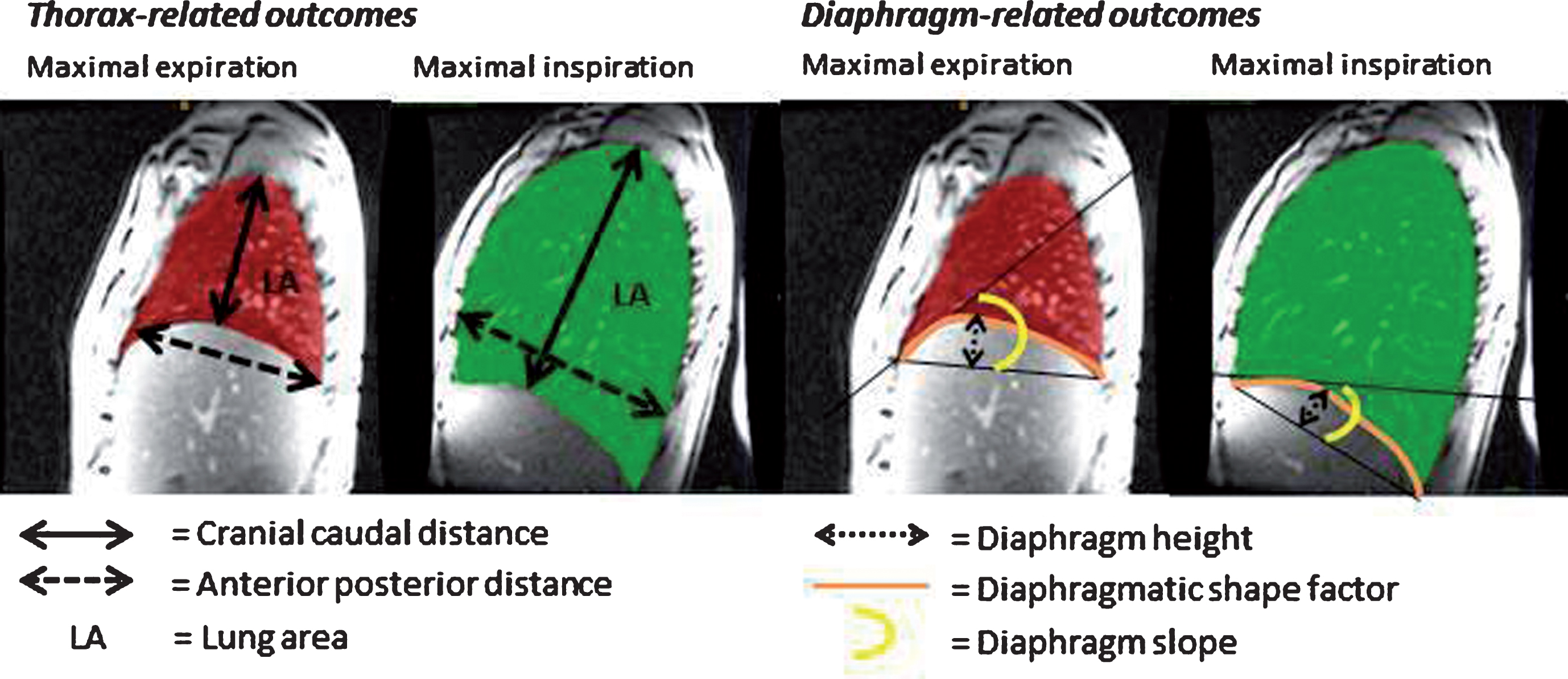
PS1Group1-041 / #478A CASE OF VLCAD DEFICIENCY MYOPATHY WITH NEW MUTATION AND FAVORABLE RESPONSE TO L-CARNITINE, RIBOFLAVIN, AND COQ10
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Farzad Fatehi1, Yalda Nilipour2, Shahriar Nafissi1
1Neurology, Tehran Univeristy of Medical Sciences, Tehran, IR;2Pathology, Mofid Hospital, Tehran, IR
Background: Very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency is a very rare condition preventing the body from transforming very long-chain fatty acids (VLCAs) to energy, principally during periods of fasting. Signs and symptoms of VLCAD deficiency classically manifest during infancy or early childhood with hypoglycemia, lethargy, muscle weakness, and episodes of dark urine. There is no approved treatment for such patients. Herein, we describe a patient with a new mutation favorably responding to the treatment. Methods: A 38-year-old man was referred for a very slowly progressive lower limb weakness since childhood with the inability to climb upstairs since two years before. He reported episodes of dark urine after exercise. On examination, he had the weakness of neck flexion, bilateral arms abduction (MRC grade 3), bilateral elbows flexion (MRC grade 4), bilateral hips flexion (MRC grade 4), and bilateral knees extension (MRC grade 4) with normal deep tendon reflexes. The CK was 5400 IU/L; cardiac exams were normal; and electromyography demonstrated a myopathic pattern in proximal muscles of upper and lower limbs. Muscle MRI revealed moderate fat deposition in paraspinal muscles, adductor and vastus muscles as well as medial heads of gastrocnemius muscles. On muscle biopsy, myopathic atrophy of mainly type I fibers with multiple necrotic/regenerative fibers and fine lipid droplets in muscle fibers in ORO stain was seen. For genetic testing, DNA was extracted from blood cells and targeted next generation sequencing (NGS) was performed for this individual. The variants located in coding regions of all the 42 genes in this NGS panel for Metabolic Myopathies were evaluated. The obtained result was confirmed with the help of direct DNA sequencing of the candidate variant in the proband. Results: Targeted next-generation sequencing revealed a pathogenic variant defined as c.900G>A (p.Met300Ile) in exon 10 of ACADVL gene due to VLCAD deficiency (MIM#201475) not described previously. Segregation analysis for the parents confirmed the heterozygote status of both father and mother (carrier status) for ACADVL gene. We started L-carnitine 2 grams daily, CoQ10 300 mg daily and riboflavin 300 mg daily with a favorable response, and after one month the patient was able to climb upstairs without aid. Conclusion: In sum, it seems that the cocktail of L-carnitine, CoQ10, and riboflavin may be an optimal option for this very rare form of lipid storage myopathy.
PS1Group1-042 / #713IMPAIRED INSULIN SIGNALLING IN SKELETAL MUSCLE OF MYOTONIC DYSTROPHY PATIENTS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Laura V. Renna1, Francesca Bose’1, Elisa Brigonzi2, Giovanni Meola2, Rosanna Cardani3
1Laboratory Of Muscle Histopathology And Molecular Biology, IRCCS Policlinico San Donato, San Donato Milanese (MI), IT;2Department Of Biomedical Sciences For Health, University of Milan, San Donato Milanese (MI), IT;3Laboratory Of Muscle Histopathology And Molecular Biology, IRCCS-Policlinico San Donato, San Donato Milanese (MI), IT
Background: Myotonic dystrophy type 1 (DM1) and type 2 (DM2) are neuromuscular multisystemic disorders caused by expanded CTG or CCTG repeats which lead to nuclear accumulation of mutant transcripts deregulating the activity of some splicing regulators and leading to aberrant alternative splicing of different genes. DM are characterized by metabolic dysfunctions such as insulin resistance, hyperinsulinemia and a fourfold higher risk of developing Diabetes mellitus type 2 (T2DM). Since it is known that insulin resistance is a risk factor for cardiovascular disease, neuropathy and loss of muscle mass, metabolic changes in DM patients might contribute to worsen some aspects of the disease at heart, skeletal muscle and brain level. Splicing alteration of insulin receptor (IR) gene is considered one of the causes of insulin resistance in DM patients, however it cannot be excluded that post-receptor signalling abnormalities could also contribute to this feature. The aim of this study is to investigate whether post-receptor abnormalities contribute to the peripheral insulin resistance in DM patients and whether deregulated metabolism pathways may be involved in muscle alterations, thus contributing to muscle atrophy and weakness in DM patients. Methods: Insulin pathway activation has been investigated in insulin stimulated skeletal muscle biopsies obtained from 9 DM1, 2 DM2, 5 healthy subjects (CTR) and 1 T2DM patients (ex vivo analysis). Activation of IRS1, AKT, p70S6K, GSK3ß, ERK1/2 and AMPK have been analysed by western blot. ERK1/2 involvement in DM insulin resistance has been investigated in human myotubes treated with the MEK inhibitor U0126 (25 μM). Results: Our results indicate that DM skeletal muscle exhibits high basal phosphorylation of AKT, GSK3ß and ERK1/2. Moreover, ex vivo analysis of insulin pathway activation has shown that insulin action appears to be impaired in DM samples as compared to CTR. Glucose uptake performed in human myotubes in the absence or presence of U0126 has shown that the specific decrease of ERK activity can lead to an increase in insulin dependent glucose uptake in both DM1 and DM2 muscle cells. Conclusion: In conclusion, ex vivo and in vitro studies show an alteration in activation of several proteins of insulin pathway that might contribute to DM insulin resistance. Results on the correlation between metabolic dysfunction and skeletal muscle impairment will be presented at the meeting. The results of this study may contribute to the identification of novel biomarkers that could be target for therapeutic intervention on insulin resistance and thus to improve the quality of life of DM patients.
PS1Group1-043 / #843DISTAL MYOPATHIES WITH RIMMED VACUOLE IN IRAN, A CLINICAL, HISTOPATHOLOGICAL AND GENETIC REPORT OF A LARGE GROUP
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Yalda Nilipour1, Shahriar Nafissi2, Farzad Fatehi3, Yalda Ashoorian4, Nahid Beladi Moghadam5, Reza Boostani6, Mohammad Rohani7, Bahram Haghi Ashtiani8, Babak Zamani9, Keyvan Basiri10, Fereshteh Ashtari11, Davood Fathi3, Hosein Shamshiri2
1Pathology, Pediatric Pathology Research Center, Research Institute for Children Health, Shahid Beheshti University of Medical Sciences, Tehran, IR;2Neurology, Tehran Univeristy of Medical Sciences, Tehran, IR;3Neurology, Iranian center of neurological research, Neuroscience institute, Shariati Hospital, Tehran Univeristy of Medical Sciences, Tehran, IR;4Pathology, Shahid Beheshti university of medical sciences, Tehran, IR;5Neurology, Shahid Beheshti university of medical sciences, Tehran, IR;6Neurology, Mashhad University of medical sciences, Mashhad, IR;7Neurology, Hazrat Rasool Hospital, Iran university of medical sciences, Tehran, IR;8Neurology, Firoozgar hospital, Iran university of medical sciences, Tehran, IR;9Iran University of Medical Sciences, Tehran, IR;10Isfahan University of Medical sciences, Isfahan, IR;11Neurology, Isfahan University of Medical sciences, Isfahan, IR
Background: Distal myopathies are a rare group of neuromuscular disorders. Rimmed vacuoles are a feature of several myopathies, most of them predominantly affects the distal muscles. The presence of vacuoles in muscle biopsy associated with other pathological findings and the clinical phenotype of patients, age of onset, mode of inheritance and muscle imaging are important diagnostic clues. Rimmed vacuole myopathy is the most common type of distal myopathy in Iran and the highest prevalence of GNE myopathy was reported in Persians Jewish population. Methods: In this study, we retrospectively evaluate the clinical and histopathological characteristics of 75 Iranian patients (34 male and 41 female) from 75 different families with clinical suspicion of hereditary inclusion body myopathy, reported to have rimmed vacuoles in their muscle biopsies. We managed to perform direct sequencing for coding exons 1 to 12 of the GNE gene of 23 patients of this group. Results: Seventeen patients out of 75 had positive family history of muscle weakness. Age of onset of symptoms was 13 to 52 years. Nine patients did not show quadriceps sparing phenotype and 4 patients had loss of ambulation at the time of biopsy which was happened 4 to 13.5 years after onset of their symptoms. Maximum Creatine Kinase detected level was 3061 U/I. In microscopic evaluation, group atrophy was a common finding and some foci of chronic inflammation were identified in 12 cases. The number and the shape of the rimmed vacuoles was observed to be in a wide variety. Thirty-two cases showed necrotic fibers in their muscle biopsies and no congophilic inclusion was seen in 35 cases. Endomysial fibrosis and adipose tissue replacement were identified in 51 and 32 cases respectively. Intermyofibrillar network disruption as core-like lesions and moth-eaten fibers were seen in 41 cases and lobulated fibers were observed in ten muscle samples. Fiber type grouping was noted in 16 patients. No pathogenic mutation in GNE gene was found in 4 patients. Twelve patients were found to be homozygote for pathogenic mutations among which the most common one was c.2228T>C p.(Met743Thr) mutation in exon 12 observed in 8 cases and two of them had homozygote mutations in exon 4 followed by 1 in exon 3 and 1 in exon 9. One case found to be heterozygote for the GNE c.2228T>C pathogenic mutation. Four patients found to be heterozygote for one pathogenic mutation and one variant of uncertain clinical significance in different exons and 2 patients found to be putative homozygotes for same variant in exon 4 which was of unknown clinical significance. Conclusion: This study is the largest group study of 75 patients suffering from distal myopathy with rimmed vacuoles from Iran. Our findings emphasize the clinical, pathological and genetic heterogeneity of the disease. GNE myopathy is the most common early adult onset autosomal recessive distal myopathy in Iran. More extensive genetic studies such as whole exome sequencing on a larger group of patients may find new genes and will expand our knowledge.
PS1Group1-044 / #896GMPPB HOMOZYGOUS VARIANT IN ADULT ONSET LIMB GIRDLE MYASTHENIC SYNDROME: A LIKELY FOUNDER MUTATION
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Veeramani Preethish-Kumar1, Ana Töpf2, Kiran Polavarapu3, Aditi Joshi4, Sunitha Balaraju2, Andreas Roos2, Rita Horvath2, Saraswati Nashi5, Seena Vengalil6, Aradhna Mathur4, Sushmita Nayak4, Sakshi Ambawat4, Mohammed Faruq4, Atchayaram Nalini6, Hanns Lochmüller7
1Clinical Neurosciences, Neurology, National Institute of Mental Health and Neurosciences (NIMHANS), Bengaluru, IN;2John Walton Muscular Dystrophy Research Center, Newcastle University, Newcastle, GB;3Clinical Neurosciences, Neurology, National Institute of Mental health and Neurosciences (NIMHANS), Bengaluru, IN;4Institute of Genomics and Integrative Biology, New Delhi, IN;5Neurology, National Institute of Mental Health and Neurosciences (NIMHANS), Bangalore, IN;6Neurology, National Institute of Mental Health and Neurosciences, Bengaluru, IN;7Department Of Neuropediatrics And Muscle Disorders, Freiburg University - Medical Center, Faculty of Medicine, Freiburg, DE
Background: Mutations in GMPPB encoding GDP-mannose pyrophosphorylase B affects glycosylation of a-dystroglycan resulting in Congenital muscular dystrophies (CMD) and Limb girdle muscular dystrophies (LGMD). To date there are few genetically verified individuals with GMPPB-associated dystroglycanopathiess. Recently, variants in GMPPB were reported to cause congenital myasthenic syndrome. Methods: Ten patients belonging to 4 south Indian families with late onset limb girdle syndrome and easy fatigability are described. Evaluation included clinical phenotyping, serum Creatine Kinase (CK), Repetitive nerve stimulation, and muscle biopsy. Six patients underwent genetic testing. Results: All were born to consanguineous parents. Five patients were evaluated at hospital and all had gradually progressive fatigable limb girdle weakness, prominent truncal weakness with calf hypertrophy and retained tendon reflexes (Table 1). CK levels were elevated in all (650 – 4547 IU/L). Muscle biopsy in 4 patients was suggestive of muscular dystrophy. RNS showed decrement response in probands from family 3 and 4. Pyridostigmine and Salbutamol were started in family 4, and they showed significant improvement in fatigue and weakness. Genetic analysis revealed identical missense mutation c.1000G>A (p.Asp334Asn) in exon 9 of GMPPB gene in 6 patients tested of all four families. This mutation was previously reported in a compound heterozygous form in two patients of Asian origin (Pakistan and India) in published literature. However, the phenotype described in these two cases was Congenital muscular dystrophy with CNS involvement. While, in all our patients the mutation was present in homozygous form and had a milder, slowly progressive phenotype consistent with Limb girdle myasthenic syndrome. This indicates the milder effect of the mutation which may require a different mutation to cause severe phenotype. As of now this variant has not been reported from other geographical regions. Furthermore, the allele frequency of c.1000G>A among south Asian population is 0.0005 and has zero frequency in other populations in ExAC database. Hence, we suspect a possible founder affect for this mutation in South Asia and needs to be further explored. Table 1: Clinical features. Conclusion: This report further expands the emerging phenotypic spectrum of GMPPB associated dystroglycanopathies and indicates a probable South Asian founder mutation with milder effect in its homozygous form.
| Family | Year | Gender | Age | Onset | Clinical features | Creatine kinase | RNS | Family History |
| 1 | 2004 | M | 50 | 42 | Weakness: Proximal>distal, truncal weakness; Fatigue +; Scapular winging; Calf hypertrophy; Fatigue +; DTRs: 2+ | 650 | Significant decrement (>20%) | 3 siblings affected |
| 2 | 2006 | F | 35 | 30 | Weakness: Proximal; fatigue +; calf hypertrophy; DTRs 2+ | 1583 | elder sister affected | |
| 3 | 2016 | F | 32 | 22 | Weakness: Proximal; fatigue +; calf hypertrophy; DTRs 2+ | 1790 | Significant decrement (>20%) | Negative |
| 4 | 2017 | F (younger sister) | 45 | 28 | Weakness: Proximal and truncal weakness; chewing difficulty; fatigue +; mild ptosis (fatigable) and facial weakness; calf hypertrophy; DTRs 2+ | 4547 | Mild Decrement (<10%) | Maternal 1st cousin affected (M/44; onset: 18 yrs) |
| 2017 | F (elder sister) | 46 | 36 | Weakness: Proximal and truncal weakness; chewing difficulty; fatigue +; facial weakness; calf hypertrophy; DTRs 2+ | 2107 | Significant Decrement (>20%) |
PS1Group1-045 / #480NFAT5 AND P38 MAPKS INTERACT IN MUSCLE CELLS RESPONDING TO OSMOTIC AND INFLAMMATORY STRESS AND IN POLYMYOSITIS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Boel De Paepe1, Sandrine Herbelet2, Jens Schmidt3, Jan De Bleecker4
1Neuromuscular Reference Center, Ghent University Hospital, Ghent, BE;2Ghent University, Ghent, BE;3Neurology, University of Goettingen, Goettingen, DE;4Neurology, Ghent University, Ghent, BE
Background: The transcription factor Nuclear Factor of Activated T-cells 5 (NFAT5) is the key regulator of cells’ responses to osmotic stress, but is also implicated in inflammatory muscle disease. NFAT5 most likely is regulated by Mitogen-activated protein kinases (MAPKs) and the four members of the p38 family of MAPKs, termed MAPK11 (p38ß), MAPK12 (p38γ), MAPK13 (p38δ) and MAPK14 (p38α), have known associations with inflammation. Methods: We study MAPKs mRNA expression and protein activation in an in vitro muscle inflammation model, and in muscle from polymyositis patients. Results: We observe that in muscle cells in culture, exposure to increased salt concentrations and pro-inflammatory cytokines influence NFAT5 mRNA expression and translocation to the nucleus. A maximal 4-fold increase of NFAT5 messenger levels in myotubes treated with IL1ß and IFNγ+IL1ß for 24h is detected, in the latter condition accompanied by a moderate increase of MAPK12 and MAPK13 phosphorylation. Neither MAPK14 expression nor phosphorylation is substantially altered by cytokines and increased NaCl concentrations. Longer exposure of myotubes in culture to cytokines does not increase NFAT5 nor MAPK14 expression, yet hyperosmotic conditions lead to a time-dependent increase of expression, reaching 9-fold (NFAT5) and 18-fold (MAPK14) after 72h. At this stage, culture densities decrease substantially, disfavoring the differentiated multinucleated cells and leaving myoblasts as the remaining cell developmental stage. In muscle tissues, levels of phosphorylated MAPK11/12/14 relative to actin content, increase from 0.52±0.24 in normal (n=4), to 0.87±0.23 in polymyositis (n=4), but this difference does not reach statistical significance (p=0.08). Immunofluorescent localization studies show expression in blood vessel endothelium and in a subset of small, most often CD56+ (regenerating) muscle fibers. Conclusion: We identify p38 MAPKs as possible phospho-activators of NFAT5 in muscle cell’s responding to inflammatory stress, and put forward MAPK12 as a likely regulator relevant to inflammatory myopathy.
PS1Group1-046 / #551IMPLICATION OF THE BREAKPOINTS POSITION IN PATIENTS WITH THE MACRODELETION OF EXONS 45 TO 55
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Javier Poyatos Garcia1, Clara Gomis Coloma2, Nuria Muelas3, Pilar Martí4, Juan J. Vílchez4
1Neurology, Instituto de Investigación Sanitaria la Fe, Valencia, ES;2Neurology, INSTITUTO DE INVESTIGACIÓN SANITARIA LA FE, Valencia, ES;3Neurology, Hospital Universitari i Politècnic La Fe, Valencia, ES;4Neurology, Hospital Universitari i Politècnic La Fe, Universitat de València and CIBERER, Valencia, ES
Background: Duchenne muscular dystrophy (DMD) is a severe myopathy that affects to 1 out of 3500 -5000 newborn boys and it is responsible of severe disability and early death, caused by mutations in the DMD gene. Becker muscular dystrophy, is an allelic form, which manifest a benign phenotype. Sixty five per cent of the mutations in the DMD consist on intragenic deletions that disrupt the reading frame of the gene, while in BMD the reading frame is preserved. An emerging gene therapy in DMD patients consist on inducing skipping of an exon that recover the reading frame in a restrictive group of cases. Multiexon skipping is another approach that can be achieved with the new Crispr-Cas9 technology. In particular, 63% of DMD mutations are located between exons 45 to 55 and it has been observed the existence of a spontaneous deletion spanning that region in asymptomatic subjects or variable Becker phenotype. Our aim is to analyze the diverse clinical profile and to refine the underlying molecular mechanisms in a cohort of subjects with a 45-55 deletion. Our hypothesis is that the different location of the intronic breakpoint positions of these patients might be responsible of their clinical phenotype. Methods: We analyzed the exact position of the intronic breakpoints in 8 index patients with this deletion. We performed an array, and multiple PCRs in order to restrict the area of the breaking points in the introns 44-45 and 55-56, following by amplification and sequencing of the deletion junction to detect the exact breakpoint We also checked the clinical features of the patients in an attempt to correlate them with their sequences. Results: A group of 6 patients shared the same breakpoints in both introns. These patients presented variable clinical manifestation (2 asymptomatics, 2 mild BMD, 1 BMD and 1 with cardiomyopathy). In one of the other 2 patients, the deletion affected the regulatory regions of the dystrophin isoform Dp140 and in fact the patient presented ADHD (attention deficit hyperactivity disorder), dyslexia and dysgraphia. The last patient presented a proximal insertion of a stretch from a deleted region in the 44-45 intronic breakpoint. This patient showed an early onset cardiomyopathy. Conclusion: We cannot certainly affirm that t the position of the intronic breakpoints in patients with deletion 45-55 is related with the clinical features of these patients, nevertheless we found some associations. We detected a major hotspot, where 6 of 8 patients share the same breakpoints and present variable clinical manifestations, where we can find asymptomatic patients (suggesting that there are other factors that may have a greater impact on the clinical manifestation of the disease, such as age or genetic context). The impairment of the regulatory regions of Dp140 in our patient has a consequence in his cognitive status. Moreover the other type of deletion may be associated with an early apparition of cardiomyopathy, nevertheless more patients with the last 2 deletion categories must be analyzed in order to stablish these hypothesis.
PS1Group1-047 / #627THE STUDY OF ALISKIREN IN MDX DYSTROPHIC MICE
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Thainá A. Marin1, Bruno M. Bertassoli1, Giuliana Petri1, Jose F.R. Santos1, Vinicius G. Silva2, Fernando L.A. Fonseca3, Alzira A.S. Carvalho4, David Feder1
1Pharmacology, Faculdade de Medicina do ABC, Santo Andre, BR;2Faculdade de Medicina do ABC, Santo Andre, BR;3Laboratory Analysis, Faculdade de Medicina do ABC, Santo Andre, BR;4Neurology, Faculdade de Medicina do ABC, Santo Andre, BR
Background: Duchenne Muscular Dystrophy (DMD) is a genetic neuromuscular disease linked to the X chromosome, caused by mutation of dystrophin gene (DYS) that encodes the dystrophin protein, responsible for the stability of the sarcolemma. In the absence / deficiency of dystrophin, the muscle cells are susceptible to damage induced by continuous cycles of degeneration and limited regeneration. Inflammation, necrosis and fibrosis are the pathophysiological characteristics that lead to the main consequence of DMD, the loss of muscle function. Aliskiren is one of the newer drugs that interact with the Renin Angiotensin Aldosterone System (RAAS), directly inhibiting renin. High levels of circulating renin activate the pathologic signaling pathway of fibrosis in several diseases through stimulation of the pro-renin receptor, whose mechanism is completely independent of both Angiotensin II (ang II) production and stimulation of Ang II type 1 receptor. Methods: We treated 8-weeks-old male mdx mice. 8 mice received 25 mg / kg of Aliskiren daily for 5 weeks and 8 mice received 0,2ml of saline solution(0,9%), both by gavage. Measure muscle strength (Kondziela test) was performed weekly, as well as the weighing of each animal. After 5 weeks, all animals were euthanized, followed by muscles biopsy of extensor digitorum longus (EDL), tibialis anterior (TA) and diaphragm muscles in order to analyse morphological features. The creatine kinase (CK) was also performed. Results: No changes were observed in the animals weight during the study. The hold impulse calculated by the Kondziela Test did not show significant differences between treated and untreated animals. CK values between the groups did not differ significantly. Regarding the Feret’s diameter, a significant increase of fibers’diameter higher than 40um was observed in diaphragm muscle of treated group (p <0.05). In addition, TA of treated group showed a significant increase in fibers” diameter higher than 60um (p <0.05) was also demonstrated. In contrast, EDL did not show difference in fiber diameter of both groups. Conclusion: Treatment with aliskiren in mdx dystrophic mice for 5 weeks did not change the weight of animals. The strength was also unaffected in treated group. The preliminary analyzes of the morphological muscles findings not show some beneficial effect of aliskiren in this model. Long-term drug studies already under way, and measure of fibrosis and inflammatory cytokines will be very helpful to determine the real effect of aliskiren in DMD.
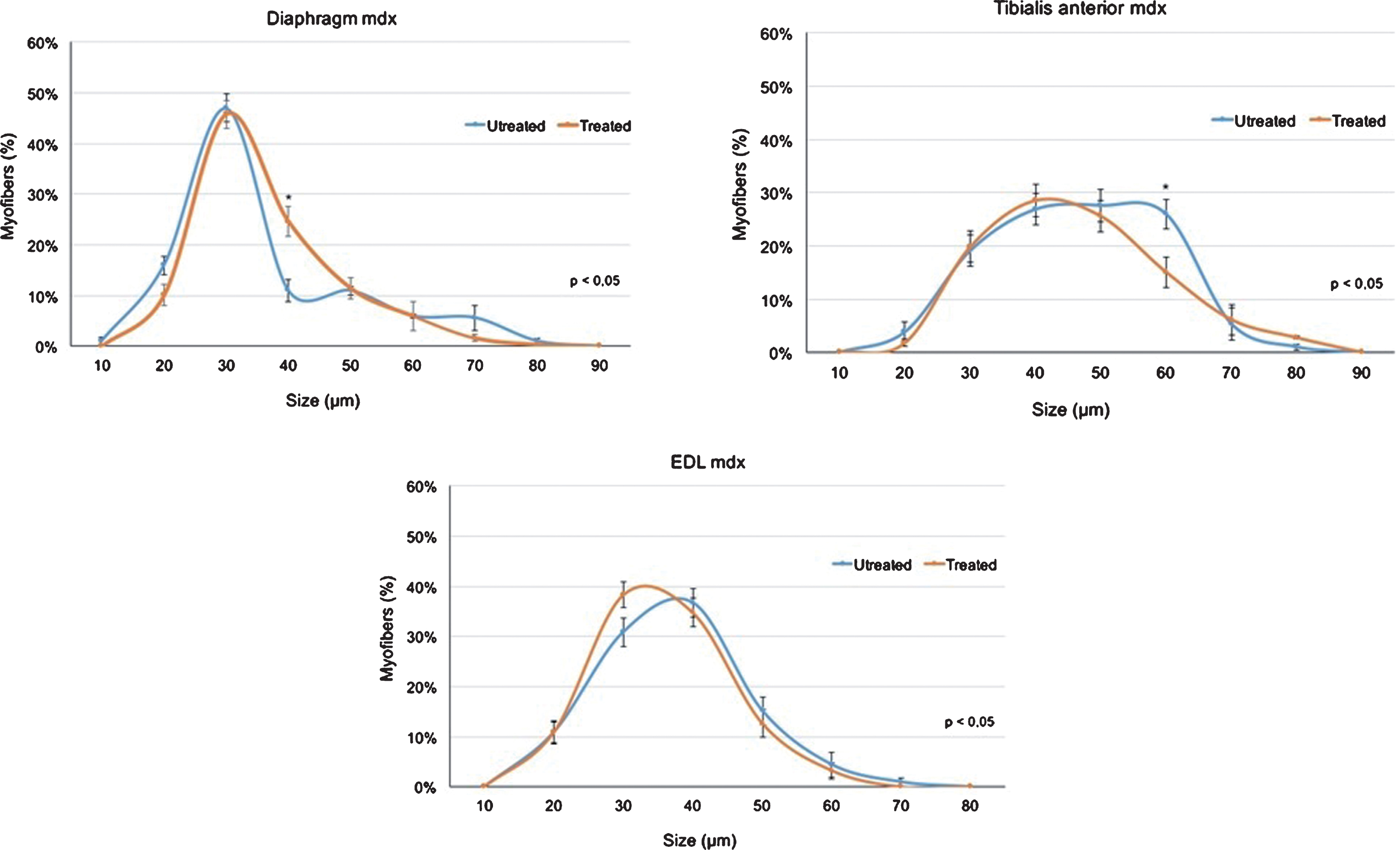
PS1Group1-048 / #691DUCHENNE MUSCULAR DYSTROPHY: DO BOYS WITH A SHORTER STATURE MAINTAIN AMBULATION LONGER?
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Léa Labove1, Magdalena Jaworski1, Cam-Tu Emilie Nguyen2
1CHU Sainte-Justine, Montreal, QC, CA;2Pediatric Neurology, CHU Sainte-Justine, Montreal, QC, CA
Background: Duchenne muscular dystrophy (DMD) is an X-linked progressive neuromuscular disorder caused by mutations in the DMD gene and affecting approximately 1:5000 male births, leading to loss of ambulation and premature death. While many promising therapies are currently being developed, corticosteroids remain the cornerstone of pharmacological management in DMD. Stunted growth is a frequent side effect of steroids in DMD patients. It is not uncommon for families and patients to voice concerns about this “visible” side effect of corticotherapy. Some clinicians have ventured that a shorter stature may confer a mechanical advantage in DMD, delaying the loss of ambulation in the shorter boys. Methods: Our aim was to study the relationship between height and the age at loss of ambulation in our population of DMD boys through a single-center retrospective chart review. Seventy boys with DMD were identified and their charts reviewed. The median age at diagnosis was 4.2 years, and median age of corticosteroid initiation was 7.5 years. Out of these 70 boys, 40 had lost ambulation at the time of this study, at a mean age of 11.6 years. We separated the boys who had lost ambulation in two groups: those who lost ambulation before 11.6 years and those who lost ambulation after 11.6 years, and compared these two groups in terms of height, using the Student’s T-test. Results: At age 10, the boys who lost ambulation after 11.6 years were on average 11.9 cm shorter than the boys who lost ambulation before 11.6 years (p<0.0001), a difference that is both statistically and clinically significant. When we looked at the height at age 7 instead of 10, the difference between the two groups was not statistically significant (+1.5 cm for the boys who lost ambulation before 11.6 years; p=0.3731). Given that the difference in average height between the two groups appears to develop between the ages of 7 and 10, we hypothesize that the height differential is due to the effect of corticosteroids, which were initiated at a median age of 7.5 years. Conclusion: In our cohort, DMD boys who lost ambulation at a later age were significantly shorter than those who lost ambulation earlier. It is not yet clear if this is due to a mechanical advantage related to the shorter stature, or if the shorter stature could represent a biomarker of optimal corticotherapy response in DMD. This important finding should be validated in multicentric DMD cohorts.
PS1Group1-049 / #898LOW LEVELS OF DYSTROPHIN PROTEIN ASSOCIATED WITH ATTENUATION OF DUCHENNE MUSCULAR DYSTROPHY PROGRESSION
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Johannes Dworzak1, Frederick Schnell2, Jay S. Charleston1, Karin Lucas2, Doug Sanders1, Diane Frank2
1Sarepta Therapeutics, Inc., Cambridge, MA, US;2Sarepta Therapeutics Inc, Cambridge, MA, US
Background: Duchenne muscular dystrophy (DMD) is a degenerative, fatal, X-linked recessive disorder affecting 1 in 3500 to 5000 male births worldwide. It is marked by an absence of dystrophin in muscle fibers stemming from DMD gene mutations. Absence of dystrophin leads to progressive loss of muscle function. The precise amount of dystrophin associated with clinically meaningful benefit in DMD patients is unclear, although numerous findings indicate that small amounts of dystrophin, associated with natural mechanisms such as spontaneous exon skipping, may lead to slower disease progression. We explored potential associations between low dystrophin levels in DMD patients and attenuation of disease progression. Methods: A literature search was conducted to identify recent relevant articles evaluating the natural occurrence of exon skipping and clinical disease progression in DMD patients. Results: A body of evidence indicates that DMD patients with mutations amenable to exon 44 skipping experience a milder disease course than other DMD subgroups. In one natural history study, patients with mutations amenable to exon 44 skipping experienced a slower rate of disease progression, with loss of ambulation (LOA) 2 years later than those with other mutations, yet traces of dystrophin were detected in only 3 of 6 patients by immunohistochemistry and in 0 of 4 patients by Western blot analysis using clinical diagnostic methods. Notably, in a subanalysis of corticosteroid-treated patients, those with mutations amenable to exon 44 skipping did not achieve a median age of LOA during follow-up, while those with mutations amenable to exon 51 skipping had a median age at LOA of 13.0 years. In a separate analysis of corticosteroid-treated DMD patients, the median age at LOA was 15.2 years in patients amenable to exon 44 skipping and 10.5 years in patients amenable to exon 51 skipping. Spontaneous low-level exon 44 skipping in these patients is thought to facilitate restoration of the messenger ribonucleic acid reading frame, facilitating dystrophin expression and/or the presence of dystrophin-positive muscle fibers. An analysis utilizing the NorthStar Ambulatory Assessment demonstrated that patients with mutations amenable to exon 44 skipping declined less rapidly over 2 years than patients with mutations amenable to exon 53 or 51 skipping. A separate study showed that patients with mutations amenable to exon 44 skipping had higher baseline values on the 6-minute walk test and more modest declines at 12 months than those with mutations amenable to skipping exons 45 or 53 (differences were not statistically significant). Some evidence suggests that DMD patients with deletions of exons 3–7 also experience a milder phenotype and express low levels of dystrophin. A recent case study described a patient aged 10 years with a nonsense mutation in exon 42 and a mild phenotype that was presumably the result of spontaneous exon 42 skipping and subsequent dystrophin production. Conclusion: These findings demonstrate that spontaneous exon skipping and production of low dystrophin levels may impact the clinical course of the disease, and offer insights regarding the interpretation of results from clinical trials evaluating therapeutic interventions designed to restore dystrophin in DMD patients.
PS1Group1-050 / #934TAMOXIFEN PROLONGS SURVIVAL, IMPROVES MOTOR FUNCTION AND REDUCES LEVELS OF DNM2 IN MTM1-NULL MICE, A MODEL OF XLCNM
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Elinam Gayi1, Laurence A. Neff1, Hesham M. Ismail1, Xènia Massana-Muñoz2, Belinda S. Cowling2, Jocelyn F. Laporte2, Leonardo Scapozza1, Olivier M. Dorchies1
1School Of Pharmaceutical Sciences, University of Geneva, Geneva, CH;2Department Of Translational Medicine And Neurogenetics, University of Strasbourg, Illkirch, FR
Background: X-linked centronuclear myopathy (XLCNM) is a rare and severe congenital myopathy characterised by generalised muscle weakness and abnormal nuclei positioning. Most affected boys die in their first year of life and survivors fail to achieve independent ambulation. It is caused by mutations in the Mtm1 gene encoding myotubularin, a ubiquitously expressed phosphoinositide phosphatase. No cure exists and very few pharmacological avenues are being explored. Here, Mtm1-null mice were treated with tamoxifen (TAM), a drug that modulates oestrogen actions and that we have previously shown to be efficacious in dystrophic (mdx5Cv) mice, a model of Duchenne muscular dystrophy (DMD). We report that TAM is also effective in Mtm1-null mice, a model of XLCNM. Methods: Wild type and Mtm1-null mice were given normal chow or a TAM supplemented chow starting at weaning. Survival was monitored, and mice underwent weekly grid-tests, weigh-ins and clinical scorings to evaluate disease progression. Mice were kept until Day 42, 84 or >200; at each of these time points in-vivo force recordings were performed in the triceps. Hind leg muscles were used for histological, ultrastructural and biochemical analyses. Results: Non-treated Mtm1-null mice died at around 45 days. By contrast, about half of the Mtm1-null mice treated with clinically relevant doses of TAM survived beyond 290 days of age. Clinical scoring showed that the motor function of the affected mice was markedly improved. In vivo force recordings performed at D42 and D84 revealed that the force of treated Mtm1-null was significantly improved after as short as 3 weeks of treatment. Histological and electron microscopy analyses show partial rescue of muscle structure and triads, consistent with improved calcium homeostasis in FDB fibres and better excitation-contraction coupling. Quantitative PCR and western blots demonstrated reduction of DNM2, a disease modifier in XLCNM. Conclusion: We found that tamoxifen extends the lifespan of Mtm1-null mice more than 6-fold, rescues their motor skills and corrects levels of known disease modifiers. These results suggest that oestrogen signalling may be a key pathway that modifies disease severity in unrelated myopathies such as XLCNM and DMD.
PS1Group1-051 / #961ARE THERE DIFFERENT CLINICAL ENTITIES WITH DISTINCT DISEASE COURSE AMONG D4Z4 REDUCED ALLELE CARRIERS?
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Giulia Ricci1, Fabiano Mele2, Lucia Ruggiero3, Elisabetta Bucci4, Lorenzo Maggi5, Monica Govi2, Francesco Sera6, Liliana Vercelli7, Lucio Santoro3, Tiziana Mongini7, Luisa Villa8, Maurizio Moggio8, Massimiliano Filosto9, Marina Scarlato10, Stefano C. Previtali10, Michelangelo Cao11, Elena Pegoraro12, Roberta Telese13, Antonio Di Muzio13, Carmelo Rodolico14, Giovanni Antonini4, Maria Grazia D’Angelo15, Angela Berardinelli16, Rachele Piras17, Maria Antonietta Maioli17, Giuliano Tomelleri2, Corrado Angelini18, Gabriele Siciliano1, Rossella Tupler2
1Department Of Clinical And Experimental Medicine, University of Pisa, Pisa, IT;2Department Of Life Sciences, University of Modena and Reggio Emilia, Modena, IT;3Department Of Neurosciences, Reproductive And Odontostomatological Sciences, University Federico II of Naples, Naples, IT;4Department Of Neuroscience, Mental Health And Sensory Organs, S. Andrea Hospital, University of Rome “Sapienza”, Rome, IT;5Besta Neurological Institute, Milan, IT;6Mrc Centre Epidemiology For Child Health, UCL Institute of Child Health, London, GB;7Department Of Neuroscience, Center For Neuromuscular Diseases, University of Turin, Turin, IT;8Fondazione Irccs Ca’ Granda Ospedale Maggiore Policlinico, Dino Ferrari Center, University of Milan, Milan, IT;9Neurology Clinic, Spedali Civili Hospital, University of Brescia, Brescia, IT;10Inspe And Division Of Neuroscience, IRCCS San Raffaele Scientific Institute, Milan, IT;11Department Of Neurosciences, University of Padova, Padova, IT;12University of Padova - Azienda Ospedaliera di Padova, Padova, IT;13Center For Neuromuscular Disease, Cesi, University “G. D’Annunzio”, Chieti, IT;14Department Of Neurosciences, Policlinico “g. Martino”,, University of Messina, Messina, IT;15Department Of Neurorehabilitation, IRCCS Institute Eugenio Medea, Bosisio Parini, Bosisio Parini, IT;16Unit Of Child Neurology And Psychiatry, IRCCS“C. Mondino” Foundation, Pavia, Pavia, IT;17ASL8, Centro Sclerosi Multipla, Cagliari, IT;18IRCCS S.Camillo Hospital, Venice, IT
Background: The phenotypic classification of patients with facioscapolohumeral muscular dystrophy (FSHD) is crucial for genetic studies and the definition of outcome measures toward trial readiness. The Italian Clinical Network for FSHD has recently proposed a new FSHD Comprehensive Clinical Evaluation Form (CCEF) that defines nine clinical categories aimed at capturing clinical diversity. In particular category A, subcategories A1-A3, identifies subjects with facial and scapular girdle weakness, subcategory B1 identifies patients showing scapular muscle weakness without facial muscle involvement. Methods: We applied the CCEF on 814 index cases carrying of contracted alleles with 1-10 D4Z4 repeats from the Italian National Registry for FSHD. Results: Our analysis suggests that the facial-sparing FSHD phenotype (subcategory B1), representing 10,2% of index cases, is associated with milder motor disability and slower progression of muscle weakness when compared with the classical FSHD phenotype (category A), regardless the size of the D4Z4 contracted allele. Conclusion: These results highlight the importance of building a precise phenotypic classification of patients and families for a correct stratification of patients to identify the best for trials.
PS1Group1-052 / #285A PH1/2 STUDY OF ENA® ANTISENSE OLIGONUCLEOTIDE (DS-5141B) WITH EXON 45 SKIPPING ACTIVITY IN PATIENTS WITH DMD
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Masafumi Matsuo1, Hiroyuki Awano2, Setsuo Hasegawa3, Satoru Inoue4, Naoyuki Maeda4, Hirofumi Komaki5
1Department Of Physical Therapy, Faculty Of Rehabilitation, Kobe Gakuin University, Kobe, JP;2Pediatrics, Kobe University Graduate School of Medicine, Kobe, JP;3Pharmaspur Inc., Tokyo, JP;4Daiichi Sankyo Co., Ltd., Tokyo, JP;5Child Neurology, National Center of Neurology and Psychiatry, Tokyo, JP
Background: Duchenne muscular dystrophy (DMD) is a progressive, lethal neuromuscular disorder caused by the absence of dystrophin due to mutation of the DMD gene. Antisense oligonucleotide (AO)-mediated exon skipping allows restoration of the dystrophin reading frame, resulting in the synthesis of partially functional dystrophin for DMD patients who have out-of-frame deletions. This exon skipping treatment strategy for DMD is now being studied using various modified nucleic acids. The most important drawback of AOs that target DMD exon skipping is not delivered into cardiac muscle. DS-5141b is an AO consisting of 2’-O,4’-C-ethylene-bridged nucleic acids (ENA®) and 2’-O-methyl RNA (2’-OMe) and induces DMD exon 45 skipping. Our ENA® oligonucleotides have suitable characteristic for nucleic acid therapeutics. They can form more thermodynamically stable duplexes with complementary RNA than other oligonucleotides and are highly stable in plasma. In pre-clinical studies, the potent efficacy and safety of DS-5141b was observed. DS-5141b at doses over 3 mg/kg induced distinct exon 45 skipping in anterior tibial muscles, diaphragm and heart in mdx mice. On the other hand, 2’-OMe AO and a phosphorodiamidate morpholino oligomer (PMO) AO with the same nucleotide sequence as DS-5141b did not show the clear exon-skipping activity at doses up to 30 mg/kg. It was remarkable that the exon 45 skipping was detected in heart at such a low dose suggesting cardiac improvement (Neuromuscular Disorders Vol. 27 Suppl. 2, 2017, Page S216). Methods: To assess the safety, tolerability, efficacy, and pharmacokinetics of DS-5141b in patients with DMD, a phase 1/2 study has been conducted as a multicenter, uncontrolled, open-label study in Japan (NCT02667483). This study was a first-in-human study consisted of 2 parts. Part 1 was a sequential dose escalation part to examine 4 dose levels (0.1, 0.5, 2.0 and 6.0 mg/kg). DS-5141b was administered subcutaneously at 2 dose levels each in 2 cohorts and administration of each dose level was once weekly for 2 weeks (Cohort 1: 0.1 and 2.0 mg/kg, Cohort 2: 0.5 and 6.0 mg/kg). In Part 2, subcutaneous administration of DS-5141b was conducted for 12 weeks at 2 dose levels (2.0 and 6.0 mg/kg). Eligible subjects were ambulant DMD male patients aged 5-10 years with a mutation amenable to exon 45 skipping. Safety assessments include adverse event, laboratory tests, change of weight, vital sign, and ECG. The primary efficacy endpoint of this study is dystrophin expression and the secondary is detection of in-frame DMD mRNA harboring exon 45 skipping in muscle tissue. Results: A total of 7 subjects had received DS-5141b and all of them had completed administration for 12 weeks in Part 2. No serious adverse events and adverse events resulting in discontinued administration have been reported. Conclusion: Results of the Ph1/2 study and properties of DS-5141b will be presented.
PS1Group1-053 / #517MOST LGMD2L MUTATIONS IN THE ANO5 GENE RESULT IN DECREASED PROTEIN LEVELS IN ANOCTAMINOPATHY PATIENTS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Anna Vihola1, Helena Luque2, Marco Savarese2, Sini Penttilä3, Giorgio Tasca4, Bjarne Udd5
1Medical Genetics, Medicum, Folkhalsan Institute of Genetics, Uni Helsinki, Helsinki, FI;2Medical Genetics, Medicum, Folkhalsan Institute of Genetics, Uni Helsinki, Helsinki, FI;3Neuromuscular Research Center, University and University Hospital of Tampere, Tampere, FI;4Fondazione Policlinico Universitario “a. Gemelli”, Istituto di Neurologia, Università Cattolica del Sacro Cuore, Rome, IT;5Department Of Neurology, Vaasa Central Hospital, Vaasa, FI
Background: ANO5 (TMEM16E) is an eight-pass, 107 kDa membrane protein, possibly playing a role in membrane dynamics. Mutations in the ANO5 gene are a frequent cause of recessive limb-girdle muscular dystrophy, LGMD2L, and Miyoshi muscular dystrophy -like disease MMD3, with distal phenotype. More than 70 different mutations, dispersed all over the ANO5 gene have been identified. The most frequent mutation in European populations is the truncating c.191dupA in the N-terminal domain of the ANO5 protein, whereas in Finland, the point mutation c.2272C>T is the most common. In addition, there are numerous patients with phenotypes compatible with anoctaminopathy, but with just one confirmed ANO5 mutation found in genetic analysis, leaving the diagnosis unsolved with conventional approach. Methods: Studying the effects of ANO5 mutations in patient biopsies at protein level for diagnostic purposes has become possible only recently, as we developed a novel western blotting method, using isolated membrane fractions, suitable for endogenous ANO5 detection in human muscle tissue. Anoctaminopathy patients in this study were confirmed by genetic analysis. Results: Using this technique, a commercial mouse monoclonal antibody detected a single 107 kDa ANO5 protein in normal muscle biopsy membrane fractions. The ANO5 band was clearly decreased in most anoctaminopathy patients tested, except for a patient with a very N-terminal point mutation, located in the soluble cytoplasmic domain of ANO5. Conclusion: We conclude that ANO5 has low tolerance for mutations, leading in most cases to protein destabilization and degradation. The method described here allows direct analysis of human ANO5 protein, which can be used in diagnostics, for evaluating the pathogenicity of the potentially harmful ANO5 variants of uncertain significance.
PS1Group1-054 / #628CRISPR/CAS GENE EDITING IN DUCHENNE MUSCULAR DYSTROPHY CULTURES TO TEST NEW TREATMENTS FOR THE DISEASE
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Patricia Soblechero-Martin1, Iker García-Jiménez2, Estibaliz Ruiz-Del-Yerro2, Edurne Albiasu-Arteta2, Virginia Arechavala-Gomeza1
1Neuromuscular Disorders, Biocruces Health Research Institute, Barakaldo, ES;2Neuromuscular Disorders, Biocruces Health Research Institute, Barakaldo, ES
Background: Gene editing techniques have the possibility of becoming a permanent cure to many genetic disorders, but the delivery hurdle of these techniques will delay their practical application, particularly in neuromuscular disorders where muscle represents an extremely large and widespread target. However, these techniques have a more immediate application in the generation of better disease models in a field where not many good human myogenic cultures are available. Methods: The aim of our studies is to use gene editing to generate a specific DMD deletion in healthy control myoblasts cultures and then correct this deletion also by gene editing, the first objective will provide us with a model to test many possible mutation-specific treatments, such as antisense oligonucleotides, while the second is a proof of concept of a possible therapeutic gene editing option. Results: For our first objective, we have designed specific guide RNAs to edit the DMD gen removing exon 52 in order to reproduce a common deletion in Duchenne patients. We have confirmed the efficacy of our designs and selected the best candidates in HEK293 cells. Once selected, the best candidate gRNAs have been transfected in a control myoblast culture to create this new “DMD-like” culture. To complete our second objective, we have designed gRNAs targeting exon 51 and followed the same methods, this time on a DMD culture missing exon 52, to restore dystrophin expression. Conclusion: Issues such as safe and efficient delivery to target regions stop gene editing from being an immediate therapeutic option, but, as shown in this poster, gene editing is a useful tool to create better disease models to help in the development of other treatments and could be a feasible option to ex vivo treatment of cultures before autologous transplant.
PS1Group1-055 / #693EXTRA-SKELETAL MUSCLE MANIFESTATIONS OF FACIOSCAPULOHUMERAL MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Cecilia Kelly, Elie Naddaf
Mayo Clinic, Rochester, MN, US
Background: Facioscapulohumeral muscular dystrophy (FSHD) is the third most common cause of muscular dystrophy. It is inherited in an autosomal dominant pattern. Over 95% of FSHD cases are type 1, due to a contraction in the D4Z4 repeats on chromosome 4p. In its classic form, FSHD is characterized by weakness of facial and shoulder girdle muscles, and can be associated with hearing loss and rarely, retinal vascular abnormalities. However, the extra-skeletal muscle manifestations of FSHD remain poorly explored. In this study, we aimed to better characterize the spectrum of such manifestations in a large, single center cohort of FSHD patients. Methods: We performed a retrospective chart review of patients seen between 2000 and 2017 carrying the diagnosis of FSHD. We only included patients with genetically-confirmed FSHD type 1 or 2. We reviewed medical records for evidence of extra-skeletal muscle manifestations in the following domains: 1) hearing loss, captured by audiometry and/or hearing aid use; 2) cardiac disease, captured by electrocardiogram, Holter, and/or echocardiography; 3) dysphagia, captured by video swallow study; 4) retinopathy, captured by ophthalmologic exam; and 5) respiratory dysfunction, captured by pulmonary function tests (PFTs) and/or overnight oximetry. Results: We identified 88 patients, 39 of whom (44%) were female. At time of evaluation, median age was 44 years (range 1.5-89), and 69 patients (78%) were adults (>18 years old). Hearing loss was present in 10 of 15 (67%) tested patients, 8 of whom were diagnosed in adulthood. Ocular abnormalities were found in 5 of 25 (20%) patients; however, none were noted to have retinal telangiectasias or exudates. Electrocardiography was abnormal in 29 of 56 patients (52%) and echocardiography was abnormal in 18 of 35 tested patients (51%). Oropharyngeal dysphagia was documented in 8 of 17 (47%) patients who underwent video swallow study, most commonly classified as mild to moderate. PFTs were abnormal in 25 of 27 patients (93%): 8 had a restrictive pattern and 23 had reduced maximal respiratory pressures. Overnight oximetry showed sleep-related disordered breathing in 14 of 24 patients (58%). A single patient had focal epilepsy diagnosed in adulthood. We will analyze the correlation between the various extra-skeletal muscle manifestations and the number of D4Z4 repeats, and present it at the time of the conference. Conclusion: Hearing loss, cardiac abnormalities, dysphagia, and respiratory insufficiency were relatively common in our cohort. However, retinal vascular abnormalities were not encountered. Increased awareness regarding the various extra-skeletal muscle manifestations of FSHD allows timely screening, and subsequently providing better patient care.
PS1Group1-056 / #717ANALYSIS OF CARDIAC TROPONIN T ALTERNATIVE SPLICING IN SKELETAL MUSCLE OF DM1 PATIENTS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Francesca Bose’1, Laura V. Renna1, Nicola Ferrari2, Valentina Labate3, Michele Cavalli4, Giovanni Meola5, Rosanna Cardani6
1Laboratory Of Muscle Histopathology And Molecular Biology, IRCCS Policlinico San Donato, San Donato Milanese (MI), IT;2Department Of Biomedical Sciences For Health, University of Milan, Milan, IT;3Department Of Cardiology Heart Failure Unit, IRCCS Policlinico San Donato, San Donato Milanese (MI), IT;4Department Of Biomedical Sciences For Health, IRCCS Policlinico San Donato, Milan, IT;5Department Of Biomedical Sciences For Health, University of Milan, San Donato Milanese (MI), IT;6IRCCS-Policlinico San Donato, San Donato Milanese (MI), IT
Background: Myotonic dystrophy type 1 (DM1) is an autosomal dominant neuromuscular disorder caused by expanded CTG repeats in the DMPK gene. Nuclear accumulation of the mutant RNA leads to aberrant alternative splicing of different genes that have been linked to the multiorgan involvement that characterized the pathology. Alteration of cardiac Troponin T (cTnT) alternative splicing observed in DM1 patients leads to the co-expression of both adult (excluding-exon 5) and fetal (including-exon 5) isoform in cardiac tissue and it is considered one cause of cardiac dysfunctions in DM1. Recently, cTnT aberrant splicing pattern has been observed also in skeletal muscle of DM1 patients. The aim of this work is to study the cTnT alternative splicing in biceps brachii (BB) muscle biopsies and in tibialis anterior (TA) samples from DM1 patients and to investigate if it is related to cardiac dysfunctions observed in these patients. Methods: cTnT fetal isoform expression was evaluated by RT-PCR using primers flanking exon 5 on both BB (n=12) and TA samples (n=11) from DM1 patients and healthy subjects (CTR; n=6 BB, n=4 TA). Patients were divided in two main groups on the basis of cardiac evaluation performed by ECG, echocardiogram and 24h ECG-Holter. Patients with cardiac involvement present at least one out of four altered parameters (increased PR, QRS, QTc, reduced %FE). Results: Both in BB and TA samples, a significantly higher expression of cTnT fetal isoform was observed in DM1 patients presenting cardiac involvement than in CTR. Moreover, in TA samples a correlation between the expression of fetal cTnT with QTc and %FE was present. In order to verify if the alteration of cTnT splicing is related to skeletal muscle alterations, we analysed aberrant alternative splicing of muscle specific genes (CLCN1, CACNA1S, NFIX, SERCA1, TNNT3, DMD1, LDB3, RYR1 and BIN1) and histopathological parameters of muscle damage (atrophic and hypertrophic factor, % of fibers with central nuclei, % of MHC embrional and neonatal positive fibers) on both BB and TA muscle biopsies. No correlation was found between cTnT fetal isoform expression and the parameter of skeletal muscle impairment. Conclusion: In conclusion, fetal isoform expression both in BB and TA skeletal muscle of DM1 patients seems to be related to cardiac involvement and could represent a possible biomarker of heart dysfunction in these patients.
PS1Group1-057 / #784IMPACT OF IDEBENONE ON RESPIRATORY BURDEN, INCLUDING RISK OF BRONCHOPULMONARY COMPLICATIONS, IN DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Oscar H. Mayer1, Mika Leinonen2, Shabir Hasham3, Thomas Meier2, Thomas Voit4, Gunnar Buyse For The Delos Study Group5
1Children’s Hospital of Philadelphia, Philadelphia, PA, US;2Santhera Pharmaceuticals, Pratteln, CH;3Santhera Pharmaceuticals, Pratteln, CH;4UCL Great Ormond Street Institute of Child Health, London, GB;5University Hospitals Leuven, Leuven, BE
Background: In Duchenne muscular dystrophy (DMD), respiratory function declines due to progressive muscle weakness, and accelerates once patients become non-ambulatory. As well-described clinical thresholds are crossed (based on forced vital capacity, expressed as a percentage of the predicted value; FVC%p), the risk and severity of respiratory complications, including bronchopulmonary adverse events (BAEs),1 increases, presenting an ever worsening disease burden. In the 52 week, Phase 3 clinical trial in DMD patients not taking glucocorticoids (DELOS), we examined the potential for idebenone to reduce the rate of respiratory function decline and the rate of BAEs. Methods: Using a combination of pre-specified and post-hoc analyses, we assessed data from the DELOS trial, which enrolled 64 DMD patients aged 10-18 years and already in respiratory decline (peak expiratory flow %p < 80%), to determine the potential of idebenone (900mg/day) to reduce disease burden by delaying the time to reaching clinically relevant respiratory function thresholds, and the frequency of BAEs. Clinically relevant thresholds for FVC%p, were compiled following a review of standard of care guidelines. We compared the number of patients (idebenone vs placebo) crossing these thresholds (FVC%p of 50%, 40%, 30%) or experiencing BAEs. Results: Treatment with idebenone reduced the risk of crossing the clinically relevant FVC%p threshold of 50% compared to placebo (hazard ratio = 0.34, (95% CI: 0.10 – 1.18). A similar trend was observed for patients crossing any clinically relevant FVC%p threshold (50, 40 or 30%) (hazard ratio = 0.51, 95% CI: 0.23 – 1.14). Patients in the idebenone group were also at a lower risk of experiencing a BAE than those in the placebo group (hazard ratio = 0.28, 95% CI: 0.12 – 0.64). Conclusion: In the DELOS trial, idebenone treatment significantly reduced the proportion of patients crossing any clinically relevant threshold of FVC and also reduced the risk of bronchopulmonary adverse respiratory events, further supporting the efficacy and utility of treatment with idebenone in patients with DMD in established respiratory function decline and not taking glucocorticoid therapy. 1. Mayer OH, et al. US Neurology 2017;13:35-41.
PS1Group1-058 / #811CLINICAL AND MOLECULAR FEATURES OF DISTAL MYOPATHIES IN A PORTUGUESE COHORT: REPORTING NOVEL GENE MUTATIONS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Catarina Falcão De Campos1, Miguel Oliveira Santos1, Isabel Conceição2, Mamede De Carvalho2
11- Departamento de Neurociências e Saúde Mental, CHLN- Hospital Santa Maria. Lisboa, Portugal 2- Instituto de Fisiologia, Universidade de Lisboa, Faculdade de Medicina, IMM. Lisboa, Portugal, Lisbon, PT;21- Departamento de Neurociências e Saúde Mental, CHLN- Hospital Santa Maria. Lisboa, Portugal 2- Instituto de Fisiologia, Universidade de Lisboa, Faculdade de Medicina, IMM. Lisboa, Portugal, Lisbon, PT
Background: Distal myopathies (DM) are a clinically and genetically heterogeneous group, which selectively or disproportionally affect distal muscles. DM are classified according to age of onset, inheritance pattern, clinical features and molecular diagnosis. Recent developments in identifying the underlying gene defects have increased the number of disorders, to more than 20 entities. Some forms of DM have higher prevalence in certain regions, such as Welander Myopathy in Sweden and Udd Myopathy in Finland. We describe the phenotypic and molecular features of the first cohort of Portuguese patients with DM, reporting novel gene associations. Methods: We retrospectively assessed patients with DM and molecular diagnosis followed in our center. Demographic and clinical data, familiar history, neurophysiological and pathological findings as well as gene mutations were collected. Results: We identified 123 patients with myopathy followed in our center which only 9 patients (7,3%) were classified as DM. Two thirds of the patients with DM had molecular genetic diagnosis, including four novel mutations. The demographic and clinical features are described in table 1. Three patients were diagnosed as tibial muscular dystrophy (Udd myopathy) associated with two likely pathogenic titin (TTN) gene mutations not previously described in the literature. Two patients were father and son and presented with the same TTN gene mutation. One young patient had diagnosis of Miyoshy myopathy, with a likely pathogenic novel mutation in the dysferlin gene. An older patient presented with progressive upper and lower limbs distal weakness. Five years later the patient developed cognitive deterioration suggestive of a frontotemporal dementia, without associations with Paget disease. A novel gene mutation in the valosin-contain-protein (VCP) was diagnosed. Lastly, we report a patient with lower limb distal weakness and hypertrophic cardiomyopathy associated with MYH7 gene mutation. Conclusion: We summarize the first cohort of Portuguese patients with DM associated with genetic mutations, including 4 novel mutations. To our knowledge, we describe for the first time a Portuguese patient with IBMPFD diagnosis associated with a novel VCP gene mutation. Molecular genetic investigation was essential since some of our patients are sporadic cases.
| Patient | Gender, Age (y) | Diagnosis | Age of onset | Onset of weaknes | CK | EMG | Muscle Biopsy | Other features | Family History | Molecular diagnosis |
| 1+ | M, 66 | Udd Myopathy | 54 | Legs Anterior compartment | 101U/L (normal) | Myopathic | Variation in fiber size, splitting and nuclear centralization | Ø | Yes (son) | TTN c.63652T>C* heterozygous |
| 2+ | M, 39 | Udd Myopathy | 30-35 | Legs Anterior compartment | N/A | Myopathic Myotonic discharge | N/A | Ø | Yes (father) | TTN c.63652T>C* heterozygous |
| 3 | M, 61 | Udd Myopathy | 50 | Legs Anterior compartment | 427U/L (increased) | Myopathic | Unspecific changes | Ø | Yes (father and brother) | TTN c.74366C>G* heterozygous |
| 4 | M, 32 | Miyoshi Myopathy | 25 | Legs Posterior compartment | 9090U/L (increased) | N/A | Dystrofic changes Absence of dysferlin | Ø | No | DYSF c.3367–3368del* homozygous |
| 5 | F, 61 | IBMPFD | 50-55 | Hands | N/A | Myopathic | Unspecific changes | Fronto-temporal dementia | No | VCP c.265C>T* heterozygous |
| 6 | F, 63 | Laing Distal Myopathy | 45 | Legs Anterior compartment | 174U/L (normal) | Myopathic Myotonic discharges | N/A | Hypertrophic cardiomyopathy (HCM) | Yed (son with HCM) | MYH7 c.2539-2542del c.3056C>A c.322C>T heterozygous |
+ Same family; * Novel gene mutation, likely pathogenic. CK - Creatine kinase; EMG - Electromyography; N/A - Non available IBMPFD - Inclusion body myopathy with Paget disease and fronto-temporal dementia TTN - Titin gene; DYSF - Dysferlin gene; VCP - Valosin-containing-protein gene; MYH7 - Myosin heavy chain 7 gene
PS1Group1-059 / #965DUCHENNE MUSCULAR DYSTROPHY AND THE HEART - HOW TO VISUALIZE BETTER? CASE SERIES REPORT
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Ruza Kalicanin Milanovic1, Ana Kosac2, Stefan Djordjevic3, Maja Bjelic3, Milan Djukic3, Jelena Mladenovic2, Jelena Glumac2, Tamara Ristic2, Majda Kiklic2, Ida Jovanovic3, Vedrana Milic Rasic2
1Health Centar Novi Pazar, Novi Pazar, RS;2Neurology, Clinic of neurology and psychiatry for children and youth, Belgrade, RS;3University children’s hospital, Belgrade, RS
Background: During the progressive course of disease of Duchenne muscular dystrophy monitoring of heart function and diagnosing the very first signs of potential heart failure is of vital importance. Aim: To correlate functional measures of patient achievements to different visualized procedures of the heart made at the same time period. Methods: We performed complete cardiological assessment, echocardiography, cardiac magnetic resonance imaging, biochemical parameters of patients diagnosed with Duchenne muscular dystrophy, confirmed by molecular genetic testing, in uniform fashion by three pediatric cardiologists educated in the same manner. All neurological assessment was performed by pediatric neurologists. Results: We included 16 boys with Duchenne muscular dystrophy, age range from 6 to 12 years (mean 9,2 +-1,7), from whom 87,5% were on stable doses of corticosteroid therapy and 81,2% were mobile, with six minute walk test in rage 182 to 579 m. Normal sinus rhythm (85-124/min) and cardiac auscultatory findings were found in all patients, with QTc interval from 0,36 to 0,43 seconds, slightly increased R/S in V1 in 9 (56,2%) patients and NT-proBNP from 7.69 to 231.6 pg/ml. In echocardiography parameters shortening fraction and ejection fraction were lower than 28% and 55% only in one patient (6,25%) 8 years old, independently mobile and on corticosteroid therapy. Cardiac magnetic resonance imaging revealed thin right ventricular myocardial wall and presence of left ventricular fibrosis in 25% of our patients, all mobile, on corticosteroid therapy and without echocardiography parameters of heart failure, with one patient having unexpectedly large fields of fibrosis despite excellent motor performances and normal echocardiography findings. Conclusion: Although echocardiography is a more available and easier accessible method, cardiac magnetic resonance imaging is becoming a superior diagnostic tool in the field of visualizing the first signs of potential cardiac dysfunction and failure in patients with Duchenne muscular dystrophy.
PS1Group1-060 / #753RATIONALE FOR EDASALONEXENT DOSE SCHEDULE IN PHASE 2 OF THE MOVEDMD® TRIAL
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Hanlan Liu1, Richard S. Finkel2, H.Lee Sweeney3, Krista Vandenborne4, Maria Mancini5, John F. Reilly6, Pradeep Bista6, Derek Wachtel7, Sachin Chandran8, Rafif Dagher9, Dominic Picarella9, Angelika Fretzen8, Andy Nichols10, Joanne Donovan5
1Dmpk And Clinical Pharmacology, Catabasis Pharmaceuticals, Inc., Cambridge, MA, US;2Nemours Children’s Hospital, Orlando, FL, US;3Myology Institute, University of Florida Health Myology Institute, Gainesville, FL, US;4Physical Therapy, 4University of Florida Health Physical Therapy, Gainesville, FL, US;5Clinical Operations, Catabasis Pharmaceuticals, Inc., Cambridge, MA, US;6Biology, Catabasis Pharmaceuticals, Inc., Cambridge, MA, US;7Biology Analytical, Catabasis Pharmaceuticals, Inc., Cambridge, MA, US;8Product Development, Catabasis Pharmaceuticals, Inc., Cambridge, MA, US;9Pharmacology & Toxicology, Catabasis Pharmaceuticals, Inc., Cambridge, MA, US;10Research And Development, Catabasis Pharmaceuticals, Inc., Cambridge, MA, US
Background: Duchenne muscular dystrophy (DMD) is a progressively debilitating and ultimately fatal inherited neuromuscular disorder caused by mutations in the gene encoding dystrophin. NF-kB is activated in DMD and drives inflammation and fibrosis, muscle degeneration and suppresses muscle regeneration. Edasalonexent is an oral small molecule that inhibits NF-κB. In the Phase 2 and the open-label extension (OLE) portions of the MoveDMD trial enrolling 4- to 7-year-old boys with DMD, edasalonexent given 33 mg/kg three times daily (TID) (100 mg/kg/day) demonstrated a preservation of muscle function and slowing of DMD disease progression through 60 weeks as compared to the rates of change during the control period prior to edasalonexent treatment. Methods: The MoveDMD Trial was designed to evaluate efficacy, safety/tolerability, pharmacokinetics (PK), pharmacodynamics (PD), and dose response of edasalonexent. The Phase 1 portion of the MoveDMD trial was a 1-week study to evaluate the safety, PK, and PD at 33 and 67 mg/kg/day, given in 2 divided doses, and 100 mg/kg/day given in 3 divided doses. Phase 2 was a 12-week placebo-controlled study with two cohorts given 33 mg/kg twice daily (BID) (67 mg/kg/day) and 33 mg/kg TID (100 mg/kg/day). Patients in the OLE continued with either dose regimen of edasalonexent. The dose schedule for the Phase 2 of the MoveDMD trial was selected based on nonclinical and clinical data including (1) the exposure/efficacy relationship observed in animal models; (2) the Phase 1 safety, tolerability, and PD in pediatric DMD patients; and (3) human PK. In the mdx mouse model of DMD, dose efficacy was driven by sustained systemic exposure. An oral dose of edasalonexent given TID showed greater pharmacological effects than the same dose given all at once daily despite similar AUC. Results: In the Phase 1 of MoveDMD trial, all three doses were well tolerated with no safety signals observed. Decreases in NF-kB driven gene expression were observed at total daily doses of 67 and 100 mg/kg, both of which were selected for the Phase 2 of MoveDMD trial. In Phase 1 PK studies in healthy volunteers, edasalonexent exhibited increasing exposure with food. Exposure plateaued at 33 mg/kg when administered with a low-fat meal. Patients typically eat meals three times per day, which as a practical matter limits dosing frequency to TID. To validate 33 mg/kg TID that provides sustained systemic exposure, a population PK model was developed and used to simulate the plasma concentration-time profile of 33 mg/kg TID and BID to reflect the regimen of Phase 2 of the MoveDMD trial. Coverage over the concentration range at which pharmacological activity was observed in the nonclinical model was greater with TID than with BID. Conclusion: In conclusion, data from preclinical studies highlight the importance of time above exposure threshold achieved by more frequent dosing. Consistent with nonclinical studies and human PK characteristics of edasalonexent, 33 mg/kg TID dose schedule in the Phase 2 and OLE portions of the MoveDMD trial showed positive dose response vs. 33 mg/kg BID and exhibited numerical improvements in physical function assessments vs. the off-treatment control period.
PS1Group1-061 / #816REPURPOSING TAMOXIFEN FOR SEVERE MYOPATHIES: FROM PRECLINICAL EVALUATION IN ANIMAL MODELS TO CLINICAL TRIALS IN PATIENTS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Olivier M. Dorchies1, Elinam Gayi2, Hesham M. Ismail2, Laurence A. Neff2, Xènia Massana-Muñoz3, Belinda S. Cowling3, Jocelyn F. Laporte3, Talya Dor4, Dirk Fischer5, Urs T. Ruegg2, Leonardo Scapozza2
1School Of Pharmaceutical Sciences, University of Geneva, Geneva, CH;2School Of Pharmaceutical Sciences, University of Geneva, Geneva, CH;3Department Of Translational Medicine And Neurogenetics, University of Strasbourg, Illkirch, FR;4Hadassah Medical Centre, Jerusalem, IL;5Department of Neurology, University of Basel Hospital, Basel, CH
Background: Duchenne muscular dystrophy (DMD), X-linked centronuclear (myotubular) myopathy (XLCNM/XLMTM) and congenital muscular dystrophy type 1A (MDC1A) are rare and fatal muscular diseases for which no cure exists. DMD is caused by the lack of dystrophin, a large protein that connects the extracellular matrix to the cytoskeleton in muscle fibres. DMD patients develop progressive muscle wasting, leading to cardiac and respiratory failure and death usually in their third decade. XLCNM is due to loss-of-function mutations in MTM1, a gene encoding the lipid phosphatase myotubularin. XLCNM patients show generalized hypotonia from birth and most of them die before the age of 2. The pathogenic mechanisms of XLCNM involve imbalance of phosphoinositide levels and of proteins involved in formation and trafficking of membrane vesicles. MDC1A is due to reduced levels or lack of laminin alpha-2, an extracellular matrix protein that plays critical roles in the stability of skeletal muscles and signalling to the myofibres. MDC1A patients show generalized hypotonia from birth and severe respiratory insufficiency. Methods: Tamoxifen, a selective oestrogen receptor modulator, is used for nearly 40 years to treat hormone-dependent breast cancers. We have tested the efficacy of tamoxifen in mdx5Cv mice, Mtm1-null mice and dyW mice, models of DMD, XLCNM and MDC1A, respectively. Results: We previously published that long-term tamoxifen improved the symptoms of DMD in mdx5Cv mice, of which force generation and fibrosis in the diaphragm and in the heart. Follow-up studies showed that tamoxifen doses as low as 0.3 mg/kg/day were efficacious. In 2017, tamoxifen was granted an Orphan Drug Designation for DMD by the European Medicines Agency. Based on our robust pre-clinical data, Talya Dor offered tamoxifen as a compassionate treatment to 3 DMD boys for over 24 months. Because of encouraging outcomes, she is now conducting a larger open-label trial with 30 DMD boys. In parallel, Dirk Fischer is leading “TAMDMD”, a multicentre, randomized, placebo-controlled, phase 3 clinical trial for assessing tamoxifen efficacy and safety in 100 DMD boys. Using the murine model of XLCNM, we found that clinically relevant doses of tamoxifen (3 mg/kg/day) corrected hallmarks of the disease, resulting in dramatic extension of the lifespan of XLCNM mice from around 40 days to >200 days. In dyW mice, the murine model of MDC1A, preliminary findings suggest that tamoxifen modestly increases the lifespan of affected mice, although it significantly enhances motor function as evaluated with a grid hanging test. Conclusion: Our work reveals the efficacy of tamoxifen in a variety of severe muscular diseases that have independent pathogenic mechanisms, as well as previously unrecognized roles of estrogen signalling as a disease modifier pathway in these fatal diseases. Our preclinical evaluation of tamoxifen in mdx5Cv mice paved the way to compassionate use and clinical trials for DMD boys. Hopefully, the launch of TAMDMD may increase the interest of clinicians for tamoxifen and accelerate clinical evaluation of that drug in patients with XLCNM. Further work is ongoing in our lab for evaluating the efficacy of tamoxifen for MDC1A.
PS1Group1-062 / #862THE CLINICAL VARIATION OF RYR1 GENE IN A LARGE FAMILY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Stacey Jankelowitz
Central Clinical School, University of Sydney, Sydney, AU
Background: RYR1 gene mutations are many and their clinical presentations are varied. There are at least 217 mutations of the gene that can result in malignant hyperthermia. The RYR1 gene codes for the ryanodine receptor which permits the passage of calcium ions in cells. Methods: This study describes the various manifestations of a single RYR1 gene mutation in a large family, including the proband, her children, her mother and her siblings. The presenting features and severity while linked and described are varied within the one family. Results: A single gene mutation may result in mild scoliosis, malignant hyperthermia and miscarriage as well as ptosis, kyphoscoliosis and weakness. The study of epigenetics will hopefully aid in the understanding of these disorders and their variable presentation. Conclusion: While gene tests are useful in determining the mutation responsible for clinical disorders, there is more to be learnt from gene expression to explain why a single gene disorder can have such varied presentation and differ in the severity of the illness within one family.
PS1Group1-063 / #940CLINICAL OUTCOME STUDY OF DYSFERLINOPATHY: WHAT ARE THE BEST FUNCTIONAL AND STRENGTH OUTCOME MEASURES FOR THIS POPULATION?
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Volker Straub1, Meredith James1, Anna Mayhew1, Marni Jacobs2, Jia Feng2, Simone Spuler3, Kate Bushby1, John Day4, Kirsti J. Jones5, Diana X. Bharucha-Goebel6, Emmanuelle Salort-Campana7, Alan Pestronk8, Maggie C. Walter9, Carmen Paradas10, Tanya Stojkovic11, Madoka Mori-Yoshimura12, Elena Bravver13, Jordi Diaz-Manera14, Elena Pegoraro15, Jerry Mendell16
1John Walton Muscular Dystrophy Research Centre, Newcastle University, Institute of Genetic Medicine, Newcastle Upon Tyne, GB;2Division Of Biostatistic And Study Methodology, Children’s National Health System, Centre for Translational Science, DC, WA, US;3Charite Muscle Research Unit, Experimental and Clinical Research Center, Berlin, DE;4Department Of Neurology, Stanford University, Palo Alto, CA, US;5Institute For Neuroscience And Muscle Research, Children’s Hospital At Westmead, University of Sydney, Sydney, AU;6Department Of Neurology, Children’s National Health System, Washington DC, US;7Neuromuscular And Als Center, La Timone Hospital, Aix-Marseille Universite, Marseille, FR;8Department Of Neurology, Washington University School of Medicine, St. Louis, MO, US;9Dept. Of Neurology, Ludwig-maximilians-university, Friedrich-Baur-Institute, Munich, DE;10Neuromuscular Unit, Department Of Neurology, Hospital U. Virgen del Rocio/Instituto de Biomedicina de Sevilla, Sevilla, ES;11Ap-hp, G.h. Pitié-salpêtrière 47-83, Boulevard De L’hôpital, Institut de Myologie, Paris, FR;12Department Of Neurology, National Center Hospital, National Center of Neurology and Psychiatry, Tokyo, JP;13Carolinas Healthcare System Neurosciences Institute, Charlotte, NC, US;14Neuromuscular Disorders Unit, Hospital de la Santa Creu i Sant Pau, Barcelona, ES;15Department Of Neuroscience, University of Padova, Padova, IT;16Center For Gene Therapy, Nationwide Children’s Hospital, Columbus, OH, US
Background: Dysferlinopathy, an autosomal recessive form of limb girdle muscular dystrophy, clinically presents with a variable spectrum of muscle weakness. The Clinical Outcome Study is an international 3 year natural history study of dysferlinopathy patients, which aims to gain a better understanding of the disease and to improve clinical trial readiness. Diagnostic criteria for inclusion in this study were a) two predicted pathogenic mutations or b) one predicted pathogenic mutation and absent mutation or c) one predicted pathogenic mutation and dysferlin protein level less than 20% as determined by blood monocyte testing. The cohort is 52% female 48% male. At baseline the ration of ambulant to non-ambulant was 3:1. The mean age was 40 years (range 12 to 88 years). The median years disease duration was 17 years (range 3-52 years). Here we aim to describe changes in functional outcome measures and muscle strength over a 2 year period in individuals with dysferlinopathy. Methods: 197 subjects across 15 sites in 8 countries underwent physiotherapy assessments including muscle strength (manual muscle testing, MMT; hand held dynamometry, HHD); and functional performance using the North Star Assessment for Dysferlinopathy (NSAD), Brooke score and Performance of Upper Limb, as well as timed tests (rise from floor, 10 metre walk / run, four stair climb and descend; Timed Up and Go, TUG; Six Minute Walk Distance), and respiratory function testing (forced vital capacity, FVC). Change was defined as significant difference in paired medians by Wilcoxon Signed Rank-Sum test (p<0.05). Results: Change is captured in this population consistently from baseline to year 1 and from year 1 to year 2 across a range of functional outcome measures. Motor performance assessed by NSAD demonstrated slow but consistent and significant deterioration in scores and confirmed NSAD as a dysferlin specific scale. All timed tests except rise from floor showed significant consistent decline between year 1 and year 2. Of the muscle groups assessed by HHD that showed significant change, the plantar flexors were the muscles with the highest mean differences (3.14 lbs, SD: 8.22). We detected a mild but significant decrease in FVC (mean difference 0.09L) Conclusion: Progression in dysferlinopathy can be demonstrated using functional outcomes and dynamometry as measured by physiotherapists over two years. Change between baseline, year 1 and year 2 were slow but relatively consistent and significant. Results will help to power future clinical trials.
PS1Group1-064 / #294BREATHING IS LIFE! INSPIRATORY MUSCLE TRAINING IN CHILDREN AND ADOLESCENTS LIVING WITH NEUROMUSCULAR DISEASES
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Anri Human1, Jennifer Jelsma2, Brenda M. Morrow3
1Physiotherapy, Sefako Makgatho Health Sciences University, Garankuwa, ZA;2Division Physiotherapy, University of Cape Town, Observatory, ZA;3Paediatrics And Child Health, University of Cape Town, Rondebosch, ZA
Background: People with neuromuscular diseases (NMD) have a high risk of morbidity and mortality caused by underlying respiratory muscle weakness and an ineffective cough. Inspiratory muscle training (IMT) aims to preserve or improve respiratory muscle strength; reduce morbidity; optimise ventilation and ultimately improve health-related quality of life (HRQoL). Inspiratory muscle training among children and adolescents with NMD is controversial, owing to differences in pathophysiology and potential risk of muscle damage in some conditions. Despite reports of potential benefits, there is insufficient evidence to guide clinical practice regarding the use of IMT in this sub-population. Methods: A pre-experimental, observational pre-test post-test study was used to determine changes in measures of spirometry, peak expiratory cough flow (PECF), inspiratory muscle strength , upper limb function and coordination (using the Motor Function Measurement (MFM) scale), adverse events and HRQoL (using the PedsQL tool). Eight participants (n=4 boys; mean age 12.71 ± 3.53 years) with a variety of NMD were included. Training consisted of 30 breaths, twice daily, five days a week, for six weeks with an electronic threshold device (Powerbreathe K3, HaB International Ltd, Southam, UK). Results: There were no significant changes in spirometry measures, PECF or HRQoL. Upper limb function and coordination (MFM score) improved from pre-to post-intervention (p<0.04). Measures of inspiratory muscle strength also improved significantly: maximal inspiratory mouth pressure (Pimax) (p<0.02), strength-index (p<0.02) and peak inspiratory flow (p<0.03). No adverse events occurred during the intervention period. Overall patient satisfaction with the IMT programme, on a 10-point visual analogue scale, was extremely high, with a median (IQR) of 9 (9-10). Conclusion: This study suggests that short term IMT may improve inspiratory muscle strength; as well as improving upper limb function and coordination. IMT may be a safe and effective adjunct intervention for children and adolescents with NMD, but larger, long-term randomised controlled trials are recommended.
PS1Group1-065 / #490PERSONALIZED MOLECULAR THERAPY IN A NEW TRANSLATIONAL LARGE ANIMAL MODEL FOR DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Tina Donandt1, Simone Reichert1, Maria Schmuck1, Claudia Kalbe2, Barbara Kessler3, Andreas Blutke4, Eckhard Wolf5, Sabine Krause1, Maggie C. Walter1
1Dept. Of Neurology, Ludwig-maximilians-university, Friedrich-Baur-Institute, Munich, DE;2Institute Of Muscle Biology And Growth, Leibniz Institute for Farm Animal Biology (FBN), Dummerstorf, DE;3Department Of Biochemistry, Molecular Animal Breeding And Biotechnology, Ludwig-Maximilians-University, Munich, DE;4Institut Für Tierpathologie, Ludwig-Maximilians-University, Munich, DE;5Gene Center And Department Of Biochemistry, Molecular Animal Breeding And Biotechnology, Ludwig-Maximilians-University, Munich, DE
Background: The development of efficient and safe gene therapy approaches in muscular dystrophies is a major challenge. Cell culture models derived from our large animal model, the DMD pig, provide an in vitro system to qualitatively and quantitatively evaluate therapeutic restoration of dystrophin. The DMD pig carries an exon 52 deletion in the dystrophin gene and mimics the most frequent human DMD mutation. A major hurdle is the low degree of myogenic differentiation in porcine satellite cells. As compared to other species, terminal differentiation of porcine myoblasts is very inefficient. Moreover, full-length dystrophin (Dp427) is not expressed until after the myoblasts begin myogenic differentiation. Methods: Primary satellite cells were isolated from pig skeletal muscle by consecutive enzymatic digestion. The fusion medium and the surface coating were optimised to facilitate myotube formation. Differentiation medium containing Ultroser G and matrigel/gelatin coated culture dishes promoted myotube surface adherence and maturation. Low levels of dystrophin protein in differentiating porcine cells demanded also an improvement of the Western blot technique. To enrich for myotubes, differential trypsination was successfully employed to yield sufficient differentiated cells for dystrophin Western blot detection. Results: Among key factors which affect myogenesis in vitro are the extracellular matrix proteins chosen to coat the culture plates and the choice of growth and differentiation media. Notably, a preplating step for 1-2 h did not improve the yield of myogenic cells. By means of optimised fusion media and culture dish coating, we significantly improved myogenic differentiation in terms of fusion index and dystrophin protein expression. It is well established that dystrophin is a low-abundance protein and constitutes less than 0.01% of total striated muscle protein. For protein extraction and Western blot detection in the porcine cell culture model, we enriched for myotubes to increase the dystrophin specific signal. Conclusion: Differentiated dystrophin-deficient porcine myoblasts provide a primary exon 52-deficient cell culture model ideally suited for evaluation of targeted RNA- or DNA-based molecular therapy. The efficiency of dystrophin restoration, potential toxicity, and additional side effects may be assessed by our preclinical approach and provides excellent translational impact for future treatment options in affected DMD patients.
PS1Group1-066 / #605DILATED CARDIOMYOPATHY AND LIMB-GIRDLE MUSCULAR DYSTROPHY-DYSTROGLYCANOPATHY REVEALING NEW DPM3 GENE MUTATIONS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Juliette Svahn1, Pascal Laforet2, Christophe Vial3, Nathalie Streichenberger4, Norma Romero5, Céline Bouchet-Séraphin6, Nathalie Seta7, Rita Menassa8, Françoise Bouhour3, Guillemette Jousserand3, Tanya Stojkovic2
1Electroneuromyography And Neuromuscular Department, Hospices Civils de Lyon, Bron, FR;2Sorbonne Universités, UPMC Univ Paris 06, INSERM UMRS974, CNRS FRE3617, Center of Research in Myology, Myology Institute, Paris, FR;3Electroneuromyography And Neuromuscular Department, Hospices Civils de Lyon, Bron, FR;4Centre de Neuropathologie Est - Hospices Civils de Lyon, Université Claude Bernard Lyon1, Institut NeuroMyogène - CNRS UMR 5310 - INSERM U1217, Bron, FR;5Unité De Pathologies Neuromusculaires, Institut De Myologie, UMRS974, Institut de Myologie, Paris, FR;6Département De Biochimie Et De Génétique, AP-HP, Hôpital Bichat, Paris, FR;7Biochemistry Laboratory, AP-HP, Hôpital Bichat, Paris, FR;8Department Of Molecular Endocrinology And Rare Diseases, Hospices Civils de Lyon, Bron, FR
Background: DPM synthase catalyzes the formation of DPM from Dolichol-phosphate and GDP-mannose(Man). The deficiency of Dolichol-P-mannose (DPM) synthase subunit 3 (DPM3) affects two particular glycosylation pathways: N-glycosylation and O-mannosylation which are respectively involved in congenital disorders of glycosylation (CDG) and alpha-dystroglycanopathies. Aberrant glycosylation of alpha-dystroglycan (aDG) leads to alpha-dystroglycanopathies. We describe novel pathogenic DPM3 mutations previously unreported in the literature in two unrelated male patients presenting limb-girdle muscular dystrophy (LGMD) and dilated cardiomyopathy. Methods: Clinical examination, cardiac and biologic investigations, muscle imaging, muscle biopsy and transferrin isoelectric focusing were performed. Next generation sequencing of 18 alpha-dystroglycanopathies genes was performed (B3GALNT2, B3GNT1, DAG1, DOLK, DPM1, DPM2, DPM3, FKRP, FKTN, GMPPB, ISPD, LARGE, POMGNT1, POMGNT2, POMK, POMT1, POMT2, TMEM5). Results: The two patients presented early and severe dilated cardiopathy, which turned out to be the main symptom in one of our patient. They developed limb-girdle muscular dystrophy predominating in pelvic girdle, in childhood and adulthood respectively. Laboratory investigations revealed elevated CK level. The deltoid muscle biopsies of both patients revealed moderate muscle dystrophy with fiber-size variation, multiple internal nuclei, and interstitial fibrosis. Immunolabelling of aDG (VIA4-1 antibody, Millipore) showed reduced alpha-dystroglycan immunostaining in patient 1 and thin and irregular sarcolemmal staining in patient 2. Muscle imaging displayed diffuse fatty degeneration in gluteus muscles, rectus femoris muscles and lower limb muscles with severe fatty replacement in the adductors, hamstrings and triceps surae muscles. Transferrin isoelectric focusing of both patients revealed an abnormal profile suggesting a congenital disorder of glycosylation (CGD) type 1 pattern, consistent with defective N-glycosylation (DPM3-CGD1). Next generation sequencing uncovered in the two patients a hemizygous deletion of DPM3 and a hemizygous pathogenic missense mutation in exon 2 c.41T>C, p.Leu14Pro, predicted to be pathogenic by in silico tools. Conclusion: Previously, defects in N-glycosylation and O-mannosylation were considered to cause separate groups of diseases but cases have been since described which linked congenital disorders of glycosylation (CDG) type I with dystroglycanopathies. DPM3 mutations have been previously reported in two patients. Lefeber et al., described a female child with mild LGMD2 and a homozygous missense mutation, who developed dilated cardiomyopathy in early adulthood (Lefeber et al., 2009). Recently, an adult female patient with isolated pelvic girdle muscle weakness and a homozygous missense mutation was reported (Van Den Bergh et al., 2017). In conclusion, patients with DPM3 mutations may present with a particular phenotype: mild to severe limb-girdle muscular dystrophy predominating in the pelvic girdle, early dilated cardiomyopathy and biochemical features of deficient protein N-glycosylation. These observations underline the importance of considering a DPM synthase defect in unsolved alpha-dystroglycanopathy patients and of performing serum N-glycosylation screening.
PS1Group1-067 / #637DYSFERLINOPATHY: PHENOTYPIC ASPECTS IN 24 MOROCCAN PATIENTS AND BENEFITS FROM MUSCLE EXERCISE AND CORTICOIDS IN SOME CASES
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Nazha Birouk1, Sana Sefiani2, Bouchra Kably3, Halima Belaïdi3, Reda Ouazzani3
1Clinical Neurophysiology Rabat, Speciality Hospital, Rabat, MA;2Neuropathology, Speciality Hospital, Rabat, MA;3Clinical Neurophysiology, Speciality Hospital, Rabat, MA
Background: Dysferlin deficiency is responsible for different phenotypes of muscle dystrophies including limb girdle syndrome and distal myopathies. This work aims to give a phenotype description of dysferlin deficient patients belonging to 22 Moroccan families and to describe the benefit from regular muscle exercise and corticosteroid treatment in some patients. Methods: All patients underwent clinical evaluation for muscle impairment distribution and orthopedic signs, CK dosage, electroneuromyography and muscle biopsy with immunocytochemistry for dystrophin, sarcoglycans and dysferlin. 3 patients received oral prednisone and 5 patients had a regular follow up with muscle exercise. Results: Twenty four patients aged 14 to 57 years were examined. There were 15 males and 9 females. Age at onset was 20,2±6,7 years (range: 12 to 35). 16 families had an established consanguinity, 11 families had more than 1 affected member. The phenotypes were as follows: Myoshi type in 7 patients, limb girdle syndrome was isolated in 5, associated to lower limbs distal muscles involvement in 7, to biceps brachialis muscles deficit in 4 and to tibialis anterior impairment in 1 patient. CK level was 1.5 to 6 X normal value. 3 patients had myalgia and inflammatory pattern on muscle biopsy. 1 patient had asymptomatic high CKemia. All patients had dystrophic changes on muscle biopsy with absence of dysferlin where as dystrophin and sarcoglycans were normal on immunocytochemistry. Functional disability was variable, 3 patients were wheel chair bound at the ages of 23, 25 and 38 years respectively. 3 patients received treatment with oral prednisone with a drastic benefit on functional disability. 5 patients had a sustained functional benefit from regular muscle exercise that was adapted to their deficits. Conclusion: Dysferlin deficiency was identified in 41.5% of muscle dytrophy families with limb girdle and or distal muscles impairment in our series. It seems to represent the second cause of muscle dystrophies in Morocco after sarcoglycanopathies. A screening for the responsible dysferlin gene mutations is necessary to identify a probable founder mutation in our population. We should consider the potential benefit from corticosteroids and regular muscle exercise in some patients.
PS1Group1-068 / #614LGMD DUE TO γ-SARCOGLYCAN DEFICIENCY (LGMD2C): STUDY OF NATURAL HISTORY IN 77 PATIENTS BELONGING TO 69 MOROCCAN FAMILIES
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Nazha Birouk1, Armel Ghislain Mpandzou1, Bouchra Kably2, Sana Sefiani3, Abdelaziz Sefiani4, Halima Belaïdi2, Reda Ouazzani2
1Clinical Neurophysiology Rabat, Speciality Hospital, Rabat, MA;2Clinical Neurophysiology, Speciality Hospital, Rabat, MA;3Neuropathology, Speciality Hospital, Rabat, MA;4Genetic, National Health Institut, Rabat, MA
Background: Sarcoglycanopathies (SGP) are autosomal recessive progressive limb-girdle muscular dystrophies, due to α-, β-, γ- and δ-sarcoglycan gene mutations, respectively leading to LGMD (limb girdle muscular dystrophy) 2D, E, C and F. In North Africa, γ-SG is the most common. We report a study of 77 patients from 69 Moroccan families with highlights to the disease progression. Methods: Throughout a 21-year time lapse, 77 patients, among 133 SGP, were regularly followed-up during an average time of 4.1 ± 3.4 years (SD) [Max:Min; 1-14]. Diagnosis of γ-SGP was established on protein immunostaining and/or genetic analysis (n = 69) and by findings obtained from siblings (n = 8). Disease progression was assessed annually in all patients by frequency, age and delay of the occurrence of the main functional incapacities, skeletal deformities, cardiac and respiratory impairments and by survival curve of the loss of ambulation. Functional scores (Research Medical Council, Vignos, modified Gardner-Medwin-Walton, walking distance and climbing stairs) were calculated in 35 patients regularly followed-up during five years. Results: Consanguinity rate was 69.6% (48/69 families), 40 (51.9%) patient were sporadic cases and 37 (48.1%) had positive familial history. Sex ratio was 0.8. Immunostaining analysis showed isolated γ-SG deficiency in 57/67 (85.1%) patients investigated and combined γ-SG deficiency with reduction of one or more other SG in 10/67 (14.9%). Genetic analysis showed Δ525T mutation in 15/23 (65.2%) patients investigated. Mean age at onset was 7.2 ± 2.8 years [1-19]. Loss of ambulation occurred after 7.5 ± 4.6 years [2-22] of disease duration; almost concomitant with scoliosis. From Kaplan Meier survival curve, median of loss ambulation was 8 ± 0.5 years (95% CI 7.03-8.96 years) with probability of 44.4%. Comparison tests of delay of the loss of ambulation showed no significant difference for gender, family history, parental consanguinity, and expression profile of γ-SG deficiency. Cardiac impairment was found in 10/53 (18.8%) patients at a mean age of 18.8 ± 10.6 years [8-45]. Main cardiac anomalies were left ventricular dysfunction either isolated or associated with dilated cardiomyopathy in 7/10 patients, incomplete right bundle branch block in 5/10, premature ventricular contraction in 3/10, and right ventricular dysfunction in 1/10. Restrictive respiratory syndrome was found in 21/24 (87.5%) patients at a mean age of 18.9 ± 8.2 years [9-43]; this syndrome became severe requiring non-invasive ventilation in 10 patients, at a mean age of 21.5 ± 9.4 years [11-43]. In the 35 patients selected for the functional assessment, scores measures changes were most significant in patients with loss of ambulation or in patients with early age at onset between 5-9 years. Conclusion: The analysis of SG natural history highlights disease prognosis and can thus help in anticipating medical management to improve patient’s quality of life.
PS1Group1-069 / #659DOES A NORMAL DYSTROPHIN STAINING IN MUSCLE BIOPSY RULE1 OUT THE MOLECULAR DIAGNOSIS OF DUCHENNE / BECKER MUSCULAR DYSTROPHY?
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Mira Ginzberg, Tally Lerman-Sagie, Dorit Lev, Ron Dabby, Menachem Sadeh, Esther Leshinsky-Silver
E. Wolfson M.D, Holon, IL
Background: Molecular genetic testing of the Dystrophin gene can confirm the clinical diagnosis of a dystrophinopathy without muscle biopsy in most individuals with Dhchenne uscular Dystrophy / Becker Muscular Dystrophy (DMD/BMD). The multiplex ligation-dependent probe amplification (MLPA) assay is the most powerful tool in screening for deletions and duplications in the dystrophin gene in patients with (DMD/BMD). Deletions of one or more exons account for about 65 % of DMD and about 85 % of BMD cases. MLPA is a quantitative method enabling the detection of deletions and duplications of all 79 DMD exons. MLPA can determine the size of the deletion or duplication and predict, whether the mutation disrupt the reading frame Methods: We describe two male siblings, born to healthy non consanguineous parents. Both have suffered since a young age from muscle weakness and recurrent episodes of rhabdomyolisis with high CK values. The muscle weakness has been improving constantly and now as grownup males, they have minimal selective proximal girdle muscle weakness. However, the older brother experiences recurrent episodes of severe rhabdomyolysis and atrial fibrillation. The younger brother, experiences fatigability and lately dilated cardiomyopathy was diagnosed. Their maternal grandfather, who died at the age of 39 years, because of cardiomyopathy, did not suffer from myopathy. Results: We describe two male siblings, born to healthy non consanguineous parents. Both have suffered since a young age from muscle weakness and recurrent episodes of rhabdomyolisis with high CK values. The muscle weakness has been improving constantly and now as grownup males, they have minimal selective proximal girdle muscle weakness. However, the older brother experiences recurrent episodes of severe rhabdomyolysis and atrial fibrillation. The younger brother, experiences fatigability and lately dilated cardiomyopathy was diagnosed. Their maternal grandfather, who died at the age of 39 years, because of cardiomyopathy, did not suffer from myopathy. Conclusion: Exon 1 deletion in the Dystrophin gene is the cause for BMD in these two brothers and probably the cause for cardiomyopathy in their grandfather. If the clinical presentation and the pattern of inheritance is suspicious enough, only molecular analysis of the dystrophin gene by sequencing and MLPA can lead us to the correct diagnosis. However, there are some pitfalls of MLPA in detecting DMD single exon deletion or duplication, because of false-positives due to sequence variations.
PS1Group1-070 / #820ESYN- AND CK9- PROMOTORS DRIVE MUSCLE-SPECIFIC EXPRESSION OF TRANSGENES IN VITRO AND IN VIVO
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Jinhong Meng1, John Counsell2, Simon Waddington3, Francesco Muntoni4, Jennifer Morgan5
1UCL Great Ormond Street Institute of Child Health, London, GB;2UCL Great Ormond Street Institute of Child Health, London, GB;3EGA Institute for Women’s Health, London, GB;4Dubowitz Neuromuscular Unit, UCL Great Ormond Street Institute of Child Health, London, GB;5University College London, London, GB
Background: Lentivirally-mediated dystrophin expression is a promising therapeutic approach for Duchenne muscular dystrophy (DMD). Lentivirus coding either full-length or mini- dystrophin can transduce patient-derived muscle stem cells, providing a valuable cell source for autologous transplantation. However, due to the large size of the dystrophin gene and the limited packaging capacity of the lentiviral vector, the promoter driving the transgene expression needs to be small. Furthermore, as dystrophin is mainly expressed in muscle fibres, the promoter must be active in differentiated muscle cells. Methods: In the present study, we tested the expression of a reporter gene driven by either ESyn- (547bp) or CK9- (429bp) promoters in comparison to the ubiquitous SFFV promotor, by transducing lentiviruses coding luciferase driven by the ESyn- , CK9- or SFFV- promoter into cultured human cells derived from lung, liver, kidney, neuroblastoma and skeletal muscle. Furthermore, these viruses were injected systemically into newborn outbred mice and the expression of luciferase was examined one month later using the IVIS Lumina III in vivo imaging system. Results: We found that cells transduced with ESyn- and CK9- lentivirus express significantly lower levels of luciferase in comparison to those transduced with SFFV- lentivirus. Interestingly, the expression of luciferase was dramatically increased in Esyn- and CK9- lentivirus-transduced muscle cells when they were induced to undergo myogenic differentiation. In mice injected with SFFV- lentivirus, luciferase was expressed in organs including liver, spleen and skeletal muscle, while in mice injected with Esyn- or CK9- lentivirus, luciferase was expressed mainly in skeletal muscles. Conclusion: our data show that both ESyn- and CK9- promoters drive skeletal muscle specific expression of a transgene, indicating that they are good candidates for driving expression of proteins within skeletal muscle.
PS1Group1-071 / #354E-CADHERIN IS ECTOPICALLY EXPRESSED IN THE MUSCLE FIBER OF INCLUSION BODY MYOSITIS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Chiseko Ikenaga, Hiroyuki Ishiura, Hidetoshi Date, Akatsuki Kubota, Shoji Tsuji, Tatsushi Toda, Jun Goto, Jun Shimizu
Department Of Neurology, The University of Tokyo, Tokyo, JP
Background: Both inclusion body myositis (IBM) and polymyositis (PM) are characterized by the endomysial infiltrates of CD8+ cytotoxic T-lymphocytes invading or surrounding major histocompatibility class 1 expressing non-necrotic muscle fibers (CD8-MHC-1 complex). Patients with IBM have rimmed vacuoles or abnormal protein accumulation and they are refractory to treatments, while patients with PM generally respond well to treatments. It seems to be important to clarify genes or pathways, which contribute to the difference of these two disease’s phenotypes. Methods: We analyzed frozen muscle biopsy samples from 62 patients with CD8-MHC-1 complex (43 IBM, 9 PM, and 10 unclassifiable) and 9 normal controls (NCs) by RNA-sequencing (Illumina Hiseq 2500). We detected the differentially expressed genes (DEGs) and performed pathway analysis by edgeR. In addition, we compared the gene expressions of cell surface antigens (CD1~CD371) among these groups. In order to evaluate the protein level of these DEGs and cell surface antigens, we performed immunohistochemistry on frozen sections of muscle biopsy samples from patients with IBM, PM, dermatomyositis (DM), immune-mediated necrotizing myopathy (IMNM), and NCs, respectively. Results: Compared with NCs, there were 8588 DEGs in IBM (FDR<0.05) and the sets of genes concerning cancer, immune system, and NFkB/TNF/PI3K-Akt/JAK-STAT/TGF-ß signaling pathways were upregulated, while the sets of genes concerning translation, transcription, and mitochondrial function were downregulated in IBM. Compared with NCs, there were 4744 DEGs in PM (FDR<0.05) and the sets of genes, which were upregulated or downregulated in PM, were the same as those in IBM. When we compared IBM with PM, there were 1419 DEGs (FDR<0.05) and the gene sets of the innate and adaptive immune systems were more upregulated in IBM than those in PM. Among these DEGs, there were 28 genes, in which the expressions of IBM were twice as much as those of PM (FDR<0.05) and 12 genes of cell surface antigens including E-cadherin (FDR<0.01). In immunohistochemistry, the muscle fibers of IBM, which were diffusely positive for p62, were also positive for E-cadherin. On the other hand, the muscle fibers, which were positive for dense p62 aggregates, were not stained with E-cadherin antibody. Although there were some muscle fibers, which were diffusely positive for p62 in the samples of IMNM and DM, they were not stained with E-cadherin antibody, except for the partial staining of perifascicular fibers with atrophy in DM. Conclusion: IBM and PM shared most of the upregulated/downregulated pathways. Among them, the gene sets of the innate and adaptive immune systems were upregulated in the muscle of IBM, compared with those of PM. Among the DEGs, E-cadherin, which is known to be expressed in the epidermis, was highly expressed in the muscle of IBM. Considering the ectopical expression and the unique staining pattern of E-cadherin in the muscle of IBM, it may have some roles in the pathogenesis of IBM.
PS1Group1-072 / #399CARE EVALUATION OF DUCHENNE MUSCULAR DYSTROPHY PATIENTS IN BRAZIL
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Karine C. Turke, Alzira A.S. Carvalho, David Feder
Faculdade de Medicina do ABC, Santo André, BR
Background: The Duchenne Muscular Dystrophy (DMD) is the most frequent genetic lethal disease in early childhood of boys. International protocols for surveillance and treatment are broadly diffused and our objective was to assess if the patients with DMD in Brazil receive adequate treatment accordingly to those protocols. Methods: Through social networks, parents and responsible person were invited to answer to an online form. The questionnaire addressed conditions of the diagnosis, exams and treatments. Results: 54 patients were included in the study. The average age was 13,0 +1,2 years. 79,62% of the boys was medicated with corticosteroids, where 76,7% used deflazacort; 18,1% prednisolone and 4,65% prednisone. In the diagnosis, 31,4% were submitted to a skeletal muscle biopsy, and 79,6% knew the genetic mutation. 40,7% had cardiac alteration. The first electrocardiogram (ECG) and echocardiogram (Echo) happened at 8,4 and 8,8 years old respectively. The ECG was not performed yearly in only 23,5% of the cases, and the Echo in 26,4%. 50% of the patients went through the holter, and 9,2% was submitted to a cardiac magnetic resonance image. Only 31,4% performed a strength test every 6 months. Just 22,11% of the studied cases attended rehabilitation programs. 79,5% of the patients over 6 years of age performed spirometry, with an average of 10,5 years old. In the assessment of coughing effort only 34,7% of the patients over 6 years old submitted to the exam. The annual spirometry was performed by 48,1% of the participants. 37% had an appointment with occupational therapist and 31,4% with a nutritionist in the last year. 59,2% of the patients had done bone densitometry, and 37,5% of those were diagnosed with osteoporosis. Only 57,4% of the participants was vaccinated against influenza in the last year. Conclusion: It is concluded that the international protocols of surveillance and management of the patients with DMD are not being followed in Brazil. This could contribute to a greater mortality and morbidity. Enhancing medical education about DMD through disclosure protocols as well as providing patient access to specialized health care and medication adherence would be meaningful to improve the quality of life of DMD patients.
PS1Group1-073 / #891GENOTYPE AND PHENOTYPE FEATURES OF BRAZILIAN PATIENTS WITH MCARDLE DISEASE
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Alzira A.S. Carvalho1, Vinicius G. Silva2, Miriam E. Koch3, Pamela O. Delgado4, Bruno M. Bertassoli2, Roseli Corazzini4, Karine C. Turke4, Denise M. Christofolini5, Itatiana Rodart5, Fernando L.A. Fonseca6, David Feder2
1Neurology, Faculdade de Medicina do ABC, Santo Andre, BR;2Pharmacology, Faculdade de Medicina do ABC, Santo Andre, BR;3Faculdade de Medicina do ABC, Santo Andre, BR;4Faculdade de Medicina do ABC, Santo André, BR;5Genetics, Faculdade de Medicina do ABC, Santo Andre, BR;6Laboratory Analysis, Faculdade de Medicina do ABC, Santo Andre, BR
Background: Glycogenosis Type V(GTV) also known as McArdle disease, is a rare autosomal recessive disorder characterized by exercise intolerance, myalgia and painful cramping since childhood or adolescence associated to persisted elevated creatine kinase(CK). Mutations in the gene cause the disorder causing inability to produce the enzyme known as phosphorylase or myophosphoryase in muscle. The objective was to analyze clinical, epidemiological and genetic aspects as well as, to demonstrate the morphological muscular findings in 18 patients with McArdle disease. Methods: The clinical parameters evaluated were: cramps, myalgia, fatigue and dark urine. CK, modified ischemic exercise test (TI), hand-grip strength (using dynamometer) were also analyzed. The muscular morphological aspects analyzed were the percentage of internal nuclei, vacuoles and Feret’s using H&E stain. The PYGM gene mutation was analyzed by screening exons 1, 15 and 17 followed by next generation sequencing (TruSight Inherited Disease panel from Illumina Inc.) when the mutation was not found. Results: The total of 18 patients were analyzed: performed muscle biopsy: n=15; CK: n=18; ischemic exercise test(IET) n=16; hand-grip strength: n=16; genetic test: n=14 (the remaining four patients were siblings belonging to three distinct families and presented the same symptoms, elevated CK and muscle biopsy compatible with GTV the four). 56% men; 44% women. Considering clinical symptoms, 66,7% of them refer cramps, 72.2% myalgia, 100% fatigue and 66,7% dark urine. Fibers with internalized nuclei (FIN): 10%; vacuoles: 16,1%; type 2 fiber predominance (T2FP): 62,3%. Feret’s diameter ranged from 21.5 to 83.5 um (type 1) and 34.6-78.2 um (type 2). CK at rest: 1767 U / L (median). IET was consisted with GTV in 100% of patients without side effects. Hand-grip strength was 44.3 kg (median). The morphological parameters vacuoles and FIN were not statistically significant when correlated. The T2FP and Feret’s diameter were significant, p = 0.0000 and p = 0.0007 respectively). The most frequent mutation was R50X in homozygous (50%). The second most frequent was association of R50X and c.1967 A>C (17%). One patient had 4 mutations that “in silica” showed to be pathogenic. There was no correlation among the parameters evaluated, even among members of the same family. Conclusion: 1. CK is not related to the histological alterations analyzed. 2. IT is a good screening test in the differential diagnosis of patients with IE and without risks. 3. The T2FP suggests a compensatory mechanism in the use of fast fiber energy. 4. The R50X Arg50Ter mutation was the most frequent in our sample. Our study was the pioneer in the analysis of the histological phenotype versus genotype. 5. The results regarding the heterogeneity of the parameters analyzed suggest that GTV presents a clinical, histological and molecular individual phenotype. Therefore, genotype and histological phenotype are not prognostic factors of the disease.
PS1Group1-074 / #571PROPOSED CUT-OFF FOR REACTIVITY OF ANTI-HMGCR AND ANTI-SRP ANTIBODIES IN PATIENTS STATIN-EXPOSED AND STATIN-UNEXPOSED
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Alzira A.S. Carvalho1, Thaiane F. Vieira1, Miriam E. Koch1, Vinicius G. Silva1, Roseli Corazzini1, Pamela O. Delgado1, Karine C. Turke1, Fernando L.A. Fonseca2, David Feder3
1Faculdade de Medicina do ABC, Santo André, BR;2Laboratory Analysis, Faculdade de Medicina do ABC, Santo Andre, BR;3Pharmacology, Faculdade de Medicina do ABC, Santo Andre, BR
Background: Introduction: Few reports have been published demonstrating that anti-HMGCR and anti-SRP are specific to immune-mediated necrotizing myopathy (IMNM). The therapeutic approach with statins is widely used in the control of dyslipidemias. However, there is no laboratory evaluation to elect patients to make use of this class of therapeutic drugs. Aim: To analyze the prevalence of anti-SRP and anti-HMGCR antibodies in a heterogeneous cohort of 85 patients to determine cut-off reference values for these antibodies. Methods: Serum samples from 85 patients were screened for the presence of anti-HMGCR and anti-SRP autoantibodies by ELISA. The demographic, clinical and morphological features were also correlated with anti-HMGCR and anti-SRP antibodies. The patients were divided in two groups; A: statin-exposed and B: statin-unexposed. Results: The mean age of groups A and B were 61.1 and 39.5 years, respectively. Anti-HMGCR and anti-SRP antibodies were present in 12.6% and 14.1% of patients, respectively. 57 statin-exposed and 28 statin-unexposed. There were no significant associations of anti-HMGCR and anti-SRP titers with age, gender, exposure to statins or symptoms; however, significantly more symptomatic patients had been exposed to statins (p = 0.003). There was no significant difference in anti-HMGCR levels between asymptomatic and symptomatic patients (p = 0.994). Conclusion: Our results defined antibody cut-off points for a heterogeneous population with different diagnoses. We also demonstrated that anti-HMGCR and anti-SRP antibodies are not 100% specific for IMNM. Eleven (12.9%) patients were positive for anti-HMGCR antibodies and 12 (14.1%) were positive for anti-SRP antibodies in this heterogeneous cohort.
PS1Group1-075 / #573NECROTIZING MYOPATHY AFTER DENGUE: CASE REPORT
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Alzira A.S. Carvalho1, Karine C. Turke1, David Feder2
1Faculdade de Medicina do ABC, Santo André, BR;2Pharmacology, Faculdade de Medicina do ABC, Santo Andre, BR
Background: Muscle involvement is rare in dengue. Usually just myalgia and rarely as myositis, hypokalemic paralysis, or rhabdomyolysis. Methods: We report a case of necrotizing myopathy occurring in a 38-year-old Caucasian woman was referred to emergency unit care complaining of low grade fever for 2 days and severe myalgia in lower limbs followed by a skin rush and thrombocytopenia. Results: The diagnosis of the dengue viral infection was confirmed on the basis of the positive serum IgM antibody. Creatine kinase levels persisted elevated after 1 year from diagnosis with minimal complains. Her first assessment revealed muscle weakness, which was proximal and asymmetric in the lower limbs accompanied by mild muscle atrophy of left lower limb. A muscle biopsy of the deltoid showed myopathic changes, including moderate variation in fiber size and necrosis, suggesting a necrotizing myopathy. Protein immunohistochemistry studies showed normal immunoexpression except a diffuse MHC class I over-expression. A genetic panel of Next Generation Sequencing containing ANO5, DYSF, GAA, SGCB, SGCG, CAPN3, FKRP, SGCA, SGCD, and TCAP genes was done and with no mutations found. Conclusion: This report is the first one to describe dengue associated with necrotizing myopathy. We should to think in infection disease in patients presenting persisted creatine kinase levels with almost no symptoms. We also suggest that reviews of creatine levels and muscle biopsy in a large cohort of patients with dengue fever would be necessary to confirm this impression.
PS1Group1-076 / #492EXPRESSION OF DYSTROPHIN ISOFORMS IN NEW DUCHENNE MUSCULAR DYSTROPHY MICE MODEL
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Tatiana Egorova1, Dmitry Vlodavets2, Denis Reshetov3, Evgenia Zotova3, Anna Polikarpova3, Svetlana Vassilieva3, Alexei Deikin1
1Core Facility, Institute of Gene Biology RAS, Moscow, RU;2Russian Children Neuromuscular Center, Veltischev Scientific Research Clinical Pediatric Institute, Pirogov Russian National Research Medical University, Moscow, RU;3Research Department, Marlin Biotech LLC, Moscow, RU
Background: New Duchene muscular dystrophy mice model was created in Russia using CRISPR/Cas9 gene editing system. Two sgRNAs targeted to introns 7 and 34 respectively were injected into mice zygotes with Cas9 protein. Cas9 introduces double-strand breaks which are repaired by cell machinery with random indel formation. As a result of microinjections into zygotes 20 mice were born with desired deletion of exons 8-34 (DMDdel8-34). Sequence verification of deletion borders revealed several groups with different mutations. Methods: Three mice representing different deletion borders with 3, 8 and 18 base pair gaps between target sites were used for breeding. For all of them sgRNAs targeted intronic regions and they have same mutations on mRNA level. Third and fourth mice generations after their breeding were analyzed using histology, physiology and molecular biology methods. Results: Here we present some features of the model. New model demonstrates representative dystrophy symptoms on muscle histology such as central nucleation in myofibers, fibrosis and calcinosis formation in striated muscles. Great difference was shown between control (CBA/C57Bl6) and experimental group (DMDdel8-34) in hanging test. Quantitative RT-PCR was used to check dystrophin mRNA presence in knockout mice. It was expected that in this case dystrophin-coding RNA should be destroyed by nonsense-mediated decay because deletion of exons 8-34 leads to frameshift in 35 exon. Surprisingly there was no difference in mRNA expression level between control and experimental groups when using primers to dystrophin exons 1-7. Dystrophin mRNA expression can be explained by alternative splicing. To verify that there is no dystrophin protein expression western blotting assay was performed on primary myoblasts derived from murine model muscles using polyclonal antibodies raised against C-terminus of dystrophin. Absence of full-length (427kDa) dystrophin form was verified but 70kDa band was detected which corresponds to Dp71 dystrophin C-terminal isoform normally expressed in non-muscle tissues such as lung or brain. Therefore, we can suggest the presence of alternative dystrophin isoforms in DMDdel8-34 muscle cells. Conclusion: The finding needs further investigation because any treatment strategy tested on the model require functional restoration of dystrophin. Short dystrophin isoforms with noncanonical expression profile existing in model muscles can occupy dystrophin binding sites on cell membrane. Even in the case of restored expression of proper muscular isoform, functionality of the protein should be investigated.
PS1Group1-077 / #759MYOTONIC DYSTROPHY TYPE 2 AS A MULTISYSTEM DISEASE
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Ivo Bozovic1, Stojan Peric1, Jovan Pesovic2, Bogdan Bjelica1, Milos Brkusanin2, Ivana Basta1, Ana Marjanovic1, Marija Brankovic1, Aleksandra Kacar1, Dusanka Savic-Pavicevic3, Vidosava Rakocevic-Stojanovic1
1Neurology Clinic, Clinical Center of Serbia, School of Medicine, University of Belgrade, Belgrade, Serbia, Belgrade, RS;2Faculty of biology, Belgrade, RS;3Faculty of Biology, Center for Human Molecular Genetics, University of Belgrade, Belgrade, RS
Background: Myotonic dystrophy type 2 (DM2) is a multisystem disorder, mostly presented with mild but heterogeneous spectrum of symptoms. The aim of this research was to provide detailed sociodemographic, clinical and laboratory data of a large DM2 cohort from the Serbian registry. Methods: In 2008, we started to prospectively enter data of all DM patients. We also retrospectively collected data of patients hospitalized from 1990 until 2008. At the end of 2017, Serbian registry comprised 87 (68%) of 128 genetically confirmed DM2 patients in Serbia, i.e. 1.2 registered cases per 100,000 inhabitants. Results: Female subjects were more prevalent in the registry (63%). The diagnostic delay was 12±11 years and mean age at entry in the Registry was 51±13 years. The most common first symptom in our patients was leg weakness, followed by handgrip myotonia and pain, although some patients presented with cataracts or extrapyramidal syndrome. In 75% of patients lens opacities were present. Severe ECG abnormalities were noted in 8% and pacemaker was implanted in 5% of DM2 subjects. Pulmonary restriction was observed in 10% of DM2 patients. Insulin resistance and diabetes mellitus were frequent in our DM2 cohort (21% and 17%, respectively). Majority of them had hyperlipidemia. Male subjects more frequently had snoring, baldness, sterility and polyneuropathy, while waddling gait and brisk muscle reflexes were more common in females. Conclusion: This registry offers a spectrum of different DM2 features presented in the Serbian population, which could be at service of earlier diagnosis and better follow-up of these patients.
PS1Group1-078 / #672HEART INVOLVEMENT IN MYOTONIC DYSTROPHY TYPE 2
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Bogdan Bjelica1, Stojan Peric1, Edita Cvijan1, Gorana Mandic-Stojmenovic1, Masa Kovacevic1, Ksenija Aleksic1, Jovan Pesovic2, Dusanka Savic-Pavicevic2, Ivana Basta1, Dragana Lavrnic1, Vidosava Rakocevic-Stojanovic1
1Neurology Clinic, Clinical Center of Serbia, School of Medicine, University of Belgrade, Belgrade, Serbia, Belgrade, RS;2Faculty of Biology, Center for Human Molecular Genetics, University of Belgrade, Belgrade, RS
Background: Myotonic dystrophy type 2 (DM2) is a slowly progressive, autosomal-dominant disease. This is a multisystemic disorder that affects the heart, which may be one of the main causes of morbidity and mortality in DM2. The aim of this study was to define cardiac impairments in patients with DM2 and its association with sociodemographic and clinical features of the patients. Methods: This retrospective study comprised 62 patients with genetically confirmed DM2, hospitalized at the Neurology Clinic, Clinical Center of Serbia, from 2013 until 2018. All patients were analyzed by electrocardiography (ECG) and transthoracic echocardiography. Results: Hypertension was observed in 42% of DM2 patients. One fifth of the DM2 patients had bradycardia while other conduction and rhythm impairments were rare, including left bundle branch block (3%), ventricular extrasystoles (5%), and supraventricular extrasystoles (2%). Only one patient had implanted pacemaker because of the first degree AV block and incomplete left bundle branch block. Valve fibrosis was common: aortic 64%, tricuspid 30%, and mitral 19%. Mitral regurgitation was seen in 27% of DM2 subjects. Diastolic dysfunction was observed in 44% of patients, while systolic dysfunction was found in 4%. Cardiomyopathy was observed in 18% of patients, of whom three fourths had dilated type. Conclusion: Cardiac conduction and rhythm defects were rare in DM2, but sometimes even pacemaker implantation is needed. Diastolic dysfunction of the left ventricle was common. This suggest that regular ECG and echocardiography screening is needed in DM2. We suppose that early treatment may prevent development of cardiac complications and early death.
PS1Group1-079 / #673VARIANT REPEATS STABILIZE EXPANSION AND MODIFY AGE AT ONSET IN MYTONIC DYSTROPHY TYPE 1
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Jovan Pesovic1, Stojan Peric2, Milos Brkusanin1, Goran Brajuskovic1, Vidosava Rakocevic-Stojanovic3, Dusanka Savic-Pavicevic1
1Faculty of biology, Belgrade, RS;2Neurology Clinic, Clinical Center of Serbia, School of Medicine, University of Belgrade, Belgrade, Serbia, Belgrade, RS;3Neurology Clinic, Belgrade, RS
Background: Myotonic dystrophy type 1 (DM1) is a variable multisystem disease with age at onset ranged from in utero to late decades. Underlying mutation is the expansion of CTG repeats at the 3¢ untranslated region of DMPK gene, characterized by a pronounced somatic instability biased towards further expansions over time. Expansion size and level of somatic instability explain ~60% of variation in age at onset. Variant repeats at DMPK expansions, observed in 3-5% of patients, have been associated with later age at onset. We aimed to study an effect of variant repeats on dynamics of somatic instability at DMPK locus and age at onset in seven patients. Methods: Four patients with pure CTG repeats served as controls. Repeat-primed PCR profiles and Sanger sequencing indicated the stable structure of different patterns of CCG (CCGCTG hexamers, small and large CCG blocks) and CTC repeats in blood cells over time ranging the period of two and a half to four years. We performed single cell small-pool PCR to size approximately 5000 individual alleles in analysed samples and thus quantify somatic instability of variant DMPK expansions as whole tracks. Results: By using the published correlation models and data set of individually sized alleles in the reference group of patients [Martorel et al. 1998; Morales et al. 2012] we obtained the following results: (1) a lower level of somatic instability of variant expansions then expected based on the expansion size and age at sampling (V=0, p=0.016 for the first time point and V=0, p=0.008 for the second time point), and a less mean residual variation in somatic instability not accounted for by the expansion size and age at sampling in patients with variant expansions than in the reference group (W=871, p=2.2e-4for the first time point and W=1004, p=6.135e-5 for the second time point); (2) a lower expansion increment over time than expected based on the expansion size and time interval between sampling (V=0, p=0.016); (3) a less mean residual variation in age at onset not accounted for by the expansion size and level of somatic instability in patients with variant expansions than in the reference group (W=172, p=0.04 for the first time point and W=192, p=0.02 for the second time point). Conclusion: Our results provide a firm evidence for a general stabilizing effect of different patterns of variant repasts at the DMPK locus in blood cells. This feature of variant repeats most likely positively modulate age at onset of DM1 symptoms and, therefore, has the clinical importance in terms of disease course prediction and new potential therapeutic approaches.
PS1Group1-080 / #674FEATURES OF THE SERBIAN COHORT OF PATIENTS WITH CALPAINOPATHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Stojan Peric1, Ana Kosac1, Marija Brankovic1, Ana Marjanovic1, Bojan Banko2, Sanja Milenkovic3, Milena Jankovic1, Dragana Lavrnic1, Ruzica Maksimovic2, Vedrana Milic Rasic4, Vidosava Rakocevic-Stojanovic1
1Neurology Clinic, Clinical Center of Serbia, School of Medicine, University of Belgrade, Belgrade, Serbia, Belgrade, RS;2Center for Radiology and Magnetic Resonance Imaging, Clinical Center of Serbia, Belgrade, Serbia, Belgrade, RS;3Department of Pathology, Clinical Hospital Centre Zemun, Belgrade, Serbia, Belgrade, RS;4Clinic for Neurology and Psychiatry for Children and Youth, Belgrade, Serbia, Belgrade, RS
Background: In majority of countries the most common form of limb-girdle muscular dystrophy is a type 2A (LGMD2A) caused by mutation in CAPN3 gene coding calpain 3 protein. Aim of this research was to analyze genetic and phenotypic features of Serbian patients with calpainopathy. Methods: This study comprised 19 patients (42% males) from 18 families with genetically confirmed LGMD2A. Here we describe their clinical and laboratory features, magnetic resonance imaging (MRI) of lower legs and histopathological (HP) findings in muscle biopsy samples. Results: Eighteen (95%) patients in this cohort had c.550delA mutation, with nine being homozygous. The age at onset ranged between 7 and 40 years (median 14 years). Only one patient had disease onset after age of 22 (at age of 40). Symptoms usually started as a proximal leg weakness and in three (16%) patients as a proximal arm weakness. Majority of patients were symptomatic at time of diagnosis, but in two of them the disease was discovered during analysis of asymptomatic hyperCKemia, while one was diagnosed as an asymptomatic family member. Serum CK levels were elevated in all patients from about four to 100 times higher than normal. EMG showed a myopathic pattern in all subjects. Mean age at the last examination was 25 ± 10 years with mean disease duration of 12 ± 5 years. Proximal leg and (to lesser degree) proximal arm muscle wasting and weakness was the most prominent feature. Two patients started to use wheelchair at the age of 19 and 27, one used a walker and one cane. Scapular winging was present in 16 (84%) patients. Ankle contractures were present in all CAPN3 subjects, while three (16%) also had elbow contractures. Muscle hypertrophy was detected in ten (53%) patients: seven had calf, two proximal leg and one lower arm hypertrophy. The facial and bulbar muscles were not affected. None of the patients had significant conduction defects nor cardiomyopathy. Four (21%) patients exhibited spirometry restriction (FVC<90%). Muscle MRI showed more severe affection of the proximal leg muscles, with triceps surae muscle being the most affected among distal muscles. Muscle HP showed dystrophic pattern with eosinophilic infiltrations in some patients. Conclusion: Calpainopathy is a common form of LGMD in Serbia and it has specific clinical, laboratory, MRI and HP features.
PS1Group1-081 / #408MALIGNANT HYPERTHERMIA AND MH-LIKE REACTIONS IN NEUROMUSCULAR DISEASES
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Bram De Wel1, Kristl G. Claeys2
1Neurology, University Hospitals Leuven And Ku Leuven, Leuven, BE;2University Hospitals Leuven And Ku Leuven, Leuven, BE
Background: Malignant Hyperthermia (MH) is a life-threatening hypermetabolic reaction in susceptible patients caused by an excessive calcium release from the sarcoplasmic reticulum in skeletal muscles in response to primarily volatile halogenated anesthetics and the depolarizing muscle relaxant succinylcholine. Mutations in the gene encoding the ryanodine receptor 1 (RYR1) are the most frequently identified underlying genetic defects, but mutations in other genes have also been described such as CACNA1S, STAC3, SCN4A and CACNL2A. Over the years, case reports have linked an increasing number of muscular phenotypes to MH susceptibility. This can cause uncertainty in the clinical neurological practice, sometimes leading to excessive labeling of patients as MH susceptible, or even worse, missing the diagnosis. MH-like reactions in response to succinylcholine (and possibly volatile anesthetics) also exist and are associated with severe hyperkalemia and rhabdomyolysis, but are thought to be of a different etiology than true MH. The aim of our study is to explore all muscular disorders that can possibly be linked with MH or MH-like reactions and to suggest an appropriate approach to interpret the accompanying risks. Methods: We performed a systematic and comprehensive search using Pubmed for studies published from the 1960’s up to January 2018. Search terms such as ‘Malignant Hyperthermia’, ‘Malignant Hyperthermia susceptibility’, ‘Malignant Hyperthermia genetics’ and ‘RYR1 myopathies’ were used. We included case reports, reviews and retrospective cohort studies. In addition, we incorporated data published on the ‘European Malignant Hyperthermia Group’ website. Results: Throughout the literature (n = 1012 papers), all patients with myopathies caused by RYR1 mutations are assumed MH susceptible, unless an in vitro contracture test has proven otherwise. This is a good practical assumption, although theoretically it is estimated that only about 30% of these patients are actually MH susceptible. The spectrum of myopathies and clinical syndromes linked to MH through RYR1 mutations now includes - amongst others - central core disease, multiminicore myopathy, congenital myopathy with cores and rods, King-Denborough syndrome, centronuclear myopathy, and nemaline myopathy. In addition, idiopathic hyperCKemia and exertional rhabdomyolysis are also part of the RYR1-disease spectrum. Mutations in CACNA1S mainly cause periodic paralysis and mutations in STAC3 are known to be the cause of Native American myopathy. Finally, there are muscle diseases that were long presumed to be at an increased risk of developing MH, such as Myotonic Dystrophy type I, that are now known to be at an equal risk of developing MH in response to volatile anesthetics compared to the general population. However, succinylcholine should still be avoided in these patients as there can occur unpredictable muscle contractures after administration and sometimes even an exaggerated hyperkalemic response (i.e. MH-like reaction). The muscular dystrophies such as Duchenne and Becker muscular dystrophy can also be associated with a MH-like reaction to succinylcholine. Conclusion: We provide a summary of current evidence linking certain neuromuscular diseases to MH or MH-like reactions peri-operatively. Our study results in a guide for the practicing clinician, in order to know when peri-operative precautions need to be taken or advised in patients with diverse muscular diseases.
PS1Group1-082 / #493ORO-PHARYNGEAL DYSPHAGIA IN MYOTONIC DYSTROPHY TYPE 1 (DM1): IDENTIFICATION OF SENSORY CHANGES IMPACTING SWALLOWING FUNCTION
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Jodi E. Allen1, Chris Turner2
1Therapy And Rehabilitation, The National Hospital for Neurology and Neurosurgery , 3BG, GB;2Institute Of Neurology, University College London Hospital MRC Centre for Neuromuscular Diseases, BG, GB
Background: Aspiration pneumonia is the leading cause of mortality in adults living with Myotonic Dystrophy Type 1 (DM1). Swallowing disturbance (oro-pharyngeal dysphagia, OPD) contributes to this profile of recurrent respiratory infection, hospital admission and premature death. Patients with DM1 rarely report alterations in their swallowing despite its high prevalence. It is currently unclear as to why this lack of complaint occurs but absence of patient report does have implications for identification of OPD and its management. This study explored the possibility of sensory alterations in DM1 as a reason for poor patient reporting by studying patient responses to post-swallow residues, aspiration and/or penetration which normally provoke an overt set of clinical signs and symptoms. Methods: A retrospective review of swallowing reports was undertaken for all adult patients with genetically confirmed DM1 referred for instrumental evaluation of swallowing via Videofluoroscopy (VFS) or Fibreoptic Endoscopic Evaluation of Swallowing (FEES) from a tertiary neuromuscular centre between May 2015-February 2018. Reports were analysed for presence of pooled secretions (FEES only), post swallow residues, awareness of residues and presence and response to aspiration and penetration. Results: A total of 39 VFS reports and 8 FEES reports were reviewed. Videofluoroscopy: 38/39 (97%) patients had post-swallow residues in the pharynx; 9 (24%) were not aware, 21 (55%) were partially aware, 7 (18%) had good awareness and 1 was not reported. 6/39 (15%) aspirated, 5 showed no response. 12/39 (31%) had food and/or drink penetrating the airway, 9 showed no response. FEES: 7/8 (88%) had pooled saliva at rest, 5 of whom had secretions in the laryngeal vestibule. 8/8 (100%) had post-swallow residues; 2 showed no awareness, 4 showed partial awareness, 2 were not reported. 2/8 aspirated, both without response. 5/8 penetrated without response. Conclusion: Post-swallow residues occur frequently as part of the OPD profile in DM1. In this study, over three quarters of patients with residues were not fully aware of them. Reduced awareness of these residues plus absence of response to aspirated and penetrated material strongly suggests an underlying sensory deficit. Altered sensation has not previously been explored in DM1 but could provide one explanation for poor reporting of OPD symptoms. Altered sensation is of clinical significance in both the identification and management of OPD. Reliance on patient report, in the presence of sensory deficits, is likely to lead to delayed or inaccurate diagnosis; putting patients at risk of unexpected chest infections and unplanned hospital admissions. Effective OPD management frequently relies on good sensory awareness in the pharynx and larynx. In the absence of this, patients may not recognise the benefits of swallow management advice, nor self-monitor its effectiveness. This is the first step in better understanding non-motor changes in OPD associated with DM1. Further research will explore the nature and aetiology of these sensory changes as well as relationships between OPD and other clinical markers with the aim of identifying more reliable tools for detection of OPD in this population. In the meantime clinicians should exercise caution when relying solely on patient report in the detection and management of swallowing problems.
PS1Group1-083 / #609HMGCR-MYOPATHY. A RARE BUT STILL A SEVERE DISEASE
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Josep M. Grau1, Pedro J. Moreno1, Sergio Prieto-González1, Albert Selva-O’Callaghan2, Odette Viñas3, José C. Milisenda1
1Internal Medicine, Hospital Clinic de Barcelona, Barcelona, ES;2Internal Medicine, Hospital Vall d’Hebrón, Barcelona, ES;3Immunology, Hospital Clinic de Barcelona, Barcelona, ES
Background: The term HMGCR-myopathy has been recently coined in order to define the cases presenting with subacute proximal muscle weakness, high values of CK, necrotizing myopathy with minimal or no inflammation in muscle biopsy and the detection of the anti HMGCR antibodies. Nevertheless the clinical picture, associated manifestations as well as its management remain controversial. Methods: Our aim was to present our series of HMGR-myopathy patients observed in a single Center at the Muscle Research Group In the Hospital Clínic of Barcelona. Results: Fourteen cases were identified. They came from a series of 1,200 muscle biopsies in the past 10 years. Relevant information is expressed in table 1. Table 1. Patient’s relevant data. CMP: cardiomyopathy; PR:partial reponse: CR: complete response. Conclusion: After a median follow-up of 38,7 months, only 4 patients are in complete remission without any immunosuppressive drug. Fifty percent required IV Ig and 30% RTX for their refractority to prednisone plus an immunosuppressive agent such as MTX, AZA or TK. All patients but one were exposed to statins. Of note was the severe dysphagia in two patients and the simultaneous and reversible cardiomyopathy in other two cases. Although necrotizing immunomediated myopathy was the pathological diagnosis in 12 cases, one case exhibited only a few regenerating fibers (untreated patient) and typical features of dermatomyositis were found in another case. The poorest outcome was observed in younger patients.
| Muscle patholoy | Gender | Age | Associations | CK | Follow-up (months) | Treatment PDN | AZA/MTX/TC | IG. IV. | RTX | Outcome |
| IMNM | M | 57 | Paravertebral | 6200 | 42 | Yes | Yes | 6 cycles | Yes | PR |
| IMNM | M | 79 | No | 7420 | 18 | Yes | Yes | No | No | Dead.Unrelated cause |
| IMNM | F | 71 | No | 4230 | 72 | Yes | Yes | No | No | PR |
| IMNM | F | 81 | No | 620 | 16 | Yes | No | No | No | PR |
| IMNM | F | 70 | No | 1240 | 20 | Yes | Yes | No | No | PR |
| IMNM | F | 84 | No | 579 | 15 | Yes | No | No | No | CR |
| IMNM | F | 51 | No | 5340 | 12 | Yes | Yes | 6 cycles | Yes | PR |
| IMNM | M | 66 | Dysphagia | 5204 | 13 | Yes | Yes | 6 cycles | No | PR |
| Regeneration | M | 77 | No | 2890 | 8 | No | No | No | No | CR |
| DM | M | 48 | Dysphagia | 7660 | 108 | Yes | Yes | 12 cycles | Yes | Poor response |
| IMNM | F | 64 | CMP | 5980 | 50 | Yes | Yes | 8 cycles | Yes | Poor response |
| IMNM | M | 60 | No | 11330 | 72 | Yes | Yes | 12 cycles | Yes | PR |
| IMNM | M | 66 | No | 1650 | 60 | Yes | Yes | No | No | CR |
| IMNM | M | 73 | Dysphagia and CMP | 6600 | 36 | Yes | Yes | 6 cycles | No | CR |
PS1Group1-084 / #639VALUE OF DIAGNOSTIC ALGORITHM IN ASYMPTOMATIC HYPER-CK-EMIA AND ITERATIVE RHABDOMYOLYSIS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Josep M. Grau1, José C. Milisenda2, Francesc Cardellach2, Judith García3
1Internal Medicine, Hospital Clínic, Barcelona, ES;2Internal Medicine, Hospital Clinic de Barcelona, Barcelona, ES;3Biochemistry, Hospital Clinic de Barcelona, Barcelona, ES
Background: Asymptomatic HiperCkemia (AHCK-emia) as well as iterative rhabdomyolysis (IR) still represent a diagnostic challenge. After the comprobation (three different samples, a week apart) of the raised CK values, and after rulling out the common causes of raised CK (strenuous exercise, illicit drugs, alcoholism, hypothyroidism, acute viral infection, seizures, surgery, trauma, dyselectrolitemia and apnea-hypopnea syndromes, among others) we propose a diagnostic algorithm. Methods: The first step in the algorithm includes a simultaneous triple approach. First the Ischemic forearm test (grip test), second, dried blood spot for excluding Pompe disease and third acylcarnitines profile (beta-oxidative dysfunctions). If the diagnosis is not yet reached, the next steps include whole-body muscle MRI and muscle biopsy, with genetic studies, mitochondrial respiratory chain activity and biochemical studies in fibroblasts culture, when indicated. Results: From January 2016 to December 2017, 25 well-selected patients were included for either AHCK-emia or IR. After sequential tests as indicated a definite diagnosis was obtained in 13 cases. The diagnosis were as follows: muscle dystrophies 5, glucogenosis 3, acylcoA multiple dehydrogenase deficiency 2, congenital myopathies 2 and myoadenilate deficiency 1. In the remaining 12 cases, all the tests, including genetics were normal. Conclusion: The proposed algorithm allows the diagnosis in only 50% of the cases. Of note is that in near 50% of the cases a definite diagnosis was not achieved.
PS1Group1-085 / #760NEXT GENERATION SEQUENCING OF DYSTROPHIN GENE IN A COHORT OF NON-DELETION/DUPLICATION DMD/BMD EGYPTIAN PATIENTS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Nagia Fahmy1, Nasser Elhawary2, Nermin Elsayed1, Sabin Schröder3, Martin Werber3
1Ain Shams University, Ain Shams University, Cairo, EG;2Umm El-qura University, Umm El-Qura University, Makkah, SA;3Centogene Ag, Centogene AG, Rostock, DE
Background: The diagnosis of Duchenne/Becker muscular Dystrophy (DMD/BMD) is still challenging owing to the large size of the gene and the occurrence of many mutations. The clinical phenotype, X-linked inheritance and high creatine kinase (CK) serum level are the corner stones in the diagnosis of DMD/BMD patients. The confirmed genetic diagnosis and accurate detection of the type and site of mutation of dystrophin gene became mandatory for the recent therapies and genetic interventions. Methods: We studied 32 patients with DMD/BMD phenotype (including 8 families with 2 affected members of each family) that had no deletion/duplication of MLPA of dystrophin gene. Patients were selected from those attending the outpatient myology and Neurology clinics, Neuromuscular unit, Faculty of Medicine, Ain Shams University, Cairo, Egypt. They had laboratory assessment included serum creatine kinase, nerve conduction and electromyography of both upper and lower limbs, Echocardiography and Multiplex Ligation-dependent Probe Amplification (MLPA) of dystrophin gene to diagnose large deletion/duplication. Muscle biopsy with histochemistry and immunohistochemistry was done to 6 patients and other patients preferred to continue further genetic study without biopsy. The dystrophin gene was analyzed in Centogene laboratories by PCR and next generation sequencing of DNA strands of the entire coding region and the highly conserved exon-intron splice junctions. Results: We found 11 patients with non-sense mutation (four of them were brothers; three of these mutations were not described before). A missense variant was detected in two brothers that were not described before, Small deletions were present in 8 patients, four of them were not described before and a patient with small duplication that was not described before. We found one patient with a hemizygous deletion encompassing intron 11 that was not covered by MLPA and another patient with a pathogenic variant predicted to disrupt the highly conserved donor splice site of exon 55. Another patient showed failure of sequencing exons 48-50, so MLPA was repeated and showed deletion of those exons. No clinically relevant variants were found in 7 patients, 3 of them had the BMD phenotype and diagnosed by immunohistochemistry and western blot study. Conclusion: Next generation sequencing of dystrophin gene is mandatory to all DMD/BMD patients with no deletion/duplication of MLPA. Muscle biopsy and immunohistochemistry of dystrophin protein and dystrophin associated proteins is still highly recommended before doing detailed genetic study. RNA sequencing is needed for those patients who are pathologically proved to have dystrophinopathy and their DNA sequencing didn’t show any mutations.
PS1Group1-086 / #823NATURAL HISTORY OF DISEASE-RELATED COMPLICATIONS IN PATIENTS WITH DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Jessie Lynch, Katherine Tsai, John Lu, Chris Mix, Andre M. York
Sarepta Therapeutics Inc, Cambridge, MA, US
Background: Duchenne muscular dystrophy (DMD) is a rare, X-linked recessive, fatal disorder in which DMD gene mutations inhibit production of dystrophin, leading to progressive muscle weakness. A current therapeutic approach for DMD involves use of phosphorodiamidate morpholino oligomers (PMOs) for exon skipping. These neutrally charged oligonucleotides target specific exons within the DMD pre-messenger ribonucleic acid (mRNA), induce skipping and exclusion of the targeted exon, and restore the DMD mRNA reading frame. Collectively, these actions facilitate translation and production of an internally shortened dystrophin protein. Clinical trials are ongoing to evaluate PMOs for use in DMD. Individuals diagnosed with DMD develop numerous complications throughout disease progression. Within the clinical trial setting, it is critical to differentiate complications associated with the disease and use of concomitant medications, such as corticosteroids, from adverse events potentially related to treatment. Methods: A review of the published literature was conducted to identify relevant articles evaluating the natural history of disease-related complications in DMD patients. Results: A majority of the literature focused on complications representing the two primary causes of death in DMD: cardiac failure, with cardiomyopathy as a significant factor, and respiratory failure, which stems from functional declines in the diaphragm and other respiratory muscles. The prevalence of cardiomyopathy (shortening fraction <28% and/or ejection fraction <55%) increased with age and disease progression, impacting 5% of patients aged 0–5 years, 38% of patients aged 14–17 years, and >60% of patients aged ≥18 years. Age was an important predictor of cardiomyopathy presence, regardless of corticosteroid use. Similarly, impairments in respiratory function also worsened with DMD progression; the majority of data showed a linear decline of ≥5% annually in percent predicted forced vital capacity (FVC%p). In some studies, decline in pulmonary function was largely unaffected by corticosteroid treatment regimen or ambulatory status. Clinical evidence suggested that DMD patients were also prone to numerous other complications, including nephrolithiasis, low bone mass density, rhabdomyolysis, and comorbid psychiatric conditions. In a logistic regression analysis, DMD was an independent risk factor for development of symptomatic nephrolithiasis (odds ratio, 14.26 vs nonambulatory, non-DMD control patients). Increased risk of low bone mass density or fracture was frequently reported and related to progressive muscle weakness, inflammatory cytokine release, continuous corticosteroid regimens, immobility, and vitamin D insufficiency, among other factors. Rhabdomyolysis is a less common but possibly fatal complication that may result from use of specific types of anesthesia in DMD patients. However, unprompted, acute episodes of rhabdomyolysis also occur in DMD patients, and many clinicians perceive this complication as a component. Finally, DMD progression was linked to comorbid psychopathologies, including intellectual disability, autistic spectrum disorder, attention deficit hyperactivity disorder, and behavioral problems. Conclusion: In conclusion, numerous comorbid conditions parallel DMD disease progression. Thorough understanding and awareness of these disease-related comorbidities is critical to differentiation from potential side effects of disease-modifying therapies, especially in the context of clinical investigation.
PS1Group1-087 / #790GOLODIRSEN LEADS TO SARCOLEMMAL DYSTROPHIN EXPRESSION IN PATIENTS WITH GENETIC MUTATIONS AMENABLE TO EXON 53 SKIPPING
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Francesco Muntoni1, Diane Frank2, Jennifer Morgan3, Joana Domingos3, Frederick Schnell2, J. G. Dickson4, Michela Guglieri5, Andreea Seferian6, Mauro Monforte7, Laurent Servais6, Volker Straub8, Eugenio Mercuri7, Skip-Nmd Study Group9
1Dubowitz Neuromuscular Unit, UCL Great Ormond Street Institute of Child Health, London, GB;2Sarepta Therapeutics Inc, Cambridge, MA, US;3University College London, London, GB;4Royal Holloway, University of London, London, GB;5ewcastle University and Royal Victoria Infirmary Hospital, Newcastle upon Tyne, GB;6Institute of Myology, Paris, FR;7Catholic University of the Sacred Heart, Rome, IT;8Newcastle University, Institute of Genetic Medicine, Newcastle Upon Tyne, GB;9SKIP-NMD Study Group, Cambridge, MA, US
Background: Golodirsen (formerly SRP-4053) is a phosphorodiamidate morpholino oligomer (PMO) that binds to dystrophin mRNA, and targets and excludes exon 53 during mRNA processing, facilitating the production of internally shortened dystrophin protein. A 2-part, first-in-human, multicenter trial (ClinicalTrials.gov Identifier: NCT02310906) is evaluating the safety, tolerability, and efficacy of golodirsen in patients with Duchenne muscular dystrophy (DMD) and genetic mutations amenable to exon 53 skipping. We report dystrophin production, exon 53 skipping, and dystrophin cellular localization at baseline and after 48 weeks of treatment with golodirsen. Methods: Part 1 of this trial, which has been completed, included a randomized, double-blind, placebo-controlled, 12-week dose escalation of golodirsen. Part 2, which is ongoing, is a 168-week open-label evaluation of once-weekly golodirsen 30 mg/kg. In preplanned interim analyses, eligible patients had paired muscle biopsies of the biceps brachii at baseline (part 1) and at Week 48 of once-weekly treatment with golodirsen (part 2). Patients newly enrolled in part 2 had the same biopsies at baseline of part 2 and at Week 48. A validated Western blot method quantified dystrophin production (primary biological end point). Exon 53 skipping was evaluated using reverse transcription polymerase chain reaction (RT-PCR). Immunohistochemistry assessed dystrophin localization and sarcolemma fiber intensity. Results: Biopsies were obtained and analyzed from all 25 patients. Mean percent of normal dystrophin protein increased from 0.095% at baseline to 1.019% at Week 48, a significant mean change of +0.924% (P<0.001). Muscle biopsy samples from all patients displayed a significant increase from baseline in exon 53 skipping via RT-PCR at Week 48 (P<0.001), demonstrating the intended mechanism of action. A positive correlation between exon 53 skipping and de novo dystrophin production was observed (Spearman r=0.500; P=0.011). Mean fiber intensity analysis showed a significant increase from baseline in de novo dystrophin production (P<0.001) and confirmed correct dystrophin localization to the sarcolemma. Conclusion: Increased mean dystrophin protein production, exon 53 skipping, and correct sarcolemmal localization of dystrophin were observed in muscle biopsies from golodirsen-treated patients. Golodirsen is the second PMO shown to increase dystrophin protein expression and demonstrate membrane localization following initiation of exon skipping. These findings add to the growing body of evidence of a role for the PMO technology platform in the treatment of DMD.
PS1Group1-088 / #945POMPE DISEASE IN FRANCE: MOLECULAR FEATURES, EPIDEMIOLOGY AND CLINICAL CORRELATIONS FROM A FOURTY-FIVE YEAR NATIONWIDE STUDY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Claudio Semplicini1, Pascaline Letard2, Marie De Antonio3, Nadjib Taouagh4, Barbara Perniconi4, Françoise Bouhour5, Andoni Echaniz-Laguna6, David Orlikowski7, Sabrina Sacconi8, Emmanuelle Salort-Campana9, Guilhem Solé10, Fabien Zagnoli11, Dalil Hamroun12, Roseline Froissart13, Catherine Caillaud14, Pascal Laforet15
1University of Padova - Azienda Ospedaliera di Padova, Padova, IT;2Laboratoire de Biochimie Métabolomique et Protéomique, Hôpital Necker Enfants Malades, Paris, France, FR;3Centre de référence des maladies neuromusculaires, CHU Henri-Mondor, AP-HP, Créteil, France., Paris, FR;4Institut de Myologie, Hôpital La Pitié-Salpétrière, AP-HP, Paris, France, Paris, FR;5Service ENMG et pathologies neuromusculaires, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Lyon, FR;6Department of Neurology, University Hospital Strasbourg, Strasbourg, FR;7Pôle de ventilation à domicile, AP-HP, Hôpital Raymond Poincaré,, Garches, FR;8Centre de référence des Maladies Neuromusculaires, Hôpital Archet, France. CNRS UMR7277, INSERM U1091, IBV - Institute of Biology Valrose, UNS Université Nice Sophia-Antipolis, Faculté de Médecine, Nice, FR;9Reference Center for Neuromuscular Diseases and ALS, La Timone University Hospital, Aix-Marseille University, Marseille, FR;10Department of Neurology, Nerve-Muscle Unit, CHU Bordeaux (Pellegrin Hospital), University of Bordeaux, Bordeaux, FR;11CHRU Cavale-Blanche,, Brest, FR;12Direction de la Recherche et de l’Innovation, CHRU de Montpellier, Hôpital Arnaud de Villeneuve, Montpellier, FR;13Service de Biochimie et Biologie Moléculaire, Centre de Biologie et Pathologie Est, Hospices civils de Lyon, Lyon, FR;14INSERM U1151, Institut Necker Enfants Malades, and Université Paris Descartes, Sorbonne Paris Cité, Paris, FR;15Centre de référence des maladies neuromusculaires Nord-Est-Ile de France, Service de Neurologie, CHU Raymond Poincaré, AP-HP, Garches, France. INSERM U1179, END-ICAP, équipe Biothérapies des Maladies du Système Neuromusculaire, Université Versailles Saint, Garches, FR
Background: Pompe disease (PD) is an autosomal-recessive disorder caused by the deficiency of the lysosomal enzyme acid a-glucosidase (GAA) due to mutations in GAA gene. The clinical spectrum of disease ranges from rapidly fatal multisystemic disorder (classical PD) to a milder adult onset myopathy (late onset PD). There are no straightforward correlations between clinical features, rate of enzymatic reduction and type of mutations. Methods: Aim of the study was to identify all patients diagnosed with non classical PD in France since 1970s, in order to characterize molecular diagnoses and clinical features in a large nationwide study. This was possible by collecting data from the two main French biochemical laboratories (Paris and Lyon) and from the French Pompe registry, that includes since 2004 virtually all French PD patients in a structured long-term observational study. Results: We included 246 late onset PD patients diagnosed since 1972 (130F, 116M; mean age at diagnosis 43.1+17.5 years). Eighty-three different variants were identified, spread all over the sequence and including all types of mutations, and 28 (34%) were novel. The common c.-32-13T>G mutation was detected in 151/170 index cases (89%). Other recurrent mutations included deletion exon 18 (7.5%), c.1927G>A, p.Gly643Arg (3.7%), c.655G>A, p.Gly219Arg (2.8%) and the small deletion c.525del, p.Glu176fs*15 (2.5%). The incidence of non-classic PD in France could be estimated around 1/80,000. Correlations between genotype and age at disease onset were weak. Patients with IVS1 mutation (n=131) presented a later onset compared to 18 non-IVS1 patients (36.2 ±13,9 vs 24,7 ± 14,8 ; p <0,01). However, patients presenting identical genotype could manifest at very different ages. Conclusion: In conclusion, in this study we present the molecular and epidemiologic data from the largest described cohort of non classic PD patients, in a nationwide study covering more than 40 years of this challenging diagnosis. The high molecular and clinical variability we described suggest the need of more population studies to identify potential disease modifiers.
PS1Group1-089 / #987A SPANISH MYOTONIC DYSTROPHY TYPE I FAMILY CARRYING INTERRUPTIONS SHOWING A MILDER AND ATYPICAL PHENOTYPE
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Emma A. Koehorst1, Alfonsina Ballester-Lopez2, Miriam Almendrote3, Alba R. Fransi3, Alicia Martínez-Piñeiro3, Giuseppe Lucente3, Ian Linares3, Judith Nuñez3, Nicolau Guanyabens4, Alejandro Lucia5, Antoni Cano5, Teresa Casadevall6, Agustí Rodríguez-Palmero3, Laura Monlleó-Neila7, Guillem Pintos-Morell3, Jaume Coll-Cantí3, Gisela Nogales-Gadea3
1Neuromuscular And Neuropaediatrics Group, IGTP Research Center, Badalona, ES;2Centro De Investigación Biomédica En Red De Enfermedades Raras (ciberer), Instituto de Salud Carlos III, Madrid, ES;3Neuromuscular And Neuropaediatrics Group, IGTP research center, Badalona, ES;4Neurology Unit, Neuroscience Department, Hospital de Mataró, Barcelona, ES;5Universidad Europea, Instituto de Investigación Hospital 12 de Octubre (i+12), Madrid, ES;6Neuromuscular Unit. Neurology Service. Neuroscience Department, Hospital Comarcal Sant Jaume de Calella, Barcelona, ES;7Neuropediatric Unit. Pediatric Service., Hospital Universitari Germans Trias i Pujol, Barcelona, ES
Background: In recent years, patients with Myotonic Dystrophy type I (DM1) carrying variant repeat patterns in the CTG expansion, have been described. The phenotypical consequences of the interrupted CTG expansion are still highly controversial. The variant repeat patterns have been associated with a complex co-segregated neurological phenotype, including an intermediate Charcot-Marie-Tooth neuropathy, deafness and encephalopathy. While others have found a milder phenotype with a later age of onset, absence of muscle atrophy and CNS involvement or an atypical phenotype with symptoms more resembling Myotonic Dystrophy type II. The number of DM1 patients described with these interruptions is low, with an estimated prevalence of 3-5%. In this study, a cohort of 50 Spanish DM1 patients has been analyzed with the aim of finding new patients carrying interruptions and determining their clinical phenotypes. Methods: Blood DNA was obtained from the participants and triplet-primed PCR (at the 5’ and 3’ sites), Acil digestion and sequencing was used to determine the presence of these interruptions and the type of interruption present (CCG, CTC or GGC). A patients’ registry was built, containing their clinical information. Results: Out of our fifty DM1 patients, four patients were found to have CCG interruptions in the 3’ end of the CTG expansion. Our clinical database revealed that these patients belong to the same family, in which patient 1, 2 and 3 are sisters, who paternally inherited the disease, and patient 4 is the son of patient 3. Mother and son showed the exact same interruption pattern, while the other 2 sisters both had a different interruption pattern. The male interrupted patient (P4, aged 37 years old) was completely asymptomatic upon clinical examination, despite the 108 CTG progenitor allele. The 3 sisters showed most of the typical features of DM1, although predominant distal weakness was absent. The most remarkable thing is that in the 3 sisters’ symptoms started at a later age (>50 y.o.), classifying them as late-onset DM1, while their symptoms resemble more of a classic/adult phenotype. Conclusion: Here, we report the presence of four interrupted cases in a cohort of 50 DM1 patients, with a prevalence of 3.4 % in our DM1 families. Inheritability of the interruption pattern was shown in our cohort, with an exact replica on maternal transmission. Our study contributes to the observation of a milder and/or atypical clinical phenotype of variant repeat carrying DM1 patients, with a later age of onset than expected and predominantly proximal weakness. The latter resembles more a DM2 phenotype.
PS1Group1-090 / #977MERRF CLASSIFICATION: IMPLICATIONS FOR DIAGNOSIS, AND CLINICAL TRIALS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Josef Finsterer1, Sinda Zarrouk-Mahjoub2, John M. Shoffner3
1Krankenanstalt Rudolfstiftung, Vienna, AT;2Pasteur Institute Of Tunis, University of Tunis El Manar and Genomics Platform, Tunis, TN;3Molecular Genetics, Neurology, Biochemical Genetics, Atlanta, US
Background: Given the etiologic heterogeneity of disease classification using clinical phenomenology, we employed contemporary criteria to classify variants associated with myoclonic epilepsy with ragged-red fibers (MERRF) syndrome and to assess the strength of evidence of gene-disease associations. Standardized approaches are used to clarify the definition of MERRF which is essential for patient diagnosis, patient classification, and clinical trial design. Methods: Systematic literature and database search with application of standardized assessment of gene-disease relationships using modified Smith criteria and of variants reported to be associated with MERRF using modified Yarham criteria. Results: Review of available evidence supports a gene-disease association for two MT-tRNAs and for POLG. Using modified Smith criteria, definitive evidence of a MERRF gene-disease association is identified for MT-TK. Strong evidence gene-disease evidence is present for MT-TL1 and POLG. Functional assays that directly associate variants with oxidative phosphorylation impairment were critical to mtDNA variant classification. In silico analysis was of limited utility to the assessment of individual MT-tRNA variants. Using contemporary classification criteria, several mtDNA variants previously reported as pathogenic/possibly pathogenic are reclassified as neutral variants. Conclusion: MERRF is primarily a MT-TK disease with pathogenic variants in this gene accounting for ~90% of MERRF cases. Although MERRF is phenotypically and genotypically heterogeneous, myoclonic epilepsy is the clinical feature that distinguishes MERRF from other categories of mitochondrial disorders. Given its low frequency in mitochondrial disorders, myoclonic epilepsy is not explained simply by an impairment of cellular energetics. Although MERRF phenocopies can occur in other genes, additional data is needed to establish a MERRF disease-gene association. This approach to MERRF emphasizes standardized classification rather than clinical phenomenology, thus improving patient diagnosis and clinical trials design.
PS1Group1-091 / #978MITOCHONDRIAL MULTI-ORGAN DISORDER SYNDROME SCORE GENERATED FROM DEFINITE MITOCHONDRIAL DISORDERS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Josef Finsterer1, Sinda Zarrouk-Mahjoub2
1Krankenanstalt Rudolfstiftung, Vienna, AT;2Pasteur Institute Of Tunis, University of Tunis El Manar and Genomics Platform, Tunis, TN
Background: Mitochondrial disorders(MIDs) frequently present as mitochondrial multi-organ disorder syndrome(MIMODS) already at onset or evolve into MIMODS during the course. This study aimed to find which are the organs/tissues most frequently affected in MIMODS, which are the most frequent abnormalities within an affected organ, if there are typical MIMODS patterns, and to generate a MIMODS-score to assess the diagnostic probability for a MID. Methods: Retrospective evaluation of clinical, biochemical, and genetic investigations of adult, definite MIDs. Results: Included were 36 definite MID patients, 19 males, 17 females, aged 29-82y. The diagnosis was based on genetic testing (n=21), on biochemical investigations (n=17), or on both (n=2). The number of organs most frequently affected was four ranging from 1-9. MIMODS was diagnosed in 97% of patients. The organs most frequently affected were the muscle (97%), central nervous system (CNS) (72%), endocrine glands (69%), heart (58%), intestines (55%), and peripheral nerves (50%). The most frequent CNS abnormalities were leucencephalopathy, prolonged visually-evoked potentials and atrophy. The most frequent endocrine abnormalities included thyroid dysfunction, short stature, and diabetes. The most frequent cardiac abnormalities included arrhythmias, systolic dysfunction, and hypertrophic cardiomyopathy. The most frequent MIMODS patterns were encephalomyopathy, encephalo-myo-endocrinopathy, and encepalo-myo-endocrino-cardiopathy. The mean±2SD MIMODS score was 35.97±27.6 (range: 11-71). A MIMODS score >10 was regarded as indicative of a MID. Conclusion: Adult MIDs manifest as MIMODS in the vast majority of the cases. Organs most frequently affected in MIMODS are the muscle, CNS, endocrine glands, and heart. A MIMODS score >10 suggests a MID.
PS1Group1-092 / #238CLINICAL PRACTICE WITH STEROID THERAPY FOR DUCHENNE MUSCULAR DYSTROPHY, A CLINICIAN SURVEY IN ASIAN AND OCEANIA
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Fumi Takeuchi1, Harumasa Nakamura2, Hirofumi Komaki2, Ichizo Nishino2, Shin’Ichi Takeda2, Ikuya Nonaka2, Naohiro Yonemoto3
1The John Walton Muscular Dystrophy Research Centre, Newcastle University, Newcastle upon Tyne, GB;2National Center of Neurology and Psychiatry, Tokyo, JP;3Department Of Biostatistics, Kyoto University School of Public Health, Kyoto, JP
Background: Several studies investigating current clinical practice for Duchenne muscular dystrophy (DMD) have been conducted in Western countries. However, there have been few studies in Asia and Oceania. This study aims to elucidate clinical practice for DMD related to steroid therapy among local experts in Asia and Oceania. Methods: The Asian and Oceanian Myology Center (AOMC) was established in 2001 to facilitate scientific communication and collaboration in neuromuscular disease field in Asian and Oceanian countries. As of 2016, the AOMC executive board members comprised 37 experts from 15 countries in Asia and Oceania (Australia, China, Hong Kong, India, Japan, Malaysia, Myanmar, New Zealand, Pakistan, Philippines, Singapore, South Korea, Taiwan, Iran, and Thailand). We enrolled local experts and/or clinicians who manage DMD patients from the regions by asking AOMC executive board members to nominate potential participants who may meet our inclusion criteria. We included clinicians who: 1) had experience in treating DMD patients; 2) were able to reply to the questionnaire in English. Responses were collected electronically with SurveyMonkey or by mail between December 2015 and June 2016. Responses by mail were inputted into SurveyMonkey on receipt. Data analysis was conducted in Microsoft Excel. The survey consisted of 17 questions regarding the respondent’s personal information (name, gender, age, speciality, and working medical site) and their usual clinical practice for patients with DMD. Results: Among the 15 countries, the executive board members in 13 countries agreed with the study and nominated 148 clinicians. We sent our questionnaire to the 148 clinicians via email in December 2015, and 87 clinicians (Australia: 3, China: 5, Hong Kong: 8, India: 13, Japan: 20, South Korea: 5, Malaysia: 2, Myanmar: 8, Pakistan: 8, Philippines: 2, Singapore: 6, Taiwan: 5, and Thailand: 2) replied by July 2016 (total response rate 62%). Among the 87 clinicians, 79 clinicians (91%) were aware of DMD care recommendations from Bushby et al. (2010). Moreover, 83 clinicians (95%) reported currently managing patients with DMD (i.e., between 2011 and 2015) and 4 clinicians (5%) reported managing patients with DMD only before 2011. The number of DMD patients appeared to differ depending on clinicians, as well as on country. In China, there were more patients with DMD at ages 0-9 and 10-19 years old, while there were more patients ≥20 years old in Japan. Daily prednisolone/prednisone was the most popular treatment regimen at initiation (n=45, 64%), but was less popular at maintenance (n=39, 56%). Steroid therapy for patients after loss of ambulation and medication for bone protection seemed to be controversial. Conclusion: Our results from an international survey of clinicians increase our knowledge of the epidemiology of DMD, as well as current clinical practice for DMD as reported by local experts in Asia and Oceania. It is important to expand registries of patients with DMD in Asia and Oceania and to accumulate real-world longitudinal patient data. These strategies will aid the study of the epidemiology and natural history and could improve treatment and care for patients with DMD worldwide.
PS1Group1-093 / #570EXPERIENCES WITH BARIATRIC SURGERY IN PATIENTS WITH FSHD AND DM1
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Nicol Voermans
Neurology, Radboudumc, Nijmegen, NL
Background: Overweight and obesity are common in patients with facioscapulohumeral dystrophy (FSHD) and myotonic dystrophy type 1 (DM1). Lifestyle change is often challenging for patients with neuromuscular diseases, especially to increase physical activity. Bariatric surgery is a treatment option for adults with severe obesity in whom lifestyle interventions have not been effective. However, very little is known about the benefits and risks in patients with neuromuscular disorders. This study therefore aims to obtain insight into the patients’ perspectives and experiences, the outcome, effects and risks of bariatric surgery in patients with FSHD and DM1. This is expected to improve counseling of patients with FSHD and DM1 who consider bariatric surgery. Methods: A qualitative study, consisting of 14 in-depth interviews with six patients (three FSHD and three DM1), four relatives, three bariatric surgeons and one general practitioner. The study used a qualitative descriptive method. Thematic analysis was used to analyze the data. Results: Four themes were formulated: 1) Overweight as burden; 2) Bariatric surgery as last option; 3) Not your standard patient; and 4) A different life, a different me. Conclusion: This qualitative study shows that bariatric surgery has beneficial physical and mental effects for most patients with FSHD and DM1, and does not influence the muscular disease course. It shows that bariatric surgery is feasible in patients with FSHD and DM1, but specific precautions and a suitable follow-up including tailored dietary and training advices are required. A guideline for the patient and the procedure would be welcome.
PS1Group1-094 / #612AN UNUSUAL PRESENTATION OF GNE MYOPATHY WITH PROMINENT AXIAL MUSCLE WEAKNESS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Jin-Mo Park1, Jin-Sung Park2
1Neurology, Dongguk University Gyeongju Hospital, Gyeongju, KR;2Department Of Neurology, Kyungpook National University Chilgok Hospital, Daegu, KR
Background: GNE myopathy is an autosomal recessive myopathy which is previously known as distal myopathy with rimmed vacuoles (OMIM 605820) or hereditary inclusion body myopathy (OMIM 600737). It is caused by biallelic mutation in GNE which is a rate-limiting, bifunctional enzyme in the sialic acid pathway (Eisenberg I 2001). GNE myopathy is clinically characterized by early involvement of tibialis anterior muscle leading to early foot drop, with relatively spared quadriceps muscles. Pathologic study of GNE myopathy frequently shows “rimmed vacuoles” with fiber size variation (Argov Z 1984). However, diagnosis of GNE myopathy can be challenging due to phenotypic variability (Park YE, de Dios JK). We report a GNE myopathy patient with axial muscle involvement, probably triggered by repeated usage of truncal muscles. Methods: A twenty-three year-old man presented with back pain. He was serving in the military service for 6 months and his back pain started after strenuous military activities that require usage of back muscles. He had no past medical history or delay in the milestones. Results: The initial physical examination revealed axial muscle weakness, and mild ankle dorsiflexion weakness. He had hypoactive deep tendon reflexes and no pathologic reflexes. The initial laboratory data were unremarkable except for elevated creatinine kinase level of 1146 IU/L. The electromyography showed evidence of active myopathy with active denervation potentials observed in right tibialis anterior, medial gastrocnemius and thoracic paraspinal muscles. The T1-weighted muscle MRI scans revealed mild fatty infiltrative and asymmetric atrophic changes in bilateral hamstring and soleus muscles. There were marked fatty infiltrations in bilateral lumbar paraspinal muscles. The muscle pathology in the left tibialis anterior muscle showed mild fiber size variation without significant inflammatory infiltrates, but showed rimmed vacuoles that were suggestive of GNE myopathy. The genetic test revealed a pair of compound heterozygous missense mutations in GNE gene, p.V572L and p.A591T, previously reported (NP–005467.1), that are one of the most common GNE mutations found in Korean and Japanese ethnicities. Conclusion: The decreased sialic acid synthesis due to bialleic mutation in UDP N-Acetylglucosamine 2-epimerase/N-Acetylmannosamine kinase leads to decreased GNE activity is known to cause GNE myopathy. A recent study reported a young male patient who presented with asymmetric hand weakness (De Dios JK, 2014). This report suggested that muscle overuse may be a potential environmental factor that can influence atypical clinical presentation in GNE myopathy. Our patient showed not only unusual onset of symptom but only unique manifestations. The most severe damage site was lumbar paraspinal muscles that is almost complete fatty replaced and atrophied. Meanwhile, anterior compartment of lower legs including tibialis anterior muscle was normal. Several reported axial involvements of GNE myopathy (Argov Z, Chu CC, Park YE). A subset of myopathies that show significant involvement of the axial muscles are known as axial myopathy currently. GNE myopathy is not a disease that is addressed for differential diagnosis (Witting 2016). GNE myopathy has phenotypic variability. In GNE myopathy, environmental factor such as repeated muscle overuse might contribute disease onset and onset site.
PS1Group1-095 / #761HIGHER MRI MUSCLE FAT FRACTION AT SIMILAR AGE IS ASSOCIATED WITH EARLIER LOSS OF AMBULATION IN DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Karin J. Naarding1, Harmen Reyngoudt2, Erik W. Van Zwet3, Melissa T. Hooijmans1, Brenda L. Wong4, Cuixia Tian4, Irina Rybalsky4, Karen C. Shellenbarger4, Julien Le Louër2, Pierre G. Carlier2, Hermien E. Kan5, Erik H. Niks6
1Neurology, Leiden University Medical Center, Leiden, NL;2Aim & Cea Nmr Laboratory, Neuromuscular Investigation Center, Institute of Myology, Paris, FR;3Department Of Biostatistics, Leiden University Medical Center, Leiden, NL;4Neurology, Cincinnati Children’s Hospital Medical Center, Cincinatti, OH, US;5C.j. Gorter Center For High Field Mri, Leiden University Medical Center, Leiden, NL;6Neurology, Leiden University Medical Center, Leiden, NL
Background: Duchenne muscular dystrophy (DMD) is characterized by progressive replacement of muscle with fat. Full market-approved drugs are lacking. Clinical trials are challenging because of the low prevalence and practical and ethical issues in performing long placebo-controlled studies in children. Muscle fat fraction (FF), measured by quantitative MRI, is considered a potential surrogate endpoint in trials since it is objective and non-invasive. However, such biomarkers are only acceptable as endpoints to regulatory agencies if they are related to clinically meaningful milestones. Therefore, we aimed to assess the additive predictive value of vastus lateralis (VL) FF to age on the loss of ambulation (LoA). Methods: 3-point gradient-echo Dixon images of the right upper leg of DMD patients were acquired at two centers, LUMC and CCHMC, between 2013 and 2016. At the LUMC, images were acquired at baseline, 12 and 24 months on a 3T MR scanner. At CCHMC, scans were acquired on a 1.5T MR scanner at baseline, 6, 12 and 18 months. Weighted mean VL FF was calculated using five slices at the mid-upper leg. FF’s were fitted to a logistic growth-model to predict FF from 0-20 years of age. Slopes of FF curves differed per patient based on FF. Month and year of LoA were determined through a detailed interview with patients and parents, either during regular clinical follow-up, or by telephone between July 2017 and February 2018. This phone call also defined last follow-up for ambulant patients. We fitted a Cox model for the time to LoA with predicted VL FF as the only (time-varying) covariate. Results: At least one VL datapoint was available for 19 LUMC DMD patients (median baseline age 8.9 years, range 5.6-16.1, seven non-ambulant), and for 16 from CCHMC (median baseline age 11.2 years, range 7.6-15.1, all ambulant). Between first MRI and February 2018 LoA occurred in ten DMD patients. VL FF data in relation to age of 35 DMD patients and their superimposed logistic growth-curves are presented in Figure 1. The hazard ratio of a percent increase in VL FF for the time to LoA was estimated at 1.13 (95% confidence interval 1.07-1.18) which was highly significant (Wald test, p<0.001). Conclusion: Vastus lateralis fat fraction has a significant additive predictive value to age on the date of loss of ambulation. This direct relation supports the use of quantitative MRI as an outcome measure for future clinical trials in DMD.
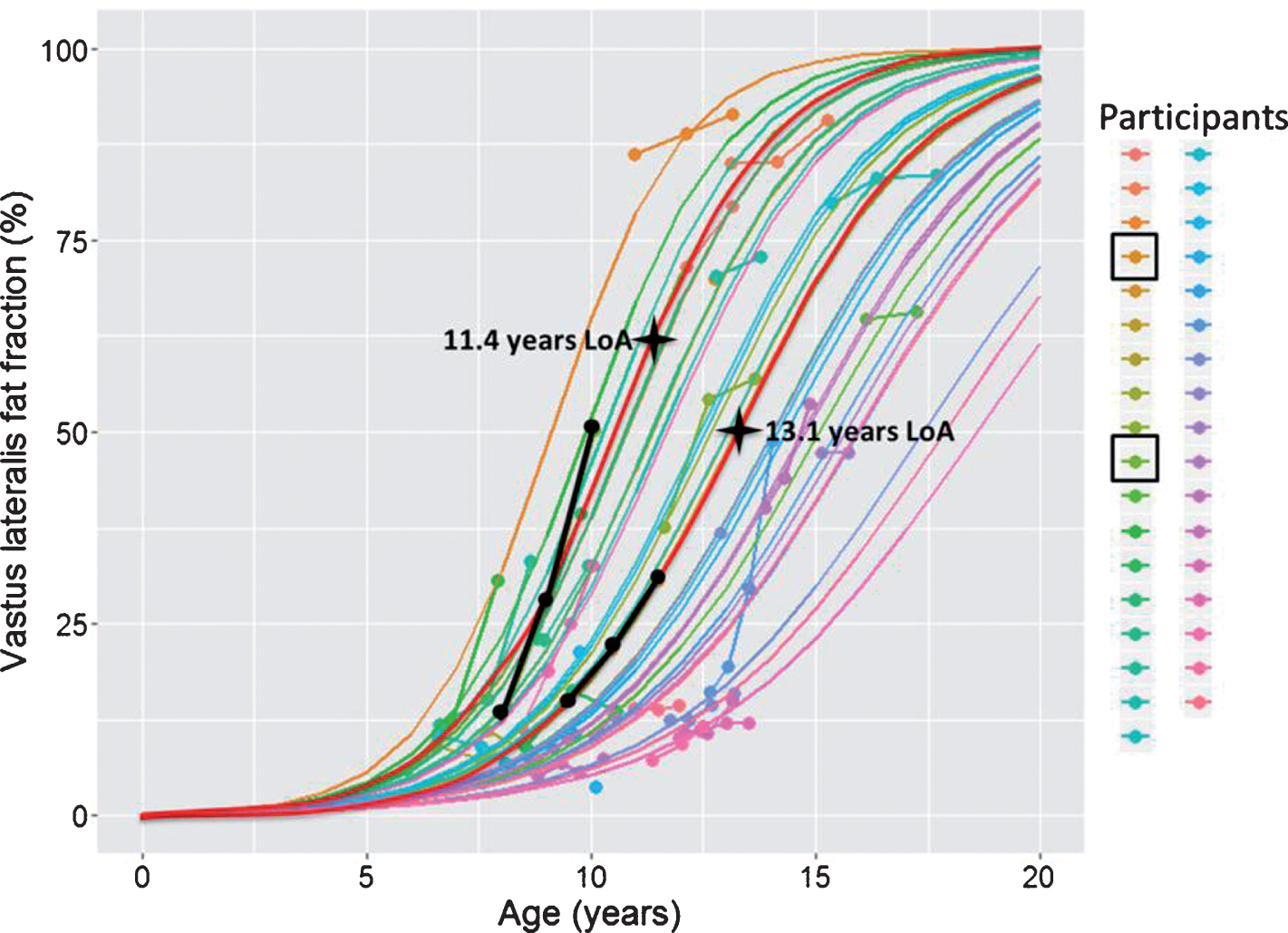

PS1Group1-096 / #1018TOR1A VARIANTS CAUSE A SEVERE ARTHROGRYPOSIS WITH DEVELOPMENTAL DELAY, STRABISMUS AND TREMOR
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Ariana Kariminejad1, Martin Dahl-Halvarsson2, Gianina Ravenscroft3, Fariba Afroozan1, Elham Keshavarz4, Hayley Goullée3
1Kariminejad-Najmabadi Pathology & Genetics Center, Tehran, IR;2Department Of Pathology, University of Gothenburg, Sahlgrenska University Hospital, Gothenburg, SE;3Centre For Medical Research, The University of Western Australia and the Harry Perkins Institute for Medical Research, Nedlands, AU;4Department Of Radiology, Mahdieh Hospital, Shahid Beheshti University of Medical Science, Tehran, IR
Background: Autosomal dominant torsin dystonia 1 is a disease with incomplete penetrance most often caused by an in-frame GAG deletion (p.Glu303del) in the endoplasmic reticulum luminal protein torsinA encoded by TOR1A. Methods: 25 cases of AMC with congenital contractures in more than two joints and in multiple body areas were collected through international collaboration and medical history, physical examination and imaging were performed. Next-generation sequencing was performed on DNA from patients. Results: We report an association of the homozygous dominant disease-causing TOR1A p.Glu303del mutation, and a novel homozygous missense variant (p.Gly318Ser) with a severe arthrogryposis phenotype with developmental delay, strabismus and tremor in three unrelated families. All parents who were carriers of the TOR1A variant showed no evidence of neurological symptoms or signs, indicating decreased penetrance similar to families with autosomal dominant torsin dystonia 1. The results from cell assays demonstrate that the p.Gly318Ser substitution causes a redistribution of torsinA from the endoplasmic reticulum to the nuclear envelope, similar to the hallmark of the p.Glu303del mutation. Conclusion: Our study highlights that TOR1A mutations should be considered in patients with severe arthrogryposis and further expands the phenotypic spectrum associated with TOR1A.
PS1Group1-097 / #877NDUFAF3 VARIANTS THAT DISRUPT MITOCHONDRIAL COMPLEX I ASSEMBLY CAUSE ASSOCIATE WITH CAVITATING LEUKOENCEPHALOPATHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Akihiko Ishiyama1, Kazuhiro Muramatsu2, Shunpei Uchino3, Chika Sakai3, Yuichi Matsushima3, Nishiki Makioka2, Tomomi Ogata2, Eriko Suzuki2, Hirofumi Komaki1, Masayuki Sasaki1, Masakazu Mimaki4, Yu-Ichi Goto3, Ichizo Nishino5
1Department Of Child Neurology, National Center Hospital, National Center Of Neurology And Psychiatry, National Center Hospital, National Center of Neurology and Psychiatry, Kodaira, JP;2Department Of Pediatrics, Gunma University Graduate School Of Medicine, Gunma University Graduate School of Medicine, Maebashi, JP;3Department Of Mental Retardation And Birth Defect Research, National Institute Of Neuroscience, National Center Of Neurology And Psychiatry, National Institute of Neuroscience, National Center of Neurology and Psychiatry, Kodaira, JP;4Department Of Pediatrics, Faculty Of Medicine, Teikyo University, Faculty of Medicine, Teikyo University, itabashi, JP;5Department Of Neuromuscular Research, National Institute Of Neuroscience, National Center Of Neurology And Psychiatry, National Institute of Neuroscience, National Center of Neurology and Psychiatry, Kodaira, JP
Background: Genetic abnormalities in mitochondrial complex assembling factors are associated with leukoencephalopathy. However, genotype-phenotype correlations remain elusive. Methods: We present a one-year-old girl with consciousness disturbance after a respiratory infection. Brain MRI revealed leukoencephalopathy with bilaterally symmetrical hyperintensity in the substantia nigra, medial thalamic nuclei, and basal nuclei, as well as cavities in the cerebral white matter and corpus callosum. Lactate levels in the spinal fluid were high, while magnetic resonance spectroscopy of the cerebral white matter and basal nuclei showed high peak lactate levels, suggesting mitochondrial dysfunction. After 2 months of hospitalization, her developmental status returned to that before admission. She acquired the ability to speak meaningful words at 2 years and 6 months and walked independently at 3 years. However, there was no deterioration in language comprehension, but her motor function deteriorated: her sitting posture became slightly unstable, and she could not walk without assistance at 4 years. Results: A muscle biopsy was performed on the biceps brachii at 3 years, revealing no pathological findings characteristic of mitochondrial-related diseases. Mitochondrial respiratory chain complex activities were analyzed in the skeletal muscle. Complex I activity levels, normalized to the activities of complex II and citrate synthase, were 17% and 21%, respectively, but complex III and V activity levels were mildly decreased. Whole exome sequencing identified compound heterozygous variations in NDUFAF3, involved in the assembly of mitochondrial complex I (c.342–343insGTG:p.117 Valdup, c.505C>A:p.Pro169Thr). Two-dimensional blue native polyacrylamide gel electrophoresis (PAGE) and sodium dodecyl sulfate-PAGE revealed reductions in Q-module (NDUFS2, NDUFS3, and NDUFA9) and P-module (NDUFB10 and NDUFB11) subunits, indicating disruption of mitochondrial complex I assembly. Conclusion: Few studies on the assembly of mitochondrial respiratory chain complexes have been conducted, and the phenotypes and pathologies of associated diseases are not completely understood. Our result suggests leukoencephalopathy with cavities associates with NDUFAF3 mutations and expands the clinical phenotypes associated to impaired assembly factor for mitochondrial complex I.
PS1Group1-098 / #979RESTRICTION ENZYME CLEAVAGE OF PCR PRODUCTS ALLOWS GENOTYPING MDX3CV, MDX4CV AND MDX5CV ALLELES WITHOUT SEQUENCING
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Laurence A. Neff, Elinam Gayi, Leonardo Scapozza, Olivier M. Dorchies
School Of Pharmaceutical Sciences, University of Geneva, Geneva, CH
Background: Duchenne muscular dystrophy (DMD) is due to the absence of dystrophin. It is characterized by progressive muscle wasting and premature death. The exploration of the pathogenic mechanisms and the evaluation of therapeutic options rely mostly on dystrophic mice. The mdx mouse originates from a spontaneous mutation that arose in a colony of C57BL/10 mice in the 1980’s. Since then, it has become the most commonly used animal model of DMD. In the 1990’s, several allelic variants of the mdx mouse, namely the mdx2Cv, mdx3Cv, mdx4Cv, and mdx5Cv, have been recovered from chemical mutagenesis in the C57BL/6J background. The different strains are characterized by differential expression of naturally occurring shorter isoforms (C-terminus truncated) of dystrophin in a tissue-dependent manner: The mdx2Cv, mdx3Cv and mdx4Cv strains are useful for studying the roles of dystrophin and dystrophin-associated glycoprotein complex in non-muscle tissues such as the retina, kidney, and brain. The mutations carried by the mdx4Cv and mdx5Cv mice are characterized by the absence of revertant fibres, an advantage for evaluating gene-, virus, or cell-mediated dystrophin restoration. Until now genotyping of this strains requires sequencing of PCR product. Here we propose a simple restriction base method allowing genotyping without sequencing. Methods: Sequence analysis revealed that the mdx3Cv and mdx5Cv alleles bear restriction sites for AluI, and HphI, respectively, which are absent from the wild type alleles. The mdx4Cv mutation disrupts a restriction site for Alw26I (BsmAI), which is normally present on the wild type allele. Based on these findings, we designed new strategies for genotyping these dystrophic strains using PCR amplification followed by enzymatic restriction of the PCR products. Results: Here we show that PCR followed by restriction digestion provides an easy mean to discriminate wt, heterozygous and homozygous mice bearing mdx3-5Cv alleles. We demonstrated the excellent stability to freeze-thaw cycles of the PCR and restriction enzyme master mixes and we optimized a number a protocol steps. The resulting protocols are user-friendly and allow straightforward genotyping at minimum cost and hands-on time. Conclusion: The mdx3Cv, mdx4Cv, and mdx5Cv strains all carry point mutations. Conventional genotyping involves PCR amplification, yielding amplicons of same sizes, followed by allele identification by sequencing, a step that is often outsourced, causing extra cost and delay in analysis. By contrast, our procedure is time and cost effective: genotyping is achieved within a day using standard molecular biology techniques and eliminates extra cost for sequencing.
PS1Group1-099 / #462CLINICO-PATHOLOGICAL CORRELATIONS IN IDIOPATHIC INFLAMMATORY MYOPATHIES
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Endre Pál1, Katinka Gulyas2, Tünde Minier2, Timea Berki3, Laszlo Czirjak2, Cecilia Varju2
1Neurology, University of Pécs, Pécs, HU;2Rheumatology And Immunology, University of Pécs, Pécs, HU;3Immunology And Biotechnology, University of Pécs, Pécs, HU
Background: Idiopathic inflammatory myopathies (IIM) are heterogeneous systemic autoimmune diseases, characterised by muscle weakness, various organ involvements and the presence of specific autoantibodies. Our aims were to classify patients according to the clinical features, muscle histology findings and autoantibody profiles. Methods: 82 patients with muscle biopsy proven IIM were included in the study. Muscle biopsy, myositis specific (MSA) and myositis associated (MAA) antibody tests were performed in each patient before starting the immunomodulatory treatment. The MSAs and MAAs (Jo-1, PL-7, PL-12, Mi-2, SRP, Pm-Scl, Ku, ribosomal, AMA-M2) were tested with Western-blot kit. The ANA, dsDNA, ENA, Sm, Sm/RNP, SS-A and SS-B antibody examinations were performed with ELISA tests. Dermatomyositis/DM/, polymyositis /PM/, juvenile PM/DM, inclusion body myositis /IBM/, overlap myositis /OM/ and immune mediated necrotizing myopathy /IMNM/ were identified based on clinical data, immunserology and muscle biopsy. Survival analysis was perfomed by Kaplan Meier test. Results: 59 women and 23 men with a mean age of 49.3 ± 14.6 years were included. Mean follow-up of the patients was 7.5 ± 4.5 years. The distribution of myositis subsets was: 30.5% (n=25) DM, 26.8% (n =22) PM, 22% (n=18) OM, 11% (n=9) IMNM, 8.5% (n=7) IBM, and 1.2% (n=1) juvenile PM/DM. Lung fibrosis (51.2%), arthritis (51.2%), Raynaud’s phenomenon (42.7%), skin symptoms (45.1%), dysphagia (24.4%) and significant cardiac involvement (15.9%) were the most prevalent disease-manifestations. Fifteen cases were associated with malignancies, mjority of them was classified as IMNM (7 out of 9 patients). 18 patients died of myositis related events: 8 patients died of malignancies (4 in the IMNM, 2 in the PM and 2 in the DM group), 6 died related to cardiac conditions, 2 in lung fibrosis and 2 caused by infections. The worst prognosis with a 10 year survival of approximately 31.1% of patients was seen in the IMNM subgroup, followed by patients with PM (68.4%) and DM (85.3%). Patients with antisynthetase antibody-positivity had worse prognosis compared with patients with other antibodies or no identifiable antibodies. The worst prognosis was seen in patients with both MSA and MAA positivity and the best in patients with no identifiable MSA and MAA. Conclusion: Complex clinicopathologic assessment is recommended for classification and management of idiopathic inflammatory myopathy cases. Muscle histology is still an important diagnostic tool in IIMs. IMNM and PM are associated with worse prognosis compared to other types of IIMs.
PS1Group1-100 / #537FIRST REPORTED VARIANT AT THE TRIM32 RING DOMAIN IN A PEDIGREE WITH LIMB-GIRDLE MUSCULAR DYSTROPHY AND BARDET-BIEDL SYNDROME
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Ana L. Pelayo-Negro1, Miguel A. Fernández-García2, Patricia Blanco-Arias2, Elena Gallardo-Agromayor3, Gerardo Gutierrez-Gutierrez4, José Berciano1
1Neurology, University Hospital Marqués de Valdecilla, Santander, ES;2Neurology, Health in Code, A Coruña, ES;3Radiology, University Hospital Marqués de Valdecilla, Santander, ES;4Neurology, University Hospital Infanta Sofía, Madrid, ES
Background: Pathogenic variants in TRIM32 have been related to limb-girdle muscular dystrophy type 2H (LGMD2H, OMIM#254110). To date, most reported variants lead to a truncated protein and are clustered at the C-terminal NHL-repeat domain. Whole gene deletions have also been described. Additionally, a missense change in the B-box motif was found in a family with Bardet-Biedl syndrome (BBS11, OMIM#615988). Methods: A 60-year-old woman began in the 4th decade with LL weakness, inability to run or stand from sitting or lying position and slow progression with difficulties in walking, fatigue and frequent falls. She had mild atrophy of pelvic and thigh muscles, CK 250UI/L, myopathic EMG pattern; not willing to perform muscle biopsy to date. She was born of healthy consanguineous parents and had two affected siblings with a similar phenotype and hypogonadism. Additionally, she had a healthy brother and a daughter, aged 37, that recently complained of fatigue and clumsiness with normal EMG and CK levels. Next-generation sequencing of 34 LGMD-related genes identified a homozygous 1bp-insertion in TRIM32, predictably originating a frameshift (NM–012210.3:c.115–116insT; NP–036342.2:p.Cys39Leufs*17). Both affected siblings were also confirmed as homozygous; father, healthy brother and daughter were heterozygous carriers. Results: This is the first reported pathogenic variant at the TRIM32 N-terminal RING domain. Homozygous whole gene deletions confirmed that a total loss of protein function resulted in LGMD2H without greater clinical severity. Despite TRIM32 recessive behaviour, some descriptions of heterozygous carriers found mild clinical symptoms and vacuolar changes in muscle biopsies, suggesting a mild dominance pattern. In our family, father and brother of the index case (aged 92 and 50, respectively) had no neuromuscular abnormalities; in contrast, the daughter (37yo) complained about mild symptoms, suggesting the possibility of some light dominancy of the TRIM32 variant. On the other hand, TRIM32 has been linked to BBS11: a syndromic form of obesity with pigmentary retinopathy, renal anomalies, cognitive problems and hypogonadism. To date, only a Bedouin family carrying the p.Pro130Ser missense variant, located at the N-terminal B-box motif, has been described. However, none of the patients with complete TRIM32 deletion showed systemic features. In our family, two siblings have hypogonadism, suggesting the possibility of an association with BBS11. Conclusion: This would be the first report of BBS features in a LGMD2H affected family and should highlight the importance of a thorough phenotypic evaluation of TRIM32 patients to further delineate this association, particularly if other RING-domain pathogenic variants shall be identified.
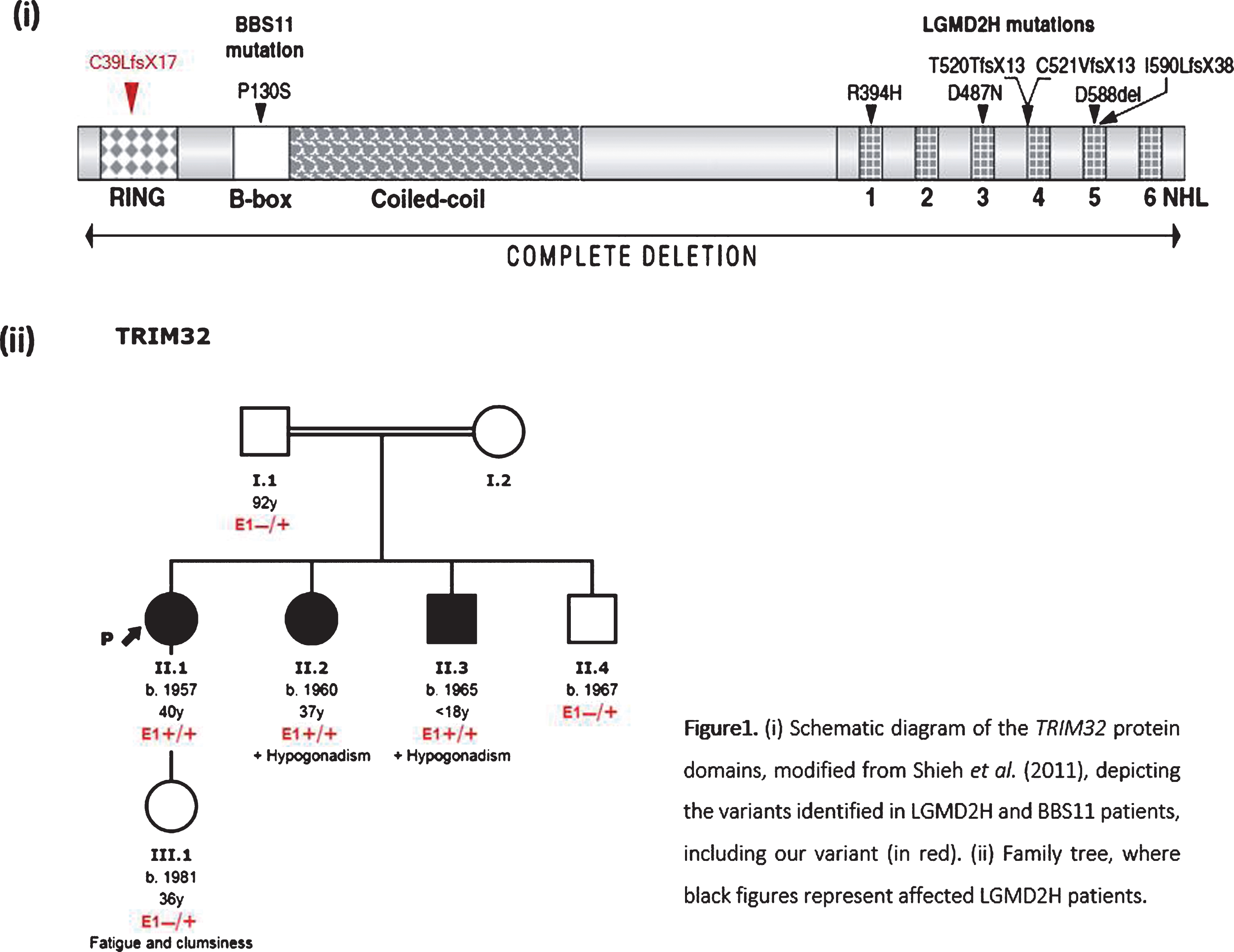
PS1Group1-101 / #572MUSCLE SPECIFIC KINASE PROTECTS MDX MOUSE MUSCLES AGAINST ECCENTRIC CONTRACTION-INDUCED LOSS OF STRENGTH
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
William D. Phillips, Joanne Ban, Sofie Trajanovska
Physiology & Bosch Institute, The University Of Sydney, Sydney, NSW, AU
Background: Muscles of mdx mice lack dystrophin, providing a mouse model for Duchenne muscular dystrophy (DMD). Eccentric contractions applied to mdx muscles cause an acute drop in force, reflecting the vulnerability of dystrophin-deficient muscles to injury. It has been reported that mdx muscles express less Muscle Specific Kinase (MuSK) and that this might predispose their neuromuscular junctions to eccentric contraction-induced damage. Methods: To increase the level of expression of MuSK we injected the tibialis anterior muscles of 8-week old male mdx and C57BL10 (wild-type background) mice with recombinant adeno-associated viral vector (AAV) encoding MuSK fused to green fluorescent protein (MuSK-GFP). Contralateral control muscles were injected with empty AAV vector. One month later the mice were anaesthetized with 2-3% inhaled isoflurane/oxygen and maximum tetanic force was recorded in response to direct muscle stimulation or stimulation via the nerve. Eccentric contractions (forced lengthening to 1.25 time optimal length) were imposed during maximum (directly evoked) activation of the muscle in situ. Maximum isometric force was recorded before, and one hour after, a series of four such eccentric contractions. Results: Consistent with previous studies mdx muscles were more vulnerable to injury compared to C57BL10 wild-type mice. After the series of four eccentric (stretch) contractions, mdx muscles retained only 73 ± 2% (mean ± SEM) of their original (pre-stretch) maximum isometric force. In contrast, mdx muscles expressing MuSK-GFP retained significantly more of their original force after identical ECs (84 ± 1%; p<0.01). Rapsyn, an established downstream effector of MuSK signaling, conferred similar protection to mdx muscles. The protective effect was evident even when isometric force was elicited by direct muscle stimulation. Conclusion: These results indicate that enhanced MuSK signaling can protect dystrophin-deficient muscle fibers from acute eccentric contraction-induced loss of force. Moreover they suggest that the MuSK/rapsyn signaling pathway can act downstream of neuromuscular transmission to protect the sarcolemma of from tension-induced injury.
PS1Group1-102 / #645NOVEL LMNA GENE MUTATION PRESENTING WITH DILATED CARDIOMYOPATHY AND LIMB-GIRDLE MUSCULAR DYSTROPHY TYPE 1B
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Martha Spilioti1, Konstantinos Notas1, George Stavropoulos2, Georgios Efthimiadis3, Kleita Michaelidou4, Ioannis Zaganas4, Maria Moschou1, Marianthi Arnaoutoglou1, Magdalini Tsolaki1
11st Department Of Neurology, Ahepa General Hospital, Aristotle University of Thessaloniki, Thessaloniki, GR;22nd Cardiology Department, Hippokration General Hospital, Aristotle University of Thessaloniki, Thessaloniki, GR;31st Cardiology Department, Ahepa University Hospital, Aristotle University of Thessaloniki, Thessaloniki, GR;4Department Of Neurology, School Of Medicine, University of Crete, Heraklion, Crete, GR
Background: Lamins A and C, encoded by the lamin A/C gene (LMNA), are major components of the nuclear lamina, a fibrillar network that sustains the structural integrity and stability of the nuclear envelope. Additionally, lamins are involved in multiple cellular processes, such as chromatin organization, DNA replication and gene regulation. LMNA gene mutations have been shown to cause several inherited diseases known as laminopathies, which include cardiac and skeletal myopathies, lipodystrophy, metabolic disorders, peripheral neuropathy and premature aging syndromes. Heart involvement counts as the most common feature, regardless of the presence of a myopathy and is characterized by conduction system disorders, arrhythmias, dilated cardiomyopathy and arrhythmogenic right ventricular cardiomyopathy. Many LMNA mutation carriers are at risk of sudden cardiac death, due to ventricular arrhythmias, high-degree atrioventricular block and end-stage heart failure. LMNA-related myopathies represent a subgroup with different phenotypes, based on the distribution of muscle weakness or age at onset: limb-girdle muscular dystrophy type 1B (LGMD1B), autosomal dominant Emery-Dreifuss muscular dystrophy and congenital muscular dystrophy. LGMD1B typically causes progressive muscle weakness in the proximal muscles of the lower limbs. Methods: We herein present a 47 years old female patient, with a medical history of arrhythmias, second degree atrioventricular block, dilated cardiomyopathy (confirmed via magnetic resonance imaging) and weakness of the proximal muscles, with distinct myopathic features in the electromyography. Cardiac and skeletal muscle involvement developed gradually during her third decade of life. Family history was positive for sudden cardiac deaths and dilated cardiomyopathy (Figure 1), indicating a possible autosomal dominant manner. Genomic DNA was extracted from patient’s peripheral blood and Whole Exome Sequencing (WES) was performed on Illumina HiSeq 2000/2500 platform. A comprehensive bioinformatics analysis for data interpretation was done using Ingenuity software (Qiagen).
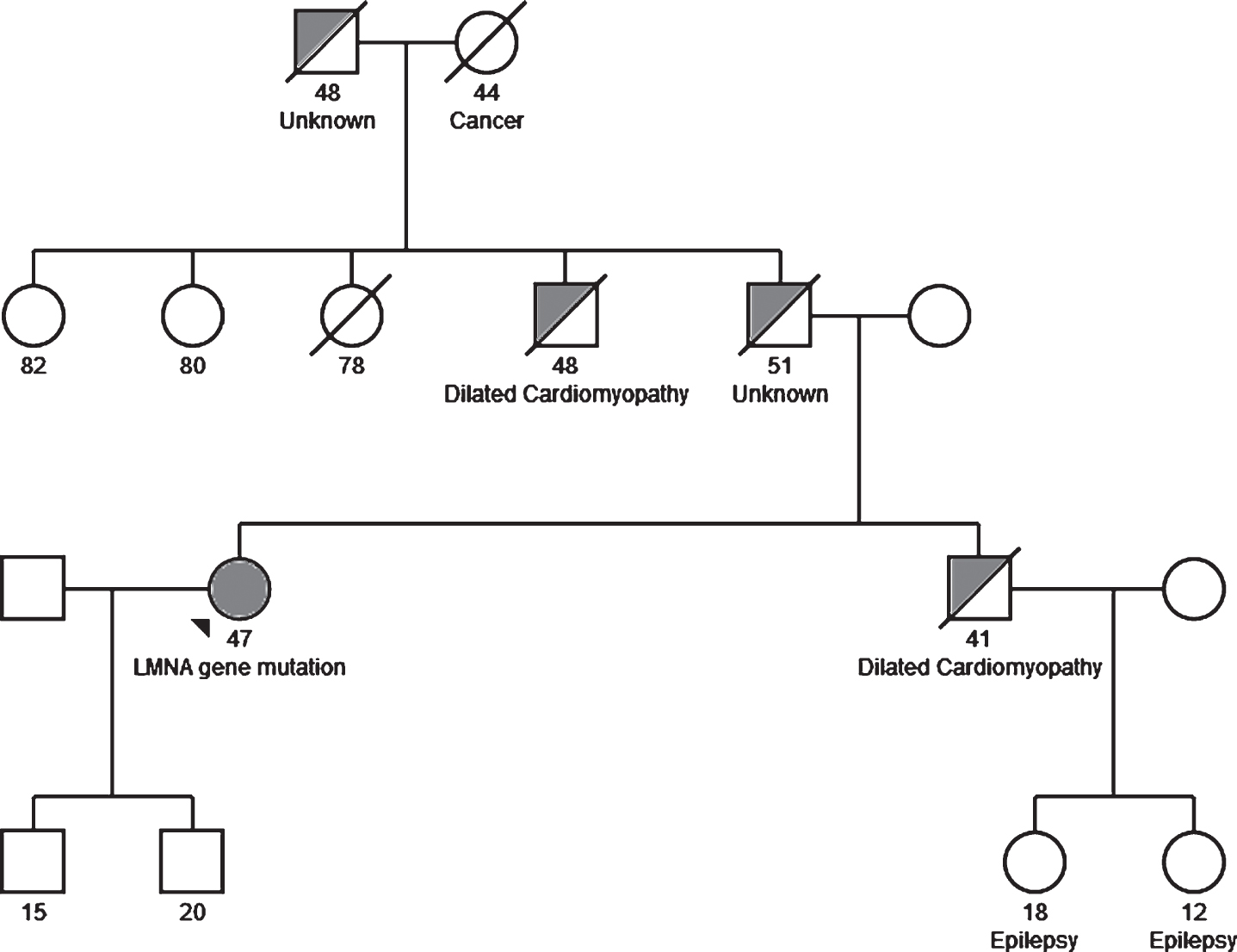
Results: We identified a novel splice site variant (c.1272+2T>C) in LMNA gene in the heterozygous state in our patient. This genetic variant results in splice site loss and in silico analysis using different bioinformatic tools predicted that it exerts a pathogenic effect. In order to verify segregation of this genetic variant in LMNA gene with disease, several affected and unaffected members of the family were tested with pending results. Conclusion: Laminopathies can present with a vast clinical spectrum and should be suspected in patients with conduction system disorders, dilated cardiomyopathy and muscle weakness. We report a novel LMNA gene mutation in a female patient, presenting with dilated cardiomyopathy and myopathy, underlining the genetic diagnostic value and cost effectiveness of WES.
PS1Group1-103 / #498LONGTERM APPLICATION OF HUMAN IMMUNOGLOBULIN G FOR EXPERIMENTAL TREATMENT OF DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Jana Zschüntzsch1, Pia V. Jouvenal1, Yaxin Zhang2, Florian Klinker3, Malte Tiburcy4, David Liebetanz3, Heinrich Brinkmeier2, Dörthe Malzahn5, Jens Schmidt1
1Neurology, University medical Center Göttingen, Göttingen, DE;2Institute Of Pathophysiology, University Medicine Greifswald, Karlsburg, DE;3Clinical Neurophysiology, University medical Center Göttingen, Göttingen, DE;4Institute Of Pharmacology And Toxicology, University medical Center Göttingen, Göttingen, DE;5mzBiostatistics, Göttingen, DE
Background: Duchenne muscular dystrophy (DMD) is a progressive, muscle wasting, inherited X-linked muscle disease caused by mutations in the dystrophin gene. The pathology consists of structural instability of the dystrophin-glycoprotein-complex (DGC), muscle degeneration, and various degrees of myoinflammation depending on the disease stage. Current treatments have targeted the genetic defect or the inflammatory response. So far, the standard treatment by glucocorticosteroids is limited because of numerous side effects. A previous study addressed the early disease phase of the well-established mdx mouse model for DMD and demonstrated an improvement of clinical and paraclinical parameters upon treatment with human immunoglobulins (IgG). The aim of the present study was to confirm the effectiveness of human IgG in a long-term preclinical study in mdx mice. Methods: Human IgG (Privigen, CSL Behring) at a dosage of 2g/kg BW or saline (NaCl) as control was administered monthly by intraperitoneal injections in mdx mice over 18 months beginning at 4 weeks of age. Advanced voluntary wheel running parameters were recorded continuously over the entire study period. Cardiac ultrasound was performed every three months and grip strength measurements every week. After 18 months, animals were sacrificed. Blood and muscle were sampled for ex vivo muscle contraction tests, quantitative PCR and histological analysis. Results: IgG compared to NaCl significantly improved the voluntary running performance and grip strength. The muscle fatigability and specific tetanic force in an ex vivo muscle contraction test remained unchanged upon IgG. A significant benefit of cardiac function was evidenced by improved fractional area shortening and ejection fraction upon human IgG. These results were supported by a reduced cardiac fibrosis and infiltration of T- cells as well as macrophages. Despite an unchanged mRNA expression of the relevant inflammatory markers TGF-beta, SPP1 and MCP1, a significant reduction of T- cells and macrophages in representative muscles (diaphragm, M. tibialis anterior, M. gastrocnemicus and M. quadriceps) was observed by immunohistochemistry in mdx mice treated with human IgG compared to controls. These findings were underlined by reduced myopathic changes upon human IgG in the same muscles. Conclusion: Taken together, this study demonstrates a beneficial effect of 18 months of IgG treatment in mdx mice, evidenced by improved voluntary wheel running parameters, cardiac function and reduced histopathological changes of the skeletal muscle, diaphragm and heart. Our long- term study underlines the importance of myoinflammatory contribution to the disease progression of DMD/ mdx and shows the therapeutic effectiveness of human IgG in the mdx mouse. Human IgG is usually well tolerated and might be a useful treatment option beside mutation-based therapies for DMD patients. The data call for a clinical trial with IgG in DMD.
PS1Group1-104 / #675BODY COMPOSITION ANALYSIS IN PATIENTS WITH MYOTONIC DYSTROPHY TYPE 1 AND 2
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Stojan Peric1, Milorad Vujnic2, Tanja Nisic3, Marija Banovic4, Bogdan Bjelica5, Ivo Bozovic5, Jovan Pesovic6, Dusanka Savic-Pavicevic6, Ivana Basta5, Zorica Stevic5, Dragana Lavrnic5, Vidosava Rakocevic-Stojanovic5
1Neurology Clinic, Clinical Center of Serbia, School of Medicine, University of Belgrade, Belgrade, Serbia, Belgrade, RS;2Faculty of Medicine, Banja Luka, BA;3Center for Obesity, Clinic of Endocrinology, Diabetes and Metabolic Disease, Belgrade, RS;4School of Medicine, Belgrade, RS;5Neurology Clinic, Clinical Center of Serbia, School of Medicine, University of Belgrade, Belgrade, Serbia, Belgrade, RS;6Faculty of biology, Belgrade, RS
Background: Myotonic dystrophies (DM1) are inherited, slowly progressive, multisystem diseases. There are only several reports about body composition in DM1 and no data for DM2. The aim of this research was to analyze the body composition of patients with DM1 and DM2, and also to examine the association between body composition and socio-demographic/clinical features of patients. Methods: Body composition was assessed by DEXA (Dual-Energy X-ray Absorptiometry) in 22 DM1 and 12 DM2 patients. A tree compartment model was used: bone mineral content (BMC), fat mass (FM) and lean tissue mass (LTM). Results: Patients with DM1 and DM2 had similar total body mass, BMC, FM and LTM. Patients with DM1 had higher trunk limb fat index (TLFI) that indicated higher accumulation of fat mass in abdominal area (visceral obesity) (1.16 ± 0.32 for DM1 vs. 0.87 ± 0.23 for DM2, p<0.05). Right ribs bone mineral density was worse in DM2 group (0.68 ± 0.07 g/cm2 vs. 0.61 ± 0.09 g/cm2, p<0.05). The percentage of FM in legs of DM1 subjects showed negative correlation with the strength of proximal leg muscles (ρ=-0.47, p<0.05). The strength of muscles in DM2 was in correlation with bone density (ρ=+0.62, p<0.05 for upper arm muscles, ρ=+0.87, p<0.01 for lower arm muscles, ρ=+0.72, p<0.05 for lower leg muscles). Conclusion: Patients with DM1 had visceral obesity and percentage of fat mass correlated with a degree of muscle weakness. In patients with DM2 lower bone mineral content was noticed. Higher degree of muscle weakness was in correlation with lower bone density in DM2.
PS1Group1-105 / #795EVALUATING THE EFFECTS OF BASELINE VARIABLES ON THE RESPIRATORY FUNCTION BENEFIT OF IDEBENONE IN DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Shabir Hasham1, Thomas Meier1, Mika Leinonen1, Thomas Voit2, Oscar H. Mayer3, Gunnar Buyse For The Delos Study Group4
1Santhera Pharmaceuticals, Pratteln, CH;2UCL Great Ormond Street Institute of Child Health, London, GB;3Children’s Hospital of Philadelphia , Philadelphia, PA, US;4University Hospitals Leuven, Leuven, BE
Background: Respiratory function decline is a major cause of morbidity and mortality in patients with Duchenne muscular dystrophy (DMD). The randomized, placebo-controlled Phase 3 DELOS trial assessed the safety and efficacy of idebenone in DMD patients already in respiratory decline and not taking concomitant glucocorticoids (GCs). This trial demonstrated that idebenone significantly slowed respiratory function loss, measured as peak expiratory flow (expressed as a percentage of predicted, PEF%p) – the primary endpoint. The objective of this analysis is to evaluate the effect of idebenone in various patient subgroups of the DELOS trial. Methods: Respiratory function data were prospectively collected from 64 DMD patients aged 10-18 years and already in respiratory decline (PEF%p < 80%). The consistency of the effect of idebenone in DELOS was investigated by analyzing change in PEF%p from baseline to week 52 (study end) in subgroups defined by prior GC use (naïve vs previous user), age (<14 years vs ≥14 years), baseline PEF%p (<50% vs ≥50%), weight (<58 kg vs ≥58 kg) and ambulatory status. Each subgroup was analyzed using a mixed model for repeated measures, a similar approach to that used in the primary analysis. Results: The observed treatment difference in PEF%p at week 52 between idebenone and placebo was 6.3% in the total intention to treat population (p=0.03). Similar PEF%p treatment differences in favor of idebenone were observed regardless prior GC use, age, baseline PEF%p, weight or ambulatory status – varying from 4.2% to 7.3%. Of note, the treatment difference in prior GC users was 6.73% compared to 6.19% in GC-naïve patients. No significant interactions between the treatment group and subgroup factors were observed. Conclusion: The previously reported treatment effect of idebenone on PEF%p was consistent in a broad population of DMD patients, regardless of previous GC use, age, weight, ambulatory status or severity of respiratory decline at treatment initiation. These results suggest that none of these factors significantly influence the respiratory function benefits of idebenone in patients with DMD.
PS1Group1-106 / #984NOVEL RYR-CALSTABIN STABILIZERS WITH THERAPEUTICAL POTENTIAL FOR DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Garazi Aldanondo1, Haizpea Lasa-Fernandez1, Jaione Lasa-Elgarresta1, Jose Ignacio Miranda2, Jesus Maria Aizpurua2, Adolfo López De Munain1, Ainara Vallejo-Illarramendi1
1Biodonostia Institute, Donostia-San Sebastian, ES;2UPV/EHU, Donostia-San Sebastian, ES
Background: Several studies indicate that calcium is dysregulated in the muscles from patients with Duchenne and Becker muscular dystrophy. In the mdx mouse model of Duchenne muscular dystrophy (DMD), the sarcoplasmic reticulum ryanodine receptor RyR1 and RyR2 are abnormally nitrosylated and this leads to calstabin depletion from the protein complex and subsequent calcium leak through these channels. RyR modulators enhance RyR-Calstabin binding preventing calcium leak, reducing muscle damage and improving muscle function. Methods: We have generated a family of small molecules, namely Ahulken compounds (AHK), which bind RyRs and modulate their activity. In this work we have analyzed the in vivo and in vitro effect of the novel RyR stabilizers AHK1 and AHK2, in the mdx mouse model and in human immortalized myotubes. Results: Treatment of human myotubes with AHK compounds for 12 hours enhanced RyR-calstabin binding, and normalized resting cytosolic calcium levels. Moreover, no toxicity was observed in myotubes treated with high concentration (2 mM) of AHK compounds. Treatment of mdx mice with AHK modulators during 5 weeks normalized intracellular calcium levels in muscle fibers, reduced biochemical and histological evidence of muscle damage and improved muscle function. Moreover, AHK2 also improved specific CNS and cardiac muscle deficiencies observed in mdx mice. Conclusion: Our study shows that AHK modulators ameliorate dystrophic phenotype in cellular and animal models of Duchenne muscular dystrophy. In addition, our results consolidate RyR-calstabin complex as a useful therapeutic target for drug development against muscular dystrophies as well as other disorders with subjacent nitro-oxidative stress.
PS1Group1-107 / #501THE EFFECT OF UNCARIA TOMENTOSA IN DIAPHRAGM MUSCLE OF MDX DYSTROPHIC MICE
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
David Feder1, Alzira A.S. Carvalho2, Bruno M. Bertassoli1, Lucas P. Giordani1, Giuliana Petri1, Fabio F. Perazzo3, Beatriz C.A. Alves4, Matheus M. Perez4, Fernando L.A. Fonseca4
1Pharmacology, Faculdade de Medicina do ABC, Santo Andre, BR;2Neurology, Faculdade de Medicina do ABC, Santo Andre, BR;3Pharmaceutical Sciences, Universidade Federal de São Paulo, Santo Andre, BR;4Laboratory Analysis, Faculdade de Medicina do ABC, Santo Andre, BR
Background: Duchenne Muscular Dystrophy (DMD) is a severe muscle wasting disease caused by mutations in dystrophin gene. This deficiency leads to sarcolemma instability, inflammation, muscle degeneration and fibrosis. Uncaria tomentosa (UT) is a medicinal plant with anti-viral, anti-mutagenic, anti-inflammatory and antioxidant properties. The objective of study was evaluating the effect of UT in diaphragm muscle of mdx dystrophic mices. Methods: 8-weeks-old male mdx mice were treated, 10 mice with 200 mg/kg of UT extract orally during 30 days (UT group) and 6 mice received saline solution orally during 30 days (control group). Creatine kinase (CK) was measured on the day of euthanasia. H&E stained frozen section of the muscle biopsy of diaphragm (DIA) was performed in order to analyze number of internal nuclei and minimal Feret diameter. In addition, gene expression of transforming growth factor-ß1 (TGF-ß1), tumor necrosis factor-a (TNF-a), osteopontin (OSP) and Myostatin (Myo) were determined by quantitative real time polymerase chain reaction (qRT-PCR). Data are presented as mean ± standard error of mean. One-way or two-way ANOVA and Bonferroni test were used for analysis of variance to determine the statistical significance, followed by the least significant difference (LSD) analysis, using Prism Statical software (Graphpad, San Diego, CA). P < 0.05 was considered statistically significant. Results: CK levels were significantly higher in UT group. UT treatment did not prevent muscle damage, indicated by increase of central nucleated fibers with a concomitant increase in number of small fibers (0-30 μM). By other side, It had an effect on the inflammatory process demonstrated by decrease in TNF-a, TGF and osteopontin expression (figure 1)
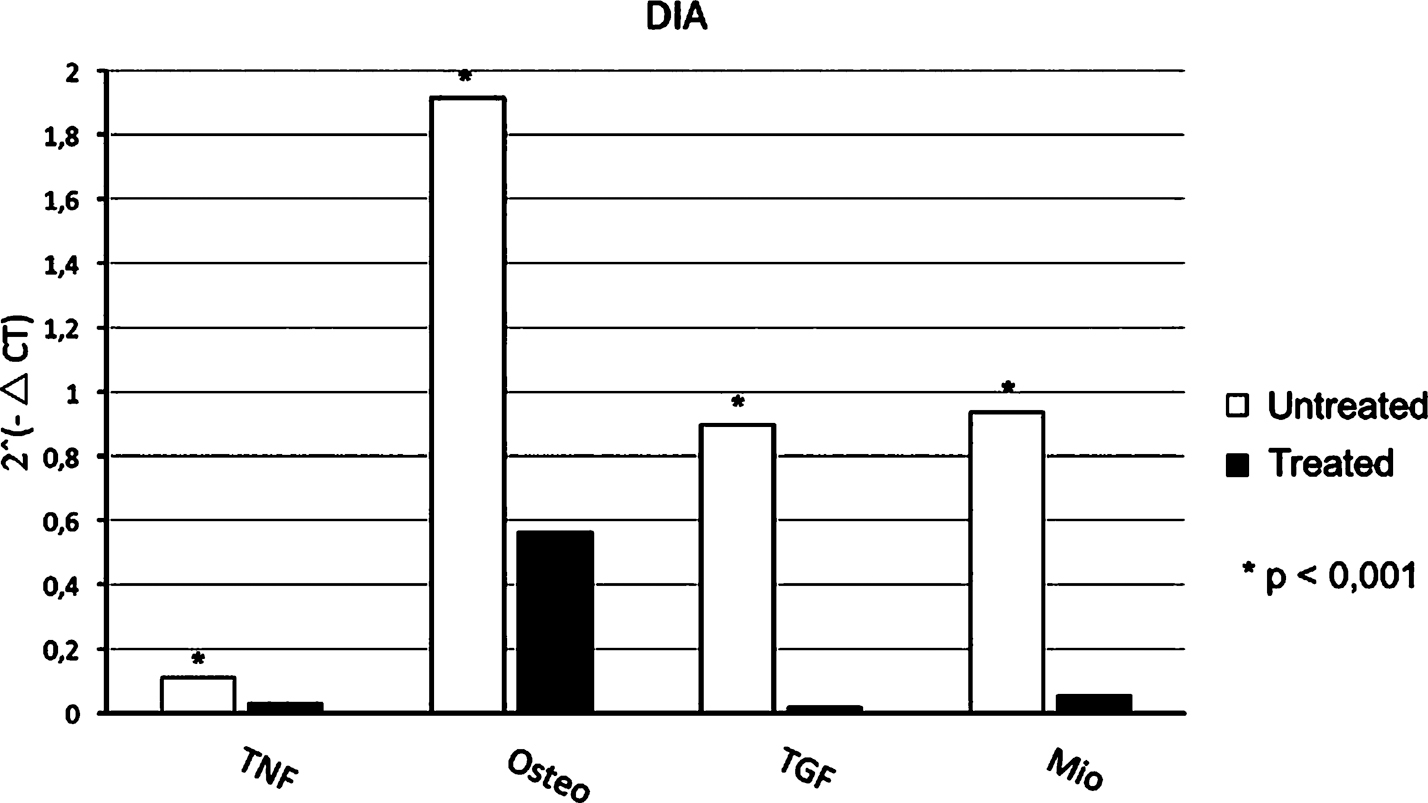
Conclusion: So, in this study, we evaluated the anti-inflammatory properties of Uncaria tomentosa against myonecrosis and inflammatory response in the diaphragm muscle of mdx mice. Uncaria tomentosa downregulates the TNF-a, TGF and osteopontin expression indicating improving of inflammatory role however, there was a worsening of the dystrophic phenotype in the diaphragm muscle, indicated by increase of CK level, number of central nucleated fibers and small fibers diameter. Perhaps, as many inflammatory pathways are triggered in muscular dystrophy, it is possible that Uncaria tomentosa, alone, is insufficient to halt the whole process.
PS1Group1-108 / #620MYTOBLOTS FOR THE EVALUATION OF NEW TREATMENTS IN NEUROMUSCULAR DISORDERS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Estibaliz Ruiz-Del-Yerro1, Patricia Soblechero-Martin2, Irene Larrañaga1, Edurne Albiasu-Arteta1, Virginia Arechavala-Gomeza3
1Neuromuscular Disorders, Biocruces Health Research Institute, Barakaldo, ES;2Neuromuscular Disorders Group, Biocruces Helath Research Institute, Barakaldo, ES;3Neuromuscular Disorders, Biocruces Health Research Institute, Barakaldo, ES
Background: New therapies for neuromuscular disorders often require to be studied in patient’s cell cultures. Many potential therapies for Duchenne Muscular Dystrophy (DMD) aim to alter the expression of key proteins, such as dystrophin or utrophin, but, as muscle cell cultures from DMD patients are scarce and do not grow or differentiate well, only a limited number of candidate drugs are tested. Moreover, dystrophin and utrophin quantification by western blotting requires a large number of cultured cells; so fewer compounds are evaluated as thoroughly as it is desirable. Methods: We have developed a quantitative assessment tool using fewer cells to contribute in the screening of better drug candidates: an “in-cell-western”, also known as cytoblot, is a quantitative immunofluorescence assay performed in microplates that makes possible the quantification of proteins directly in cell culture, combining the specificity of western blotting with the reproducibility and throughput of ELISA. Results: Our group has recently optimized the assay (‘myoblot’) to quantify several muscle proteins in differentiated myoblast cultures and we are using this method to assess dystrophin restoration treatments, drugs that aim to increase the expression of utrophin and others that alter the differentiation of cultures. Conclusion: We expect that the use of myoblots will accelerate the development and validation of new therapies.
PS1Group1-109 / #622PHENOTYPE-GENOTYPE RELATIONS IN FACIOSCAPULOHUMERAL MUSCULAR DYSTROPHY TYPE 1
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Karlien Mul1, Richard J. Lemmers2, Corinne G. Horlings3, Patrick J. Van Der Vliet2, Marianne A. Jonker4, Nicol C. Voermans3, Silvere M. Van Der Maarel2, Baziel G. Van Engelen3
1Neurology, Radboud University Medical Center, Nijmegen, NL;2Human Genetics, leiden University medical Center, Leiden, NL;3Neurology, Radboud University Medical Center, Nijmegen, NL;4Health Sciences, Radboud University Medical Center, Nijmegen, NL
Background: Facioscapulohumeral muscular dystrophy (FSHD) is characterized by large variability in disease severity and penetrance, that is only partially explained by the current knowledge on the disease mechanism. This study integrates previous findings on phenotype-(epi)genotype relations to assess how much of the clinical variability can be explained. Additionally, clinical scores per body region are used to refine the interpretation of (epi)genetic data. Methods: We included 148 FSHD1 patients from 49 different families and performed genetic testing (D4Z4 repeat array size, haplotype, and D4Z4 methylation), extensive clinical evaluation (clinical severity scores, manual muscle testing, motor function measure, facial weakness score), and muscle MRI of the leg muscles. Results: The explained variability in disease severity by familial factors, including the D4Z4 repeat array size, was approximately 40%. Asymptomatic gene carriers had longer repeat sizes compared to symptomatic individuals (7.3 vs 6.2 units, p=0.001). The D4Z4 methylation (Delta1 value) was not useful as a prognostic factor for disease severity and penetrance on an individual level. The D4Z4 repeat array size explained approximately 29% and 15% of the variability in the involvement of the facial muscles and upper extremity muscles respectively. Involvement of the leg muscles was influenced by age, but only to a very limited extent by the repeat array size (explained variance approximately 3%). Conclusion: This study provides additional evidence that other disease modifying factors play an important role in the clinical variability of FSHD patients, since only 40% of the variability can be attributed to familial factors. Possibly, the facial muscles are more sensitive to DUX4, whereas involvement of the leg muscles is highly dependent on disease modifying factors.
PS1Group1-110 / #708SCN4A CAN ACT AS MODIFIER GENE IN PATIENTS WITH MYOTONIC DYSTROPHY TYPE 2
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Anna Binda1, Laura V. Renna2, Francesca Bose’2, Elisa Brigonzi3, Annalisa Botta4, Rea Valaperta5, Barbara Fossati6, Ilaria Rivolta1, Giovanni Meola3, Rosanna Cardani7
1School Of Medicine And Surgery, University of Milano Bicocca, Monza, IT;2Laboratory Of Muscle Histopathology And Molecular Biology, IRCCS Policlinico San Donato, San Donato Milanese (MI), IT;3Department Of Biomedical Sciences For Health, University of Milan, San Donato Milanese (MI), IT;4Department Of Biomedicine And Prevention, Tor Vergata University of Rome, Rome, IT;5Research Laboratories, IRCCS Policlinico San Donato, San Donato Milanese (MI), IT;6Department Of Neurology, IRCCS Policlinico San Donato, San Donato Milanese (MI), IT;7Laboratory Of Muscle Histopathology And Molecular Biology, IRCCS-Policlinico San Donato, San Donato Milanese (MI), IT
Background: Myotonic dystrophy type 2 (DM2) is caused by a CCTG repeat expansion in intron 1 of the CNBP/ZNF9 gene located on chromosome 3q21. The mutant RNA transcripts containing CCUG repeats alter the activity of specific RNA binding proteins involved in alternative splicing regulation leading to splicing defects that are considered the primary cause of DM1 symptoms. Usually myotonia is mild and inconsistent in DM2 even by electromyography and it has been correlated with the disruption of the alternative splicing of the muscle chloride channel CLCN1. It is noteworthy that mutations in CLCN1 and SCN4A can act as modifier genes in patients with DM2 disease leading to an intensification of their myotonia. We report a 32 years old DM2 patient who presented an atypical phenotype characterized by an early severe myotonia since he was 12 years old. Mexiletine treatment resulted ineffective in reducing myotonia. While no mutation on CLCN1 gene was found, the genetic analysis of SCN4A gene showed a c.2717G>C base exchange in exon 14 predicting an S906T substitution. This variant is considered a benign polymorphism however electrophysiological studies revealed that it affects the fast and slow gating processes. Methods: To investigate the atypical DM2 phenotype of our patient, the combined effects of DM2 mutation and S906T substitution have been studied performing whole-cell patch-clamp in voltage and current clamp mode in myoblasts and myotubes obtained from the skeletal muscle biopsy of the patient. Results have been compared to those obtained in muscle cells derived from his mother, who is also affected by DM2, but did not present the S906T polymorphism. Results: A faster decay of the inactivation kinetics and a +5mV shift in the availability curve (p<0.05) were found in the sodium current recorded in myoblasts derived from the proband compared to his mother. In myotubes showing a stable resting membrane potential, instead, the rheobase current was significantly lower in the proband respect to the mother while the overshoot and the maximum slope of the depolarizing phase of the action potential were higher in the proband respect to the mother. Conclusion: These findings suggest that SCN4A polymorphism may be responsible for a higher excitability of DM2 patients sarcolemma, supporting the severe myotonic phenotype observed. We suggest that SCN4A may be considered a modifier factor and its screening should be performed in DM2 patients with uncommon clinical features.
PS1Group1-111 / #834AAV-SERCA2A EXPRESSION AMELIORATED CARDIOMYOPATHY IN THE MDX MOUSE MODEL OF DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Yongping Yue1, Nalinda Wasala1, William Lostal1, Lakmini Wasala1, Nandita Niranjan2, Roger Hajjar3, Gopal Babu2, Dongsheng Duan1
1Molecular Microbiology And Immunology (medical Science Bldg), University of Missouri, Columbia, MO, US;2Cell Biology And Molecular Medicine, New Jersey Medical School, Rutgers, The State University of New Jersey, Newark, US;3Mount Sinai School of Medicine, New York, US
Background: Loss of dystrophin leads to Duchenne muscular dystrophy (DMD). Increased cytosolic calcium levels play a critical pathogenic role in the development DMD cardiomyopathy. Cytosolic calcium can be removed by the sarco/endoplasmic reticulum calcium ATPase (SERCA). SERCA activity is reduced in dystrophic muscle. Methods: To test whether SERCA gene transfer can improve calcium recycling and mitigate dystrophic phenotype, we delivered an adeno-associated virus (AAV)-9 SERCA2a vector to 3-m-old mdx mice at the dose of 6E12 viral genome particles/mouse via the tail vein. Whole-body performances were assessed at 8 and 18 months post-treatment using treadmill running and forelimb grip force assays. Serum creatine kinase (CK) activity and heart function (ECG and left ventricular catheterization) were examined at 18 months post-treatment. Results: Western blots and immunostaining confirmed robust SERCA2a expression in heart and skeletal muscle. Forelimb grip strength and treadmill running were significantly improved in treated mice. AAV-9 SERCA2a treatment also reduced the serum CK level, a marker of muscle damage. Importantly, SERCA2a treatment significantly improved heart function. Specifically, all ECG parameters were normalized to the wildtype levels. Several measures of systolic function were normalized in the catheter assay. Treatment also significantly improved ejection fraction, the rate of cardiac relaxation and the end diastolic volume. Conclusion: Our results suggest that AAV-9 SERCA2a gene therapy is a promising approach to treat DMD cardiomyopathy
PS1Group1-112 / #623QUANTITATIVE ANALYSIS OF THIGH MUSCLE BUNDLES OF PATIENTS WITH MYOTONIC DYSTROPHY TYPE 1 (DM1), USING CT IMPAIRMENT RATIO
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Takahiro Nakayama1, Satoshi Kuru2
1Department Of Neurology, Division Of Neuromuscular Diseases, Yokohama Rosai Hospital, Yokohama, JP;2Neurology, National Hospital Organization Suzuka National Hospital, Suzuka, JP
Background: It was reported that the atrophy of cervical and thoracic paravertebral, vastus lateral and medial head of gastrocnemius muscle were commonly affected in the patients with myotonic dystrophy type 1 (DM1), however, that the time dependent deterioration had not been confirmed until now. We wanted to know the affected pattern of their thigh muscle and estimating function of their deterioration. Methods: Subjects: CT images of 7 DM1 patients (3 males and 4 females), who were examined once a year, enrolled in this study. Their ages at first visit to hospitals were from 17 to 43, and the number of CTG repeats were from 100 to 1200. Their CT data was collected from 3 to 11 years. Method: The Impairment ratios of each muscle bundle were plotted in each patient as long as possible. The muscle impairment ratio is the ratio of affected muscle fibers in muscle bundles, and calculation formulas were developed by our group. The ratio was determined by following formula, and we used the previously reported CT values of 58 and -98 HU for muscle and fat, respectively. impairment ratio=(“muscle CT value” - “pixel value”)/(“muscle CT value” - “fat CT value”) This investigation was carried out in accordance with the Human Research Guidelines of the Institutional Ethics Review Board of Yokohama Rosai Hospital. Results: In DM1 patients, firstly there is a period of slight muscle impairment. After this period, the muscle bundles deteriorate, and this deterioration was well estimated by cumulative normal distribution function. At the end of deterioration stage, we deduced the intermediate vastus thigh muscle is most commonly severely impaired. Most cases felt difficulty to walk at the age when the most affected muscle was impaired 10-20 %. There was no correlation between the CTG repeats and the deterioration. Conclusion: We inferred the muscle will begin to be impaired after the increase of CTG repeats in muscles. The intermediate vastus muscle will be more commonly and more severely affected than surrounding thigh muscles. We concluded that the cumulative normal distribution function was suitable for estimating their muscle deterioration.
PS1Group1-113 / #740LIMB-GIRDLE MUSCULAR DYSTROPHY IN THE CZECH REPUBLIC
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Kristyna Stehlikova1, Jana Zidkova2, Kamila Reblova3, Radim Mazanec4, Stanislav Vohanka5, Jana Haberlova6, Lenka Fajkusova7
1Centre Of Molecular Biology And Gene Therapy, University Hospital Brno, Brno, CZ;2Centre Of Molecular Biology And Gene Therapy, University Hospital Brno, Brno, CZ;3Ceitec, Masaryk University Brno, Brno, CZ;4Neurology, Second Faculty of Medicine, Charles University and University Hospital Motol, Prague, CZ;5Neurology, University Hospital Brno, Brno, CZ;66 Department Of Child Neurology, Second Faculty of Medicine, Charles University and University Hospital Motol, Prague, CZ;7Masaryk University Brno, Brno, CZ
Background: Limb-girdle muscular dystrophy (LGMD) is a group of disorders, which first affect muscles of the hip and shoulder areas. Genes associated with LGMD normally encode proteins that play vital roles in the muscle function, cell signalling, cell membrane repair, or removal of potentially toxic wastes from cells. 32 genes have been identified so far, 7 with autosomal dominant inheritance (LGMD1) and 25 with autosomal recessive inheritance (LGMD2). LGMD1 is relatively rare and represents less than 10% of all LGMD. LGMD2 is much more common, having a cumulative prevalence of 1:15,000, with some differences among countries. Methods: We perform molecular genetic diagnostics of neuromuscular disorders (NMD). At present, it is based on gene panel sequencing, pulsed-field gel electrophoresis and Southern analysis (for diagnostics of FSHD1), and repeat-primed PCR (for diagnostics of myotonic dystrophy) in case of muscular dystrophies and myopathies. Differential diagnosis of the LGMD remains quite large and may include LGMD, FSHD, Emery-Dreifuss muscular dystrophy, manifesting carriers of DMD, congenital myasthenic syndromes, congenital myopathies presenting later in childhood or adulthood, and others. Genes associated with mentioned diseases (except FSHD1 which is tested separately) are analysed in parallel with LGMD obligatory genes. Results: We present results of molecular genetic diagnostic of Czech LGMD patients determined by gene panel sequencing (used since 2012) or Sanger sequencing within the gene-by-gene approach (by 2011). In LGMD, we have 166 patients with confirmed genetic diagnosis – 94 patients with variants in CAPN3, 17 patients with variants in FKRP, 14 patients with variants in ANO5, 14 patients with variants in SGCA, 8 patients with a variant in LMNA, 6 patients with variants in DYSF, 4 patients with a variant in CAV3, 3 patients with variants in POMT2, 2 patients with variants in TRIM32, and 1 patient with variants/variant in each of ISPD, SGCG, SGCB, and TNPO3. Conclusion: Besides patients with genetically confirmed LGMD, we have LGMD patients without genetic diagnosis although all so far known muscular dystrophy/myopathy related genes were analysed. These patients are candidates for whole exome sequencing. This study was supported by the project of the Technology Agency of the Czech Republic (TE2000058) and the project of the Grant Agency of the Czech Republic (GA16-11619S/2016).
PS1Group1-114 / #837PULMONARY FUNCTION IN ADVANCED DUCHENNE MUSCULAR DYSTROPHY: ETEPLIRSEN-TREATED PATIENTS VERSUS A NATURAL HISTORY COHORT
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Heather Gordish-Dressman1, Erik Henricson2, Lixin Han3, Ashish Dugar3, Craig Mcdonald2, Cooperative International Neuromuscular Research Group (Cinrg)4
1George Washington University School of Medicine and Health Sciences, Washington, DC, US;2University of California Davis, Davis, CA, US;3Sarepta Therapeutics, Inc., Cambridge, MA, US;4. US
Background: Eteplirsen is a phosphorodiamidate morpholino oligomer that binds to and excludes exon 51 from dystrophin pre-mRNA, allowing translation and production of internally shortened dystrophin protein. It is approved in the US for treatment of patients with Duchenne muscular dystrophy (DMD) with a confirmed genetic mutation amenable to exon 51 skipping. We evaluated pulmonary function in patients with advanced DMD in a phase 2, open-label, multicenter study of eteplirsen. Methods: Eligible males aged 7-21 years with advanced DMD were enrolled and received once-weekly intravenous eteplirsen 30 mg/kg for 96 weeks. Eligible patients must have been receiving either no corticosteroids or a stable oral corticosteroid dose for ≥24 weeks prior to study entry, with no change in dosage anticipated. Pulmonary function, a predefined exploratory outcome, was assessed by mean change from baseline in percent predicted forced vital capacity (FVC%p). Safety assessments related to pulmonary function included mean change from baseline in respiratory rate (breaths/min) by Week 96. FVC%p outcomes were also evaluated in a Cooperative International Neuromuscular Research Group (CINRG) DMD Natural History Study cohort (patients aged 8-19 years) receiving standard of care treatment with glucocorticoids. Results: Of 24 patients enrolled, 23 (96%) completed 96 weeks of eteplirsen treatment (baseline median age, 13.0 y [range: 8-19]; mean FVC%p, 68.7). Eteplirsen-treated patients had a mean (standard error [SE]) change from baseline in FVC%p of -6.4 (2.16) at Year 1. Mean (SE) change in FVC%p from Year 1 to Year 2 for eteplirsen-treated patients was -0.5 (2.07). Mean respiratory rates were 19.0 at baseline, 20.5 at Week 48, and 20.5 at Week 96 for eteplirsen-treated patients. A comparable CINRG natural history subset (n=10) had similar demographic and clinical characteristics, with mean change in FVC%p of -8.2 over a time frame of 1 year. Conclusion: Eteplirsen treatment slowed the decline of FVC%p in patients with advanced DMD relative to a comparable natural history cohort. The rate of decline for eteplirsen-treated patients slowed most appreciably in Year 2, suggesting a greater impact on disease progression with prolonged eteplirsen treatment.
PS1Group1-115 / #931CLINICAL OUTCOME STUDY OF DYSFERLINOPATHY: TEENAGE EXERCISE AS A POTENTIAL MODIFIER OF DISEASE SEVERITY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Claudio Semplicini1, Ursula Moore2, Marni Jacobs3, Roberto Fernández-Torrón4, Jiji Jang3, Meredith James2, Anna Mayhew2, Laura Rufibach5, Plavi Mittal5, Michele Eagle2, Avital Cnaan6, Pierre G. Carlier7, Andrew Blamire8, Heather Hilsden2, Hanns Lochmuller2, Ulrike Grieben9, Simone Spuler10, Carolina Tesi Rocha11, John W. Day11, Kirsti J. Jones12, Diana X. Bharucha-Goebel13, Emmanuelle Salort-Campana14, Matthew Harms15, Alan Pestronk15, Sabine Krause16, Olivia Schreiber-Katz16, Maggie C. Walter16, Carmen Paradas17, Jean-Yves Hogrel18, Tanya Stojkovic19, Shin’Ichi Takeda20, Madoka Mori-Yoshimura21, Elena Bravver22, Susan Sparks22, Jordi D. Manera23, Luca Bello24, Elena Pegoraro1, Jerry R. Mendell25, Kate Bushby2, Volker Straub26
1University of Padova - Azienda Ospedaliera di Padova, Padova, IT;2The John Walton Muscular Dystrophy Research Centre, Newcastle Hospitals Trust, Newcastle University, Newcastle Upon Tyne, GB;3Division Of Biostatistic And Study Methodology, Children’s National Health System, Centre for Translational Science, DC, WA, US;4Neuromuscular Area, Biodonostia Health Research Institute, Neurology Service, Donostia University Hospital, Donostia University Hospital, Donostia-San Sebastian, ES;5Jain Foundation, Seattle, US;6Pediatrics, Epidemiology And Biostatistics, George Washington University, Washington DC, US;7Aim & Cea Nmr Laboratory, Neuromuscular Investigation Center, Institute of Myology, Paris, FR;8Magnetic Resonance Centre, Institute For Cellular Medicine, Newcastle University, Newcastle Upon Tyne, GB;9Charite Muscle Research Unit, A Joint Cooperation Of The Charité Medical Faculty And The Max Delbrück Center For Molecular Medicine, Experimental and Clinical Research Center, Berlin, DE;10Charite Muscle Research Unit, Experimental and Clinical Research Center, Berlin, DE;11Department Of Neurology And Neurological Sciences, Stanford University School of Medicine, Stanford, CA, US;12Institute For Neuroscience And Muscle Research, Children’s Hospital At Westmead, University of Sydney, Sydney, AU;13Department Of Neurology, Children’s National Health System, Washington DC, US;14Neuromuscular And Als Center, La Timone Hospital, Aix-Marseille Universite, Marseille, FR;15Department Of Neurology, Washington University School of Medicine, St. Louis, MO, US;16Friedrich-baur Institute, Departement Of Neurology, Ludwigs-Maximilians University of Munich, Munich, DE;17Neuromuscular Unit, Department Of Neurology, Hospital U. Virgen del Rocio/Instituto de Biomedicina de Sevilla, Sevilla, ES;18Institut De Myologie, Boulevard de l’Hôpital, Paris, FR;19Ap-hp, G.h. Pitié-salpêtrière 47-83, Boulevard De L’hôpital, Institut de Myologie, Paris, FR;20National Center of Neurology and Psychiatry, Tokyo, JP;21Department Of Neurology, National Center Hospital, National Center of Neurology and Psychiatry, Tokyo, JP;22Carolinas Healthcare System Neurosciences Institute, Charlotte, NC, US;23Neuromuscular Unit, Neurology Department, Hospital de la Santa Creu i Sant Pau, Barcelona, ES;24Department Of Neuroscience, University of Padova, Padova, IT;25Center for Gene Therapy Nationwide Children’s Hospital, Columbus, US;26John Walton Muscular Dystrophy Research Centre, Newcastle University, Institute of Genetic Medicine, Newcastle Upon Tyne, GB
Background: The Clinical Outcome Study of Dysferlinopathy (COS) is an international 3 year natural history study of dysferlinopathy patients. Dysferlinopathy, a genetic muscular dystrophy caused by DYSF mutations, demonstrates a variable age of presentation and progression rate, suggesting disease modifying factors. Here we investigate teenage exercise intensity and subsequent disease progression in 182 patients. Methods: Participants were asked about the type, level and frequency of all physical activity prior to symptom onset. Self-reported age of first symptoms, first wheelchair use and full-time wheelchair use was taken from screening questionnaires. Exercise was classified based on metabolic equivalents (METs) as moderate (MET 3–6) or vigorous (MET >6). Participants were coded, based on the maximum frequency of activity reported between ages 10 and 18 years, as 0—no physical activity; 1—vigorous activity occasionally/monthly, or moderate activity once weekly; 2— moderate activity multiple times per week or vigorous activity once weekly; and 3—vigorous activity multiple times per week. Age of symptom onset was compared by analysis of variance (ANOVA) with least squares means for individual group differences. Risk of symptom onset, occasional wheelchair use and full-time wheelchair requirement over time were compared for exercise groups 1, 2 and 3 against group 0 using Cox proportional hazards regression. Interaction between teen exercise level, gender and clinical diagnosis was also assessed by two-way ANOVA. Subgroups of limb girdle muscular dystrophy 2B (LGMD2B), Miyoshi myopathy (MM) or ‘other’ (all genetically confirmed dysferlinopathies) were used for analysis. Results: Estimated mean age of symptom onset differed by group (P=0.03) and was later in group 0 (mean 24.8 years; 95% CI 22.3 to 27.2 years) compared with groups 2 (mean 20.2 years; CI 18.1 to 22.3 years, P=0.006) and 3 (mean 20.6 years, CI 18.4 to 22.8 years, P=0.01), but not group 1 (mean 21.7years; CI 17.7 to 25.7 years, P=0.20). Cox regression analysis suggested that groups 2 (Hazard ratio 1.56 95% CI 1.06 to 2.30)) and 3 (Hazard ratio 1.54 (1.04 to 2.30)) were at increased risk of earlier symptom onset than group 0 (figure 1). This was not significant for group 1 (Hazard ratio R 1.38 (0.78 to 2.45)). Conclusion: This study raises implications for early diagnosis and clinical care, however as we did not look at the effects of exercise once symptoms began, we would not advocate that symptomatic patients stop exercising.
PS1Group1-116 / #507GLYCOGENOSIS TYPE V (MCARDLE DISEASE): THERAPY WITH VITAMIN B6
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Annika Saak, Heinz Reichmann, Jochen Schaefer
Neurology, University Hospital Dresden, Dresden, DE
Background: The glycogen storage disease type V or McArdle disease is one of the metabolic myopathies and is characterized by a stress intolerance with myalgia, myopathy and muscle cramps. The autosomal-recessive disorder is in Europe usually caused by a stop mutation (P-R50X) in the PYGM gene. There is no causal form of therapy so far, so that patients are advised of symptomatic therapy with creatine or cane sugar before physical exertion. Positive effects of vitamin B6 on the subjective muscular resilience have been described in individual case reports. Our patient, in which a previously undescribed genetic defect (c. 1456G > A) could be detected in the PYGM gene, was treated with vitamin B6 for over 4 years. The subjective improvement reported by the patient should be clinically and laboratory-chemically objectified. Methods: We performed recurrent clinical examinations to find a subjective symptom improvement. In addition, the laboratory-chemical determination of the lactate and ammonia with and without vitamin B6 therapy was carried out during the work test with 2-minute ischemia. Results: The patient reports a progressive improvement in muscular strength and endurance and negates myalgia under the vitamin B6 therapy. A doubling of the walking distance could be achieved under vitamin B6. Under the vitamin B6 substitution occurred a drawdown of the creatine kinase. In the work test with vitamin B6 supplementation exposed a normal increase in lactate and ammonia, while without vitamin B6 only a slight increase in lactate was recorded. Conclusion: We report on a patient with a previously undescribed homozygous, pathogenic mutation in the PYGM gene, which is located in the vicinity of the PLP binding site of the myophosphorylase and thus explains the good response to vitamin B6. The daily substitution of vitamin B6 could help to improve clinical and laboratory chemical muscular symptoms. In our view, a therapy attempt seems justified in the case of low costs and side effects of vitamin B6 in the case of proven McArdle disease, particularly in the case of a mutation in the immediate vicinity of the PLP binding site of the myophosphorylase.
PS1Group1-117 / #782MASSIVE INCREASE IN CARDIAC TROPONIN T WITHOUT CARDIAC INVOLVEMENT IN NECROTIZING MYOPATHY WITH ANTI-HMGCR-ANTIBODIES
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Annika Saak1, Jochen Schaefer2, Sandra Jackson2
1Neurology, University Hospital Dresden, Dresden, DE;2Neurology, University Hospital Dresden, Dresden, DE
Background: Immune-mediated necrotizing myopathies may be caused by antibodies to anti-SRP or anti-HMG CoA reductase. They are characterized by muscle weakness, pronounced hyperCKemia and myalgias and may be associated with malignancies. Therapeutically, various immunosuppressants such as steroids, immunoglobulins or cytotoxic drugs are used. Methods: A 19-year-old statin-naïve patient developed acute tetraplegia with muscle tenderness and high antibody titers to HMG-CoA reductase. Besides marked hyperCKemia (up to 308μCat / L; norm<3.2) a massive increase in cardiac troponin T (up to 2580 ng / L; norm <14) was noted in the absence of overt cardiac involvement. A musculoskeletal origin of cardiac troponin T was suspected.We determined cardiac troponin T (cTnT) and cardiac troponin I (cTnI) under various therapeutic regimens and investigated for cardiac involvement due to the underlying necrotizing myopathy. In addition, the expression of cTnT and cTnI is currently being analyzed in undifferentiated (myoblasts) and differentiated (myotubes) cultured muscle cells. Results: Extensive cardiac diagnostic investigations (ECG, TTE, cardiac CT and MRI, heart rate variability) were unremarkable. However, despite massively elevated cTnT levels, cardiac troponin I values always remained normal. Alongside the clinical improvement under intravenous immunoglobulin therapy, there was a concomitant decrease in creatine kinase, cTnT and anti-HMG-CoA reductase antibody titers. Conclusion: We report a statin-naive patient with severe anti-HMGCR-positive necrotizing myopathy, who had massively elevated cTnT levels. After exclusion of cardiac involvement, which was corroborated by normal concentrations of the entirely cardiospecific cTnI, we interpret the massive increase of cTnT in the patient as of exclusively musculoskeletal origin. Although cTnT may be mildly elevated (10fold) in various myopathies, it has – to our knowledge – not been described to rise to such an extent (200fold) in myopathies without cardiac involvement. Compared to mature myocytes, HMGCR expression is up-regulated in regenerating muscle fibres which may therefore undergo an accelerated turnover in an anti-HMGCR-positive necrotizing myopathy. Moreover, some evidence suggests that cTnT which is expressed only in low amounts in mature myocytes, is expressed in much higher amounts in regenerating skeletal muscle cells, thus explaining particularly high levels of cTnT in our patient. This is to be verified in muscle cell cultures at various stages of differentiation.
PS1Group1-118 / #688RESULTS OF NORTH STAR AMBULATORY ASSESSMENTS IN THE ACT DMD TRIAL IN IN PATIENTS WITH NMDMD
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Francesco Muntoni1, Panayiota Trifillis2, Gary L. Elfring2, Stuart W. Peltz3, Craig Mcdonald4
1Dubowitz Neuromuscular Unit, UCL Great Ormond Street Institute of Child Health, London, GB;2PTC Therapeutics, Inc, South Plainfield, NJ, US;3PTC Therapeutics Inc, South Plainfield, NJ, US;4Physical Medicine And Rehabilitation, UC Davis Medical Center, Sacramento, CA, US
Background: Ataluren is the first drug to treat the underlying cause of nonsense mutation Duchenne muscular dystrophy (nmDMD) by promoting readthrough of a premature stop codon to produce full-length functional dystrophin. ACT DMD was a phase 3, randomized, double-blind, placebo-controlled trial of ataluren that enrolled males aged 7–16 years with nmDMD and baseline six-minute walk distance (6MWD) ≥150m and ≤80%-predicted. Methods: Eligible patients were randomized 1:1 to receive ataluren 10, 10, 20 mg/kg or placebo orally three times daily for 48 weeks. The NSAA is a validated functional scale to measure disease progression specifically in ambulant boys with DMD. It consists of 17 activities ranging from standing from a chair to jumping. Each activity is scored as 0, 1, or 2; the sum of these 17 scores forms the total score, which is linearized to a 0 (worst)-100 (best) score. The intent-to-treat population of ACT DMD consisted of 228 patients (ataluren, n=114; placebo, n=114). Results: Overall, patients who received ataluren gained a 1.5-point advantage in NSAA observed score compared with patients who received placebo (mean NSAA scores, ataluren: -7.0; placebo: -8.5; p=0.270). In addition, fewer patients who received ataluren shifted from a score of 1 or 2 (able to perform function) to 0 (unable to perform function) across all 17 activities compared with those who received placebo. Between-group differences across each of the 17 activities consistently favored ataluren, ranging from 1% (lift head) to 11% (jump) to 12% (rise from chair). Conclusion: In conclusion, ataluren is the first drug to demonstrate a benefit to patients with nmDMD compared with placebo as assessed by NSAA scores.
PS1Group1-119 / #932NONSENSE AND SINGLE NUCLEOTIDE FRAMESHIFT MUTATIONS IN BECKER MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Monica Traverso1, Chiara Panicucci1, Michela Catteruccia2, Paolo Broda1, Francesca Madia3, Giulia Pozzolini3, Maria Derchi4, Annalaura Torella5, Claudio Bruno6, Federico Zara3, Vincenzo Nigro5, Carlo Minetti1, Adele D’Amico2, Chiara Fiorillo7
1Pediatric Neurology And Muscular Disorders, Gaslini Children Hospital, Genoa, IT;2Unit Of Neuromuscular Disorders, Laboratory Of Molecular Medicine, Bambino Gesu’ Children’s Hospital,, Rome, IT;3Neurogenetic Laboratory, University of Genoa-Gaslini Children Hospital, Genoa, IT;4Cardiology, University of Genoa-Gaslini Children Hospital, Genoa, IT;5Precision Medicine, Università degli Studi della Campania “Luigi Vanvitelli”, Naples, IT;6Centre Of Myology, University of Genoa-Gaslini Children Hospital, Genoa, IT;7Neuroscience, University of Genoa, Genoa, IT
Background: Nonsense and frameshift mutations in DMD gene potentially determine the interruption of protein synthesis and degradation of a truncated dystrophin, for this reason they are usually associated with severe Duchenne phenotype (DMD). Conversely, in-frame mutations lead to reduced or abnormal protein synthesis and are associated to milder Becker muscular dystrophy (BMD). Exceptions of this rule are found in a small percentage of cases. In these uncommon cases, the exact location of the mutation is considered an important factor. Nevertheless other factors, such as presence of splicing regulatory elements, can influence the ultimate outcome and they are largely unexplored in routine diagnostic procedures. Methods: We present clinical, molecular and immunohistochemistry study of a sample of 9 Italian BMD patients (age range 7-30 years) carrying nonsense or single nucleotide mutations in DMD gene. Patients were evaluated with North-Star Ambulatory Assessment (NSAA) and muscle MRI. Muscle biopsies were obtained from 8 cases. Routine histological procedures, immunofluorescence and western blot were performed. We also analysed the specific transcripts involving the different mutations via RT-PCR. Results: Most mutations are nonsense, localised between exon 1-29 and in exon 74. One mutation is a splice site variant leading to absence of exon 11 and subsequent loss of reading frame. Another mutation is a single nucleotide duplication leading to downstream frameshift in exon 73. Six mutations are described and have been associated either with DMD or BMD. Three variants are novel. From a clinical point of view, all patients are still ambulant, however 4 presented moderate phenotype (NSAA below 30) and the oldest patient was not able to rise from the floor autonomously. CK was significantly elevated (range 3000-15000 U/l, normal value < 200). The 2 oldest patients also displayed signs of heart involvement and were in therapy with ACE inhibitors. No patient was taking corticosteroids. Muscle sections showed variable degree of necrosis and degeneration of muscle fibres, which correlates with age at biopsy and CK level. Dystrophin expression was only partially reduced with immunofluorescence from moderate severe, to mild reduction. Interesting in most patients staining for alpha-dystroglycan was also markedly reduced. Western blot showed either absent dystrophin or reduced amount of protein of normal size. In three cases a lower weight dystrophin band was detected with Western Blot. RT-PCR documented presence of normal size products in nonsense mutations and of shorter products in the case of splice site mutation. Conclusion: In this small cohort, we observed nonsense and single nucleotide frameshift mutations in BMD patients, few presenting a moderate phenotype when compared with the most common BMD patient with multiple exon deletions. Additionally, we showed that dystrophin mRNA and protein are still retained in affected muscles, suggesting an altered function of the protein. We argue that this can be highlighted by marked reduction of the distrophyn-dystroglycans complex. These cases underscore the importance of a complete analysis of DMD gene and its transcript in order to provide better prognostic information and genotype-phenotype correlation. Additionally presence of nonsense mutations in BMD patients raise question on the possible therapeutic approaches.
PS1Group1-120 / #959DEVELOPMENT OF A PROGNOSTIC MODEL FOR 1-YEAR CHANGE IN TIMED 4 STAIR-CLIMB IN DUCHENNE PATIENTS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Nathalie Goemans1, Marleen Vanden Hauwe1, James Signorovitch2, Gautam Sajeev2, Zhiwen Yao2, Madeline Jenkins2, Erin Mcdonald2, Imbrahima Dieye2, Susan J. Ward3
1Neuromuscular Reference Centre, Department Of Paediatrics And Child Neurology, University Hospitals Leuven, Leuven, BE;2Analysis Group, Boston, US;3cTAP, Cambridge, US
Background: Longitudinal progression of disease is inherently heterogeneous among individuals with Duchenne muscular dystrophy (DMD). The resulting variation in outcome measures can complicate clinical trial design and potentially cloud interpretation of results. The collaborative Trajectory Analysis Project (cTAP) is a pre-competitive coalition of academic clinicians, drug developers, and patient foundations; cTAP was formed in 2015 to identify biostatistical approaches to overcome the challenges of high variation in clinical trials in DMD. We previously reported a prognostic score that explained 60% of variation in the change in 6 minute walk distance (6MWD) over 1 year, and significantly improved upon prognosis from baseline age, 6MWD and steroid use by also incorporating timed function tests, height and weight [Goemans N, vanden Hauwe M, Signorovitch J, Swallow E, Song J, CollaborativeTrajectory Analysis Project (cTAP) (2016). Individualized Prediction of Changes in 6-Minute Walk Distance for Patients with Duchenne Muscular Dystrophy. PLoS ONE 11(10): e0164684. doi:10.1371/journal.pone.0164684] The aim of the present study was to develop a prognostic model for 1-year change in 4-stair climb (4SC) among DMD patients, and to assess the additional predictive value of the model compared to commonly used factors (i.e., age, baseline 6MWD and steroid use). Methods: Natural history data were collected from DMD patients approximately every 6 months over the course of 2 to 5 years during routine clinical practice at the Universitaire Ziekenhuizen pediatric neurology clinic in Leuven, Belgium. Patient demographics, treatment experience and ambulatory outcomes were recorded at each visit. Annualized changes in 4SC were studied between all pairs of visits separated by ~1 year (8-16 months). Prediction models were developed using multivariable regression for repeated measures. Generalized estimating equations (GEE) with an exchangeable covariance structure were used to account for the use of multiple pairs of visits from individual patients. Results: A total of n=235 ~1-year follow-up intervals from n=81 boys were included. Mean age was 9.1 years and mean 4-SC was 3.84 s at the start of these intervals; average duration of steroid use was 29.11 months. During the subsequent ~1-year, mean annualized change in 4SC was 0.71 s with a standard deviation (SD) of 2.20. Predictions based on age, baseline 4SC and steroid use explained 13% of variation in annualized 4SC changes (R-squared = 0.13). A broadened prognostic model, adding timed 10-meter walk/run, and rise from supine, as well as height and weight, significantly improved prediction, explaining 34% of variation in annualized 6MWD changes (R-squared=0.34). In the broadened model, parameters that were most strongly correlated with 1 year change in 4SC were steroid use > 1 year (p <0.001), baseline 4SC (s)(p < 0.001), baseline rise from supine (s)(p < 0.001) and baseline 10 meter walk run (s) (p<0.05). Conclusion: A prognostic model incorporating timed function tests significantly improved prediction of 1-year changes in 4SC. Explained variation was more than doubled compared to predictions based only on age, baseline 4SC and steroid use, indicating significant potential for broader prognostic models to inform clinical trial design and interpretation in DMD
PS1Group1-121 / #989AMBULATORY ELECTROCARDIOGRAPHIC LONGITUDINAL STUDY IN THE GRMD DOG MODEL OF DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Inès Barthélémy, Xavier Cauchois, Stephane Blot
U955 - Imrb, Inserm, Ecole Nationale Vétérinaire D’alfort, Upec, U955 - IMRB, Inserm, Ecole Nationale Vétérinaire d’Alfort, UPEC, Maisons-Alfort, FR
Background: Duchenne muscular dystrophy (DMD) is a progressive muscle disease leading to motor, respiratory and cardiac disabilities. Electrocardiographic (ECG) monitoring is part of the standards of care for DMD patients, and reveals tachycardia, elongated QT, deep Q-waves, premature ventricular beats (PVBs), and early decreased heart rate variability (HRV). The preclinical research of treatments targeting DMD is supported by a translational chain, which ultimate link is the GRMD dog (Golden Retriever Muscular Dystrophy), since it faithfully reproduces the DMD disease course in a large animal context. This includes the occurrence of a cardiomyopathy, with onset of myocardial fibrosis in the 6-12 months of age period, followed by a decrease of heart contractility, and ultimately in some dogs a death from decompensated dilated cardiomyopathy. Since this model is used at the very last preclinical step, there is a need to evaluate the effect of candidate treatments using methods analog to those used in patients. Few studies have focused on ECG in GRMD, but no one longitudinally using ambulatory Holter recordings, and investigating HRV. We aimed to describe the sequential ECG alterations in GRMD dogs in order to provide relevant indices to be used in preclinical studies. Methods: Holter ECG recordings were performed monthly overnight in 15 GRMD and 4 healthy littermates from the age of 2 months until the age of 24 months. The lead II was used for quantitative analysis, which first focused on four timepoints: 6, 12, 18, and 24 months of age. The heart rate, QT interval, Q/R ratio, and the occurrence of arrythmias were quantified. A HRV analysis was also performed including time-domain analysis and frequency domain analysis. Results: One of the 4 healthy dogs presented an increased heart rate, a markedly reduced HRV, and had transiently elevated serum cTpnI. We thus chose to exclude this dog from the analysis. The heart rate was found increased in GRMD dogs, this difference being significant from the age of 12 months. There was a trend towards elongated QT at 24 months. We found a significantly increased Q/R ratio only at the age of 6 months. PVBs were observed in 5 over the 8 dogs at 24 months of age. Two of them already presented such arrhythmias at 6 months of age, and most of the others started to exhibit PVBs between 12 and 18 months of age. These PVBs were polymorphic in two dogs, and could be seen either isolated, in doublets or triplets; transient episodes of ventricular tachycardia could also be detected. HRV analysis revealed significantly decreased pNN50 at 12 months of age, and a decrease of the LF/HF ratio from the age of 12 months. Conclusion: This study provides new indices to monitor the cardiac disease in GRMD dogs, and again demonstrates similarities between GRMD and DMD cardiomyopathies. These similarities and the availability of a comprehensive evaluation set including ECG, echocardiography, cardiac NMRI, paralleling the one used in DMD patients, make the GRMD dog an optimal context to anticipate therapeutic effects expectable in DMD patients, measured with the same tools and indicators.
PS1Group1-122 / #652MIMICS OF INCLUSION BODY MYOSITIS: CASE PRESENTATIONS AND IDENTIFICATION OF TYPICAL PITFALLS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Rachel Zeng, Stefanie Glaubitz, Karsten Schmidt, Per-Ole Carstens, Jens Schmidt, Jana Zschüntzsch
Department Of Neurology, University of Göttingen, Göttingen, DE
Background: Inclusion body myositis (IBM) is an idiopathic inflammatory myopathy with typical clinical symptoms und histological features. However, due to a range of acquired or genetically determined myopathies with similar presentations, IBM is often missed and diagnosed in error as another neuromuscular disease. Methods: Along with dermatomyositis, polymyositis, overlap myositis and (immune mediated) necrotizing myopathy, IBM belongs to the group of idiopathic inflammatory myopathies (IIM). The typical clinical presentation of IBM involves slowly progressive muscle atrophy and weakness with a preference of long finger flexors, knee and foot extensors, often with an asymmetric distribution. Other symptoms may include myalgia and dysphagia, the latter reported in over the half of the patients with IBM. The diagnosis of IBM is currently based on a defined composition of clinical, serological and muscle histologic criteria. Additionally, electromyography, muscle MRI and muscle ultrasound may help to confirm the diagnosis. A muscle biopsy is required to diagnose a clinico-pathologically defined IBM, showing the co-existence of myodegenerative and inflammatory pathologic features. Nevertheless, the diagnosis of IBM or the differentiation from other neuromuscular diseases can be difficult due to atypical clinical presentations. Histopathological findings can also lead to confusion since inflammatory infiltrates can also be found in hereditary myopathies like dysferlinopathy or facioscapulohumeral dystrophy. The myodegenerative characteristics of myofibrillar myopathies like vacuoles and abnormal accumulations of filamentous proteins can also be mistaken. Only a correct and reliable diagnosis of IBM allows for recruitment to clinical studies and helps to find novel, exploratory treatment modalities and prevent unnecessary treatments. Results: Here, we provide typical case reports of hereditary inclusion body myopathy (hIBM), titinopathy (TMD/LGMD2J), facioscapulohumeral muscular dystrophy (FSHD), polymyositis, chronic inflammatory axonal polyneuropathy (CIAP) and other neuromuscular diseases that can mimic IBM. Conclusion: The data are carefully discussed and typical clinical pitfalls are summarized.
PS1Group1-123 / #690LONG-TERM PULMONARY FUNCTION IN NON-AMBULATORY PATIENTS WITH NMDMD TREATED WITH ATALUREN
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Craig Mcdonald1, Eugenio Mercuri2, Francesco Muntoni3, Kathryn Selby4, Fengbin Jin5, Gianina Panaghie-Meltzer6, Panayiota Trifillis5, Marcio Souza6, Stuart W. Peltz6, Mar Tulinius7
1Physical Medicine And Rehabilitation, UC Davis Medical Center, Sancramento, CA, US;2Università Cattolica del Sacro Cuore, Rome, IT;3Dubowitz Neuromuscular Unit, UCL Great Ormond Street Institute of Child Health, London, GB;4British Columbia Children’s Hospital, Vancouver, BC, CA;5PTC Therapeutics, Inc, South Plainfield, NJ, US;6PTC Therapeutics Inc, South Plainfield, NJ, US;7Gothenburg University, Gothenburg, SE
Background: Duchenne muscular dystrophy (DMD), an X-linked, recessive disease affecting ~ 1 in every 3600–6000 live male births, is caused by dystrophin gene mutations. Dystrophin is critical for myofiber structural stability and function; its absence leads to progressive muscle dysfunction, loss of ambulation, and early death from respiratory/cardiac failure. Forced vital capacity (FVC) < 1 L raises mortality risk in DMD patients. About 10–15% of patients have a dystrophin gene nonsense mutation (nonsense mutation DMD [nmDMD]), where a premature stop codon halts translation to generate truncated, nonfunctional dystrophin. Ataluren promotes ribosomal readthrough of the premature stop codon to produce a full-length dystrophin protein, treating the underlying cause of nmDMD. Methods: FVC data from non-ambulatory patients at study entry (unable to run/walk 10 m in ≤ 30 seconds) aged 9 to 18 years receiving oral ataluren (40 mg/kg/day [10, 10, and 20 mg/kg for morning, midday and evening doses, respectively]) were obtained from Study 019 (NCT01557400; begun in 2012; data cut-off Jan. 31, 2017), an international, multicenter, open-label trial that enrolled patients from previous ataluren PTC-sponsored trials at non-US study sites. Data for age-matched, non-ambulatory patients (wheelchair-bound) in subjects receiving SOC (not ataluren) were obtained from an ongoing natural history study (CINRG, NCT00468832; from 2012 through Nov.18, 2016) and compared to 019 FVC < 1L by Kaplan-Meier analysis. Results: Subgroups included 38 ataluren- and 58 SOC-treated patients. At data cut-off fewer ataluren-treated vs SOC-treated patients had FVC <1 L (7.9% vs 39.7%, respectively). FVC < 1L was reached by 50% of SOC-treated patients by age) age 19.3 years (95% CI, 18.8–22.6). At age 19.3 years, only 16% of ataluren-treated patients had FVC < 1 L. FVC <1L was reached by 50% of SOC-treated patients after 7.1 years (5.3–9.4) with SOC (log-rank test p = 0.067); 10% of ataluren-treated patients had FVC <1L after 4 years. Most adverse events (AEs) on ataluren were mild (29.5% of patients) or moderate (31.8% of patients). The most common AEs were nasopharyngitis (45.5%), headache (27.3%), vomiting (27.3%), and gastroenteritis (22.7%). Conclusion: Findings suggest that ataluren preserves lung function in non-ambulatory patients with nmDMD. Safety and tolerability were consistent with previous findings.
PS1Group1-124 / #701USE OF A >/= 5-SECOND THRESHOLD IN BASELINE TIME TO STAND FROM SUPINE TO PREDICT PROGRESSION IN DMD
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Craig Mcdonald1, Marcio Souza2, Gary L. Elfring3, Panayiota Trifillis3, Joseph Mcintosh2, Stuart W. Peltz2, Eugenio Mercuri4
1Physical Medicine And Rehabilitation, UC Davis, Sacramento, CA, US;2PTC Therapeutics Inc, South Plainfield, NJ, US;3PTC Therapeutics, Inc, South Plainfield, NJ, US;4Università Cattolica del Sacro Cuore, Rome, IT
Background: Time to stand from supine is a predictor for loss of clinically meaningful Duchenne muscular dystrophy (DMD) milestones. A ≥5-second threshold in this functional test can predict disease progression over 48 weeks; conversely, a <5-second threshold suggests functional stability. This analysis examined the usefulness of a ≥5-second threshold as a subset analysis for DMD trials. Methods: A post hoc analysis was conducted using data from the subset of patients with a baseline time to stand from supine ≥5 seconds from two 48-week, randomized controlled trials examining the efficacy and safety of ataluren in patients with nonsense mutation DMD: a phase 2b (Study 007/NCT00592553; boys ≥5 years) and a phase 3 trial (Study 020/NCT01826487; boys ≥7–≤16 years). Results: In Study 007, 37 ataluren- and 29 placebo-treated patients met the ≥5-second threshold. Their respective mean (standard deviation, SD) baseline 6-minute walk distances (6MWD) were 311.3 (91.1) and 312.2 (82.2) m. In Study 020, 56 ataluren- and 46 placebo-treated patients met the ≥5-second threshold: mean (SD) baseline 6MWD, 349.9 (60.2) vs 336.6 (63.8) m, respectively. A beneficial ataluren response vs placebo was observed across multiple endpoints in both trials in the ≥5-second threshold subgroup. Study 007 (ataluren vs placebo, respectively); 48-week change in: 6MWD (–35.9 vs –85.6 m, p=0.01), 10-m run/walk (2.5 vs 5.1 seconds, p=0.10), 4-stair climb (3.5 vs 7.5 seconds, p=0.02). Study 020 (ataluren vs placebo, respectively); 48-week change in: 6MWD (–48.7 vs –100.1 m, p<0.01), 10-m run/walk (2.3 vs 4.8 seconds, p=0.03), 4-stair climb (4.7 vs 7.6 seconds, p=0.04). Conclusion: Consistent with published natural history data, placebo-treated patients with a baseline time to stand from supine ≥5 seconds showed declining ambulatory ability over 48 weeks. A ≥5-second threshold may represent a useful subset analysis for DMD trials; this post hoc analysis highlights an ataluren treatment benefit vs placebo.
PS1Group1-125 / #780NOVEL THERAPEUTIC PERSPECTIVES FOR SARCOGLYCANOPATHY, IN VITRO AND IN VIVO STATE OF THE ART
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Dorianna Sandona1, Roberta Sacchetto2, Elisa Bianchini2, Marcello Carotti2, Michela Soardi3, Chiara Fecchio4
1University of Padova, Padova, IT;2university of padova, Padova, IT;3university of padova, padova, IT;4Biomedical Science, University of Padova, Padova, IT
Background: Sarcoglycans (SG) are glycosylated proteins (alpha-, beta-, gamma- or delta-SG) forming a key structural complex, essential for the sarcolemma integrity of striated muscles during contraction. In sarcoglycanopathies, defects in any one of the sarcoglycan genes lead to the strong reduction or even the loss of the SG-complex. Most of the reported cases are due to missense mutations originating a full length but folding-defective proteins. We proved that the primary pathological event in sarcoglycanopathy occurs in the Endoplasmic Reticulum, where the quality control system, by proof-reading newly synthesized sarcoglycans, recognizes and delivers to proteasomal degradation the folding-defective mutants. This results in the secondary loss of the wild-type partners. We also observed that many missense mutants retain their function as the entire complex can be properly rescued by reducing the mutant degradation. These findings opened new therapeutic perspectives for this neglected disease allowing to design small molecule-based approaches aimed either to inhibit sarcoglycan mutants degradation, or to help their folding so that, skipping disposal, they can assemble and traffic at the proper site of action. Methods: Culture and differentiation of primary myogenic cells of LGMD2D patients. Biochemical and molecular analysis of sarcoglycan expression. Primary myotubes treatment with small molecules. AAV transduction of newborn alpha-SG KO mice. CRISPR-Cas9 genome editing of zebrafish. Results: We tested several small molecules known as CFTR correctors which successfully recovered different mutants of alpha-sarcoglycan in cellular models and, notably, the whole SG-complex in primary myotubes from a patient suffering of alpha-sarcoglycanopathy. Moreover, we performed a functional test by measuring CK release from patient’s myotubes under stressful conditions, showing membrane stabilization when cells were pretreated with CFTR correctors. Interestingly, cytotoxicity tests highlighted absent or low toxicity of these compounds on cells. To confirm in vivo this successful strategy we need animal models expressing folding-defective sarcoglycans. As the available SG-KO and KI mice are unsuitable to our purposes, and considering the large number of reported sarcoglycan missense mutants, our aim is now the generation and characterization of novel and unconventional alpha-sarcoglycanopathy vertebrate models. To this purpose, we have planned to generate transiently “humanized” mice, by the transduction of the null mice with rAAVs (recombinant adeno associated viruses) expressing different missense mutants of the human alpha-SG. On the other hand, we are focusing on the small vertebrate zebrafish, also considering that the available SG KI mice failed to develop a dystrophic phenotype. Taking advantage of state of the art genome editing techniques such as the CRISPR-cas9 system, we have obtained the beta- and delta-SG KO lines, at present under characterization, whereas the generation of SG KI zebrafish is ongoing. Conclusion: While mice are usually the vertebrate model of election for pharmacological studies, zebrafish represents a valuable and recognized vertebrate for modelling muscle diseases and presents advantages over mammals especially for quicker and wider drug screening procedures. Together, these two complementary approaches will allow us to evaluate efficacy and safety of the promising molecule identified in vitro and will foster the development of a cure for sarcoglycanopathy.
PS1Group1-126 / #807ATALUREN IN PATIENTS AGED ≥ 2 TO <5 YEARS WITH NMDMD: 28-WEEK RESULTS FROM A PHASE 2 STUDY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Cuixia Tian1, Robert Kong2, Fengbin Jin3, Edward O’Mara3, Panayiota Trifillis3, Joseph Mcintosh2, J B. Renfroe4
1Neurology, Cincinnati Children’s Hospital Medical Center, Cincinatti, OH, US;2PTC Therapeutics Inc, South Plainfield, NJ, US;3PTC Therapeutics, Inc, South Plainfield, NJ, US;4Child Neurology Center of NW Florida, Gulf Breeze, FL, US
Background: Nonsense mutation Duchenne muscular dystrophy (nmDMD) is a rare, X-linked genetic disorder that relts in a progressive decline in function, loss of ambulation and early death due to respiratory or cardiac complications. Ataluren enables production of full-length dystrophin protein by promoting ribosomal readthrough of a premature stop codon in the mRNA transcribed from the dystrophin gene. The goal of dystrophin restoration therapy is to slow or stabilize disease progression in patients with nmDMD. Ataluren is conditionally approved by the European Medicines Agency for the treatment of ambulatory patients aged ≥ 5 years with nmDMD. Initiation of ataluren therapy for dystrophin restoration at a younger age, prior to substantive muscle loss, may maximize its therapeutic benefit. Methods: Ataluren study 030 was an observational, open-label Phase 2 study designed to evaluate the safety and pharmacokinetics (PK) of ataluren (10, 10, and 20 mg/kg) in patients aged ≥2 to <5 years with nmDMD. The study included a 4-week treatment period, a 48-week extension period, and a 4-week follow-up period. Secondary objectives in Study 030 evaluated changes in timed function tests (TFTs) and the 3-part and 8-part North Star Ambulatory Assessment (NSAA) scales, adapted for children <5 years of age. All patients were male (N=14) with genotypic confirmation of nmDMD. Results: Two patients were excluded from the current analysis: one patient did not have reported functional assessment at Week 28; and one patient did not have baseline measurement all for post line evaluations, resulting in N=12. Seven out of the fourteen patients in the safety population (50%) reported ≥1 treatment-emergent adverse event (TEAE) during the extension phase, all of which were deemed unrelated to the study drug; there were no serious TEAEs or discontinuations due to a TEAE. Pyrexia, ear infection, and nasopharyngitis were the most common TEAEs, each occurring in 2 patients (14.3%). The interim analysis presented (Table) represents a preliminary assessment of ataluren’s efficacy after 28 weeks of treatment. Conclusion: These interim data represent preliminary assessments of ataluren’s safety and efficacy after 28 weeks of treatment in patients with nmDMD aged ≥ 2 to < 5 years.

PS1Group1-127 / #456A FEMALE CARRIER OF BECKER MUSCULAR DYSTROPHY PRESENTING WITH A MYOPATHY WITH PIPESTEM CAPILLARIES
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Giuseppe Cosentino1, Filippo Brighina2, Brigida Fierro2, Laura Pilati2, Massimiliano Mirabella3, Carmelo Rodolico4
1University of Palermo, Palermo, IT;2University of Palermo, Palermo, IT;3Cattolica University, Roma, IT;4University of Messina, Messina, IT
Background: Myopathy with pipestem capillaries represents a very rare entity of uncertain classification with a poor prognosis, characterized by necrosis of muscle fibers, minimal cellular infiltration, and vessel wall thickening with luminal narrowing. Methods: We describe the case of a carrier woman of Becker muscular dystrophy (BMD), who presented at the age of 61 yr with acute onset of neck and shoulder pain followed by proximal weakness of the upper limbs and the neck. Over the next 4 years, weakness progressively worsened also involving the distal muscles of the upper limbs and the pelvic girdle muscles. The electromyographic evaluation showed myopathic signs with minimal denervation in the affected muscles. Serum levels of creatine kinase were slightly increased, and a myositis-specific antibody panel was negative. A muscle biopsy of the right tibialis anterior muscle showed abnormal variation in fiber size with thin atrophic fibers, central nuclei, presence of numerous pipestem capillaries, and minimal necrosis of muscular fibers. Some cytochrome-oxidase (COX) negative muscle fibers compatible with slight mitochondrial abnormalities were also observed. As autoimmunity is considered to play a role in the development of myopathy with pipestem capillaries, patient was treated with oral corticosteroids, which determined a slight improvement (especially as regards pain). Patient also underwent a trial with azathioprine, that was stopped after several months due to inefficacy, and subsequently a trial with methotrexate, which was also soon discontinued due to side effects. At the moment the patient refuses any immunosuppressive therapy except for prednisone, and its clinical status is quite stable. Results: This case suggests that, albeit rarely, a necrotizing myopathy with pipestem capillaries could occur in previously asymptomatic patients with dystrophin gene mutations. Conclusion: The present case has having a more favorable outcome than previously reported in the literature. The role of dystrophinopathy as a triggering factor for this rare form of myopathy and/or as a disease modifier could be hypothesized though it remains questionable.
PS1Group1-128 / #459URINARY TITIN IS A NON-INVASIVE BIOMARKER TO DIAGNOSE DUCHENNE MUSCULAR DYSTROPHY EVEN IN ADVANCED STAGE
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Hiroyuki Awano1, Yuka Ishikawa2, Yukitoshi Ishikawa2, Masaaki Matsumoto3, Masashi Nagai3, Taku Shirakawa4, Nobuhiro Maruyama5, Yo-Ichi Nabeshima6, Kazumoto Iijima3, Masafumi Matsuo4
1Pediatrics, Kobe University Graduate School of Medicine, Kobe, JP;2Yakumo National Organization Hospital, Yakumo, JP;3Pediatrics, Kobe University Graduate School of Medicine, Kobe, JP;4Physical Therapy, Kobe Gakuin University, Kobe, JP;5Immuno-Biological Laboratories Co., Ltd., Fujioka, JP;6Laboratory Of Molecular Life Science, Foundation for Biomedical Research and Innovation, Kobe, JP
Background: Duchenne muscular dystrophy (DMD) is a fatal muscle-wasting disease caused by lack of dystrophin. As the disease progresses, muscle mass decreases. Loss of muscle mass is supposed to reduce leaking enzymes including creatine kinase (CK), a most common-used biomarker for DMD. Therefore, reliable biomarker regardless of muscle loss is required to evaluate the status of dystrophic muscles, especially in advanced stage of DMD patients Titin/connectin is the largest protein in human and functions as molecular spring in the sarcomere. We previously reported titin-fragment in urine was a novel disease progression biomarker correlated with serum CK in young DMD patients (Awano et al. 2017). Urinary titin in advanced DMD remains unknown. In this study, we measured urinary titin level of advanced DMD patients and examined the association between urinary titin and serum CK. Methods: Sixty-two DMD patients from aged 21 to 50 years and 15 healthy male controls were enrolled in this study. Titin was quantified using Titin N-Fragment Assay Kit – IBL (Immuno-Biological Laboratories Co, Ltd., Fujioka, Japan). Titin was normalized to urine creatinine concentration and was expressed as pmol/mg creatinine (Cr). Statistical analysis was analyzed by Mann-Whitney U test, ANOVA, and logistic regression analysis. Receiver operating characteristic (ROC) curve was generated and area under the curve (AUC) was calculated by GraphPad PRISM 7.02 (Graphpad Software, CA, USA). Values of p <0.05 were concidered statistically significant. The protocol of this study was approved by the ethical committee. Results: Urinary titin was determined in 62 and 15 samples in advanced DMD patients and controls, respectively. In advanced DMD patients, median concentration of urinary titin was 57.7 pmol/mg Cr. This was significantly higher than those of controls (1.8 pmol/mg Cr) (p<0.0001). The AUC in the ROC curve was 0.99, indicating that urinary titin was a diagnostic biomarker in progressed stage of DMD. We previously reported that urinary titin decreased by aging and was significantly correlated to serum CK concentration in DMD patients (Awano et al.2017). An association between concentration of urinary titin, and age or serum CK was investigated by liner regression analysis. The analysis revealed that urinary titin showed a significant decline with age (p<0.0001) and a positive correlation to serum CK (p<0.001). It was noted that 46 out of 62 patients (74.2%) had normal or low CK level. On the other hand, all except 1 patients showed higher titin concentration than controls. Conclusion: Urinary titin was first disclosed to be higher than healthy males in advanced DMD patients and demonstrated to be an accurate biomarker for DMD diagnosis even in elder patients. Notably, urinary titin was elevated in DMD patients who had normal or low CK level. This suggested that titin fragmentation was still active in the progressed-stage patients. It was indicated that urinary titin was a better biomarker than serum CK in advanced DMD patients. Reference; Awano H, et al. Diagnostic and clinical significance of the titin fragment in urine of Duchenne muscular dystrophy patients. Clin Chim Acta. 476.111-116.2018
PS1Group1-129 / #830A NOVEL MUSCLE PHENOTYPE IN A PATIENT WITH TROPOMYOSIN-RECEPTOR KINASE-FUSED GENE (TFG) DISEASE
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Nicolas N. Madigan1, Jennifer A. Tracy1, William J. Litchy1, Zhiyv Niu2, Margherita Milone1
1Neurology, Mayo Clinic, Rochester, MN, US;2Laboratory Genetics & Genomics, Mayo Clinic, Rochester, MN, US
Background: The tropomyosin-receptor kinase-fused gene (TFG) protein is ubiquitously expressed across tissues, including muscle, and is predominantly present in neural tissue. Autosomal dominant TFG mutations were first identified in Okinawa and Kansai prefecture patients with proximal weakness and distal sensory loss, who were diagnosed with a hereditary sensorimotor neuropathy (HSMNO). Pathological studies later demonstrated that HSMNO is a motor neuronopathy with associated sensory ganglionopathy. Limited muscle pathological findings showing fiber size variability and fiber type grouping have been reported. TFG-related neurological disorders are now understood to encompass a spectrum of phenotypes which include Charcot-Marie-Tooth disease type 2 (CMT2) and hereditary spastic paraplegia. Recently there have been several other genes that are recognized to cause motor neuron disease, neuropathy and myopathy. Methods: Review of clinical and laboratory data in a patient with TFG-related neurological disease. Results: A 44-year-old Caucasian male presented with a 7 year history of upper and lower limb muscle fasciculation and cramps, progressive asymmetric proximal more than distal weakness, muscle atrophy and length-dependent sensory loss. He was areflexic. He had bilateral pes cavus. He could not walk on either his toes or heels. Serial measurements of serum creatine kinase ranged between 1897-2400 U/L (normal <336). Neurophysiologic studies demonstrated findings suggestive of a diffuse disorder of motor neurons in association with a sensory neuronpathy. Muscle biopsy of the quadriceps showed fiber type grouping and atrophic fibers of either histochemical type, but also several fibers with rimmed and non-rimmed vacuoles, increased numbers of internalized nuclei, fiber splitting, and necrotic and regenerating fibers. In addition there were several foci of perivascular inflammatory cells. Next generation exome sequencing detected a known pathogenic heterozygous missense variant in exon 8 (c.854C>T, p.Pro285Leu) of theTFG gene, affecting the carboxy-terminal proline-glutamine rich (P/Q) protein domain. Conclusion: Our patient presented with a classic clinical phenotype of HSMNO and a known pathological genotype. Prominent rimmed and unrimmed vacuoles were present in several muscle fibers, which suggests an associated myopathic component separate from neurogenic changes. Of interest, the function of the TFG protein relates to vesicle trafficking of unfolded and misfolded proteins through the endoplasmic reticulum and the ubiquitin-proteasome system (UPS). The P/Q domain specifically regulates the interaction with the UPS, and disruption of this domain may lead to accumulation of abnormal protein in motor neurons as well as in muscle.
PS1Group1-130 / #264GLYCOGEN STORAGE DISEASE TYPE IV PRESENTING AS CONGENITAL MYOPATHY WITH CONTRACTURES AND RIGID SPINE
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Maggie C. Walter1, Stephan Wenninger1, Angela Abicht2
1Dept. Of Neurology, Ludwig-maximilians-university, Friedrich-Baur-Institute, Munich, DE;2Medical Genetic Center, Munich, DE
Background: Glycogen storage disase type 4 (GSD4) ist an autosomal recessive disorder caused by branching enzyme deficiency. The phenotype is very heterogeneous, ranging from a congenital or infantile neuromuscular disorder to late-onset polyglycosan body disease. We report on a 5-year-old girl from Ukraine, presenting with elongated facial bones, proximal weakness, atrophy of the shoulder girdle muscles, severe hip, knee and ankle contractures, hyperlaxity of elbow joints and rigid spine. First symtoms were noted at birth in form of hypotonia and congenital arthrogryposis with bilateral hip luxation, requiring splint treatment for six months. Motor milestones were mildly delayed, turning around at 5 months of age, sitting at 7 months, crawling at 2,5 years, standing on knees from age 3; however, walking was never achieved. The parents are non-consanguineous, the patient is the only child, there are no other affected family members. Methods: We perfomed a clinical and electrophysiological examination, timed function test, oximetry, lung function test, cranial MRI, cardiac and abdominal ultrasound, along with a blood test for CK and multigene panel diagnostics for congenital myopathies. Results: Clinical testing revealed normal cranial nerves, no facial weakness, no high palate, but elongated facial bones, no macroglossia, atrophy of shoulder girdle muscles and proximal paresis of arm and leg muscles. The arms could be elevated up to 45°. Severe contractures in hips, knees and ankles along with pes equinus was detected, while elbow, hand and finger joints showed hyperlaxity. Manual muscle strength showed bilaterally MRC 3/5 in arm elevation, biceps brachiii 4+/5, triceps 4+/5. Head flexor and extensor 4-/5, quadriceps right 4-/5, left 3+/5, iliopsoas 5-/5 and gluteus 4+/5 bilaterally. Hand grip dynamometry revealed 2 kg bilaterally. The patient can sit independently, and gets up from lying position with the help of the arms. Independent standing or walking is not possible. Bilateral scapular winging, hyperlordosis, and rigid spine, but no scoliosis or stiff neck was seen. Tiimed test 6.43 sec. from lying to sitting position, displaying beevor’s sign. Coordination and sensibility was intact, nerve conduction velocities were normal. Electromyography in proximal and distal leg muscles showed mild myopathic changes if any, CK levels were normal. Cranial MRI, cardiac and abdominal ultrasound showed normal results. Puls oximetry was normal, while vital capacity was reduced with 0.57l and 66% of normal in sitting, and 58% of normal in lying position. However, the little patients cooperation was compromised by the language barrier. At age 5, height was 115 cm, weight 15 kg. Multigene panel diagnostics for congential myopathies, including collagen-6 deficiency, which we had initially suspected, did not reveal any pathogenic mutations. Therefore, we broadened the panel and detected two mutations in the GBE1 gene: c.691+2T>C and c.708G>C, both previously described as pathogenic in GSD4. Segregation analysis in the parents was in line with the autosomal recessive trait. Conclusion: Our studies provide new insights in the heterogeneity of genotype/phenotype correlations in glycogen storage diseases and congenital myopathies. Even though GSD4 represents a rare condition, testing should be included into the diagnostic work-up of congenital myopathies with contractures, rigid spine and/or joint hyperlaxity.
PS1Group1-131 / #475STATIN-ASSOCIATED AUTOIMMUNE NECROTIZING MYOPATHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
So Hyun Ahn1, Jung-Joon Sung2, Yoon-Ho Hong3, Kyoung Hyun Kwun1, Jin Ah Kim1, Je-Young Shin4, Sung Min Kim1, Ah Won Kim1
1Neurology, Seoul National University Hospital, Seoul, KR;2Neurology, Seoul National University College of medicine, Seoul University Hospital, Seoul, KR;3Seoul National University College of medicine, SMG-SNU Boramae Medical Center, Seoul, KR;4Seoul National University Hospital, Seoul, KR
Background: Necrotizing autoimmune myopathy presents with subacute proximal limb muscle weakness and a high serum creatine kinase (CK) level. Statins are generally well tolerated, but recently, progressive necrotizing myopathy leading to profound weakness has been directly linked to statin therapy. Methods: Case1 : A 63-year-old woman presented to our hospital with progressive weakness on both upper and lower extremities. She had a history of taking statin medication 10 months ago. Needle electromyography demonstrated features consistent with an active myopathy. Her serum CK level was 13237 IU/L. She treated with IV steroid pulse therapy suspected as r/o dermatomyositis or polymyositis. After then, initially she showed responsiveness of steroid therapy, but her symptom was exacerbated while steroid tapering. At January 2016, She presented dysphagia and newly appeared both arm weakness. Muscle biopsy showed some necrotizing and regenerating myofibers without infiltration of inflammatory cells in endomysium or perivascular area. She treated with oral steroid and immunosuppressive agents, and her symptoms was improved and serum CK level was also decreased.
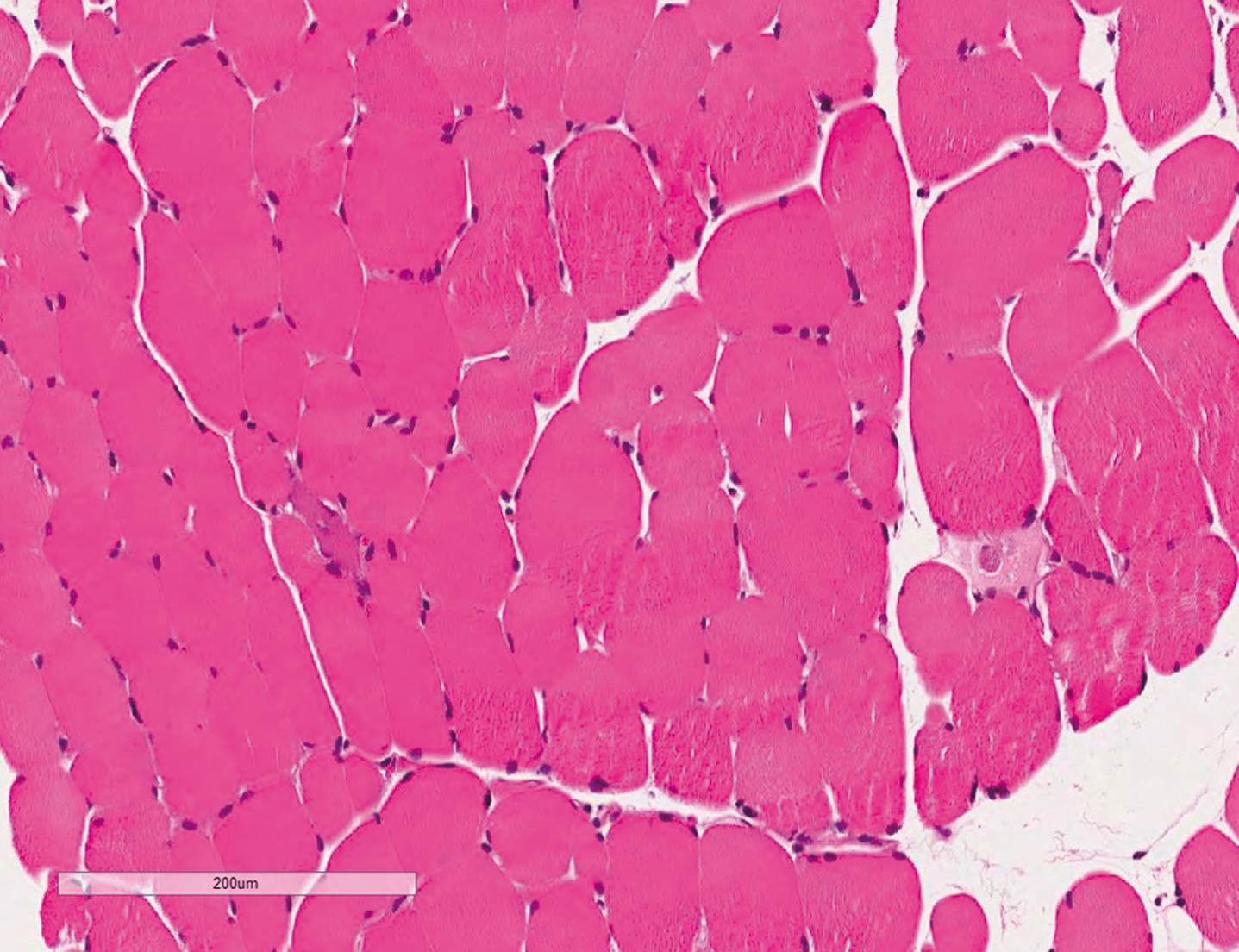
Results: Case 2: A 65-year-old woman presented to the neurology department with progressive weakness on both legs. She took stains for two years for preventing atherosclerosis. The weakness of both legs worsened and she could not climb the stairs and could not walk without assistance. Serum CK level was 1994 IU/L. Muscle biopsy showed some necrotizing and regenerating myofibers without infiltration of inflammatory cells in endomysium or perivascular area. She treated with IV methylprednisolone, and followed by oral prednisolone and tacrolimus.
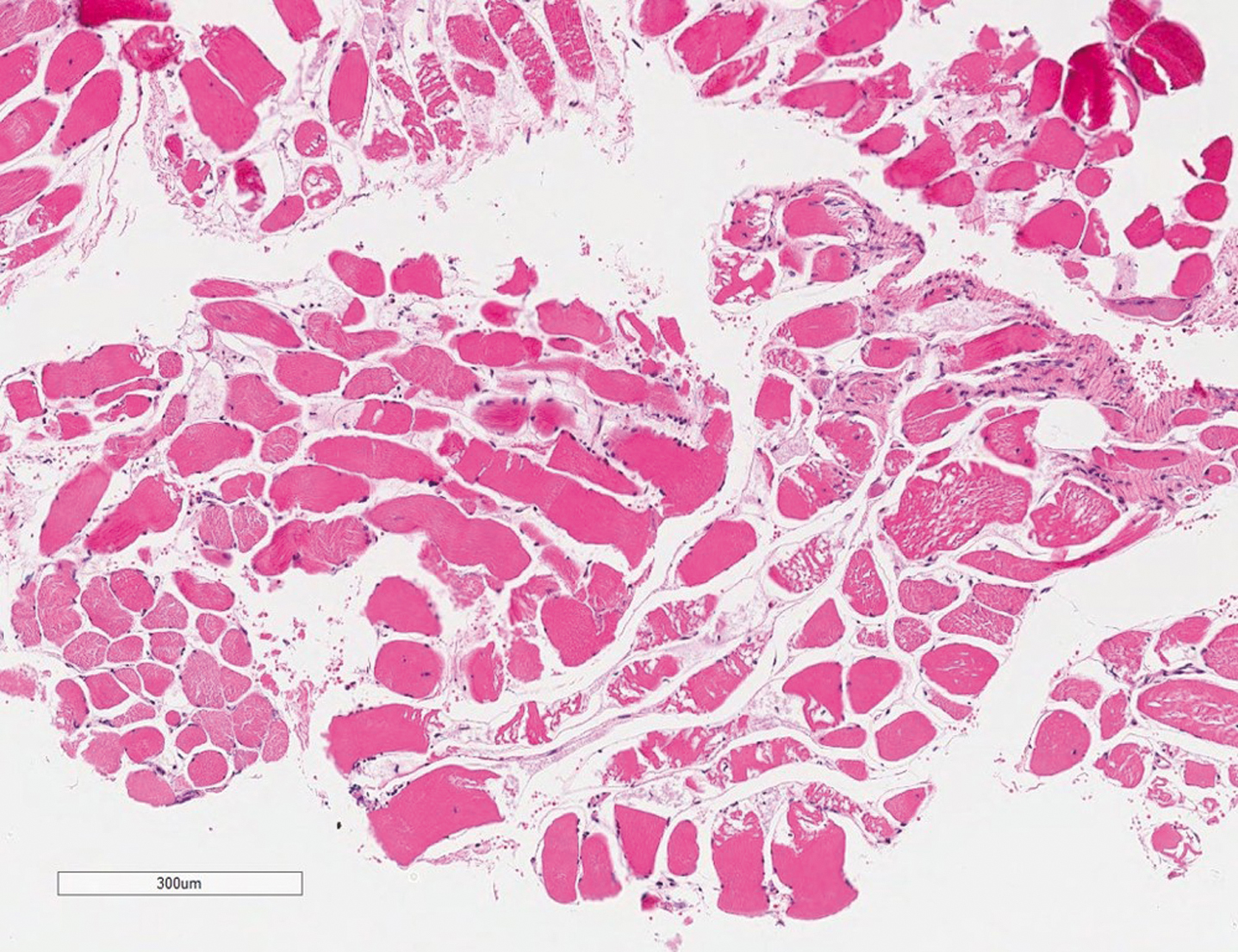
Conclusion: We reported two cases of statin-associated autoimmune necrotizing myopathy. The distinct histologic profile consists of profound necrotic, degenerating, or regenerating muscle fibers without inflammation. It may occur anytime and even long after initial exposure or statin cessation. Proper awareness of this rare complication is important to prevent further damage from statin use and treatment plan.
PS1Group1-132 / #881PHENOTYPIC HETEROGENEITY IN THREE PATIENTS WITH M.3243A>G MUTATION
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Birute Burnyte1, Kristina Grigalioniene1, Arunas Vaitkevicius2, Donatas Petroska3, Loreta Cimbalistiene1, Vaidutis Kucinskas1, Algirdas Utkus1
1Institute Of Biomedical Sciences Of The Faculty Of Medicine, Vilnius University, Vilnius, LT;2Institute Of Clinical Medicine Of The Faculty Of Medicine, Vilnius University, Vilnius, LT;3Department Of Pathology, Forensic Medicine and Pharmacology of the Faculty of Medicine of Vilnius University, Vilnius, LT
Background: Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) syndrome is the most frequent syndromic manifestation of the m.3243A>G mutation. We aim to describe the phenotypic variability of three adult patients harboring the m.3243A>G mutation. Methods: Patients were identified in the research project during years 2015-2017. All three unrelated adult patients underwent extended clinical, laboratory, imaging and neurophysiological evaluations. An open muscle biopsy was performed in two patients for diagnostic purposes prior to the molecular study. Mitochondrial DNA (mtDNA) was investigated to determine disease mechanisms. The whole-length Sanger sequencing for detection of pathogenic point variants and long PCR and Multiplex Ligation-dependent Probe Amplification (MLPA) methods for detection of mtDNA deletions/duplications were applied. Results: We identified three female patients with the m.3243A>G mutation. The average age at onset was 21.3 and at diagnosis 40.3 years. All three patients had short stature, ptosis, hearing impairment, dysarthria, dysphonia, feeding difficulties, exercise intolerance and muscle weakness as well gastrointestinal dysmotility, diarrhoea, constipation and abdominal distension. One patient manifested with ataxic syndrome, one with acute severe weakness after exposure to chemical agents at work, and one with slowly progressive exercise intolerance. Pigmentary retinopathy was detected in one patient. Liver impairment was observed in one patient, and liver biopsy showed macrovesicular steatosis. Only one patient showed persistent mild lactic acidosis. Endocrine dysfunction was observed in two patients, and was represented by diabetes mellitus type I and hypogonadism, and diabetes type II, respectively. Cardiac involvement was detected in one patient. Respiratory involvement was not observed in all three patients. No one referred a migraine. Sudden neurological deterioration was observed in two patients. Electroneurography in two patients showed decreased axonal amplitudes of sensory nerves. Electromyography was myopathic in two patients. Muscle biopsies were performed in two patients and showed ragged red fibers and COX deficiency until 10% of myocites only in one patient’s biopsy (from m. guadriceps femori). Neuroimaging showed diffuse cerebral and cerebellar atrophy in one patient, and subcortical focal abnormalities in two patients. EEG abnormalities were not observed in all patients. One patient presented lack of social interactions and emotional instability. Pathogenic variant m.3243A>G was detected in all the patients and heteroplasmy levels were less than 50% for pathogenic variant. Large-scale mtDNA deletions or duplications were not detected in all three cases. Conclusion: We report detailed phenotypic and genetic characteristics of three unrelated Lithuanian patients with the m.3243A>G mutation. Although the patients harbor the same mutation, they demonstrate a heterogeneous phenotype. Since the heteroplasmy level is not the main consideration explaining the phenotypic variability, our study supports the previous research findings that nuclear genomic factors also contribute to the phenotypic expression of MELAS. Supported by grant TAP LLT-02/2015.
PS1Group1-133 / #485DOUBLE TROUBLE: A CHILD WITH DUCHENNE MUSCULAR DYSTROPHY AND NOONAN SYNDROME
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Yun Jeong Lee1, Heamin Jang2, Ji Young Park3, Jin Sung Park4
1Pediatrics, School of Medicine, Kyungpook National University, Daegu, KR;2Pediatrics, Kyungpook national university children’s hospital, Daegu, KR;3Pathology, School of Medicine, Kyungpook National University, Daegu, KR;4Neurology, School of Medicine, Kyungpook National University, Daegu, KR
Background: Duchenne muscular dystrophy (DMD) is an X-linked disorder, caused by mutation in the DMD gene, which encodes the dystrophin protein and affects 1 in 3600-6000 live male births. Noonan syndrome (NS) is a multiple congenital anomaly syndrome with a prevalence of 1:1000. It is clinically characterized by facial dysmorphism, growth retardation and congenital heart defects. In addition skeletal, genitourinary and skin manifestations can be also observed. Recent literatures show that approximately 50% of NS is caused by heterozygous missense mutations in PTPN11 gene. Herein, we describe a rare case of a child with coinheritance of DMD and NS based on pathology study and genetic analysis using the next generation sequencing. Methods: 8 year-old boy was referred to our hospital due to elevated serum creatine kinase level (CK level; 8250 U/L) and progressive proximal limb weakness. He had a past history of pulmonary stenosis and aterial septal defect diagnosed at 1 year of age. The neuropsychological test reflected language, and cognitive developmental delay compared to his age. The patient showed waddling gait, Gower sign and calf muscle hypertrophy. The morphology of the patient was typical of NS, including short stature and dysmorphic facial features such as webbed neck, low posterior hair line, triangle shaped face, small chin, wild forehead, prominent nasolabial fold, low set posteriorly rotated ear and down slanting palpebral fissures. The needle electromyography study showed short amplitude, polyphasic motor unit potentials that were compatible with a myopathy. However clinical feature of proximal limb weakness and hyperCKemia are not common features of NS. Results: Therefore, we proceeded with the muscle biopsy that showed severe dystrophic alterations with marked size variation and perimysial fibrosis in the Hematoxylin-Eosin stain and complete loss of dystrophin was observed in dystrophin staining, that were consistent with dystrophinopathies. DNA analysis by multiplex ligation-dependant probe amplification (MLPA) analysis of the DMD gene was negative. Therefore, next generation sequencing (NGS) analysis was utilized and hemizygous c.3433-13T>A in DMD gene was found and this was predicted to disrupt the highly conserved acceptor splice site of exon 26. In addition, a known pathogenic heterozygous missense mutation in c.236A>G in PTPN11 gene was also detected. With the above clinical, pathologic features and genetic results, the patient was diagnosed to have a double trouble of DMD and NS. Conclusion: There are reports on double trouble of two different monogenetic diseases. Previous studies have described coexistence of NS with 22q11.2 deletion syndrome, retinitis pigmentosa, neurofibromatosis and Charcort-marie-tooth disease. A recent report described a double trouble found in Becker muscular dystrophy and NS via muscle biopsy and genetic testing in accordance to our study. Our study is of significance as this is the first report of double trouble of DMD and NS and currently we have proceeded with the functional study to verify the pathogenicity of our novel mutation.
PS1Group1-134 / #926THE HETEROZYGOUS R155C VCP MUTATION: TOXIC IN HUMANS, HARMLESS IN MICE
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Christoph Clemen1, Lilli Winter2, Karl-Heinz Strucksberg3, Carolin Berwanger1, Matthias Türk4, Ludwig Eichinger3, - The German Mouse Clinic Consortium5, Rolf Schröder6
1Department Of Neurology, Heimer Institute For Muscle Research, Ruhr-University Bochum, Bochum, DE;2Center For Anatomy And Cell Biology, Medical University of Vienna, Vienna, AT;3Institute Of Biochemistry, Medical Faculty, University of Cologne, Cologne, DE;4Department Of Neurology, Friedrich-Alexander University Erlangen-Nürnberg (FAU), Erlangen, DE;5The German Mouse Clinic, Munich, DE;6Neuropathology, University Hospital Erlangen, Erlangen, DE
Background: VCP, a very abundant and evolutionarily highly conserved member of the Triple-A ATPase family, is involved in a plethora of cellular processes such as membrane dynamics, protein quality control, cell cycle, apoptosis, and DNA damage response. Mutations in the VCP gene cause Inclusion Body Myopathy associated with Paget disease of bone and Fronto-temporal Dementia (IBMPFD). Moreover, four more neurodegenerative disorders, Amyotropic Lateral Sclerosis (ALS), Parkinson’s disease, Hereditary Spastic Paraplegia, and Charcot-Marie-Tooth disease type 2 (HMSN2) have been attributed to VCP missense mutations. The exact molecular mechanisms by which VCP mutations cause these late-onset disorders remain elusive. In the present study, we report on the generation and comprehensive analysis of a R155C VCP knock-in mouse model, which harbors the ortholog of one of the most frequently occurring human pathogenic VCP mutations. Methods: i) generation of R155C VCP knock-in mice; ii) comprehensive phenotypic and morphological analyses at the German Mouse Clinic Consortium in Munich; iii) VCP mRNA and protein expression analyses; iv) comparative myopathological analysis of human and murine R155C VCP mutant skeletal muscle tissue. Results: We successfully generated a R155C VCP knock-in mouse strain (B6J.B6J-Vcptm2(R155C)Ccrs) using an appropriate targeting vector for homologous recombination. To restore the original VCP gene structure, mice were bred to C57BL/6J mice expressing Cre recombinase for removal of the neomycin selection cassette. Correct gene targeting was confirmed by Southern blotting, PCR genotyping, and sequencing of both genomic and complementary DNA. Breeding of heterozygous R155C VCP knock-in mice did not yield in the generation of mice homozygous for the R155C VCP mutation. VCP immunoblotting indicated unchanged VCP protein levels in skeletal muscle and brain tissue derived from heterozygous mice as compared to wild-type littermates. Notably, our comprehensive phenotypic and morphological analyses of heterozygous R155C VCP knock-in mice did not reveal any signs of a previously described mutant VCP-related striated muscle, bone or central nervous system pathology. In comparison to wild-type littermates, heterozygous mice showed aberrant parameters for plasma lactate, fasting cholesterol, serum albumin, oxygen consumption, platelets, CD8+/Ly6C+ T-cells, and liver weight. The pathological significance of these findings is currently unclear. Conclusion: While the heterozygous R155C VCP mutation has been shown in several reports to cause IBMPFD and ALS in humans, our analysis of corresponding R155C VCP knock-in mice did not detect any signs of a striated muscle, bone or central nervous system pathology. Though the failure to generate mice homozygous for the R155C VCP missense mutation clearly denotes a toxic effect not compatible with life, the lack of any clear-cut VCP-related pathology in heterozygous R155C knock-in mice is somewhat unexpected and may be attributed to an R155C VCP expression level below a disease-causing threshold. This issue is currently addressed in further experiments.
PS1Group1-135 / #497ADAPTIVE PROTEIN QUALITY CONTROL RESPONSE IN DESMINOPATHY SKELETAL MUSCLE CELLS AND TISSUE
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Christoph Clemen1, Lilli Winter2, Carolin Berwanger3, Marina Spörrer4, Ursula Schlötzer-Schrehardt5, Rolf Schröder6
1Department Of Neurology, Heimer Institute For Muscle Research, Ruhr-University Bochum, Bochum, DE;2Center For Anatomy And Cell Biology, Medical University of Vienna, Vienna, AT;3Department Of Neurology, Heimer Institute For Muscle Research, Ruhr-University Bochum, Bochum, DE;4Department Of Physics, Friedrich-Alexander-University Erlangen-Nuremberg, Erlangen, DE;5Department Of Opthalmology, University Hospital Erlangen, Erlangen, DE;6Neuropathology, University Hospital Erlangen, Erlangen, DE
Background: Mutations of the human desmin gene on chromosome 2q35 cause autosomal-dominant and -recessive myopathies and cardiomyopathies with marked clinical variability. For pathophysiological and therapeutic studies, we generated and characterized hetero- and homozygous R349P desmin knock-in mice, which serve as patient-mimicking disease models for dominant and recessive desminopathies with maintained protein expression. By crossbreeding our desminopathy mouse strain with p53 knock-out mice, we now generated immortalized desminopathy myoblast cell lines. We used these immortalized desminopathy myoblasts in conjunction with skeletal muscle tissue from the corresponding desminopathy mice to study the effects of R349P mutant desmin on the ubiquitin-proteasome system, bulk autophagy, chaperone-assisted selective autophagy, and heat shock proteins. Methods: i) R349P desmin knock-in mice; ii) p53 knock-out mice; iii) immortalized R349P desmin knock-in skeletal muscle myoblasts and myotubes; iv) indirect immunofluorescence microscopy; v) determination of proteasomal activity; vi) immunoblotting addressing autophagy rate, chaperone-assisted selective autophagy (CASA), and heat shock protein levels. Results: We crossbred our R349P desmin knock-in mouse line with p53 knock-out mice, prepared muscles from offspring with the following genotypes, hom R349P desmin knock-in/hom p53 knock-out, het R349P desmin knock-in/hom p53 knock-out, and wild-type littermates/hom p53 knock-out, and finally generated immortalized satellite cell-derived myoblast cultures. Desmin immunoblots and immunostains of differentiated myotubes revealed that these desminopathy cell models closely mirror central aspects of the desmin pathology observed in our desminopathy mice. Proteasomal activity measurements in muscle tissue lysates revealed a 1.23-fold and a 1.47-fold increase in hetero- and homozygous R349P desmin knock-in mice, respectively, as compared to the wild-type. Notably, treatment of the desminopathy myotubes with the proteasome inhibitor MG132 did not alter the levels of both wild-type and R349P mutant desmin protein. Immunoblotting of tissue and myotube lysates demonstrated markedly increased levels of the serin/threonin protein kinase Ulk-1 and sequestosome 1 (p62/SQSTM1) as well as of p62/SQSTM1 and LC3-II, which are classical autophagy-related markers. Furthermore, we detected increased protein levels of filamin-C and BAG-3, which are essential components of the CASA system. Finally, we assessed the protein levels of aB-crystallin, Hsp27, and Hsp90, and found increased signal intensities in skeletal muscle homogenates from homozygous mice, while only aB-crystallin and Hsp27, but not Hsp90, were increased in both desminopathy myotube genotypes. Conclusion: Our analyses in the newly generated immortalized desminopathy muscle cell cultures in conjunction with their corresponding desminopathy mouse models revealed a complex pattern of adaptive protein quality control response to the presence of R349P mutant desmin. Key findings are the upregulation of proteasomal activity, autophagy, CASA, and heat shock protein levels, which were predominantly seen in the homozygous and, to a lesser extent, in the heterozygous R349P desmin knock-in skeletal muscle tissues and cultured immortalized muscle cells.
PS1Group1-136 / #514CLINICAL BACKGROUND OF 94 ADULT PATIENTS WHO RECOGNIZED NEMALIN RODS IN MUSCLE TISSUE
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Akihiro Hashiguchi1, Kento Kodama2, Itsuro Higuchi3, Hiroshi Takashima4
1Neurology, Neurological Disease Center, Kagoshima University Hospital , Kagoshima city, JP;2Department Of Neurology And Geriatrrics, Kagoshima University Graduate School of Medical and Dental Sciences, Kagoshima, JP;3Physical Therapy Foundation Course, Kagoshima University School of Health, Kagoshima, JP;4Department Of Neurology And Geriatrrics, Kagoshima University Graduate School of Medical and Dental Sciences, Kagoshima, JP
Background: It is natural to recognize nemaline rods in muscle tissue in congenital nemaline myopathy patients. On the other hand, we sometimes find unexpected nemaline rods in adult muscle tissue. From the muscle biopsy record of the past 40 years, we will clarify the background of adult patients with nemaline rods. Methods: We extracted adult cases with nemaline rods from the muscular pathological reports analyzed in our facility over the past 40 years. We compared the clinical background and pathology of cases. Results: We extracted 94 adult cases with nemaline rods. In the pathological classification of 94 cases, myopathy was 72 cases, neuropathy was 16 cases, and merger of both was 6 cases. The age at examination was widely distributed in the 20s to 80s, and no characteristic was seen by age. In myopathy 72 cases, inflammatory myopathy was included in 13 cases (18%).Eight cases of 16 cases of neuropathy were motor neuron disease such as amyotrophic lateral sclerosis, hereditary motor neuropathy, Kugelberg-Welander disease. Many cases coexisting with changes of mitochondrial abnormality, rimmed vacuole and cores. Conclusion: There is no disease specificity in the adult neemarin rod, not only in various myopathies but also in neuropathy.
PS1Group1-137 / #720P62 IMMUNOSTAINING COULD HELP IN DIFFERENTIATING POLYMYOSITIS FROM SIBM
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
José C. Milisneda1, Cristina Jou2, Iago Pinal-Feranndez3, Josep M. Grau4
1Internal Medicine, Hospital Clínic de Barcelona, Barcelona, ES;2Hospital Sant Joan de Déu, Barcelona, ES;3National Institute Of Arthritis And Musculoskeletal And Skin Diseases, National Institutes of Health, Bethesda, Maryland, AL, US;4Internal Medicine Service, Hospital Clínic de Barcelona, Barcelona, ES
Background: The sensitivity and specificity of the existing criteria for sIBM have been reviewed recently, with high specificity but variable sensitivity. In consequence major problems arise for the diagnosis in early stages or to predict when a polymyositis is going to become a sIBM. Our objective was to investigate the utility of p62 immunostaining to predict when a PM is going to become a sIBM. Methods: This retrospective study enrolled a sample from the longitudinal muscle unit cohort of Hospital Clínic de Barcelona. We compared 4 muscle biopsies from (clinico-pathologically well-defined sIBM), with seven patients classified as having PM, based on European Neuromuscular Center (ENMC) criteria, and with normal muscle biopsies (control group). The PM cases were divided in two groups, because four of them had an associated disease (autoimmune or cancer) and the rest had an isolated PM. Clinical data was review from clinical charts, and p62 histopathological analysis was made with double immunofluorescence. In addition the amount of p62 in muscle biopsies was quantified using “Image J” software. Results: We observed a significant increase of p62 expression (expressed in % of muscle area) in the isolated PM group, compared with the other PM cases (associated to a SLE, systemic sclerosis, HLAB27+ artrhopathy, and non-Hogkin lymphoma) and normal group. Also the expression of p62 was higher in the sIBM group. Of note was the poor response to immunesuppresive therapy in the four isolated PM cases, and furthermore one of them developing a typical clinical phenotype of sIBM, confirmed by a second muscle biopsy six years later. Picture. Double immunofluorescence for p62. A) Normal patient. B) PM with associated disease. C) Isolated PM. D) sIBM. Conclusion: Despite the limitations of the present study, it seems that p62 positivity immunostaining could help in the identification of refractory isolated PM cases, with high probability to evolve to classical sIBM.
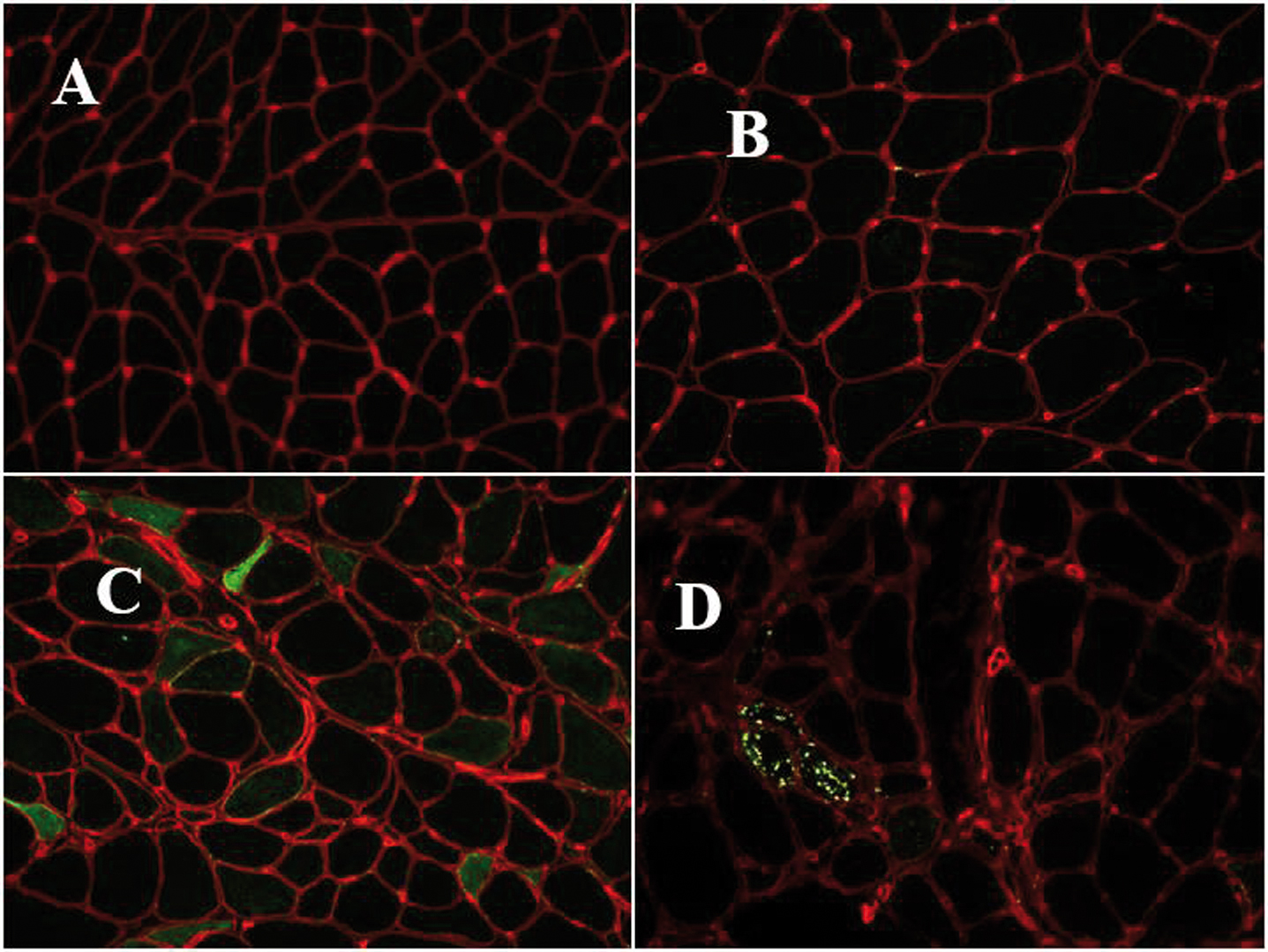
PS1Group1-138 / #347HOMOZYGOSITY OF THE AUTOSOMAL DOMINANT VCP P.ARG159HIS MUTATION
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Willem De Ridder1, Tine Deconinck2, Peter De Jonghe2, Katherine Johnson3, Ana Töpf3, Marta Bertoli3, Lauren Phillips3, Daniel Macarthur4, Jonathan Baets2
1Neurogenetics Group, Vib-uantwerp Center For Molecular Neurology, University of Antwerp, Antwerp, BE;2Neurogenetics Group, Vib-uantwerp Center For Molecular Neurology, University of Antwerp, Antwerp, BE;3John Walton Muscular Dystrophy Research Center, Newcastle University, Newcastle upon Tyne, GB;4Broad Institute of Harvard and MIT, Cambridge, MA, US
Background: Mutations in valosin containing protein (VCP) are linked to a dominantly inherited multisystem proteinopathy (MSP1) with inclusion body myopathy (IBM), Paget disease of bone (PDB) and frontotemporal dementia (FTD). Additional phenotypes have been described recently. More than 40 different missense mutations have been reported to date. Many gaps remain in our knowledge of this intriguing disorder with regard to molecular mechanisms of the disorder, genotype-phenotype correlations and penetrance. Methods: We studied a family in which the currently 33-year-old male index patient and his father exhibited progressive limb-girdle weakness. Whole-body muscle magnetic resonance imaging (MRI) was performed on a 1.5T MRI platform. Muscle biopsies were obtained from quadriceps muscle and analysed following standard histologic and immunohistochemical techniques for light microscopy. Whole exome sequencing of leucocyte DNA of the index patient was performed. Genes known to be associated with limb-girdle weakness, including VCP, were analysed for pathogenic variants. A candidate variant in VCP was confirmed by Sanger sequencing and segregation analysis was performed with available DNA-samples of family members. Results: We describe clinical, radiological and anatomopathological details for the index patient and his father. Intriguingly, on the maternal side of the family, many individuals had been diagnosed with a dementia phenotype in the past. To our surprise, the index patient was homozygous for the known p.Arg159His mutation in VCP. When studying the familial history in detail, we identified distant consanguinity between the parents of the index patient. Comprehensive baseline studies were performed to evaluate potential subclinical bone or central nervous system involvement. Conclusion: We identified the known VCP p.Arg159His mutation in homozygosity in a patient exhibiting a MSP1 phenotype. The phenotype of the index patient does not seem to be strikingly worse compared to heterozygous patients. Homozygous mutations in inherited autosomal dominant disorders are rare events. The case yields insightful and intriguing observations and incites to reflect on the presumably complex molecular mechanisms of VCP-linked disease.
PS1Group1-139 / #742GENOTYPE CHARACTERIZATION OF CZECH PATIENTS WITH FACIOSCAPULOHUMERAL DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Jana Zidkova1, Stanislav Vohanka2, Radim Mazanec3, Lenka Fajkusova4
1Centre Of Molecular Biology And Gene Therapy, University Hospital Brno, Brno, CZ;2Neurology, University Hospital Brno, Brno, CZ;3Neurology, Second Faculty of Medicine, Charles University and University Hospital Motol, Prague, CZ;4University Hospital Brno and Masaryk University Brno, Brno, CZ
Background: Facioscapulohumeral dystrophy (FSHD) is an autosomal dominant muscle disorder characterized by progressive weakness of facial, scapular and humeral muscles. FSHD is commonly associated with contraction of the D4Z4 macrosatellite repeat on chromosome 4q35 (FSHD1) or with mutations in the SMCHD1 or DNMT3B gene (FSHD2). Both situations result in a partial opening of the D4Z4 chromatin structure and transcription of D4Z4-encoded DUX4 mRNA in muscles. A specific haplotype is necessary for stabilization of the DUX4 transcript by providing the polyadenylation signal and is called 4qA haplotype. Methods: The number of D4Z4 macrosatellite repeats was determined by pulsed-field gel electrophoresis, Southern blotting and hybridization with specific probes, and analysis of genes SMCHD1 and DNMT3B by gene panel sequencing. Results: The aim of our study was to confirm FSHD on molecular level. We tested FSHD1 in 400 patients and FSHD1 was confirmed in 210 of them. We correlated the available clinical data with detected number of repeats and found out that small number of repeats (1-3; 21% of probands) is connected with earlier onset and more severe phenotype. Most FSHD1 patients (69%) have medium number of repeats (4-8 repeats) and border number of repeats was detected only in 10% of probands. Conclusion: We detected two pathogenic variants in the SMCHD1 gene and so confirmed FSHD2. Both patients have 13 D4Z4 repeats and 4qA haplotype. This study was supported by the project of the Technology Agency of the Czech Republic (TE02000058) and the project of the Grant Agency of the Czech Republic (GA16-11619S/2016).
PS1Group1-140 / #362OCULOPHARYNGEAL MUSCLE WEAKNESS AFTER TREATMENT WITH CHECKPOINT INHIBITORS: PATHOLOGY IS BEYOND THE NEUROMUSCULAR JUNCTION
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Anahit C. Mehrabyan1, Aiesha Ahmed2, Charles Specht3
1Neurology, University of North Carolina at Chapel Hill, Chapel Hill, US;2Neurology, PennState Hershey Medical Center, Hershey, PA, US;3PennState Hershey Medical Center, Hershey, PA, US
Background: Oculopharyngeal weakness related to checkpoint inhibitors became a well-known phenomenon over the last several years. The condition has been reported in patients with pre-existing myasthenia gravis, as well as de novo diagnosis after the initiation of treatment with checkpoint inhibitors. It has been reported after the use of programmed-death 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) inhibitors, and combination of these agents. Methods: We report a case of progressive oculopharyngeal muscle weakness with detailed clinical follow up over the two months after the initial clinical presentation, laboratory, electrodiagnostic, immunologic and pathologic findings. Results: 69 yo man was diagnosed with recurrence of malignant melanoma with metastasis and was treated with combination of checkpoint inhibitors: Nivolumab (PD-1 inhibitor) and Ipilimumab (CTLA-4 inhibitor). He presented with an acute onset of right proximal leg weakness, which progressed to extraocular muscle weakness, asymmetric bilateral ptosis, pharyngeal, tongue and low facial muscle weakness, with subsequent resolution of right leg weakness and progression of oculopharyngeal symptoms. He was diagnosed with immune mediated myopathy based on clinical, laboratory, electrodiagnostic and histopathology findings. Conclusion: An isolated subacute acquired oculopharyngeal weakness in an adult population is the most common presentation of autoimmune mediated dysfunction of neuromuscular junction, myasthenia gravis. Here we present an unusual case of subacute onset of severe oculopharyngeal weakness due to autoimmune myopathy secondary to checkpoint inhibitors. Despite of unusual clinical presentation the cases of severe progressive muscle weakness with high suspicion for primary muscle pathology, including elevated creatine kinase (CK), negative/normal acetylcholine receptor antibodies, progression of symptoms despite immunotherapy, need thorough evaluation to exclude immune mediated myopathies, including detailed electrodiagnostic testing and muscle biopsy.
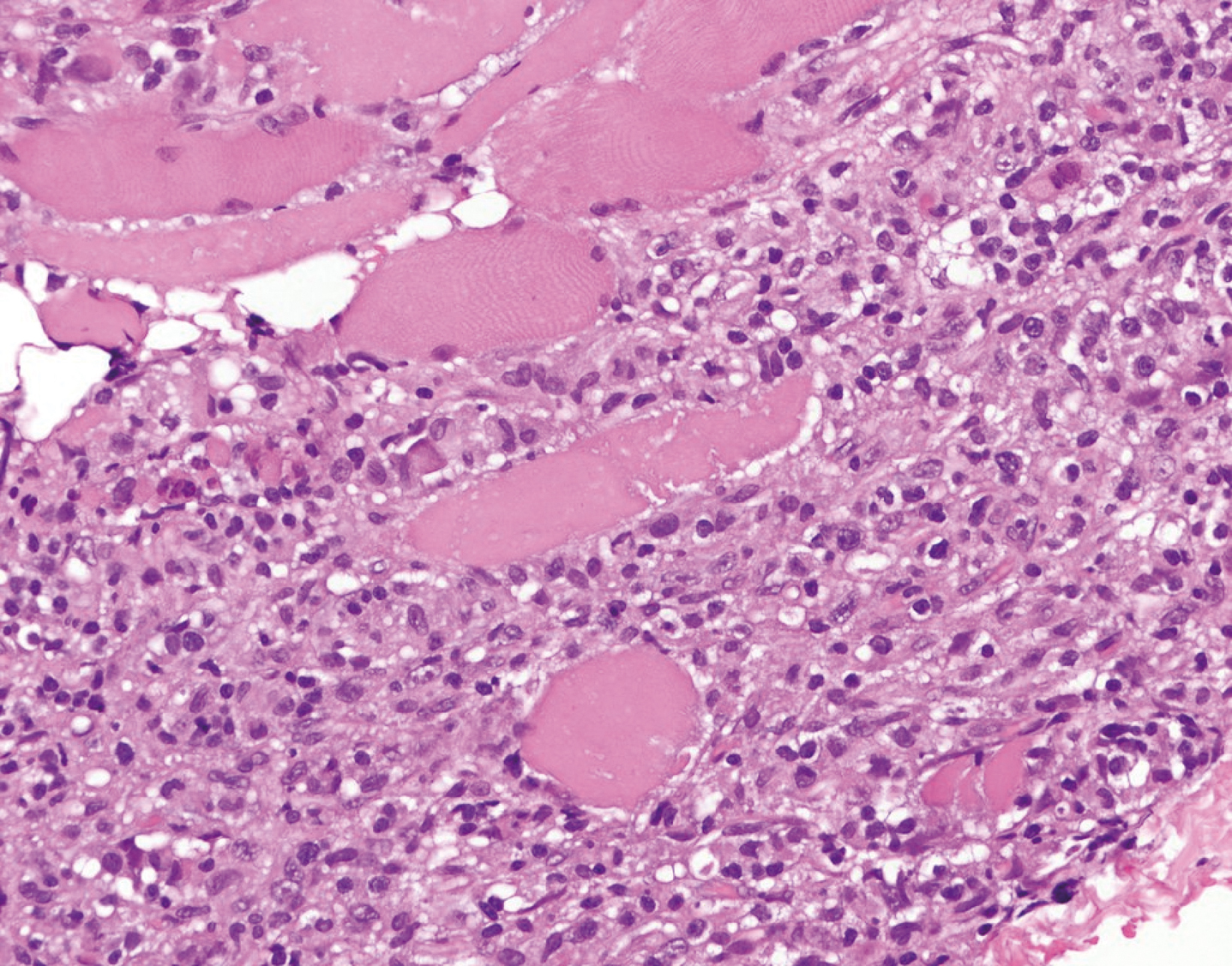
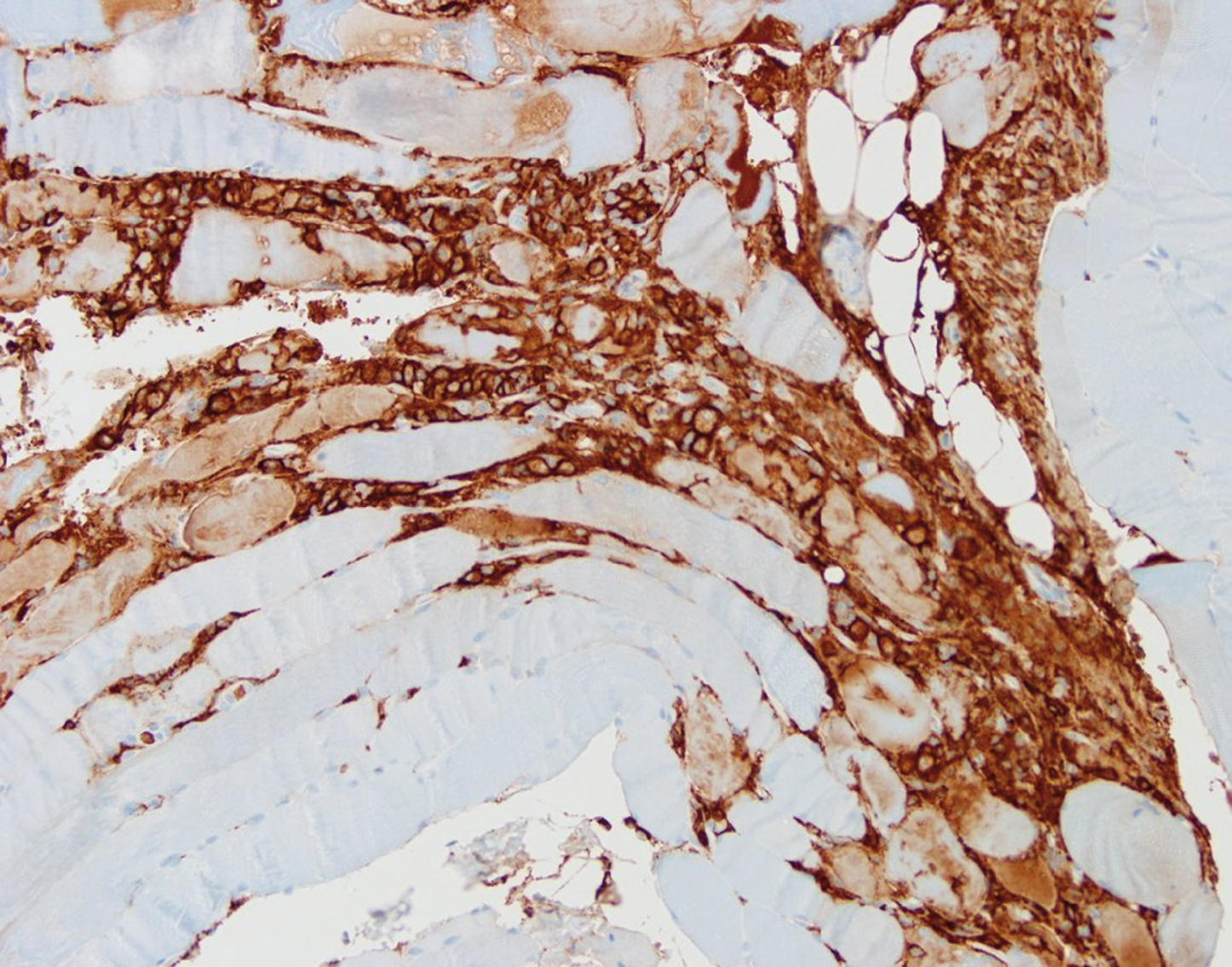
PS1Group1-141 / #755A CASE OF CENTRAL CORE DISEASE WITH NOVEL RYR1 MUTATION IN KOREAN PATIENT
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Je-Young Shin1, Chanmi Huh2, So Hyun Ahn3, Kyoung Hyun Kwun4, Jin Ah Kim4, Ah Won Kim4, Yoon-Ho Hong5, Jung-Joon Sung6
1Neurology, Seoul National University Hospital, Seoul, KR;2Seoul National University Hospital, Seoul, KR;3Neurology, Seoul National University hospital , Seoul , KR;4Neurology, Seoul National University Hospital, Seoul, KR;5Seoul National University College of medicine, SMG-SNU Boramae Medical Center, Seoul, KR;6Neurology, Seoul National University College of medicine, Seoul University Hospital, Seoul, KR
Background: Central core disease (CCD), or central core myopathy, is a congenital myopathy with non-progressive muscle weakness, mainly inherited in an autosomal dominant pattern. The gene responsible for CCD is type I ryanodine receptor (RYR1). The C-terminal domain mutation of RYR1 is relatively well known for its genotype-phenotype correlation due to its typical clinical and pathologic features. Methods: In this case report, we present a Korean patient with de novo missense mutation of a C-terminal domain of RYR1 gene. The purpose of this study is to broaden genetic mutational spectrum and understanding of genetic-phenotype correlation in CCD. Results: A 31-year-old man presented with non-progressive bilateral gait disturbance which lasted for about fifteen years. The patient showed musculoskeletal problems. Serum creatinine phosphokinase level was normal. There was no specific finding in other laboratory tests including creatinine phosphokinase. Electromyography (EMG) findings showed myopathic patterns. The muscle biopsy revealed the typical histopathologic features of CCD. The sequence analysis showed de novo missense mutation [c.14815G>T (p.Asp4939Tyr)] of RYR1 gene in the proband. We found no genetic variant or mutation among other family members. Conclusion: We report one Korean patient with CCD in whom de novo missense mutation in the C-terminal transmembrane region of RYR1 gene was identified. This mutation is expected to be the ‘(likely) pathogenic variants.’ Although mutations in the C-terminal of RYR1 are known to have significant muscle weakness since neonatal period, the index case did not present muscle symptoms until his adolescent period. On the other hand, the patient exhibited non-progressive muscle symptoms with proximal weakness and musculoskeletal deformities; his histopathologic findings were consistent with typical CCD. In this sense, even though patients do not show clinical symptoms of typical CCD, we still need muscle biopsy and genetic analysis, especially when we cannot find other causes. We hope that this case report helps us to broaden our clinical spectrum of CCD in association with RYR1 gene mutation.
PS1Group1-142 / #999MYOADENILATE DEAMINASE DEFICIENCY IN PATIENTS WITH MYALGIA
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Gabriele Siciliano, Costanza Simoncini, Giulia Ricci
Department Of Clinical And Experimental Medicine, University of Pisa, Pisa, IT
Background: Myoadenylate deaminase deficit (MAD) is the most common cause of metabolic myopathies. Methods: In our Center, we routinely use forearm ischemic exercise testing for blood lactate and ammonium in evaluation of patients with exercise intolerance, myalgias, muscle cramps and/or hyperCKemia. Results: In the last 5 years, we have detected lack of increase in the blood ammoniun concentration during exercise in twelve unrelated patients, all affected by exercise intolerance with muscle cramps and myalgia. In these patients, genetic test confirmed the presence of mutations in AMPD1 gene, suggestive of myopathy due to myoadenylate deaminase deficiency (MAD). Conclusion: It has been reported that the spectrum of the MAD deficiency can range from asymptomatic carriers to patients who manifest exercise-induced muscle pain, fatigue, idiopathic hyperCKemia and, occasionally, rhabdomyolysis. MAD deficiency is not currently considered a well proven disease entity. Nevertheless, in people with muscle symptoms, AMPD1 gene mutations seem to be significantly more frequent than healthy population, suggesting a possible clinical significance of this defect.
PS1Group1-143 / #365A PHASE IIA STUDY OF TAS-205, A NOVEL INHIBITOR OF HEMATOPOIETIC PROSTAGLANDIN D SYNTHASE, IN DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Tsuyoshi Matsumura1, Hirofumi Komaki2, Satoshi Kuru3, Takahiro Nakayama4, Shinnichi Takeda5
1Neurology, National Hospital Organization Toneyama National Hospital, Toyonaka, JP;2Child Neurology, National Center Hospital, National Center of Neurology and Psychiatry, Kodaira, JP;3Neurology, National Hospital Organization Suzuka National Hospital, Suzuka, JP;4Neurology, Yokohama Rosai Hospital, Yokohama, JP;5National Institute of Neuroscience, National Center of Neurology and Psychiatry, Kodaira, JP
Background: Inflammation has been included in the pathological process of Duchenne muscular dystrophy (DMD). Prostaglandin D2 (PGD2) is involved in inflammation, and hematopoietic PGD synthase (HPGDS) is expressed in myonecrotic areas in DMD. A phase I study demonstrated that TAS-205, a novel HPGDS inhibitor, was highly safe and tolerable in patients with DMD when given at 1.67 to 13.33 mg/kg as a single- and repeated- doses twice daily for 7 days. TAS-205 also decreased urinary excretion of tetranor-PGDM, the pharmacodynamics marker to show HPGDS inhibition. Based on the results of a phase I study, a phase IIa study was carried out. Methods: The primary objective of this double-blind randomized study was to evaluate the efficacy of TAS-205 orally administered twice daily for 24 weeks compared with placebo in ambulatory patients with DMD in an exploratory manner. The secondary objective was to evaluate the safety and dose response relationship in efficacy and safety, and also the effect on the urinary excretion of tetranor-PGDM. Subjects were randomized to one of 3 groups; low-dose group (6.67 to 13.33 mg/kg/dose), high-dose group (13.33 to 26.67 mg/kg/dose), and placebo group evenly. This study was carried out at 11 sites in Japan. Results: Thirty-six subjects were enrolled and 35 subjects were treated with the investigational product. The full analysis set consisted of 34 subjects, and the per protocol set consisted of 32 subjects where the main efficacy evaluation was conducted. The primary endpoint was the change from baseline in the measured 6-minute walk distance (6MWD) at week 24. It was -17.0 ± 55.6 m (mean value ± SD) in the placebo group, -3.5 ± 67.3 m in the low-dose group, and -7.5 ± 37.3 m in the high-dose group. The difference of the mean value from the placebo group was 13.5 m (p = 0.625) in the low-dose group and 9.5 m (p = 0.646) in the high-dose group. The decrease in 6MWD tended to be less in the TAS-205 groups as compared with the placebo group. The muscle volume index (%MVI) by skeletal muscle CT in the thigh and lower leg, as the secondary endpoint for efficacy, tended to be less decreased in the TAS-205 group as compared with the placebo group. Additionally, there were several subjects where muscle volume of the thigh and lower leg increased in high-dose group. The change rate in the urinary excretion of tetranor-PGDM corrected by creatinine from baseline significantly decreased in the high-dose groups as compared with the placebo group. No adverse drug reactions specific to the TAS-205 were observed, suggesting proper tolerability. Conclusion: From the above results, it was suggested that TAS-205 had a possibility of being effective for ambulatory patients with DMD.
PS1Group1-144 / #767USE OF HUMAN INDUCED PLURIPOTENT STEM CELLS FOR MODELLING SKELETAL MUSCULAR DEFECTS ASSOCIATED TO MYOTONIC DYSTROPHY TYPE 1
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Julie Tahraoui1, Lea Lesueur2, Christian Pinset3, Xavier Nissan2, Denis Furling4, Cecile Martinat3
1I-stem, Inserm Ueve Umr861, CORBEIL ESSONNES, FR;2I-stem, CECS, CORBEIL ESSONNES, FR;3I-stem, INSERM UEVE UMR861, CORBEIL ESSONNES, FR;4Institut De Myologie, Centre de Recherche en Myologie, UMRS974, PARIS, FR
Background: Myotonic Dystrophy type I (DM1) is a rare neuromuscular disease that is mainly characterized by myotonia, progressive muscle weakness and wasting. DM1 is an autosomal dominant disorder caused by an expanded CTG repeat in the 3’ UTR of DMPK gene with the size of the expanded repeats globally correlates with disease severity. This abnormal expansion leads to a toxic gain-of-function of the mutated mRNAs which aggregate within the nucleus in association with different RNA binding proteins such as the splicing factor MBNL1. Consequently, several alternative splicing defects have been identified in skeletal muscular biopsies from DM1 patients. Up to now, cellular models available for DM1 rely mainly on primary cultures of skeletal muscle cells derived from DM1 patients. However, several difficulties, such as a limited in vitro proliferation, may limit the use of primary cells, especially considering drug screening approaches for this incurable disease. Methods: Based on recent protocols allowing the conversion of human pluripotent stem cells into skeletal muscle cells without genetic modifications, our goal was to evaluate the potential offered by DM1 specific hiPSCs to better understand the effect of DM1 mutation on the generation and the maintenance of skeletal muscle cells. Results: Altogether, our results demonstrate the potential offered by DM1 specific hiPSC-derived skeletal muscle cells to phenocopy and decipher new molecular pathophysiological mechanisms. Conclusion: Furthermore, this new cellular system holds great promises to identify new compounds capable to target the RNA toxicity associated with DM1 mutation.
PS1Group1-145 / #367A CASE OF FACIOSCAPULOHUMERAL MUSCULAR DYSTROPHY TYPE 2 WITH NOVEL FRAMESHIFT MUTATION OF SMCHD1 GENE IN KOREA
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Jung Hwan Lee1, Moon-Woo Seong2, Chang-Seok Ki3, Young Chul Choi4
1Department Of Neurology, Ewha Womans University Mokdong Hospital, Seoul, KR;2Department Of Laboratory Medicine, Seoul National University Hospital, Seoul, KR;3Department Of Laboratory Medicine And Genetics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, KR;4Department Of Neurology, Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul, KR
Background: Facioscapulohumeral muscular dystrophy 2 (FSHD2) is a genetic muscular disorder characterized by DNA hypomethylation on the 4q-subtelomeric macrosatellite repeat array, D4Z4. DNA hypomethylation is caused by heterozygous mutations in the gene encoding structural maintenance of chromosomes flexible hinge domain containing 1 (SMCHD1). In Korea, there are no reports of patient with FSHD2. We reported a first case of FSHD2 in Korea. Methods: A 40-year-old male visited to our clinic for asymmetric upper and lower extremity weakness. He said that His father, dead due to liver cirrhosis, and elder brother were similar with him. He was normal at birth and presented normal development. He presented asymmetric arm weakness after the age of 10 and leg weakness after the age of 24. And, he also presented winged scapula and Popeye arm. On neurologic examination, we could observe bilateral facial weakness, asymmetric proximal dominant arm and leg weakness. Results: First, we performed conventional Southern blot analysis but D4Z4 repeat size was 12, which means normal. Then, we performed whole exome sequencing and found novel frameshift mutation (c.3801delG, p.Glu1267Aspfs*5) of SMCHD1 gene. Then, we re-checked size of D4Z4 repeat with 4qA haplotype and performed hypomethylation assay of D4Z4 repeat region. As a result, he has homozygous 4qA haplotype and mild reduced D4Z4 repeat size (41kb/39kb), and reduced D4Z4 methylation ratio (14.6%). So, we report a first case of FSHD2 in Korea. Conclusion: For rapid and accurate diagnosis of FSHD2 patients, the genetic analysis of D4Z4 haplotype and methylation is very important and needed to add for diagnostic step for FSHD.
PS1Group1-146 / #1012ASYMMETRIC WEAKNESS IN HMGCOA REDUCTASE ANTIBODY NECROTIZING MYOPATHY
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Madhu Soni
Neurological Sciences, Rush University Medical Center, Chicago, IL, US
Background: Objectives: To report a case of asymmetric weakness in statin-associated HMGCoA reductase antibody necrotizing myopathy, and to heighten awareness of the potential etiology. Background: Necrotizing myopathy due to statins and other drug-induced or immune-mediated triggers typically presents with symmetric, proximal weakness that can be abrupt in onset and aggressive in its course, requiring prompt recognition to stop the offending agent, institute immunotherapy if necessary and provide the appropriate supportive care. A case is described here of an elderly gentleman with prominent, asymmetric proximal upper extremity weakness that did not improve with immunotherapy. Methods: Case Report: A 74- year-old, previously active, man was brought in for progressive weakness over 6 weeks that started abruptly. Symptoms started with symmetric, painless proximal lower extremity weakness limiting ambulation and use of stairs, and contributed to falls. The weakness progressed to involve the upper extremities, starting on the left where there was self-limited pain in the shoulder associated with weakness. Weakness in the right upper extremity was painless. Dysphagia and dyspnea also developed. The patient had been on daily atorvastatin for 10 years. Exam was notable for asymmetric right arm abduction weakness of 0/5 compared to 3/5 contralaterally. He had moderate weakness of neck flexion and bilateral hip flexion, and was unable to stand without assistance. Work up included a left quadriceps muscle biopsy that revealed an active, necrotizing myopathy. HMGCoA reductase Ab was elevated. The patient was admitted for IVIG treatment and received prednisone 60 mg orally, along with physical and occupational therapy. Results: After two courses of IVIG, a month apart, his strength markedly improved with resolution of weakness in the neck and left upper extremity and mild residual weakness in the proximal lower extremities. Right arm abduction remained 0/5. He denied pain, including with passive movement of the arm at the shoulder. Shoulder xray revealed no significant abnormalities. Due to the discrepancy in right arm abduction, despite improvement in the remainder of his strength, a shoulder MRI was obtained and revealed a large tear of the right supraspinatus tendon and a moderate tear in the biceps. Conclusion: Asymmetric weakness is atypical in statin-associated HMGCoA reductase necrotizing myopathy and should trigger consideration for alternative, non-neurologic causes that may warrant an alternative treatment approach. In this case, a previously undiagnosed painless rotator cuff tear was the cause of the right arm abduction weakness that was refractory to immunotherapy for the underlying necrotizing myopathy. Heightened awareness and identification of potential co-exising orthopedic conditions and complications is essential to guide appropriate treatment and avoid unnecessary additional immunotherapy in immune-mediated myopathies.
PS1Group1-147 / #809CAN NECK FLEXION WEAKNESS PREDICT CHANGES IN SWALLOWING AND COUGH PEAK FLOW IN PATIENTS WITH MYOTONIC DYSTROPHY TYPE 1?
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Charlotte Massey1, Jodi E. Allen2, Umesh Vivekananda3, Nikoletta Nikolenko4, Cecilia Jimenez-Moreno4, Chris Turner5
1Therapy And Rehabilitation, The National Hospital for Neurology and Neurosurgery, BG, GB;2Therapy And Rehabilitation, The National Hospital for Neurology and Neurosurgery , 3BG, GB;3University College London Hospitals, BU, GB;4John Walton Muscular Dystrophy Research Centre, BZ, GB;5Institute Of Neurology, University College London Hospital MRC Centre for Neuromuscular Diseases, BG, GB
Background: Recurrent pneumonia secondary to bulbar and respiratory muscle weakness is the leading cause of morbidity and mortality in patients with Myotonic Dystrophy Type 1 (DM1). Early detection of cough and swallow impairment enables prophylactic measures such as cough augmentation and swallow management to be put in place to prevent acute respiratory decline. Measuring neck flexor strength using the MRC muscle score is a simple, efficient and inexpensive measure that can be used in the outpatient or domicillary setting without specialist equipment. We hypothesised that alterations in neck flexion strength could be used to predict deterioriation in swallowing and cough ability and therefore has potential utility as a tool to identify the need for early physiotherapy and speech and language therapy assessment and management. Methods: 52 adult patients with genetically confirmed DM1 were enrolled as part of the PHENO DM1 natural history study. Neck flexion strength was assessed using the MRC muscle scale by a trained physiotherapist with the patient in lying. Cough Peak Flow (CPF) was assessed in sitting using a Mini-Wright peak flow meter fitted with a face mask. Swallow function was assessed using the Sydney Swallow Questionnaire (SSQ), a validated patient self-report tool of swallowing symptoms. Spearman’s rank was used to establish correlations between neck flexion strength, SSQ and CPF. Results: Significant correlations were found between MRC neck scores and CPF (p=0.01) and MRC neck and SSQ (p=0.04). Conclusion: This is the first study to consider relationships between neck flexion strength, swallow function and cough. Correlations have been identified. It is possible that MRC neck flexion scores could be used to help predict changes in swallowing and cough function; however further research needs to explore this in more detail, including analysis of instrumental measures of swallowing ability, respiratory and mobility parameters. The ongoing search for tools to support identication of patients at increased risk of respiratory decline secondary to bulbar weakness and poor cough would allow prophylactic measures to be put in place to prevent unplanned hospital admissions with chest infection or pneumonia.
PS1Group1-148 / #778THE ADDITIONAL VALUE OF HISTOPATHOLOGICAL FASCIA EXAMINATION IN DIAGNOSING MYOSITIS
Topic: Group 1 – Muscle Diseases of Genetic Origin and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy
Anneke Van Der Kooi
Neurology, AMC, Amsterdam, NL
Background: Early diagnosis of idiopathic inflammatory myopathies or myositis may prevent irreversible disease damage and disability, but is often complicated. Needle muscle biopsy is widely used as its diagnostic accuracy appears generally comparable to that of the more invasive en-bloc biopsy. However, en-bloc biopsy including skin, fascia and muscle allows additional histopathological examination of the fascia. We investigated whether additional histopathological fascia examination might contribute to early diagnosis of myositis. Methods: Fifty-five en-bloc biopsies from patients who were diagnosed with myositis were retrospectively reviewed: dermatomyositis (DM; n = 9), non-specific/overlap myositis (NM/OM; n = 17), immune-mediated necrotizing myopathy (n = 21), and anti-synthetase syndrome (n = 8). Biopsies were scored for skin, muscle, and fascia involvement and re-evaluated in case of presumed isolated fascia involvement. Results: In DM and NM/OM, muscle analysis alone led to the diagnosis of myositis in 89% and 94%, respectively. One patient (11%) with DM and one (6%) with (NM/OM) had isolated histopathological fascia involvement, which contributed to the diagnosis. Conclusion: Our findings indicate that en-bloc biopsy allowing histopathological fascia examination may contribute to an early diagnosis of DM and NM/OM.
PS2Group3-001 / #268A TALE OF TWO CHARCOT’S JOINTS: LONDON AND DAR-ES-SALAAM
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Chin Yong Kok
Mrc Neuromuscular Centre, National Hospital of Neurology, London, GB
Background: We report two cases of long-standing neuropathy presenting with predominantly foot deformities. Methods: Case reports. Results: Case 1: A 32 year old man from Tanzania had twisted his ankle whilst playing football. He experienced no pain but developed localised swelling. A CT of the ankle and foot revealed a shattered navicular bone with anterior subluxation. He was referred as a case of leprosy for nerve biopsy. However, further history revealed that at the age of eight while climbing Mount Kilimanjaro his father commented that he walked with bilateral foot drop. He denied any sensory symptoms. Examination showed pes cavus on the right and a Charcot’s joint of the left ankle. He had evidence of a length dependant motor neuropathy with no sensory abnormalities. No thickened nerves. Neurophysiology showed a length dependant sensory- motor axonal neuropathy. The CMT2/HSN panel revealed heterozygous mutation in Ras-related protein Rab-7a gene. Case 2: A 74 year old lady with a 17 years history of a painful neuropathy with ulceration and Charcot’s joints. She was a longstanding diabetic on oral hypoglycaemic agents. Neurophysiological studies showed n axonal sensory – motor neuropathy with abnormal thermal thresholds. The CMT2/HNS panel revealed heterozygous mutation in serine palmitoyltransferase, long chain base subunit 2 gene (SPTLC2). Conclusion: The use of next-generation sequencing makes it easy to undertake genetic tests. These two cases highlight the need to consider genetic diagnoses which obviate the need for nerve biopsies.
PS2Group3-002 / #908HETEROGENEITY OF THE PATIENT PATHWAY AND CHARACTERISTICS FOR HATTR AMYLOIDOSIS: PERSPECTIVES FROM CENTRAL AND EASTERN EUROPE
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Yesim Parman1, Ivailo Tournev2, Daniel Coriu3, Michael Arad4, Marta Lipowska5, Sergei Nikitin6, Stayko Sarafov2, Janez Zidar7
1Istanbul University, Istanbul, TR;2University Hospital Aleksandrovska, Sofia, BG;3University of Medicine, Bucharest, RO;4Chaim Sheba Medical Center, Tel-Hashomer, IL;5Medical University of Warsaw, Warsaw, PL;6Institute of General Pathology and Pathophysiology, Moscow, RU;7University Medical Center, Ljubljana, SI
Background: Hereditary transthyretin (hATTR) amyloidosis is an autosomal dominant disease, caused by a range of mutations in the transthyretin gene. As such, hATTR presents in many different forms and with considerable variation in signs and symptoms, particularly across geographic locations. Such heterogeneity, in combination with its rarity, can result in a sub-optimal patient pathway to diagnosis and, ultimately, management. As such, it is important that we share our experiences to understand the challenges and obstacles to enhancing the patient journey. The aim of this project was to therefore assess the current clinical picture for patients with hATTR amyloidosis across Central and Eastern Europe (CEE). Methods: In November 2017, 8 experts, from 7 countries in CEE, involved in the diagnosis and care for patients with hATTR amyloidosis, presented insights from their respective countries. The panel was asked to share their experiences in terms of: how patients enter the healthcare system; who is typically responsible for diagnosing patients with hATTR amyloidosis; common misdiagnoses; availability of multidisciplinary networks involved in diagnostic work-up; availability of, and access to, Centres of Excellence; and, capabilities for genetic testing and counselling for family members. Additionally, the typical patient profile in CEE, including genetic background, age of disease onset and rate of disease progression were discussed. Results: Through the sharing of experiences country-by-country, it emerged that there is a high degree of heterogeneity of clinical practice and patient characteristics in CEE. Factors that drove such diversity included: • Range of different genotypes encountered, including: Asp38Ala, Gly47Glu, Glu54Gln, Glu89Gln, Glu89Lys, Ile107Phe, Phe33Leu, Ser77Phe, Ser77Tyr, Thr40Lys, Val122Ala, Val30Met, Val71Ala, among other rare mutations • Subsequent variability in symptomatology, especially in the age of onset and presenting symptoms, which are often non-neurologic • Natural history of hATTR amyloidosis within this region • Status of disease awareness outside of Centres of Excellence, including of key red-flag signs and symptoms that should raise suspicion • Availability and use of specific diagnostic tests for hATTR amyloidosis It emerged that, although there is a need for a global approach to identifying and managing patients with hATTR amyloidosis, such heterogeneity of disease and practice in CEE might necessitate a more localised approach in this region. The expert panel discussed options that might facilitate optimal patient care, including: the sharing of expertise nationally and internationally through improved communication networks; development and further optimisation of Centres of Excellence; and, improved disease awareness among clinicians who are likely to encounter patients with hATTR amyloidosis. Conclusion: Clinical practice across CEE is highly heterogeneous, partly due to the broad spectrum of hATTR amyloidosis genotypes and phenotypes, as well as the varying structures of healthcare systems. Improved communication is key to heightening the urgency of identifying and managing this complex disease for clinicians across the region; sharing of such local experiences with colleagues, nationally and internationally, may help in facilitating the delivery of optimal patient care.
PS2Group3-003 / #966AUTONOMIC AND SENSORY NEUROPATHY: CHALLENGES IN THE ETIOLOGY AND TREATMENT OF A PEDIATRIC CASE
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Mafalda Sampaio1, Cristina Garrido1, Manuela Santos1, Ana Sousa2, Sónia Figueiroa1
1Neuropediatrics, Centro Hospitalar do Porto, Porto, PT;2Neurophysiology, Centro Hospitalar Porto, -, PT
Background: The acute presentation of severe dysautonomia, sometimes with additional neuromuscular or central nervous system manifestations, may reflect impairment of sympathetic, parasympathetic and/or enteric functions. Autoimmune autonomic ganglionopathy (AAG) is an acquired, immunomediated, dysautonomia. It has a wide clinical spectrum and antibodies against the alpha-3 subunit of the nicotinic acetylcholine receptor (a3-AChR Ab) on autonomic ganglia have been found acutely only in about 50% of patients. The low prevalence of the disease in the pediatric population and the difficulty in recognizing the symptoms often lead to a significant delay in the diagnosis. The differential diagnosis, especially in children, encompass immune, paraneoplastic, genetic, metabolic and mitochondrial disorders. Methods: (Not applicable). Results: CASE REPORT A 15 months girl was admitted for vomiting during an acute pyelonephritis. During the first week, she developed irritability, a rash with periorbital edema and severe mucositis with loss of a primary tooth. Additionally, she developed dysphonia, hemodynamic instability, and bladder and bowel dysfunction. On the second week, she presented paroxysmal events of bradycardia and cyanosis, and developed hypotonia associated with poor spontaneous limb movements (but offering some resistance when manipulated) and arreflexia. Brain and spinal cord MRI disclosed gadolinium enhancement of the cauda equina’s roots, mainly dorsal roots. Cerebrospinal fluid analysis showed albuminocytologic dissociation. Guillain-Barré syndrome (GBS) was hypothesized and IVIG treatment started. On the 4th week, EMG revealed absent sensory potentials and normal motor potentials. Because of paroxysms of staring, cyanosis and oromandibular movements, EEG was performed and disclosed an encephalopathic pattern. Corticotherapy was started. The extensive metabolic, immune, paraneoplastic and infectious investigation was unremarkable. Throughout the first months, she had intermittent hyperthermia and bladder dysfunction. Since then, she had a slowly progressive neurological improvement, being able to walk by the age of 3 years old. She has intermittent vomiting, diarrhea, with failure to thrive associated with polydipsia and intermittent hyperthermia. Arreflexia persists. The last neurophysiological evaluation, at age 4, was consistent with a sensory ganglionopathy, and sympathetic and parasympathetic dysautonomia. Conclusion: This clinical case illustrates an acute presentation of prominent pandysautonomia with additional clinical manifestations secondary to sensory ganglionopathy, leading to an initial diagnosis of a sensory variant of GBS. Like in GBS, the acute presentation, a previous infection, the albuminocytological dissociation and a partial response to immunomodulatory treatment, suggest an immune-mediated mechanism. In our case, an extensive etiology workup was unremarkable. Although very rarely described in pediatric age, we consider the hypothesis of AAG, and testing for a3-AChR Ab is ongoing. We would also like to discuss further treatment approach, since there are case reports in which some functional improvement is reported with treatment, even in seronegative patients. Further studies will be useful to establish treatment protocols for acute autoimmune neuropathies, and to do a better characterization of seronegative cases in order to determine other relevant autonomic nervous system antibodies.
PS2Group3-004 / #228EXTENSIVE GENETIC ANALYSIS IN A TAIWANESE COHORT WITH CHARCOT-MARIE-TOOTH DISEASE
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Kon-Ping Lin1, Yi-Chu Liao2, Yi-Chung Lee3
1Taipei Veterans General Hospital, Taipei, TW;2Neurology, Taipei Veterans General Hospital, Taipei, TW;3Neurology, Taipei Veterans General Hospital, Taipei, TW
Background: Charcot-Marie-Tooth disease (CMT) comprises a clinically and genetically heterogeneous group of inherited neuropathies. Mutations in at least 90 genes have been described to cause CMT. The aim of this study was to determine the frequency and spectrum of mutations in different genes in a Taiwanese CMT cohort of Han Chinese origin. Methods: We recruited 427 unrelated CMT patients from the Neurology Service of Taipei Veterans General Hospital, Taiwan. A series of genetic testing was performed, including a real-time PCR-based method to investigate PMP22 duplication, sanger sequencing to check GJB1 mutations, a targeted sequencing panel covering all known CMT genes and further genes causing neuropathy (124 genes in total), and whole exome sequencing in several selected large pedigrees. Results: Mutations have been identified in 315 of the 427 patients (73.8%), including 275 with demyelinating CMT (85.7% of all demyelinating CMT cases; 275/321), 38 with axonal CMT (36.5% of all axonal CMT cases; 38/104) and 2 with dominant intermediate CMT (100%; 2/2). Among the 315 patients with clear genetic diagnosis, 208 were found to have a PMP22 duplication (48.7% of total CMT cases; 208/427), 40 had a GJB1 mutation (9.4%; 40/427), 14 had a MPZ mutation (3.3%; 14/427), 14 had a MFN2 mutation (3.3%; 14/427) and 8 had a NEFL mutation (1.9%; 8/427). Thirty-one patients carry a mutation in 15 different rare CMT disease genes, including LITAF, EGR2, FBLN5, SH3TC2, GARS, HSPB1, GDAP1, AARS, LRSAM1, IGHMBP2, MORC2, BSCL2, KIF5A, TFG and GNB4. Fifteen novel mutations were identified in this cohort. Conclusion: This study clearly demonstrates the distribution and frequency of mutations in a Taiwanese CMT cohort of Han Chinese origin, and expands the spectrum and supports their global presence of mutations in the causative genes of CMT.
PS2Group3-005 / #566IMPACT OF PATISIRAN ON AUTONOMIC NEUROPATHY IN HEREDITARY TRANSTHYRETIN-MEDIATED AMYLOIDOSIS PATIENTS
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Michelle Mauermann1, David Adams2, Alejandra Gonzalez-Duarte3, Teresa Coelho4, Chih-Chao Yang5, Michael Polydefkis6, Arnt Kristen7, Ivaylo Tournev8, Hartmut Schmidt9, John Berk10, Kon-Ping Lin11, Pritesh Gandhi12, Marianne Sweetser12, Matthew White12, Jared Gollob12, Ole Suhr13
1Mayo Clinic, Rochester, US;2Neurology Department, LFB, Le Kremlin Bicetre, FR;3Instituto Nacional de Ciencias Medicas y Nutricion, Salvador Zubiran, MX;4Hospital de Santo Antonio, Porto, PT;5National Taiwan University Hostpital, Taipei, TW;6Johns Hopkins, Baltimore, MD, US;7Heidelberg University Hospital, Heidelberg, DE;8University Multiprofile Hospital for Active Treatment, Sofia, BG;9University Hospital Muenster, Muenster, DE;10Boston Universty, Boston, MA, US;11Neurology, Taipei-Veterans General Hospital, Taipei, TW;12Alnylam Pharmaceuticals, Cambridge, MA, US;13Department Of Public Health And Clinical Medicine, Umea University, Umea, SE
Background: Hereditary transthyretin-mediated (hATTR) amyloidosis is a rare, multisystemic, rapidly progressive, life-threatening disease with heterogeneous clinical presentation including sensory, motor and autonomic neuropathy, as well as cardiac involvement. Clinical manifestations of autonomic dysfunction may include chronic diarrhea and nausea/vomiting, orthostatic hypotension, and urinary tract infections. In the Phase 3 APOLLO study, patisiran, an investigational RNAi therapeutic, showed improvement in primary, secondary and exploratory endpoints compared to placebo and was generally well tolerated in hATTR amyloidosis patients. Here, all data on measurements of autonomic symptoms from APOLLO are presented. Methods: APOLLO was a multi-center, international, randomized (2:1), double-blind study of patisiran 0.3mg/kg or placebo IV q3W in hATTR amyloidosis patients with polyneuropathy (NCT01960348). The composite autonomic symptom score 31 (COMPASS-31): a 31-item questionnaire (range 0-100 points) used to evaluate patient reported autonomic neuropathy symptoms on the following six domains: orthostatic intolerance, vasomotor, secretomotor, gastrointestinal, bladder, and pupillomotor. Subjective perceptions of autonomic nerve function were captured within the Norfolk quality of life diabetic neuropathy (Norfolk QOL-DN), a 35-item patient-reported scale. Postural blood pressure (PBP) was captured in the modified neuropathy impairment score (mNIS+7 score). Among all 3 tools, higher scores are indicative of worsening autonomic symptoms. Results: APOLLO enrolled 225 patients: mean age 61 years (24-83), 74% males and 43% V30M. Overall, COMPASS-31 showed a least squares (LS) mean decrease (improvement) of -5.29 points with patisiran compared to an LS mean increase (worsening) of 2.24 points with placebo, for an overall treatment difference of -7.53 points at 18-months (p=0.0008). Patisiran treated patients experienced a benefit compared to placebo across all COMPASS-31 domains, and showed an improvement relative to baseline for the orthostatic intolerance and gastrointestinal domains (Figure 1). Similarly, autonomic improvement with patisiran and worsening with placebo was observed in autonomic domain of Norfolk QOL-DN and PBP component of mNIS+7 with an LS mean change (SEM) compared to baseline for patisiran, placebo: [-0.3 (0.19), 0.8 (0.39)] and [-0.2 (0.06), 0.1 (0.11)], respectively. These findings were accompanied by a lower frequency of adverse events of syncope, nausea, and urinary tract infection in the patisiran group compared to placebo group. Conclusion: In APOLLO, multiple measures of autonomic symptoms and function showed a consistent benefit of patisiran over placebo as well as an improvement compared to baseline resulting in lowering of GI symptoms (e.g. diarrhea and constipation) and orthostatic intolerance (e.g. inability to stand upright).
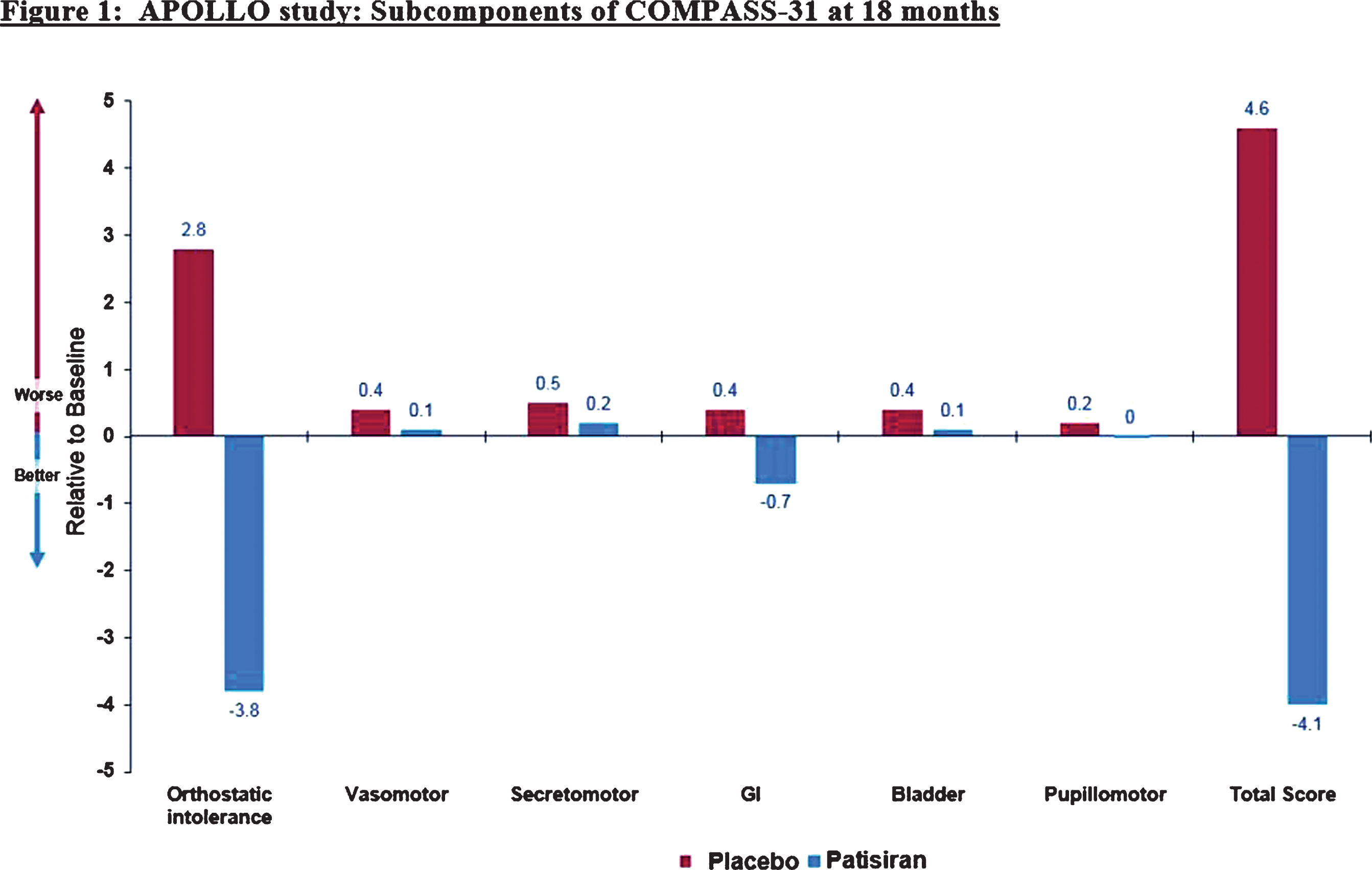
PS2Group3-006 / #705THE FUNCTIONAL AND STRUCTURAL EVALUATION OF SMALL FIBERS IN ASYMPTOMATIC PATIENTS WITH VAL30MET MUTATION
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Ezgi Yilmaz1, Can Ebru Bekircan-Kurt2, Sibel Kocabeyoglu3, Fatma Gokcem Yildiz1, Murat Irkec3, Ersin Tan1, Sevim Erdem-Ozdamar1
1Neurology, Hacettepe University, Ankara, TR;2Neurology, Hacettepe University, Ankara, TR;3Ophtalmology, Hacettepe University, Ankara, TR
Background: Transthyretin (TTR)-related hereditary amyloidosis usually presents as a progressive, axonal sensory autonomic and motor neuropathy called familial amyloid polyneuropathy (FAP). Distinct polyneuropathies including sensorimotor polyneuropathies, entrapment neuropathies, autonomic neuropathy and small fiber neuropathy can manifest in TTR-FAP patients. Moreover, small fiber neuropathy (SFN) could be the first manifestation of FAP. Here, we investigated the structural and functional properties of small fibers in asymptomatic carriers of Val30Met mutation. Methods: A family with Val30Met mutation, whom index case was diagnosed with FAP 20 years ago, has been followed in our department. Five asymptomatic family members with Val30Met mutation were included to the study. The neurological examination, DN4 questionnaire, body mass index (BMI), 24-hour urine parameters plasma brain natriuretic peptide levels were assessed. Nerve conduction studies, sympathetic skin response test, heart rate variability (HRV) were done. were performed to detect cardiac involvement. Quantitative sensory testing (QST) was assessed to determine cold and warm sensation thresholds. Skin punch biopsies were performed and intraepidermal nerve fiber densities (IENFD) were calculated. Moreover subbasal corneal nerve densities, long nerve fiber (LNFs) count per frame, long nerve fiber length (LNFL) and the total number of nerve branches/frame (TNF) were evaluated by in vivo confocal microscopy (IVCM). Results: The mean age was 39.6 years (18-60) and four of the carriers were female. The mean BMI was 26 kg/m2 (23.2-29.4). Only one carrier defined burning in her feet upon questioning and his DN4 questionnaire score was 2. The neurological examination of all carriers was normal. Quantitative sensory testing (QST), 24-hour urine parameters, plasma brain natriuretic peptide levels, echocardiogram were normal in all carriers. One had Mobitz type 1 atrioventricular block detected by Holter monitoring. The neurophysiological studies including nevre conduction studies, sympathetic skin responses and heart rate variability tests were all normal compared to age and sex matched reference values. In vivo corneal confocal microscopic evaluation revealed normal basal epithelial cell density. LNFL (mean=2315.9),LNFs (mean=3),TNF(mean=6.8) were lower in all carriers than age and sex matched controls. One carrier had grade 3 subbasal nevre tortuosity. The mean IENFD was 3.23 fibers/mm (1.16-4.75) and all IENFD of the patients were lower than the age and sex matched normative values. Conclusion: In this pilot study small fiber loss was determined by skin punch biopsy and IVCM in all of our asymptomatic carriers with Val30Met mutation. However the functional diagnostic tests such as QST, sympathetic skin responses did not show any functional loss in this early stage in these asymptomatic carriers. It is open to debate if a carrier should be considered as a patient in such a condition. But it is for sure that the carriers must be followed by morphological as well as functional studies. As they develop symptoms comparison of these tests with the presymptomatic basal values will be very valuable for diagnosing a carrier as a patient and starting treatment.
PS2Group3-007 / #954CLINICAL AND ELECTROPHYSIOLOGICAL FEATURES OF AMAN FROM THE SONORAN OUTBREAK
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Suraj A. Muley, Shafeeq Ladha
Neurology, Barrow Neurological Institute, Phoenix, AZ, US
Background: Acute motor axonal neuropathy (AMAN) is a rare variant of Guillain-Barré syndrome. Patients with AMAN have an axonal electrophysiology affecting motor nerves with sparing of the sensory nerves. Even though the clinical features and electrophysiology are suggestive of axonal degeneration, there is increasing evidence of a spectrum of axonal changes from reversible axonal conduction failure on the one extreme to severe axonal degeneration on the other. Patients may fall along this spectrum. Thus far, the majority of information about AMAN is based on outbreaks of this disease in China and Mexico. Given the rare nature of this disease, the pathophysiological basis of this disease can become clearer if future outbreaks of this disease are studied systematically, both clinically and electrophysiologically. Methods: There was an outbreak of AMAN in the Sonoran state of Mexico and the adjoining area of the United States in June 2011. We studied the clinical features and electrophysiological findings of 14 patients from this outbreak in August 2011 and again in a follow-up visit in August 2013. The clinical characteristics that were studied included antecedent illness, time from onset of symptoms to nadir, severity of disability at nadir including ability to ambulate, need for mechanical ventilation, treatments administered, time to improvement, degree of improvement in terms of independent ambulation and severity of residual deficits. Electrophysiological features that were studied included median, ulnar, peroneal and tibial motor amplitudes, conduction velocities, distal latencies and F-wave latencies. Median, ulnar and sural sensory responses were also studied. Electromyography of tibialis anterior, vastus lateralis, first dorsal interosseous and biceps brachii muscles was studied. Results: The average age of patient’s was 50.3 years. All patients had a rapidly progressive course with an average of 3.15 days from symptom onset to nadir. 11 of 14 (78.5%) patients became nonambulatory and 4 of 14 (29%) needed mechanical ventilation. Eight patients were treateed with IVIG, 2 patients were treated with both IVIG and plasma exchange and 4 patients received no immunotherapy. Severely reduced CMAP amplitudes were seen in the motor nerves along with normal sensory responses. Four of the 11 nonambulatory patients required mechanical ventilation. Three of the 4 patients who were nonambulatory at follow-up had required mechanical ventilation.One of 4 untreated and one of 8 IVIG-treated patients were nonambulatory. All patients who received both IVIG and plasma exchange were nonambulatory. 4 of 14 patients reached nadir within 1-2 days and 10 of 14 patients within 3-6 days. From the first group 1 was intubated and 1 was nonambulatory at follow-up and from the second group 4 patients were intubated and 3 were nonambulatory at follow-up. Mean median, ulnar, peroneal and tibial CMAP amplitudes were 0.175, 0.25, 0.23 and 0.66 mV in non-ambulatory patients versus 3.11, 2.98, 2.17 & 4.63 mV in ambulatory patients. Conclusion: Despite severe axonal features most patients improved in a time course suggestive of reversible axonal failure. Poor prognostic factors included the need for mechanical ventilation and severity of CMAP amplitude reduction. Shorter time from onset to nadir may be a good prognostic sign.
PS2Group3-008 / #980METABOLIC SYNDROME COMPONENTS AND NEUROLOGIC OUTCOMES IN A BARIATRIC SURGERY POPULATION
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Brian Christopher Callaghan, Emily Villegas-Umana, Ericka Chant, Evan Reynolds, Mousumi Banerjee, Eva Feldman
University of Michigan School of Public Health, Ann Arbor, US
Background: Previous studies demonstrate that diabetes, pre-diabetes, and obesity are the main metabolic drivers of polyneuropathy. Whether these same factors are associated with other neurologic outcomes is unknown. The aim of this study is to investigate the association of MetS components with polyneuropathy, retinal and cognitive function in a bariatric surgery cohort prior to surgery. Methods: Patients were recruited from the Bariatric Surgery Clinic at the University of Michigan and lean controls from a research website (no MetS components based on NCEP/ATPIII definition). Participants underwent extensive metabolic phenotyping including a glucose tolerance test and fasting lipid profile. Polyneuropathy was assessed using the Toronto consensus definition of probable clinical neuropathy. Retinal function was measured with frequency doubling technology perimetry (average mean deviation), and cognitive function with the NIH Toolbox (composite score). Multivariate regression models were used to evaluate the association between MetS components and neurologic outcomes adjusted for age, gender, and height. Results: We have recruited 109 bariatric surgery participants and 35 lean controls. In the bariatric population, the mean (SD) age was 45.3 (11.1). In the bariatric group, 33.9% had diabetes, 38.5% pre-diabetes, and 27.5% normoglycemia. The DSP prevalence was 3% in lean controls, 10.0% in normoglycemic, 7.1% in pre-diabetic, and 40.5% in diabetic bariatric participants (p<0.001 for trend). Retinal function was 0.01 (2.61), -0.10 (2.68), -2.11 (4.28), and -1.62 (4.56) (p=0.01 for trend), and cognitive function was 117.2 (14.5), 108.3 (17.5), 106.8 (18.6), 103.7 (19.3) (p=0.001 for trend) in these same groups. Waist circumference (OR=1.36, 95%CI 1.07-1.73), triglycerides (OR=1.44, 95%CI 1.01-2.05), and systolic blood pressure (OR=2.18, 95%CI 1.12-4.27) were associated with polyneuropathy. Diabetes (PE=-2.5, p=0.03) and Pre-diabetes (PE=-2.3, p=0.03) were associated with retinal function, and waist circumference (PE=-1.5, p<0.001) was associated with cognitive function. Conclusion: Polyneuropathy, retinal and cognitive function all decline with worsening glycemic status. Obesity is significantly associated with polyneuropathy and cognitive dysfunction, whereas pre-diabetes and diabetes were significantly associated with retinal dysfunction. Interestingly, while clinical DSP is common in this population, clinical retinopathy and dementia are not, indicating that DSP may be the first metabolic complication in the morbidly obese.
PS2Group3-009 / #412A SUCCESSFUL TREATMENT OF IDIOPATHIC BRACHIAL NEURITIS WITH SONO-GUIDED INJECTION AND LOW DOSE STEROID THERAPY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Chang-Hwan Kim
Physical & Rehabilitation Medicine, InHa University Hospital, inchon, KR
Background: Brachial neuritis has been conventionally treated with high dose oral corticosteroid (1mg/kg) for relieving pain or help improve functional recovery. However, the higher therapeutic dose could have various systemic side effects. A significant number of brachial neuritis patients suffer from persistent pain, weakness, fatigue and leading to an impairment. We intended to reduce side effects and achieve better recovery with effective pain control, we have used low-dose steroid therapy with ultrasound-guided local steroid injection (brachial plexus block). Methods: Case: A fourty-one-year old right-handed woman has got a common cold and this has gradually got better. About two weeks later, she felt pain and sudden weakness on left upper arm without trauma. Her history was negative for hereditary or metabolic neuropathies. On physical examination, she could not elevate arm; shoulder flexion and abduction (MRC 2/5), elbow flexion (MRC 4/5). A laboratory study and shoulder X-rays showed no abnormalities. The nerve conduction study and electromyography showed left brachial plexopathy involving C5 radiculopathy. The brachial plexus MRI showed lesions involving left divisions and lateral, posterior cord with mild swelling on post-ganglionic level of C5 nerve root, suggesting radiculoplexus neuropathy. Results: We started low-dose steroid therapy (0.5 mg/kg, 0.4mg/kg for 3 days each following 0.2mg/kg for maintaining dose 4 weeks course). Concurrently, 2-week interval, 3-times brachial plexus injection was performed. We used MRC grades of following 3 muscle pairs comprising MRC sum score for brachial plexitis: the three weakest muscles (shoulder flexor, abductor and elbow flexor). The MRC sum score were increased from 8 to 13 in one month, and further improved to 15 in 2nd month. And pain scale was graded from 5 to 2 and finally 0 in the same periods. Conclusion: Although precise pathophysiology of idiopathic brachial plexitis is thought to be associated with localized inflammatory-immune attack. Therefore, ultrasound-guided injection would reduce systemic side effects and improve therapeutic outcome.
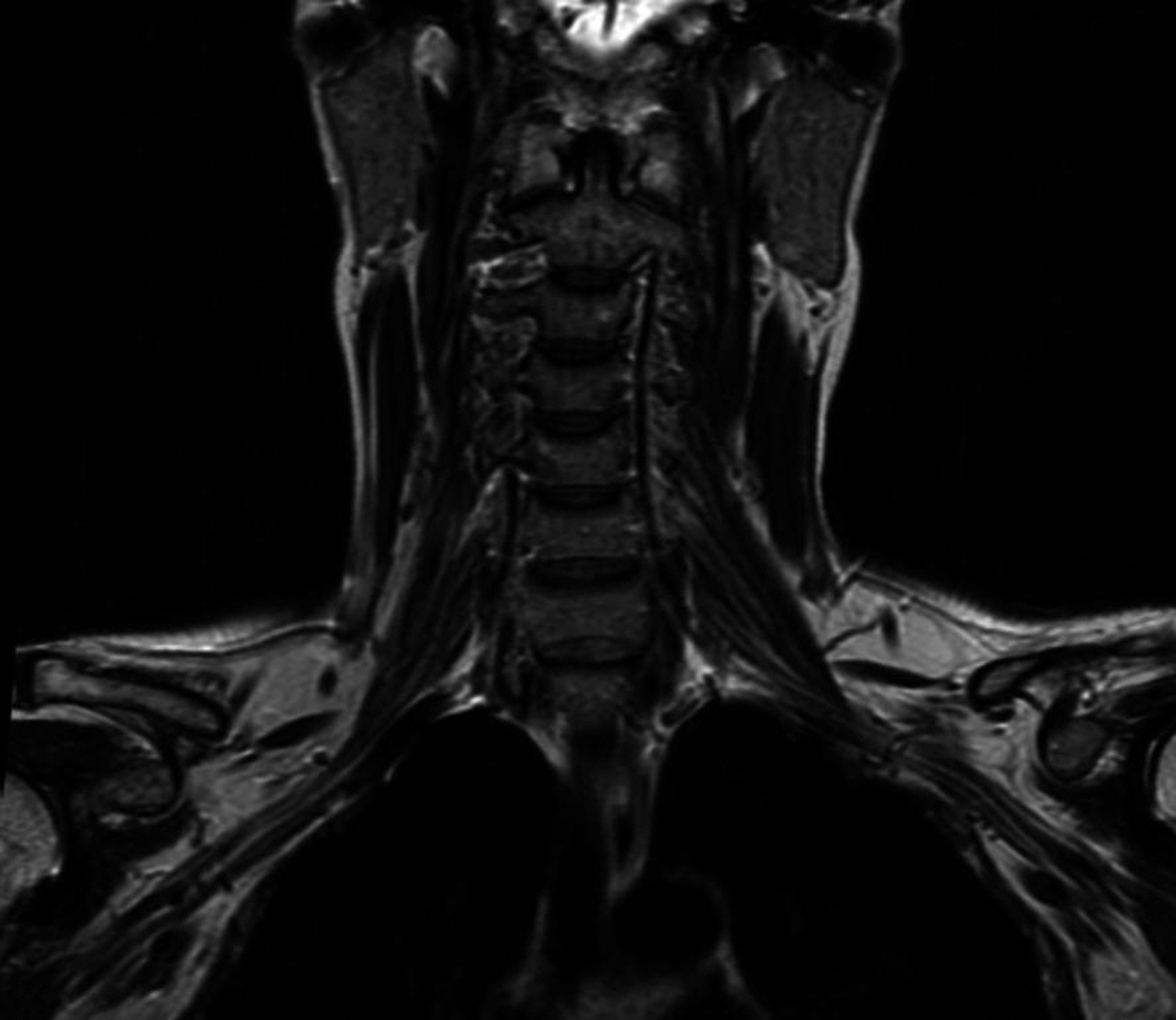
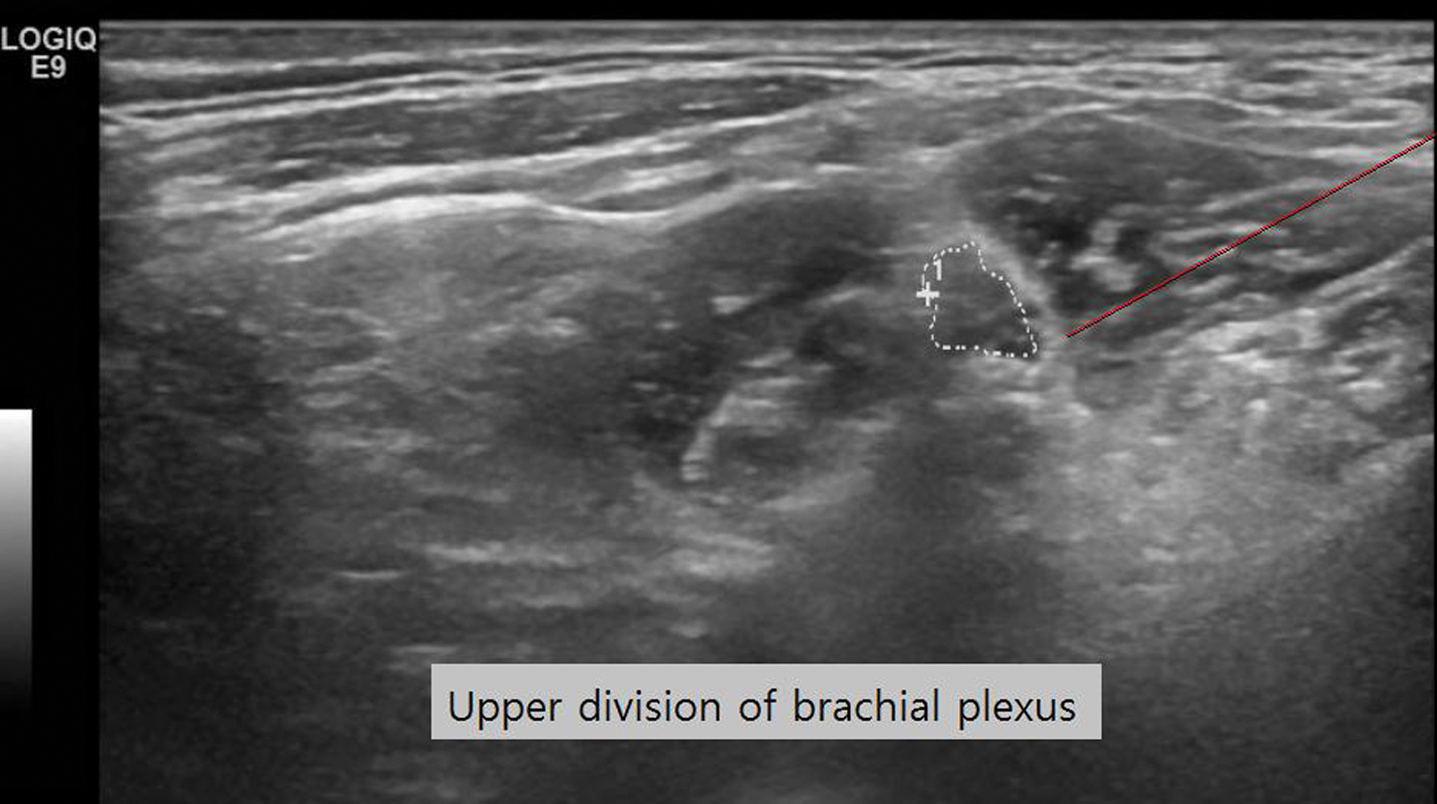
PS2Group3-010 / #455APOLLO PHASE 3 STUDY: IMPACT OF BASELINE NEUROPATHY SEVERITY ON RESPONSE TO PATISIRAN
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Laura Obici1, Teresa Coelho2, David Adams3, Alejandra Gonzalez-Duarte4, William O’Riordan5, Chih-Chao Yang6, Arnt Kristen7, Ivaylo Tournev8, Hartmut Schmidt9, John Berk10, Kon-Ping Lin11, Pritesh Gandhi12, Marianne Sweetser12, Tim Lin12, Jared Gollob12, Ole Suhr13
1Fondazione IRCCS Policlinico San Matteo, Pavia, IT;2Hospital de Santo Antonio, Porto, PT;3Neurology Department, LFB, Le Kremlin Bicetre, FR;4Instituto Nacional de Ciencias Medicas y Nutricion, Salvador Zubiran, MX;5eStudy Site, La Mesa, NM, US;6National Taiwan University Hostpital, Taipei, TW;7Heidelberg University Hospital, Heidelberg, DE;8University Multiprofile Hospital for Active Treatment, Sofia, BG;9University Hospital Muenster, Muenster, DE;10Boston Universty, Boston, MA, US;11Taipei Veterans General Hospital, Taipei, TW;12Alnylam Pharmaceuticals, Cambridge, MA, US;13Department Of Public Health And Clinical Medicine, Umea University, Umea, SE
Background: Hereditary transthyretin-mediated (hATTR) amyloidosis is a rare, multi-systemic, rapidly progressive, life-threatening disease caused by mutations in the transthyretin (TTR) gene resulting in multi-organ TTR amyloid deposition. Clinical manifestations include sensorimotor and autonomic neuropathy, gastrointestinal symptoms, and cardiomyopathy. The modified neuropathy impairment score (mNIS+7), a sensitive quantitative measure to assess somatotopic distribution of muscle weakness, sensory loss, and autonomic symptoms, has been utilized in clinical trials to evaluate polyneuropathy progression in hATTR amyloidosis patients. In the phase 3 APOLLO study, patisiran, an investigational RNAi therapeutic, resulted in a statistically significant improvement in mNIS+7 compared to placebo in patients with hATTR amyloidosis, and was generally well tolerated. Here, we present an analysis of the mNIS+7 response by baseline NIS quartile in order to demonstrate the impact of patisiran across a broad range of neuropathy severity. Methods: APOLLO was a multi-center, international, randomized (2:1), double-blind, placebo-controlled study of patisiran 0.3 mg/kg or placebo IV q3W in adult patients with hATTR amyloidosis with polyneuropathy (NCT01960348). The primary endpoint was change in mNIS+7 from baseline to 18-months. Patients were grouped into quartiles by baseline NIS scores (Figure 1). The mean change from baseline in mNIS+7 over time was calculated for each quartile by treatment group. Results: APOLLO enrolled 225 patients: mean age 65 years (24-83), 74% males and 43% V30M. Compared to baseline, patients treated with patisiran had an improvement in neuropathy as measured by mNIS+7 (LS mean change: -6.0 points), while placebo patients worsened (LS mean change: +28.0 points) resulting in a LS mean difference of -34.0 points favoring patisiran at 18-months (p=9.3 x 10-24). Effect was seen as early as 9 months. Within each baseline NIS quartile (Figure 1), mNIS+7 worsened in the placebo group, with mean changes from baseline at 18-months of 20.0, 27.4, 31.9 and 32.5 points going from lowest (less severe disease) to highest (more severe disease) quartiles. In contrast, in the patisiran group mNIS+7 improved or remained stable within each quartile going from lowest (less severe disease) to highest (more severe disease) quartiles with (-2.8, -6.6, -1.0, and -6.0.) points at 18-months (Figure 1). Conclusion: APOLLO patients on placebo presenting with either early or advanced neuropathy demonstrated significant disease progression over 18-months as measured by change in mNIS+7. These findings were consistent regardless of the severity of polyneuropathy at study entry indicating that treatment with patisiran can improve or stabilize the course of the disease.
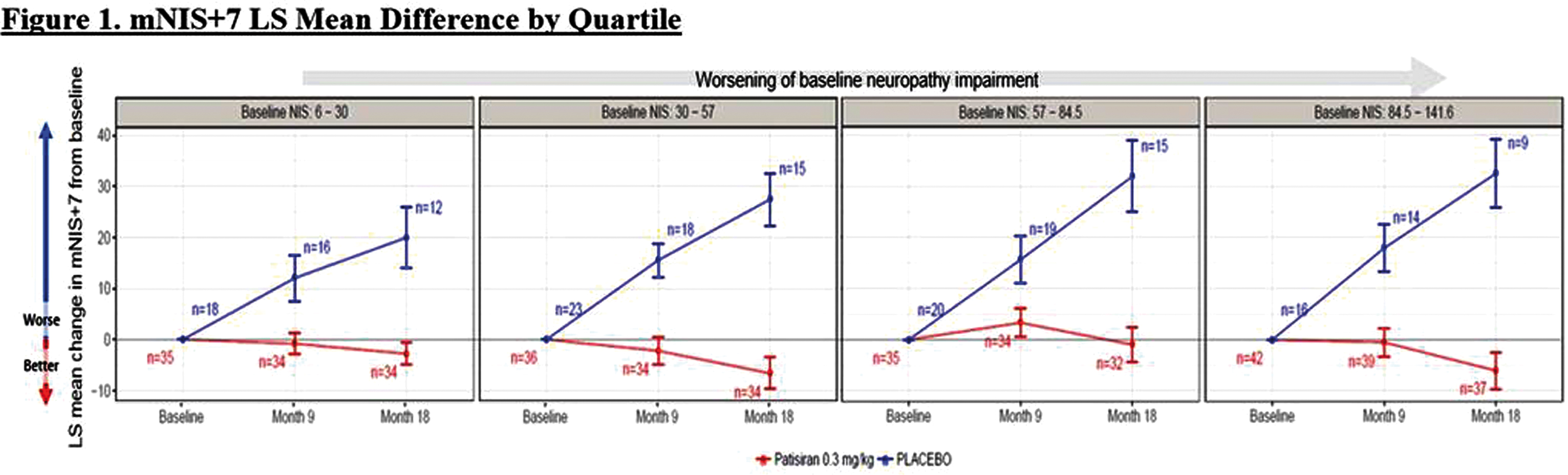
PS2Group3-011 / #575CLINICAL SPECTRUM OF HEREDITARY SPASTIC PARAPARESIS BY MUTATION IN KIF1A GENE
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Carlos Ortez1, Debora Itzep2, Laura Carrera-García2, Daniel Natera De Benito3, Anna Lia Frongia4, Delia Yubero2, Giovanni Stevanin5, Jaume Colomer2, Andres Nascimento2
1Neuromuscular Unit, San Joan de Deu Hospital, Barcelona, ES;2Hospital Sant Joan de Déu, Barcelona, ES;3Neuromuscular Disorders Unit, Hospital Sant Joan de Déu, Barcelona, ES;4Neuromuscular Unit, Sant Joan de Deu Hospital, Barcelona, ES;5Institut National de la Santé et de la Recherche Medicale (INSERM), Paris, FR
Background: KIF1A is a member of the kinesin family and functions as an anterograde motor protein that transports membranous organelles along microtubules. The phenotypic spectrum of hereditary spastic paraparesis (HSP) related to KIF1A is broad. Families with HSP with dominant and recessive inheritance have been described. Methods: Retrospective study of a cohort of patients followed in the Neuromuscular Unit of our hospital with a diagnosis of HSP with mutations in KIF1A gene. Results: Five cases of patients affected by HSP with mutations in KIF1A with an average age of 14 years (range 12-19) are reported. One of them has a pure HSP and four have complex forms, two with a family history of HSP. In the complex forms (three patients), axonal motor neuropathy was associated to mild-moderate intellectual disability, optic atrophy and cerebellar atrophy was observed in one patient. Clinical debut at an average age of 11.5 months (range: 1-18 months) in the form of hypertonia, equine gait and / or frequent falls. Progressive development of spasticity of proximal predominance of lower extremities. Currently all maintain autonomous spastic walk. All patients are carriers of heterozygous mutations in KIF1A gene. Conclusion: In our series, the HSP by mutations in KIF1A gene predominate the complex form associated with neuropathy and intellectual disability. Our data reaffirm the existence of heterozygous de novo mutations related to a more severe phenotype of complex HSP of early onset and autosomal dominant inheritance.
PS2Group3-012 / #607PLACEBO EFFECT IN THE PATH STUDY OF SUBCUTANEOUS IMMUNOGLOBULIN IN CHRONIC INFLAMMATORY DEMYELINATING POLYNEUROPATHY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Richard A. Lewis1, Vera Bril2, Nan Van Geloven3, Hans-Peter Hartung4, Gen Sobue5, Orell Mielke6, John-Philip Lawo6, Billie L. Durn7, David R. Cornblath8, Ingemar Merkies9, Ivo Van Schaik10
1Department Of Neurology, Cedars-Sinai Medical Center, Los Angeles, US;2Department Of Medicine (neurology), University Health Network, University of Toronto, Toronto, CA;3Department Of Medical Statistics And Bioinformatics, Leiden University Medical Center, Leiden, NL;4Department Of Neurology, Heinrich-Heine University, Duesseldorf, DE;5Department Of Neurology, Nagoya University Graduate School of Medicine, Nagoya, JP;6CSL Behring, Marburg, DE;7CSL Behring, Fuquay Varina, NC, US;8Department Of Neurology, Johns Hopkins University, Baltimore, MD, US;9Department Of Neurology, Maastricht University Medical Center, Maastricht, NL;10Department Of Neurology, Academic Medical Center, University of Amsterdam, Amsterdam, NL
Background: Placebo response is a recognised variable that is important in the design of clinical trials. In most clinical trials, ‘placebo response’ refers to unexpected improvement while on placebo. However, in trials involving chronic inflammatory demyelinating polyneuropathy (CIDP), it can refer to a lack of clinical deterioration on placebo when such deterioration was expected. This lack of deterioration may also be in part due to the remitting-relapsing nature of CIDP. The PATH study investigated subcutaneous immunoglobulin (SCIG) versus placebo in the treatment of CIDP and had two periods in which the placebo response could be evaluated. In the open-label Ig dependency period, intravenous immunoglobulin (IVIG) was withdrawn from subjects stated by their physicians to be IVIG-dependent. In the randomisation period, subjects were randomised to SCIG maintenance therapy or placebo. Here we detail the placebo response rates from PATH and compare these with other studies of CIDP and IVIG therapy from the literature. Methods: PATH investigated SCIG (IgPro20, CSL Behring) as maintenance therapy for CIDP. Before randomisation to SCIG or placebo, subjects underwent IVIG withdrawal and, upon clinical deterioration, were restabilised with IVIG (IgPro10, CSL Behring). IVIG restabilisation comprised an initial dose of 2 g/kg followed by 3–4 doses of 1 g/kg at 3-week intervals. Subjects were then randomised to weekly SCIG maintenance therapy (0.2 or 0.4 g/kg) or placebo for 24 weeks. The primary endpoint was CIDP relapse or withdrawal (relapse: ≥1 point increase in total adjusted Inflammatory Neuropathy Cause and Treatment [INCAT] score). Placebo responses in other studies were identified through a literature review of articles published on PubMed (2000-2017). Results: Of the 245 subjects who entered the Ig dependency period, 28 (11.4%) did not deteriorate despite being thought IVIG-dependent. A further 10 subjects did not continue to the restabilisation period due to other reasons. Of the 207 who entered the IVIG restabilisation period, 172 subjects demonstrating CIDP stability were randomised to placebo (n=57) or to SCIG 0.2 g/kg (n=57) or 0.4 g/kg (n=58). In the placebo group, 21 (37%) did not relapse or withdraw, compared with 61% and 67% of the two SCIG arms, despite all having deteriorated in the Ig dependency phase. Reports in the literature of placebo-controlled trials in patients with CIDP indicate placebo response or non-relapse rates of 10–55%, using a range of outcome measures (e.g., Medical Research Council sum score, INCAT, reduction in mean dose) and a variety of active treatments (e.g., IVIG, methotrexate). Conclusion: Among CIDP patients thought to be dependent on IVIG, 11.4% were able to stop IVIG without deterioration. For those shown to be IVIG dependent, when given placebo in a blinded fashion, 37% did not relapse or withdraw. This study has important implications for clinical practice and CIDP clinical trials in highlighting both placebo effect and the remitting-relapsing nature of CIDP as confounding factors when assessing efficacy. Comparisons with other studies raise questions as to the reasons for this effect and whether study design can help mitigate these effects.
PS2Group3-013 / #936THE EFFECT OF REHABILITATION ON SLEEP, RESPIRATORY AND LIFE QUALITY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Filiz Meryem Sertpoyraz1, Figen Baydan2, Mehtap Turanoglu1
1Physical Therapy And Rehabilitation Clinic, Izmir Tepecik Education and Research Hospital, Izmir, TR;2Pediatric Neurology, Tepecik Egitim ve Arastirma Hastanesi, Izmir, TR
Background: Polynoropathy usually starts symmetrically, sensibly, in lower limb limbs. Paresthesia, pain and motor involvement are accompanied by weakness We wanted to share the developments that occurred after rehabilitation in our patient with sensoromotor mix type polyneuropathic diagnosis. Methods: A 28-year-old male patient was admitted to Izmir Hospital Neuromuscular Diseases Unit with complaints of walking, unable to use his hands, and quadriplegic weakness. The patient was born with a low birth weight (2 kg) in the story, and there was retardation in motor development compared to their peers. He walked after a 2 year old. He was evaluated orthopedic physician at the age of 4. He underwent tendon lengthening operation due to deformity in his right foot. He was diagnosed polyneuropathy. During this period, she continued her intensive rehabilitation program. At the age of 7, his long-legged walking device was used to mobilize. His walk was completely disturbed due to his progress and he was able to mobilize with a wheelchair at the age of 15. 2 times orthopedic surgery. No known muscle and nervous system disease in the family. Functional Ambulance Scale: 0, using a wheelchair. Barthel index :90 Advanced dependent, Dorsolomber scoliosis present. Bilateral upper extremity elbows, wrists, flexion contractures in the hips, knees and ankles in the lower extremities. Upper limb proximal muscle branch 4/5, distal 3/5, lower extremity proximal muscle branch 2-3 / 5, distal 2/5. Atrophy was present in quadriceps, gastrocnemius, and hand interosseal muscles. EMG:Findings compatible with advanced stage sensorial dominance, sensoromotor axonal and demyelinating polyneuropathy in chronic process Bone Densitometry: Hip Femur neck T score: -3,2 Respiratory function test: Severe restrictive type of respiratory failure X ray: Dorsolomber scoliosis Polysomnography: BIPAP was recommended due to Obstructive Sleep Apnea Syndrome in the middle Alendronate and calcium + cholecalciferol were started for osteoporosis. A regular rehabilitation program has started.The basis of the rehabilitation program was stretching, joint range of motion and breathing exercises. After rehabilitation ,In the pulmonary function test were significantly improved compared to previous findings. The findings of mild obstructive apnea syndrome were present on control polysomnography. Sleep therapy was significantly improved with rehabilitation therapy. Joint range of motion and muscle strength increased. Results: a Polyneuropathy rehabilitation is the most important method in the treatment of patients. Conclusion: It is possible for the patient to be able to perform his / her daily life activities independently, in order to participate in community life by rehabilitation.

PS2Group3-014 / #379MULTIFOCAL MOTOR NEUROPATHY WITH CONDUCTION BLOCK: A CASE SERIES
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Aysha A. Alshareef
Neurology, King Abdulaziz University , jeddah , SA
Background: Motor neuropathies and multifocal motor neuropathy with conduction block are treatable causes of neuropathy that present with the clinical syndrome of asymmetric motor weakness with atrophy,. The diagnosis of the disorder relies on the typical clinical presentation as well as electro diagnostic findings of conduction block at non compression Site, in this article we reviewed the clinical characteristics, electrophysiological findings and treatment response of six patients with diagnosis of multifocal motor neuropathy with conduction clock in Saudi Arabia ;king Abdulaziz University hospital. Methods: Data of 6 patients of multifocal motor neuropathy with conduction block, who were diagnosed and treated from 2015 to August 2018 in Department of Neurology, king Abdulaziz university hospital at Saudi Arabia , were analyzed retrospectively, including clinical features, electrophysiological studies, and response to immunotherapy. Results: The 6 cases included 2 females and 4 males, 4 cases was above age of 50y ,the oldest case was 60 y old and the youngest was 28y old. The follow-ups after treatment were between 6 months to 2y.all from Arab population, the duration of symptoms prior presentation to our department were variable (10y,5y,3y,6m,2m,1m) All 6 cases were presented asymmetrically , 4 patients presented with right hand weakness predominantly finger drop ,one case presented with right claw hand, the commonest nerve affected was the radial nerve followed by ulnar nerves , and the distal part of limbs was initially affected at all patients. Only 3 patients have wasting of small muscle of the hand predominately first dorsal interosseous muscle,Two patients has mild paresthesia of the hand. Electrophysiological studies demonstrated temporal dispersion and conduction blocks on non-entrapment sites in more than one nerves in all patients , asymptomatic limb nerve conduction study revealed conduction block in five patients.sensory nerve conduction study were normal in all 6 patients, fibrillation and fasciculation were recorded in two patients , rapid firing rate ,decreased recruitment and large motor unit potential were recorded in all patients , anti GM1 antibodies were done for 4 patients and I was negative,4 cases showed significant improvement after IV Ig infusion while one patient did not improve and one missed follow up. Conclusion: asymmetrical pure motor nerve damage is the most important manifestation of multifocal motor neuropathy with conduction block, conduction block is the most important diagnostic criteria and it is found in symptomatic and asymptomatic limb ,radial nerve is the commonest nerve affected ,. IV Ig is an effective treatment for this disease
PS2Group3-015 / #560PHARMACOKINETICS AND SAFETY OF A SELECTIVE ANTIBODY-SCAVENGING GLYCOPOLYMER FOR THE TREATMENT OF ANTI-MAG NEUROPATHY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Delphine Demeestere1, Butrint Aliu1, Wolf Heusermann1, Beat Ernst1, Pascal Hänggi2, Ruben Herrendorff2
1Institute Of Molecular Pharmacy, University of Basel, Basel, CH;2R&d, Polyneuron Pharmacetuicals AG, Basel, CH
Background: Anti-myelin-associated glycoprotein (MAG) neuropathy is a demyelinating polyneuropathy that is caused by pathogenic monoclonal IgM antibodies against MAG. These autoantibodies specifically recognize the human natural killer-1 (HNK-1) glycoepitope, which is highly expressed on MAG. Patients suffer from paresthesia, sensory ataxia, tremor and a varying degree of sensorimotor deficits, but no safe and efficacious treatment is available. Therefore, we developed a glycopolymer, PPSGG, which presents a mimetic of the HNK-1 epitope in a multivalent manner on a biodegradable polymer backbone, and demonstrated its therapeutic potential as a selective scavenger for anti-MAG autoantibodies in an immunological mouse model. Methods: In this study, we explored the pharmacokinetic properties of PPSGG and some toxicological parameters by using imaging tools (2-photon tomography, confocal microscopy) and immunoassays (ELISA, Luminex). Results: Tissue distribution was explored after injection of a fluorescently labeled PPSGG in wild type mice and revealed a predominant presence of the drug in the liver and spleen. The presence of PPSGG in the marginal zone of the spleen, co-localization with the macrophages marker F4/80+ in the liver and the short half-life (t1/2, 16.9 ± 5.5 min) of the drug suggest a fast uptake and metabolism by the mononuclear phagocyte system. Additionally, PPSGG did not induce any obvious systemic inflammation or anti-drug antibody formation in vivo in mice, and no hepatoxicity in a human liver microtissue model. Conclusion: In conclusion, all obtained results so far are in favor of the clinical use of PPSGG in selective autoantibody removal.
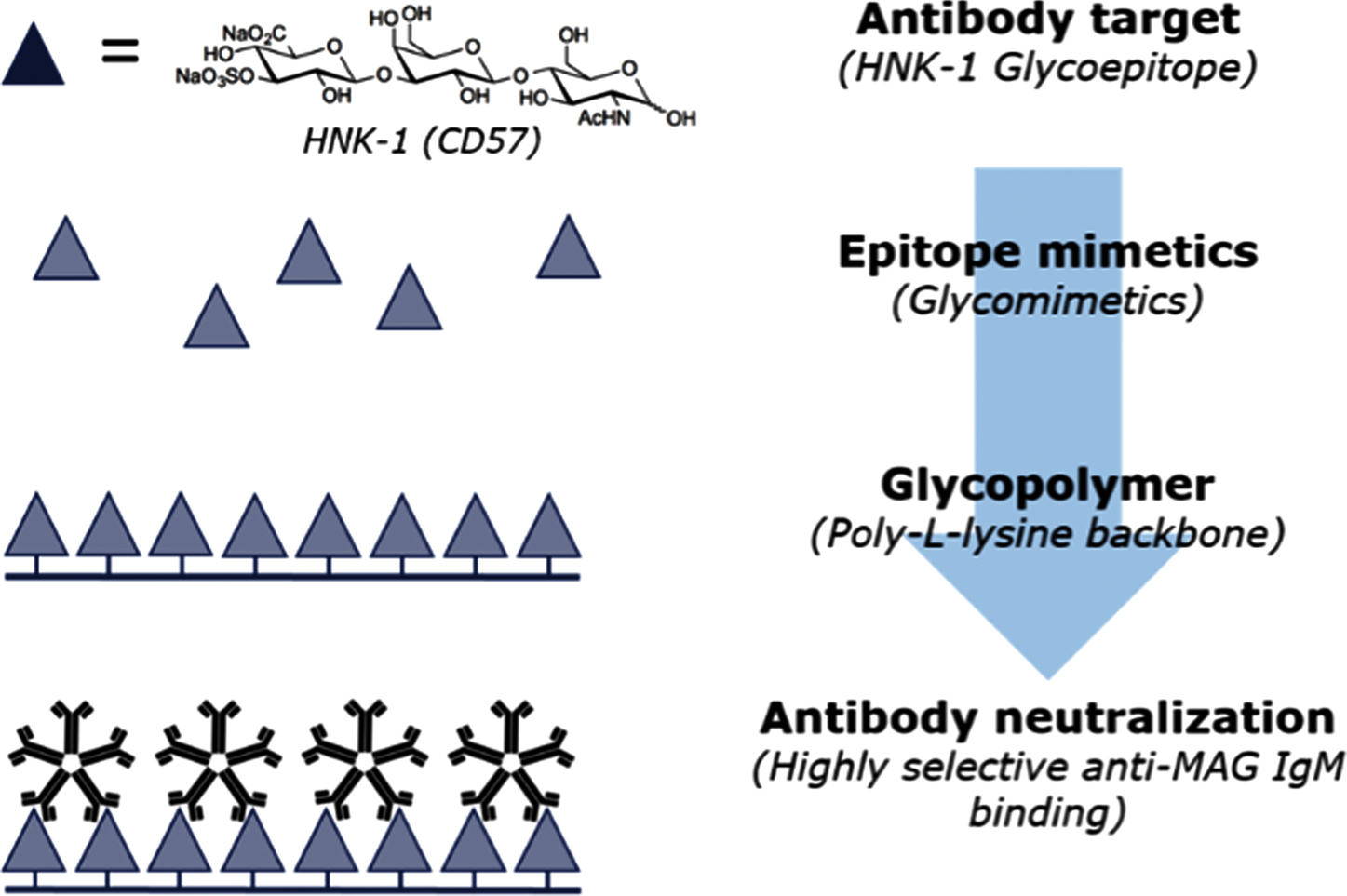
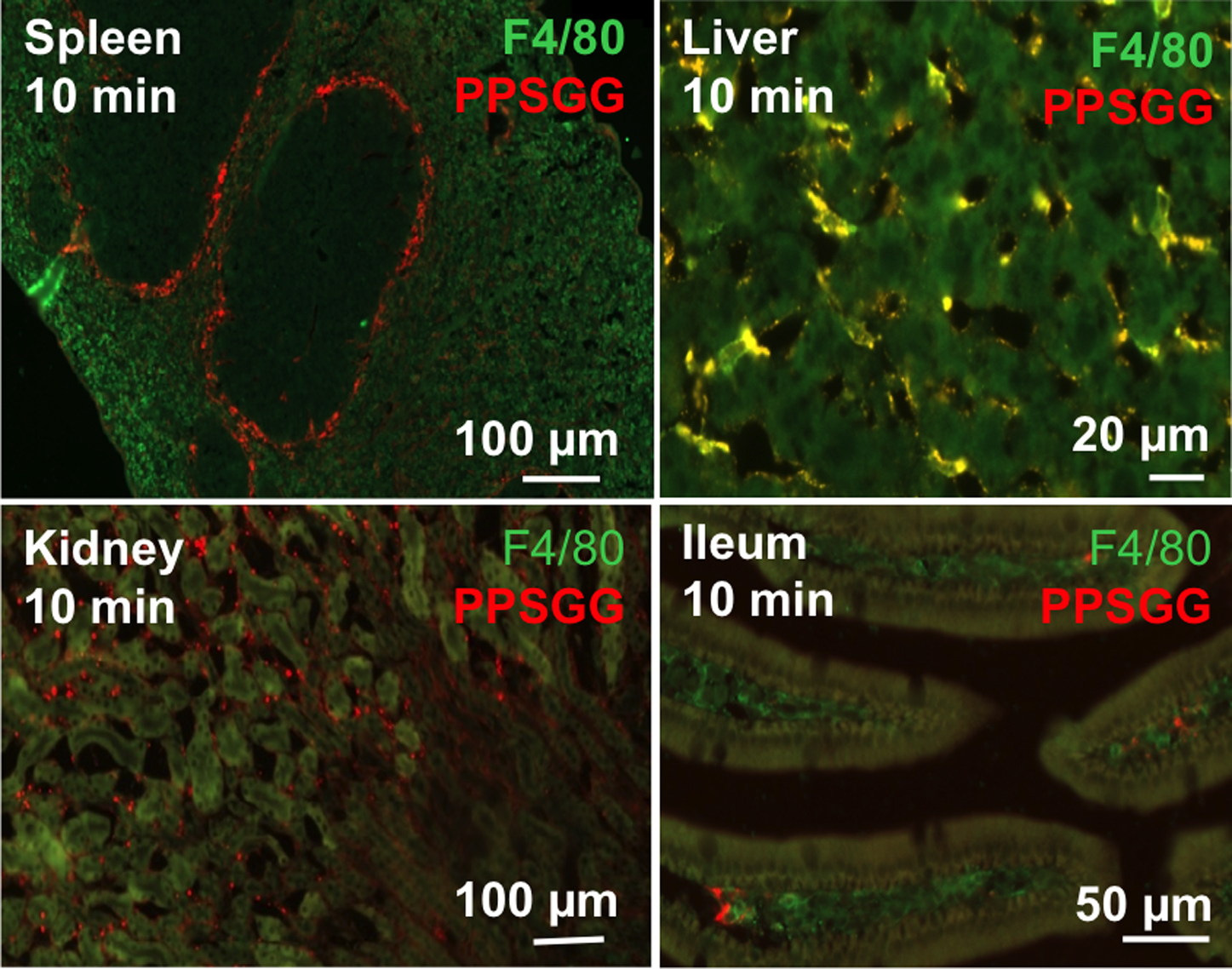
PS2Group3-016 / #587A NOVEL MISSENSE MUTATION OF TRANSTHYRETIN CAUSING AMYLOIDOSIS
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Hua-Chuan Chao1, Yo-Tsen Liu2, Yi-Chung Lee3, Kon-Ping Lin4
1Medicine, Taipei Veterans General Hospital, Taoyuan Branch, Taoyuan, TW;2Neurology, Taipei-Veterans General Hospital, Taipei, TW;3Neurology, Taipei Veterans General Hospital, Taipei, TW;4Taipei Veterans General Hospital, Taipei, TW
Background: Hereditary transthyretin amyloidosis (ATTR) is a disorder caused by dominantly inherited transthyretin (TTR) mutations, and is characterized as a progressive peripheral neuropathy and autonomic neuropathy associated with systemic involvement. The Taiwanese population is unique that majority of subjects carry the Ala97Ser missense mutation in TTR. We reported a patient with generalized neuropathy and cardiac involvement, caused by TTR Glu89Asp, which is a novel variant with heterozygous missense mutation. Methods: This male patient presented with distal paresthesia from his toes without impaired moto function since his age 59. Intermittent chest tightness, diarrhea, sensitive to light while driving and erectile dysfunction developed at the age of 64. In addition, progressively ascending numbness, tingling pain with loss of body weight up to 12 kg were reported in two years. Progressively weakness and muscular atrophy developed, resulting wheelchair-bound at his age 68. The neurological evaluation including nerve conduction study, SSR/RRIV and HUT were obtained, which revealed axonal polyneuropathy and autonomic dysfunction. Echocardiography and Tc-PYP scan were collected for cardiac evaluation, and cardiac amyloidosis was documented. He received colonoscopy and rectum biopsy, and the focal amyloid deposition was confirmed pathologically. Results: Transthyretin (TTR) gene analysis showed one point mutation (G>T) in the third base of codon 327 with the corresponding amino acid substitution of Glu for Asp. Glu89Asp (c.327G>T) was absent in the public database Mutations in Hereditary Amyloidosis (http://amyloidosismutations.com/cdna-attr.html) and Genome Aggregation Database (gnomAD). This putative pathogenic variant was also absent in 997 Taiwanese control exomes from Taiwan biobank database (http://taiwanview.twbiobank.org.tw/index). In silico predictions of pathogenicity via MutationTaster (http://www.mutationtaster.org), and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), Glu89Asp (c.327G>T) was predicted as a disease-causing variant. According to American College of Medical Genetics and Genomics (ACMG) standards and guideline, Glu89Asp was considered as a likely pathogenic variant. Conclusion: We reported here a patient with Glu89Asp TTR mutation, who presented with painful peripheral neuropathy, early autonomic dysfunction and cardiac involvement. This is a novel missense change at the 89 amino acid residue which is different from the previously reported variant.
PS2Group3-017 / #619CHRONIC MOTOR AXONAL NEUROPATHIES: A CHALLENGING DIAGNOSIS
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Yaacov Anziska1, Ilana Lasner2
1Neurology, SUNY-Downstate Medical Center, Brooklyn, NY, US;2Neurology, Kings County Hospital, Brooklyn, AL, US
Background: Chronic motor axonal neuropathies (CMAN) are rarely described in the literature and are usually thought to be immune-mediated, associated with anti-GM antibodies. Typically, these neuropathies are treated with immunomodulatory therapies, including IVIG or plasma exchange (PLEX), with varying degrees of success. However, there are instances of CMAN which are not primarily immune-mediated and are associated with various systemic diseases. Methods: In our case series, we reviewed the inpatient charts for four inpatients diagnosed with CMAN. Data included nerve coduction studies/Electromyography, laboratory tesing, and clinical examination, as documented on daily notes. Results: We describe 4 cases of chronic motor axonal neuropathies, in which neither anti-GM1 antibodies nor conduction block were present on laboratory and electrophysiologic testing. One of the cases was a patient with newly diagnosed lupus, one with newly diagnosed sacrocidosis, one with multiple myeloma (discovered 8 months after onset of motor weakness), and one with a chronic IgG gammopathy. Only the multiple myeloma patient responded temporarily to PLEX, eventually relapsing, and none of the other patients responded to various immunomodulatory therapies, including IVIG, PLEX, or high-dose steroids. Three of the patients had severe ventilatory weakness, bulbar symptoms (dysphagia, dysarthria), and vocal cord paralysis/dysfunction. Only the multiple myeloma patient recovered permanently after receiving a combination of Bendamustine and Rituximab treatment; the lupus patient improved mildly on Plaquenil, Cytoxan, Prednisone, and Imuran. Conclusion: We propose that for patients with CMAN, besides testing for various antibodies (GM-1, GD1a, GD1b, and GQ1b), a broader workup should include rheumatologic, oncologic, and infiltrative processes. Unfortunately, clinical recovery may still be limited for these patients, but might be improved if treatment could focus on the underlying systemic disease, rather than a general immunomodulatory approach administering only IVIG or PLEX.
PS2Group3-018 / #730QUALITY OF LIFE IN A CLINICAL STUDY OF MAINTENANCE TREATMENT OF CIDP WITH IGPRO20: THE PATH STUDY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Hans-Peter Hartung1, Vera Bril2, Ingemar Merkies3, Ivo Van Schaik4, Richard A. Lewis5, Nan Van Geloven6, David R. Cornblath7, Gen Sobue8, Orell Mielke9, Rajiv Mallick10, Billie L. Durn11, John-Philip Lawo9, Alphonse Hubsch12
1Department Of Neurology, Heinrich Heine University, Düsseldorf, DE;2Department Of Medicine (neurology), University Health Network, University of Toronto, Toronto, ON, CA;3Maastricht University Medical Center, Maastricht, NL;4Department Of Neurology, Academic Medical Center, University of Amsterdam, Amsterdam, NL;5Department Of Neurology, Cedars-Sinai Medical Center, Los Angeles, CA, US;6Department Of Medical Statistics And Bioinformatics, Leiden University Medical Center, Leiden, NL;7Department Of Neurology, Johns Hopkins University, Baltimore, MD, US;8Department Of Neurology, Nagoya University Graduate School of Medicine, Nagoya, JP;9CSL Behring, Marburg, DE;10CSL Behring, King of Prussia, PA, US;11CSL Behring, Fuquay Varina, NC, US;12CSL Behring, Bern, CH
Background: Subcutaneous immunoglobulin (SCIG) offers an alternative administration option to intravenous immunoglobulin (IVIG), with anticipated improvements in patient convenience and autonomy. As part of an international randomized clinical study evaluating two doses of IgPro20 (a 20% SCIG solution) versus placebo in the maintenance treatment of patients with CIDP, we evaluated patient-reported quality of life (QoL). Methods: Following stabilization on IgPro10 (a 10% IVIG solution), patients received 0.2 or 0.4 g/kg IgPro20 weekly, or placebo. The primary outcome was percentage of patients with CIDP relapse (determined by adjusted Inflammatory Neuropathy Cause and Treatment score) or withdrawal during 24 weeks of treatment. Patient-reported quality of life was assessed using the EuroQoL (EQ)-5D instrument. The EQ-5D comprises five health dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression, each rated in terms of three possible health status levels: No problems (assigned 0 points), Some/Moderate problems (1 point), and Extreme problems (2 points). A change from baseline to last post-dose observation on each of the health dimensions was classified in terms of improvement (decline of 1–2 points), maintenance (no numeric score change) and deterioration (increase of 1–2 points). Consistent with the study’s primary outcome, the ‘maintenance’ and ‘improvement’ categories, were collapsed, post-hoc, to show improvement/maintenance versus deterioration. In addition to the five health dimensions, the EQ-5D also assessed overall patient-reported health status on a Visual Analog Scale (VAS) ranging from 0 (worst possible health) to 100 (best possible health). Results: A higher proportion of SCIG treated subjects improved or maintained health status in each of the 5 dimensions of the EQ-5D, compared to placebo (Table 1). Additionally, compared to a decline in median VAS scores in the placebo group (-10.0 points; Q1, Q3: -25.0, 0.0 points), median VAS score declined marginally in the 0.2 g/kg IgPro20 group (-5.0 points; Q1, Q3: -15.0, 6.0 points) and was stable in the 0.4 g/kg IgPro20 group (0.0 points, Q1, Q3: -7.5, 5.5) (p<0.005, across treatment groups). Table 1: Numbers and percentages of patients maintained or improved versus worsened on the five health dimensions of the EuroQol-5D: IgPro20 dose groups and placebo. Conclusion: Patient-reported QoL was maintained in a higher proportion of patients treated with either dose of subcutaneous IgPro20, compared to placebo. Differences with placebo were greatest for the domains of mobility and usual activities, consistent with corresponding differences in prevention of neuromuscular relapse impacting physical function in CIDP.
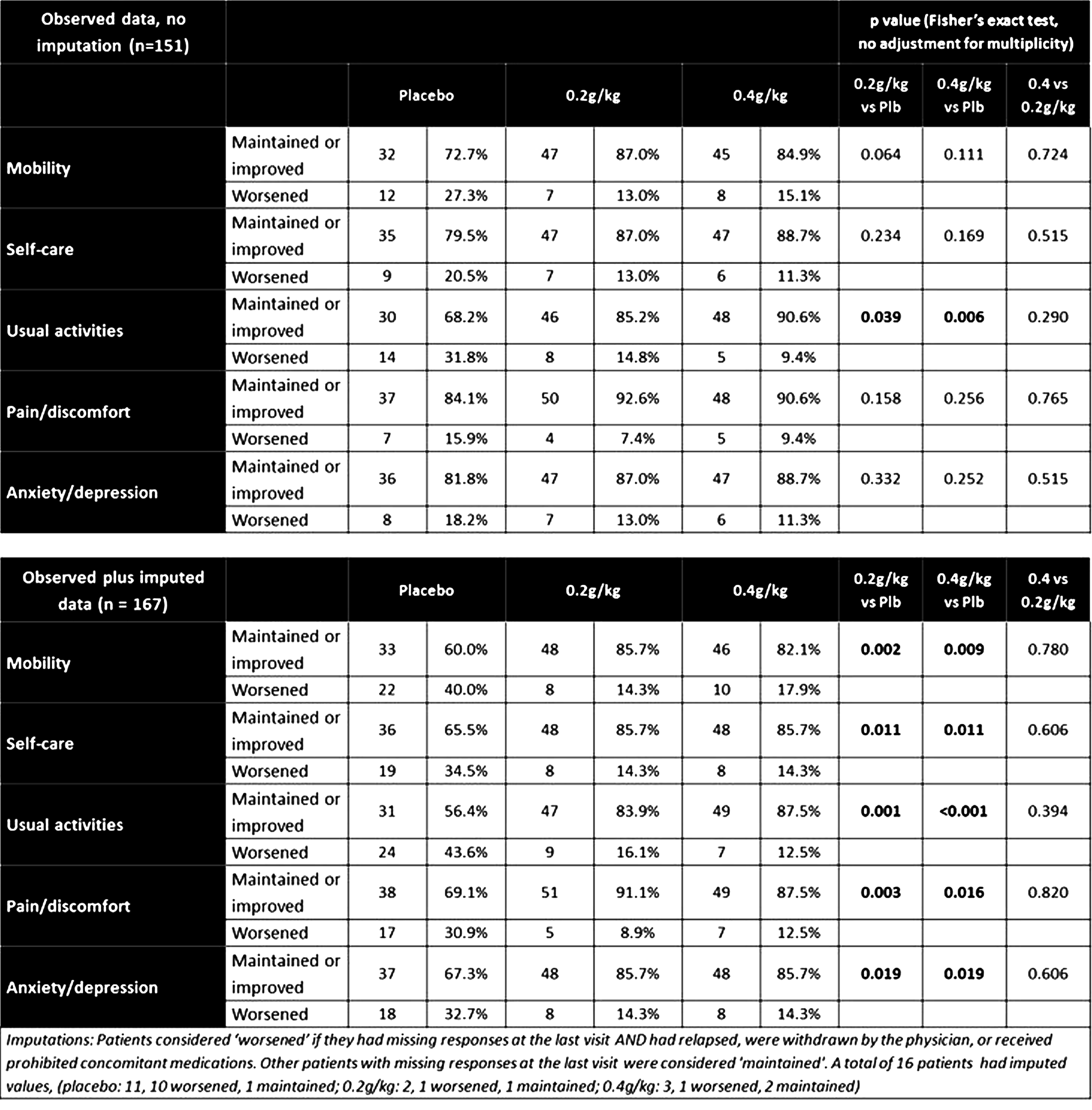
PS2Group3-019 / #651FEASIBILITY OF SWITCHING FROM INTRAVENOUS TO SUBCUTANEOUS IG THERAPY IN CIDP: PATH TRIAL RESULTS VERSUS CLINICAL EXPERIENCE
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Dario Cocito1, Erdita Peci1, Alberto Romagnolo1, Vera Bril2, Nan Van Geloven3, Hans-Peter Hartung4, Richard A. Lewis5, Gen Sobue6, John-Philip Lawo7, Orell Mielke7, Billie L. Durn8, David R. Cornblath9, Ingemar Merkies10, Ivo Van Schaik11
1University of Turin, Turin, IT;2Department Of Medicine (neurology), University Health Network, University of Toronto, Toronto, CA;3Department Of Medical Statistics And Bioinformatics, Leiden University Medical Center, Leiden, NL;4Department Of Neurology, Heinrich-Heine University, Duesseldorf, DE;5Department Of Neurology, Cedars-Sinai Medical Center, Los Angeles, US;6Department Of Neurology, Nagoya University Graduate School of Medicine, Nagoya, JP;7CSL Behring, Marburg, DE;8CSL Behring, Fuquay Varina, NC, US;9Department Of Neurology, Johns Hopkins University, Baltimore, MD, US;10Department Of Neurology, Maastricht University Medical Center, Maastricht, NL;11Department Of Neurology, Academic Medical Center, University of Amsterdam, Amsterdam, NL
Background: In chronic inflammatory demyelinating polyneuropathy (CIDP), immunoglobulin (Ig) therapy is typically administered intravenously (IVIG). Subcutaneous Ig administration (SCIG) enables independence from hospitals and increased convenience. The phase 3 PATH study showed efficacy of SCIG in CIDP. Here we evaluate the feasibility of switching from IVIG to SCIG in CIDP patients by comparing PATH data with clinical experience. Methods: In PATH, subjects with CIDP were switched to SCIG (0.2 or 0.4 g/kg/week) or placebo after IVIG induction. Adverse events (AEs), quality of life (QoL; EuroQol - 5 Dimension questionnaire) and patient preference were assessed. Observational studies of switching from IVIG to SCIG in CIDP and multifocal motor neuropathy (MMN), with assessment of safety, QoL (Life Quality Index [LQI] questionnaire) and patient preference are detailed. Results: The percentage of PATH subjects who experienced ≥1 AE with IVIG was 48% (rate: 0.175/infusion). Corresponding percentages for SCIG-0.2 and SCIG-0.4 were 58% and 52% (0.08 and 0.05/infusion). The most common AE was headache for IVIG (16%, 0.033/infusion), and local infusion site reactions for SCIG (19% [0.03/infusion] for SCIG-0.2; 29% [0.02/infusion] for SCIG-0.4). Most subjects (88%) felt SCIG was easier to use versus IVIG. Significantly more subjects (P-values <0.005) improved/maintained QoL health status with SCIG versus placebo. In observational studies, switching from IVIG to SCIG was associated with increased QoL and reduced systemic AEs (Table 1). Conclusion: The randomised PATH study and observational studies comprising large cohorts of subjects have documented the feasibility, safety and efficacy of SCIG therapy in CIDP.
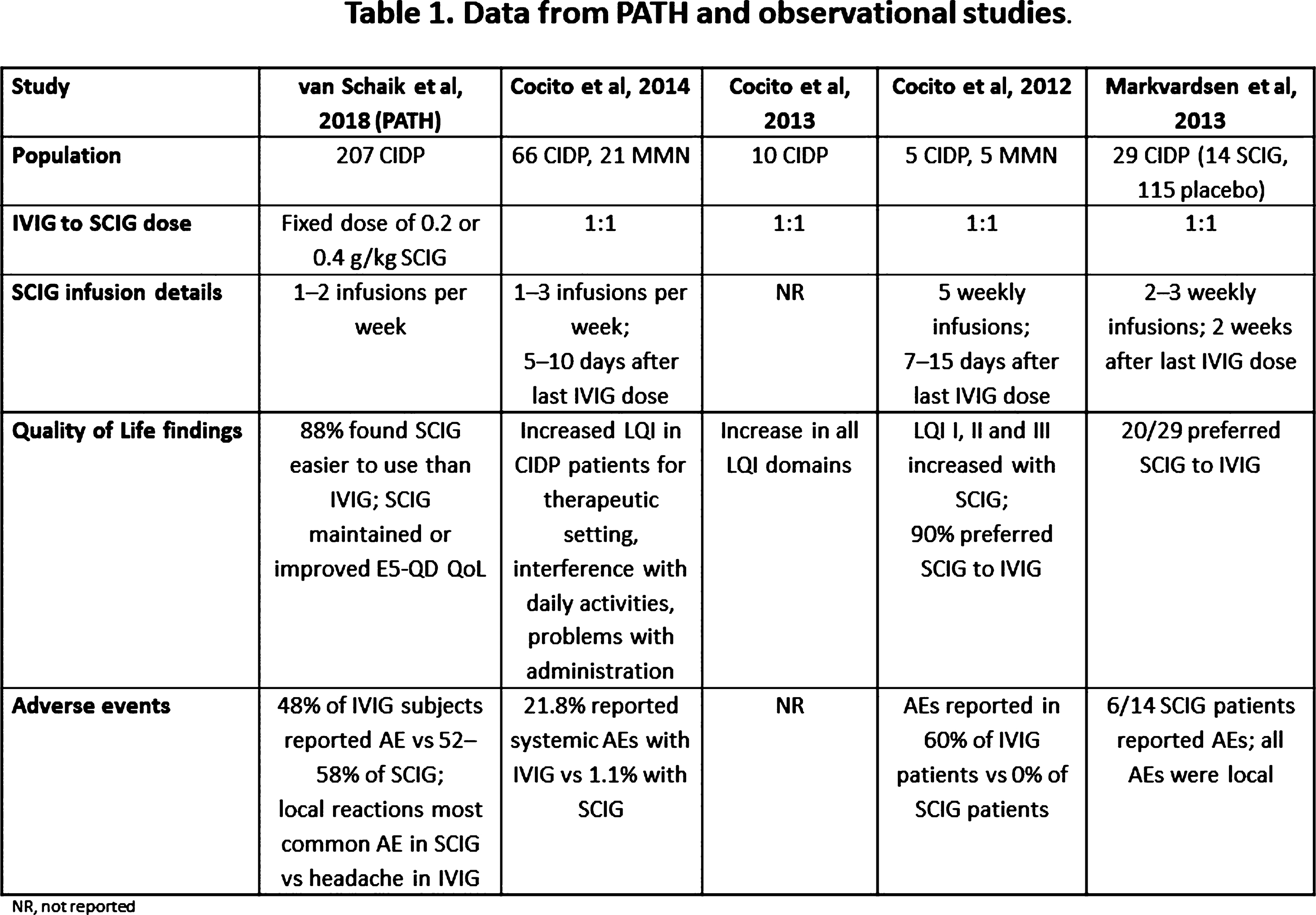
PS2Group3-020 / #924TWO NOVEL VARIANTS IN THE SLC25A46 GENE CAUSING OPTICAL ATROPHY AND PERIPHERAL NEUROPATHY IN CZECH SIBLINGS
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Marie Šedivá1, Petra Laššuthová2, Jirina Kanáková3, Jana Haberlova4, Lucie Sedlácková2, Markéta Vlcková5, Pavel Seeman6
1Department Of Paediatric Neurology, 2nd Faculty of Medicine, Charles University in Prague and Motol University Hospital, Prague, CZ;2Department Of Paediatric Neurology, Dna Laboratory, 2nd Medical Faculty, Charles University and University Hospital Motol, Prague, CZ;3Paediatric Department, Hospital Klatovy, Klatovy, CZ;4Department Of Paediatric Neurology, 2nd Faculty of Medicine, Charles University in Praue and Motol University Hospital, Prague, CZ;5Department Of Biology And Medical Genetics, 2nd Faculty of Medicine, Charles University in Prague and Motol University Hospital, Prague, CZ;6Department Of Paediatric Neurology, Dna Laboratory, 2nd Medical Faculty, Charles University and University Hospital Motol, Prague, Prague, CZ
Background: Hereditary polyneuropathies are a heterogeneous group of diseases with all modes of inheritance and variable phenotype. There are 2 main groups according to impairment of either myelin sheets (HMSN I) or axons (HMSN II). Polyneuropathy is sometimes connected with optical neuropathy. SLC25A46 gene was identified to be responsible for autosomal recessive hereditary optic atrophy in combination with peripheral axonal neuropathy and cerebellar atrophy. (1) The patients from the original study belonged to 4 families and were all compound heterozygotes for variants of SLC25A46, different in each family. (1) Methods: We present a clinical case of a 19-year-old male with primarily axonal motor and sensitive peripheral polyneuropathy and optical atrophy who has a similarly affected sister, and a healthy mother. The father is not in contact with the family. The patient was examined by whole exome sequencing. Results: By whole exome sequencing (WES), two novel variants in compound heterozygous state where found in the patient and his older sister. One is a nonsense variant in the SLC25A46 (NM–138773.2):c.1208T>G (p.Leu403*) and the second is a missense variant c.1075G>A (p.Val359Met), affecting a highly-conserved amino-acid residue and predicted as pathogenic. The healthy mother is a heterozygous carrier of the variant p.Leu403*, the father was not available for testing. The patient was born from 2nd uneventful pregnancy to healthy parents, with no perinatal complications. He first presented with impairment of visual function and atrophy of optic nerves at the age of 3 years. Since the age of 6 years he had walking difficulties and progressive deformities of feet (pedes excavates). Since the age of 10 years ataxia was noticed. The condition is slowly progressive, with a similar course in both affected siblings. Brain and spine MRI were normal. EEG was with abnormal basal activity, without any specific findings. EMG showed peripheral axonal motor and sensory polyneuropathy. Genetically there was coincidental cytogenetic finding of karyotype 47,XXY. No variants in MFN2 and GDAP genes, neither in the gene panel associated with hereditary polyneuropathies, that could explain the cause of the disease, were found. Finally, the two above-mentioned variants were detected by WES. Conclusion: To conclude, two novel likely pathogenic variants of SLC25A46 were found in 2 siblings presenting with optic neuropathy associated with polyneuropathy and ataxia, with autosomal recessive inheritance pattern. So far, the association of variants in this gene has been described in only four families by Abrams (2015).(1) Our case supports the role of SLC25A4 gene in etiology of hereditary optic atrophy associated with peripheral neuropathy. References: ABRAMS, Alexander J, Robert B HUFNAGEL, Adriana REBELO, et al. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nature Genetics [online]. 2015, 47(8), 926-932 [cit. 2018-03-15]. DOI: 10.1038/ng.3354. Supported by: AZV 16-30206, MH CZ – DRO, University Hospital Motol, Prague,Czech Republic 00064203
PS2Group3-021 / #950GENOME-WIDE DNA METHYLATION PROFILING OF HUMAN DIABETIC PERIPHERAL NEUROPATHY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Eva Feldman
University of Michigan, Ann Arbor, US
Background: DNA methylation is an important epigenetic regulator in both health and disease. However, to date there are no studies investigating possible alterations in DNA methylation in the peripheral nerves of human patients with diabetic neuropathy. Methods: In the current study, we assessed DNA methylation profiles of 12 patients with diabetic peripheral neuropathy (DPN). Examination of sural nerve biopsies from these individuals revealed that 6 patient biopsies had significant nerve regeneration whereas 6 had significant nerve degeneration over a period of 52 weeks. These samples underwent reduced representation bisulfite sequencing (RRBS) for analysis of DNA methylation between patients who exhibited sural nerve regeneration and those who exhibited degeneration. Results: There were a total of 3,460 differentially methylated CpGs (DMCs) and 246 differentially methylated regions (DMRs) between the two patient cohorts. Analyses of genes associated with the DMCs identified pathways highly enriched in neuron development and differentiation, as well as pathways associated with cancer. Conclusion: These results suggest that DNA methylation has an important role in regulating nervous system development and cellular proliferation in the progression or regression of DPN. Additionally, these novel results provide insights into possible epigenetic regulation of DPN and offer useful information for future DPN research.
PS2Group3-022 / #948TRANSCRIPTIONAL SIGNATURE OF DIABETIC PERIPHERAL NEUROPATHY CONSERVED ACROSS HUMAN AND MOUSE
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Eva Feldman
University of Michigan, Ann Arbor, US
Background: Aims/hypothesis: Diabetic peripheral neuropathy (DPN) is one of the most common complications of diabetes. Despite extensive research, the underlying mechanisms leading to DPN are not fully understood. While multiple murine models and patient samples have been examined, a unified systematic approach has yet to compare observed changes between groups which may help identify a possible conserved mechanism of DPN. In this study, we aimed to identify DPN-related transcriptional pathways conserved across human and various murine models of diabetes by using a systems biology approach. Methods: Eight microarray datasets were collected on peripheral nerve samples from murine models of type 1 (streptozotocin-treated) and type 2 (db/db and ob/ob) diabetes of different ages and human subjects with non-progressive and progressive DPN. Differentially expressed genes (DEGs) were identified between control and diabetic samples in murine models, and non-progressive and progressive human samples using a unified analysis pipeline. A transcriptional network for each DEG set was constructed based on literature-derived gene-gene interaction information and shared sub-networks between the human and murine networks were identified using a network-comparison program. Seven pairwise human-vs-murine comparisons resulted in sub-networks including 46 to 396 genes, which were further merged into a single network of 688 genes. Results: Functional analyses on the identified shared networks between murine and human datasets revealed that genes involved in LXR/RXR activation and adipogenesis were highly affected by diabetes and conserved across both species. Centrality analysis detected the most highly connected genes that may play important roles in this cross-species DPN shared network, including PIK3CA, MAPK8, CD44, MAPK1, CREB1, LEP, CCL2, JUN, ESR1, FOS, CD36, IL1B, HGF, and PLAT. These genes indicate enrichment of different pathways with the most significant including glucocorticoid receptor signaling, multiple cytokine pathways, and chemokine signaling. Conclusion: s/interpretations: Our systems biology approach identified highly conserved pathways across human and murine models such as LXR/RXR activation and multiple immune signaling pathways. These pathways likely play a role in DPN pathogenesis and provide new possible mechanism-based targets for DPN therapy.
PS2Group3-023 / #260RESTABILISATION TREATMENT AFTER IVIG WITHDRAWAL IN CHRONIC INFLAMMATORY DEMYELINATING POLYNEUROPATHY: THE PATH STUDY RESULTS
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Orell Mielke1, Vera Bril2, Nan Van Geloven3, Hans-Peter Hartung4, Richard A. Lewis5, Gen Sobue6, John-Philip Lawo1, Billie L. Durn7, David R. Cornblath8, Ingemar Merkies9, Amgad Shebl1, Ivo Van Schaik10
1CSL Behring, Marburg, DE;2Department Of Medicine (neurology), University Health Network, University of Toronto, Toronto, CA;3Department Of Medical Statistics And Bioinformatics, Leiden University Medical Center, Leiden, NL;4Department Of Neurology, Heinrich-Heine University, Duesseldorf, DE;5Department Of Neurology, Cedars-Sinai Medical Center, Los Angeles, US;6Department Of Neurology, Nagoya University Graduate School of Medicine, Nagoya, JP;7CSL Behring, Fuquay Varina, NC, US;8Department Of Neurology, Johns Hopkins University, Baltimore, MD, US;9Department Of Neurology, Maastricht University Medical Center, Maastricht, NL;10Department Of Neurology, Academic Medical Center, University of Amsterdam, Amsterdam, NL
Background: In patients with chronic inflammatory demyelinating polyneuropathy (CIDP), the dose or frequency of intravenous immunoglobulin (IVIG) administration is recommended to be periodically reduced to assess the need for ongoing therapy. Little is known about the effectiveness of IVIG restabilisation in patients who worsen after IVIG withdrawal. Methods: In PATH, a randomised, double-blind study of subcutaneous immunoglobulin in CIDP, IVIG therapy was withdrawn before randomisation. Subjects not deteriorating within 12 weeks of IVIG withdrawal discontinued the study as Ig dependency was not confirmed. Upon deterioration (increase in Adjusted Inflammatory Neuropathy Cause and Treatment [INCAT] score), subjects received IVIG restabilisation with IgPro10 (CSL Behring): induction dose 2 g/kg bw, maintenance doses 1 g/kg every 3 weeks for up to 10–13 weeks. Results: Of 245 subjects in whom IVIG was withdrawn, 28 (11.4%) did not deteriorate within 12 weeks. Another 10 subjects withdrew for other reasons, leaving 207 in the restabilisation phase. Of these 91% improved in at least 1 efficacy measure (improvement: 1 point in adjusted INCAT score, 4 points in RODS score, 8kPa in mean grip strength or 3 points in MRC score) by 10–13 weeks. Adjusted INCAT score improved in 72.9% with ~21% of subjects improving beyond their status at study entry. Improvements were seen in all secondary scores (Figure 1). Post-study follow-up of non-improving subjects revealed most subjects had improved. Conclusion: IVIG withdrawal was effective in detecting subjects not requiring IVIG therapy. For IVIG-dependent subjects, restabilisation with IgPro10 was effective in reversing observed deteriorations within 12 weeks.
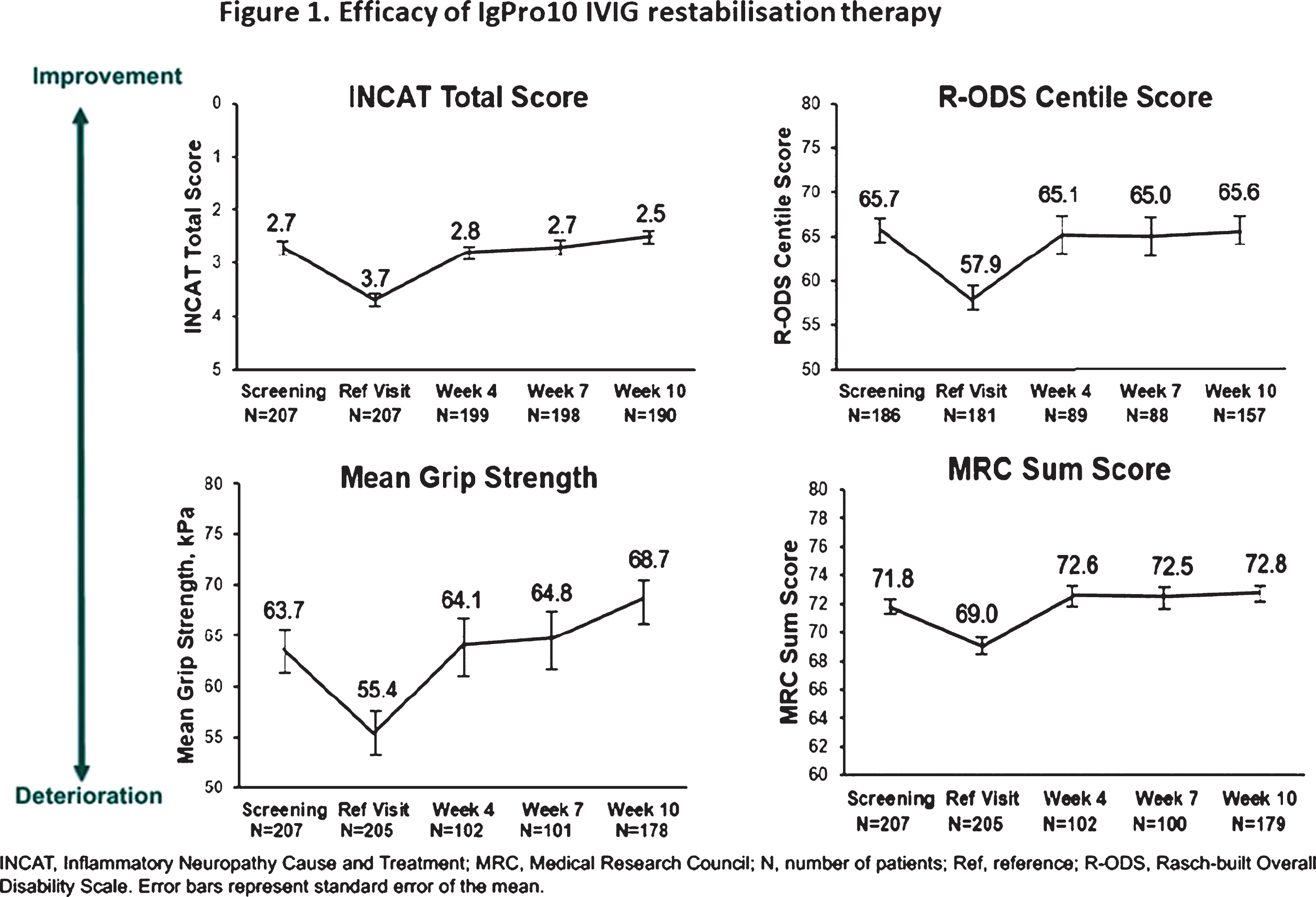
PS2Group3-024 / #625EFFICACY AND SAFETY OF INTRAVENOUS IMMUNOGLOBULIN IGPRO10 IN CIDP: COMBINED ANALYSIS OF THE PRIMA AND PATH STUDIES
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Ingemar Merkies1, Jean-Marc Léger2, Vera Bril3, Nan Van Geloven4, Hans-Peter Hartung5, Richard A. Lewis6, Gen Sobue7, John-Philip Lawo8, Billie L. Durn9, David R. Cornblath10, Jan De Bleecker11, Claudia Sommer12, Wim Robberecht13, Mika Saarela14, Jerzy Kamienowski15, Zbigniew Stelmasiak16, Bjorn Tackenberg17, Orell Mielke8, Ivo Van Schaik18
1Department Of Neurology, Maastricht University Medical Center, Maastricht, NL;2National Referral Center For Neuromuscular Diseases, University Hospital Pitié-Salpêtrière, Paris, FR;3Department Of Medicine (neurology), University Health Network, University of Toronto, Toronto, CA;4Department Of Medical Statistics And Bioinformatics, Leiden University Medical Center, Leiden, NL;5Department Of Neurology, Heinrich-Heine University, Duesseldorf, DE;6Department Of Neurology, Cedars-Sinai Medical Center, Los Angeles, US;7Department Of Neurology, Nagoya University Graduate School of Medicine, Nagoya, JP;8CSL Behring, Marburg, DE;9CSL Behring, Fuquay Varina, NC, US;10Department Of Neurology, Johns Hopkins University, Baltimore, MD, US;11Neurology, AZ St-Lucas, Ghent, BE;12Neurologische Klinik Und Poliklinik, Universitätsklinikum Würzburg, Würzburg, DE;13UZ Leuven, Leuven, BE;14Department Of Neurology, Helsinki University Central Hospital, Helsinki, FI;15Dolnoslaski Szpital Specjalistyczny, Wroclaw, PL;16Samodzielny Publiczny Szpital Kliniczny, Lublin, PL;17Department Of Neurology, Philipps University, Marburg, DE;18Department Of Neurology, Academic Medical Center, University of Amsterdam, Amsterdam, NL
Background: Efficacy and safety of intravenous immunoglobulin IgPro10 (CSL Behring) were investigated in CIDP subjects in two studies: PRIMA and PATH. Methods: PRIMA was a prospective, open-label, single-arm study in 28 CIDP subjects investigating IgPro10 for induction (2g/kg) and maintenance therapy (1g/kg every 3 weeks for 21 weeks). This regimen was also used in 207 IVIG-pretreated subjects during the 10–13 week restabilisation period of PATH (before randomisation to subcutaneous immunoglobulin vs placebo). Both studies investigated response (defined as ≥1-point decrease in adjusted Inflammatory Neuropathy Cause and Treatment [INCAT] score), changes in mean grip strength and Medical Research Council (MRC) score, and safety. We analysed separate and pooled results from both studies. Results: INCAT response rate at last observation was 76.9% (95% confidence interval [CI]: 49.7–91.8) in PRIMA IVIG-pretreated subjects (60.7% in all PRIMA subjects) and 72.9% (95% CI: 66.5–78.5) in PATH. In the pooled cohort (n=235), INCAT response rate was 71.1% (95% CI: 65.0–76.5; Table 1). Most responders improved by week 4; additional responses occurred up to week 13 (Table 1). No clear differences in characteristics were noticed between early (responding before Week 7) and late responders. Change from baseline to last observation in efficacy parameters is shown in Table 2. In the pooled cohort, 271 adverse drug reactions (ADRs) were reported in 105 subjects (44.7%), a rate of 0.144 ADRs per infusion.
Conclusion: Pooled PRIMA/PATH data showed improvement in disability with IgPro10 in a large cohort of CIDP subjects. Improvement was seen at up to 10–13 weeks, suggesting some subjects may need multiple Ig doses to respond.
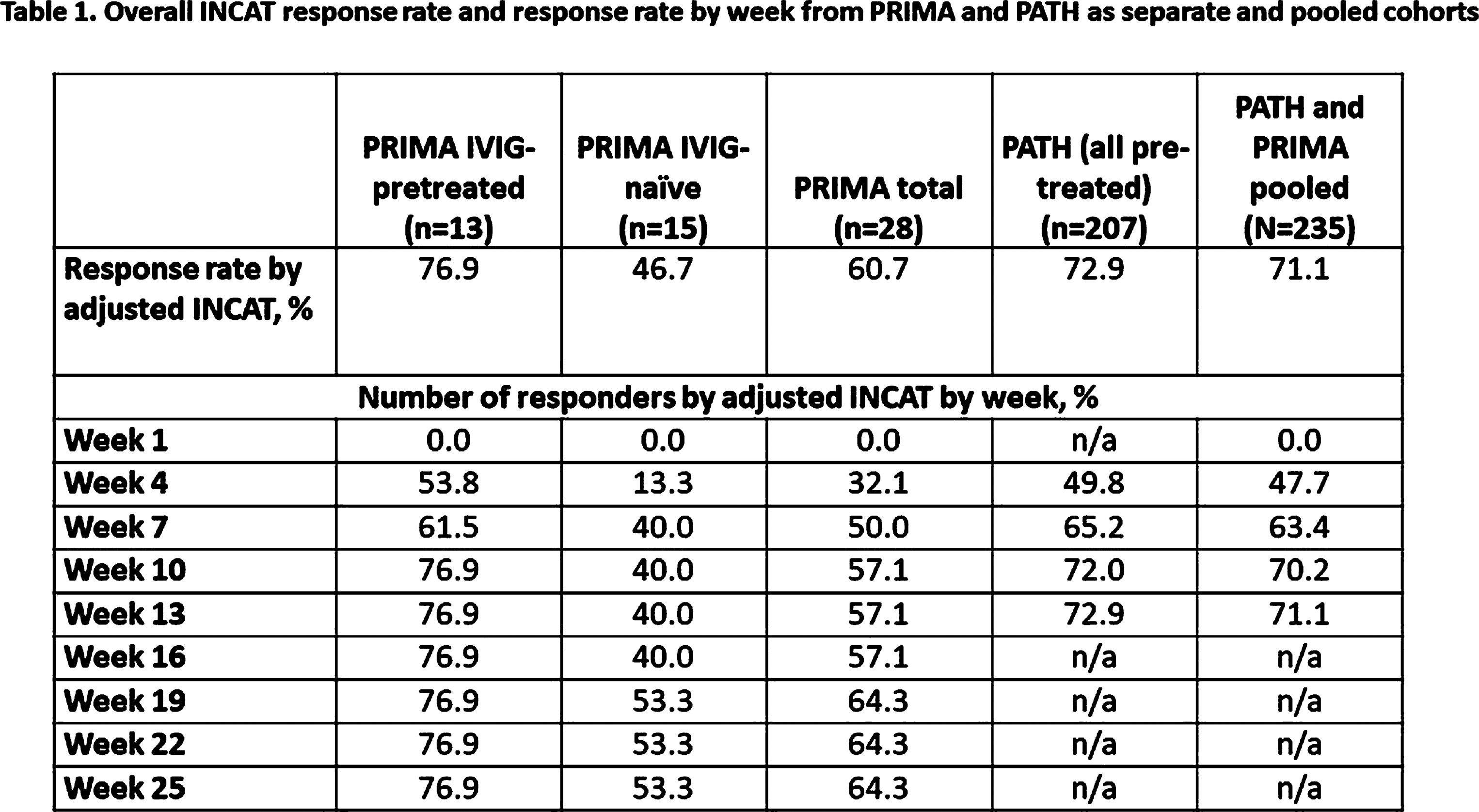
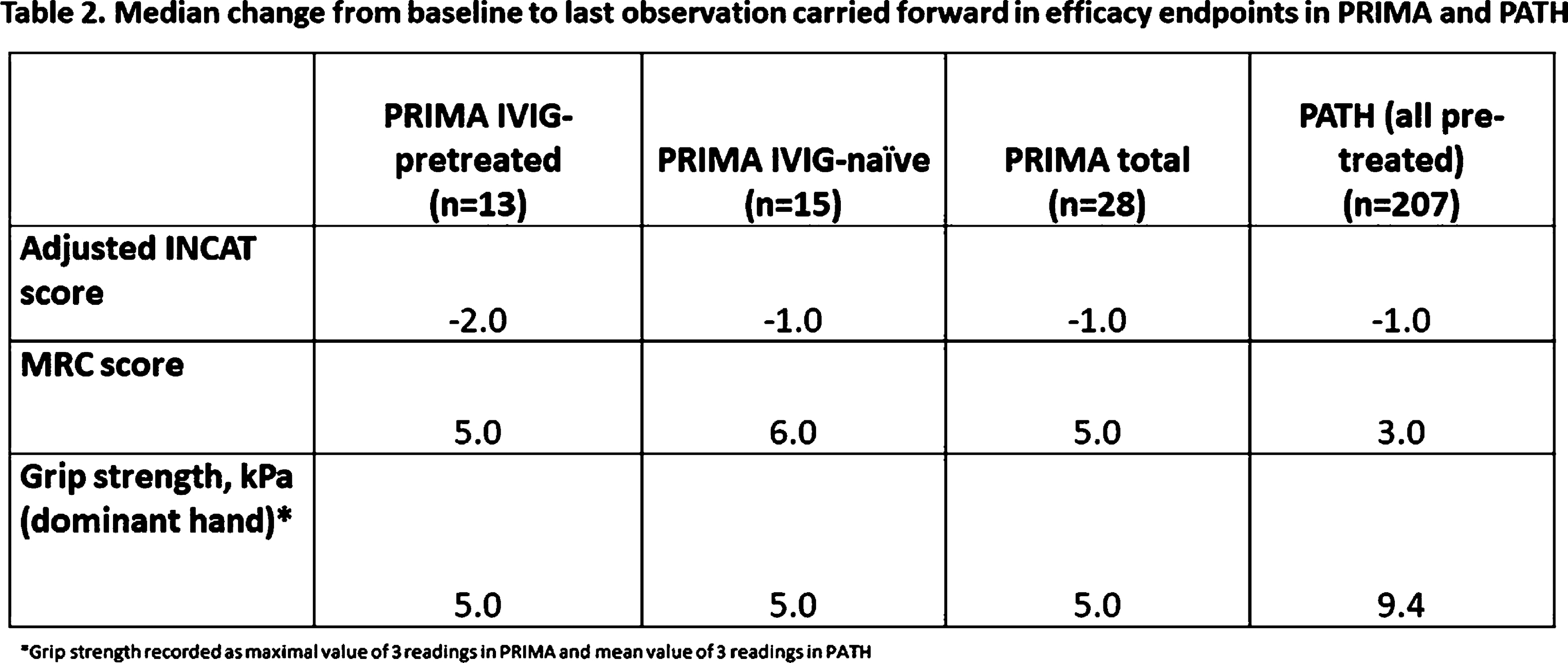
PS2Group3-025 / #538A FATAL CASE OF HEPATITIS E-ASSOCIATED GUILLAIN-BARRE SYNDROME
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Ho Ming June Wong1, Wai Sze Katie Kwan2, Chun Yu Say2, Wing Yin Winnie Wong2
1Medicine And Geriatrics, Caritas Medical Centre, Hong Kong, HK;2Medicine & Geriatrics, Caritas Medical Centre, Hong Kong, HK
Background: Extra-hepatic manifestations of hepatitis E infection have been increasingly recognised. Guillain-Barre syndrome (GBS) is one of the most commonly reported neurological complication. The mechanism of neurological involvement was believed to be due to molecular mimicry. Favorable neurological outcome was generally observed in hepatitis E-associated GBS while fatality was uncommonly encountered. Methods: We hearby report an 85-year-old Chinese lady who presented with classical form of GBS and acute hepatitis E infection. Results: An 85-year-old Chinese lady presented with unsteady gait and numbness of the four extremities for one day. She also reported nausea and reduced appetite for five days. Her past medical history was notable for hypertension, type 2 diabetes, hyperlipidemia and ischemic heart disease. On examination, she was fully conscious and oriented. She had jaundice and was apyretic. Her blood pressure was 172/63 mmHg and the pulse was 84/min. Cranial nerve examination was unremarkable. There were symmetrical distal weakness over the upper and lower limbs with Medical Research Council (MRC) muscle grading of 4/5. Generalised areflexia and glove-and-stocking sensory loss were noted. Cardiovascular, respiratory and abdominal examinations were unremarkable. Laboratory investigation revealed elevated liver enzymes (alkaline phosphatase (ALP) 292U/L, alanine transferase (ALT) 1085U/L, aspartate aminotransferase (AST) 333U/L) and elevated total bilirubin 74umol/L (direct bilirubin 70umol/L). Complete blood picture, renal function test, electrolytes, glucose and clotting profile were unremarkable. Anti-hepatitis A virus (HAV) IgM, hepatitis B surface antigen and anti-hepatitis C antibody were negative. Anti-hepatitis E virus (HEV) IgM was positive. Cerebrospinal fluid (CSF) analysis revealed albuminocytological dissociation with protein of 0.70g/L and white blood cell (WBC) of 6/mm3 while red blood cell (RBC) was 2000/mm3. Nerve conduction study was suggestive of sensorimotor polyneuropathy with demyelinating features. A diagnosis of hepatitis E-associated GBS was made. Intravenous immunoglobulin (IVIg) was started since the second day of admission. The patient’s condition deteriorated, however, with progressive ascending weakness, bulbar dysfunction, dysautonomia and type II respiratory failure requiring invasive mechanical ventilation on day 5. Two days later, she developed ventricular fibrillation requiring repeated defibrillation and cardiopulmonary resuscitation. Despite maximal support, she finally succumbed after 7 days of hospitalization. Conclusion: Severe GBS with autonomic dysfunction may occur in the setting of acute hepatitis E infection and can be fatal. Any neurological symptoms in patients with concurrent hepatitis E infection should be carefully looked into to ensure timely diagnosis and effective treatment. Whereas in patients who are diagnosed with GBS, deranged liver function may indicate an associated hepatitis E infection.
PS2Group3-026 / #899PUTATIVE DIGENIC INHERITANCE OF CHARCOT-MARIE-TOOTH DISEASE INVOLVING TWO NOVEL HETEROZYGOUS MUTATIONS IN MARS AND HARS
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Martin Krenn1, Anna Grisold1, Gudrun Zulehner1, Jakob Rath1, Hakan Cetin1, Matthias Tomschik1, Bojan Zagrovic2, Lukas Bartonek2, Tomislav Kokotovic3, Vanja Nagy3, Matias Wagner4, Tim M. Strom4, Fritz Zimprich1
1Department Of Neurology, Medical University of Vienna, Vienna, AT;2Department Of Structural And Computational Biology, Max F. Perutz Laboratories, University of Vienna, Vienna, AT;3Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases, Vienna, AT;4Institute Of Human Genetics, Technical University Munich, Munich, DE
Background: Charcot-Marie-Tooth disease is a clinically and genetically highly heterogeneous disorder. All subtypes together represent the most prevalent form of inherited polyneuropathies. The genetic spectrum of Charcot-Marie-Tooth disease currently involves more than 70 causative genes. Amongst many others, mutations in genes encoding various aminoacyl-tRNA synthetases have been reported to be involved in the pathogenesis of the disease. In recent years, the two genes HARS (histidyl-tRNA synthetase) and MARS (methionyl-tRNA synthetase) have been shown to cause inherited peripheral neuropathies in few cases with variable clinical phenotypes. Methods: We provide a detailed clinical characterization and the findings of diagnostic whole-exome sequencing in a patient with Charcot-Marie-Tooth disease. Results: This female patient first noticed a weakness for toe dorsiflexion and then developed a bilateral weakness for plantar and dorsiflexion of the foot at the age of 24 years. The symptoms were reported to progress slowly. On neurological examination at the age of 30, she revealed a mild weakness of hand muscles (MRC grade 4 for wrist extension), whereas distal strength in both lower limbs was severely impaired (MRC grade 2 for foot and MRC grade 1-2 for great toe dorsiflexion, MRC grade 3-4 for plantar flexion). Deep tendon reflexes were absent throughout. The lower limbs were distally affected by symmetric paraesthesia and reduced sensation for light touch and vibration. There was also a significant atrophy of the calf muscles. Nerve conduction studies indicated a severe sensory and motor polyneuropathy. CSF analysis showed mildly increased protein levels (44 mg/dl), while the cell count was normal. Serum electrophoresis and testing for ganglioside antibodies (anti-GM1, anti-GQ1b) as well as serological testing for autoimmune antibodies (e.g. ANA, ENA, ANCA) was all negative. The family history was unremarkable for any neurological disorders. After having excluded acquired causes, whole-exome sequencing was conducted as the next diagnostic step and showed two novel mutations in the two aminoacyl-tRNA synthetase genes HARS and MARS, each present in a heterozygous carrier state. The missense mutation in HARS (NM–002109.4): c.1488G>T, p.(Glu496Asp) was predicted to lead to an exchange of an evolutionary conserved amino acid. The detected MARS variant (NM–004990.3): c.181–183del, p.(Ser61del) causes a deletion of a highly conserved amino acid. Both mutations were absent from the in-house database including more than 9,000 exomes as well as from more than 120,000 alleles of the Exome Aggregation Consortium (ExAC)-Browser (http://exac.broadinstitute.org) and therefore considered to be pathogenic. Conclusion: To our knowledge, this is the first case suggesting a digenic inheritance pattern in Charcot-Marie-Tooth disease involving two new mutations in the functionally related genes MARS and HARS. However, the extent of pathogenicity of each individual mutation remains unclear and highlights the need for functional analyses beyond the clinical application of whole-exome sequencing.
PS2Group3-027 / #960A RANDOMIZED, BLINDED, LIFESTYLE INTERVENTION STUDY IMPROVES THE EXPIRATION:INSPIRATION RATIO IN DIABETIC NEUROPATHY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
James Russell1, Lindsay Zilliox2, Pranith Kumar1
1University of Maryland School of Medicine, Baltimore, US;2University of Maryland School of Medicine, Baltimore, MD, US
Background: Diabetic autonomic neuropathy is a common complication of type 2 diabetes mellitus (T2DM) that may affect up to 73% of patients. Currently, there is a need for therapies that will reduce progression or reverse diabetic autonomic neuropathy. The expiration:inspiration (EI) ratio is one of the most sensitive and reproducible measures of cardiac autonomic neuropathy (CAN). Methods: A blinded, randomized, intention to treat, parallel group intervention study was performed to examine the effect of a yearlong lifestyle intervention program in subjects with impaired glucose tolerance or T2DM and neuropathy. The lifestyle intervention program consisted of a dietary weight loss intervention that was tailored to the participants’ caloric expenditure and a graded, moderate intensity aerobic physical activity intervention that was tailored to the participants’ baseline physical activity (TDPA). The TDPA intervention group was compared to a standard care (SC) group receiving general dietary and exercise advice. Results: Data was obtained from 30 subjects in the TDPA and 31 on the SC group. Most participants had impaired glucose tolerance (89% -duration 6.91 + 1.31 months) and 11% had T2DM (duration 20.70 + 8.26 months). Both groups were matched at baseline for age, gender, BMI, and glycemic control. Gender distribution was 69% male and 31% female). There was an interval improvement in the EI ratio in 80% of the subjects in the TDPA, compared to SC group, over the duration of the intervention (P<0.05). The mean relative improvement in the EI ratio was greatest between 0 and 6 months in the TDPA group (0.15) compared to a decline in the SC group (-0.04). Conclusion: In this randomized, blinded, intention to treat study of a lifestyle intervention in diabetic neuropathy, there is improvement in CAN in the intervention group. This is a very important finding because of the strong association between impaired CAN and sudden death.
PS2Group3-028 / #668PREDICTORS OF EARLY RETIREMENT IN PATIENTS WITH CHRONIC INFLAMMATORY DEMYELINATING POLYRADICULONEUROPATHY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Bogdan Bjelica1, Ivo Bozovic1, Ivana Basta1, Aleksandra Kacar1, Ana Nikolic1, Aleksandra Dominovic-Kovacevic2, Zoran Vukojevic2, Vesna Martic3, Aleksandar Stojanov4, Gordana Djordjevic4, Milutin Petrovic5, Miroslav Stojanovic5, Stojan Peric1
1Neurology Clinic, Clinical Center of Serbia, School of Medicine, University of Belgrade, Belgrade, Serbia, Belgrade, RS;2Neurology Clinic, Clinical Center Banja Luka, Banja Luka, Republic of Srpska, Bosnia and Herzegovina, Banja Luka, BA;3Neurology Clinic, Military Medical Academy, Belgrade, Serbia, Belgrade, RS;4Neurology Clinic, Clinical Center Nis, Nis, Serbia, Nis, RS;5Neurology Clinic, Clinical Center Kragujevac, Kragujevac, Serbia, Kragujevac, RS
Background: It has been previously shown that being unemployed or retired is one of the independent predictors of worse quality of life (QoL) in chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). The aim of this study was to assess predictors of early retirement in CIDP subjects. Methods: We included 107 patients diagnosed with CIDP. Following measures were used: questionnaire on employment status, Medical Research Council (MRC) Sum Score, Inflammatory Neuropathy Cause and Treatment (INCAT) disability and sensory scores, Beck Depression Inventory (BDI), and Krupp’s Fatigue Severity Scale (FSS). Results: Fifteen percent of CIDP patients were employed, nine percent were unemployed due to CIDP, while 28% were retired early. Mean age when patients got retired due to CIDP was 50.0±8.2 years. Mean time from CIDP onset to retirement was 2.7±2.3 years. Older age at onset, lower education, and more severe motor/sensory deficit appeared as significant independent predictors of being retired due to CIDP. Retired patients compared to employed patients were in 12 times higher risk to suffer from depression (OR=12.2, 95%CI=1.41-100), and in eight-time higher risk of having fatigue (OR=8.2, 95%CI=1.89-35.82). Conclusion: Older patients with lower education, more severe weakness and sensory impairment were most likely to be retired due to CIDP. Early-retirement was highly associated with depression and fatigue. Therefore, maintaining employment should be important aim in treatment of CIDP patients.
PS2Group3-029 / #699NEUROPATHY AS AN ADVERSE EFFECT OF VASCULAR ENDOTHELIAL GROWTH FACTOR TYROSINE KINASE INHIBITORS, A META-ANALYSIS
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Bhaskar Roy1, Avash Das2, Abhishek Maiti3, Dhrubajyoti Bandyopadhyay4, Kumar Ashish3
1Neurology, Yale University, New Haven, CT, US;2Cardiovascular Medicine, Massachusetts General Hospital, Boston, MA, US;3MD Anderson Cancer Center, Houston, TX, US;4St Luke Roosevelt, New York, NY, US
Background: Targeted therapies like vascular endothelial growth factor tyrosine kinase inhibitors (VEGF-TKI) is the first-choice treatment in several types of cancers. Neuropathy as an adverse effect of VEGF-TKI is not yet established. In this pairwise and network meta-analysis, we explored neuropathy with VEGF-TKI use. Methods: Published data search up to March 2017 identified 26 randomized clinical trials (RCTs) reporting neuropathy in cancer patients treated with VEGF-TKI. The primary outcome was presence of neuropathy at the end of the trial. VEGF-TKI was compared either with combination chemotherapy or with placebo. Meta-analysis was performed using frequentist’s approach to estimate Relative Risk (RR) of individual treatment. Network meta-analysis based on random effects model estimating RR was performed. Results: Twenty-six RCTs including 11,971 cancer patients were included in this analysis. Six studies compared VEGF-TKI with placebo and others in combination. VEGF-TKI was associated with higher risk of neuropathic events (RR 1.12; 95% CI, 1.01-1.24, p<0.01). High grade (III and above) neuropathy (RR 1.23; 95% CI, 1.01-1.48) and sensory neuropathy (RR 1.23; 95% CI, 1.05-1.31) were noted more frequently with VEGF-TKI but did not reach statistical significance. When compared against placebo, VEGF-TKI was strongly associated with neuropathy (RR 1.88; 95% CI, 1.16-3.06, p= 0.01). Network meta-analysis did not show increased risk of neuropathy with any particular VEGF-TKI therapy compared to others. There was moderate heterogeneity among the included studies (I2=44%).
Conclusion: VEGF-TKI is possibly associated with increased risk of neuropathy.
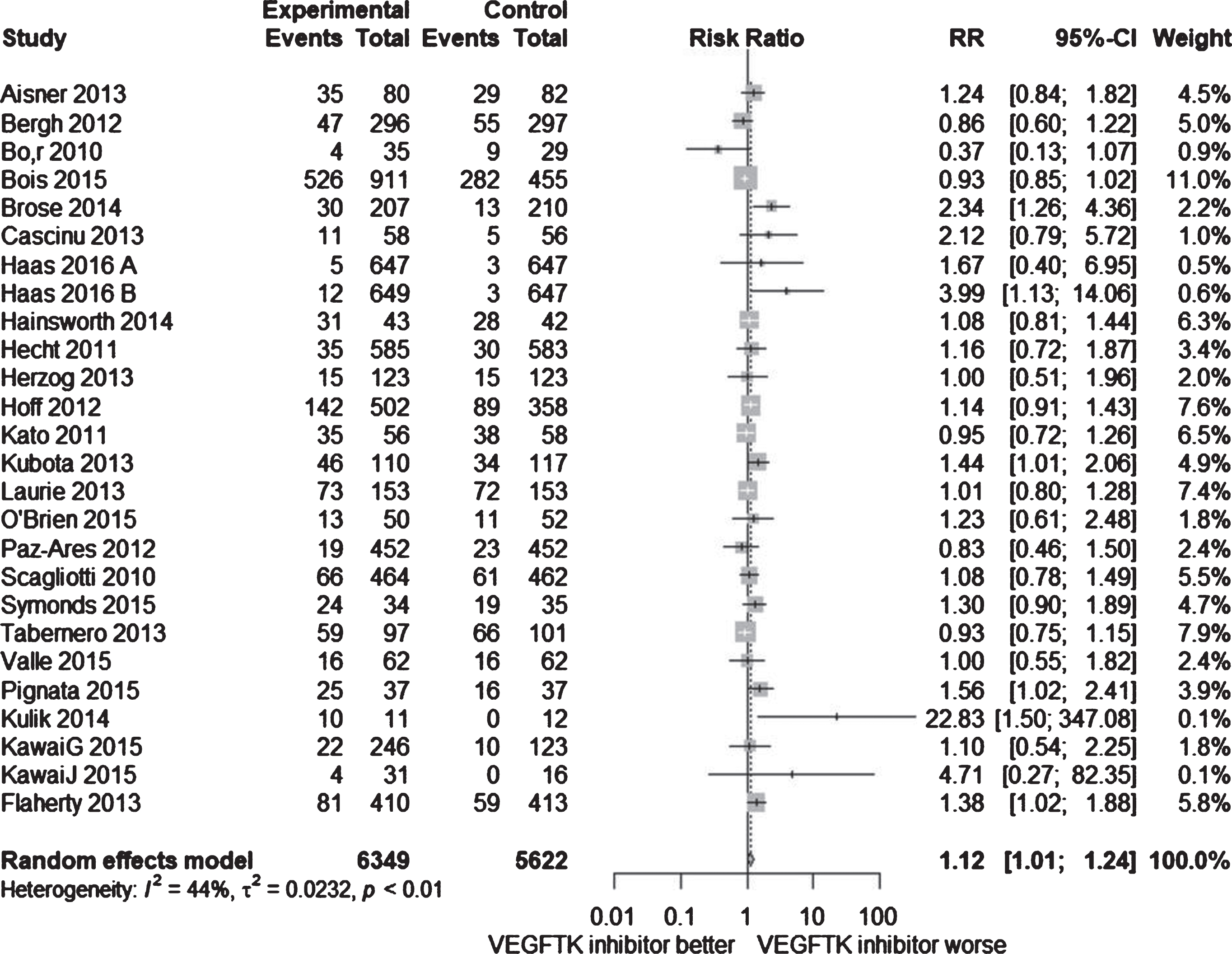
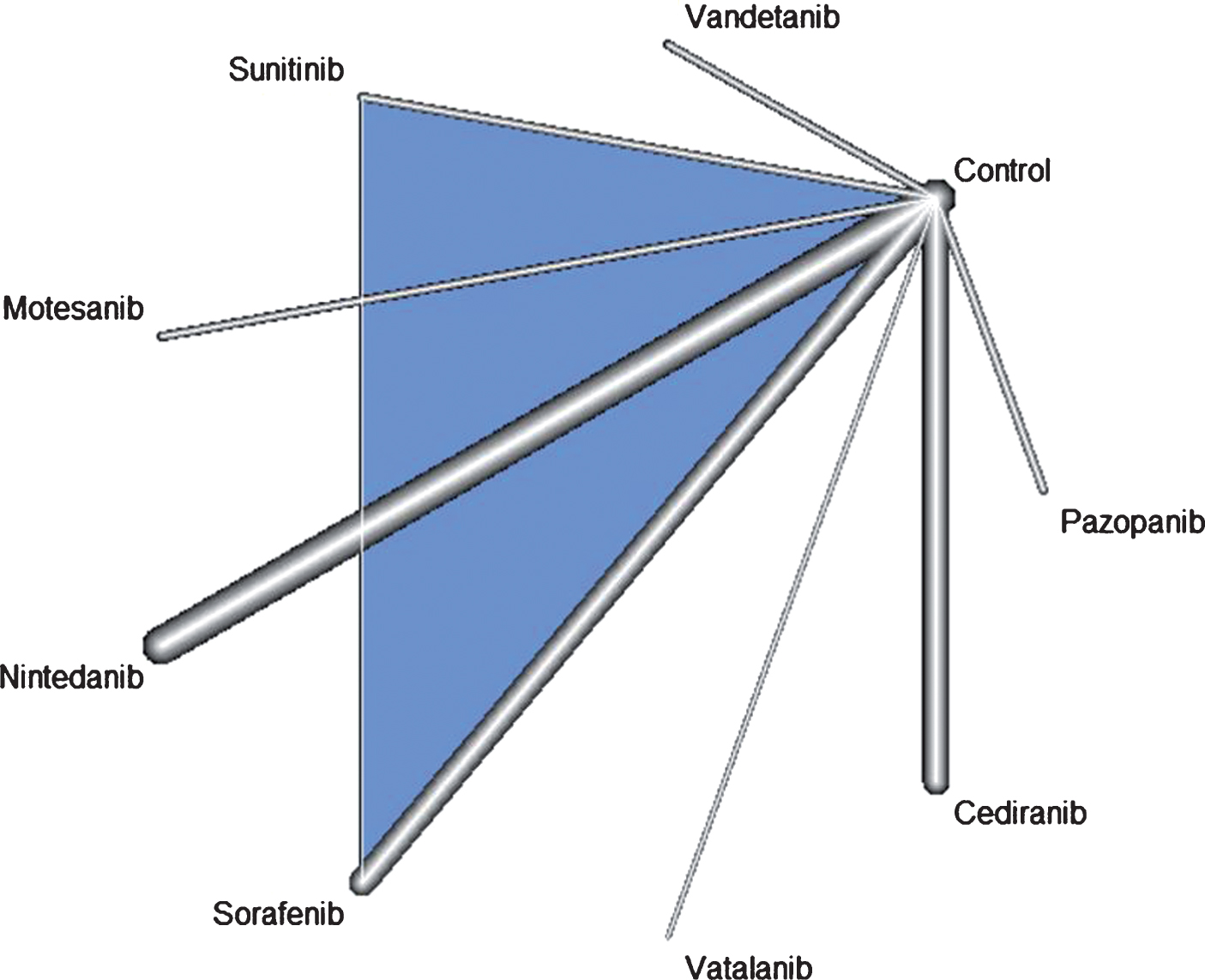
PS2Group3-030 / #727AXONAL FUNCTION PREDICTS RESPONSE TO SUBCUTANEOUS IMMUNOGLOBULIN IN CIDP: THE PATH STUDY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Vera Bril1, Hans-Peter Hartung2, Gen Sobue3, John-Philip Lawo4, Orell Mielke4, Billie L. Durn5, Ingemar Merkies6
1Department Of Medicine (neurology), University Health Network, University of Toronto, Toronto, CA;2Department Of Neurology, Heinrich-Heine University, Duesseldorf, DE;3Department Of Neurology, Nagoya University Graduate School of Medicine, Nagoya, JP;4CSL Behring, Marburg, DE;5CSL Behring, Fuquay Varina, NC, US;6Department Of Neurology, Maastricht University Medical Center, Maastricht, NL
Background: Chronic Inflammatory Demyelinating Polyneuropathy (CIDP) is an immune mediated disease starting with functional impairment and demyelination; in later stages, axonal degeneration may occur. Methods: PATH was a randomised, double-blind study investigating 0.2 (low) and 0.4 g/kg (high) weekly doses of maintenance SCIG IgPro20 (Hizentra, CSL Behring) versus placebo (N=172). After Ig dependency testing, and IVIG restabilisation, patients were randomised to SCIG or placebo for 25 weeks or until early termination. Nerve conduction studies (NCS) were performed before study drug administration and at end of study visit. Exploratory comparisons of relapse rates (defined as a >=1 point increase by adjusted Inflammatory Neuropathy Cause and Treatment score) were undertaken on patients with assumed non-axonal damage (amplitude > 1) versus assumed axonal damage (amplitude <=1) at the peroneal ankle stimulation site. Results: Patients with assumed non-axonal damage who received placebo had a 73% relapse rate versus 39% on low-dose and 19% on high-dose SCIG. Patients with assumed axonal damage had relapse rates of 25%, 30% and 19% for placebo, low-dose and high-dose SCIG, respectively (Table 1). Conclusion: CIDP patients with assumed non-axonal damage had a high relapse rate when switched from IVIG to placebo that was significantly lower in patients switched to SCIG therapy. Relapse rates were lower in assumed axonal damage patients and were not influenced by SCIG. These findings could help in redesigning future trials including maintenance regimens based on NCS categorisation of patients.
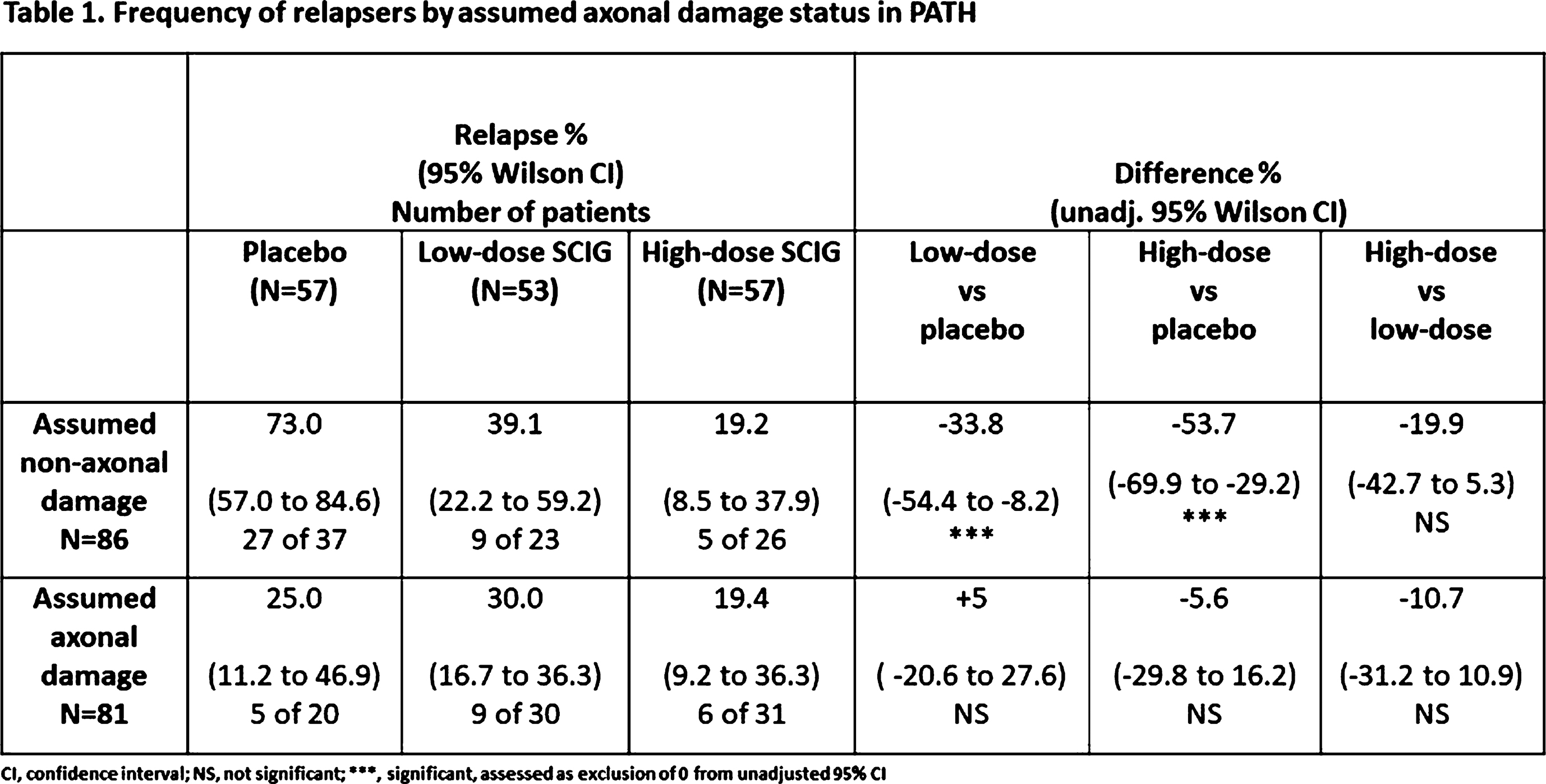
PS2Group3-031 / #722ELECTROPHYSIOLOGICAL TESTING IN PATIENTS WITH CIDP TREATED WITH SUBCUTANEOUS IMMUNOGLOBULIN: THE PATH STUDY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Vera Bril1, Ivo Van Schaik2, Nan Van Geloven3, Hans-Peter Hartung4, Richard A. Lewis5, Gen Sobue6, John-Philip Lawo7, Orell Mielke7, Billie L. Durn8, Ingemar Merkies9
1Department Of Medicine (neurology), University Health Network, University of Toronto, Toronto, CA;2Department Of Neurology, Academic Medical Center, University of Amsterdam, Amsterdam, NL;3Department Of Medical Statistics And Bioinformatics, Leiden University Medical Center, Leiden, NL;4Department Of Neurology, Heinrich-Heine University, Duesseldorf, DE;5Department Of Neurology, Cedars-Sinai Medical Center, Los Angeles, US;6Department Of Neurology, Nagoya University Graduate School of Medicine, Nagoya, JP;7CSL Behring, Marburg, DE;8CSL Behring, Fuquay Varina, NC, US;9Department Of Neurology, Maastricht University Medical Center, Maastricht, NL
Background: Nerve conduction studies (NCS) form an essential part of the diagnostic criteria for chronic inflammatory demyelinating polyneuropathy (CIDP) and are objective measures of peripheral nerve function independent of confounders that may interfere with clinical scores. The objective of this analysis was to determine whether NCS remain stable in patients with CIDP during study drug administration with either low or high dose subcutaneous immunoglobulin (SCIG) or placebo. Methods: This was a randomised, double-blind trial investigating 0.2 (low) and 0.4 g/kg (high) weekly doses of maintenance SCIG IgPro20 (CSL Behring) versus placebo in 172 subjects. After testing for dependency on Ig therapy, patients were randomised to SCIG or placebo, and NCS were performed at the start and end of the study drug administration period at 25 weeks or early termination. Electrophysiological data from each patient was averaged to give average proximal latency, conduction velocity, conduction block (30, 40 and 50%), compound muscle action potential amplitude, and we compared the mean changes in these averaged parameters from baseline to completion visit. Results: The majority of NCS parameters did not change; however, averaged proximal latency increased in the placebo group (+1.1 ms) but remained stable in both IgPro20 groups (low dose: +0.1 ms; high dose: -0.1 ms). The averaged motor nerve conduction velocity (MNCV) decreased in the placebo group (-1.6 m/s) but increased in both IgPro20 groups (low dose: +0.2 m/s; high dose: +1.0 m/s). Conclusion: Most NCS parameters did not change during the SCIG treatment phase of the PATH study aside from those reflecting speed of conduction (averaged proximal latency and MNCV) which went in a positive direction in SCIG treated patients compared with placebo patients.
PS2Group3-032 / #921CHRONIC RELAPSING INFLAMMATORY OPTIC NEUROPATHY (CRION): A MANIFESTATION OF MYELIN OLIGODENDROCYTE GLYCOPROTEIN ANTIBODIES
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Haeng-Jin Lee1, Boram Kim2, Patrick Waters3, Sarosh Irani3, Mark Woodhall3, So Hyun Ahn2, Jung-Joon Sung2, Seong-Joon Kim1, Sung Min Kim2
1Ophthalmology, Seoul National University Hospital, Seoul, KR;2Neurology, Seoul National University Hospital, Seoul, KR;3Nuffield Department Of Clinical Neurosciences, John Radcliffe Hospital, Oxford, GB
Background: The key clinical features of chronic relapsing inflammatory optic neuropathy (CRION) include relapsing inflammatory optic neuritis (ON) and steroid-dependency, both of which also have been reported among patients with myelin oligodendrocyte glycoprotein antibodies (MOG-Abs). We aimed to investigate the relevance of the presence of serum MOG-Ab with the current diagnostic criteria for CRION among patients with idiopathic inflammatory optic neuritis (iON). Methods: Retrospective reviews of the prospectively collated database between 2011 and 2017 were performed in a tertiary referral multiple sclerosis / neuromyelitis optica center. Sixty-four patients with iON, who did not meet the diagnostic critera for multiple sclerosis, neuromyelitis optica (NMO) spectrum disorder with/without NMO-IgG, or acute disseminated encephalomyelitis, and who had no symptomatic central nervous system (CNS) lesion other than the optic nerve were included from the cohort of 615 patients with inflammatory demyelinating diseases of the CNS. Fulfillment for the current diagnostic criteria of CRION, assay result for the serum IgG1 MOG-Ab, and the characteristics of CRION patients compared to those of non-CRION with MOG-Ab. Results: Twelve patients with iON fulfilled the current diagnostic criteria for CRION all of whom tested positive for MOG-Ab. Among the other 52 patients, 14 had relapsing disease courses and 38 had monophasic course, of which group the MOG-Ab positivity were 0% and 29%, respectively. Though the CRION patients with MOG-Ab had more relapsing disease courses and poorer OCT outcomes at follow up than non-CRION patients with MOG-Ab, they did not differ in terms of age at onset, sex, duration of the steroid treatment after initial attack. The CRION patients with MOG-Ab experienced their first steroid-dependent worsening/relapse in 2.2 (ranged from 0.43 – 6.97) months from their disease onset. Conclusion: The CRION, according to the current diagnostic criteria, appears to be mostly a manifestation of the relapsing optic neuritis associated with MOG-Ab. Among iON patients with MOG-Ab, absence of the steroid-dependent attack in an early stage of the disease can be associated with a long term non-relapsing disease courses and better outcomes.
PS2Group3-033 / #304PREVALENCE AND RISK FACTORS OF CARPAL TUNNEL SYNDROME IN OYSTER-SHUCKERS
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Kee Hong Park1, Rock Bum Kim2, Eun Bin Cho3, Ki-Jong Park3
1Neurology, Gyeongsang National University Hospital, Jinju, KR;2Regional Cardiocerebrovascular Center, Gyeongsang National University Hospital, Jinju, KR;3Neurology, Gyeongsang National University Changwon Hospital, Changwon, KR
Background: To investigate the prevalence of carpal tunnel syndrome (CTS) in oyster-shuckers and to reveal the risk factors of CTS. Methods: One hundred and forty-seven oyster-shuckers were recruited. All the subjects had worked for more than 10 years. CTS was diagnosed based on nerve conduction study. Subjects with abnormal ulnar nerve study were excluded to rule out polyneuropathy (n = 17). Degree of CTS was graded as normal, mild (decreased sensory nerve conduction velocity with normal distal motor latency), moderate (decreased sensory nerve conduction velocity with prolonged distal motor latency), and severe (absent sensory nerve action potential with prolonged distal motor latency). Risk factors analyzed included age, sex, BMI, medical history, work time, exercise time, and laboratory data. Results: CTS was seen in 66.9% (n = 87) of the whole population (n = 130). In a multivariate logistic regression analysis, risk of CTS was increased with longer work time (odds ratio, 1.43; 95% confidence interval, 1.18–1.73) and higher BMI (odds ratio, 1.27; 95% confidence interval, 1.05–1.53). Degree of CTS showed statistically significance only with work time (p = 0.005). Conclusion: CTS is prevalent in oyster-shuckers. In order to reduce the risk of CTS, it may be necessary to adjust the working time and reduce the BMI.
PS2Group3-034 / #469NEXT GENERATION SEQUENCING TECHNOLOGIES IN THE GENETIC DIAGNOSIS OF EARLY ONSET HEREDITARY SPASTIC PARAPLEGIAS
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Laura Carrera-García1, Daniel Natera De Benito1, Debora Itzep1, Anna Lia Frongia1, Andrea Sariego1, Delia Yubero2, Elena Maqueda1, Loreto Martorell3, Carlos Ortez1, Jaume Colomer3, Giovanni Stevanin4, Andres Nascimento1
1Neuromuscular Disorders Unit, Hospital Sant Joan de Déu, Barcelona, ES;2Department Of Genetics, Hospital Sant Joan de Déu, Barcelona, ES;3Hospital Sant Joan de Déu, Barcelona, ES;4Institut National de la Santé et de la Recherche Medicale (INSERM), Paris, FR
Background: Hereditary spastic paraplegias (HSP) are a genetically heterogeneous group of diseases that are characterized by progressive symmetric spasticity of lower extremities. Mutations in SPAST represent the most frequent molecular etiology. Today, more than 60 genes related to hereditary spastic paraplegias are recognized. The aim of our work is to analyse the utility of Next-generation-sequencing (NGS) diagnostics in patients with HSP and describe the clinical and molecular genetic findings associated with specific genes. Furthermore, we also describe phenotypes and distinguish clinical signs associated with the various HSP-related genes, which might help to guide diagnosis. Methods: A cohort of children followed-up in our hospital with clinical diagnosis of HSP was recruited. Metabolic or structural origin was discarded in all of them. A NGS-based gene panel of 74 genes involved in HSP was performed. Demographic and phenotype data were collected from medical records. The study complies with the ethical guidelines of the institutions involved. Results: Sixty-seven unrelated index cases were analysed by the NGS-HSP panel. In some of them, several genetic tests, including SPAST, ALT1, MFN2, MPZ, GDAP1 sequencing, had previously been performed with negative results. The NGS-HSP panel diagnostic yield was 31.3%, since 21 out of 67 patients reached a molecular genetic diagnosis. Among index cases with a confirmed genetic diagnosis, 7 presented with pure and 14 with complicated HSP. The mean age at first symptoms was 26 months; a male/female ratio was 15/6. Mutations were found in 12 different genes. The genes most frequently involved were KIF1A (5/21 patients), SPAST (4/21), and ALT1 (3/21). Additionally, 9 cases were detected in family members. Conclusion: The use of next generation sequencing technologies has increased the number of genes known and has expanded both the phenotype and genotype of HSP. Molecular diagnosis was obtained in a third of patients using a NGS-HSP panel. Knowledge of the genes involved in HSP is now opening possibilities to build the functional pathways related with these disorders and could hopefully lead to a potential treatment for HSP patients.
PS2Group3-035 / #710A CASE OF SEROPOSITIVE NEUROMYELITIS OPTICA IN A PATIENT WITH CO-EXISTING MYASTHENIA GRAVIS AND SYSTEMIC LUPUS ERYTHEMATOSUS
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Il-Han Yoo1, So-Hyun Park1, Suk-Won Ahn2
1Chung-Ang University Hospital, Heukseok-ro, Dongjak-gu, Seoul, KR;2Neurology, ChungAng University Hospital, Seoul, KR
Background: Neuromyelitis optica (NMO) is an immune related disease involving central nervous system (CNS) which has been previously reported in association with other autoimmune diseases. Herein, we report an unusual case of a 51-year-old female with NMO, myasthenia gravis (MG) and systemic lupus erythematosus (SLE) who initially presented with optic neuritis, transverse myelitis and abnormality in the brain magnetic resonance image (MRI).
Methods: A 51-year-old female presented with visual disturbance, gait disturbance, slurred speech and dysesthesia at lower limbs which has developed over several times. Results: Brain MRI revealed high signal intensity lesion at bilateral hemispheres, pons and both optic nerve sheath with focal gadolinium enhancement. The serum aquaporin-4 (APQ4) receptor antibody level was positive; therefore she was initially diagnosed as NMO by the NMO diagnostic criteria and has been treated with high-dose steroid and immunosuppressants. Furthermore, the immune-serologic studies yielded positive titers for acetylcholine receptor (AchR) antibodies, antinuclear antibodies (ANA), anti-double stranded DNA antibodies (anti-dsDNA), lupus anticoagulant and anti-cardiolipin antibodies. And repetitive nerve stimulation test (RNST) revealed typical decremented CMAP amplitude by the low-frequency nerve stimulation. Collectively, in addition to NMO, she was also diagnosed as seropositive MG and SLE on the basis of diagnostic criteria. Conclusion: We consider that this patient had a seropositive neuromyelitis optica (NMO) spectrum disorder which was associated with systemic autoimmune disease including MG and SLE, therefore the possibility of NMO should be considered in patients with autoimmune diseases who have similar clinical manifestations, and anti-AQP4 antibody should be assessed.
PS2Group3-036 / #275NERVE ULTRASOUND ASSESSMENT IN A NOVEL MUTATION C.379DELG (PALA127LEUFS*52) IN DRP2 GENE
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Stephan Wenninger1, Ralf Jankovits2, Beate Schlotter-Weigel1
1Dept. Of Neurology, Ludwig-maximilians-university, Friedrich-Baur-Institute, Munich, DE;2Neurozentrum Prien, Prien, DE
Background: Mutations in the gene “Dystrophin-related protein 2” (DRP2) cause an X-linked intermediate Charcot Marie Tooth (CMT) neuropathy. The protein contributes to the periaxin-DRP2-dystroglycan (PDG) complex that leads to the creation of Cajal bands. Disruption of the PDG-complex is followed by hypermyelination and destabilization of the Schwann cell-axon. This report is about a 67-year old man who noticed first neuropathic symptoms at age of 65. The symptoms included numbness, tingling and burning in the feet, a slight gait disturbance and recurrent muscle cramps in distal legs. There were no similar affected individuals in his family. A sensorimotor, predominantly demyelinating polyneuropathy in lower limbs was diagnosed previously and a slight elevation of proteins in cerebrospinal fluid was found. Suspecting CIDP, the patient received immunotherapy with oral corticosteroids and one course of IVIg 0,2mg/kg. Both therapies did not improve symptoms convincingly. Methods: A clinical and electrophysiological examination, nerve ultrasound, along with immunological tests for antibodies and multigene panel diagnostics for hereditary neuropathies were performed. Results: Clinical examination in our clinic revealed a predominant symmetric numbness in lower limbs and a slight sensory ataxia. Walking on tiptoes was normal, but walking on heels was mildly impaired, combined with decent distal muscular atrophy. Distal muscle tendon reflexes on lower limbs were absent and tendon reflexes of proximal muscles of lower limbs as well as tendon reflexes on upper limbs were normal.Sensory deficits of all qualities were present in a stocking distribution distal symmetric. CMTNSv2-Score was 6, indicating a mild clinical affection. Electrophysiological examination revealed an intermediate, mainly axonal sensorimotor neuropathy of predominantly lower limbs with slowed NCVs and severely reduced amplitudes. There was no conduction block or temporal dispersion. For both sural nerves there were no responses. EMG revealed a chronic neurogenic pattern and a reduction in recruitment in distal muscles and slight neurogenic changes in lateral vastus and first dorsal interosseous muscles. Nerve ultrasound showed generalized thickened peripheral nerves in upper and lower limbs as well as plexus brachialis, measured as cross-sectional area (CSA) and diameter. Concerning the combination of severely affected peripheral nerves with thickening of peripheral and proximal nerves including plexus brachialis in ultrasound, remarkably good clinical compensation and barely notably improvement after immunotherapy, we performed a genetic testing for demyelinating polyneuropathies. A novel hemizygous stop mutation c.379delG (pAla127Leufs*52) in the DRP2 gene was found. This mutation was not described in the literature so far. Conclusion: Combined examinations in electrophysiology and high-resolution nerve ultrasound can help to identify and characterize hereditary neuropathies in a cost-effective manner. Characteristically, a higher peripheral nerve CSA can be found in patients with CMT 1A, but there are also reports of mild to moderate CSA elevations in CMT 2 and X-linked CMT patients. Based on the patient’s data and presentation, we conclude that this patient has a DRP2-related intermediate CMT with late onset and mild clinical affection. This case contributes to the knowledge of DRP2-associated neuropathies with new insights in nerve ultrasound in intermediate CMT.
PS2Group3-037 / #370PHRENIC NERVE DEMYELINATION CAUSING ACUTE RESPIRATORY FAILURE IN MADSAM
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Chantal Baldwin1, Karl Ng2
1Neurology, Royal North Shore Hospital, Sydney, NSW, AU;2Neurology, Royal North Shore Hospital, ST LEONARDS, NSW, AU
Background: A 72-year-old man with an eight year history of Multifocal acquired demyelinating sensory and motor neuropathy (MADSAM) presented with acute worsening of left sided weakness and new right upper limb weakness in an asymmetrical pattern. He was managed with an induction dose of intravenous immunoglobulin (IVIG) 2g/kg over five days. Initial nerve conduction studies demonstrated a generalised demyelinating neuropathy affecting the upper limbs to a greater degree than the lower limbs, of note motor conduction block was seen in the right median forearm segment. Methods: He developed acute type 2 respiratory failure requiring intubation and ventilation. IVIG was continued with marked improvement in respiratory parameters and limb weakness. Repeat nerve conduction studies demonstrated resolution of the previously seen motor conduction block and phrenic nerve studies showed prolonged latencies: right 16.3ms and left 14.8ms with amplitudes of 0.7 and 0.9mV respectively (normal latency 5.53-7.72ms and amplitude 0.66-1.46mV). Results: Respiratory involvement is considered atypical in CIDP and rarely presents acutely. A review of 67 consecutive CIDP patients by Gorson et al. suggests this perception may be inaccurate. Nine percent of their cohort developed respiratory failure at some point during the course of their illness requiring ventilatory support. Stojkovic et al subsequently reported four cases of respiratory failure secondary to phrenic nerve palsy in CIDP. Phrenic nerve involvement was confirmed on neurophysiological testing of the phrenic nerve which revealed reduced CMAP amplitudes or prolonged latencies. Conclusion: Macia et al. performed a prospective study evaluating the phrenic nerve involvement in 14 patients with multifocal motor neuropathy (MMN) and 14 patients with CIDP. Electrophysiological phrenic nerve involvement was common in CIDP and rare in MMN. Specifically relating to MADSAM Verschueren et al reported in their retrospective cohort of 13 MADSAM patients, two presenting with a relapsing remiting course developed respiratory failure requiring intensive care unit admission.
PS2Group3-038 / #545BACLOFEN, NALTREXONE AND SORBITOL ALL CONTRIBUTE TO THE EFFICACY OF PXT3003 IN CMT1A RATS
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Thomas Prukop1, Stephanie Wernick1, David Ewers1, Anthony Brureau2, Julien Laffaire3, Nathalie Cholet2, Klaus A. Nave1, Rodolphe Hajj2, Daniel Cohen4, Michael W. Sereda1
1Neurogenetics, Max-Planck-Institute of Experimental Medicine, Göttingen, DE;2Experimental Biology And Pharmacology, Pharnext, Issy Les Moulineaux, FR;3Biostatistics, Pharnext, Issy Les Moulineaux, FR;4Pharnext, Issy Les Moulineaux, FR
Background: The most common type of Charcot-Marie-Tooth disease is caused by a duplication of PMP22, leading to dysmyelination, axonal loss and progressive muscle weakness (CMT1A). Currently, no approved treatment for CMT1A patients is available. The novel polytherapeutic drug PXT3003, a low-dose combination of Baclofen, Naltrexone and Sorbitol, was previously shown to correct Pmp22 overexpression, improve dysmyelination and axonal loss in Pmp22 transgenic rats, a known animal model of CMT1A. Here, we performed a long-term treatment study in adult CMT1A rats with PXT3003 to test the superior effect of PXT3003 compared to its three binary combinations of Baclofen/Naltrexone, Baclofen/Sorbitol and Naltrexone/Sorbitol. Additionally, CMT1A and wildtype rats were included as placebo controls. Methods: Treatment was performed in adult rats starting at postnatal week 6, after the manifestation of symptoms commonly observed in CMT1A rats, and was continued until week 18. Behavioral analyses were performed at the age of 6, 10, 14 and 18 weeks (grip strength, bar test and inclined plane test). Furthermore, electrophysiological measurements and a hot plate test were carried out at the end of the study. Post-mortem analyses included mRNA, protein and histological analyses of peripheral nerves. Results: No significant effect on motor endpoints was observed with any of the binary combinations of Baclofen/Naltrexone, Baclofen/Sorbitol or Naltrexone/Sorbitol. In contrast, PXT3003 proved its superior efficacy to the binary combinations on the behavioral level in CMT1A rats. Conclusion: This data suggests a synergistic effect of the three drug components contained in PXT3003 for CMT1A in vivo and clearly demonstrates the contribution of baclofen, naltrexone and sorbitol to the overall positive efficacy of PXT3003. These findings highlight the value of pleiotropic combinational repurposing of drugs at low doses for rapid development of highly safe combinations.
PS2Group3-039 / #638IDEAL TIME FOR A REPEAT CONDUCTION TO SUBTYPE VARIANTS IN GUILLAIN BARRE SYNDROME
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
A K. Meena1, Ramesh Chepuru2, Sailaja Sarva2, Sireeesha Yareeda2, Niharika Mathukomali2
1Neurology, Nizam’s Institute of Medical Sciences, HYDERABAD, IN;2Neurology, Nizam’s Institute of Medical Sciences, HYDERABAD, IN
Background: Guillain Barre Syndrome (GBS) is the most common severe form of peripheral neuropathy with rapid onset and progression. There are several variants of GBS. Many studies have suggested that serial electrophysiology is essential for accurate diagnosis of GBS. We aim to study the usefulness of serial nerve conduction studies (NCS) and at what time a repeat conduction is needed to subtype GBS. Methods: We reviewed the records of patients discharged with the diagnosis of GBS from our institute retrospectively from Jan 2011 to Dec 2016. We included patients with at least 2 electrophysiological studies. At least 4 motor nerve conduction and 3 sensory nerve conduction studies were done. We applied 3 sets of criteria at various time period in disease course: Hadden et al (1988), Rajabally et al (2014) and Uncini et al (2017). Approval was taken from institutional ethical board. Statistical analysis was done using Fisher’s exact tests, McNemar’s test, Excel 2013. Results: Most of the patients presented to our hospital at the end of 1st (50.56%) and 2nd week (34.18%) of disease onset. Out of 354 patients 136 had complete data with repeat NCSs. Hadden’s and Uncini’s criteria were almost equally sensitive (37% vs 35%) in diagnosing AIDP as compared to Rajabally’s criteria (26%) in the 1st week of the illness amongst 77 patients. Equivocal findings not fulfilling any criteria was seen more frequently with Rajabally’s criteria (38% vs 30%). The 3 criteria were equally sensitive to diagnose axonal GBS in first week of illness in all 77 patients. Repeat conduction study showed was done in 1st week of disease course. Using Hadden’s and Uncini criteria 6.5% (5 patients out of 77 patients) converted from AIDP to axonal at 2nd week of illness and equivocal to axonal 9% (7 patients out of 77).When Rajabally criteria was used the conversion of equivocal to axonal was 13% (10 patients out of 77). Shifting rate was lower where initial NCS was done at 3rd week and later (2% & 8%). Forty three out of 53 patients who had repeat conduction at 5 weeks did not show any changes in NCS as compared to that at 3 weeks. Eleven percent(15 patients) of patients had equivocal findings at the end of 4 weeks not fulfilling any criteria. Conclusion: Single study is sufficient to subtype axonal variant of GBS if 1st study shows axonal subtype irrespective of the time course. Repeat conduction study should be done to further subtype patients in whom initial study showed AIDP/ Equivocal/not fulfilling as 5-10% of patients with initial conduction study showing AIDP changed to axonal on repeated conduction. As there is insignificant change in electrophysiological findings at 3rd week and beyond to subtype GBS probable time to repeat conduction study is 3rd week in disease course. A significant proportion of may not fulfill any of the criteria even after 4 weeks wherein only clinical criteria for GBS , evidence of peripheral neuropathy and exclusion of other causes is important to diagnose GBS.
PS2Group3-040 / #662COST OF ILLNESS IN CHARCOT-MARIE-TOOTH NEUROPATHY: RESULTS FROM GERMANY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Simone Thiele1, Elisabeth Schorling2, Peter Reilich1, Sabine Krause1, Klaus Nagels2, Maggie C. Walter1
1Friedrich-baur Institute, Departement Of Neurology, Ludwigs-Maximilians University of Munich, Munich, DE;2Institute Of Management In Medicine And Health Care Sciences, University of Bayreuth, Bayreuth, DE
Background: Charcot Marie-Tooth (CMT) neuropathies are the most common inherited neurological diseases with a prevalence of 1 in 2500 individuals. In CMT, the quality of life (QoL) is reduced compared to the general population. No causative treatment is currently available but new therapeutic strategies are being investigated and various innovative clinical trials are expected within the next years. Objective Increasing demands on efficacy and safety of newly developed drugs are likely to increase costs for the health care system. However, innovative therapies may have the potential to reduce the cost of illness (COI) by improving the clinical phenotype. Our study aimed to estimate the total annual cost of illness in CMT. Methods: The study enrolled 397 patients with genetically confirmed diagnosis of CMT; subtypes of the disease were differentiated. A micro-cost method was used to determine the COI. Direct and indirect costs were assessed to determine the economic burden of CMT. Results: The most frequent genetic diagnoses in our cohort were CMT1A (PMP22 duplication; n=246), HNPP (PMP22 deletion; n=36), CMT1X (GJB1 mutation; n=33), CMT1B (MPZ mutation; n=24), CMT2A (MFN2 mutation, n=16) and CMT4C (SH3TC2 mutation; n=12), accounting for 92,4% of the genetically confirmed mutations. The remaining subtypes accounted for 1% each. Patients were classified as slightly, moderately or severely affected by the CMT neuropathy score (CMTNS of <10, 11–20 or >20, respectively). The economic burden of CMT patients was assessed and analyzed with regard to the actual CMT standard of care. Conclusion: Our results significantly contribute to a more comprehensive insight into the current burden of CMT and quality of life status as related to CMT health services in Germany. In the light of innovative therapeutic interventions, our results suggest a notable potential for a reduction in overall COI and improvement of HRQOL if the therapeutic intervention leads to a less severe course of the disease. Our results could help accelerating the implementation of innovative CMT therapies in standard CMT clinical care.
PS2Group3-041 / #697CLINICAL HETEROGENEITY OF ANTI-GM2-GANGLIOSIDE ANTIBODY SYNDROME
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Jong Seok Bae1, Sun Min Yoon2, Soon Kyung Shim2
1Neurology, Kangdong Sacred Heart Hospital, Seoul, KR;2Neurology, Kangdong Sacred Heart Hospital, Seoul, KR
Background: Antiganglioside antibody is known to play a pathogenic role in Guillain-Barré syndrome (GBS). Either an immunoglobulin (Ig)G- or IgM-type anti-GM2 antibody is detected in rare cases in GBS patients. However, the specific pathogenic role of these antibodies in GBS has not been reported previously. This study aimed to define and characterize the clinical spectrum of GBS with anti-GM2 positivity. Methods: We reviewed the database of the DAUNIT (Dong-A University Neuroimmunology Team), which has collected sera of GBS and its variants from more than 40 general and university-based hospitals in Korea. Detailed information about the involved patients was often obtained and corrected by the charge doctor applying additional questionnaires. Results: Four patients with acute monophasic peripheral neuropathy or cranial neuropathy with isolated IgM-type anti-GM2-antibody positivity were recruited. In addition, IgG-type anti-GM2 antibody was solely detected in the sera of another four patients. The IgM-positive group comprised heterogeneous syndromes: two cases of acute motor axonal neuropathy, one of acute inflammatory demyelinating polyneuropathy, and one of isolated facial diplegia. In contrast, all of the cases enrolled in the IgG-positive group manifested with dizziness with or without oculomotor palsy due to cranial neuropathy syndrome. Conclusion: This study identified that anti-GM2 antibody can be found in various subtypes of GBS in rare cases. Compared to the clinical heterogeneity of the IgM-positive group, the IgG-positive group can be characterized by cranial-dominant GBS presenting mainly with oculomotor and vestibular dysfunctions.
PS2Group3-042 / #798LATE-ONSET AXONAL CHARCOT-MARIE-TOOTH ASSOCIATED WITH AUTOSOMAL RECESSIVE MME MUTATIONS
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Tania García-Sobrino1, Maria Pacifica Vidal-Lijo2, Elena Pintos3, Vicenzo Lupo4, Carmen Espinós5, Teresa Sevilla6, Julio Pardo7
1Neurology, Hospital Clínico Santiago de Compostela, Santiago de Compostela, ES;2Neurophysiology, Hospital Clínico Santiago de Compostela, Santiago de Compostela, ES;3Pathology, Hospital Clínico Santiago de Compostela, Santiago de Compostela, ES;4Centro de Investigación Príncipe Felipe. INCLIVA & IIS La Fe Rare Diseases Joint Units, Valencia, ES;5Unit Of Genetics And Genomics Of Neuromuscular And Neurodegenerative Disorders And Service Of Genomics And Traslational Genetics, Centro de Investigación Príncipe Felipe (CIPF), and Joint Unit for research on Rare Diseases, CIPF-IISLa Fe, Valencia, ES;6Neurology, 4Neurology Department. Hospital Universitari i Politecnic La Fe. Neuromuscular Research Unit, Instituto de Investigación Sanitaria La Fe. Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER). Valencia. Spain, Valencia, ES;7Neurology, Hospital Clínico Santiago de Compostela, Santiago de Compostela, ES
Background: Charcot-Marie-Tooth (CMT) disease is the most common inherited peripheral neuropathy. The disease onset usually occurs in the first two decades of life and shows a slow progression. Clinically CMT phenotype is characterized by distal weakness, wasting and sensory loss with foot deformities. Axonal CMT (CMT2) with preserved nerve conduction velocities (MMMNCV > 38 m/s) are a group of clinically and genetically heterogeneous subtypes. A high rate of CMT2 remain without genetic diagnosis. In 2016 MME mutations were identified in patients with CMT2 linked to an autosomal dominant or autosomal recessive inheritance pattern. We report a family with an autosomal recessive late-onset CMT2 associated with MME mutations. Methods: A 57-year-old man without medical or family history and no consanguinity. He developed a progressive gait disturbance, cramps and distal lower limb weakness with foot drop at the age of 53. Neurological examination revealed pes cavus, gynecomastia, fasciculations and wasting in lower limbs, slight left hand weakness, proximal lower limb weakness (4/5) and distal lower limb weakness (3/5), areflexia and steppage gait. Clinical progression was evident in the first year of evolution and patient associated distal lower limb sensory loss. Results: Laboratory investigations showed a high serum CK (600-1500UI/L). Spinal cord MRI was normal. Nerve conduction study are consistent with an axonal motor neuropathy. Needle electromyography showed acute denervation and fasciculations in lower limbs with reduced recruitment pattern in upper and lower limb muscles. After six month a nerve conduction study revealed an axonal motor and sensory neuropathy. Lower limb muscle MRI showed a bilateral fatty infiltration of the gastrocnemius, soleus and in the anterolateral group. A myopathic and neuropathic pattern was evident in the muscle biopsy. Molecular diagnosis revealed that our patient was a homozygous for the c.466delC mutation in the MME gene. Neurological examination and nerve conduction studies were normal in his relatives. Molecular study revealed that his parents and two sisters were heterozygous for the same mutation in MME gene. Conclusion: CMT2 are a clinically and genetically heterogeneous group of neuropathies. The screening of MME mutations should be performed in patients with late-onset neuropathies with or without a family history in wich acquired causes are ruled out and the phenotype is compatible with CMT2.
PS2Group3-043 / #567POPULATION PHARMACOKINETICS (PK) OF INVESTIGATIONAL PATISIRAN IN HEALTHY VOLUNTEERS AND IN PATIENTS
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Varun Goel1, Claudia Jomphe2, Nathalie Gosselin2, Husain Attarwala3, Xiaoping Zhang4, Jean-Francois Marier2, Gabriel Robbie3
1Alnylam Pharmaceuticals, Cambridge, MA, US;2Cetera, Montreal, QC, CA;3Alnylam Pharmaceuticals, Cambridge, AL, US;4Clinical Pharmacology And Pharmacometrics, Alnylam Pharmaceuticals Inc, Cambridge, MA, US
Background: Patisiran drug product is composed of drug substance ALN-18328 (siRNA) and lipid excipients formulated in lipid nanoparticle in isotonic phosphate buffer saline. Population PK models were developed to describe plasma PK profiles of ALN-18328 and the 2 novel lipids (DLin-MC3-DMA and PEG2000-C-DMG) and to identify sources of variability. Methods: PK data collected were pooled from 5 clinical studies following single or multiple doses (0.01 to 0.5 mg/kg) to healthy volunteers (N=22) or patients with Hereditary Transthyretin-Mediated (hATTR) amyloidosis (N=177). Nonlinear mixed-effects modeling was used to identify PK compartmental models. Covariate-parameter relationships were explored using a pre-specified full model approach. The clinical relevance of covariate effects was assessed based on statistical significance and impact on steady-state PK exposures. Results: PK of ALN-18328 was described with a semi-mechanistic model with linear elimination from the liver (Figure 1). PK of the 2 lipids were described by 3-compartment mammillary models with linear elimination. All models resulted in acceptable goodness-of-fit. Estimated PK parameters are shown in Table 1. For patients in phase 3 study (N=148), post-hoc plasma clearance (geometric mean) and associated inter-patient variability (% CV) of ALN-18328, DLin-MC3-DMA, and PEG2000-C-DMG were estimated to be 0.422 L/h (47.5%), 0.128 L/h (25%), and 0.128 L/h (19.6%) respectively. None of following covariates affected PK exposures: baseline age, sex, race (Caucasian/non-Caucasian), mild and moderate renal impairment, mild hepatic impairment, anti-drug antibody status, and concomitant administration of moderate or strong CYP3A inhibitors or inducers. There was a trend of increasing PK exposures with increasing body weights; however, this did not result in differences in TTR lowering. Conclusion: Population analyses adequately described plasma PK profiles of patisiran components across a wide range of dose levels. Overall PK of patisiran components was linear, dose-proportional, time-invariant and predictable with chronic dosing of 0.3 mg/kg q3w patisiran over 2 years.
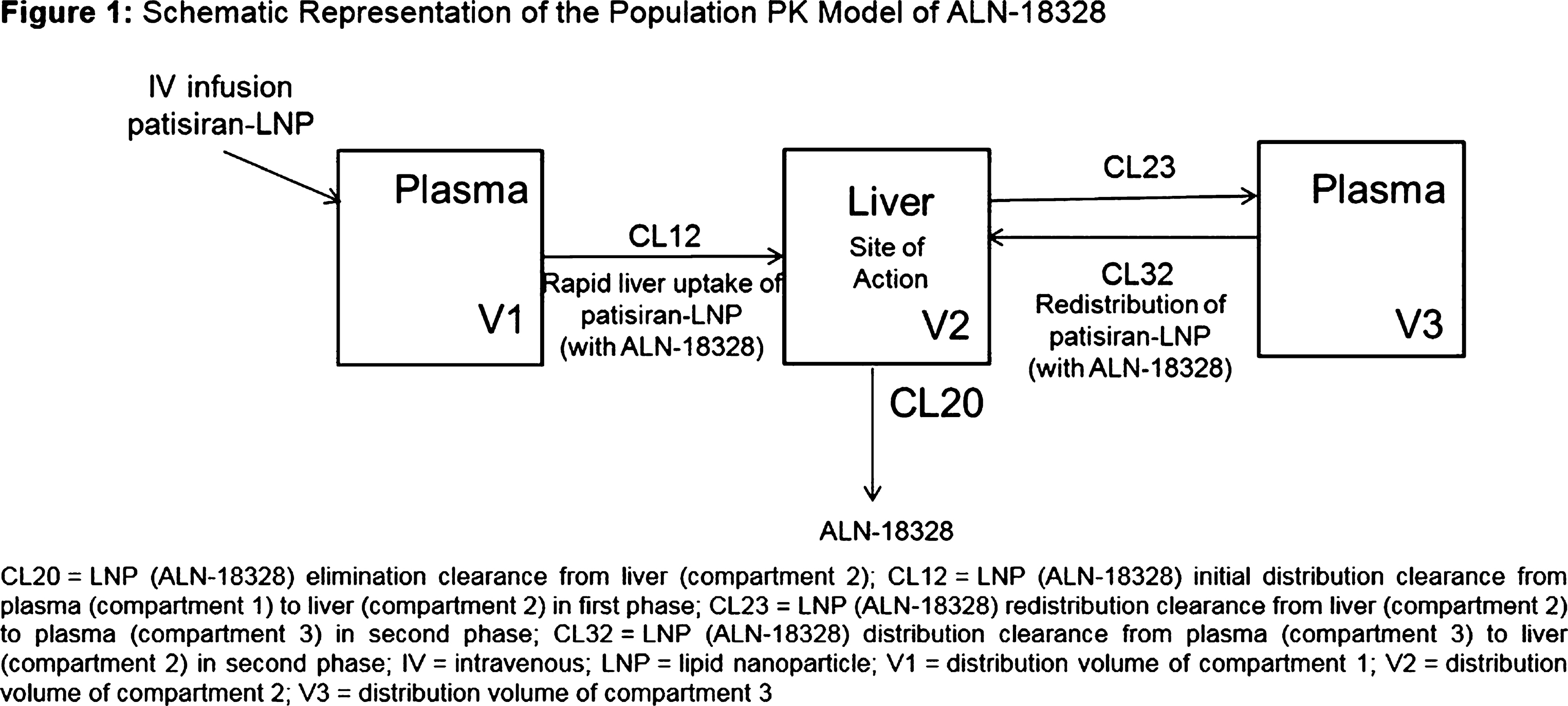
PS2Group3-044 / #788CHARACTERIZATION OF NEURONAL MOLECULAR MECHANISMS UNDERLYING CMT2Z NEUROPATHY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Paula Sancho1, Luca Bartesaghi2, Olivia Miossec2, Francisco García-García3, Laura Ramírez-Jiménez1, Vincenzo Lupo4, Roman Chrast2, Carmen Espinós1
1Unit Of Genetics And Genomics Of Neuromuscular And Neurodegenerative Disorders And Service Of Genomics And Traslational Genetics, Centro de Investigación Príncipe Felipe (CIPF), and Joint Unit for research on Rare Diseases, CIPF-IISLa Fe, Valencia, ES;22department Of Neuroscience And Department Of Clinical Neuroscience, Karolinska Institutet, Stockholm, SE;3Unidad De Bioinformática Y Bioestadística, Centro de Investigación Príncipe Felipe, Valencia, ES;4Unit Of Genetics And Genomics Of Neuromuscular And Neurodegenerative Disorders And Service Of Genomics And Traslational Genetics, Centro de Investigación Príncipe Felipe (CIPF), and Joint Unit for Research on Rare Diseases, CIPF-IISLa Fe, Valencia, ES
Background: Mutations in MORC2 lead to an axonal form of Charcot-Marie-Tooth neuropathy (CMT2Z). To date, thirty-two families have been described with mutations in MORC2, indicating that this gene is relatively frequently involved in CMT cases. While the genetic data clearly established the causative role of MORC2 in CMT2Z, its phenotypic consequences in patients and role in neuronal biology remains to be clarified. Methods: RNA and protein expression of MORC2 was tested at different development stages in several neural tissues: sciatic nerve, dorsal root ganglion (DRG) and brain. In order to determine the effect of the most common MORC2 clinical mutations, p.S87L and p.R252W in a neural context, we used an in vitro culture system based on virally mediated overexpression of MORC2 in sensory neurons. ATPase activity of MORC2 was measured by using colorimetric assay. A rat gene expression microarray was used for transcriptomics studies. Results: Here, we show that the full-length form of MORC2 is predominantly expressed in both adult and embryonic human neural tissue and that the expression of Morc2 is dynamically regulated in the developing and maturing peripheral and central nervous systems. We observed that in particular the overexpression of p.S87L mutant in sensory neurons induced significant axonal phenotype. In addition, by using a colorimetric assay we observed that p.S87L mutant has an impaired ATPase activity which may contribute to its pathogenicity. Since MORC2 has recently been identified as an essential gene required for epigenetic silencing by the human silencing hub (HUSH) complex, we decided to look with a transcriptomic approach in sensory neurons for genetic and biochemical pathways altered in presence of mutated forms of MORC2. Preliminary data from this analysis indicate complex changes affecting transcripts involved in neuronal and axonal functions. Conclusion: Collectively, our data provide an important insight into the pathomechanisms underlying CMT2Z and the role of MORC2 in nervous system.
PS2Group3-045 / #955INCREASED DEACETYLATION OF PROTEINS BY SIRTUIN 1 PROTEIN OVER EXPRESSION REVERSES T2D PERIPHERAL NEUROPATHY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
James Russell, Krish Chandrasekaran, Cheng-Yeng Ho, Pranith Kumar, Sai Sruthi Reddy
University of Maryland School of Medicine, Baltimore, US
Background: Sirtuin1 (SIRT1) protein uses NAD+ as a substrate to deacetylate transcription factors, cofactors, and histones to enhance mitochondrial function. The sirtuins use NAD+ as a co-substrate to catalyze the deacetylation and/or mono-ADP ribosylation of target proteins. Overall, there is evidence that SIRT1 is neuroprotective. Furthermore, there is evidence that activation of SIRT1 by NAD+ is important in axonal regeneration. In part, SIRT1 neuroprotection may be because of improved energy homeostasis. SIRT1 activity in the nucleus deacetylates histone, non-histone transcriptional and co-transcriptional factors that regulate glucose homeostasis and fat oxidation. Although SIRT1 isoforms are primarily present in the nucleus and cytosol and are very important in neuroprotection, there is a distinct Mt isoform. This mitochondrial regulatory function may be critical in reducing diabetic injury to mitochondria within the peripheral nervous system. Methods: We tested the hypothesis whether increased expression of SIRT1 protein in dorsal root ganglion (DRG) neurons would rescue mice from peripheral neuropathy induced by a high fat diet (HFD). Neuron-specific, doxycycline (DOX)-inducible, SIRT1 protein over expressing C57BL6 transgenic mouse (nSIRT1OE Tg) were generated. Expression of SIRT1 protein was observed in small to medium sized DRG neurons. nSIRT1OE was shut off by feeding, weaned Tg mice, with DOX in the diet for 12 weeks. The Tg mice were divided into 3 groups. Group # 1: nSIRT1OE Tg mice fed with standard diet (STD) plus DOX for 4 months; Group # 2: nSIRT1OE Tg mice fed with HFD plus DOX for 4 months; Group # 3: nSIRT1OE Tg mice fed with HFD plus DOX for 2 months until they develop neuropathy and then switched to HFD minus DOX for additional 2 months. Neuropathy was determined by mechanical allodynia thresholds (MAT) and nerve conduction velocity (NCV) at 0, 2 and 4 months. Intraepidermal nerve fiber density (IENFD) was measured at 4 months. Results: MAT, NCV and IENFD were decreased in HFD-fed (Group # 2) mice compared to STD-fed mice (Group # 1) at 2 and 4 months. In Group # 3 mice, 2 months after turning on nSIRT1OE, we observed a reversal of mechanical allodynia, NCV and attenuation of IENFD (Group # 3 compared to Group # 2). Western blot of protein extracts from DRG neurons showed that HFD increased acetylation of proteins and SIRT1OE abolished the HFD-induced acetylation of proteins. The mitochondrial bioenergetics profile of cultured adult DRG neurons showed that hyperglycemia induced a decrease in the spare respiratory capacity in wild type (WT) DRG neurons. This was corrected in nSIRT1OE Tg DRG neurons. Conclusion: In type 2 diabetic neuropathy, altered acetylation of proteins and reduced mitochondrial function via a defective SIRT1 pathway may contribute to developing distal axonopathy. In contrast, overexpression of SIRT1 is able to reverse experimental diabetic neuropathy. SIRT1 is amenable to activation by therapies that increase NAD+ in diabetic neuropathy.
PS2Group3-046 / #953PGC-1a REGULATES NEURONAL RESPITRATORY CHAIN SUPERCOMPLEXES IN MITOCHONDRIA
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
James Russell, Joungil Choi, Mohammad Salimian, Sai Sruthi Reddy
University of Maryland School of Medicine, Baltimore, US
Background: Loss of PGC-1a signaling is important in the development of experimental diabetic neuropathy (DN) [Choi J et al. Neurobiology of Disease, 2014. 64; 118–130]. In PGC- a knockout (KO) mice there is exacerbation of DN with loss of large and small myelinated fibers, loss of mitochondria (Mt) and DNA content, and increased protein oxidation. We have shown potential important functions of 35-kDa PGC-1a, a novel PGC-1a isoform in regulating Mt integrity. Methods: The focus of the present work was to analyze the respiratory chain complex integrity in neurons of PGC-1 a KO mice. Results: In the respiratory supercomplexes of 1,030, 980, and 840 kDa, the levels of complexes 1,030 and 980 kDa are significantly decreased, while the level of 840-kDa complex is significantly increased in PGC-1 a KO mice compared to wild type (WT) mice. Mass spectrometry analysis identifies; (1) subunits of complex I, III, and IV in the 1,030-kDa band, 2) subunits of complex I and III in the 980-kDa band, and (3) subunits of complex I in the 840-kDa band. Quantification results reveal that the activities of complexes I, III, and IV within the 1,030-kDa protein complex are significantly decreased in PGC-1a KO mice. In contrast, we observe little complex I, III, and IV activities either in the 980- or 840-kDa complex. We observed altered Mt morphology and impaired respiration in PGC-1 a KO mice. Co-immunoprecipitation and mass spectrometry analyses reveal that the 35-kDa PGC-1a is associated with NADH dehydrogenase (ubiquinone) flavoprotein 2 (NDUFV2), a subunit of respiratory complex I in the Mt. Immunoblot analysis combined with two-dimensional-blue native gel show the presence of both 35-kD a PGC-1a and NDUFV2 proteins in the 1,030-kDa protein band. Conclusion: These results are consistent with a cooperative role of 35-kDa PGC-1a and NDUFV2 in the organization of Mt respiratory supercomplex assembly. The study provides evidence that 35-kDa PGC-1a is present in the Mt where it could directly regulate Mt function and affect response to diabetes and in diabetic neuropathy.
PS2Group3-047 / #330THE VALIDITY OF SUDOSCAN IN SCREENING OF UPPER EXTREMITY NEUROPATHIES: A PILOT STUDY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Geun-Young Park, Sun Im, Hung Jung Koo, Yongjun Jang, Choong Sik Chae
Rehabilitation Medicine, Bucheon St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Bucheon-si, KR
Background: SUDOSCAN (Impeto Medical, France) is a recently used device in the quantitative assessment of sudomotor function to screen peripheral polyneuropathy (PPH), and Sudomotor function was assessed through values of electrochemical skin conductance (ESC) by two pairs of electrodes for feet and hands. Conventionally, nerve conduction study(NCS) and needle electromyography (EMG) are regarded as a golden standard in the evaluation of large nerve fiber function, but there is a limitation to evaluate small nerve fiber and sweat glands dysfunction. Interestingly, there are a few reports suggesting that neuropathies involving lower extremities, such as lumbosacral radiculopathy, can also induce sudomotor dysfunction. However, there has been no report targeting upper extremity neuropathy. The objectives of this study was to determine the diagnostic validity of SUDOSCAN in screening CTS and cervical radiculopathy, which are one of the most common pathologies of dysesthesia on upper extremities, in an outpatient clinic with suspected patients of sudomotor dysfunction. Methods: Medical record of 48 patients with dysesthesia on upper extremities, who underwent SUDOSCAN, NCS and EMG, between May 1st, 2015 and August 31st, 2015, were analyzed retrospectively. Based on NCS and EMG results, these subjects were allocated to three different groups: Normal findings(Group A, 16 patients), CTS(Group B, 16 patients) and cervical radiculopathy(Group C, 9 patients). ANOVA and Kruskal-Wallis analysis were used to compare the differences in demographic features, average ESC values on feet and hands of SUDOSCAN and pain severity measured by Michigan neuropathy screening instrument(MNSI). Results: There were demographic differences about age and proportion on sex among those groups and no differences for the proportion of DM and alcoholics history. Group A was younger than B and C, and male proportion is higher than female in only in group C. There were no demographic differences about DM and alcoholic history among those groups. ESC values on feet and hands in CTS diagnosed-group B(Mean±SD 61.56±14.70 μS, 52.17±15.37 μS) smaller than Group A(62.63±19.61 μS, 60.43±17.43 μS) and group C(64.88±24.15 μS, 61.33±25.01 μS), respectively. However, there were no significant differences among three groups in statistical analysis by using ANOVA. Pain severity using MNSI were each 2.88±0.99(group A), 3.73±1.89(group B) and 2.40±1.28(group C), all of which showed no statistical significance among these groups. Conclusion: Due to the limited number and uneven distribution of the subjects, our result failed to reveal the usefulness of SUDOSCAN in screening of CTS or cervical radiculopathy among patients with dysesthesia on upper extremities. However, because we have seen the tendency of decrease in ESC value on feet and hands by SUDOSCAN among CTS patients, large population study for SUDOSCAN should be considered in order to validate our findings.
PS2Group3-048 / #415IPSC MOTOR NEURONS FROM AN X-LINKED CMT TYPE 6 PATIENT AS A MODEL FOR STUDYING AXONAL DEGENERATION AND DEVELOPING THERAPIES
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Gonzalo Perez Siles1, Rebecca Screnci2, Anthony Cutrupi1, Melina Ellis1, Carolyn Ly1, Garth A. Nicholson3, Marina L. Kennerson1
1Northcott Laboratory (neurobiology), ANZAC Research Institute, Sydney, NSW, AU;2School Of Life Sciences, University of Technology Sydney, Sydney, NSW, AU;3Concord Hospital, Molecular Medicine Laboratory, Sydney, NSW, AU
Background: A missense mutation (c.G473A; p.R158H) in the pyruvate dehydrogenase kinase 3 (PDK3) gene causes an X-linked form of Charcot-Marie-Tooth neuropathy (CMTX6). PDK3 negatively regulates the pyruvate dehydrogenase complex (PDC) by reversible phosphorylation. Mitochondrial PDC catalyses the oxidative decarboxylation of pyruvate to acetyl CoA and links glycolysis to the critical energy producing Krebs (TCA) cycle. We have shown the p.R158H mutation causes hyperactivity of PDK3 and hyperphosphorylation of the E1 subunit of PDC, leading to reduced PDC activity. As a result, CMTX6 patient fibroblasts show mitochondrial abnormalities, lactate acidosis and reduced ATP production Methods: We have generated a line of induced pluripotent stem cells (iPSCs) by re-programming CMTX6 patient fibroblasts carrying the PDK3 p.R158H mutation (iPSCCMTX6). We also have engineered an isogenic control using the CRISPR/Cas9 system (iPSCisogenic). iPSCCMTX6 and iPSCisogenic have been differentiated into spinal cord motor neurons following a 28 days differentiation protocol and L1CAM magnetic bead enrichment to increase the percentage of mature motor neurons. Results: Our preliminary experiments demonstrate the iPSCCMTX6 line shows E1-hyperphosphorylation, confirming the reprogramed cells retain the pathogenic molecular signature found in the CMTX6 patient fibroblasts. Treating the iPSCCMTX6 cells with the pan-PDK inhibitor dichloroacetate (DCA) reduced the levels of phosphorylation in the patient-derived cells, demonstrating E1-hyperphosphorylation is a reversible process and an ideal pharmacological target for the development of treatment therapies. Gene editing of the mutated residue in PDK3 completely restores phosphorylation levels. This result undoubtedly indicates the p.R158H mutation is responsible of the disease phenotype. Conclusion: iPSC lines re-programmed from CMTX6 patient fibroblasts are an ideal resource to investigate molecular mechanisms underlying axonal degeneration in the relevant neuronal tissue affected in patients. Future studies culturing patient and isogenic control motor neurons will assess the therapeutic effect of compounds that can revert E1 hyperphosphorylation and ameliorate classical features of axonal degeneration affecting axon morphology and function
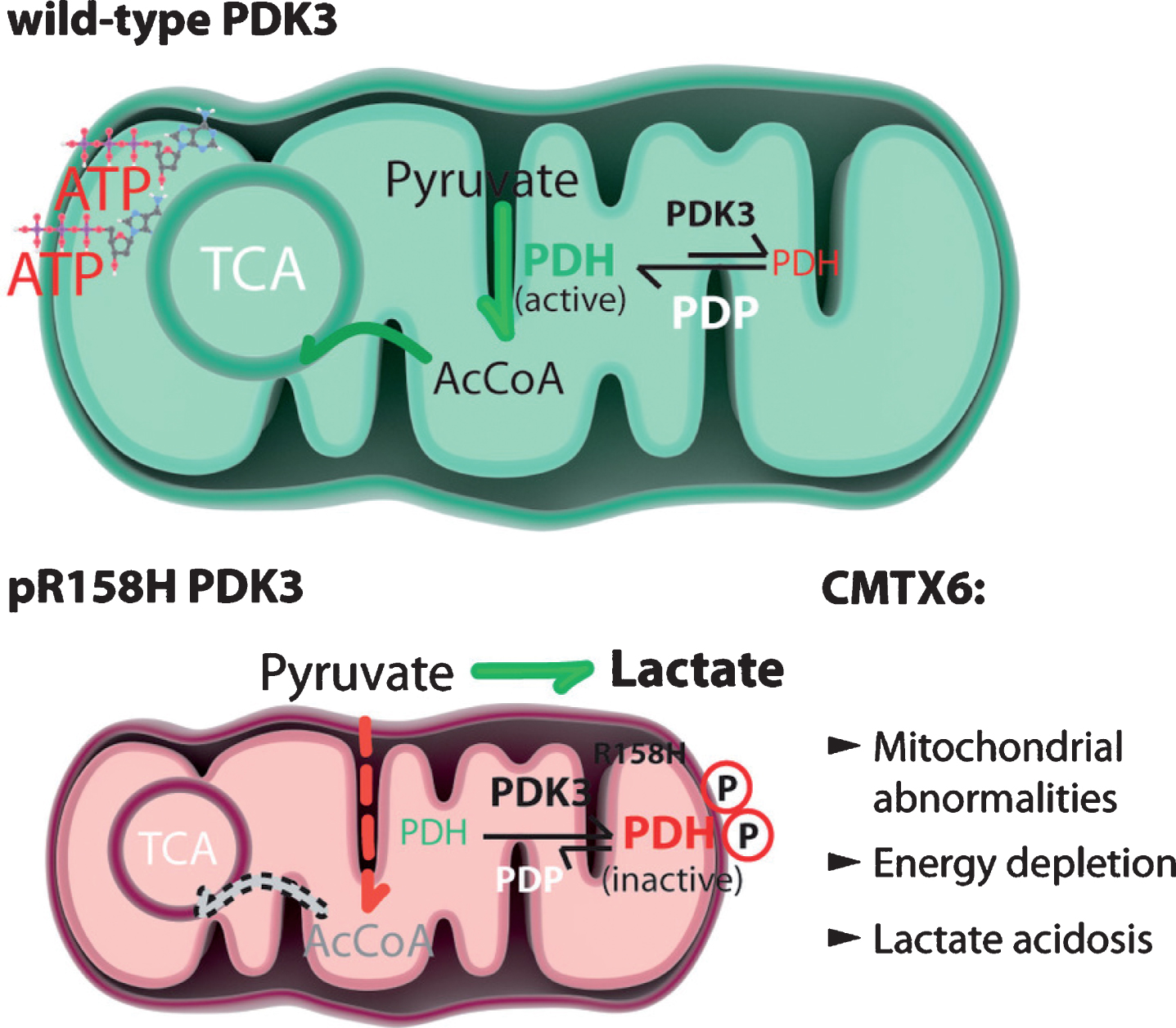
PS2Group3-049 / #542PARANODAL ANTIBODIES IN AUSTRIAN PATIENTS WITH ACUTE ONSET INFLAMMATORY NEUROPATHY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Desiree De Simoni1, Elisabeth Lindeck-Pozza2, Harald Hegen3, Wolfagang Löscher3, Friedrich Zimprich4, Romana Hoeftberger5, Julia Wanschitz3
1Neurology, University Hospital St. Poelten, St. Poelten, AT;2Sozialmedizinisches Zentrum Süd, Vienna, AT;3Medical University of Innsbruck, Innsbruck, AT;4Department Of Neurology, Medical University of Vienna , Vienna, AT;5Medical University of Vienna, Vienna, AT
Background: Nodal, paranodal and juxtaparanodal proteins have been identified as antigens in peripheral inflammatory neuropathies, however the frequency and clinical relevance of antibody responses against these targets remain poorly investigated in Guillain-Barré-Syndrome. Methods: Patients with acute onset inflammatory neuropathies were identified by exploration of the local databases of the Departments of Neurology and the Institute of Neurology of the Medical Universities in Innsbruck and Vienna. Patient data, electrophysiological classification and presence of anti-ganglioside antibodies were retrospectively retrieved by review of patient records. Only patients meeting Brighton criteria certainty level one or two were included in the study. We so far identified 100 patients who were admitted to the hospital between 2004 and 2017 with typical clinical presentation and electrophysiological results consistent with one of the subtypes of GBS, amongst them AIDP, AMAN, AMSAN, MFS, and pharyngo-cervico-brachial GBS variant. Some patients with the initial suspicion of AIDP had disease duration of more than 6 months and were reclassified as acute onset CIDP. Sera, and if available CSF, from all 100 patients and sera from twenty control individuals with non-inflammatory polyneuropathy were screened by an optimized tissue based assay using rat brains and teased nerve fibers for immune responses against surface antigens and by cell-based assays with transfected HEK cells for antibodies against contactin1 (CNTN1), contactin2 (CNTN2), contactin-associated-protein1 (CASPR1), and neurofascin-155 (NF155). Results: In the teased nerve fiber assays, some of the patients showed specific antibody staining to the node of ranvier, some of which were also positive in the brain tissue assay. None of the GBS patients’ sera had antibody reaction to CNTN1, CNTN2, CASPR1 or NF155 in cell-based assays. Among the CIDP patients, two patients demonstrated reactivity against CNTN1 with similar clinical presentation as previously described. None of the controls had any antibody reaction in the performed tests. Conclusion: Our results suggest that antibody responses to CNTN1, CNTN2, CASPR1 or NF155 are absent in Austrian GBS patients, although analysis of more patients is needed to confirm these results. Furthermore, it remains to be established whether antibodies against CNTN1 may predict a chronic course in acute onset inflammatory neuropathies.
PS2Group3-050 / #695ULNAR NERVE CONDUCTION STUDY USING THE FIRST DORSAL INTEROSSEOUS MUSCLE RECORDING IN HEALTHY KOREAN SUBJECTS
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Joong-Yang Cho1, Hye Jung Lee2
1Neurology, Ilsan Paik Hospital, Inje University College of Medicine , Goyang-si, Gyeonggi-do, KR;2Neurology, Ilsan Paik Hospital, Inje University College of Medicine, Goyang-si, Gyeonggi-do, KR
Background: Ulnar neuropathy at the wrist (UNW) can be difficult to localize by nerve conduction studies. The standard approach has been the demonstration of prolonged distal motor latency to first dorsal interosseous (FDI), in conjunction with a normal dorsal ulnar sensory response and denervation on needle EMG of ulnar innervated hand muscles with sparing of ulnar innervated muscles proximal to the wrist. However, none of these electrophysiologic abnormalities in isolation clearly differentiate patients with UNW from the more numerous patients with ulnar neuropathy at the elbow (UNE). Recently, conduction block (CB), along with conduction slowing, of FDI motor fibers across the wrist using a simplified version of the short segment incremental study (SSIS) were reported in patients with UNW. The purpose of this study was to describe normative values for ulnar nerve conduction study using the FDI recording before a simplified version of the short segment incremental study (SSIS) was applied in patients with UNW. Methods: Ulnar nerve motor conduction study with FDI and abductor digiti minimi muscle (ADM) recording was performed in 132 hands of 66 healthy subjects. Ulnar NCS was performed with 2 different recording electrode montages (ADM-base of 5th finger; FDI-thumb) and differences in latency and amplitude were compared. The ulnar nerve was stimulated at the wrist (5 cm proximal to the active recording electrode for ADM). Results: The maximal values for terminal motor latency to the ADM and FDI muscle were 2.5 msec and 4.3 msec, respectively. The maximal side-to-side terminal motor latency difference to FDI was 0.4 msec. In addition, the maximal side-to-side terminal motor latency difference to ADM was also 0.4 ms. The maximal ipsilateral latency difference between ADM and FDI was 1.9 msec. There was a tendency for the terminal motor latency of FDI to increase with advancing age. Conclusion: We obtained the normative values for ulnar nerve conduction study using the FDI recording, which is useful in applying a simplified version of the SSIS in patients with UNW.
PS2Group3-051 / #907PATIENT ASSISTED INTERVENTION FOR NEUROPATHY: COMPARISON OF TREATMENT IN REAL LIFE SITUATIONS (PAIN-CONTROLS)
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Richard J. Barohn1, Byron Gajewski2, Mamatha Pasnoor1, Lexie Brown2, Laura Herbelin1, Kim Kimminau3, Omar Jawdat1, Tina Liu1, Chad Parks1, Pam Shlemon4, Mazen Dimachkie5, P Pain-Control’S Study Team1
1Neurology, University of Kansas Medical Center, Kansas City, KS, US;2Department Of Biostatistics, The University of Kansas Medical Center, Kansas City, US;3Family Medicine, The University of Kansas Medical Center, Kansas City, US;4The Foundation of Peripheral Neuropathy, Kansas City, US;5Neurology, The University of Kansas Medical Center, Kansas City, KS, US
Background: Cryptogenic sensory polyneuropathy (CSPN) is a common slowly progressive neuropathy that affects adults and presents with significant neuropathic pain for which multiple medications have been tried including antiepileptics, antidepressants, topicals and narcotics. A web based survey among neuromuscular experts suggested pregabalin as being more effective than other medications, however there are presently no comparative studies to assess the most effective medication. Objectives: To determine which of the 4 pharmaceutical therapies (pregabalin, duloxetine, nortriptyline or mexiletine) is most effective for neuropathic pain and best tolerated with least side effects in cryptogenic sensory polyneuropathy. Methods: We performed a prospective randomized open labelled comparative effectiveness study of CSPN patients through the Patient Centered Outcomes Research Institute (PCORI). The study used a Bayesian adaptive design, which included response adaptive randomization. At each interim analysis a decision was made to either continue enrolling patients or to stop the trial for success. CSPN patients who fulfilled the inclusion and exclusion criteria were enrolled into this study. Patients underwent a baseline neurological evaluation and randomly assigned to one of the 4 neuropathic medications for 12 weeks. The co-primary outcomes are the change in Likert-like pain scale and quit rates. The outcome measures were performed at baseline, week 4, 8 and 12. Results: There were a total of 402 CSPN patients with 134, 126, 73, and 69 randomized to nortriptyline, duloxetine, pregabalin, and mexiletine, respectively. The posterior probability each treatment was best were 0.52, 0.43, 0.05, and 0.00, with efficacy rates 25.4%, 23.0%, 15.1%, 20.3% and quit rates of 38.1%, 37.3%, 42.5%, 58.0%, respectively. Conclusion: Mexiletine met our criteria for being a loser, primarily due to side effects. While there was no clear winner, overall nortriptyline and duloxetine outperformed pregabalin and mexiletine.
PS2Group3-052 / #938A NEW MUTATION IN MORC 2 CAUSES SCAPULOPERONEAL CHARCOT-MARIE-TOOTH NEUROPATHY: DAVIDENKOW SYNDROME
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Marina Frasquet1, Fernando Mas2, Juan Francisco Vázquez-Costa3, Carmen Espinós4, Vincenzo Lupo5, Teresa Sevilla6
1Neuromuscular And Ataxia Research Group, Instituto de Investigación Sanitaria La Fe, Valencia, ES;2Radiology, Eresa, Hospital Universitari i Polècnic La Fe, Valencia, ES;3Neurology, Hospital Universitari i Politècnic La Fe, Valencia, ES;4Unit Of Genetics And Genomics Of Neuromuscular And Neurodegenerative Disorders And Service Of Genomics And Traslational Genetics, Centro de Investigación Príncipe Felipe (CIPF), and Joint Unit for research on Rare Diseases, CIPF-IISLa Fe, Valencia, ES;5Unit Of Genetics And Genomics Of Neuromuscular And Neurodegenerative Disorders And Service Of Genomics And Traslational Genetics, Centro de Investigación Príncipe Felipe (CIPF), and Joint Unit for Research on Rare Diseases, CIPF-IISLa Fe, Valencia, ES;6Neurology, Hospital Universitari i Politècnic La Fe, Universitat de València and CIBERER, Valencia, ES
Background: Davidenkow (1927) described the clinical picture of a neuropathy which was characterized by onset of weakness in legs’ anteroexternal group, pes cavus, involvement of the scapular girdle, and distal hypoesthesia in limbs. He called this syndrome “scapuloperoneal amyotrophy”. Our objective is to report clinical, muscle imaging, neurophysiological and molecular features of a patient with a new mutation in MORC2 (c.1031C> T; p.A344V) associated with a scapuloperoneal phenotype. Methods: Clinical evaluation, electrophysiological study and muscular magnetic resonance imaging (MRI) of a patient who presented progressive muscle weakness since the second decade of life and with a positive family history (affected father). Genetic study was reached by means of a gene panel. The proband, his affected father and two healthy brothers were genetically studied. Results: A 45-year-old patient presented inability to walk on heels since childhood. He developed progressive distal weakness in the lower limbs at age 12 with frequent falls, and then in upper extremities with consequent difficulty in raising arms and in fine object manipulation with hands. Clinical examination showed weakness in proximal and distal muscle of upper and lower limbs, with relative preservation of flexo-extension of elbows and knees. Pinprick and vibration sensation were reduced in distal lower and upper limbs. Electrophysiological studies were compatible with an asymmetric axonal motor and sensory neuropathy, which was in concordance with the disease severity. Muscle MRI showed fatty infiltration in muscles of the shoulder and pelvis girdle, and also in thighs, legs and feet. Fatty infiltration had a multifocal pattern with preservation of some intrinsic muscles of the feet and affecting some muscles of the shoulder girdle such as the deltoid and subscapular muscles. Conclusion: This patient presents a phenotype compatible with Davidenkow Syndrome. The clinical picture of all patients with mutations in MORC2 is reviewed. FUNDING: Instituto de Salud Carlos III (ISCIII) – FEDER [Grants no. PI15/00187 and PI12/00946]; Fundación para la Investigación del Hospital Universitari La Fe [2015/0085]. The Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) is an initiative from the ISCIII.
PS2Group3-053 / #522AUTOPHAGIC NEUROMYOPATHY CAUSED BY P.R140G HEAT SHOCK PROTEIN B1 (HSPB1) MUTATION
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Marina Frasquet1, Nuria Muelas2, Vincenzo Lupo3, Inmaculada Azorín4, Roger Vílchez1, Maria Jose Chumillas5, Ricard Rojas-García6, Carmen Espinós7, Teresa Sevilla8, Juan J. Vílchez8
1Neuromuscular And Ataxia Research Group, Instituto de Investigación Sanitaria La Fe, Valencia, ES;2Neurology, Hospital Universitari i Politècnic La Fe and CIBERER, Valencia, ES;3Unit Of Genetics And Genomics Of Neuromuscular And Neurodegenerative Disorders And Service Of Genomics And Traslational Genetics, Centro de Investigación Príncipe Felipe (CIPF), and Joint Unit for Research on Rare Diseases, CIPF-IISLa Fe, Valencia, ES;4Neuromuscular And Ataxia Research Group, Instituto de Investigación Sanitaria La Fe and CIBERER, Valencia, ES;5Clinical Neurophysiology, Hospital Universitari i Politècnic La Fe and CIBERER, Valencia, ES;6Neuromuscular Diseases Unit, Hospital de la Santa Creu i Sant Pau and Universitat Autònoma de Barcelona, Barcelona, ES;7Unit Of Genetics And Genomics Of Neuromuscular And Neurodegenerative Disorders And Service Of Genomics And Traslational Genetics, Centro de Investigación Príncipe Felipe (CIPF), and Joint Unit for research on Rare Diseases, CIPF-IISLa Fe, Valencia, ES;8Neurology, Hospital Universitari i Politècnic La Fe, Universitat de València and CIBERER, Valencia, ES
Background: Mutations in the heat shock protein B1 (HSPB1) gene are responsible of a variety of neuromuscular disorders including diverse forms of neuropathies and rare cases of distal myopathy, the latter not being fully characterized yet. The mechanism responsible of these variable phenotypic manifestations reminds unrevealed. We report a large cohort of patients from different families harboring the same mutation in the HSPB1 gene. Our aim is to provide novel data that contribute to the understanding of HSPB1 –related disorders. Methods: We analyze patients’ clinical, neurophysiological and muscle MRI features. We provide pathological data from muscle and sural nerve biopsies that were studied by conventional, immune-fluorescence and electron-microscopic methods. Genetic diagnosis was performed in some cases by targeted Sanger sequencing and in others by means of an in-home designed gene panel for testing Charcot-Marie-Tooth (CMT) and distal hereditary motor neuropathy (dHMN) genes. Results: The cohort comprises 18 patients (6 families) harboring the HSPB1 p.R140G mutation and living in Eastern Spain. Twelve subjects from 5 families developed a neuropathy that started as a dHMN in 10 of them and as an axonal Charcot-Marie-Tooth (CMT2) in the other two. The additional six patients, all belonging to the same family, presented a myopathic picture that was characterized by raised serum creatine-kinase (CK) and progressive distal weakness in three cases, a long-lasting asymptomatic hyperCKemia in two others, while the sixth case resulted to be an asymptomatic mutation carrier. Pathologically the myopathy was characterized as an autophagic vacuolar myopathy (AVM) defined by the occurrence of membrane lined vacuoles with sarcolemma features, proliferation of autophagosomes and the presence of aggregates containing the autophagic markers LC3-II and p62. The nerve pathology showed loss of myelinated and unmyelinated fibres, axonal atrophy, myelin remodeling, neurofilament disarrangement and accumulation of autophagic vacuoles in axoplasm and Schwann cell cytoplasm. Conclusion: This study gives detailed insights into the clinical presentation of HSPB1 gene mutations and expands the list of causes of AVMs. Our observations may also contribute to guide futures research approaches because of the resemblance of HSPB1-related neuro-myopathy to colchicine myopathy caused by an alteration of b-tubuline, a molecule that is involved in the experimental neuropathy of HSPB1 transgenic models. FUNDING: Instituto de Salud Carlos III (ISCIII) – FEDER [Grants no. PI15/00187, PI12/00946 and PI16/00316]; Fundación para la Investigación del Hospital Universitari La Fe [2015/0085]; Fundación Isabel Gemio; The Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) is an initiative from the ISCIII.
PS2Group3-054 / #227HEREDITARY TRANSTHYRETIN AMYLOIODOSIS MIMICKING REFRACTORY CHRONIC INFLAMMATORY DEMYELINATING POLYRADICULONEUROPATHY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Kyomin Choi1, Jeeyoung Oh2
1Department Of Neurology, Konkuk University Medical Center, Seoul, KR;2Department Of Neurology, Konkuk University Medical Center, Seoul, KR
Background: Transthyretin (TTR) amyloidosis is a life-threatening systemic amyloidosis caused by autosomal dominant mutation of the TTR gene. Amyloid deposition in peripheral nerves causes polyneuropathy known as familiar amyloidotic polyneuropathy (TTR-FAP). Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is clinically defined as a ‘chronically progressive, stepwise or recurrent proximal and distal weakness and sensory dysfunction of all extremities, developing over at least 2 months. Patients with CIDP were treated by immunotherapy. However, 20–33% of CIDP patients remain refractory to these therapies. The refractory CIDP is not completely understood, incorrect diagnosis is considered one of causes of therapeutic failure. To report this issue, we performed retrospective review of 3 patients who were misdiagnosed refractory CIDP and finally confirmed TTR-FAP by genetic analysis. Methods: We reviewed three patients who were considered refractory CIDP and finally confirmed alternate diagnosis of TTR-FAP. All of patients underwent intensive neurologic examination, and serial electrophysiological studies. Other types of primary and secondary amyloidosis and paraproteinemic polyneuropathy were excluded by laboratory testing of protein immune electrophoresis in serum and urine. TTR gene mutations and types were identified by polymerase chain reaction and direct sequencing of TTR gene after informed consent. Results: Patient 1. This male patient initially presented paresthesia of bilateral lower limbs at the age of 38 years. In electrophysiologic study, conduction block and temporal dispersion of compound muscle action potential (CMAP) were shown on three nerves in bilateral upper limbs. He treated with prednisolone and immunoglobulin but no improvement. After the presentation of severe autonomic symptoms, he diagnosed amyloidosis from the biopsy of colon and genetically confirmed to have Lys35Asn mutation in TTR gene. Patient 2. This female patient initially presented urinary incontinence at age of 45 years. She was referred to neurology department due to progressive generalized weakness. In electrophysiological study, delayed terminal latencies, slow nerve conduction velocities(NCVs) and extended duration of CMAP suggested demyelinating neuropathy. Despite administration of steroid, her symptoms kept worsening and she complained of decreased visual acuity. Considering cataracts caused by steroid, she was performed ophthalmologic evaluation. Vitreous opacity associated to amyloid deposit was found. Lys35Asn heterozygous mutation of TTR gene was demonstrated. Patient 3. A 46-year-old man complained weakness of distal lower limbs. His body weight decreased 11 kilograms for a year due to chronic diarrhea. Following electrophysiological study, temporal dispersion, delayed terminal latencies and slow NCVs were presented on three nerves of bilateral upper limbs. He was treated by steroid, but there was no clinical improvement. The gastrointestinal symptoms of amyloidosis are demonstrated by tissue biopsy performed in colon. On genetic analysis, heterozygous mutation of Ala36Pro was detected in TTR gene. Conclusion: Our findings showed that the main reason of inadequate treatment outcome might be incorrect diagnosis of CIDP. These three patients had significant features indicating clues of alternative diagnosis. First, coexisting symptoms including gastrointestinal or cardiac diseases and autonomic dysfunctions. Second, these three patients did not show elevation of CSF protein. Third, these 3 patients presented distal dominant weakness. Clinician should be aware that electrophysiological findings of TTR-FAP sometimes can mimic the demyelinating diseases.
PS2Group3-055 / #565TRANSTHYRETIN REDUCTION WITH PATISIRAN IN THE APOLLO PHASE 3 STUDY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Teresa Coelho1, David Adams2, Alejandra Gonzalez-Duarte3, William O’Riordan4, Chih-Chao Yang5, Michael Polydefkis6, Arnt Kristen7, Ivaylo Tournev8, Hartmut Schmidt9, John Berk10, Kon-Ping Lin11, Pritesh Gandhi12, Marianne Sweetser12, Christine Powell12, Jared Gollob12, Ole Suhr13
1Hospital de Santo Antonio, Porto, PT;2Neurology Department, LFB, Le Kremlin Bicetre, FR;3Instituto Nacional de Ciencias Medicas y Nutricion, Salvador Zubiran, MX;4eStudy Site, La Mesa, NM, US;5National Taiwan University Hostpital, Taipei, TW;6Johns Hopkins, Baltimore, MD, US;7Heidelberg University Hospital, Heidelberg, DE;8University Multiprofile Hospital for Active Treatment, Sofia, BG;9University Hospital Muenster, Muenster, DE;10Boston Universty, Boston, MA, US;11Taipei Veterans General Hospital, Taipei, TW;12Alnylam Pharmaceuticals, Cambridge, MA, US;13Department Of Public Health And Clinical Medicine, Umea University, Umea, SE
Background: Mutations in transthyretin (TTR), whose physiological roles include binding to retinol binding protein (RBP) which then binds retinol (also known as vitamin A), and thyroxin (T4), can result in multi-organ TTR amyloid deposition causing a life-threatening disease known as hereditary transthyretin-mediated (hATTR) amyloidosis. hATTR manifests in a heterogeneous manner with clinical presentations including sensorimotor and autonomic neuropathy, gastrointestinal symptoms, as well as cardiomyopathy. Based on the hypothesis that the reduction of circulating TTR levels will reduce TTR amyloid deposition leading to stabilization or improvement of disease, patisiran, an investigational RNAi therapeutic, was evaluated in patients with hATTR amyloidosis. Patisiran resulted in improvement in primary, secondary, as well as exploratory endpoints and was generally well tolerated in the APOLLO study. Furthermore, patisiran resulted in rapid, sustained and durable TTR reduction. Given the wide range of TTR genotypes observed in this patient population, we investigated the degree of reduction for TTR, as well as additional pharmacodynamic (PD) assessments RBP and vitamin A, achieved in patisiran patients in APOLLO on a mutational basis. Methods: APOLLO was a Phase 3, randomized (2:1), double-blind, study of patisiran 0.3mg /kg or placebo IV q3W in hATTR amyloidosis patients with polyneuropathy (NCT01960348). Males or females (18 to 85 years of age) with a diagnosis of hATTR amyloidosis with a documented TTR mutation, with a NIS of 5 to 130 and a PND score of ≤3b at baseline were eligible. Serum TTR levels were measured serially by ELISA and serum RBP was quantified using nephelometry. Results: APOLLO enrolled 225 patients with a mean age of 61 years; 45% and 55% had FAP Stage 1, or 2, respectively; 76% had PND>1; 56% had cardiac involvement. Enrolled patients had one of 39 different mutations: 43% V30M and 58% non-V30M mutations. At 18 months, mean TTR reduction was 4.8% (n=47) in the placebo group and 84.3% (n=130) for the patisiran group. Among patients with the V30M and non-V30M mutation groups, mean TTR reduction in the patisiran groups was 79.7% (n=56) and 76.5% (n=92), respectively. Further, a similar effect on TTR was observed across the 29 different non-V30M mutations evaluated in the patisiran group. As expected based on the mechanism of action, trends at 18 months for RBP and vitamin A were consistent with patisiran TTR reduction in each population for RBP: overall 45.3% (n=146), V30M 60.2%,(n=56), non-V30M 36.0% (n=90) and for vitamin A: overall 62.4% (n=145), V30M 68.9% (n=56), non-V30M 58.3% (n=89). Safety events were similar between treatment arms with no clinical manifestations of vitamin A deficiency or thyroid disorder. Detailed data, including earlier timepoints, to be presented. Conclusion: Patisiran resulted in robust and durable serum TTR reduction throughout 18 months of dosing in V30M and non-V30M patients. These data show that patisiran had a similar effect on TTR in patients with hATTR amyloidosis regardless of TTR genotype.
PS2Group3-056 / #589INFLAMMATORY NEUROPATHY AFTER IMMUNE CHECKPOINT THERAPY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Min-Xia Wang1, Jeannette Lechner-Scott2, Toan Nguyen1, Judith M. Spies1
1University of Sydney, Royal Prince Alfred Hospital, Sydney, NSW, AU;22. University of Newcastle, John Hunter Hospital, Newcastle, NSW, AU
Background: The immune checkpoint inhibitor therapies, anti-programmed cell death-1 (PD-1) and anti-CTLA4 monoclonal antibodies, are among the most promising new therapies for a variety of cancers. Despite significant clinical benefits, these therapies can lead to immune-related adverse events in a small number of patients. These include gastrointestinal, dermatologic, hepatic, endocrine, pulmonary and neurological complications. Severe neurological complications, although rare, require early recognition and management to prevent irreversible neurological deficit. A variety of peripheral neuropathies have been described after checkpoint inhibitor therapy, including inflammatory demyelinating neuropathies. Here we present the nerve pathology in two cases of inflammatory axonal neuropathy following anti-PD1 therapy. Methods: Case 1. A 64 year-old woman developed subacute asymmetric weakness and sensory disturbance in the lower limbs, and paraesthesia at hands after 4 cycles of anti-PD1 (pembrolizumab) treatment for mesothelioma. MRI showed no evidence of lumbosacral root or plexus pathology. A sural nerve biopsy showed a marked reduction in myelinated fibre density, scattered nerve fibres undergoing active axonal degeneration and an epineurial vessel obliterated by intense inflammatory cell infiltrates (figure), suggestive of a vasculitic neuropathy. Case 2. A 73 year-old man presented with myalgia in lower limbs, left foot drop, speech and swallowing difficulties after the second dose of pembrolizumab for metastatic melanoma. He had a left foot drop, symmetric mild proximal upper limb weakness and distal loss of pin and temperature sensation in the feet. Electrophysiology revealed an axonal peripheral neuropathy with widespread denervation. A right sural nerve biopsy showed a chronic active axonal neuropathy with mild inflammation (inflammatory cells adjacent to endoneurial vessels). Results: Both patients responded well to cessation of anti-PD1 treatment and high dose steroid. Conclusion: A variety of immune mediated neurological complications have been described with immune checkpoint inhibitor therapy including peripheral neuropathy, myasthenia gravis, myositis, aseptic meningitis, CNS demyelination and encephalitis. Peripheral neuropathy is the most common neurological complication and is estimated to occur in perhaps 1% of patients. Distal sensorimotor neuropathy, GBS, CIDP, polyradiculitis and enteric neuropathy have been reported. In one series, 50% of cases were mild and responded to cessation of therapy +/- steroids. In more severe cases further immunomodulatory therapy is required. These two cases provided histopathological evidence of vasculitic/inflammatory neuropathy with ani-PD1 therapy. With increased use of immune checkpoint therapy in cancer, it is important for clinicians to remain vigilant in order to recognise symptoms of immune mediated adverse events, and to initiate prompt management.
PS2Group3-057 / #646BENEFIT-RISK PROFILE OF INTRAVENOUS IMMUNOGLOBULIN (IVIG) AND SUBCUTANEOUS IMMUNOGLOBULIN (SCIG) IN CIDP: THE PATH STUDY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Amgad Shebl1, Billie L. Durn2, Vera Bril3, Ingemar Merkies4, Hans-Peter Hartung5, Richard A. Lewis6, Gen Sobue7, John-Philip Lawo1, David R. Cornblath8, Orell Mielke1, Ivo Van Schaik9
1CSL Behring, Marburg, DE;2CSL Behring, Fuquay Varina, NC, US;3Department Of Medicine (neurology), University Health Network, University of Toronto, Toronto, CA;4Department Of Neurology, Maastricht University Medical Center, Maastricht, NL;5Department Of Neurology, Heinrich-Heine University, Duesseldorf, DE;6Department Of Neurology, Cedars-Sinai Medical Center, Los Angeles, US;7Department Of Neurology, Nagoya University Graduate School of Medicine, Nagoya, JP;8Department Of Neurology, Johns Hopkins University, Baltimore, MD, US;9Department Of Neurology, Academic Medical Center, University of Amsterdam, Amsterdam, NL
Background: PATH investigated efficacy and safety of SCIG (IgPro20, CSL Behring) as maintenance therapy for chronic inflammatory demyelinating polyneuropathy (CIDP). Before randomisation to IgPro20 or placebo, subjects underwent IVIG withdrawal and, upon clinical deterioration, were re-stabilised with IVIG (IgPro10, CSL Behring). IgPro10 and IgPro20 have the same manufacturing process with the only differences between products being the final immunoglobulin concentration and administration route. The benefit-risk profiles of IgPro10 IVIG and IgPro20 SCIG are evaluated here. Methods: IVIG re-stabilisation comprised an initial dose of 2g/kg followed by 3–4 doses of 1g/kg at 3-week intervals. Subjects were then randomised to weekly SCIG maintenance therapy (0.2 or 0.4 g/kg) or placebo for 24 weeks. The adverse event (AE) profile and relapse (change in adjusted Inflammatory Neuropathy Cause and Treatment [INCAT] score) were analysed for both products. Results: A total of 207 subjects received 1894 IVIG infusions and 115 of these subjects subsequently received 4225 SCIG infusions. The most frequent AEs were headache (IVIG) and local reactions (SCIG). The majority of headaches (65%) and local reactions (95%) were mild. With IVIG, 9 haemolysis AEs occurred (all non-serious and resolved without transfusion); none occurred with SCIG. No thromboembolic events, renal failures or deaths were reported (Tables 1&2). A total of 83% of subjects re-stabilised on IVIG; subsequently, 81% of subjects did not relapse on SCIG 0.4 g/kg (0.2 g/kg: 67%; placebo: 44%). Conclusion: Both IVIG and SCIG had the expected effectiveness reported in literature, while rates of systemic AEs were lower with both SCIG doses.
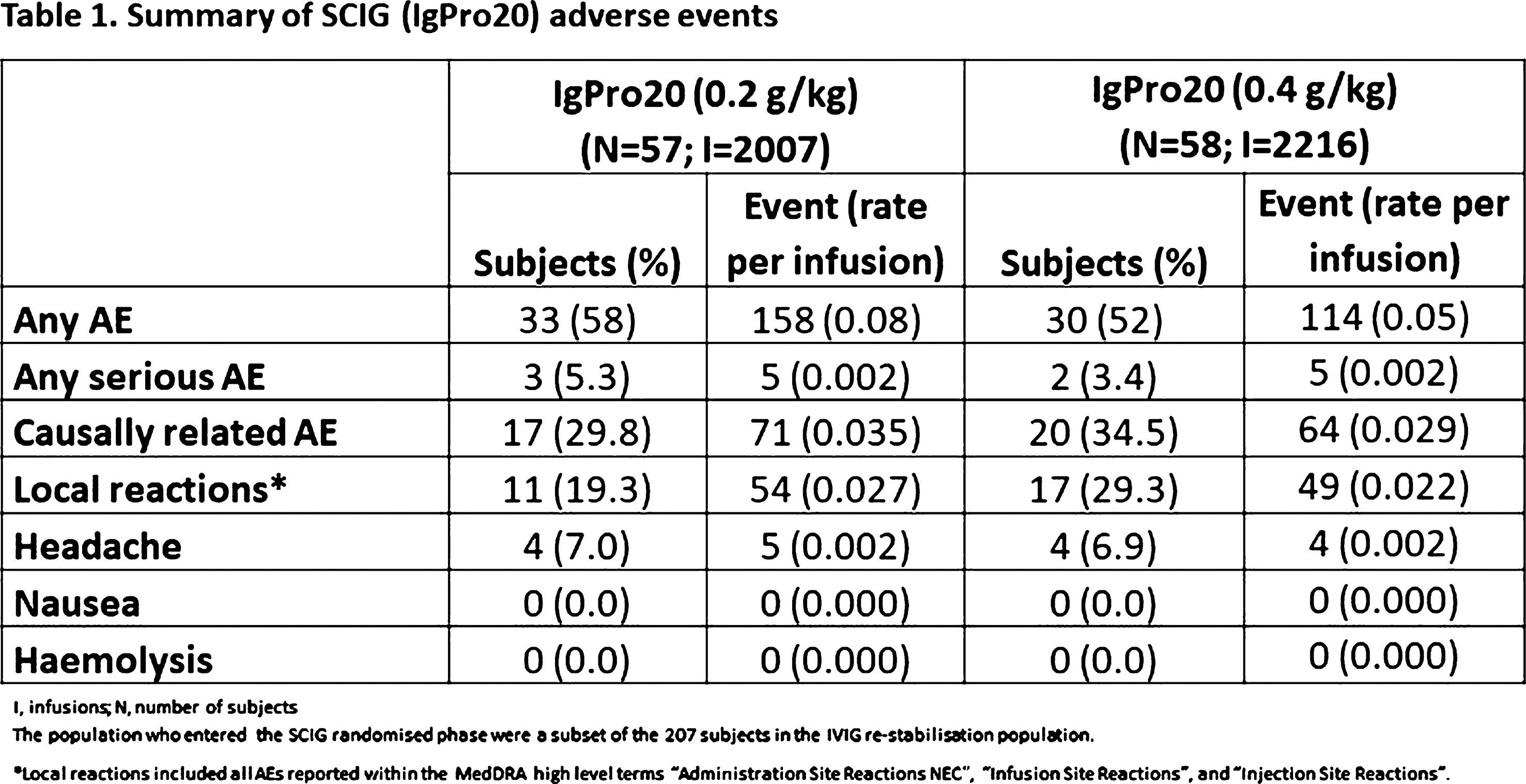
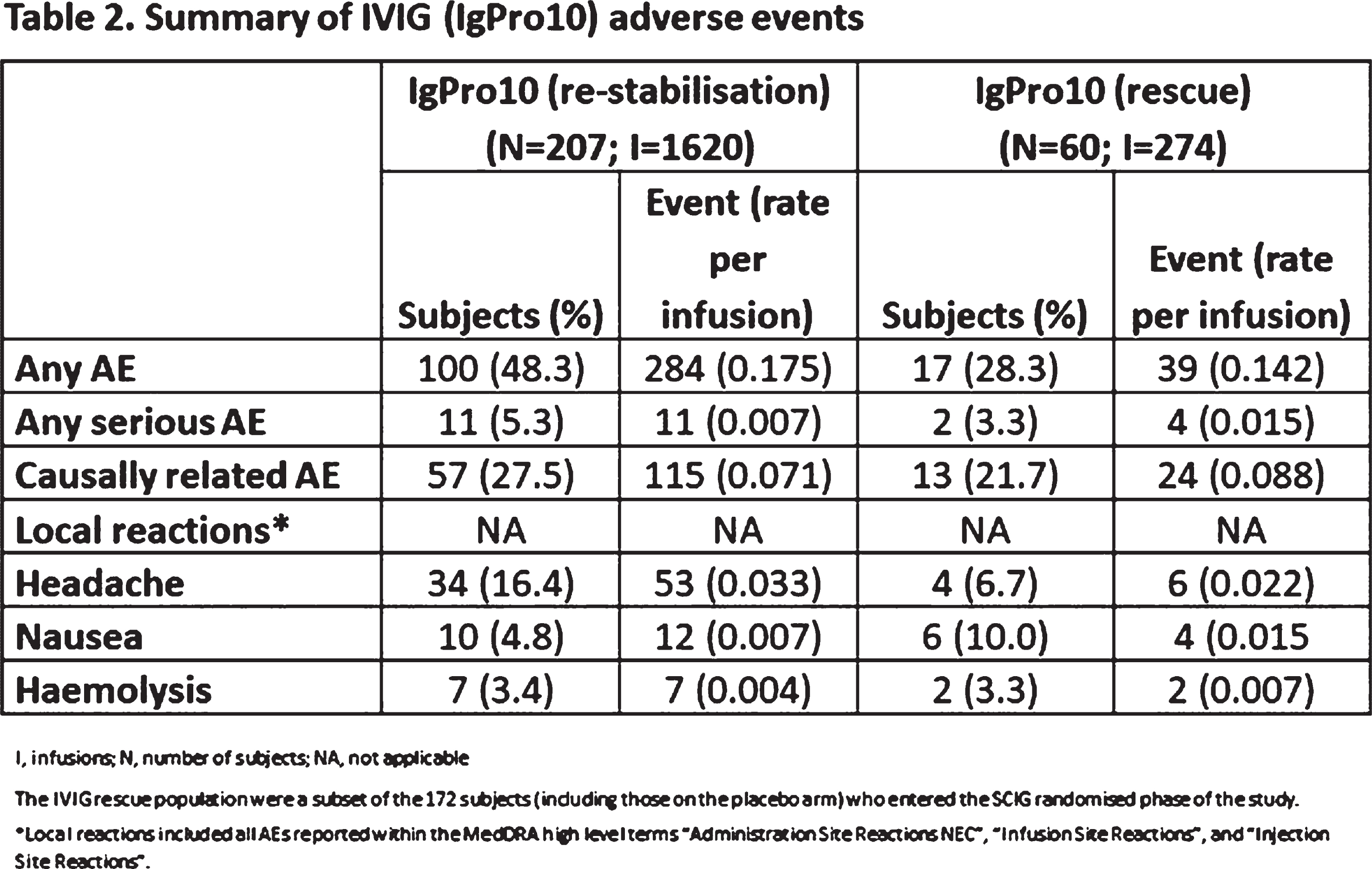
PS2Group3-058 / #703EPIDEMIOLOGY OF HEREDITARY MOTOR-SENSORY NEUROPATHIES IN THE POPULATION OF THE REPUBLIC OF BASHKORTOSTAN
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Elena Saifullina1, Rim Magzhanov2, Irina Khidiyatova3, Elsa Khusnutdinova3
1Neurology And Medical Genetics, Bashkir State Medical University, Ufa, RU;2Bashkir State Medical University, Ufa, RU;3Institute of Biochemistry and Genetics, Ufa, RU
Background: Hereditary motor-sensory neuropathies (HMSN, Charcot-Marie-Tooth disease) are the most common inherited disorders of the peripheral nervous system, with an overall prevalence estimated at 9.37 - 20.1/ 100 000 [Barreto L.C. et al., 2016]. The aim of our study was to estimate the prevalence of HMSN in the population of the Republic of Bashkortostan (Russian Federation). Methods: The study covers the 46-years period from 01 January 1970 to 01 January 2016 in the territory of Bashkortostan where, according to the 01.01.2016, the population was 4,071,064 (1,907,815 males and 2,164,249 females). Data on the patients with HMSN were obtained from registry “Hereditary motor-sensory neuropaties in the Republic of Bashkortostan”. The prevalence of the disease was calculated per 100,000 population. Crude rates were standardized by age using the method of direct standardization according to the world population. Results: We registered 540 patients with HMSN in Bashkortostan: 330 males and 210 females. The most frequent type was HMSN 1 (HMSN 1 : HMSN 2 = 3 : 1). The crude prevalence of HMSN by 01.01.2016 was 13.3/100,000 for all subtypes. Gender-specific prevalence was 17.3/100,000 for males and 9.7/100,000 for females. The highest age-specific prevalence was registered in the oldest age male group (60+years; 29.3/100,000). The highest age-specific prevalence among females was registered in the group of 40-59 years old (14.7/100,000). Conclusion: The prevalence of HMSN in the Republic of Bashkortostan appeared higher than it was established in our previous study (10.6/100,000).
PS2Group3-059 / #785INFLUENCE OF THE EXTRACT OF SUCUPIRA BRANCA (PTERODON EMARGINATUS VOGEL) IN SURAL NERVE OF DIABETIC RATS
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Bruno M. Bertassoli1, Aline S. Faiola1, José Armando Jr1, Giuliana Petri1, Jose F.R. Santos1, Fernando L.A. Fonseca2, Alzira A.S. Carvalho3, David Feder1
1Pharmacology, Faculdade de Medicina do ABC, Santo Andre, BR;2Laboratory Analysis, Faculdade de Medicina do ABC, Santo Andre, BR;3Neurology, Faculdade de Medicina do ABC, Santo Andre, BR
Background: The sucupira branca plant (SB) (Pterodon emarginatus Vogel) is used in tradicional medicine treatment as antirheumatic, analgesic, anti-inflammatory and anti-parasitic agent. The tuber portion of sucupira root, also known as “sucupira potatoes” has been used as alternative treatment for diabetes mellitus (DM). The purpose of this study is to evaluate the effect of root extract of SB in the sural nerve of diabetic rats. Methods: In this study, Wistar rats (Ratus norvegicus), were selected with age ranging from 14 to 16 weeks, weighting 250-350g were used. Diabetes was induced by single, subcutaneous injection of aloxane (120mg / kg). 7 days after induction, blood glucose was dosed. We considered diabetic rat those one with glycemia higher than 200mg/dL. The animals were divided into 3 groups. Control group (GC) (n = 7): treated with physiological solution; (n = 8): treated with 100 mg / kg / day of SB extract and incremental dose group (GD) (n = 8): treated with 100 mg / kg / day SB extract of 20% of the dose every week. All groups were treated via gavage for 6 weeks. The animals were then anesthetized followed by glycemia analysis. Then, they were euthanized by anesthetic overdose, followed by collection of right sural nerve. Semithin sections stained with toluidine blue for axonal morphometric and fiber density. The images were obtained at a magnification of 40x under an optical microscope using the imageJ program. For statistical analysis, the program Graph prism6 was used, with significance level p <0.05. Results: Glycemia levels (mg/dL): GC 744.7 ± 110.4; GDC 523.4 181.6; GDP 610.4 ± 147.8 with significant difference between GC and GDC groups. Morphometric analysis: mean fiber density of myelinazed axons - GC 3236, GDC 3454 and GDP 3535. We obtained a significant diference (p<0,05) between GC and GDP group. The Feret diameter of myelinated fibers: GC 6,37+1,63um; GDC 5,36+1,73um; GDP 4,87+ 1,65um. Feret diameter of axons: GC 4,54+ 0,97um;GDC 4,02+1,18um; GDP 3,99+1,29um. No significant diference was observed among the groups. Conclusion: Although sucupira branca demonstrated to be hypoglycemiant agent, we did not find significant difference among the three groups in relation to Feret diameter of myelinated fibers and axons. However the fiber density was significant difference between the GC and GDP groups. That could suggest a possible positive effect of sucupira branca in peripheral nerves.
PS2Group3-060 / #925EXPANDING THE PHENOTYPIC SPECTRUM OF GARS MUTATIONS
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Giulia Ricci, Veronika Boczonadi, Boglarka Bansagi, Rita Horvath
Institute Of Genetic Medicine, Newcastle University, Newcastle, GB
Background: Mutations in the GARS gene have been associated with autosomal dominant disorders such as Charcot-Marie-Tooth 2D (CMT2D), distal hereditary motor neuropathy V ((dHMN-V) or distal spinal muscular atrophy V (dSMAV), generally starting in the second decade of life with predominant distal extremity weakness and atrophy of upper limb with slow progression. Methods: We identified two families with novel heterozygous GARS mutations, respectively c.647A>G p.(His216Arg) and c.1528A>C p.(Lys510Gln). Results: The c.647A>G variant of GARS gene was detected in the index case (male, 36 years old) and in his mother (66 years old). They showed the classical clinical picture characterized by weakness and atrophy of hand muscles since teenage and, subsequently, by mild and slow progressive distal lower limb weakness, with loss of toe walking ability but without sensory impairment. Electrophysiological examination was consistent with axonal motor neuropathy. In the other family, in which the GARS c.1528A>C variant segregated, we identified three affected members (index case, her daughter and her granddaughter). They showed a different phenotype with a predominant involvement of lower limbs started in early childhood and characterized by tiptoeing and ankle contractures. The index case, at the age of 52, complained of distal wasting in her feet and feet deformities with clawing toes and reduced deep tendon reflexes; she required a stick to aid her walk. The daughter, aged 30, also reported disabling pain Electrophysiological examination detected signs of axonal neuropathy, moderate inactive neurogenic change and neuromuscular junction defect. Conclusion: In the last years, several case descriptions have expanded the phenotypic spectrum associated with mutations in the GARS gene, ranging from almost asymptomatic carriers or patients with a late adult onset disease and mild clinical symptoms to very disabling infantile/childhood forms with diffuse weakness of both upper and lower limbs. Our cases further confirm the clinical variability of GARS mutations.
PS2Group3-061 / #262REDDISH SKIN COLOR CHANGE IN A PATIENT WITH COMPRESSIVE RADIAL NEUROPATHY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Byung-Nam Yoon1, Ji-Won Yang2, Jung-Joon Sung3, Kee-Hong Park4, Suk-Won Ahn5
1Neurology, Inje University College of Medicine, Seoul Paik Hospital, Seoul, KR;2Neurology, Gachon University, Gil medical Center, Incheon, KR;3Neurology, Seoul National University College of medicine, Seoul University Hospital, Seoul, KR;4Neurology, Kyung Sang University Hospital, Jinju, KR;5Neurology, ChungAng University Hospital, Seoul, KR
Background: The motor and sensory symptoms caused by compressive radial neuropathy are well known, but the involvement of the autonomic nervous system or the dermatologic symptoms is not well known. In carpal tunnel syndrome (CTS), diverse dermatologic symptoms, including ulceration, blistering, hypohidrosis, Raynaud’s phenomenon, and irritant contact dermatitis, have been reported, but these symptoms are rare in radial neuropathy. Here, we report an unusual case of compressive radial neuropathy with reversible reddish skin color change. Methods: Case presentation A 42-year-old male was referred for left wrist drop, finger drop and a tingling sense over the lateral dorsum of the left hand. The patient reported that he was well until 4 days prior, when he was drunk and awoke with the symptoms. For 4 days, slight improvement of weakness occurred. Neurologic examination revealed weakness of the left wrist and finger extension (Medical Research Council grade II). Finger abduction appeared weak, but strength was much better when the hand was passively extended to the neutral position. Wrist and finger flexion was intact. On sensory examination, there was a well-demarcated area of hypoesthesia and a tingling sensation over the lateral dorsum of the left hand between the thumb and index fingers extending into the proximal phalanges of the 2nd finger. In addition, a reddish skin color change was observed in the similar area. According to the clinical information and neurologic examination, he was diagnosed with compressive radial neuropathy. Results: In this case, the patient presented with a reddish skin color change. The reddish skin color change suggested autonomic nervous abnormality caused by vasodilation. These kinds of autonomic dysfunctions are reported in CTS but rarely reported in radial neuropathy. A reddish skin color change is also observed in patients with acute phase complex regional pain syndrome (CRPS). In CRPS, inhibition of sympathetic vasoconstrictor activity leads to vasodilation and skin warming. In our case, similar to CRPS, sympathetic dysfunction caused by radial nerve compression may induce a vasodilation and reddish color of skin innervated by the radial nerve. As the radial motor and sensory symptoms improve, the reddish skin color change would improve. With a reddish skin color change, an elevated skin temperature could be suspected but not evident, and the skin thermometer test was not performed. Conclusion: Autonomic symptoms in radial neuropathy are very unusual. Vasomotor innervation to the skin of the radial aspect of the dorsum of the hand is normally provided by the median nerve, whereas sensory and sudomotor innervation is received via the radial nerve. In a study using local anesthetic nerve block, only one subject of 18 is revealed to have vasomotor innervation via the radial nerve. To the best of our knowledge, this is the first report of compressive radial neuropathy with a reversible reddish skin color change. Compressive radial neuropathy is presented with not only motor and sensory symptoms but also autonomic symptoms; therefore, careful examination and inspection are needed at diagnosis.
PS2Group3-062 / #453UNCLASSIFIED CONGENITAL AXONAL NEUROPATHY IN GIPSY FAMILLIES IN PORTUGAL
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Teresa Moreno1, Raquel Siva2, Isabel Conceição3, José Castro3, Isabel D. Castro3, Fatima Furtado4, Oana Moldovan2
1Unidade De Neuropediatria, Hospital Santa Maria, Lisboa, PT;2Serviço Genética, Hospital Santa Maria, Lisbon, PT;3Laboratorio Neurofisiologia, Hospital Santa Maria, Lisboa, PT;4Serviço De Pediatria, Hospital Joaquim Fernandes, Beja, PT
Background: Autosomal recessive forms of Charcot-Marie-tooth (CMT) represents around 10% of all CMT cases and are more frequent in consanguineous populations. Gypsies are a minority with an estimated frequency of 10-14 million (8 million in Europe), with a high prevalence of intrafamilial marriages and consanguinity. Autosomal recessive forms are mainly represented by demyelinating forms of neuropathies, with 3 known genes associated (NDRG1, HK1, SH3TC2), but axonal forms are more rare and less known. Methods: In our tertiary, university hospital (specialized pediatric neuromuscular consultation), 61 patients were observed by possible hereditary neuropathies in the last 15 years, from whom 16 are from Roma origin. The neuropathy was not confirmed in 2, despite familial history. From 14 patients, 8 presented axonal forms, 7 with an unusual congenital phenotype. Results: We present 7 patients, from 3 families, all consanguineous. Both sexes were represented with no gender predominance. All presented at birth or in first weeks of life, with hypotonia, feeding difficulties, congenital hip luxation, artrogriposis and equinovarus feet. An important scoliosis was present in the first year of life. EMG showed an axonal form of predominantly sensitive neuropathy. Clinical evolution was with severe hand and feet deformities, hallux valgus and adducted thumbs, peroneal atrophy and severe scoliosis. Muscular strength was long preserved and didn’t represent a major clinical feature. Absent osteotendinous reflexes. Independent walk was achieved between 4 and 10 years old. Sensitive ataxia is also present. Conclusion: From the genetic point of view, strongly suggestive of recessive hereditary transmission, only EGRD was done in one patient and recently a NGS panel was done in another – with no identified genetic cause. We are planning to pursue eventually by WES for a common genetic cause of an unusual, severe, very homogeneous phenotype.
PS2Group3-063 / #504ETIOLOGY AND OUTCOME IN NEUROMUSCULAR PATIENTS PRIMARILY PRESENTING WITH DIAPHRAGMATIC DYSFUNCTION
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Matthias Türk1, Irina Weber2, Gernot Vogt-Ladner2, Rolf Schröder3, Martin Winterholler2
1Department Of Neurology, Friedrich-Alexander University Erlangen-Nürnberg (FAU), Erlangen, DE;2Department Of Neurology, Hospital Rummelsberg, Schwarzenbruck, DE;3Neuropathology, University Hospital Erlangen, Erlangen, DE
Background: In neuromuscular disorders uni- or bilateral diaphragmatic dysfunction is caused by affection of the phrenic nerves, the neuromuscular junctions or the diaphragm muscle. Diaphragmatic dysfunction leading to respiratory problems is common in later stages of neuromuscular disorders, but there is little knowledge about its presence as the first symptom of neuromuscular disorders. The aim of this study was to analyze the etiology and clinical outcome in patients presenting with uni- or bilateral diaphragmatic dysfunction but no prior history or diagnosis of a specific neuromuscular disease. Methods: Retrospective analysis of the medical records of patients, who had been treated at the Department of Neurology of the University Hospital Erlangen (1995 – 2001), Germany, and the Department of Neurology of the Rummelsberg Hospital (2002 – 2014), Germany. Inclusion criteria were documented neurophysiological evidence of diaphragmatic dysfunction and absence of previous medical history or diagnosis explaining diaphragmatic dysfunction. Patients suffering from critical illness neuropathy/myopathy were excluded from this study. Medical records were analyzed in terms of medical history, neurological examination, laboratory investigations, cerebrospinal fluid analysis, pulmonary function tests, neurophysiological examinations, and clinical outcome. Results: In total, 30 patients with diaphragmatic dysfunction (17 unilateral; 13 bilateral) were included in this study. Analysis of pulmonary function tests showed that decline of the predicted forced vital capacity (FVC%pred) in supine position, which was pathologic in all tested patients, is a very sensitive parameter to detect diaphragmatic dysfunction. Neurophysiological examination revealed phrenic neuropathy in 28 patients and disorder of the neuromuscular transmission as well as myopathy in one patient each. Extensive clinical, neurophysiological, radiologic and laboratory testing lead to a definite diagnosis in 71% of patients with phrenic neuropathy comprising iatrogenic lesion (n=6), amyotrophic lateral sclerosis (n=5), neuralgic amyotrophy (n=2), neuroborreliosis (n=2), multifocal motor neuropathy (n=1), chronic inflammatory demyelinating neuropathy (n=1), post-polio syndrome (n=1), spondylosis affecting the nerve root C4/5 (n=1) and diabetes mellitus (n=1). In 29% of patients, the etiology of phrenic neuropathy (6 unilateral, 2 bilateral) remained elusive despite extensive work-up. The patients with neurophysiological evidence of a neuromuscular transmission disorder and myopathy were diagnosed with myasthenia gravis and Pompe´s disease, respectively. Outcome analysis of all patients with follow-up data revealed full recovery in 19%, partial recovery in 41%, stable course in 15% and progressive deterioration in 26% of patients. Latter group comprised all patients with amyotrophic lateral sclerosis. Conclusion: Our study highlights that extensive clinical, respiratory, neurophysiological, radiologic and laboratory testing is obligatory to unravel the diagnosis and etiology of diaphragmatic dysfunction especially in consideration of treatable conditions.
PS2Group3-064 / #540IQYMUNE® IS EFFECTIVE AS MAINTENANCE TREATMENT FOR MMN: A RANDOMISED, DOUBLE-BLIND, CROSS-OVER STUDY VERSUS KIOVIG®
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Rabye Ouaja1, Ousmane Alfa Cissé2, Eduardo Nobile-Orazio3, Jean-Marc Léger4
1Immunology Franchise, LFB, LES ULIS , FR;2Global Medical Affairs Unit, LFB, Les Ulis, FR;3Humanita Research Hospital, Neurology Unit, Rozzano, IT;4National Referral Center For Neuromuscular Diseases, University Hospital Pitié-Salpêtrière, Paris, FR
Background: To demonstrate the non-inferiority of Iqymune® versus Kiovig® in the maintenance therapy of MMN, LFB conducted a phase III, multicenter, randomized, double-blind, active-comparator-controlled, cross-over study. Methods: Twenty-two adult MMN patients, treated with any IVIg (except Kiovig® or Iqymune®) at an individualized and stable maintenance dose within the range of 1 to 2 g/kg every 4 to 8 weeks, were enrolled. They were randomized to receive either Kiovig first and then Iqymune (sequence A) or Iqymune first, then Kiovig (sequence B). Each product was administered for 24 weeks. The primary endpoint was the difference between Iqymune® and Kiovig® in the mean assessments of MMRC 10 sum score (strength of 5 muscle groups in upper limbs and 5 muscle groups in lower limbs, both sides, making a score range from 0 to 100) during the evaluation period (non-inferiority margin of Δ = 2). Secondary efficacy endpoints were assessed through 3 other MRC sum scores, INCAT disability score, grip strength, clinical global impression and discontinuation of study treatment. Exploratory objectives were assessed through MMN-Rasch-built Overall Disability Scale (R-ODS), Total serum IgG trough levels, IgM anti-GM1 and anti-GM2 antibodies levels and change in grip strength 2 weeks after the last course compared to the grip strength just before this specific course. Safety was also assessed. Results: Based on a linear mixed-model, the non-inferiority of Iqymune compared to Kiovig was demonstrated, independently from any of the covariates (value at baseline, treatment period and treatment sequence. The difference ‘Iyqymune - Kiovig’ estimate was -0.01 with a 95% CI of [-0.51 to 0.48] that was centered around zero and entirely above the non- inferiority margin of -Δ = -2. No difference was found between products for all secondary endpoints. For categorical efficacy endpoints, analysis was purely descriptive (no statistical test). The number of adverse reactions (AR) and the percentage of affected patients were similar for both products: 39 AR in 45.5% of patients with Iqymune® vs 32 AR in 52.4% of patients with Kiovig®. No thromboembolic event was observed. Biological tests were performed before and after each treatment course. No IVIg-associated hemolysis or renal function impairment was observed. A marked decrease (-25%) in mean neutrophils count was observed after courses with both products, causing sometimes mild and transitory neutropenia. Conclusion: This is the first clinical trial comparing the efficacy and safety of two IVIg in the maintenance treatment of MMN. Iqymune, a novel 10% IVIg, was found non-inferior to the comparator with regards to primary and secondary endpoints.
PS2Group3-065 / #361ANTI-MAG TITERS PRE/POST DEGLYCOSYLATION IN PATIENTS WITH IGM NEUROPATHY: CORRELATION WITH CLINICAL PHENOTYPE IN 8 CASES
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Katherine Beadon1, Jean Neil2, Emilien Delmont3, B Haghi4, L Musset2, S Attarian3, José Boucraut3, Jean-Marc Léger4
1Neurology, University Hospital Pitié-Salpêtrière, Paris, FR;2Immunology, University Hospital Pitié-Salpêtrière, Paris, FR;3University Hospital Timone, Marseille, FR;4Neurology, University Hospital Pitié-Salpêtrière, Paris, FR
Background: Anti myelin associated glycoprotein (MAG) demyelinating neuropathy associated with IgM monoclonal gammopathy (MG) is a dysimmune disorder resulting in symmetric, distal, sensory neuropathy with variable degrees of ataxia and tremor. There is evidence that anti-MAG antibodies are pathogenic including the induction of demyelination in animal models. However, titers of IgM and anti-MAG antibodies do not seem to correlate with clinical severity, evolution or response to treatment in some cases. MAG is a transmembrane glycoprotein located on periaxonal Schwann cells. The target antigen in anti-MAG neuropathy is HNK-1, a carbohydrate epitope on MAG. MAG shows a high affinity for alpha-2,3-linked sialic acid (2,3-SA). Human monoclonal IgM has 5 heavy chain glycosylation sites. IgM may bind to MAG via these glycan epitopes without clinical effect, resulting in overestimation of anti-MAG titers in patient sera. By removing the clinically neutral interactions, there may be an improvement in the correlation with clinical phenotype and evolution. Methods: Serum was analysed in 8 patients with anti-MAG neuropathy and also in 2 patients with IgM MG and anti-MAG antibodies without anti-MAG neuropathy (Non anti-MAG Neuropathy). Clinical features, Overall Neuropathy Limitations Scale (ONLS) and response to treatment were recorded. For 3 patients, serial measurements of anti-MAG activity were performed 1 or more years apart. IgM was extracted and purified by affinity chromatography. Digestion of IgM was performed by Jack bean alpha-mannosidase. Digestion was assessed using lectin blot with Concanavalin A. IgM extracts were then tested for anti-MAG activity pre and post-deglycosylation using ELISA and Indirect Immunofluorescence (IIF). Results: For the 8 first patients, mean duration of illness was 9 years (1-25). Seven had pure sensory and 1 sensorimotor deficits. All had demyelinating neuropathy. Seven out of 8 had an IgM MG and all anti-MAG antibodies. Five patients received immunosuppressant treatment and 3 showed clinical improvement after treatment. Deglycosylation resulted in decrease in anti-MAG activity in all but 1 patient. Only 1 of 6 patients (12.5%) showed a saturation of activity (>70,000 BTU) after deglycosylation vs 6 of 8 patients before (75%). In the two patients with non anti-MAG Neuropathy, deglycosylation resulted in a decrease in titers by a factor of 4.1-6.5 (mean 5.4, 20,500-39,000 BTU pre and 3700-8400 BTU post). No correlation was seen between anti-MAG titers pre or post-deglycosylation and ONLS score or stability of the disease. For the patient with IgM MG, neuropathy and serial serum samples, post-deglycosylation results correlated with IgM titers (IgM 3.7→7.5 g/L, anti-MAG 22,000→55,000 BTU). Conclusion: No correlation between clinical phenotype and anti-MAG activity before or after deglycosylation was found in patients with anti-MAG neuropathy. However, deglycosylation showed interesting results in patients with anti-MAG activity without anti-MAG neuropathy. The activity of anti-MAG antibodies is multifactorial and also depends on the affinity and avidity of the antibody-target interaction. After deglycosylation, the titers may better represent pathogenic activity and help to follow a given patient’s clinical status prospectively.
PS2Group3-066 / #606ULTRASONOGRAPHY IS USEFUL IN EVALUATION FOR ULNAR NEUROPATHIES WITH NO LOCALIZATION IN ELECTROPHYSIOLOGICAL STUDIES
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Jae Young An1, Dae Woong Bae2
1Department Of Neurology, St. Vincent Hospital, The Catholic University of Korea, Suwon, KR;2St. Vincent Hospital, The Catholic University of Korea, Suwon, KR
Background: Ulnar neuropathy is the second most common peripheral nerve entrapment neuropathy. Most common compression site of the nerve is within cubital tunnel. Nerve conduction studies (NCS) are the gold standard for localization of nerve compression site. However, there may be false negative findings of NCS. Mild nerve involvement, improper position, nerve dislocation or Martin-Gruber anastomosis (MGA) may explain some false negative results of NCS. Methods: We describe NCS and ultrasonography (US) findings in two patients with pain and numbness in their right 4th and 5th fingers. They complained of minimal weakness of fourth and fifth finger flexion. Results: Nerve conduction studies showed no definite results about localization of ulnar neuropathy except MGA at the forearm. Electromyography revealed high amplitude in Adductor digiti minimi. US showed an increased cross-sectional area of right ulnar nerve just below and at the medial epicondyle. Conclusion: This case indicates that US may be another important diagnostic tool for evaluation compressive neuropathy, especially in ulnar neuropathy with MGA.
PS2Group3-067 / #990CLINICAL AND GENETIC ANALYSIS OF HEREDITARY PERIPHERAL NEUROPATHY IN EGYPTIAN POPULATION
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Nourelhoda A.A. Haridy1, Jana Vandrovcova2, Mohamed A. Abd El-Hamed3, Sherifa A. Hamed3, Henry Houlden2
1Department Of Neurology And Psychiatry, Assuit University Hospitals, Assiut, EG;2Department Of Molecular Neuroscience, UCL Institute of neurology, London, GB;3Department Of Neurology And Psychiatry, Assuit university hospitals, Assiut, EG
Background: Hereditary neuropathies are the most common inherited neurological diseases. They encompass a range of diseases from those in which the neuropathy is the predominant feature as in Charcot-Marie-Tooth (CMT) and related neuropathies, to those in which the neuropathy occurs as part of a multisystem disease as in Friedreich’s ataxia and leukodystrophy. With the advent of next-generation sequencing, more than 80 genes and loci have been described in causing inherited neuropathies. A lot of studies for inherited neuropathies were carried out worldwide with no studies so far from Egypt. The aim of our study is to characterize the clinical and genetic features of Egyptian patients with hereditary neuropathy. Methods: We recruited 79 patients from 45 unrelated Egyptian families with a clinical diagnosis suggestive of hereditary neuropathy. After an informed consent, all patient were examined neurologically and systemically. Nerve conduction studies were performed to determine the type of neuropathy (Demylinating or axonal). Acquired causes of neuropathies as nutritional were excluded. Blood samples from all probands and first degree relatives (parents and siblings) were obtained when available. Genomic DNA was extracted according to the standard protocol. Multiplex ligation detection probe amplification (MLPA) was performed as the first genetic testing to exclude the commonest type (CMT1A). Whole exome sequencing was performed for negative MLPA results. Validation of variants was carried out by traditional Sanger sequencing and the detected mutations were segregated in other family members when available. Results: Interestingly, no case with CMT1A was diagnosed in our cohort in spite of the clear autosomal dominant pattern in some families along with confirmed and demyelinating features in the nerve conduction studies. We have reached the molecular diagnosis for 24 patients from 12 families out of 45 so far (30.37% patients, 26.66% families). All detected variants were novel in known neuropathy genes like; NEFL, SH3TC2, FGD4, MFN2, KIF1B, FBLN5, ABHD12, LMNA and GDAP1 with one known variant in SPTLC1. Conclusion: From our study, we conclude that PMP22 duplication is not a common cause for Egyptian inherited neuropathy. Other causes like NEFL, MFN2, and SPTLC1 were found as a cause for autosomal dominant neuropathies. SH3TC2 mutations were detected to be the cause of neuropathy in three families so far which suggest that this gene mutation is a common cause of CMT4 in Egypt. Rare gene mutations like FGD4 were described. Work is still progressing for diagnosis of the other families.
PS2Group3-068 / #427THE USEFULNESS OF NERVE CONDUCTION STUDIES IN CHEIRALGIA PARESTHETICA
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
In Soo Joo
Neurology, Ajou University School of Medicine, Suwon, KR
Background: Cheiralgia paresthetica (CP) is an isolated sensory mononeuropathy of the superficial radial nerve. The SRN could be easily damaged by prolonged external compression or diverse procedures. The diagnosis of CP basically relies on the characteristic sensory symptoms, but nerve conduction studies (NCS) may be useful for identifying the exact site or the extent of nerve injury. Methods: Sixteen CP patients having a relevant clinical history and typical physical findings were recruited for last 7 years. The sensory nerve action potentials and sensory nerve conduction velocities were recorded bilaterally using a modified Shirali’s method. Results: Of the 16 patients, 13(81%) were female. Mean age of onset was 47.7 years. Left side was more commonly involved (left : right : both = 8, 6, 2). Symptom duration was 1 day to 1 year. The most common cause of CP was venipuncture (7, 44%), followed by prolonged external compression (4, 25%). Only 5 patients (31.3%) revealed abnormal NCS findings. The prognosis of CP was relatively good, and did not show any significant correlation with NCS findings. Conclusion: For the diagnosis of CP, taking a careful history and performing a thorough physical examination are more important than the NCS, at least in such case of external compression or minor nerve injury.
PS2Group3-069 / #728BACLOFEN, NALTREXONE AND SORBITOL ALL CONTRIBUTE TO PXT3003-INDUCED MYELINATION IN CMT1A DRG CO-CULTURES
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Nathalie Cholet1, Julien Laffaire2, Rodolphe Hajj3, Daniel Cohen4
1Experimental Biology, Pharnext, Issy-les-Moulineaux, FR;2Biostatistics, Pharnext, Issy Les Moulineaux, FR;3Experimental Biology, PHARNEXT, Issy-les-Moulineaux, FR;4Ceo, PHARNEXT, Issy-les-Moulineaux, FR
Background: Charcot-Marie-Tooth type 1A disease (CMT1A) is due to the duplication of the gene encoding the peripheral myelin protein of 22 kDa (PMP22). CMT1A is characterized by a dys/demyelination of peripheral nerves associated with a reduced nerve conduction velocity of both motor and sensory fibers, all leading to a progressive muscle weakness and loss of proprioception. Although CMT1A is the most common inherited neuropathy, there is currently no approved treatment. We previously showed that a combination of 3 drugs, baclofen, sorbitol and naltrexone (PXT3003) was effective in pre-clinical CMT1A models as well as in a Phase 2 clinical study in CMT1A patients. The aim of this work was to i) demonstrate the ability of PXT3003 to induce myelination of CMT1A neurons, and ii) prove that baclofen, naltrexone and sorbitol all contribute to this myelination induction by comparing the activity of PXT3003 to the drugs alone or combined two by two (duos). Methods: Transgenic rodent models based on Pmp22 overexpression develop a demyelinating neuropathy mimicking the human disease and offer the possibility to modelize CMT1A features in vitro. Hence, we developed an in vitro model of sensory neurons and Schwann cells co-culture derived from CMT1A transgenic rats that was validated and adapted to 96-well culture plates for drug testing. Total myelin length was quantified with an automatic image analyzer after PMP22 immunostaining. This model allows us to mimic the physiological process of in vivo myelination. Results: We confirmed myelination impairment of CMT1A neurons, as assessed by a decrease of total myelin length in CMT1A cultures compared to wild-type. We subsequently performed a full dose-response analysis of single drugs and showed their own promyelinating dose-dependent activities. We then tested 27 combinations of the three drugs at their sub-active concentrations to assess their synergistic activity when put all together (PXT3003). We found that 5 out of these 3 drug combinations induced myelination whereas none of the 3 possible 2X2 combinations revealed a significant effect at the same sub-active concentrations, suggesting a synergy between the 3 drugs. Conclusion: We validated a sophisticated co-culture system mimicking the pathophysiological myelination process in CMT1A disease. We confirmed PXT3003 activity on myelination induction and clearly demonstrated the contribution of the 3 drugs to the effect of PXT3003 for the treatment of CMT1A. These findings highlight the value of combinational repurposing of drugs at low doses to act synergistically on pleiotropic pathways and represent an important novel approach for rapid drug development and safety improvement.
PS2Group3-070 / #922AN AUTOSOMAL DOMINANT FAMILY WITH UNCOMPLICATED SPASTIC PARAPLEGIA DUE TO A STOP MUTATION IN HARS
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Nicolas Dubuisson1, Michel Dupuis2, Laurent Servais3, V Benoit4, Christine Verellen-Dumoulin4
1Neurology, Avenue Hippocrate 10,, Bruxelles, BE;2Clinique Saint-Pierre Ottignies, Ottignies, BE;3Institute of Myology, Paris, FR;4Institut de Pathologie et de Génétique (IPG) Gosselies, Gosselies, BE
Background: The hereditary spastic paraplegias (HSPs) are clinically and genetically heterogeneous disorders. HSPs are classified as «uncomplicated» if neurological impairment is limited to lower extremity spastic weakness and as «complicated» if the impairment of the uncomplicated HSP is accompanied by other systemic or neurologic abnormalities such as peripheral neuropathy. Methods: A 54 years old woman has been suffering since her thirties from HSP and one of her sons developed the same clinical condition before 10 years of age.The mother is still able to walk after 20 years of evolution. Both patients have brisk reflexes with clonus and Babinski sign. There are no clinical nor electrophysiological signs of peripheral involvement. Results: For the 2 patients, various sets of HSP associated genes were tested negative by Sanger sequencing and NGS. Mendeliome sequencing of 2742 genes (Capture SureSelect Inherited Disease , Agilent followed by sequencing Illumina) performed on the mother ,her husband and their son has shown a stop mutation in HARS (NM–002109.05) to the HSP patients.This mutation c.499C>T p.(Arg167*) has not been described yet. Conclusion: Hars mutation has been reported in 4 families harbouring autosomal dominant axonal and demyelinating motor and sensory neuropathies (HMSN) (Brozkova et al 2015). This gene encodes a histidyl-tRNA synthetase and all the described variations were missense variants.This family is the fifth described family with a HARS variant and the uncomplicated HSP clinical presentation is unique and expands the clinical symptoms from HMSN to autosomal dominant SP. For genetic counseling this gene should be added to the known 41 HSP related genes.
PS2Group3-071 / #259CONDUCTION STUDIES OF PHRENIC NERVE IN HEALTHY CHILDREN
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Vladislav Voitenkov1, Evgenia Ekusheva2, Natalia Skripchenko3
1Clinical Neurophysiology, Pediatric Research and Clinical Center for Infectious Diseases, Saint-Petersburg, RU;2Advanced Training Institute of the Federal Medical Biological Agency, Moscow, RU;3Pediatric Research and Clinical Center for Infectious Diseases, Saint-Petersburg, RU
Background: In adult population conduction studies parameters of phrenic nerve (its CMAP latency and amplitude) are quite well established. In children, on the contrary, these parameters change with the age and tend to disperse widely. Our goal was to establish our own set of normative data for latency and amplitude of phrenic nerve CMAP in healthy children of different age. Methods: 48 healthy children (mean age 9,19±5,43, range 1-18 years, 28 females, 20 males) were enrolled. Conduction studies were performed with active electrode at xiphoid process and reference at the VII intercostal space, with stimulation at the external edge of the lower third of sternocleidomastoid muscle, with maximum stimulation at 40 mA. Additional electrodes were placed on m.Deltoideus and m.Serratus Anterior to control the amount of ecitatory output to the ce rvical plexus (Fig.1). Results: Mean CMAP latency in the group was 5,64±1,25 ms and amplitude was 0,66±0,34 μV. As the group was subdivided on age subgroups as follows: 1-2 years (n=7), 3-5 years (n=9), 6-12 years (n=14) and 13-18 years (n=17), CMAP latencies were 4,96±1,94; 5,01±1,13; 5,42±0,84 and 6,44±1,43 ms and CMAP amplitudes were 1,01±0,37; 0,87±0,31; 0,61±0,24 and 0,45±0,21 μV. CMAP amplitudes in very young children aged 1-2 years were significantly (p<0.05) higher than in children aged 6-12 and 13-18 years. Typical CMAP is presented on Fig. 2. Conclusion: Conduction study of the phrenic nerve, if performed properly, is a simple and reliable method. In pediatric population, though, age dynamic and wide range of normative parameters has to be taken into the consideration. In children CMAP amplitude is significantly higher at the age under 2 years than in older ones (6-18 years).
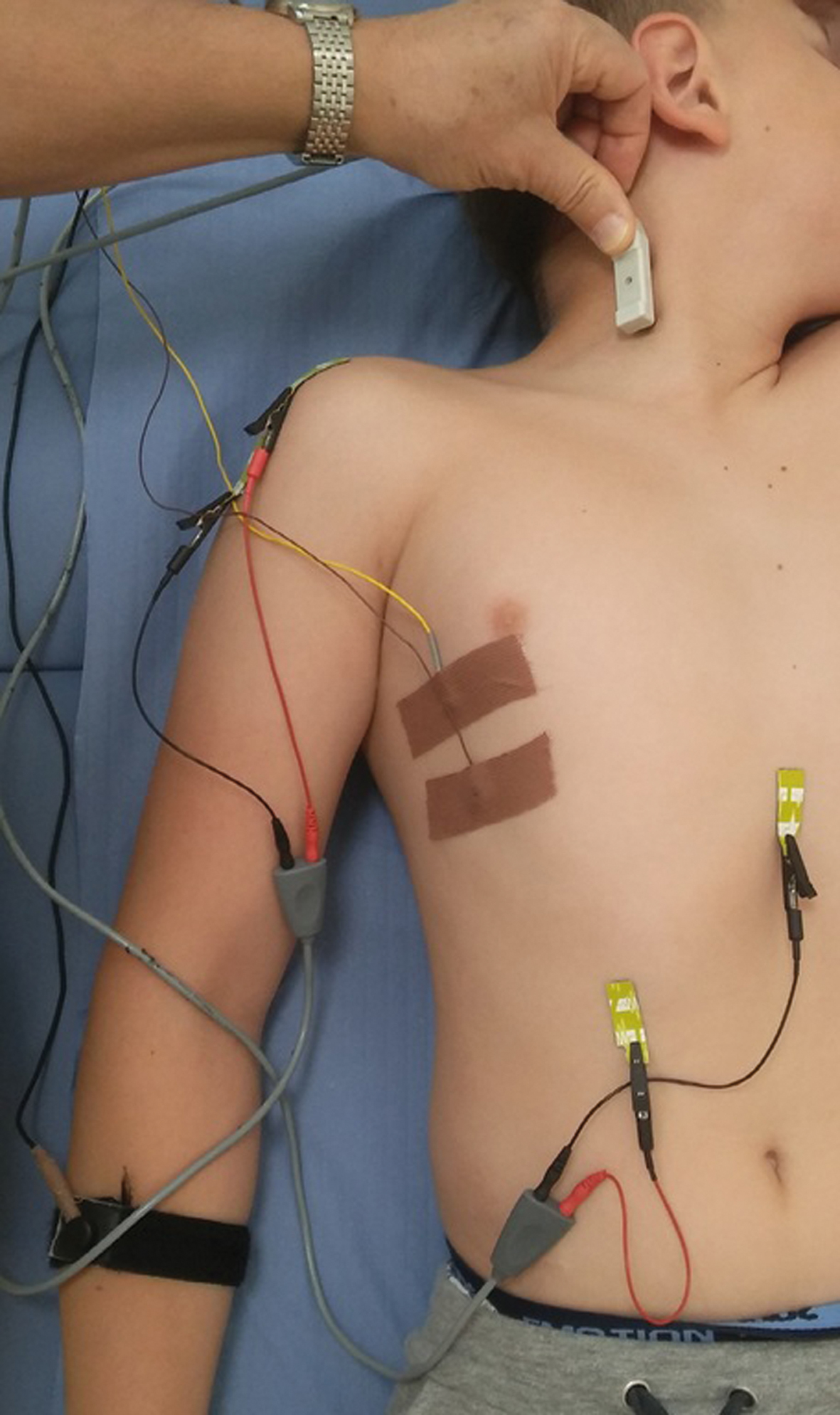
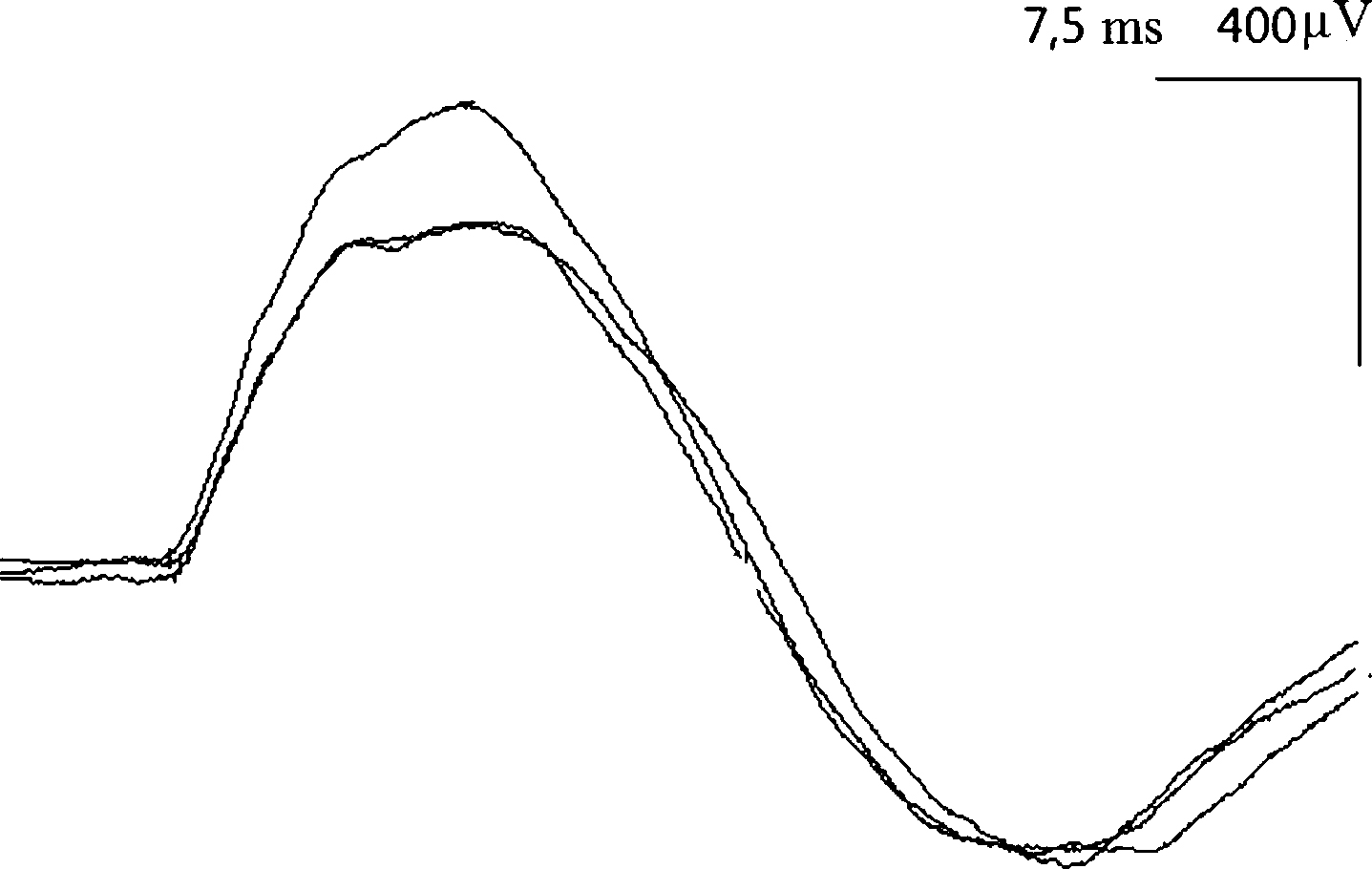
PS2Group3-072 / #310FIBER TRACTOGRAPHY OF FACIAL NERVE: IMPLICATIONS FOR HEMIFACIAL SPASM
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Yeow Hoay Koh1, Ling Ling Chan2, Ee Wei Lim2
1Neurology, Singapore General Hospital, Singapore, SG;2Singapore General Hospital, Singapore, SG
Background: Diffusion tensor imaging and fiber tractography of the facial nerve is particularly challenging due to its slenderness and anatomic position in an area fraught with susceptibility and CSF pulsation artifacts. Nevertheless, there is potential utility in surgical navigation and semi-quantitative disease monitoring of response to treatment in various 7-8th CN pathologies including hemifacial spasm. Methods: Sub-mm inplane resolution DTI scans over the facial nerves were acquired. 3D MR angiography and heavily T2-weighted high resolution scans were also obtained. Fiber tracking of the 7-8th nerves was performed using commercially available software. The FA and MD of the 7-8th nerves tracked are recorded and the DTI tractography quality is compared to that reported in the literature. Results: 21 subjects (9 men, 12 women) with mean age of 61 years (37-84) were scanned. The 7-8th nerves were successfully tracked in 11 subjects. The 7th nerve can be seen separately from the 8th nerve in 5 subjects. The mean DTI parametrics are: fractional anisotropy 0.301 ± 0.026 (0.271-0.341) and mean diffusivity 4820 ± 492 x 10-6 mm2/sec (4000-5600 x 10-6 mm2/sec). The DTI tractography is compatible with the expected DTI findings of the 7-8th nerves, supporting the validity of our imaging methodology. Conclusion: Our initial data demonstrate that DTI fiber tracking of the facial nerve is clinically feasible and would be useful for evaluation of clinical conditions such as hemifacial spasm.
PS2Group3-073 / #860NEUROMYOPATHY CAUSED BY LONG TERM COLCHICINE THERAPY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Min Young Chun, Jung Hwan Lee, Kee-Duk Park
Neurology, Mokdong Hospital, Ewha Womans University College of Medicine, Seoul, KR
Background: Colchicine is commonly used for the treatment of gout, familial Mediterranean fever, Behcet’s disease, and recurring pericarditis with effusion. The use of colchicine is usually safe, but several adverse effects such as gastrointestinal intolerance are known. Neuromyopathy is rare complication and can develop in the setting of acute overdose or chronic administration in therapeutic doses. Two cases of colchicine induced neuromyopathy has been previously reported as acute toxicity in Korea. We reported on a case of neuromyopathy after long term colchicine therapy in Korea. Methods: A 72-year-old man was referred to our institution due to 5 months history of proximal muscle weakness and myalgia. He had angina pectoris and Behcet’s disease, leading to the treatment of colchicine (1.2mg daily for about 6 years), cyclosporine, methylprednisolone, simvastatin, and aspirin. Neurologic examination revealed proximal muscle weakness and muscle tenderness on palpitation. Hypesthesia was only present in the tiptoes. Deep tendon reflexes are absent. Results: Laboratory studies for common causes of neuropathy were normal. The serum cyclosporine level was low (45.10 ng/mL). The serum creatine kinase was elevated (1130 IU/L). The serum creatinine level was in normal rang. Chest and abdominopelvic CT scans did not show any malignant lesions. Nerve conduction studies showed axonal sensorimotor polyneuropathy and needle electromyography revealed a myopathic pattern. Muscle biopsy of biceps brachii demonstrated myofibers with mild variation of size and shape and occasionally noted degenerated or necrotic myofibers. On electron microscopic examination, degenerative or necrotic myofibers show disruption of intermyofibrillar pattern and degenerated organelles with frequently noted autophagic vacuoles. Myofibers with prominent subsarcolemmal accumulation of autophagic vacuoles were also noted. In addition, there were myofibers with smeared Z line material and myofilament disruption. He was initially suspected with inflammatory myopathy and was treated with steroid pulse therapy. However, muscle weakness did not improve. So, we considered colchicine-induced neuromyopathy and discontinued the colchicine. Then, muscular weakness and myalgia improved steadily, and serum creatine kinase level became decreased into normal range (25 IU/L). Three months later, his muscle power improved, and he could walk without a cane. Conclusion: Colchicine induced neuromyopathy is a rare toxic complication. However, we should consider ‘colchicine induced neuromyopathy’ for the patients, who present muscular weakness of unknown origin in a condition of treatment with colchicine because that clinical suspicion is the most important for the diagnosis and treatment is just only the termination of colchicine. In conclusion, this is the first report of the neuromyopathy after long term colchicine therapy in Korea, and we should be cautious to use colchicine.
PS2Group3-074 / #372CHARCOT MARIE TOOTH DISEASE TYPE 2CC DUE TO A MUTATION IN THE NEUROFILAMENT-HEAVY GENE IN A GERMAN FAMILY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Elena Ikenberg1, Peter Reilich2, Corinna Heller3, Angela Abicht4, Benedikt Schoser2, Maggie C. Walter4
1Friedrich-baur Institute, Departement Of Neurology, Ludwigs-Maximilians University of Munich, Munich, DE;2Friedrich-baur Institute, Departement Of Neurology, Ludwigs-Maximilians University of Munich, Munich, DE;3CeGaT GmbH und Praxis für Humangenetik, Tübingen, DE;4Dept. Of Neurology, Ludwig-maximilians-university, Friedrich-Baur-Institute, Munich, DE
Background: Neurofilaments are structural components of motor axons. Accumulation of neurofilament occurs in various pathological conditions. Recently different variants resulting in translation of a cryptic amyloidogenic element (CAE) in the 3' untranslated region (UTR) of the neurofilament-heavy polypeptide (NFH) gene have been described to cause charcot marie tooth disease type 2CC (CMT2CC) by forming amyloidogenic toxic protein aggregation. Here we describe the first German pedigree suffering from CMT2CC and give an overview about the features of CMT2CC described so far. Methods: Patients: This study examined a German autosomal dominant CMT2 family with two affected members. Patient 1: Around the age of 8 years the patient experienced difficulties in running and squatting. At the age of 16 a muscle biopsy showing myopathic changes in line with muscular atrophy was performed when he underwent Achilles tendon surgery due to a neurogenic clubfoot. Neurological examination at age 34 displayed marked symmetric atrophy and weakness of the ventral thighs and extensor and flexor muscles of the feet. Tendon reflexes were absent. A distal hypoesthesia from the ankles to the toes was noted. Electromyography revealed a severe neuropathic pattern and pseudomyotonic runs in tibialis anterior muscle. Nerve-conduction-velocity studies were consistent with a motor and sensory axonal neuropathy of the lower limbs. Patient 2: The father of the patient developed a weakness of the lower limbs when he was 16. At the age of 34 he noticed a mild weakness of the upper limbs. The father is 74 years old and wheelchair bound since 64. A muscle biopsy in 1978 showed features suggestive of denervation. The electromyogram of the lower limbs showed a chronic neuropathic pattern,. Nerve-conduction-velocity studies of the motoric medianus were mildly elongated. Genetic analysis: Genomic DNA was purified from peripheral blood. Next generation sequencing identified a heterozygous mutation c.3057dupG (p.K1020Efs*43) in the NFH gene. Results: We identified a mid-adult onset family of two generations carrying a mutation of c.3057dupG (p.K1020Efs*43) in NFH. Until now, only five pedigrees have been reported worldwide. Our patients presented with a similar symptom profile to that of the previously reported. Clinical features include progressive muscle weakness and muscle wasting, predominantly affecting the lower limbs. Mild sensory deficits and absent deep tendon reflexes are also common. Electrophysiological studies show a symmetrical, distal and proximal sensorimotor axonal neuropathy and can be pathological even before clinical symptoms appear. In addition to classic CMT features some patients are also reported to have increased serum creatine kinase, myopathic changes in electromyographic studies and pyramidal signs. Conclusion: Here we report of the first German pedigree suffering from CMT2CC due to an autosomal dominant mutation of c.3057dupG (p.K1020Efs*43) in NFH. The father of our patient was misdiagnosed with spinal muscular atrophy, while our patient initially presented with a distal myopathy phenotype; therefore, the clinical spectrum can mimic motoneuron and muscle phenotypes. Our observation further expands the phenotypic spectrum of NFH mutations and indicates that NFH mutations should be considered in the differential diagnosis of patients presenting with gait disturbances, muscular weakness and muscle wasting predominantly affecting the lower limbs.
PS2Group3-075 / #550ISOLATED ABDUCENS PALSY AS TOLOSA-HUNT SYNDROME IN A SYSTEMIC LUPUS ERYTHEMATOSUS PATIENT
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Valeria Serban
Neurology Clinic, Cluj, RO
Background: Tolosa Hunt Syndrome (THS) is a rare occurrence (one case per million per year) first described by Tolosa in 1954 and explored further by Hunt publishing six cases of remittent unilateral retro-orbital pain and extra-ocular nerve palsies (third, fourth, sixth) leading to diplopia, responding well to steroids. Frontal parenthesis can be present as well due to fifth cranial nerve involvement. It is caused by idiopathic granulomatous inflammation. We report a rare case of THS presenting as new onset isolated sixth nerve palsy in a young patient known with systemic lupus erythematosus (SLE). Methods: Twenty seven year-old right-handed lady presented with new onset frontal headache and diplopia persisting for the past several days. No previous similar symptoms. She had no ptosis, limb weakness nor parenthesis. She carried a diagnosis of lupus treated with Plaquinil and low dose steroids for several years On exam there was isolated sixth nerve palsy on the right. However, third, fourth and fifth nerves on the right were not clinically affected. The rest of the exam was unremarkable. Results: AAN was positive in high titers as well as anti-phospholipid antibodies. Auto-antibodies to AchR, MuSK, titin, striated muscle were all absent. In addition, ANCA and rheumatoid factor were not present. TSH was normal. Cerebrospinal fluid (CSF) had normal values and infectious etiology was ruled out. MRI brain and MRA head with and without contrast revealed increased density in the right cavernous sinus suggestive for inflammatory tissue. There was a fair clinical response to high dose steroids. Diplopia improved in several days. Plaquinil was continued, as well. Repeat MRI and MRA head revealed partial resolution of the inflammation present in the right cavernous sinus. Conclusion: This case is unique due to several features. First, the clinical presentation was not the classic painful ophthalmoplegia involving third, fourth, sixth nerves plus frontal parenthesis due to fifth cranial nerve involvement. Instead patient presented with isolated sixth nerve palsy and frontal headaches. Second, the response to steroids was not as spectacular as previously reported. And third, our patient already had an auto-immune condition (SLE) treated with steroids and immunosuppression. Possible lupus to have a role in the pathogenesis of the inflammation developed in the right cavernous sinus. Infectious component was ruled out. Further investigation is needed for a better understanding.
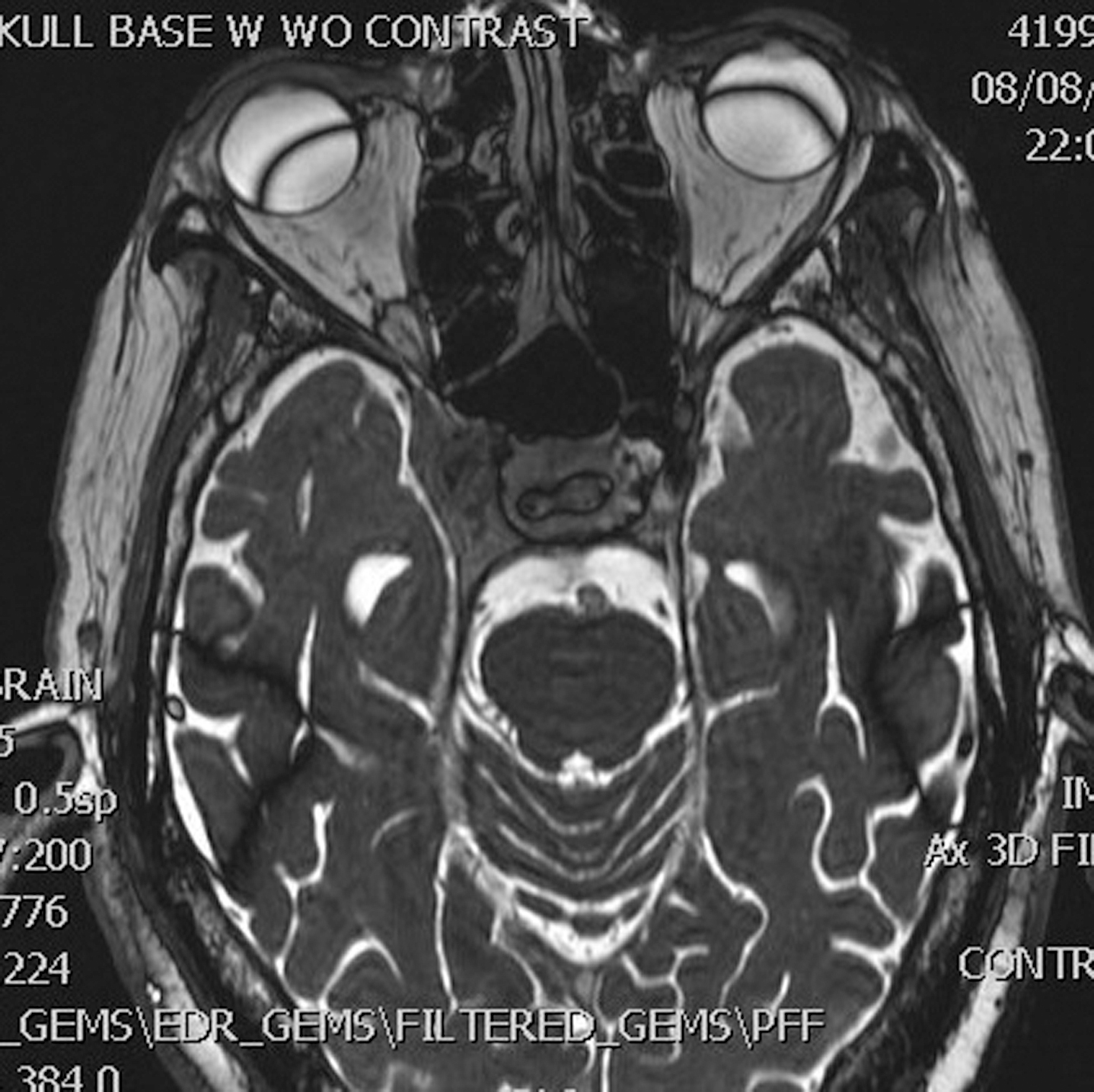
PS2Group3-076 / #751MULTIFOCAL MOTOR NEUROPATHY IN AUSTRIA: A NATIONWIDE SURVEY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Eva-Maria Oberreiter1, Stefan Quasthoff2, Marcus Erdler3, Raffi Topakian4, Susanna Grinzinger5, Friedrich Zimprich6, Karl Stieglbauer7, Julia Wanschitz1, Stephan Höger8, Klaus Berek9, Claudia Thaler-Wolf10, Norbert Embacher11, Julia Jecel12, Isolde Heß-Eberle13, Waltraud Kleindienst5, Michael Huemer14, Eva Laich15, Dierk Oel16, Wolfgang Oertl17, Eva Lenzenweger17, Wolfagang Löscher1
1Medical University of Innsbruck, Innsbruck, AT;2Medical University Graz, Graz, AT;3SMZ Ost, Vienna, AT;4Neurology, Klinikum Wels-Grieskirchen, Wels, AT;5Paracelus University Salzburg, Salzburg, AT;6Department Of Neurology, Medical University of Vienna, Vienna, AT;7Private Practice, Linz, AT;8LKH Graz Süd-West, Graz, AT;9BKH Kufstein, Kufstein, AT;10Private Practice, Hall iT, AT;11University Clinic St. Pölten, St. Pölten, AT;12Krankenhaus Hiezing, Vienna, AT;13Private Practice, Salzburg, AT;14Kardinal Schwarzenberg Klinikum, Schwarzach im Pongau, AT;15LKH Steyr, Steyr, AT;16Neurology, Klinikum Wels-Grieskirchen, Wels, AT;17Kepler-Universitätsklinikum Linz, Linz, AT
Background: Multifocal Motor Neuropathy (MMN) is a rare immune-mediated neuropathy, which leads to slowly progressive, asymmetric muscle weaknesses. An electrophysiological motor conduction block (CB) is the hallmark of this disorder. With early diagnosis and treatment with intravenous immunoglobulins (IVIG) MMN is a treatable disorder, at least during the first years of disease. Due to its rarity, data on clinical features and response to long-term treatment are sparse and limited to a few studies. The aim of this study was to provide a survey of Austrian MMN patients and a comprehensive review of epidemiological and clinical features of MMN cases in Austria and their response to treatment. Also, we compared our results with previously published studies. Methods: Austrian neurologists were contacted via the Austrian Neurological Society (ÖGN) asked to provide anonymized data on MMN patients under their care. A total of 57 patients were reported in Austria which have been diagnosed between 1993 and 2017. We performed an exploratory analysis of clinical, electrophysiological and laboratory data, as well as IVIG use and treatment outcome. Results: The point prevalence of MMN in Austria is 0,65 / 100.000. The male to female ratio was 2,2:1 and the median age of symptom onset was 45 years. Mean diagnostic delay was 5,4 ± 8,33 ys., but there was a decline in diagnostic delay in relation to symptom onset; i.e. mean diagnostic delay was 17 ys, when symptoms began between 1982-86 and was 0,9 ys when symptom onset was between 2012-2017. MMN typically began in the upper extremity (77,2%) and in distal muscles (87,7%). CB was found in 95 % of the patients, most frequently in the ulnar and median nerves. 43 % showed IgM antibodies against GM1. 51 of 57 patients were treated with IVIG at median intervals of 4 weeks with a mean dosage of 1,55g/kg. High muscle strength was a favorable prognostic factor, while elevated serum igM anti-GM1 antibody titer, muscle atrophy and long intervals of treatment were negative prognostic factors. Duration of treatment correlated with outcome: the longer the treatment, the worse the outcome. IVIG improved muscle strength, but frequently lost its effectiveness after treatment duration of > 7 years. Conclusion: These results show that the prevalence of MMN in Austria is similar to the few reports from other countries. Also, the present study corroborates previous results on clinical, electrophysiological and laboratory features in MMN. Intravenous Immunoglobulins are still the most effective treatment; however, deterioration of symptoms during long-term treatment though occurs in most cases.
PS2Group3-077 / #973AUTOSOMAL RECESSIVE CHARCOT MARIE TOOTH DISEASE: CLINICAL, ELECTROPHYSIOLOGY AND GENETIC SPECTRUM IN A TUNISIAN SERIES
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Imen Kacem1, Emna Ellouz2, Mouna Ben Djebara1, Eric Leguern3, Amina Gargouri1, Riadh Gouider4
1razi hospital, tunis, TN;2Neurology, Gabes Hospital, ibn khaldoun street gabes, TN;3salpetriere, paris, FR;4Neurology, Razi Hospital, Manouba, TN
Background: Charcot Marie Tooth disease is a genetically heterogeneous group of hereditary motor and sensory neuropathies. Autosomal Recessive forms (AR CMT) are known to be the least common forms worldwide but still frequent in the Mediterranean basin because of high rate of consanguinity. Objectives We aimed to investigate clinical, electrophysiological and genetic features of AR CMT in a Tunisian cohort. Methods: Patients and Methods We performed a prospective study of a cohort of AR CMT Tunisian patients followed in the neurology department of Razi Hospital, Tunisia from June 2002 to December 2017. Clinical and electrophysiological data were analyzed. Neuropathy was classified in axonal or demyelinating form according to nerve conduction velocities in upper limb motor nerves NCVs. Disability was assessed by the Overall Neuropathy Limitations Scale (ONLS) and CMT neuropathy scale (CMTNS). Genetic study was made to all probands in Neurogenetic Department of Pitié Salpêtrière-France. Written informed consent was obtained from all of the participants. Results: We included 65 patients belonging to 43 families. Mean age at diagnosis was 20.8 years. Consanguinity was found in 94 %. Mean age of onset was 3.7 years. Delay of milestone was found in 11% of cases. Classical phenotype was found in the majority of patients and uncommon presentation as pyramidal syndrome or glaucoma were found in two families.Axonal neuropathy was found in 48.5% cases and demyelinating one in 51.5% cases. Mean ONLS score was 4.7 and mean CMTNS one was 12.2%. Axonal polyneuropathy was more frequently associated to an early onset (90.9%)(p<0.001), with more severe weakness (p=0.04) and more severe ONLS score. Gene mutations were identified in 15 families with predominance of demyelinating forms. The GDAP1gene mutations were the most frequent (26.3%).Four novel mutations were detected and characterized phenotypically. Conclusion: Discussion We reported a large AR CMT cohort with a high consanguinity rate. Recessive CMT forms, are more severe and have an earlier onset than the dominant forms. They usually show pure CMT clinical phenotype; however, some forms can be associated to cyphoscoliosis, deafness or glaucoma. Demyelinating forms (CMT 4) were more frequent but axonal ones (AR-CMT 2) were more severe with rapid progress. In concordance with literature, Mutations in GDAP1 are a frequent cause of AR CMT. Conclusion AR CMT are common in Tunisia. Comparatively to dominant forms, AR ones were characterized by an earlier onset, more severe clinical features and with important handicap. Electrophysiological results and associated signs must be considered to guide molecular study. GDAP1 gene mutations still the most frequent mutation in these cases.
PS2Group3-078 / #257COLCHICINE INDUCED NEUROMYOPATHY IN A PATIENT USING CONCOMITANT DIURETICS
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Young-Do Kim
Neurology, Incheon St. Mary’s Hospital, The Catholic University of Korea, Incheon, KR
Background: Colchicine is commonly used to treat gout. The common side effects of colchicine include gastroenteritis, blood dyscrasias, and dermatitis; the neuromuscular toxicity associated with colchicines is under-recognized. The characteristic neuromuscular complication is a neuromyopathy, presenting with subacute proximal lower extremity weakness, elevated serum creatine kinase (CK) and electromyography (EMG) showing an ‘‘irritable myopathy’’ and a length-dependent sensorimotor peripheral neuropathy. We present a patient with colchicine-induced neuromyopathy using comcomitant diuretics. Methods: Case : An 70-year-old woman with a significant past medical history including hypertension, heart failure with pericardial effusion, liver cirrhosis with ascites, hypothyroidism and hyperuricemia presented to our clinic for evaluation of general weakness for 1 month. The weakness started in the both thighs and lower legs about 4 weeks prior to presentation and progressively worsened. She complained of myalgia in the arms and legs with mild paresthesia and hypoesthesia. She denied cranial nerve deficits or bladder and bowel difficulty. But, she compained of continous diarrhea for 3 weeks. She was on colchicine 1.2 mg per day, which she had been taking for several months for pericardial effusion and hyperuricemia. Also, she had been taking diuretics (furosemide 80mg, spironolactone 25mg) for 2 months due to generalized edema and pleural effusion. Results: On examination, cognition and cranial nerves were normal. She had both proximal and distal upper limb weakness (Medical Research Council [MRC] grade 3/5); severe hip flexor and dorsiflexor weakness (MRC grade 2–3/5) and diffuse lower limb weakness otherwise (MRC grade 3/5). She had graded sensory loss to pain and cold sensation up to the knees,reduced vibration sensation at the ankles and reduced joint position sensation at the toes. Laboratory investigations revealed high creatinine (1.3 mg/dL; normal range 0.6–1.1). Serum CK (1581 U/mL; normal range 52–336) and aldolase (9.4 U/mL; normal range 1–7.5) were both elevated. Nerve conduction studies revealed sensorimotor polyneuropathy of axonal type. Needle electrode examination demonstrated fibrillation, postitive sharp and myotonic potentials in distal extremity muscles; motor unit potentials were of varying duration and amplitudes in various muscles diffusely. These clinical, laboratory and electrophysiologic findings were consistent with colchicine-induced neuromyopathy aggravated by diuretics. Colchicine was discontinued. Two months later the patient had no weakness in the arms with mild weakness in the lower limbs (MRC grade 4/5). Conclusion: The neuromuscular complications associated with colchicine are commonly under-recognized. Chronic renal dysfunction or diuretic use makes patients more susceptible to this neuromuscular complication at the usual dose range. Co administration of any drug that is metabolized by the CYP3A4 system can induce colchicine myopathy. Laboratory investigations usually show mild to marked elevation of CK. The characteristic electrodiagnostic findings include myopathic motor unit potentials with features of ‘‘membrane irritability’’ as may be seen in inflammatory myopathy and a coexistent sensorimotor axonal polyneuropathy. The common diagnostic consideration in patients with colchicine myoneuropathy is polymyositis because of predominant proximal weakness, elevation of CK and irritable myopathy on EMG.
PS2Group3-079 / #389A CASE OF MULTIFOCAL MOTOR NEUROPATHY: COMPLEMENTARY ROLE OF ULTRASOUND
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Jung Im Suk1, Hae Joo Rha2
1Neurology, Catholic University of Daegu, School of Medicine, Daegu, KR;2Catholic University of Daegu, School of Medicine, Daegu, KR
Background: Multifocal motor neuropathy (MMN) is an uncommon, asymmetric motor neuropathy. As MMN is a treatable disorder, its differentiation from lower motor neuron disease is important. Evidence of conduction block (CB) or positive IgM anti-GM1 is considered one of important markers for the diagnosis. However, some patients with atypical MMN have no detectable CB or anti-GM1 antibodies.
Methods: A 40-year-old man presented with two-year history of progressive left-sided weakness without sensory symptoms. We experienced a case of MMN with focal nerve enlargement on ultrasound. Results: Nerve conduction study revealed partial conduction block in two nerves. Anti-GM1 antibody is negative and other laboratory studies were unremarkable. Ultrasound showed focal nerve enlargement of right median nerve at the forearm and left ulnar nerve at the elbow (Fig. 1). Weakness improved completely after intravenous immunoglobulin. Ultrasound image shows focal enlargement of median and ulnar nerve (arrow). (A and B) The cross-sectional area (CSA) of right (A) and left (B) median nerve at the wrist is normal. (C and D) The CSA of right (C) median nerve at the forearm is increased at 15.2 mm2 and left (D) median nerve is normal (5.9 mm2). (E and F) The CSA of right (E) and left (F) median nerve at the upper arm is normal. (G and H) The CSA of right (G) and left (H) ulnar nerve at the wrist is normal. (I and J) The CSA of right (I) ulnar nerve at the elbow is normal (3.5 mm2), but left (J) ulnar nerve is increased at 11.1 mm2. t; tendon, c; carpal bone, fds; flexor digitorum superficialis, fdp; flexor digitorum profundus, b; brachial artery, br; brachialis, u; ulnar artery, me; medial epicondyle. Conclusion: Ultrasound can be a valuable tool in supporting the diagnosis of MMN.
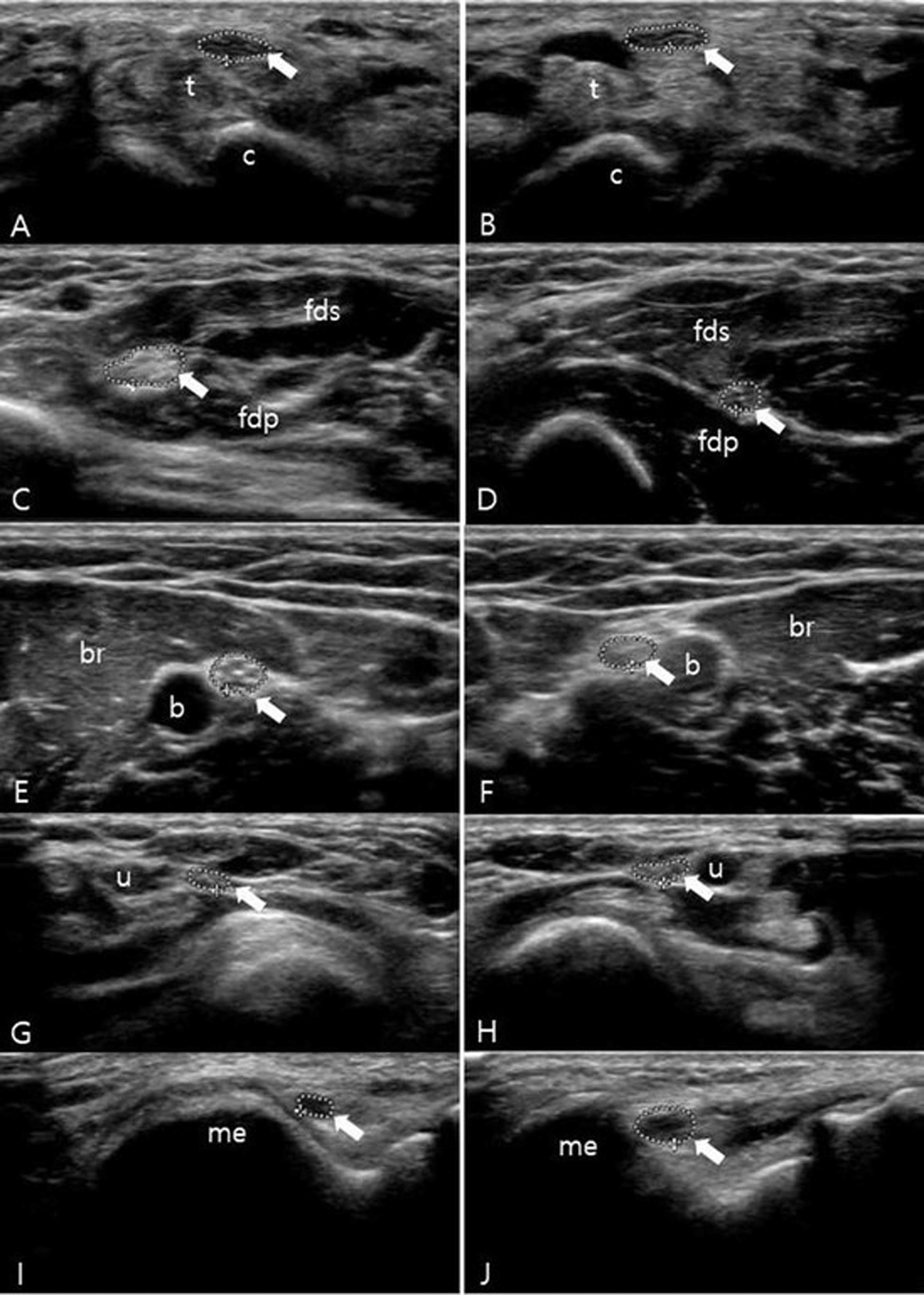
PS2Group3-080 / #568PHARMACOKINETICS OF PATISIRAN IN PATIENTS WITH HEREDITARY TRANSTHYRETIN-MEDIATED AMYLOIDOSIS
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Xiaoping Zhang, Varun Goel, Gabriel Robbie
Alnylam Pharmaceuticals, Cambridge, MA, US
Background: Patisiran is an investigational RNA interference therapeutic in development for the treatment of hereditary transthyretin-mediated amyloidosis (hATTR). Early clinical development has demonstrated that patisiran was generally well tolerated and elicited robust, rapid, and sustained lowering of TTR protein levels in healthy volunteers and in patients. Here, we report pharmacokinetic (PK) data from the long-term, open-label Phase 2 study in patients (NCT01961921). Methods: Adult patients (N=27) received patisiran 0.3mg/kg intravenously (IV) q3w over 24 months. Intensive plasma PK sampling was performed on Weeks 1, 34, and 106 for the characterization of PK properties of ALN-18328 (siRNA) and 2 novel lipid excipients (DLin-MC3-DMA and PEG2000-C-DMG). Plasma PK parameters were computed using non-compartmental analysis. Achievement of steady state was also assessed by measuring maximum plasma concentration at the end of infusion (Cmax) and pre-dose concentration (Ctrough) over 24 months of treatment. Results: After IV infusion of patisiran in patients, all 3 analytes exhibited multiphasic plasma profiles (Figure 1).PK parameters are summarized in Table 1. PK profiles from all 3 analytes were characterized by a short half-life (t1/2a) in the first phase and a relatively long terminal half-life (t1/2b). Cmax and/or Ctrough increased over time and approached steady state by week 24. Accumulation of area under the curve at steady state compared to the first dose (AUCtau) was 3.2- fold for ALN-18328 and 1.7- fold for DLin-MC3-DMA, whereas no accumulation for PEG2000-C-DMG. The mean volume of distribution (Vss) for ALN-18328, DLin-MC3-DMA, and PEG2000-C-DMG was 3.43-, 6.33-, and 1.75-fold higher, respectively, than the blood volume in a typical 70-kg human, indicating distribution outside the vasculature. Conclusion: After chronic dosing of patisiran 0.3 mg/kg q3w, steady state was reached by week 24 with moderate accumulation. PK of patisiran components were stable over time.
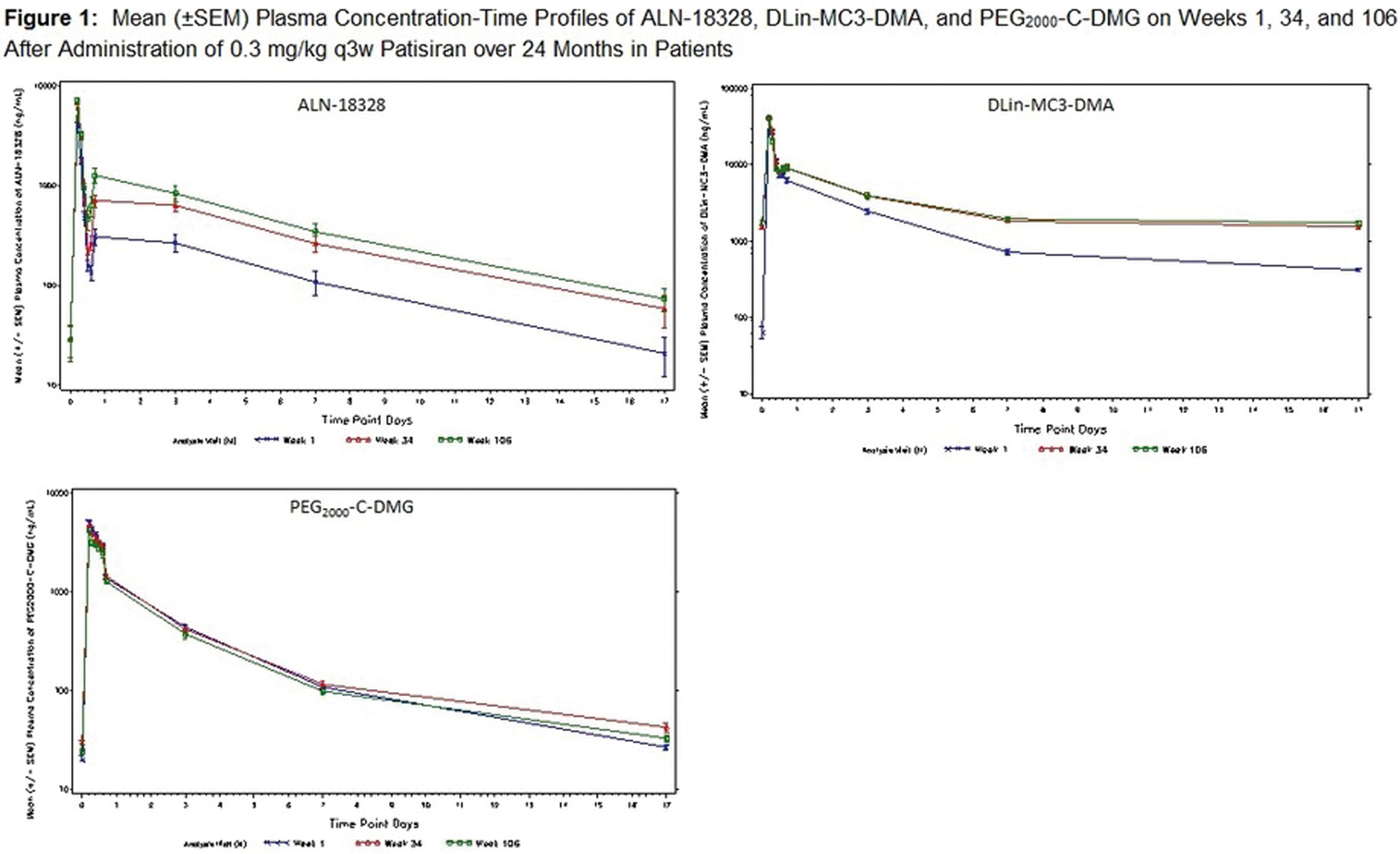
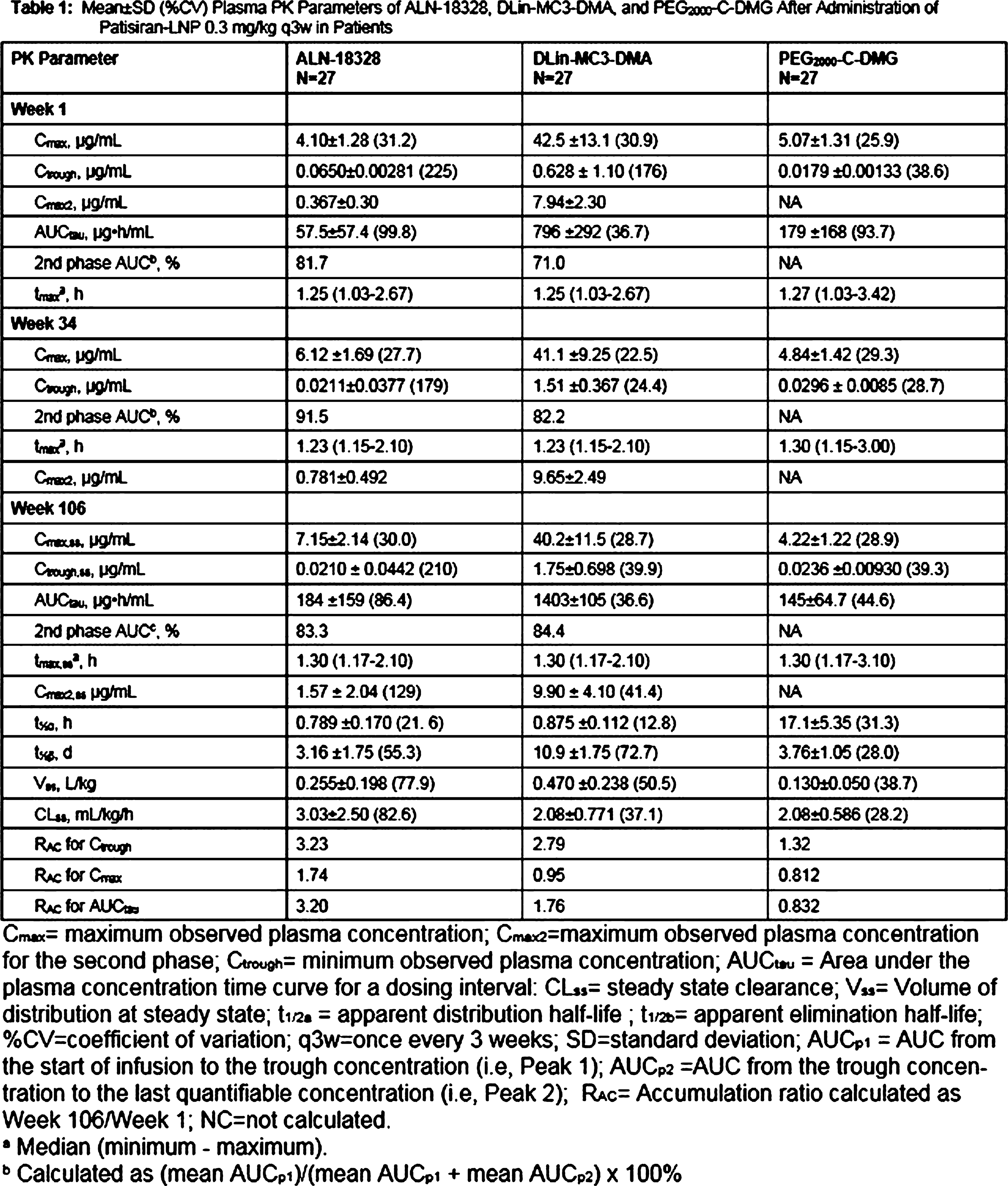
PS2Group3-081 / #735ROLE OF THE ER STRESS TRANSCRIPTION FACTOR XBP1 IN CHARCOT-MARIE-TOOTH DISEASE TYPE 1B
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Thierry Touvier1, Cinzia Ferri2, Rosa Mastrangelo2, Cristina Rivellini3, Laurie Glimcher4, Lawrence Wrabetz5, Maurizio D’Antonio2
1Division Of Genetics And Cell Biology, Ospedale San Raffaele, Milan, IT;2Division Of Genetics And Cell Biology, Ospedale San Raffaele, Milan, IT;3Division Of Neuroscience, Ospedale San Raffaele, Milan, IT;4Department Of Cancer Immunology And Virology, Dana-Farber Cancer Institute, Boston, MA, US;5Hunter James Kelly Research Institute, Hunter James Kelly Research Institute, Buffalo, NY, US
Background: Mpz protein is the most abundant protein in myelin of peripheral nerves. The mutant MpzS63del causes Charcot-Marie-Tooth (CMT) 1B disease in humans and a similar demyelinating neuropathy in transgenic mice. MpzS63del protein provokes an endoplasmic reticulum (ER) stress in myelinating Schwann cells, which mount an unfolded protein response (UPR) characterized by activation of PERK, ATF6 and XBP1 pathways. We have previously reported that activation of CHOP and GADD34, two downstream mediators of PERK, is pathogenetic in MpzS63del mice (Pennuto, 2008; D’Antonio, 2013) but the role of the other UPR branches remains to be investigated. Methods: We generated a new mouse model with Schwann cells-specific ablation of XBP1 and in parallel we exploited MpzS63del dorsal root ganglia (DRG) explant cultures in which XBP1 signaling is modulated by gain/loss of function approaches. Results: We observed that absence of XBP1 dramatically worsens hypomyelination and electrophysiological/locomotor parameters in young and adult S63del neuropathic animals. Interestingly we observed that a strong upregulation of PERK and IRE1-mediated RIDD signalings in neuropathic animals lacking XBP1. This suggests that activation of XBP1 targets has an essential role in limiting MpzS63del toxicity, which cannot be compensated by other stress responses. Moreover, we demonstrated in S63del DRG cultures that inhibition of XBP1 pathway decreases myelination whereas activation of XBP1 signaling slightly ameliorates myelination. Conclusion: Altogether, these data demonstrate that the XBP1 pathway has a critical adaptive role in MpzS63del neuropathy and suggest that activating this pathway may be beneficial for CMT1B and perhaps for other neuropathies characterized by UPR activation.
PS2Group3-082 / #1032OBINUTUZUMAB, A NEW ANTI-CD20 ANTIBODY, IS ACTIVE AND EFFECTIVE IN ANTI-MAG ANTIBODY POLYNEUROPATHY
Topic: Group 3 – Peripheral Neuropathy, Cranial Nerves, Mononeuropathies: Clinical Features, Pathophysiology, Therapy
Chiara Briani1, A Visentin2, A Salvalaggio1, M Ruiz1, M Cacciavillani3
1Neurology Unit, Department Of Neuroscience, University of Padova, Padua, IT;2Hematology And Clinical Immunology Unit, Department Of Medicine, University of Padova, Padua, IT;3CEMES, Data Medica Group, Padova, IT
Background: Rituximab, a chimeric anti-CD20 monoclonal antibody (mAb), has been used in polyneuropathy associated with anti-myelin-associated-glycoprotein (MAG) antibodies with controversial results. Obinutuzumab, a new glycoengineered humanized anti-CD20 mAb, in combination with chemotherapy, induces longer progression-free survival in chronic lymphocytic leukemia (CLL) and follicular lymphoma, as compared with rituximab. We describe two patients with anti-MAG antibody neuropathy who were successfully treated with Obinutuzumab. Patient 1. A 82-yr-old man presented with severe demyelinating sensory-motor neuropathy. At our first evaluation, he used wheelchair to travel outdoors, was incapable of standing and walked few steps only with bilateral support. He had distal weakness at lower limbs (2/5 MRC), tactile hypoestesia and loss of vibration up to knees, areflexia. He had a clonal B lymphocytosis CD5+ CD23+, compatible with CLL, IgM lambda paraprotein and anti-MAG titre was >70,000BTU. The presence of favorable CLL prognostic markers (mutated IGHV gene and absent of TP53 deletions or mutations) prompt us to use chlorambucil+obinutuzumab (obinutuzumab iv at 1000mg on day 1, 8 and 15 of cycle 1 and day 1 of cycles 2-6; chlorambucil os at 0.5mg/kg on day 1 and 15 of cycles 1-6). At cycle 6 the patient was able to stand, gait was possible with monolateral support, tactile hypoestesia was limited to feet, distal strength and vibration improved. M-protein decreased from 15.8g/L to 11.61 at cycle 6. Similarly, IgM level (14.8vs8.7g/L), lambda free-light chain (145vs59mg/L) and lymphocytes (6,340vs900/μl) decreased. Anti-MAG still was >70,000BTU. Patient 2. A 84-yr-old female presented mild demyelinating sensory-motor neuropathy complained of distal sensory impairment at four limbs (painful paresthesia) and subjective gait instability. Areflexia at lower limbs was observed. She had clonal B lymphocytosis compatible with CLL and with IgM kappa paraprotein at immunofixation. Anti-MAG titre was >70,000BTU. Chlorambucil + obinutuzumab (obinutuzumab iv at 1000mg on day 1, 8 and 15 of cycle 1 and day 1 of cycles 2-3; chlorambucil os at 0.5mg/kg on day 1 and 15 of cycles 1-3) were administered. After 3 cycles monoclonal component was absent and the patient reported dramatic improvement of symptoms with reduction of pain and gait normalization. Neurological evaluation was unremarkable, except for absent Achilles reflexes. Our data indicate that Obinutuzumab may be effective in anti-MAG antibody polyneuropathy. CLL might have had a role in the response to therapy, but a possible role of chlorambucil+obinutuzumab in anti-MAG antibody polyneuropathy, regardless of the associated hematological condition, should be considered in future trials.
PS2Group5-001 / #544LATE-ONSET POMPE DISEASE ASSOCIATED WITH POLYNEUROPATHY
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Marta Lamartine Monteiro1, Gauthier Remiche2
1Neurology, Erasme Hospital, Brussels, BE;2Centre De Référence Neuromusculaire, Service De Neurologie, Hôpital Erasme, Université Libre de Bruxelles, Brussels, BE
Background: This study aimed to assess if late-onset Pompe disease (LOPD) could be associated with polyneuropathy. LOPD is a rare autosomal recessive disease caused by alpha-glucosidase lysosomal enzyme deficiency. This strikes mainly striated muscle but also involves many other tissues. To the best of our knowledge an involvement of large nerve fibers was never depicted. However it has been showed in mice models of Pompe’s disease, glycogen accumulation in Schwann cells and in the perineurium of peripheral nerves. Moreover two LOPD patients were reported as having a small fiber neuropathy. Methods: We retrospectively assessed medical records of LOPD patients followed in our center in order to assess potential indicative clinical and neurophysiological data suggesting a polyneuropathy. Moreover we systematically looked for the presence of potential etiologies for polyneuropathy. LOPD and polyneuropathy international diagnostic criteria were applied and ethic committee agreement was obtained. Results: Four of the six LOPD patients found in our database had clinical evidence for polyneuropathy and it was confirmed by electroneuromyography for three of them. None had other common known cause of polyneuropathy. Conclusion: Polyneuropathy seems overrepresented in LOPD patients. Pathophysiological basis may be evoked to explain this potential disabling additional condition in these patients.
PS2Group5-002 / #825CORRELATION BETWEEN ELECTRICAL IMPEDANCE MYOGRAPHY AND TWO QUANTITATIVE ULTRASOUND PARAMETERS IN DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Bhaskar Roy1, Basil Darras2, Craig Zaidman3, Jim Wu4, Kush Kapur2, Seward Rutkove5
1Neurology, Yale University, New Haven, CT, US;2Boston Children’s Hospital, Harvard Medical School, Boston, MA, US;3Neurology, Washington University at St Louis, St Louis, MO, US;4Neurology, Beth Israel Deaconess Medical Center, Boston, MA, US;5Neurology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, US
Background: Quantitative ultrasound (QUS) parameters, such as grayscale levels (GSL), quantitative backscatter analysis (QBA) and electrical impedance myography (EIM) are potential non-invasive biomarkers of muscle health in Duchenne muscular dystrophy (DMD). This study explores correlation between GSL and QBA values with EIM parameters. Methods: GSL, QBA values along with EIM parameters, 50 kHz phase, 100kHz/300kHz phase ratio, and least-squares fit of 100 to 500 kHz phase (slope) at baseline, 6 months, and 12 months from 6 muscles were recorded from an ongoing study of QUS and EIM in 36 DMD and 29 healthy boys, aged 2–14 years. Results: Among the DMD patients, GSL values showed significant correlation with all EIM parameters in biceps brachii. Particularly, strong correlation was noted with EIM 100/300 ratio and EIM-slope at 12-months; spearman rho was -0.67 and 0.7 respectively, p-values <0.01. Weak-to-moderate but consistent correlation was noted between GSL and all EIM parameters in deltoid except at baseline for EIM-slope (p-value was 0.06). Significant correlation was noted between GSL values with EIM-50kHz at every time point in the forearm muscles. Among lower extremity muscles, quadriceps showed significance or near significance at 6 months and 12 months with every EIM parameters. In gastrocnemius, weak correlation was noted with EIM 100/300 ratio reaching significance or near significance. No consistent correlation was noted in tibialis anterior. QBA values showed a similar trend. Consistently significant correlation was noted between QBA and all EIM parameters in biceps and deltoid. Correlation was strong for biceps at 12 months; rho for EIM 100/300 ratio, EIM-slope were -0.716, and 0.731 respectively (p-values <0.01). Strong correlation was also noted at baseline and 6-months with EIM-50kHz (rho values -0.72 and -0.78 respectively, p-values <0.01). Forearm muscles showed moderate to strong correlation with EIM-50kHz at every time points. Among lower extremity muscles, gastrocnemius showed consistent correlation most of the time. In quadriceps, significance was reached at 6 months with all EIM parameters. Again, tibialis anterior did not show any consistent correlation. When considered in groups, proximal muscles, upper extremity muscles, and all six-muscles grouped together showed significant correlation throughout with all EIM parameters for both GSL and QBA values. In majority of the occasions, spearman rho was > 0.5 and p-values were <0.05. Correlations noted in the group of distal muscles showed statistical significance or near significance most of the time. Lower extremity muscles taken together did not show significant correlation consistently. No consistent correlation between the QUS and EIM parameters was noted, either in individual muscle or group of muscles among the healthy subjects. Changes in GSL and QBA values from baseline to 12 months were correlated with changes in different EIM parameters from baseline to 12 months. No consistent correlation was noted when these changes were considered in individual muscles, except in gastrocnemius. However, when averaged muscle groups were considered, more consistent correlation pattern emerged. Conclusion: Moderate correlation between GSL and QBA with EIM parameters probably reflects on the distinct nature of the muscle-health information provided by these non-invasive methods.
PS2Group5-003 / #868PHYSIOLOGICAL DIFFERENCES IN SARCOLEMMAL EXCITABILITY MEASUREMENTS BETWEEN HUMAN MUSCLES
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Robert Boland-Freitas, James Lee, Karl Ng
Neurology, Royal North Shore Hospital, ST LEONARDS, AU
Background: The sarcolemmal resting membrane potential (RMP) affects muscle excitability, contractility and force generation[1]. There is however limited in vivo data on the normal RMP of the human sarcolemma. We hypothesize that the in vivo RMP may differ between human muscles with different physiological roles. Methods: EMG recording of muscle velocity recovery cycles (MVRC) allows direct in vivo measurement of sarcolemmal excitability measures. MVRC were recorded from a proximal antigravity muscle, rectus femoris (RF), and compared to paired recordings from a distal non-antigravity muscle, tibialis anterior (TA) in thirty normal individuals. Results: Significant differences in muscle relative refractory period (TA:3.97ms vs RF:3.67ms, P=0.006), early supernormality (TA: 10.4% vs RF:14.3%, P<0.001) and late supernormality (TA:3.35% vs RF:5.46%, P<0.001) were observed. Conclusion: The results are suggestive of a relatively depolarized RMP in tibialis anterior versus rectus femoris and differences in t-tubule function between these muscles. This novel result lends support to inter-muscle differences in normal excitability and physiology. Mechanisms underlying this include differences in fiber type make-up[2] and physiological function[3] between muscles in normal individuals. References 1. Clausen, T., Na+-K+ pump regulation and skeletal muscle contractility. Physiol Rev, 2003. 83(4): p. 1269-324. 2. Bottinelli, R. and C. Reggiani, Human skeletal muscle fibres: molecular and functional diversity. Prog Biophys Mol Biol, 2000. 73(2-4): p. 195-262. 3. Tirrell, T.F., et al., Human skeletal muscle biochemical diversity. J Exp Biol, 2012. 215(Pt 15): p. 2551-9.
PS2Group5-004 / #384LABEL-FREE IMAGING OF ABNORMAL LIPID ACCUMULATION IN PERIPHERAL NERVES FROM FABRY DISEASE PATIENTS USING RAMAN SPECTROSCOPY
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Yu Nagashima1, Atsushi Iwata1, Kosuke Yoshioka2, Junko Omachi3, Jun Shimizu1, Tatsuhi Toda1, Junji Yumoto3, Makoto K. Gonokami4
1Department Of Neurology, The University of Tokyo Hospital, Tokyo, JP;2School Of Engineering, The University of Tokyo, Tokyo, JP;3School Of Science, The University of Tokyo, Tokyo, JP;4The University of Tokyo, Tokyo, JP
Background: Fabry disease is an X-linked genetic disorder resulting from deficient activity of alpha-galactosidase A. It causes systemic accumulation of globotriaoslyceramide (Gb3) in the plasma and cellular lysosomes of tissues and organs throughout the body. In routine histopathological studies, toluidine blue staining usually demonstrates abnormal lipid accumulation, but it is not molecular specific and the other lipids in addition to Gb3 are potentially stained at the same time. The problem is that there is no guarantee that the accumulated lipid is really Gb3. In this study, we measured molecular vibrational spectra of Gb3. Vibrational spectra obtained using Raman spectroscopy provides vibrational information characteristic of chemical groups or bonds in a molecule without any labeling procedures. Measuring vibrational spectra enables us to locate and quantitate Gb3 within tissues in a molecular specific manner. Methods: Vibrational spectra of a 10μm-thick frozen sections of sural and femoral nerves biopsied or autopsied from three Fabry disease patients were obtained using spontaneous Raman microspectroscopy and compared with the spectra of Gb3, phosphatidylcholine (PC) and sphingomyelin (SM) in vitro. We also measured sural nerve sections from vasculitis patients as disease control using the same procedure. Results: The Raman shift in 1064cm-1 was more prominent in PC and SM than in Gb3, whereas the Raman shift in 1088cm-1 only existed in Gb3 (Figure 1(a)). The ratio of them (1088cm-1 to 1064cm-1) enabled us to contrast Gb3 in the perineurium with PC- and SM-rich nerve fibers in Fabry disease patients (Figure 1(c)). This ratio was not elevated in the sections from the disease control cases. Next, in order to visualize spatial distribution of these lipid species quantitatively, the spectral image data were linearly decomposed into 16 independent bases using sparse coding. The most highly correlated basis with Gb3, PC and SM spectra in vitro were selected and spatially mapped. These mappings visualized a streak-like accumulation of Gb3 within the perineurium, whereas PC and SM showed relatively ubiquitous distribution including the myelinated nerve fibers (Figure 1(d)). Conclusion: The Raman shifts worked as a molecular specific marker of Gb3 within the peripheral nerves from the Fabry disease patients. This is a label-free and lipid-specific visualization technique of Gb3 and is potentially useful even within the tissues other than peripheral nerves.
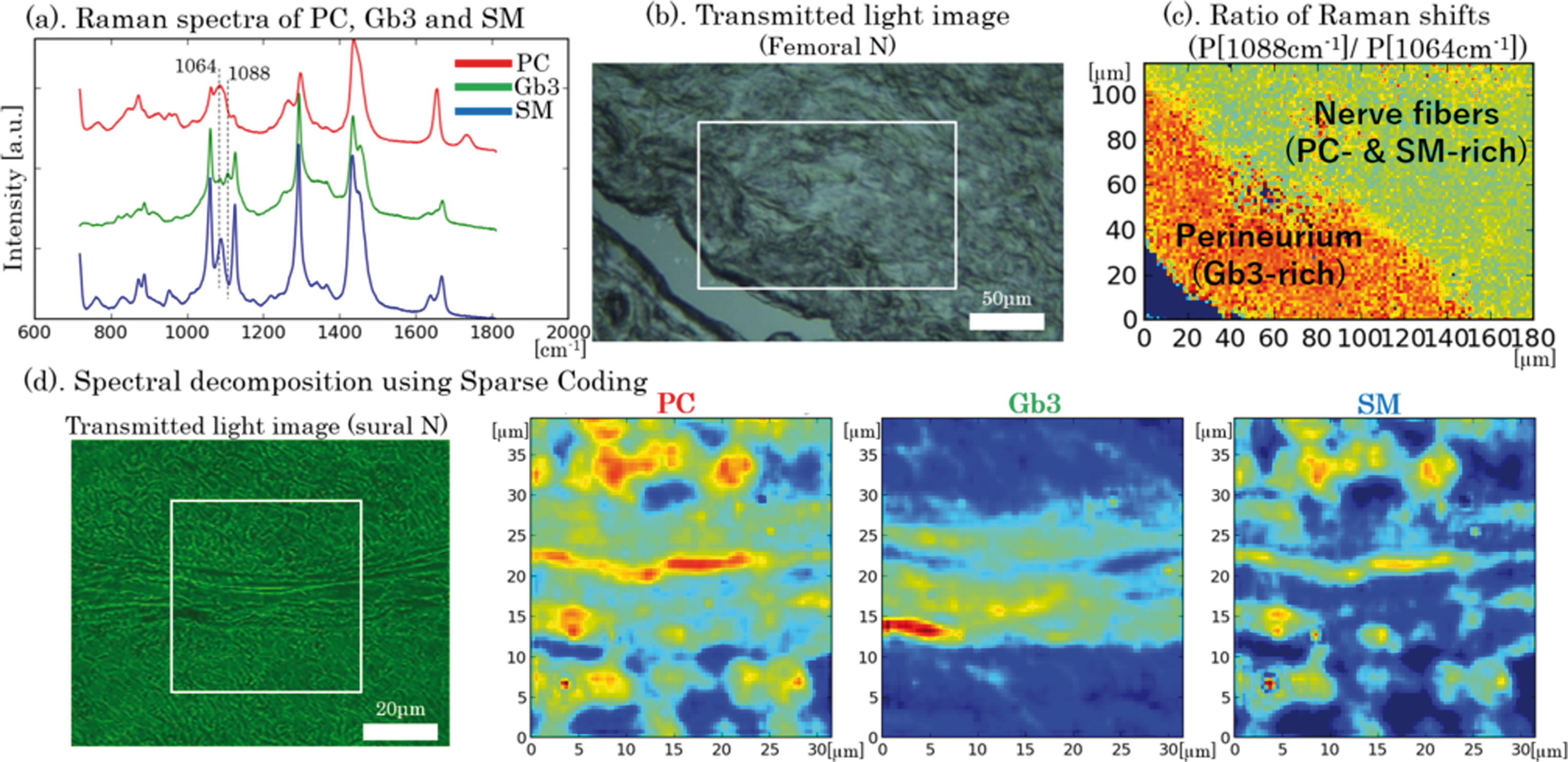
PS2Group5-005 / #433MODULATING EFFECTS OF UNSATURATED FATTY ACIDS ON GENE EXPRESSION OF MYOSIN HEAVY CHAIN CLASS I AND IIB IN C2C12 MYOCYTES
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Junko Yamaji1, Yoshiaki Mori2
1Department Of Nutrition Sciences, Kansai University of Welfare Sciences, Kashiwara, JP;2 Department Of Rehabilitation Sciences, Kansai University of Welfare Sciences, Kashiwara, JP
Background: Oleic acid (OA) and linoleic acid (LA) belong to long-chain unsaturated fatty acids and present in nuts, seeds, fishes, and other plants. It has been reported that OA and LA had act as a calcineurin activator, but the effects on skeletal muscle still remains almost elusive. Interleukin-6 (IL-6) is a cytokine from immune cells and is also known as a myokine, an anti-inflammatory cytokine from muscle. Recently, we reported that IL-6- and/or calcineurin-activation increased messenger RNA (mRNA) expression of myosin heavy chain class I (MyHC I), IL-6 and heat shock protein (HSP) 70 during the myotube differentiation of C2C12 cells, murine skeletal muscle myoblast cell line. In the present study, we examined the effects of unsaturated fatty acids including oleic acid and linoleic acid on expression levels of MyHC I, myosin heavy chain class IIb (MyHC IIb) and IL-6 mRNA in C2C12 cells. Methods: C2C12 cells were used after induction of differentiation from myoblast cells to myotubes. At the beginning of differentiation of C2C12 cells, the cells were incubated in D-MEM containing 2% serum with or without agents, which are oleic acid and linoleic acid and a calcineurin inhibitor, namely cyclosporine A, and IL-6 and anti-IL-6 antibody as a IL-6 receptor antagonist. After 24-hr incubation, the medium supplemented with agents was removed from cell culture dish and cells were maintained in D-MEM containing 2% serum for 3 days. The mRNA expression levels were measured by quantitative RT-PCR method using Taqman probes. Quantitative RT-PCR method was performed by a LightCycler Nano system (Roche Applied Science). Quantitative RT-PCR data was analyzed by the comparative CT method. The mRNA expression levels of MyHC I, MyHC IIb, and IL-6 mRNAs expressed as the cycle time (Ct) values normalized by the Ct of 18S as reference gene expression level. Statistical analyses were performed by the Tukey’s test. Results: First, we assessed the effect of unsaturated fatty acids and calcineurin inhibitor on MyHC I and MyHC IIb mRNA expression. MyHC I and MyHC IIb mRNA expression levels were upregulated by unsaturated fatty acids such as oleic acid or linoleic acid. These mRNA levels of MyHC I and MyHC IIb were inhibited by cyclosporine A with oleic acid or linoleic acid. Second, we tested the effects of unsaturated fatty acids, and anti-IL-6 antibody on MyHC I and MyHC IIb mRNA expression. The upregulation of MyHC I and MyHC IIb mRNA expression by oleic acid and linoleic acid was decreased by anti-IL-6 receptor antibody. Third, we examined the effects of unsaturated fatty acids and calcineurin inhibitor on IL-6 mRNA level. Then, the expression level of IL-6 mRNA were significantly upregulated by oleic acid and linoleic acid. The upregulation of IL-6 mRNA expression by oleic acid and linoleic acid was decreased by cyclosporine A. Conclusion: These results indicated that secretion of IL-6 induced by calcineurin activation increases MyHC I and MyHC IIb mRNA in C2C12 cells. COI:No
PS2Group5-006 / #716ELECTROPHYSIOLOGICAL DIAGNOSTIC OF NEUROMUSCULAR DISEASES IN NEW-BORNS, INFANTS AND TODDLERS
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Philip J. Broser1, Oswald Hasselmann2, Oliver Maier3, Jürg Lütschg3
1Neuropediatrics, Children`s Hospital of Eastern Switzerland, St Gallen, CH;2Neuropediatrics, Children`s hospital of Eastern Switzerland, St. Gallen, CH;3Neuropediatrics, Children`s hospital of Eastern Switzerland, St Gallen, CH
Background: Recognizing neuromuscular disease in newborns, infants and toddlers is even for the experienced neuro-pediatrician a challenging task. Many neuromuscular diseases present in the beginning only with very minor symptoms, other cases are quite severe with i.e. generalized muscular hypotonia requiring mechanical ventilation and making a proper clinical assessment difficult. However, early treatment of neuromuscular conditions can alter the prognosis significantly. Therefore precise diagnosis is important. Methods: The children`s hospital of Eastern Switzerland has become a major centre for neurophysiological diagnostic in children. We have established a workup to precisely assess the neuromuscular system in newborns and toddlers. Our assessment protocol starts with the basic study of sensory and motor nerves and typically a EMG of the M. tibialis anterior. Depending on the results of the basic study the examination is extended based on the clinical picture. In the case of a Floppy infant with a suspicion of a myasthenia a single fibre EMG is conducted typically at one of the muscles innervated by the cranial nerves. If an asymmetrical weakness is present the examination focuses on the affected muscle and its contralateral counterpart. Results: Based on this assessment protocoll we can diagnose a significant number of neuromuscular diesease. In this presentation we will show examples how spinal muscular atrophy can be diagnosed early by recording pathological discharges during wake and sleep, how a congenital myasthenic syndrome can be diagnosed by single fibre EMG techniques and how especially in clinical apparent myotonia an examination of the mother can show myotonic discharges and lead to the diagnosis of myotonic dystrophy in the child. Conclusion: We conclude our presentation with an outlook of the current developments in pediatric electorneuromyography and especially foucs on latets developments in the application of single fibre EMG techniques in the pediatric population.
PS2Group5-007 / #964DISCOVERY OF PROTEIN BIOMARKERS FOR DYSFERLINOPATHY
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Laura Rufibach1, Zsuzsanna Hollander2, Darlene Ly Dai2, Virginia Chen2, Amrit Singh2, Douglas Albrecht1, Bradley Williams1, Hillarie Windish1, Elaine Lee1, Plavi Mittal1, Anna Mayhew3, Marni Jacobs4, John W. Day5, Kirsti J. Jones6, Diana X. Bharucha-Goebel7, Matthew Harms8, Alan Pestronk8, Maggie C. Walter9, Tanya Stojkovic10, Susan Sparks11, Elena Bravver11, Jordi Diaz-Manera12, Elena Pegoraro13, Carmen Paradas14, Jerry Mendell15, Hanns Lochmuller3, Kate Bushby3, Volker Straub3, Sara Assadian2, Janet E. Wilson-Mcmanus16, Derek S. Smith17, Christoph H. Borchers18, Bruce Mcmanus2, Raymond Ng19
1Jain Foundation, Seattle, WA, US;2Prevention of Organ Failure (PROOF) Centre of Excellence, Vancouver, BC, CA;3John Walton Muscular Dystrophy Research Centre, Newcastle University, Institute of Genetic Medicine, Newcastle Upon Tyne, GB;4Division Of Biostatistic And Study Methodology, Children’s National Health System, Centre for Translational Science, DC, WA, US;5Department Of Neurology And Neurological Sciences, Stanford University School of Medicine, Stanford, CA, US;6Institute For Neuroscience And Muscle Research, Children’s Hospital At Westmead, University of Sydney, Sydney, AU;7Department Of Neurology, Children’s National Health System, Washington DC, US;8Department Of Neurology, Washington University School of Medicine, St. Louis, MO, US;9Friedrich-baur Institute, Departement Of Neurology, Ludwigs-Maximilians University of Munich, Munich, DE;10Institut de Myologie, Paris, FR;11Carolinas Healthcare System Neurosciences Institute, Charlotte, NC, US;12Neuromuscular Disorders Unit, Hospital de la Santa Creu i Sant Pau, Barcelona, ES;13Department Of Neuroscience, University of Padova, Padova, IT;14Neuromuscular Unit, Department Of Neurology, Hospital U. Virgen del Rocio/Instituto de Biomedicina de Sevilla, Sevilla, ES;15Center For Gene Therapy, Nationwide Children’s Hospital, Columbus, OH, US;16Netcad, Canadian Blood Services, Vancouver, BC, CA;17Genome Bc Proteomics Centre, University of Victoria, Victoria, BC, CA;18Proteomics Centre, Segal Cancer Centre, Lady Davis Institute, Jewish General Hospital, McGill University, Montreal, QC, CA;19Department Of Computer Science, University of British Columbia, Vancouver, BC, CA
Background: Dysferlinopathy is a rare neuromuscular disease caused by deficiency in the dysferlin protein and results in progressive skeletal muscle weakness of the limbs, which often leads to the inability to walk. The most common clinical presentations of dysferlinopathy are limb-girdle muscular dystrophy type 2B (LGMD 2B) and Miyoshi Myopathy type 1 (MMD1). Due to the rare nature of the disease, the molecular signatures that differentiate patients from healthy people, as well as different levels of severity, have not been defined. Methods: Blood-based biomarker discovery was performed to identify proteins that can be used to assess disease state and progression, with the goal of eventually using these signatures to assess the efficacy of emerging therapies. For the pilot phase, patients were split into highly ambulant (N=25) and non-ambulant (N=25) groups to assess profiles associated with disease progression and to also identify profiles when compared to healthy age and sex matched controls (N=25). Ensembling was applied to combine protein biomarkers to improve upon the performance of individual proteins. In the technical replication phase, the best performing protein panels were recalibrated in the same subjects using a targeted platform called multiple reaction monitoring mass spectrometry (MRM-MS). In the external replication phase, the MRM panels with the best performance were analyzed in 145 baseline visit samples of the dysferlinopathy natural history study called the Clinical Outcome Study for Dysferlinopathy (COS). In the longitudinal phase, MRM data and clinical metrics from 3 timepoints from COS (baseline, year 1, and year 2) are being evaluated in 120 samples per visit to explore whether longitudinal MRM protein data can be used to stratify dysferlinopathy patients with respect to their future clinical outcomes. Results: In the patients versus healthy control comparison, the best performing protein biomarker panel using MRM had an area under the receiver operating characteristics curve (AUC) performance of AUC=0.92 with 92% sensitivity, and 76% specificity. When this panel was tested in the external COS replication cohort it had an AUC=0.89. In the ambulatory versus non-ambulatory comparison, the best performing panel using MRM had an AUC=0.91 with 92% sensitivity and 79% specificity. The external replication performance of this MRM panel in the COS subjects yielded an AUC of 0.77. Conclusion: We identified protein biomarkers that show promise for evaluating and monitoring disease states in dysferlinopathy, and potentially, to predict the effect of therapeutics during clinical trials. The upcoming analysis of the longitudinal COS samples will help us correlate clinical outcomes with protein biomarkers.
PS2Group5-008 / #992NERVE ULTRASOUND FOR THE IDENTIFICATION OF TREATMENT-RESPONSIVE CHRONIC NEUROPATHIES WITHOUT NERVE CONDUCTION ABNORMALITIES
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Stephan Goedee1, Ingrid Herraets1, Leo Visser2, Thies Van Asseldonk2, Hessel Franssen1, Ludo Van Der Pol1, Leonard Van Den Berg1
1Neurology, Brain Centre Rudolph Magnus, UMC Utrecht, Utrecht, NL;2Neurology, Elisabeth-Tweesteden Hospital, Tilburg, NL
Background: Diagnostic consensus criteria for inflammatory neuropathies primarily rely on NCS results that indicate multifocal demyelination. However, NCS can be inconclusive or even normal in patients with neuropathies that respond to immune-modulating treatment. Methods: We analysed 240 incident patients with a clinical phenotype compatible with chronic inflammatory demyelinating polyneuropathy (CIDP), Lewis Sumner Syndrome (LSS) or multifocal motor neuropathy (MMN). Work-up consisted of routine ancillary investigations recommended in the diagnostic consensus guidelines and a standardized sonographic protocol assessing arm nerves and brachial plexus. All patients without any demyelinating features on NCS, but with sonographic abnormalities fitting inflammatory neuropathy underwent standard treatment with IVIg. Treatment effect was evaluated by routine clinical examination and hand grip or myometry. Results: We found 17 (7%) patients with a clinical phenotype compatible with CIDP (n=10), LSS (n=1) and MMN (n=6, with 3 positive for anti-GM1 auto-antibodies), sonographic nerve enlargement of proximal median nerve segments and/or cervical (nerve) roots, yet negative NCS (2 only axonal loss, 15 normal). In contrast, MRI of brachial plexus was normal in 8/17 (47%) and protein content of cerebrospinal fluid in 6/17 (35%). All 17 patients showed objective improvement on IVIG treatment. Conclusion: Our study suggests that nerve ultrasound may help to improve detection of treatment-responsive chronic neuropathies, even in patients not fulfilling the diagnostic NCS and other supportive criteria, including MRI. Therefore, future revisions of the diagnostic consensus criteria may include sonography as complementary technique to NCS and MRI.
PS2Group5-009 / #810MOTOR-UNIT NUMBER ESTIMATION IN THE ABDUCTOR POLICIS BREVIS MUSCLE OF PATIENTS WITH CARPAL TUNNEL SYNDROME
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Bahram Haghiashtiani1, Fahimeh Akhondi2, Zahra Mirza Asgari2, Mostafa Almasi2, Babak Zamani2, Mohammad Reza Motamed2, Masood Mehr Pour2
1Neurology, Iran University of Medical Sciences, IR;2Iran University of Medical Sciences, Tehran, IR
Background: Median nerve entrapment at wrist is the most common of all entrapment neuropathies and, consequently, one of the most frequent reasons for referral for an electrodiagnostic (EDX) study. In any individual patient with CTS, there may be little correlation between the frequency and severity of clinical symptoms and signs and the abnormalities seen on routine nerve conduction studies. Also, decreased compound Muscle action potentials (CMAPs) is only a late finding in the EDX study of CTS patients and becomes apparent when the thenar eminence is actually atrophic. Motor Unit Number Estimation (MUNE) is a unique EDX technique that enables us to estimate the number of axons innervating a given muscle. A motor unit (MU) is defined as a single motor neuron, and all muscle fibers innervated by it. Activation of a motor neuron causes contraction of all the muscle fibers innervated by it; all of the motor units that innervate a single muscle are considered to be its motor unit pool. A number of MUNE methods have been described that include the Incremental Technique, Multiple Point Stimulation Spike Triggered Statistical method, F wave MUNE, and MUNIX. In this study we opted for the incremental technique to evaluate the number and amplitude of active motor units in the Abductor Policis Brevis (APB) muscle of patients presenting with symptoms and signs typical of CTS. Methods: We evaluated 42 hands of 21 patients (2 males and 19 females), who presented with clinical features indicative of CTS. We performed routine Median nerve conduction studies, and divided our subjects to mild, moderate, and severe CTS based on routine Electrodiagnostic (EDX) findings. We estimated the number and the amplitude of motor units by the Incremental stimulation technique, and compared our findings to normal values. For MUNE, we used the Incremental stimulation technique originally described by McComas. The recording and stimulating electrodes were placed as described above. First, we set the gain at 50 microvolts (μVs) and the sweep at 5 milliseconds, and using supramaximal stimulation, we measured the distal Median CMAP amplitude in two ways: Base-to-Peak (from the baseline to the first negative peak) and Peak-to-Peak (from the first negative peak to the following positive peak). Finally, we divided the supramaximal CMAP amplitude to the mean amplitude of a single motor unit to yield a MUNE value. Results: MUNE values were as follows: 95.5 ± 28.9 in males and 100.1 ± 32.4 in females by the Base-to-Peak method, and 98.7 ± 20.6 in men and 91.3 ± 40.9 in women by the Peak-to-Peak way. These values were significantly lower than the normal population. We also observed a significant correlation between MUNE values and the severity of CTS. Conclusion: MUNE is a valid tool to assess CTS patients grading even in the early stages of the disease.
PS2Group5-010 / #739HIGH RESOLUTION NEUROSONOGRAPHY IN PATIENTS WITH CARPAL TUNNEL SYNDROME AND NORMAL CONTROLS
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Atchayaram Nalini1, Saraswati Nashi2, Veeramani Preethish-Kumar1, Nishtha Yadav3, Kajari Bhattacharya3, Kiran Polavarapu1, Seena Vengalil1
1Neurology, National Institute of Mental Health and Neurosciences, Bengaluru, IN;2NIMHANS, Bengaluru, IN;3Neuro Imaging And Interventional Radiology, National Institute of Mental Health and Neurosciences, Bengaluru, IN
Background: Carpal tunnel syndrome (CTS) is the commonest entrapment neuropathy with a prevalence of 3.8%. Ultrasonography (USG) measurements for the diagnosis of CTS has sufficient accuracy and allows differential diagnosis and supports the surgeon. The objective was to compare the sensitivity of electrodiagnosis and high resolution nerve US in CTS. Methods: Nerve US was performed with Philips Epiq 7G, linear 5 - 18Hz transducer. Median nerve (MN) cross sectional area (CSA) was recorded transversely at level of pisiform and scaphoid bones parallel to each other (carpal tunnel inlet).Distal to proximal ratio was calculated 4cm from the distal crease in forearm. Nerve conductions were performed as per standard procedure. MR Neurography (axial and sagittal images using a temporomandibular surface coil, fat saturation, and flow suppression). Results: Total number of patients studied were 30. F:M=4:1. Mean age at presentation was 41.6 ± 10.5(27-61) years. Clinically, 28(93.3%) had bilateral CTS [symmetrical-4; asymmetrical-24] and two had unilateral involvement. Duration of illness varied from 2 months to 8 years(mean 35.8 ± 25.5). All had sensory complaints while 9 patients (30%) also had motor deficits. Sensitivity of Tinel sign / Phalen’s maneuver was 40% and 63.3% respectively. By USG, cross sectional area (CSA) at pisiform bone level was >0.1cm2 in 27 patients. Distal/proximal ratio was ≥1.5 bilaterally in 16 patients and unilaterally in 5(right -3, left -2). Mean distal CSA of RMN=1.44±0.42 cm2 (0.8-2.5), left=1.31±0.3cm2 (0.7-2.0). By comparison, in age and sex matched controls[37 cases; 75 hands, F = 39; M = 6)], the mean distal CSA was 0.79±0.13cm2(0.59 – 0.11) and 0.77±0.1cm2(0.53 – 0.1) in right and left median nerves respectively. Mean CSA of patients compared to controls was significantly higher. Nerve conduction studies in 27 patients revealed absent evoked motor response in 4 patients (right–3, left –1). Mean distal motor latency and compound muscle action potential of right median nerve (RMN) in 24 cases was 5.8±1.4s(4.2-11.4) and 8.65±3.0mv(3.3 - 10.3) respectively. On the left(26 cases), it was 5.5±1.8 s(3.3-10.3) and 8.3±3.7mv(0.6-15.8). Median nerve SNAPs were absent in 16 patients (bilateral –10, right–4, left-2). Mean sensory latencies were 4.5±2.4 s(2.5-12) and 3.4±0.7s(2-4) in right(13) and left(14) sides respectively with corresponding mean sensory amplitudes[Right: 7.2±5.3mv(1.5-23) Left: 9.9±5.9mv(2.3-22.1) and conduction velocities[Right: 35.6±11m/s(11.7- 58) Left: 43.7±13m/s (22.2-68.6)]. MRI in 19 patients showed mean distal CSA of RMN of 1.55±0.49 cm2 (range, 1-2.7) and LMN =1.57±0.4 cm2(range, 1-2.7). Conclusion: This study emphasizes the importance of USG as sensitive, cheaper and effective alternate to MRI in the confirmation of CTS. CSA of the proximal MN could be used as a predictor of severity of CTS. Moreover, the mean CSA and inlet CSA may be valid and easy-to-acquire parameters for routine clinical use in confirming CTS.
PS2Group5-011 / #793CAN WE EARLY DETECT CARDIAC DYSFUNCTION IN PATIENTS WITH DUCHENNE MUSCULAR DYSTROPHY?
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Michal Marchel, Janusz Kochanowski, Agnieszka Serafin, Bozena Truszkowska, Grzegorz Opolski
Department Of Cardiology, Medical University of Warsaw, Warsaw, PL
Background: Duchenne muscular dystrophy (DMD) is characterized by musculoskeletal abnormalities due to the mutations in dystrophin coding gene (dystrophinopathy), frequently accompanied by cardiac dysfunction. Methods: The aim of the study was to analyse both ventricular function in adult DMD patients (pts) with the use of among other techniques global longitudinal strain assessed by speckle-tracking echocardiography. The group of 33 adult male pts with genetically confirmed dystrophinopathy as well as 25 healthy volunteers were subjected to echocardiography with the assessment of global longitudinal strain (GLS) [Philips Epiq 7]. Results: The mean left ventricular ejection fraction (LVEF) was 48,0 +/- 15,3% and 65,3 +/- 3,7 for DMD pts and for controls respectively (p<0,05). We identified three subgroups of DMD patients: 1 – significant systolic dysfunction - LVEF< 40% (n = 12), 2 – mid-range and preserved systolic function - LVEF>=40% (n = 21) as well as 3 – preserved systolic function - LVEF>=50% (n = 19) accompanied by group 4 (healthy controls, n=25). In group 1 we observed lower GLS as compared to group 4 (11,8 +/- 2,6 vs. 20,6 +/- 3,4; p=0,01), but also pts with persistent LVEF (group 2) had lower GLS than controls (16,6 +/- 2,0 vs. 20,6 +/- 3,4; p=0,01). Pts with preserved LVEF (group 3, mean EF 59,4 +/- 7,3) had also lover GLS compering to control group (16,8 +/- 2,1 vs. 20,6 +/- 3,4; p=0,02). Conclusion: Severe left ventricular systolic dysfunction is common in DMD pts, (12/33, 36%). The quantitative assessment of global longitudinal myocardial deformation can be helpful in detection of systolic abnormalities in DMD pts with normal LVEF. Subclinical systolic dysfunction is frequent (13/19, 66%) in DMD pts with LVEF still in normal range.
PS2Group5-012 / #197PERIPHERAL NERVOUS SYSTEM DISORDERS IN MULTIPLE MYELOMA: RABAT CLINICAL NEUROPHYSIOLOGY DEPARTMENT EXPERIENCE
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Laila Belarabi1, Nezha Birouk2, Bouchra Kably2, Leila Errguig2, Halima Belaïdi2, Reda Ouazzani2
1Clinical Neurophysiology, Speciality Hospital, Rabat, MA;2Clinical Neurophysiology, Speciality Hospital, Rabat, MA
Background: Multiple myeloma is a cancer formed by malignant plasma cells. Peripheral neuropathy has been considered as mainly secondary to the plasma cell dyscrasia itself, or following a direct compression. The association of neuropathy with multiple myeloma apart of these mecanisms is rare, often asymptomatic and discovered during the pre-chemotherapy assessment. With the advent of new drugs such as thalidomide and bortezomide for chemotherapy, the iatrogenic neurotoxicity has become the leading cause of peripheral neuropathy in multiple myeloma. Methods: We conducted a retrospective study from January 2014 to September 2017 including all patients referred to the department for an electroneuromyogram (ENMG) as part of a pre-chemotherapy assessment or in post-thalidomide follow-up, whether the patient was symptomatic or not. Results: We collected a serie of 32 cases, which we divided into two groups : patients before treatment with thalidomide (18 cases) and under thalidomide (14 cases). In the first group before treatment, the mean age was 58.6 years, with a sex ratio of 1.6. Four axonal sensory motor polyneuropathy (25%), and one demyelinating sensory motor polyneuropathy was discovered at the ENMG without any clinical manifestation. In the second group, the mean age was 61 years with a sex ratio of 1.3. Eight patients (57%) had peripheral neuropathy predominant to lower limb in six cases and painful in five cases. Conclusion: Polyneuropathy in multiple myeloma is well recognized, most often with compressive involvement, secondary to plasma cell dyscrasia or autoimmune mechanism. In subclinical neuropathies, it is often a sensory motor axonal polyneuropathy, predominant in distal and sensitive. Thalidomide is responsible for length-dependent sensory axonal polyneuropathies requiring close clinical and ENMG follow-up, in order to stop treatment before causing irreversible damage. Finally, early detection of peripheral neuropathy and the use of dose adjustment algorithms should help reduce the side effects while maintaining anti-tumor efficacy
PS2Group5-013 / #343CRAMPS, FASCICULATIONS AND MUSCLE FATIGABILITY DUE TO VITAMIN D DEFICIENCY
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Laila Belarabi, Nazha Birouk, Bouchra Kably, Reda Ouazzani
Clinical Neurophysiology, Ibn Sina Hospital, Rabat, MA
Background: The clinical signs of vitamin D deficiency are well known, dominated by the manifestations of hypocalcemia and secondary hyperparathyroidism. In adults, this syndrome is mostly represented by proximal muscle weakness, associated in some cases with cramps, fasciculations, and paresthesia. Patients can be misdiagnosed as having a motor neuron disease or muscle intolerance to effort, the ENMG being often normal or showing only fasciculations or neurogenic pattern.
Methods: We report the clinical and outcome data in 5 patients with muscles cramps, fasciculation’s and fatigability. Results: 5 patients were included in this study, 2 women and 3 men, aged from 20 to 59 years. All patients suffered from muscle weakness and exercise induced muscle pain, with a reduction in the walking distance in 2 cases, cramps in 3 cases, and fasciculations in 2 cases. The usual biological investigations were normal in all cases, except elevated CK (3xN) in 1 case. The ENMG was normal in 3 cases, and revealed diffuse fasciculations in 1 patient. 2 patients underwent muscle biopsy that was normal. Vitamin D level was reduced in all patients. After correction with vitamin D supplementation, 3 patients are totally asymptomatic, with a reduction of the symptoms in the others. Conclusion: Vitamin D deficiency, despite its classic manifestations, must be included in the causative diagnosis of muscle fatigability, cramps and fasciculations, in order to prevent the patient from delayed diagnosis and in some cases from invasive examinations.
PS2Group5-014 / #617NEXT-GENERATION SEQUENCING FOR MOLECULAR DIAGNOSIS IN NEUROMUSCULAR DISORDERS. RESULTS FROM 151 PATIENTS
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Daniel Natera De Benito1, Jessica Exposito Escudero2, Delia Yubero2, Lidia González Quereda3, Pía Gallano Petit3, Ana Töpf4, Carlos Ortez5, Laura Carrera-García2, Anna Lia Frongia6, Cristina Jou2, Cecilia Jimenez Mallebrera2, Anna Codina2, Jaume Colomer2, Andres Nascimento2
1Neuromuscular Disorders Unit, Hospital Sant Joan de Déu, Barcelona, ES;2Hospital Sant Joan de Déu, Barcelona, ES;3Hospital Sant Pau, Barcelona, ES;4John Walton Muscular Dystrophy Research Center, Newcastle University, Newcastle, GB;5Neuromuscular Unit, Hospital Sant Joan de Déu, Barcelona, ES;6Hospital Sant Joan de Déu, Barcelona, ES
Background: Inherited neuromuscular disorders (NMD) are chronic genetic diseases posing a significant burden on patients and the health care system. Despite tremendous research and clinical efforts, the molecular causes remain unknown for a significant proportion of patients, due to genetic heterogeneity and the use of a conventional gene-by-gene approach until relatively recently. Massively parallel sequencing using next generation sequencing (NGS) technologies has emerged as a successful approach to interrogate multiple genes simultaneously. It has proven to be an efficient and cost-effective tool to accelerate patient diagnosis. Herein, we describe the results obtained in an extensive study of 151 genetically undiagnosed patients presenting clinical signs of muscular dystrophies, congenital myopathies, or other neuromuscular conditions. We report the huge improvement observed after NGS aplication into the clinical field. Methods: Patients with clinical criteria of a neuromuscular disease and that had been studied using different NGS strategies were collected. The 151 patients had been evaluated with three different methodologies, due to the quick evolution of NGS in this field. The first group of patients (n=58) was studied with a custom panel (Nextera Rapid Capture, Illumina) including 118 genes, sequenced on a MiSeq instrument (Illumina) and analysed with the Variant Studio and DNA Nexus softwares. The second group (n=40) had sequenced 4813 genes associated to human disease (commercial TruSight One Sequencing Panel (Illumina) and Illumina sequencing on a NextSeq500), with an in-house bioinformatic analysis developed in the bioinformatics unity. The third group (n=53) followed a broader strategy, performing an Illumina exome capture (38 Mb baited target) to interrogate the whole exome, and after sequencing and bioinformatics analysis, only 169 genes associated with neuromuscular disorders were studied to check for candidate mutations. Data interpretation and candidate variants selection was performed according to the ACMG guidelines, and sanger sequencing in cases and progenitors was done in cases where strong candidates appeared. Results: Patients were classified, according to their clinical phenotype, as affected by congenital myopathy (52%), muscular dystrophies (29%), congenital myasthenic syndromes (6%), neuropathies (5%), or other clinical conditions (8%), including, among others, metabolic myopathy and hereditary spastic paraplegia. Most patients were sporadic cases. The majority of samples collected had previously been tested unsuccessfully, according to the observed phenotype. Eighty cases (53%) obtained a diagnosis, showing known disease-associated variants, or variants of a likely pathogenicity, or variants predicted to affect function in genes corresponding to clinical suspicions. Genetic cause of the disease was elucidated in 24/40 samples (60%) by the clinical exome, in 30/58 samples (51%) by the custom panel, and in 26/53 samples (49%) by the selected-WES analysis. Conclusion: In recent years, NGS has totally transformed the approach to the study of neuromuscular disorders. Our experience indicates that about half of the patients with a suspected neuromuscular disease are solved with NGS technology. Despite this, the molecular causes remain unknown for 47% of patients. Some of the unsolved cases could in fact be attributable to the kind of NGS strategy applied, to previously undescribed causative genes, or to genetic variation in areas that are out of the rule.
PS2Group5-015 / #258TRANSCRANIAL MAGNETIC STIMULATION AS A DIAGNOSTIC AND PROGNOSTIC TOOL IN CHILDREN WITH SEQUELAE OF ACUTE TRANSVERSE MYELITIS
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Vladislav Voitenkov1, Natalia Skripchenko2
1Clinical Neurophysiology, Pediatric Research and Clinical Center for Infectious Diseases, Saint-Petersburg, RU;2Pediatric Research and Clinical Center for Infectious Diseases, Saint-Petersburg, RU
Background: Diagnostic process and especially prognosis in acute transverse myelitis in children may be challenging. Diagnostic TMS may be useful additional tool in this process. Goal of our study was to evaluate motor pathways in children with viral myelitis using TMS. Methods: 20 healthy children (7-14 years old, average 12) without signs of spinal disorders were enrolled as controls. Main group consisted of 24 patients (8-16 years old, average 11) with acute viral transverse myelitis, in 4 cases with cervical localization of the lesion, in 7 – thoracic and in 13 – lumbar. Etiology was enterovirus (n=20) Epstein-Barr (n=4) viruses. 18 children were unable to walk, lower paresis was seen in other 6 cases; ventilation was not required in any cases. We used single-pulse TMS protocol. Neiro-MS D magnetic stimulator was used. Neiro-MVP EMG/EP system was used for data registration. Results: Three neurophysiologic patterns were observed in the myelitis group: Presence of both cortical MEP (although disperse and low-amplitude) and spinal MEP (n=15). Presence of only spinal MEP without cortical one (n=6)). Total absence of both spinal and cortical MEP (n=3). (Fig. 1). In the follow-up period (2-5 years) 10 children (42%) were unable to walk 50 meters and required a walker, 11 (46%) had normal motor function and 3 (13%) had total paraplegia without any movement of the lower limbs, no signs of recovery. Among those 11 who fully recovered only the first TMS pattern (presence of both cortical and spinal MEPs) was registered. Those who recovered with sequelae had first or second pattern and in 3 patients who did not recover only the third pattern was seen. ROC-analysis data revealed significant correlation between the long-term (3-5 years) recovery of walk in patients with consequences of myelitis and CMCT value≥28,7 ms. Conclusion: Acute transverse myelitis in 96% of the cases causes neurophysiologic changes which may be detected by diagnostic TMS. Method may be used as a predicting tool: absence of the cortical and spinal MEP may be considered as a sign of highly probable poor clinical outcome.

PS2Group5-016 / #762TARGETED METHYL-SEQ QUANTIFICATION BY NGS TECHNOLOGY FOR ROUTINE DIAGNOSTIC OF FSHD
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Stefanie Bulst1, Florentine Scharf1, Anna Benet-Pagès1, Peter Reilich2, Sibylle Jakubiczka3, Martin Zenker3, Maggie C. Walter4, Elke Holinski-Feder1, Angela Abicht1
1Medical Genetic Center, Munich, DE;2Friedrich-baur Institute, Departement Of Neurology, Ludwigs-Maximilians University of Munich, Munich, DE;3Institut Für Humangenetik, Otto-von-Guericke-University of Magdeburg, Magdeburg, DE;4Dept. Of Neurology, Ludwig-maximilians-university, Friedrich-Baur-Institute, Munich, DE
Background: Facioscapulohumeral Muscular Dystrophy (FSHD) is associated with hypomethylation of the D4Z4 microsatellite, leading to an increased expression of the DUX4 gene which is suggested to cause progressive muscle atrophy and ultimately FSHD pathology. Chromatin changes are caused by contractions to less than 11 units of the D4Z4 repeat (FSHD1) or by pathogenic variants in chromatin regulatory proteins (FSHD2). We developed a novel NGS-based method in an attempt to diagnose FSHD and distinguish FSHD1 from FSHD2 in a single approach. Methods: Enrichment of 4qA (A) and 4qAL (AL) associated D4Z4 repeats as well as all 4q35 D4Z4 repeat units (DUX4) was performed using nested PCRs after bisulfite conversion of genomic DNA (EpiTect). DNA libraries were prepared with the NEBNext Ultra DNA Library Prep Kit and sequenced on an Illumina MiSeq System. Reads were mapped to A, AL and DUX4 using bwameth. Mean methylation levels were calculated with an in-house script. Results: We analyzed 21 patients with a confirmed FSHD diagnosis and 21 negative controls. We could confirm the hypomethylation status in all patients. Low methylation levels tend to correlate with a more severe clinical disease status. Due to a chromatin dysregulation DUX4 is hypomethylated in FSHD2 as well; as expected we could confirm lower methylation levels in DUX4 in FSHD2-patients compared to FSHD1. Conclusion: In conclusion, sensitive detection of methylation levels in the D4Z4 array may give insight into the clinical progression levels more exactly than currently available methods. Additionally, a much higher yield and throughput in FSHD diagnostics may be achieved.
PS2Group5-017 / #579FACIAL NERVE ULTRASOUND IN BELL’S PALSY
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Seol-Hee Baek1, Ye-Ji Kwon1, Yoo Hwan Kim2, Hung Youl Seok3, Byung-Jo Kim1
1Department Of Neurology, Korea University Medical Center, Korea University College of Medicine,, Seoul, KR;2Department Of Neurology, Hangang Sacred Heart Hospital, Hallym University Medical Center, Seoul, KR;3Department Of Neurology, Keumyung University Dongsan Medical center, Daegu, KR
Background: Bell’s palsy is one of the causes of acute paralysis of the facial nerve. However, the etiology of Bell’s palsy is not clearly understood. Ultrasound is the useful tool for evaluating neuromuscular disease. This study is aimed to investigate the usefulness of facial nerve ultrasound in Bell’s palsy. Methods: We prospectively recruited patients with Bell’s palsy who visited Korea University Anam Hospital from January 2016 and November 2017. Ultrasonography was performed using a 15 MHz linear array transducer (HD15 system, Philips Ultrasound, Bothell, WA, USA). Facial nerves were scanned bilaterally along its longitudinal axis inside the parotid gland. The diameters of facial nerve with and without sheath were measured at the proximal and distal portion. Results: Eighty-one patients (43 males, mean age 43.9 ± 15.9 years) were enrolled. At the proximal portion of the facial nerve, the diameters with and without sheath on the affected side were significantly larger than the unaffected side (0.97mm vs. 0.85mm, p<0.001; 0.61mm vs. 0.32mm; p = 0.003, respectively). At the distal portion of the facial nerve, the diameter with sheath was significantly different between the affected and unaffected side (0.86mm vs. 0.80mm, p=0.004). However, the diameter without sheath was not a significant difference (0.31mm vs. 0.29mm; p= 0.633). Among 81 patients, 34 patients have tested follow-up sonography after 2 months. At the proximal portion of the facial nerve, the diameter with sheath on the affected side was significantly decreased after 2 months later (p=0.037). However, there were no significant changes in diameters with the sheath at a distal portion of the facial nerve between initial and follow-up test. Also, the diameters without sheath did not significantly change at both proximal and distal portion of the facial nerve. Conclusion: The diameters of facial nerve were enlarged on the affected side, especially at the proximal portion. Ultrasonography may be helpful for assessment of facial nerve in Bell’s palsy.
PS2Group5-018 / #385CGRP UPREGULATES MYOSIN HEAVY CHAIN TYPE I MESSENGER RNA THROUGH CALCINEURIN-IL-6-INDEPENDENT MANNER IN C2C12 CELLS
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Yoshiaki Mori1, Junko Yamaji2
1Department Of Rehabilitation Sciences, Kansai University of Welfare Sciences, Kashiwara, JP;2Department Of Nutrition Sciences, Kansai University of Welfare Sciences, Kashiwara, JP
Background: Our previous study using differentiated C2C12 cells indicated that myosin heavy chain type I (MyHC I), myosin heavy chain type IIb (MyHC IIb), and interleukin-6 (IL-6) mRNA expression levels were significantly increased by the application of calcineurin activators. In this study, we examined the effects of calcitonin gene-related peptide (CGRP) on mRNA levels of MyHC I, MyHC IIb, and IL-6 in C2C12 cells. CGRP is a 37 amino acid peptide which belongs to a family of related peptides including calcitonin, amylin, and adrenomedullin. CGRP peptides are mainly localized in sensory and central neurons and have been implicated in a variety of physiological processes. CGRP has also been identified in spinal motoneurons of several species and in the nerve terminals of the rodent neuromuscular junction. Effects of CGRP have been studied in two skeletal muscle cell lines, rat L6 cells and mouse C2C12 cells. These cell lines appear to express CGRP receptors coupled to adenylyl cyclase activity. However, the effects of CGRP on expression of MyHC mRNA have not been reported in the skeletal muscle cells. Methods: C2C12 cells were maintained until semi-confluence in D-MEM containing 15%FBS. The cells were incubated in D-MEM containing 2%FBS with or without agents at a beginning of differentiation. After 24h. incubation, the medium was removed and the cells were maintained in differentiation medium for 3 days. MyHC I, MyHC IIb, and IL-6 mRNA expression levels were measured by the real-time PCR method. Results: First, we confirmed the effect of calcineurin activators, such as chlorogenic acid and oleic acid, on mRNA expression levels of MyHC I, MyHC IIb, and IL-6 in C2C12 myocytes. These mRNA levels were significantly increased by the administration of calcineurin activators, and the effects of calcineurin activators on upregulation of these mRNA levels were attenuated by the co-administration of cyclosporine A. The effects of calcineurin activators on upregulation of MyHC I and MyHC IIb mRNA levels were significantly attenuated by the co-administration of anti-IL-6 receptor antibody, although IL-6 mRNA levels were not affected by co-administration of the antibody. These results indicate that the upregulation of MyHC I and MyHC IIb mRNA by the administration of calcineurin activators would be the result of autocrine/paracrine secretion of IL-6 in C2C12 myocytes. Second, we tried to examine the effects of CGRP on mRNA expression levels of MyHC I, MyHC IIb, and IL-6. Administration of CGRP to the culture medium did not affect to the MyHC IIb and IL-6 mRNA levels. However, MyHC I mRNA levels were significantly increased by the administration of CGRP. Conclusion: These results suggest that the effects of CGRP on upregulation of MyHC I mRNA levels do not depend on calcineurin-IL-6-mediated mechanisms. Further experiments need to be done to clarify the CGRP-mediated intracellular signaling in the upregulation of MyHC I mRNA.
PS2Group5-019 / #600ANALYSIS OF EUROPEAN EARLY-ONSET MYASTHENIA GRAVIS GWAS SIGNALS IN AFRICAN GENOMES
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Jeannine M. Heckmann1, Melissa Nel2, Nicola Mulder3
1Medicine (neurology), University of Cape Town, Cape Town, ZA;2Neurology Research Group, University of Cape Town, Cape Town, ZA;3Computational Biology Division, University of Cape Town, Cape Town, ZA
Background: Genome wide association studies (GWAS) in myasthenia gravis (MG) have only been performed in European populations to date. The follow-up of these putative susceptibility variants in African populations, whose genomes have substantially increased genetic diversity and shorter linkage disequilibrium blocks, has the potential to fine-map true causal variants. We performed a cross-racial analysis to determine whether previously identified European GWAS signals in individuals with early-onset acetylcholine receptor (AChR) antibody positive myasthenia gravis (Eur-EOMG) are replicated in African genomes with the same disease phenotype. Methods: 25 individuals with African genetic ancestry from South Africa and early-onset AChR-MG (SA-EOMG), underwent whole genome sequencing. HLA allele group assignment was inferred through “best-match” alignment of reads against the IMGT/HLA database (v3.29.0.1, 2017) using HLA Explore software (Omixon). Ancestry-matched controls were from a local dataset (SA-controls, n=94) where available, or the African sample from the 1000 Genomes Project (AFR-controls, n=1322). Association signals in genetic loci, which showed positive association with Eur-EOMG from two published GWAS studies (HLA class I and II regions, TNIP1 and PTPN22), were interrogated in the SA-EOMG data. Results: An HLA class I region variant (rs7750641), which is strongly associated with Eur-EOMG (minor allele frequency (MAF) 0.41; odds ratio (OR) 6.3) and tags the HLA-B*08 allele in this population group, was lower in SA-EOMG compared to SA-controls (MAF 0.10; p=0.069). However, the HLA-B*08 allele was associated with SA-EOMG (MAF 0.22; OR 4.1, p<1x10-3) which is consistent with previous reports in EUR-EOMG (MAF 0.41, OR 6.4), despite a lower frequency amongst Africans with EOMG. This was also true for an HLA-DQA1 variant (rs601006) which was lower in SA-EOMG compared to Eur-MG, but higher compared to AFR-controls (MAF 0.40, OR 2.3, p=0.007). Due to its proximity to HLA-B, we interrogated the frequency of TNFA -308 G>A in SA-EOMG but our data did not replicate previously reported associations with EOMG (OR 1.7, p=0.151). GWAS signals for Eur-EOMG outside the HLA region, were not replicated in our cohort: TNIP1 (rs2233287, OR 1.2, p=0.681), PTPN22 (rs2476601, minor allele not observed in SA-EOMG dataset and SA-control MAF 0.01). Conclusion: This cross-racial analysis using an African EOMG dataset, showed that the GWAS association signals previously found outside the HLA region among Europeans with EOMG, could not be replicated. Furthermore, the LD structure in the African HLA class I region may contribute a stronger association signal with HLA-B*08 in SA-EOMG individuals compared to variants close to HLA-B such as rs7750641 (upstream) or TNFA -308 G>A (downstream). These results suggest that African genomes can be an informative resource to fine map association regions. Funding: South African (SA) National Research Foundation (113416), SA MRC, AFM Telethon (20049), Novartis Africa CRG Mobility fellowship, Rare disease foundation and BC Children’s Hospital Foundation (2016).
PS2Group5-020 / #648RNASEQ IN URINE-DERIVED STEM CELLS IDENTIFIED THE EXPRESSION OF 264 NEUROMUSCULAR GENE TRANSCRIPTS
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Maria Sofia Falzarano1, Hana Osman1, Rachele Rossi1, Rita Selvatici1, Marcella Neri1, Francesca Gualandi2, Mingyan Fang3, Zhiyuan Lu3, Andrea Grilli4, Silvio Bicciato4, Alessandra Ferlini1
1Medical Sciences, University of Ferrara, Ferrara, IT;2Medical Sciences, University of Ferrara, Ferrara, IT;3BGI-shenzhen, Shenzhen, CN;4Dipartimento Di Scienze Della Vita, University of Modena, Modena, IT
Background: Urine specimen has become a valuable source for isolating urine stem cells (USCs), a novel, non-invasive and powerful modelling tool for studying human specific diseases as well as for generating pluripotent patient-specific cell lines. Few studies have investigated the applicability of USCs for studying neuromuscular diseases. In a previous work, we demonstrated that native USCs and differentiated myogenic USCs are able to recapitulate the specific DMD phenotype, and provide a suitable in vitro environment for AONs mediated dystrophin gene splicing modulation. Methods: In this study we have analyzed the whole transcriptome of native USCs in order to propose them as an in vitro model for studying other neuromuscular diseases. The expression of 398 genes involved in neuromuscular diseases (http://www.musclegenetable.fr/) was investigated by RNAseq analysis on USCs isolated from three healthy donors. Results: We found that 264 out of 398 neuromuscular interrogated genes are expressed in wild type native USCs. All considered disease groups (hereditary motor and sensory neuropathies (85 genes), motor neuron diseases (58 genes), hereditary paraplegias (58 genes), hereditary ataxias (51 genes), distal myopathies (16 genes), congenital myopathies (32 genes), muscular dystrophies (45 genes), congenital muscular dystrophies (34 genes), ion channel muscle diseases (13 genes), myotonic syndromes (6 genes), with the only exception of ion channel genes, are represented with a different rate of expression. Indeed 50-60% of analyzed genes causing muscular dystrophies, congenital muscular dystrophies, distal myopathies, hereditary ataxias, hereditary paraplegias, hereditary neuropathies are detectable at an appreciable level. Up to 70% of genes associated to motor neuron diseases are expressed in USCs.Based on RNAseq data, gene specific analysis can disclose several gene transcript behaviours as novel isoforms or splicing choices. As an example, COL6A1, COL6A2 and COL6A3 messengers show novel isoforms due to skipping events. Conclusion: Our results demonstrate that USCs may represent a robust tool for studying pathogenic mechanisms underlining neuromuscular diseases, proving functional meaning of novel DNA variations, testing drug profiling, and identifying novel biomarkers. Acknowledgment A grant of The Duchenne Parent Project Italy has funded this research.
PS2Group5-021 / #706NEURONOPATHY IN SPINOCEREBELLAR ATAXIA TYPE 2: A NERVE ULTRASOUND STUDY
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Luciana Pelosi1, Rosa Iodice2, Antonella Antenora2, Dean Kilfoyle3, Eoin Mulroy3, Miriam Rodrigues3, Richard Roxburgh3, Alessandro Filla2, Fiore Manganelli2, Lucio Santoro3
1Neurology And Clinical Neurophysiology, Bay of Plenty District Health Board, Auckland, NZ;2Neurosciences, Reproductive Sciences And Odontostomatology, University Federico II, Naples, Italy, naples, IT;3Neurology And Clinical Neurophysiology, 3Auckland District Health Board (ADHB, Auckland, NZ
Background: Somatosensory impairment is not uncommon in autosomal dominant spinocerebellar ataxia (SCA), but it is unclear to what extent this is a neuronopathy from neuron degeneration in dorsal root ganglia, or a primary peripheral nerve pathology, as typically seen in length-dependent neuropathy. We have recently shown that patients with cerebellar ataxia neuropathy vestibular areflexia syndrome (CANVAS), who have sensory impairment from neuronopathy, have pathologically small nerves on ultrasound. This is significantly different from the ultrasound findings in patients with primary axonal neuropathy, whose nerves are either normal or enlarged. In this study, we wished to see if the nerve ultrasound abnormality of CANVAS neuronopathy was also present in patients with SCA type 2. Methods: The ultrasound cross-sectional area of median, ulnar, sural and tibial nerves of 18 patients (mean age 51.9, 11 females) with SCA2 (confirmed by ATXN2 gene mutation) were compared to the reference values of our control population. All patients underwent objective assessment of sensory deficit using the Inflammatory Neuropathy Cause and Treatment (INCAT) sensory sum score (ISS), ataxia assessment using the scale for the assessment and rating of ataxia (SARA) and, disability quantification using the INCAT disability scale. All patients had motor (median, ulnar, peroneal and tibial nerves) and antidromic sensory (median, ulnar, radial and sural nerves) nerve conduction studies. Results: All but one patient had clinical evidence of sensory impairment with ISS score ≥ 1 (mean 3.88). The ultrasound nerve cross-sectional area was significantly reduced at three or more sites in 13 patients and increased at three sites in one patient. Electrophysiology showed reduced amplitude sensory action potentials in 11 patients. The distribution of these abnormalities was non-length dependent in eight patients who also had reduced nerve cross-sectional area on ultrasound, length-dependent in two patients with normal ultrasound, and not clearly defined in one patient with reduced nerve cross-sectional area. Pearson’s correlation analysis showed weak to moderate inverse correlation (p < 0.05) between SARA scores and nerve cross sectional area at two upper limb sites. Conclusion: By analogy with the CANVAS study, the small nerves in patients with SCA2 are likely to reflect neuronopathy with axonal loss secondary to degeneration of the nerve cells. Neuronopathy was identified by nerve ultrasound in 13 (72%) patients, thus representing the most common type of nerve impairment in our SCA2 patient population. In addition, electrophysiology and/or ultrasound findings consistent with axonal neuropathy were present in 3 (16%) patients. Nerve ultrasound had higher sensitivity and specificity than electrophysiology for neuronopathy in our patient cohort, suggesting that it can be a useful complementary tool in the investigation of neuronopathy in spinocerebellar ataxia syndromes.
PS2Group5-022 / #743NERVE ULTRASOUND IN POEMS REVEALS PROXIMAL AND DISTAL SWELLING OF NERVES SIMILAR TO CIDP
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Anne-Kathrin Peyer Kauffmann1, Einar Wilder-Smith2
1Neurozentrum, Luzerner Kantonsspital, Luzern, CH;2Neurologie, Universität, Bern, CH
Background: The electrophysiological features of the polyneuropathy seen in POEMS syndrome are well established. Little is known about the imaging of the nerves in this rare and severe paraneoplastic polyneuropathy. Methods: We describe the clinical, electrophysiological and sonographic features of the peripheral nerves and cervical roots of a 50-year old patient according to Ultrasound Pattern Sum Score (UPSS). Results: A 50 year old female patient was newly diagnosed with POEMS syndrome according to the diagnostic criteria of the International Myeloma Working Group (IMWG). On nerve conduction studies, no motor and sensory nerve recordings were recordable at the lower extremities and upper extremities showed signs of primary demyelination. High resolution ultrasound of median, ulnar, common and superficial peroneal, tibial and sural nerve showed hypoechoic enlargement of the nerves and their fascicles of variable degree. Moreover, longitudinal diameter and cross sectional areas of cervical roots were markedly enlarged. In summary, the total UPSS was 12/20 points, the distal UPSA 9/12, the proximal UPSB 3/3 compatible with the typical features of CIDP showing proximal and distal enlargement of the nerves. Conclusion: Sonographic findings in POEMS resemble the features seen in CIDP.
PS2Group5-023 / #796COMPARISON OF HOME-BASED VERSUS HOSPITAL-BASED SPIROMETRY MEASUREMENTS IN DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Christian Rummey1, Thomas Meier2, Mika Leinonen3, Shabir Hasham2, Thomas Voit4, Oscar H. Mayer5, Gunnar Buyse For The Delos Study Group6
1Clinical Data Science GmbH, Basel, CH;2Santhera Pharmaceuticals, Pratteln, CH;3Santhera Pharmaceuticals, Pratteln, CH;4UCL Great Ormond Street Institute of Child Health, London, GB;5Children’s Hospital of Philadelphia , Philadelphia, PA, US;6University Hospitals Leuven, Leuven, BE
Background: Respiratory function decline in Duchenne muscular dystrophy (DMD) is caused by progressive weakening of respiratory muscles and leads to a high disease burden and early mortality. Standard of care guidelines recommend routine respiratory function monitoring to guide patient management, anticipate complications and improve outcomes. We compared respiratory function measurements obtained using a hand-held device allowing frequent home-based measurements to hospital-based spirometry measures in patients with DMD taking part in the randomized, controlled, Phase 3 DELOS trial. Methods: Respiratory function data were prospectively collected from 64 DMD patients aged 10-18 years, and not taking concomitant glucocorticoids. All patients were required to have established respiratory function decline (peak expiratory flow <80%p) at baseline. Patients were treated with idebenone (900 mg/day) or placebo. Spirometry was conducted during hospital visits at baseline and at 3 month intervals thereafter over the 52 week study period. Patients also measured peak expiratory flow (PEF) and forced expiratory volume in 1 second (FEV1) weekly at home using the ASMA-1 device (Vitalograph). Data were analyzed using a mixed model for repeated measures method. Results: Patients enrolled in the DELOS study were in an advanced stage of disease, with a mean age of 14.3 years. A majority (92%) were already non-ambulatory and showed signs of significant upper limb weakness at baseline (59% had a Brooke’s score of 5 or above). The weekly changes in PEF measured at home (expressed as percent of predicted, PEF%p) compared well with those from hospital-based measurements, showing a 5.6% (p = 0.002) overall treatment difference in favor of idebenone across all weekly visits, compared to a 6.27% difference at last visit using standard hospital spirometry (p = 0.031). Similarly to the hospital based data, the weekly ASMA-1 data indicated a stabilization of PEF%p in the idebenone group versus a natural history consistent decline in the placebo group. Conclusion: Home-based recording of commonly used respiratory function measures is reliable in pediatric and adolescent patients with DMD, and may prove useful and convenient when used in combination with hospital-based monitoring. These data further support the efficacy of idebenone in slowing respiratory function decline in DMD.
PS2Group5-024 / #867AGE-RELATED DIFFERENCES IN MUSCLE MEMBRANE POLARIZATION AS ASSESSED BY VELOCITY RECOVERY CYCLES
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
James Lee1, Robert Boland-Freitas2, Karl Ng3
1Neurology, Royal North Shore Hospital, SYDNEY, AU;2Neurology, Royal North Shore Hospital, Sydney, NSW, AU;3Neurology, Royal North Shore Hospital, ST LEONARDS, AU
Background: The exact mechanisms underlying the loss of skeletal muscle bulk and power with normal human ageing are not well established. The aim of this study was to examine the effect of age on skeletal muscle membrane properties in normal patients as assessed by muscle velocity recovery cycles (MVRC). Methods: Informed consent was obtained from 74 healthy control patients (aged 18 to 84yo, median age 35 and interquartile range 29-55yo) with intact neuromuscular strength (MRC score 5). MVRC recordings were obtained from tibialis anterior (N=74) and rectus femoris (N=32) muscles as representative distal and proximal muscles. Data was captured using QTRACS threshold tracking software employing the M3RC3 recording protocol with subsequent data analysis through the MAnal8 program. Results: Regression analysis on tibialis anterior MVRC recordings demonstrated that increasing age was associated with longer muscle relative refractory period (MRRP: r2= 0.38, P<0.001). There was a reduction in early supernormality (r2= 0.38, P<0.001) and a smaller reduction in additional late supernormality with multiple conditional stimuli as age increased (r= -0.45, p<0.001). Analysis of rectus femoris MVRCs revealed a similar association of increased age with longer MRRP (r2= 0.30, P = 0.002) and reduced early supernormality (r2= 0.19, P = 0.01), but no significant association with late supernormality was apparent. Conclusion: The changes suggest resting muscle membrane potential appears relatively more depolarized with increasing age in adulthood. Possible reasons for this finding include the increasing predominance of type 1 muscle fibres with age (1), or age-related Na+/K+ ATPase and mitochondrial dysfunction (2). These findings may represent an important mechanism in explaining age-related muscle decline. REFERENCES 1. St-Jean-Pelletier F, Pion CH, Leduc-Gaudet JP, Sgarioto N, Zovile I, Barbat-Artigas S, et al. The impact of ageing, physical activity, and pre-frailty on skeletal muscle phenotype, mitochondrial content, and intramyocellular lipids in men. J Cachexia Sarcopenia Muscle. 2016. 2. Miljkovic N, Lim JY, Miljkovic I, Frontera WR. Aging of skeletal muscle fibers. Ann Rehabil Med. 2015;39(2):155-62.
PS2Group5-025 / #885EFFECTIVE DIAGNOSTIC AND TREATMENT METHODS IN VASCULAR PARKINSONISM AND PARKINSON’S DISEASE: TEMPORHYTHMAL CORRECTION
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Dilshoda T. Akramova
Neurology, Tashkent Medical Academy, Tashkent city, UZ
Background: It is known that, in vascular parkinsonism and Parkinson’s disease it is observed shortening of steps, bradi-, oligokinesis, and also tremor disturb such kind of patients. Methods: order to early and effective differential diagnostics, also to properly treat of tremor and short steps in vascular parkinsonism and Parkinson’s disease it is used method of tempo-rythmal correction 80 patients have participated in our research and the mean age of them was 62.3 ± 4.7 years. All patients were divided into 4 group. 1st group - patients who have vascular parkinsonism and they have received both medicamentous treatment and temporythmal correction. In the group 2 were patients with Parkinson’s disease, they have also received both medicamentous treatment and temporythmal correction. Group 3 was a group of patients with vascular parkinsonism they have received only medicamentous treatment. 4th group of patients with Parkinson’s disease, they have received also only medicamentous treatment. First of all, there is measured height, weight and body mass index. Calculated and scheduled common length of steps, amount of steps passed in 500 m and sum of spent kcal. Patients were observed during 10 days: was selected quiet music and have measured amount of steps and length of passed distance for 3 times during the 10 day. Patients walked in the morning under quiet music, on the midday under quickened and on the evening under fast rythm music. The results were recorded while there were walking. All patients were evaluated by the Parkinson’s Disease Questionnaire (PDQ-39) scale. Results: According to the results of our 10-day observations. In 1st group the maximal positive result according to PDQ-39 was on “vital activity” and was 2.87 ± 1.36. Patients said that they had felt easiness while dressing, bathing, eating and serving to themselves. In normal people the average length of step is 40% of the height, in the first days of tempo-rythmal correction in all patients this index was 25 or 28% and 30 or 32% on the last day of correction. In patients of 2nd group according to PQD-39 the maximal positive outcome was on “vital activity” and is 2.23 ± 1.16. Patients of this group said that they felt more easiness on dressing and eating. The average length of steps in patients with Parkinson’s disease was 21 or 23% on the first day of the temporythmic correction, and 22,24% in the last days of the temporythmal correction. Groups 3 and 4 received only medicamentous therapy, and correction was not conducted in group patients. The PDQ-39 consisted in 3rd group - 1.9 ± 2.16 and 4th group - 2.03 ± 1.37. Conclusion: conclusion, we can say that temporythmal correction is method of treatment and rehabilitation, which is effective in each type of vascular parkinsonism and Parkinson’s disease also in economical aspect that patient can use both in the hospital and in the home The average length of steps in Parkinson’s disease is more shorter than in the vascular parkinsonism.Temporitmal correction is an effective and cost-effective method of differential diagnosis of vascular parkinsonism and Parkinson’s disease
PS2Group5-026 / #316KIF5A, MUTATION CAN LEAD TO SPASTIC PARAPLEGIA (SPG10); GABAARS-VESICLE TRANSPORT MOTOR INTERACTS WITH G3BP2 AND GABARAPS
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Dae-Hyun Seog1, Sangjin Kim2
1Biochemistry, Inje University College of Medicine, Busan, KR;2Neurology, Inje University College of Medicine, Busan, KR
Background: Spastic paraplegia type 10 (SPG10) is a rare form of autosomal dominant hereditary spastic paraplegia due to mutations in kinesin superfamily proteins 5A (KIF5A). KIF5A mutations have been associated with a wide clinical spectrum, ranging from pure hereditary spastic paraplegia to isolated peripheral nerve involvement or complicated hereditary spastic paraplegia phenotypes. Most KIF5A mutations are clustered in the motor domain of the protein that is necessary for microtubule interaction. Molecular motor protein KIF5A is involved in the gamma-aminobutyric acid type A receptors (GABAAR) containing vesicle transport. Deletion in the KIF5A can impair intracellular transport of GABAA receptors. However, the binding partners or regulation proteins of KIF5A have not yet been fully identified. Methods: We were used the yeast two-hybrid system to identify the binding proteins that interact with the C-terminal 73 amino acids of KIF5A. The binding affinity was quantified by measuring ß-galactosidase activity in liquid cultures of yeast transformed cells. Direct interaction between binding proteins and KIF5A in mammalian cells as well as in vitro was assayed using the co-immunoprecipitation with the antibodies. The cellular co-localization in cells was used the immunocytochemistry. Results: We revealed an interaction between the C-terminal region of KIF5A and GABARAPs, and G3BP2, which is involved in stress granules (SGs) formation and mRNA-protein (mRNP) localization. KIF5s are a heterotetrameric protein composed of two heavy chains (KIF5s), and two light chains (KLCs). The KIF5s possess a motor domain at the amino-terminal region connected by a long stalk domain to a carboxyl (C)-terminal tail domain. The motor domain, which binds to microtubules, is responsible for the motor activity of kinesin 1, and the C-terminal tail domain is responsible for interacting to cargoes. KIF5s have been revealed to consist of three closely related subtypes: KIF5A, KIF5B, and KIF5C. KIF5B is expressed ubiquitously in many tissues, whereas KIF5A and KIF5C are expressed in nervous tissue. KIF5A can form homodimer or heterodimer with KIF5B, or KIF5C. GABARAPs and G3BP2 bound to the C-terminal 73 amino acids of KIF5A and did not interact with the other KHCs (KIF5B, and KIF5C) in the yeast two-hybrid assay. However, other G3BP isoform, G3BP1 did not interact with KIF5A, suggesting a specific interaction of KIF5A with GABARAP and G3BP2. The KIF5A-GABARAP, G3BP2 interaction was also assessed in mammalian cells. When the lysate of HEK-293T cells transiently expressing myc-KIF5A and FLAG-GABARAP, FLAG-G3BP2 was immunoprecipitated with anti-FLAG antibody, KLC1 and KIF5A, but not KIF3A were co-precipitated with GABAR, GABARAP and G3BP2. This result indicates that GABARAP and G3BP2 associate with kinesin 1. In order to further confirm whether KIF5A and G3BP2 co-localize in cells, KIF5A was co-expressed with EGFP-GABARAP, EGFP-G3BP2 in HEK-293T cells. KIF5A and GABAR, GABARAP and G3BP2 co-localized at the same region in cells. Conclusion: These results suggest that KIF5A may transport of GABAAR containing vesicles and SGs or mRNP granules. Furthermore, these results strengthened the link between the cargos trafficking and Spastic paraplegia phenotypes.
PS2Group5-027 / #394VANGL2, A CORE COMPONENT OF THE WNT PCP PATHWAY CONTRIBUTES TO NEUROMUSCULAR JUNCTION FORMATION
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Myriam Boex1, Julien Messéant2, Stéphanie Bauché3, Laure Strochlic4, Bertrand Fontaine4
1Team “gen-phys: Neugenetics And Physiology” Umr–s1127, Brain and Spine Institute, Paris, FR;2Team “gen-phys: Neugenetics And Physiology” Umr–s1127, Brain and Spine Institute, Paris, FR;3Equipe Neurogenetic And Physiology, Institut du Cerveau et de la Moelle epinière, Paris, FR;4Institut du Cerveau et de la Moelle épiniere, Paris, FR
Background: The development of the neuromuscular junction (NMJ), a cholinergic synapse between the axon terminal of a motoneuron and a specialized region of the skeletal muscle fiber requires a dynamic communication via various reciprocal signaling processes between the two elements that are only partially understood. Several studies have recently highlighted the emerging and complex role of Wnt signaling at the NMJ. Secreted Wnt ligands (notably Wnt4 and Wnt11) signal through interaction with the Frizzled-like cysteine rich domain (CRD) of the muscle-specific tyrosine kinase MuSK to activate both canonical/b-catenin dependent and core Planar Cell Polarity (PCP) pathways. First evidence for a role of Wnt PCP signaling during NMJ formation comes from studies in Zebrafish showing that both Wnt4a and Wnt11r cooperate to initiate NMJ formation likely by stimulating PCP-dependent MuSK endocytosis. In mice, we previously demonstrated that one of the key PCP core component, namely Van Gogh-Like protein 2 (Vangl2), is expressed in both pre- and postsynaptic compartments, interacts with extracellular Wnt4 and Wnt11 and is required to shape the neuromuscular synapse. However, the molecular determinants of Vangl2 signaling at the NMJ still largely remains unknown. Methods: Using experimental mouse models and a large array of cell biology technics we investigated Vangl2 function during NMJ development. Results: In this study, we first examined in vivo the NMJ phenotype of diaphragms from Vangl2 knock-out (KO; Vangl2-/-) mouse embryos and showed that the absence of Vangl2 induces pre- and postsynaptic differentiation defects characterized by a decrease in the number and volume of acetylcholine receptors (AChRs) and overgrowth of motor axon within the target field. Moreover, loss of Vangl2 function inhibited Wnt11-mediated AChRs clustering in primary muscle cells. We further demonstrated that only muscle (-HSA) but not motoneuronal (-HB9) specific conditional KO (cKO) of Vangl2 recapitulates both pre- and postsynaptic defects of the Vangl2-/- mice and Vangl2 forms a complex with MuSK independently of its CRD. Conclusion: All together, these results demonstrate that a Wnt/PCP signaling pathway in the muscle relying on MuSK/Vangl2 interaction contributes to the establishment of mammalian neuromuscular connectivity.
PS2Group5-028 / #604A 3D SYSTEM FOR MECHANICAL CHARACTERIZATION OF ARTIFICIAL SKELETAL MUSCLE MICROTISSUE
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Marina Spörrer1, Delf Kah2, Sandra Wiedenmann2, Ingo Thievessen2, Rolf Schröder3, Wolfgang H. Goldmann2, Ben Fabry2
1Department Of Physics, Biophysics Group, Friedrich-Alexander University Erlangen-Nuremberg, Erlangen, DE;2Department Of Physics, Biophysics Group, Friedrich-Alexander University Erlangen-Nuremberg, Erlangen, DE;3Neuropathology, University Hospital Erlangen, Erlangen, DE
Background: Artificial skeletal muscle tissues grown from cells that are isolated from patients with myofibrillar myopathies offer the prospect to study otherwise hard to measure processes such as muscle contractility, the occurrence of microlesions after exercise, or the response to drugs. However, previous attempts to grow artificial muscle have suffered from insufficient differentiation of myoblast precursor cells and poor maturation into an in-vivo-like, highly aligned, and contractile tissue. Methods: Here, we develop a method for growing muscle tissue in 4x2x2 mm microwells containing two flexible silicone pillars that serve as force sensors. Myofibroblasts are seeded in a collagen/matrigel suspension. Upon polymerization of the collagen/matrigel mixture into a soft 3-D hydrogel, the myoblasts spread, adhere, and compact the matrix, and form an aligned tissue that spans between the pillars. When cultured in a differentiation medium containing insulin, transferrin and selenium (ITS), muscle tissues start to contract upon electrical pacing 6-7 days after seeding. A Stretchable device with 18 wells. Inset: close-up view of a single well. B Top view of a microtissue containing 7500 myoblasts. Inset: staining of aligned myotubes. Green: alpha-actinin, blue: dapi. C Side view of a well with tissue. D Contractile force of a tissue in response to electrical pacing at 1 Hz. Results: We find an optimal tissue formation and force generation (2 kPa static tension, 400 Pa contractile tension) for a collagen concentration of 0.6 mg/ml and a Matrigel concentration of 1.5 mg/ml. At lower or higher Matrigel concentrations, contractile tension dramatically decreases. This is attributable to absence of matrix-mediated signaling at lower Matrigel concentrations, and poor alignment of collagen fibers and hence of the differentiated myotubes at higher Matrigel concentrations. Conclusion: These findings suggest that addition of different matrix components and fibroblasts to primary myoblasts critically influence tissue formation and contraction, and thus need to be tuned for optimal function of the microtissues.
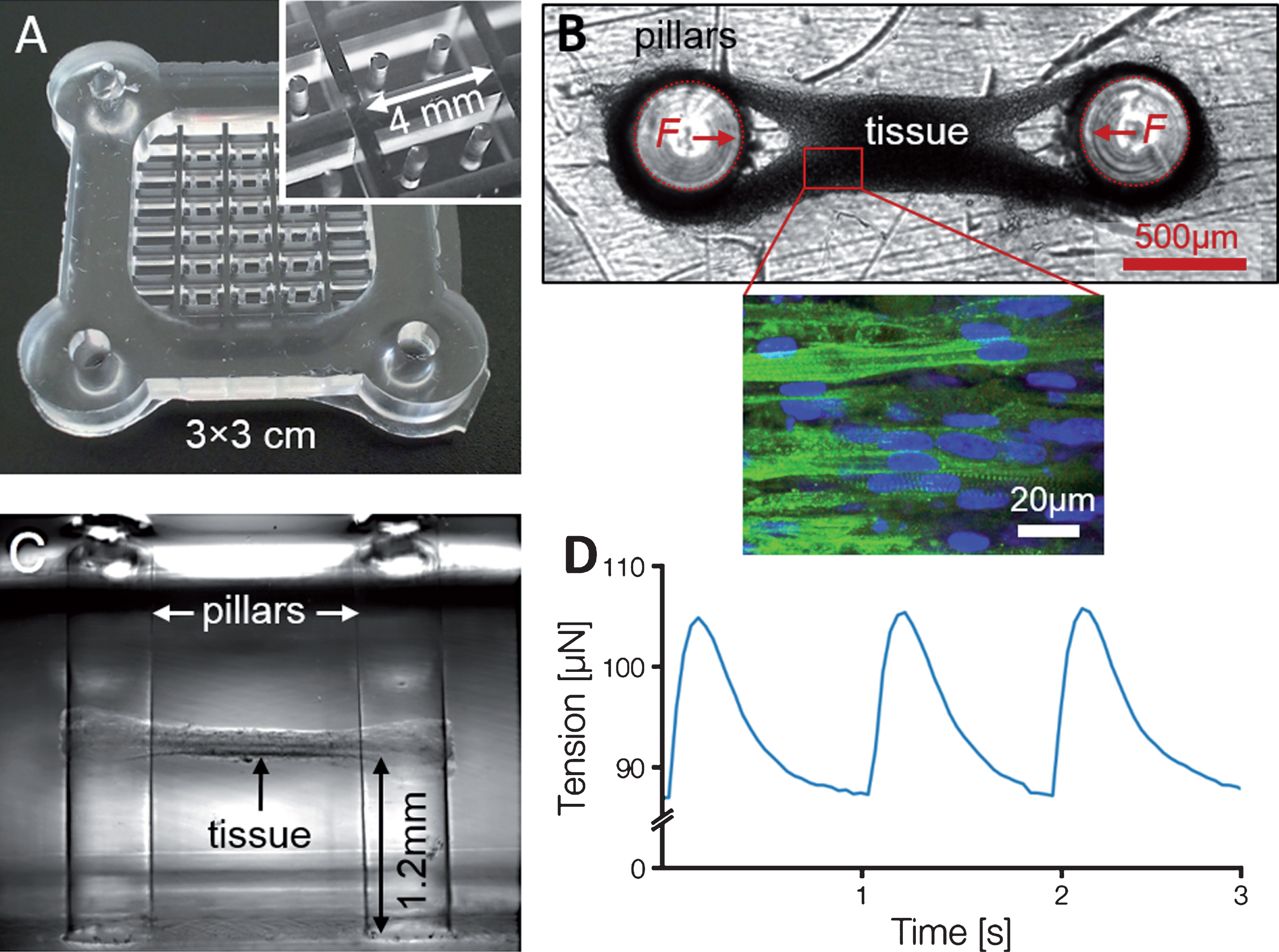
PS2Group5-029 / #27POTENTIOALLY CONFOUNDING VARIABLES OF GDF-15: MITOCHONDRIA DISEASE AND OTHER NEUROLOGICAL DISEASES
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Akiko Ishii1, Seitaro Nohara2, Zenshi Miyake2, Naoki Tozaka2, Sho Okune2, Hayato Takeda2, Hiroshi Tsuji2, Yasushi Tomidokoro2, Kiyotaka Nakamagoe2, Kazuhiro Ishii2, Masahiko Watanabe2, Akira Tamaoka2, Shuichi Yatsuga3, Yasutoshi Koga3
1Neurology, University of Tsukuba, Tsukuba, JP;2Neurology, University of Tsukuba, Tsukuba, JP;3Pediatrics, Kurume University, kurume, JP
Background: GDF-15 (Growth Differentiation Factor 15) is one of transforming growth factor beta superfamily and regulate inflammation and apoptosis in various disease, such as heart failure, kidney dysfunction, and tumor. Yatsuga et al. reported that GDF-15 was a new biomarker of mitochondria diseases. The aim of this study is to clarify potentially confounding variables of GDF-15 and demonstrate as mitochondria biomarker. Methods: We used serum from 14 patients of MELAS, 15 patients of limbic encephalitis (LE), 10 patient of multiple sclerosis (MS), and 19 patients of ALS. GDF-15 and FGF-21 are measured using ELISA. Clinical features of patients are also evaluated retrospectively. Results: Average of GDF-15 and FGF-21 in mitochondria diseases are 2860.7 pg/ml and 855 pg/ml, respectively. Average of GDF-15 and FGF-21 in LE are 786.2 pg/ml and 238.2 pg/ml, respectively. GDF-15 and FGF-21 in MS are 575.4 pg/ml and 384.0 pg/ml, respectively. Average of GDF-15 and FGF-21 in ALS are 509.6 pg/ml and 119.3 pg/ml, respectively. Although, two patients with ALS showed high FGF-21, there were no correlation with age, severity, and phenotype. GDF-15 and FGF-21 are significantly elevated in mitochondria diseases.There is no correlation between GDF-15 and clinical features, such as ADL level, number of cells in cerebrospinal fluid (CSF), and brain MRI findings. No GDF-15 elevation is found in three patients having cancer. There are tendency of correlation between GDF-15 and protein concentration of CSF in LE patients. GDF-15 shows positive correlation between EDSS in MS patients (Pearson correlation coefficient 0.74). Conclusion: GDF-15 is a useful biomarker of mitochondria disease and potentially influenced by severity of diseases. Age is the most potentially confounding variables of GDF-15.
PS2Group5-030 / #726QUANTITATIVE ESTIMATES OF ULTRASOUND IMAGING IN MUSCLE PATHOLOGY
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Dimitra Veltsista1, Elisabeth Chroni2
1Neurology, University Hospital of Patras, Patras, GR;2Neurology, University of Patras, Patras, GR
Background: There is a growing tendency to employ ultrasound examination for neuromuscular investigation. This study aimed to explore potential markers for quantitative assessments of muscle ultrasound appearance in order to increase the test reliability and objectivity. Methods: Ten patients (6 men; mean±sd age 50.1±8.5 years) with neurogenic weakness (NE), diagnosed with spinal muscular atrophy, amyotrophic lateral sclerosis or motor neuronopathy and 10 patients (8 men; mean±sd age 36.2±16.2 years) with a primary myopathy (MY) i.e. Duchenne, Becker, limb-girdle or facioscapulohumeral muscular dystrophy were examined. The control group (C) consisted of 14 healthy subjects (8 men; 37.4±14.0 years). Data were obtained from ultrasound transverse image of the biceps brachii muscle at the 2/3 distance from acromion to antecubital fossa using a linear array probe. Images were exported and the entire region of muscle was manually selected and analyzed using ImageJ fiji, processing program. A histogram of echo intensity was created depicting the counts (pixels) for each bin on a gray scale from 0 (black) to 255 (white). The mean (sd) gray scale, minimum, maximum and mode value as well as the full width at half maximum value were estimated. Muscles were scored for strength on MRC and their images on Heckmatt’s rating scale. Results: Group comparison, employing one-way ANOVA post hoc Dunnett t-test , showed significantly increased values of mean gray scale for the patient groups as opposed to control (NE 60.6±15.4 / MY 67.6±16.3 / C 47.6 ±6.9 and corresponding p-values= 0.032, 0.001). Full width at half maximum was also significantly higher for patients (NE 47.3± 9.2 / MY 50.6±9.5 / C 33.1±5.9 and both p-values =0.000). Figure illustrates the group histogram shifting and broadening in diseased muscles. No statistical differences between patient groups were found. There was a linear correlation of mean gray scale values with MRC score (R2=0.5, p-value= 0.000) but not with Heckmatt’s score. Conclusion: Our findings suggested that creation of gray scale histograms followed by estimation of full width at half maximum value, which seemed to be the most sensitive measurement, may serve as a reliable index of muscle pathology reflecting the severity though not the type of the primary pathogenic process.
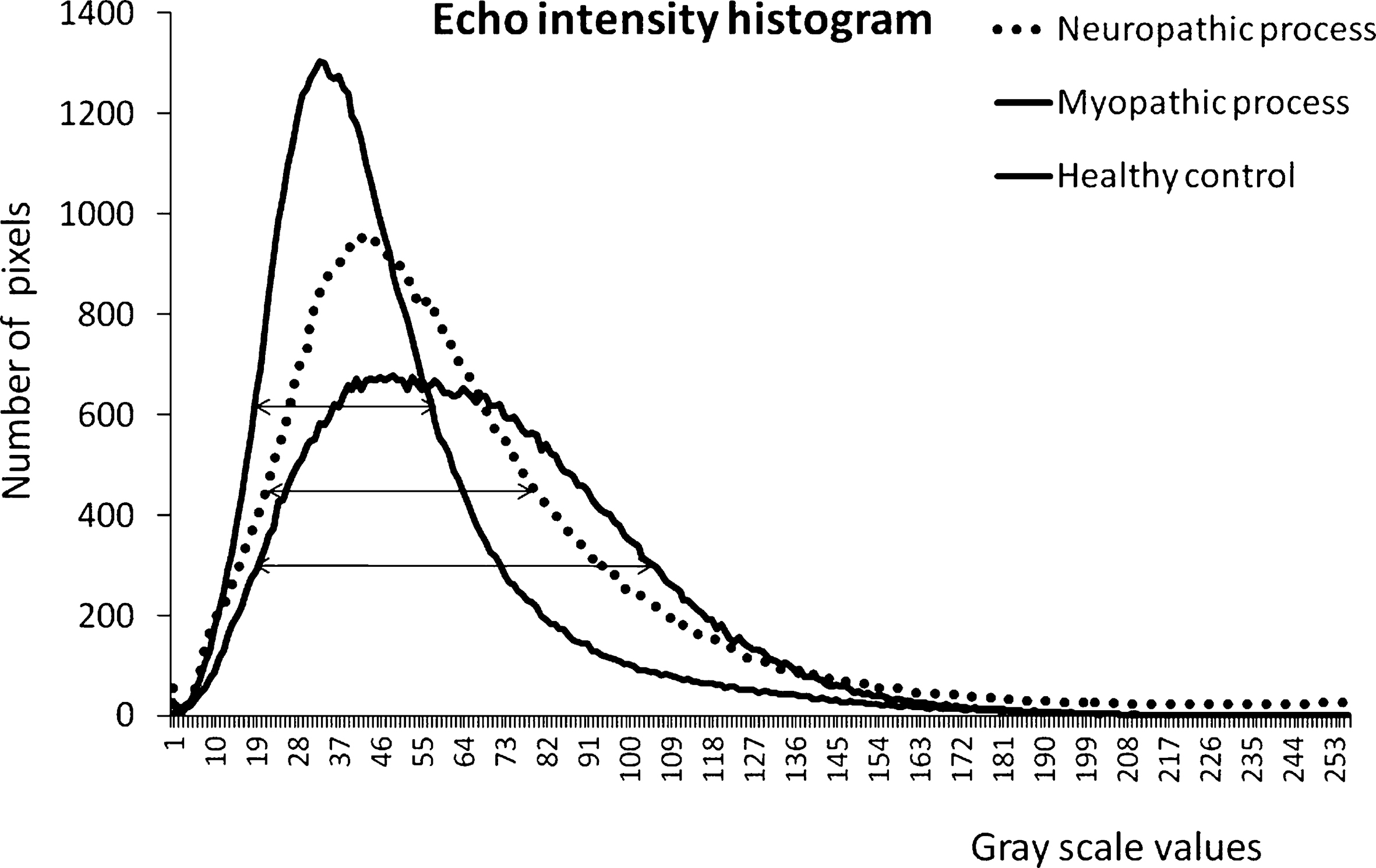
PS2Group5-031 / #828CIRCULATING KV1.3+ CELLS IN PATIENTS WITH SPORADIC INCLUSION BODY MYOSITIS
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Tahseen Mozaffar1, Marie Wencel2, Karissa Munoz3, Jenna Kastenschmidt3, S. Armando Villalta3
1Neurology, University of California, Irvine, Orange, CA, US;2Neurology, University of California, Irvine, Orange, US;3Physiology And Biophysics, University of California, Irvine, Irvine, CA, US
Background: We had recently shown that muscle biopsies from patients with sIBM express large numbers of CD3+ cells (22.4% to 53.1%) that co-localized Kv1.3, a voltage-gated potassium channel seen on effector memory T-cells. We examined if circulating Kv1.3+ cells can be detected in unstimulated samples from sIBM patients. Methods: Blood collection was done at the annual patient conference of the Myositis Association in San Diego, CA with immediate processing for PBMCs. Multichannel flow cytometry was done, with a panel containing the following markers: CD3, CD8, CD4, CD244, CD57, CD28 viability marker, and Kv1.3. Descriptive statistics and student t-tests were used for statistical purposes. Results: PBMCs were collected from 22 patients with a diagnosis of IBM, 9 patients with PM, 9 patients with DM and 5 healthy controls. CD4+CD57+CD28null cells were seen in 3.8 ± 0.9% of sIBM patients (p=0.02 vs. HC) while CD8+CD57+CD28null cells were seen in 50 ± 3.6% of sIBM patients (p=0.01 vs. HC). The percentage of these cells was not statistically different in DM and PM patients. Percentage of Kv1.3+ cells was statistically increased in both CD4+ as well as CD8+ cell populations (CD4+CD244+Kv1.3+ = 0.53 ± 0.15%; p<0.02 vs HC and CD8+CD244+Kv1.3+ = 1.70 ± 0.21%; p=0.02). Conclusion: Circulating Kv1.3+ cells were statistically increased in sIBM compared to healthy controls and other inflammatory myopathies. The numbers of these cells are small, probably representing a small percentage of the large T-cell repertoire in circulating blood. We next plan to isolate live T-cells from skeletal muscle biopsies from sIBM patients to quantitate presence of Kv1.3+ cells in diseased muscle.
PS2Group5-032 / #467ULTRASOUND IMAGING OF GENIOGLOSSUS MUSCLE FUNCTION WITH CONTRAST AGENT
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Orest M. Semeryak
Lviv Regaional Clinical Hospital Center of NMD and Disease of PNS, Lviv, UA
Background: INTRODUCTION. the human upper airway has many important physiological functions including speech, swallowing and breathing. The human tongue forms an important part of the upper airway and plays a key role in the act of swallowing. Among these, the genioglossus (GG) is the largest dilator of the upper airway and main swallowing muscule. There is a large group of neuromuscular disorders, which can cause dysphagia. In patients with neuromuscular diseases, a gradual dystrophy of the muscle of the tongue with a violation of the act of swallowing, sweating and breathing disorder is observed. Therefore, in this situation it is necessary to find an adequate, safe screening method of dynamic visualization for the evaluation of structural changes and motor disorders of the muscles of the tongue. Methods: METHODS. Ultrasound does not emit radiation, is non-invasive, gives good resolution of soft tissue structures, can be used as a screening method and dynamically during the treatment process, and can display cross-sectional anatomy and tissue motion in real time. It also has been used to demonstrate dynamic upper airway motion during swallowing and speech. Ultrasonography can be used to quantify GG movement. Ultrasound images from the suprahyoid region and from the surface of the tongue were made in 25 healthy subjects aged 10 to 30 years to determine the normal structure and movements; that data were compared with 5 patients with motoneuron disease. To detect manifestations of dysphagia and to study the nature of the movements of the tongue when swallowing with normal and pathology, we used contrast media - a solution of sugar saturated with small gas bubbles. Results: RESULTS. All measurements were feasible in all subjects and patients with good reproducibility. Our findings suggest gradual involvement of oral muscles in motoneuron disease. The use of contrast shows the mechanism of movement of the muscles of the tongue and the movement of the epiglottis while swallowing. Conclusion: CONCLUSION. This study demonstrates a novel use of ultrasound to quantify the GG movement in awake humans during swalloving act. It is a simple, reproducible, safe technique that does not emit radiation and allows use during the treatment process without restrictions. Use the ultrasound in oral muscles is feasible in healthy children, adults and patients with neuromuscular disorders. These data show that it is possible to differentiate between healthy persons and patients with neuromuscular disorders. The use of contrast provides an additional understanding of the pathological process, gives additional data on swallowing biomechanics, and clearly illustrates problems with dysphagia.
PS2Group5-033 / #535COMPUTATIONAL SPEECH ANALYSIS AS A TOOL FOR EARLY DETECTION OF BULBAR DYSFUNCTION IN ALS PATIENTS
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Bernat B. Recasens1, Diego C. Lahoz2, Anna G. Solà3, Maria S. Martinez4, Francisco A. Cabrera5, Juana M. Llorens6, Ana B. Corberó6, Montserrat V. Moreno7, Miguel Angel R. Perez8
1Neurology, Hospital del Mar, Barcelona, Barcelona, ES;2Hospital del Mar, Barcelona, ES;3Physical Medicine And Rehabilitation Department, Hospital del Mar, Barcelona, ES;4Neurology, Hospital del Mar, Barcelona, ES;5Pompeu Fabra University, Barcelona, ES;6Pneumology Department, Hospital del Mar, Barcelona, ES;7Endocrinology Department, Hospital del Mar, Barcelona, ES;8Neurology Department, Hospital del Mar, Barcelona, ES
Background: In amyotrophic lateral sclerosis (ALS) the initial symptoms of dysarthria are subtle and there is no objective method for assessing them, which can be difficult to early detection. We propose the computational analysis of speech as a method for the early detection of bulbar impairment in ALS patients. Methods: An analysis of different speech acoustic variables using Praat software was performed. ALS patients with no apparent bulbar impairment, according to El Escorial criteria, were compared to healthy controls (H). Lingual strength, measured with IOPI system, was also included as alternative measure of bulbar involvement expression. Results: 9 patients were evaluated and compared to 20 healthy volunteers. ALS patients compared to volunteer group presented a greater number of pauses (p=0.016), longer duration during the reading of a text (p = 0.005), as well as a lower verbal fluency (p <0.001). In addition, they presented a significant decrease in the lingual movement in the horizontal plane (p <0.001), with a trend towards a lingual anterior position (p=0.005) and inferior (p=0.003) during phoneme emission. The decrease in lingual strength was related (r=0.80) with a decrease in lingual movement (p=0.003). Conclusion: ALS patients without apparent bulbar dysfunction had an affectation of the lingual movements in the horizontal plane. The computational analysis of speech is a simple and objective tool that seems useful for an early detection of bulbar dysfunction in ALS and even allows to distinguish from other etiologies of dysarthria.
PS2Group5-034 / #985THE IMPORTANCE OF A NON-INVASIVE SCREENING IN PROXIMAL MYOPATHIES
Topic: Group 5 – Novel Diagnostic Methods in Neuromuscular Diseases and Basic Sciences in Neuromuscular Diseases
Giorgia Bruno, Lia Allegorico, Luca Lombardi, Giuseppe Di Iorio, Simone Sampaolo
Second Neurology, University of Campania “Luigi Vanvitelli”, Naples, IT
Background: The Late-Onset Pompe Disease (LOPD) is caused by compound mutations of the acid alpha-glucosidase gene (GAA) which lead to deficiency of GAA enzyme activity and accumulation of glycogen within autophagic vacuoles. LOPD affects primarily proximal muscle, as in a limb-girdle muscolar dystrophy and polymyosities. Methods: We describe unusual cases of three patients presenting hyperCKemia, proximal muscle weakness, autoimmunity nonspecific indices slightly above the reference range, that received inflammatory myopathy diagnosis. So they underwent to steroid and immunosuppressor therapy, that worsened the clinical picture. Therapeutic failure imposed clinical and instrumental revaluation. Results: The first evaluation was made with Lymphocytes on blood smears that disclosed PAS positive glycogen granula in their cytoplasm and Dried Blood Spot (DBS) that revealed reduced GAA enzyme activity. Elettromyography extended to paraspinal muscles revealed a myopathic pattern with complex repetitive discharges. For this reason we decided to repeat muscle biopsy: histological examination revealed a myopathy pattern characterized by consistent glycogen storage at intermyofibrillary and sub- sarcolemmal levels and numerous PAS and acid phosphatase positive vacuoles in many fibres. In suspect of LOPD, patients also performed an ANGIO-MRI that disclosed dolicho-ectasis of basilar trunk. Genetic investigations confirmed LOPD. Fig. 1. a) PAS positive glycogen granula in the Lymphocytes cytoplasm on blood smears b) Histology and histochemistry of quadriceps muscle biopsy revealed a myopathic pattern characterized by presence of numerous vacuolary formations of variable shape, number and extension. These findings are suggestive of vacuolar myopathy, like glycogenosis. Conclusion: Proximal muscle weakness and hyperCKemia should be screened at first, with rapid and non-invasive methods like peripheral blood smear and DBS to avoid, despite a rise in aspecific inflammatory markers, immunomodulatory therapies able to worse, potentially, the clinical features of a metabolic myopathy.
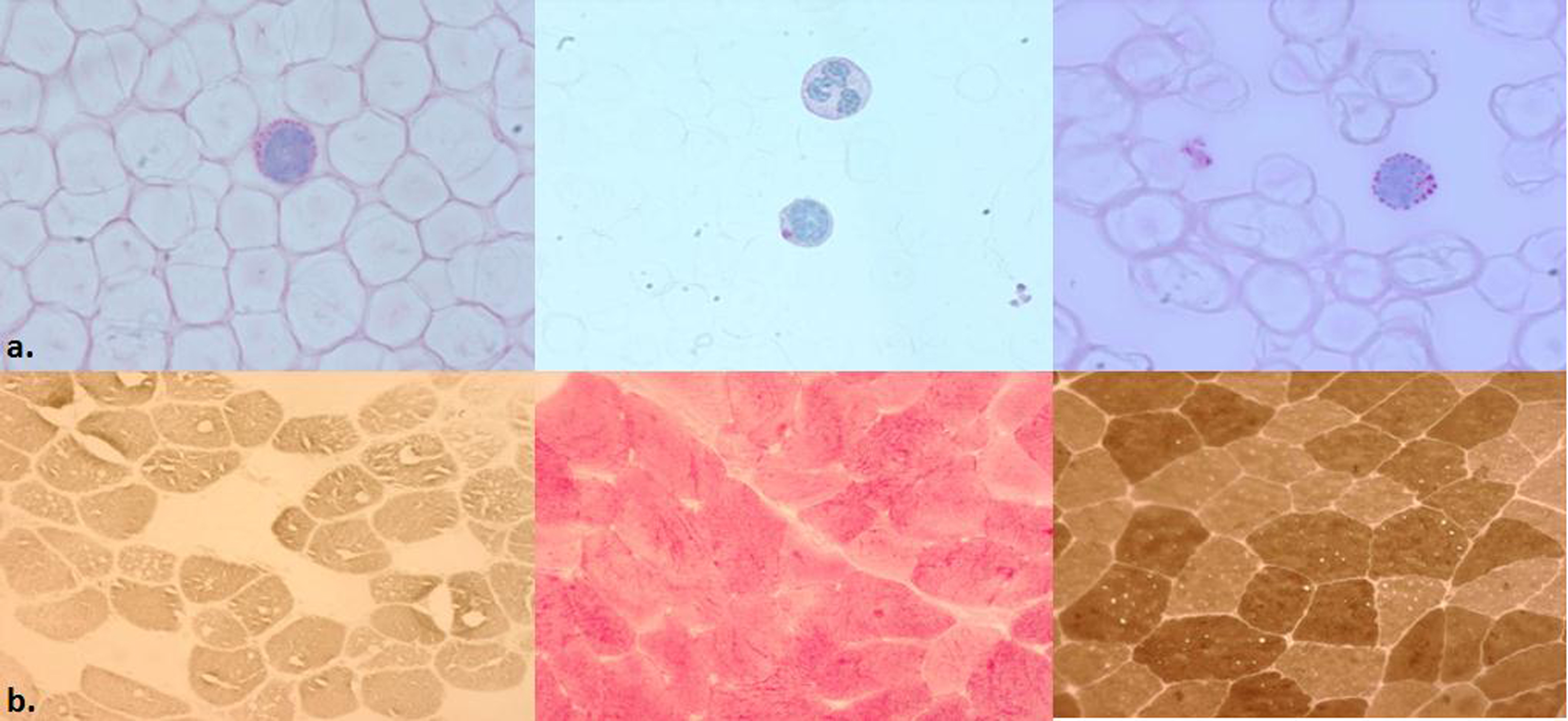
PS2Group6-001 / #349CLINICAL DIVERSITY OF P/Q-TYPE CALCIUM CHANNEL ANTIBODY-ASSOCIATED PARANEOPLASTIC DISORDERS
Topic: Group 6 – Cancer related Disorders and General Diseases
Takashi Irioka1, Yuko K. Takahashi2, Susumu Igarashi1, Takamasa Majima2, Hirokazu Shiraishi3, Shunsuke Yoshimura3, Hiroko Kitanosono3, Takanori Yokota2, Masakatsu Motomura4
1Department Of Neurology, Yokosuka Kyosai Hospital, Kanagawa, JP;2Department Of Neurology And Neurological Science, Tokyo Medical and Dental University, Graduate School, Tokyo, JP;3Department Of Neurology And Strokology, Nagasaki University Hospital, Nagasaki, JP;4Department Of Electrical And Electronics Engineering, Faculty of Engineering, Nagasaki Institute of Applied Science, Nagasaki, JP
Background: P/Q-type voltage-gated calcium channel antibodies (P/Q-VGCC abs) are not only pathogenic in Lambert-Eaton myasthenic syndrome (LEMS) but are also associated with diverse paraneoplastic neurologic disorders (PNDs). Methods: To clarify clinical characteristics of PNDs associated with small cell lung cancer (SCLC) and P/Q-VGCC abs. We encountered 3 adult male patients with SCLC- and P/Q-VGCC ab-associated PNDs. We reviewed their medical records to investigate the clinical phenotypes of the PNDs, autoantibody profiles, treatments, and therapeutic responses. Results: 1) PND phenotypes. Patient 1: A 65-year-old man suffered from dry mouth for several months. Thereafter, he presented with proximal limb weakness and bilateral ptosis. Tendon reflexes were depressed, and electrophysiologic studies confirmed LEMS. Patient 2: A 69-year-old man developed subacute pancerebellar syndrome, which was compatible with paraneoplastic cerebellar degeneration (PCD). Neurologic and electrophysiologic examinations did not reveal complicated LEMS. Patient 3: A 71-year-old man presented with unstable gait for months. Subsequently, dysesthesia in the limbs appeared. Neurologic examination revealed dysarthria and kinesthetic sensory loss with Romberg sign. Electrophysiologic studies were consistent with sensory polyneuropathy (sensory neuronopathy) without LEMS. 2) Autoantibody profiles. Radioimmunoprecipitation assays for P/Q-VGCC abs were positive in all 3 patients. Paraneoplastic autoantibody evaluation of the serum (Mayo Clinic) revealed N-type VGCC antibody and anti-glial nuclear antibody (AGNA-1, Sox1 antibody) in Patient 2, but no coexisting autoantibodies were found in Patients 1 and 3. 3) Treatments and responses. All 3 patients were treated with chemotherapy against SCLC. The neurologic symptoms were resistant to chemotherapy in Patients 1 and 3, but the cerebellar symptoms in Patient 2 improved robustly. Improvement of the ataxia was correlated with decreases of anti-P/Q-VGCC antibody titers in the serum. Additionally, Patient 2 was treated with immunotherapies (plasma exchange [PLEX] and intravenous immunoglobulins [IVIG]) after the chemotherapy. Again, the ataxia improved immediately (the next day after the first PLEX or after IVIG infusion for 3 consecutive days). Conclusion: P/Q-VGCC abs are associated with various types of PNDs and can affect both the central and peripheral nervous systems. Although it remains uncertain whether P/Q-VGCC abs have pathogenic, functional effects on PCD, the immediate clinical response of Patient 2 to PLEX, which removes plasma-soluble factors including antibodies, could support such a hypothesis.
PS2Group6-002 / #290CLINICAL FEATURES AND TREATMENT OUTCOMES OF NEUROLYMPHOMATOSIS IN KOREA
Topic: Group 6 – Cancer related Disorders and General Diseases
Duk Hyun Sung
Department Of Physical And Rehabilitation Medicine, Samsung Medical Center, Seoul, KR
Background: Neurolymphomatosis (NL) is a disease entity characterized by infiltration of malignant lymphocytes into the peripheral nervous system. The aim of the present study was to analyze clinical features, radiographic evaluation, treatment and outcomes of patients with primary and secondary NL. Methods: We retrospectively investigated 10 patients with NL diagnosed at one tertiary hospital from June 2013 to October 2016. We extracted data including clinical presentations, Magnetic resolution imaging (MRI), Positron emission tomography (PET) scans, Cerebrospinal fluid (CSF) cytology findings, results of electrodiagnostic study, treatments, and outcomes. We defined response of NL to therapy as clinical improvement of neurologic deficits. Results: There were 8 men and 2 women, and mean age at onset was 54.1 years (range: 33-83). Nine patients had secondary NL and one patient had primary NL. All patients were related to non-Hodgkin lymphoma. Of presenting symptoms, pain was reported in all patients. MRI was abnormal in 7 out of 9 patients with enhancement of spinal nerve roots (9 patients), plexus (5 patients), peripheral nerve (4 patients), and cranial nerve (1 patient). PET better identified NL than MRI and was positive in all patients. Electrodiagnostic studies demonstrated plexoradiculopathy (4 patients), radiculopathy (1 patient), sensorimotor polyneuropathy (3 patient), mononeuropathy multiplex (1 patients). One patient underwent nerve biopsy which confirmed primary NL in the S2 root. Eight patients received systemic chemotherapy, 4 intrathecal chemotherapy. Neurological improvement was observed in 3 patients. Seven cases who underwent the multimodality treatments of cancer eventually deteriorated and died. Conclusion: NL should be considered in the differential diagnosis of any neuropathy in patients with lymphoma or leukemia as well as no previous comorbidity in the setting of a painful and motor weakness. Because it could be confused with other neuropathies in lymphoma and various musculoskeletal diseases, a high index of suspicion and familiarity with clinical manifestation of NL is key. The diagnosis of NL is challenging but contemporary imaging tools such as PET/CT frequently detect the relevant neural invasion.
PS2Group6-003 / #916PEDIATRIC EXTRADURAL COMPRESSIVE MYELOPATHY SECONDARY TO GANGLIONEUROBLASTOMA; NERVE ROOTS AND BRACHIAL PLEXUS BEWARE
Topic: Group 6 – Cancer related Disorders and General Diseases
Amanda Yaworski1, Ratika Srivastava1, Vivek Mehta2, Atilano Lacson3, Hanna Kolski1
1Pediatric Neurology, University of Alberta, Edmonton, CA;2Neurosurgery, University of Alberta, Edmonton, CA;3Pathology, University of Alberta, Edmonton, CA
Background: We present a four-year-old boy with long-standing history of bilateral hand weakness, whose symptoms went unnoticed for years prior to diagnosis. Methods: Our patient presented with a one week history of abnormal gait and frequent falls. Further inquiry found lifelong bilateral hand weakness, described as weak pincer grasp since infancy and the inability to close hands around objects. These symptoms had been progressively worsening over six months. He was medically healthy and otherwise active. Developmentally he reached gross motor milestones at an appropriate time but regressed to the point of having trouble going down stairs. His fine motor skills had always been delayed using a fisted pencil grasp and was unable to use buttons or shoelaces. Physical exam showed decreased muscle bulk and atrophy in both hands and feet. A contracture at the left heel cord was noted but otherwise he had normal range of motion. He had a predominantly distal pattern of weakness with weak hand grip and ankle dorsiflexion bilaterally (left > right), with weak shoulder girdle muscles bilaterally. Reflexes were slightly brisk with upgoing plantars. His gait showed reduced left foot clearance, and he was unable to heel walk or tandem walk. Results: An MRI brain and spine found a left hemithorax superior sulcus tumour encasing the great vessels and left brachial plexus, extending to the left side of the neck. The mass extended into the intervertebral foramina from C3 to T4 and caused cord compression and edema in the spinal cord. The patient received one round of chemotherapy (carboplatin and etoposide) for presumed neuroblastoma, and high dose dexamethasone which resulted in clinical improvement of his gait and hand weakness. The first biopsy report revealed a ganglioneuroblastoma after Canadian Oncology Group (COG) review. Follow-up MRI suggested no change in mass size. Tumour debulking took place through a multilevel laminoplasty, following which our patient showed substantial improvement in gait. The post-operative MRI also showed improvement in cord compression. The second tissue sample confirmed a ganglioneuroblastoma, intermixed. The tumour showed focal necrosis and degenerative changes among the ganglionic component with no immature neuroblastic components, which could represent therapy effect. Full tumour resection was deferred due to the precarious location surrounding the vessels and left brachial plexus. Conclusion: This case demonstrates the variation in presentation of pediatric neurological diseases. The relatively mild and slow progression of this patient’s symptoms caused delay in seeking medical attention. It reminds us that a high index of suspicion is necessary for accurate localization and timely diagnosis of cervical myelopathies. Our case is unique because to the best of our knowledge, this is the first reported case of an extradural compressive myelopathy secondary to ganglioneuroblastoma in a pediatric patient. This case also highlights the need for ongoing clinical, radiographic, and pathologic correlations.
PS2Group9-001 / #417PSYCHOLOGICAL ASPECTS IMPACTING QUALITY OF LIFE OF PATIENTS WITH MYOPATHY
Topic: Group 9 – Miscellaneous
Amandine Rohmer Cohen1, Virginia Noel2, Michèle Mane2, Sylvie Zorgani2, Delphine Delorme2, Juan Rangel Escribano2, Catherine Bungener1
1Université Paris Descartes, Boulogne-Billancourt, FR;2Hôpital Rothschild, Paris, FR
Background: Literature shows that the patient’s quality of life is affected by depression, fatigue, and anxiety. The aim of this study was to assess the quality of life using a new evaluation scale: the quality of life in genetic neuromuscular disease (QOL-gNMD, Dany, 2017) and its link with three psychological variables: depression, anxiety and fatigue. Methods: 31 patients with adult-form of myopathy were included: 14 patients with myotonic dystrophy type 1, 9 with facioscapulohumeral dystrophy, 4 with limb girdle muscular dystrophy, 1 with mitochondrial myopathy, 1 with Central Cores myopathy, 1 with Ullrich muscular dystrophy, 1 with inclusion body myositis (population description in table 1).
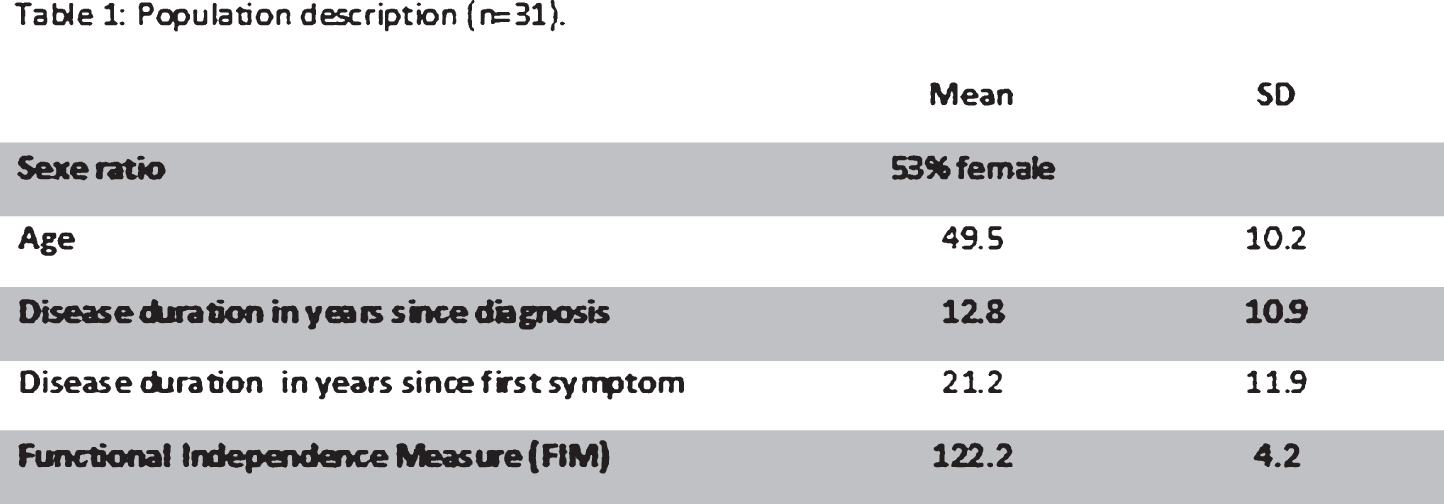
Results: Spearman correlations showed that depression and fatigue were the most correlated with the quality of life, and its three under-score. State anxiety was only correlated with “impact of physical symptom”. Disease duration since diagnosis was correlated with under-score “activities and social participation”. There was no correlation with trait anxiety, disease duration since first symptom, Functional Independence Measure score, age or genre.
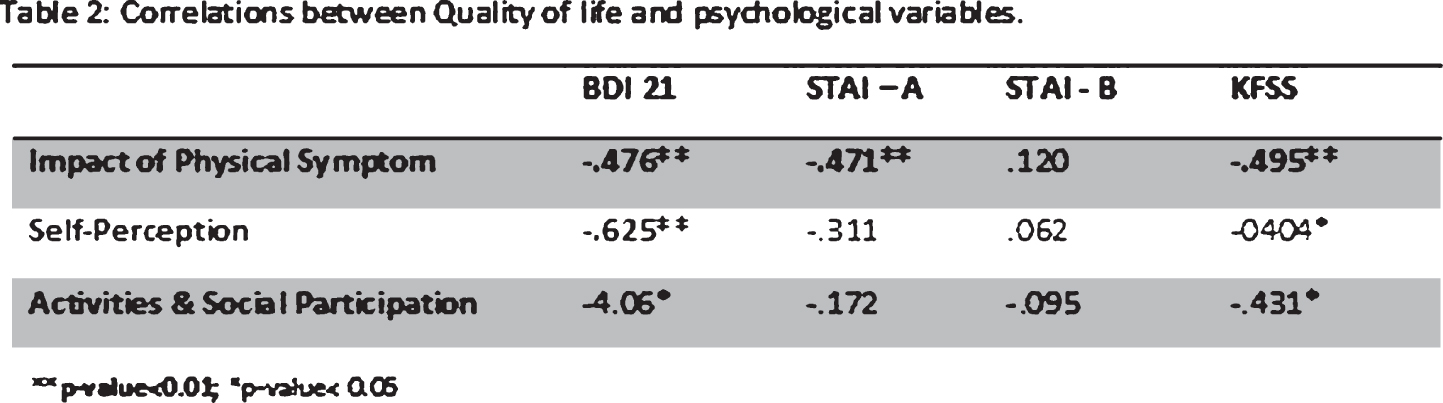
Conclusion: Results are consistent with previous studies which described a strong influence of patient’s mood on quality of life. Depression and fatigue affect the different aspects of the quality of life of patients with myopathy. Different ways to preserve the quality of life are discussed, for example: therapeutic workshop to prevent fatigue or regular assessment of depression.
PS2Group9-002 / #610AN UNCOMMON CO-EXISTENCE OF INHERITED AND CHRONIC INFLAMMATORY DEMYELINATING POLYNEUROPATHY
Topic: Group 9 – Miscellaneous
Ieva Glazere1, Signe Šetlere2, Inese Kazaine2, Guntis Rozentāls2
1Neurology, Pauls Stradins Clinical University Hospital, Riga, LV;2Pediatric Neurology, Children’s Clinical University Hospital, Riga, LV
Background: Chronic inflammatory demyelinating polyneuropathy (CIDP) is an autoimmune neuropathy with a low prevalence between pediatric patients. Diagnosis of CIDP can be very challenging due to its ability to mimic other peripheral nerve disorders such as Charcot-Marie-Tooth neuropathy (CMT). CMT1A is the most common subtype of CMT with highly variable phenotype and age of onset. CIDP and CMT may clinically manifest not only with similar symptoms but in rare occasions also co-exist. Accurate and detailed examination is crucial to confirm both overlapping diagnosis and start the appropriate treatment. Methods: Retrospective analysis of patient’s history and data. Results: We present a case of previously healthy 13-year-old boy with slowly progressive gait difficulties, inability to lift his feet since November 2016. In January 2017 symptoms rapidly progressed until he was not able to walk properly and stand up from a sitting position. History data revealed no infections or trauma before, he has 3 healthy brothers with no clinical symptoms. His clinical examination was remarkable for severe muscle atrophy and weakness in legs (MRC grade 3/5 proximally, 2/5 distally), and generalized hyporeflexia. No sensory impairment. Lumbar puncture was performed, cerebrospinal fluid (CSF) workup showed elevated protein level (0.73 g/L), with normal CSF cell counts. Further, a nerve conduction study demonstrated a severe motor - sensory axonal demyelinating polyneuropathy in lower extremities, mild in upper extremities; MRI scan of spine revealed nerve root enhancement in cervical and lumbosacral regions. He received intravenous immunoglobulins (IVIG) totally 2g/kg over 5 days. As clinically patient has pes cavus, DNA analysis was ordered and approved diagnosis of CMT1A neuropathy by detecting 17p11.2±12 duplication. Antiganglioside antibodies were negative. The patient recovered and was discharged with a mild weakness (MRC grade 4/5) in lower limbs. In October 2017 he was hospitalized again because of rapidly progressive leg weakness. The second MRI scan revealed nerve root enhancement and enlargement in cauda equina nerve roots, CSF showed elevated protein level (0.82 g/L). He responded poorly to the second course of IVIG, he was discharged with moderate distal and mild proximal paresis in both legs. Conclusion: The main purpose of the report was to demonstrate an uncommon combination of inherited neuropathy and acquired inflammatory polyneuropathy. Both diseases were confirmed by laboratory, electrophysiological and genetic tests, as well as clinical course and MR imaging. There have been only few publications discussing a possible predisposition to develop an inflammatory neuropathy for patients with CMT. Our case could support this possibility as the patient has a negative family history and no antecedent events. By identifying this association there is a potential to determine a rate of response to immune therapy. According to the literature (Rajabally et al., 2016) approximately 50 percent of CMT1A patients respond to CIDP treatment, which is less then patients without hereditary neuropathies. To conclude, patients with hereditary neuropathies could experience a rapid clinical deterioration, in that case an overlap with inflammatory neuropathies should be considered to establish adequate treatment; further research may unveil the immune processes associated with CMT and its overlap in inflammatory neuropathy pathogenesis.
PS2Group9-003 / #797ASSESSING IDEBENONE’S IMPACT ON RESPIRATORY FUNCTION IN DUCHENNE MUSCULAR DYSTROPHY: META-ANALYSIS OF TWO CLINICAL TRIALS
Topic: Group 9 – Miscellaneous
Christian Rummey1, Mika Leinonen2, Shabir Hasham2, Thomas Voit3, Oscar H. Mayer4, Gunnar Buyse For The Delos Study Group5
1Clinical Data Science GmbH, Basel, CH;2Santhera Pharmaceuticals, Pratteln, CH;3UCL Great Ormond Street Institute of Child Health, London, GB;4Children’s Hospital of Philadelphia , Philadelphia, PA, US;5University Hospitals Leuven, Leuven, BE
Background: Patients with Duchenne muscular dystrophy (DMD) experience respiratory function decline that accelerates after they become non-ambulatory, leading to significant disease burden and, in many cases, death. The ability of idebenone to slow respiratory function decline in patients with DMD has been investigated in two randomized, placebo-controlled trials (the phase 2 DELPHI trial and phase 3 DELOS trial). DELPHI included patients (aged 8 to 16 years) irrespective of baseline respiratory function status, while DELOS required patients (aged 10 to 18 years) to be in the respiratory decline phase. DELPHI allowed the randomization of both glucocorticoid users and non-users, whilst DELOS included only glucocorticoid non-users. Methods: Source data from both trials were pooled and analyzed for treatment effects on peak expiratory flow and forced vital capacity, expressed as a percentage of predicted (PEF%p and FVC%p). Change in PEF%p and FVC%p from baseline to weeks 26 and 52 (end of study) were analyzed using a mixed model for repeated measures, with study as a random effect. Results: In total, 76 patients (DELPHI: 12; DELOS: 64) were in the respiratory function decline phase (PEF%p < 80%), of which 72 were not using glucocorticoids. Across all patients, the difference between the idebenone and placebo groups in PEF%p from baseline to week 52 was 8.0%p (p = 0.003). When omitting glucocorticoid users, this difference was 7.7%p (p = 0.005). For FVC%p, the treatment difference was 3.6%p (p = 0.046) in all patients, with a comparable difference of 3.5%p (p = 0.051) when omitting glucocorticoid users. Conclusion: Results from a meta-analysis with data from two randomized, placebo-controlled trials (DELPHI and DELOS) demonstrate that idebenone slows respiratory function decline in patients with DMD. Additionally, the magnitude of treatment difference observed was unaffected by concomitant glucocorticoid use.
PS2Group9-004 / #812DEMANDS FROM THE PSYCHOLOGY OF HEALTH FOR THE CARE OF PEOPLE WITH STEINERT€
Topic: Group 9 – Miscellaneous
Alejandro J. Solernou Ferrer1, Alexis L. Ruiz2, Tatiana Z. Vaillant3, Carlos E. Oyola Valdizán4
1Department Of Neurogenetics, Institute of Neurology and Neurosurgery, Havana, CU;2Faculty Of Psychology, University of Havana, Havana, CU;3Institute of Neurology and Neurosurgery, Havana, CU;4Management in Health Services, Lima, PE
Background: The psychology of the health faces the challenge from the attention to people with illnesses and its support groups to guarantee its quality of life and psychological well-being, like it is in the case of the neurodegenerative illnesses. On Steinert’s disease from a literature search, it was observed that only the neuropsychological and psychopathological characteristics of psychology are described. Then, the psychosocial characteristics that these people can present are not described and the authors of this work consider that it is one more element, necessary to better understand the reality that people with this disease go through. It is not a matter of neglecting the neuropsychological and psychopathological elements, but of incorporating into this understanding the psychosocial elements so that it is a more comprehensive understanding, which can lead to an improvement in the multidisciplinary care that these people need to receive, not only from psychology. Methods: This work presents a proposal of what the authors have denominated “demands for the support boarding to people that have the illness of Steinert”, starting from a series of necessities identified in a previous work with mixed methodology that consisted on the bibliographical revision, the application of a semiestructuredinterview and the familiogram, to a group of 15 patients with this illness that they are assisted in the Institute of Neurology and Neurosurgery. Results: The demands were contained in different levels according to people that contributed them and they will be good to guide the work with those same people. Among the results of the study prior to this work, some of the characteristics found were the lack of knowledge in the different levels of health care about the existence of the disease which delays the diagnosis of the disease. Also the lack of prior knowledge about the disease in these patients stands out as a worrying element, even though many of them have relatives with the same condition. Based on the characteristics found in the previous study, a series of demands were made based on the needs identified of the patients and of the different socialization groups in which they can be found. They were grouped as follows: 1. Patients with Steinert’s Myotonic Dystrophy 2. Their families 3. Friends, neighbors, co-workers. 4. Institutions and social organizations 5. Society Some common demands were identified in all groups, among them is to have more information about the disease. In addition, the need to develop actions to keep as long as possible in the different activities of life in society to people with this disease, this work shows some examples. Conclusion: The characterization made evidences aspects necessary for attention with the humanistic approach that characterizes Cuban health. The elaborated demands are an integrative and systemic proposal from the Psychology of Health that should support the accompaniment of people with this disease and their relatives.
PS2Group9-005 / #263POMPHOLYX AND ECZEMATOUS SKIN REACTIONS AFTER INTRAVENOUS IMMUNOGLOBULIN THERAPY: CASE REPORTS AND REVIEW OF THE LITERATURE
Topic: Group 9 – Miscellaneous
Byung-Nam Yoon1, Ji-Won Yang2
1Neurology, Inje University College of Medicine, Seoul Paik Hospital, Marrenarero Gung-gu , KR;2Gachon University, Gil medical Center, Incheon, KR
Background: Intravenous immunoglobulin (IVIGs) has had a major impact on the treatment of neurological disorders including Guillain-Barre syndrome, chronic inflammatory demyelinating polyneuropathy, myasthenia gravis, and inflammatory dermatositis. IVIG administration is generally considered safe and well tolerated, except for several adverse effects such as anaphylactic reactions, thromboembolic events and acute renal failure. Most of the adverse effects include headache, fever, flu-like symptom, chest tightness, blood pressure change and dyspnea. The skin adverse reactions are very uncommon. Recently we experienced two cases with pompholyx associated with IVIG therapy. Methods: Case presentations ; We report 2 cases of characteristic extensive eczematous reaction that occurred approximately 7 days after administration of IVIG for Guillain-Barre Syndrome (GBS) and Miller Fisher Syndrome(MFS). The onset was characterized by dyshidrotic lesions on the palms, rapidly followed by pruriginous maculopapular lesions involving the whole body. They were treated with topical and/or systemic steroids, and complete resolution of skin lesions was observed within 1 month. Results: Severe eczematous skin reaction with a characteristic lesions on palms that then extends to the rest of the body is a rare but characteristic adverse effect of high dose IVIG therapy. A similar report has been consistently reported. We could not fully understand the pathophysiology and the precautions for the side effects are not clear. Fortunately the side effects are easily treated. Conclusion: With the use of IVIG increasing, it is important for neurologists or dermatologists to recognize this cutaneous side effect and its clinical course.
PS2Group9-006 / #541GLOBAL VS INDIVIDUAL MUSCLE FATTY DEGENERATIVE CHANGES TO MONITOR DISEASE PROGRESSION IN IMMUNE-MEDIATED NECROTIZING MYOPATHY
Topic: Group 9 – Miscellaneous
Cédi Koumako1, Harmen Reyngoudt2, Jean-Marc Boisserie3, Océane Landon-Cardinal4, Yves Allenbach4, Pierre G. Carlier2
1Aim & Cea Nmr Laboratory, Neuromuscular Investigation Center, Institute of Myology, Paris, FR;2Aim & Cea Nmr Laboratory, Neuromuscular Investigation Center, Institute of Myology, Paris, FR;3Nmr Laboratory, Neuromuscular Investigation Center, Institute of Myology, Paris, FR;4Department Of Internal Medicine And Clinical Immunology, APHP Pitié-Salpêtrière Hospital, Paris, FR
Background: Fat fraction (FF), as calculated from water-fat (Dixon) NMR images, is a largely accepted, if not fully validated, muscle imaging biomarker, which has been proposed as an outcome measure in most neuromuscular disorders these last few years. The question remains, however, as to whether specific muscle or muscle group should be taken into consideration for longitudinal evaluation in specific neuromuscular diseases. Most often all muscles are segmented and evaluated over a number of slices. Here, we looked into a cohort of patients suffering from immune-mediated necrotizing myopathy (IMNM), an inflammatory neuromuscular disease characterized by severe muscular lesions and muscle fatty replacement. The aim of this work was to compare whole segment FF with individual muscle and muscle group FF and identify the most efficient procedure to quantify disease progression. Methods: Fourteen IMNM patients (46.4±17.7 years, 18.9-74.8 age range, 5 male) were scanned twice within a one-year interval on a clinical 3T NMR system (Siemens). A fat/water 3-point Dixon NMR sequence was performed at the level of the thigh and the leg (right and left). FF was calculated both in the different individual muscles, muscle groups and in the central slice of the whole leg and thigh. The changes in FF (ΔFF) using all methods (individual, group or whole) were compared using paired t-tests. Standardized response means (SRM) were also calculated. Results: ΔFF was highly significant (P<0.01) for all investigated separate muscles, muscle groups and the whole segment. In the leg, ΔFF was only highly significant in gastrocnemius medialis muscle (P<0.001) and non-significant in muscle groups and the whole segment. ΔFF from the whole thigh segment resulted in an SRM of 0.80, which was similar when combining quadriceps and hamstring muscle groups (0.8). Lower SRMs were found when evaluating separate muscle groups (hamstring and quadriceps: 0.7) or individual muscles (vastus lateralis: 0.6, rectus femoris: 0.6, biceps femoris: 0.7). In leg, all SRM values were smaller than 0.5. Conclusion: Using the SRMs to evaluate the ability of various FF estimation procedures to monitor disease progression, it appeared that the simplest, mono-slice and global, estimate of thigh FF was also the most sensitive in IMNM patients. This suggests that multi-slice individual muscle segmentation might not be a mandatory tedious step in using quantitative NMR imaging to characterize muscle structural changes. There has also been a recurrent quest by investigators for the most appropriate muscle on which to focus the analysis. Our results indicate that this question is likely to be pointless. Other yet unpublished data in LGMD2B and LGMD2I patients showed that global approaches similar to the one we propose were also associated with superior performance. Still, generalization of the concept is premature in the present stage. Other conditions need to be revisited. Applicability to contractile tissue indices and water T2 distribution has yet to be investigated. Because the global approach takes into account the intermuscular fat, changes in patient nutritional status will likely bias the evaluation of the muscular FF and the impact has to be carefully determined.
PS2Group9-007 / #632KCNK9 IMPRINTING SYNDROME WITH CONGENITAL HYPOTONIA, DYSMORPHISM AND DEVELOPMENTAL DELAY DUE TO NOVEL MUTATION P.ALA237ASP
Topic: Group 9 – Miscellaneous
Pavel Seeman1, Jana Haberlova2, Lucie Sedlácková3, Marie Šedivá4, Petra Laššuthová4
1Child Neurology, Dna Laboratory, 2nd Medical Faculty, Charles University and University Hospital Motol, Prague, Praha , CZ;26 Department Of Child Neurology, Second Faculty of Medicine, Charles University and University Hospital Motol, Prague, CZ;3Child Neurology, Dna Laboratory, 2nd Medical Faculty, Charles Universityl, Prague, Praha, CZ;4Child Neurology, Dna Laboratory, 2nd Medical Faculty, Charles University and University Hospital Motol, Prague, Praha, CZ
Background: KCNK9 imprinting syndrome also called Birk Barel mental retardation dysmorphism syndrome is a very rare neuromuscular disorder which is caused by always only one amino acid exchange p.Gly236Arg in the KCNK9 gene, which is silenced on the paternal chromosome. Patients with this mutation demonstrate congenital hypotonia, cleft palate, delayed development, feeding problems and normal MRI and EEG. We report a novel cause of this syndrome in a Czech girl with congenital neurogenic hypotonia, weakness and developmental delay. Methods: Whole exome sequencing (WES) of patients DNA from blood and Sanger sequencing of the exon 2 of the KCNK9 gene were used in the DNA sample from the patient and from the healthy father. Neurological and electrophysiological and muscle biopsy examination of the patient were done. Results: A novel mutation, only the second at the amino acid level in this gene, namely p.Ala237Asp (c.710C>A) in exon 2 of KCNK9 gene was detected in heterozygous state by WES and later confirmed by Sanger sequencing in the patient, but not in her healthy father, the mother is not alive anymore. This mutation is not in ExAC database and affects a highly conserved aminoacid and is predicted to be deleterious/disease causing. DNA samples from maternal grandparents were not available at the time of abstract preparation. The patient is now 17 year old female, who is followed in our clinic for congenital hypotonia and generalized weakness of unknown cause since her age of 8 months. Birth weight and length were normal (3200 g/ 50 cm). She had a cleft palate, which was corrected and the motor development was delayed: walking at the age of 4 years. The patient has also mild mental retardation – mental developmental delay, she spoke first words at the age of 5 years. There was a feeding problem and swallowing problem. EMG showed pure motor primary axonal peripheral neuropathy and decreased number of motor units. Muscle biopsy at the age of 1,5 year showed mild neurogenic changes with selective atrophy of type II fibers and hypertrophy of type I fibers. She has fasciculations on the tongue. The muscle weakness is slowly progressive, she is still able to walk several hundreds meters independently, but for longer distances she uses wheelchair since the age of 12 years. Several DNA tests were done without clarification, incl. SMN 1 gene. Brain MRI at the age of 11 was normal. We have not yet tried the therapy by flufenamic acid (FFA) in this patient, but this is now available after the clarification of the cause of the disease. Conclusion: Birk Barel mental retardation dysmorphism syndrome or KCNK9 imprinting syndrome may be caused not only by always the same mutation p.Gly236Arg, but also by the mutation affecting residue p.Ala237 and the new causal genotype is associated with the same or very similar phenotypic expression.
PS2Group9-008 / #655QUANTITATIVE NMR OUTCOME MEASURES FOR SKELETAL MUSCLE CHARACTERIZATION OF DUCHENNE MUSCULAR DYSTROPHY PATIENTS
Topic: Group 9 – Miscellaneous
Teresa Gerhalter1, Lena V. Gast1, Benjamin Marty2, Regina Trollmann3, Stephanie Schüssler3, Frank Roemer1, Frederik B. Laun1, Michael Uder1, Rolf Schröder4, Pierre G. Carlier2, Armin M. Nagel1
1Institute Of Radiology, University Hospital Erlangen, Erlangen, DE;2Aim & Cea Nmr Laboratory, Neuromuscular Investigation Center, Institute of Myology, Paris, FR;3Department Of Pediatrics, University Hospital Erlangen, Erlangen, DE;4Neuropathology, University Hospital Erlangen, Erlangen, DE
Background: DMD is a hereditary neuromuscular disease caused by mutations in the dystrophin gene associated with inflammation, ionic homeostasis dysregulation, and exhaustion of regenerative capacities which leads ultimately to replacement of muscle by fatty and fibrotic tissue. Quantitative NMR has been used to describe the natural history of the disease and has been suggested as an outcome measure in clinical trials for DMD. Most of these studies evaluated the fat fraction (FF) and the 1H transverse relaxation time (water T2) to assess disease severity and progression. Both methods have their limitations: water T2 changes are non-specific events influenced by the usually prescribed steroid therapy and fatty degenerative changes, while picking up the extreme phenotypic variability, have a poor predictive value. Thus, there is a need to identify NMR variables as potential early sensitive indicators of dystrophic muscle response to treatment. Higher total sodium concentrations (TSC) and intracellular weighted sodium (ICW) signal have been measured by 23Na MRI. Here, we evaluated the sensitivity of 23Na MRI in DMD in comparison to the commonly used water T2 and fat fraction. Methods: MRI of the right calf was performed in twelve DMD patients (age 9.0±3.1 years) and twelve age-matched controls (age 9.6±2.3 years) on a 3T whole-body MR system (Siemens Healthcare). Water T2 was determined using a multi-slice-multi-echo sequence (TR 3000ms, 32 echoes: TEs 9.5-304ms, Tacqu 3 min 41 sec) and a tri-exponential fitting procedure. FF was measured using a 3-point DIXON method (TR 10 ms, TEs 2.75/3.95/5.15ms, Tacqu 3 min 12 sec). TSC was derived from an density-adapted 3D-radial UTE sequence (TE/TR 0.3/50ms, Tacqu 6 min 53 sec). An inversion-recovery (IR) sequence was used to achieve an ICW signal (TE/TR 0.3/124ms, TI 34ms, Tacqu 9 min 50 sec). ROIs were drawn on the gastrocnemius medialis (Gas.m.), soleus (Sol), tibialis anterior (TA) and posterior (TP) muscles. Results: Mean water T2 and FF were elevated in DMD patients compared to healthy controls (38.6±2.8 ms vs 36.1±1.5 ms and 0.078±0.072 vs 0.024±0.012, p=0.046 and p=0.007, respectively). Patients showed an increased TSC of 24.7±4.45 mM and an elevated ICW of 27.26±3.73 a.u. (controls: 16.66±2.46 mM TSC and 19.11±2.58 a.u. ICW, both p<0.0001). Moreover, the 23Na indices are frequently abnormal in DMD even when water T2 or FF remained in the normal range. DMD patients at all ages exhibited abnormally elevated sodium indices, whereas FF increased with age and the water T2 fluctuated during the course of disease. Conclusion: Sodium anomalies seemed to be systematically present in patients with DMD compared to controls and precede fatty degenerative changes. Sodium alterations may also occur before water T2 increases, however more investigations are needed to confirm this observation. Moreover, muscles that were relatively spared, such as the TA and TP, showed increased TSC or ICW in all participating DMD patients. Although this study, which is still in progress, has currently its limitation in the small number of subjects, the data supports that 23Na MRI could serve as an early and sensitive biomarker to investigate ion channel leakage and membrane integrity.
PS2Group9-009 / #772ASSESSMENT OF LIVER SAFETY USING AN EMERGING LIVER BIOMARKER, GLUTAMATE DEHYDROGENASE
Topic: Group 9 – Miscellaneous
Sumita Chowdhury1, Ruth Osahon2, David Peters1, Gary Layton2, Anne Heatherington1, David Roblin2, Francesco Muntoni3
1Summit Therapeutics Inc., Cambridge, MA, US;2Summit Therapeutics Inc., Abingdon, GB;3Dubowitz Neuromuscular Unit, UCL Great Ormond Street Institute of Child Health, London, GB
Background: To evaluate hepatic safety of ezutromid, a first-in-class small molecule utrophin modulator, using standard hepatic monitoring in conjunction with the emerging biomarker, glutamate dehydrogenase (GLDH). Thorough assessment of hepatic safety during drug development is essential; however, it is challenging in Duchenne muscular dystrophy (DMD) as traditional enzymes (ALT and AST) are not liver-specific and are markedly elevated due to muscle injury in DMD. GLDH has emerged as a potentially more sensitive and specific exploratory liver injury biomarker. With normal levels in DMD patients, it may be a better marker of ongoing acute liver injury compared to ALT. Recent consortia efforts (D-RSC) have resulted in regulatory support for GLDH as a drug induced liver injury (DILI) biomarker to complement the usual liver parameters, potentially replacing AST/ALT in assessing serious liver injury (e.g. Hy’s Law case) as an indication of hepatocellular necrosis. Methods: PhaseOut DMD is a Phase 2 open-label study of ezutromid administered to 40 ambulatory patients with DMD. In addition to other safety monitoring, hepatic safety was monitored by standard liver enzymes (ALT, AST, GGT, ALP) and functional markers (bilirubin, coagulation times) as well as GLDH, evaluated at screening, baseline, and throughout ezutromid dosing to monitor for evidence of liver safety signal. The Roche analytical method used to measure serum GLDH is based on UV measurement of GLDH enzymatic activity associated with NADH oxidation in the presence of substrate with an accuracy of 0.0 to 1.8%, precision of 0.6 to 1.9%, sensitivity of 0.5 U/L, and reference range of 0 to 6.4 U/L. The assay is robust and repeatable. Results presented include up to the Week 24 interim analysis, by which time a DILI effect would typically be observed. Results: During 24 weeks of treatment, two patients exceeded protocol-specified GLDH threshold of ≥ 3x ULN prompting ezutromid to be withheld: 20 U/L at Week 4 in one case, and 22.1 U/L at Week 24 in the other. After drug withholding, GLDH in both cases dropped below threshold within 3 weeks. Ezutromid was successfully resumed in both cases. There were no recurrences of GLDH elevation despite re-challenge. The bilirubin, INR, GGT, and ALP, were in the clinically normal range with no overt clinical signs of liver disease, both at the time of GLDH elevation and upon re-initiation of ezutromid. The totality of the data suggests no hepatic safety issues in either of these subjects; both have successfully continued long-term ezutromid therapy. Implementing Hy’s Law (with GLDH > 2.5x ULN replacing ALT/AST), would have prevented potentially unnecessary actions resulting from using GLDH > 3x ULN as a stand-alone criterion for hepatic safety. Conclusion: 1) Rigorous assessment of the hepatic safety profile of ezutromid over 24 weeks has indicated no clinically meaningful concerns. 2) The emerging liver-specific biomarker GLDH may be of particular value in monitoring liver injury in DMD. However, to avoid potentially unnecessary actions, it should be evaluated in the context of functional assessment of the liver with bilirubin and/or coagulation, and not as a stand-alone parameter for hepatic safety.
PS2Group9-010 / #536PERCEIVED QUALITY OF LIFE AND MOTOR SCALES OUTCOMES IN TYPE 2 AND 3 SMA PATIENTS: ARE THEY RELATED?
Topic: Group 9 – Miscellaneous
Anna Lia Frongia1, Irene Zschaeck1, Macarena M.A. Alarcon Cornejo1, Daniel Natera De Benito2, Laura Carrera-García3, Joaquin F. Mata1, Nuria Padros1, Obdulia Moya1, Judit Armas1, Julita Medina1, Carlos Ortez1, Jaume Colomer4, Andres Nascimento1
1Neuromuscular Disorders Unit, Hospital Sant Joan de Déu, Barcelona, ES;2Neuromuscular Disorders Unit, Hospital Sant Joan de Déu, Barcelona, ES;3Unidad De Patología Neuromuscular., Hospital Sant Joan de Déu, Barcelona, ES;4Hospital Sant Joan de Déu, Barcelona, ES
Background: Spinal muscular atrophy (SMA) related to SMN1 gene mutations is the most common genetic cause of mortality in childhood. It presents with atrophy and progressive muscular weakness due to motor neuron degeneration that restricts mobility and increases physical dependence, affecting quality of life. Patients with later-onset SMA (SMA type 2 and 3) became symptomatic after 6 months and life expectancy is shortened by early onset scoliosis, joint contractures, and restrictive lung disease, requiring a multidisciplinary approach. To date, there is no cure for this neuromuscular disorder, thus it becomes important to determine how far the disease and its treatment compromise a child’s QoL. Methods: To describe the correlation between the functional motor status with the perceived quality of life by families and patients we compare the outcomes scores from PedsQL with scores of functional motor scales (EK2, HFMSE and RULL) in a cohort of type 2 and 3 SMA patients. Results: Fifty-one patients with SMA (42 with type 2 and 9 with type 3) were enrolled in the study. Mean age was 10 years (range: 3-18.5). Mean score in patients and parents PedsQl was 67,32 (range 13-91) and 61,25 (range 22-90) respectively. HFMS score of 0 was reported in 12 SMA 2 patients and in 1 SMA 3 type, in this group mean RULL score was 10 (range 0-21). 4 out of 42 parents of children with SMA 2 and 3 out of 9 parents of SMA type 3 patients reported scores >80 in PedsQL. When comparing patients and parents PedsQL scores, a moderate or large discrepancy was found in 5 out of 51 families. In this group, in-depth unstructured interviews were conducted and analyzed with a qualitative approach. Conclusion: Regardless of the functional motor outcome, children and adolescents with SMA, as well as their parents, have a perception of relatively good quality of life. These findings are similar to those reported in several other studies.
PS2Group9-011 / #791DEVELOPMENT OF A PSYCHOSOCIAL INTERVENTION PROGRAMME FOR MOTOR NEURON DISEASE
Topic: Group 9 – Miscellaneous
Priya T. Thomas1, Manjusha Warrier2, Atchayaram Nalini3
1Department Of Psychiatric Social Work, National Institute of Mental Health and Neuro Sciences, Bangalore, IN;2Department Of Psychiatric Social Work, National Institute of Mental Health and Neuro Sciences, Bangalore, IN;3Department Of Neurology, National Institute of Mental Health and Neuro Sciences, Bangalore, IN
Background: Background: Amyotrophic Lateral Sclerosis is a progressive neuromuscular disorder that can have significant and debilitating impact on the affected patient and families. Few studies have looked in to the neuropsychiatric and other psychosocial dimensions in this group especially in the sociocultural scenario that is unique to a developing country like India. Methods: Aim: To assess develop a structured psychosocial intervention program for persons living with Motor Neuron Disease and their families. Method A quasi experimental, pre-test post-test research study was performed among the patients and family members of 80 patients diagnosed with ALS (Both Males and females) who were receiving treatment from a national tertiary referral care centre for Neurological disorders in South India. All the patients were evaluated for their complaints and were diagnosed as Definite ALS according to the El Escorial Criteria. Mean age at onset of illness in this group was 51.6. Mean duration of illness at the time of presenting to hospital was eleven months. ALSFRS was used to assess the functional limitations due to the illness. All patients were assessed with Beck’s Depression Inventory (BDI), Beck’s Anxiety Inventory (BAI), ALSQoL (R) and COPE. Caregivers completed Caregiver Burden Assessment (Zarit Burden Inventory) and COPE. Structured psychosocial intervention was developed, pilot tested, and was delivered by a trained Psychiatric Social Worker. All the patients and caregivers were re assessed on the same variables immediately after the intervention. Results: The preliminary analysis showed that psychosocial intervention is effective in bringing down the mental health concerns in the patients. Caregiver Burden and Quality of life showed positive changes after the intervention. Coping strategies most commonly adopted were emotional ventilation and instrumental support. Further results and the implications of the study will be presented. Conclusion: Diagnosis of a degenerative illness that is progressive and debilitative has significant impact on the mental health and coping in the affected individual. The treatment plan should take into account the various dimensions of living with the illness, the tasks that might arise during the advanced stages and periodic support during the course of the illness.
PS2Group9-012 / #746HTLV-1-SPECIFIC CTLS IN THE SPINAL CORDS OF ASYMPTOMATIC HTLV-1 CARRIERS
Topic: Group 9 – Miscellaneous
Eiji Matsuura
Neurology And Geriatrics, Kagoshima University Graduate School of Medical and Dental Sciences, Kagoshima city, JP
Background: HTLV-1 is an exogenous human retrovirus that is the causative agent of adult T-cell leukemia/lymphoma (ATL) and chronic inflammatory neurological disease, HTLV-1-associated myelopathy/Tropical spastic paraparesis (HAM/TSP). The major route of HTLV-1 infection is from mothers by a breastfeeding. Symptoms of HAM/TSP, however, typically manifest in patients later in life (50-60 years old in age). Some patients have just urinary dysfunction for several decades without motor symptom. The etiology of the urinary dysfunction in HTLV-1 carriers remains unclear. Little is known about the inflammation in the spinal cord lesion of HTLV-1 carrier (AC) before the onset of motor symptom. Methods: Aim: To elucidate onset of inflammation in the spinal cord of AC, we performed immunohistochemical study with the spinal cord of AC and tried to detect HTLV-1-specific CTL. Methods: We obtained autopsied spinal cords and brain from the patient with HAM/TSP and HTLV-1 carriers after obtaining written, informed consent from their family. One brain sample and four spinal cords were for use in this study. This study was approved by the Kagoshima University Ethics Committee. Results: CD8+ lymphocytes and HTLV-1-specific CTLs were detected in a brain and the four spinal cords. Interestingly, inflamed lesions of HTLV-1 carrier were distributed mainly in the lower thoracic level of the spinal cords like the spinal cord of HAM/TSP. Conclusion: Our data suggest that latent inflammatory change occurs in the spinal cords of ACs.
PS2Group9-013 / #941AAV-ASSOCIATED SENSORY GANGLIONITIS AND AXONOPATHY IN LABORATORY ANIMALS: A RETROSPECTIVE STUDY
Topic: Group 9 – Miscellaneous
Juliette Hordeaux, Elizabeth L. Buza, Laura Richman, Christian Hinderer, Nathan Katz, Peter Bell, James M. Wilson
Gene Therapy Program, Department Of Medicine, University of Pennsylvania, Perelman School of Medicine, Philadelphia, PA, US
Background: Adeno-associated viral (AAV) vectors are increasingly being used in clinical trials to treat neurologic disorders. Neurotropic serotypes such as AAV9 have been demonstrated to transduce spinal alpha motor neurons and dorsal root ganglia sensory neurons efficiently when administered intravenously to juvenile animals or intrathecally in both juveniles and adults. As more nonclinical safety and tolerability studies are performed using these vectors, we were confronted with unexpected toxicity targeting dorsal root ganglia neurons and their corresponding central and peripheral axons. Previous studies have reported efficient transduction of dorsal root ganglia utilizing systemic or intrathecal AAV delivery. Dorsal root ganglia are outside of the blood brain barrier, directly bathed into cerebrospinal fluid, and contain a cell body rich area that presents a high density of fenestrated capillaries. Those anatomic considerations likely explain why they are so efficiently targeted following both systemic and intrathecal administration. Methods: The toxicity was observed in adult rhesus macaques after intrathecal administration of 1 – 5x1013 Genome Copies of AAV (1 – 5x1011 Genome Copies per gram of brain) and in juvenile macaques and piglets after high dose intravenous administration (2x1014 GC/kg). It was not observed in dogs, cats, or rodents dosed in the cerebrospinal fluid with up to 2x1011 GC/g brain. Results: The toxicity was characterized by neuron cell body degeneration with mononuclear cell infiltration in the dorsal root ganglia, and axonopathy with dilated myelin sheaths and occasional macrophages in the corresponding central and peripheral axons. Rhesus macaques were less severely affected with subclinical lesions only detected on histological examination whereas piglets demonstrated proprioceptive deficits and ataxia. Conclusion: The significance of the DRG, spinal cord and peripheral nerve nonclinical findings is not currently known and for many monogenic neurologic diseases without a standard of care, the benefit-risk profile is still in favor of AAV-mediated gene therapy. For the first-in-human trials, informed consent, electrophysiological recordings as well as careful monitoring of sensory symptoms such as paresthesias or changes in sensation of pain, temperature, touch or proprioception will be essential.
PS2Group9-014 / #813OSTEOPOROSIS IN DUCHENNE MUSCULAR DISTROPHY PATIENTS
Topic: Group 9 – Miscellaneous
Filiz Meryem Sertpoyraz1, Figen Baydan2
1Physical Therapy And Rehabilitation Clinic, Izmir Tepecik Education and Research Hospital, Izmir, TR;2Pediatric Neurology, Tepecik Egitim ve Arastirma Hastanesi, Izmir, TR
Background: Osteoporosis is a progressive metabolic bone disease that results in increased bone fragility and fracture tendency as a result of low bone mass and disruption of the micro-architecture of bone tissue. Duchenne Muscular Dystrophy (DMD) is a progressive, junctional type of muscle disease characterized by a distroflex syndrome defect that passes through X. Muscle weakness, immobilization and steroid use in DMD patients accelerate the development of osteoporosis and are implicated in studies that lead to osteoporosis-related complications. Objective: To evaluate the status of osteoporosis in patients with DMD. Methods: Retrospective, descriptive study In the Neuromuscular Disorders unit, files of 48 male patients who had received a definite DMD diagnosis and signed the voluntary form were examined. Serum 25 (OH) vitamin D, calcium, phosphorus, alkaline phosphatase and parathormone levels were recorded in the first visit. Demographic information, functional activity levels, steroid use, fracture story, fracture site and first time measurements were recorded. Lomber and hip z scores were evaluated. The number of patients is 48 and all are male. Inclusion criteria: 1. Patients signed voluntary consent form 2. Patients between the ages of 8 and 18 3 Men 4. Patients with definite DMD Exclusion criteria : 1. Patients who do not sign the voluntary consent form 2. Osteoporosis caused by endocrine, malignant disease, rheumatic disease 3. Those who do not have definite DMD diagnosis. Results: Forty eight (48) male patients with DMD were included in the study. The mean age of DMD-diagnosed patients was 14.17 ± 4.4 (8-24) years, The mean duration of diagnosis was 9.8 ± 4.11 (1-20) years. Mean DMD 25 (OH) vitamin D level: 15 ± 7,8
| Serum 25 (OH) vitamin D levels | Number of patients: n percent (%) |
| D vitamin deficiency | 32 (67%) |
| D vitamin insufficiency | 15 (31%) |
| D vitamin sufficient | 1 (2%) |
There was no statistically significant difference in vitamin D levels among patients with and without steroids in DMD patients. P = 0.720 There was no significant difference between vitamin D levels in patients with and without fractures in DMD patients. P = 0.833 The average Lomber Z score in DMD patients: -2.0 ± 1.5, hip Z score: -3 ± 1,3 The decrease in hip bone density was statistically significant. Conclusion: We have found vitamin D deficiency-inadequacy at a high rate in our patients. Loss of bone density in the hip is thought to be due to early gait loss. Considering high prevalence of vitamin D deficiency and osteoporosis in DMD patients, it seems vitamin D supplementation can improve vitamin D status and osteoporosis in these patients.
PS2Group9-015 / #635NEUROMUSCULAR GENETIC REGISTRY IN RUSSIA
Topic: Group 9 – Miscellaneous
Dmitry Vlodavets1, Anastasya Monakhova1, Denis Reshetov2, Svetlana Artemyeva1, Irina Shulyakova1, Olga Shidlovskaya1, Elena Belousova1
1Russian Children Neuromuscular Center, Veltischev Scientific Research Clinical Pediatric Institute of Pirogov Russian National Research Medical University, Moscow, RU;2Science Advice, Moscow, RU
Background: Neuromuscular disorders are widespread all over the globe. In the most developed countries of Europe and North America, extensive work is underway to identify these patients suffering from neuromuscular diseases, aimed both at identifying individual needs and improving the quality of life, and on stimulating the development of basic science and molecular genetic technologies, which in the near future will be able to cure such patients. The main ways of working are to concentrate efforts and knowledge of patients, clinicians, scientists on a specific problem (for a particular disease). After another round of accumulation of knowledge, a breakthrough takes place - there is a technology that allows solving the problem with specific diseases. So, for example, it happened with Pompe’s disease and SMA. One way of initial accumulation of knowledge is to create registers for specific diseases to unite patients into unique groups. This allows to determine the incidence and prevalence of diseases in specific populations. Methods: About 3 years ago we started to collect blood and data of the DMD and SMA patients in Russia and collect them in 2 Genetic Registers. Results: At that moment most of the patients were not provided with genetic testing. To the date we collected more than 500 patients with DMD and BMD; more than 550 patients with different types of SMA. Now most of them are genotyped and few patients with sarcoglycanopathies were surprisingly found in DMD group. During this work, we identified more than 80 new point mutations in DMD gene. About half of the SMA patients now have not only deletion of 7 exon test but also a copy number of SMN2 gene testing. And since the 2018 we started the project Neuromuscular Genetic Registry in Russia. The aim is to collect different data of patients (clinical, genetics, muscle MRI, etc.) also with collecting patient’s blood. We have concentrated on several diseases that are most common in Russia: congenital muscular dystrophy type 1A, collagen 6 disorders (Bethlem and Ullrich CMD), FKRP patients with LGMD2I, CAPN3 patients with LGMD2A, patients with dysferlinopathies, patients with Landousy muscular dystrophy, patients with RYR1 mutations and other rare conditions. Conclusion: With the total population in Russia about 150 million of people there are hundreds of patients that still have no genetics and proved diagnosis. This is making them very far from the future treatment. Those patients who have been tested genetically could be included in our Neuromuscular Genetic Registry and provide additional information about the natural history of the disease that possibly in future will help scientists for better understanding pathogenesis and other factors that could influence on the course of the disease. And also collecting the representative groups of patients with different disorders will allow our site to take part in clinical trials providing to our patients advanced treatment.
PS2Group9-016 / #792ALTERNATIVE ANALYSES OF RESPIRATORY FUNCTION IN DUCHENNE MUSCULAR DYSTROPHY: CONSISTENT TREATMENT BENEFIT OF IDEBENONE
Topic: Group 9 – Miscellaneous
Oscar H. Mayer1, Mika Leinonen2, Christian Rummey3, Thomas Meier4, Shabir Hasham4, Thomas Voit5, Gunnar Buyse For The Delos Study Group6
1Children’s Hospital of Philadelphia, Philadelphia, PA, US;2Santhera Pharmaceuticals, Pratteln, CH;3Clinical Data Science GmbH, Basel, CH;4Santhera Pharmaceuticals, Pratteln, CH;5UCL Great Ormond Street Institute of Child Health, London, GB;6University Hospitals Leuven, Leuven, BE
Background: In Duchenne muscular dystrophy (DMD), progressive weakness of respiratory muscles leads to life-threatening respiratory complications. In a randomized placebo-controlled, Phase 3 clinical trial (DELOS) in 64 DMD patients aged 10-18 years and not taking concomitant glucocorticoids, idebenone (900 mg/day) reduced the rate of respiratory function decline, as measured by peak expiratory flow (PEF), forced vital capacity (FVC) and forced expiratory volume in 1 sec (FEV1) – as percent predicted (%p). Additional analyses were undertaken using alternative statistical models to determine the consistency of magnitude of efficacy of idebenone. Methods: We compared the efficacy of idebenone vs placebo on PEF%p, FVC%p and FEV1%p in a pre-specified analysis, and also carried out two additional post hoc analyses. Analysis 1 (pre-specified): Change from baseline (BL) to last visit (week 52), was analyzed by mixed model for repeated measures, with treatment group, visit and treatment group by visit interaction as fixed factors, and the BL as a covariate. Analysis 2 (post-hoc): Similar to above, but taking into account all post-BL visits (weeks 13, 26, 39 and 52). Analysis 3 (post-hoc): The slope across all visits was analyzed using a random coefficient regression model. Results: Using analyses 1 and 2, the estimated difference between idebenone and placebo for PEF%p was 6.3%p (p = 0.031) at the last visit. The differences at weeks 13, 26 and 39 were 3.9%p (p = 0.104), 8.3%p (p < 0.001) and 7.6%p (p = 0.016), respectively, with an overall difference (all post-BL visits combined) of 6.5% (p = 0.006). The estimated difference at last visit for FVC%p was 3.3%p (p = 0.082), with week 13, 26 and 39 differences of 3.3%p (p = 0.021), 4.7%p (p < 0.002) and 5.0%p (p = 0.019), respectively, and an overall difference of 4.1% (p=0.005). For FEV1, the results were 6.4% at the last visit, with week 13, 26 and 39 differences of 5.2%p (p = 0.010,), 9.3%p (p < 0.001,) and 6.9%p (p = 0.017), respectively, and an overall difference of 7% (p = 0.0011). Using analysis 3, the estimated annual slope difference across all visits was 5.8%p (p = 0.007) for PEF%p, 4.1%p (p < 0.001) for FVC%p and 6.9%p (p < 0.001) for FEV1%p. Conclusion: Based on results from three different analyses for PEF%p, FVC%p and FEV1%p data, idebenone consistently slowed the rate of respiratory function decline compared with placebo in patients with DMD.
PS2Group9-017 / #423MISDIAGNOSIS OF DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 9 – Miscellaneous
Isabella D.L. Kalil1, Julia D.L. Fernandes2, Arnaldo J. Godoy2
1University City of Sao Paulo, Alto de Pinheiros - São Paulo, BR;2UNIVERSITY CITY OF SAO PAULO, Alto de Pinheiros - São Paulo, BR
Background: Becker muscular dystrophy (BMD) was first described 65 years ago as a benign variant of Duchenne muscular Dystrophy (DMD). BMD is clinically more heterogeneous, with a wide spectrum of clinical presentations. About 50% of the patients become symptomatic by the age of 10. We report the case of a boy diagnosed as DMD but who was, in fact, a BMD patient. Methods: We asked permission for the mother to report her child condition. The patient was seen this February at our university clinic in Sao Paulo City. Results: The patient is 10 years old and is attending the fourth grade of an elementary school in the rodeo city of Barretos, State of Sao Paulo, Brazil. His mother told us he presented speech difficulty a year ago. The boy went to see a child neurologist, who found his calves enlarged and considered the diagnosis of DMD. His mother noticed a “different” way of walking and her son fell frequently during the past few months. He has been taking prednisolone for 7 months. During the first 5-6 months he was on an intermittent regimen. For about a month, he has been taking the medicine every day. In 2017, March, his molecular test showed deletion of the exons 18 to 41. CK levels were 10,719 in May of 2017 and 4,931 in January of 2018. Echocardiogram was normal this February. Physical examination showed a cushingoid facies. Muscle strength in the upper limbs graded 4, proximally and 5, distally and in the lower limbs graded 4 globally. Pseudohypertrophy of the calves was detected. He could walk and run for 20 meters without showing tiredness. Gowers sign was positive. It took 5.5 seconds for him to stand from the supine position. We told his mother his son was a BMD patient and not DMD and recommended her to give prednisolone intermittently again and come back in 3 months. By that time, if no significant progression of the disease will be detected, we intend to interrupt the use of steroids. Conclusion: This report stresses the difficulty of making a correct diagnosis in case of rare diseases. As BMD is much less frequent compared to DMD, the physician attending our patient considered DMD as the likely diagnosis. Because of that, the child was treated with steroids. He is already presenting significant side effects. Changing his diagnosis to BMD allowed us to reduce the amount of that medicine to evaluate in a safer manner the course of the illness. Although a combination of clinical and molecular genetic evaluations may be sufficient for accurate diagnosis of BMD and DMD, in many cases, the contribution of a histopathological test still remains an important component of evaluation. We are trying to convince the mother to accept us to do a muscle biopsy. In spite of the spread of the knowledge about neuromuscular rare diseases, it is very important to emphasize the subtle differences between similar conditions such as DMD and BMD. Otherwise, patients will be put in risk of mistreatment.
PS2Group9-018 / #679ENSURING HIGH QUALITY MUSCLE BIOPSIES AND MAGNETIC RESONANCE BIOMARKERS IN THE PHASEOUT DMD STUDY
Topic: Group 9 – Miscellaneous
Jon Tinsley1, Yalin Hasoglu2, Michael A. Boss3, Diane Frank4, Crystal Faelan5, Krista Vandenborne6, Francesco Muntoni7
1Summit Therapeutics Plc, Abingdon, GB;2Summit Therapeutics plc, Abingdon, GB;3Summit Therapeutics plc, Cambridge, MA, US;4Sarepta Therapeutics Inc, Cambridge, MA, US;5Flagship Biosciences, Westminster, CO, US;6Department Of Physical Therapy, University of Florida, Gainesville, FL, US;7Dubowitz Neuromuscular Unit, UCL Great Ormond Street Institute of Child Health, London, GB
Background: Utrophin is a naturally occurring protein that may act as a functional replacement for dystrophin. Increasing the production of utrophin in mature muscle fibres protects and improves outcome in preclinical models, and could modify the course of Duchenne Muscular Dystrophy (DMD). Methods: PhaseOut DMD (NCT02858362) is a Phase 2 open-label study of ezutromid administered to 40 ambulatory patients with DMD. Primary endpoints (week 48) are magnetic resonance spectroscopy (MRS) assessments, with an interim analysis conducted after 24 weeks treatment (recently reported elsewhere). Quantitative magnetic resonance imaging (spin-echo and 8 point Dixon) and localized proton spectroscopy is performed at baseline, 12, 24, 36 and 48 weeks to measure the intramuscular fat fraction (FF) and transverse relaxation time (bulk T2 and water T2) in lower extremity muscles. Key secondary endpoints from muscle biopsy include analyses of utrophin protein levels (estimate of target engagement) and developmental myosin heavy chain (MHCd; a biomarker for muscle fibre regeneration). Biopsies were to be collected from the biceps at baseline (n=40) with a second biopsy taken either after 24 weeks (n=25) or 48 weeks (n=15) of ezutromid dosing. Results: PhaseOut DMD sites have undergone a robust training and certification process to ensure high quality and reproducible quantitative MRI/MRS data acquisition. All images and spectra are acquired by standardized procedures and are centrally reviewed and processed. Quantification of MRS endpoints are performed by an automated blinded process with regular, scheduled data transfers. Quantification of the images is performed by 2 independent MRI experts; a 3rd adjudicating reviewer is used in the event of discordance. All images and spectra, except 1, up to week 24 have yielded data of sufficient quality for the quantification of MRI/MRS biomarkers in the vastus lateralis and soleus. High quality muscle biopsies allow early evaluation of muscle health biomarkers and target engagement. A thorough biopsy process, including training of all sites (surgeons and histotechnicians) has been developed. Biopsy samples from all UK and US sites were shipped to a centralised laboratory for analysis. All biopsy samples taken at baseline and week 24 have been deemed of suitable quality for processing, as confirmed by a pathologist’s assessment of the stained cryosection. Utrophin, MHCd, and fibre morphometrics were quantified in 3,000-15,000 fibres/section using analytically validated immunohistochemistry and image analysis. Analyses demonstrated high reproducibility between replicates, with all biopsies yielding data for at least one biomarker. Conclusion: Implementation of robust methodology for the collection of biomarkers in the PhaseOut DMD study has facilitated the evaluation of both target engagement and early indicators of disease progression at the 24-week interim timepoint.
PS2Group9-019 / #892TREAT-NMD: ADVANCING DIAGNOSIS, TREATMENT AND CARE IN NEUROMUSCULAR RARE DISEASES
Topic: Group 9 – Miscellaneous
Anne Oyewole1, Joanne Lee1, Rebecca Leary1, Katharine Bushby1, Annemieke Aartsma-Rus2, Janbernd Kirschner3, Kevin Flanigan4
1The John Walton Muscular Dystrophy Research Centre, Newcastle University, Newcastle, GB;2Human Genetics, Leiden University Medical Center, Leiden, NL;3Department of Neuropediatrics and Muscle Disorders, Medical Center-University of Freiburg, Freiburg, DE;4Nationwide Children’s Hospital, Columbus, US
Background: The rarity and heterogeneity of neuromuscular disorders contributes to some of the challenges faced both for providing good quality healthcare and for conducting translational research in the neuromuscular field. TREAT-NMD (Translational Research in Europe: Assessment and Treatment of Neuromuscular Disorders) is a global network of world-class experts within the neuromuscular community. Our mission is to accelerate the process of developing new treatments and improve the health and quality of life of people around the world with NMDs, by supporting all stages of translational research. Over the last decade we have identified major barriers in the diagnosis, treatment and care for people with NMD and have developed essential ‘go-to’ tools and resources to overcome these challenges. Methods: Since 2007, TREAT-NMD has played a central role in bringing together experts within the NMD community (patients, advocacy organisations, scientists, clinicians and industry) to develop models, projects and resources to meet the challenges for NMD along the translational pathway, from the interpretation of preclinical research to resources for clinical trial readiness. Results: As part of a large scale international effort (CARE NMD project) with the involvement of many experts and patient organisations, TREAT-NMD has contributed to the development and dissemination of care standards for Duchenne muscular dystrophy (DMD) and spinal muscular atrophy (SMA) and the creation of ‘family friendly’ guides. The DMD family guide, which is now available in 35 languages, has been downloaded over 17,500 times since 2011 and continues to ensure best practice care is more widespread across the world. TREAT-NMD is now working with clinicians, physiotherapists, nutritionists and patient organisations, to begin work on updating the SMA and DMD family guides, based on the recently published standard of care recommendations for these disorders. Over the last decade the TREAT-NMD network has played a pivotal role in successfully bringing together partners for numerous grant applications. The resources that were started or supported by TREAT-NMD have contributed to more than 20 funded national and international projects, including RD-Connect, OPTIMISTIC, BIO-NMD and most recently IMI2-PREFER and the European Reference Network (ERN) on NMD (EURO-NMD). Since 2009, the TREAT-NMD advisory committee for therapeutics (TACT), a unique multi-disciplinary international group of experts, have reviewed over 46 applications for promising drug candidates, providing guidance on the translation and development path of therapeutics programs in NMDs. Two other important tools that have helped to accelerate trial planning and remove potential barriers to getting trials established to test potential therapies, are the TREAT-NMD affiliated Patient Registries and the Care and Trial Sites Registry (CTSR). Conclusion: TREAT-NMD has fostered an environment in which collaborative initiatives have and continue to support translational research efforts. The resources generated by TREAT-NMD have contributed heavily across several areas of activity including care standards, preclinical research and clinical trial readiness. Furthermore, TREAT-NMD Global Patient Registries, the CTSR and TACT experience has been invaluable in the field. A great many lessons have been learned and opportunities for improved care and treatments for people living with NMD are closer than ever before.
PS3Group2-001 / #336THYMECTOMY LOWERS THE MYASTHENIA GRAVIS BIOMARKERMIR-150-5P
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Carl Johan Molin1, Anna R. Punga2, Liis Sabre2, Tanel Punga2, Cleo-Aron Weis3
1Neuroscience, Uppsala University, Uppsala, SE;2Uppsala University, Uppsala, SE;3Pathology, University Medical Centre Mannheim/University of Heidelberg, Mannheim, DE
Background: The aim of the study was to analyze the effect of thymectomy on the proposed disease-specific microRNA (miRNA) biomarkers miR-150-5p and miR-21-5p in Myasthenia Gravis (MG) patients. For this purpose we examined serum samples from the prospective Randomized Trial of Thymectomy in Myasthenia Gravis (MGTX) and evaluated the longitudinal changes in clinical pattern compared to these miRNA levels. Methods: Serum samples were obtained from 80 acetylcholine receptor positive (AChR+) MG patients included in the MGTX trial. 38 patients were randomized to thymectomy plus prednisone treatment and 42 patients were randomized to prednisone treatment. Serum samples were analyzed for the expression of miR-150-5p and miR-21-5p with qRT-PCR at baseline and 1, 2 and 3 years after randomization. Results: Patients treated with thymectomy had lower levels of miR-150-5p at 1-year post thymectomy, both compared to baseline values and to the prednisone group. No change in miRNA-levels was found in the prednisone group. Levels of miR-21-5p displayed a negative correlation with prednisone dose within the prednisone group.
Conclusion: Thymectomy lowers the levels of the proposed biomarker miR-150-5p, which strengthens its position as a potential disease-specific biomarker for AChR+ MG.
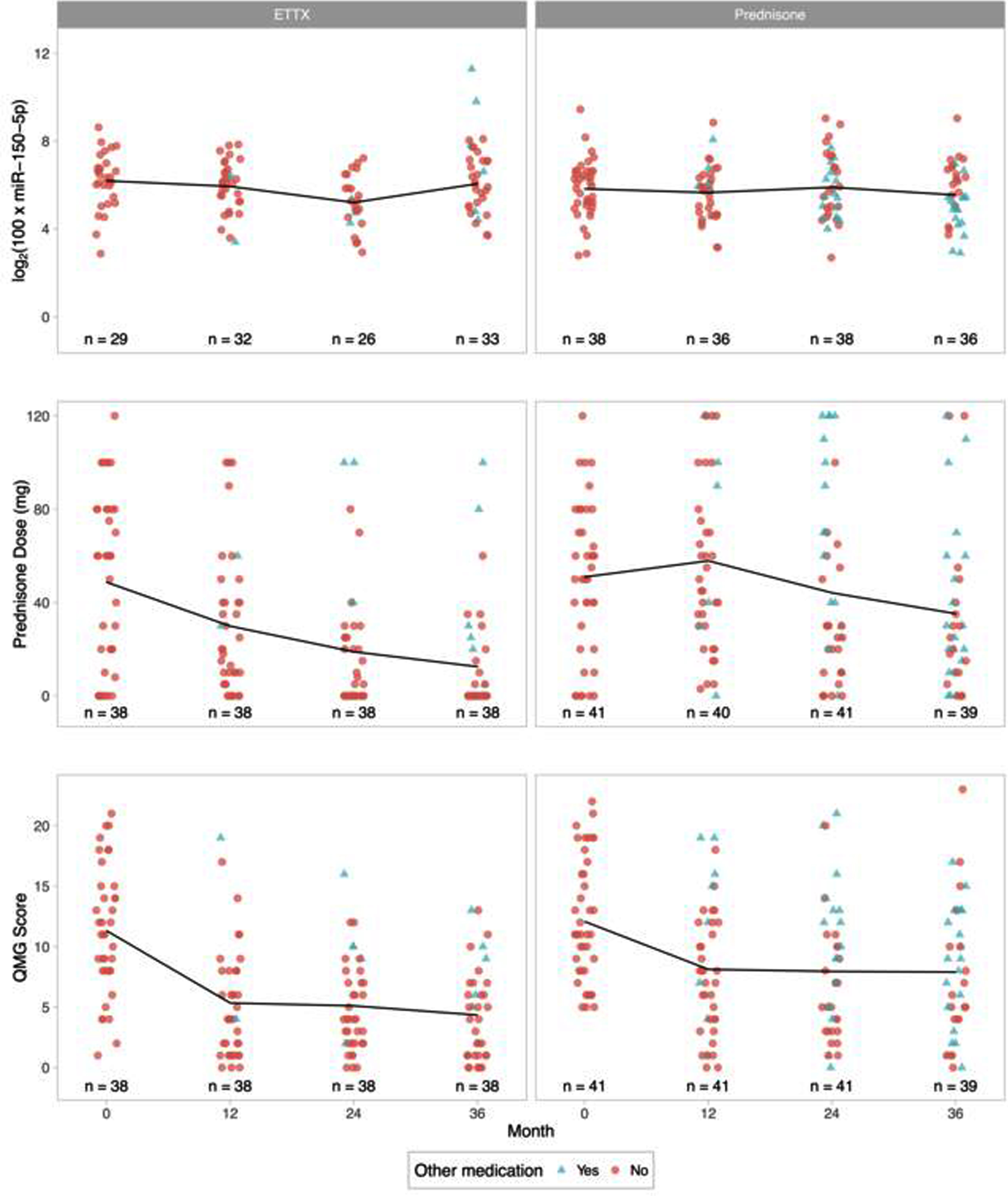
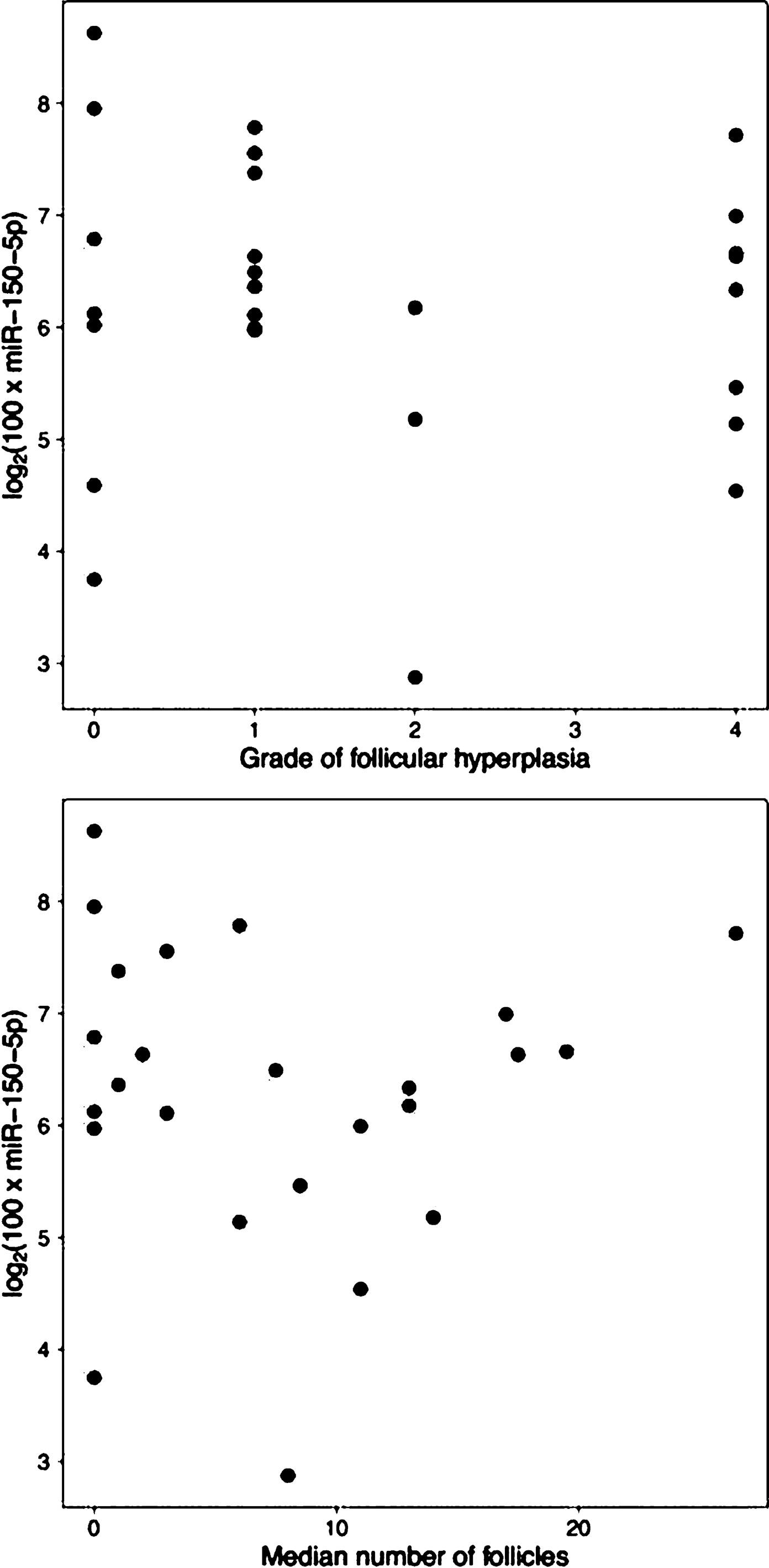
PS3Group2-002 / #457EFFICACY OF SUBCUTANEOUS IMMUNOGLOBULIN IN MYASTHENIA GRAVIS EXACERBAION
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Zaeem A. Siddiqi1, Grayson Beecher2, Dustin Anderson3
1Neurology/medicine, University Of Alberta, Edmonton, CA;2Neurology/medicine, University of Alberta, Edmonton, AB, CA;3University of Alberta, Edmonton, AB, CA
Background: Intravenous immunoglobulin (IVIg) is an effective therapy for patients with worsening MG. Recently, the subcutaneous form of immunoglobulin (20% SCIG; Hizentra) has been used in neuromuscular conditions amenable to IVIg therapy. SCIG appears to be associated with fewer adverse effects, comparable efficacy and the added advantage of flexibility due to self infusion by the patients. The efficacy of SCIG has not been systematically studied in patients with MG. Methods: This is an investigator-initiated, single phase, open label, phase II clinical trial in patients with moderately severe exacerbation in MG (MGFA Grade II and III). Enrolled subjects self-infused 20% SCIg (Hizentra) at 2gm/kg over 3 to 4 weeks in a dose escalating manner dosing regimen. The primary endpoint was the change in quantitative MG (QMG) score from baseline to study end at 6 weeks. Secondary endpoints included change in manual muscle testing (MMT), MG activities of daily living (MG-ADL), and MG composite (MGC) scores. The trial was approved by Health Canada and the local institution ethics board. Results: Of the 29 patients who were screened 23 were enrolled in the trial and 22 (female =14; male=8; mean age 53.0 + 19.6 years) completed the study. At the end of study period the QMG score decreased from 14.9 + 4.1 to 9.8 + 5.6 (p< 0.0001), MMT score decreased from 16.8 + 9.5 to 5.2 + 4.5 (p<0.0001), MG-ADL score decreased from 9.5 6 3.0 to 4.6 6 3.0 (p<0.0001), and MGC score decreased from 17.4 + 5.0 to 5.6 + 4.5 (p<0.0001). Transient worsening of the MG occurred in two patients initially but neither had to be withdrawn. Conclusion: SCIg is effective in the treatment of mild to moderate MG exacerbation. Comparative efficacy must be established with larger randomized controlled trials.
PS3Group2-003 / #461FEASIBILITY AND SAFETY OF SUBCUTANEOUS IMMUNOGLOBULIN IN MYASTHENIA GRAVIS EXACERBATION
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Zaeem A. Siddiqi1, Grayson Beecher2, Dustin Anderson3
1Neurology/medicine, University Of Alberta, Edmonton, CA;2Neurology/medicine, University of Alberta, Edmonton, AB, CA;3University of Alberta, Edmonton, AB, CA
Background: The University of Alberta Myasthenia Gravis (MG) program is the primary site of investigator initiated clinical trial to assess the feasibility, safety and efficacy of 20% SCIG in MG exacerbation. This study describes the results in the safety and feasibility domains, while the efficacy of SCIG is described in a separate study. Methods: A dose of 2gm/kg of SCIG was infused in a flexible regimen over 4 weeks and outcome measures assessed at 6 weeks. The safety of infusions was assessed through patient reported adverse effects, infusion site reactions and incidence of hemolysis. Serum Ig levels were serially monitored during the study period. Patient compliance and tolerability were assessed via Treatment Satisfaction Questionnaire for Medication (TSQM; a score of 0 represents not satisfied and 100 represents fully satisfied). Results: Of the 23 patients enrolled 22 patients completed the trial, one discontinued due to MG worsening. Three patients had subsequent worsening and were re-enrolled resulting in a total of 25 SCIG infusions being analyzed. Mean total dose infused was 180.8gms (range: 104 to 204gms), mean volume 904ml per patient (range 520–1020 ml). All patients were able to infuse the full-prescribed SCIG dose and needed only a single training session. Automated pump infusions resulted in better compliance than manual infusions. There was no incidence of hemolysis or any other serious adverse event. Two patients previously intolerant to IVIg were able infuse the full SCIg dose. Minor infusion related site reactions (mild redness, and itching), a mild flu like syndrome and headache were reported with some infusions. None caused discontinuation of infusions. Serum IgG levels gradually increased by about 200% at study end. The TSQM showed satisfaction scores in the domains of effectiveness, side effects, and convenience of 70.9 +19.0, 94.0+13.9, and 75.0+19.0, respectively. Conclusion: This study provides Class IV evidence that despite its large volume SCIG appears to be well tolerated at standard doses for neuromuscular diseases i.e. 2 gm/kg, when infused over four weeks. Infusion site reactions and systemic side effects appear to be mild. SCIG may be better tolerated than IVIg. Majority of patients report high levels of overall satisfaction with the effectiveness, convenience, and side effect profile of SCIG.
PS3Group2-004 / #578A DOUBLE-BLIND PLACEBO-CONTROLLED STUDY TO EVALUATE SAFETY AND EFFICACY OF FCRN ANTAGONIST ARGX-113 IN GENERALIZED MG
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Jan Verschuuren1, Vera Bril2, Renato Mantegazza3, Andrej Szczudlik4, Said R. Beydoun5, Francisco Rodriguez De Rivera6, Maria Rosa Rottoli7, Fredrik Piehl8, Philip Van Damme9, Tuan Vu10, Peter Ulrichts11, Katrien Verschuuren12, Antonio Guglietta12, Hans De Haard12, Nicolas Leupin12, James F. Howard Jr13
1Leiden University Medical Center, Leiden, NL;2University of Toronto, Toronto, CA;3Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, IT;4Krakowska akademia neurologii, Krakow, PL;5USC Keck School of Medicine, Los Angeles, CA, US;6Hospital Universitario La Paz, Madrid, ES;7Azienda Ospedaliera Papa Giovanni XXIII, bergamo, IT;8Karolinska university, Stockholm, SE;9UZ Leuven, Leuven, BE;10USF health, Tampa, FL, US;11argenx, Zwijnaarde, BE;12argenx, Zwijnaarde, BE;13University of North Carolina, Chapel Hill, NC, US
Background: Myasthenia gravis (MG) is an autoimmune disease driven by IgG autoantibodies targeting the neuromuscular junction hence leading to muscle weakness. ARGX-113 (efgartigimod) is an Fc fragment antagonizing the IgG salvage receptor FcRn thereby accelerating the degradation of IgGs and reducing their serum levels. This effect may be beneficial in IgG-driven autoimmune diseases such as MG. Methods: A phase 2 randomized, double-blind, placebo-controlled, multicenter study was conducted in 24 patients with generalized MG (gMG). Primary objective of the study was to evaluate the safety and tolerability of ARGX-113 and secondary objectives included an efficacy analysis using four established clinical scales (MG-ADL, QMG, MGC and MG-QoL-15r). Adult anti-AChR+ gMG patients with MG-ADL ≥ 5 were enrolled. Patients were randomized 1:1 to receive four once-weekly infusions of 10 mg/kg ARGX-113 or placebo and were monitored 8 additional weeks after last treatment. Results: ARGX-113 was well tolerated in all patients, with most adverse events characterized as mild and deemed unrelated to the study drug. No serious or severe adverse events were reported. Treatment with ARGX-113 resulted in rapid and strong clinical improvement over placebo lasting for the entire duration of the study as measured by all four efficacy scales (MG-ADL, QMG, MGC and MG-QoL-15r). More specifically, 75% of patients treated with ARGX-113 had a clinically meaningful improvement in MG-ADL scores (≥2-point reduction from baseline) for a period of at least 6 consecutive weeks versus 25% of placebo patients (p = 0.0391). All patients in the treatment arm showed a rapid and deep reduction of their total IgG levels and disease improvement correlated with reduction in pathogenic IgG levels. Conclusion: These results validate ARGX-113 as a potential novel therapeutic option for gMG patients and warrant further clinical evaluation.
PS3Group2-005 / #696WHEN IS BETTER GOOD ENOUGH? PATIENT ACCEPTABLE STATES IN MYASTHENIA GRAVIS
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Carolina Barnett1, Meg Mendoza2, Hans Katzberg1, Vera Bril2
1University of Toronto, toronto, ON, CA;2University of Toronto, Toronto, CA
Background: Clinical trials are often aimed at detecting the minimally important difference (MID)—that is, the smallest change in scores that patients consider significant. While important, the MID does not reflect the overall disease impact. For example, some patients might experience a significant improvement, but still have severe disease. The Patient Acceptable Symptom State (PASS) is an absolute threshold in an outcome measure where patients in general feel well or “well enough”. PASS thresholds have not been estimated for Myasthenia Gravis (MG) measures. The main objective of this study was to estimate PASS scores for some commonly used MG measures, including the MGII, MGC, QMGS and MG-ADL. Methods: We analyzed data on 257 patients with MG who participated in the validation of the MGII. As anchor of acceptable state, we used the global health VAS from the EQ-5D, considering 95% values as cut-points to define PASS. We estimated PASS scores for each measure using the 75th percentile method—the 75% of the distribution of patients who were PASS positive. We also built receiver operating characteristic curves to find the points of best sensitivity and specificity to classify a patient as PASS positive. Results: The 75th percentile PASS scores for the MGII, MGC, QMGS and MG-ADL were: 19, 4, 7 and 4 points respectively. The ROC method PASS scores were: 11.5, 3.5, 4.5 and 2.5 points respectively. Conclusion: These PASS estimates might help inform clinical trials, to determine acceptable outcomes in MG. Further studies are needed using MG-specific anchors to determine PASS and also to understand which factors beyond disease severity—such as disease duration or age—are associated with achieving PASS.
PS3Group2-006 / #888BIOMARKER DISCOVERY IN CHILDHOOD REFRACTORY OCULAR MYASTHENIA GRAVIS
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Chaiyos Khongkhatithum, Kannikar Srisuwan, Somchai Chutipongtanate, Lunliya Thampratankul, Anannit Visudthibhan
Pediatrics, Faculty of Medicine Ramathibodi Hospital, Bangkok, TH
Background: Childhood ocular myasthenia gravis (OMG) is characterized by selective weakness of ocular muscles. It’s response to treatment varies from one the other. The OMG children who were unresponsive to steroid and treated with the other immunosuppressive drug, called refractory OMG. Refractory to therapy was observed from 15-30% of patients with OMG. Identification of the relevant biomarkers in children with OMG may be useful for determining of the prognosis and for appropriate treatment strategies.This study is aimed to identify the potential biomarkers for prediction the clinical severity and response to treatment in childhood OMG patients. Methods: This cross-sectional study was conducted from March 2016 to February 2017 at Department of Pediatrics, Faculty of Medicine Ramathibodi Hospital. Seven children with refractory OMG and seven aged-matched children with non-refractory OMG were recruited. In addition, four aged-matched children with other autoimmune neurological diseases being treated with the similar immunosuppressive drug to those with refractory OMG, were also included to the control group. Plasmas were collected and these specimens underwent proteomic analysis using two-dimensional gel electrophoresis (2-DE). Protein spots, that showed significant difference, were further analyzed by matrix- assisted laser desorption/ionization time of flight/time of flight (MALDI-TOF/TOF) mass spectrometry. Results: Four protein spots with significantly different intensity among the three groups were identified on 2-DE. All of them had increased expression (3.8, 2, 2.4 and 1.8 folds respectively) in the refractory OMG group compared to the others. Three proteins with their isoform were identified from these 4 spots, including transthyretin, apolipoprotein A-I (Figure 1) and two isoforms of fibrinogen alpha chain (Figure 2). Conclusion: Transthyretin, apolipoprotein A-I and fibrinogen alpha chain were the potential candidate biomarker of refractory ocular myasthenia gravis in children. The validation of these proteins with immunoassay technique (western blot) and the clinical implications of these proteins will be elucidated by further studies.
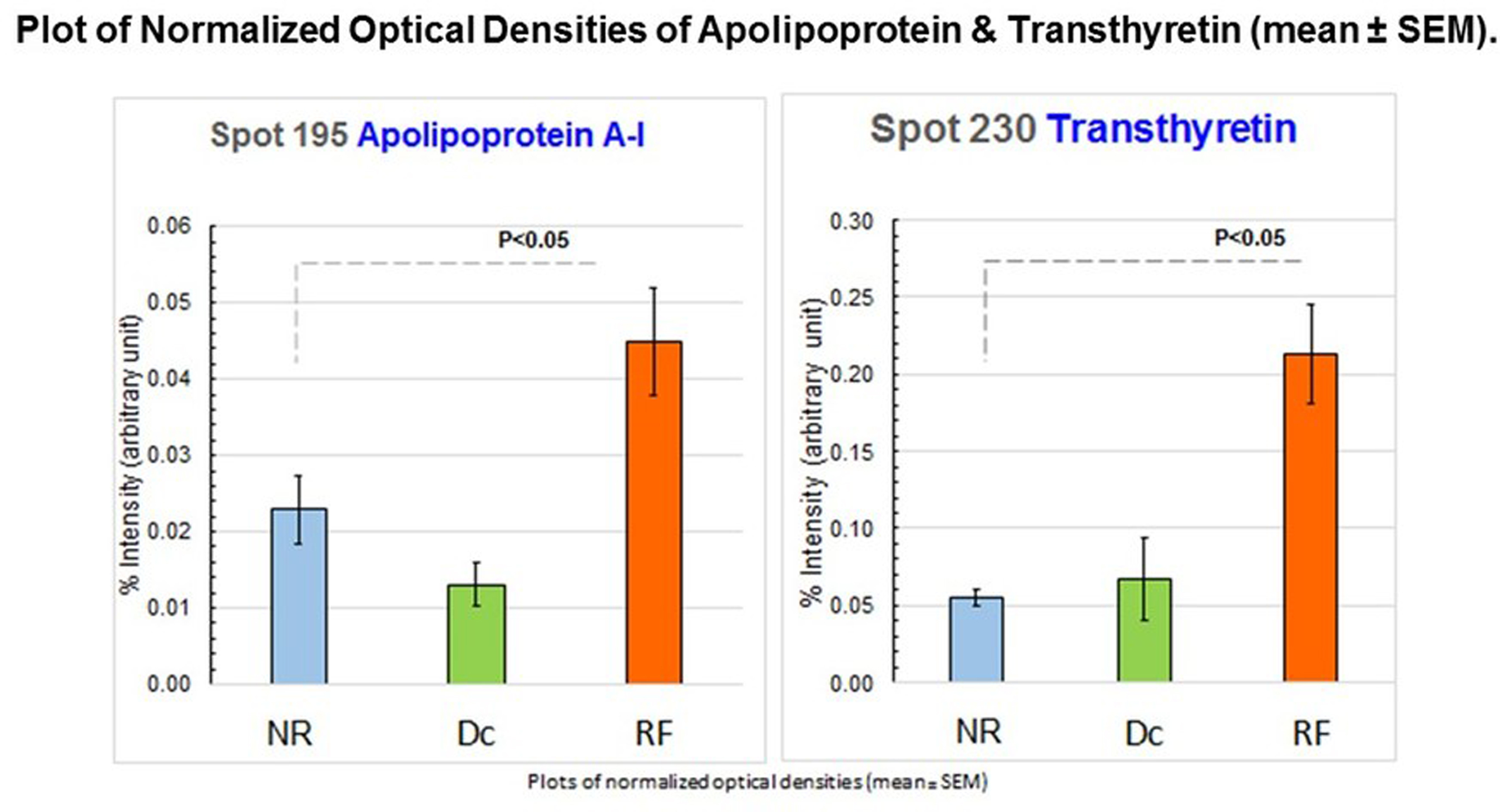
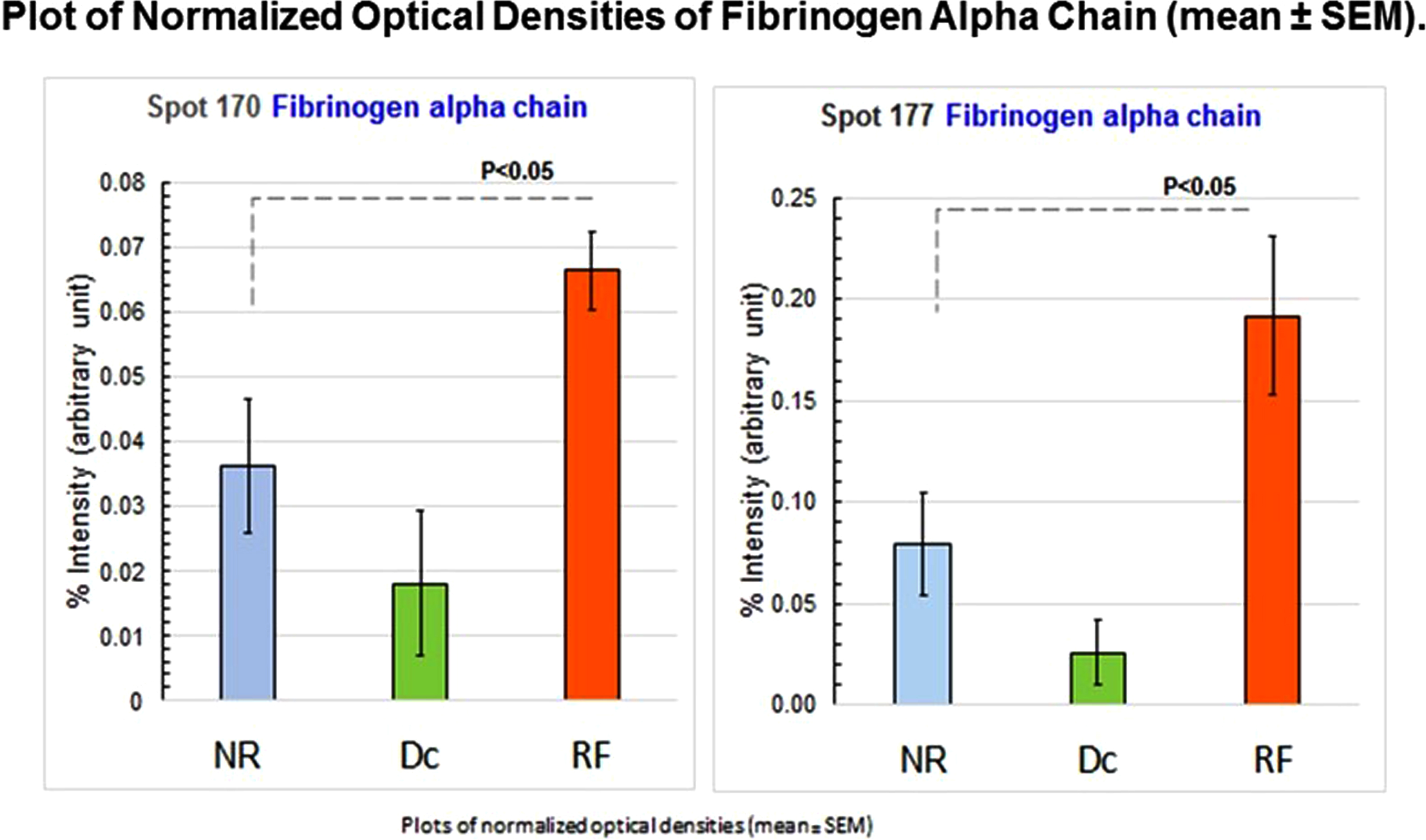
PS3Group2-007 / #863DIFFERENCES BETWEEN THYMOMATOUS AND NON-THYMOMATOUS MYASTHENIA GRAVIS
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Florencia Aguirre1, Analisa Manin2, Andres Villa1
1Neurology, Ramos Mejia Hospital, Buenos Aires, AR;2Neurology, Ramos Mejia Hospital, Buenos Aires City, AR
Background: Myasthenia Gravis (MG) is an autoimmune disease mediated by autoantibodies against post-synaptic components of the neuromuscular junction. The thymus gland is considered to play a rol in the pathogenesis of MG, with evidence of thymic abnormalities in a large proportion of patients. Pathological abnormalities of the thymus are described in about 80% of MG patients and thymomas are detected in around 15% of all MG cases. The aim of the study was to investigate epidemiological, clinical and serological characteristics and treatment responses of individuals diagnosed as having thymoma associated MG (tMG) and compare these parameters with non-thymomatous MG patients (non-tMG). Methods: We conducted a retrospective observational study in a population of patients with diagnosis of MG. Records of MG patients assisted at a public hospital in Buenos Aires City between 2008 and 2018 were reviewed. According chest imaging findings, patients were classified in tMG, those patients with images compatible with thymomas, and non-tMG, those with findings suggestive of thymic hyperplasia and normal thymus. Results: Out of 166 MG patients who were evaluated for thymus involvement with a computarized tomography scan or magnetic resonance imaging scan of the chest, 117 (70,5%) had normal thymus, 30 (18.05%) had thymic hyperplasia and 20 (12.05%) were compatible with thymomas. In tMG 75% were women vs 61.22% in non-tMG patients. The mean MG age of onset was 47.45 years in tMG, older than patients with non-thymomatous MG (non-tMG), which mean onset age was 37,63 (p= 0,031). AChR-ab seropositivity was detected in 100% of tMG. In non-tMG 73% were AChR-ab seropositivity, out of 32 AChR-ab negative patients, 5/22 (22.7%) were MuSK-ab positive. Related to clinical severity, tMG patients showed more severe disease according to maximal MGFA score achieved, 90% tMG patients had III, IV or V grade vs 67.35% in non-tMG (p= 0.038). Also, myasthenic crisis were more frequent in tMG patients than non-tMG: 30% vs 10.88%respectively (p =0.018). However, no differences were found related to immunossupresion requirements or remission rate between groups. There were no differences in the risk of autoimmune comorbidity between tMG and non-tMG patients. Autoimmune thyroid disease was the most frequent association in both groups. Conclusion: Thymoma-associated myasthenia gravis represented 12.02 % Patients with tMG showed a female predominance and started their disease at an older age than non-tMG. All tMG cases were associated to AChR-antibody. tMG patient had a more severe course according to maximal grade of MGFA achieved. However, no differences were found related to immunossupresion requirements between groups.
PS3Group2-008 / #861THYMOMA-ASSOCIATED MYASTHENIA GRAVIS IN ARGENTINA €
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Florencia Aguirre1, Analisa Manin2, Villa Andres1
1Neurology, Ramos Mejia Hospital, Buenos Aires, AR;2Neurology, Ramos Mejia Hospital, Buenos Aires City, AR
Background: Thymoma is a tumor originating from the epithelial cells of the thymus gland. Myasthenia Gravis (MG) is the commonest paraneoplastic disorder associated with this tumor, occurring in around 20 to 65% of the cases. Thymomas are traditionally considered to have a benign behavior, but it ranges from an indolent growth pattern to highly invasive. The aim of this study was to analyze histological characteristics, tumoral invasiveness, and prognosis of thymomas occurring in patients with diagnosis of MG in an argentinian population. Methods: We conducted a retrospective observational study reviewing records of patients with diagnosis of Myasthenia gravis (MG) who underwent thymectomy at the “Ramos Mejia Hospital”, a public hospital in Buenos Aires City. Patients with diagnosis of thymoma and MG were analized. All pathological specimens were classified histologically according to the WHO thymoma classification (2004 revision) and pathological staging was based on the Masaoka system. We also, analized the clinical evolution using MGFA Post Interventios Status (MGFA-PIS) score at last visit. Results: Amongst 166 MG patient routinely evaluated for thymus involvement with chest imaging, 20 had findings compatible with thymoma. Of these, fifteen patients accepted to undergo a thymectomy. The mean age at thymectomy was 45.87 years (range 27-66 years) and the mean follow-up duration post-thymectomy was 8.4 years (range 1-27 yeears). Tumor extension evaluated by Masaoka system showed 6/15 in stage I, 7/15 patients in stage II, 2 patients in stage III and 1 patient in stage IV. According to histopathological examination, 6/14 patients had mixed type thymoma (WHO type AB), 5/14 had cortical type thymoma (WHO type B), and 2 patients thymic carcinoma (WHO type C). One patient had thymolipoma. Specimen of one patient was lost. After surgical resection, 5/15(27.8%) required radiotherapy and 2/15 (11.1%) chemotherapy. 1/15 developed late recurrence of thymoma, it occurred 13 years after initial thymectomy. One patient had other extrathymic neoplasm (breast cancer). No patient died during follow-up. Concerning Clinical outcome according to MGFA-PIS score, 6 patients improved, 7 patients were on minimal manifestation stage and 2 patients in pharmacological remission. Interestingly, in 2/15 patient thymoma was detected >5 years after the onset of MG with a normal initial thoracic imaging screening, one of them developed an invasive type C thymoma. Conclusion: Twenty cases of thymoma were identified in an Argentinian population of myasthenic patients. Of these, 15 underwent a thymectomy. Concerning histological subtypes we found a prodminance of mixed type thymomas, and a 13% of type C, higher than previously reported in other series. We found a favorable clinical outcome with 86.6% of patients in pharmacological remission or minimal manifestation stage. Two patients developed thymoma many years after myasthenia gravis was diagnosed, highlighting the importance to keep searching this neoplasia in the follow up of myasthenic patients.
PS3Group2-009 / #211EPIDEMIOLOGICAL FEATURE OF MYASTHENIA GRAVIS IN SOUTHERN CHILEAN POPULATION
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Mario Fuentealba1, Manuel Padilla2, Fabian Maturana3
1Medicine, University of Concepcion, concepcion, CL;2hospital de arauco, concepcion, CL;3university of concepcion, concepcion, CL
Background: Introduction: Myasthenia gravis (MG) is a infrequent neuromuscular disease of autoimmune origin, with a prevalence of 1.5-17.9 per 100000 around the world. In Chile there is only one epidemiologic study of estimated prevalence using the hospital record of the prescription of pyridostigmine whose clinical use is exclusive in MG, and that through a the statistical technique of capture and recapture is obtaining a estimated prevalence 8.36 x 1000000 (1) The Regional Hospital of Concepcion and Hospital of Arauco are the only centers of the public health system of two southern chilean provinces (Concepcion and Arauco) that dispense pyridostigmine for the patients with MG Methods: Method In Concepcion and Arauco hospitals was obtained the registry of patients who received the prescription of this drug in the 2013-2016 period. Were excluded those under 15 years of age, deceased and those residing in communes not assigned to the corresponding health service. The diagnosis of MG was confirmed by carrying out a review of the clinical record (capture and recapture). the prevalence was calculated using official data from the Ministry of Health with a with a population coverage older than 15 years of 511718 people in the 2014 latest year available (2). Results: 49 patients were registered who met the inclusion and exclusion criteria. reason women / men of 5.1 (41 women and 8 men) / 1 with average age of 55.23 years. the estimated prevalence was 9.6 per 100000 population Conclusion: The estimated prevalence of MG in these southern provinces of Chile is comparable to the national series published using the capture and recapture statistical technique (1) Journal of Neuromuscular Diseases 3 (2016) S185 (2) www.fonasa.cl
PS3Group2-010 / #210MYASTHENIA GRAVIS: THYMECTOMY IN CHILE (MGTC)
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Mario Fuentealba1, Roberto Gonzalez2, Alejandra Riquelme2
1Medicine, University of Concepcion, concepcion, CL;2university of concepcion, concepcion, CL
Background: The historical controversy of the efficacy of Thymectomy in myasthenia gravis (MG) was resolved favorably with Randomized Trial of Thymectomy in Myasthenia Gravis (MGTX) awhat did he show a benefit of thymectomy in patients with myasthenia gravis with elevated anti nicotinic acetylcholine antibodies (AChR) with respect to clinical outcomes and requirements and of immunosuppressive drugs. Methods: descriptive analytic study, period 1990-2015, at Regional Clinical Hospital of Concepción: Review of databases, surgical protocols, clinical records and interviews. Description perioperative clinical characteristics, anticholinesterasic treatment, anatomopatological study and immediate and remote results. Follow up to 10 years with the next classifications: Masaoka, Zielinski, DeFilippi and Myasthenia Gravis Foundation of America Post-Intervention State (MGFA-PIS). Application of poll: Myasthenia Gravis Daily Life Activities (MGADL) and Myasthenia Gravis Quality of Life scale 15 (MGQOL 15). Results: 58 patients; 42 (72,4%) women, mean age 35,4 ± 13,9 years. The most frequent initial symptoms were ptosis, diplopia and dysphagia (67%,48%,39%),Preoperative classification (Osserman) grades: I 9 (15,5%), IIA 8 (13,8%), IIB 40 (69%). ETT 100%. Anatomopathological study: thymic hyperplasia 39 (67,2%). Postoperative morbidity 4 (6,9%), without mortality. Follow-up at 1, 3, 5, 8 and 10 years: Masaoka palliation rate (71,7%/77,5%/67,7%/70,0%/70,6%), remission rate (13,0%/ 15,0%/19,4%/35,0%/ 35,3%); Zielinski positive results (79,6%/ 87,5%/ 87,1%/ 90,0%/ 82,4%); DeFilippi improved (80,4%/87,5%/87,1%/90,0%/82,4%); MGFA-PIS improved (67,4%/ 77,5%/ 77,5%/ 75,0%/ 70,6%).(figure) Application of surveys: Myasthenia Gravis Activities of daily living (MGADL) and Myasthenia Gravis Quality of life scale 15 (MGQOL 15) group average (1,65 / 6,31) respectively. There was a significant decrease in the average dose of oral anticholinesterase (piridostigmine) between the preoperative dose and the follow-up (271mg/201mg/146mg/158mg/124mg/115mg). Conclusion: One limitation of the work is not to count antibodies for the selection of patients, Nevertheless our results being comparable with series that used this criterion. One possible explanation is the high frequency of thymic hyperplasia in not thymomatous MG (82 %) since a direct correlation has been reported between the level of AChR and the degree of thymic hyperplasia. however, it is mandatory to do so when accessibility to this diagnostic method is available Conclusion: In clinically selected patients with classical phenotype of MG , ETT has good immediate and long term outcomes
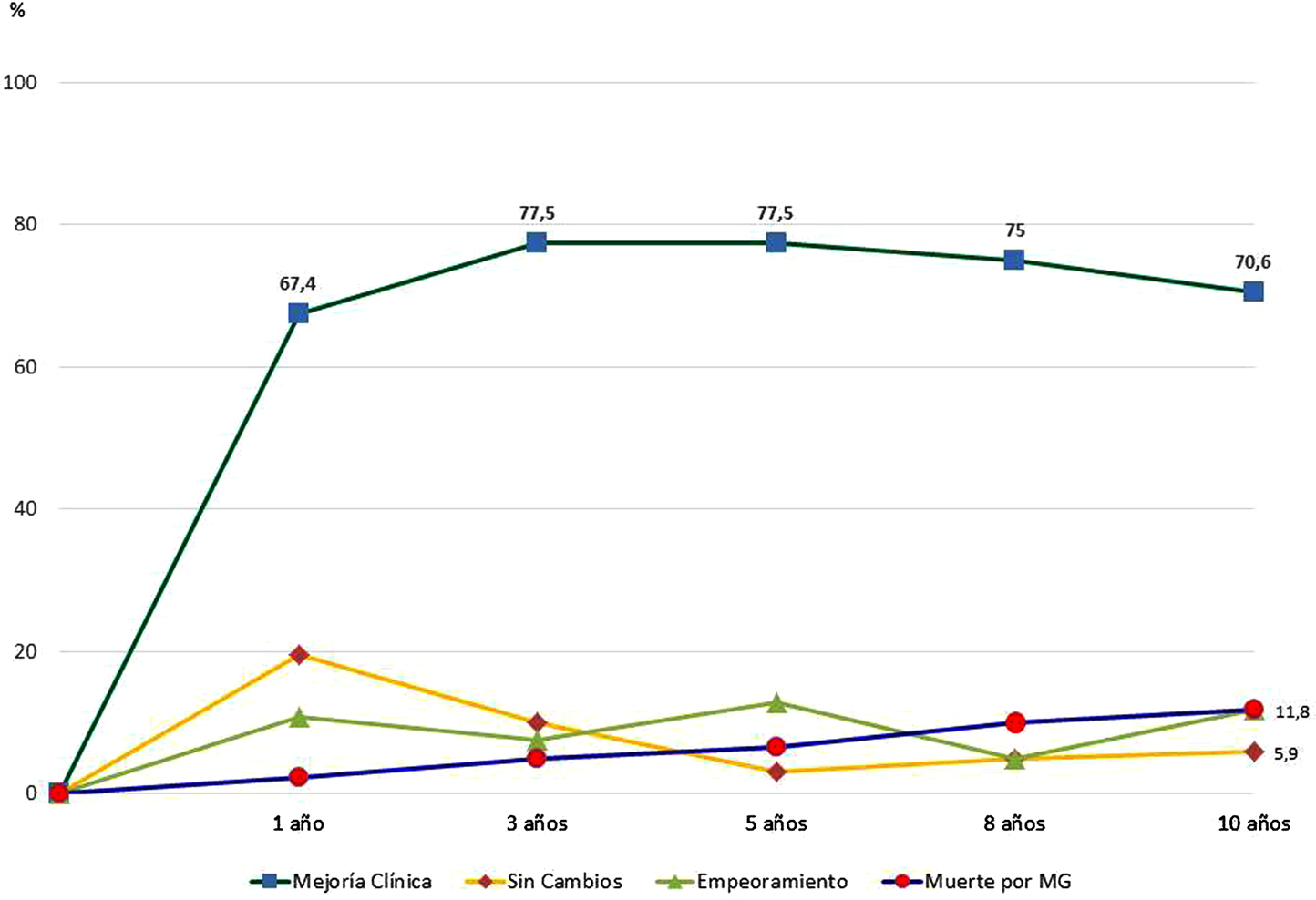
PS3Group2-011 / #430RESPONSE TO ECULIZUMAB IN PATIENTS WITH ACHR+ REFRACTORY MYASTHENIA GRAVIS RECENTLY TREATED WITH CHRONIC IVIG
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Saiju Jacob1, Hiroyuki Murai2, Kimiaki Utsugisawa3, Richard J. Nowak4, Heinz Wiendl5, Kenji P. Fujita6, Fanny O’Brien6, James F. Howard Jr7
1University Hospitals of Birmingham, Birmingham, GB;2International University of Health and Welfare, Narita, JP;3Hanamaki General Hospital, Hanamaki, JP;4Yale University School of Medicine,, New Haven, CT, US;5University of Münster, Münster, DE;6Alexion Pharmaceuticals, New Haven, CT, US;7University of North Carolina, Chapel Hill, NC, US
Background: Patients with refractory myasthenia gravis (MG) may require chronic intravenous immunoglobulin (IVIg) to treat their disease. The phase 3, randomized, double-blind, placebo-controlled REGAIN study evaluated the efficacy and safety of eculizumab in patients with anti-acetylcholine receptor antibody-positive refractory generalized MG. This subgroup analysis investigated response to eculizumab in patients who received chronic IVIg before entry into REGAIN, compared with placebo. Methods: The use of concomitant IVIg was not permitted during REGAIN other than as rescue therapy, and patients previously treated with IVIg participated in a washout period of 4 weeks prior to randomization. Patients who had previously received chronic IVIg were included if they received IVIg ≥4 times in 1 year, with at least one dose occurring during the 6 months prior to the first dose of REGAIN study drug. Results: Of the 18 patients (eculizumab n=9, placebo n=9) evaluated in this subgroup analysis, four experienced exacerbations during REGAIN; of these, one received eculizumab and three received placebo (Table). Eculizumab-treated patients had numerically larger clinically relevant improvements (mean [SD]: MG activities of daily living score [MG-ADL], -5.3 [4.0]; quantitative MG score [QMG], -4.1 [6.1]) compared with placebo (mean [SD]: MG-ADL, -2.1 [2.8]; QMG, -1.3 [3.5]) (Table). The majority of patients who received eculizumab (7/9) had clinically meaningful improvements in their MG (by ≥3 points for MG-ADL and/or ≥5 points for QMG), compared with placebo (3/9). The safety profile of eculizumab was consistent with its known profile. An open-label extension of REGAIN is ongoing, and interim data (December 2017 cut-off) will also be presented for this subgroup. Conclusion: Eculizumab treatment results in a trend toward meaningful clinical improvements and fewer disease exacerbations, compared with placebo, for patients who previously received chronic IVIg (NCT01997229, NCT02301624).
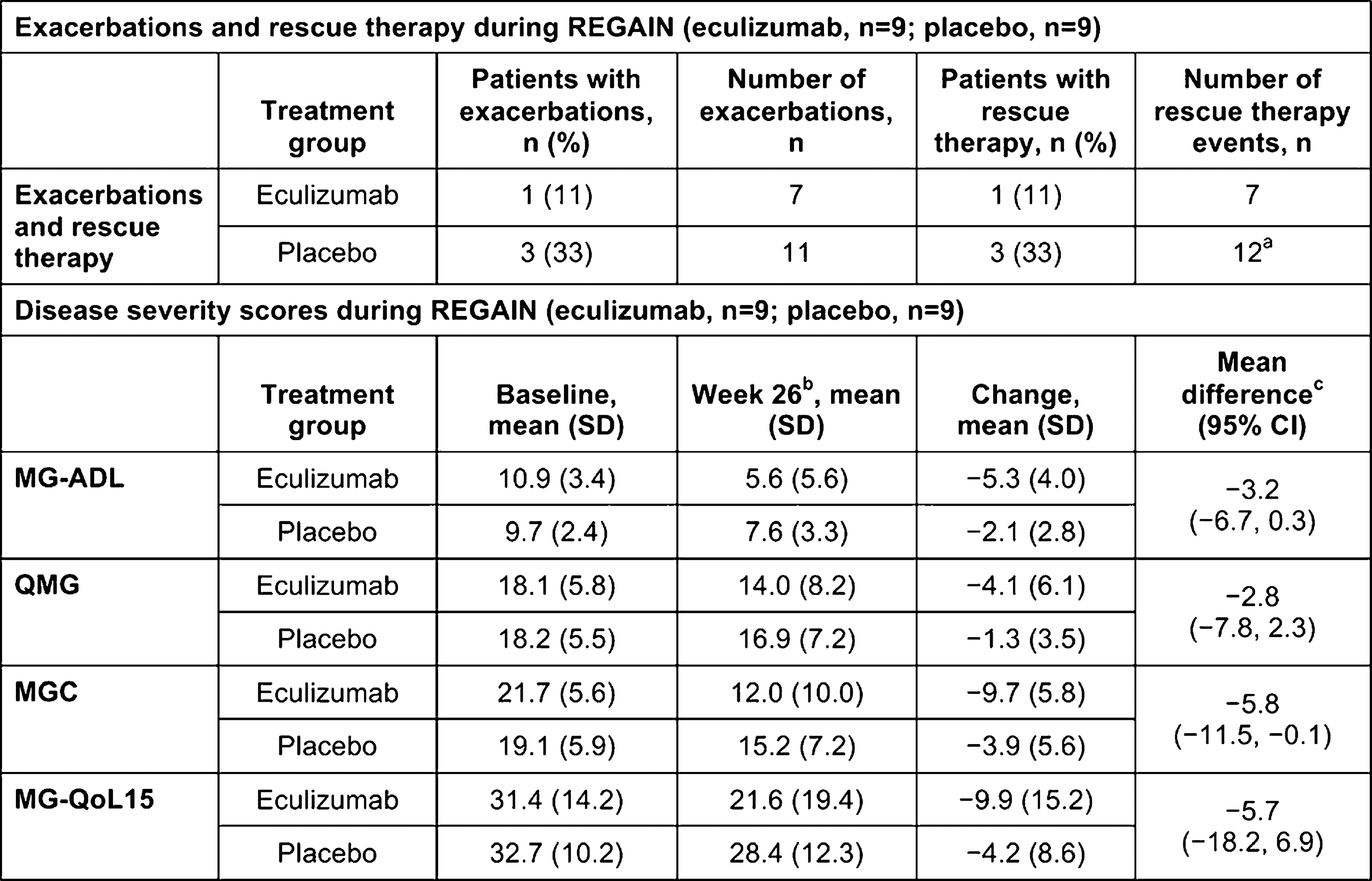
PS3Group2-012 / #432ECULIZUMAB REDUCES EXACERBATION RATES IN PATIENTS WITH ACHR+ REFRACTORY GENERALIZED MYASTHENIA GRAVIS
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Saiju Jacob1, Jeffrey T. Guptill2, Andreas Meisel3, Kenji P. Fujita4, Kaushik Patra4, James F. Howard Jr5
1University Hospitals of Birmingham, Birmingham, GB;2Duke University School of Medicine, Durham, NC, US;3Charité Universitätsmedizin Berlin, Berlin, DE;4Alexion Pharmaceuticals, New Haven, CT, US;5University of North Carolina, Chapel Hill, NC, US
Background: Exacerbations are a major burden for patients with anti-acetylcholine receptor antibody-positive (AChR+) refractory generalized myasthenia gravis (gMG). In AChR+ gMG, complement activation and membrane attack complex (MAC)/terminal complement complex formation are central to neuromuscular junction pathology and contribute to chronic disease morbidities. Eculizumab uniquely targets complement and specifically inhibits MAC formation. We analyzed the effect of eculizumab on exacerbation rates for patients from the REGAIN study and interim data from the open-label extension (September 2016 cut-off). Methods: REGAIN was a 26-week phase 3, randomized, double-blind, placebo-controlled study assessing the efficacy and safety of eculizumab in patients with AChR+ refractory gMG. Patients who completed REGAIN could enroll in the open-label extension. Patient–years- adjusted exacerbation rates were estimated for the year prior to study entry (REGAIN, N=125), during REGAIN (placebo, N=63), and during REGAIN and the open-label study (all eculizumab exposure combined; N=123 [56/62 eculizumab and 61/63 placebo-treated patients entered the extension]). Comparisons of event rates were based on a Poisson regression model. Exacerbations during the studies were defined as all reported events of clinical worsening/deterioration, MG crises or requirement for rescue therapy. Exacerbations/MG crises before REGAIN were as defined in patient records. Results: Exposure to eculizumab resulted in a 56% relative reduction in exacerbation rates compared with placebo (p=0.0409), and a 66% relative reduction compared with the year prior to REGAIN entry (p=0.0008) (Figure). The long-term safety profile of eculizumab was consistent with its known profile. REGAIN and updated interim (December 2017 cut-off) open-label study data will be presented. Conclusion: Eculizumab reduced exacerbation rates in patients with refractory gMG, suggesting that elevated complement activation contributes to risk of exacerbation in this patient population (NCT01997229, NCT02301624).
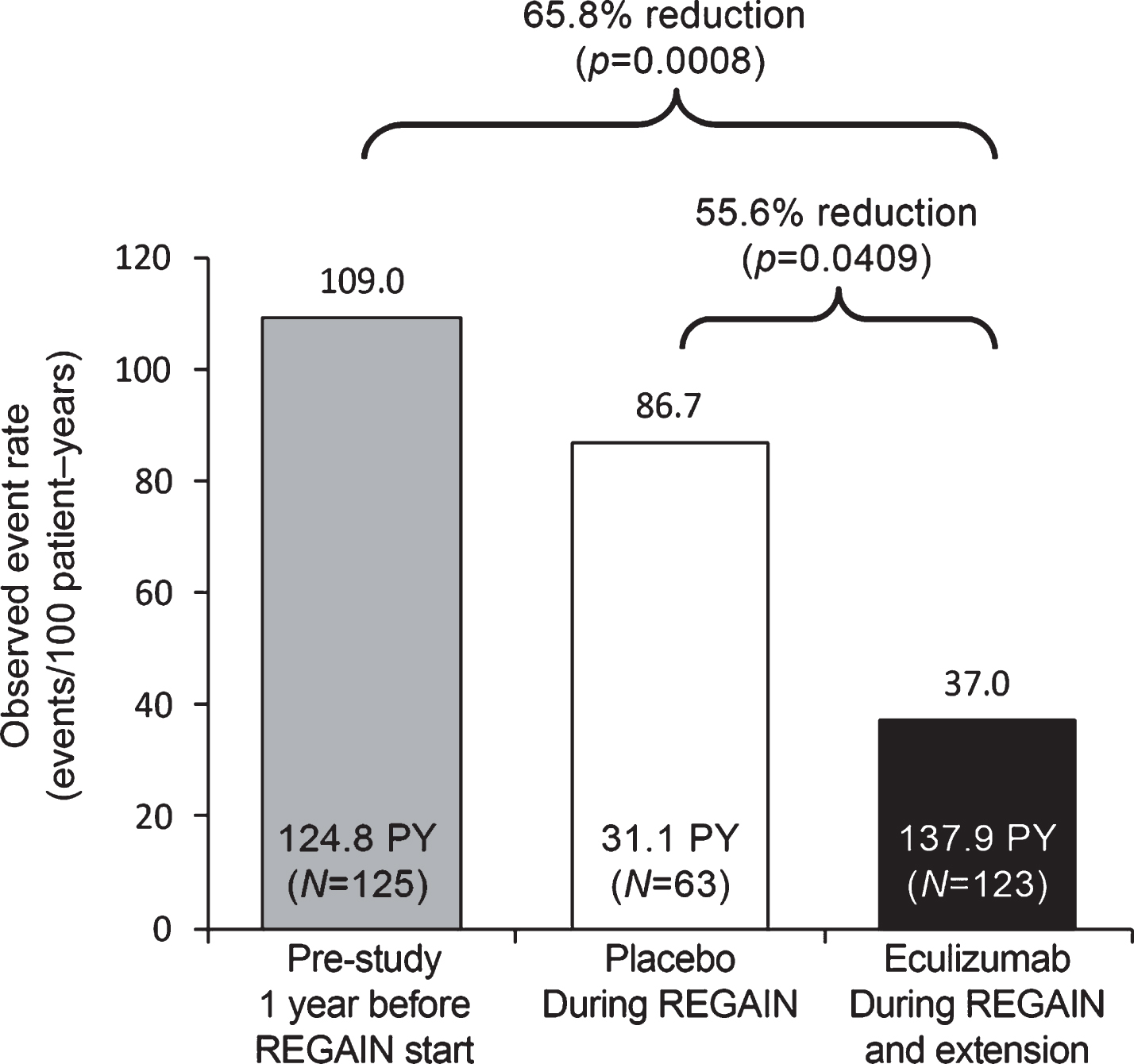
PS3Group2-013 / #486NONLETHAL CHRNA1-RELATED CONGENITAL MYASTHENIC SYNDROME CAUSED BY NOVEL HETEROZYGOUS MISSENSE MUTATIONS
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Daniel Natera De Benito1, Laura Carrera-García2, Anna Lia Frongia3, Lidia González Quereda4, Pía Gallano Petit4, Cristina Jou3, Anna Codina3, Cecilia Jimenez Mallebrera3, Carlos Ortez3, Jaume Colomer3, Andres Nascimento3
1Neuromuscular Disorders Unit, Hospital Sant Joan de Déu, Barcelona, ES;2Unidad De Patología Neuromuscular., Hospital Sant Joan de Déu, Barcelona, ES;3Hospital Sant Joan de Déu, Barcelona, ES;4Hospital Sant Pau, Barcelona, ES
Background: Congenital myasthenic syndromes (CMSs) are a heterogeneous group of genetic disorders affecting neuromuscular junction (NMJ). Postsynaptic NMJ defects encompass up to 75% of CMS. Defects in the acetylcholine receptor (AChR) can be primary, resulting from a decreased expression of one of the subunits, or alternatively can result from kinetic abnormalities of the ion channel that give rise to slow and fast channel syndromes. Most cases of primary AChR deficiency involve mutations in the CHRNE gene, because the epsilon subunit can be substituted by the foetal gamma subunit, thus rescuing the phenotype. Compound heterozygous or homozygous mutations in the CHRNA1, CHRNB or CHRND gene do not benefit from the gamma subunit rescue and are frequently lethal. We herein present a nonlethal case of a severely affected CMS patient that harbors two missense mutations in CHRNA1. Methods: One patient with genetically confirmed diagnosis of CHRNA1-related CMS followed-up in our hospital is described. Medical record of the patient with demographic information, clinical data, electrophysiological studies, and muscle biopsy findings were compiled. Results: We describe a patient previously misdiagnosed as congenital myopathy. He was born from unaffected non-consanguineous parents out of an uneventful pregnancy with reduced foetal movements. Immediately after birth, the patient required orotracheal intubation due to respiratory insufficiency and was admitted to the neonatal ICU, where he stayed for a year and a half. He was discharged to continue home care with gastrostomy and tracheostomy, requiring BiPAP support. Despite having gradually improved, he remained severely impaired, being able to walk but never having acquired the ability to stand up from floor. Physical exam demonstrated bilateral ptosis with partial ophthalmoplegia, facial hypomimia, and proximal greater than distal weakness in all four limbs. His serum CK level has always remained within the normal range. Muscle biopsy showed variability in fiber size with occasional small and round fibers, a marked predominance of type 1 fibres, and a partial disruption of the intermyofibrillar network. Decremental response following repetitive stimulation at 3 Hz was not detected. Electromyography revealed myopathic features. Mutation analysis revealed two heterozygous missense variants in the CHRNA1 gene, c.119G>A (p.Arg40Gln) and c.257G>A (p.Arg86His). None of these changes had been previously reported in LOVD database and they were both absent in a population of 120,000 control chromosomes. Both missense changes were predicted pathogenic or disease causing by in silico prediction tools. Both asymptomatic parents were heterozygous carriers. Based on exon trapping analysis studies, c.257G>A (p.Arg86His) seems to be a critical splicing mutation that causes exclusive inclusion of exon P3A in patient muscle, which is known to produce a non-functional AChR. A significant beneficial response after treatment with pyridostigmine was observed: he was able to stand from floor for the first time in his life after few days of treatment. Conclusion: Recessive CHRNA1 mutations are rare. Our case expands the number of mutations associated with CHRNA1-related CMS. Genetic comprehensive studies may be required to differentiate CMS from congenital myopathies. Reaching a diagnosis is particularly important in CMS given the existence of accessible and effective treatments.
PS3Group2-014 / #521THE FIRST KOREAN CASE OF COLQ-MUTANT MYASTHENIC SYNDROME
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Sun-Hye Jung, Jin-Hong Shin, Dae-Seong Kim
Neurology, Pusan National University Yangsan Hospital, Kyung sang Nam Do, KR
Background: The COLQ gene encodes a collagen-like strand that anchors acetylcholinesterase upon MuSK molecule at the endplate of neuromuscular junction. Homozygous or compound heterozygous mutation in this gene causes reduction in acetylcholinesterase activity, which in turn leads to an overloading of cations at the synaptic space. It is also pathogenically similar to anti-MuSK myasthenia gravis. Many cases could have been missed before the advent of next generation sequencing, as the clinical presentation is often non-specific. Methods: A 34-year-old woman presented with one year of progressive limbs weakness. She has not suffered difficulties in her daily activity before, though she had not been good at sports since her childhood. She first noticed weakness in her both legs when she stands up from squatting. She also felt difficulty raising heavy objects, which she could manage. The weakness progressed in a few months that she could not keep up with the walking pace with her friend. In 6 months, she could not stand up from sitting on a chair, nor walk without aid. Repetitive nerve stimulation test revealed marked decremental response on her trapezius muscle, indicative of postsynaptic neuromuscular transmission defect. Needle electromyography showed low amplitude, short duration MUPs and rapid recruitment in several muscles, while nerve conduction study was unremarkable. Muscle imaging as well as muscle pathology showed mild myopathy. Blood chemistry findings were all within normal limits. Results: Custom panel myopathic genes detected compound heterozygous mutation in COLQ, c.1195+1G>A (rs755782087) and c.1354C>T (p.R452C, rs368932156), which was reported to be pathogenic in Japanese patients with congenital myasthenic syndrome. She started to take bambutrol 10mg qd, which significantly improved her limbs weakness. Conclusion: Here we report the first Korean case of congenital myasthenic syndrome by COLQ mutations. Higher level of awareness is required for this ultra-rare disease, as some forms of congenital myasthenic syndromes including COLQ-mutant one can be alleviated by use of beta agonist.
PS3Group2-015 / #583PREVALENCE AND CHARACTERISTICS OF OBSTRUCTIVE SLEEP APNEA IN KOREAN MYASTHENIA GRAVIS PATIENTS
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Jin-Sung Park1, Sung-Jae Heo2, Jin-Mo Park3, Yun Jeong Lee4
1Department Of Neurology, Kyungpook National University Chilgok Hospital, Daegu, KR;2Department Of Otorhinolayngology-head & Neck Surgery, Kyungpook National University, School of Medicine, Daegu, KR;3Department Of Neurology, Dongguk University Gyeongju hospital, Gyeongju, KR;4Pediatrics, Kyungpook national university children’s hospital , Daegu , KR
Background: Myasthenia gravis (MG) is an autoimmune disease caused by antibodies that bind against nicotinic acetylcholine receptor of the postsynaptic membrane. It is characterized by progressive and diurnal muscle weakness and sometimes with respiratory difficulty. There a only few studies on obstructive sleep apnea (OSA) as one of the clinical symptom in MG which is caused by weakness of the oropharyngeal muscles, diaphragm and muscles that are involved in moving the chest wall. The prevalence of OSA between the age of 40 and 65 years was 3-7% in male and 2-5% in females in western countries. The prevalence of OSA in MG needs to be elucidated but according to a few studies, approximately 20-60% of the MG patients had OSA. The aim of our study was to find the prevalence of OSA in stable Korean MG patients and analyze the characteristics of OSA in these patients. Methods: 18 MG patients were enrolled (9 males and 9 females). All the patients consisted of MGCS 0 or 1 and all the patients had no change in their medication for at least 6 months. All the patients gave their informed consent to access their clinical data and perform an overnight PSG. The patients also underwent Korean Epworth sleepiness scale (ESS). The clinical data constituted of body mass index (BMI), disease follow up duration, post synaptic receptor antibody status, thymectomy status, pulmonary function test, and current medication usage. Results: The mean age of the enrolled MG patients was 49.6±11.1 with a mean 6.7±5.3 years of follow up. The mean body mass index was 23.7±3.4 and all the patients showed MGFA score of less than 1. 14 patients were acetylcholine receptor antibody positive, 1 MuSK antibody positive and 3 double seronegative patients. Only 5 patients were on low dose steroids (<20mg) and 8 patients (44.4) underwent thymectomy. The Korean ESS showed a prevalence of 27.8% (5 out of 18). The polysomnography performed in all stable MG patients showed an OSA prevalence of 7 out of 18 patients (38.9%) (AHI > 5), and of these patients 2 patients were mild, 3 patients were moderate and 2 patients were severe. AHI in MG patients correlated with disease duration (r=0.979, p=0.007) and BMI (r=0.728, p=0.001), but it showed no correlation to acetylcholine receptor antibody level, thymectomy status, pulmonary function or steroid dosage. Conclusion: In conclusion, prevalence of OSA in stable Korean MG patients was 38.9% which was higher than the normal population. Sleep apnea showed statistically significant correlation to disease duration and BMI. Further well designed longitudinal studies are warranted to understand the pathophysiology of OSA in neuromuscular disease which is one of the under recognized clinical symptom in MG.
PS3Group2-016 / #1002PHARMACOLOGICAL TREATMENT ADHERENCE IN PATIENTS WITH MYASTHENIA GRAVIS: ASSOCIATED FACTORS AND CLINICAL CONSEQUENCES
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Bruno K. Vitturi, Ada Pellegrinelli, Berenice C.O. Valério
Neurology, Faculdade de Ciências Médicas da Santa Casa de São Paulo, São Paulo, BR
Background: Several recent studies are devoted to therapeutic advances of Myasthenia Gravis (MG). However, therapeutic success is impossible without a good therapeutic adherence. Medication adherence is a public health problem and this has not been previously studied in MG patients. Methods: Consecutive patients from our MG Clinics were included in a cross-sectional study. Medication adherence was assessed with the Morisky Medication Adherence Scale (MMAS) and patients were classified in “adherent” or “non-adherent”. Participants were assessed for myasthenia gravis severity with the MG Composite Scale (MGC) and myasthenia gravis-related quality of life with the MG-QOL15. Patients were screened for depression with the Hospital Anxiety and Depression Scale (HADS). Univariate and multivariate statistical analysis were employed to compare clinical characteristics of the two groups of patients and regression models were used to assess predictors of adherence. Results: We enrolled 58 patients and 42 (72.4%) were women. The mean age was 46.6 (± 17.0) years. Only 26 (44.8%) of patients were adherent to treatment. Patients who were not adherent to medication had more weakness (p<0.05) and had a worse quality of life (p<0.05). Determinants of low adherence to treatment were: low educational level, multiple comorbidities and higher medication doses (p<0.05). Duration of the disease and MGC scale were not statistically associated with adherence to MG medication. Adverse effects of medications were the cause of poor adherence only in 4 (15.4%) patients. Higher likelihood of adherence was associated with female sex (OR 1.20; 95% CI: 1.03–1.37; p<0.05) and age groups older than 38 years (ORs 1.220–1.331; p<0.01). Depression was associated with a lower likelihood of adherence (OR 0.66; 95% CI: 0.2–0.78; p<0.0001) as well. Conclusion: Medication adherence should be assessed routinely in patients with myasthenia gravis. Our study may help healthcare professionals identify patients at greatest risk of poor adherence.
PS3Group2-017 / #1000SOCIAL CONSEQUENCES AND QUALITY OF LIFE OF PATIENTS WITH MYASTHENIA GRAVIS
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Bruno K. Vitturi, Ada Pellegrinelli, Berenice C.O. Valério
Neurology, Faculdade de Ciências Médicas da Santa Casa de São Paulo, São Paulo, BR
Background: Several papers are devoted to the pathophysiology and treatment of Myasthenia Gravis (MG). However, very few studies have taken into consideration the social and professional consequences of MG as well as its effects on patient’s quality of life and on their personal lives. Methods: Consecutive patients with MG with at least 1-year of clinical follow-up were included in a cross-sectional study. Baseline clinical characteristics were recorded, a social questionnaire was applied to all patients and the social support was assessed using Medical Outcome Study Social Support Survey (MOS-SSS). Clinical severity at the worst condition was graded according to the MG Foundation of America classification, and that at the current condition was determined according to the quantitative MG composite (MGC). Quality of life was evaluated with the 15-item Myasthenia Gravis Quality of Life Scale (15-MGQoL). Statistical analysis of these variables included t-tests, Chi-square tests and linear and multivariable regression. Results: We included 54 patients in our study. The patients’ mean age was 46.6 years and the mean disease duration was 10.8 years. About 52.5% of patients had experienced unemployment, 4.0 % had been unwillingly transferred, 18% had retired and 78.0% had experienced a decrease in income, 38.2% of whom reported that the decrease was ≥50% of their previous total income. In addition, 76.0% of the patients reported feeling reduced social positivity. Factors promoting social disadvantages were advanced age, severity of illness, dose of prednisone, low educational level and marital status (p<0.05). Time of disease onset and gender didn’t predict social outcomes. However, severe forms of the disease and a worse performance in MGC were correlated with a worse quality of life and increased social disadvantages (p<0.05). Furthermore, quality of life was directly related to social support (p<0.01). Conclusion: Patients with MG are often evaluated according to the presence and the severity of neurological symptoms. However, this study shows that the disease may be accompanied by numerous social problems and by a significant reduction of quality of life. The investigation of these aspects is indispensable for an integral approach of patients with MG.
PS3Group2-018 / #682HIGH EFFICACY AND SAFETY OF RITUXIMAB FOR MYASTHENIA GRAVIS: A NATIONWIDE STUDY BY AUSTRIAN ADULT NEUROLOGISTS
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Raffi Topakian1, Friedrich Zimprich2, Stephan Iglseder3, Norbert Embacher4, Michael Guger5, Karl Stieglbauer6, Dieter Langenscheidt7, Jakob Rath8, Stefan Quasthoff9, Philipp Simschitz10, Julia Wanschitz11, Petra Müller1, Dierk Oel1, Stefan Einsiedler1, Günther Schustereder1, David Windisch12, Wolfagang Löscher11
1Neurology, Klinikum Wels-Grieskirchen, Wels, AT;2Department Of Neurology, Medical University of Vienna , Vienna, AT;3Neurology, Hospital St. John of God, Linz, AT;4University Clinic St. Pölten, St. Pölten, AT;5Department Of Neurology Ii, Kepler University Hospital, Linz, AT;6Consultant neurologist in private practice, Linz, AT;7Landeskrankenhaus Rankweil, Rankweil, AT;8Department Of Neurology, Medical University of Vienna, Vienna, AT;9Medical University Graz, Graz, AT;10Klinikum Klagenfurt, Klagenfurt, AT;11Medical University of Innsbruck, Innsbruck, AT;12Landeskrankenhaus Bruck, Bruck, AT
Background: Most patients with myasthenia gravis (MG) do need long-term immunosuppressive therapy. However, conventional steroid-sparing agents may have intolerable side effects, fail, or take too long to achieve sufficient control of disease activity. Rituximab (RTX), a monoclonal antibody that rapidly depletes B cells, has emerged as an off-label treatment option. The aim of this nationwide retrospective study was to assess the use, efficacy and safety of RTX for MG. Methods: All Austrian neurologists, neuromuscular specialists and departments of neurology were contacted via the Austrian Neurological Society to provide anonymized data of all adult MG patients treated with RTX with a minimum follow-up of 3 months. The Myasthenia Gravis Foundation of America Clinical Classification and Postintervention Status scales were used to assess disease severity and outcomes, respectively. Results: A total of 56 patients were reported. 34 (60.7%) patients were women. Median age at diagnosis of MG and at start of RTX were 41.5 (24.3; 65.8) years and 47.5 (33.0; 71.0) years, respectively. Median length of follow-up after start of RTX was 20 (10; 53.5) months. 39 (69.6%) patients had anti-acetylcholine receptor antibodies (AchR ab+), 14 (25%) patients had anti-muscle-specific tyrosine kinase antibodies (MuSK ab+), and 3 patients were seronegative. Eight (14.3%) patients had thymoma-associated MG. Protocols for RTX induction and maintenance therapy varied widely across centers. Before the start of RTX, 47 (83.9%) patients had already received high cost therapies (plasma exchange, immune adsorption or immunoglobulins), and 33 (58.9%) patients had “severe MG” (grade 3b or worse). Three months after RTX, 14 of 53 (26.4%) patients had already achieved pharmacological remission. At last follow-up, 24 of 56 (42.9%) patients had pharmacological or complete stable remission, and another 14 (25%) patients had minimal manifestations. At last follow-up, remission was achieved in 71.4% of MuSK ab+ patients and 35.9% of AchR ab+ patients, respectively. In 33 of the 39 (84.6%) patients still taking steroids before RTX, steroids could be stopped altogether or the dose tapered to a dose 50% less than the initial dose. RTX was well tolerated and safe. One patient with highly refractory AchR ab+ MG died 4.5 months after RTX from a cardiac cause. Multivariate logistic regression analysis identified presence of MuSK ab as the only baseline variable independently predicting remission at last follow-up. Conclusion: In this retrospective study on RTX for MG, the largest to date on the topic, RTX was found to be safe, efficacious and relatively fast-acting. Benefit from RTX was greatest in MuSK ab+ MG.
PS3Group2-019 / #876DIFFERENTIAL RESPONSE TO RITUXIMAB IN ACHR AND MUSK ANTIBODY POSITIVE MYASTHENIA GRAVIS: A SINGLE-CENTER RETROSPECTIVE STUDY
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Tess D. Litchman1, Bhaskar Roy1, Valentine Njike2, Aditya Kumar1, Aditi Sharma3, Richard J. Nowak1
1Neurology, Yale University School of Medicine, New Haven, CT, US;2Yale-Griffin Prevention Research Center, Derby, US;3Neurology, Yale University, New Haven, CT, US
Background: Response to immunomodulatory therapies in myasthenia gravis (MG) can be variable. A subset of MG patients remains refractory to conventional agents. B-cell targeted therapy with rituximab has shown durable response in treating refractory MG. This study compares the response to rituximab therapy between acetylcholine receptor autoantibody positive (AChR+) and muscle-specific kinase autoantibody positive (MuSK+) MG. Methods: This retrospective study included patients with either AChR+ or MuSK+ MG, who were treated with rituximab between 05/2003 to 05/2017. Results: Thirty-three patients were identified (17 AChR+, 16 MuSK+; mean age 35.9 ± 15.6 years; 24 women and 9 men) with a mean follow-up of 1861 ± 953.4 days. The mean baseline MGFA Clinical Class was 1.76 ± 0.75 for the entire group, which improved to 0.61 ± 0.88 at 12-months and to 0.63 ± 0.79 at last follow-up (p-values <0.001) after rituximab treatment. The mean MFGA Clinical Class for AChR+ patients improved from a baseline of 1.7 ± 0.77 to 0.56±0.81 at 12-months (p-value 0.005) and 0.63 ± 0.8 at last visit (p-value 0.008) after rituximab treatment. Similarly, MuSK+ patients improved from a mean baseline class of 1.81 ± 0.75 to 0.66 ± 0.98 at 12-months (p-value 0.004) and 0.63±0.8 at last follow-up visit (p-value 0.003). Symptom-free state was attained at 12-months in 62.5% and 66.7% of AChR+ and MuSK+ patients, respectively. Twenty-one patients achieved clinical remission (12 AChR+, 9 MuSK+) with time to remission of 441.4 ± 336.6 days for AChR+ versus 230 ± 180.8 days for MuSK+ patients (p-value 0.049). The mean prednisone dose requirement decreased in both groups following rituximab treatment (AChR+: baseline 46.8 ± 20.8 mg/day, 12-months 11.8 ± 15.3 mg/day, last follow-up 17.9 ± 15.6; MuSK+: baseline 30.9 ± 24.9 mg/day, 12-months 9.5 ± 8.7 mg/day, last follow-up 2.8 ± 3.8 mg/day), however, there was no statistically significant difference between the two groups (p-values between the groups at baseline 0.057, at 12-months 1, and at last follow-up 0.75). The relapse rate was 58.8% and 37.5% for AChR+ and MuSK+ patients, respectively. AChR+ patients required more frequent hospitalization for exacerbation even after rituximab therapy (p-value 0.046). The number of maintenance IVIg or PLEX treatments were reduced in MuSK+ patients post-rituximab (p-value 0.0153 and 0.0033, respectively), but not in AChR+ patients. Similarly, MuSK+ patients required rescue therapy less frequently following rituximab (p-value 0.029). The number of post-rituximab IVIg rescue therapy cycles in the MuSK+ patients was lower than in the AChR+ group (p-value 0.0198). Conclusion: Rituximab therapy is observed to have both a clinical benefit and durable response in the majority of AChR+ and MuSK+ MG patients with refractory disease, however, there is no significant difference between these groups in terms of MGFA Clinical Class improvement, symptom-free state and prednisone burden. Overall, MuSK+ patients may experience greater benefits, including earlier time to remission, lower relapse rate, fewer hospitalizations, and require less IVIg/PLEX therapy post-treatment.
PS3Group2-020 / #915OUTCOMES IN GENERALIZED MYASTHENIA GRAVIS PATIENTS IN THE NEW MILLENIUM: DATA FROM A SINGLE-CENTER RETROSPECTIVE STUDY
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Matthias Tomschik, Jakob Rath, Gudrun Zulehner, Hakan Cetin, Eva Hilger, Anna Paul, Friedrich Zimprich
Department Of Neurology, Medical University of Vienna, Vienna, AT
Background: Myasthenia gravis (MG) is an autoimmune disorder that when generalized can lead to life threatening complications and often requires immunosuppression. Based on clinical characteristics, serologic findings and thymus pathologies, patients are divided into subgroups with differing disease characteristics, responses to treatments, and prognoses. The 21st century has seen important discoveries in the pathophysiology of myasthenia and new immunosuppressive agents have been available for treatment. In light of these advances, we aim to provide an update on the disease course of generalized myasthenia gravis with data from the new millennium. Methods: We retrospectively analyzed electronic patient records of all MG patients diagnosed and treated after Jan. 1st 2000 until 2016 at our neurology department. Symptom severity and pharmacologic therapies were recorded at yearly follow up visits for all patients who presented with generalized myasthenic symptoms within the first year after disease onset. Additionally, we collected data on baseline characteristics, antibody status, occurrence of life threatening events, date of thymectomy, and thymus histology whenever applicable. Cases were organized into five distinct subgroups: early onset AChR antibody positive, late onset AChR antibody positive, MuSK antibody positive, double-seronegative, and thymoma associated. A dichotomized outcome variable “sustained remission” was defined as freedom from symptoms for more than two consecutive years and until last follow-up. Differences in distribution between groups were analyzed for statistical significance by Fisher’s exact test. Results: We identified 153 patients matching our inclusion criteria, who were followed for one year or longer. Follow up visits at three years were recorded for 103 patients, five years for 80 and 10 years for 38 patients. Elevated AChR antibody levels were found in 129 patients (84%), 52 of which had an onset before fifty years of age and 77 thereafter. MuSK antibodies were found in seven patients (5%) and 17 patients were double seronegative (11%). Seventy-three patients (47%) underwent thymectomy and thymoma was confirmed histologically in 19 patients (12%). Twenty-four myasthenia-related life threatening events occurred in 21 patients. Sixteen of these events occurred in late onset AChR antibody positive patients. Symptom severity, treatment regimens, and intensity of immunosuppression differed significantly between subgroups over the years. MuSK antibody positive patients in this cohort were treated aggressively, including use of monoclonal antibodies, but in turn had milder disease severity and no life threatening events. Thirty of 103 patients followed for three years or longer achieved sustained remission. Patients with sustained remission achieved freedom from symptoms early and needed less immunosuppression, even though 40% required intensive immunosuppression in their first year. No patients with thymoma achieved sustained remission regardless of therapeutic regiment. Conclusion: We provide an update on the outcome of generalized myasthenia gravis patients and their prognostic factors from a single-center retrospective study. Use of modern immunosuppressive agents led to low morbidity in younger seropositive patients. Seronegative patients appeared more treatment resistant while late onset AChR antibody positive patients were at the highest risk for life threatening complications. Thymoma associated MG remains a therapeutic challenge and highlights the need for further expansion of treatment options.
PS3Group2-021 / #967B CELL TARGETED TREATMENT IN MYASTHENIA GRAVIS (BEATMG) - A PHASE 2 TRIAL OF RITUXIMAB IN MG: TOPLINE RESULTS
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Richard J. Nowak1, Christopher Coffey2, Jonathan M. Goldstein3, Mazen Dimachkie4, Michael Benatar5, Safawa Huq6, Brenda Pearson7, Jon W. Yankey2, Liz Uribe2, Laura Herbelin8, Ted M. Burns9, Kevin C. O’Connor1, Robin Conwit10, John T. Kissel11, Gil I. Wolfe12, David A. Hafler1, Merit E. Cudkowicz13, Richard J. Barohn14
1Neurology, Yale University School of Medicine, New Haven, CT, US;2Clinical Trials Statistical Data Management Center, University of Iowa, Iowa City, US;3Neurology, Hospital for Special Surgery, New York City, US;4Neurology, The University of Kansas Medical Center, Kansas City, KS, US;5Neurology, University of Miami, Miami, US;6Massachusetts General Hospital, Boston, US;7University of Iowa, Iowa City, US;8Neurology, University of Kansas, Kansas City, KS, US;9Department Of Neurology, University of Virginia, Charlottesville, US;10Neurology, National Institute of Neurological Disorders and Stroke, Bethesda, US;11Neurology, Ohio State University, Columbus, US;12Department Of Neurology, University of Buffalo, Buffalo, US;13Neurology, Massachusetts General Hospital, Boston, US;14Neurology, University of Kansas Medical Center, Kansas City, KS, US
Background: Despite available therapies, some myasthenia gravis (MG) patients continue to have uncontrolled disease or intolerable adverse effects. Autoreactive B cells appear to play an important role in the immunopathogenesis of MG and represent a rational target for therapeutic development. Additionally, encouraging preliminary findings with rituximab strengthened rationale for this trial. The primary objective of the BeatMG study is to determine whether rituximab is a safe and beneficial therapeutic for MG that warrants further study in an efficacy trial. Methods: We conducted a randomized, double-blind, placebo-controlled, multicenter phase 2 clinical trial utilizing a futility analysis for the primary outcome. Participants 21-90 years of age, with confirmed acetylcholine receptor (AChR) antibody positive generalized MG (Myasthenia Gravis Foundation of America Class II-IV) were eligible for enrollment if they were taking ≥15 mg of prednisone per day. Participants were randomized 1:1 to receive a two-cycle rituximab regimen (375 mg/m2 IV weekly x 4 weeks, repeated at Week 24) versus matching placebo; randomization was stratified based on baseline immunotherapy regimen. Study duration was 52 weeks, and enrolled a total of 52 participants. A forced steroid taper, beginning at Week 8, was guided by clinical stability or improvement. The primary outcome was a measure of steroid sparing effect, defined as the proportion of participants achieving a ≥75% reduction in mean daily prednisone dose in the 4 weeks prior to week 52 and with clinical improvement or no significant worsening. Safety evaluations included adverse events, vital signs, and laboratory assessments. Secondary outcomes included MG-specific clinical assessments: MG Composite (MGC) and Quantitative MG (QMG) scores, among others. Results: Primary and secondary outcome results from the study will be available for presentation and discussion. Conclusion: We will discuss the role of rituximab in the treatment of patients with AChR antibody positive disease based on the findings of this study.
PS3Group2-022 / #295CONGENITAL MYASTHENIC SYNDROME: AN INDIAN SCENARIO
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Sunitha Balaraju1, Veeramani Preethish Kumar2, Ana Töpf1, Seena Vengalil2, Kiran Polavarapu2, Saraswati Nashi2, Andreas Roos1, Rita Horvath1, Nalini A2, Hanns Lochmüller3
1John Walton Muscular Dystrophy Research Center, Newcastle University, Newcastle, GB;2NIMHANS, Bengaluru, IN;3Institute Of Genetic Medicine, Newcastle University, International Centre for Life, Newcastle Upon Tyne, US
Background: Congenital myasthenic syndromes (CMS) are a heterogeneous group of inherited neuromuscular disorders, caused by mutations in genes coding for proteins involved in the maintenance of structure and function of the neuromuscular junction (NMJ). Treatment is available and the choice of medical treatment varies with the CMS subtype, thus patients can be effectively treated and prognosis can be improved only when the precise genetic cause is known. In addition, the frequency of the different CMS-causing genes varies with ethnic and geographic origin. There are only a few reports from India describing the occurrence of DOK7, RAPSN and COLQ mutations, and the prevalence of various gene mutations in Indian population is unknown, hence, limiting the effective prognosis and access to treatments in these patients. Methods: Bloods from 74 clinically suspected CMS families (total of 193 samples) were collected and DNA was isolated. Samples were subjected to screening of the four most common CMS mutations, namely DOK7 c.1124–1127dupTGCC, CHRNE c.130insG, CHRNE c.1327delG and RAPSN c.264C>A. Cases where there was clinical suspicion of a specific causative gene (e.g. CHRNE and GMPPB) were subjected to Sanger sequencing of the whole gene including all exons and exon-intron boundaries. Results: A total of 85 samples comprising 74 index cases and 11 affected family members were subjected to screening of common mutations. All cases were negative for the RAPSN c.264C>A European founder mutation. We identified six homozygous cases with the common CHRNEc.1327delG mutation. In addition we identified seven cases with heterozygous DOK7 mutations, including the common c.1124–1127dupTGC (n=5), a novel c.1127–1128insTGCC (p.A378Sfs*30) and a novel c.1083–1086delCGTG (p.V362Gfs*93) variant. Since the phenotype of these patients carrying heterozygous mutations was suggestive of DOK7-CMS, the full coding sequence of DOK7 was Sanger sequenced but no second mutation was detected. Similarly, further sequencing of CHRNE (n=21) and GMPPB (n=3) full coding region in patients where the phenotype was suggestive of these genes identified 16/21 cases with CHRNE and 3/3 with GMPPB mutations. The overall solved rate was 37.6% and this was higher in consanguineous pedigrees (50%). Conclusion: Mutations in CHRNE gene are the most common cause of CMS at a hot-spot screening in our large Indian patient cohort. This approach may help to provide quick genetic diagnosis in at least a third of all clinically suspected cases. Clinical diagnosis further aids in providing clues for specific gene screening. The remaining patients will undergo further testing with next-generation sequencing technologies.
PS3Group2-023 / #435‘MINIMAL SYMPTOM EXPRESSION’ IN ACETYLCHOLINE RECEPTOR-POSITIVE REFRACTORY MYASTHENIA GRAVIS PATIENTS TREATED WITH ECULIZUMAB
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
John Vissing1, Saiju Jacob2, Kenji P. Fujita3, Fanny O’Brien3, James F. Howard Jr4
1Rigshospitalet, Copenhagen, DK;2University Hospitals of Birmingham, Birmingham, GB;3Alexion Pharmaceuticals, New Haven, CT, US;4University of North Carolina, Chapel Hill, NC, US
Background: REGAIN was a 26-week, phase 3, randomized, double-blind, placebo-controlled study of eculizumab versus placebo for patients with anti-acetylcholine receptor antibody-positive (AChR+) refractory generalized myasthenia gravis (gMG). Patients completing REGAIN could participate in an open-label extension study. We evaluated achievement of ‘minimal symptom expression’ in REGAIN and from an interim analysis of the open-label study, as measured by two patient-reported outcomes. Methods: ‘Minimal symptom expression’ was defined as achievement of myasthenia gravis activities of daily living (MG-ADL) total score of 0–1 or myasthenia gravis quality of life 15 (MG-QOL15) total score of 0–3. The proportions of placebo- and eculizumab-treated patients achieving ‘minimal symptom expression’ in REGAIN, and after 26 weeks of eculizumab treatment in the open-label study for the placebo/eculizumab and eculizumab/eculizumab groups were calculated. Results: At week 26 of REGAIN, a higher proportion of patients receiving eculizumab than placebo achieved ‘minimal symptom expression’: MG-ADL (21.4% vs 1.7%, respectively; difference = 19.7%, 95% CI: 8.5, 31.0); MG-QOL15 (16.1% vs 1.7%, respectively; difference = 14.4%, 95% CI: 4.2, 24.5). After 26 weeks of eculizumab treatment in the open-label study, ‘minimal symptom expression’ in MG-ADL was achieved by similar proportions of the placebo/eculizumab and eculizumab/eculizumab groups (23.6% vs 24.5%, respectively; difference = 0.9%, 95% CI: –15.6, 17.3). For MG-QOL15, the proportion achieving ‘minimal symptom expression’ in the placebo/eculizumab group increased to 12.5% (vs 23.4% in the eculizumab/eculizumab group; difference = 10.9%, 95% CI: –4.0, 25.8) (Figure). The long-term safety profile of eculizumab was consistent with its known profile. Conclusion: Eculizumab was associated with a higher proportion of patients with AChR+ refractory gMG achieving ‘minimal symptom expression’ compared with placebo, assessed using MG-ADL and MG QOL15 (NCT01997229, NCT02301624).
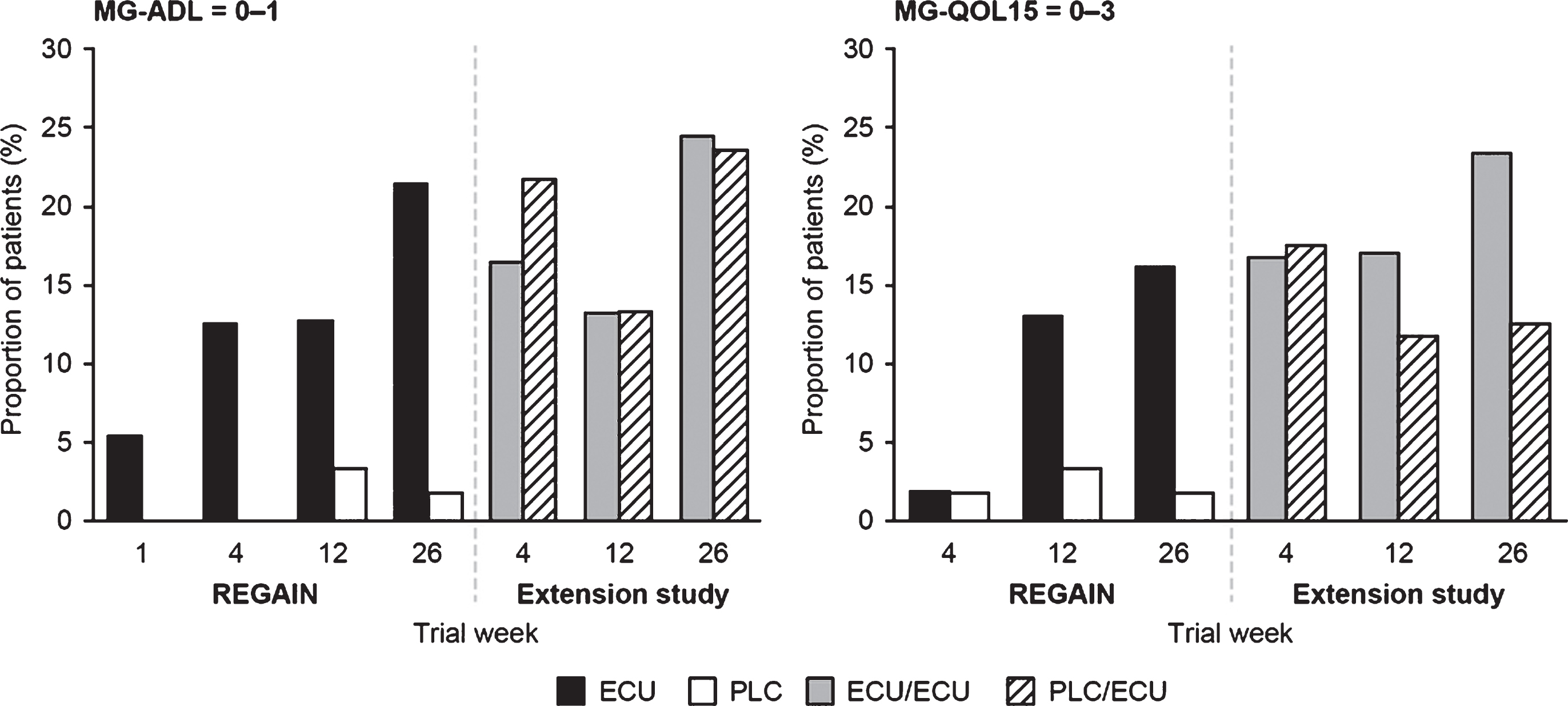
PS3Group2-024 / #466THE PLACEBO EFFECT IN MYASTHENIA GRAVIS: A META-ANALYSIS
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Elisa Frisaldi1, Aziz Shaibani2, Jan Vollert3, Bruno Ferrero4, Roberta Carrino4, Hayan Ibraheem2, Lene Vase5, Fabrizio Benedetti4
1Department Of Neuroscience “rita Levi Montalcini”, University of Turin Medical School, Turin, IT;2Nerve & Muscle Center Of Texas, Nerve & Muscle Center, Houston, TX, US;3Center Of Biomedicine And Medical Technology Mannheim Cbtm, Heidelberg University, Medical Faculty Mannheim, Mannheim, DE;4Department Of Neuroscience “rita Levi Montalcini”, University of Turin Medical School, Turin, IT;5Department Of Psychology And Behavioural Sciences, School of Business and Social Sciences, Aarhus, DK
Background: This meta-analysis aims to determine the magnitude of placebo effect for generalized myasthenia Gravis (MG) in a pool of eligible clinical trials. Depending on clinical conditions and outcome measures examined, placebo effects in neuromuscular disorders show a substantial variability and most of our knowledge about them comes from painful neuropathies. In MG, the clinical improvement attributable to a placebo effect seems to be smaller than the improvement due to active treatments. MG represents quite an interesting model to investigate placebo effects because drug and placebo responses can be quantified by both subjective and objective measurements. Methods: A comprehensive literature search via PubMed, Scopus, Web of Science, Cochrane Controlled Trial Register and Embase provided the basis of our meta-analysis. Any restrictions for year of publication, duration of treatment, or sample size were applied. Eligible for inclusion were only randomized clinical trials (RCTs) including a placebo control group and setting the Quantitative Myasthenia Gravis (QMG) score as primary or secondary outcome, since QMG test has demonstrated high inter-rater reliability, concurrent validity, and responsiveness. Main outcome variable was number of patients improving the QMG score of ≥3 points per group, or, when this information could not be retrieved, QMG change from baseline per group. On this basis, odds ratios and 95% confidence intervals or drug effect sizes were calculated. Results: Twelve studies, covering a total of 579 participants, met the inclusion criteria. Tirasemtiv, eculizimab, tacrolimus, mycophenolate mofetil, intravenous immunoglobulin, methotrexate, cyclosporine and terbutaline were the different interventions compared to placebo for the treatment of MG. Improvement in the QMG score for groups receiving placebos were generally low in most studies, but responsiveness varied broadly between studies between (from 0% to 57%), which should be attributed to random sampling bias. The magnitude of the verum effect was consistently above these numbers (from 25% to 86%), leading to odd’s ratios and effect sizes favoring drug over placebo, as a forest plot revealed (Figure 1). Since none of the RCTs analyzed in this meta-analysis include a natural history group, it was impossible to disentangle improvements due to the active drug or to the real placebo component from improvements due to other phenomena, such as spontaneous reductions of symptoms severity. Conclusion: In accordance with previous studies, our meta-analysis showed little improvements attributable to placebo interventions in clinical trials for MG management. All active treatments show a significantly higher impact on QMG scores compared to placebos, except from one study (Wolfe 2002).
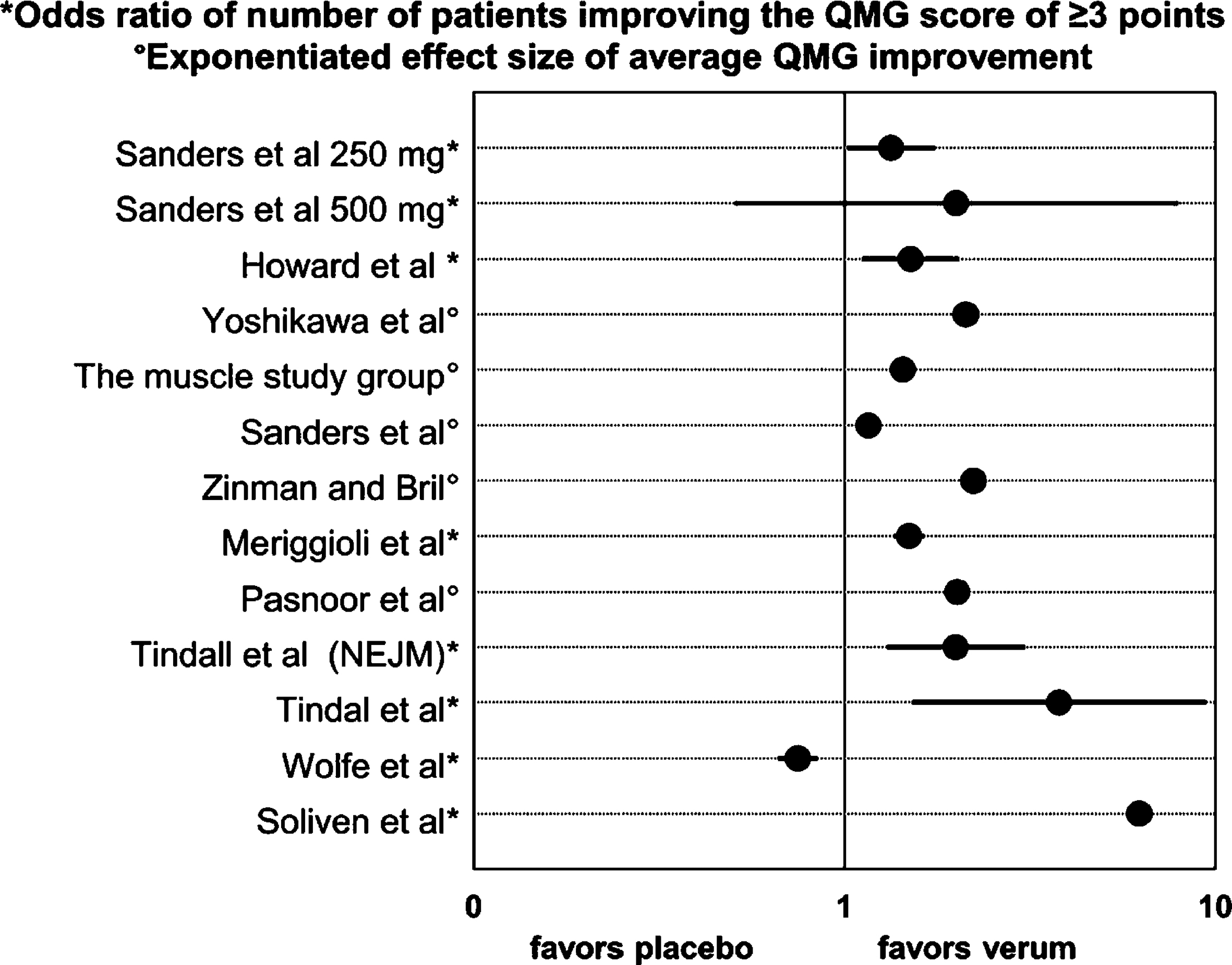
PS3Group2-025 / #678CONGENITAL MYASTHENIC SYNDROMES IN A SUBPOPULATION OF THE NORTH OF PORTUGAL
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Luísa Sousa1, Ernestina Santos1, João Martins2, Ana Sousa3, Marcio Cardoso3, Fernando Silveira4, Goreti Nadais4, Ricardo Maré5, Andreia Veiga6, Catarina S. Santos7, Teresa Coelho3, Manuela Santos8
1Neurology, Centro Hospitalar do Porto, Porto, PT;2Neurology, Hospital Pedro Hispano - Unidade Local de Saúde de Matosinhos, Matosinhos, PT;3Neurophysiology, Centro Hospitalar Porto, Porto, PT;4Neurology, Centro Hospitalar São João, Porto, PT;5Neurology, Hospital de Braga, Braga, PT;6Neurology, Centro Hospitalar Trás-os-Montes e Alto Douro, Vila Real, PT;7Neurology, Centro Hospitalar Entre Douro e Vouga, Santa Maria da Feira, PT;8Neuropediatrics, Centro Hospitalar Porto, -, PT
Background: Congenital myasthenias are rare diseases of the neuromuscular junction that usually present with ophthalmoparesis, ptosis and motor fatigability. Nevertheless, the clinical and genetic presentations are heterogeneous. The aim of this work was to analyse the characteristics of a group of patients with congenital myasthenic syndromes from a region of Portugal. Methods: We conducted a retrospective review of medical charts of patients diagnosed with congenital myasthenic syndromes from a convenience sample. Cases were identified from Neurology and Neuropediatrics consultations from six different hospitals in the north of Portugal. Demographic, clinical, electromyographic and genetic tests’ results were reviewed and described. Results: 23 patients from 16 families were identified, including 7 pairs of siblings. Median age was 37 years old (range 13-54). The first manifestation of disease was related to ocular involvement (n=11), motor weakness or gait disturbance (n=9), and bulbar or respiratory involvement (n=2). In most cases, symptoms were noted in the first two years of life, with one patient developing life-threatening symptoms in the neonatal period. The most frequent mutation was of the epsilon subunit of the acetylcholine receptor gene, CHRNE (n=16, 69%), followed by DOK-7 (n=6, 26%) and rapsyn mutations (n=1). The median time from symptom onset to genetic testing was 37.5 years. The most severe phenotypes were observed in two brothers with DOK-7 mutations and the patient with the rapsyn mutation, with periods of severe respiratory insufficiency or dysphagia requiring a gastrostomy tube placement. Regarding treatment, 19 patients were started on pyridostigmine, out of which 14 had at least partial improvement and 3 deteriorated (all with DOK-7 mutations). All DOK-7 cases improved with salbutamol or ephedrine. One of the patients with CHRNE mutation also benefited with salbutamol. Conclusion: All cases had a post-synaptic neuromuscular junction defect. Clinical characteristics are congruent with what is described in the literature for these patients. In this convenience sample we found a high relative frequency of DOK-7 mutations, superior to what is reported in larger published cohorts. The early identification of the genetic mutation is important since it may impact treatment decisions.
PS3Group2-026 / #577FETAL ACETYLCHOLINE RECEPTOR INACTIVATION SYNDROME: A RARE, BUT POTENTIALLY TREATABLE CAUSE OF FAMILIAL MYOPATHY
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Doris Nguyen1, Stephan Botez2, Cam-Tu Emilie Nguyen3
1CHU Sainte-Justine, Montreal, QC, CA;2Neurology, CHUM, Montreal, QC, CA;3Pediatric Neurology, CHU Sainte-Justine, Montreal, QC, CA
Background: Fetal Acetylcholine Receptor Inactivation Syndrome (FARIS) is a recently recognized congenital myopathy due to in utero exposure to maternal acetylcholine receptor (AChR) antibodies directed against the fetal AChR gamma subunit. Unlike transient neonatal myasthenia gravis, the manifestations of FARIS extend well beyond the neonatal period. In children who survive the neonatal period, the myopathic features of FARIS typically improve. The diagnosis of FARIS is particularly challenging when the mother is not known for myasthenia gravis (MG). We report the second case of FARIS born to an asymptomatic mother and review the literature. Methods: Case report and review of the literature. Results: We describe a term female, with polyhydramnios, congenital weakness, hypotonia, arthrogryposis, respiratory and bulbar insufficiency requiring intubation and gastrostomy, followed by marked clinical improvement in childhood. At age 9, she is able to play tennis, but still has significant facial weakness and velopharyngeal insufficiency. Brother had a similar, but more severe phenotype and passed away at age 6 weeks. Extensive investigations in the siblings, including whole exome sequencing, were non diagnostic. The mother, who is completely asymptomatic for MG, was found to have elevated AChR antibodies and postexercice exhaustion on repetitive nerve stimulation. We found 12 other FARIS cases from 6 different families. Only one other mother was asymptomatic for MG. FARIS begins antenatally, with polyhydramnios and reduced fetal movements, and can result in lethal arthrogryposis multiplex congenita. In children who survive the neonatal period, the myopathic features of FARIS typically improve, but often there is persistent myopathic facies and velopharygeal insufficiency. The risk of recurrence in subsequent pregnancies is extremely high. More aggressive MG treatment during pregnancy appears to lessen the severity of FARIS. Conclusion: FARIS is a rare, early onset myopathy caused by in utero exposure to maternal AChR antibodies directed against the fetal AChR gamma subunit. The diagnosis of FARIS is particularly challenging when the mother is not known for myasthenia gravis (MG), but has important clinical implications for future pregnancies as MG treatment during pregnancy appears to impact the severity of FARIS. Therefore, FARIS should be included in the differential diagnosis of congenital myopathies.
PS3Group2-027 / #463CLINICAL AND SEROLOGICAL PREDICTORS OF THYMOMA RECURRENCES IN MYASTHENIA GRAVIS
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Anna De Rosa1, Roberta Ricciardi2, Michelangelo Maestri2, Melania Guida2, Stefania Rizzo2, Antonio Chella3, Franca Melfi4, Marco Lucchi5, Ubaldo Bonuccelli2, Gabriele Siciliano2
1Department of Clinical and Experimental Medicine, Neurology Unit, University of Pisa, Italy, Pisa, IT;2Department of Clinical and Experimental Medicine, Neurology Unit, University of Pisa, Italy, Pisa, IT;3Division of Pneumonology, Cardio Vascular and Thoracic Department, University Hospital of Pisa, Pisa, Italy, Pisa, IT;4Multidisciplinary Center of Robotic Surgery, University of Pisa, Italy, Pisa, IT;5Cardiothoracic and Vascular Surgery Department, University of Pisa, Italy, Pisa, IT
Background: TAMG (Thymoma-associated MG) represents one of the subtypes of MG associated with autoantibodies against the acetylcholine receptor (AChR-Ab). We analyzed the clinical and serological features of patients with thymoma and relapsed thymoma, at different time points, in order to identify a possible relationship among relapses, clinical features and changes in AChR-Ab titres. Methods: We enrolled 349 MG patients with AChR-Ab and thymoma: 317 with TAMG and 32 (9%) who experienced one or more recurrences of thymoma. We then retrospectively assessed: age of MG onset, MG clinical status according to MGFA (Myasthenia Gravis Foundation of America), time of thymectomy, surgical approach, post-thymectomy status and oncological features (according to histological classifications: WHO and Masaoka-Koga). AChR-Ab serum titres have been closely monitored overtime. P-values < 0.05 were considered statistically significant. Results: Patients with relapsed thymoma were younger than those without recurrences (p<0,0001) with an average neoplastic disease-free time of about 3.7 years. In relapsed thymomas symptoms worsened immediately after the first thymectomy (p<0,0001). However, MG symptoms did not worsen by the time of recurrence of thymoma. Relapsed thymomas had a Masaoka stage more aggressive than those without recurrences (p<0,0001). Patients with relapses achieved a better MG status than those with thymoma (p<0,0115). Overall, AChR-Ab titres in patients with thymoma decreased immediately after thymectomy (thymomas: p<0,0001; relapsed thymomas: p<0,02) and remained unchanged overtime. There was no statistical difference in AChRAb titres before and immediately after thymectomy between patients with relapsed thymoma and patients without relapses. Conclusion: Thymoma recurrences are not associated with an increase of AChR-Ab titres or worsening of MG symptoms but with the histological grading. Clinical presentations are similar in thymoma and relapsed thymoma, although symptoms can get worse immediately after thymectomy in the second group of patients. In the long-term, pharmacological and complete stable remission is achieved by a high percentage of patients with relapsed thymoma. Although rare, relapsed thymoma does occur and further studies would need to be carried out to identify possible biomarkers of recurrence.
PS3Group2-028 / #631THE EFFECTS OF NEUROTROPHIC FACTORS ON HUMAN MUSCLE CELLS: A COMPARISON WITH MURINE MUSCLE CELLS
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Susie Barbeau1, Fannie Semprez2, Claire Legay1
1CNRS UMR8119, Université Paris Descartes, Sorbonne Paris Cité, Paris cedex , FR;2CNRS UMR8119, Université Paris Descartes, Sorbonne Paris Cité, Paris cedex, FR
Background: We aim at establishing mature human neuromuscular junction (NMJ) in vitro models representative of different neuromuscular diseases (NMD) and to understand the mechanisms of cross-talks between muscle cells and motor neurons at the synapse in normal and pathological conditions. NMD correspond to a vast group of diseases that perturbs or even progressively blocks the control and the force of muscle voluntary movement by affecting NMJs structure or activity. To date, no efficient curative treatments have been identified for NMD. Progresses towards identification of new treatments have been hampered by the incomplete understanding of the molecular mechanisms that control synapse formation and maintenance and the paucity of informations concerning the human NMJ. There are two types of NMDs, autoimmune diseases and congenital myasthenic syndromes (CMS). Our team focus on CMS and more specifically on CMS with Acetylcholinesterase (AChE) deficiciency due to mutations in ColQ, a collagen secreted by muscle cells. In the past, we have analyzed in details the phenotype of transgenic mice deficient for ColQ and although these transgenic mice represent a good model for the CMS, it remains that the entire ColQ gene is deleted in these mice when in human, point mutations or small deletions have been described. In addition, in vivo analysis are not allowing us to determine the impact of ColQ mutations on muscle cells independently of the motor neuron. Methods: Cell culture, immunocytochemistry, morphological analyses, epifluorescence and confocal imaging. Results: Therefore in a first step, we focus on human muscle cells and their sensitivity to neurotrophic factors in normal conditions in vitro. We will present the morphological characteristics of human myotubes obtain from human primary myoblasts on micropatterned culture dishes designed to optimize myoblasts fusion and myotubes differentiation. We also explored the impact of various proteins secreted at the NMJ junction on AChR clustering in these human myotubes. We compare these results with murine myotubes cultured in the same conditions. Preliminary results emphazise the differences and similarities between the 2 species. Future experiments will test the same factors in human muscle cells from patients suffering from CMS with AChE deficiency. Conclusion: These preliminary results are a necessary step to understand and characterize at the muscle cells and NMJ levels the deficiencies engender by the mutations involved in ColQ-CMS and develop new therapeutics.
PS3Group2-029 / #900A PILOT STUDY OF ENGINEERED AGRIN AND ITS EFFECT ON THE SEVERITY OF EXPERIMENTAL AUTOIMMUNE MYASTHENIA GRAVIS
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Zhiguo Li1, Minshu Li2, Kristofer Wood1, Steffan Hettwer3, Suraj Muley4, Fu-Dong Shi2, Qiang Lui2, Shafeeq Ladha4
1Neurobiology, Barrow Neurological Institute, Phoenix, US;2Tianjin Neurological Institute, Tianjin, CN;3Neurotune AG, Schlieren-Zurich, CH;4Neurology, Barrow Neurological Institute, Phoenix, US
Background: Since standard immunotherapy for MG involves reducing the pathogenic effects of disease causing antibodies, strategies that enhance repair of these changes are attractive as potential adjunctive therapies to immunosuppression treatments. Activation of agrin and its downstream pathways may accomplish this by promoting repair and organization of the NMJ. Therefore, we attempted to determine the effects of a soluble C-terminus fragment of neural agrin (NT-1654) on the severity of experimental autoimmune myasthenia gravis (EAMG). Compared to prior agrin agonists which had poor tissue penetration, NT-1654 is resistant to degradation and highly soluble owing to induced mutations at the neurotrypsin and heparin binding sites, respectively. Methods: EAMG was induced in female Lewis rats by immunization with the Torpedo acetylcholine receptor (tAChR) and complete Freund’s adjuvant. A total of forty-eight rats used in this study were assigned to three EAMG groups receiving vehicle phosphate-buffered saline (disease control), 2 mg/kg NT-1654, or 6 mg/kg NT-1654, and a naïve control group (n =12 per group). NT-1654 was dissolved in PBS and injected daily s.c. into tAChR immunized rats during the first 10 days after immunization, and then every other day for the following 20 days. Performing serum agrin antibody assays assessed the immunogenicity of NT-1654. EAMG clinical scores, body weight, repetitive nerve stimulation, and muscle histopathology were assessed as readouts. Results: NT-1654 reduced clinical severity of EAMG in the rats as evidenced by higher EAMG clinical scores and improved weight gain. Administration of NT-1654 also effectively promoted the clustering of AChRs at NMJs and alleviated the neurophysiologic impairment of NMJ transmission. Increased staining of muscle-specific kinase (MuSK) protein was seen although alterations in MuSK mRNA expression was not. Conclusion: These findings suggest that NT-1654 may be a promising agent to induce repair and augment synaptic strength at NMJs in EAMG. Agrin antibodies were not seen suggesting that there is no significant immunogenicity of NT-1654. Although these findings suggest that NT-1654 could have a beneficial effect in EAMG, future investigations are needed to validate these results and test NT-1654 in the setting of treatments typically used in MG (corticosteroids) and at the time of symptom onset.
PS3Group2-030 / #375MUTATIONS IN GFPT1-RELATED CMS UNDERLIE A TUBULAR AGGREGATES MYOPATHY WITH SYNAPTOPATHY
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Stéphanie Bauché1, Geoffroy Vellieux1, Damien Sternberg2, Maris-Josephine Fontenille1, Guy Brochier3, Julien Messéant1, Michel Fardeau3, Norma Romero3, Emmanuel Fournier4, Nathalie Streichenberger5, Veronique Manel6, Arnaud Lacour7, Aleksandra Nadaj-Pakleza8, Sylvie Sukno9, Françoise Bouhour10, Pascal Laforet11, Bertrand Fontaine1, Laure Strochlic1, Bruno Eymard1, Frederic Chevessier12, Tanya Stojkovic11, Sophie Nicole1
1Neurogenetic And Physiology, Inserm U 1127, CNRS UMR 7225, Sorbonne Universités, UPMC Université Paris 06 UMR S 1127, Institut du Cerveau et de la Moelle épinière, ICM, Paris, FR;2Cardiogénétique Et Myogénétique, APHP, Paris, FR;3Unité De Pathologies Neuromusculaires, Institut De Myologie, UMRS974, Institut de Myologie, Paris, FR;4Institut du Cerveau et de la Moelle epinière, Paris, FR;5Centre de Neuropathologie Est - Hospices Civils de Lyon, Université Claude Bernard Lyon1, Institut NeuroMyogène - CNRS UMR 5310 - INSERM U1217, Lyon, FR;6Service d’Epileptologie Clinique, des Troubles du Sommeil et de Neurologie Fonctionnelle de l’Enfant, Hôpital Femme Mère Enfant, Lyon, FR;7Clinique neurologique et centre de référence des maladies rares neuromusculaires, Hôpital Roger-Salengro, CHRU de Lille, Lille, FR;8Centre de Référence des Maladies Neuromusculaires Nantes-Angers, Service de Neurologie, Centre Hospitalier Universitaire d’Angers, Angers, FR;9Centre Hospitalier Béthune Beuvry, Service des consultations externes pédiatrie, Béthune, FR;10Hospices Civils de Lyon, Hôpital Neurologique Pierre Wertheimer, Service d’ENMG-Pathologies neuromusculaires, Bron, FR;11Sorbonne Universités, UPMC Univ Paris 06, INSERM UMRS974, CNRS FRE3617, Center of Research in Myology, Myology Institute, Paris, FR;12Institute of Neuropathology, University-Hospital Erlangen, schwabachanlage 6, Erlangen, DE
Background: Congenital myasthenic syndromes (CMS) are a clinically and genetically heterogeneous group of inherited disorders that share common clinical features characterized by fluctuation of muscle weakness and fatigability resulting from defective synaptic transmission at the neuromuscular junction (NMJ). The distribution of symptoms and treatment’s efficiency diverge according to the localization of the mutated proteins at the NMJ. Mutations in GFPT1 (glutamine-fructose-6-phosphate transaminase 1), a gene encoding an enzyme involved in glycosylation of ubiquitous proteins, cause a limb-girdle congenital myasthenic syndrome (LG-CMS) with tubular aggregates (TAs) characterized predominantly by affection of the proximal skeletal muscles and presence of highly organized and remodeled sarcoplasmic tubules in patients’ muscle biopsies. GFPT1 is the first and rate-limiting enzyme of the hexosamine biosynthetic pathway (HBP) involved in ubiquitous glycosylation processes. Since 2011, a total of 52 patients with GFPT1 mutations have been clinically reported. In this study, we report the first retrospective clinical description and the molecular investigations of 11 individuals from 9 unrelated families with LG-CMS linked to recessively-inherited mutations in GFPT1. Interestingly, GFPT1 is expressed in various tissues, including skeletal muscle and motor nerve, raising the intriguing question of why mutations in such a ubiquitous gene would specifically lead to synaptic and muscular disorganization. Methods: Yet, none of studies was dedicated to a long-term clinical follow-up, and investigation of the endplate was only reported for 7 patients. In this study, we report the retrospective clinical description and the molecular investigations of LG-CMS individuals with GFPT1 mutations. We analyzed the synaptic features and glycosylation level in muscle biopsies from 4 patients. Results: The retrospective clinical evaluation of LG-CMS individuals stresses an evolution toward a myopathic weakness that occurs concomitantly to ineffectiveness of usual CMS treatments. Analysis of neuromuscular biopsies from 3 unrelated individuals demonstrates that the maintenance of NMJs is dramatically impaired with loss of post-synaptic junctional folds and evidence of denervation-reinnervation processes affecting the 3 main NMJ components. Moreover, molecular analyses of the human muscle biopsies confirm glycosylation defects of proteins with reduced O-glycosylation and show reduced sialylation of transmembrane proteins in extrajunctional area. Conclusion: In this study, we identified 9 new GFPT1 mutations, further stressing the high number of mutations reported in this gene associated with CMS and reinforcing that GFPT1 is the first genetic cause of ubiquitous CMS. We highlight a profound remodeling of NMJs characterized by a loss of junctional folds as well as abnormal nerve endings when GFPT1 is decreased and impaired N- and O-glycosylation processes not only at the synapse but also in the extrajunctional area. Together with the clinical evolution of LG-CMS linked to GFPT1 towards a myopathic weakness phenotype with progressive ineffectiveness of usual CMS treatments, these histological and molecular results lead us to consider individuals with GFPT1 mutations as suffering from a “tubular aggregates myopathy with synaptopathy” rather than from the usual ubiquitous CMS due to reduced post-synaptic density of AChRs linked to hypoglycosylation.
PS3Group2-031 / #553THE POTENTIAL OF A NOVEL ANTIGEN-SPECIFIC GLYCOPOLYMER IN THE TREATMENT OF PATIENTS WITH MULTIFOCAL MOTOR NEUROPATHY
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Delphine Demeestere1, Horst Prescher1, Beat Ernst1, Pascal Hänggi2, Ruben Herrendorff2
1Institute Of Molecular Pharmacy, University of Basel, Basel, CH;2R&d, Polyneuron Pharmacetuicals AG, Basel, CH
Background: Multifocal motor neuropathy (MMN) is a rare inflammatory peripheral neuropathy that causes asymmetric muscle weakness and atrophy. The pure motor neuropathy is characterized by conduction block and, in many cases, pathogenic autoantibodies against the glycan epitope of the GM1 ganglioside. Current treatment is unspecific, leading to severe side effects, and lacks efficacy. For this reason, we are developing an antigen-specific approach with a potentially better safety and long-term efficacy, i.e. glycopolymers that selectively bind anti-GM1 autoantibodies. These glycopolymers comprise the natural GM1 glycoepitope, or mimetic thereof, that is presented in a multivalent way on a biodegradable polymer backbone to increase avidity. Methods: Both serum from MMN patients and plasma from GM1 immunized and non-immunized mice were used for enzyme-linked immunosorbent assay (ELISA) analysis. Results: Serum samples from MMN patients were tested for the presence of anti-GM1 antibodies and revealed the presence of both anti-GM1 IgG and IgM in, respectively, 57 % (8/14) and 14 % (2/14) of the cases. According to literature, mainly anti-GM1 IgM antibodies are linked to the pathogenesis of MMN, but our results indicate an underestimated role for anti-GM1 antibodies of the IgG isotype. As it is well accepted that the local glycolipid environment is important in facilitating or inhibiting antibody binding to GM1 in immunoassays, a combined GM1/galactocerebroside (GalC) assay was performed. Indeed, the percentage of patients positive for IgM antibodies increased to 36% (5/14) of the MMN sera tested, but the opposite was true for IgG antibodies with 43% (6/14). Furthermore, some preliminary experiments performed with a glycopolymer containing the natural GM1 epitope showed the successful blocking of patient anti-GM1 IgM antibodies. Interestingly, a combination of both the GM1 and GalC epitope on the polymer backbone gave a similar inhibitory effect. An active immunization model of MMN, in which BALB/c mice were immunized with GM1, did not generate robust high anti-GM1 IgM levels. Moreover, administration of the glycopolymer carrying the natural epitope in GM1-immunized mice only lowered anti-GM1 IgM levels to a limited extent. It is well known that healthy subjects have low affinity anti-GM1 IgM antibodies in the circulation, which we also observed in non-immunized mice. When injecting these wild type mice with the same GM1 glycopolymer, the same limited response was observed. So, we believe that the presence of natural low affinity anti-GM1 IgM antibodies explains the high variability of anti-GM1 IgM antibody induction in the active immunization model and the limited response upon drug administration. Therefore, we plan to introduce a humanized mouse MMN model and a human motor neuron model to further characterize the therapeutic value of the different GM1 glycopolymers. Conclusion: In conclusion, these preliminary findings suggest a potential role for anti-GM1 IgG antibodies in MMN and support the use of GM1 glycopolymers in scavenging anti-GM1 antibodies from patient sera. However, there is an urgent need for a better in vivo mouse model for MMN, in which high affinity anti-GM1 IgM antibodies are present.
PS3Group2-032 / #584RELATIONSHIP OF ACETYLCHOLINE RECEPTOR ANTIBODY TITER AND MYASTHENIA GRAVIS SEVERITY IN CIPTO MANGUNKUSUMO HOSPITAL
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Fika T. Widayati1, Ahmad Y. Safri2, Fitri Octaviana2, Luh Ari Indrawati2, Dewi Wulandari3, Tony Loho3, Manfaluthy Hakim2
1Neurology, Universitas Indonesia, Faculty of Medicine, Jakarta, ID;2Neurology, universitas Indonesia, faculty of medicine, Jakarta, ID;3Clinical Pathology, universitas Indonesia, faculty of medicine, Jakarta, ID
Background: Acetylcholine receptor antibody (antiAChR) is main antibody in pathogenesis of myasthenia gravis (MG) and important for diagnostic test. MG clinical features can be devided into ocular MG, generalisata MG and bulbar MG and each individual can have different severity assesed with MG composite scale (MGCS). Aim this study was to evaluate relationship between antiAChR titer with MG severity based on MGCS in Cipto Mangunkusumo Hospital,Jakarta. Methods: This was a descriptive-analytic cross sectional study in Cipto Mangunkusumo Hospital during January 2017 to November 2017. Inclusion criteria were 18-75 years old MG patients. The concentration of antiAChR was measured by ELISA method (Euroimun,Germany). MG severity was assessed using MGCS and blood sample for antiAChR was withdrawn at the same time. MGCS value ranging from 0-52. Results: There were 72 subjects MG met inclusion criteria and ratio between female and male was 2.5:1. MG with ocular onset was found in 57 subjects (79.2%) and proportion of early onset MG (age of onset < 50 years old) was 77.8% (56 subjects). Median of MG duration was 3 (0.25-29) years. Fifty eight (80.6%) subjects had generalized MG, thirteen (18.1%) had ocular MG and only one (1.4%) subject had bulbar MG. MGCS mean 7.82±3.91 and ptosis is the most common finding. About 59.7% subject had seropositif antiAChR and 3 among them had titer >800nmol/L (exceeded quantification limit). There were no association between antiAChR titers and MGCS (p 0.727). Additional analysis, there were no significant difference between seropositivity of antiAChR and gender, age of onset, type of MG, MGFa classification and imunosupresant therapy. Conclusion: AntiAChR titer was not related to MG severity.
PS3Group2-033 / #833THE IMPACT OF REFRACTORY MYASTHENIA GRAVIS (MG) ON PATIENT HEALTH-RELATED QUALITY-OF-LIFE (QOL)
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Audra N. Boscoe1, Haichang Xin2, Gilbert J. L’Italien3, Linda Harris4, Gary R. Cutter5
1Agios Pharmaceuticals, Cambridge, MA, US;2University of Alabama, Birmingham, AL, US;3Alexion Pharmaceuticals , New Haven, US;4Alexion Pharmaceuticals, New Haven, US;5Biostatistics, University of Alabama at Birmingham, Birmingham, AL, US
Background: Myasthenia gravis (MG) is a debilitating disease that can substantially interfere with patients’ activities of daily living, particularly among the subset of patients whose disease is refractory to conventional therapies. The impact of refractory MG on health-related quality-of-life (QOL) and disease burden has not been fully characterized. This study assessed both QOL and disease burden in patients with refractory MG compared to patients with non-refractory MG. Methods: The study population was derived from the enrollment survey of the Myasthenia Gravis Foundation of America’s (MGFA) MG Patient Registry. The study included MG patients aged 18 years or older who had been diagnosed as having MG for two or more years prior to completing the enrollment survey. Patients were classified as refractory or non-refractory based on the number and type of treatments prescribed and their level of symptomatology as measured by the Myasthenia Gravis Activities of Daily Living Scale (MG-ADL) total score. Refractory was defined as those who received at least two prior and one current MG treatment at the time of enrollment along with an MG-ADL score of ≥6. Patients who met the treatment criteria but had an MG-ADL score lower than 6, were not included in the study. All other patients were classified as having non-refractory MG. QOL was assessed by comparing the overall and individual item scores of the Myasthenia Gravis Quality of Life 15-item Scale (MG-QOL15) between refractory and non-refractory patients. Results: This analysis included 56 refractory and 717 non-refractory patients; 26 patients were excluded because they met the treatment criteria for refractory MG but not the MG-ADL criteria and thus could not be cleanly classified as either refractory or non-refractory. At the time of enrollment, refractory patients were younger [48.5 years (SD: 12.1) vs. 55.4 years (SD: 14.6), p<.01], had a younger mean age of MG onset [36.5 years (SD: 15.7) vs. 45.8 years (SD: 17.5), p<.01], and were more likely to be female (82.1% vs. 65.3%, p<.01) than non-refractory patients. Refractory patients were significantly more likely than non-refractory patients to have had 2 or more clinical exacerbations (37.5% vs. 21.1%, p<.01), ≥1 emergency room visit (42.9% vs. 22%, p<.01), and ≥1 hospitalization (37.5% vs. 18.4%, p<.01) in the prior 6 months. Refractory patients also exhibited significantly higher mean MG-QOL15 total scores (representing worse QOL) than patients with non-refractory MG [31.4 (SD 11.1) vs. 20.8 (SD: 15.0) p<0.01]. Furthermore, all individual item scores differed significantly between refractory and non-refractory patients (p≤.01). Conclusion: These findings demonstrate that refractory MG patients exhibit greater clinical burden compared with non-refractory MG patients and that this increased burden is correlated with a detrimental impact on patient QOL as measured by MG-QOL15 total and individual item scores. The MG-QOL15 was shown in this study to be a sensitive marker of patient-centered, QOL issues that reflect the debilitating nature of refractory MG.
PS3Group2-034 / #281VOCAL CORD PARALYSIS: A RARE PRESENTATION OF MYASTHENIA GRAVIS €
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Vera Montes, Sandra Sousa, Rui Guerreiro, Cátia Carmona, Fernando Pita
Neurology, Cascais Hospital, Alcabideche (Cascais) - Lisboa, PT
Background: Involvement of vocal cords as an initial presentation of myasthenia gravis (MG) in adults is extremely rare. Methods: We describe a case of vocal cord paralysis as initial manifestation of MG and perform a review of the cases of MG presenting with vocal cord paralysis reported in literature, using the Pubmed’s MeSH database to localize the MeSH terms “Vocal Cord Paralysis” and “Myasthenia Gravis”. Results: A 80-year-old caucasian woman, who was admitted to the emergency department for acute respiratory failure requiring invasive mechanical ventilation. Examination revealed dysphonia, dysphagia, dyspnea and an inspiratory stridor caused by an airway obstruction because of a bilateral paralysis in vocal cord abduction, confirmed by nasolaryngofibroscopy. A tracheostomy was performed. At that time muscle strength was normal. Cranial nerve examination failed to disclose ophthalmoparesis or ptosis. Sensory modalities were intact. After one month she developed limb muscle weakness with fatigability and a bilateral ophthalmoparesis. Her history included hypertension, type 2 diabetes and hypothyroidism. She had no previous exposure to alcohol and tobacco. There was no family history of neurologic or autoimmune disorders. Laboratory investigations revealed normal renal, liver and thyroid functions. Serum antibodies against acetylcholine receptors (AchR), muscle-specific kinase (MuSK) and low-density lipoprotein receptor-related protein (LRP4) were negative. Magnetic resonance of the head and neck, and chest computed tomography were unremarkable. Edrophonium test was negative. Repetitive nerve stimulation didn´t show abnormal decremental response but single fiber electromyography confirmed dysfunction of the neuromuscular junction. She received intravenous immunoglobulin therapy and corticosteroid, without clinical benefit. Pyridostigmine was also ineffective. She was discharged, tracheostomized, with prednisolone 60 mg a day and pyridostigmine 60 mg four times a day. For lack of improvement, she started azathioprine three months later. After two months, there was a relapse with worsening of laryngopharyngeal symptoms. She initiated plasmapheresis with a partial improvement of dysphonia and dysphagia and muscle power, despite the need to maintain tracheostomy. She repeats the AchR and MuSK antibodies that were negative. Nine months after the initial symptoms, the patient died by asphyxia caused by obstruction of the upper airway by alimentary content. From the systematic review, we found 15 published cases of MG that presented with vocal cord paralysis, 60% female and 40% male, with a mean age of 57.0 ± 18.0 years. Acute respiratory failure was the form of presentation in 87% of patients, dysphonia in 47% and dysphagia in 47% of the cases. 33% eventually developed ocular symptoms and in 27% MG was generalized. Antibodies associated with MG were detected in 33% of the cases and 53% did not respond to pyridostigmine therapy. 87% of the patients required invasive mechanical ventilation and 67% tracheostomy, of which 50% maintained tracheostomy for more than 6 months. Conclusion: Orolaryngopharyngeal manifestations cause by upper airway obstruction, such as dysarthria, dysphonia or dysphagia are uncommon in MG, especially as presenting symptoms. This case demonstrates the importance of considering neuromuscular junction disorders in the approach to new onset acute respiratory failure due to with vocal cord paralysis.
PS3Group2-035 / #581SERONEGATIVE MYASTHENIA GRAVIS WITH ANTI-LRP4 ANTIBODIES
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Jee Eun Kim1, Kee Hong Park2, Hye Seon Seo3, Jung-Joon Sung4, Yoon-Ho Hong5
1Department Of Neurology, Seoul Medical Center, Seoul, KR;2Neurology, Gyeongsang National University Hospital, Jinju, KR;3Medical Research Institute, Seoul Medical Center, Seoul, KR;4Neurology, Seoul National University College of medicine, Seoul University Hospital, Seoul, KR;5Seoul National University College of medicine, SMG-SNU Boramae Medical Center, Seoul, KR
Background: Myasthenia gravis (MG) is an autoimmune disorder characterized by fluctuating muscle weakness. 85-90% of MG patients have auto-antibodies to postsynaptic proteins against the nicotinic acetylcholine receptor (AChR) or muscle-specific tyrosine kinase (MuSK). Recently, anti-low-density lipoprotein receptor-related protein 4 (LRP4) antibodies were known to associated with MG pathogenesis in double-seronegative (seronegative to AchR and MUSK) patients. Methods: Here, we report one Korean patient who was double-seronegative, but had anti-LRP4 antibodies. Results: The 57-year-old man with history of hypertension and coronary artery disease was admitted for limb weakness predominantly in lower part and fluctuating binocular diplopia which aggravated when tired. A neurological examination revealed mild lower extremities weakness in proximal and distal similarly (Medical Research Council grade 4+). There were no signs of weakness in the ocular, bulbar, neck muscles. His deep tendon reflexes were preserved with no upper motor neuron sign. In the result of routine blood tests, creatine kinase is slightly increased (276U/L; normal range 24-204 U/L) but other results including thyroid function and tumor marker were normal. The serum anti-AChR antibodies and anti-MuSK antibodies were negative. A chest computer tomography (CT) showed 1cm sized nodule in anterior mediastinum suspecting thymoma. Repetitive nerve stimulation test (RNST) of the right orbicularis oculi, trapezius, abductor digiti quinti and flexor carpi ulnaris muscles showed abnormal decremental responses which is consistent with MG. Nerve conduction study was normal. Electromyography showed evidence of active myopathy and muscle biopsy from the left vastus lateralis muscle was consistent with mitochondrial myopathy. Anti-LRP4 antibody was tested with his serum by cell based assay based on human LRP4 expressing HEK293 cells and revealed to positive. The administration of an oral cholinesterase inhibitor and steroid was initiated, and his weakness moderately improved. Conclusion: Clinical characteristics of anti-LRP4 antibody associated MG are not well known until now. Several studies report that anti-LRP4 antibody positive MG patients to have late-onset age, mild symptoms, good therapeutic response, and no thymic changes. Our case with double-seronegative, anti-LRP4 antibody positive MG patient also showed symptom onset in late age and mild weakness without ocular symptom. Testing for anti-LRP4 antibodies need to be considered when the clinical suspicion for myasthenia gravis with mild symptom, but negative conventional diagnostic tests.
PS3Group2-036 / #666INHIBITION OF ACETYLCHOLINE RECEPTOR FUNCTION BY MYASTHENIA GRAVIS SERA DEPENDS ON RECEPTOR CLUSTERING
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Hakan Cetin1, Richard Webster2, Wei Liu2, Inga Koneczny3, Friedrich Zimprich4, Judith Cossins2, David Beeson2, Angela Vincent2
1Department Of Neurology, Medical University of Vienna, Vienna, AT;2Nuffield Department Of Clinical Neurosciences, University of Oxford, Oxford, GB;3Institute Of Neurology, Medical University of Vienna, Vienna, AT;4Department Of Neurology, Medical University of Vienna , Vienna, AT
Background: Myasthenia gravis (MG) is an autoimmune disorder of the neuromuscular junction clinically associated with fatigue and weakness. Autoantibodies directed against the acetylcholine receptor (AChR) can be detected in about 80% of MG cases. Other antibodies (Abs) bind only to AChRs that are clustered on a cell surface, or to MuSK, LRP4 or unidentified antigens (i.e. seronegative MG, SNMG). Some of the sera from MG patients including those from patients with seronegative MG were shown to inhibit AChR function. However, all studies were performed before the identification of MuSK- or clustered AChR-Abs. Our aim here was to perform a comprehensive functional evaluation of the effects of sera with AChR-, clustered AChR- or MuSK-Abs, or of sera from SNMG patients, first on unclustered AChRs and then on clustered AChRs. Methods: The CN21 muscle cell line expresses unclustered AChR and was transfected with rapsyn to enable receptor clustering. The whole-cell patch clamp technique was used for electrophysiological analyses. Serum application and rapid AChR stimulation was achieved using a fast perfusion system. Five AChR stimulations with the cells in physiological solution were followed by five AChR stimulations in the presence of serum (with one stimulation every 25s). The reduction of AChR current amplitudes during the perfusion with MG serum was measured and compared to healthy controls (HC). For MG sera, any current reduction higher than 3 standard deviations from HC mean values was considered to denote a functionally blocking serum. Results: The mean current reduction of five HC sera was -8.8 ± 1.2% in unclustered AChRs and -8.0 ± 1.9% in clustered AChRs (mean ± SEM). Unclustered AChRs were blocked by 40% of the sera in the AChR-Ab group (n=5), by 16.7% of the sera in the clustered AChR-Ab group (n=6), by 20% of the sera in the MuSK-Ab group (n=5) and also by 40% of the sera in the SNMG group (n=5). Clustered AChRs were only blocked by AChR-Ab sera (40%) and clustered AChR-Ab sera (66.7%) and by none of the MuSK or SNMG sera. Conclusion: We provide evidence that sera with clustered AChR-Abs exhibit a blocking effect predominantly on clustered AChRs, with 50% of the sera inhibiting AChR function only when the receptors are clustered. Surprisingly, a significant proportion of SNMG sera were also associated with a blocking effect but only when unclustered AChRs were examined. This suggests a yet unknown pathogenic serum factor in some of the SNMG sera.
PS3Group2-037 / #601ECULIZUMAB SHOWS CONSISTENCY OF IMPROVEMENT ACROSS MUSCLE GROUPS IN PATIENTS WITH ACHR-POSITIVE REFRACTORY MYASTHENIA GRAVIS
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Renato Mantegazza1, Kenji P. Fujita2, Fanny O’Brien2, James F. Howard Jr3
1Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, IT;2Alexion Pharmaceuticals, New Haven, CT, US;3University of North Carolina, Chapel Hill, NC, US
Background: The quantitative myasthenia gravis (QMG) test was a key prospectively-defined efficacy measure in REGAIN, a 26-week, phase 3, randomized, double-blind, placebo-controlled study of patients with anti-acetylcholine receptor antibody-positive refractory generalized myasthenia gravis (MG). Ocular and generalized weakness have previously shown different degrees of response to various therapies (prednisone has shown the most benefit for ocular symptoms; intravenous immunoglobulin/plasma exchange has shown most benefit for generalized symptoms). In REGAIN, eculizumab showed a consistent trend toward rapid and sustained improvement across all four domains (bulbar, respiratory, limb and ocular) in the patient-reported MG activities of daily living (MG-ADL) scale. We examined the effect of eculizumab on different muscle groups (bulbar, respiratory, gross motor and ocular) during REGAIN using QMG, an objective, physician-reported measure of muscle strength. Methods: Changes from baseline to week 26 in QMG domain scores were measured in patients with abnormal QMG domain scores at baseline. Repeated-measures analyses of QMG scores for bulbar (swallowing and speech), respiratory (forced vital capacity), gross motor (limb and axial motor items) and ocular (ocular and facial muscles) domains were performed. Results: Eculizumab-treated patients showed improvements in all QMG domain scores from baseline to week 26 in REGAIN (Figure). Repeated-measures analyses (least-squares means, standard error of mean) showed rapid, sustained improvements across all four QMG domains, with a trend towards significant differences between eculizumab and placebo at week 26 (bulbar, p=0.0628; respiratory, p=0.0682; gross motor, p=0.0114; ocular, p=0.0017). The safety profile of eculizumab was consistent with its known profile. Conclusion: In contrast to other therapies reporting differential effects for different muscle groups, eculizumab showed a consistent clinical response across all muscle groups (bulbar, respiratory, gross motor and ocular) using QMG assessments. This is consistent with previously reported MG-ADL results (NCT01997229, NCT02301624).
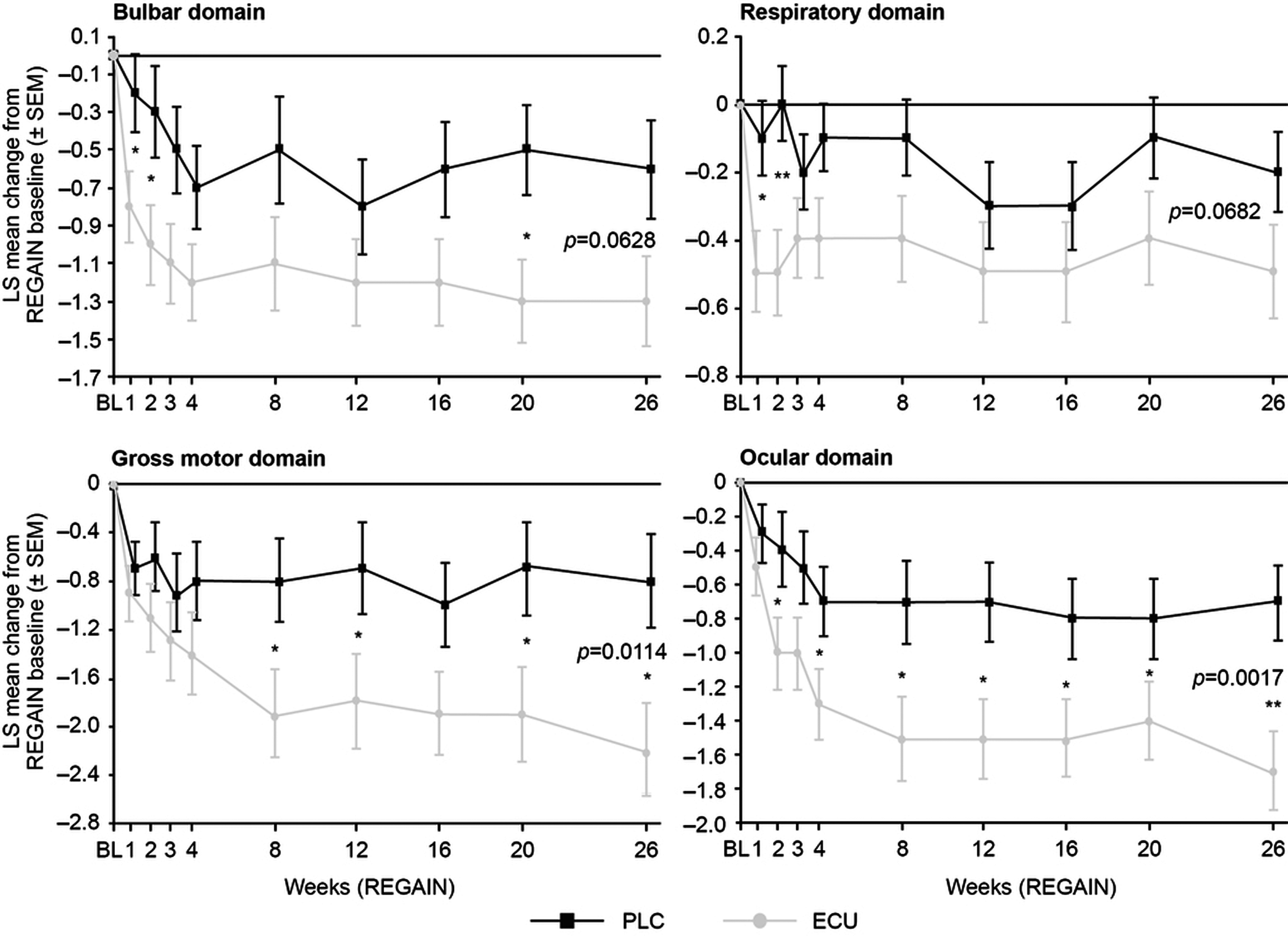
PS3Group2-038 / #649ACHIEVEMENT OF MINIMAL MANIFESTATIONS IN ECULIZUMAB-TREATED ACHR-POSITIVE REFRACTORY MYASTHENIA GRAVIS PATIENTS
Topic: Group 2 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy
Renato Mantegazza1, Gil Wolfe2, Srikanth Muppidi3, Heinz Wiendl4, Kenji P. Fujita5, Fanny O’Brien5, James F. Howard Jr6
1Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, IT;2Jacobs School of Medicine and Biomedical Sciences, University at Buffalo, Buffalo, NY, US;3Stanford University School of Medicine, Stanford, CA, US;4University Hospital Münster, Münster, DE;5Alexion Pharmaceuticals, New Haven, CT, US;6University of North Carolina, Chapel Hill, NC, US
Background: Patients with anti-acetylcholine receptor positive (AChR+) refractory generalized myasthenia gravis (gMG) continue to experience significant unresolved disease symptoms and increased risk of exacerbations and hospitalizations, despite multiple therapies. We investigated if eculizumab helps this population achieve Minimal Manifestations as evaluated by the MG Foundation of America (MGFA) Post-interventional Status.
Methods: Patients were enrolled in REGAIN, a 26-week, phase 3, randomized, double-blind, placebo-controlled study of the safety and efficacy of eculizumab in patients with AChR+ refractory gMG. Patients who completed REGAIN could enroll into the open-label extension study. Post-interventional Status, including achievement of Minimal Manifestations, was assessed at visits 4, 12 and 26 of REGAIN and week 26 of the open-label study. The sub-categories of Minimal Manifestations (MM-0, 1, 2 or 3) were not used. Results: At week 26 of REGAIN, 34/56 eculizumab-treated patients (61%) had a Post-interventional Status of Improved and 14 (25%) achieved Minimal Manifestations; 25/59 placebo-treated patients (42%) had a status of Improved, with 8 (14%) achieving Minimal Manifestations (Figure). Improvements from REGAIN baseline to week 26 of the open-label study (September 2016 cut-off) were similar between the placebo/eculizumab and eculizumab/eculizumab groups. A Post-interventional Status of Improved was achieved by 75/104 (72%) patients (eculizumab/eculizumab, 35/48 [73%]; placebo/eculizumab, 40/56 [71%]). Minimal Manifestations status was achieved by 48/104 (46%) patients (eculizumab/eculizumab, 21/48 [44%]; placebo/eculizumab, 27/56 [48%]). The long-term safety profile of eculizumab was consistent with that previously reported. Conclusion: In REGAIN, eculizumab treatment was associated with a higher proportion of patients achieving Minimal Manifestations, compared with placebo. By week 26 of the open label study, a similar proportion of patients had achieved Minimal Manifestations in the eculizumab/eculizumab and placebo/eculizumab groups (NCT01997229, NCT02301624).
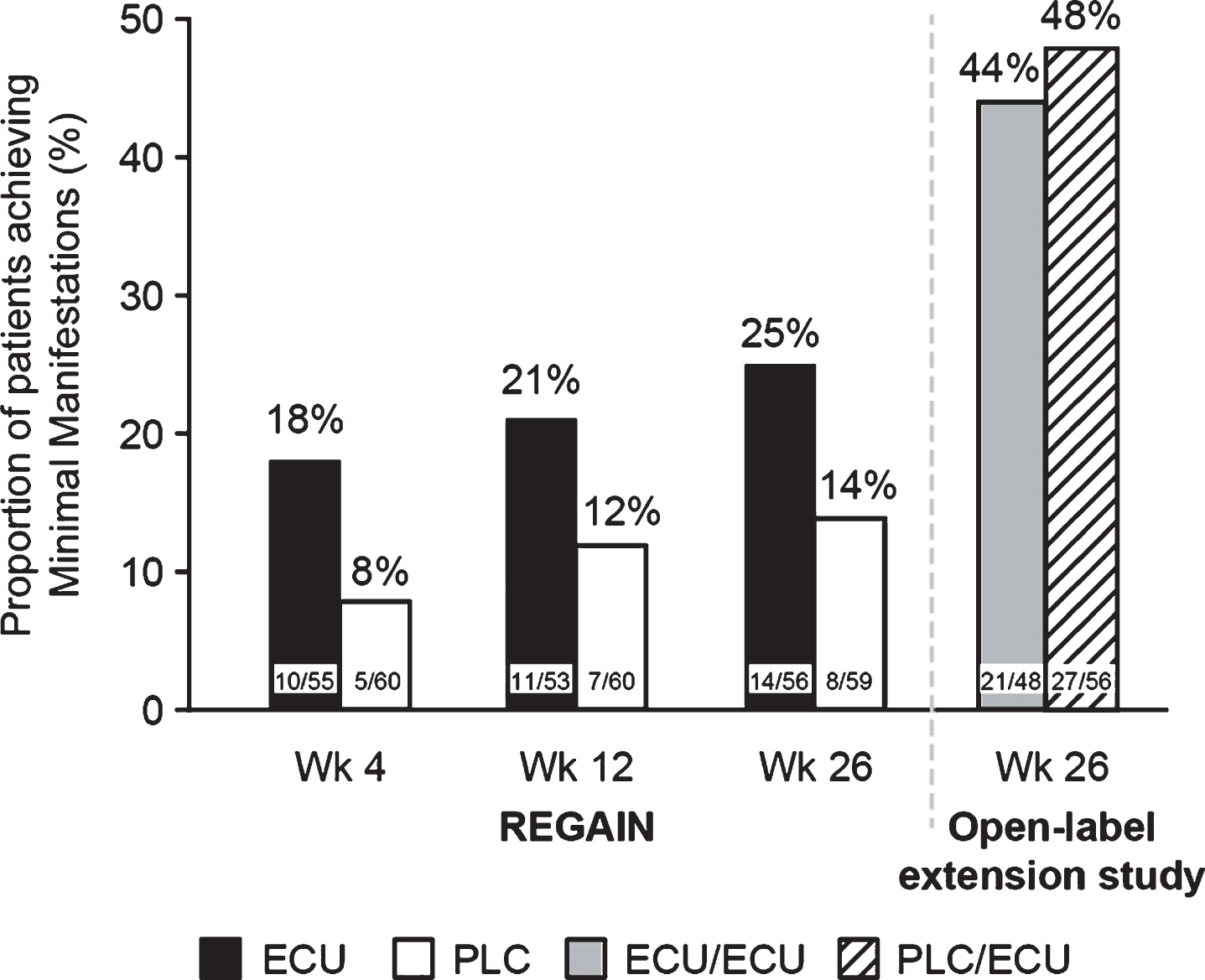
PS3Group4-001 / #512INCREASED GLUCOSE FLUCTUATION IN MOTOR NEURON DISEASE DURING ENTERAL NUTRITION
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Hiroto Takada1, Seiko Kon1, Yumiko Ogasawara2, Toshiko Ota3
1Neurology, Aomori National Hospital, National Hospital Organization, Aomori, JP;2Medical Technology, Aomori National Hospital, National Hospital Organization, Aomori, JP;3Nursing, Aomori National Hospital, National Hospital Organization, Aomori, JP
Background: Skeletal Muscle is a primary site for glucose uptake and storage, and the muscle loss influences glucose metabolism. Increased glucose excursion is reported during enteral nutrition (EN) therapy. Patients with motor neuron disease (MND) in advanced stage develop severe muscle atrophy and require EN. The aim of the study was to elucidate daily glucose profiles in MND patients with EN minutely using continues glucose monitoring (CGM). Methods: Five patients with MND were participated (the mean age; 63.0 years, the mean duration of disease; 10.4 years, five male patients). All patients were bedridden and under the artificial ventilator management with tracheotomy, having EN therapy with gastrogavage. None had a past history of diabetes mellitus (DM). CGM was performed for 72 hours. Oral glucose tolerance test (OGTT) was carried out at the last part of the CGM recording. Results: Three of five patients were diagnosed as DM and one as borderline of glucose intolerance by OGTT, in spite of normal HbA1c (the mean; 5.0%)and fasting plasma glucose (the mean; 90.4 mg/dl) level. Two patients without DM and one DM patient showed hypoglycemia. Regarding CGM profiles, hypoglycemia was observed like excessive reaction subsequently to the peak of the sharp rising glucose value after the infusion of enteral nutrient, frequenty from about four hours after the administration in the evening to midnight. Conclusion: In MND patients with severe muscle atrophy during EN management, glucoe fluctuation is increased, and hyperglycemia or hypoglycemia is not rare. Careful observation is necessary for the nutritional management in advanced patients with NMD.
PS3Group4-002 / #745PD AND SAFETY DATA FROM JEWELFISH, A STUDY OF RG7916 IN SMA PATIENTS PREVIOUSLY ENROLLED IN A SMN2-SPLICING MODIFIER STUDY
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Claudia A. Chiriboga1, Eugenio Mercuri2, Dirk Fischer3, Dominik Kraus4, Norman Thompson4, Gillain Armstrong5, Heidemarie Kletzl6, Marianne Gerber6, Yumi Cleary6, Tobias Bergauer6, Ksenija Gorni6, Omar Khwaja6
1Department Of Neurology, Columbia University Medical Center, New York, US;2Paediatric Neurology And Nemo Center, Catholic University and Policlinico Gemelli, Rome, IT;3Department of Neurology, University of Basel Hospital, Basel, CH;4Roche Pharmaceutical Research and Early Development, Roche Innovation Center, Basel, CH;5Roche Products Ltd, Welwyn Garden City, GB;6Roche Pharmaceutical Research and Early Development, Roche Innovation Center, Basel, CH
Background: Spinal muscular atrophy (SMA) is characterized by motor neuron loss and muscle atrophy due to reduced levels of survival of motor neuron (SMN) protein from loss of function of the SMN1 gene. RG7916 (RO7034067) is an investigational, orally administered, centrally and peripherally distributed small molecule that is being evaluated for its effect to modulate SMN2 pre-mRNA splicing towards the production of full-length SMN2 mRNA and increase of SMN protein. JEWELFISH (NCT03032172) is a multi-center, open-label, exploratory study evaluating the safety, tolerability and pharmacokinetics of daily oral RG7916 in patients with SMA Type 2 or 3, aged 12–60 years, who previously participated in a study with therapies targeting SMN2 splicing. The pharmacodynamic effects on SMN2 mRNA and SMN protein are also assessed. Methods: At the time of abstract submission, 10 patients have received RG7916 for a minimum of 2 months, and up to 11 months, duration as part of the JEWELFISH study. Results: To date, no drug-related adverse events leading to withdrawal have been reported. Preliminary pharmacodynamic data in whole blood from these 10 patients receiving RG7916 showed a rapid increase in the full-length SMN2/SMN2Δ7 mRNA ratio after treatment initiation, with up to a 4-fold increase from baseline over 4 weeks. This resulted in an up to 4-fold SMN protein increase versus baseline after 4 weeks of treatment. A JEWELFISH update, including safety and biomarker data, will be presented. Conclusion: In JEWELFISH, oral RG7916 treatment modulated SMN2 mRNA and increased SMN protein. While the number of patients in the JEWELFISH study is limited, the magnitude of the SMN protein increase and the safety observations thus far are comparable to what is seen in the SUNFISH Part 1 study (NCT02908685) with patients who have not previously received an SMN2-targeting therapy. JEWELFISH is currently recruiting in sites in Europe and the US. Acknowledgments The authors would like to thank all individuals enrolled in the RG7916 studies, their families and the site staff involved. Study sponsored by F. Hoffmann-La Roche.
PS3Group4-003 / #774MLPA BASED SMN1 DELETION ANALYSIS: CLINICAL CORRELATION IN INDIAN PATIENTS WITH SPINAL MUSCULAR ATROPHY
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Hansashree Padmanabha1, Veeramani Preethish-Kumar2, Kiran Polavarapu2, Saraswati Nashi3, Seena Vengalil2, Deepah Sekar4, Atchayaram Nalini5, Priya Thomas6, Krishna Gk6
1Neurology, Nimhans, Bengaluru, Bengaluru, IN;2Neurology, National Institute of Mental Health and Neurosciences, Bengaluru, IN;3NIMHANS, Bengaluru, IN;4NIMHANS, Bengaluru, Bengaluru, IN;5Neurology, National Institute of Mental Health and Neurosciences, IN;6Neurology, NIMHANS, Bengaluru, Bengaluru, IN
Background: SMA is caused due to mutation of survival motor neuron 1(SMN 1) gene located on chromosome 5q13. About 95 - 98% are homozygous for SMN1 deletion or gene conversion of SMN1 to SMN2 and 2-5% are compound heterozygotes or have an intragenic SMN1 mutation. The objective of this study is to describe the utility of multiplex ligation-dependent probe amplification technique (MLPA) in the genetic diagnosis of SMA and correlate it clinically in a large cohort from India. Methods: It was a retrospective study conducted from a single quaternary care center for neurological disorders. Medical records of the patients who attended the neuromuscular clinic, during October 2012 to December 2018, and genetically confirmed to have SMA based on MLPA, were included. MLPA was used to detect deletions in exon 7 and exon 8 of the SMN1 gene using SALSA MLPA P034 and P035 probe sets(MRC Holland, Netherlands) as per manufacturer’s instructions. Patient’s demographics, clinical features, type of SMA as per international SMA consortium criteria, and the site of deletion(exon 7, exon 8 or both) were recorded. Results: During the study period a total of 96 patients were genetically confirmed to have SMA based on MLPA. It comprised of 55 boys and 41 girls. Thirty-nine patients(40.6%) belonged to type 1; twenty-one(22%) to type 2, fourteen(14.6%) to type 3a, nineteen(19.8%) to type 3b and three patients(3.1%) with ages 26, 31 and 44years belonged to type 4-SMA. Consanguinity was noted in twenty-eight families, along with affected sibling in eight patients. Tongue fasciculation was prominent in type 1, type 2 and type 3a SMA; whereas calf hypertrophy was marked in type 3b and type 4 SMA patients. MLPA analysis revealed combined deletions of exon 7 and 8 in seventy-four patients (77%), and isolated exon 7 deletions in twenty-four patients (25%). Isolated exon 8 deletion wasn’t present in any case. The detailed results are described in table 1. Conclusion: In the study cohort, majority of type 1 and type 2 SMA patients had combined deletion of exon 7 plus exon 8. Nonetheless, in a minority of patients with type 1 and type 2 SMA, isolated exon 7 deletion was found. The mutational spectrum in type 3 SMA differed in the sense, those who had isolated exon 7 deletion had a milder type 3b SMA, those with combined had type 3a SMA. Isolated exon 8 deletions probably aren’t pathogenic and doesn’t cause SMA.
| Variables | SMA 1 (N=39) | SMA 2 (N=21) | SMA 3a (N=14) | SMA 3b (N=19) | SMA 4 (N=3) |
| Age at presentation in years (Mean ± SD) | 3.38 ± 4.21 | 5.52 ± 4.45 | 11.14 ±6.07 | 18.68 ± 9.06 | 33.67 ±9.29 |
| Age at onset in months (Mean ± SD) | 0.21 ± 0.92 | 12.05 ± 3.95 | 30.43 ± 5.98 | 104.2 ± 43.4 | 268 ± 30.1 |
| Total duration of illness (Mean ± SD) | 33.85 ± 47.7 | 56.24 ± 51.62 | 104.57 ± 72.02 | 117.47 ± 100 | 132 ± 135.2 |
| Sex (M: F) | 15:24 | 11 : 10 | 11:3 | 15:4 | 3:0 |
| Initial presentation Bulbar [N (%)] Hypotonia [N (%)] Gait disturbances [N (%)] | 14 (36%) 25 (64%) None | 1 (4.8%) 20 (95.2%) None | None 14 (100%) None | None 17 (89.5%) 2 (10.5%) | None None 3 (100%) |
| Attained sitting [N(%)] | 20 (51%) | 18 (85.7%) | 14 (100%) | 19 (100%) | 3 (100%) |
| Attained walking [N(%)] | None | 13 (62%) | 14 (100%) | 19 (100%) | 3 (100%) |
| Tongue fasciculation’s [N(%)] | 12 (31%) | 7 (33%) | 7 (50%) | 2 (11%) | None |
| Calf hypertrophy [N(%)] | 4 (10.3%) | 2 (9.5%) | 5 (35.7%) | 12 (63.2%) | 2 (66.7%) |
| Kyphoscoliosis [N(%)] | 3 (7.7%) | 3 (14.3%) | 5 (35.7%) | 3(15.8%) | None |
| Arthrogryposis [N(%)] | None | 1 (4.8%) | None | None | None |
| Elbow contractures [N(%)] | 1 (2.6%) | 3 (14.3%) | None | 1 (5.3%) | None |
| Wrist contractures [N(%)] | 1 (2.6%) | None | None | None | None |
| Hip contractures [N(%)] | 5 (12.8%) | 2 (9.5%) | None | 2 (10.5%) | None |
| Ankle contractures [N(%)] | 17 (43.6%) | 14 (66.7%) | 11 (78.6%) | 15 (78.9%) | 2 (66.7%) |
| Creatine kinase (Mean ± SD) | 177.67 ± 74.8 | 175.2 ± 85.3 | 305.9 ±174.4 | 714 ± 708.4 | 220 |
| Isolated exon 7 deletion [N(%)] | 7 (18%) | 5 (24%) | 2 (14%) | 8 (42%) | 2 (67%) |
| Isolated exon 8 deletion [N(%)] | None | None | None | None | None |
| Combined exon 7 plus exon 8 deletion [N(%)] | 33 (84.6%) | 16 (76.2%) | 13 (93%) | 11 (58%) | 1 (33%) |
PS3Group4-004 / #464ACUTE MOTOR NEURONOPATHY ASSOCIATED TO AN INFECTION BY TICK-BORNE ENCEPHALITIS VIRUS
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Lisa Bauer1, Marie–Line Harlay2, Andoni Echaniz-Laguna1, Raoul Herbrecht3, Vincent Castelain2, Jean-Baptiste Chanson4
1Neurologie, Hôpitaux Universitaires de Strasbourg, STRASBOURG, FR;2Réanimation, Hôpitaux Universitaires de Strasbourg, STRASBOURG, FR;3Hématologie, Hôpitaux Universitaires de Strasbourg, STRASBOURG, FR;4Neurologie, Hôpitaux Universitaires de Strasbourg, STRASBOURG, FR
Background: The infection by the Tick-Borne Encephalitis (TBE) flavivirus is usually associated with various brain lesions. We report here a case of TBE infection resulting in a motor neuronopathy. Methods: A 60-year-old man, often bitten by ticks in France and Germany, was addressed for fever and headaches followed by left hemiparesis and disorder of consciousness. His medical history only included an untreated mild chronic lymphocytic leukemia. Brain MRI showed a diffuse leptomeningeal contrast enhancement and analysis of cerebrospinal fluid (CSF) found a lymphocytic meningitis (595 lymphocytes per mm3, N<5) and an increased proteinorachy (1,51g/L, N<0,45). IgM but not IgG antibodies against TBE virus were discovered in CSF and serum. Borrelia burgdorferi serology was negative. A few days later, a tetraparesis with abolition of tendinous reflexes but preserved sensory function appeared. A first electroneuromyographic examination (ENMG) found markedly decreased compound muscle action potential (CMAP) amplitudes in four limbs without alteration of sensory parameters. Acute denervation was present in needle examination of proximal and distal muscles. Level of consciousness progressively improved but respiratory impairment and motor deficit predominant in upper limbs persisted. A new ENMG examination one month later found a worsening of CMAP amplitudes and denervation in proximal and distal muscles of upper limbs. A spinal cord MRI showed a hypersignal of cervical and thoracic grey matter. Results: The association of purely motor clinical and ENMG abnormalities involving proximal and distal muscles suggested an acute motor neuronopathy. Inflammation of grey matter in spinal cord confirmed that the ingoing TBE infection resulted in anterior horn damages. This case was reminiscent of abnormalities caused by poliomyelitis (polio= grey) virus or West Nile virus. Anterior horn damages have been rarely reported with other viruses. Up to now, no case associated with TBE virus was described. Conclusion: TBE virus infection may be associated with an acute motor neuronopathy and should be evoked in the context of acute motor deficit with abolished reflexes during a lymphocytic meningitis.
PS3Group4-005 / #647ASSOCIATION OF AMYOTROPHIC LATERAL SCLEROSIS AND MULTIPLE SCLEROSIS: A CASE REPORT
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Bruno K. Vitturi1, Charles P. Tilbery2, Wilson L. Sanvito1
1Neurology, Faculdade de Ciências Médicas da Santa Casa de São Paulo, São Paulo, BR;2Centro De Atendimento E Tratamento De Esclerose Múltipla (catem), Faculdade de Ciências Médicas da Santa Casa de São Paulo, São Paulo, BR
Background: Amyotrophic lateral sclerosis (ALS) and multiple sclerosis (MS) are two different idiopathic neurological disorders. Occurrence of both ALS and MS in the same patient is described as extremely rare in literature; its likelihood is estimated to be 3 : 10 billion. Methods: We present a case of man diagnosed with concurrent multiple sclerosis and amyotrophic lateral sclerosis. Results: The case we present concerns a 48-year-old male patient that in 2011 presented a left lower limb weakness with distal onset associated with a low back pain. Two years later the patient evolved with weakness and pain in right lower limb and finally in 2015 the patient was admitted to our hospital presenting an upper left limb paresis with a proximal onset associated with dysphagia and dysphonia. Neurological examination also found monoplegia in left lower limb and weakness grade 2/5 in lower right limb. There was a substantial hand hypotrophy as well. No deficit of sensation or coordination was present. No accompanying or relevant previous diseases were described by the patient and no history of neurological disorders was known in his family. Serologies and blood analysis were normal. Brain MRI revealed a typical MS pattern with numerous inflammatory bilateral lesions characterized by T2-weighted hypersignal foci with slight post-contrast enhancement spread through fronto-parietal white matter, brain stem, midbrain, cerebellum and thalamus. Cerebrospinal fluid (CSF) analysis showed 4 leucocytes/μL (88% lymphocytes), protein 49 mg/dl, glucose 71 mmol/L and positive oligoclonal bands (OCB). A diagnosis of MS was entertained. EMG showed chronic signs of bilateral myotome involvement corresponding to bulbar, cervical, thoracic and lumbosacral segments that associated with normal sensory conduction localized the involvement of the lower motor neuron, what supported the diagnosis of ALS. The patient was treated with Riluzole and Glatiramer Acetate. Our patient developed clinical and pathological features consistent with separate diagnoses of MS and ALS. MRI abnormalities exceed substantially the signal alterations usually described in ALS. Detection of OCB and the clinical course of the neurological deficits suggest MS as well. On the other hand, our patient didn’t present sensory deficits, what sustain the diagnosis of ALS since in MS it is one of the most common symptoms. In addition, high-grade peripheral paresis and amyotrophy are not supposed to be relevant in MS, as MS is considered to be limited to central nerve system. Coexistence of ALS and MS may be due to random coincidence, common pathway in both diseases or genetic and exogenous factors, leading to an increased predisposition for neurodegeneration. Conclusion: The association of multiple sclerosis and amyotrophic lateral sclerosis is very rare and represents a diagnostic and therapeutic challenge. This case may indicate that these two neurodegenerative diseases may have some point in common in pathophysiology. Further studies are needed for better understanding this association.
PS3Group4-006 / #733EXPLORATION OF THE REPOSITIONING POTENTIAL OF MARKETED DRUGS FOR NEUROPATHIES
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Jacqueline Gide1, Florine Roussange2, Hélène Polvèche1, Didier Auboeuf3, Anne Boland4, Jean-François Deleuze4, Borckman Knut5, Johanna Tournois1, Marc Peschanski6, Cecile Martinat7, Sandrine Baghdoyan2
1I-stem, CECS, CORBEIL ESSONNES, FR;2Motor Neurone Disease, INSERM/UEVE UMR 861, I-STEM, Corbeil-Essonnes, FR;3Laboratoire De Biologie Et Modélisation De La Cellule,, ENS de Lyon, Lyon, FR;4Centre National De Genotypage, CEA, EVRY, FR;5Interdisciplinary Pediatric center for children with developmental disabilitiesand severe chronic disorders,, Gottingen, DE;6I-stem Cecs, INSERM UEVE UMR861, CORBEIL ESSONNES, FR;7I-stem, INSERM UEVE UMR861, CORBEIL ESSONNES, FR
Background: Human embryonic stem cells and their progenies are powerful models for drug discovery studies. Their amplification and differentiation allow reaching a large amount of well characterized cells in cell type of interest. Taking advantage of this property, we proceed to the screeing of 50 marketed drugs in derivatives from human pluripotent stem cells in order to identfy drugs that could have a potential therapeutic effect for rare monogenic diseases. Methods: A human pluripotent stem cell line was differentiated in Mesenchymal Stem Cells (MSC) and treated with 50 marketed drugs at the dose of 10μM for 24 hours in triplicates. The modulation of genes after drug treatment was analyzed by deep RNA sequencing approach. Among the regulated genes, we focussed on those whose modulation could present a potential therapeutic effect for rare monogenic diseases. Results: In MSC, we have identified drugs that are able to induce the expression of SQSTM1 whose mutation is responsible of Amyotrophic lateral sclerosis (ALS), and to decrease the level of expression of LMNB1 and PLP1 genes whose duplication is causative for Adult-onset autosomal dominant leukodystrophy (ALDL) and Pelizaeus-Merzbacher disease (PMD) respectively. We then undertook the validation of these regulations in the cell type affected in the disease and in a pathological cellular model. Conclusion: By the use of an unbiaised RNA deep sequencing approach and human pluripotent stem cell lines, we have identified two marketed drugs with a therapeutic potential for three rare diseases through the modulation of LMNB1, PLP1 and SQSTM1 genes expression.
PS3Group4-007 / #527DISSECTING UBA5 DYSFUNCTION IN NERVOUS SYSTEM DISORDERS USING THE ZEBRAFISH
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Rita Serrano1, Viola Oorschot2, Georg Ramm2, Robert Bryson-Richardson1
1School Of Biological Sciences, Monash University, Melbourne, VIC, AU;2Ramaciotti Centre For Cryo-electron Microscopy, Monash University, Melbourne, VIC, AU
Background: Mutations in the ubiquitin-like modifier activating enzyme 5 (UBA5) gene were recently identified to cause childhood-onset cerebellar ataxia and infantile-onset epileptic encephalopathy, disorders predominantly associated with the central nervous system. Cerebellar ataxia patients show cerebellar atrophy, motor dysfunction, and mild peripheral demyelination; whereas, the clinical symptoms associated with epileptic encephalopathy include reduced motor skills, developmental delay, intellectual disability, microcephaly, delayed brain myelination, and recurrent seizures, ultimately resulting in the death of most patients within the first 2 decades of life. The function of Uba5 in the nervous system and how mutations in uba5 result in disease remains to be elucidated. Methods: To understand the pathophysiology associated with uba5 mutation and to identify pathways that could reduce disease progression we are using the zebrafish. Using Crispr-Cas9 methodology we generated novel uba5 alleles to investigate the consequences of uba5 loss-of-function on zebrafish locomotion, survival, and nervous system development and homeostasis. Results: We found that uba5 mutation results in a significant reduction of locomotor activity that recapitulates the mobility impairment in uba5 patients. Notably, the motor impairment is not preceded by damage to muscle structure or neuronal network, as demonstrated by immuno-labelling. However, electron microscopy reveals signs of myelin degeneration, a hallmark of neuropathies, in the nerves innervating the skeletal muscle of uba5 mutants. This observation suggests that myelin synthesis or maintenance is impaired in uba5 mutants in the peripheral nervous system and may be an initial trigger to the resulting phenotype. Finally, we show that uba5 mutants have a reduced lifespan. Conclusion: These models provide a new tool for characterising the function of Uba5 in the nervous system and determining the role of Uba5 in causing neurological disease.
PS3Group4-008 / #702BRANAPLAM IN TYPE 1 SPINAL MUSCULAR ATROPHY: RESPIRATORY SUPPORT AND FEEDING
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Nicolas Deconinck1, Ap Born2, G Baranello3, Enrico Bertini4, Nathalie Goemans5, R Pingili6, J Praestgaard7, R Roubenoff8, U Schara9, Emilie Voltz10
1Department Pediatric Neurology, HUDERF, Brussels, BE;2Rigshospitalet, Copenhagen,, DK;3Istituto Neurologico “Carlo Besta”, Milano, IT;4Unit Of Neuromuscular Disorders, Laboratory Of Molecular Medicine, Bambino Gesu’ Children’s Hospital,, Rome, IT;5Treat-NMD Neuromuscular Network, Leuven, BE;6Novartis Pharmaceuticals, East Hanover, NJ, US;76Novartis Pharmaceuticals, Novartis Pharmaceuticals, East Hanover, NJ, US;8Novartis Inst for Biomedical Research, Basel, CH;9Universitaetsklinikum, Essen, DE;10Novartis Institutes for BioMedical Research, Basel, CH
Background: Introduction: LMI070X2201 (NCT02268552) is an open-label, multi-part, first-in-human study of oral branaplam (formerly known as LMI070) in infants with Type 1 spinal muscular atrophy (SMA) with 2 SMN copies. Part 1 of this study evaluated the safety, tolerability, pharmacokinetics, pharmacodynamics, and efficacy of branaplam after 13 weeks of treatment and estimated the Maximum Tolerated Dose (MTD) and optimal dosing regimen of enterally administered branaplam in patients with Type 1 SMA. Methods: Methods: Safety and efficacy data were collected following completion of the initial 13-week treatment period for as long as the patients remained in the study. Treatment extensions and dose escalation were also allowed after the initial 13-week treatment period. This report focuses on respiratory support and feeding; updated safety information will also be presented. Results: Results: 14 patients enrolled in the trial. One patient failed screening. 13 patients were treated with branaplam starting at 2.2 to 7.6 months of age. All continued into the treatment extension period. Five patients died during the course of the trial from disease progression as assessed by the investigators and the Data Monitoring Committee. No patients withdrew from treatment for reasons other than death. Eight patients continue on branaplam with current ages of 27.5 to 34 months and they have been followed for between 24 and 31 months. No MTD has been reached; the emerging safety profile of branaplam shows adverse events that are mild, reversible and manageable. The most commonly reported events include pyrexia, pneumonia, constipation, vomiting, and diarrhea. Pulmonary events were the most frequent cause of hospital admissions. No dose dependent toxicities were observed across all the dose groups. As of the most recent evaluation on February 5, 2018, four (50%) surviving patients required no ventilatory support and four (50%) patients required some ventilatory support (Figure). Transition to oral administration from initial enteral tube administration has been successful; five (62.5%) patients are exclusively orally fed and three (37.5%) patients are managed with a combination of oral and tube feeding. Four of the 5 patients fed only by mouth also required no ventilatory support. Conclusion: Conclusions: Results from this ongoing open-label, first-in-human trial of branaplam in SMA Type 1 demonstrate good safety and tolerability. Over long-term follow-up, 50% of patients required no ventilatory support and 62.5% were exclusively orally fed. Figure
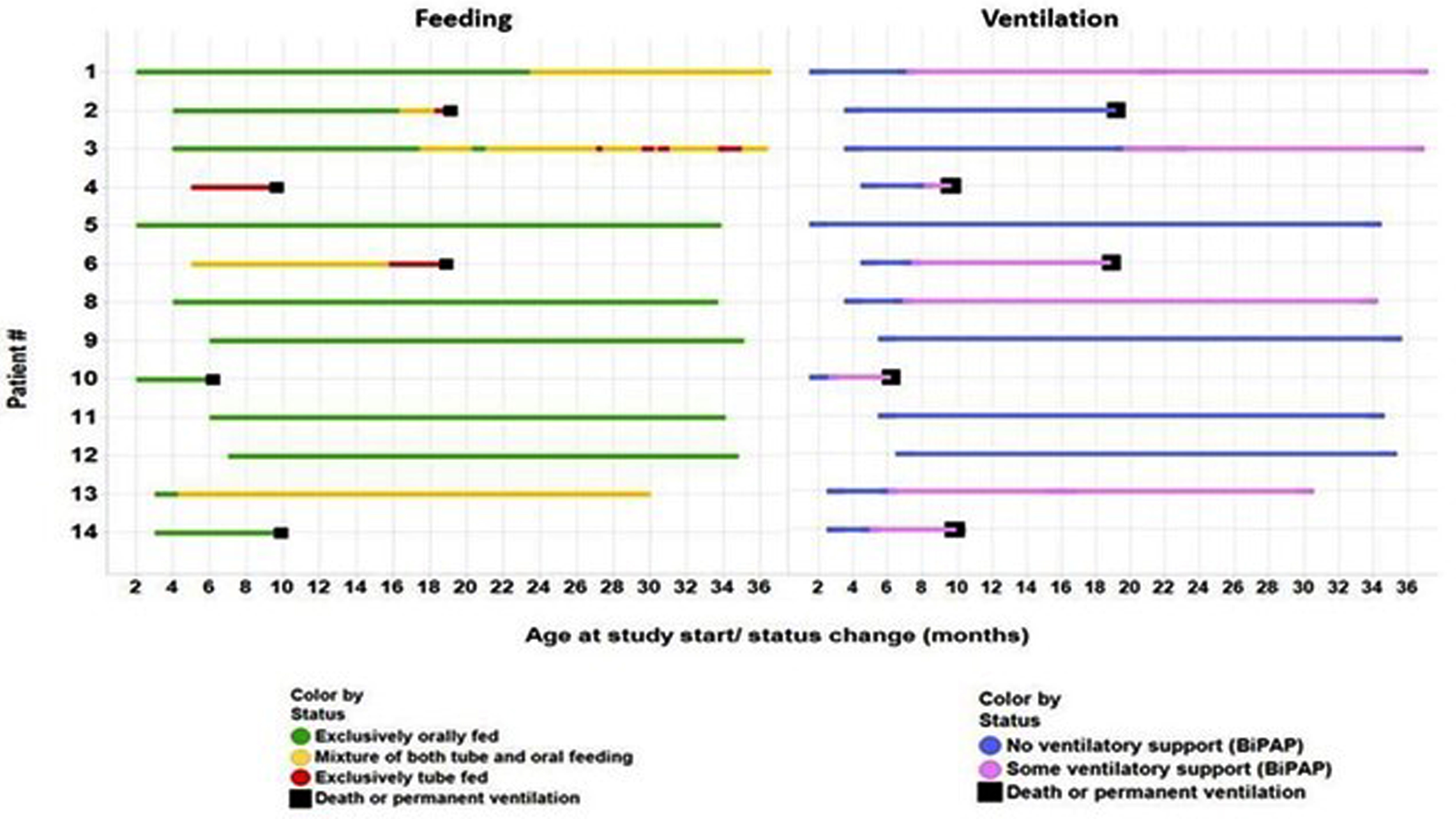
PS3Group4-009 / #502NUSINERSEN TREATMENT IN LONGSTANDING ADULT SMA TYPE 3 PATIENTS
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Anne J. Stauber1, Natalie Hackel1, Eva Greckl1, Benedikt Schoser2, Maggie C. Walter1
1Dept. Of Neurology, Ludwig-maximilians-university, Friedrich-Baur-Institute, Munich, DE;2Friedrich-baur Institute, Departement Of Neurology, Ludwigs-Maximilians University of Munich, Munich, DE
Background: Spinal muscular atrophy (SMA) is a progressive autosomal recessive motor neuron disease caused by loss of the survival motor neuron 1 gene (SMN1), and is characterized by loss of motor neurons in the spinal cord and lower brain stem, resulting in severe and progressive muscular atrophy and weakness. SMA is clinically heterogeneous and has been categorized into four types (0-3) based on age of onset, severity of motor decline and life expectancy. With greater understanding of the molecular basis of SMA, a major focus of therapeutic developments has been on increasing the full-length SMN protein by increasing the inclusion of exon 7 in SMN2 transcripts, enhancing SMN2 gene expression, stabilizing the SMN protein or replacing the SMN1 gene. In June 2017, the antisense oligonucleotide Nusinersen has been approved as first treatment for all types of SMA by the EU, based on results from two pivotal multicenter, controlled studies. Nusinersen alters the splicing of SMN2 pre-mRNA in order to increase production of full-length SMN protein. Methods: 12 adult patients with SMA type 3 between 18 and 54 years of age and a disease duration ranging from 6 to 40 years were treated with intrathecal loading doses at day 1, 14, 28 and 63, followed by maintenance dose every 4 months. None of the patients suffered from severe scoliosis or contractures. Three Patients were wheelchair-dependent at baseline. Patients were monitored within the SMArtCARE initiative for disease progression, side effects and treatment efficacy by clinical examination, monitoring of respiratory function, assessment of the Functional Rating Scale (FRS-SMA), 6-Minute-Walk-Test (6-MWT), Revised Upper Limb Module (RULM) and Hammersmith Functional Rating Scale-Expanded (HFSME). Results: Intrathecal administration was well tolerated in general; in some patients, back pain and headache was noted. Laboratory tests did not reveal nephrotoxicity. At visit 4 (day 63), a mild clinical benefit in form of improved endurance, reduced frequency of falls and better motor function was reported by the patients. Conclusion: Although the SMA research field is rapidly expanding with all the new therapeutic opportunities, there are still several questions that remain unsolved. The timing for optimal intervention for all these approaches is not clear in the human, and in particular at which point there is irreversible pathology that precludes any meaningful therapeutic response. Early diagnosis will be crucial for therapeutic success. However, treatment response should not be based on improvement of motor function alone, but focus on preventing disease progression. The clinical benefit in patients with long-standing disease will have to be further evaluated regarding efficacy and side effects in the years to come.
PS3Group4-010 / #770DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS OF SPINAL MUSCULAR ATROPHY
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Bakhytkul Myrzaliyeva1, Marzhan Lepessova2
1Medical Faculty, International Kazakh-Turkish University, Turkistan, KZ;2Child Neurology And Medical Genetics, Kazakh Medical University of Continuing Education, Almaty, KZ
Background: Introduction: Spinal Muscular Atrophy (SMA) - a heterogeneous group of hereditary diseases that occur with lesion / loss of motor neurons of the anterior horns of the spinal medulla and are characterized by the syndromic complex of the “floppy infant”. SMA is one of the common causes of child mortality caused by hereditary diseases. Children’s forms of the disease have a progressive nature and form an early disability. Methods: Patients and methods: clinical case of the syndrome “floppy infant” in the boy 9 months. Results: Results: complaints of weakness in the legs and hands, a delay in motor development: poorly poises the head, does not turn over, does not sit. Symptoms appeared after 3 months of life: poor sucking, poor weight gain, muscle weakness. Burdened obstetric mother anamnesis: antenatal fetal death in the first pregnancy at week 24, undeveloped 2 pregnancy; this pregnancy against a background of toxicosis, the threat of interruption and thrombophilia. Clinical data: fasciculation of the tongue, diffuse muscle hypotension, more pronounced in the hands, a dough-like consistency of muscles, muscle strength reduced to 1 point, areflexia, decreased abdominal reflexes, “frog” posture, chest deformation, beginning contractures in the elbow joints. Delay in motor development, psycho-emotional development does not suffer. Diagnostic findings: the level of Creatinephosphokinase is within normal limits. Genetic diagnosis: deletion of exons 7-8 of the SMN gene in the homozygous state is not detected; analysis of the number of copies of SMN: 2 copies of the SMN1 gene, 2 copies of the SMN2 gene. Analysis of the carrier of a married couple - parents are not carriers of deletions of exons 7-8 of the SMN gene. EMG: Neuronal type - signs of lesion of the anterior horns of the spinal medulla. Conclusion: Conclusion: for genetic confirmation of the diagnosis of spinal muscular atrophy, it is necessary to perform SMN sequencing in search of a point mutation. In the absence of a mutation, the diagnosis of proximal SMA is not confirmed. It is necessary to exclude SMA, not related to SMN-gene, distal forms, X-linked SMA, SMARD.
PS3Group4-011 / #933A CASE OF HEXANUCLEOTIDE REPEAT EXPANSION CAUSING AMYOTROPHIC LATERAL SCLEROSIS
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Priya D. Shanmugarajah, Judy O.S. Archer
Neurology, Pinderfields General Hospital, Wakefield, GB
Background: Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative condition involving large motor neurone of the brain and spinal cord, with an incidence of 1 in 100,000. Majority of ALS cases are sporadic, however autosomal dominant inheritance are seen in 5-10% of cases, with C9orf72 being the most common. This hexanucleotide repeat expansion of GGGGCC (G4C2) is commonly associated with bulbar onset ALS and behavioural variant frontotemporal dementia (FTD). We describe a case of probable motor neurone disease(MND) with positive gene test for C9orf72. Methods: 69 year old right handed man presented with a year history of progressive swallowing difficulties, slurred speech, weight loss and weakness of his limbs particularly his right arm. He also noticed muscle wasting and twitching. He denied any sensory symptoms, sphincter disturbance, personality change or psychiatric symptoms in the form of hallucination or delusions. He has poorly controlled diabetes, fibromyalgia and previous shoulder fracture. His mother died from MND and his maternal aunt was affected with parkinsonism. On examination, he had wasting of the intrinsic small muscle of the hand with flattening of the thenar eminence and first dorsal interosseous, particularly affecting the right more than the left hand. Pectoralis major, thoracic paraspinal muscles and triceps were wasted with fasciculation. Deep tendon reflexes were easily obtainable. He had dysarthria and tongue fasciculation with normal jaw jerk. Sensory examination were normal with flexor plantar response. Results: His blood test was normal for thyroid function test, vitamin D, B12 and folate. Autoimmune screening bloods were negative for myelin associated glycoprotein, ganglioside antibodies, protein electrophoresis, ANA and ANCA. His HbA1c was elevated at 75 mmol/mol. MRI brain scan showed mild cortical and cerebellar atrophy. MRI spine showed degenerative changes affecting the cervical spine. Neurophysiology showed chronic neurogenic changes on needle EMG affecting muscles from the cervical, thoracic and lumbosacral region however active denervation was only seen in thoracic paraspinal muscles and right upper limb giving the diagnosis of probable motor neurone disease (MND). Given the family history of MND and parkinsonism, genetic testing was done which showed positive C9orf72 hexanucleotide repeat expansion confirming the diagnosis of hereditary ALS Conclusion: C9orf72 carriers are commonly associated with bulbar onset ALS and behavioural variant FTD. This heterogeneous hexanucleotide expansion of G4C2 is not restricted to this phenotype as it’s also implicated in parkinsonism in ALS, corticobasal syndrome, Huntington-like disorder and primary lateral sclerosis. Our patient with a family history of MND and parkinsonism, presented with bulbar onset ALS without features of FTD. Neurophysiology supported a diagnosis of probable MND with 2 regions depicting active and chronic denervation changes. Awaji-shima criteria have 60.7% sensitivity in reaching a diagnosis of MND compared to the revised El-Escorial criteria of 28%. Both criteria had specificity of 95.9%. A definite diagnosis of hereditary ALS was achieved in this patient via genetic testing. Clinicians should have a low threshold for genetic testing in ALS as it has pertinent implications for patient’s progeny as well as decoding the molecular mechanism of MND, allowing enhanced disease modelling to identify therapeutic targets
PS3Group4-012 / #426A DE NOVO HETEROZYGOUS POINT MUTATION IN THE DYNC1H1 GENE CAUSING SPINAL MUSCULAR ATROPHY WITH LOWER EXTREMITY DOMINANT
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Kabelo J. Thusang1, Huiyuan Jiang2
1Neurology, Children’s Hospital of Michigan, Wayne State University Scholl of Medicine, Detroit, MI, US;2Neurology, Children’s Hospital of Michigan, Wayne State University scholl of medicine, Detroit, MI, US
Background: Several mutations of DYNC1H1 gene have been identified, which have been associated with variable neurological and neurodegenerative phenotypes including autosomal dominant spinal muscular atrophy with lower extremity dominant (SMA-LED), intellectual disability, Charcot-Marie-Tooth (CMT) disease, brain malformation and cataract. We have one patient who was found to have a novel mutation of DYNC1H1. Methods: We evaluated a 5-year-old boy who presented for evaluation of global developmental delay and hypotonia at the age 3 years. Electromyography (EMG), muscle biopsy and whole exome sequencing (WES) were performed which led to the diagnosis. Results: The patient presented with hypotonia and weakness of bilateral lower extremity and mild cognitive disability. He later developed fatigability with chewing and dysphagia. His work up showed normal chromosomal microarray, normal Fragile X DNA testing and normal serum creatine kinase levels. He had a normal nerve conduction studies and electromyogram as well as normal magnetic resonance imaging of the brain. Muscle biopsy demonstrated severely atrophic muscles with grouping and panfascicular atrophy and increased esterase staining in atrophic fibers suggestive of advanced neurogenic atrophy. He had a normal SMN1 gene test. WES revealed a de novo, heterozygous point mutation (p.L5681I) in the DYNC1H1 gene. The parents have no such mutation. Conclusion: The mutations in the DYNC1H1 gene have been associated with variable neurological and neurodegenerative phenotypes. We presented a novel de novo heterozygous mutation (p.L5681I) in the DYNC1H1 gene causing SMA-LED. This patient developed bulbar symptoms, which is also unique. It will add onto the spectrum of dyneinopathy. EMG may not be a sensitive test for the early stage of the disease. WES is a confirmative test.
PS3Group4-013 / #460SMARD1: A RARE CAUSE OF HYPOTONIA AND RESPIRATORY FAILURE IN INFANCY
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Cristina Garrido1, Jorge Oliveira2, Ana Sousa3, Marcio Cardoso4, Rosario Santos5, Manuela Santos6
1Neuropediatria, Centro Hospitalar Porto, 011, PT;2Genetic Service, Centro hospitalar Porto, -, PT;3Neurofisiology Service, Centro Hospitalar Porto, -, PT;4Centro Hospitalar Porto, -, PT;5Genetic Service, Centro Hospitalar Porto, -, PT;6Neuropediatrics, Centro Hospitalar Porto, -, PT
Background: Spinal muscular atrophy (SMA) with respiratory distress type 1 (SMARD1) is a rare genetic condition caused by bi-allelic defects in the immunoglobulin MU-binding protein 2 (IGHMBP2) gene, leading to irreversible degeneration of motor neurons in the anterior horns and in spinal root ganglia. In contrast to the SMA associated with 5q13.2 locus, the sensory and autonomic nervous system can also be affected in SMARD1, manifesting as a distal predominant muscular weakness with chief phrenic nerve involvement causing diaphragmatic paralysis and early respiratory distress. Previous genotype-phenotype correlations suggest that the amount of functional protein being produced from the mutated gene may modulate the SMARD1 phenotype. The combination of different pathogenic variants in IGHMBP2 gene can also give rise to an axonal Charcot-Marie-Tooth disease type 2S (CMT2S), another less severe neurologic phenotype. Methods: Review of SMARD cases of a tertiary centre in the last 10 years. Results: Clinical cases Patient P1 presented as a 2-month-old male baby with apneic events and cyanotic spells which evolved to a progressive respiratory insufficiency. On physical examination at that time, he had a weak cry and lower limb distal weakness. Severe axonal motor and sensory neuropathy were documented on EMG. The genetic analysis of IGHMBP2 revealed two compound heterozygous pathogenic variants: c.958C>T (p.Arg320*) and c.1060+1G>T. The variant affecting the donor spice site was further characterized at mRNA level. Two different out-of-frame transcripts (due to exon-skipping or partial intronic retention events) were identified. Clinically, he developed a global progressive muscle weakness predominantly in lower limbs and after an anesthetic procedure he became dependent on ventilator support. Currently, he is 12 month-old and had gastrostomy for feeding and a tracheostomy for invasive ventilator support (24 hours per day). Patient P2 was a 9-month-old female baby admitted to Intensive care due to a respiratory infection needing ventilatory support (apneic events). At that time, she presented early motor delay and distal lower limb weakness, axial hypotonia, and hyporeflexia. In this patient two novel compound heterozygous pathogenic variants were also identified in IGHMBP2: c.1327C>T (p.Arg443Cys) and c.1757-1G>A. The variant affecting splicing was shown to disrupt the native site which leads to the use of an exonic cryptic splice-site in exon 13 (r.1757–1762del, p.Gly586–Glu587del). She died in the sequence of a respiratory infection at10 months of age. Conclusion: In our tertiary neuromuscular center only two cases of SMARD1 have been diagnosed in the last ten years confirming the rarity of this clinical entity. Electrophysiological investigation in one of the patients (P1), showed a severe axonal sensorimotor neuropathy consistent with the descriptions in literature. Respiratory distress resulting from irreversible phrenic denervation requires, as in our patient, lifelong mechanical ventilation. Unusual prolonged survival (>4 years) has been reported in some patients upon mechanical ventilation. SMARD1 should be considered in infants with a distal limb predominance weakness even before evolving to a respiratory distress.
PS3Group4-014 / #640NUSINERSEN EXPERIENCE IN INDIVIDUALS WITH SPINAL MUSCULAR ATROPHY TYPE III: A CASE SERIES
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Janbernd Kirschner1, Kathryn J. Swoboda2, Eugenio Mercuri3, Basil T. Darras4, Claudia A. Chiriboga5, Richard Foster6, Ishir Bhan7, Wildon Farwell7
1Klinik für Neuropädiatrie und Muskelerkrankungen, Freiburg, DE;2Massachusetts General Hospital, Boston, MA, US;3Università Cattolica del Sacro Cuore, Rome, IT;4Boston Children’s Hospital, Boston, MA, US;5Columbia University Medical Center, New York, NY, US;6Biogen, Maidenhead, GB;7Biogen, Cambridge, MA, US
Background: Spinal muscular atrophy (SMA) is a hereditary neuromuscular disease associated with progressive muscular weakness and loss of motor function caused by insufficient levels of survival motor neuron (SMN) protein. Treatment with nusinersen, an antisense oligonucleotide drug that modulates splicing of SMN2 pre-mRNA and promotes production of full-length SMN protein, has demonstrated significant and clinically meaningful benefits on motor function in symptomatic infants and children, and infants in the presymptomatic stage. Here, we review outcomes in children, adolescents, and young adults with SMA Type III who were treated with nusinersen. Methods: This case series includes 11 participants with SMA Type III who received their first treatment with intrathecal nusinersen (3–12mg) in the Phase 1b/2a CS2 study (NCT01703988) (age at treatment initiation: <10 years, n=5; ≥10 years, n=6), then enrolled in CS12 (NCT02052791), an open-label, ~2-year extension study of nusinersen 12 mg administered intrathecally, and had the longest follow-up. For this analysis, we assessed changes in motor function from CS2 baseline to last visit of CS12 (Day 715, n=10; Day 624, n=1; last patient last visit, June 30, 2016) using the Hammersmith Functional Motor Scale–Expanded (HFMSE), Upper Limb Module (ULM), and 6-Minute Walk Test (6MWT). Clinically meaningful improvement in HFSME was defined as a 3-point score increase. Changes in quality of life (QOL) were assessed with the PedsQL Core and Neuromuscular (NM) modules. Results: At treatment initiation in CS2, median (range) age of participants was 10.9 (3.6–16.0) years and 8/11 were ambulatory. Four participants with CS2 baseline HFMSE scores of 51–59 points achieved clinically meaningful improvements of 3–7 points from baseline at CS12 last visit. HFMSE scores remained stable or improved slightly (0- to 2-point change) in 6 participants (CS2 baseline values: 37-62) and declined in 1 participant (CS2 baseline value: 31). In the 3 nonambulatory participants, ULM scores were 18 at every study visit. In the 8 ambulatory participants, distances walked on the 6MWT increased by 50–159 meters from CS2 baseline values of 45–550 meters to last CS12 visit. Caregiver/parent-reported PedsQL Core scores improved or remained stable from CS2 baseline values of 33.7–69.6 in 10 participants. Caregiver/parent-reported NM scores improved or remained stable from CS2 baseline values of 45–81 in 9 participants. Self-reported Core scores improved or remained stable from CS2 baseline values of 55.4–66.3 in 3 participants. Self-reported NM scores improved or remained stable from CS2 baseline values of 69–86 in 5 participants. Changes in self-reported scores were not available for 3 participants who were age <5 years at CS2 baseline. The most common adverse events (AEs) were those expected following lumbar puncture. The AE pattern in this cohort was consistent with clinical trial experience. Conclusion: In contrast to the expected decline in motor function over time based on natural history, in this cohort almost all children, adolescents, and young adults with SMA Type III who were treated with nusinersen showed clinically meaningful improvements or stabilization of disease and generally improved or stable QOL over time.
PS3Group4-015 / #752RG7916 SIGNIFICANTLY INCREASES SMN PROTEIN IN SMA TYPE 1 BABIES
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Giovanni Baranello1, Laurent Servais2, John Day3, Nicolas Deconinck4, Eugenio Mercuri5, Andrea Klein6, Basil Darras7, Riccardo Masson8, Heidemarie Kletzl9, Yumi Cleary10, Gillain Armstrong11, Timothy Seabrook9, Christian Czech10, Marianne Gerber10, Fung Lee9, Kristina Gelblin9, Stephane Nave10, Ksenija Gorni10, Omar Khwaja10
1Developmental Neurology Unit, Carlo Besta Neurological Research Institute Foundation, Milan, IT;2Institute of Myology, Paris, FR;3Department Of Neurology, Stanford University, Palo Alto, CA, US;4Queen Fabiola Children’s University Hospital, Université Libre de Bruxelles, Brussels, Belgium and Neuromuscular Reference Center UZ Ghent, Ghent, Belgium, Ghent, BE;5Paediatric Neurology And Nemo Center, Catholic University and Policlinico Gemelli, Rome, IT;6University Children’s Hospital Basel, Basel, Switzerland and Inselspital, Bern, Switzerland, Bern, CH;7Boston Children’s Hospital, Harvard Medical School, Boston, MA, US;8Developmental Neurology Unit, Carlo Besta Neurological Research Institute Foundation, Milan, IT;9Roche Pharmaceutical Research and Early Development, Roche Innovation Center, Basel, CH;10Roche Pharmaceutical Research and Early Development, Roche Innovation Center, Basel, CH;11Roche Products Ltd, Welwyn Garden City, GB
Background: Spinal muscular atrophy (SMA) is characterized by motor neuron loss and muscle atrophy, due to reduced levels of survival of motor neuron (SMN) protein from a loss of function of the SMN1 gene. While SMN1 produces full-length SMN protein, a second gene, SMN2, produces only low levels of functional SMN protein. RG7916 (RO7034067) is an investigational, orally administered, centrally and peripherally distributed small molecule that is being evaluated for its effect on modulating SMN2 pre-mRNA splicing towards the production of full-length SMN2 mRNA and increase of SMN protein. FIREFISH (NCT02913482) is an ongoing, multi-center, open-label, 2-part, seamless study of RG7916 in babies aged 1–7 months with Type 1 SMA and two SMN2 gene copies. Methods: We present interim data, including SMN protein data, from FIREFISH Part 1 (n=8–24), an exploratory study assessing the safety, tolerability, pharmacokinetics and pharmacodynamics of RG7916 at different dose levels. Confirmatory Part 2 (n=40) will assess safety and efficacy of RG7916, with a primary endpoint of the proportion of infants sitting without support for 5 seconds after 12 months. Results: In an interim analysis of FIREFISH Part 1, a dose-dependent increase in SMN protein levels in blood was observed, with an up to 6.5-fold increase versus baseline after 4 weeks of treatment at the highest dose of RG7916 (range 2.0–6.5). To date, no protocol-defined safety-related stopping rules have been met. While follow-up has been limited in time, none of the following events have been reported: loss of the ability to swallow, tracheostomy or need for permanent ventilation. Updated survival data will also be presented. Conclusion: The up to 6.5-fold increase in SMN protein observed in FIREFISH Part 1 is expected to be beneficial based on naturally occurring differences in SMN protein levels between SMA severity types (e.g., Type 2 vs. Type 1 with differences up to approximately 2-fold). The clinical effects will be assessed in Part 2. All doses explored so far have been well tolerated. The FIREFISH study is currently recruiting globally. Acknowledgments The authors would like to thank all individuals enrolled in the RG7916 studies, their families and the site staff involved. Study sponsored by F. Hoffmann-La Roche.
PS3Group4-016 / #391PATHOGENIC VARIANT OF THE REEP1 GENE IN A KOREAN FAMILY WITH AUTOSOMAL DOMINANT HEREDITARY SPASTIC PARAPLEGIA
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Dong Wook Namgung1, Young Chul Choi2, Hyung Jun Park3, Bun Chun Suh4
1Neurology, Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul, KR;2Neurology, Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul, KR;3Neurology, Gangneung Asan Hospital, University of Ulsan College of Medicine, Gangneung, KR;4Neurology, Kangbuk Samsung Hospital, Sungkyunkwan University School of Medicine, Seoul, KR
Background: Hereditary spastic paraplegia (HSP) is a heterogeneous group of genetic disorders characterized by progressive lower-limb spastic paralysis due to corticospinal tract degeneration. Three genes account for up to 50% of pathogenic variants in families with autosomal dominant HSP: the causative gene is SPAST in 40%; ATL1 in 10%, and REEP1 in 4.5 to 6%. However, pathogenic variants in REEP1 gene have not been reported in Korea.
Methods: To identity a pathogenic variant, whole exome sequencing was performed. Results: The proband in the HSP family (an 8-year-old boy; Fig. 1A, III-8) presented to our clinic with gait disturbance. To identity a pathogenic variant, whole exome sequencing was performed on the proband (III-8). He first noticed an unstable gait at an age of 5 years, and his gait disturbance had progressed slowly since then. When we examined him at the age of 8 years, he could still ambulate independently. A neurological examination demonstrated muscle weakness and brisk reflexes of the lower extremities. His ankle joints were affected by contracture. However, motor function of the upper extremities was normal, and he did not exhibit cognitive decline, sensory deficits, or bladder dysfunction. Magnetic resonance images of brain and spinal cord were normal. Electrophysiological studies were normal. The family history showed that HSP was inherited in an autosomal dominant manner (Fig. 1A). His mother (a 45-year-old woman, II-10) and oldest aunt (a 52-year-old woman, II-4) also showed spastic paraplegia and ankle contracture. His younger brother (a 5-year-old boy,III-10) did not complain of gait disturbance, but showed mild spasticity and hyperreflexia of the lower extremities. However, his younger sister (a 5-year-old girl, III-11) did not show any abnormal neurologic deficits. Whole exome sequencing was performed on the proband (III-8). After screening of HSP-related genes, we identified a heterozygous pathogenic variant (c.337C>T) (p.Arg113*) in REEP1 (NM–022912). This variant was previously reported as the pathogenic variant in complicated HSP presented with both spastic paraplegia and lower motor neuron signs including pes cavus and motor neuropathy. (Hewamad-duma, et al. Neurogenetics 2009). However, the present patients did not show any electrophysiological evidence of peripheral neuropathy. In addition, the rate of clinical disease progression was relatively slow, and two adults (II-4 and II-10) could still ambulate independently at an age of 52 and 45 years, respectively. Conclusion: This is the first report of the pathogenic variant of the REEP1 gene in a Korean family with an autosomal dominant HSP using whole exome sequencing.
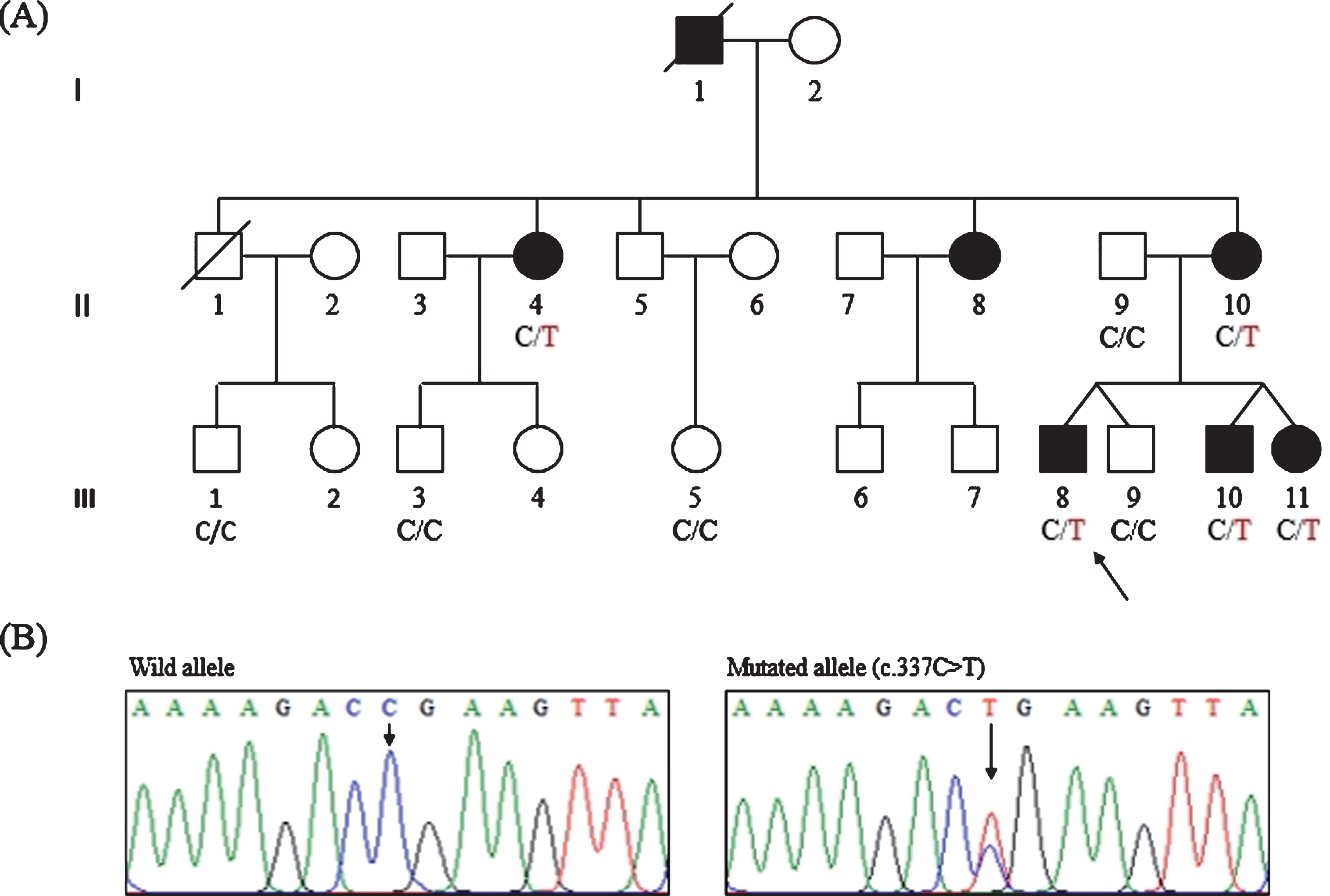
PS3Group4-017 / #626NEUROFILAMENT LIGHT CHAIN AS A POTENTIAL BIOMARKER IN SPINAL MUSCULAR ATROPHY
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Thomas Peters1, Hervé Jullien De Pommerol2, Diethilde Theil3, Marie-Anne Valentin3, Agnieszka Kieloch3, David Leppert2, Emilie Voltz3
1Translational Medicine / Biomarker Development, Novartis Institutes for BioMedical Research, Basel, CH;2Novartis Pharma AG, Basel, CH;3Novartis Institutes for BioMedical Research, Basel, CH
Background: Biomarker assessments in patients with degenerative neurologic diseases have demonstrated that neurofilament light chain (NfL) is a sensitive, blood-based, exploratory marker that can identify patients, aid in prediction of long-term outcomes, and be used for assessing effects of treatment. At present, little is known about blood NfL levels in patients with spinal muscular atrophy (SMA). Methods: Blood samples for NfL assessment were obtained from 30 healthy adult volunteers (24-55 years old), 21 healthy children (1-4 years of age), and 18 healthy children (8-10 years old). Samples were also obtained from 18 patients with SMA Type III and 2 with SMA Type II (6 males and 14 females, 21-66 years of age), and from 12 patients with SMA Type I (2.2-7.6 months of age) enrolled in an open-label, multi-part, first-in-human, proof of concept study of branaplam, a small-molecule RNA splicing modulator. Serum NfL levels were measured using the ultra-sensitive Simoa HD-I Analyzer. The lower limit of quantification was 3.38 pg/mL and the upper limit was 1128 pg/mL. Inter-run accuracy was 98.5% to 115.2% and inter-run precision was 7.0% to 16.1%. Results: The geometric mean serum level of NfL in healthy adults was 13.4 pg/mL (reference range = 4.6 – 38.9 pg/mL), that for healthy pediatric subjects 8-10 years old was 2.5 pg/mL (measured range = 1.7-6.9 pg/mL), and that for healthy pediatric subjects 1-4 years old was 4.0 pg/mL (measured range = 1.7-22.9 pg/mL). Adults with SMA Type II or III had lower NfL levels (7.2 pg/mL, range = 3.9-23.2 pg/mL) vs healthy adults, but there was substantial overlap in measurements for the two groups. The mean pre-treatment NfL level in pediatric patients with SMA Type I was 443 pg/mL (range = 177-983 pg/mL) and did not overlap with values from healthy pediatric subjects (Figure). There also appeared to be an inverse correlation between pre-treatment NfL levels and CHOP INTEND scores for the patients with SMA Type I. Conclusion: Blood NfL levels in adult SMA Type II/III patients seem to be within the normal reference range while those for pediatric SMA Type I patients are higher than those for healthy pediatric subjects. There is also a trend for an inverse relationship between pre-treatment blood NfL levels and CHOP INTEND scores. These conclusions are limited by the small number of samples evaluated and the lack of data in an age-matched healthy children. The dynamic course of blood NfL levels in SMA requires further study.
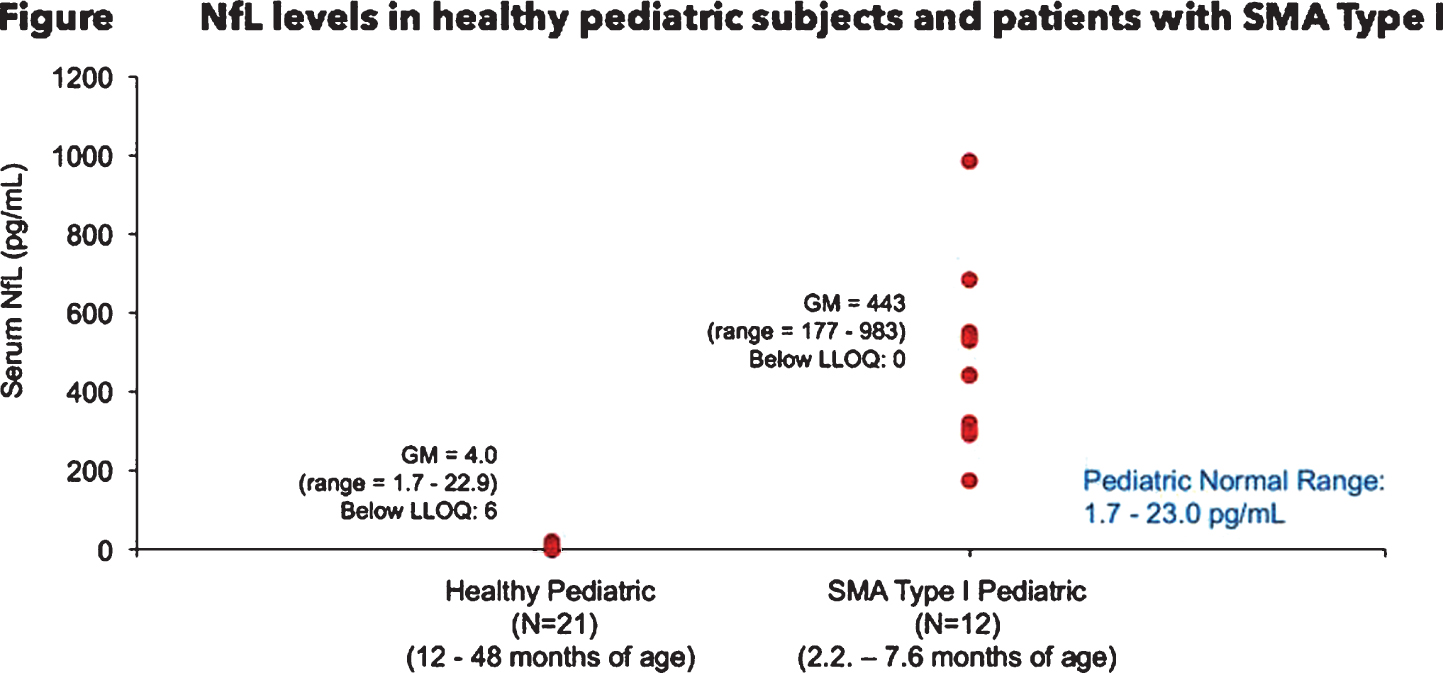
PS3Group4-018 / #337ENHANCEMENT OF BULBAR FUNCTION IN ALS: LESSONS LEARNED FROM THE NUEDEXTA TREATMENT TRIAL
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Richard Smith1, Kathleen Myers1, Eric Macklin2
1Center for Neurologic Study, La Jolla, CA, US;2Massachusetts General Hospital, Neurological Clinical Research Institute and Biostatistics Center, Boston, MA, US
Background: Following up anecdotal reports, a multi-centered placebo-controlled treatment trial was conducted to determine if Nuedexta could enhance bulbar function in ALS patients. The drug, a combination product containing dextromethorphan (DM) and quinidine, was originally approved for the treatment of emotional lability. The effect on emotionality was thought to be mediated by the action of DM on sigma-1 receptors which have recently been reported to decorate motor neurons in the brain stem. Methods: Whereas most trials in ALS are conducted over long intervals, we designed a 70-day study, placing patients on either drug or placebo for 30 days followed by a washout period and subsequent placement in the remaining limb of the trial. Another unique aspect of the study was the use of a recently validated self-report scale, the Center for Neurologic Study Bulbar Function Scale (CNS-BFS), as the primary endpoint. Secondary endpoints in the study included traditional measures, such as the ALSFRS-R, its bulbar subscale, and timed speech and swallowing. Results: The CNS-BFS proved to be a robust endpoint. Bulbar functions with Nuedexta treatment were significantly enhanced, with p-values for the overall scale as well as its speech, swallowing, and salivation subdomains ranging from 0.0003-0.009. All other measures assessing bulbar function except for the bulbar component of the ALSFRS-R failed to reach statistical significance (see Table 1). The effect of Nuedexta is dramatically illustrated in Figure 1, which compares the two treatment periods (placebo vs. drug). Conclusion: While speech, swallowing and salivation responded to drug treatment, no effects were seen on other motor functions such as breathing or walking, implying that a useful therapy may only target a subpopulation of motor neurons. Additional studies need to be undertaken to understand the duration of this treatment effect and to discern whether Nuedexta has an effect on survival in ALS patients.
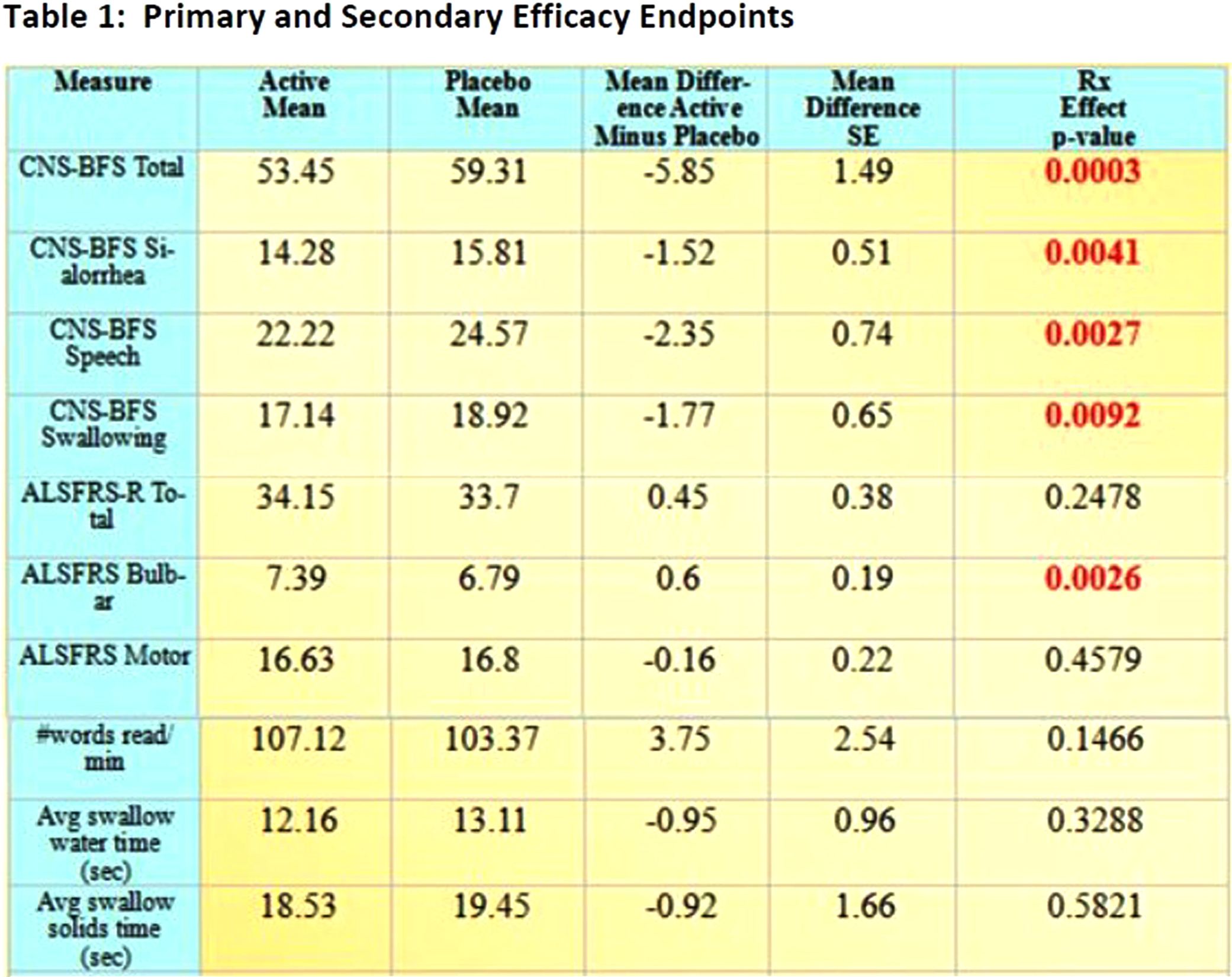
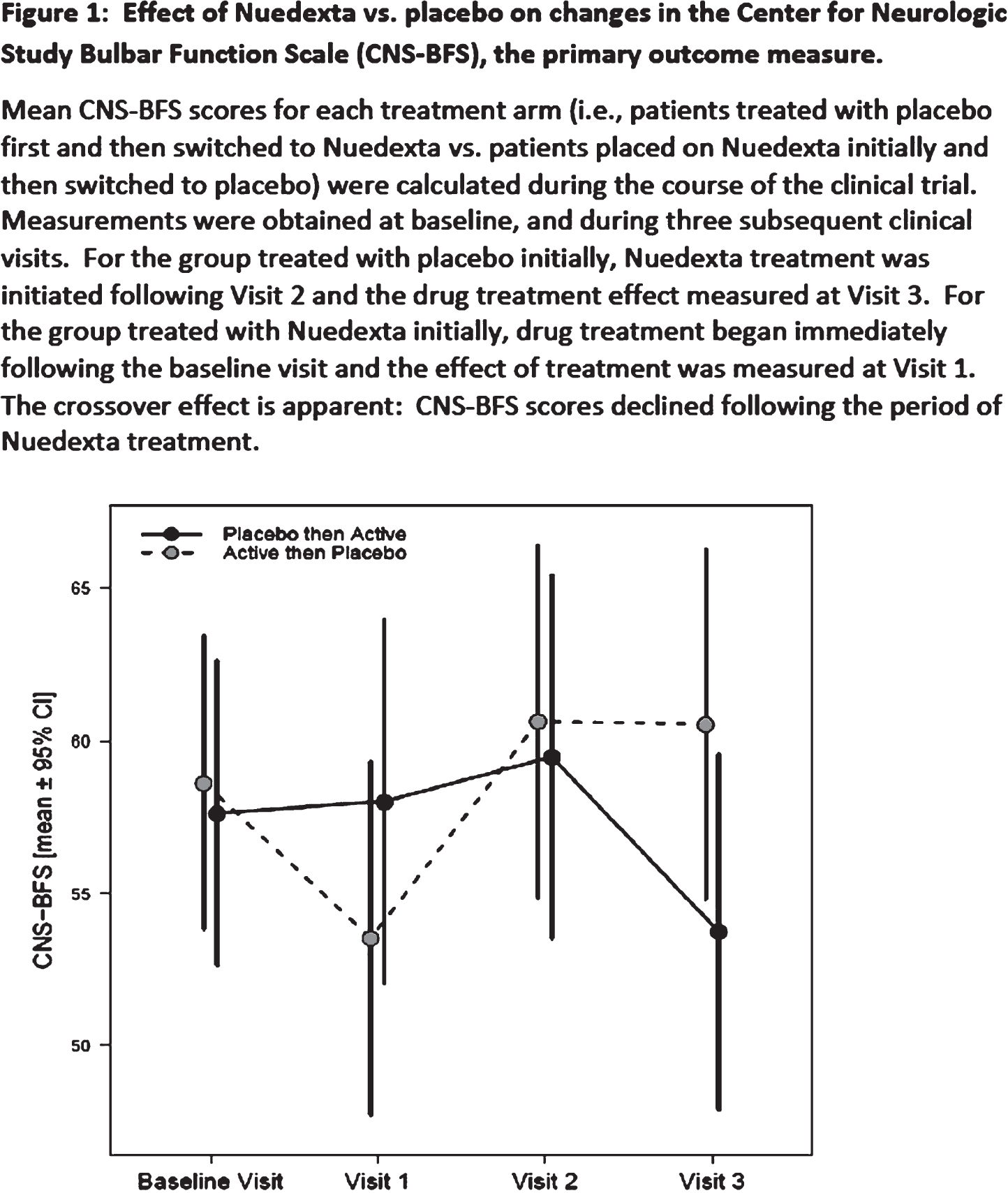
PS3Group4-019 / #533SALBUTAMOL TREATMENT IN TYPE 2 SMA PATIENTS: 18 MONTHS ASSESSMENT
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Anna Lia Frongia1, Daniel Natera De Benito2, Macarena M.A. Alarcon-Cornejo1, Laura Carrera-García3, Ariadna Borras1, Nuria Padros1, Obdulia Moya1, Judit Armas1, Julita Medina1, Meritxel Vigo4, Cristina Jou1, Cecilia Jimenez Mallebrera1, Carlos Ortez1, Jaume Colomer1, Andres Nascimento1
1Neuromuscular Disorders Unit, Hospital Sant Joan de Déu, Barcelona, ES;2Neuromuscular Disorders Unit, Hospital Sant Joan de Déu, Barcelona, ES;3Unidad De Patología Neuromuscular., Hospital Sant Joan de Déu, Barcelona, ES;4Hospital Sant Joan de Déu, Barcelona, ES
Background: Spinal muscular atrophy (SMA) is an autosomal recessive disease characterized by motor neuron degeneration, resulting in several major phenotypes defined by age at disease onset and maximal motor capacity. Patients with SMA type 2 became symptomatic between 6 and 12 months, typically they achieve the ability to sit without support, but not independent walk. Data on natural history showed a slow decline in motor milestones over time and life expectancy is shortened by early onset scoliosis, joint contractures, and restrictive lung disease. Different studies have demonstrated that salbutamol may have a beneficial effect on muscle function in patients with SMA type 2 and 3. Methods: To describe the response to salbutamol in SMA type 2 patients motor function assessment was performed before starting treatment and every 6 months. Hammersmith Functional Motor Scale Expanded (HFMSE), Revised Upper Limb Module (RULM), and Egen Klassifikation (EK2) were used. Motor scales were longitudinally evaluated over an 18-month period. Results: Fourty eight children with a mean age of 10 years (range: 2-21 years) took oral salbutamol during a period of at least 6 month. Children < 6 years presented a functional improvement during the first 6 months of treatment (HFMSE +4,29) that reduced in intensity after 12 months (HFMSE +1,14). However, amelioration in upper extremity tended to stabilize (RULM +3,67). Patients > 6 years showed a slow decrease in motor function scales similar to that described in SMA natural history. 77% of families (92% <6 years and 71% in >6years) referred a subjective positive effect. Conclusion: Salbutamol treatment may be effective in AME 2 patients, particularly during the first months of therapy, and in younger than 6 years. An apparent higher effect on upper limb is observed. Absence of a control group is the main limitation in our study.
PS3Group4-020 / #510VERY LATE-ONSET AMYOTROPHIC LATERAL SCLEROSIS IN A PORTUGUESE COHORT: WHICH DIFFERENCES?
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Miguel Oliveira Santos1, Marta Gromicho2, Susana Pinto2, Mamede De Carvalho1
11. Institute of Physiology Unit, Instituto de Medicina Molecular, Faculty of Medicine, University of Lisbon, Portugal. 2. Department of Neurosciences and Mental Health, Hospital de Santa Maria, Centro Hospitalar Lisboa Norte, Lisbon, Portugal., Lisbon, PT;2Institute of Physiology Unit, Instituto de Medicina Molecular, Faculty of Medicine, University of Lisbon, Portugal., Lisbon, PT
Background: Amyotrophic lateral sclerosis (ALS) is a rapidly progressive and fatal motor neuron disorder characterized by degenerative changes in upper and lower motor neurons. Although its incidence has been stable among Western countries, population-ageing effect will probably increase the proportion of very-old ALS patients. On the other hand, this particular population has been excluded from clinical trials. Therefore, its characterization is sparse. We aim to raise awareness of demographic and clinical profile of oldest ALS patients, including their survival. Methods: A retrospective study was performed, including 1162 ALS patients followed longitudinally at our ALS unit from January 1995 to December 2017. The patients were divided in two groups, according to whether disease onset was before or after 80 years. Demographic, clinical and survival data were compared between groups. ALS diagnosis was established based on the revised El Escorial criteria and supported by the Awaji criteria since 2008. Frontotemporal dementia (FTD) clinical diagnosis was established by personality and social behaviour changes, lack of insight for the on going clinical condition, with preserved memory, visuospatial abilities and praxis. Comparison between the very-old and younger patients was performed using chi-square and t-student tests, as appropriate. Survival analysis was done by Kaplan-Meier log-rank test and the multivariate Cox proportional hazards model. A p-value of <0.05 was considered statistically significant. Results: 54 of 1162 patients (4.65%) were aged 80 or over. From those, 29 (53.70%) were men and the mean age of onset was 83 years (SD±2.63). Contrasting with the younger group, bulbar onset was remarkably the most common presentation form in the oldest patients (55.56%, p<0.001), but with no gender preference (p=0.54). Clinical criteria for FTD were met in 4 (7.41%, p=0.36) and only 1 (1.85%, p=0.10) had a positive family history. All patients were taking riluzol, and non-invasive ventilation was started in 28 (51.85%, p=0.11). Significantly shorter mean disease duration before first visit was disclosed in the very-old group (13.38±9.24 vs. 19.98±25.0 months, p<0.001). Survival was shorter in the oldest group (28.8±23.5 vs. 44.1±41.9 months, p<0.001), which was significantly dependent on disease duration and onset form, both of each showed no interaction impacting on their survival. Conclusion: Very-old patients represented a minor but distinctive ALS group. A predominant bulbar presentation (with no gender dependence) was disclosed, and it could probably explain the shorter disease duration before first visit. Despite being oldest, it was not an exclusion factor for good health care practises, in particular multidisciplinary treatment, non-invasive ventilation and riluzol prescription.
PS3Group4-021 / #773ATAXIN-2 IN RUSSIAN ALS PATIENTS ATAXIN-2 IN RUSSIAN ALS PATIENTS
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Vera S. Demeshonok1, Tatiana Alekseeva2, Vladimir Lapin3, Vladimir Nazarov3
1Neurology, North-Western State Medical University named after I.I. Mechnikov, Russia, RU;2V.A.Almazov NMRC, Санкт-Петербург, RU;3Laboratory Of Autoimmune Diagnostics, Center For Molecular Medicine, First St. Petersburg I. Pavlov State Medical University, St-Petersburg, RU
Background: Amyotrophic lateral sclerosis is a fatal progressive neurodegenerative disorder, which is characterized by the loss of motor function, including bulbar and spinal involvement. Mutations in SOD1 and C9orf72 are the most common genetic causes of the familial and sporadic ALS and it is more often noticed in patients with the early onset of the disease. The polyQ intermediate expansions in ataxin-2 is also increased risk for ALS. Methods: We investigated the polyQ expansions in ataxin-2 in 70 patients (33 males; 37 females) with sporadic ALS and 40 unrelated controls without history of neurodegenerative disorders from St-Petersburg (Russia). There are 11 patients with early onset of symptoms (up to 46 years old), and they were tested for the presence of mutations in SOD1 and C9orf72 genes. The patients were hospitalized in Neurology Departments of V.A. Almazov NMRC or NWSMU from May to November 2017.All patients were examined and included according to El Escorial (2015). Results: We did not detect mutations in SOD1 and C9orf72 genes in the examined patients with early onset of the disease. We defined higher intermediate PolyQ repeat length expansions (27-32) for ataxin-2 in 6 patients (8,57%): 2 men with a spinal onset and 4 women (2 with a spinal and 2 with a bulbar onset of the disease). It should be noted that all 6 patients were characterized by slow progression on a ALSFRS-R Scale. PolyQ intermediate expansions were not determined in none of the control group. Conclusion: Ataxin-2 PolyQ intermediate expansions are associated with ALS in Russia.
PS3Group4-022 / #824AVXS-101 PHASE 1 GENE THERAPY CLINICAL TRIAL IN SMA TYPE 1: EVENT-FREE SURVIVAL AND ACHIEVEMENT OF DEVELOPMENTAL MILESTONES
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Jerry R. Mendell1, Samiah Al-Zaidy1, Richard Shell1, W. D. Arnold2, Louise Rodino-Klapac1, Thomas W. Prior3, Linda Lowes1, Lindsay Alfano1, Katherine Berry1, Kathleen Church1, John T. Kissel4, Sukumar Nagendran5, James L’Italien5, Douglas M. Sproule5, Courtney Wells5, Arthur H.m. Burghes2, Kevin Foust5, Kathrin Meyer1, Shibi Likhite1, Brian Kaspar1
1Center for Gene Therapy Nationwide Children’s Hospital, Columbus, OH, US;2Department of Neurology, Ohio State University, Columbus, OH, US;3Department of Pathology, Ohio State University, Columbus, OH, US;4Department of Pediatrics, Ohio State University, Columbus, OH, US;5-, Bannockburn, IL, US
Background: Spinal muscular atrophy (SMA) is a devastating, monogenic neurodegenerative disease. Children with its most severe form, SMA Type 1 (SMA1), will never sit unassisted or maintain head control. A natural history study of SMA1 reported that none achieved an Infant Test of Neuromuscular Disorders (CHOP-INTEND) score of >40 by 6 months of age (1 transient exception) and 92% died or required permanent ventilation by 20 months. This trial explores safety and efficacy of a single intravenous administration of gene replacement therapy in SMA1. AVXS-101 delivers the survival of motor neuron (SMN) gene in a single intravenous dose via the adeno-associated virus (AAV) 9 viral vector, which crosses the blood–brain barrier. Methods: In this phase 1 trial, 15 patients with SMA1 confirmed by genetic testing (with 2xSMN2 copies) were enrolled. Patients received an intravenous dose of AVXS-101 at the low dose (Cohort 1, n=3) or proposed therapeutic dose (Cohort 2, n=12). The primary objective was safety and secondary objectives included survival (avoidance of death/permanent ventilation) and ability to sit unassisted (video confirmed by external independent reviewer). CHOP-INTEND scores and other motor milestones were additional objectives. Results: AVXS-101 appeared to have a favorable safety profile and to improve survival (as of August 7, 2017). All 15 patients were alive and event-free at 20 months of age and did not require permanent mechanical ventilation. Patients in Cohort 2 demonstrated improvements in motor function: 11/12 had achieved CHOP-INTEND scores >40 points and a mean increase of 24.6 points from a mean baseline of 28.2 points; 11/12 were able to sit unassisted for at least 5 seconds, 10 for at least 10 seconds, and 9 for at least 30 seconds; 11/12 achieved head control and 9 could roll over. Two patients were able to crawl, pull to stand, stand independently, and walk independently. Asymptomatic elevated serum aminotransferase levels occurred in 4 patients and were attenuated by prednisolone. Conclusion: In contrast with the natural history, a one-time intravenous administration of AVXS-101 appeared to demonstrate a positive impact on the survival of both cohorts and a dramatic, sustained impact on motor function of Cohort 2: 11/12 patients achieved CHOP-INTEND scores and motor milestones rarely or never seen in this population. No waning of effect or clinical regression in motor function was reported up to the August 7, 2017 data cut. A clinical update of the 24-month safety follow-up will be given at the time of presentation.
PS3Group4-023 / #714AN AUTOMATED ANALYSIS OF REPEATER F-WAVES IN ALS
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Dimitra Veltsista1, Elisabeth Chroni2
1Neurology, University Hospital of Patras, Patras, GR;2Neurology, University of Patras, Patras, GR
Background: An increased frequency of repeater F-waves (Freps) has been demonstrated in neurogenic abnormalities such as amyotrophic lateral sclerosis (ALS). Analysis of repeater F-waves is rarely performed as part of routine studies, primarily because of difficulty in identifying them by simple visual inspection. The purpose of this study is to present the results of a computerized analysis of Freps and explore its clinical usefulness in ALS. Methods: Forty consecutive F-waves were recorded following supramaximal stimulation of the ulnar and peroneal nerve in 52 patients with ALS (33 males, mean age 64.1 ± 10.2 years, mean height 172.3±8.1cm) and 52, age- and height-matched, healthy subjects (33 males, mean age 63.8 ± 11.9 years, mean height 172.5±8.1cm). ALS patients were further divided into 3 groups based on the location of their initial symptoms (upper limbs, lower limbs, bulbar). The F-waves were analyzed using an automated computerized system which identifies Freps and groups them (Figure). Parameters of Freps and non repeater F-waves (NRs) were studied and compared. Results: In the ALS group, Index Freps (100xFreps/total number of F-waves) were significantly higher than the control group (52.9±25.3 vs 7.3±7.8 in ulnar nerve and 75±20.5 vs 23.6±18.4 in peroneal nerve) (P < 0.001). Likewise, the total number of repeating neurons (RNs) and Freps persistence (100xFreps/40stimuli) were significantly different between the groups. In healthy subjects, the peroneal nerve has significantly more Freps compared to the ulnar nerve (P < 0.001). None of the healthy subjects had more than 5 repetitions of any RN in the ulnar nerve, compared to ALS patients (P < 0.001). No significant differences in RNs and NRs were identified between the subgroups of the ALS patients. Conclusion: Indexes of Freps and RNs are significantly increased in ALS patients, even in non advanced stages of disease, when compound muscle action potential (CMAP) values remain normal. These measures possibly reflect the excitability state of motor neurons and might be useful in the evaluation of early motor neuron dysfunction and follow-up of disease progression. The automated analyzing system improves accuracy and facilitates repeater F- wave identification and is less time consuming, thus suitable for routine neurophysiological studies.
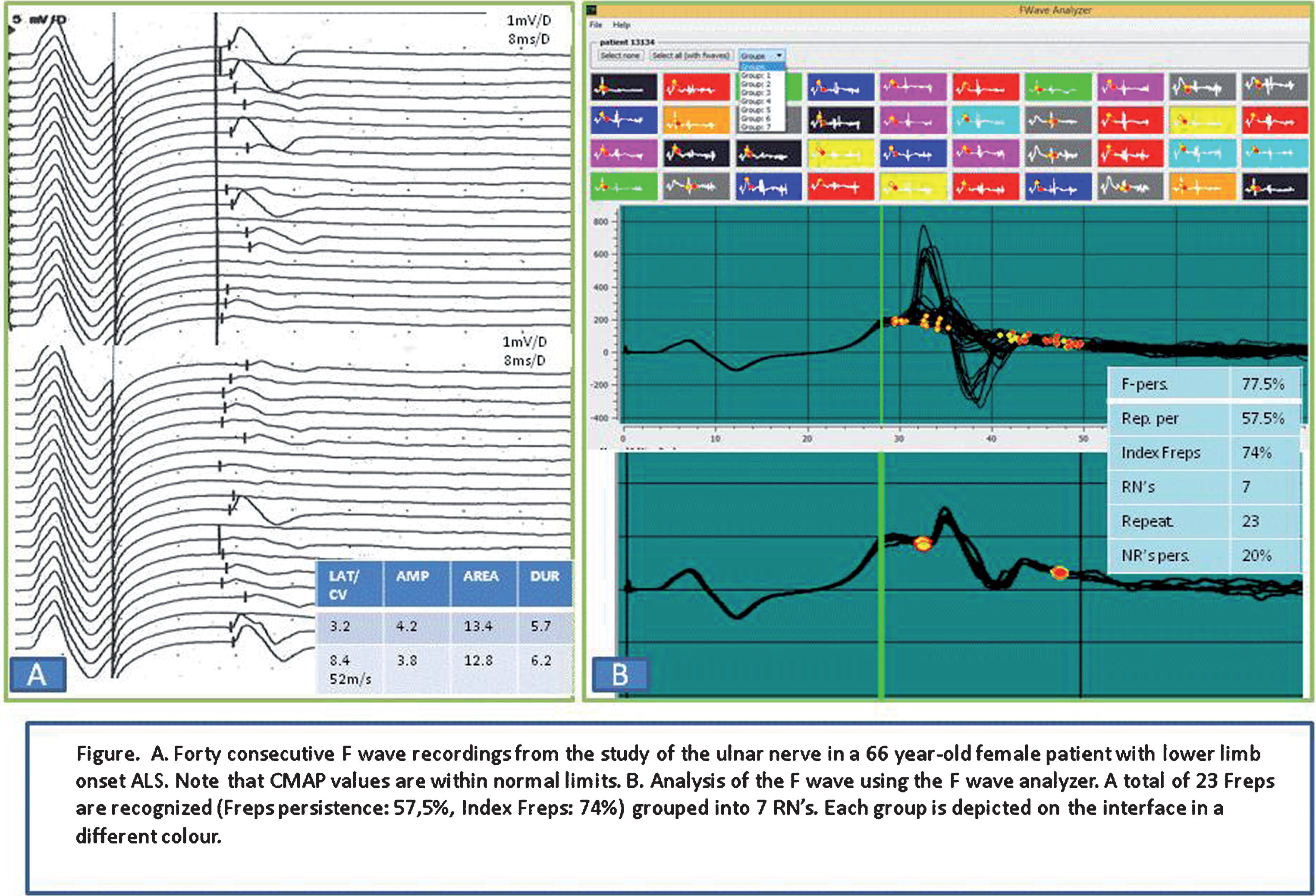
PS3Group4-024 / #771THE OLEOS TRIAL: A LONG-TERM FOLLOW-UP OF OLESOXIME-TREATED TYPE 2 AND NON-AMBULATORY TYPE 3 SMA PATIENTS
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Francesco Muntoni1, Sabine F. Recktenwald2, Enrico Bertini3, Eugenio Mercuri4, Janbernd Kirschner5, Muna El-Khairi6, Anna Lusakowska7, Giacomo P. Comi8, Jean-Marie Cuisset9, Jane Ives6, W Ludo Van Der Pol10, Carole Vuillerot11, Ksenija Gorni12, Paulo Fontoura2
1Dubowitz Neuromuscular Centre, UCL Great Ormond Street Institute of Child Health and Great Ormond Street Hospital for Children, London, GB;2Neuroscience Product Development, F. Hoffmann-La Roche Ltd, Basel, CH;3Department Of Neurosciences And Neurorehabilitation, Bambino Gesù Children’s Research Hospital IRCCS, Rome, IT;4Paediatric Neurology And Nemo Center, Catholic University and Policlinico Gemelli, Rome, IT;5Department Of Neuropediatrics And Muscle Disorders, Medical Center, University of Freiburg, Freiburg, DE;6Roche Products Limited, Welwyn Garden City, GB;7Department Of Neurology, Medical University of Warsaw, Warsaw, PL;8Dino Ferrari Center, Department of Pathophysiology and Transplantation, University of Milan, Neurology Unit, I.R.C.C.S. Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, IT;9Department Of Neuropediatrics, Neuromuscular Disease Reference Centre, Roger Salengro Hospital, Regional University Teaching Hospital (CHRU), Lille, FR;10Department Of Neurology And Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, NL;11Department Of Paediatric Physical Medicine And Rehabilitation, Hôpital Femme Mère Enfant, Centre Hospitalier Universitaire de Lyon, Lyon, FR;12Roche Pharmaceutical Research and Early Development, Roche Innovation Center, Basel, CH
Background: Olesoxime is an oral, daily administered compound that supports the function of mitochondria. In a previous randomized, double-blind, Phase 2 study (NCT01302600) in patients aged 3–25 years with Type 2 or non-ambulatory Type 3 spinal muscular atrophy (SMA), olesoxime maintained motor function over 24 months, whilst the placebo group declined. OLEOS (NCT02628743) is an open-label extension of the Phase 2 study assessing the long-term safety and efficacy of olesoxime in patients with Type 2 or non-ambulatory Type 3 SMA. Methods: One hundred and twenty-nine patients with Type 2 or non-ambulatory Type 3 SMA from the previous Phase 2 study were enrolled and treated with olesoxime (10 mg/kg), and the majority have been followed for 12 months (n=104). Primary endpoint is safety and secondary endpoints include change in Motor Function Measure (MFM) D1+D2 from baseline up to 5 years. OLEOS baseline visit occurred 2.4–5.1 years (median 3 years) after study drug discontinuation in Phase 2. Results: Consistent with previous studies, olesoxime was generally safe and well tolerated at the dose assessed. Maintenance of motor function observed over 2 years in the Phase 2 study was followed by a substantial decline in MFM D1+D2 (>2 points/year) after drug discontinuation. However, the ~2-point MFM treatment difference between olesoxime and placebo at the end of Phase 2 was maintained at OLEOS baseline. Furthermore, olesoxime open-label treatment stabilized motor function (mean change in MFM D1 + D2 from baseline: 6 months, -0.03 [standard deviation (SD), 4.79; n=124]; 12 months, -0.22 [SD, 4.74, n=104]). These data support the long-term stabilization of motor function observed in the Phase 2 study. A study update including novel 18-month data will be presented. Conclusion: Olesoxime was generally safe and well-tolerated. Data from the OLEOS open-label extension study showed that olesoxime maintained motor function over 12 months in the treated patients. A study update (including novel 18-month data) will also be presented. These data suggest that olesoxime offers the potential to provide meaningful clinical benefit to patients with SMA by preventing loss of motor function, and may play a role in the future therapeutic management of SMA. Acknowledgments The authors would like to thank all the individuals enrolled in OLEOS and the previous Phase 2 study, their families and the site staff involved. Study sponsored by F. Hoffmann-La Roche.
PS3Group4-025 / #794SMA-RTER? COGNITIVE ASSESSMENT IN SPINAL MUSCULAR ATROPHY TYPE 1-2 USING EYE TRACKING
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Lionel Paternoster1, Nicolas Deconinck2, Simon Baijot1, Gaetane Deliens3
1Department Of Pediatric Neurology, Queen Fabiola Children’s University Hospital, Brussels, BE;2Department Pediatric Neurology, HUDERF, Brussels, BE;3Ulb Neurosciences Institute, Université Libre de Bruxelles (ULB), Brussels, BE
Background: Spinal muscular atrophy is a devastating inherited disorder caused by ubiquitous deficiency in the SMN protein, with an incidence of around 1 to 6.000-10.000 live births. It is the second most common autosomal recessive disorder and the most frequent genetic cause of infant death. SMA main feature is the progressive loss of motor activity caused by motor neurons degeneration and functional and structural dysfunction of neuromuscular synapses, leading to muscle atrophy. Recent evidence suggests that SMA is a systemic disease. In the brain SMN is expressed in many cell types in addition to motoneurons, brain morphology changes have been described, circadian dysregulation, abnormal fatty acid metabolism and abnormal glucose homeostasis have been observed. Symptoms of signs of a systemic disease may likely become increasingly apparent in treated children treated, where neuromuscular symptoms are alleviated and life extended. Literature on cognitive aspects of SMA type 1-2 is poor and divergent: von Gotard et al [2002] supported a normal cognitive function of SMA patients while Polido et al [2017] demonstrated matching pairs difficulty in children with SMA type 1. Indeed, physical impairments make usual IQ tests often unusable in many SMA children. Methods: We develop an eye tracking system (Tobii®) for the assessment of cognitive functions in SMA patients belonging to the severe end of the spectrum. The set of tests contains the following subtests: - Matrix subtest of Weschler non-verbal scale of ability for the study of fluid reasoning and perceptual reasoning. - Recognition subtest of Weschler non-verbal scale of ability for the study of visuo-spatial memory. - Peabody picture vocabulary test for the study of receptive vocabulary (French). - The chimeric animal stroop test to study inhibition. - Face recognition subtest of NEPSY-II to study face encoding, discrimination and recognition. - Picture complement test to study the spatial reasoning. - Matching pair test. Fourty children from 2 to 12 years old have been enrolled: 10 SMA type 1, 10 SMA type 2, and 20 age and sex matched control patients. Tests will take place in two sessions of 30 minutes for children from 2 to 4 years and in 1 session of 30 to 60 minutes for children from 5 to 12 years. The methodology and first results will be presented. Results: The developed protocol aims at validating eye tracking assessment tool for cognitive functions in SMA type 1 and 2. Conclusion: Cognitive assessment in spinal muscular atrophy type 1-2 using eye tracking system will help to adapt the overall management of those patients and to evaluate the impact on cognition of future SMA treatments.
PS3Group4-026 / #642LONGER-TERM ASSESSMENT OF NUSINERSEN SAFETY/EFFICACY IN INFANTILE-ONSET SPINAL MUSCULAR ATROPHY: INTERIM ANALYSIS OF SHINE
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Richard S. Finkel1, Diana Castro2, Michelle A. Farrar3, Mar Tulinius4, Kristin J. Krosschell5, Kayoko Saito6, Yiwei Zhang7, Ishir Bhan7, Wildon Farwell8, Sandra P. Reyna7
1Nemours Children’s Hospital, Orlando, FL, US;2UT Southwestern Medical Center, Dallas, TX, US;3UNSW Medicine, UNSW Sydney and Sydney Children’s Hospital, Randwick, NSW, AU;4Gothenburg University, Gothenburg, SE;5Northwestern University, Chicago, IL, US;6Tokyo Women’s Medical University, Tokyo, JP;7Biogen, Cambridge, MA, US;8Clinical Development, Biogen, Cambridge, MA, US
Background: Nusinersen is an antisense oligonucleotide approved for the treatment of SMA. It has demonstrated a favorable benefit:risk profile and shown significant and clinically meaningful efficacy on motor function across a broad spectrum of SMA populations, and event-free survival (time to death or permanent ventilation) in infantile-onset SMA. The objective of the current analysis was to report interim results from the SHINE study (NCT02594124) for patients with infantile-onset SMA (most likely to develop Type I) who transitioned from ENDEAR. Methods: SHINE is an open-label extension study for infants/children who participated in the ENDEAR, CHERISH, CS12, or CS3A nusinersen trials. Nusinersen doses were administered according to the regimen and participant’s cohort from the previous trial. The primary endpoint is safety/tolerability; secondary endpoints include achievement of Hammersmith Infant Neurological Examination – Section 2 (HINE-2) motor milestones and event-free survival defined as time to death or permanent ventilation (tracheostomy or ≥16 hours ventilation/day continuously for >21 days in the absence of acute reversible event). Results: The cutoff date was June 30, 2017; 89 patients transitioned from ENDEAR, 65/81 previously randomized to nusinersen and 24/41 to sham-control. Within SHINE only, 83 patients had an adverse event (AE). There were no treatment-related serious AEs. The most frequent AEs were pyrexia and upper respiratory tract infection. Mean (95% CI) change in HINE-2 total score from nusinersen initiation to last observed visit was 1.1 (0.20–1.90) for patients who received sham-control in ENDEAR and nusinersen in SHINE (n=20/24) and 5.8 (4.58–7.04) for those who received nusinersen in ENDEAR and SHINE (n=74/81; pooled ENDEAR/SHINE data). Median (95% CI) event-free survival time among patients treated with sham-control in ENDEAR was 22.6 (13.6–31.3) weeks versus 73.0 (36.3–NA) weeks among those who received nusinersen in ENDEAR and SHINE. Conclusion: Improvements in motor function and event-free survival continued among patients who initiated nusinersen in ENDEAR and motor function improved among those who initiated nusinersen in SHINE. Further analysis of SHINE data will provide additional information on the long-term safety/tolerability and efficacy of repeated nusinersen doses across multiple SMA populations.
PS3Group4-027 / #712A CASE OF KENNEDY’S DISEASE IN A PATIENT INITIALLY PRESENTED WITH RECURRENT PERIODONTITIS AND OROMANDIBULAR PAIN
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Il-Han Yoo1, So-Hyun Park1, Suk-Won Ahn2
1Chung-Ang University Hospital, Heukseok-ro, Dongjak-gu, Seoul, KR;2Neurology, ChungAng University Hospital, Seoul, KR
Background: Kennedy disease (KD), also known as X-linked spinal bulbar muscular atrophy, is a lower motor neuron degenerative disease with onset in adulthood and is characterized by slowly progressive muscular weakness and sensory neuropathy. Typically, affected patient develop symptoms and findings in the bulbar muscles with weakness, atrophy, tongue and facial muscle fasciculation and may develop difficulty managing secretions with the risk of aspiration pneumonia. Here we present a case of SBMA initially presented as recurrent periodontitis and oromandibular pain. Methods: A 45-year-old man visited our neurological department through dental clinics and presented with chronic and recurrent periodontitis accompanied by oromandibular pain over the course of 7 years. Results: Neurological examinations revealed mildly slurred speech, gait disturbance, facial fasciculation, decreased sensory function and proximal muscle weakness with Medical Resource Council (MRC) grade 4/5 in lower limbs. Laboratory investigations revealed significantly increased a creatine kinase (CK) level and nerve conduction study (NCS) and electromyography (EMG) revealed sensory polyneuropathy with wide spreading denervation potentials. Furthermore, he showed an expanded cytosine-adenine-guanine (CAG) repeat in the androgen receptor (AR) gene analysis; therefore he was confirmatively diagnosed as Kennedy disease. Systemically, bulbar symptoms including dysphagia, orofacial muscle dysfunction, drooling and aspiration can predispose to malnutrition, social embarrassment and social isolation in addition to intraoral food retention, oral hygiene avoidance secondary to aspiration fear and perioral infections. Therefore these discomforts might result in the patient’s inability to maintain ideal oral hygiene. Conclusion: Here we present a case of SBMA initially presented as recurrent periodontitis and oromandibular pain. It is important to consider neuromuscular diseases backgrounds to severe periodontitis cases, as this may eventually be relevant to treatment and to understanding the pathogenesis of periodontitis.
PS3Group4-028 / #737UPDATED PHARMACODYNAMIC AND SAFETY DATA FROM SUNFISH PART 1, A STUDY OF ORAL RG7916 IN PATIENTS WITH TYPE 2 OR TYPE 3 SMA
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Eugenio Mercuri1, G Baranello2, Janbernd Kirschner3, Laurent Servais4, Nathalie Goemans5, Maria C. Pera1, Anne Marquet6, Gillain Armstrong7, Heidemarie Kletzl6, Marianne Gerber6, Christian Czech6, Yumi Cleary6, Margaret Chan8, Stephane Nave6, Ksenija Gorni6, Omar Khwaja6
1Paediatric Neurology And Nemo Center, Catholic University and Policlinico Gemelli, Rome, IT;2Developmental Neurology Unit, Carlo Besta Neurological Research Institute Foundation, Milan, IT;3Department of Neuropediatrics and Muscle Disorders, Medical Center-University of Freiburg, Freiburg, DE;4Institute of Myology, Paris, FR;5Neuromuscular Reference Centre, Department Of Paediatrics And Child Neurology, University Hospitals Leuven, Leuven, BE;6Roche Pharmaceutical Research and Early Development, Roche Innovation Center, Basel, CH;7Roche Products Ltd, Welwyn Garden City, GB;8Roche Pharmaceutical Research and Early Development, Roche Innovation Center New York, New York, NY, US
Background: Spinal muscular atrophy (SMA) is characterized by motor neuron loss and muscle atrophy, due to reduced levels of survival of motor neuron (SMN) protein from loss of function of the SMN1 gene. While SMN1 produces full-length SMN protein, a second gene, SMN2, produces only low levels of functional SMN protein. RG7916 (RO7034067) is an investigational, orally administered, centrally and peripherally distributed small molecule that is being evaluated for its effect on modulating SMN2 pre-mRNA splicing towards the production of full-length SMN2 mRNA and increase of SMN protein. SUNFISH (NCT02908685) is a multi-center, double-blind, placebo-controlled trial (randomized 2:1, RG7916:placebo) in patients with Type 2 or 3 SMA aged 2–25 years. Methods: SUNFISH comprises two parts: Part 1 evaluates the safety, tolerability and pharmacokinetics/pharmacodynamics of different RG7916 dose levels (n=51); the pivotal Part 2 assesses the efficacy and safety of the RG7916 dose level selected from Part 1 (n=168). Results: We have previously presented an early analysis of SUNFISH Part 1, which showed that RG7916 administration resulted in a dose-dependent increase in full-length SMN2 mRNA and a concomitant decrease in SMN2Δ7 mRNA. SMN protein levels in whole blood increased in a dose-dependent manner up to a median of 2.5-fold. To date, adverse events were mostly mild, resolved despite ongoing treatment and were reflective of the underlying disease. There have been no drug-related safety findings leading to withdrawal. The safety, tolerability and pharmacokinetic/pharmacodynamic data from Part 1 were sufficient to inform the selection of a dose level for SUNFISH Part 2, to evaluate the efficacy and safety of RG7916. We will provide a more recent and detailed SUNFISH update with biomarker, pharmacokinetic and safety results from patients included in Part 1. To date, all Part 1 patients have been treated with RG7916 for a duration of a minimum of 3 months (in the last placebo patient who switched to active treatment) and up to 16 months. Conclusion: In SUNFISH Part 1, RG7916 treatment modulated SMN2 mRNA and increased SMN protein in a dose-dependent manner. The clinical benefit of the selected dose level is being assessed in SUNFISH Part 2, which is currently recruiting globally. Acknowledgments The authors would like to thank all individuals enrolled in the RG7916 studies, their families and the site staff involved. Study sponsored by F. Hoffmann-La Roche.
PS3Group4-029 / #799MORPHINE FOR DYSPNEA IN ALS
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Lev Brylev1, Vadim Parshikov2, Yaroslava Krasnaya2, Ekaterina Dikhter2, Egor Larin3, Yana Batmanova2, Vasiliy Shtabnitskiy4, Sergey Avdeykin5, Anastasia Ataulina5, Daria Puzanok2, Anna Kasianova2
1Buyanov City Hospital, Moscow, RU;2ALS center Moscow, Moscow, RU;3Center of palliative medicine, Moscow, RU;4Chaika, Moscow, RU;5Buyanov city hospital, Moscow, RU
Background: Dyspnea affects more than half patients with amyotrophic lateral sclerosis (ALS). Management of this distressing symptom can be challenging. The most effective drug for dyspnea is morphine, but it is underused for this indication, due to lack of evidence of safety of morphine in patients with ALS and lack of information about it’s effectiveness. Methods: In ALS centre Moscow we followed 430 patients with mean age 59,3 ±10,8 years, with median ALSFRS-R 26 (17;32) and median vital capacity 51,0% (38;80) on admission. 25 ALS patients with mean age 66.3 ±12,1 years, 14 males and 11 females recieved morphine for dyspnea. We used Borg scale to measure dyspnea. In five patients we measured safety of morfine by measuring pCO2 in arterial blood during morfine infusion. Patients were divided as respontenders to morfine if they had good dyspnea control on morphine, partial responders if they had 3 points desrease on Borg scale and nonresponders. Results: Morphine didn’t cause elevation of pCO2 in arterial blood of our patients. 50% of patients were good responders, 40%- partial responders and 10% - nonresponders. Dose of morphine in the group of responders (23±8,2) mg per day was higher, than in partial responders - (11,8±3.4 mg) and nonresponders (10±2.7 mg). 48% of patients used lorasepam for concominant laringospasms. Conclusion: Morphine is highly effective in 50% of ALS patients with dyspnea and doesn’t supress respiration in doses, that are prescribed to control dyspnea. Morphine should be more widely used to control dyspnea in ALS patients.
PS3Group4-030 / #944EXTRACELLULAR VESICLES FROM ALS SPINAL CORD AND BRAIN CONTAIN DYSREGULATED MIRNAS
Topic: Group 4 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy
Eva Feldman
University of Michigan, Ann Arbor, US
Background: Aims/hypothesis: Amyotrophic lateral sclerosis (ALS) is a terminal neurodegenerative disease. Clinical and molecular observations indicate that motor neuron death and muscle atrophy in ALS proceeds in a patterned manner suggesting that ALS develops as a non-infectious prion-like disease1,2. Extracellular vesicles (EVs) maybe play a role in the spread of ALS as they facilitate transfer of molecules from cell-to-cell3-5 In this project we aim to determine the role of EVs cargo in ALS pathology. Aberrant epigenetic mechanisms such as DNA methylation and miRNAs levels have been reported in ALS6. We hypothesize that EVs may be involved in the transfer of epigenetic alterations in ALS. Methods: Methods: We used an immuno-affinity-based microfluidic device, the ExoChip, for isolation and quantification of EVs7 from frontal cortex (Ctrl=8, ALS=12), spinal cord (Ctrl=6, ALS=12), and plasma (Ctrl=9, ALS=7). Electron microscopy was used for morphological characterization of EVs. EV-contained miRNAs were profiled with NanoString technology and analyzed by nSolver and NanoStringDiff. Results: Results: Analysis of plasma, frontal cortex, and spinal cord indicates there is no significant increase in the availability of EVs in ALS, when compared to control subjects. However, we detected alterations in the levels of mature miRNAs contained in EVs from all examined tissues. Conclusion: Conclusions/interpretations: We have identified altered miRNAs packaged in EVs from ALS tissue. These miRNAs are important in biological functions and pathways previously associated with neurodegeneration. Identification of altered EVs-contained miRNAs in ALS have both diagnostic and therapeutic potential, as EVs can represent biomarkers and may be used for the delivery of drugs or genetic molecules including miRNAs.
PS3Group7-001 / #590THE IMPACT OF PARTICIPATION IN CLINICAL TRIALS ON PSYCHOSOCIAL WELLBEING OF CHILDREN WITH DMD AND THEIR PARENTS
Topic: Group 7 – Patient Related Issues
Sam Geuens1, Joanna Willen2, Corine Antonis2, Nathalie Goemans3, Liesbeth De Waele2
1Child Neurology, UZ Leuven, Leuven, BE;2Child Neurology, UZ Leuven, Leuven, BE;3Treat-NMD Neuromuscular Network, Leuven, BE
Background: Approximately 35-40 per 100 000 children are diagnosed with a neuromuscular disorder, but for most of those diseases no treatment is available. However, more and more clinical trials (CT) are experimenting with promising therapies. Participation in a CT is for most patients and their parents the only hope to receive a treatment that can have a significant positive impact on the disease. The burden of participation in a CT, however, should not be underestimated: frequent time investment, impact on family activities, absence of work and school, side effects of the medication,… Studies investigating this burden are scarce and the costs for participation for individual families are not known. The aim of this study is to measure the psychosocial impact and the burden of participation in a CT for patients with Duchenne Muscular Dystrophy (DMD). Methods: A cross-sectional research design was constructed with a group of patients who participated in a CT and an age- and gender-matched control group of patients with DMD, but not participating in a CT. Both parents of patients in both groups were asked to fulfill a Child Behavior Checklist (CBCL) reporting the wellbeing of their child and an Adult Self Report (ASR) reflecting their own wellbeing. Furthermore they were asked to complete a pediatric quality of life inventory (PedsQL). All children were asked to report their quality of life by filling in an age appropriate version of the PedsQL. If they were older than 11 years an extra questionnaire was given, namely the Youth Self Report (YSR), reflecting their own wellbeing.In the CT-group an extra explorative survey, the Clinical Trial Survey (CTS), was conducted. This instrument was developed to measure the burden of participation to a CT for patients and their families. This new instrument includes 24 questions divided into 6 different domains. Results: 25 children with DMD who participated in a CT are included. The control group consisted of 20 age-matched children. At the moment of abstract submission only preliminary data was available. 25 mothers and 22 fathers of boys with DMD who participated in a CT completed the CTS. This survey will provide us with explorative information and will allow us to estimate and quantify the impact of participating in a clinical trial for both children and parents. Secondly, we have data from behavioral questionnaires and quality of life questionnaires of boys and parents in a clinical trial and a control group and therefor able to detect differences between groups. Moreover, because we also have self-report scores from both mothers and fathers reporting on themselves, we can illustrate what the impact is on the parents and if there is a difference between them. Finally, we want to see if we can identify some factors contributing to the impact of a CT. Conclusion: Definitive results and conclusions areexpected at the end of April 2018
PS3Group7-002 / #452PHYSICAL ACTIVITY IN PEOPLE WITH LIMB-GIRDLE MUSCULAR DYSTROPHY AND CHARCOT-MARIE-TOOTH DISEASE IN NORWAY
Topic: Group 7 – Patient Related Issues
Aristomo Andries, Marleen R. Van Walsem, Jan Frich
Institute Of Health And Society, University of Oslo, Oslo, NO
Background: Active lifestyle is recommended to promote health and well-being, including in people living with disabilities. People with neuromuscular diseases (NMD), particularly limb-girdle muscular dystrophy (LGMD) and Charcot-Marie Tooth disease (CMT), may have challenges to do certain physical activities. While other studies have demonstrated benefits of physical activity in the people with NMD, there is a lack of knowledge about physical activity level in people with LGMD and CMT. The aim of this study is to investigate the self-reported physical activity levels in people with LGMD and CMT. Methods: This is a cross-sectional observational study. People who were registered in the national database for neuromuscular conditions (University Hospital of North Norway) were selected randomly and invited to participate in the study. Invitations were sent by postal mail. People who fulfilled the inclusion criteria and consented to participate filled out a set of paper-based questionnaires or online-based questionnaire in University of Oslo’s database system. Physical activity level was investigated using self-assessed International Physical Activity Questionnaire short form (IPAQ-sf). Participants with vigorous-intensity activity for at least 3 days and achieved at least 1500 metabolic equivalent (MET)-minutes/week or 7 days of any other activities with at least 3000 MET-minutes/week were classified as “health-enhancing physical activity” (HEPA). Participants outside the HEPA group were classified as either “inactive” or “minimally active” according to IPAQ-sf protocol. Socio-economic-demographic factors were also included in the questionnaire. Other factors which may influence physical activity level such as functional ability, health related quality of life, and fatigue were investigated using validated questionnaires. Data were analyzed using descriptive statistics. Researchers used IBM SPSS statistical program version 25 to perform the analysis. Results: Out of 236 potential participants were invited to the study, 145 (61.4%) participants responded to the study and filled out questionnaire either paper-based or online. 79 respondents (54.5%) had been diagnosed with CMT and 66 (45.5%) respondents had been diagnosed with LGMD. Of the respondents 61 (42.1%) were male and 84 (57.9%) were female. The median of MET-minutes/week was 1180.50 (IQR=2643.63). Mean sitting time was 518.56 minutes (SD=231.86). 48 (45.3%) respondents were classified as “inactive”, 30 (28.3%) as “minimally active”, and 28 (26.4%) as “HEPA”. The odds ratio (OR) of respondents who have HEPA compared to inactive/minimally active was 5.5-fold (95% CI 1.51-20.10, p=0.005) in people with normal activity daily living/ADL (measured by Barthel’s index) compared to respondents with decreased ADL. Conclusion: More than a quarter of study participants have fulfilled the HEPA category, while the majority of respondents were unable to achieve this. Participants with normal ADL showed greater odds of being physically active compared to those with decreased ADL. To our knowledge, this study is the first to investigate self-reported physical activity level among people with LGMD and CMT in Norway. The study findings are important to understand how to achieve optimum physical activity level for beneficiaries. Furthermore, factors which may influence physical activity need to be thoroughly examined. It may also be interesting to compare these findings to other group of neuromuscular diseases.
PS3Group7-003 / #822THE JAIN FOUNDATION DYSFERLIN PATIENT REGISTRY: ACCELERATING THE PACE OF RESEARCH AND TREATMENT
Topic: Group 7 – Patient Related Issues
Laura Rufibach, Sarah Shira, Laurie Long, Bradley Williams, Douglas Albrecht
Jain Foundation, Seattle, US
Background: Dysferlinopathy is a recessively inherited form of muscular dystrophy caused by mutations in the DYSF gene. The most common clinical forms are limb girdle muscular dystrophy type 2B (LGMD2B) and Miyoshi Myopathy type 1 (MMD1). Onset of muscle weakness generally occurs in young adulthood with loss of ambulation typically within 10-20 years. The prevalence of this disease is estimated to be between 5-8/million. The Dysferlin Registry is an important tool to facilitate and accelerate clinical research through identification of information and recruitment of participants. It also is an interactive registry that allows individuals with dysferlinopathy to communicate with one another and the members of the Jain Foundation. Aims: - Identify and recruit patients for clinical studies and trials - Empower and cultivate a trial ready patient community through education and engagement - Encourage communication and expedite our understanding of challenges patients are navigating in daily living, ultimately assisting in targeting possible outcome measures for trials - Provide patients with up to date information on research progress - Support and encourage research by providing access to data. Methods: The Dysferlin Registry is exclusively for individuals who have been confirmed to have a dysferlinopathy through the evaluation of their clinical symptoms, dysferlin protein levels, and genetic analysis. Diagnosed patients are invited to join and after accepting, they enter data by creating a password-protected account through an online portal. The patient also chooses their level of engagement within the community. These levels range from private (allowing only the registry administrators to see their presence in the registry) to fully active (where they set up an online profile; allowing for communication with other members). The registry also features a portal that allows clinicians to enter patients into the registry after obtaining the necessary consent. The registry collects contact, demographic, clinical, genetic, protein, phenotypic and familial information. Results: As of February 2018, there are currently 654 people registered from 41 countries, with the largest number residing in the United States. The ages of the people registered ranges from 10-79, with 67% being between the ages of 25-50. The average age of onset was 26 years of age with 79% reporting onset of muscle symptoms between 15-30. The majority (75%) of the registrants indicate they are still ambulant, but with differing levels of ambulation from running to walking only with assistance. The Dysferlin Registry has already supported recruitment and pre-trial design for multiple clinical studies and trials. Conclusion: Patient registries are invaluable tools in the field of rare diseases because with so few patients every one counts. Please encourage all your dysferlinopathy patients to register. The Jain Foundation Dysferlin Registry is seeking to cultivate an informed, trial ready community that has the potential to accelerate clinical trials and support research into possible treatments. Please visit www.dysferlinregistry.jain-foundation.org and if you have a patient who may be eligible, please email us at .
PS3Group7-004 / #618PATIENT PERSPECTIVES ON THE SIDE EFFECT BURDEN OF TREATMENTS FOR MYASTHENIA GRAVIS (MG) AND THEIR IMPACT ON DAILY LIFE
Topic: Group 7 – Patient Related Issues
Elizabeth D. Bacci1, Karin S. Coyne1, Linda Harris2, Jiat-Ling Poon3, Audra N. Boscoe4
1Business Unit Of Ppd, Evidera, Bethesda, MD, US;2Alexion Pharmaceuticals, New Haven, US;3Lilly Corporate Center, Eli Lilly and Company, Indianapolis, IN, US;4Agios Pharmaceuticals, Cambridge, MA, US
Background: Standard treatments for patients with myasthenia gravis are associated with side-effects; however, the frequency, severity and impact of these side effects on patients’ lives are not fully understood. The objective of this study was to characterize the side effects of standard treatment for generalized MG and their associated impact on health-related quality of life (HRQL) from the patient perspective. Methods: MG Foundation of America patient registry (managed by the coordinating center at the University of Alabama Birmingham and the MGFA Registry Committee) members were invited to participate in a web survey. Participants were asked to brainstorm side effects (Step 1) experienced due to treatments for MG and the impact of those side effects on HRQL using a group concept mapping approach. The statements provided by participants were synthesized, and participants were re- surveyed to rate the tolerability and severity of side effects (Step 2) based on the statements, and the frequency and severity of HRQL impacts on a 1 (least) to 4 (most) scale. Participants also completed sociodemographic questions, the MG Quality of Life Scale (MGQOL15), and the Morisky Medication Adherence Scale (MMAS-8). Descriptive statistics were used to characterize the study sample and summarize participants’ ratings. Results: A total of 246 participants contributed to the brainstorming exercise (63% female; mean±SD age 58.4±13.4 years); 158 (64%) participated in the rating exercise. Nearly all (92%) participants confirmed that their MG symptoms were consistent with generalized MG and not limited to ocular symptoms. The majority reported currently being on pyridostigmine (69%) and/or immunosuppressive treatment (68%). Just under half (46%) reported having previously used at least two immunosuppressive treatments and one-third reported having received IVIg and/or plasmapheresis on more than four separate occasions over the past year. The brainstorming exercise produced 874 statements, which were condensed to 35 side effects and 23 HRQL impact statements. Blood clots, infections/decreased immunity, weight gain, and diarrhea were the least tolerable and most severely rated side effects. The most frequent and severe HRQL impacts were sleep interference and reduction in physical and social activities. Subgroup analyses compared rating results between participants who had used multiple prior therapies (refractory patients) and had prior experience with side effects to non-refractory patients, and determined that the severity and tolerability of the side effects experienced were similar regardless of treatment refractory status. However, HRQL impacts were more frequent and severe for refractory patients compared to non-refractory patients. MGQOL15 results indicated moderate impact on HRQL, with most participants (79%) reporting either some or quite a bit of frustration with their MG and many (57%) reporting severe to very severe impacts on their occupational skills and job status. Per the MMAS-8, 36% of patients had low adherence and 33% had medium adherence to their MG medications. Conclusion: MG patients experience high burden of treatment side effects with existing treatment options, which potentially impacts treatment adherence and their health outcomes. Based on these findings, there appears to be a need for better and more tolerable treatment options for this patient population.
Author Index
Aartsma-Rus A. PS2Group9-019, S323
Aban I.B. SS 23.2, S39
Abd El-Hamed M.A. PS2Group3-067, S270
Abicht A. PS1Group1-130, S204, PS2Group3-074, S275, PS2Group5-016, S295
Adams D. PS2Group3-005, S220, PS2Group3-010, S225, PS2Group3-055, S261
Afroozan F. PS1Group1-096, S178
Aguirre F. PS3Group2-007, S330, PS3Group2-008, S330
Ahmed A. PS1Group1-140, S212
Ahn S.H. PS1Group1-131, S205, PS1Group1-141, S213, PS2Group3-032, S244
Ahn S.-W. PS2Group3-035, S246, PS2Group3-061, S266, PS3Group4-027, S378
Aizpurua J.M. PS1Group1-038, S132, PS1Group1-106, S186
Akhondi F. PS2Group5-009, S290
Akramova D.T. PS2Group5-025, S301
Alarcon Cornejo M.M.A. PS2Group9-010, S317
PS3Group4-019, S371
Albiasu-Arteta E. PS1Group1-054, S146, PS1Group1-108, S187
Albrecht D. PS2Group5-007, S288, PS3Group7-003, S382
Aldanondo G. PS1Group1-038, S132, PS1Group1-106, S186
Aleff R.A. PS1Group1-011, S110
Alekseeva T. PS3Group4-021, S373
Aleksic K. PS1Group1-078, S164
Alfa Cissé O. PS2Group3-064, S268
Alfano L. PS3Group4-022, S373, SS 13.4, S25
Aliu B. PS2Group3-015, S229
Allegorico L. PS2Group5-034, S307
Allenbach Y. PS2Group9-006, S314, WS 13.3, S58
Allen J.E. PS1Group1-082, S167, PS1Group1-147, S216
Almasi M. PS2Group5-009, S290
Almendrote M. PS1Group1-089, S173
Alshareef A.A. PS2Group3-014, S228
Alves B.C.A. PS1Group1-107, S186
Al-Zaidy S. PS3Group4-022, S373, SS 13.4, S25
Amato A. SS 19.2, S31, WS 4.1, S48, WS 7.3, S53
Ambawat S. PS1Group1-016, S114, PS1Group1-044, S138
A N. PS3Group2-022, S342
Anderson D. PS3Group2-002, S326, PS3Group2-003, S326
Andres V. PS3Group2-008, S330
Andries A. PS3Group7-002, S381
Angelini C. OA 3.1, S91, PS1Group1-051, S144, TC 1.3, S75 WS 16.4, S64
An J.Y. PS2Group3-066, S270
Annousamy M. PS1Group1-004, S104
Antenora A. PS2Group5-021, S298
Antonini G. PS1Group1-051, S144
Antonis C. PS3Group7-001, S380
Anziska Y. PS2Group3-017, S230
Aparicio J. PS1Group1-013, S112
Apreleva A. WS 3.4, S47
Arad M. PS2Group3-002, S218
Arahata H. PS1Group1-029, S125
Archer J.O.S. PS3Group4-011, S364
Arechavala-Gomeza V. PS1Group1-054, S146, PS1Group1-108, S187
Argov Z. OA 2.2, S90, SS 1.1, S10
Armando Jr J. PS2Group3-059, S265
Armas J. PS2Group9-010, S317, PS3Group4-019, S371
Armstrong G. PS3Group4-002, S356, PS3Group4-015, S367, PS3Group4-028, S378
Arnaoutoglou M. PS1Group1-102, S183
Arnold W.D. PS3Group4-022, S373, SS 13.4, S25
Artemyeva S. PS2Group9-015, S320
Ashish K. PS2Group3-029, S241
Ashoorian Y. PS1Group1-043, S137
Ashtari F. PS1Group1-043, S137
Assadian S. PS2Group5-007, S288
Ataulina A. PS3Group4-029, S379, WS 3.4, S47
Attarian S. PS2Group3-065, S269
Attarwala H. PS2Group3-043, S251
Auboeuf D. PS3Group4-006, S360
Auer-Grumbach M. SS 9.4, S19, TC 3.1, S79
Auradé F. PS1Group1-007, S107
Avdeykin S. PS3Group4-029, S379, WS 3.4, S47
Awano H. PS1Group1-052, S145, PS1Group1-128, S202
Azorín I. PS2Group3-053, S259
Azuma Y. SS 24.2, S40
Babacic H. PS1Group1-002, S102
Babu G. PS1Group1-111, S189
Bacci E.D. PS3Group7-004, S383
Bae D.W. PS2Group3-066, S270
Bae J.S. PS2Group3-041, S250
Baek S.-H. PS2Group5-017, S295
Baets J. PS1Group1-022, S119, PS1Group1-138, S210
Baghdoyan S. PS3Group4-006, S360
Baijot S. PS3Group4-025, S376
Balaraju S. PS1Group1-044, S138, PS3Group2-022, S342
Balcarek P. PS1Group1-025, S122
Baldwin C. PS2Group3-037, S247
Ballester-Lopez A. PS1Group1-089, S173
Bandyopadhyay D. PS2Group3-029, S241
Banerjee M. PS2Group3-008, S223
Ban J. PS1Group1-101, S182
Banko B. PS1Group1-080, S165
Banovic M. PS1Group1-104, S185
Bansagi B. PS2Group3-060, S265
Baranello G. PS3Group4-008, S361, PS3Group4-015, S367, PS3Group4-028, S378
Barbeau S. PS3Group2-028, S347
Barnett C. PS3Group2-005, S328
Barohn R.J. PS2Group3-051, S257, PS3Group2-021, S341, SS 11.2, S21, SS 22.2, S37, SS 23.1, S38, TC 6.1, S83
Bartesaghi L. PS2Group3-044, S252
Barthélémy I. PS1Group1-005, S105, PS1Group1-007, S107, PS1Group1-008, S108, PS1Group1-121, S197
Bartnik E. PS1Group1-036, S131
Bartonek L. PS2Group3-026, S239
Basiri K. PS1Group1-043, S137
Basta I. PS1Group1-077, S163, PS1Group1-078, S164, PS1Group1-104, S185, PS2Group3-028, S241
Batmanova Y. PS3Group4-029, S379, WS 3.4, S47
Battaglia D. PS1Group1-027, S123
Bauché S. PS2Group5-027, S302, PS3Group2-030, S348
Baudin P.Y. PS1Group1-004, S104
Bauer L. PS3Group4-004, S358
Baydan F. PS2Group3-013, S227, PS2Group9-014, S320
Beadon K. PS2Group3-065, S269
Bechtold C. PS1Group1-014, S112
Bednarik J. PS1Group1-037, S132
Beecher G. PS3Group2-002, S326, PS3Group2-003, S326
Beeson D. PS3Group2-036, S353, SS 24.1, S40, WS 14.2, S59
Bekircan-Kurt C.E. PS2Group3-006, S222
Beladi Moghadam N. PS1Group1-043, S137
Belaïdi H. PS1Group1-067, S157, PS1Group1-068, S157, PS2Group5-012, S292
Belarabi L. PS2Group5-012, S292, PS2Group5-013, S292
Belaraju S. SS 24.2, S40
Bello L. PS1Group1-115, S192
Bell P. PS2Group9-013, S319
Belousova E. PS2Group9-015, S320
Benatar M. PS3Group2-021, S341
Ben Djebara M. PS2Group3-077, S277
Benedetti F. PS3Group2-024, S343
Benet-Pagès A. PS2Group5-016, S295
Bennett D. SS 15.3, S27
Benoit V. PS2Group3-070, S272
Berardinelli A. PS1Group1-051, S144
Berciano J. PS1Group1-100, S181
Berek K. PS2Group3-076, S276
Bergauer T. PS3Group4-002, S356
Berini S. PS1Group1-011, S110
Berki T. PS1Group1-099, S180
Berk J. PS2Group3-005, S220, PS2Group3-010, S225, PS2Group3-055, S261
Bernert G. SS 5.1, S13, TC 4.1, S81
Berry K. PS3Group4-022, S373
Bertassoli B.M. PS1Group1-047, S141, PS1Group1-073, S161, PS1Group1-107, S186, PS2Group3-059, S265
Bertini E. PS1Group1-027, S123, PS3Group4-008, S361, PS3Group4-024, S375
Bertoli M. PS1Group1-138, S210
Berwanger C. PS1Group1-134, S207, PS1Group1-135, S208
Beydoun S.R. PS3Group2-004, S327
Bhan I. PS3Group4-014, S366, PS3Group4-026, S377
Bharucha-Goebel D.X. PS1Group1-063, S153, PS1Group1-115, S192, PS2Group5-007, S288
Bhattacharya I. PS1Group1-019, S117
Bhattacharya K. PS2Group5-010, S291
Bianchini E. PS1Group1-125, Bicciato S. PS2Group5-020, S297
Bieri O. PS1Group1-017, S116
Binda A. PS1Group1-110, S188
Birouk N. PS1Group1-067, S157, PS1Group1-068, S157, PS2Group5-012, S292, PS2Group5-013, S292, SS 8.2, S16, SS 21.3, S35
Bischoff C. TC 5.1, S82
Bista P. PS1Group1-060, S151
Bjelica B. PS1Group1-077, S163, PS1Group1-078, S164, PS1Group1-104, S185, PS2Group3-028, S241
Bjelic M. PS1Group1-059, S150
Blamire A. PS1Group1-009, S108, PS1Group1-115, S192
Blanchard-Gutton N. PS1Group1-007, S107, PS1Group1-008, S108
Blanco-Arias P. PS1Group1-100, S181
Blot S. PS1Group1-005, S105, PS1Group1-007, S107, PS1Group1-008, S108, PS1Group1-121, S197
Blutke A. PS1Group1-065, S155
Boczonadi V. PS2Group3-060, S265
Boex M. PS2Group5-027, S302
Boisserie J.-M. PS2Group9-006, S314
Boland A. PS3Group4-006, S360
Boland-Freitas R. PS2Group5-003, S285, PS2Group5-024, S300
Bonati U. PS1Group1-017, S116
Bönnemann C.G. PS1Group1-035, S130, PS1Group1-039, S133
Bonuccelli U. PS3Group2-027, S346
Boostani R. PS1Group1-043, S137
Borchers C.H. PS2Group5-007, S288
Born A. PS3Group4-008, S361
Borras A. PS3Group4-019, S371
Boscoe A.N. PS3Group2-033, S351, PS3Group7-004, S383
Bose’ F. PS1Group1-042, S136, PS1Group1-056, S148, PS1Group1-110, S188
Boss M.A. PS2Group9-018, S323
Botez S. PS3Group2-026, S345
Botta A. PS1Group1-110, S188
Bouchet-Séraphin C. PS1Group1-066, S156
Boucraut J. PS2Group3-065, S269
Bouhour F. PS1Group1-066, S156, PS1Group1-088, S172, PS3Group2-030, S348
Bozovic I. PS1Group1-077, S163, PS1Group1-104, S185, PS2Group3-028, S241
Brajuskovic G. PS1Group1-079, S164
Brankovic M. PS1Group1-077, S163, PS1Group1-080, S165
Bravver E. PS1Group1-063, S153, PS1Group1-115, S192, PS2Group5-007, S288
Briani C. PS2Group3-082, S283
Brighina F. PS1Group1-127, S202
Brigonzi E. PS1Group1-042, S136, PS1Group1-110, S188
Bril V. PS2Group3-012, S226, PS2Group3-018, S231, PS2Group3-019, S233, PS2Group3-023, S236, PS2Group3-024, S237, PS2Group3-030, S243, PS2Group3-031, S244, PS2Group3-057, S263, PS3Group2-004, S327, PS3Group2-005, S328, SS 10.1, S19
Brinkmeier H. PS1Group1-103, S184
Brkusanin M. PS1Group1-077, S163, PS1Group1-079, S164
Brochier G. PS3Group2-030, S348
Broda P. PS1Group1-119, S195
Broser P.J. PS2Group5-006, S288
Brown L. PS2Group3-051, S257
Bruno C. PS1Group1-119, S195
Bruno G. PS2Group5-034, S307
Brureau A. PS2Group3-038, S248
Brusse E. PS1Group1-030, S126, PS1Group1-040, S134
Brylev L. PS3Group4-029, S379, WS 3.4, S47
Bryson-Richardson R. PS3Group4-007, S361
Bucci E. PS1Group1-051, S144
Buchbjerg J. PS1Group1-014, S112
Bulst S. PS2Group5-016, S295
Bungener C. PS2Group9-001, S310
Burden S. PL 4.1, S6
Burghes A.H.M. PS3Group4-022, S373, SS 13.4, S25
Burns T.M. PS3Group2-021, S341, SS 20.1, S32
Burnyte B. PS1Group1-132, S206
Bushby K. PS1Group1-009, S108, PS1Group1-063, S153, PS1Group1-115, S192, PS2Group5-007, S288, PS2Group9-019, S323
Buyse For The Delos Study Group G. PS1Group1-057, S148, PS1Group1-105, S185, PS2Group5-023, S299, PS2Group9-003, S312, PS2Group9-016, S321
Buza E.L. PS2Group9-013, S319
Byrne B. PS1Group1-014, S112
Cabrera F.A. PS2Group5-033, S307
Cacciavillani M. PS2Group3-082, S283
Caillaud C. PS1Group1-088, S172
Callaghan B.C. PS2Group3-008, S223, WS 20.4, S71
Camerino G.M. PS1Group1-028, S124
Cano A. PS1Group1-089, S173
Cao M. PS1Group1-051, S144
Capogrosso R.F. PS1Group1-028, S124
Capone A. PS1Group1-017, S116
Cardani R. PS1Group1-042, S136, PS1Group1-056, S148, PS1Group1-110, S188
Cardellach F. PS1Group1-084, S169
Cardoso M. PS3Group2-025, S345, PS3Group4-013, S365
Carlier P.G. PS1Group1-004, S104, PS1Group1-005, S105, PS1Group1-009, S108, PS1Group1-095, S177, PS1Group1-115, S192, PS2Group9-006, S314, PS2Group9-008, S316
Carmes E.R. PS1Group1-006, S106
Carmona C. PS3Group2-034, S351
Carnesecchi S. PS1Group1-004, S104, PS1Group1-005, S105
Carotti M. PS1Group1-125,
Carrera-García L. PS2Group3-011, S226, PS2Group3-034, S245, PS2Group5-014, S293, PS2Group9-010, S317, PS3Group2-013, S335, PS3Group4-019, S371
Carrera L. PS1Group1-013, S112
Carrino R. PS3Group2-024, S343
Carrozzo R. PS1Group1-027, S123
Carstens P.-O. PS1Group1-122, S198
Carvalho A.A.S. PS1Group1-047, S141, PS1Group1-072, S160, PS1Group1-073, S161, PS1Group1-074, S162, PS1Group1-075, S162, PS1Group1-107, S186, PS2Group3-059, S265
Casadevall T. PS1Group1-089, S173
Castelain V. PS3Group4-004, S358
Castro D. PS3Group4-026, S377
Castro I.D. PS2Group3-062, S267
Castro J. PS2Group3-062, S267
Catteruccia M. PS1Group1-119, S195
Cauchois X. PS1Group1-121, S197
Cavaletti G. SS 6.3, S14
Cavalli M. PS1Group1-056, S148
Cetin H. PS2Group3-026, S239, PS3Group2-020, S340, PS3Group2-036, S353, SS 24.3, S40
Chae C.S. PS2Group3-047, S254
Chandran S. PS1Group1-060, S151
Chandrasekaran K. PS2Group3-045, S253
Chan L.L. PS2Group3-072, S273
Chan M. PS3Group4-028, S378
Chanson J.-B. PS3Group4-004, S358
Chant E. PS2Group3-008, S223
Chao H.-C. PS2Group3-016, S230
Charleston J.S. PS1Group1-049, S143
Chella A. PS3Group2-027, S346
Chen V. PS2Group5-007, S288
Chepuru R. PS2Group3-039, S249
Chê V. PS1Group1-004, S104
Chevessier F. PS3Group2-030, S348
Chiriboga C.A. PS3Group4-002, S356, PS3Group4-014, S366
Cho E.B. PS2Group3-033, S245
Choi J. PS2Group3-046, S254
Choi K. PS2Group3-054, S260
Choi Y.C. PS1Group1-145, S215, PS3Group4-016, S368
Cho J.-Y. PS2Group3-050, S257
Cholet N. PS2Group3-038, S248, PS2Group3-069, S271
Chowdhury S. PS2Group9-009, S316
Chrast R. PS2Group3-044, S252
Christofolini D.M. PS1Group1-073, S161
Chroni E. PS2Group5-030, S305, PS3Group4-023, S374
Chumillas M.J. PS2Group3-053, S259
Chun M.Y. PS2Group3-073, S274
Church K. PS3Group4-022, S373, SS 13.4, S25
Chutipongtanate S. PS3Group2-006, S328
Ciet P. PS1Group1-040, S134
Cimbalistiene L. PS1Group1-132, S206
Cirino R.H.D. PS1Group1-006, S106
Claeys K.G. PS1Group1-081, S166, WS 16.2, S62
Cleary Y. PS3Group4-002, S356, PS3Group4-015, S367, PS3Group4-028, S378
Clemen C. PS1Group1-134, S207, PS1Group1-135, S208
Cloutier M. PS1Group1-020, S118
Cnaan A. PS1Group1-115, S192
Cocito D. PS2Group3-019, S233
Codina A. PS2Group5-014, S293, PS3Group2-013, S335
Coelho T. PS2Group3-005, S220, PS2Group3-010, S225, PS2Group3-055, S261, PS3Group2-025, S345, WS 10.3, S56
Coffey C. PS3Group2-021, S341
Cohen D. PS2Group3-038, S248, PS2Group3-069, S271
Coll-Cantí J. PS1Group1-089, S173
Colomer J. PS1Group1-013, S112, PS2Group3-011, S226, PS2Group3-034, S245, PS2Group5-014, S293, PS2Group9-010, S317, PS3Group2-013, S335, PS3Group4-019, S371
Comi G.P. PS3Group4-024, S375
Conceição I. PS1Group1-058, S149, PS2Group3-062, S267
Conte E. PS1Group1-028, S124
Conwit R. PS3Group2-021, S341
Cooperative International (Cinrg) PS1Group1-114, S191
Neuromuscular Research Group
Coppenrath E.M. PS1Group1-009, S108
Corazzini R. PS1Group1-073, S161, PS1Group1-074, S162
Corberó A.B. PS2Group5-033, S307
Coriu D. PS2Group3-002, S218
Cornblath D. PL 2.3, S5
Cornblath D.R. PS2Group3-012, S226, PS2Group3-018, S231, PS2Group3-019, S233, PS2Group3-023, S236, PS2Group3-024, S237, PS2Group3-057, S263
Cosentino G. PS1Group1-127, S202
Cossins J. PS3Group2-036, S353
Cossu G. TC 9.3, S86
Counsell J. PS1Group1-070, S159
Cowling B.S. PS1Group1-050, S143, PS1Group1-061, S152
Coyne K.S. PS3Group7-004, S383
Cozzoli A. PS1Group1-028, S124
Crha I. PS1Group1-037, S132
Csimma C. TC 9.1, S86
Cudkowicz M.E. PS3Group2-021, S341
Cuisset J.-M. PS3Group4-024, S375
Cutrupi A. PS2Group3-048, S255
Cutter G.R. PS3Group2-033, S351, SS 23.2, S39
Cvijan E. PS1Group1-078, S164
Czech C. PS3Group4-015, S367, PS3Group4-028, S378
Czirjak L. PS1Group1-099, S180
Dabbous O. PS1Group1-020, S118
Dabby R. PS1Group1-069, S158
Da Cunha C.L.P. PS1Group1-006, S106
Dagher R. PS1Group1-060, S151
Dahl-Halvarsson M. PS1Group1-096, S178
Dai D.L. PS2Group5-007, S288
D’Amico A. PS1Group1-027, S123, PS1Group1-119, S195
D’Angelo M.G. PS1Group1-051, S144
D’Antonio M. PS2Group3-081, S283
Darras B. PS2Group5-002, S284, PS3Group4-015, S367
Darras B.T. PS3Group4-014, S366
Das A. PS2Group3-029, S241
Date H. PS1Group1-071, S159
Davies K. PS1Group1-019, S117
Davis M. PS1Group1-096, S178
Day J. PS1Group1-063, S153, PS3Group4-015, S367
Day J.W. PS1Group1-009, S108, PS1Group1-115, S192, PS2Group5-007, S288
De Antonio M. PS1Group1-088, S172
De Bellis M. PS1Group1-028, S124
De Bleecker J. PS1Group1-045, S139, PS2Group3-024, S237
De Bruijne M. PS1Group1-040, S134
De Carvalho M. PS1Group1-058, S149, PS3Group4-020, S372
Deconinck N. PS3Group4-008, S361, PS3Group4-015, S367, PS3Group4-025, S376
Deconinck T. PS1Group1-138, S210
De Fornel P. PS1Group1-008, S108
De Haard H. PS3Group2-004, S327
Deikin A. PS1Group1-076, S163
De Jonghe P. PS1Group1-138, S210
Deleuze J.-F. PS3Group4-006, S360
Delgado P.O. PS1Group1-073, S161, PS1Group1-074, S162
Deliens G. PS3Group4-025, S376
Della Valle C. PS1Group1-021, S119
Delmont E. PS2Group3-065, S269
Delorme D. PS2Group9-001, S310
De Luca A. PS1Group1-028, S124, TC 9.5, S86
Demeestere D. PS2Group3-015, S229, PS3Group2-031, S349
Demeshonok V.S. PS3Group4-021, S373, WS 3.4, S47
De Paepe B. PS1Group1-045, S139
Derchi M. PS1Group1-119, S195
De Ridder W. PS1Group1-022, S119, PS1Group1-138, S210
De Rosa A. PS3Group2-027, S346
De Rover M. PS1Group1-034, S129
De Simoni D. PS2Group3-049, S256
De Stefano N. OA 3.2, S92
Devigili G. TC 8.2, S85
De Visser M. OA 2.1, S90, SS 1.3, S10, SS 16.1, S28, TC 1.1, S74
De Waele L. PS3Group7-001, S380
De Wel B. PS1Group1-081, S166
Dias Henriques S.F. PS1Group1-015, S113
Diaz-Manera J. PS1Group1-004, S104, PS1Group1-063, S153, PS2Group5-007, S288
Dickson J.G. PS1Group1-087, S171
Dieye I. PS1Group1-120, S196
Di Iorio G. PS2Group5-034, S307
Dikhter E. PS3Group4-029, S379, WS 3.4, S47
Dimachkie M. PS2Group3-051, S257, PS3Group2-021, S341, TC 6.2, S84
Di Muzio A. PS1Group1-051, S144
Diodato D. PS1Group1-027, S123
Djordjevic G. PS2Group3-028, S241
Djordjevic S. PS1Group1-059, S150
Djukic M. PS1Group1-059, S150
Do H. PS1Group1-021, S119
Domingos J. PS1Group1-087, S171
Dominovic-Kovacevic A. PS2Group3-028, S241
Donandt T. PS1Group1-065, S155
Doneddu P. WS 5.1, S49
Donovan J. PS1Group1-060, S151
Doorenweerd N. OA 3.3, S92 PS1Group1-034, S129
Dorchies O.M. PS1Group1-026, S122, PS1Group1-050, S143, PS1Group1-061, S152, PS1Group1-098, S179
Dor T. PS1Group1-061, S152
Dowling J.J. PS1Group1-039, S133, TC 4.3, S82
Drexler W. SS 14.3, S26
Duan D. PS1Group1-111, S189
Dubuisson N. PS2Group3-070, S272
Ducci R.D. PS1Group1-006, S106
Dugar A. PS1Group1-114, S191
Dupuis M. PS2Group3-070, S272
Durn B.L. PS2Group3-012, S226, PS2Group3-018, S231, PS2Group3-019, S233, PS2Group3-023, S236, PS2Group3-024, S237, PS2Group3-030, S243, PS2Group3-031, S244, PS2Group3-057, S263
Dworzak J. PS1Group1-049, S143
Dyck P..J.B. TC 3.4, S81, WS 13.2, S58, WS 19.2, S69
Eagle M. PS1Group1-009, S108, PS1Group1-115, S192
Echaniz-Laguna A. PS1Group1-088, S172, PS3Group4-004, S358
Efthimiadis G. PS1Group1-102, S183
Egorova T. PS1Group1-076, S163
Eichhorn P. PS1Group1-002, S102
Eichinger L. PS1Group1-134, S207
Einsiedler S. PS3Group2-018, S338
Ekusheva E. PS2Group3-071, S272
Elfring G.L. PS1Group1-118, S194, PS1Group1-124, S199
Elhawary N. PS1Group1-085, S169
El-Khairi M. PS3Group4-024, S375
Ellis M. PS2Group3-048, S255
Ellouz E. PS2Group3-077, S277
Elsayed N. PS1Group1-085, S169
Embacher N. PS2Group3-076, S276, PS3Group2-018, S338
Engel A.G. PS1Group1-011, S110
England J.D. SS 21.1, S34
Erdem-Ozdamar S. PS2Group3-006, S222
Erdler M. PS2Group3-076, S276
Ernst B. PS2Group3-015, S229, PS3Group2-031, S349
Errguig L. PS2Group5-012, S292
Espinós C. PS2Group3-042, S251, PS2Group3-044, S252, PS2Group3-052, S258, PS2Group3-053, S259
Evangelista T. SS 24.2, S40
Ewers D. PS2Group3-038, S248
Exposito Escudero J. PS2Group5-014, S293
Eymard B. PS3Group2-030, S348
Fabry B. PS2Group5-028, S303
Faelan C. PS1Group1-019, S117, PS2Group9-018, S323
Fahmy N. PS1Group1-085, S169
Faiola A.S. PS2Group3-059, S265
Fajkusova L. PS1Group1-113, S190, PS1Group1-139, S211
Falcão De Campos C. PS1Group1-058, S149
Falzarano M.S. PS2Group5-020, S297
Fang M. PS2Group5-020, S297
Fardeau M. PS3Group2-030, S348
Farrar M.A. PS3Group4-026, S377
Farrugia M.E. PS1Group1-033, S128
Faruq M. PS1Group1-016, S114, PS1Group1-044, S138
Farwell W. PS3Group4-014, S366, PS3Group4-026, S377
Fatehi F. PS1Group1-041, S136, PS1Group1-043, S137
Fathi D. PS1Group1-043, S137
Faust A. PS1Group1-025, S122
Fecchio C. PS1Group1-125,
Feder D. PS1Group1-047, S141, PS1Group1-072, S160, PS1Group1-073, S161, PS1Group1-074, S162, PS1Group1-075, S162, PS1Group1-107, S186, PS2Group3-059, S265
Feldman E. PS2Group3-008, S223, PS2Group3-021, S235, PS2Group3-022, S235, PS3Group4-030, S380, SS 10.3, S20
Felice K. PS1Group1-032, S128
Feng J. PS1Group1-021, S119, PS1Group1-063, S153
Ferlini A. PS2Group5-020, S297
Fernandes J.D.L. PS2Group9-017, S322
Fernández-García M.A. PS1Group1-100, S181
Fernández-Torrón R. PS1Group1-009, S108, PS1Group1-115, S192
Ferrari N. PS1Group1-056, S148
Ferrero B. PS3Group2-024, S343
Ferri C. PS2Group3-081, S283
Fierro B. PS1Group1-127, S202
Figà Talamanca L. PS1Group1-027, S123
Figueiroa S. PS2Group3-003, S219
Filla A. PS2Group5-021, S298
Filosto M. PS1Group1-051, S144
Finkel R.S. PS1Group1-060, S151, PS3Group4-026, S377
Finsterer J. PS1Group1-090, S173, PS1Group1-091, S174
Fiorillo C. PS1Group1-119, S195
Fischer D. PS1Group1-017, S116, PS1Group1-026, S122, PS1Group1-061, S152, PS3Group4-002, S356
Flanigan K. PS2Group9-019, S323
Fominyh M. WS 3.4, S47
Fominyh V. WS 3.4, S47
Fonseca F.L.A. PS1Group1-047, S141, PS1Group1-073, S161, PS1Group1-074, S162, PS1Group1-107, S186, PS2Group3-059, S265
Fontaine B. PS2Group5-027, S302, PS3Group2-030, S348
Fontenille M.-J. PS3Group2-030, S348
Fontoura P. PS3Group4-024, S375
Fonzino A. PS1Group1-028, S124
Fossati B. PS1Group1-110, S188
Foster R. PS3Group4-014, S366
Fournier E. PS3Group2-030, S348
Foust K. PS3Group4-022, S373, SS 13.4, S25
Frank D. PS1Group1-049, S143, PS1Group1-087, S171, PS2Group9-018, S323
Fransi A.R. PS1Group1-089, S173
Franssen H. PS2Group5-008, S289
Frascella M. PS1Group1-021, S119
Frasquet M. PS2Group3-052, S258, PS2Group3-053, S259
Freeman R. SS 9.5, S19
Fretzen A. PS1Group1-060, S151
Frich J. PS3Group7-002, S381
Frisaldi E. PS3Group2-024, S343
Froissart R. PS1Group1-088, S172
Fromes Y. PS1Group1-005, S105
Frongia A.L. PS2Group3-011, S226, PS2Group3-034, S245, PS2Group5-014, S293, PS2Group9-010, S317, PS3Group2-013, S335, PS3Group4-019, S371
Fuentealba M. PS3Group2-009, S331, PS3Group2-010, S332
Fujita K.P. PS3Group2-011, S333, PS3Group2-012, S334, PS3Group2-023, S342, PS3Group2-037, S354, PS3Group2-038, S355
Funato M. PS1Group1-029, S125
Furling D. PS1Group1-144, S214
Furtado F. PS2Group3-062, S267
Fusco L. PS1Group1-027, S123
Gajewski B. PS2Group3-051, S257
Gallano Petit P. PS2Group5-014, S293, PS3Group2-013, S335
Gallardo-Agromayor E. PS1Group1-100, S181
Gandhi P. PS2Group3-005, S220, PS2Group3-010, S225, PS2Group3-055, S261
Garcia A. PS1Group1-021, S119
García-García F. PS2Group3-044, S252
García J. PS1Group1-084, S169
García-Jiménez I. PS1Group1-054, S146
García-Sobrino T. PS2Group3-042, S251
Gargouri A. PS2Group3-077, S277
Garrido C. PS2Group3-003, S219, PS3Group4-013, S365
Gast L.V. PS2Group9-008, S316
Gayi E. PS1Group1-050, S143, PS1Group1-061, S152, PS1Group1-098, S179
Geist J. PS1Group1-035, S130
Gelblin K. PS3Group4-015, S367
Genge A. SS 11.4, S23
Gerber M. PS3Group4-002, S356, PS3Group4-015, S367, PS3Group4-028, S378
Gerevini S. PS1Group1-004, S104
Gerhalter T. PS2Group9-008, S316
Geuens S. PS3Group7-001, S380
Ghaleh B. PS1Group1-005, S105
Ghezzi D. PS1Group1-027, S123
Ghislain Mpandzou A. PS1Group1-068, S157
Gicquel E. PS1Group1-015, S113
Gidaro T. PS1Group1-004, S104
Gide J. PS3Group4-006, S360
Gilhus N.E. WS 18.1, S67
Ginzberg M. PS1Group1-069, S158
Giometto B. SS 3.1, S12
Giordani L.P. PS1Group1-107, S186
Gioulbasanis I. WS 7.1, S52
Gk K. PS3Group4-003, S357
Glaubitz S. PS1Group1-122, S198
Glazere I. PS2Group9-002, S311
Glimcher L. PS2Group3-081, S283
Gloor M. PS1Group1-017, S116
Glumac J. PS1Group1-059, S150
Gocheva V. PS1Group1-017, S116
Godoy A.J. PS2Group9-017, S322
Goedee S. PS2Group5-008, S289, TC 8.1, S84
Goel V. PS2Group3-043, S251, PS2Group3-080, S280
Goemans N. PS1Group1-120, S196, PS3Group4-008, S361, PS3Group4-028, S378, PS3Group7-001, S380, SS 13.3, S25
Goh K.-J. PS1Group1-031, S127
Goldmann W.H. PS2Group5-028, S303
Goldstein J.M. PS3Group2-021, S341
Gollob J. PS2Group3-005, S220, PS2Group3-010, S225, PS2Group3-055, S261
Gomis Coloma C. PS1Group1-046, S140
Gonokami M.K. PS2Group5-004, S286
Gonzalez-Duarte A. PS2Group3-005, S220, PS2Group3-010, S225, PS2Group3-055, S261
González Quereda L. PS2Group5-014, S293, PS3Group2-013, S335
Gonzalez R. PS3Group2-010, S332
Gorbachev K. WS 3.4, S47
Gordish-Dressman H. PS1Group1-114, S191, TC 9.4, S86
Gorman G.S. PS1Group1-033, S128
Gorni K. PS3Group4-002, S356, PS3Group4-015, S367, PS3Group4-024, S375, PS3Group4-028, S378
Gosselin N. PS2Group3-043, S251
Goto J. PS1Group1-071, S159
Goto Y.-I. PS1Group1-097, S178
Gotschall R. PS1Group1-021, S119
Gouider R. PS2Group3-077, S277
Goullée H. PS1Group1-096, S178
Govi M. PS1Group1-051, S144
Grau J.M. PS1Group1-083, S168, PS1Group1-084, S169, PS1Group1-137, S210
Gray J. PS1Group1-004, S104
Greckl E. PS3Group4-009, S362
Grieben U. PS1Group1-115, S192
Grigalioniene K. PS1Group1-132, S206
Grilli A. PS2Group5-020, S297
Grimm A. TC 5.4, S83
Grinzinger S. PS2Group3-076, S276
Grisold A. PS2Group3-026, S239, SS 8.1, S16, SS 17.4, S29, WS 13.1, S57, WS 20.2, S70
Grisold W. SS 8.1, S16, SS 17.4, S29, WS 13.1, S57
Gromicho M. PS3Group4-020, S372
Grouse C.K. WS 11.1, S56
Groves K. PS1Group1-004, S104
Gruber A. PS1Group1-011, S110
Gualandi F. PS2Group5-020, S297
Guanyabens N. PS1Group1-089, S173
Guerin A. PS1Group1-020, S118
Guerreiro R. PS3Group2-034, S351
Gueven N. PS1Group1-017, S116
Guger M. PS3Group2-018, S338
Guglieri M. PS1Group1-087, S171
Guglietta A. PS3Group2-004, S327
Guida M. PS3Group2-027, S346
Gulyas K. PS1Group1-099, S180
Guptill J.T. PS3Group2-012, S334
Gutierrez-Gutierrez G. PS1Group1-100, S181
Haberlova J. PS1Group1-113, S190, PS2Group3-020, S234, PS2Group9-007, S315
Hackel N. PS3Group4-009, S362
Hafler D.A. PS3Group2-021, S341
Hafner P. PS1Group1-017, S116, PS1Group1-026, S122
Haghiashtiani B. PS2Group5-009, S290
Haghi Ashtiani B. PS1Group1-043, S137
Haghi B. PS2Group3-065, S269
Hajjar R. PS1Group1-111, S189
Hajj R. PS2Group3-038, S248, PS2Group3-069, S271
Hakim M. PS3Group2-032, S350
Hamed S.A. PS2Group3-067, S270
Hamilton L.E. PS1Group1-011, S110
Hamroun D. PS1Group1-088, S172
Hänggi P. PS2Group3-015, S229, PS3Group2-031, S349
Han L. PS1Group1-114, S191
Hanna M. TC 6.4, S84
Hardiman O. WS 12.2, S57
Haridy N.A.A. PS2Group3-067, S270
Harlaar L. PS1Group1-030, S126, PS1Group1-040, S134
Harlay M. PS3Group4-004, S358
Harms M. PS1Group1-115, S192, PS2Group5-007, S288
Harris L. PS3Group2-033, S351, PS3Group7-004, S383
Hartung H.-P. PS2Group3-012, S226, PS2Group3-018, S231, PS2Group3-019, S233, PS2Group3-023, S236, PS2Group3-024, S237, PS2Group3-030, S243, PS2Group3-031, S244, PS2Group3-057, S263
Hasegawa S. PS1Group1-052, S145
Hasham S. PS1Group1-057, S148, PS1Group1-105, S185, PS2Group5-023, S299, PS2Group9-003, S312, PS2Group9-016, S321
Hashiguchi A. PS1Group1-136, S209
Hasoglu Y. PS2Group9-018, S323
Hasselmann O. PS2Group5-006, S288
Hattori A. PS1Group1-029, S125
Hausner T. SS 2.2, S11, WS 9.3, S55
Hayhurst Bennett M. PS1Group1-023, S120, PS1Group1-024, S121
Heatherington A. PS1Group1-019, S117, PS2Group9-009, S316
Heckmann J.M. PS2Group5-019, S297
Hegen H. PS2Group3-049, S256
Heller C. PS2Group3-074, S275
Hendriksen J.G.M. PS1Group1-034, S129
Henricson E. PS1Group1-114, S191
Heo S.-J. PS3Group2-015, S336
Herbelet S. PS1Group1-045, S139
Herbelin L. PS2Group3-051, S257, PS3Group2-021, S341
Herbrecht R. PS3Group4-004, S358
Hercher D. SS 2.3, S12
Herraets I. PS2Group5-008, S289
Herrendorff R. PS2Group3-015, S229, PS3Group2-031, S349
Heß-Eberle I. PS2Group3-076, S276
Hettwer S. PS3Group2-029, S347
Heusermann W. PS2Group3-015, S229
Higuchi I. PS1Group1-136, S209
Hilger E. PS3Group2-020, S340
Hilsden H. PS1Group1-115, S192
Hilz M.J. SS 9.3, S19, TC 3.3, S80
Hinderer C. PS2Group9-013, S319
Hiroyuki A. PS1Group1-003, S103
Hobson E. SS 16.2, S28
Ho C.-Y. PS2Group3-045, S253
Hoeftberger R. PS2Group3-049, S256, SS 3.2, S12
Hofer M. WS 19.1, S68
Höger S. PS2Group3-076, S276
Hogrel J.-Y. PS1Group1-115, S192
Hoke A. SS 6.2, S14, WS 9.2, S55
Holinski-Feder E. PS2Group5-016, S295
Hollander Z. PS2Group5-007, S288
Hollingsworth K.G. PS1Group1-034, S129
Holvoet B. PS1Group1-008, S108
Hong Y.-H. PS1Group1-131, S205, PS1Group1-141, S213, PS3Group2-035, S352
Hooijmans M.T. PS1Group1-095, S177
Hordeaux J. PS2Group9-013, S319
Horlings C.G. PS1Group1-001, S102, PS1Group1-010, S109, PS1Group1-109, S188
Horn S. SS 22.1, S36
Horvath R. PS1Group1-044, S138, PS2Group3-060, S265, PS3Group2-022, S342, SS 24.2, S40
Houlden H. PS2Group3-067, S270
Howard Jr J.F. PS3Group2-004, S327, PS3Group2-011, S333, PS3Group2-012, S334, PS3Group2-023, S342, PS3Group2-037, S354, PS3Group2-038, S355
Hubsch A. PS2Group3-018, S231
Huemer M. PS2Group3-076, S276
Huh C. PS1Group1-141, S213
Hulova M. PS1Group1-037, S132
Human A. PS1Group1-064, S154
Huq S. PS3Group2-021, S341
Ibraheem H. PS3Group2-024, S343
Igarashi S. PS2Group6-001, S308
Iglseder S. PS3Group2-018, S338
Iijima K. PS1Group1-003, S103, PS1Group1-128, S202
Ikenaga C. PS1Group1-071, S159
Ikenberg E. PS2Group3-074, S275
Ilchenko M. WS 3.4, S47
Imanishi T. PS1Group1-003, S103
Im S. PS2Group3-047, S254
Inashkina I. PS1Group1-035, S130
Indrawati L.A. PS3Group2-032, S350
Inoue S. PS1Group1-052, S145
Iodice R. PS2Group5-021, S298
Irani S. PS2Group3-032, S244
Irastorza A. PS1Group1-038, S132
Irioka T. PS2Group6-001, S308
Irkec M. PS2Group3-006, S222
Ishigaki K. PS1Group1-029, S125
Ishii A. PS2Group5-029, S304
Ishii K. PS2Group5-029, S304
Ishikawa Y. PS1Group1-128, S202, PS1Group1-128, S202
Ishiura H. PS1Group1-071, S159
Ishiyama A. PS1Group1-097, S178
Ismail H.M. PS1Group1-050, S143, PS1Group1-061, S152
Itzep D. PS1Group1-013, S112, PS2Group3-011, S226, PS2Group3-034, S245
Ivanova M. WS 3.4, S47
Ivanov T. WS 3.4, S47
Ives J. PS3Group4-024, S375
Iwata A. PS2Group5-004, S286
Jackson S. PS1Group1-035, S130, PS1Group1-117, S194
Jacob S. PS3Group2-011, S333, PS3Group2-012, S334, PS3Group2-023, S342
Jacobsen L. PS1Group1-014, S112
Jacobs M. PS1Group1-063, S153, PS1Group1-115, S192, PS2Group5-007, S288
Jakubiczka S. PS2Group5-016, S295
James M. PS1Group1-009, S108, PS1Group1-063, S153, PS1Group1-115, S192
Jang H. PS1Group1-133, S206
Jang J. PS1Group1-115, S192
Jang Y. PS2Group3-047, S254
Jankelowitz S. PS1Group1-062, S153
Jankovic M. PS1Group1-080, S165
Jankovits R. PS2Group3-036, S247
Jawdat O. PS2Group3-051, S257
Jaworski M. PS1Group1-048, S142
Jecel J. PS2Group3-076, S276
Jelsma J. PS1Group1-064, S154
Jenkins M. PS1Group1-120, S196
Jiang H. PS3Group4-012, S365
Jimenez Mallebrera C. PS1Group1-013, S112, PS2Group5-014, S293, PS3Group2-013, S335, PS3Group4-019, S371
Jimenez-Moreno C. PS1Group1-147, S216
Jin F. PS1Group1-123, S198, PS1Group1-126, S200
Johnson K. PS1Group1-138, S210
Jomphe C. PS2Group3-043, S251
Jones K.J. PS1Group1-009, S108, PS1Group1-063, S153, PS1Group1-115, S192, PS2Group5-007, S288
Jonker M.A. PS1Group1-001, S102, PS1Group1-109, S188
Joo I.S. PS2Group3-068, S271
Joshi A. PS1Group1-016, S114, PS1Group1-044, S138
Jou C. PS1Group1-013, S112, PS1Group1-137, S210, PS2Group5-014, S293, PS3Group2-013, S335, PS3Group4-019, S371
Jousserand G. PS1Group1-066, S156
Jouvenal P.V. PS1Group1-103, S184
Jovanovic I. PS1Group1-059, S150
Juergen D. PS1Group1-014, S112
Jullien De Pommerol H. PS3Group4-017, S369
Jung S.-H. PS3Group2-014, S336
Kably B. PS1Group1-067, S157, PS1Group1-068, S157, PS2Group5-012, S292, PS2Group5-013, S292
Kacar A. PS1Group1-077, S163, PS2Group3-028, S241
Kacem I. PS2Group3-077, S277
Kah D. PS2Group5-028, S303
Kaji R. TC 7.1, S84
Kalbe C. PS1Group1-065, S155
Kalf J.G. PS1Group1-010, S109
Kalicanin Milanovic R. PS1Group1-059, S150
Kalil I.D.L. PS2Group9-017, S322
Kaliszewska M. PS1Group1-036, S131
Kamienowski J. PS2Group3-024, S237
Kaminska A.M. PS1Group1-036, S131
Kaminski H.J. SS 23.2, S39
Kanáková J. PS2Group3-020, S234
Kan H.E. PS1Group1-034, S129, PS1Group1-095, S177
Kapur K. PS2Group5-002, S284
Kariminejad A. PS1Group1-096, S178
Karussis D. OA 1.3, S90
Kasianova A. PS3Group4-029, S379, WS 3.4, S47
Kaspar B. PS3Group4-022, S373, SS 13.4, S25
Kastenschmidt J. PS2Group5-031, S306
Katzberg H. PS3Group2-005, S328
Katz N. PS2Group9-013, S319
Kaur S. PS1Group1-025, S122
Kay C.S.K. PS1Group1-006, S106
Kazaine I. PS2Group9-002, S311
Kelly C. PS1Group1-055, S147
Kelly M. TC 9.1, S86
Kennerson M.L. PS2Group3-048, S255
Keshavarz E. PS1Group1-096, S178
Kessler B. PS1Group1-065, S155
Khanna R. PS1Group1-021, S119
Khidiyatova I. PS2Group3-058, S264
Khongkhatithum C. PS3Group2-006, S328
Khusnutdinova E. PS2Group3-058, S264
Khwaja O. PS3Group4-002, S356, PS3Group4-015, S367, PS3Group4-028, S378
Ki C.-S. PS1Group1-145, S215
Kieloch A. PS3Group4-017, S369
Kierdaszuk B. PS1Group1-036, S131
Kiernan M.C. SS 15.2, S27, SS 18.1, S30, SS 19.3, S32
Kiklic M. PS1Group1-059, S150
Kilfoyle D. PS2Group5-021, S298
Kim A.W. PS1Group1-131, S205, PS1Group1-141, S213
Kim B. PS2Group3-032, S244
Kim B.-J. PS2Group5-017, S295
Kim C.-H. PS2Group3-009, S224
Kim D.-S. PS3Group2-014, S336
Kim J.A. PS1Group1-131, S205, PS1Group1-141, S213
Kim J.E. PS3Group2-035, S352
Kimminau K. PS2Group3-051, S257
Kim R.B. PS2Group3-033, S245
Kim S. PS2Group5-026, S301
Kim S.-J. PS2Group3-032, S244
Kim S.M. PS1Group1-131, S205, PS2Group3-032, S244
Kimura T. PS1Group1-029, S125
Kim Y.-D. PS2Group3-078, S278
Kim Y.H. PS2Group5-017, S295
Kirschner J. PS2Group9-019, S323, PS3Group4-014, S366, PS3Group4-024, S375, PS3Group4-028, S378, SS 13.1, S24
Kissel J.T. PS3Group2-021, S341, PS3Group4-022, S373, SS 13.4, S25
Kitanosono H. PS2Group6-001, S308
Klein A. PS1Group1-017, S116, PS3Group4-015, S367
Kleindienst W. PS2Group3-076, S276
Kletzl H. PS3Group4-002, S356, PS3Group4-015, S367, PS3Group4-028, S378
Klinker F. PS1Group1-103, S184
Knut B. PS3Group4-006, S360
Kobayashi M. PS1Group1-029, S125
Kocabeyoglu S. PS2Group3-006, S222
Kochanowski J. PS2Group5-011, S291
Koch M.E. PS1Group1-073, S161, PS1Group1-074, S162
Kodama K. PS1Group1-136, S209
Koehorst E.A. PS1Group1-089, S173
Koga Y. PS2Group5-029, S304
Koh Y.H. PS2Group3-072, S273
Kok C.Y. PS2Group3-001, S218
Kokotovic T. PS2Group3-026, S239
Kolb S. SS 13.4, S25
Kollmer J. SS 14.1, S26, WS 6.3, S52,
Kolski H. PS2Group6-003, S310
Komaki H. PS1Group1-029, S125, PS1Group1-052, S145, PS1Group1-092, S175, PS1Group1-097, S178, PS1Group1-143, S214
Koneczny I. PS3Group2-036, S353
Kong R. PS1Group1-126, S200
Kon S. PS3Group4-001, S356
Kontrogianni- PS1Group1-035, S130Konstantopoulos A.
Koo H.J. PS2Group3-047, S254
Korneychuk S. WS 3.4, S47
Kosac A. PS1Group1-059, S150, PS1Group1-080, S165
Kostera-Pruszczyk A. PS1Group1-036, S131
Kosuga M. PS1Group1-029, S125
Koumako C. PS2Group9-006, S314
Kovacevic M. PS1Group1-078, S164
Kralickova E. PS1Group1-037, S132
Krasnaya Y. PS3Group4-029, S379, WS 3.4, S47
Kraus D. PS3Group4-002, S356
Krause S. PS1Group1-065, S155, PS1Group1-115, S192, PS2Group3-040, S249
Krenn M. PS2Group3-026, S239
Krishnan M. PS1Group1-014, S112
Kristen A. PS2Group3-005, S220, PS2Group3-010, S225, PS2Group3-055, S261
Kroon H.M.J.M. PS1Group1-010, S109
Krosschell K.J. PS3Group4-026, S377
Kubota A. PS1Group1-071, S159
Kucinskas V. PS1Group1-132, S206
Kumar A. PS3Group2-019, S339
Kumar P. PS2Group3-027, S240, PS2Group3-045, S253
Kumar V..P. PS3Group2-022, S342
Kuntz N. PS1Group1-039, S133
Kuo H.-C. SS 23.2, S39
Kuru S. PS1Group1-029, S125, PS1Group1-112, S190, PS1Group1-143, S214
Kwan W.S.K. PS2Group3-025, S238
Kwon Y.-J. PS2Group5-017, S295
Kwun K.H. PS1Group1-131, S205, PS1Group1-141, S213
Kyriakides T. TC 1.4, S75
Labarge S. PS1Group1-024, S121
Labate V. PS1Group1-056, S148
Labolle D. PS1Group1-004, S104
Labove L. PS1Group1-048, S142
Lace B. PS1Group1-035, S130
Lacour A. PS3Group2-030, S348
Lacson A. PS2Group6-003, S310
Ladha S. PS2Group3-007, S222, PS3Group2-029, S347
Laffaire J. PS2Group3-038, S248, PS2Group3-069, S271
Laforet P. PS1Group1-066, S156, PS1Group1-088, S172, PS3Group2-030, S348
Lahoz D.C. PS2Group5-033, S307
Laich E. PS2Group3-076, S276
Laing N. PS1Group1-096, S178
Lamartine Monteiro M. PS2Group5-001, S284
Lamperti C. PS1Group1-027, S123
Landon-Cardinal O. PS2Group9-006, S314
Langenscheidt D. PS3Group2-018, S338
Lapin V. PS3Group4-021, S373
Laporte J.F. PS1Group1-050, S143, PS1Group1-061, S152
Larin E. PS3Group4-029, S379, WS 3.4, S47
Larrañaga I. PS1Group1-108, S187
Lasa Elgarresta J. PS1Group1-038, S132
Lasa-Elgarresta J. PS1Group1-106, S186
Lasa Fernandez H. PS1Group1-038, S132
Lasa-Fernandez H. PS1Group1-106, S186
Lasner I. PS2Group3-017, S230
Laššuthová P. PS2Group3-020, S234, PS2Group9-007, S315
Laugel V. PS1Group1-017, S116
Laun F.B. PS2Group9-008, S316
Lauria G. WS 19.3, S69
Laveille C. PS1Group1-004, S104
Lavrnic D. PS1Group1-078, S164, PS1Group1-080, S165, PS1Group1-104, S185
Lawlor M.W. PS1Group1-039, S133
Lawo J.-P. PS2Group3-012, S226, PS2Group3-018, S231, PS2Group3-019, S233, PS2Group3-023, S236, PS2Group3-024, S237, PS2Group3-030, S243, PS2Group3-031, S244, PS2Group3-057, S263
Layton G. PS1Group1-019, S117, PS2Group9-009, S316
Leary R. PS2Group9-019, S323
Lechner-Scott J. PS2Group3-056, S262
Lee E. PS2Group5-007, S288
Lee F. PS3Group4-015, S367
Lee H.-J. PS2Group3-032, S244
Lee H.J. PS2Group3-050, S257
Lee J. PS2Group5-003, S285, PS2Group5-024, S300, PS2Group9-019, S323
Lee J.H. PS1Group1-145, S215, PS2Group3-073, S274
Lee Y.-C. PS2Group3-004, S220, PS2Group3-016, S230
Lee Y.J. PS1Group1-133, S206, PS3Group2-015, S336
Legay C. PS3Group2-028, S347
Léger J.-M. PS2Group3-024, S237, PS2Group3-064, S268, PS2Group3-065, S269, WS 5.2, S50
Leguern E. PS2Group3-077, S277
Lehmann D. PS1Group1-012, S111
Leinonen M. PS1Group1-057, S148, PS1Group1-105, S185, PS2Group5-023, S299, PS2Group9-003, S312, PS2Group9-016, S321
Le Louër J. PS1Group1-095, S177
Lemmers R.J. PS1Group1-001, S102, PS1Group1-109, S188
Lenzenweger E. PS2Group3-076, S276
Leontyeva T. WS 3.4, S47
Lepessova M. PS3Group4-010, S363
Leppert D. PS3Group4-017, S369
Lerman-Sagie T. PS1Group1-069, S158
Leshinsky-Silver E. PS1Group1-069, S158
Lesueur L. PS1Group1-144, S214
Letard P. PS1Group1-088, S172
Leupin N. PS3Group2-004, S327
Lev D. PS1Group1-069, S158
Lewis R.A. PS2Group3-012, S226, PS2Group3-018, S231, PS2Group3-019, S233, PS2Group3-023, S236, PS2Group3-024, S237, PS2Group3-031, S244, PS2Group3-057, S263
Lewis S.L. WS 11.2, S56
Liao Y.-C. PS2Group3-004, S220
Liberatore G. WS 5.1, S49
Liebetanz D. PS1Group1-103, S184
Likhite S. PS3Group4-022, S373, SS 13.4, S25
Li M. PS3Group2-029, S347
Lim E.W. PS2Group3-072, S273
Linares I. PS1Group1-089, S173
Lindeck-Pozza E. PS2Group3-049, S256, SS 17.4, S29
Lin K.-P. PS2Group3-004, S220, PS2Group3-005, S220, PS2Group3-010, S225, PS2Group3-016, S230, PS2Group3-055, S261
Lin T. PS2Group3-010, S225
Lipowska M. PS2Group3-002, S218
L’Italien G.J. PS3Group2-033, S351
L’Italien J. PS3Group4-022, S373, SS 13.4, S25
Litchman T.D. PS3Group2-019, S339
Litchy W.J. PS1Group1-129, S203
Liu H. PS1Group1-060, S151
Liu T. PS2Group3-051, S257
Liu W. PS3Group2-036, S353
Liu Y.-T. PS2Group3-016, S230
Li Z. PS3Group2-029, S347
Llorens J.M. PS2Group5-033, S307
Lochmüller H. PS1Group1-115, S192, PS2Group5-007, S288
PL 1.3, S3, PS1Group1-044, S138, PS3Group2-022, S342, SS 24.2, S40, TC 2.4, S78
Loho T. PS3Group2-032, S350
Lombardi L. PS2Group5-034, S307
Long L. PS3Group7-003, S382
Longman C. PS1Group1-033, S128
López De Munain A. PS1Group1-038, S132, PS1Group1-106, S186
Lorenzl S. WS 3.3, S47
Lorenzoni P.J. PS1Group1-006, S106
Löscher W. PS2Group3-049, S256, PS2Group3-076, S276, PS3Group2-018, S338
Lostal W. PS1Group1-111, S189
Lowes L. PS3Group4-022, S373, SS 13.4, S25
Lucas K. PS1Group1-049, S143
Lucchi M. PS3Group2-027, S346
Lucente G. PS1Group1-089, S173
Lucia A. PS1Group1-089, S173
Ludolph A. PL 3.1, S5, SS 13.2, S25, TC 7.2, S84
Ludo Van Der Pol W. PS3Group4-024, S375
Lui Q. PS3Group2-029, S347
Lu J. PS1Group1-086, S170
Lulé D. TC 7.3, S84
Lun Y. PS1Group1-021, S119
Lupescu T. WS 20.1, S70
Lupo V. PS2Group3-042, S251, PS2Group3-044, S252, PS2Group3-052, S258, PS2Group3-053, S259
Luque H. PS1Group1-053, S146
Lusakowska A. PS3Group4-024, S375
Lütschg J. PS2Group5-006, S288
Lu Z. PS2Group5-020, S297
Ly C. PS2Group3-048, S255
Lynch J. PS1Group1-086, S170
Lyon K. PS1Group1-023, S120
Lysogorskaya E. WS 3.4, S47
Macarthur D. PS1Group1-138, S210
Macklin E. PS3Group4-018, S370
Madia F. PS1Group1-119, S195
Madigan N.N. PS1Group1-129, S203
Maeda N. PS1Group1-052, S145
Maestri M. PS3Group2-027, S346
Maggi L. PS1Group1-051, S144
Magzhanov R. PS2Group3-058, S264
Maier O. PS2Group5-006, S288
Maioli M.A. PS1Group1-051, S144
Maiti A. PS2Group3-029, S241
Majima T. PS2Group6-001, S308
Makioka N. PS1Group1-097, S178
Maksimovic R. PS1Group1-080, S165
Mallick R. PS2Group3-018, S231
Malzahn D. PS1Group1-103, S184
Mammen A. PL 1.2, S3, SS 3.3, S13, WS 4.2, S49
Mancini M. PS1Group1-060, S151
Mandic-Stojmenovic G. PS1Group1-078, S164
Manel V. PS3Group2-030, S348
Mane M. PS2Group9-001, S310
Manera J.D. PS1Group1-009, S108, PS1Group1-115, S192
Manganelli F. PS2Group5-021, S298
Manin A. PS3Group2-007, S330, PS3Group2-008, S330
Manji H. PS2Group3-001, S218, WS 17.2, S65
Mantegazza R. PS3Group2-004, S327, PS3Group2-037, S354, PS3Group2-038, S355
Mantuano P. PS1Group1-028, S124
Maqueda E. PS2Group3-034, S245
Marchel M. PS2Group5-011, S291
Maré R. PS3Group2-025, S345
Maresh K. PS1Group1-004, S104
Marier J.-F. PS2Group3-043, S251
Marini-Bettolo C. PS1Group1-034, S129
Marin T.A. PS1Group1-047, S141
Marjanovic A. PS1Group1-077, S163, PS1Group1-080, S165
Marosi C. WS 7.4, S53
Marquet A. PS3Group4-028, S378
Marsolier J. PS1Group1-015, S113
Martens K. PS1Group1-011, S110
Martic V. PS2Group3-028, S241
Martinat C. PS1Group1-144, S214, PS3Group4-006, S360
Martinez M.S. PS2Group5-033, S307
Martínez-Piñeiro A. PS1Group1-089, S173
Martins J. PS3Group2-025, S345
Martí P. PS1Group1-046, S140
Martorell L. PS2Group3-034, S245
Marty B. PS1Group1-004, S104, PS2Group9-008, S316
Maruyama N. PS1Group1-128, S202
Marx A. SS 23.2, S39, SS 23.3, S40, WS 18.3, S68
Mas F. PS2Group3-052, S258
Masrori P. PS1Group1-022, S119
Massana-Muñoz X. PS1Group1-050, S143, PS1Group1-061, S152
Massey C. PS1Group1-147, S216
Masson R. PS3Group4-015, S367
Mastrangelo R. PS2Group3-081, S283
Mata J.F. PS2Group9-010, S317
Mathukomali N. PS2Group3-039, S249
Mathur A. PS1Group1-016, S114, PS1Group1-044, S138
Matsumoto M. PS1Group1-128, S202
Matsumura T. PS1Group1-029, S125, PS1Group1-143, S214
Matsuo M. PS1Group1-003, S103, PS1Group1-052, S145, PS1Group1-128, S202
Matsushima Y. PS1Group1-097, S178
Matsuura E. PS2Group9-012, S319
Maturana F. PS3Group2-009, S331
Mauduit D. PS1Group1-008, S108
Mauermann M. PS2Group3-005, S220, SS 17.3, S29, WS 5.3, S51
Mayer O.H. PS1Group1-057, S148, PS1Group1-105, S185, PS2Group5-023, S299, PS2Group9-003, S312, PS2Group9-016, S321
Mayhew A. PS1Group1-009, S108, PS1Group1-063, S153, PS1Group1-115, S192, PS2Group5-007, S288, TC 9.8, S87
Mazanec R. PS1Group1-037, S132, PS1Group1-113, S190, PS1Group1-139, S211
Mcdermott C. TC 7.4, S84, WS 3.2, S47
Mcdonald C. PS1Group1-114, S191, PS1Group1-118, S194, PS1Group1-123, S198
PS1Group1-124, S199
Mcdonald E. PS1Group1-120, S196
Mcfarland R. PS1Group1-033, S128
Mcgovern V. SS 13.4, S25
Mcintosh J. PS1Group1-124, S199, PS1Group1-126, S200
Mcmacken G. SS 24.2, S40
Mcmanus B. PS2Group5-007, S288
Medina J. PS2Group9-010, S317, PS3Group4-019, S371
Meena A.K. PS2Group3-039, S249
Mehrabyan A.C. PS1Group1-140, S212
Mehr Pour M. PS2Group5-009, S290
Mehta V. PS2Group6-003, S310
Meier T. PS1Group1-057, S148, PS1Group1-105, S185, PS2Group5-023, S299, PS2Group9-016, S321
Meisel A. PS3Group2-012, S334
Mele F. PS1Group1-051, S144
Melfi F. PS3Group2-027, S346
Menassa R. PS1Group1-066, S156
Mendell J. PS1Group1-063, S153, PS2Group5-007, S288, SS 13.4, S25
Mendell J.R. PS1Group1-115, S192, PS3Group4-022, S373
Mendoza M. PS3Group2-005, S328
Meng J. PS1Group1-070, S159
Meng S. SS 8.1, S16, SS 14.2, S26, SS 17.4, S29, WS 6.1, S51, WS 13.1, S57
Mensova L. PS1Group1-037, S132
Meola G. PS1Group1-042, S136, PS1Group1-056, S148, PS1Group1-110, S188
Mercuri E. PS1Group1-087, S171, PS1Group1-123, S198, PS1Group1-124, S199, PS3Group4-002, S356, PS3Group4-014, S366, PS3Group4-015, S367, PS3Group4-024, S375, PS3Group4-028, S378, TC 4.2, S82
Merkies I. PS2Group3-012, S226, PS2Group3-018, S231, PS2Group3-019, S233, PS2Group3-023, S236, PS2Group3-024, S237, PS2Group3-030, S243, PS2Group3-031, S244, PS2Group3-057, S263, TC 8.3, S85, WS 8.3, S54
Meshram C. SS 21.4, S36
Messéant J. PS2Group5-027, S302, PS3Group2-030, S348
Meyer K. PS3Group4-022, S373, SS 13.4, S25
Michaelidou K. PS1Group1-102, S183
Mielke O. PS2Group3-012, S226, PS2Group3-018, S231, PS2Group3-019, S233, PS2Group3-023, S236, PS2Group3-024, S237, PS2Group3-030, S243, PS2Group3-031, S244, PS2Group3-057, S263
Milenkovic S. PS1Group1-080, S165
Milic Rasic V. PS1Group1-059, S150, PS1Group1-080, S165
Milisenda J.C. PS1Group1-083, S168, PS1Group1-084, S169
Milisneda J.C. PS1Group1-137, S210
Miller T. SS 18.2, S30
Milone M. PS1Group1-011, S110, PS1Group1-129, S203
Mimaki M. PS1Group1-097, S178
Minetti C. PS1Group1-119, S195
Minier T. PS1Group1-099, S180
Minisman G. SS 23.2, S39
Miossec O. PS2Group3-044, S252
Mirabella M. PS1Group1-127, S202
Miranda J.I. PS1Group1-038, S132, PS1Group1-106, S186
Mirza Asgari Z. PS2Group5-009, S290
Mittal P. PS1Group1-115, S192, PS2Group5-007, S288
Mix C. PS1Group1-086, S170
Miyake Z. PS2Group5-029, S304
Mladenovic J. PS1Group1-059, S150
Moggio M. PS1Group1-051, S144
Moldovan O. PS2Group3-062, S267
Molin C.J. PS3Group2-001, S324
Monakhova A. PS2Group9-015, S320
Monforte M. PS1Group1-087, S171
Mongini T. PS1Group1-051, S144
Monlleó-Neila L. PS1Group1-089, S173
Montagnese F. PS1Group1-002, S102
Montes V. PS3Group2-034, S351
Moore U. PS1Group1-115, S192
Moreno M.V. PS2Group5-033, S307
Moreno P.J. PS1Group1-083, S168
Moreno T. PS2Group3-062, S267
Morgan J. PS1Group1-070, S159, PS1Group1-087, S171
Morgese M.G. PS1Group1-028, S124
Mori Y. PS2Group5-005, S287, PS2Group5-018, S296
Mori-Yoshimura M. PS1Group1-063, S153, PS1Group1-115, S192
Morrow B.M. PS1Group1-064, S154
Morrow J.M. WS 4.3, S49
Moschou M. PS1Group1-102, S183
Motamed M.R. PS2Group5-009, S290
Motomura M. PS2Group6-001, S308
Moya O. PS2Group9-010, S317, PS3Group4-019, S371
Mozaffar T. PS2Group5-031, S306, TC 6.3, S84
Muchart J. PS1Group1-013, S112
Muelas N. PS1Group1-046, S140, PS2Group3-053, S259
Mueller O. WS 15.4, S62
Mukhametov T. WS 3.4, S47
Mulder N. PS2Group5-019, S297
Muley S. PS3Group2-029, S347
Muley S.A. PS2Group3-007, S222
Mul K. PS1Group1-109, S188
Müller-Felber W. PS1Group1-039, S133
Müller P. PS3Group2-018, S338
Mulroy E. PS2Group5-021, S298
Muni Lofra R. PS1Group1-009, S108
Munoz K. PS2Group5-031, S306
Muntoni F. PS1Group1-004, S104, PS1Group1-019, S117, PS1Group1-070, S159, PS1Group1-087, S171, PS1Group1-118, S194, PS1Group1-123, S198 PS2Group9-009, S316, PS2Group9-018, S323, PS3Group4-024, S375
Muppidi S. PS3Group2-038, S355
Murai H. PS3Group2-011, S333
Muramatsu K. PS1Group1-097, S178
Musset L. PS2Group3-065, S269
Myers K. PS3Group4-018, S370
Myrzaliyeva B. PS3Group4-010, S363
Naarding K.J. PS1Group1-095, S177
Nabeshima Y.-I. PS1Group1-128, S202
Nadais G. PS3Group2-025, S345
Nadaj-Pakleza A. PS3Group2-030, S348
Naddaf E. PS1Group1-011, S110, PS1Group1-055, S147
Nafissi S. PS1Group1-041, S136, PS1Group1-043, S137
Nagai M. PS1Group1-128, S202
Nagaraju K. SS 1.2, S10
Nagashima Y. PS2Group5-004, S286
Nagel A.M. PS2Group9-008, S316
Nagels K. PS2Group3-040, S249
Nagendran S. PS3Group4-022, S373, SS 13.4, S25
Nagy V. PS2Group3-026, S239
Nair A. PS1Group1-021, S119
Nakamachi Y. PS1Group1-003, S103
Nakamagoe K. PS2Group5-029, S304
Nakamura H. PS1Group1-092, S175
Nakayama T. PS1Group1-112, S190, PS1Group1-143, S214
Nalini A. PS1Group1-016, S114, PS1Group1-018, S116, PS1Group1-044, S138, PS2Group5-010, S291, PS2Group9-011, S318, PS3Group4-003, S357
Namgung D.W. PS3Group4-016, S368
Narayanappa G. PS1Group1-016, S114
Narayanaswami P. SS 11.3, S22
Nascimento A. PS1Group1-013, S112, PS1Group1-026, S122, PS2Group3-011, S226, PS2Group3-034, S245, PS2Group5-014, S293, PS2Group9-010, S317, PS3Group2-013, S335, PS3Group4-019, S371
Nashi S. PS1Group1-016, S114, PS1Group1-018, S116, PS1Group1-044, S138, PS2Group5-010, S291, PS3Group2-022, S342, PS3Group4-003, S357
Natali Sora M.G. PS1Group1-004, S104
Natera De Benito D. PS1Group1-013, S112, PS2Group3-011, S226, PS2Group3-034, S245, PS2Group5-014, S293, PS2Group9-010, S317, PS3Group2-013, S335, PS3Group4-019, S371
Nave K.A. PS2Group3-038, S248
Nave S. PS3Group4-015, S367, PS3Group4-028, S378
Nayak S. PS1Group1-016, S114, PS1Group1-044, S138
Nazarov V. PS3Group4-021, S373
Neff L.A. PS1Group1-050, S143, PS1Group1-061, S152, PS1Group1-098, S179
Neil J. PS2Group3-065, S269
Nel M. PS2Group5-019, S297
Neri M. PS2Group5-020, S297
Neuronext Nn103 Beatmg PS3Group2-021, S341 Study Team .
Ng K. PS2Group3-037, S247, PS2Group5-003, S285, PS2Group5-024, S300
Ng R. PS2Group5-007, S288
Nguyen C.-T.E. PS1Group1-048, S142, PS3Group2-026, S345
Nguyen D. PS3Group2-026, S345
Nguyen T. PS2Group3-056, S262
Ng Y.S. PS1Group1-033, S128
Nichols A. PS1Group1-060, S151
Nicholson G.A. PS2Group3-048, S255
Nicole S. PS3Group2-030, S348
Nigro V. PS1Group1-119, S195, WS 16.3, S63
Nikitin S. PS2Group3-002, S218
Nikolenko N. PS1Group1-147, S216
Nikolic A. PS2Group3-028, S241
Niks E.H. PS1Group1-034, S129, PS1Group1-095, S177
Nilipour Y. PS1Group1-041, S136, PS1Group1-043, S137
Niranjan N. PS1Group1-111, S189
Nishino I. PS1Group1-092, S175, PS1Group1-097, S178
Nisic T. PS1Group1-104, S185
Nissan X. PS1Group1-144, S214
Niu Z. PS1Group1-011, S110, PS1Group1-129, S203
Njike V. PS3Group2-019, S339
Nobile-Orazio E. PS2Group3-064, S268, WS 5.1, S49, WS 8.1, S53
Noel V. PS2Group9-001, S310
Nogales-Gadea G. PS1Group1-089, S173
Nohara S. PS2Group5-029, S304
Nonaka I. PS1Group1-092, S175
Notas K. PS1Group1-102, S183
Noursalehi M. PS1Group1-039, S133
Nowak R.J. PS3Group2-011, S333, PS3Group2-019, S339, PS3Group2-021, S341
Nuñez J. PS1Group1-089, S173
Oberreiter E.-M. PS2Group3-076, S276
Obici L. PS2Group3-010, S225, WS 10.1, S55
O’Brien F. PS3Group2-011, S333, PS3Group2-023, S342, PS3Group2-037, S354, PS3Group2-038, S355
O’Connor E. SS 24.2, S40
O’Connor K.C. PS3Group2-021, S341
Octaviana F. PS3Group2-032, S350
Oel D. PS2Group3-076, S276, PS3Group2-018, S338
Oertl W. PS2Group3-076, S276
Ogasawara Y. PS3Group4-001, S356
Ogata K. PS1Group1-029, S125
Ogata T. PS1Group1-097, S178
Oh J. PS2Group3-054, S260
Ohno K. PL 4.2, S6
Okune S. PS2Group5-029, S304
Okuyama T. PS1Group1-029, S125
Oliveira J. PS3Group4-013, S365
Oliveira Santos M. PS1Group1-058, S149, PS3Group4-020, S372
Oliver D.J. OA 2.3, S91, PL 3.3, S5, WS 3.1, S46
Oliver K.R. SS 11.1, S21
Omachi J. PS2Group5-004, S286
O’Mara E. PS1Group1-126, S200
Oorschot V. PS3Group4-007, S361
Opolski G. PS2Group5-011, S291
O’Riordan W. PS2Group3-010, S225, PS2Group3-055, S261
Orlikowski D. PS1Group1-088, S172
Orsini A.-L. PS1Group1-026, S122
Ortez C. PS1Group1-013, S112, PS2Group3-011, S226, PS2Group3-034, S245, PS2Group5-014, S293, PS2Group9-010, S317, PS3Group2-013, S335, PS3Group4-019, S371
Osahon R. PS1Group1-019, S117, PS2Group9-009, S316
Osman H. PS2Group5-020, S297
Ota T. PS3Group4-001, S356
Ouaja R. PS2Group3-064, S268
Ouazzani R. PS1Group1-067, S157, PS1Group1-068, S157, PS2Group5-012, S292, PS2Group5-013, S292
Oyewole A. PS2Group9-019, S323
Oyola Valdizán C.E. PS2Group9-004, S313
Ozasa S. PS1Group1-029, S125
Padberg G.W. PS1Group1-001, S102, WS 2.3, S45
Padilla M. PS3Group2-009, S331
Padmanabha H. PS3Group4-003, S357
Padros N. PS2Group9-010, S317, PS3Group4-019, S371
Pain-Control’S Study Team P. PS2Group3-051, S257
Pajusalu S. PS1Group1-035, S130
Palace J. SS 24.4, S41
Pál E. PS1Group1-099, S180
Panaghie-Meltzer G. PS1Group1-123, S198, S198
Panicucci C. PS1Group1-119, S195
Papetti L. PS1Group1-027, S123
Paradas C. PS1Group1-063, S153, PS1Group1-115, S192, PS2Group5-007, S288
Pardo J. PS2Group3-042, S251
Pareyson D. SS 7.3, S15, TC 3.2, S79, WS 10.2, S55
Park G.-Y. PS2Group3-047, S254
Park H.J. PS3Group4-016, S368
Park J.-M. PS1Group1-094, S176, PS3Group2-015, S336
Park J.-S. PS1Group1-094, S176, PS3Group2-015, S336
Park J.S. PS1Group1-133, S206
Park J.Y. PS1Group1-133, S206
Park K.-D. PS2Group3-073, S274
Park K.-H. PS2Group3-061, S266
Park K.H. PS2Group3-033, S245, PS3Group2-035, S352
Park K.-J. PS2Group3-033, S245
Parks C. PS2Group3-051, S257
Park S.-H. PS2Group3-035, S246, PS3Group4-027, S378
Parman Y. PS2Group3-002, S218
Parmova O. PS1Group1-037, S132
Parshikov V. PS3Group4-029, S379, WS 3.4, S47
Pasnoor M. PS2Group3-051, S257
Paternoster L. PS3Group4-025, S376
Paternostro-Sluga T. WS 1.2, S44
Patra K. PS3Group2-012, S334
Paul A. PS3Group2-020, S340
Pearson B. PS3Group2-021, S341
Peci E. PS2Group3-019, S233
Peduto A. PS1Group1-009, S108
Pegoraro E. PS1Group1-051, S144, PS1Group1-063, S153, PS1Group1-115, S192, PS2Group5-007, S288
Pelayo-Negro A.L. PS1Group1-100, S181
Pellegrinelli A. PS3Group2-016, S337, PS3Group2-017, S338
Pelnena D. PS1Group1-035, S130
Pelosi L. PS2Group5-021, S298
Peltier A. WS 20.3, S71
Peltz S.W. PS1Group1-118, S194, PS1Group1-123, S198, PS1Group1-124, S199
Penttilä S. PS1Group1-053, S146
Pera M.C. PS3Group4-028, S378
Perazzo F.F. PS1Group1-107, S186
Perez M.A.R. PS2Group5-033, S307
Perez M.M. PS1Group1-107, S186
Perez Siles G. PS2Group3-048, S255
Peric S. PS1Group1-077, S163, PS1Group1-078, S164, PS1Group1-079, S164, PS1Group1-080, S165, PS1Group1-104, S185, PS2Group3-028, S241
Perlingeiro R. OA 1.1, S90
Perniconi B. PS1Group1-088, S172
Peschanski M. PS3Group4-006, S360
Pesovic J. PS1Group1-077, S163, PS1Group1-078, S164, PS1Group1-079, S164, PS1Group1-104, S185
Pestronk A. PS1Group1-063, S153, PS1Group1-115, S192, PS2Group5-007, S288, SS 18.3, S31, WS 7.2, S52
Peters D. PS2Group9-009, S316
Peters T. PS3Group4-017, S369
Petri G. PS1Group1-047, S141, PS1Group1-107, S186, PS2Group3-059, S265
Petroska D. PS1Group1-132, S206
Petrovic M. PS2Group3-028, S241
Petty R.K. PS1Group1-033, S128
Peyer Kauffmann A.-K. PS2Group5-022, S299
Pfeffer G. PS1Group1-011, S110
Phillips L. PS1Group1-138, S210
Phillips W.D. PS1Group1-101, S182, TC 2.1, S76
Picarella D. PS1Group1-060, S151
Piehl F. PS3Group2-004, S327
Piétri-Rouxel F. PS1Group1-007, S107
Pilati L. PS1Group1-127, S202
Pinal-Feranndez I. PS1Group1-137, S210
Pingili R. PS3Group4-008, S361
Pinset C. PS1Group1-144, S214
Pinto S. PS3Group4-020, S372
Pintos E. PS2Group3-042, S251
Pintos-Morell G. PS1Group1-089, S173
Piras R. PS1Group1-051, S144
Pita F. PS3Group2-034, S351
Pitchforth J. PS1Group1-004, S104
Pittaro A. PS1Group1-040, S134
Pitt M. TC 4.4, S82
Pivneva I. PS1Group1-020, S118
Podborska M. PS1Group1-037, S132
Podnar S. TC 5.3, S83
Polavarapu K. PS1Group1-016, S114, PS1Group1-018, S116, PS1Group1-044, S138, PS2Group5-010, S291, PS3Group2-022, S342, PS3Group4-003, S357
Polikarpova A. PS1Group1-076, S163
Polvèche H. PS3Group4-006, S360
Polydefkis M. PS2Group3-005, S220, PS2Group3-055, S261
Ponery A.S. PS1Group1-021, S119
Pontifex C.S. PS1Group1-011, S110
Poon J.-L. PS3Group7-004, S383
Porte Thome F. PS1Group1-004, S104, PS1Group1-005, S105
Powell C. PS2Group3-055, S261
Poyatos Garcia J. PS1Group1-046, S140
Pozzolini G. PS1Group1-119, S195
Praestgaard J. PS3Group4-008, S361
Prasad S. PS1Group1-039, S133
Preethish-Kumar V. PS1Group1-016, S114, PS1Group1-018, S116, PS1Group1-044, S138, PS2Group5-010, S291, PS3Group4-003, S357
Prescher H. PS3Group2-031, S349
Previtali S. PS1Group1-004, S104
Previtali S.C. PS1Group1-051, S144
Prieto-González S. PS1Group1-083, S168
Prior T.W. PS3Group4-022, S373, SS 13.4, S25
Prukop T. PS2Group3-038, S248
Punga A.R. PS3Group2-001, S324, SS 20.2, S32, WS 18.2, S67
Punga T. PS3Group2-001, S324
Punzón I. PS1Group1-007, S107, PS1Group1-008, S108
Puzanok D. PS3Group4-029, S379, WS 3.4, S47
Quasthoff S. PS2Group3-076, S276, PS3Group2-018, S338
Querol L. SS 12.3, S24
Rabbia M. PS1Group1-014, S112
Rakocevic-Stojanovic V. PS1Group1-077, S163, PS1Group1-078, S164, PS1Group1-079, S164, PS1Group1-080, S165, PS1Group1-104, S185
Ramirez A. PS1Group1-013, S112
Ramírez-Jiménez L. PS2Group3-044, S252
Ramm G. PS3Group4-007, S361
Rangel Escribano J. PS2Group9-001, S310
Rao S.N. PS1Group1-018, S116
Rath J. PS2Group3-026, S239, PS3Group2-018, S338, PS3Group2-020, S340
Ratnasingam R..J. PS1Group1-031, S127
Ravenscroft G. PS1Group1-096, S178
Rawla J. PS1Group1-114, S191
Reblova K. PS1Group1-113, S190
Recasens B.B. PS2Group5-033, S307
Recktenwald S.F. PS3Group4-024, S375
Reddy S.S. PS2Group3-045, S253, PS2Group3-046, S254
Reichert S. PS1Group1-065, S155
Reichmann H. PS1Group1-116, S193
Reilich P. PS2Group3-040, S249, PS2Group3-074, S275, PS2Group5-016, S295
Reilly J.F. PS1Group1-060, S151
Reilly M. PL 2.1, S4, SS 19.1, S31
Relaix F. PS1Group1-007, S107
Remiche G. PS2Group5-001, S284
Renfroe J.B. PS1Group1-126, S200
Renna L.V. PS1Group1-042, S136, PS1Group1-056, S148, PS1Group1-110, S188
Reshetov D. PS1Group1-076, S163, PS2Group9-015, S320
Reyna S.P. PS3Group4-026, S377
Reyngoudt H. PS1Group1-095, S177, PS2Group9-006, S314
Reynolds E. PS2Group3-008, S223
Rha H.J. PS2Group3-079, S279
Ricciardi R. PS3Group2-027, S346
Ricci G. PS1Group1-051, S144, PS1Group1-142, S213, PS2Group3-060, S265
Richard I. PS1Group1-015, S113, TC 9.2, S86
Richman L. PS2Group9-013, S319
Rickard A. PS1Group1-023, S120
Rico S. PS1Group1-039, S133
Riquelme A. PS3Group2-010, S332
Ristic T. PS1Group1-059, S150
Rivellini C. PS2Group3-081, S283
Rivolta I. PS1Group1-110, S188
Rizzo S. PS3Group2-027, S346
Robberecht W. PS2Group3-024, S237
Robbie G. PS2Group3-043, S251, PS2Group3-080, S280
Roblin D. PS1Group1-019, S117, PS2Group9-009, S316
Rocha M. PS1Group1-012, S111
Rodart I. PS1Group1-073, S161
Rodino-Klapac L. PS3Group4-022, S373, SS 13.4, S25
Rodolico C. PS1Group1-051, S144, PS1Group1-127, S202
Rodrigues M. PS2Group5-021, S298
Rodriguez A. WS 6.2, S52
Rodriguez De Rivera F. PS3Group2-004, S327
Rodríguez-Palmero A. PS1Group1-089, S173
Roemer F. PS2Group9-008, S316
Rohani M. PS1Group1-043, S137
Rohmer Cohen A. PS2Group9-001, S310
Rojas-García R. PS2Group3-053, S259
Romagnolo A. PS2Group3-019, S233
Romero N. PS1Group1-066, S156, PS3Group2-030, S348
Roos A. PS1Group1-044, S138, PS3Group2-022, S342, SS 24.2, S40
Rosa H. PS1Group1-012, S111
Rossi R. PS2Group5-020, S297
Rottoli M.R. PS3Group2-004, S327
Roubenoff R. PS3Group4-008, S361
Roussange F. PS3Group4-006, S360
Roxburgh R. PS2Group5-021, S298
Roy B. PS1Group1-032, S128, PS2Group3-029, S241, PS2Group5-002, S284, PS3Group2-019, S339
Rozentals G. PS2Group9-002, S311
Rubino D. PS1Group1-017, S116
Rubino-Nacht D. PS1Group1-026, S122
Rudolf R. WS 14.3, S59
Rüegg M. WS 14.1, S59
Ruegg U.T. PS1Group1-061, S152
Rufibach L. PS1Group1-115, S192, PS2Group5-007, S288, PS3Group7-003, S382
Ruggiero L. PS1Group1-051, S144
Ruiz A.L. PS2Group9-004, S313
Ruiz-Del-Yerro E. PS1Group1-054, S146, PS1Group1-108, S187
Ruiz M. PS2Group3-082, S283
Rummey C. PS2Group5-023, S299, PS2Group9-003, S312, PS2Group9-016, S321
Russell J. PS2Group3-027, S240, PS2Group3-045, S253, PS2Group3-046, S254, SS 10.2, S19
Rutkove S. PS2Group5-002, S284
Rybalsky I. PS1Group1-095, S177
Saak A. PS1Group1-035, S130, PS1Group1-116, S193, PS1Group1-117, S194
Saarela M. PS2Group3-024, S237
Sabre L. PS3Group2-001, S324
Sacchetto R. PS1Group1-125,
Sacconi S. PS1Group1-088, S172, TC 1.2, S74
Sadeh M. PS1Group1-069, S158
Saegusa J. PS1Group1-003, S103
Safri A.Y. PS3Group2-032, S350
Said G. SS 21.2, S34, WS 17.1, S64
Saifullina E. PS2Group3-058, S264
Saito K. PS3Group4-026, S377
Sajeev G. PS1Group1-120, S196
Sakai C. PS1Group1-097, S178
Salimian M. PS2Group3-046, S254
Salort-Campana E. PS1Group1-063, S153, PS1Group1-088, S172, PS1Group1-115, S192
Salvalaggio A. PS2Group3-082, S283
Sampaio M. PS2Group3-003, S219
Sampaolesi M. PS1Group1-008, S108
Sampaolo S. PS2Group5-034, S307
San Antonio Arce V. PS1Group1-013, S112
Sanarica F. PS1Group1-028, S124
Sancho P. PS2Group3-044, S252
Sanders D. PS1Group1-049, S143
Sandona D. PS1Group1-125, S200
Sandroni P. SS 9.6, S19
Santhosh R. PS1Group1-016, S114
Santoro L. PS1Group1-051, S144, PS2Group5-021, S298
Santos C.S. PS3Group2-025, S345
Santos E. PS3Group2-025, S345
Santos J.F.R. PS1Group1-047, S141, PS2Group3-059, S265
Santos M. PS2Group3-003, S219, PS3Group2-025, S345, PS3Group4-013, S365
Santos R. PS3Group4-013, S365
Sanvito W.L. PS3Group4-005, S359
Sarafov S. PS2Group3-002, S218
Sariego A. PS2Group3-034, S245
Saroja M.K. PS1Group1-018, S116
Sarva S. PS2Group3-039, S249
Sasaki M. PS1Group1-097, S178
Savarese M. PS1Group1-053, S146, WS 16.3, S63
Savic-Pavicevic D. PS1Group1-077, S163, PS1Group1-078, S164, PS1Group1-079, S164, PS1Group1-104, S185
Sawyer A.M. PS1Group1-009, S108
Say C.Y. PS2Group3-025, S238
Scapozza L. PS1Group1-050, S143, PS1Group1-061, S152, PS1Group1-098, S179
Scarlato M. PS1Group1-051, S144
Schaefer A.M. PS1Group1-033, S128
Schaefer J. PS1Group1-035, S130, PS1Group1-116, S193, PS1Group1-117, S194
Schara U. PS1Group1-026, S122, PS3Group4-008, S361
Scharf F. PS2Group5-016, S295
Schlotter-Weigel B. PS2Group3-036, S247
Schlötzer-Schrehardt U. PS1Group1-135, S208
Schmidhammer R. WS 9.1, S54
Schmidt H. PS2Group3-005, S220, PS2Group3-010, S225, PS2Group3-055, S261
Schmidt J. PS1Group1-025, S122, PS1Group1-045, S139, PS1Group1-103, S184, PS1Group1-122, S198
Schmidt K. PS1Group1-025, S122, PS1Group1-122, S198
Schmidt S. PS1Group1-017, S116, PS1Group1-026, S122
Schmidt U. PS1Group1-023, S120, PS1Group1-024, S121
Schmuck M. PS1Group1-065, S155
Schnell F. PS1Group1-049, S143, PS1Group1-087, S171
Schorling E. PS2Group3-040, S249
Schoser B. PS1Group1-002, S102, PS2Group3-074, S275, PS3Group4-009, S362
Schreiber-Katz O. PS1Group1-115, S192
Schröder R. PS1Group1-134, S207, PS1Group1-135, S208, PS2Group3-063, S267, PS2Group5-028, S303, PS2Group9-008, S316, WS 15.2, S60, WS 19.1, S68
Schröder S. PS1Group1-085, S169
Schüssler S. PS2Group9-008, S316
Schustereder G. PS3Group2-018, S338
Scola R.H. PS1Group1-006, S106
Screnci R. PS2Group3-048, S255
Seabrook T. PS3Group4-015, S367
Sebastião A.M. TC 2.3, S77
Šedivá M. PS2Group3-020, S234, PS2Group9-007, S315
Sedlácková L. PS2Group3-020, S234, PS2Group9-007, S315
Seeman P. PS2Group3-020, S234, PS2Group9-007, S315
Seferian A. PS1Group1-087, S171
Sefiani A. PS1Group1-068, S157
Sefiani S. PS1Group1-067, S157, PS1Group1-068, S157
Sekar D. PS1Group1-018, S116, PS3Group4-003, S357
Selby K. PS1Group1-123, S198, S198
Selva-O’Callaghan A. PS1Group1-083, S168
Selvatici R. PS2Group5-020, S297
Semeryak O.M. PS2Group5-032, S306
Semina N. WS 3.4, S47
Semplicini C. PS1Group1-088, S172, PS1Group1-115, S192
Semprez F. PS3Group2-028, S347
Seog D.-H. PS2Group5-026, S301
Seo H.S. PS3Group2-035, S352
Seok H.Y. PS2Group5-017, S295
Seong M.-W. PS1Group1-145, S215
Sera F. PS1Group1-051, S144
Serafin A. PS2Group5-011, S291
Serban V. PS2Group3-075, S276
Sereda M. SS 7.2, S15
Sereda M.W. PS2Group3-038, S248
Serjensen T. SS 5.4, S14
Serrano R. PS3Group4-007, S361
Sertpoyraz F.M. PS2Group3-013, S227, PS2Group9-014, S320
Servais L. PS1Group1-004, S104, PS1Group1-039, S133, PS1Group1-087, S171, PS2Group3-070, S272, PS3Group4-015, S367, PS3Group4-028, S378
Seta N. PS1Group1-066, S156
Šetlere S. PS2Group9-002, S311
Sevilla T. PS2Group3-042, S251, PS2Group3-052, S258, PS2Group3-053, S259
Shaibani A. PS3Group2-024, S343
Shakir R. SS 22.3, S38
Shamshiri H. PS1Group1-043, S137
Shanmugarajah P.D. PS3Group4-011, S364
Sharma A. PS3Group2-019, S339
Shebl A. PS2Group3-023, S236, PS2Group3-057, S263
Shellenbarger K.C. PS1Group1-095, S177
Shell R. PS3Group4-022, S373, SS 13.4, S25
Shidlovskaya O. PS2Group9-015, S320
Shieh P.B. PS1Group1-039, S133
Shi F.-D. PS3Group2-029, S347
Shimizu J. PS1Group1-071, S159, PS2Group5-004, S286
Shim S.K. PS2Group3-041, S250
Shin J.-H. PS3Group2-014, S336
Shin J.-Y. PS1Group1-131, S205, PS1Group1-141, S213
Shiraishi H. PS2Group6-001, S308
Shirakawa T. PS1Group1-128, S202
Shira S. PS3Group7-003, S382
Shlemon P. PS2Group3-051, S257
Shoffner J.M. PS1Group1-090, S173
Shtabnitskiy V. PS3Group4-029, S379, WS 3.4, S47
Shulyakova I. PS2Group9-015, S320
Siciliano G. PS1Group1-051, S144, PS1Group1-142, S213, PS3Group2-027, S346
Siddiqi Z.A. PS3Group2-002, S326, PS3Group2-003, S326
Signorovitch J. PS1Group1-120, S196
Silani V. SS 16.3, S29
Silva V.G. PS1Group1-047, S141, PS1Group1-073, S161, PS1Group1-074, S162
Silveira F. PS3Group2-025, S345
Simoncini C. PS1Group1-142, S213
Simschitz P. PS3Group2-018, S338
Singh A. PS2Group5-007, S288
Siva R. PS2Group3-062, S267
Skripchenko N. PS2Group3-071, S272, PS2Group5-015, S294
Slater C. TC 2.2, S77
Smith B. PS1Group1-039, S133
Smith D.S. PS2Group5-007, S288
Smith F. PS1Group1-009, S108
Smith R. PS3Group4-018, S370
Snadden L. PS1Group1-033, S128
Soardi M. PS1Group1-125,
Soblechero-Martin P. PS1Group1-054, S146, PS1Group1-108, S187
Sobue G. PS2Group3-012, S226, PS2Group3-018, S231, PS2Group3-019, S233, PS2Group3-023, S236, PS2Group3-024, S237, PS2Group3-030, S243, PS2Group3-031, S244, PS2Group3-057, S263
Solà A.G. PS2Group5-033, S307
Solana-Guizar E. PS1Group1-024, S121
Solé G. PS1Group1-088, S172
Solernou Ferrer A.J. PS2Group9-004, S313
Sommer C. PS2Group3-024, S237, SS 15.1, S27
Sonett J.R. SS 23.2, S39
Soni M. PS1Group1-146, S216
Soska R. PS1Group1-021, S119
Sousa A. PS2Group3-003, S219, PS3Group2-025, S345, PS3Group4-013, S365
Sousa L. PS3Group2-025, S345
Sousa S. PS3Group2-034, S351
Souza M. PS1Group1-123, S198, PS1Group1-124, S199
Sparks S. PS1Group1-115, S192, PS2Group5-007, S288
Specht C. PS1Group1-140, S212
Spies J.M. PS2Group3-056, S262
Spilioti M. PS1Group1-102, S183
Spinty S. PS1Group1-026, S122
Spörrer M. PS1Group1-135, S208, PS2Group5-028, S303
Sproule D.M. PS1Group1-020, S118, PS3Group4-022, S373, SS 13.4, S25
Spuler S. PS1Group1-063, S153, PS1Group1-115, S192
Srisuwan K. PS3Group2-006, S328
Srivastava R. PS2Group6-003, S310
Srotova I. PS1Group1-037, S132
Staff N. SS 6.1, S14, WS 11.3, S57
Statland J.M. WS 2.2, S45
Stauber A.J. PS3Group4-009, S362
Stavropoulos G. PS1Group1-102, S183
Stavusis J. PS1Group1-035, S130
Steck A. WS 8.2, S54, WS 17.3, S66
Stehlikova K. PS1Group1-113, S190
Stelmasiak Z. PS2Group3-024, S237
Sternberg D. PS3Group2-030, S348
Stevanin G. PS2Group3-011, S226, PS2Group3-034, S245
Stevic Z. PS1Group1-104, S185
Stieglbauer K. PS2Group3-076, S276, PS3Group2-018, S338
Stojanov A. PS2Group3-028, S241
Stojanovic M. PS2Group3-028, S241
Stojkovic T. PS1Group1-009, S108, PS1Group1-063, S153, PS1Group1-066, S156, PS1Group1-115, S192, PS2Group5-007, S288, PS3Group2-030, S348
Stradalova P. PS1Group1-037, S132
Straub V. PL 1.1, S3, PS1Group1-009, S108, PS1Group1-034, S129, PS1Group1-063, S153, PS1Group1-087, S171, PS1Group1-115, S192, PS2Group5-007, S288
Streichenberger N. PS1Group1-066, S156, PS3Group2-030, S348
Strochlic L. PS2Group5-027, S302, PS3Group2-030, S348
Strom T.M. PS2Group3-026, S239
Strucksberg K.-H. PS1Group1-134, S207
Struhal W. SS 9.2, S18
Study Group S.-N. PS1Group1-087, S171
Sugie K. PS1Group1-029, S125
Suh B.C. PS3Group4-016, S368
Suhr O. PS2Group3-005, S220, PS2Group3-010, S225, PS2Group3-055, S261
Su J. PS1Group1-005, S105
Suk J.I. PS2Group3-079, S279
Sukno S. PS3Group2-030, S348
Sung D.H. PS2Group6-002, S309
Sung J.-J. PS1Group1-131, S205, PS1Group1-141, S213, PS2Group3-032, S244, PS2Group3-061, S266, PS3Group2-035, S352
Sutherland H. PS1Group1-009, S108
Suzuki E. PS1Group1-097, S178
Svahn J. PS1Group1-066, S156
Sweeney H.L. PS1Group1-060, S151
Sweeney L. PS1Group1-014, S112
Sweetser M. PS2Group3-005, S220, PS2Group3-010, S225, PS2Group3-055, S261
Swoboda K.J. PS3Group4-014, S366
Szczudlik A. PS3Group2-004, S327
Tackenberg B. PS2Group3-024, S237
Tahraoui J. PS1Group1-144, S214
Tajsharghi H. PS1Group1-096, S178
Takada H. PS1Group1-029, S125, PS3Group4-001, S356
Takahashi M.P. PS1Group1-029, S125
Takahashi T. PS1Group1-029, S125
Takahashi Y.K. PS2Group6-001, S308
Takashima H. PS1Group1-136, S209
Takeda H. PS2Group5-029, S304
Takeda S. PS1Group1-092, S175, PS1Group1-115, S192, PS1Group1-143, S214
Takeshita E. PS1Group1-029, S125
Takeuchi F. PS1Group1-092, S175
Tamaoka A. PS2Group5-029, S304
Tanaka N. PS1Group1-029, S125
Tan C.-Y. PS1Group1-031, S127
Tan E. PS2Group3-006, S222
Tan H.-T. PS1Group1-031, S127
Taouagh N. PS1Group1-088, S172
Tapscott S.J. WS 2.1, S45
Tasca G. PS1Group1-053, S146
Taylor R.W. PS1Group1-012, S111, PS1Group1-033, S128
Telese R. PS1Group1-051, S144
Tesi Rocha C. PS1Group1-009, S108, PS1Group1-115, S192
Thaler-Wolf C. PS2Group3-076, S276
Thampratankul L. PS3Group2-006, S328
The German Mouse Clinic PS1Group1-134, S207Consortium -.
Theil D. PS3Group4-017, S369
Thibaud J.-L. PS1Group1-008, S108
Thiele S. PS2Group3-040, S249
Thievessen I. PS2Group5-028, S303
Thomas P. PS3Group4-003, S357
Thomas P.T. PS1Group1-018, S116, PS2Group9-011, S318
Thömke F. SS 8.3, S17
Thompson N. PS3Group4-002, S356
Thusang K.J. PS3Group4-012, S365
Tian C. PS1Group1-095, S177, PS1Group1-126, S200
Tiburcy M. PS1Group1-103, S184
Tiddens H.A.W.M. PS1Group1-040, S134
Tilbery C.P. PS3Group4-005, S359
Timmerman V. SS 7.1, S15
Tinsley J. PS1Group1-019, S117, PS2Group9-018, S323
Tirucherai G. PS1Group1-014, S112
Toda T. PS1Group1-071, S159, PS2Group5-004, S286
Tomelleri G. PS1Group1-051, S144
Tomidokoro Y. PS2Group5-029, S304
Tomschik M. PS2Group3-026, S239, PS3Group2-020, S340
Tonska K. PS1Group1-036, S131
Topakian R. PS2Group3-076, S276, PS3Group2-018, S338
Topaloglu H. PS1Group1-026, S122
Töpf A. PS1Group1-044, S138, PS1Group1-138, S210, PS2Group5-014, S293, PS3Group2-022, S342, SS 24.2, S40
Torella A. PS1Group1-119, S195, WS 16.3, S63
Toscano A. SS 4.1, S13
Tournev I. PS2Group3-002, S218, PS2Group3-005, S220, PS2Group3-010, S225, PS2Group3-055, S261
Tournois J. PS3Group4-006, S360
Touvier T. PS2Group3-081, S283
Tozaka N. PS2Group5-029, S304
Trabace L. PS1Group1-028, S124
Tracy J.A. PS1Group1-129, S203
Trajanovska S. PS1Group1-101, S182
Traverso M. PS1Group1-119, S195
Trifillis P. PS1Group1-118, S194, PS1Group1-123, S198, PS1Group1-124, S199, PS1Group1-126, S200
Trollmann R. PS2Group9-008, S316
Truszkowska B. PS2Group5-011, S291
Tsai K. PS1Group1-086, S170
Tsolaki M. PS1Group1-102, S183
Tsuji H. PS2Group5-029, S304
Tsuji S. PS1Group1-071, S159
Tulinius M. PS1Group1-123, S198, PS3Group4-026, S377
Tupler R. PS1Group1-051, S144
Turanoglu M. PS2Group3-013, S227
Turke K.C. PS1Group1-072, S160, PS1Group1-073, S161, PS1Group1-074, S162, PS1Group1-075, S162
Türk M. PS1Group1-134, S207, PS2Group3-063, S267
Turnbull D.M. PS1Group1-012, S111, PS1Group1-033, S128
Turner C. PS1Group1-082, S167, PS1Group1-147, S216
Uchino S. PS1Group1-097, S178
Udd B. PS1Group1-053, S146, SS 4.3, S13
Uder M. PS2Group9-008, S316
Ulrichts P. PS3Group2-004, S327
Uncini A. SS 12.1, S23
Uribe L. PS3Group2-021, S341
Urtizberea A. WS 16.1, S62
Utkus A. PS1Group1-132, S206
Utsugisawa K. PS3Group2-011, S333
Vaillant T.Z. PS2Group9-004, S313
Vaitkevicius A. PS1Group1-132, S206
Valaperta R. PS1Group1-110, S188
Valentin M.-A. PS3Group4-017, S369
Valenzano K.J. PS1Group1-021, S119
Valério B.C.O. PS3Group2-016, S337, PS3Group2-017, S338
Vallejo Illarramendi A. PS1Group1-038, S132, PS1Group1-106, S186
Van Alfen N. TC 5.2, S82
Van Asseldonk T. PS2Group5-008, S289
Van Damme P. PS3Group2-004, S327
Van Den Bergh P. TC 8.4, S85
Van Den Berg L. PL 3.2, S5, PS2Group5-008, S289, WS 12.1, S57
Vandenborne K. PS1Group1-019, S117, PS1Group1-060, S151, PS2Group9-018, S323
Vanden Hauwe M. PS1Group1-120, S196
Van Der Beek N.A.M.E. PS1Group1-030, S126, PS1Group1-040, S134
Van Der Kooi A. PS1Group1-148, S217
Van Der Kooi E.L. PS1Group1-001, S102
Van Der Maarel S.M. PS1Group1-001, S102, PS1Group1-109, S188
Van Der Ploeg A.T. PS1Group1-030, S126, PS1Group1-040, S134
Van Der Pol L. PS2Group5-008, S289
Van Der Vliet P.J. PS1Group1-109, S188
Van Doorn P.A. PL 2.2, S4, PS1Group1-030, S126, PS1Group1-040, S134
Vandrovcova J. PS2Group3-067, S270
Van Engelen B.G. SS 4.2, S13, PS1Group1-001, S102, PS1Group1-010, S109, PS1Group1-109, S188
Van Geloven N. PS2Group3-012, S226, PS2Group3-018, S231, PS2Group3-019, S233, PS2Group3-023, S236, PS2Group3-024, S237, PS2Group3-031, S244
Van Kooten H.A. PS1Group1-030, S126, PS1Group1-040, S134
Van Schaik I. PS2Group3-012, S226, PS2Group3-018, S231, PS2Group3-019, S233, PS2Group3-023, S236, PS2Group3-024, S237, PS2Group3-031, S244, PS2Group3-057, S263
Van Walsem M.R. PS3Group7-002, S381
Van Zwet E.W. PS1Group1-095, S177
Varju C. PS1Group1-099, S180
Vase L. PS3Group2-024, S343
Vassilieva S. PS1Group1-076, S163
Vázquez-Costa J.F. PS2Group3-052, S258
Veiga A. PS3Group2-025, S345
Vellieux G. PS3Group2-030, S348
Veltsista D. PS2Group5-030, S305, PS3Group4-023, S374
Vengalil S. PS1Group1-016, S114, PS1Group1-018, S116, PS1Group1-044, S138, PS2Group5-010, S291, PS3Group2-022, S342, PS3Group4-003, S357
Vercelli L. PS1Group1-051, S144
Verellen-Dumoulin C. PS2Group3-070, S272
Verrigni D. PS1Group1-027, S123
Verschuuren J. PS3Group2-004, S327, SS 3.4, S13
Verschuuren K. PS3Group2-004, S327
Vial C. PS1Group1-066, S156
Vidal-Lijo M.P. PS2Group3-042, S251
Vidal N. PS1Group1-004, S104
Vieira T.F. PS1Group1-074, S162
Vigo M. PS3Group4-019, S371
Vihola A. PS1Group1-053, S146
Vílchez J.J. PS1Group1-046, S140, PS2Group3-053, S259
Vílchez R. PS2Group3-053, S259
Villa A. PS3Group2-007, S330
Villa L. PS1Group1-051, S144
Villalta S..A. PS2Group5-031, S306
Villegas-Umana E. PS2Group3-008, S223
Vilquin J.-T. PS1Group1-008, S108
Viñas O. PS1Group1-083, S168
Vincent A. PL 4.3, S6, PS3Group2-036, S353
Vincent A.E. PS1Group1-012, S111
Visentin A. PS2Group3-082, S283
Visser L. PS2Group5-008, S289
Vissing J. PS3Group2-023, S342
Visudthibhan A. PS3Group2-006, S328
Vitturi B.K. PS3Group2-016, S337, PS3Group2-017, S338, PS3Group4-005, S359
Vivekananda U. PS1Group1-147, S216
Vlckova E. PS1Group1-037, S132
Vlcková M. PS2Group3-020, S234
Vlodavets D. PS1Group1-076, S163, PS2Group9-015, S320
Voermans N.C. PS1Group1-001, S102, PS1Group1-093, S175, PS1Group1-109, S188
Vogt-Ladner G. PS2Group3-063, S267
Vohanka S. PS1Group1-037, S132, PS1Group1-113, S190, PS1Group1-139, S211
Voitenkov V. PS2Group3-071, S272, PS2Group5-015, S294
Voit T. PS1Group1-057, S148, PS1Group1-105, S185, PS2Group5-023, S299, PS2Group9-003, S312, PS2Group9-016, S321, SS 5.2, S13
Vollert J. PS3Group2-024, S343
Volov A. WS 3.4, S47
Voltz E. PS3Group4-008, S361, PS3Group4-017, S369
Vorobyeva A. WS 3.4, S47
Vroom E. TC 9.9, S87
Vuillerot C. PS3Group4-024, S375
Vujnic M. PS1Group1-104, S185
Vukojevic Z. PS2Group3-028, S241
Vu T. PS3Group2-004, S327
Wachtel D. PS1Group1-060, S151
Waddington S. PS1Group1-070, S159
Wagner K.R. PS1Group1-014, S112, TC 9.7, S87
Wagner M. PS2Group3-026, S239
Walter M.C. PS1Group1-009, S108, PS1Group1-063, S153, PS1Group1-065, S155, PS1Group1-115, S192, PS1Group1-130, S204, PS2Group3-040, S249, PS2Group3-074, S275, PS2Group5-007, S288, PS2Group5-016, S295, PS3Group4-009, S362
Wang M.-X. PS2Group3-056, S262
Wanschitz J. PS2Group3-049, S256, PS2Group3-076, S276, PS3Group2-018, S338
Ward S.J. PS1Group1-120, S196
Warrier M. PS2Group9-011, S318
Wasala L. PS1Group1-111, S189
Wasala N. PS1Group1-111, S189
Watanabe M. PS2Group5-029, S304
Waters P. PS2Group3-032, S244
Weber I. PS2Group3-063, S267
Webster R. PS3Group2-036, S353
Weis C.-A. PS3Group2-001, S324, SS 23.2, S39
Wells C. PS3Group4-022, S373, SS 13.4, S25
Wencel M. PS2Group5-031, S306
Wenninger S. PS1Group1-130, S204, PS2Group3-036, S247
Wenning G. SS 9.1, S18
Werber M. PS1Group1-085, S169
Wermelinger A.C.C. PS1Group1-006, S106
Werneck L.C. PS1Group1-006, S106
Wernick S. PS2Group3-038, S248
Whitaker C. PS1Group1-032, S128
White M. PS2Group3-005, S220
Whittaker R. SS 24.2, S40
Wiche G. WS 15.1, S59
Widayati F.T. PS3Group2-032, S350
Wieben E. PS1Group1-011, S110
Wiedenmann S. PS2Group5-028, S303
Wielopolski P.A. PS1Group1-040, S134
Wiendl H. PS3Group2-011, S333, PS3Group2-038, S355
Wilder-Smith E. PS2Group5-022, S299
Willen J. PS3Group7-001, S380
Williams B. PS2Group5-007, S288, PS3Group7-003, S382
Willison H.J. SS 12.2, S24
Wilson J.M. PS2Group9-013, S319
Wilson-Mcmanus J.E. PS2Group5-007, S288
Wimmer K. SS 17.1, S29
Windebank A. SS 2.1, S11, WS 1.1, S44
Windisch D. PS3Group2-018, S338
Windish H. PS2Group5-007, S288
Winterholler M. PS2Group3-063, S267
Winter L. PS1Group1-134, S207, PS1Group1-135, S208, WS 15.3, S61
Wohlgemuth M. PS1Group1-001, S102
Wolf E. PS1Group1-065, S155
Wolfe G.I. PS3Group2-021, S341, PS3Group2-038, S355, SS 20.3, S33, SS 23.2, S39
Wong B.L. PS1Group1-014, S112, PS1Group1-095, S177
Wong H.M.J. PS2Group3-025, S238
Wong W.Y.W. PS2Group3-025, S238
Woodhall M. PS2Group3-032, S244
Wood K. PS3Group2-029, S347
Wrabetz L. PS2Group3-081, S283
Wright N. PS1Group1-035, S130
Wu E.Q. PS1Group1-020, S118
Wu J. PS2Group5-002, S284
Wulandari D. PS3Group2-032, S350
Wu Q. PS1Group1-032, S128
Xin H. PS3Group2-033, S351
Xu S. PS1Group1-021, S119
Yadav N. PS2Group5-010, S291
Yamaji J. PS2Group5-005, S287, PS2Group5-018, S296
Yamamoto T. PS1Group1-003, S103
Yang C.-C. PS2Group3-005, S220, PS2Group3-010, S225, PS2Group3-055, S261
Yang J.-W. PS2Group3-061, S266, PS2Group9-005, S313
Yankey J.W. PS3Group2-021, S341
Yao Z. PS1Group1-120, S196
Yareeda S. PS2Group3-039, S249
Yatsuga S. PS2Group5-029, S304
Yaworski A. PS2Group6-003, S310
Yildiz F.G. PS2Group3-006, S222
Yilmaz E. PS2Group3-006, S222
Yokota T. PS2Group6-001, S308
Yonemoto N. PS1Group1-092, S175
Yoo I.-H. PS2Group3-035, S246, PS3Group4-027, S378
Yoon B.-N. PS2Group3-061, S266, PS2Group9-005, S313
Yoon S.M. PS2Group3-041, S250
York A.M. PS1Group1-086, S170, PS1Group1-087, S171
Yoshimura S. PS2Group6-001, S308
Yoshioka K. PS2Group5-004, S286
Yubero D. PS1Group1-013, S112, PS2Group3-011, S226, PS2Group3-034, S245, PS2Group5-014, S293
Yue Y. PS1Group1-111, S189
Yumoto J. PS2Group5-004, S286
Zadeh G. SS 17.2, S29
Zaganas I. PS1Group1-102, S183
Zagnoli F. PS1Group1-088, S172
Zagrovic B. PS2Group3-026, S239
Zaidman C. PS2Group5-002, S284
Zakharova M. WS 3.4, S47
Zakroyshikova I. WS 3.4, S47
Zamani B. PS1Group1-043, S137, PS2Group5-009, S290
Zambon A. PS1Group1-004, S104
Zara F. PS1Group1-119, S195
Zarrouk-Mahjoub S. PS1Group1-090, S173, PS1Group1-091, S174
Zatz M. OA 1.2, S90
Zeng R. PS1Group1-122, S198
Zenker M. PS2Group5-016, S295
Zhang X. PS2Group3-043, S251, PS2Group3-080, S280
Zhang Y. PS1Group1-103, S184, PS3Group4-026, S377
Zidar J. PS2Group3-002, S218
Zidkova J. PS1Group1-113, S190, PS1Group1-139, S211
Zierz S. PS1Group1-012, S111
Zilliox L. PS2Group3-027, S240
Zimprich F. PS2Group3-026, S239, PS2Group3-049, S256, PS2Group3-076, S276, PS3Group2-018, S338, PS3Group2-020, S340, PS3Group2-036, S353, SS 8.4, S18
Zorgani S. PS2Group9-001, S310
Zotova E. PS1Group1-076, S163
Zschaeck I. PS2Group9-010, S317
Zschüntzsch J. PS1Group1-103, S184, PS1Group1-122, S198
Zulehner G. PS2Group3-026, S239, PS3Group2-020, S340
Zumbrunn T. PS1Group1-017, S116


